
Identification of an Auto-regulatory Feedback Pathway Involving IL … · 2003-12-16 ·...
40
Identification of an Auto-regulatory Feedback Pathway Involving IL-1α in Induction of Constitutive NF-κB Activation in Pancreatic Cancer Cells Jiangong Niu ‡,§ , Zhongkui Li ‡ , Bailu Peng ‡ and Paul J. Chiao ‡,§,#,¶ ‡ Departments of Surgical Oncology and # Molecular and Cellular Oncology, The University of Texas M.D. Anderson Cancer Center, Houston, TX 77030 § Program of Cancer Biology, Graduate School of Biomedical Sciences, The University of Texas-Houston Health Science Center, Houston, TX 77030 ¶ To whom correspondence should be addressed: Department of Surgical Oncology, Unit 107 The University of Texas M.D. Anderson Cancer Center 1515 Holcombe Blvd. Houston, TX 77030 Tel.: 713-794-1030 Fax: 713-794-4830 Email: [email protected] Running Title: NF-κB activation in metastatic pancreatic cancer cells 1 by guest on August 30, 2018 http://www.jbc.org/ Downloaded from
Transcript of Identification of an Auto-regulatory Feedback Pathway Involving IL … · 2003-12-16 ·...

Identification of an Auto-regulatory Feedback Pathway Involving IL-1α in Induction of
Constitutive NF-κB Activation in Pancreatic Cancer Cells
Jiangong Niu‡,§, Zhongkui Li‡, Bailu Peng‡ and Paul J. Chiao‡,§,#,¶
‡Departments of Surgical Oncology and #Molecular and Cellular Oncology, The University of
Texas M.D. Anderson Cancer Center, Houston, TX 77030
§ Program of Cancer Biology, Graduate School of Biomedical Sciences, The University of
Texas-Houston Health Science Center, Houston, TX 77030
¶ To whom correspondence should be addressed: Department of Surgical Oncology, Unit 107 The University of Texas M.D. Anderson Cancer Center 1515 Holcombe Blvd. Houston, TX 77030 Tel.: 713-794-1030 Fax: 713-794-4830 Email: [email protected]
Running Title: NF-κB activation in metastatic pancreatic cancer cells
1
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Summary
We previously reported that NF-κB is constitutively activated in most human
pancreatic cancer tissues and cell lines but not in normal pancreatic tissues and
immortalized pancreatic ductal epithelial cells. IκBαM-mediated inhibition of constitutive
NF-κB activity in human pancreatic cancer cells suppressed tumorigenesis and liver
metastasis in an orthotopic nude mouse model, suggesting that constitutive NF-κB
activation plays an important role in pancreatic tumor progression and metastasis.
However, the underlying mechanism by which NF-κB is activated in pancreatic cancer
remains to be elucidated. In this study, we found that an autocrine mechanism accounts
for the constitutive activation of NF-κB in metastatic human pancreatic cancer cell lines.
Further investigation showed that interleukin-1α was the primary cytokine secreted by
these cells that activates NF-κB. Neutralization of interleukin-1α activity suppressed the
constitutive activation of NF-κB and the expression of its downstream target gene, uPA, in
metastatic pancreatic cancer cell lines. Our results demonstrate that regulation of
interleukin-1α expression is primarily dependent on AP-1 activity, which is in part induced
by signaling pathways dependent and independent of EGF receptors. In conclusion, our
findings suggest a possible mechanism for the constitutive activation of NF-κB in metastatic
human pancreatic cancer cells and a possible missing mechanistic links between
inflammation and cancer.
2
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Introduction
NF-κB is a family of pleiotropic transcription factors that control the expression of
numerous genes involved in growth, tumorigenesis, tumor metastasis, differentiation, embryonic
development, apoptosis, and inflammation (1-4). Five members of mammalian NF-κB have
been described: NF-κB1 (p50 and its precursor, p105), NF-κB2 (p52 and its precursor, p100), c-
Rel, RelA (p65) and RelB, each of which has a 300-residue-long Rel homology domain (RHD)
(2,3,5). The interaction of c-Rel, RelA and RelB with their inhibitors, the IκBs, results in
inactive complexes in the cytoplasm by masking the nuclear localization signal (6,7). The
inhibitor proteins IκBα, IκBβ, IκBγ, IκBε, Bcl-3, and the Drosophila protein Cactus have been
described and characterized (2,3,5). In most cell types, NF-κB proteins are sequestered in the
cytoplasm by the inhibitor IκB in an inactive form(2,6,7). On stimulation, IκB is phosphorylated
by IκB kinase (IKK), which triggers its rapid degradation (8-10). Consequently, NF-κB proteins
are released and translocated into the nucleus, where they activate the expression of target genes
(1-3,5). One of the key target genes regulated by NF-κB is its inhibitor IκBα. A feedback
inhibition pathway for control of IκBα gene transcription and downregulation of transient
activation of NF-κB activity has been described (11-13).
Members of NF-κB family are involved in the development of cancer. For instance, v-
Rel, which is carried by a highly oncogenic retrovirus, causes an aggressive tumor in young birds
and is able to transform avian lymphoid cells and fibroblasts (1,14). The mutated c-rel oncogene
also transforms cells. Many reports demonstrated that chromosomal amplification,
3
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

overexpression, and recurrent genomic rearrangement in the genes encoding c-Rel, Bcl-3, p105
(p50), and p100 (p52) have been identified in many human hematopoietic cancers and several
types of solid tumor such as human non-small cell lung carcinomas, squamous cell carcinomas
of the head and neck, and adenocarcinomas of breast (1,15). Many tumors have acquired genetic
alterations in the signaling pathways that regulate NF-κB activation. For example, defective
IκBα led to constitutive nuclear NF-κB activity, which in turn conferred a growth advantage of
Hodgkin's disease tumor cells (16). The elevated IKK activities were also found in some of the
tumor cells, suggesting that IKK was activated by as yet unidentified aberrant upstream signaling
cascades (16). We previously reported that RelA, the p65 subunit of the NF-κB transcription
factor, was constitutively activated in most pancreatic cancer tissues and human pancreatic
cancer cell lines but not in normal pancreatic tissues and immortalized pancreatic ductal
epithelial cells (17,18). Our more recent work showed that inhibition of constitutive NF-κB
activity by a mutant IκBα (S32, 36A) completely suppressed the liver metastasis of the
pancreatic cancer cell line ASPC-1, and the tumorigenic phenotype of a nonmetastatic pancreatic
tumor cell line, PANC-1, suggesting that constitutive RelA activity plays a key role in pancreatic
cancer metastasis and tumor progression (19,20). However, the mechanisms by which NF-κB
transcription factors are constitutively activated in pancreatic cancer still remain to be elucidated.
Several studies have shown that secretion of pro-inflammatory cytokines are increased in
cancers (21-24). These pro-inflammatory cytokines, such as tumor necrosis factor α (TNF-α)
and interleukin-1 (IL-1), are the potent activators of NF-κB that induce phosphorylation and
rapid degradation of IκB, exposing the nuclear localization sequence of NF-κB and resulting in
NF-κB nuclear translocation (25,26). Some of these cytokines, such as IL-1 and TNF have been
reported to be regulated by NF-κB in some cancer cell lines (27-30). Therefore, it is possible
4
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

that autocrine secretion of these cytokines forms a positive feedback loop that induces the
constitutive NF-κB activation. The involvement of autocrine stimulation mechanisms of these
cytokines in cancer progression has been suggested (31-35). For example, overexpression of IL-
1 as an autocrine growth factor has been reported in a number of tumors in which activation of
Raf plays an important role in transformation, suggesting that blockade of IL-1 signaling may be
an approach to limiting the growth of certain tumors (31). Both exogenous and endogenous IL-
1α contribute to the transcriptional activation of NF-κB and AP-1, induce the expression of IL-8,
and promote cell survival and the growth of head and neck squamous cell carcinoma cell lines in
vitro (32-34). A recent study also suggested that overexpression of interleukin-1β (IL-1β) might
confer the chemoresistance in certain pancreatic cancer cell lines through NF-κB activation (35).
However, the mechanism, by which the regulation of the expression of these inflammatory
cytokines is altered, is unclear. Whether the overexpression of inflammatory cytokines such as
TNF-α and IL-1 activate NF-κB or the constitutive activity of NF-κB induces the overexpression
of these cytokines in cancer cells is not known.
To determine the mechanisms by which NF-κB is constitutively activated in metastatic
pancreatic cancer cells, we sought to delineate the signaling cascades that lead to constitutive
NF-κB activation. We generated a number of pancreatic cancer cell lines that express a
phosphorylation-defective IκBα mutant to inhibit constitutive NF-κB activity. Our results show
that overexpression of IL-1α is primarily induced by AP-1 activity, which is partially dependent
on the overexpression of epidermal growth factor receptor (EGFR) in several metastatic
pancreatic cancer cell lines but not in nonmetastatic pancreatic cancer cell lines. Thus, autocrine
stimulation of IL-1α, but not IL-1β, induces the constitutive activation of NF-κB, which in turn
induces the expression of its downstream target genes.
5
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Experimental Procedures
Cell lines and Reagents The human pancreatic cancer cell lines ASPC-1, PANC-1, Capan-
1, and CFPAC-1 were purchased from the American Type Culture Collection (ATCC)
(Manassas, VA). Human pancreatic cancer cell lines MDAPanc-3 and MDAPanc-28 were
established by Marsha Frazier and Douglas B. Evans (The University of Texas M. D. Anderson
Cancer Center) (36,37). The human pancreatic cancer cell lines ASPC-1/IκBM and MDAPanc-
28/IκBM were constructed as previously described by Dong et al. (18). Wild-type Mouse
Embryonic Fibroblast (WT-MEF) cells were established in our laboratory according to the report
by Beg et. al. (38). All cell lines were grown in the original cell culture medium specified by the
American Type Culture Collection (Manassas, VA) and DMEM containing 10% FCS.
Neutralizing IL-1α, IL-1β, TNFα, TNFβ, and IL-1 receptor type I (IL-1RI) antibodies were
obtained from R&D Systems (Minneapolis, MN). EGFR neutralizing antibody C225 was
obtained from Dr. Zhen Fan (The University of Texas M. D. Anderson Cancer Center).
The preparation of conditioned medium (CM): Human pancreatic cancer cells were
cultured in serum-free medium for 48 h. The medium was then harvested and centrifuged at
1000rpm for 5 min. Four milliliters of supernatant were then used as conditioned medium and
added to WT-MEF cells.
Nuclear Extract Preparation and Electrophoretic Mobility Shift Assay (EMSA) The
human pancreatic cancer cells were cultured in serum-free medium for 48 h. The harvested
conditioned media were used to treat WT-MEFs at different intervals. Two microgram per ml
6
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

(2µg/ml) neutralizing antibodies were used to block the corresponding cytokines. EGFR
antibody C225 was used as 20 ng/ml for 48 h prior to the experiment. The nuclear extracts were
prepared according to the method of Andrews and Faller (39). DNA binding assays for NF-κB
proteins were performed with 15 µg of nuclear extracts as described by Chiao et al (11). 32P-
labeled double-stranded oligonucleotides (5′- CTCAACAGAGGGGACTTTCCGAGAGGC
CAT - 3′) containing the κB site found in the HIV long terminal repeat were used as probes. The
mutant κB site for HIV long terminal repeat (5′- CTCAACAGAGTTGACTTTTCGAGAG
GCCAT - 3′) was used for competition studies. The competition was performed with a 50-fold
excess of unlabeled wild-type or mutant κB oligonucleotides. Supershift experiments were
performed with anti-RelA and anti-p50 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA).
The reactions were analyzed on 4% polyacrylamide gels containing 0.25 X TBE
(Tris/Borate/EDTA) buffer.
Western Blot Analysis Cytoplasmic extracts were used for the detection of IκBα protein.
Total protein extracts from human pancreatic cancer cell lines were isolated for the detection of
IL-1α and IL-1β level. For detection of IL-1α and IL-1β levels in conditioned medium, the
conditioned medium from human pancreatic cancer cell cultures was harvested at the same ratio
of medium volume to cell number and dialyzed for 24 h, 1 ml of dialyzed medium was then
dried to a volume of 30 µl. The concentrated medium or 100 µg of protein extracts was resolved
by SDS-PAGE, transferred to nylon membranes (Immobilon-P. Millipore, Bedford, MA), and
detected with IκBα, IL-1α, IL-1β antibodies (Santa Cruz Biotechnology), uPA antibody
(American Diagnostica, Greenwich, CT), or β-actin antibody (Sigma Chemical, St. Louis, MO).
7
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Subsequent Western blot analyses were carried out with Lumi-light Western blot substrate
(Roche Diagnostics, Indianapolis, IN).
Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Northern Blot Analysis
Total RNA was extracted using Trizol reagent according to the manufacturer’s protocol
(Invitrogen Life Technologies, Carlsbad, CA). The RNA was then reverse-transcribed into
cDNA. The primers used for IL-1α were 5′-GGTAGTAGCAACCAACGGGA-3′ and 5′-
TGGGTATCTCAGGCATCTCC -3′, and the primers used for IL-1β are 5′-AGCTGATGGCC
CTAAACAGA-3′ and 5′-TCTTTCAACACGCAGGACAG-3′. The PCR products were 419bp
for IL-1α and 498bp for IL-1β. The PCR conditions were as follows: 94 °C for 5 min, then
perform 30 cycles at 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min. Finally samples were
extended at 72 °C for 7 min. The PCR products were cloned into a pCR2.1-TOPO vector
(Invitrogen Life Technologies) and were subsequently sequenced to confirm their identity. The
IL-1α (BamHI and EcoRV-digested) and IL-1β (EcoRI for IL-1β-digested) cDNAs were
purified and used as the probes for northern blot analysis.
Total RNA (30 µg) was subjected to electrophoresis through a 1.2% agarose gel containing
formaldehyde, transferred to a Hybond nylon filter (MSI), and ultraviolet cross-linked. The blots
were hybridized with 32P-labeled human IL-1α or IL-1β cDNA and exposed to display the
results.
Primer Extension and DNA Sequencing Total RNA (25 µg) from ASPC-1 and MDAPanc-28
cells were preheated at 65 °C for 5 min and then put on ice for 5 min. After that, 200 ng of 32P-
end-labeled primer (5′-GCTGT AGTTG TGTTC TGGCT GA- 3′), 5 units of RNase inhibitor,
2.5 mM dNTP mix, 2.5mM MgCl2, 1µl of Improm-II reverse transcriptase and 1X RT reaction
buffer (Promega, Madison, WI) were added. The mixture were incubated at 25 °C for 15 min,
8
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

and then transferred to 42 °C for 1 h. The reaction products were treated with RNase H for 30
min, boiled for 5 min, chilled on ice, and analyzed in 8% denaturing polyacrylamide gels with
DNA sequencing reaction products of human IL-1α genomic clone using the same 32P-end-
labeled primer and DNA size marker, φX174 (32P-end-labeled). DNA sequencing was performed
using a Sequenase kit (United States Biochemical, Cleveland, OH).
Mutagenesis of AP-1 sites in IL-1a promoter IL-1α promoter was cloned by PCR. The
right primer was 5′ -CGCTCGAGGCTGTAGTTGTGTTCTGGCTGA- 3′ (the sequence
underlined is the restriction site for Xho I), and the left primer was 5′ -CGGAGCTCTGGGAA
CCCAAAACA TTCAT- 3′ (the sequence underlined is the restriction site for Sac I). PCR
products were cloned into the pGL2 Firefly luciferase vector (Promega). The mutagenesis was
performed using the mutagenesis kit from Stratagene (La Jolla, CA). The primer used for AP1-1
(site 1) is 5′ -CCAT TCCCAAACTTGTCCACTGGCTTCTGGCTGAGGCC- 3′, for AP-1-2
(site 2) is 5′ -CCCTCT TCAGAAAAGATGGCACATTTTCCCTC- 3′ (AP-1 sites were
underlined and mutated sites were in bold letters). The mutated AP-1 sites were verified by
sequencing.
Transient Transfection and Luciferease Reporter Gene Analysis One microgram of IL-1α
reporter gene (1µg) constructs were cotransfected into relA-/- MEF cells with an internal control,
p-TK Renilla luciferase (pRL-TK) using lipotransfection method (FuGENE 6, Roche
Diagnostics) in triplicate. The activity of both Firefly and Renilla luciferase were determined 48
h after transfection using a dual-luciferase reporter assay system (Promega).
9
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Results
Conditioned Medium from Metastatic Human Pancreatic Cancer Cells Induces NF-κB
Activation We previously reported that NF-κB was constitutively activated in most human
pancreatic cancer tissues and cell lines (17) and that inhibition of constitutive NF-κB activation
suppresses tumorigenesis and liver metastasis of pancreatic cancer cell lines (19,20).
Constitutive NF-κB activity was greatly reduced in the metastatic pancreatic cancer cell lines
when the fresh medium was replenished 1-2 h before the EMSA (Niu et al., unpublished
observation). One possible mechanism is that the constitutive NF-κB activity was induced
through autocrine stimulation. To test whether human pancreatic cancer cells secret an inducer
for NF-κB activation, we cultured the human metastatic pancreatic cancer cell lines ASPC-1 and
MDAPanc-28, and the nonmetastatic pancreatic cancer cell line PANC-1 in serum-free medium
for 48 h, and then isolated the conditioned media to stimulate WT-MEF cells at different times.
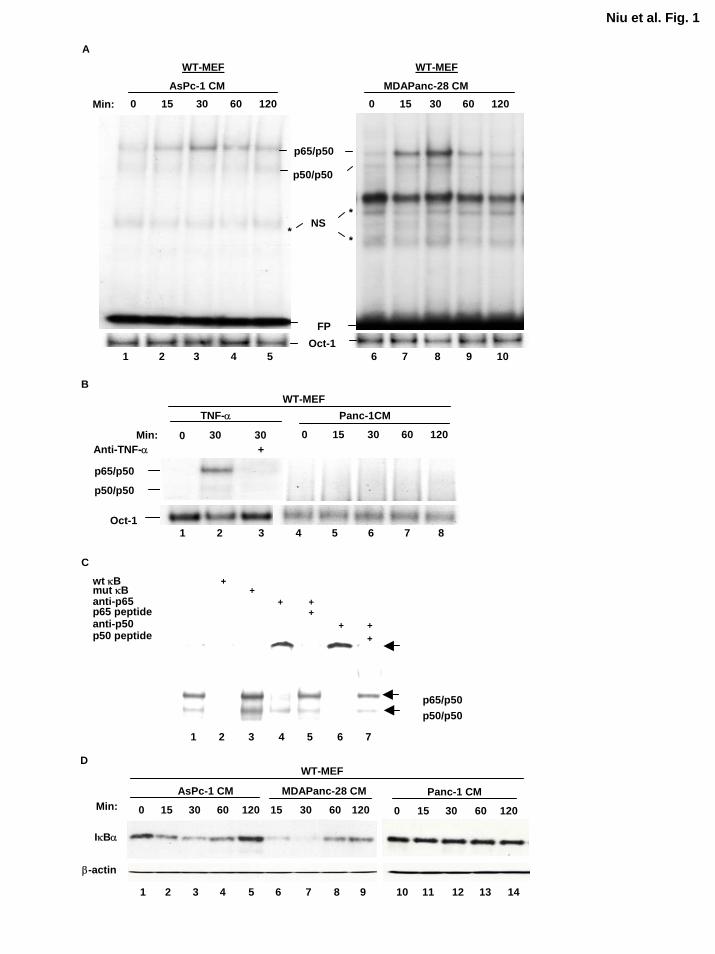
Our results showed that the conditioned medium from ASPC-1 and MDAPanc-28 pancreatic
cancer cell lines induced NF-κB activation in a time-dependent mode but did not increase Oct-1
DNA binding activity (Fig. 1A). TNF-α induced NF-κB activation in WT-MEF cells, but the
conditioned medium from PANC-1 cells did not (Fig. 1B). These results suggest that metastatic
but not nonmetastatic human pancreatic cancer cell lines secrete the activators of NF-κB, which
may in turn induce NF-κB constitutive activity in these cells. The competition and supershift
assays confirmed the specificity and identity of the κB DNA binding activity as p65(RelA)/p50
heterodimers (Fig. 1C). Conditioned medium-stimulated NF-κB activation was confirmed by
the conditioned medium-induced and time-dependent degradation of IκBα (Fig. 1D).
Furthermore, the conditioned media from the other human metastatic pancreatic cancer cell lines,
Capan-1, CFPAC-1 and MDAPanc-3, also induced NF-κB DNA binding activity in WT-MEF
10
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

cells (data not shown). These results suggest that the secretion of NF-κB inducer is a common
phenomenon in metastatic pancreatic cancer cells.
Interleukin-1α Is the Primary Cytokine Secreted by Human Metastatic Pancreatic Cancer
Cell Lines To identify which inducer is secreted by these pancreatic cancer cell lines, we first
screened a panel of neutralizing antibodies of common inducers of NF-κB, including IL-1α, IL-
1β, TNFα, and TNFβ before we performed biochemical purification and identification
approaches. The conditioned media from these cell lines were isolated and incubated with IL-
1α, IL-1β, TNFα, and TNFβ neutralizing antibodies. Subsequently, these media were used to
stimulate WT-MEF cells for 15, 30, 60, and 120 min, and nuclear extracts were then isolated for
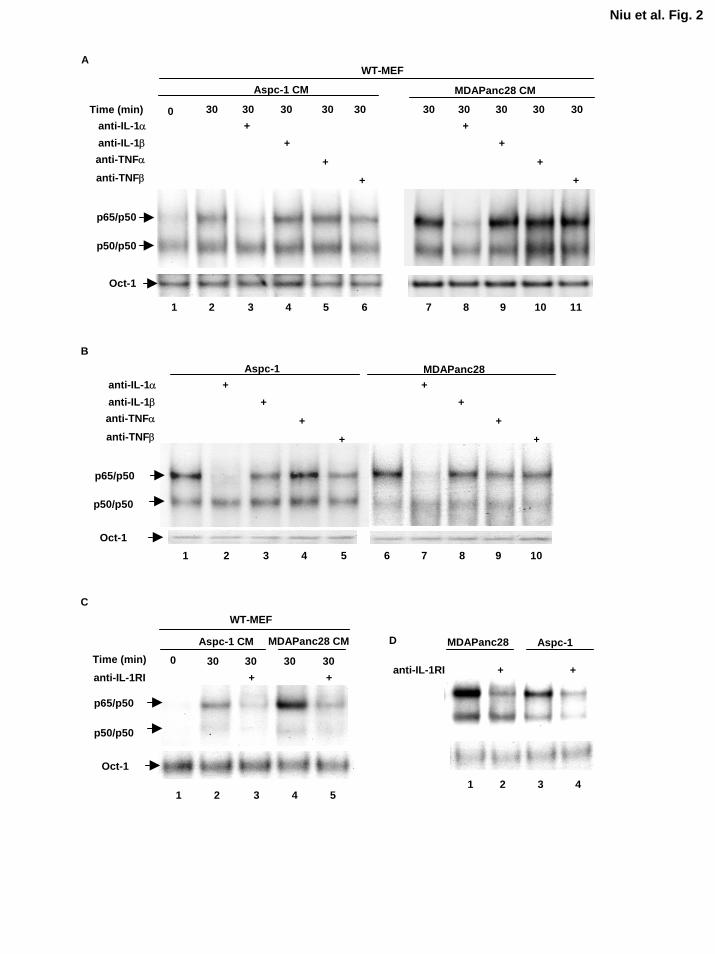
EMSA. We found that the IL-1α neutralizing antibody blocked NF-κB-inducing activity in the
ASPC-1 and MDAPanc-28 conditioned media, but the other neutralizing antibodies did not have
much effect (Fig. 2A). Consistently, IL-1α neutralizing antibody blocked the NF-κB inducing
activity from the conditioned media of other human metastatic pancreatic cancer cell lines,
Capan-1, CFPAC-1 and MDAPanc-3 (data not shown). This implies that IL-1α is the major
cytokine and NF-κB inducing activity secreted by these cells.
To determine whether IL-1α autocrine stimulation induces constitutive NF-κB activity in
ASPC-1 and MDAPanc-28 cells, we treated these cells directly with neutralizing antibodies
against IL-1α, IL-1β, TNFα and TNFβ for 1 h and isolated the nuclear extracts for EMSA.
Anti-IL-1α neutralizing antibody completely inhibited constitutive NF-κB activity in these cells,
but anti-IL-β, TNFα and TNFβ neutralizing antibodies only had minimal effects on constitutive
NF-κB activity (Fig. 2B). To provide further evidence of the role of IL-α in autocrine
stimulation, we treated WT-MEFs and MDAPanc-28 and ASPC-1 cells with anti-IL-1 receptor
type 1 antibodies for 120 min to block their IL-1 receptors. The results showed that blocking the
11
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

IL-1 receptor in these cells inhibited IL-1α-induced NF-κB activation (Fig. 2C and 2D).
Together, these results suggest that IL-1α is the principal cytokine that contributes to the
induction of constitutive NF-κB activation in MDAPanc-28 and ASPC-1 cells.
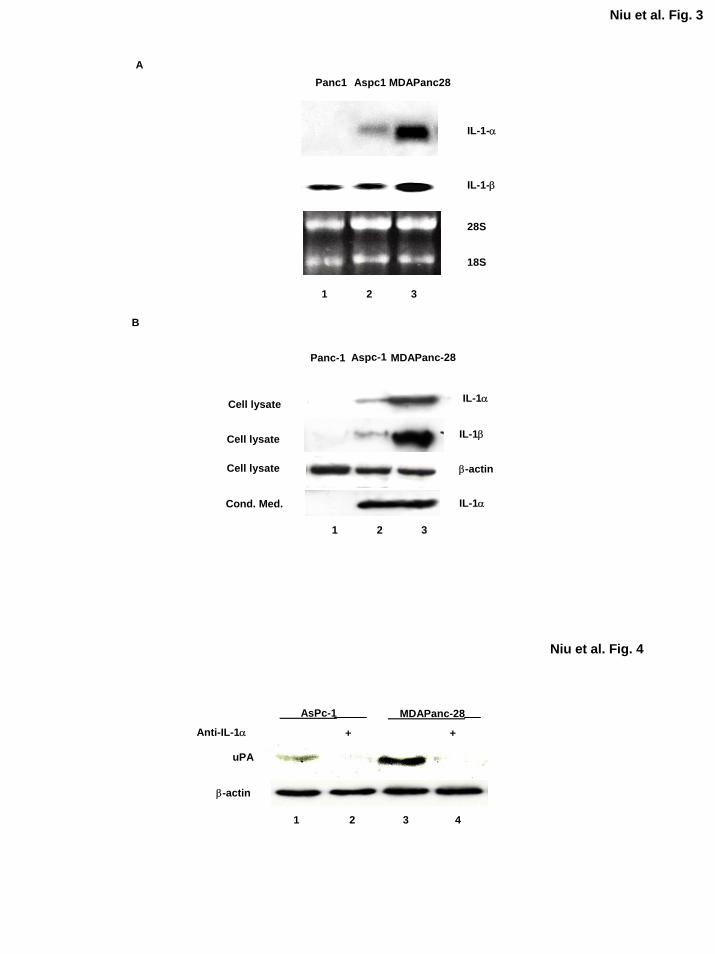
To determine the expression and secretion of IL-1α in MDAPanc-28, ASPC-1, and PANC-1
cells, we performed Northern and Western blot analyses. In MDAPanc-28 and ASPC-1 cells,
high levels of IL-1α mRNA were expressed, but its expression was not detectable in PANC-1
cells (Fig. 3A). On Western blot analysis, high levels of IL-1α protein were detected in both
cell extracts (50 µg) and conditioned medium from the metastatic human pancreatic cancer cell
lines ASPC-1 and MDAPanc-28 but not detected in those from PANC-1 cells (Fig. 3B).
Although IL-1β mRNA was detected in these pancreatic cancer cell lines, much higher level of
IL-1β mRNA was detected in MDAPanc-28 cells than those in ASPC-1 and PANC-1 cells (Fig.
3A). The level of IL-1β protein was very low and barely detectable in ASPC-1 cells and
undetectable in PANC-1 cells using 200 µg protein extracts with the anti-IL-1β antibody,
consistent with the expression patterns of IL-1β in these two cell lines detected in Northern blot
analysis. However, none of the conditioned medium from these three cells had detectable IL-1β
neither (data not shown). Much lower levels of IL-1β as compared with the level of IL-1α
expressed in ASPC-1 and MDAPanc-28 is consistent with the minor reduction of constitutive
NF-κB activities in anti-IL-1β neutralizing antibody completely treated conditioned media from
these cells (Fig. 2B). Together, these results demonstrate that IL-1α is the primary cytokine
secreted by metastatic human pancreatic cancer cells and induces constitutive NF-κB activity in
these cells.
12
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Neutralizing Antibody of IL-1α Inhibits the Expression of NF-κB Downstream Target Gene
We previously reported that urokinase-type plasminogen activator (uPA), which plays important
roles in tumor invasion, is regulated by constitutive NF-κB activation in pancreatic cancer cells
(40). To determine whether the inhibition of IL-1α-induced NF-κB activation decreases the
expression of uPA, we treated ASPC-1 and MDAPanc-28 cells with IL-1α neutralizing antibody.
Western blot analysis showed that inhibition of the IL-1α signaling cascade in these cells
blocked the expression of uPA (Fig. 4). These results further suggest that IL-1α-regulated NF-
κB activity plays a key role in controlling the expression of uPA.
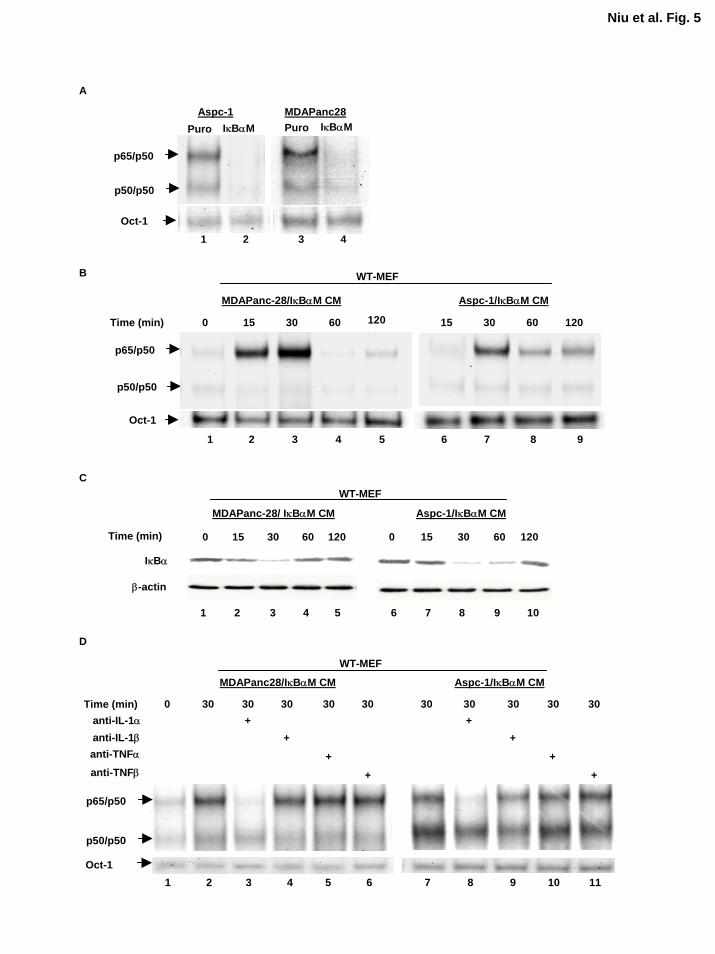
Inhibition of NF-κB by IκBαM Does Not Alter the NF-κB-Inducing Activity of IL-1α in the
Conditioned Medium Our data suggest that IL-1α is the primary cytokine secreted by metastatic
human pancreatic cancer cell lines. Since our results show that IL-1α autocrine stimulation
activates NF-κB activity constitutively in metastatic pancreatic cancer cell lines, and the
previous reports showed that IL-1α was not only one of the potent NF-κB inducers but also one
of the NF-κB regulated genes, these led us to ask: which one comes first in these cells, IL-
1α overexpression or NF-κB constitutive activation? We had previously established ASPC-
1/IκBαM and MDAPanc-28/IκBαM cells by pooling ASPC-1 and MDAPanc-28 cells infected
with a retrovirus encoding the phosphorylation mutant of IκBα (S32, 36A) (IκBαM) to
specifically inhibit NF-κB activity (18). As shown in Figure 5A, the NF-κB constitutive activity
in these cells was completely inhibited by IκBαM. However, conditioned medium from ASPC-
1/IκBαM and MDAPanc-28/IκBαM cells still activated NF-κB and induced the degradation of
IκBα in WT-MEF cells in a time-dependent manner (Fig. 5B and C). Importantly, anti-IL-1α
13
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

neutralizing antibody still inhibited IL-1α-induced NF-κB activity, indicating that IL-1α is the
primary cytokine in the conditioned media from ASPC-1/IκBαM and MDAPanc-28/IκBαM
cells that activates NF-κB in WT-MEF cells (Fig. 5D). These results suggest that inhibition of
NF-κB did not inhibit the expression of IL-1α which is regulated by an NF-κB-independent
mechanism.
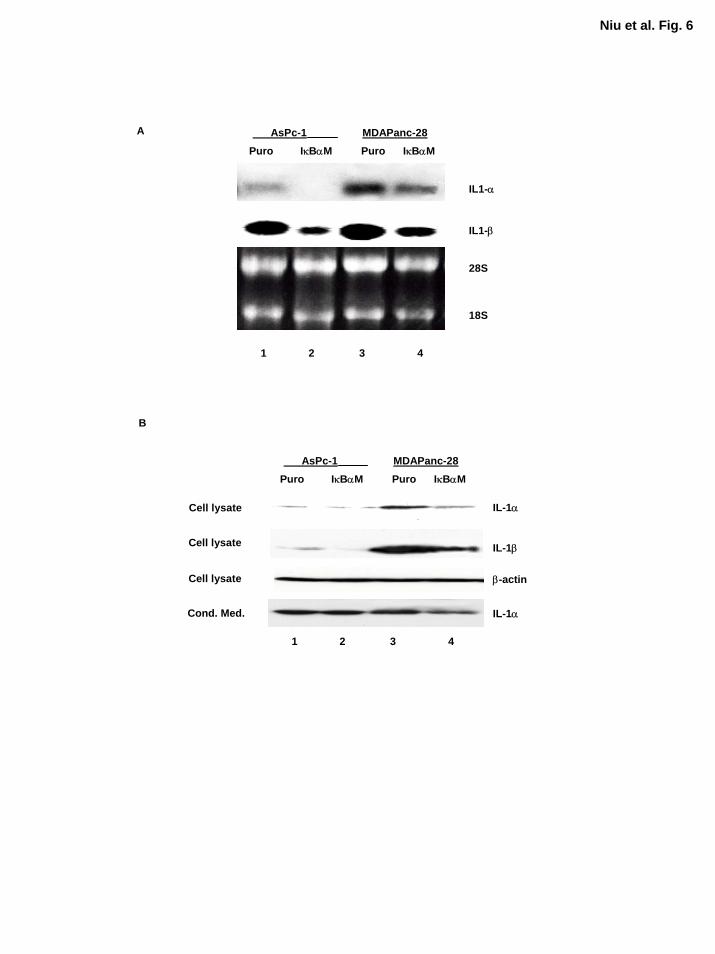
Inhibition of NF-κB Decreased but Does Not Completely Inhibit Expression of IL-1α
Although IκBαM-mediated inhibition of NF-κB did not inhibit IL-1α expression completely and
did not diminish the ability of IL-1α to induce NF-κB activation as determined by EMSA, it was
of great interest to determine whether the expression of IL-1α was decreased in MDAPanc-
28/IκBαM and ASPC-1/IκBαM cells. Northern blot analysis showed that the expression of IL-
1α and IL-1β at mRNA level were decreased in MDAPanc-28/IκBαM and ASPC-1/IκBαM
cells (Fig. 6A). However, the sufficient levels of IL-1α protein and activity are remained in
ASPC-1/IκBαM and MDAPanc-28/IκBαM cells for activating NF-κB (Fig. 6B). IL-1β was
barely detected in ASPC-1 protein extract and was not detected in the conditioned media from
these pancreatic cancer cells (Fig. 6B and data not shown). These results suggest that
transcription factors other than NF-κΒ primarily regulate the expression of IL-1α, which induces
constitutive NF-κB activation in these cancer cells and constitutive NF-κB activity then
enhances the expression of IL-1α. However, the signaling pathway and transcription factors that
regulate IL-1α expression in metastatic pancreatic cancer cells remain to be determined.
14
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

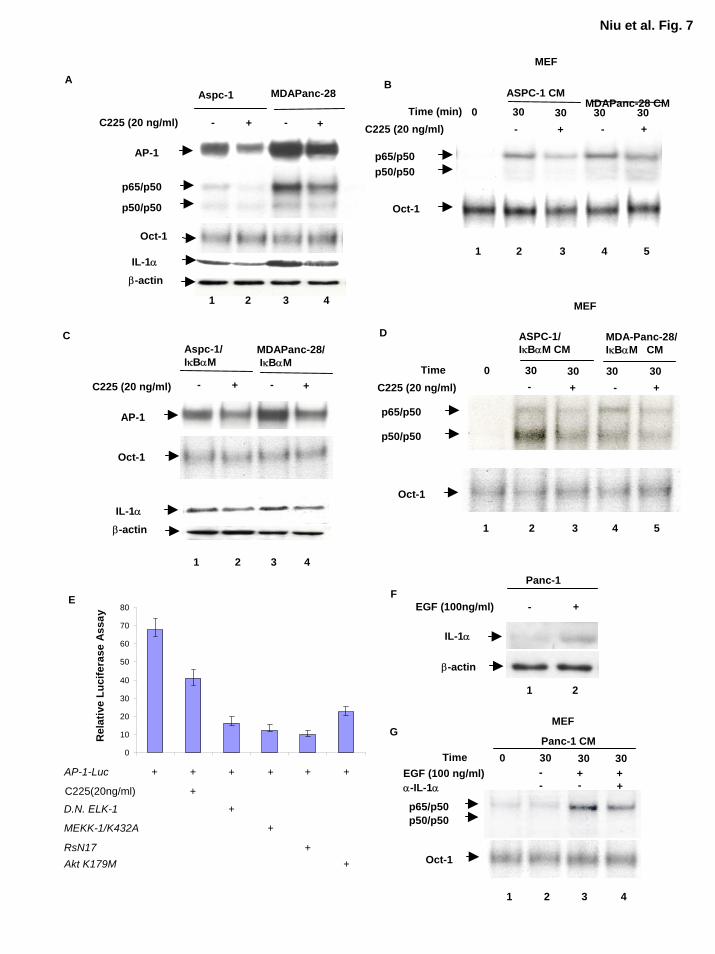
AP-1 Activity Modulated by Overexpression of EGFR Plays a Key Role in Regulating IL-
1α Expression Very little is known about regulation of the IL-1α gene despite importance of IL-
1α in cytokine signaling cascades and the reports that IL-1α is one of the downstream target
genes regulated by NF-κB and that the human IL-1α promoter contains an LPS-inducible AP-1-
binding site composed of Jun and Fos proteins (30,41). AP-1 is regulated by many stimuli, most
notably serum and growth factors (42,43). Overexpression of EGFR, which occurs in
approximately 90% of human pancreatic tumors, may be the genetic alteration with the most
significant clinical correlation (44,45). On the basis of these experimental findings, we sought to
determine whether the EGFR signaling pathway and AP-1 play a role in regulating IL-1α
expression in these pancreatic cancer cells. Our results showed that EGFR blockade by anti-
EGFR antibody (C225) reduced AP-1 and NF-κB but not the Oct-1 DNA binding activities in
ASPC-1, MDAPanc-28, ASPC-1/IκBαM, and MDAPanc-28/IκBαM cells (Fig. 7A and C).
Inhibition of EGFR activation in ASPC-1, MDAPanc-28, ASPC-1/IκBαM, and MDAPanc-
28/IκBαM cells also decreased the levels of IL-1α expression because the conditioned medium
from these cells treated with anti-EGFR antibody (C225) reduced the NF-κB-inducing activity in
the WT-MEF cells (Fig. 7B and D). Together, these results suggest that the expression of IL-1α
is partially regulated by EGFR signaling cascades through AP-1 transcription factors.
AP-1 transcription factors are regulated by multiple signaling cascades. As shown in
Figure 7C and D, AP-1 activity was already elevated in the absence of growth factor stimulation
and anti-EGFR antibody only partially inhibited this elevated AP-1 activity. To determine
whether EGF-induced AP-1 activity can be inhibited by the mutant of EGFR downstream
signaling molecules, AP-1 activity was determined using AP-1 luciferase reporter gene assays
with anti-EGFR antibody (c225) treated AsPc-1 cells and AsPc-1 cells transfected with dominant
15
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

negative Elk-1 (∆Elk-1) and ras (RsN17) and kinase-dead AKT and MEKK1. As shown in
Figure 7E, These results suggest that growth factor dependent and/or independent activation of
MAP kinase, PI-3 kinase, and other signaling pathways are involved in the stimulation of AP-1,
which in turn induce IL-1α expression in the pancreatic cancer cell line.
To determine whether EGF stimulation induces expression of IL-1α, we stimulated Panc-
1, a nonmetastatic pancreatic cancer cell line with EGF (100 ng/ml) for 18 hours and analyzed
the expression of IL-1α. As shown in Figure 7F, expression of IL-1α induced by EGF
stimulation in Panc-1 cells. The conditioned media from Panc-1 cells with or without EGF-
stimulation were used to stimulate wild-type MEF cells and the results showed that NF-κB was
activated by the conditioned medium from EGF-stimulated Panc-1 cells and anti-IL-1α
neutralizing antibody only partially inhibited the conditioned medium-induced NF-κB activation
(Fig 7G). The partial inhibition of NF-κB activation by the IL-1α neutralizing antibody may be
due to the presence of either residual EGF or additional cytokines in conditioned medium.
Taken together these results suggest that the IL-1α expression was induced by EGF in a
nonmetastatic pancreatic cancer cell line Panc-1.
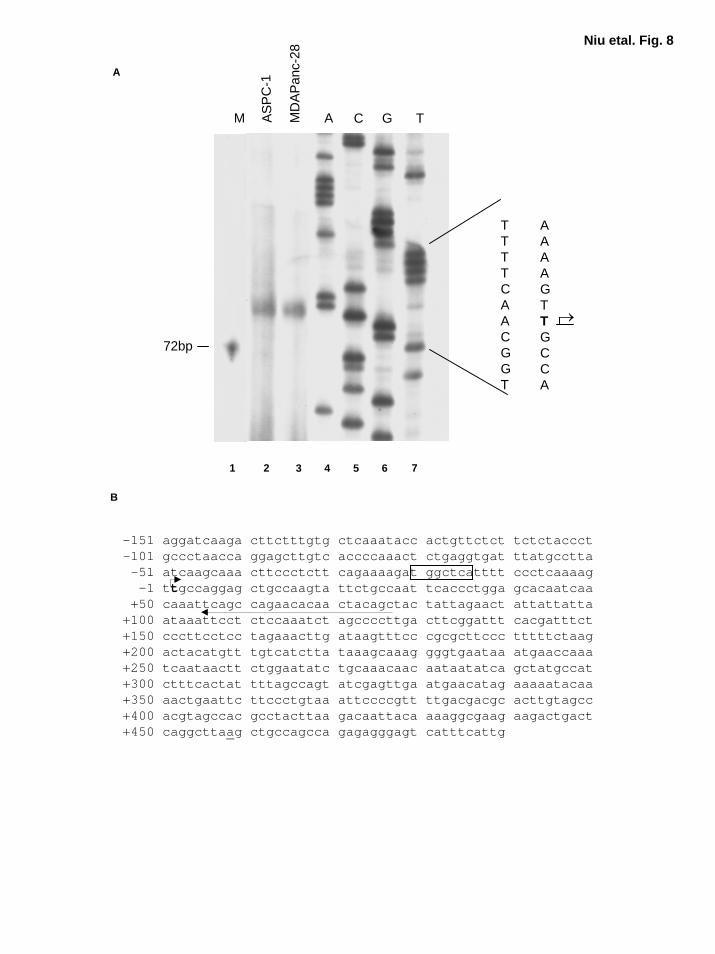
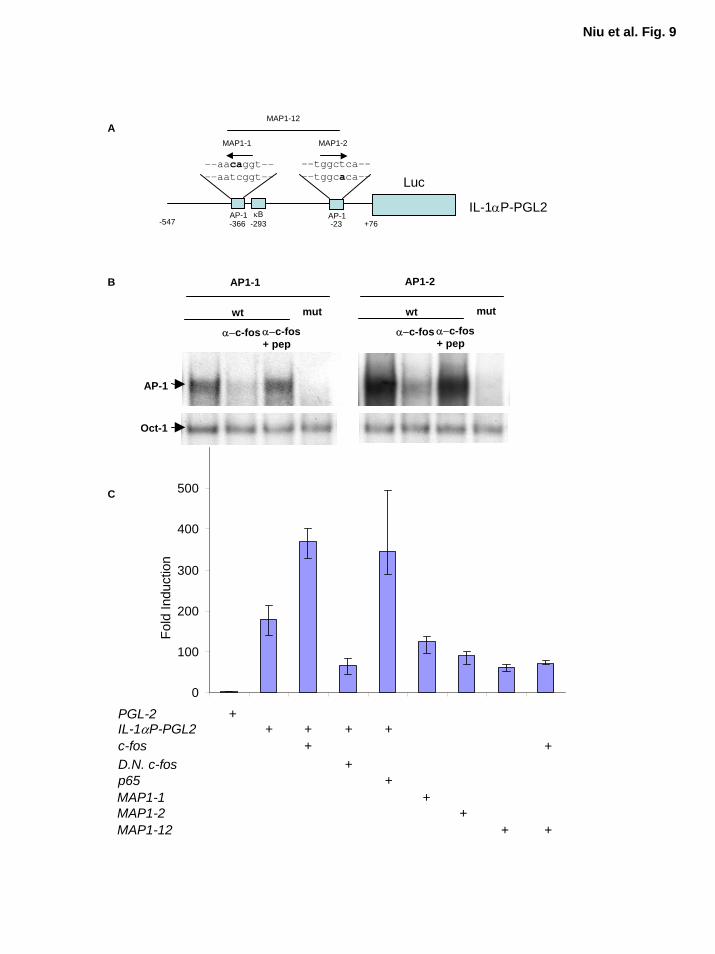
Expression of IL-α Is Regulated by AP-1 and NF-κB To determine whether both AP-1
and NF-κB take part in regulating IL-1α expression, we sought to clone human IL-1α promoter
regions. To date, most studies of the IL-1α promoter have been based on a previously reported
IL-1α cDNA sequence (NCBI accession number X02851) (46). However, our RT-PCR results
suggested that the IL-1α transcription starting site is farther upstream than the previously
identified site (data not shown) (46). To verify this finding, we performed primer extension
assays. Our results show that the newly identified transcription starting site was actually 458 bp
16
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

upstream of the previous reported starting site (Fig. 8A and 8B) (46). There was one κB-like site
at -293, and two AP-1 binding sites at -367, and -22 in this fragment of the 623 bp newly
identified IL-1α promoter region (Fig. 8A and B). TATA box was not identified in this region
(Fig. 8A and B). Schematic diagram of IL-α promoter and reporter gene constructs with wild-
type and mutant AP-1 sequences indicated in Figure 9A. To determine whether these two AP-1
elements (AP1-1 and AP1-2) in IL-1 promoter interact with Fos/AP-1, we performed EMSA
with wild-type and mutant AP1-1 and AP1-2 probes. The competition and supershift assays
showed the presence of c-Fos in the AP-1 DNA binding activity (Fig. 9B). These results suggest
that IL-α is likely to be one of the target genes regulated by AP-1 proteins. On the basis of these
results, we performed IL-1α promoter luciferase reporter gene assays to determine whether these
two AP-1 sites identified in IL-1α promoter are functional. Since pancreatic cancer cells have
poor efficiency for transient transfection, and to better study the effect of AP-1 and NF-κB on
IL-1α transcriptional activity, we selected relA-/- MEF cells for the reporter gene analysis. We
found that RelA (p65) activated this IL-1α promoter reporter gene construct, whereas dominant-
negative c-Fos inhibited IL-1α promoter activity (Fig. 9C). Overexpression of c-Fos induced
strong luciferase reporter gene activity in the reporter gene construct with wild-type AP-1 sites,
but not in the reporter gene constructs with the double mutant AP-1 sites, MAP1-12 (Fig. 9C).
The basal IL-1α promoter activity was also inhibited by the presence of the mutant AP-1 sites,
MAP1-1, MAP1-2, and MAP1-12 (Fig. 9C). These results demonstrate that both AP-1 and NF-
κB regulate IL-1α expression. Thus, our findings suggest that the constitutive NF-κB activation
induced by IL-1α autocrine stimulation in these pancreatic cancer cell lines may be initiated by
EGFR dependent or independent signaling cascades mediated AP-1 activity.
17
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Discussion
Pancreatic adenocarcinoma is the fourth leading cause of adult cancer death in the United
States. The 5-year survival rate continues at 1-3% (47). At the time of diagnosis, most patients
with pancreatic cancer have advanced and metastatic disease (48). Studies have suggested that a
genetic profile for pancreatic cancer is emerging based on the most frequently detected mutations
in this disease (49). K-ras mutation is an early event in pancreatic carcinogenesis that has been
detected in 80-95% of pancreatic cancers; overexpression of EGFR occurs in approximately 90%
of human pancreatic tumors; and inactivation of Smad4, Ink4a/Arf, and p53 tumor suppressor
genes has been identified in approximately 50-75% of pancreatic cancers (49). However, the
role of specific genetic alterations that initiate tumorigenesis and mediate its cardinal clinical
features of locally aggressive growth, metastasis, and chemotherapy resistance remains to be
elucidated.
Recently, we demonstrated that RelA, the p65 subunit of NF-κB transcription factors, is
constitutively activated in most human pancreatic cancer tissues and cell lines (17), and this
activation has been shown to play key roles in pancreatic tumorigenesis and liver metastasis of
pancreatic adenocarcinoma (19,20). Therefore, understanding the mechanisms by which NF-κB
is constitutively activated in pancreatic cancer cells will elucidate the signaling cascades that
directly or indirectly regulate NF-κB activation.
In this study, we found that the autocrine mechanism is responsible for the constitutive
activation of NF-κB in metastatic human pancreatic cancer cell lines but not in nonmetastatic
ones. Subsequent studies showed that IL-1α is the primary NF-κB-activating cytokine secreted
by these cell lines. IL-1α has been shown to play important roles in pro-inflammatory responses,
cancer cell growth, and metastasis (15,50-52), and has been suggested that it participates in
18
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

cancer cell invasion and metastasis by regulating the expression of some adhesion molecules.
For instance, IL-1α could increase the expression of MMP-9, E-selectin, integrin-1, and IL-8 and
inhibit plasminogen activator inhibitor-1 (34,50-53). A recent report suggested that IL-1α
enhances the adhesion of metastatic human pancreatic cancer cells to extracellular matrix (ECM)
proteins, but not nonmetastatic ones (53). Our findings showed that IL-1α is the primary
cytokine that causes constitutive NF-κB activation in metastatic human pancreatic cancer cells
and that blocking IL-1α in these cells inhibited the expression of urokinase-type plasminogen
activator (uPA) (Fig. 4). Together with our previous findings that constitutively activated NF-
κB plays key roles in pancreatic tumorigenesis and metastasis and that overexpression of uPA in
pancreatic adenocarcinoma is regulated by constitutively activated RelA (19,20,40), one possible
mechanism by which IL-1α participates in pancreatic tumorigenesis and cancer metastasis is the
IL-1α-regulated expression of a number of key determinants for tumorigenic and metastatic
phenotype through NF-κB.
IL-1α is a key mediator of the inflammatory response (54). It is not only a potent inducer of
NF-κB but also a downstream target gene of NF-κB (25,30). This led us to an interesting
question: Which of the molecular alteration, overexpression of IL-1α or constitutive activation
of NF-κB, precedes in the development of pancreatic cancer cells? In this study, we
demonstrated that expression and secretion of IL-1α were decreased but not completely blocked
by the IκBαM-mediated inhibition of NF-κB, and that IL-1α present in the conditioned medium
isolated from both ASPC-1/IκBαM and MDAPanc-28/IκBαM cells is still capable of activating
NF-κB in WT-MEF cells, similar to that isolated from ASPC-1 and MDAPanc-28 cells. These
findings suggest that expression of IL-1α is primarily regulated by NF-κB-independent
19
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

mechanisms in human pancreatic cancer cells. The constitutive NF-κB activity triggered by IL-
1α autocrine stimulation may enhance the IL-1α expression. Thus, this may initiate a possible
formation of a positive feedback loop and connect inflammatory responses and cancer
development.
Previous studies have shown that the expression of IL-1α is regulated by NF-κB and AP-1
transcription factors (25,42). In our newly identified IL-1α promoter region, there are multiple
NF-κB and AP-1 sites, and our reporter gene analysis confirmed that IL-1α is regulated by both
NF-κB and AP-1. IL-1α has also been found to induce AP-1 activity (32). These findings
suggest that IL-1α autocrine stimulation may be initiated through AP-1 activation.
The recently proposed progression model of pancreatic adenocarcinoma suggests a profile of
genetic alterations that includes overexpression of EGFR and mutations of the K-ras gene (49).
EGFR and K-ras may induce AP-1 activation and IL-α overexpression in pancreatic cancers.
Studies have shown that EGF induced AP-1 activity, and both EGFR tyrosine kinase activity and
autophosphorylation at Y (1173) play a critical role in EGF-induced AP-1 activation (55).
Several reports have suggested that the Ras signaling pathway induces AP-1 activation and
regulates IL-1α expression (56-58). Consequently, autocrine production of IL-1α induces the
transcriptional activation of both NF-κB and AP-1. These findings imply that these genetic
alterations in the development of pancreatic adenocarcinoma possibly initiate the overexpression
of IL-1α, which, in turn, induces a large number of the downstream target genes that encodes the
key determinants of tumorigenic and metastatic phenotypes through activation of NF-κB and
AP-1 transcription factors. The working model for constitutive NF-κB activation in metastatic
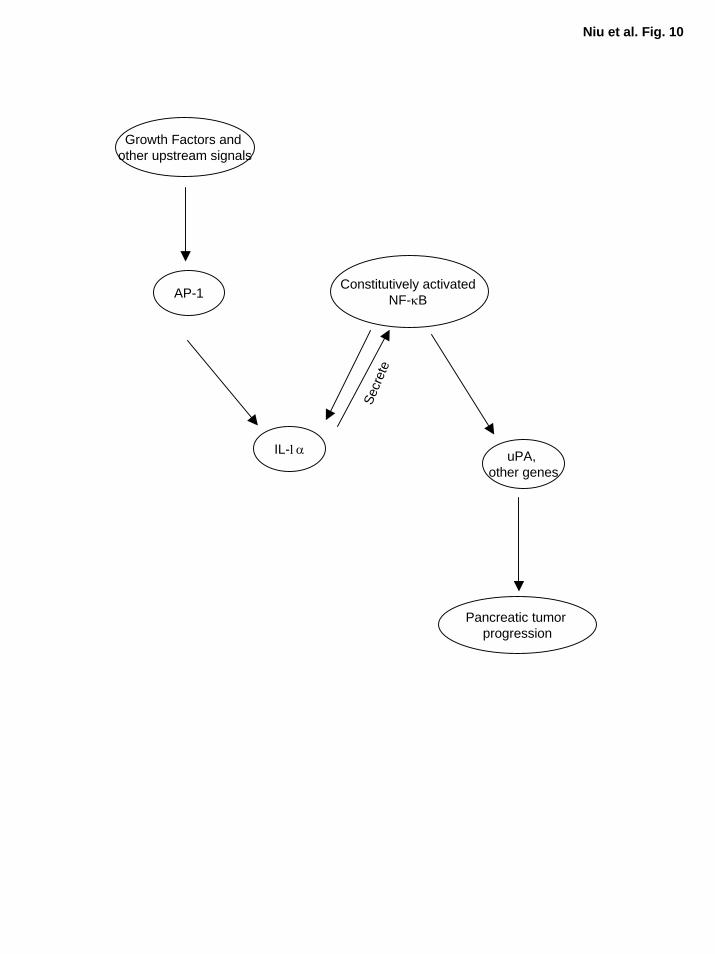
pancreatic cancer cells is illustrated in Fig. 10.
20
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

In summary, we identified the pro-inflammatory cytokine IL-1α as the primary cytokine that
induces constitutive NF-κB activation in metastatic human pancreatic cancer cell lines. The
constitutive NF-κB activity in turn enhances the expression of IL-1α, suggesting that this
autocrine mechanism plays an important role in the development of the tumorigenic and
metastatic phenotype, making a positive feedback loop possible. Autocrine production of IL-1α
may be primarily induced by AP-1 activity, and AP-1 is a potential target for IL-1α, suggesting
that an additional positive feedback loop may be possible between IL-1α expression and AP-1
activity. Many studies have shown that both NF-κB and AP-1 are activated in response to
growth factors and inflammatory stimuli, and constitutive activation of NF-κB and AP-1 has
been shown to be critical in the development of cancer. Therefore, our results also suggest that
NF-κB and AP-1 might be the missing mechanistic links between inflammation and cancer.
Acknowledgement We are grateful to Dr. Inder M. Verma at Salk Institute for the generous gift of RelA-/- mouse
embryonic fibroblasts, Dr. Zhen Fan at MDACC for the anti-EGFR antibody (C225), and Dr.
Christian Schmidt for the wild-type mouse embryonic fibroblasts and critical reading of the
manuscript. We also thank Ann Sutton (Department of Scientific Publications) for editorial
assistance. The work was supported by grants from National Cancer Institute (CA73675-01,
R21PA-98-029, CA097159 and CA78778-01).
21
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

References
1. Gilmore, T. D., Koedood, M., Piffat, K. A., and White, D. W. (1996) Oncogene 13, 1367-
1378 2. Li, Q., and Verma, I. M. (2002) Nat Rev Immunol 2, 725-734 3. Ghosh, S., and Karin, M. (2002) Cell 109 Suppl, S81-96 4. Karin, M., and Lin, A. (2002) Nat Immunol 3, 221-227 5. Baldwin, A. S., Jr. (1996) Annu Rev Immunol 14, 649-683 6. Sen, R., and Baltimore, D. (1986) Cell 47, 921-928 7. Baeuerle, P. A., and Baltimore, D. (1988) Science 242, 540-546 8. Baeuerle, P. A., and Baltimore, D. (1996) Cell 87, 13-20 9. Chen, Z., Hagler, J., Palombella, V. J., Melandri, F., Scherer, D., Ballard, D., and
Maniatis, T. (1995) Genes Dev 9, 1586-1597 10. Brown, K., Gerstberger, S., Carlson, L., Franzoso, G., and Siebenlist, U. (1995) Science
267, 1485-1488 11. Chiao, P. J., Miyamoto, S., and Verma, I. M. (1994) Proc Natl Acad Sci U S A 91, 28-32 12. Sun, S. C., Ganchi, P. A., Ballard, D. W., and Greene, W. C. (1993) Science 259, 1912-
1915 13. Rice, N. R., and Ernst, M. K. (1993) Embo J 12, 4685-4695 14. Moore, B. E., and Bose, H. R., Jr. (1988) Virology 162, 377-387 15. Karin, M., Cao, Y., Greten, F. R., and Li, Z. W. (2002) Nat Rev Cancer 2, 301-310 16. Emmerich, F., Meiser, M., Hummel, M., Demel, G., Foss, H. D., Jundt, F., Mathas, S.,
Krappmann, D., Scheidereit, C., Stein, H., and Dorken, B. (1999) Blood 94, 3129-3134 17. Wang, W., Abbruzzese, J. L., Evans, D. B., Larry, L., Cleary, K. R., and Chiao, P. J.
(1999) Clin Cancer Res 5, 119-127 18. Dong, Q. G., Sclabas, G. M., Fujioka, S., Schmidt, C., Peng, B., Wu, T., Tsao, M. S.,
Evans, D. B., Abbruzzese, J. L., McDonnell, T. J., and Chiao, P. J. (2002) Oncogene 21, 6510-6519
19. Fujioka, S., Sclabas, G. M., Schmidt, C., Niu, J., Frederick, W. A., Dong, Q. G., Abbruzzese, J. L., Evans, D. B., Baker, C., and Chiao, P. J. (2003) Oncogene 22, 1365-1370
20. Fujioka, S., Sclabas, G. M., Schmidt, C., Frederick, W. A., Dong, Q. G., Abbruzzese, J. L., Evans, D. B., Baker, C., and Chiao, P. J. (2003) Clin Cancer Res 9, 346-354
21. Voronov, E., Shouval, D. S., Krelin, Y., Cagnano, E., Benharroch, D., Iwakura, Y., Dinarello, C. A., and Apte, R. N. (2003) Proc Natl Acad Sci U S A 100, 2645-2650
22. Apte, R. N., and Voronov, E. (2002) Semin Cancer Biol 12, 277-290 23. Wilson, J., and Balkwill, F. (2002) Semin Cancer Biol 12, 113-120 24. Lotem, J., and Sachs, L. (2002) Oncogene 21, 3284-3294 25. Osborn, L., Kunkel, S., and Nabel, G. J. (1989) Proc Natl Acad Sci U S A 86, 2336-2340 26. Israel, A., Le Bail, O., Hatat, D., Piette, J., Kieran, M., Logeat, F., Wallach, D., Fellous,
M., and Kourilsky, P. (1989) Embo J 8, 3793-3800 27. Shakhov, A. N., Collart, M. A., Vassalli, P., Nedospasov, S. A., and Jongeneel, C. V.
(1990) J Exp Med 171, 35-47 28. Collart, M. A., Baeuerle, P., and Vassalli, P. (1990) Mol Cell Biol 10, 1498-1506
22
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

29. Hiscott, J., Marois, J., Garoufalis, J., D'Addario, M., Roulston, A., Kwan, I., Pepin, N., Lacoste, J., Nguyen, H., Bensi, G., and et al. (1993) Mol Cell Biol 13, 6231-6240
30. Mori, N., and Prager, D. (1996) Blood 87, 3410-3417 31. Vale, T., Ngo, T. T., White, M. A., and Lipsky, P. E. (2001) Cancer Res 61, 602-607 32. Yasumoto, K., Okamoto, S., Mukaida, N., Murakami, S., Mai, M., and Matsushima, K.
(1992) J Biol Chem 267, 22506-22511 33. Ondrey, F. G., Dong, G., Sunwoo, J., Chen, Z., Wolf, J. S., Crowl-Bancroft, C. V.,
Mukaida, N., and Van Waes, C. (1999) Mol Carcinog 26, 119-129 34. Wolf, J. S., Chen, Z., Dong, G., Sunwoo, J. B., Bancroft, C. C., Capo, D. E., Yeh, N. T.,
Mukaida, N., and Van Waes, C. (2001) Clin Cancer Res 7, 1812-1820 35. Arlt, A., Vorndamm, J., Muerkoster, S., Yu, H., Schmidt, W. E., Folsch, U. R., and
Schafer, H. (2002) Cancer Res 62, 910-916 36. Frazier, M. L., Fernandez, E., de Llorens, R., Brown, N. M., Pathak, S., Cleary, K. R.,
Abbruzzese, J. L., Berry, K., Olive, M., Le Maistre, A., and Evans, D. B. (1996) Int J Pancreatol 19, 31-38
37. Frazier, M. L., Pathak, S., Wang, Z. W., Cleary, K., Singletary, S. E., Olive, M., Mackay, B., Steck, P. A., and Levin, B. (1990) Pancreas 5, 8-16
38. Beg, A. A., Sha, W. C., Bronson, R. T., and Baltimore, D. (1995) Genes Dev 9, 2736-2746
39. Andrews, N. C., and Faller, D. V. (1991) Nucleic Acids Res 19, 2499 40. Wang, W., Abbruzzese, J. L., Evans, D. B., and Chiao, P. J. (1999) Oncogene 18, 4554-
4563 41. Bailly, S., Fay, M., Israel, N., and Gougerot-Pocidalo, M. A. (1996) Eur Cytokine Netw
7, 125-128 42. Shaulian, E., and Karin, M. (2001) Oncogene 20, 2390-2400 43. Sclabas, G. M., Fujioka, S., Schmidt, C., Fan, Z., Evans, D. B., and Chiao, P. J. (2003) J
Gastrointest Surg 7, 37-43 44. Smith, J. J., Derynck, R., and Korc, M. (1987) Proc Natl Acad Sci U S A 84, 7567-7570 45. Matsuda, K., Idezawa, T., You, X. J., Kothari, N. H., Fan, H., and Korc, M. (2002)
Cancer Res 62, 5611-5617 46. Furutani, Y., Notake, M., Fukui, T., Ohue, M., Nomura, H., Yamada, M., and Nakamura,
S. (1986) Nucleic Acids Res 14, 3167-3179 47. Jemal, A., Murray, T., Samuels, A., Ghafoor, A., Ward, E., and Thun, M. J. (2003) CA
Cancer J Clin 53, 5-26 48. Breslin, T. M., Hess, K. R., Harbison, D. B., Jean, M. E., Cleary, K. R., Dackiw, A. P.,
Wolff, R. A., Abbruzzese, J. L., Janjan, N. A., Crane, C. H., Vauthey, J. N., Lee, J. E., Pisters, P. W., and Evans, D. B. (2001) Ann Surg Oncol 8, 123-132
49. Hruban, R. H., Goggins, M., Parsons, J., and Kern, S. E. (2000) Clin Cancer Res 6, 2969-2972
50. Panozzo, M. P., Basso, D., De Paoli, M., Carraro, P., Burighel, D., and Plebani, M. (1996) Int J Clin Lab Res 26, 240-244
51. Nguyen, M., Corless, C. L., Kraling, B. M., Tran, C., Atha, T., Bischoff, J., and Barsky, S. H. (1997) Am J Pathol 150, 1307-1314
52. Andersen, K., Maelandsmo, G. M., Hovig, E., Fodstad, O., Loennechen, T., and Winberg, J. O. (1998) Anticancer Res 18, 3299-3303
23
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

53. Sawai, H., Yamamoto, M., Okada, Y., Sato, M., Akamo, Y., Takeyama, H., and Manabe, T. (2001) Pancreas 23, 399-405
54. Arend, W. P. (2002) Cytokine Growth Factor Rev 13, 323-340 55. Li, J., Ma, C., Huang, Y., Luo, J., and Huang, C. (2003) Oncogene 22, 211-219 56. Demetri, G. D., Ernst, T. J., Pratt, E. S., 2nd, Zenzie, B. W., Rheinwald, J. G., and
Griffin, J. D. (1990) J Clin Invest 86, 1261-1269 57. Castelli, C., Sensi, M., Lupetti, R., Mortarini, R., Panceri, P., Anichini, A., and Parmiani,
G. (1994) Cancer Res 54, 4785-4790 58. Schmid-Alliana, A., Menou, L., Manie, S., Schmid-Antomarchi, H., Millet, M. A.,
Giuriato, S., Ferrua, B., and Rossi, B. (1998) J Biol Chem 273, 3394-3400
Footnotes
The abbreviations used are: TNF, tumor necrosis factor; IL, interleukin; MEF, mouse embryonic
fibroblast; EGFR, epidermal growth factor receptor; PCR, polymerase chain reaction; EMSA,
electrophoretic mobility shift assay; uPA, urokinase-type plasminogen activator.
24
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Figure Legends
Figure 1. Conditioned media from metastatic human pancreatic cancer cell lines induce
NF-κB activation. EMSA was performed to determine the NF-κB activity in WT-MEF cells
stimulated by the conditioned media isolated from A, the human metastatic pancreatic cancer cell
lines ASPC-1 and MDAPanc-28 and B, the nonmetastatic pancreatic cancer cell line PANC-1 at
different time intervals as indicated. The TNFα-induced NF-κB activity in WT-MEF cells was
determined as a control (B, lanes 1-3). The Oct-1 DNA binding activities were determined as
loading controls. C, The nuclear extract from WT-MEF cells treated with MDAPanc-28-
conditioned medium was used for the competition and supershift assays as indicated. D, The
cytoplasmic extracts isolated from WT-MEF cells stimulated with the conditioned media in A
and B were used for western blot analysis for detecting the levels of IκBα proteins. NS, non-
specific bands; FP, free probe; CM, conditioned-medium.
Figure 2. IL-1α is the primary cytokine in the conditioned media from metastatic
pancreatic cancer cells that induced NF-κB activation. A, EMSA was performed to
determine the NF-κB activity in WT-MEF cells stimulated with anti-IL-1α, -IL-1β, -TNFα, or –
TNFβ antibodies (2 µg/ml for 2 h) neutralized conditioned media from ASPC-1 and MDAPanc-
28 cells for 30 min, as indicated. B, EMSA was performed to determine the NF-κB activity in
ASPC-1 and MDAPanc-28 cells treated with anti-IL-1α, -IL-1β, -TNFα, or -TNFβ neutralizing
antibodies (2 µg/ml) for 1 h, as indicated. C, EMSA was performed to determine the NF-κB
activity in WT-MEF cells treated with and without anti-IL-1RI antibody (2 µg/ml for 2 h), and
stimulated with the conditioned media from ASPC-1 and MDAPanc-28 cells for 30 min, as
25
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

indicated. D, EMSA was performed to determine the NF-κB activity in ASPC-1 and MDAPanc-
28 cells treated with and without anti-IL-1RI antibody (2 µg/ml for 2 h), as indicated. The Oct-1
DNA binding activities were determined as loading controls in both C and D.
Figure 3. IL-1α is differentially expressed in human pancreatic cancer cell lines. A,
Northern blot analysis was performed with the total RNA isolated from PANC-1, ASPC-1 and
MDAPanc-28 cells using either IL-1α or IL-1β cDNA as the probe as indicated. As a loading
control, 28S and 18S ribosomal RNA were visualized by ethidium staining. B, Western blot
analysis was performed to detect the levels of IL-1α and IL-1β using 50 µg and 200 µg whole
cell extracts, respectively, and conditioned media from PANC-1, ASPC-1, and MDAPanc-28
cells. The blots were re-probed with anti-β-actin antibody for loading controls, and the
conditioned media were collected in the same volume:cell number ratio, dialyzed, and
concentrated before the analysis.
Figure 4. The expression of the NF-κB downstream target gene uPA was inhibited by the
IL-1α neutralizing antibody. The levels of uPA expression in ASPC-1 and MDAPanc-28 cells
treated with or without anti-IL-1α-neutralizing antibody (2 µg/ml) for 1 h as indicated. The
levels of β-actin were determined as loading controls.
Figure 5. Inhibition of NF-κB alone is not sufficient to reduce the levels of IL-1α
expression and NF-κB-inducing activity in conditioned media. A, NF-κB activity in ASPC-
1/Puro, MDAPanc-28/Puro, ASPC-1/IκBαM, and MDAPanc-28/IκBαM cells were confirmed
by EMSA. B, The NF-κB-inducing activities in the conditioned media from ASPC-1/IκBαM
26
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

and MDAPanc-28/IκBαM cells were analyzed by EMSA using WT-MEF cells at various time
points as indicated. C, The levels of IκBα protein were determined by Western blot analysis
using cytoplasmic extracts isolated from WT-MEF cell stimulated by the conditioned media in
B. The levels of β-actin were determined with anti-β-actin antibody as loading control. D,
EMSA was performed to determine the NF-κB activity in WT-MEF cells stimulated with the
conditioned media from ASPC-1/IκBαM and MDAPanc-28/IκBαM cells treated with anti-IL-
1α, -IL-1β, -TNFα, or -TNFβ-neutralizing antibodies (2 µg/ml for 2h) for 30 min, as indicated.
The Oct-1 DNA binding activities were determined as loading controls.
Figure 6. Inhibition of NF-κB decreased but not completely blocked IL-1α expression and
secretion. A, Northern blot analysis of the levels of IL-1α and IL-1β expression in ASPC-
1/Puro, MDAPanc-28/Puro and ASPC-1/IκBαM, MDAPanc-28/IκBαM cells. As a loading
control, the levels of 28S and 18S ribosomal RNA were determined in these cells. B, Western
blot analysis of whole cell extracts and conditioned media from ASPC-1/Puro, MDAPanc-
28/Puro and ASPC-1/IκBαM, MDAPanc-28/IκBαM cells. 50 µg and 200 µg whole cell extracts
were used for IL-1α and IL-1β, respectively. The levels of β-actin were determined with an anti-
β-actin antibody as loading control. The conditioned media were collected in the same volume:
cell number ratio, dialyzed, and concentrated before the analysis as described in Experimental
Procedures.
Figure 7. Downregulation of EGFR activity reduced AP-1 activity and levels of IL-1α
expression. A, AP-1 and NF-κB activities were determined by EMSA in the nuclear extracts
isolated from ASPC-1 and MDAPanc-28 cells treated with and without an anti-EGFR antibody
27
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

(20 ng/ml) for 48 h. Oct-1 DNA activity was determined for the loading control. The levels of
IL-1α and β-actin expression were determined by Western blot analysis using cytoplasmic
extracts isolated from the same ASPC-1 and MDAPanc-28 cells used in EMSA as described in
Experimental Procedures. B, NF-κB and Oct-1 activities were analyzed by EMSA in the WT-
MEF cells stimulated with the conditioned media from ASPC-1 and MDAPanc-28 cells treated
with or without an anti-EGFR antibody (20 ng/ml) for 48 h. C, AP-1 and Oct-1 activities were
determined by EMSA in the nuclear extracts isolated from ASPC-1/IκBαM and MDAPanc-
28/IκBαM cells treated with and without an anti-EGFR antibody (20 ng/ml). The levels of IL-
1α and β-actin expression were determined by Western blot analysis using cytoplasmic extracts
isolated from the same ASPC-1/IκBαM and MDAPanc-28/IκBαM cells used in EMSA as
described in Experimental Procedures. D, NF-κB and Oct-1 activities were analyzed by EMSA
in the WT-MEF cells stimulated with the conditioned media from ASPC-1/IκBαM and
MDAPanc-28/IκBαM cells treated with or without an anti-EGFR antibody (20 ng/ml). E, AP-1
reporter gene assays. 1.0 µg AP-1 reporter gene constructs and the control p-TK Renilla
luciferase were cotransfected into Aspc-1 cells with 1.5 µg of expression vectors encoding
dominant-negative ELK-1, Ras, Akt, and MEKK-1 or with anti-EGFR antibody (C225) treatment
in 12-well plates. The experiments were repeated in triplicate. F, Panc-1 cells were kept in the
media with 0% serum for 48 h, then stimulated with 100ng/ml EGF for 18 h. The whole cell
lysates were used to detect IL-1α and β-actin expression. G, The conditioned medium from
Panc-1 cells in F treated with or without neutralizing IL-1α antibody (2µg/ml, 2h) were used to
stimulate wild-type MEF cells. Nuclear extracts were used for EMSA to determine the NF-κB
DNA binding activity as indicated.
28
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Figure 8. A novel transcription start site for human IL-1α was identified. A, Primer
extension analysis for determining the start site of human IL-1α transcription start site. The
primer sequence is indicated in B. Lane 1, molecular marker; lane 2-3, primer extension
products; lane 4-7, sequence for IL-1α genomic DNA using the same primer. B, Partial genomic
DNA sequence for human IL-1α ( NCBI accession number AF536338). Arrow indicates the
primer used for primer extension. The nucleotide sequence is numbered with the newly identified
transcription start site (position +1, in bold letter and an arrow). One of the two AP-1 sites was
boxed. The previously identified mRNA starting site is underlined (position +458).
Figure 9. IL-1α promoter is regulated by AP-1 activity. A, The fragment of newly identified
623 bp IL-1α promoter region was cloned into pGL2 Firefly luciferase reporter gene vector, the
two AP-1 sites were mutated as indicated by bold letter. The arrows indicate the orientation of
the AP-1 sites. B, EMSA was performed using MDAPanc-28 nuclear extracts with
oligonucleotides encoding the wild-type and mutant AP-1 sties in IL-1 promoter (AP1-1 and
AP1-2) as shown in A. The Fos supershift assays with and without competing peptide showed
the presence of c-Fos in the AP-1 DNA binding activity and the Oct-1 DNA binding activities
were determined as loading controls. C, Reporter gene analysis for IL-1α promoter. 1.0 µg
wild-type or mutant IL-1α reporter gene constructs were cotransfected with 1.5 µg of wild-type
c-fos, relA(p65) or dominant-negative c-fos into relA-/- cells in a 12-well plate using p-TK
Renilla luciferase as a control as indicated. The experiments were repeated in triplicate. The
activities of both Firefly and Renilla luciferase were determined using the Dual-Luciferase
Reporter Assay System (Promega). The luciferase activities were normalized to the Renilla
29
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

luciferase activity of the internal control. Data represent the mean ± standard error from three
different experiments performed in triplicate.
Figure 10. A working model for constitutive NF-κB activation in pancreatic cancer. In
metastatic pancreatic cancer cells, growth factors and/or other upstream signal cascades trigger
AP-1 activation, which in turn induces the expression of its downstream target genes such as IL-
1α. The IL-1α autocrine stimulation activates NF-κB, which further elevates the expression of
its downstream target gene, IL-1α, thus, resulting a positive feedback loop and causing NF-κB to
be constitutively activated. The constitutively activated NF-κB regulates its downstream target
genes, promoting pancreatic tumor progression and metastasis.
30
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Niu et al. Fig. 1
A
p65/p50
p50/p50
FP
WT-MEF
0Min: 15 30 60 120 0 15 30 60 120AsPc-1 CM MDAPanc-28 CM
*
*
*
1 2 3 4 5 6 7 8 9 10Oct-1
NS
WT-MEF
C
wt κBmut κBanti-p65p65 peptideanti-p50p50 peptide
++
+ +
+
+++
p65/p50p50/p50
1 2 3 4 5 6 7
D
IκBα
β-actin
WT-MEF
0Min: 15 30 60 120 15 30 60 120
AsPc-1 CM MDAPanc-28 CM
1 2 3 4 5 6 7 8 9
0 15 30 60 120
Panc-1 CM
10 11 12 13 14
BWT-MEF
p65/p50
p50/p50
0Min: 15 30 60 120
Panc-1CMTNF-α
Anti-TNF-α30
+300
1 2 3 4 5 6 7 8Oct-1
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Niu et al. Fig. 2
AWT-MEF
Aspc-1 CM MDAPanc28 CM
Time (min)anti-IL-1αanti-IL-1βanti-TNFα
anti-TNFβ
++
+
+
++
+
+
p65/p50
p50/p50
0 30 30 30 30 30 30 30 30 30 30
1 2 3 4 5 6 7 8 9 10 11
Oct-1
BAspc-1 MDAPanc28
anti-IL-1αanti-IL-1βanti-TNFα
anti-TNFβ
++
+
+
++
+
+
p65/p50
p50/p50
1 2 3 4 5 6 7 8 9 10Oct-1
C
MDAPanc28 CM D MDAPanc28
+ +
1 2 3 4
Aspc-1
anti-IL-1RI
WT-MEF
Aspc-1 CMTime (min)anti-IL-1RI + +
0 30 30 30 30
p65/p50
p50/p50
Oct-1
1 2 3 4 5
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Niu et al. Fig. 3
APanc1 Aspc1 MDAPanc28
IL-1-α
IL-1-β
1 2 3
28S
18S
B
Panc-1 Aspc-1 MDAPanc-28
IL-1αCell lysate
Cond. Med.
Cell lysate
Cell lysate β-actin
IL-1α
IL-1β
1 2 3
Niu et al. Fig. 4
uPA
β-actin
AsPc-1 MDAPanc-28
Anti-IL-1α + +
1 2 3 4
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Niu et al. Fig. 5
A
Aspc-1 MDAPanc28
p65/p50
p50/p50
Puro IκBαM Puro IκBαM
1 2 3 4Oct-1
B WT-MEF
Aspc-1/IκBαM CMMDAPanc-28/IκBαM CM
Time (min)
p65/p50
p50/p50
1 2 3 4 5 6 7 8 9
0 15 30 60 120 15 30 60 120
Oct-1
CWT-MEF
IκBα
Aspc-1/IκBαM CMMDAPanc-28/ IκBαM CM
Time (min) 0
β-actin
15 30 60 120 0 15 30 60 120
1 2 3 4 5 6 7 8 9 10
D
WT-MEF
Aspc-1/IκBαM CMMDAPanc28/IκBαM CM
Time (min)anti-IL-1αanti-IL-1βanti-TNFα
anti-TNFβ
++
+
+
++
+
+
p65/p50
p50/p50
0 30 30 30 30 30 30 30 30 30 30
1 2 3 4 5 6 7 8 9 10 11Oct-1
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Niu et al. Fig. 6
A
IL1-α
IL1-β
Puro IκBαM Puro IκBαMAsPc-1 MDAPanc-28
1 2 3 4
28S
18S
B
IL-1αCell lysate
Cond. Med.
Cell lysate
Cell lysate β-actin
IL-1α
IL-1β
1 2 3 4
Puro IκBαM Puro IκBαMAsPc-1 MDAPanc-28
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Niu et al. Fig. 7
Aspc-1 MDAPanc-28
C225 (20 ng/ml) - + +-
p65/p50
p50/p50
AP-1
A
IL-1αβ-actin
Oct-1
MDAPanc-28 CM
1 2 3 4
C225 (20 ng/ml)
p65/p50p50/p50
ASPC-1 CM
- +-+
MEF
Time (min) 0 30 30 30 30
Oct-1
B
1 2 3 4 5
C
AP-1
Oct-1
IL-1αβ-actin
Aspc-1/IκBαM
MDAPanc-28/IκBαM
- + +-C225 (20 ng/ml)
1 2 3 4
p65/p50
p50/p50
Oct-1
ASPC-1/IκBαM CM
MDA-Panc-28/IκBαM CM
C225 (20 ng/ml) - +-
MEF
Time 0 30 30 30 30+
D
1 2 3 4 5
Panc-1
EGF (100ng/ml) - +
IL-1α
β-actin
1 2
E
1 2 3 4
p65/p50p50/p50
Oct-1
GMEF
Panc-1 CM
EGF (100 ng/ml) - +Time 0 30 30 30
+α-IL-1α +- -
Rel
ativ
e Lu
cife
rase
Ass
ay
AP-1-Luc
D.N. ELK-1
MEKK-1/K432A
RsN17Akt K179M
+ + + + +
+
+
++
F
0
10
20
30
40
50
60
70
80
+
C225(20ng/ml) +
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Niu etal. Fig. 8
A
MD
APan
c-28
72bp
M ASPC
-1
A C G T
TTTTCAACGGT
AAAAGTTGCCA
→
1 2 3 4 5 6 7
-151 aggatcaaga cttctttgtg ctcaaatacc actgttctct tctctaccct –101 gccctaacca ggagcttgtc accccaaact ctgaggtgat ttatgcctta –51 atcaagcaaa cttccctctt cagaaaagat ggctcatttt ccctcaaaag –1 ttgccaggag ctgccaagta ttctgccaat tcaccctgga gcacaatcaa +50 caaattcagc cagaacacaa ctacagctac tattagaact attattatta+100 ataaattcct ctccaaatct agccccttga cttcggattt cacgatttct +150 cccttcctcc tagaaacttg ataagtttcc cgcgcttccc tttttctaag +200 actacatgtt tgtcatctta taaagcaaag gggtgaataa atgaaccaaa+250 tcaataactt ctggaatatc tgcaaacaac aataatatca gctatgccat +300 ctttcactat tttagccagt atcgagttga atgaacatag aaaaatacaa +350 aactgaattc ttccctgtaa attccccgtt ttgacgacgc acttgtagcc +400 acgtagccac gcctacttaa gacaattaca aaaggcgaag aagactgact +450 caggcttaag ctgccagcca gagagggagt catttcattg
B by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Niu et al. Fig. 9
IL-1αP-PGL2
Luc
-547 +76AP-1 κB AP-1-366 -293 -23
--tggctca----tggcaca--
--aacaggt----aatcggt--
MAP1-1 MAP1-2
MAP1-12A
B
AP-1
Oct-1
AP1-1 AP1-2
wt mut
α−c-fos α−c-fos+ pep
wt mut
α−c-fos α−c-fos+ pep
PGL-2IL-1αP-PGL2 c-fosD.N. c-fosp65
Fold
Indu
ctio
n
++ ++
+
+
++
MAP1-1MAP1-2MAP1-12
++
+
C
0
100
200
300
400
500
+
+
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Niu et al. Fig. 10
Growth Factors and other upstream signals
AP-1
IL-1α
Constitutively activated NF-κB
Secr
ete
uPA, other genes
Pancreatic tumor progression
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Jiangong Niu, Zhongkui Li, Bailu Peng and Paul J. Chiaoinduction of constitutive NF-kappaB activation in pancreatic cancer cells
Identification of an auto-regulatory feedback pathway involving IL-1alpha in
published online December 16, 2003J. Biol. Chem.
10.1074/jbc.M309789200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on August 30, 2018
http://ww
w.jbc.org/
Dow
nloaded from


















