Glucose Orientation and Dynamics in α-, β-, and γ-Cyclodextrins
7
Glucose Orientation and Dynamics in r-, -, and γ-Cyclodextrins Kevin J. Naidoo,* ,† M. Riedaa Gamieldien, † Jeff Yu-Jen Chen, † Go ¨ran Widmalm, ‡ and Arnold Maliniak § Department of Chemistry, UniVersity of Cape Town, Rondebosch, 7701, South Africa., Department of Organic Chemistry, Arrhenius Laboratory, Stockholm UniVersity, S-106 91 Stockholm, Sweden., DiVision of Physical Chemistry Arrhenius Laboratory, Stockholm UniVersity, S-106 91 Stockholm, Sweden ReceiVed: June 12, 2008; ReVised Manuscript ReceiVed: September 11, 2008 We investigate, using molecular dynamics (MD) computer simulations, the conformational behavior of R-, -, and γ-cyclodextrins (CDs). Our analysis of a 30 ns trajectory of CD solution dynamics reveals the underlying conformational behaviours of the CDs that explain their relative flexibility. The distributions of the torsion angles related to the glycosidic linkages, P(φ,ψ) were calculated for the three CDs. Most noticeable is the limited range in φ torsion rotations compared with ψ rotations for all the CDs. This difference between the three CDs is amplified in the motion and dynamics of their glucose monomers when we monitor their orientational and librational positions relative to the macrocyclic mean plane. The relaxation times of the monomers to their equilibrium orientations follow the pattern γ-CD >R-CD > -CD. The root-mean-square deviations of the motion of the monomer centers of mass from the mean macrocyclic planes exhibit the same trend. Introduction Cyclodextrins(CDs) are cyclic R-(1f4)-linked carbohydrate oligomers constructed from glucose units. Three of the most important nonmodified cyclodextrins are the R-, -, and γ-CDs (see Figure 1) which comprise 6, 7, and 8 units of a glucopy- ranose monomer, respectively. 1,2 Cyclodextrins are produced from starch materials, and due to their unique structural features these molecules are invaluable in a variety of industrial applications. The most important industrial sectors include pharmaceuticals, agricultural chemi- cals, cosmetics, and foods. Almost all applications involve the ability of CDs to alter the physical, chemical, and biological properties of guest molecules, through the formation of inclusion complexes. This effect is achieved by the encapsulation of guest molecules into the cones of the CDs. Small and larger aromatic substances complex best with - and γ-CDs, respectively, whereas aliphatic chains are more suited to R-CDs. The choice of CD used in drug delivery depends on requirements of the dosage form, the route of delivery, and the solubilizing capacity required for handling the drug load. A vast amount of pharmaceuticals consist of low molecular weight aromatic moieties that are too large to fit into the R-CD cavity and while they do fit into the of γ-CD cavity it is often economically not viable. Therefore, -CD has been traditionally identified as the preferred drug carrier. However, an inhibiting property of -CD has been its relatively low solubility in water (18.8 mg/mL at 25 °C) compared with R-CD (145.0 mg/mL at 25 °C) and γ-CD (232.0 mg/mL at 25 °C). 3 Consequently, although -CD is best suited in most cases, due to its cavity size its low intrinsic solubility in water makes it impractical to use. Because of commercial interest as well as the puzzling anomalous solubility trend of R-, -, and γ-CDs, investigations into the underlying molecular reasons for these continue to attract substantial scientific attention. 4,5 In general, CDs are only able to adopt a limited number of conformations in the aqueous medium due to the restrictions imposed on the rotation of sugar units about the interglycosidic bonds. The allowed conformations of CDs are also dependent on the intra- and intermolecular forces. Thus, the most stable conformations of these cyclic oligomers will be those that balance these molecular interactions. More than 20 years ago, Koehler et al. published a series of articles, using computational methods, addressing the phenom- ena of the unexpectedly low solubility of -CD. 6 They in- vestigated the characteristics of asymmetry motion of the macrocycles, mobility of the water molecules, and the distribu- tion of a variety of CD structural geometries. 6 More recently, other computational investigations of the hydration effects and conformational properties of the CDs were reported. 4,7 We reported earlier that the order of water structure around the -CD is higher than in the case of the other two cycloamyloses and the differential solute solvent interactions of these three cyclic sugars may be due to differences in their conformational be- havior. 8 The conformations of CDs can be analyzed on the molecular and monomeric length scales as illustrated in Figure 2. Studies of cyclodextrin conformation generally focus only on the molecular length scales and the associated variables that govern the CD shapes. The most important of these are the rotations about the glycosidic bonds (the top row middle il- lustration in Figure 2) and the hydroxyl groups (top row last illustration in Figure 2). At the monomeric length scale the vertical and angular motion of the glucose rings relative to the overall macrocyclic ring are the two fundamental variables that fully describe the monomer contributions to the overall CD conformations. Here we set out to address the underlying mole- cular and monomeric conformational reasons for the relative inflexibility of -CD. The conformational behavior of the three CDs in water was investigated using a 30 ns trajectory generated from a molecular dynamics (MD) computer simulation. * To whom correspondence should be addressed. E-mail: Kevin.Naidoo@ uct.ac.za. Fax: +27-21-686-4333. † University of Cape Town. ‡ Department of Organic Chemistry, Stockholm University. § Division of Physical Chemistry, Stockholm University. J. Phys. Chem. B 2008, 112, 15151–15157 15151 10.1021/jp805174y CCC: $40.75 2008 American Chemical Society Published on Web 11/01/2008
Transcript of Glucose Orientation and Dynamics in α-, β-, and γ-Cyclodextrins

Glucose Orientation and Dynamics in r-, �-, and γ-Cyclodextrins
Kevin J. Naidoo,*,† M. Riedaa Gamieldien,† Jeff Yu-Jen Chen,† Goran Widmalm,‡ andArnold Maliniak§
Department of Chemistry, UniVersity of Cape Town, Rondebosch, 7701, South Africa., Department of OrganicChemistry, Arrhenius Laboratory, Stockholm UniVersity, S-106 91 Stockholm, Sweden., DiVision of PhysicalChemistry Arrhenius Laboratory, Stockholm UniVersity, S-106 91 Stockholm, Sweden
ReceiVed: June 12, 2008; ReVised Manuscript ReceiVed: September 11, 2008
We investigate, using molecular dynamics (MD) computer simulations, the conformational behavior of R-,�-, and γ-cyclodextrins (CDs). Our analysis of a 30 ns trajectory of CD solution dynamics reveals the underlyingconformational behaviours of the CDs that explain their relative flexibility. The distributions of the torsionangles related to the glycosidic linkages, P(φ,ψ) were calculated for the three CDs. Most noticeable is thelimited range in φ torsion rotations compared with ψ rotations for all the CDs. This difference between thethree CDs is amplified in the motion and dynamics of their glucose monomers when we monitor theirorientational and librational positions relative to the macrocyclic mean plane. The relaxation times of themonomers to their equilibrium orientations follow the pattern γ-CD > R-CD > �-CD. The root-mean-squaredeviations of the motion of the monomer centers of mass from the mean macrocyclic planes exhibit the sametrend.
Introduction
Cyclodextrins(CDs) are cyclic R-(1f4)-linked carbohydrateoligomers constructed from glucose units. Three of the mostimportant nonmodified cyclodextrins are the R-, �-, and γ-CDs(see Figure 1) which comprise 6, 7, and 8 units of a glucopy-ranose monomer, respectively.1,2
Cyclodextrins are produced from starch materials, and dueto their unique structural features these molecules are invaluablein a variety of industrial applications. The most importantindustrial sectors include pharmaceuticals, agricultural chemi-cals, cosmetics, and foods. Almost all applications involve theability of CDs to alter the physical, chemical, and biologicalproperties of guest molecules, through the formation of inclusioncomplexes. This effect is achieved by the encapsulation of guestmolecules into the cones of the CDs. Small and larger aromaticsubstances complex best with �- and γ-CDs, respectively,whereas aliphatic chains are more suited to R-CDs.
The choice of CD used in drug delivery depends onrequirements of the dosage form, the route of delivery, and thesolubilizing capacity required for handling the drug load. A vastamount of pharmaceuticals consist of low molecular weightaromatic moieties that are too large to fit into the R-CD cavityand while they do fit into the of γ-CD cavity it is ofteneconomically not viable. Therefore, �-CD has been traditionallyidentified as the preferred drug carrier. However, an inhibitingproperty of �-CD has been its relatively low solubility in water(18.8 mg/mL at 25 °C) compared with R-CD (145.0 mg/mL at25 °C) and γ-CD (232.0 mg/mL at 25 °C).3 Consequently,although �-CD is best suited in most cases, due to its cavitysize its low intrinsic solubility in water makes it impractical touse. Because of commercial interest as well as the puzzlinganomalous solubility trend of R-, �-, and γ-CDs, investigations
into the underlying molecular reasons for these continue toattract substantial scientific attention.4,5
In general, CDs are only able to adopt a limited number ofconformations in the aqueous medium due to the restrictionsimposed on the rotation of sugar units about the interglycosidicbonds. The allowed conformations of CDs are also dependenton the intra- and intermolecular forces. Thus, the most stableconformations of these cyclic oligomers will be those thatbalance these molecular interactions.
More than 20 years ago, Koehler et al. published a series ofarticles, using computational methods, addressing the phenom-ena of the unexpectedly low solubility of �-CD.6 They in-vestigated the characteristics of asymmetry motion of themacrocycles, mobility of the water molecules, and the distribu-tion of a variety of CD structural geometries.6 More recently,other computational investigations of the hydration effects andconformational properties of the CDs were reported.4,7 Wereported earlier that the order of water structure around the �-CDis higher than in the case of the other two cycloamyloses andthe differential solute solvent interactions of these three cyclicsugars may be due to differences in their conformational be-havior.8 The conformations of CDs can be analyzed on themolecular and monomeric length scales as illustrated in Figure2. Studies of cyclodextrin conformation generally focus onlyon the molecular length scales and the associated variables thatgovern the CD shapes. The most important of these are therotations about the glycosidic bonds (the top row middle il-lustration in Figure 2) and the hydroxyl groups (top row lastillustration in Figure 2). At the monomeric length scale thevertical and angular motion of the glucose rings relative to theoverall macrocyclic ring are the two fundamental variables thatfully describe the monomer contributions to the overall CDconformations. Here we set out to address the underlying mole-cular and monomeric conformational reasons for the relativeinflexibility of �-CD. The conformational behavior of the threeCDs in water was investigated using a 30 ns trajectory generatedfrom a molecular dynamics (MD) computer simulation.
* To whom correspondence should be addressed. E-mail: [email protected]. Fax: +27-21-686-4333.
† University of Cape Town.‡ Department of Organic Chemistry, Stockholm University.§ Division of Physical Chemistry, Stockholm University.
J. Phys. Chem. B 2008, 112, 15151–15157 15151
10.1021/jp805174y CCC: $40.75 2008 American Chemical SocietyPublished on Web 11/01/2008

Computer Simulations
Atom names follow the convention for glucose, prefixed bythe residue number. The torsion angles are defined by φxy,xH1-xC1-yO4-yC4 and ψxy, xC1-yO4-yC4-yH4 for theR(1f4)-linkages where x and y identify the atoms on adjacentglucose monomers respectively.
We follow the same computational procedure as previouslyreported;8 consequently here we only summarize the main fea-tures of the MD simulations.
The program CHARMM was used for all the molecularmechanics computations9 and a carbohydrate solution force field(CSFF)10 was used to model the CDs. The SPCE water model11
implemented with periodic boundary conditions was used todescribe water interactions. The MD simulations were run for
500 ps to equilibrate the system and extended to 30 ns for datacollection using a 1 fs integration time step with the bondsinvolving hydrogens fixed via the restraint algorithm SHAKE.12
All solution simulations were run using an isothermal-isobaricensemble (NPT) with P ) 1 bar and T ) 300 K. Configurationsof the molecules were stored at intervals of 0.5 ps in allsimulations and analyzed over the entire 30 ns trajectory.
Results and Discussion
In this paper, we assess the connection between the molecularand monomer degrees of freedom, illustrated in Figure 2, andthe flexibilities of R-, �-, and γ-CD. The principal molecularrotors affecting the internal motion of a cyclic system such asthe CDs in an aqueous media are rotations about glycosidicbonds linking the glucose monomeric units and the rotation ofthe exocyclic hydroxyl groups. These motions determine therelative orientations of the glucopyranose monomer units.Since the exocyclic groups found on each CD are identi-cal, the three CDs should exhibit analogous characteristics.Therefore, the differences between the rms fits among them,as shown by us previously,8 should be owing to their dif-ferences in conformational flexibility.
Glycosidic Linkage Analysis. In linear saccharides, the majordegrees of freedom are related to bond-rotations at the glycosidictorsion angles φ and ψ defined as H1′-C1′-O4-C4 andC1′-O4-C4-H4 that report the flexibility about the two bonds(C1′-O4 and O4-C4) connecting the component glucosemonomers. These rotations are significantly restricted in mac-rocyclic compounds such as the cyclodextrins. A powerful NMRtechnique for investigation of molecular conformations is themeasurement of scalar spin-spin 3JCH couplings. The couplingsrelated to the φ torsion angle, Jφ, are identical for the threemolecules, whereas Jψ values increase as a function of themacrocyclic size. In contrast to the NMR experiments, computersimulations provide direct information about the average mo-lecular conformations, i.e., φ and ψ torsion angles.
From the NMR coupling constants the CDs can be character-ized by a single conformational state and no major conforma-tional difference between the molecules can be observed. Boththe averages and the standard deviations are similar for all threeCDs.13 The CD torsion angles can be translated to scalar 3JCH
coupling constants using a Karplus-type relationship14
Figure 1. Schematic of R- �- and γ-cyclodextrins (CDs) containing 6, 7 and 8 glucose monomers, respectively. The torsion angles φ and ψ aredefined by the following atoms H1′-C1′-O4-C4 and C1′-O4-C4-H4, respectively.
Figure 2. Conformational levels of abstraction. The top row showsthe primary molecular rotors that affect the CD conformation. Thebottom row shows the fundamental monomeric motion that mostinfluences the CD macrocycle flexibility.
TABLE 1: Long-Range trans-Glycosidic 1H, 13C ScalarCoupling Constants Reported in Hz for the CyclodextrinsWhere Experimental Values Are Taken from ref 13
molecule
experimental simulation
Jφ Jψ Jφ Jψ
R-CD 5.2 5.0 6.4 6.4�-CD 5.2 5.2 6.6 6.6γ-CD 5.2 5.4 6.6 6.6
3JCOCH ) 7.49 cos2κ - 0.96 cos κ + 0.15 (1)
15152 J. Phys. Chem. B, Vol. 112, No. 47, 2008 Naidoo et al.
where κ is the torsion angle (φ or ψ). The couplings calculatedfrom the trajectory are included in Table 1. The agreementbetween the simulated and experimental values is satisfactory.In particular, the trends for the three CDs from our computersimulations correlate with the NMR experimental results13
indicating that the simulations are of sufficient length and theCSFF force field10 is accurate enough to investigate the detailsof R-, �-, and γ-CD conformational space.
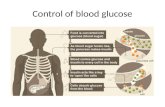
We analyze the glycosidic linkages of the series of CDs interms of the first primary molecular conformational variable(φ,ψ) as shown in Figure 2. The average φ and ψ torsion anglesfor all three CDs are close to zero degrees which significantlydeviates from the average conformation of a maltose solution15
(φ ) -47.5° and ψ ) -35.0°). However, the probabilitydistribution P(φ,ψ) (Figure 3a-c) reveals much more about theconformational space explored by each CD and the extent oftheir relative conformational mobility. The entire P(φ,ψ) isshown in contours of 10% where the innermost concentricrepresents the smallest conformational deviation from the ave-rage φ and ψ torsion angles about zero degrees and is thereforethe most probable population occurrence. It is immediatelynoticeable that �-CD samples a region of φ,ψ space that iscentered about the (-8, 4°) most probable conformation (Figure3b) compared with the two most probable conformations (-15,
-14°) and (-17, 37°) for R-CD and (-16, -13°) and (9, 17°)for γ-CD (Figure 3b,c, respectively). These compare reasonablywell with the average φ,ψ values observed in crystal structuresof (-10.2, 7.6°) for �-CD, (-10.8, 8.8°) for R-CD and (-11.1°,7.1°) for γ-CD.16 In Figure 3, we note that the �-CD distribution(Figure 3b) exhibits only one minimum, whereas the other two(Figures 3a,c) indicate two states or possibly plateaux. This isin agreement with experimental NMR (NOEs, J- and dipolarcouplings) results obtained for the R-CD.13 In fact, the NOEsand J-couplings determined in ref 13 were not consistent witha single conformational state, i.e., a distribution function withat least two states was required.
This difference can be better contextualized when we describethe results in terms of a fundamental repeat unit of the CDsnamely the disaccharide maltose (see Figure 2). The glycosidiclinkage free energy landscape is a primary metric used todescribe the conformational preferences of oligosaccharides.Previously, we calculated the free energy/potential of mean force(PMF) for maltose as a function of φ and ψ torsion angles15
The PMF surface for maltose in explicit aqueous solutionwas calculated using an iterative adaptive umbrella samplingmethod,17 as implemented by us in CHARMM,18 combined witha weighted histogram analysis method.10,19 Here we show asection of the (φ,ψ) free energy surface, contoured at 2 kcalmol-1 (Figure 4), that is relevant to CD solution dynamics atambient temperatures. We mark on the surface the furthestsampled points (W, X, Y, and Z) seen in R- and γ-CD as theysample the largest φ,ψ surface. The �-CD samples a band ofsyn conformations along the X (0.4 kcal mol-1) T Y (6.0 kcalmol-1) leg and remains well within an energetic envelope of 6kcal mol-1 about the global minimum for maltose. The gly-cosidic rotations in R-CD explores a similar area along the XT Y line but in addition it expands along the Y (6.0 kcal mol-1)T Z (9.2 kcal mol-1) path almost reaching the conformationaltransition state conformation leading to the anti conformation.The γ-CD also has low conformational strain on its componentdimer glycosidic linkages since it samples conformations wellwithin a 7 kcal mol-1 energetic envelope about the maltose freeenergy global minimum.
Notably, the variation in the highest probability distributions(inner contours) for the three CDs indicate that there is
Figure 3. The normalized probability densities P(φ,ψ) for (a) R-, (b) �-, and (c) γ-CDs calculated over the 30 ns trajectory. The probabilities arecontoured at probability density intervals of 10% with the outer concentric indicating a 90% probability density. The area bounded by the pointsW, X, Y, and Z marks the maximum range of the probability density seen for R- and γ-CD.
Figure 4. The maltose glycosidic linkage potential of mean force(PMF) as a function of the φ,ψ torsion angles. The minimum andmaximum are at 0 and 20 kcal mol-1, respectively. The plot is contouredat 2 kcal mol-1. The area bounded by the points W, X, Y, and Z markthe maximum range of the glycosidic φ,ψ torsion angles for R- andγ-CD that is shown in Figure 3c.
W(φ, ψ) ) -kT ln P(φ, ψ) (2)
Cyclodextrin Conformational Flexibility. J. Phys. Chem. B, Vol. 112, No. 47, 2008 15153
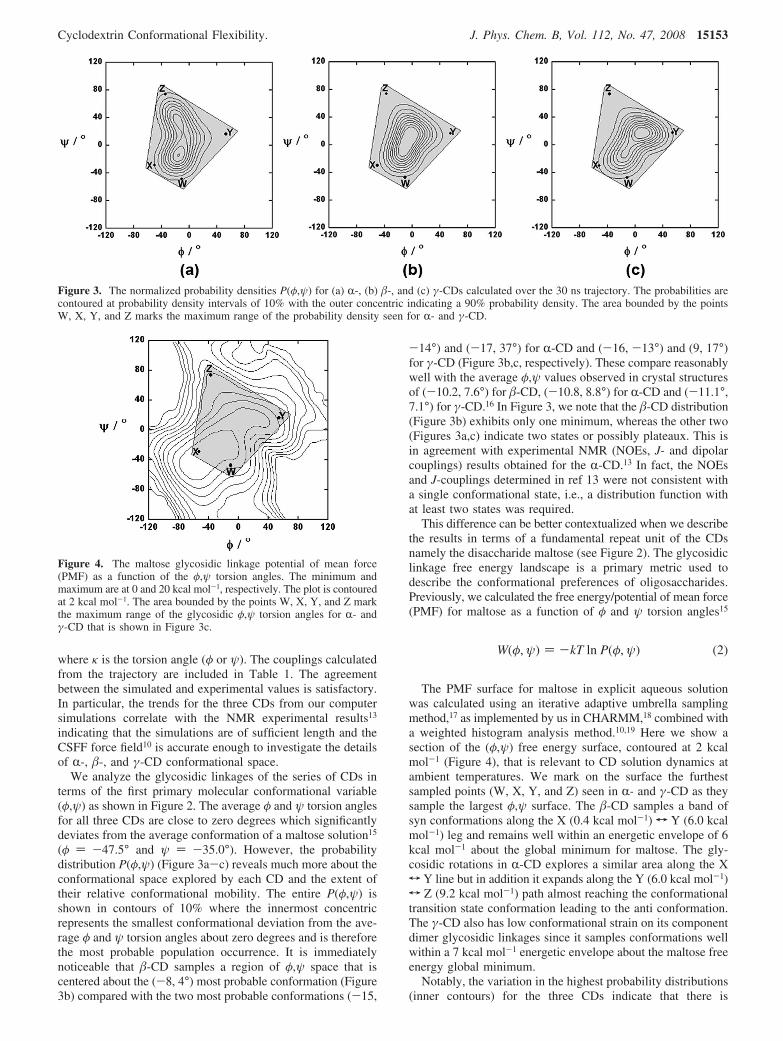
significantly more deviation in the ψ than the φ torsion angles.Although the CD molecules are strained, the macrocyclicrestriction on the component maltose dimers still allows themto fluctuate on the periphery of their primary equilibrium energywell. The time scale for these fluctuations is defined by the timecorrelation functions (TCFs) calculated for the two torsion anglesψ and φ, and displayed in Figure 5. All the TCFs exhibit verysimilar behavior and the characteristic correlation times for thesedynamical processes are of the order of 0.1-0.2 ns. We observehowever that Cφ(t) decay in the order τγ > τR ) τ�, where τ isthe characteristic correlation time, and the corresponding orderfor Cψ(t) is τγ > τR > τ�. This could be the most important factorcontributing to the relative internal dynamics of �-CD whencompared to R- and γ-CD. The motion and distribution of theφ and ψ torsions therefore indicate that the relative flexibilityof the CDs originates mostly from the ψ torsion angle andprovides a partial explanation of the rms TCF trends describingthe relative rigidity of the CD structures observed in our previouswork.8
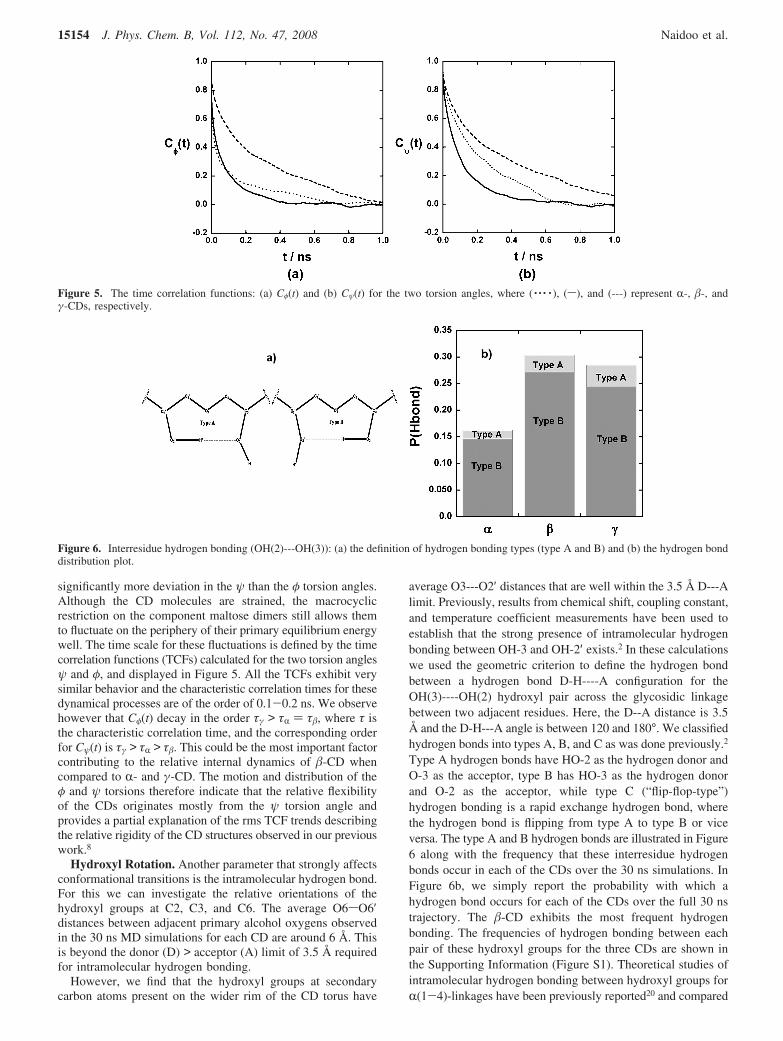
Hydroxyl Rotation. Another parameter that strongly affectsconformational transitions is the intramolecular hydrogen bond.For this we can investigate the relative orientations of thehydroxyl groups at C2, C3, and C6. The average O6sO6′distances between adjacent primary alcohol oxygens observedin the 30 ns MD simulations for each CD are around 6 Å. Thisis beyond the donor (D) > acceptor (A) limit of 3.5 Å requiredfor intramolecular hydrogen bonding.
However, we find that the hydroxyl groups at secondarycarbon atoms present on the wider rim of the CD torus have
average O3---O2′ distances that are well within the 3.5 Å D---Alimit. Previously, results from chemical shift, coupling constant,and temperature coefficient measurements have been used toestablish that the strong presence of intramolecular hydrogenbonding between OH-3 and OH-2′ exists.2 In these calculationswe used the geometric criterion to define the hydrogen bondbetween a hydrogen bond D-H----A configuration for theOH(3)----OH(2) hydroxyl pair across the glycosidic linkagebetween two adjacent residues. Here, the D--A distance is 3.5Å and the D-H---A angle is between 120 and 180°. We classifiedhydrogen bonds into types A, B, and C as was done previously.2
Type A hydrogen bonds have HO-2 as the hydrogen donor andO-3 as the acceptor, type B has HO-3 as the hydrogen donorand O-2 as the acceptor, while type C (“flip-flop-type”)hydrogen bonding is a rapid exchange hydrogen bond, wherethe hydrogen bond is flipping from type A to type B or viceversa. The type A and B hydrogen bonds are illustrated in Figure6 along with the frequency that these interresidue hydrogenbonds occur in each of the CDs over the 30 ns simulations. InFigure 6b, we simply report the probability with which ahydrogen bond occurs for each of the CDs over the full 30 nstrajectory. The �-CD exhibits the most frequent hydrogenbonding. The frequencies of hydrogen bonding between eachpair of these hydroxyl groups for the three CDs are shown inthe Supporting Information (Figure S1). Theoretical studies ofintramolecular hydrogen bonding between hydroxyl groups forR(1-4)-linkages have been previously reported20 and compared
Figure 5. The time correlation functions: (a) Cφ(t) and (b) Cψ(t) for the two torsion angles, where ( · · · · ), (s), and (---) represent R-, �-, andγ-CDs, respectively.
Figure 6. Interresidue hydrogen bonding (OH(2)---OH(3)): (a) the definition of hydrogen bonding types (type A and B) and (b) the hydrogen bonddistribution plot.
15154 J. Phys. Chem. B, Vol. 112, No. 47, 2008 Naidoo et al.
the competing intermolecular hydrogen bonds that are formedbetween the hydroxyl groups at C2′ and C3 and individual watermolecules.21
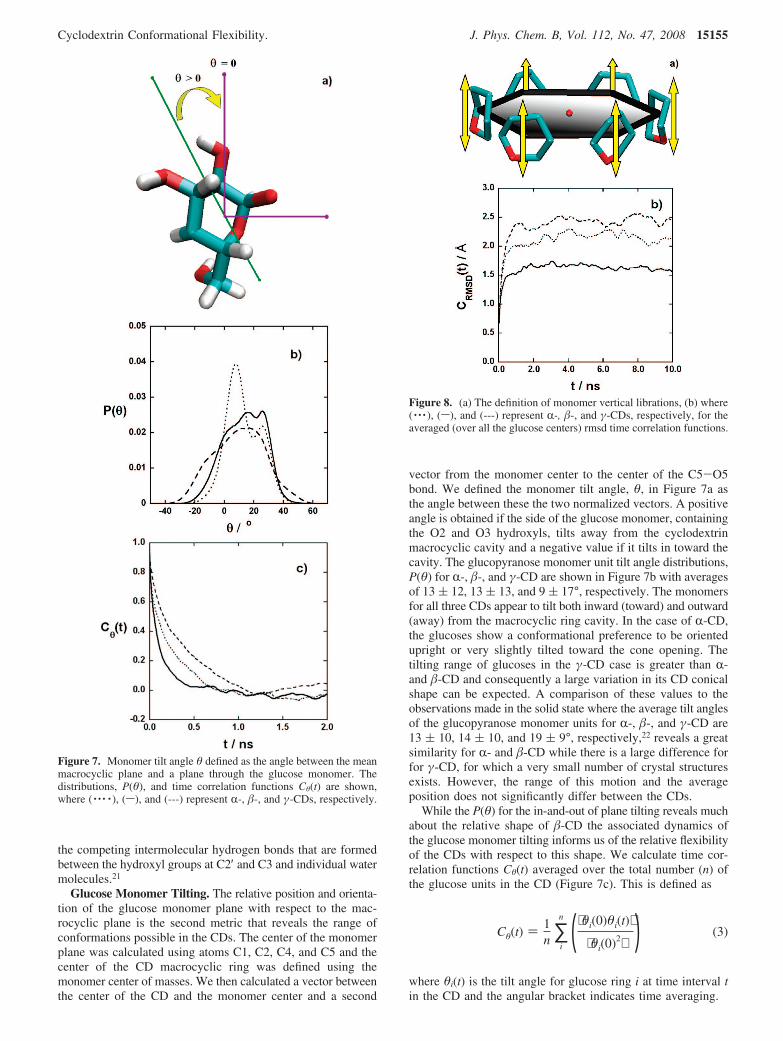
Glucose Monomer Tilting. The relative position and orienta-tion of the glucose monomer plane with respect to the mac-rocyclic plane is the second metric that reveals the range ofconformations possible in the CDs. The center of the monomerplane was calculated using atoms C1, C2, C4, and C5 and thecenter of the CD macrocyclic ring was defined using themonomer center of masses. We then calculated a vector betweenthe center of the CD and the monomer center and a second
vector from the monomer center to the center of the C5-O5bond. We defined the monomer tilt angle, θ, in Figure 7a asthe angle between these the two normalized vectors. A positiveangle is obtained if the side of the glucose monomer, containingthe O2 and O3 hydroxyls, tilts away from the cyclodextrinmacrocyclic cavity and a negative value if it tilts in toward thecavity. The glucopyranose monomer unit tilt angle distributions,P(θ) for R-, �-, and γ-CD are shown in Figure 7b with averagesof 13 ( 12, 13 ( 13, and 9 ( 17°, respectively. The monomersfor all three CDs appear to tilt both inward (toward) and outward(away) from the macrocyclic ring cavity. In the case of R-CD,the glucoses show a conformational preference to be orientedupright or very slightly tilted toward the cone opening. Thetilting range of glucoses in the γ-CD case is greater than R-and �-CD and consequently a large variation in its CD conicalshape can be expected. A comparison of these values to theobservations made in the solid state where the average tilt anglesof the glucopyranose monomer units for R-, �-, and γ-CD are13 ( 10, 14 ( 10, and 19 ( 9°, respectively,22 reveals a greatsimilarity for R- and �-CD while there is a large difference forfor γ-CD, for which a very small number of crystal structuresexists. However, the range of this motion and the averageposition does not significantly differ between the CDs.
While the P(θ) for the in-and-out of plane tilting reveals muchabout the relative shape of �-CD the associated dynamics ofthe glucose monomer tilting informs us of the relative flexibilityof the CDs with respect to this shape. We calculate time cor-relation functions Cθ(t) averaged over the total number (n) ofthe glucose units in the CD (Figure 7c). This is defined as
where θi(t) is the tilt angle for glucose ring i at time interval tin the CD and the angular bracket indicates time averaging.
Figure 7. Monomer tilt angle θ defined as the angle between the meanmacrocyclic plane and a plane through the glucose monomer. Thedistributions, P(θ), and time correlation functions Cθ(t) are shown,where ( · · · · ), (s), and (---) represent R-, �-, and γ-CDs, respectively.
Figure 8. (a) The definition of monomer vertical librations, (b) where( · · · ), (s), and (---) represent R-, �-, and γ-CDs, respectively, for theaveraged (over all the glucose centers) rmsd time correlation functions.
Cθ(t) ) 1n ∑
i
n (⟨θi(0)θi(t)⟩
⟨θi(0)2⟩ ) (3)
Cyclodextrin Conformational Flexibility. J. Phys. Chem. B, Vol. 112, No. 47, 2008 15155

By fitting the TCFs to single exponentials, we obtaincorrelation times of 0.2, 0.1, and 0.4 ns for R-, �-, and γ-CD.The relaxation time of �-CD for the glucose tilting motion istherefore shorter than that of R- and γ-CD, which is inagreement with the argument that it is conformationally the leastflexible. These TCFs decay in the same order for the three CDsas they do for the Cψ(t), which is expected since the ψ torsionangle is the fundamental molecular cause for the differences inthe CD glucose monomers tilting toward and away from theirmacrocyclic cones.
Glucose Monomer Vertical Librations. A general approachto investigate the internal motion of the cyclodextrins in solutionis to calculate a TCF, say Crms(t) ) ⟨rms(0,t)⟩ . Here, rms(t1,t2)denotes the best root-mean-square fit obtained between structuresat times t1 and t2 using a standard fitting procedure. Prior tocalculation of the rms fit, the orientational and translationaldynamics of the CDs should be removed. These TCFs werecalculated for all three CDs in our previous communication.8
Initially a perfect correlation between the molecular structuresis observed, and therefore Crms(0) ) ⟨rms(0,0)⟩ ) 0. In ourinterpretation,8 the long time limit of the TCF corresponds to ageneralized amplitude of the internal motion, whereas the timeto reach the plateau reflects the relaxation process of themacrocyclic ring toward the equilibrium structure. The rms fitTCF for the �-CD indicates the most restricted macrocyclic ringmotion and relaxes significantly faster than the other CDs. Inthat study we observed rms fits for each macromolecule in itsentirety that implied an inflexibility pattern, which follows thesolubility trend for the three CDs.
We now consider trajectories of the glucose monomer centersof masses in relation to the mean macrocyclic ring plane in thelaboratory frame (i.e., removing translation and orientationmotion). Every structure 0.05 ps apart along the trajectory forthe full 30 ns was superimposed onto a macrocyclic ringreference plane. This was constructed using the macrocyclicgeometric center (red dot in Figure 8a) and all the glucosecenters of mass. The vertical displacement (δ) of the center ofmass of each glucose monomer in the CD ring relative to themean macrocyclic ring plane is illustrated in Figure 8a. The δfor each glucose monomer was then calculated for the wholetrajectory. A positive value was assigned if a glucose monomerwas found to have a positive z coordinate and likewise anegative value for the opposite case.
We then calculated the root mean square deviation (rmsd)monomer displacement (δ) time correlation function Crmsdδ(t)averaged over the total number (n) of monomer vertical lib-rations in each CD. This is defined as
where δrmsd(i)(t) is the rmsd of the center of mass displacementfor glucose ring i at time interval t in the CD.
The rmsd TCFs for these vertical librations, Crmsdδ(t), aredisplayed in Figure 8b. These TCFs approximate an exponentialfunctional form where at the start there is perfect correlationbetween the molecular structures with δrmsd(0,0) ) 0. After this,the difference between the structures increase and reach a plateauin the long time limit. Two types of information can be derivedfrom the rmsd TCFs: (i) a value of Crmsdδ(t) corresponding tothe plateau, and (ii) a characteristic (relaxation) time requiredto reach the plateau. The value of the displacement read fromthe plateau can be interpreted as a generalized amplitude of the
monomer motion, whereas the time to reach the plateau reflectsthe relaxation process of the glucose monomer unit towardthe average distance from the mean macrocyclic ring plane. Theaverage levels of the plateau are 2.2, 1.6, and 2.4 Å for R-, �-,and γ-CDs, respectively, while the estimated relaxation timesare 1.4, 1.0, and 1.7 ns for R-, �-, and γ-CDs, respectively. Wesee from this that �-CD undergoes the most restricted macro-cyclic ring motion and relaxes significantly faster than the otherCDs. Furthermore, there are much less fluctuations in Crmsdδ(t)compared with R- and γ-CD. This leads us to conclude that themonomers in �-CD undergo small amplitude fast verticallibrations when reaching their librational equilibrium.
Conclusions
We have studied aqueous solutions of R-, �-, and γ-CDs usingMD computer simulations. The agreement between experimentaland calculated spin-spin couplings is satisfactory and wetherefore conclude that the conformational behavior of the CDsis adequately described in our computer simulations. The generalstructural behavior, monitored by the time correlation function,Crmsd(t) indicated that �-CD is more rigid than the other CDs.A possible molecular origin for this observation is the differ-ences in their glycosidic conformational space P(φ,ψ) whichwas evaluated from the trajectory. The conformational space isslightly more limited in the �-CD case compared with the othertwo CDs.
The ranges of the angular tilting, θ, of the glucose monomerstoward and away from the macrocyclic center are relativelysimilar for the three CDs. However, the time taken to relax intoan equilibrium state follows the pattern where �-CD relaxesfastest followed by R-CD and γ-CD. Similarly the generalizedamplitude of the monomer motion relative to the mean mac-rocyclic plane have magnitudes in the order γ-CD > R-CD >�-CD. These monomer librations relax fastest for �-CD followedby R-CD with γ-CD being the slowest to reach the meandisplacement from the macrocyclic plane. This finding confirmsthat at the monomeric length scale �-CD is the least flexible ofthe three CDs.
We indicated in Figure 2 that the most influential molecularparameters that could affect oligosaccharide conformations arethe torsion rotations about the glycosidic bonds and the ori-entations of the hydroxyl groups that may form intermolecularhydrogen bonds. In this study, we find that the molecularparameters that track the solubility trend are the distribution ofthe ψ torsion angles and the associated relaxation times withit. This leads us to conclude that the range and rate of rotationof the ψ torsions in the CDs are the basis of their glucosemonomer units’ orientational and librational motion and dynam-ics. This in turn underlies the order of macrocyclic inflexibility.
We have investigated 30 ns solution simulations of R-, �-,and γ-CDs, which resulted in a detailed comparative descriptionof their molecular and monomeric conformations. We canspeculate that the reason for the rigidity of the �-CD is simplydue to the inability of the uneven number of constituent maltosedimers making up the �-CD ring, to traverse the favored synfree energy φ,ψ well as easily as even numbered R- and γ-CDs.This difference is most apparent for transitions that requireglycosidic linkage rotations about the ψ torsion angle. Finally,we speculate that the conformational behavior of the CDsobserved in this study may have important consequences forcyclodextrins forming inclusion complexes.
Acknowledgment. This work is based upon research sup-ported by the South African Research Chairs Initiative (SARChI)
Crmsdδ(t) ) 1n ∑
i
n (⟨δrmsd(i)(0)δrmsd(i)(t)⟩
⟨δrmsd(i)(0)2⟩ ) (4)
15156 J. Phys. Chem. B, Vol. 112, No. 47, 2008 Naidoo et al.

of the Department of Science and Technology and the NationalResearch Foundation (NRF) awarded to K.J.N. In addition, itwas supported by the NRF, the Swedish Development Coopera-tion Agency (SIDA), the Swedish Research Council (VR) andthe Carl Trygger Foundation.
Supporting Information Available: Hydrogen bondinganalysis for the individual glucose fragments is displayed inFigure S1 and described in the text. This material is availablefree of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) (a) Dodziuk, H. Cyclodextrins and Their Complexes: Chemistry,Analytical Methods, Applications; Wiley: London, 2006. (b) Lipkowitz,K. B. Chem. ReV 1998, 98, 1829.
(2) Schneider, H. J.; Hacket, F.; Rudiger, V.; Ikeda, H. Chem. ReV1998, 98, 1755.
(3) French, D. AdV. Carbohydr. Chem. 1957, 12, 189.(4) (a) Jozwiakowski, M. J.; Connors, K. A. Carbohydr. Res. 1985,
143, 51. (b) Starikov, E. B.; Brasicke, K.; Knapp, E. W.; Saenger, W. Chem.Phys. Lett. 2001, 336, 504.
(5) (a) Linert, W.; Margl, P.; Renz, F. Chem. Phys. 1992, 161, 327.(b) Momany, F. A.; Willet, J. L. Carbohydr. Res. 2000, 326, 210. (c) Varady,J.; Wu, X. W.; Wang, S. M. J. Phys. Chem. B 2002, 106, 4863. (d) Funasaki,N.; Ishikawa, S.; Neya, S. J. Phys. Chem. B 2002, 106, 6431. (e) Heine,T.; Dos Santos, H. F.; Patchkovskii, S.; Duarte, H. A. J. Phys. Chem. A2007, 111, 5648. (f) Snor, W.; Liedl, E.; Weiss-Greiler, P.; Karpfen, A.;Viernstein, H.; Wolschann, P. Chem. Phys. Lett. 2007, 441, 159. (g) Karpfen,A.; Liedl, E.; Snor, W.; Viernstein, H.; Weiss-Greiler, P.; Wolschann, P.Monatsh. Chem. 2008, 139, 363.
(6) (a) Koehler, J. E. H.; Saenger, W.; van Gunsteren, W. F. J. Mol.Biol. 1988, 203–241. (b) Koehler, J. E. H.; Saenger, W.; van Gunsteren,
W. F. Euro. Biophys. J. 1988, 16, 153. (c) Koehler, J. E. H.; Saenger, W.;van Gunsteren, W. F. Euro. Biophys. J. 1987, 15, 197. (d) Koehler, J. E. H.;Saenger, W.; van Gunsteren, W. F. Euro. Biophys. J. 1987, 15, 211.
(7) (a) Bonnet, P.; Jaime, C.; Morin-Allory, L. J. Org. Chem. 2002,67, 8602. (b) Pereira, C. S.; de Moura, A. E.; Freitas, L. C. G.; Lins, R. A.J. Braz. Chem. Soc. 2007, 18, 951.
(8) Naidoo, K. J.; Chen, J. Y.-J.; Jansson, J. L. M.; Widmalm, G.;Maliniak, A. J. Phys. Chem. B 2004, 108, 4236.
(9) Brooks, B. R.; Bruccoleri, R. E.; Olafson, B. D.; States, D. J.;Swaminathan, S.; Karplus, M. J. Comput. Chem. 1983, 4 (2), 187.
(10) Kuttel, M. M.; Brady, J. W.; Naidoo, K. J. J. Comput. Chem. 2002,23, 1236.
(11) Berendsen, H. J. C.; Grigera, R. J.; Straatsma, T. P. J. Phys. Chem.1987, 91, 6269.
(12) van Gunsteren, W. F.; Berendsen, H. J. C. Mol. Phys. 1977, 34,1311.
(13) Thaning, J.; Stevensson, B.; Ostervall, J.; Naidoo, K. J.; Widmalm,G.; Maliniak, A. J. Phys. Chem. B 2008, 112.
(14) Cloran, F.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 1999,121, 9843.
(15) Kuttel, M. M.; Naidoo, K. J. J. Phys. Chem. B 2005, 109, 7468.(16) Saenger, W. R.; Jacob, J.; Gessler, K.; Steiner, T.; Hoffmann, D.;
Sanbe, H.; Koizumi, K.; Smith, S. M.; Takaha, T. Chem. ReV. 1998, 98,1787.
(17) (a) Schmidt, R. K.; Brady, J. W.; Teo, B. J. Phys. Chem. 1995, 99,11339. (b) Bartels, C.; Karplus, M. J. Comput. Chem. 1997, 18, 1450.
(18) Naidoo, K. J.; Brady, J. W. J. Am. Chem. Soc. 1999, 121, 2244.(19) Kumar, S.; Bouzida, D.; Swendsen, R. H.; Kollman, P. A.;
Rosenberg, J. M. J. Comput. Chem. 1992, 13, 1011.(20) Chen, Y.-J.; Naidoo, K. J. J. Phys. Chem. B 2003, 107, 9558.(21) Naidoo, K. J.; Chen, Y.-J. Mol. Phys. 2003, 101, 2687.(22) Harata, K. Chem. ReV. 1998, 98, 1803.
JP805174Y
Cyclodextrin Conformational Flexibility. J. Phys. Chem. B, Vol. 112, No. 47, 2008 15157