
EGF AND HYPOXIA-INDUCED EXPRESSION OF Instituto di Ricerche Farmacologiche ‘Mario...
24
EGF AND HYPOXIA-INDUCED EXPRESSION OF CXCR4 ON NON-SMALL CELL LUNG CANCER CELLS IS REGULATED BY THE PI3-KINASE/PTEN/AKT/mTOR SIGNALING PATHWAY AND ACTIVATION OF HIF-1α* Roderick J. Phillips ‡ , Javier Mestas ‡ , Mehrnaz Gharaee-Kermani ‡ , Marie D. Burdick ‡ , Antonio Sica § , John A. Belperio ‡ , Michael P. Keane ‡ , and Robert M. Strieter ‡,¶ ‡ Division of Pulmonary and Critical Care Medicine and ¶ Departments of Pathology and Pediatrics, David Geffen School of Medicine at UCLA, Los Angeles, CA 90095 § Instituto di Ricerche Farmacologiche ‘Mario Negri’, 20157, Milan, Italy. Running Title: Regulation of CXCR4 expression in NSCLC cells Address correspondence to: Robert M. Strieter, Division of Pulmonary and Critical Care Medicine, David Geffen School of Medicine at UCLA, 900 Veteran Avenue, 14-154 Warren Hall, Los Angeles, CA 90095-1786, Tel. 310-794-1999; Fax. 310-794-1998; E- mail: [email protected] Non-small cell lung cancer (NSCLC) expresses a particularly aggressive metastatic phenotype and patients with this disease have a poor prognosis. CXCR4 is a cell surface receptor that has been shown to mediate the metastasis of many solid tumors including lung, breast, kidney and prostate. In addition, overexpression of the epidermal growth factor receptor (EGFR) is associated with the majority of NSCLC and has been implicated in the process of malignant transformation by promoting cell proliferation, cell survival and motility. Here, we show for the first time that activation of the EGFR by EGF increases CXCR4 expression and the migratory capacity of NSCLC cells. Furthermore, many solid tumors are associated with low oxygen tension and when NSCLC cells were cultured with EGF under hypoxic conditions, CXCR4 expression was dramatically enhanced. A molecular analysis of these events indicated that augmented CXCR4 expression was regulated by the PI3- kinase/PTEN/AKT/mTor signal transduction pathway, activation of HIF- 1α, and ultimately HIF-1-dependent transcription of the CXCR4 gene. Thus, a combination of low oxygen tension and overexpression of EGFR within the primary tumor of NSCLC may provide the microenvironmental signals necessary to upregulate CXCR4 expression and promote metastasis. Non–small cell lung cancer (NSCLC) 1 is one of the leading causes of malignancy-related mortality in the United States – indeed fewer than 15% patients survive beyond 5 years after diagnosis. The virulence of this cancer is mediated in part by the specific and aggressive metastatic pattern of primary neoplastic cells to regional lymph nodes, liver, adrenal glands, contralateral lung, brain, and the bone marrow (1-4). In this respect, we, and others have now demonstrated that the metastatic propensity of tumors from several different types of cancer including lung, breast, ovarian, renal, and prostate is related to the expression of the chemokine receptor CXCR4 (5-12). In fact, in human NSCLC- SCID mouse chimera we have observed that the neoplastic cells present at the sites of the secondary metastases express dramatically upregulated levels of this chemokine receptor in comparison with the cancerous cells present in the primary tumor (11). Furthermore, in both NSCLC and breast cancer it has been shown that the ligand for CXCR4, CXCL12, exhibited peak levels of expression in organs that were the preferred 1 JBC Papers in Press. Published on March 31, 2005 as Manuscript M500963200 Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc. by guest on May 22, 2018 http://www.jbc.org/ Downloaded from
Transcript of EGF AND HYPOXIA-INDUCED EXPRESSION OF Instituto di Ricerche Farmacologiche ‘Mario...

EGF AND HYPOXIA-INDUCED EXPRESSION OF CXCR4 ON NON-SMALL CELL LUNG CANCER CELLS IS REGULATED BY
THE PI3-KINASE/PTEN/AKT/mTOR SIGNALING PATHWAY AND ACTIVATION OF HIF-1α*
Roderick J. Phillips‡, Javier Mestas‡, Mehrnaz Gharaee-Kermani‡, Marie D. Burdick‡, Antonio Sica§, John A. Belperio‡, Michael P. Keane‡, and Robert M.
Strieter‡,¶
‡ Division of Pulmonary and Critical Care Medicine and ¶Departments of Pathology and Pediatrics, David Geffen School of Medicine at UCLA, Los Angeles, CA 90095
§Instituto di Ricerche Farmacologiche ‘Mario Negri’, 20157, Milan, Italy. Running Title: Regulation of CXCR4 expression in NSCLC cells
Address correspondence to: Robert M. Strieter, Division of Pulmonary and Critical Care Medicine, David Geffen School of Medicine at UCLA, 900 Veteran Avenue, 14-154 Warren Hall, Los Angeles, CA 90095-1786, Tel. 310-794-1999; Fax. 310-794-1998; E-mail: [email protected]
Non-small cell lung cancer (NSCLC) expresses a particularly aggressive metastatic phenotype and patients with this disease have a poor prognosis. CXCR4 is a cell surface receptor that has been shown to mediate the metastasis of many solid tumors including lung, breast, kidney and prostate. In addition, overexpression of the epidermal growth factor receptor (EGFR) is associated with the majority of NSCLC and has been implicated in the process of malignant transformation by promoting cell proliferation, cell survival and motility. Here, we show for the first time that activation of the EGFR by EGF increases CXCR4 expression and the migratory capacity of NSCLC cells. Furthermore, many solid tumors are associated with low oxygen tension and when NSCLC cells were cultured with EGF under hypoxic conditions, CXCR4 expression was dramatically enhanced. A molecular analysis of these events indicated that augmented CXCR4 expression was regulated by the PI3-kinase/PTEN/AKT/mTor signal transduction pathway, activation of HIF-1α, and ultimately HIF-1-dependent transcription of the CXCR4 gene. Thus, a combination of low oxygen tension and overexpression of EGFR within the
primary tumor of NSCLC may provide the microenvironmental signals necessary to upregulate CXCR4 expression and promote metastasis.
Non–small cell lung cancer (NSCLC)1 is one of the leading causes of malignancy-related mortality in the United States – indeed fewer than 15% patients survive beyond 5 years after diagnosis. The virulence of this cancer is mediated in part by the specific and aggressive metastatic pattern of primary neoplastic cells to regional lymph nodes, liver, adrenal glands, contralateral lung, brain, and the bone marrow (1-4).
In this respect, we, and others have now demonstrated that the metastatic propensity of tumors from several different types of cancer including lung, breast, ovarian, renal, and prostate is related to the expression of the chemokine receptor CXCR4 (5-12). In fact, in human NSCLC-SCID mouse chimera we have observed that the neoplastic cells present at the sites of the secondary metastases express dramatically upregulated levels of this chemokine receptor in comparison with the cancerous cells present in the primary tumor (11). Furthermore, in both NSCLC and breast cancer it has been shown that the ligand for CXCR4, CXCL12, exhibited peak levels of expression in organs that were the preferred
1
JBC Papers in Press. Published on March 31, 2005 as Manuscript M500963200
Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

destination for their respective metastases (5,11). Moreover, when the CXCR4/CXCL12 biological axis was perturbed in these systems using either neutralizing anti-CXCR4 or neutralizing anti-CXCL12 antibodies, the host’s metastatic burden was significantly reduced, while the size of the primary tumor was unaffected (5,11). Thus, it appears that the normal physiology of CXCR4 and CXCL12 has been usurped by several different types of cancer to promote the specific metastasis of neoplastic cells to distant organs.
Tumors such as NSCLC typically require neovascularization to mediate growth and promote metastasis, yet paradoxically the most malignant tumors have been found to prosper under conditions of low oxygen tension or hypoxia (13-18). This paradox occurs because the tumor vasculature is structurally and functionally abnormal, resulting in perfusion that is characterized by marked spatial and temporal heterogeneity. Thus tumor progression requires an increased adaptation to hypoxia and the master switch that appears to regulate this phenomenon is the transcription factor, Hypoxia Inducible Factor-1 (HIF-1) (14,15,19,20). Indeed, an extensive body of work has already shown that HIF-1 regulates the transcription of several gene clusters that are crucial to tumor progression including angiogenesis, cell survival, glucose metabolism and invasion/metastasis (14,18,21).
HIF-1 is a heterodimer comprising a constitutively expressed HIF-1β subunit and a highly regulated HIF-1α subunit (14-16). Classically, ambient oxygen tension regulates the rate at which HIF-1α protein is degraded: under normoxic conditions specific proline residues in the HIF-1α protein are hydroxylated, facilitating the binding of the von Hippel-Lindau (VHL) tumor-suppressor protein (22,23). VHL is the recognition component of the E3 ubiquitin-protein ligase and ubiquitylation of HIF-1α targets the protein for rapid degradation by the 26S proteasome. By contrast, under hypoxic conditions the rates
of proline hydroxylation decreases, thus preventing the binding of VHL to HIF-1α and promoting HIF-1 mediated transcription of target genes (13,14,24).
Oxygen-independent regulation of HIF-1α has also been shown to occur, although this is thought to be cell-type specific. Here, growth factors stimulate HIF-1α synthesis via activation of the phosphatidylinositol 3-kinase (PI3-kinase) and mitogen-activated protein kinase (MAP-kinase) pathways (14,25,26). PI3-kinase activates the downstream serine/threonine kinase AKT and the mammalian target of rapamycin, mTOR (27-30). PI3-kinase, itself, is regulated by the phosphatase activity of the tumor suppressor gene PTEN (27,30,31). Both the PI3-kinase and MAP-kinase pathways converge on the p70 S6-kinase, which initiates a cascade of events that ultimately leads to an increase in the rate at which HIF-1α mRNA is translated into protein (14). Other studies have also suggested the potential involvement of NF-κB (32,33).
In an effort to address the mechanisms governing the upregulation of CXCR4 in NSCLC we have used an in vitro model system to study this pivotal chemokine receptor. Our data indicate that exposure of NSCLC cells to hypoxia or EGF results in a significant upregulation of CXCR4 expression and chemotactic behavior. In addition, both hypoxia and EGF activate HIF-1α, and this in turn increases transcription at the CXCR4 promoter. The PI3-kinase inhibitors wortmannin and LY294002 and the mTOR inhibitor, rapamycin, inhibit activation of HIF-1α and hence upregulation of CXCR4 expression. Moreover, introduction of wildtype PTEN into NSCLC cells also inhibits hypoxia-induced upregulation of CXCR4 expression. Taken together, therefore, these data suggest that dysregulated signal transduction through the PI3-kinase pathway in NSCLC leads to activation of HIF-1α, upregulation of CXCR4 and increased metastatic potential.
2
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

EXPERIMENTAL PROCEDURES Human NSCLC cell lines - The
H157 and A549 non-small cell lung cancer cell lines were obtained from ATCC. These cell lines were cultured in RPMI 1640 media (Whitaker Biomedical Products, Whitaker, CA) together with 1 mM L-glutamine, 25 mM HEPES buffer, 100 U/ml penicillin, 100 ng/ml streptomycin and 10% FCS (RPMI complete media). Prior to assay the cells were transferred to RPMI starvation media, which comprises 1 mM L-glutamine, 25 mM HEPES buffer, 100 U/ml penicillin, 100 ng/ml streptomycin and 1% FCS or 0.25% HSA. Where applicable the PI3-kinase inhibitors, LY294002 (20 – 50 mM), wortmannin (100 – 250 nM) and the mTOR inhibitor, Rapamycin (10 ng/ml) were preincubated with cells for two hours prior to exposure to hypoxia and stimulation with EGF (20 ng/ml)
RNA isolation and Real Time PCR - Total RNA was isolated from both A549 and H157 cells using TRIzol (Life Technologies, Rockville, MD) and by following the manufacturer’s instructions. Briefly, cells were lysed in TRIzol and then mixed with chloroform. The lysate was then centrifuged to separate RNA, DNA and protein. Total RNA was recovered, precipitated with isopropanol, washed in 75% ethanol to remove impurities and finally dissolved in water. Next, 1.5 µg of RNA was taken and DNase treated to remove contaminating DNA prior to reverse transcription to cDNA using a ProSTAR First Strand RT-PCR kit (Stratagene) and by following the manufacturer’s instructions. Subsequently, the cDNA was assayed for changes in CXCR4 expression by real-time PCR using the ABI Prism 7700 sequence detector and SDS analysis software (Applied Biosystems, Foster City, CA) as previously described (34).
Antibody staining and FACS analysis - Cells from each cell line were taken and resuspended in ice-cold staining buffer (PBS + 2 % FCS + 0.1 % sodium azide) and incubated with Fc block for 5 minutes at 4oC. Subsequently, the cells
were stained with FITC-conjugated anti-CXCR4 antibodies or the appropriate isotype control at 4oC for 20 minutes, after which time they were washed twice with staining buffer. Samples were finally analyzed on a FACScan flow cytometer (Becton Dickinson) using Cellquest 3.2.1f1 software.
Hypoxia treatment and extract preparation - Cells were cultured to a density of approximately 80% in complete media and then transferred to starvation media. Next, A549 and H157 cells were exposed to either normoxia (ambient oxygen tension) or hypoxia (94% nitrogen, 5% carbon dioxide and 1% oxygen) in Modular Incubator Chambers (Billups-Rothenberg, Inc., Del Mar CA) for the times indicated. Subsequently, whole cell extracts (WCE) or nuclear (N) and cytoplasmic (C) extracts of A549 and H157 cells were prepared. Briefly, WCE lysis buffer comprised 20 mM HEPES pH 7.9, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, and 0.2 mM EDTA, plus a panel of protease and phosphatase inhibitors (PMSF, DTT, and NaF at 1 mM; aprotinin, leupeptin, pepstatin, and β-glcerophophate at10 µg/ml. Buffer A for extraction of cytoplasmic fractions comprises 10 mM HEPES, pH 7.9, 10 mM KCl and 0.1 mM EDTA (plus the above panel of protease and phosphatase inhibitors), while buffer C for extraction of the nuclear fraction comprised 20 mM HEPES, pH 7.9, 400 mM NaCl, and 1 mM EDTA (plus the protease and phosphatase inhibitors described above).
Western Blotting - Immunoblotting was performed on 40 µg of total protein from either WCE or N and C extracts. Following SDS-PAGE, the proteins were electrophoretically transferred to a PVDF membrane at 100 V for one hour at room temperature and then blocked in BLOTTO for 30 minutes. Subsequently, the membranes were incubated overnight at 4oC with either a mouse anti-human HIF1-α (1:500; BD, San Diego, CA), rabbit anti-human CXCR4 (1:500; Oncogene Research Products, Cambridge, MA), or rabbit anti-
3
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

human phosphoAKT (1:1000; Cell Signaling Technology, Beverly, MA). Subsequently, the blots were washed in TTBS and then incubated with either donkey anti-rabbit or goat anti-mouse horseradish peroxidase-conjugated secondary antibodies for 45 minutes at room temperature. After washing in TTBS (x3, 15 minutes each wash), the immunoreactive proteins were finally visualized using ECL Plus (Amersham Pharmacia Biotech) and by following the manufacturer’s instructions. To demonstrate equal loading of each lane, the membranes were then reprobed with a GAPDH antibody (1:500; Abcon) or total AKT antibody (1:1000; Cell Signaling Technology, Beverly, MA)
Chemotaxis - A549 cells and H157 cells previously exposed to either hypoxia or normoxia for 24 hours were harvested by trypsinization, counted, and resuspended in RPMI 1640 media containing 10% FCS at a concentration of 106/ml. Neuroprobe filters (5 µm diameter) pretreated with 5 µg/ml fibronectin and 12-well chemotaxis chambers were used for these assays. CXCL12 (30 ng/ml; Peprotech, Rocky Hill, NJ) was added to the lower wells and 105 cells were added to each of the upper wells. The chemotaxis chambers were then incubated for 6 hours at 37oC. After fixing in methanol and staining in 2% Toluidine blue, the number of cells that had migrated through to the underside of the filters was calculated by counting the total number of cells in 5 separate fields of view under 400X magnification. In similar experiments A549 cells were either left untreated or pretreated with the PI3-kinase inhibitors wortmannin (100 nM; Upstate Biotechnology) and LY294002 (20 µM; Cell Signal Technology) for 2 hours prior to chemotactic analysis.
EMSA - A549 cells were either exposed to hypoxia or normoxia for 6 hours and then nuclear extracts were prepared. Oligonucleotide probes for HIF-1α were generated by 5’ end labeling of the sense strand with γ-32P ATP (Amersham Biosciences; Piscataway, NJ) and T4 polynucleotide kinase. Subsequently,
labeled wildtype (WT; 5’agcttGCCCTACGTGCTGTCTCAg3’) and mutant (M; 5’agcttGCCCTAAAAGCTGTCTCAg3’) probes (20) were purified using the MERmaid kit (Bio 101 Systems, Irvine, CA) and by following the manufacturer’s instructions. Binding reactions were performed in a total volume of 20 µl containing 10 µg of nuclear extract and 0.5 µg poly (dI-dC)• (dI-dC), in 10 mM Tris-HCl (pH 7.5), 50 mM KCl (pH 7.5), 50 mM NaCl, 1 mM MgCl2, 1mM EDTA, 5mM DTT, and 5% glycerol. Probe (5 x 104 cpm) was then added to the reaction mixture and incubated for 10 minutes at room temperature prior to loading onto a 5% non-denaturing polyacrylamide gel. Electrophoresis was performed at 175 volts in 0.5x TBE, pH 8.3 (1x TBE comprises 89 mM Tris-HCl, 89 mM boric acid, and 5 mM EDTA) at 4oC for 3 hours. Gels were then vacuum dried and exposed to film with intensifying screens for 24 hours. Excess (100 fold) unlabeled WT oligonucleotide was preincubated with the reaction mixture for 15 minutes prior to addition of γ-32P ATP WT probe.
Transient Transfections - A549 and H157 cells were cultured to a density of approximately 80% in complete media in 6-well plates. Transfections were performed with Lipofectamine 2000 and Optimem media (Invitrogen, Carlsbad, CA) and by following the manufacturer’s instructions. Briefly, 2.5 µg of either WT- or C124S-PTEN constructs (35) were mixed with 4 µl of Lipofectamine and 1 ml of Optimem media for 20 minutes at room temperature. Subsequently, this mixture was added to the NSCLC cells, which were then incubated at 37oC for 90 minutes. Finally, the transfection mixture was removed and replaced with either DMEM plus 10% FCS (H157 cells) or RPMI1640 plus 10% FCS (A549 cells). The cells were then cultured for 24 hours and 37oC prior to exposure to normoxia or hypoxia for a further 24 hours. Upon exposure to normoxia or hypoxia cultures were returned to media containing
4
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

only 1% FCS. Subsequently, RNA was prepared and subjected to real-time PCR analysis of CXCR4 expression as described above. In similar experiments A549 and H157 cells were cotransfected with a 2.6 kb sequence of either the WT-CXCR4 promoter (36,37) or a mutant form of CXCR4 promoter, where the HIF-1α binding site had been mutated upstream of a luciferase reporter, together with 0.5 µg of the of the renilla control construct (pRL-SV40; Promega, Madison, WI), and a HIF-1α construct. A GFP construct was to equalize the DNA transfection load. The cells were then cultured for 48 hours and 37oC prior to analysis. After the 48-hour incubation period, cell extracts were made using the luciferase reporter lysis buffer (Promega). Each lysate was subsequently assayed in the dual luciferase reporter assay (Promega) following the manufacturer’s instructions; luciferase activity was determined using a Monolight series 2010 luminometer (Analytical Luminescence Laboratory) and then normalized to the renilla control.
Statistical Analysis - Comparisons
were evaluated by Student’s unpaired t–test. Results were considered statistically significant if P values were 0.05 or less.
RESULTS
Hypoxia promotes upregulation of
CXCR4 expression in non-small cell lung cancer cells. We have previously shown that upregulation of CXCR4 expression is a key component in the metastasis of NSCLC cells in vivo (11), but the mechanisms that regulate expression of this chemokine receptor are unclear. Initial evidence in other cancerous cells has indicated that hypoxia-induced HIF-1 activation may be involved (10,36). To address this phenomenon in non-small cell lung cancer, therefore, we exposed tumor cells (cultured in RPMI starvation media) to a hypoxic environment (94% N2, 5% CO2, 1% O2) and performed a kinetic analysis to examine changes in CXCR4 expression and function
(Figure 1). Using real-time PCR, our data revealed that the expression of CXCR4 mRNA was strongly elevated in hypoxia-exposed A549 and H157 NSCLC cells by 6 hours when compared to the normoxic control (Figure 1A). This expression remained elevated until at least 24 hours (Figure 1A).
Next, we wanted to determine whether the increase in CXCR4 mRNA correlated with an increase in protein levels of CXCR4. We examined this both at the level of intracellular expression (Figure 1B) and cell surface expression (Figure 1C). Our results indicated that NSCLC cells exposed to hypoxia showed a significant increase in intracellular CXCR4 protein levels when compared to the normoxic control by 6 hours and these levels remained elevated until at least 24 hours (Figure 1B). Indeed, by 24 hours both A549 cells and H157 cells showed significantly greater expression of CXCR4 at the cell surface (Figure 1C).
To determine whether this increased expression of CXCR4 was functional, we performed chemotaxis assays (Figure 1D). Here, NSCLC cells were exposed to hypoxia or normoxia for 24 hours and then treated with CXCL12 for 6 hours. Although both A549 and H157 cells demonstrated chemotactic behavior in response to CXCL12 under normoxic conditions, the magnitude of these responses was dramatically enhanced in those cells exposed to hypoxia for 24 hours (Figure 1D). Thus, we have demonstrated that hypoxia not only increases expression of CXCR4 on NSCLC cells, but also enhances the migratory ability of these cells in response to CXCL12.
Hypoxia activates HIF-1α expression in NSCLC cells and promotes HIF-1-mediated transcription at the CXCR4 promoter. Having established that a physiological event such as hypoxia is capable of upregulating CXCR4 expression in NSCLC cells, we next wanted to examine the underlying biochemistry that mediates this phenomenon. It has been well-established that hypoxia regulates the
5
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

expression of HIF-1α, which is a key component of the transcription factor HIF-1 (14-16). And HIF-1, itself, is thought to regulate the transcription of several gene clusters crucial to tumor progression including angiogenesis, cell survival, glucose metabolism and invasion/metastasis (13,14,18,21). Thus, we exposed A549 cells and H157 cells to normoxia or hypoxia for the times indicated and then examined intranuclear HIF-1α expression by western analysis (Figure 2A). Under normoxic conditions little or no intranuclear expression of HIF-1α was observed. This is in keeping with known data, which has suggested that under normal ambient conditions the tumor suppressor gene, VHL, binds to HIF-1α and targets it for degradation (22,23). However, under hypoxic conditions, strong intranuclear expression of HIF-1α was observed within two hours and this expression remained elevated for a total of 6 hours; by 24 hours, HIF-1α expression had returned to background levels (Figure 2A).
Next, we wanted to determine whether hypoxia promoted an increase in the binding of the HIF-1 transcription factor (which comprises inducibly expressed HIF-1α and constitutively expressed HIF-1β) to its cognate DNA-binding motif (Figure 2B). Therefore, we exposed A549 cells to normoxia or hypoxia for 4 hours and then prepared nuclear extracts for analysis by EMSA. Our results indicate that under normoxic conditions there is little or no inducible binding of HIF-1 to its cognate binding motif (Figure 2B, lane 1), whereas four hours exposure to hypoxia mediated a strong signal (Figure 2B, lane 2). To demonstrate the specificity of this binding activity, we added nuclear extract from hypoxia-treated cells to a labeled probe containing a mutated form of the core binding motif (5’-AAAAG-3’ instead of 5’-ACGTG-3’; figure 2B, lane 3), or included an excess of cold WT probe with the 32P-labeled WT probe (Figure 2B, lane 4). Under both conditions specific binding of
the HIF-1 transcription factor was abrogated.
To further verify that HIF-1 contributed to the transcription and upregulation of CXCR4 gene expression, we transfected a luciferase reporter construct containing a 2.6 kb fragment of the wildtype CXCR4 promoter (WT-CXCR4) into A549 cells and H157 cells (Figure 2C). In addition, we co-transfected either a random control cDNA (GFP; figure 2C, lanes 1 and 5) or HIF-1α cDNA (Figure 2C, lanes 2 and 6). Under these conditions, significant transactivation of the CXCR4 promoter was only observed in those cells receiving the HIF-1α cDNA, but not the GFP cDNA (compare lanes 1and 2, and 5 and 6). Next, we mutated a consensus HIF-1 binding site in the CXCR4 promoter (M-CXCR4) and repeated the transfections described above. On this occasion, HIF-1α failed to mediate transcriptional activation of the CXCR4 promoter (Figure 2C, compare lanes 4 and 5, and 7 and 8). These data, therefore, suggest that the HIF-1 transcription factor regulates expression of the CXCR4 gene.
The PI3-kinase pathway contributes to the regulation of CXCR4 expression mediated by HIF-1. To more fully elucidate the signaling pathways involved in the regulation of CXCR4 expression we treated A549 cells and H157 cells with the PI3-kinase inhibitors wortmannin and LY294002. Previous studies in prostate cancer cells and glioblastoma cells have implicated a role for PI3-kinase hypoxia-induced HIF-1α activation (26,38). To that end, we pretreated NSCLC cells with either wortmannin (250 nM) or LY294002 (50 µM) for 2 hours and then subjected these cell lines to either normoxic or hypoxic conditions for a further 6 hours (Figure 3A(i) and 3B(i)). Subsequently, we examined intranuclear HIF-1α levels by western analysis. We observed that in the absence of PI3-kinase inhibitors, a strong HIF-1α signal was obtained after 6 hours of hypoxia treatment (Compare figure 3A(i) lane 4 and 3B(i) lane 4). However, in the presence of the PI3-kinase inhibitors (and
6
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LY294002 in particular) HIF-1α activation was strongly inhibited (Compare figure 3A(i) lanes 4 and 6, and 3B(i) lanes 4 and 6).
Next, we wanted to know whether these same PI3-kinase inhibitors that blocked HIF-1α activation would also block expression of CXCR4. To do this we exposed A549 cells and H157 cells to either wortmannin (250 nM) or LY294002 (50 µM) for 24 hours in the presence of either normoxia or hypoxia (Figure 3A(ii) and (iii), and 3B(ii) and (iii)). Under these conditions, the upregulation of CXCR4 expression mediated by hypoxia was indeed abrogated by the PI3-kinase inhibitors, although some basal expression of CXCR4 remained (Compare Figure 3A(ii) lanes 4-6, and 3B(ii) lanes 4-6). This data was further verified at the level of CXCR4 mRNA expression. Here, LY294002 strongly inhibited hypoxia-induced CXCR4 expression (Figure 3C and 3D).
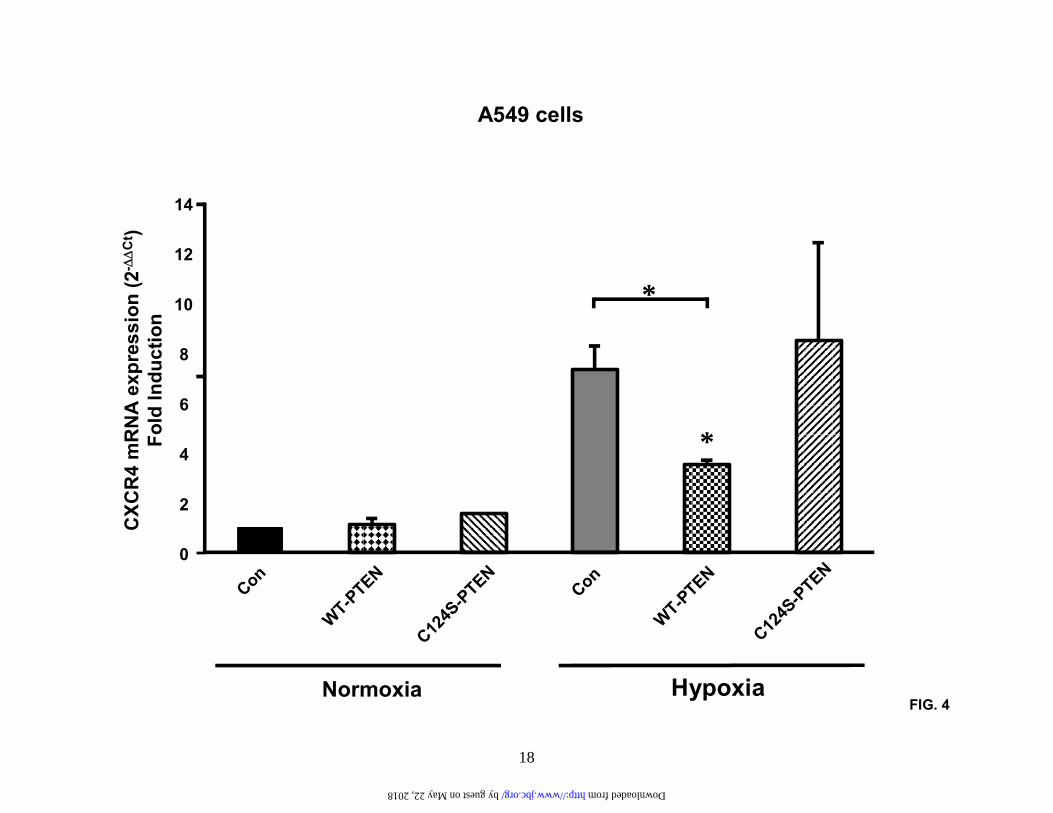
The tumor suppressor gene PTEN is also known to be a key regulatory component in the PI3-signaling cascade (26,27,30). We therefore transfected A549 cells with either wildtype PTEN (WT-PTEN) or a catalytically inactive form of PTEN containing a C124S missense mutation (C124S-PTEN) (35). Next, we exposed these transfected cells to either normoxia or hypoxia for 24 hours and then extracted RNA to examine changes in CXCR4 mRNA expression by real-time PCR (Figure 4). Our results indicate that overexpression of WT-PTEN, but not the catalytically inactive form, C124S-PTEN, significantly curtailed hypoxia-induced activation of CXCR4. Taken together, these data suggest that hypoxia-induced HIF-1α activation and CXCR4 expression are regulated, at least in part, by PI3-kinase and the PTEN tumor suppressor gene.
Epidermal Growth Factor-activated PI3-kinase signaling synergizes with hypoxia treatment to dramatically upregulate CXCR4 expression. Gain of function mutations in receptor tyrosine kinases (RTK) such as the epidermal growth
factor receptor are known to be a feature of the majority of non-small cell lung cancers (39-41). Furthermore, these RTK are also known to signal through the PI3-kinase pathway (39-42). Therefore, we decided to treat A549 cells and H157 cells with EGF in the presence or absence of LY294002 and under normoxic and hypoxic conditions to assess changes in CXCR4 mRNA expression by real-time PCR (Figure 5A). Under normoxic conditions, EGF alone induced a 5 – 10 fold induction in CXCR4 mRNA expression (Compare Figure 5A panels (i) and (ii)) alone. In both cell lines the observed induction of CXCR4 mRNA by EGF was strongly inhibited by LY294002. Hypoxia-induced CXCR4 mRNA expression was of a similar magnitude to that observed by EGF treatment under normoxic conditions, and although LY294002 abrogated hypoxia-induced CXCR4 mRNA expression, the inhibition was not complete (Compare Figure 5A panels (i) and (ii) and also Figure 3C and 3D). The combined treatments of EGF plus hypoxia produced a dramatic increase in CXCR4 mRNA expression in both NSCLC cell lines, and this synergistic increase was also susceptible to inhibition by LY294002 (Compare Figure 5A panels (i) and (ii)). In addition, treatment with either EGF or hypoxia or EGF plus hypoxia resulted in a significant upregulation of CXCR4 protein levels in both A549 cells and H157 cells (Figure 5B and data not shown); moreover, pretreatment with LY294002 modulated the EGF-induced increase in CXCR4 protein expression (Figure 5C).
The substrate for PI3-kinase is phosphatidylinositol-4,5-bisphosphate (PIP2) to generate the second messenger phosphatidylinositol-3,4,5-triphosphate (PIP3), which activates phosphatidylinositol-dependent kinase (PDK), which in turn phosphorylates and activates the serine/threonine kinase, AKT (protein kinase B) (28,29). To further characterize the signaling pathway that mediates EGF-induced upregulation of
7
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

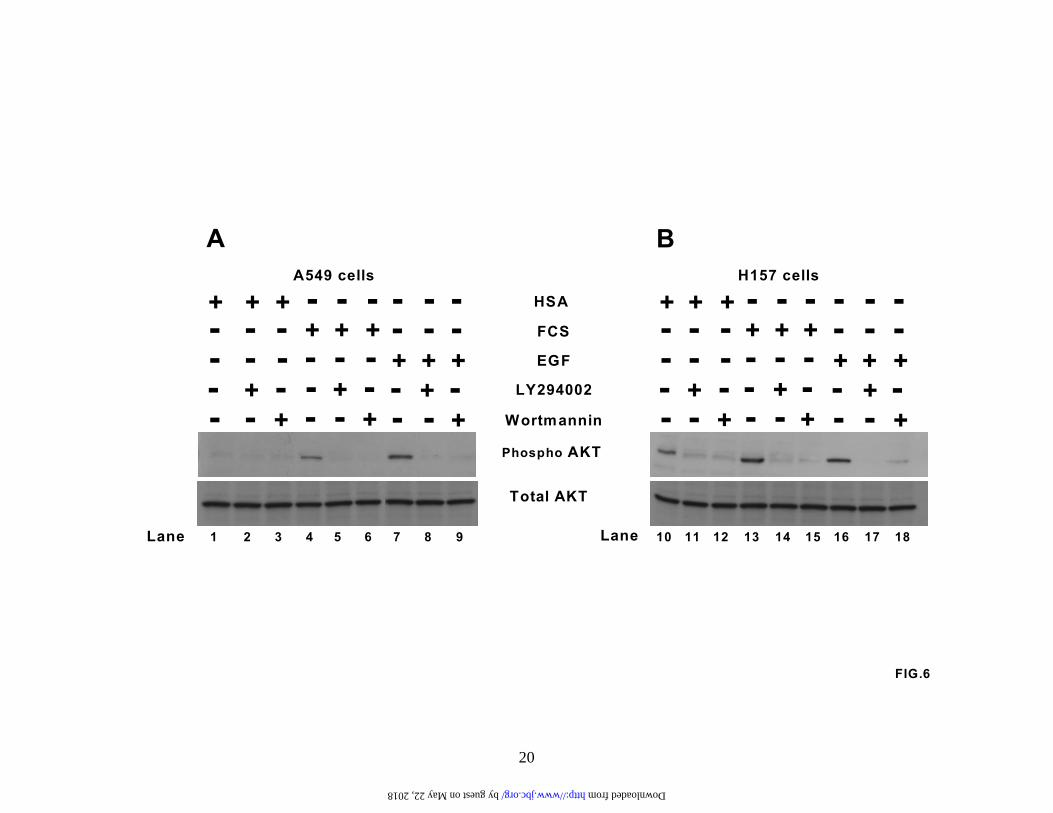
CXCR4, we therefore examined changes in the activation state of AKT (Figure 6). Cells were cultured in RPMI starvation media with 0.25% human serum albumin (HSA) for 24 hours, pretreated with either LY294002 (50 µM; lanes 2, 5, 8, 11, 14, and17) or wortmannin (250 nM; lanes 3, 6, 9, 12, 15, and 18) for 2 hours and then either left unstimulated (lanes 1-3 and 10-12) or stimulated with 10% FCS (lanes 4-6 and 13-15) or 20 ng/ml EGF (lanes 7-9 and 16-18) for 10 minutes. We then examined AKT activity by western analysis. Our results showed that in unstimulated A549 cells there was no constitutive AKT phosphorylation (lane 1), whereas both EGF and FCS induced strong phosphorylation and activation of AKT (compare lanes 4 and 7). These phosphorylation events were strongly inhibited by the PI3 kinase inhibitors (lanes 5 and 6 and 8 and 9). By contrast, constitutive phosphorylation of AKT was still observed in H157 cells despite prior culture in RPMI starvation media with 0.25% HAS for 24 hours (lane 10). However, both the constitutive AKT phosphorylation and the augmented signaling mediated by FCS and EGF was completely modulated by the PI3-kinase inhibitors (Figure 6B).
Downstream of the serine/threonine kinase AKT is another key component of the PI3-kinase signaling cascade – namely, mTOR (43). Therefore, to further establish the signaling sequence through which EGF upregulates expression of CXCR4, we serum-starved A549 cells for 24 hours, pretreated with the mTOR-specific inhibitor, rapamycin (10 ng/ml) and then treated with the EGF (20 ng/ml) for an additional 24 hours. Subsequently, we prepared RNA, and analyzed changes in CXCR4 expression by real-time quantitative PCR (Figure 7A). EGF-mediated upregulation of CXCR4 mRNA under both normoxic conditions and hypoxic conditions was strongly inhibited in the presence of rapamycin (Figure 7A).
Next, we wanted to determine whether EGF, itself, was capable of activating HIF-1α in NSCLC cells under normoxic conditions. Previous studies have
already suggested that growth factors and cytokines are capable of activating HIF-1α in the absence of low oxygen tension (14,32,33,38). Therefore, serum-starved A549 cells were treated with EGF (20 ng/ml) for 6 hours under normoxic or hypoxic conditions and extracts prepared to examine intranuclear HIF-1α activity by western analysis (Figure 7B). Our results revealed that EGF was able to induce HIF-1α, although the extent of the activation was more modest than that observed by hypoxia. Furthermore, in the presence of rapamycin, EGF-induced HIF-1α activation was inhibited under both normoxic and hypoxic conditions (Figure 7B).
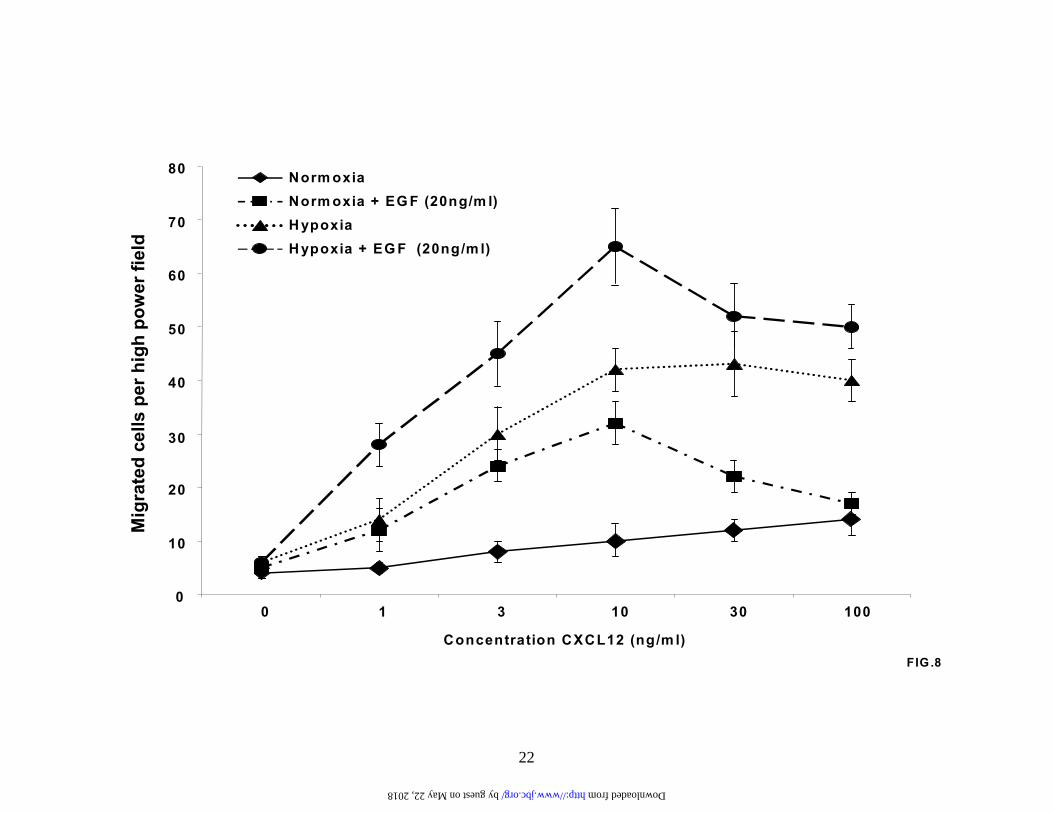
Finally, we wanted to determine whether the increased expressed of CXCR4 observed in the presence of EGF and particularly EGF plus hypoxia lead to an increase in function. Therefore, serum-starved A549 cells were exposed to EGF under normoxic and hypoxic conditions and then stimulated with varying concentrations of CXCL12 to measure the chemotactic potential of the cells (Figure 8). As described previously (Compare Figure 1D and Figure 8), hypoxia strongly upregulated chemotaxis in response to CXCL12. Remarkably, the combination of hypoxia and EGF promoted an even greater increase in A549 chemotaxis. In addition, under normoxic conditions pretreatment with EGF alone mediated a significant increase in migration in response to CXCL12, when compared to the normoxic control (Figure 8). Moreover, this response was maximal at a lower concentration of CXCL12, suggesting that EGF sensitizes the cells in response to CXCL12. Thus the growth factor EGF is capable of upregulating CXCR4 expression and chemotactic potential in NSCLC cells via a pathway that involves PI3-kinase, AKT, mTOR, and HIF-1α.
PI3-kinase also regulates CXCL12-induced chemotaxis in NSCLC cells. As CXCR4 is an important component of the metastatic pathway in non-small cell lung cancer, we wanted to determine whether the
8
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

PI3-kinase pathway modulated the chemotactic behavior of NSCLC cells in response to its cognate ligand, CXCL12. Therefore, serum-starved A549 cells were pretreated with either LY294002 (20 µM) or wortmannin (100 nM) for two hours, and then stimulated for a further 6 hours in the presence of CXCL12 (30 ng/ml). Our data revealed that both PI3-kinase inhibitors strongly inhibited CXCL12-induced chemotaxis, whereas the MEK1/2 inhibitor, UO126, had no effect on the CXCR4/CXCL12 chemotactic axis (Figure 9 and data not shown). This suggests that the PI3-kinase signaling pathway is capable of regulating both the expression of CXCR4 and the CXCR4/CXCL12 chemotactic axis in non-small cell lung cancer cells.
DISCUSSION
This study provides, to our
knowledge, the first indication that both hypoxia and the EGF regulate expression of CXCR4 on non-small cell lung cancer cells. Moreover, EGFR activation in the presence of hypoxia further augments CXCR4 expression. Having identified two key physiological signals that regulate CXCR4 expression, we then examined the molecular signaling pathways involved. The EGFR is a RTK that activates PI3-kinase and subsequent downstream targets including AKT and mTOR. In the presence of either PI3-kinase inhibitors or mTOR inhibitors, we found that we could block both activation of HIF-1α and increases in CXCR4 expression. We further showed that HIF-1 directly transactivates CXCR4 gene expression. Finally, inhibitors of PI3-kinase also prevent chemotaxis of NSCLC cells in response to CXCL12, the cognate ligand for CXCR4. Thus, the PI3-kinase pathway abrogates increased expression of CXCR4 induced by hypoxia and the EGFR, and CXCL12-mediated chemotaxis (see Figure 8 for an illustration of these pathways).
HIF-1α is constitutively expressed in most cells, but it normally undergoes a post-translational modification that targets it for degradation by the 26S proteasome (14-
16). The key regulatory molecule involved in this process is the tumor suppressor gene, VHL (22,23). This protein moiety is the recognition component of the E3 ubiquitin-protein ligase and it is the VHL-mediated ubiquitination of HIF-1α that targets the transcription factor for degradation. Hypoxia prevents VHL from binding to HIF-1α, thus stabilizing the expression of this transcription factor. Active HIF-1 then regulates a host of genes involved in cellular processes such as proliferation, survival, glucose metabolism, and angiogenesis (14,16,18,21). Our data and that of two other recent studies (10,36) now suggest that HIF-1 also regulates CXCR4-mediated metastasis. In particular, Staller and colleagues have shown that a common mutation in clear cell renal carcinoma is loss (or functional inactivation) of pVHL, resulting in persistent activation of HIF-1 and a dramatic upregulation of CXCR4 expression (10). We have demonstrated an analogous pathway in NSCLC cells that regulates CXCR4 expression. Here, hypoxia functionally inactivates VHL, albeit temporarily, and facilitates accumulation of HIF-1α and increased CXCR4 transcription. Interestingly, little is known about the regulation of the VHL gene, itself, despite the fact that the promoter was cloned 10 years ago (44,45). A better understanding of the regulatory elements governing the expression of this important tumor suppressor gene may yield vital clues in the ongoing search for effective therapeutics to abrogate the aggressive metastasis associated with NSCLC.
Although permanent functional inactivation of VHL is not a common phenotype in NSCLC, overexpression of EGFR is strongly correlated with disease progression in squamous carcinomas, large cell and adenocarcinomas. Furthermore, we have demonstrated for the first time that under ambient oxygen tension the EGF/EGFR biological axis activates the HIF-1 transcription factor, and this in turn upregulates CXCR4 expression and function. The underlying biochemistry associated with this phenomenon involves
9
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

activation of the PI3-kinase/PTEN/AKT/mTOR pathway. Indeed, overexpression of wildtype PTEN effectively inhibits upregulation of CXCR4 expression. In similar studies Zundel and colleagues reported that overexpression of wildtype PTEN in glioblastoma cells that lacked a functional PTEN ablated hypoxia and IGF induction of HIF-1 regulated genes (26). Further evidence in support of the notion that growth factors such as EGF stabilize and activate HIF-1α is provided by a study in prostate cancer cells where EGF-mediated HIF-1α activation resulted in the induction of VEGF gene expression – a gene known to be under the control of HIF-1(38).
Other signaling molecules including TNF-α and IL-1β have also been shown to activate HIF-1α expression, although in these studies it has been suggested that the cytokines act indirectly through NF-κB (32,33).
Indeed, the role of VHL in the growth factor and cytokine-mediated activation of HIF-1α remains unclear. Jung and colleagues (33) have indicated that an unknown, TNF and NF-κB regulated factor that interferes with VHL binding to HIF-1α is involved. Moreover, ligation of the EGFR also activates the MAP-kinase pathway, which appears to share at least some of the same downstream elements that are found in the PI3-kinase signaling pathway, including p70 S6 kinase and the eukaryotic translation initiating factor, 4E-
BP1 (14,43). It is again unclear at this time what role, if any, the MAP-kinases play in EGF/EGFR-dependent expression of CXCR4. Thus, in future studies we will examine the potential contribution of both the NF-κB and MAP-kinase signal transduction pathways to EGFR-mediated activation of HIF-1α.
In addition to binding EGF, the EGFR also binds Transforming Growth Factor-α (TGF-α) a gene whose expression is regulated by HIF-1 (46). This observation introduces the intriguing possibility that an autocrine signaling pathway may develop in the malignant progression of NSCLC cells involving activation of HIF-1 (either via the EGFR or via hypoxia), persistent upregulation of TGF-α expression, and chronic activation of the EGFR. This in turn would lead to the continuous expression of CXCR4 and ultimately the genesis of a highly metastatic tumor cell.
Taken together, our data identifies several key signaling molecules necessary for the development of a metastatic phenotype in NSCLC. These include EGFR, PI3-kinase/PTEN, mTor, HIF-1α/VHL and CXCR4. The chemokine receptor, CXCR4, represents the final common mediator of these pathways, and therefore provides an attractive therapeutic target for treatment of not only NSCLC, but also other highly metastatic cancers such as breast and kidney.
REFERENCES
1. Fidler, I. J. (1990) Cancer Res 50, 6130-6138 2. Smith, W., and Khuri, F. R. (2004) Semin Oncol 31, 11-15 3. Fidler, I. J. (1999) Cancer Chemother Pharmacol 43 Suppl, S3-10 4. Devesa, S. S., Blot, W. J., Stone, B. J., Miller, B. A., Tarone, R. E., and Fraumeni,
J. F., Jr. (1995) J Natl Cancer Inst 87, 175-182 5. Muller, A., Homey, B., Soto, H., Ge, N., Catron, D., Buchanan, M. E.,
McClanahan, T., Murphy, E., Yuan, W., Wagner, S. N., Barrera, J. L., Mohar, A., Verastegui, E., and Zlotnik, A. (2001) Nature 410, 50-56
6. Belperio, J. A., Phillips, R. J., Burdick, M. D., Lutz, M., Keane, M., and Strieter, R. (2004) Chest 125, 156S
7. Zlotnik, A. (2004) Semin Cancer Biol 14, 181-185
10
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8. Taichman, R. S., Cooper, C., Keller, E. T., Pienta, K. J., Taichman, N. S., and McCauley, L. K. (2002) Cancer Res 62, 1832-1837
9. Strieter, R. M., Belperio, J. A., Phillips, R. J., and Keane, M. P. (2004) Semin Cancer Biol 14, 195-200
10. Staller, P., Sulitkova, J., Lisztwan, J., Moch, H., Oakeley, E. J., and Krek, W. (2003) Nature 425, 307-311
11. Phillips, R. J., Burdick, M. D., Lutz, M., Belperio, J. A., Keane, M. P., and Strieter, R. M. (2003) Am J Respir Crit Care Med 167, 1676-1686
12. Scotton, C. J., Wilson, J. L., Milliken, D., Stamp, G., and Balkwill, F. R. (2001) Cancer Res 61, 4961-4965
13. Vogelstein, B., and Kinzler, K. W. (2004) Nat Med 10, 789-799 14. Semenza, G. L. (2003) Nat Rev Cancer 3, 721-732 15. Semenza, G. L. (2000) Respir Res 1, 159-162 16. Semenza, G. L. (1999) Annu Rev Cell Dev Biol 15, 551-578 17. Ryan, H. E., Lo, J., and Johnson, R. S. (1998) Embo J 17, 3005-3015 18. Hanahan, D., and Folkman, J. (1996) Cell 86, 353-364 19. Wang, G. L., and Semenza, G. L. (1995) J Biol Chem 270, 1230-1237 20. Semenza, G. L., and Wang, G. L. (1992) Mol Cell Biol 12, 5447-5454 21. Wood, S. M., Wiesener, M. S., Yeates, K. M., Okada, N., Pugh, C. W., Maxwell,
P. H., and Ratcliffe, P. J. (1998) J Biol Chem 273, 8360-8368 22. Jaakkola, P., Mole, D. R., Tian, Y. M., Wilson, M. I., Gielbert, J., Gaskell, S. J.,
Kriegsheim, A., Hebestreit, H. F., Mukherji, M., Schofield, C. J., Maxwell, P. H., Pugh, C. W., and Ratcliffe, P. J. (2001) Science 292, 468-472
23. Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., Salic, A., Asara, J. M., Lane, W. S., and Kaelin, W. G., Jr. (2001) Science 292, 464-468
24. Leung, S. K., and Ohh, M. (2002) J Biomed Biotechnol 2, 131-135 25. Fukuda, R., Hirota, K., Fan, F., Jung, Y. D., Ellis, L. M., and Semenza, G. L.
(2002) J Biol Chem 277, 38205-38211 26. Zundel, W., Schindler, C., Haas-Kogan, D., Koong, A., Kaper, F., Chen, E.,
Gottschalk, A. R., Ryan, H. E., Johnson, R. S., Jefferson, A. B., Stokoe, D., and Giaccia, A. J. (2000) Genes Dev 14, 391-396
27. Sulis, M. L., and Parsons, R. (2003) Trends Cell Biol 13, 478-483 28. Fresno Vara, J. A., Casado, E., de Castro, J., Cejas, P., Belda-Iniesta, C., and
Gonzalez-Baron, M. (2004) Cancer Treat Rev 30, 193-204 29. Brognard, J., Clark, A. S., Ni, Y., and Dennis, P. A. (2001) Cancer Res 61, 3986-
3997 30. Goberdhan, D. C., and Wilson, C. (2003) Hum Mol Genet 12 Spec No 2, R239-
248 31. Sansal, I., and Sellers, W. R. (2004) J Clin Oncol 22, 2954-2963 32. Jung, Y. J., Isaacs, J. S., Lee, S., Trepel, J., and Neckers, L. (2003) Faseb J 17,
2115-2117 33. Jung, Y., Isaacs, J. S., Lee, S., Trepel, J., Liu, Z. G., and Neckers, L. (2003)
Biochem J 370, 1011-1017 34. Belperio, J. A., Dy, M., Burdick, M. D., Xue, Y. Y., Li, K., Elias, J. A., and
Keane, M. P. (2002) Am J Respir Cell Mol Biol 27, 419-427
11
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

35. Myers, M. P., Stolarov, J. P., Eng, C., Li, J., Wang, S. I., Wigler, M. H., Parsons, R., and Tonks, N. K. (1997) Proc Natl Acad Sci U S A 94, 9052-9057
36. Schioppa, T., Uranchimeg, B., Saccani, A., Biswas, S. K., Doni, A., Rapisarda, A., Bernasconi, S., Saccani, S., Nebuloni, M., Vago, L., Mantovani, A., Melillo, G., and Sica, A. (2003) J Exp Med 198, 1391-1402
37. Caruz, A., Samsom, M., Alonso, J. M., Alcami, J., Baleux, F., Virelizier, J. L., Parmentier, M., and Arenzana-Seisdedos, F. (1998) FEBS Lett 426, 271-278
38. Zhong, H., Chiles, K., Feldser, D., Laughner, E., Hanrahan, C., Georgescu, M. M., Simons, J. W., and Semenza, G. L. (2000) Cancer Res 60, 1541-1545
39. Hendler, F. J., and Ozanne, B. W. (1984) J Clin Invest 74, 647-651 40. Franklin, W. A., Veve, R., Hirsch, F. R., Helfrich, B. A., and Bunn, P. A., Jr.
(2002) Semin Oncol 29, 3-14 41. Salomon, D. S., Brandt, R., Ciardiello, F., and Normanno, N. (1995) Crit Rev
Oncol Hematol 19, 183-232 42. Schlessinger, J. (2000) Cell 103, 211-225 43. Bjornsti, M. A., and Houghton, P. J. (2004) Nat Rev Cancer 4, 335-348 44. Kuzmin, I., Duh, F. M., Latif, F., Geil, L., Zbar, B., and Lerman, M. I. (1995)
Oncogene 10, 2185-2194 45. Zatyka, M., Morrissey, C., Kuzmin, I., Lerman, M. I., Latif, F., Richards, F. M.,
and Maher, E. R. (2002) J Med Genet 39, 463-472 46. Krishnamachary, B., Berg-Dixon, S., Kelly, B., Agani, F., Feldser, D., Ferreira,
G., Iyer, N., LaRusch, J., Pak, B., Taghavi, P., and Semenza, G. L. (2003) Cancer Res 63, 1138-1143
FOOTNOTES
*We should like to thank Dr. Charles Sawyers for kindly providing the C124S-PTEN and WT-PTEN mammalian expression vectors. This work was supported by NIH Grants HL66027, CA87879, and P50CA90388. 1The abbreviations used in this manuscript are: NSCLC, Non-small cell lung cancer; CXCR4, CXC chemokine receptor 4; HIF-1, Hypoxia Inducible Factor-1; VHL; Von Hippel-Lindau; PI3-kinase, phosphatidylinositol 3-kinase; PTEN, phosphatase and tensin homolog deleted on chromosome 10; HAS, human serum albumin; EMSA, electrophoretic mobility shift assay; RTK, receptor tyrosine kinase; EGF, epidermal growth factor; RLU, relative light units; mTOR, mammalian target of rapamycin.
FIGURE LEGENDS
Fig.1. Hypoxia induces CXCR4 expression and enhances CXCR4 function. (A). A549 cells and H157 cells were serum-starved and exposed to either normoxia or hypoxia (94% N2, 5% CO2 and 1% O2) for the times indicated. Changes in gene expression were then determined by real-time quantitative PCR. Fold induction represents increases in CXCR4 expression under hypoxic conditions compared to the normoxic controls. Results are representative of five separate experiments. (B). A549 cells and H157 cells were treated as in (A) and then subjected to western analysis to examine changes in CXCR4 protein levels. Results are representative of five separate experiments. (C). A549 cells and H157 cells were serum-starved and exposed to either normoxia or hypoxia for 24 hours and stained for cell surface expression of CXCR4. Surface expression was determined by flow cytometry using a rabbit anti-human CXCR4 monoclonal antibody.
12
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Results are representative of three separate experiments. (D). A549 cells and H157 cells were treated as in (C) and then subjected to chemotaxis in response to the indicated concentrations of CXCL12 for 6 hours. Data represent the mean ± SEM from 5 high power fields. *P < 0.05. Fig.2. Hypoxia induces activation of the HIF-1 transcription and transactivation of the CXCR4 promoter. (A). A549 cells and H157 cells were serum-starved and exposed to either normoxia or hypoxia (94% N2, 5% CO2 and 1% O2) for the times indicated. Subsequently, nuclear extracts were prepared and analyzed by SDS-PAGE and western blotting with a specific mouse-anti human HIF-1α monoclonal antibody. Results are representative of six separate experiments. (B). A549 cells and H157 cells were serum-starved and exposed to either normoxia or hypoxia for 4 hours. Nuclear extracts were then prepared and analyzed by EMSA (Lane 1, normoxic extract; lanes 2-4 hypoxic extracts). Binding studies were performed using a wildtype 32P-labeled oligo (lanes 1, 2, and 4) or a mutant 32P-labeled oligo (lane 3). Specific, inducible binding is represented by HIF-1. NS, non-specific binding; C, constitutive binding. (C). A549 cells and H157 cells were transfected with a reporter construct comprising a 2.6 kb fragment of the wildtype CXCR4 promoter (WT-CXCR4; lanes 1 and 2, and 5 and 6) or a CXCR4 promoter containing a mutation in the HIF-1 binding site (M-CXCR4; lanes 3 and 4, and 7 and 8) upstream of a luciferase gene and a separate renilla control reporter. Cells were co-transfected with either a control vector expressing GFP (lanes 1, 3, 5, and 7) or a vector expressing HIF-1α (lanes 2,4,6, and 8). After 48 hours in culture extracts were prepared and analyzed in a luminometer. RLU (relative light units) were normalized to the renilla control. Data represent the mean ± SEM. *P < 0.05. Fig.3. PI3-kinase inhibitors block HIF-1α induction and CXCR4 mRNA expression. (A). and (B). A549 cells and H157 cells were serum-starved, left untreated or pretreated with the PI3-kinase inhibitors LY294002 (50 µM) and wortmannin (250 nM) for 2 hours and exposed to normoxia or hypoxia (94% N2, 5% CO2 and 1% O2) for either 6 hours (A (i) and B (i)) or 24 hours (A (ii) and (iii) and B (ii) and (iii)). Subsequently, nuclear ((A (i) and B (i)) or cytoplasmic (A (ii) and (iii) and B (ii) and (iii)) extracts were prepared and analyzed by western blotting for HIF-1α (A (i) and B (i)), CXCR4 (A (ii) and B (ii)), and GAPDH (A (iii) and B (iii)). Results are representative of three separate experiments. (C) and (D). A549 cells and H157 cells were serum-starved, left untreated or pretreated with LY294002 (50 µM) for 2 hours and then exposed to either normoxia or hypoxia for 24 hours. Changes in gene expression were then determined by real-time quantitative PCR. Fold induction represents increases in CXCR4 expression under hypoxic conditions compared to the normoxic controls. Results are representative of three separate experiments. Data are the mean ± SEM. *P < 0.05. Fig.4. Wildtype PTEN, but not mutant PTEN, blocks hypoxia-induced CXCR4 expression. A549 cells were transfected with mammalian expression vectors containing either wildtype PTEN (WT-PTEN) or a catalytically inactive form of PTEN (C124S-PTEN). Transfected cells were then exposed to either normoxia or hypoxia (94% N2, 5% CO2 and 1% O2) for 24 hours and RNA prepared. Changes in CXCR4 mRNA expression were then determined by real-time quantitative PCR. Fold induction represents increases in CXCR4 expression under hypoxic conditions compared to the normoxic controls. Data are the mean ± SEM. *P < 0.05. Fig.5. EGF induces CXCR4 expression via a pathway that involves PI3-kinase and HIF-1α. (A). A549 cells and H157 cells were serum-starved, left untreated or pretreated with LY294002 (50 µM) for 2 hours and then exposed to either normoxia or hypoxia (94% N2, 5% CO2 and 1% O2) for 24 hours in the presence or absence of EGF (20 ng/ml). RNA was then prepared and changes in gene expression determined by real-time quantitative PCR. Fold induction represents increases
13
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

in CXCR4 expression under hypoxic conditions compared to the normoxic controls. Results are representative of three separate experiments. (B). A549 cells were serum-starved for 24 hours and then exposed to either normoxia or hypoxia for the times indicated in the presence or absence of EGF (20 ng/ml). Subsequently, cytoplasmic extracts were prepared and analyzed by immunoblotting for CXCR4 and GAPDH protein levels. Results are representative of three separate experiments. (C). A549 cells were serum-starved, left untreated or pretreated with LY294002 (50 µM) for 2 hours and then stimulated with EGF (20 ng/ml) for 24 hours under normoxic conditions. Next, cytoplasmic extracts were prepared and analyzed by western for CXCR4 and GAPDH protein levels. Results are representative of three separate experiments. Fig. 6. Activation of AKT in NSCLC cells. (A) A549 cells and (B) H157 cells were serum-starved, left untreated (lanes 1 and 10) or pretreated with the PI3-kinase inhibitors LY294002 (50 µM; lanes 2, 5, 8, 11, 14,and 17) and wortmannin (250 nM; 3, 6, 9, 12, 15, and 18) for 2 hours. Cells were then either left unstimulated (lanes 1-3 and 10-12) or stimulated with 10% FCS (lanes 4-6 and 13-15) and 20 ng/ml EGF (lanes 7-9 and 16-18) for 10 minutes. Whole cell extracts were then prepared and phosphorylated AKT and total AKT levels determined by immunoblotting. Results are representative of three separate experiments. Fig. 7. Rapamycin inhibits EGF induced CXCR4 and HIF-1a under both normoxic and hypoxic conditions. (A). A549 cells were serum starved, pretreated with rapamycin (10 ng/ml) for 2 hours and then stimulated with EGF (20 ng/ml) for a further 24 hours. Subsequently, RNA was prepared and changes in CXCR4 gene expression determined by real-time quantitative PCR. Fold induction represents increases in CXCR4 expression under hypoxic conditions compared to the normoxic controls. Results are representative of three separate experiments. (B). Serum-starved A549 cells in normoxia or hypoxia were pretreated with rapamycin (10 ng/ml) for 2 hours and then stimulated with EGF (20 ng/ml) for an additional 6 hours. Next, nuclear extracts were prepared and activation of HIF-1α determined by immunoblotting. Fig.8. EGF pretreatment augments chemotaxis of CXCR4-bearing A549 cells. A549 cells were serum starved and then exposed to normoxia or hypoxia in the absence or presence of EGF (20 ng/ml) for 24 hours. The cells were then subjected to chemotaxis in response to CXCL12 (1, 3, 10, 30, 100ng/ml) for 6 hours. Data represent the mean ± SEM from 10 high power fields. Fig.9. CXCL12-mediated chemotaxis of A549 cells is inhibited by PI3-kinase inhibitors. A549 cells were either left untreated (Lane 1) or pretreated with the PI3-kinase inhibitors wortmannin (100 nM; lanes 3 and 5) and LY294002 (20 µM; lanes 4 and 6) for 2 hours. Cells were then examined for chemotactic behavior in the presence or absence of CXCL12 (30 ng/ml; lanes 2, 5 and 6). Data represent the mean ± SEM from 5 high power fields. *P < 0.05.
14
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from
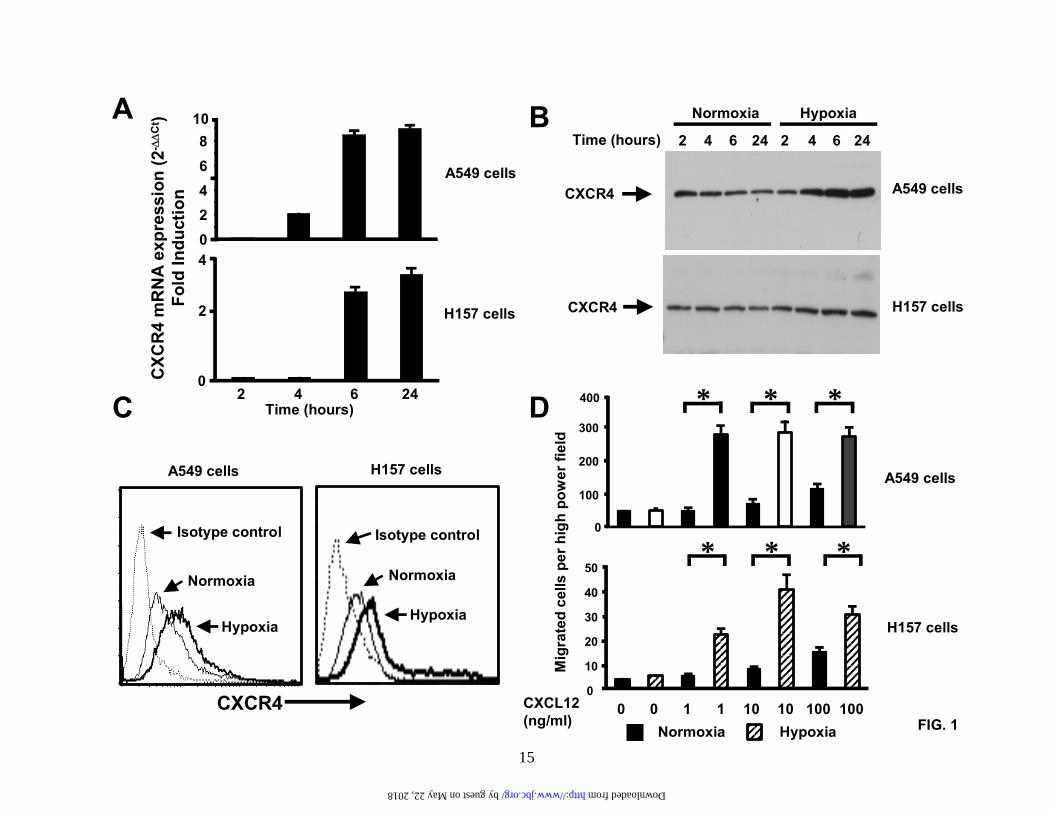
FIG. 1
CXC
R4
mR
NA
exp
ress
ion
(2-∆
∆C
t )Fo
ld In
duct
ion
02468
10
Time (hours)
0
2
4
2 4 6 24
A549 cells
H157 cells
A
D
0
10
20
30
40
50
0
100
200
300
400
CXCL12(ng/ml)
0 0 1 1 10 10 100 100Normoxia Hypoxia
A549 cells
H157 cells
C
A549 cells
Isotype control
Normoxia
Hypoxia
H157 cells
Isotype control
Normoxia
Hypoxia
CXCR4
* * *
* * *
B
CXCR4 A549 cells
CXCR4 H157 cells
2 4 6 24 2 4 6 24Time (hours)
Normoxia Hypoxia
Mig
rate
d ce
lls p
er h
igh
pow
er fi
eld
15
by guest on May 22, 2018 http://www.jbc.org/ Downloaded from
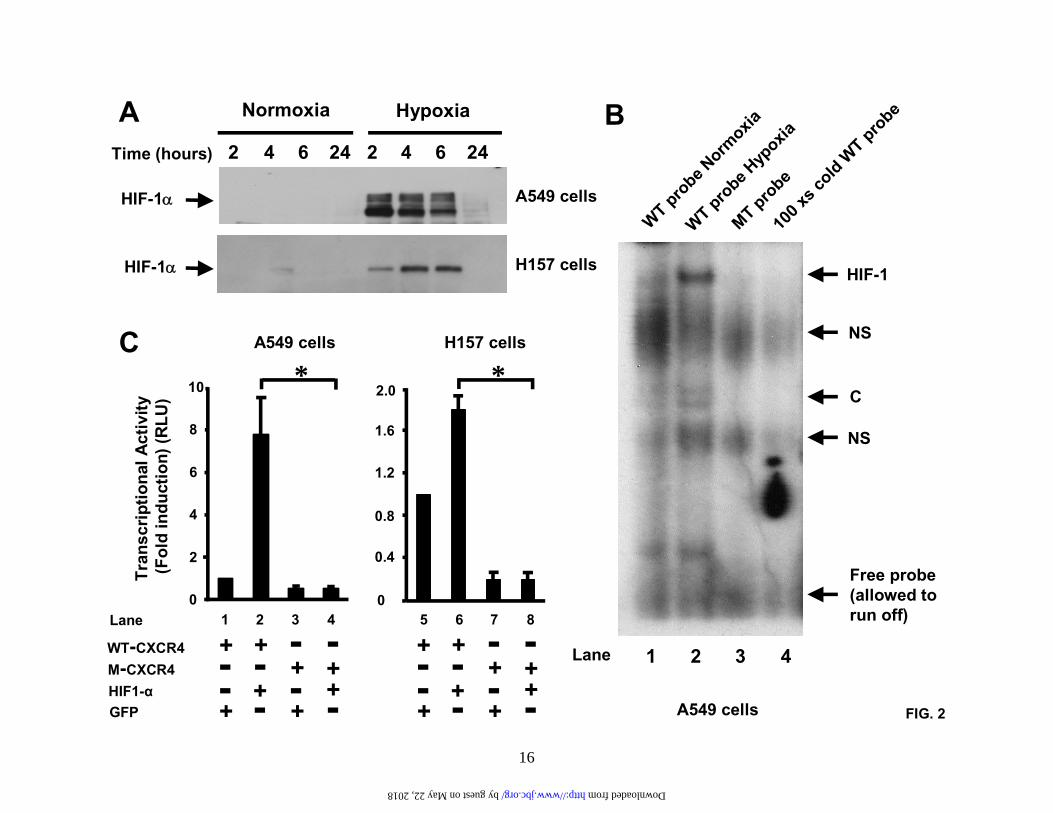
FIG. 2
A B
HIF-1α
Time (hours) 2 4 6 24 2 4 6 24
Normoxia Hypoxia
HIF-1α HIF-1
NS
NS
C
Free probe(allowed torun off)
WT probe N
ormoxia
WT probe H
ypoxia
MT probe
100 x
s cold
WT probe
WT-CXCR4M-CXCR4
GFPHIF1-α
A549 cells
H157 cells
A549 cells
0
2
4
6
8
10
Tran
scrip
tiona
l Act
ivity
(Fol
d in
duct
ion)
(RLU
)
0
0.4
0.8
1.2
1.6
2.0
C* *
Lane 1 2 3 4
A549 cells H157 cells
Lane 1 2 3 4 5 6 7 8
+ ++ +
+ +++
- --- -- -- + +
+ ++ +
++
- --- -- --
16
by guest on May 22, 2018 http://www.jbc.org/ Downloaded from
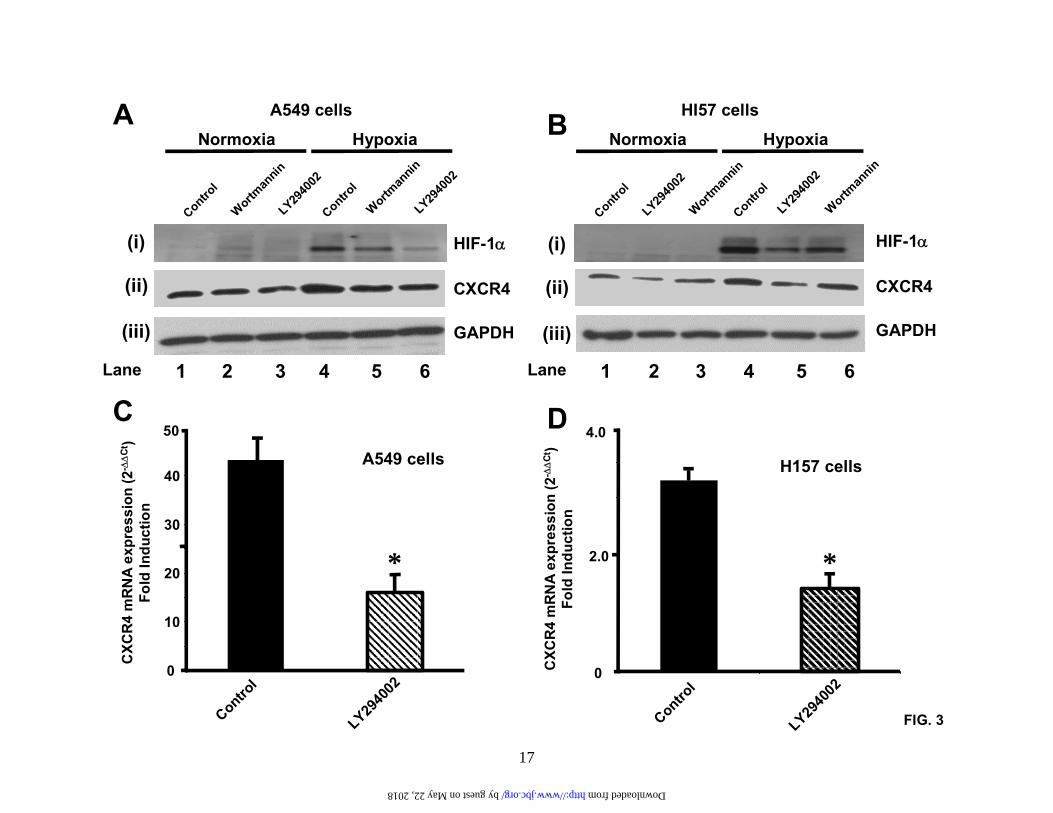
FIG. 3
HIF-1α
CXCR4
GAPDH
A
Control
Wortman
ninLY29
4002
Control
Wortman
ninLY29
4002
A549 cells
HypoxiaNormoxia
Control
LY2940
020
10
20
30
40
50
CXC
R4
mR
NA
expr
essi
on (2
-∆∆
Ct)
Fold
Indu
ctio
n
*
A549 cells
C
0
2.0
4.0
Control
LY2940
02
H157 cells
*
D
CXC
R4
mR
NA
expr
essi
on (2
-∆∆
Ct)
Fold
Indu
ctio
n
Lane 1 2 3 4 5 6
(i)
(ii)
(iii)
B
Control
LY2940
02Wortm
annin
Control
LY2940
02Wortm
annin
HI57 cells
HypoxiaNormoxia
Lane 1 2 3 4 5 6
(i)
(ii)
(iii)
HIF-1α
CXCR4
GAPDH
17
by guest on May 22, 2018 http://www.jbc.org/ Downloaded from
0
2
4
6
8
10
12
14
Con
WT-PTEN
C124S
-PTEN
Con
WT-PTEN
C124S
-PTEN
CXC
R4
mR
NA
exp
ress
ion
(2-∆
∆C
t )Fo
ld In
duct
ion
Normoxia Hypoxia
*
*
A549 cells
FIG. 4
18
by guest on May 22, 2018 http://www.jbc.org/ Downloaded from
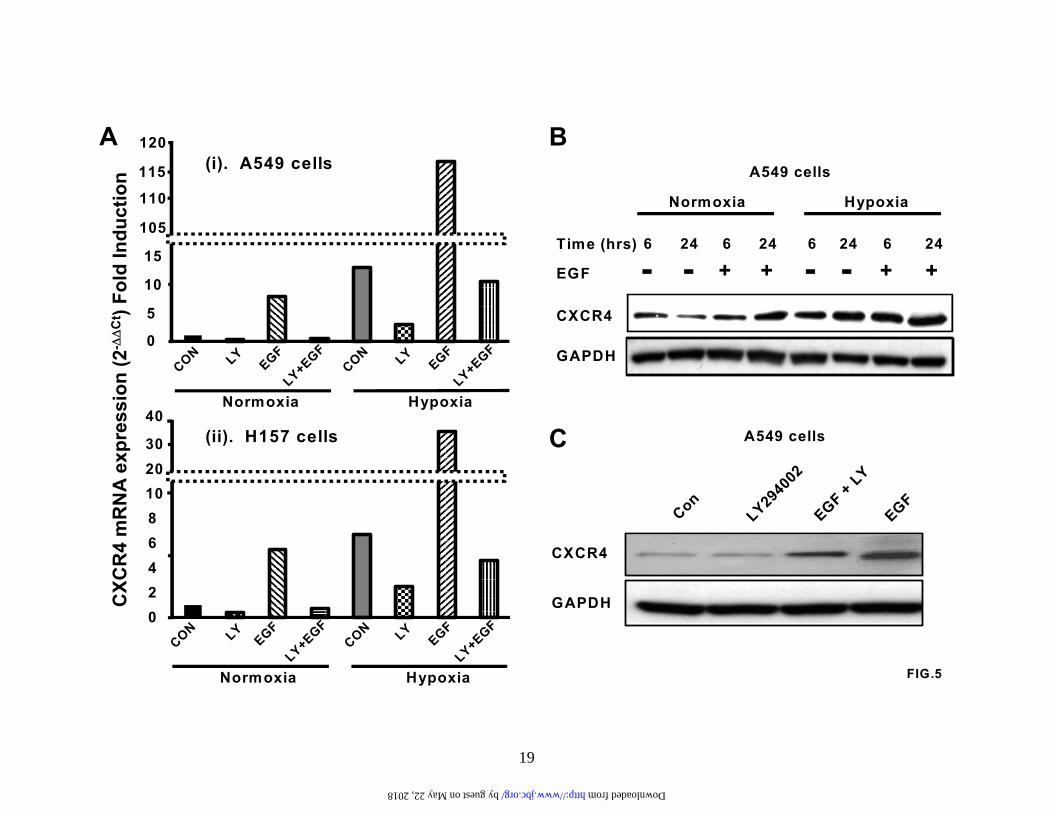
FIG.5
A
02468102030
40
CON
EGF LY+EGF
LY
CON
EGF LY+EGF
LY
Normoxia Hypoxia
0
5
10
15
105
110
115
120C
XCR
4 m
RN
A ex
pres
sion
(2-∆
∆C
t ) Fo
ld In
duct
ion
CON
EGF LY+EGF
LY
CON
EGF LY+EGF
LY
Normoxia Hypoxia
(i). A549 cells
(ii). H157 cells
CXCR4
GAPDH
Time (hrs)
BA549 cells
HypoxiaNormoxia
EGF
6 24 246246246
- - - -+ + + +
C
Con
LY29400
2EGF + LY
EGF
A549 cells
CXCR4
GAPDH
19
by guest on May 22, 2018 http://www.jbc.org/ Downloaded from
-- - - - -+ + +-- - -- -+ + +-- - ++- - --- + -- +- + -+- - +- -- +
Total AKT
Phospho AKT
HSA
EGF
FCS
LY294002
Wortmannin
-- - - - -+ + +-- - -- -+ + +-- - ++ +- - -+-- + -- +- + -+- - +- -- - +
1 2 3 4 5 6 987
-
A549 cells H157 cells
Lane Lane 11 12 13 14 15 1610 1817
FIG.6
A B
20
by guest on May 22, 2018 http://www.jbc.org/ Downloaded from
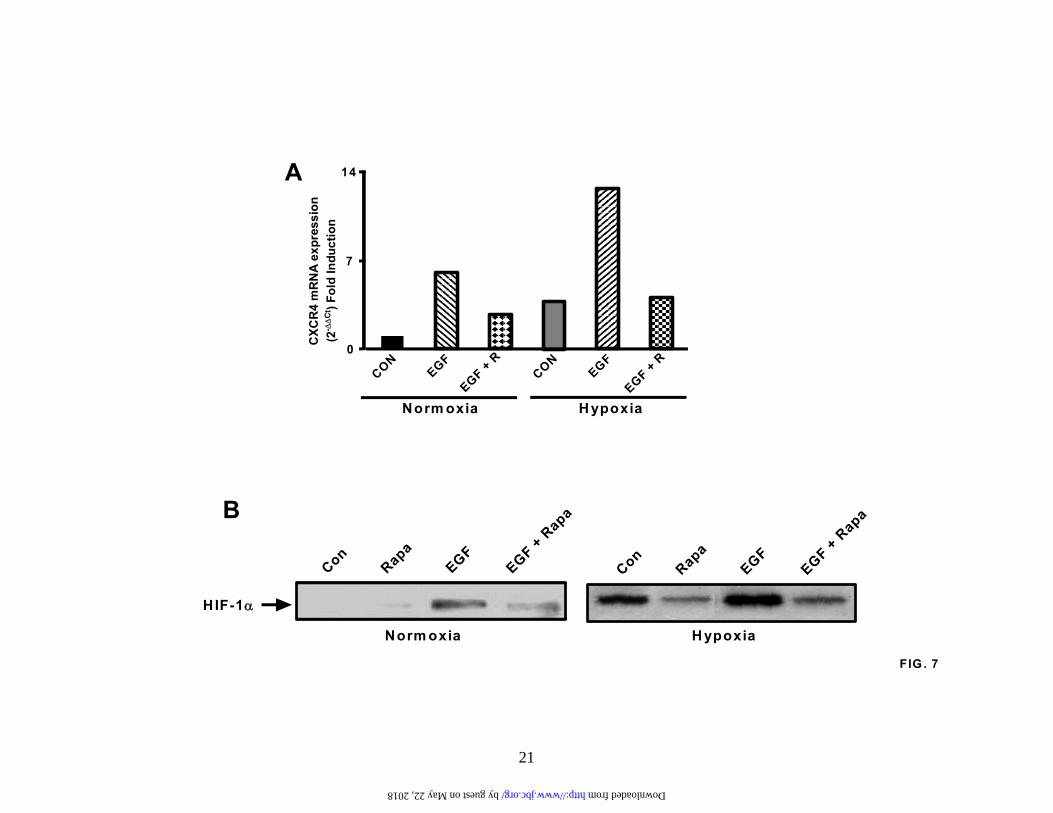
FIG . 7CON
EGFEGF + R
CON
EGF
EGF + RNorm oxia H ypoxia
0
14
7
CXC
R4
mR
NA
expr
essi
on(2
-∆∆
Ct) F
old
Indu
ctio
n
A
B
Con
Rapa
EGF
EGF + Rapa
HIF-1α
Con
Rapa
EGF
EGF + Rapa
Norm oxia H ypoxia
21
by guest on May 22, 2018 http://www.jbc.org/ Downloaded from
Norm oxiaNorm oxia + EG F (20ng/m l)H ypoxiaH ypoxia + EG F (20ng/m l)
0
10
20
30
40
50
60
70
80
0 1 3 10 30 100
Mig
rate
d ce
lls p
er h
igh
pow
er fi
eld
Concentration CXCL12 (ng/m l)FIG .8
22
by guest on May 22, 2018 http://www.jbc.org/ Downloaded from
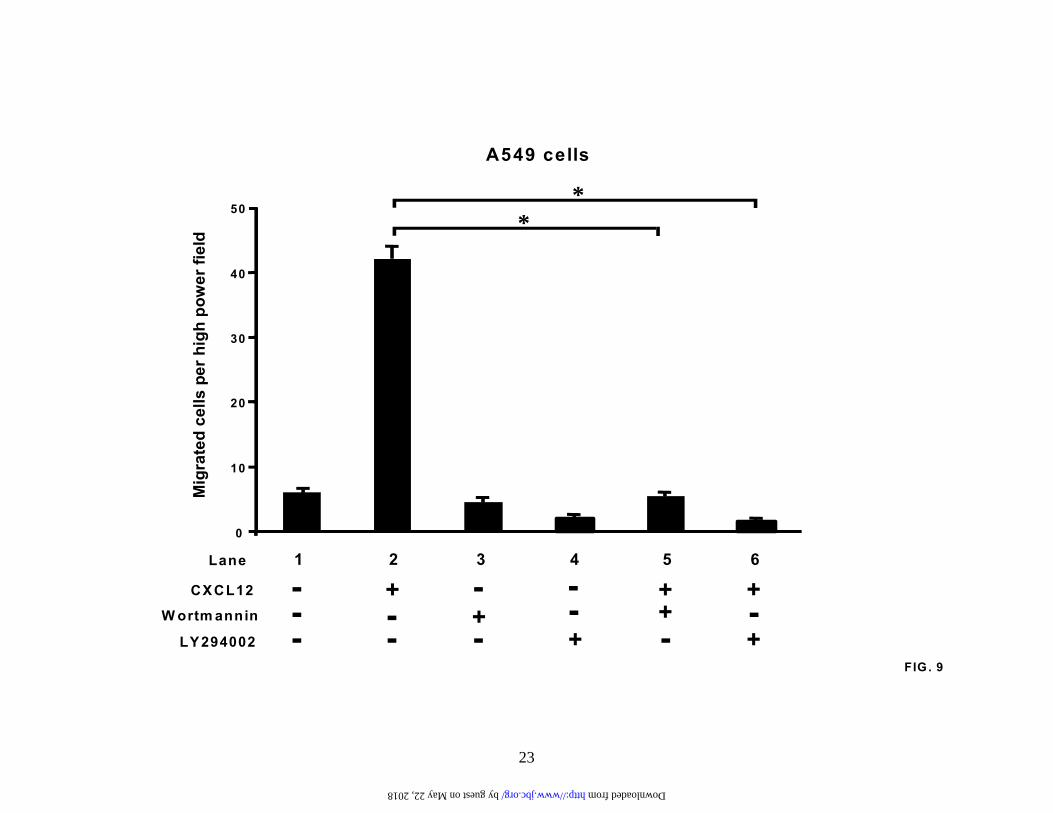
FIG . 9
0
10
20
30
40
50
CXCL12
W ortm annin
LY294002
--- -
- ---
--
-++
++
++
+
**
A549 cells
Mig
rate
d ce
lls p
er h
igh
pow
er fi
eld
Lane 1 2 3 4 65
23
by guest on May 22, 2018 http://www.jbc.org/ Downloaded from

Sica, John A. Belperio, Michael P. Keane and Robert M. StrieterRoderick J. Phillips, Javier Mestas, Mehrnaz Gharaee-Kermani, Marie D. Burdick, Antonio
αHIF-1regulated by the PI3-kinase/PTEN/AKT/mTOR signaling pathway and activation of
EGF and hypoxia-induced expression of CXCR4 on non-small cell lung cancer cells is
published online March 31, 2005J. Biol. Chem.
10.1074/jbc.M500963200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on May 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from














![CLNS 09/2049 CLEO 09-02 D and K mesons - arXiv · The CLEO-c detector has been described in detail elsewhere [8, 9, 10]. The 53-layer track-ing system, composed of two drift chambers](https://static.fdocument.org/doc/165x107/60586a9e2e7ec550e00b44f9/clns-092049-cleo-09-02-d-and-k-mesons-arxiv-the-cleo-c-detector-has-been-described.jpg)



