Crystal Structure of Glucose: Placing Hydrogen Atoms · PDF fileMaterials Design Application...
3
Click here to load reader
Transcript of Crystal Structure of Glucose: Placing Hydrogen Atoms · PDF fileMaterials Design Application...

APPLICATION NOTE
Copyright © Materials Design 2009
Crystal Structure of Glucose: Placing Hydrogen Atoms by Computations
The crystal structure of a purely organic, hydrogen-bonded molecular crystal is very well described by density functional theory with a gradient-corrected Perdew-Becke-Ernzerhof potential. The computations were preformed with the VASP program using the projector augmented wave method with a plane wave basis set. The agreement between computed and experimental lattice parameters is better than 2% with a tendency of the calculations to overestimate the bond lengths. The calculations provide equilibrium positions for the hydrogen atoms, which are difficult to place based on x-ray diffraction data.
Keywords: Glucose crystal, computed structure, hydrogen bonds
Experimental Facts The sugar α-glucose is an extremely important building block of organic materials. In 1952 McDonald and Beevers [1] reported the crystal structure of this organic crystal. This was one of the first structure determinations of crystalline compounds containing a sugar, just one year before Watson and Crick published their work on the structure of DNA.
A network of hydrogen bonds provides the intermolecular bonding resulting in a crystal with the space group P212121. The density obtained from the x-ray diffraction data is reported to be 1.56 g/cm-3. In their work, McDonald and Beevers were unable to determine the positions of the H-atoms involved in the hydrogen bonding and thus they placed these H-atoms simply in the middle of the O-H…H motifs.
Computed results The computed equilibrium lattice parameters are in good agreement with experimental data as can be seen in Table 1. In fact the slight overestimation of bond lengths is fairly typical for the gradient-corrected density functional calculations.
Table 1. Computed and experimental lattice parameters for crystalline α-glucose. The computed density is 1.54 g/cm-3 compared with 1.56 g/cm-3 obtained from experiment [1].
a (Å) b (Å) c (Å)
Experiment [1] 10.36 ± 0.02 14.84 ± 0.02 4.97 ± 0.02
Computed (DFT-PBE, VASP) 10.36 14.85 5.05
Deviation 0.0 % 0.1 % 1.7 %

Materials Design Application Note Crystal structure of α-glucose
- 2 -
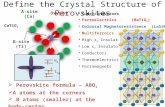
Figure 1. Crystal structure derived from x-ray diffraction [1] and from ab-initio computations using MedeA-VASP.
Figure 2. Detail of the experimental and computed crystal structure of α-glucose. In the experimental structure H-atoms were simply placed in the middle of O…O pairs. The computed structure gives the asymmetric arrangements typical of hydrogen bonds.

Materials Design Application Note Crystal structure of α-glucose
- 3 -
Significance VASP with its plane wave basis is very well suited for purely organic molecular crystals, which are bonded by a network of H-bonds. In fact the precision in describing bonding geometries and total energies of molecules with VASP has been demonstrated for the G2-1 test set of molecules [2]. VASP achieves a precision which can only be reached by very large Gaussian (quintuple) basis sets. This illustrates the universality and performance of the projector augmented plane wave method as implemented in VASP.
It should be noted that the present DFT-GGA level of theory does not describe those molecular crystals properly, which are bonded by weak van-der-Waals interactions (such as solid methane). For these systems, a special treatment of the dispersive forces is required to obtain good intermolecular distances.
MedeA modules used for this application The present calculations were performed with the MedeA platform using the following integrated modules of the MedeA software environment
• Standard MedeA framework including crystal structure builders and geometric analysis tools
• JobServer and TaskServers
• VASP 5.2 and its graphical user interface as integrated in MedeA
References 1. T. R. R. McDonald and C. A. Beevers, Acta
Cryst. 5, 654 (1952)
2. J. Paier, R. Hirschl, M. Marsman, and G. Kresse, J. Chem. Phys. 122, 234102 (2005)
For further information please contact Materials Design, Inc. PO Box 2000, 3465 Mountain View Blvd. #17 Angel Fire, NM 87710, USA T +1 760 495-4924 F +1 760 897-2179 [email protected] www.materialsdesign.com