
Chung Han Wang. Voltage-gated channelopathies Voltage-dependent Na+ channels nine genes encode the...
12
Chung Han Wang
-
Upload
bryce-stephens -
Category
Documents
-
view
223 -
download
0
Transcript of Chung Han Wang. Voltage-gated channelopathies Voltage-dependent Na+ channels nine genes encode the...

Chung Han Wang

Voltage-gated channelopathies
• Voltage-dependent Na+ channelsnine genes encode the pore-forming α subunit, with four genes encoding the ancillary β subunitsgene subunit Clinical syndrome mutatuin
SCN1A α1 GEFS+
Dravet syndromR1648HW1204RD188YD1866Y
SCN2A α2 BFNISDravet syndrom
R188WR102XL1563V
SCN1B β1 GEFS+ C121WR85CR85H
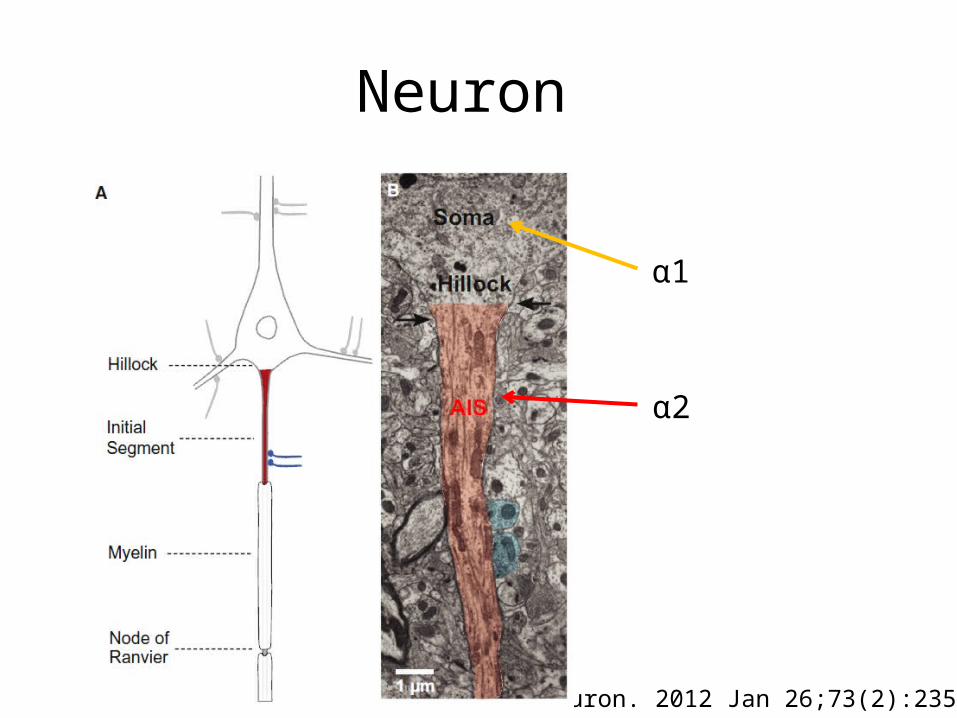
Neuron
Neuron. 2012 Jan 26;73(2):235-47.
α1
α2

SCN1A• Forms a fast inactivating voltage-dependent Na+ channel.
Clinical syndrome and molecular findings• Since first discovered in 2000 over 100 mutations in SCN1A have been
described in epilepsy.• Missense mutations at various sites in SCN1A protein are associated with
GEFS+.• While most individuals have mild self-limited phenotypes, the devastating
disorder of Dravet syndrome can also be seen in patients with SCN1A mutations.
• More often, Dravet syndrome occurs as a sporadic disorder with de novo mutations.
• Both missense mutations and nonsense mutations are seen.• Recently, deletions of whole exons, multiple exons or the whole gene have
been found in Dravet syndrome.• Vaccination, particularly with pertussis vaccine, was previously alleged to
cause a severe epileptic encephalopathy.• Most cases of GEFS+ appear to have complex inheritance, likely due to a
number of genes.

Functional impact of mutations and potential neuronalmechanisms.
• 1.R1648H accelerated recovery from inactivation possibly due to an impairment in a secondary ‘latch’ that stabilizes inactivation.2.W1204R showed a negative shift in the voltage-dependence of activation and steady-state inactivation.3. D188Y showed less use-dependent decline in Na+ channel amplitude.4. D1866Y mutation also decreased the modulation by β1 subunit of the SCN1A protein resulting in an increased persistent current.
• Increases in persistent Na+ current will contribute to depolarisation, thereby reducing the voltage required to fire action potentials.
• Persistent Na+ current can also contribute to the shaping of repetitive firing, the generation of rhythmicity and the amplification of both excitatory and inhibitory currents.
• More recent studies have not supported the simple view that gain-of-function mutations in Na+ current should be responsible for hyperexcitability.
• A number of SCN1A mutations (e.g., V1353L,I1657C, R859C) linked to GEFS+ exhibit loss-of-function.
• This argues that both increases and decreases in channel activity can underlie neuronal hyperexcitability that causes GEFS+.

Animal models• The Scn1a knock-out mouse models mimic the loss of-function mutations
found in most cases of this disease.• In some genetic backgrounds, heterozygous mice developed spontaneous
seizures and sporadic death reflecting the severity of the disease in humans.
• Interestingly, although Na+ currents were essentially unchanged in hippocampal excitatory pyramidal neurons, the current was substantially reduced in inhibitory interneurons.
• This was due to differential compensation by other Na+ channel subunits within the different cell types.
• A significant reduction in interneuron function was postulated to underlie hyperexcitability in these animals.
• A heterozygous knock-in mouse carrying the R1407X truncation mutation found in humans also displayed epileptic seizures early in life.
• This study structurally isolates a SCN1A protein deficit to the AIS of a subpopulation of inhibitory interneurons in the developing neocortex.
• pronounced action potential attenuation during continuous firing in fast-spiking parvalbumin-positive interneurons.

• Therefore, a selective loss of inhibitory neuron contribution during network activity may explain the severe excitable phenotype noted in Dravet syndrome.
• It is therefore possible that a loss- and gain-of function can result in an excitable phenotype, via impact on different neuron types.

SCN2A• They are important for action potential initiation, propagation and the
generation of repetitive firing.
Clinical syndrome and molecular findings• Missense mutations in this gene have been most frequently associated
with BFNIS,a mild, self-limited epilepsy syndrome of the first year of life.• Earlier, a small Japanese family with complex inheritance contributing to
febrile and afebrile seizures was observed with a missense R188W mutation.
• Similarly to that observed with the SCN1A mutations, where missense mutations often have mild phenotypes and truncations more severe ones.
• A de novo nonsense mutation in SCN2A (R102X) has been found in a single subject with more a severe epileptic phenotype that has similarities with Dravet syndrome.

Functional impact of mutations and potential neuronalmechanisms
• The R188W mutation slow the inactivation of the channel and an augmentation of the Na+ current was predicted to underlie neuronal excitability.
• L1563V mutation associated with BFNIS revealed an increase in excitability due to a depolarised shift in fast inactivation.
• The mutation increased excitability of the neonatal isoform of the SCN2A channel but not the adult isoform.
• The greater excitability of the adult relative to the neonatal isoform suggests a role for developmentally regulated splicing in controlling neuronal excitability.
• Analysis of the R102X mutant protein, linked to a more severe epileptic phenotype, co-expressed with the wild-type (WT) channel revealed that mutated protein shifted the voltage dependence of inactivation of the WT protein in a hyperpolarizing direction.
• SCN2A channels harbouring the L1330F, L1563V, R223Q, and R1319Q mutations have also been transfected into pyramidal and bipolar neocortical neuronal cultures.

SCN1B• SCN1B encodes the β1 ancillary subunit. • Na+ channelβ subunits are multifunctional, modulating channel gating,
regulating the level of channel expression and potentially acting as a cell adhesion molecule.
Clinical syndrome and molecular findings• The first mutation in SCN1B was described in an Australian family with
GEFS+.• A missense mutation was found at a highly conserved cysteine residue
(C121W) in an extracellular domain.• This mutation disrupts a disulfide bridge involved in the stabilisation of the
immunoglobulin-like domain.• Families with SCN1B mutations have been recently described including
two novel mutations (R85C and R85H) .• Five patients with the C121W mutation had confirmed temporal lobe
epilepsy.

Functional impact of mutations and potential cellularmechanisms
• Electrophysiological studies of co-expressed mutant b (C121W) subunit and pore-forming a subunit, in both mammalian and Xenopus oocycte expression systems, are consistent with loss of b subunit function.
• The result is a potential increase in Na+ current due to slowing of inactivation, increased availability of channels at hyperpolarized potentials, and reduction in the channel rundown during high-frequency activation.
• A similar loss of modulatory effects of the β subunit on SCN2A channels was noted for the R85C and R85H mutations. A net increase in Na+ current is predicted to increase neuron excitability.
• The impact of the C121W (or other) mutation may not be limited to its effect on Na+ channel kinetics. A cell adhesion assay demonstrated a disruption in the ability of the mutated β subunit to mediate protein–protein interactions.
• The homozygous Scn1B knock-out mouse displays spontaneous generalised seizures.
• An abnormal expression pattern for the SCN1A and SCN3A protein in hippocampal neurons, a disruption in axo glial communication and/or a developmental disruption in neurite growth are present in the knock-out animal.



















