
c3gc40136g 1095..1111 - stuba.skszolcsanyi/education/files/Chemia... · DOI: 10.1039/c3gc40136g...
17
Green Chemistry TUTORIAL REVIEW Cite this: Green Chem., 2013, 15, 1095 Received 18th January 2013, Accepted 28th February 2013 DOI: 10.1039/c3gc40136g www.rsc.org/greenchem Hydrolysis of cellulose to glucose by solid acid catalysts Yao-Bing Huang and Yao Fu* As the main component of lignocelluloses, cellulose is a biopolymer consisting of many glucose units connected through β-1,4-glycosidic bonds. Breakage of the β-1,4-glycosidic bonds by acids leads to the hydrolysis of cellulose polymers, resulting in the sugar molecule glucose or oligosaccharides. Mineral acids, such as HCl and H 2 SO 4 , have been used in the hydrolysis of cellulose. However, they suffer from problems of product separation, reactor corrosion, poorcatalyst recyclability and the need for treatment of waste effluent. The use of heterogeneous solid acids can solve some of these problems through the ease of product separation and good catalyst recyclability. This review summarizes recent advances in the hydrolysis of cellulose by different types of solid acids, such as sulfonated carbonaceous based acids, polymer based acids and magnetic solid acids. The acid strength, acid site density, adsorption of the sub- stance and micropores of the solid material are all key factors foreffective hydrolysis processes. Methods used to promote reaction efficiency such as the pretreatment of cellulose to reduce its crystallinity and the use of ionic liquids or microwave irradiation to improve the reaction rate are also discussed. 1. Introduction The discovery and utilization of fossil resources has changed the energy supplement of the whole world. Current energy systems are mainly based on these fossil resources such as coal, petroleum and natural gas. However, the rapid consump- tion of these resources, together with the resulting global warming caused by CO 2 emissions, raises sustainability issues regarding the existing energy systems based on fossil resources. To solve this problem, people are forced to explore renewable resources to substitute the fossil resources in order to meet increasing energy demands. 1–4 Until now, various forms of energy systems have been developed as excellent alternatives such as solar, wind, biomass, hydroelectric and geothermal. 5,6 Among these new forms of energy sources, biomass is the only sustainable source of organic carbon on Earth, which is considered as one part of the solution for pro- ducing fuels and chemicals. 7–9 Biomass can be obtained all over the world and it generally occurs in the form of organic materials such as grass, wood, agricultural crops and their residues and waste. These Yao-Bing Huang Yao-Bing Huang has studied at the University of Science of Tech- nology of China (USTC) since 2004. After obtaining his Bache- lor’s Degree in 2008, he contin- ued to study green organic synthesis under the supervision of Prof. Qiang-Xiang Guo and Yao Fu. Now, he works on the development of efficient hetero- geneous catalysts for organic reactions and biomass conver- sion. Yao Fu Dr Yao Fu obtained his PhD degree from the Department of Chemistry at USTC in 2005. He then began his academic career at the Anhui Province Key Lab- oratory of Biomass Clean Energy. He is presently a professor of organic chemistry at USTC. His research interests cover tran- sition-metal catalyzed organic reactions, the synthesis of hetero- geneous catalysts for green chem- istry processes and the selective conversion of biomass derived compounds into value-added chemicals and biofuels. Department of Chemistry, Anhui Province Key Laboratory of Biomass Clean Energy, University of Science and Technology of China, Hefei 230026, P. R. China. E-mail: [email protected] This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 1095–1111 | 1095 Downloaded by University of Oxford on 30/04/2013 15:22:23. Published on 28 February 2013 on http://pubs.rsc.org | doi:10.1039/C3GC40136G View Article Online View Journal | View Issue
Transcript of c3gc40136g 1095..1111 - stuba.skszolcsanyi/education/files/Chemia... · DOI: 10.1039/c3gc40136g...

Green Chemistry
TUTORIAL REVIEW
Cite this: Green Chem., 2013, 15, 1095
Received 18th January 2013,Accepted 28th February 2013
DOI: 10.1039/c3gc40136g
www.rsc.org/greenchem
Hydrolysis of cellulose to glucose by solid acid catalysts
Yao-Bing Huang and Yao Fu*
As the main component of lignocelluloses, cellulose is a biopolymer consisting of many glucose units
connected through β-1,4-glycosidic bonds. Breakage of the β-1,4-glycosidic bonds by acids leads to the
hydrolysis of cellulose polymers, resulting in the sugar molecule glucose or oligosaccharides. Mineral
acids, such as HCl and H2SO4, have been used in the hydrolysis of cellulose. However, they suffer from
problems of product separation, reactor corrosion, poor catalyst recyclability and the need for treatment
of waste effluent. The use of heterogeneous solid acids can solve some of these problems through the
ease of product separation and good catalyst recyclability. This review summarizes recent advances in the
hydrolysis of cellulose by different types of solid acids, such as sulfonated carbonaceous based acids,
polymer based acids and magnetic solid acids. The acid strength, acid site density, adsorption of the sub-
stance and micropores of the solid material are all key factors for effective hydrolysis processes. Methods
used to promote reaction efficiency such as the pretreatment of cellulose to reduce its crystallinity and
the use of ionic liquids or microwave irradiation to improve the reaction rate are also discussed.
1. Introduction
The discovery and utilization of fossil resources has changedthe energy supplement of the whole world. Current energysystems are mainly based on these fossil resources such ascoal, petroleum and natural gas. However, the rapid consump-tion of these resources, together with the resulting globalwarming caused by CO2 emissions, raises sustainability issues
regarding the existing energy systems based on fossilresources. To solve this problem, people are forced to explorerenewable resources to substitute the fossil resources in orderto meet increasing energy demands.1–4 Until now, variousforms of energy systems have been developed as excellentalternatives such as solar, wind, biomass, hydroelectric andgeothermal.5,6 Among these new forms of energy sources,biomass is the only sustainable source of organic carbon onEarth, which is considered as one part of the solution for pro-ducing fuels and chemicals.7–9
Biomass can be obtained all over the world and it generallyoccurs in the form of organic materials such as grass, wood,agricultural crops and their residues and waste. These
Yao-Bing Huang
Yao-Bing Huang has studied atthe University of Science of Tech-nology of China (USTC) since2004. After obtaining his Bache-lor’s Degree in 2008, he contin-ued to study green organicsynthesis under the supervisionof Prof. Qiang-Xiang Guo andYao Fu. Now, he works on thedevelopment of efficient hetero-geneous catalysts for organicreactions and biomass conver-sion. Yao Fu
Dr Yao Fu obtained his PhDdegree from the Department ofChemistry at USTC in 2005. Hethen began his academic careerat the Anhui Province Key Lab-oratory of Biomass Clean Energy.He is presently a professor oforganic chemistry at USTC. Hisresearch interests cover tran-sition-metal catalyzed organicreactions, the synthesis of hetero-geneous catalysts for green chem-istry processes and the selectiveconversion of biomass derived
compounds into value-added chemicals and biofuels.
Department of Chemistry, Anhui Province Key Laboratory of Biomass Clean Energy,
University of Science and Technology of China, Hefei 230026, P. R. China.
E-mail: [email protected]
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 1095–1111 | 1095
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6G
View Article OnlineView Journal | View Issue

materials come from biological photosynthesis from CO2,water and sunlight, thus making them sustainable and greenfeedstocks with zero carbon emission.10 The first generation ofbiofuels are mainly bioethanol and biodiesel which are pro-duced from sugars, starches and vegetable oils. However, con-cerns were raised that the utilization of these materialscompeted with food for feedstocks.11 Moreover, their potentialavailability is limited by the amount of fertile soil.11
Due to the above limitations, the second generation of bio-fuels has mainly concentrated on the utilization of lignocellu-losic biomass. Lignocellulose can be grown in combinationwith food or on barren land. Every year, sees the formation ofabout 220 billion dry tons of available biomass (ca. 45 EJ ofenergy content),12,13 and lignocellulose forms about 70–95%of it.14 Lignocelluloses are composed of mainly three com-ponents, cellulose (40–50%), hemicelluloses (25–35%) andlignin (15–20%) (percentages of the components vary fromdifferent biomass resources).15,16 A schematic biorefinery forthe utilization of lignocellulose is shown in Scheme 1.17
Among these biopolymers, cellulose is the most valuable onewhich can be converted into glucose and subsequently fermen-ted into bioethanol or dehydrated into platform molecules.18
Besides, cellulosic biofuels have been considered as the mostpromising candidates for the replacement of current pet-roleum based fuels.19
The first step for cellulose utilization is to depolymerize itinto soluble oligosaccharides and glucose. However, thenatural polymer forms a robust crystal structure with highchemical stability, thus making the depolymerization pro-cesses more difficult. The established methods for cellulosehydrolysis are known to be catalyzed by cellulose enzymes. Butlimitations of the enzymes systems are obvious in that the pro-cesses are always low efficiency and have a high enzymecost.20–22 At the same time, much work has been concentratedon the hydrolysis of cellulose to glucose with mineral acids.Sulfuric acid is known as a typical acid for this reaction.However, the large-scale use of acid suffers from several
problems such as reactor corrosion, catalyst recovery andrequires treatment of the acid residue, producing lots ofwaste.23,24 From the industrial point of view, the above limit-ations must be taken into account when designing productionprocesses. In addition, several aspects also need to be ofconcern such as economy, simplicity, efficiency and environ-mental friendliness.
The utilization of heterogeneous acids has the potential toovercome some of the above limitations due to the ease of sep-aration of catalysts. Significant development of this transform-ation has been made by using various types of catalysts withlarge pore size and strong acid strength.25–28 Associated withthese solid acids, many pretreatment technologies have alsobeen developed to reduce the crystallinity of cellulose andincrease its surface area in order to improve the reactionefficiency and selectivity, for example, ball-milling,29,30 solubil-ization/precipitation in ionic liquids,31 liquid acids/alkalinesolutions,32,33 the non-thermal atmospheric plasma method34
and so on. It is worth noting that most hydrolysis processeswith solid acids require the pretreatment of cellulose as shownin Table 1. Apart from that, microwave irradiation inducedassistance of the hydrolysis of cellulose is an effective heatingmethod compared to conventional oil heating.35–37
In this review, we summarize the recent advances in thehydrolysis of cellulose into glucose with solid acids. The reviewis divided into the following parts: (1) structure of cellulose;(2) hydrolysis of cellulose by liquid acids; (3) hydrolysis of cel-lulose by solid acids. The final part of this review presents anoutlook of the future of this domain.
2. Structure of cellulose
Early exploration work on the structure of cellulose was carriedout by Anseleme Payen.38 He obtained a resistant fibrous solidmaterial with the formula C6H10O5 after the treatment ofvarious plant tissues with acids and ammonia. It was the
Scheme 1 Utilization of lignocelluloses to produce chemicals and fuels.
Tutorial Review Green Chemistry
1096 | Green Chem., 2013, 15, 1095–1111 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online
earliest “cellulose”.39 The structure of cellulose was elucidatedin 1920 by Hermann Staudinger.40 He found that cellulose wascomposed of D-glucose units, linked to each other to form longchains (Scheme 2). The structure of cellulose was made up ofmany glucose moieties linked in the form of β-D-anhydrogluco-pyranose units (AGUs). The AGUs are linked to each otherthrough glycosidic bridges on the C1 and C4 carbon atoms.41
The number of repeating AGUs is defined as the degree ofpolymerization (DP) of cellulose. Generally, the DP of cellulosevaries according to the original type of materials, for example,the common range of DP for wood pulp is about 300–1700 andis 800–10 000 for cotton or other plant fibers.41 Due to thehuge number of hydroxyl groups and linkage of β-1,4-glyco-sidic bonds, it is easy to form intra- and intermolecule hydrogenbonds which make the cellulose resistant during chemical andbiological treatment and make it highly insoluble in commonsolvents.42
According to the crystal features, cellulose has at least sevenforms, Iα, Iβ, II, IIII, IIIII, IVI, IVII.
43,44 In nature, most cellulose
occur in the state of Iα, Iβ. They can be found in the samematerial from almost every biomass source. The ratio of thetwo crystalline forms depends on the source of cellulose. Gen-erally, cellulose Iα is rich in the cell walls of algae and bacterialcellulose while cellulose Iβ is abundant in cotton, wood andtunicate cellulose.45 Cellulose Iα,β is thermodynamically notthe most stable form in nature.46 When cellulose I swells inconcentrated sodium hydroxide, it easily forms the thermody-namically more stable cellulose II after the removal of the swel-ling agent.42,43 Cellulose III can be obtained by treatment ofcellulose Iα, Iβ with the ammonia fiber explosion process. Cel-lulose IV can be obtained by treating cellulose III with glycerolat 206 °C.42,43 However, cellulose III, IV are not common andare thus ignored in our discussion. The more generally knownand discussed forms of cellulose are microcrystalline Avicelcellulose and α-cellulose which come from pure bacterial cellu-lose and the treatment of wood with alkali extraction.47
3. Hydrolysis of cellulose by liquid acids
The hydrolysis of cellulose is a process to break the β-1,4-glyco-sidic bonds of the polymer which is an essential step for theconversion of cellulose and opens the possibility of sub-sequent catalytic transformations. Direct hydrolysis of ligno-cellulosic biomass with acids goes back to the early 19th century
Table 1 Different performances of solid acid catalysts in the hydrolysis of cellulose
CatalystAcidity(mmol g−1)
Pretreatmentmethod Solvents
Assistancemethod
Temp.(K)
Time(h)
Glucose andTRSa yield/% Ref.
HNbMoO6 1.90 — H2O — 403 12 8.5 63Zn–Ca–Fe — — H2O — 433 20 29 64Amberlyst-15 — — [BMIm]Cl/H2O 373 5 11 68CP–SO3H 0.067 — H2O 393 10 93 74Nafion NR50 — [BMIm]Cl H2O 403 2 35 76Nafion SAC50 — — H2O 463 24 11 77PCPs–SO3H 1.80 — H2O — 393 3 5.3 78AC–SO3H 1.90 — H2O — 373 3 64 75BC–SO3H 1.98 — H2O Microwave 363 1 19.8 83CSA–SO3H
b 1.76 Milled/sieved H2O Microwave 403 1 34.6 84SC–SO3H 2.15 [BMIm]Cl/H2O — 383 4 63 85AC–N–SO3H-250 2.23 Milled H2O — 423 24 62.6 86CMK-3-SO3H 2.39 Milled H2O — 423 24 74.5 86SimCn–SO3H 0.37 Milled H2O — 423 24 50.4 87H3PW12O40 — Milled H2O — 423 2 18 97H3PW12O40 — — H2O — 453 2 50.5 98H5BW12O40 — — H2O — 333 6 77 99H3PW12O40 — Milled H2O Microwave 363 3 75.6 100Micellar HPA — — H2O — 443 8 39.3 101[MIMPSH]nH3−nPW — — H2O/MIBK — 413 5 36 102CsH2PW12O40 — — H2O — 433 6 27 103H-beta 1.05 Milled H2O — 423 24 12 105HY zeolite — Milled [C4mim]Cl/H2O Microwave 373 0.13 37 106HY zeolite 1.36 — [BMIm]Cl/H2O — 403 2 50 107Fe3O4–SBA–SO3H 1.09 — H2O — 423 3 26 108Fe3O4–SBA–SO3H 1.09 Solvent activation H2O — 423 3 50 108MNPs@SiO2–SO3H 0.5 — H2O — 423 3 30.2 112Ru/CMK-3 — Milled H2O — 503 24 34.2 116CaFe2O4 0.068 Milled H2O — 423 24 36 117HT–OHCa 1.17 Milled H2O — 423 24 40.7 118
a TRS: total reducing sugars. b Corncob was used as the starting material.
Scheme 2 Structure of cellulose (degree of polymerization n = DP).
Green Chemistry Tutorial Review
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 1095–1111 | 1097
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online

and several processes were reported to be effective in the 20thcentury.48 The first technology for the acid hydrolysis of cellu-lose was developed by Faith in 1923.49 In the process, sulfuricacid solution (0.5 wt%) was used to treat wood waste in brick-lined percolators. After about 45 min at 170 °C, a dilute sugarsolution was obtained. The solution was transferred for fermen-tation after neutralization. A 50% yield of sugars was obtainedaccording to the theoretical yield of fermentable sugars.
After the work, many processes were reported to be effectivein the hydrolysis of cellulose. They used different kinds ofacids such as HCl, H2SO4, HF and organic acids (oxalic,maleic, furmaric). In 1937, Bergius reported the hydrolysis ofcellulose with 40 wt% HCl (ca. 12 mol L−1) at room temp-erature.50 Cellulose was solubilized in concentrated acid togetherwith hemicelluloses, leaving insoluble lignin. The hydrolysisproducts were glucose and oligosaccharides without dehy-dration products. Concentrated HCl (6–7 mol L−1) was alsoeffective in the hydrolysis of cellulose in the presence of CaCl2and LiCl as the additives.51 The results showed that the saltshad a swelling effect by increasing the hydrolysis rate, leadingto an 85% yield of glucose at 90 °C. Additionally, concentratedacids had an apparent swelling effect on cellulose. Celluloseswells when the sulfuric acid concentration is above 50%.52
Camacho et al.53 studied the effect of H2SO4 concentration(from 31%–70%, (w/v)) on the solubilization rate of microcrys-talline cellulose, revealing that acid promoted the total solubil-ization of cellulose when the concentration was above 62%.Although hydrolysis with concentrated acids operates at lowtemperature and atmospheric pressure, these processes hadstrict requirements on water content and displayed severecorrosion.50
Hydrolysis of cellulose with dilute acid usually requireshigh temperature. Madison et al.54 reported a treating processin which wood was treated with 0.5 wt% H2SO4 in a continu-ous reaction. The degradation of products was minimized forthe short residence time of cellulose in the reactor. Anothersuccessful process using an isothermal plug flow reactor wasreported by Grethlein and Thompson.55 They used 1 wt%H2SO4 in the continuous process at 240 °C with a short resi-dence time of 0.22 min; 50% glucose was obtained at last. Afew years later, Harris et al.56 used a two stage system contain-ing dilute H2SO4 in the saccharification of wood. After thehemicellulose was extracted, cellulose was transferred forhydrolysis and a high purity of glucose was obtained.
The hydrolysis of cellulose was achieved with gaseous HClas reported by the Wilke group.57 The HCl stream wasadsorbed on the dried wood particles and initiated thehydrolysis of hemicelluloses. After extraction of the hemicellu-lose sugars, the residue was dried and mixed with HCl gas at45 °C, achieving 90% conversion of the available carbohydratecontent of wood to sugars. High pressure HCl systems werealso reported for the hydrolysis reaction.57,58 Apart fromgaseous HCl, anhydrous HF was employed in the hydrolysis ofcellulose. The main advantage of using anhydrous HF is itslow boiling point (19.5 °C) which facilitated the acid recovery.The yield of sugars was about 45% at 0 °C.59 The recovery
experiment showed that only 0.05–0.1% of HF remained in thesystem, opening the possibility of reusing mineral acids in thehydrolysis of cellulose.57
Current strategies for the hydrolysis of cellulose with acidsare mainly accomplished with the acids mentioned above.Although homogeneous-based processes are efficient, themineral acid systems still suffer from major problems inproduct/catalyst separation, reactor corrosion, catalyst recy-cling and the treatment of waste effluent. For example, in theH2SO4 based system, the acid in the reaction mixture has to beneutralized with a mixture of CaO/CaSO4 which forms lots ofgypsum as waste.56 For the HCl system it is difficult to reusethe liquid acid while the HF system is very toxic when utilizedin large-scale processes.57,60 In the search for greener pro-cesses for hydrolyzing cellulose, solid acids were developed toaddress some of the problems and have some unique catalyticproperties that mineral acids do not possess.
4. Hydrolysis of cellulose by solid acids
The hydrolysis of cellulose into sugars with solid acids hasreceived increasing attention. Solid acid catalysts have variousadvantages over liquid acid catalysts: ease of product separ-ation, recyclability, less damage to the reactor. Besides, the useof solid acid catalysts can reduce the pollutants which willhave a minimal impact on the environment. Up till now,numerous reviews have been published on the transformationof cellulose with solid acids.25–28 Herein, we would like to sum-marize the recent advances in this subject. Several types ofsolid acids are included in the following part and their cata-lytic performance is summarized in Table 1.
4.1 Metal oxides
Metal oxides are a type of solid catalyst with many Lewis acidsites. Metal oxides are always prepared with high specificsurface and pore sizes which are easy for the reactants toaccess and contact with the active sites inside the metal oxidepores. These metal oxides can be used for the hydrolysis ofsucrose, cellobiose and even cellulose.
As a type of strong solid acid, mesoporous transition-metaloxides have been prepared and used in organic chemical trans-formations.61 Recently, Domen et al.62 reported that meso-porous Nb–W oxide could be used as a solid catalyst for thehydrolysis of sucrose and cellobiose. The rate of glucose pro-duction and the turnover frequency (TOF) for the hydrolysis ofsucrose were higher than those of other solid acids (i.e. Amber-lyst-15, Nb2O5). The acid strength increased gradually with theaddition of W, reaching the highest reaction rate with meso-porous Nb3W7 oxide. The high catalytic performance of Nb3W7
oxide was attributed to the high surface area mesoporousstructure and strong acid sites. However, the Nb–W oxide cata-lyst had a lower activity for the cellobiose hydrolysis due to thelow Brønsted acid sites.
In order to solve the problem, Domen et al. also reporteda layered transition-metal oxide HNbMoO6 that exhibited
Tutorial Review Green Chemistry
1098 | Green Chem., 2013, 15, 1095–1111 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online
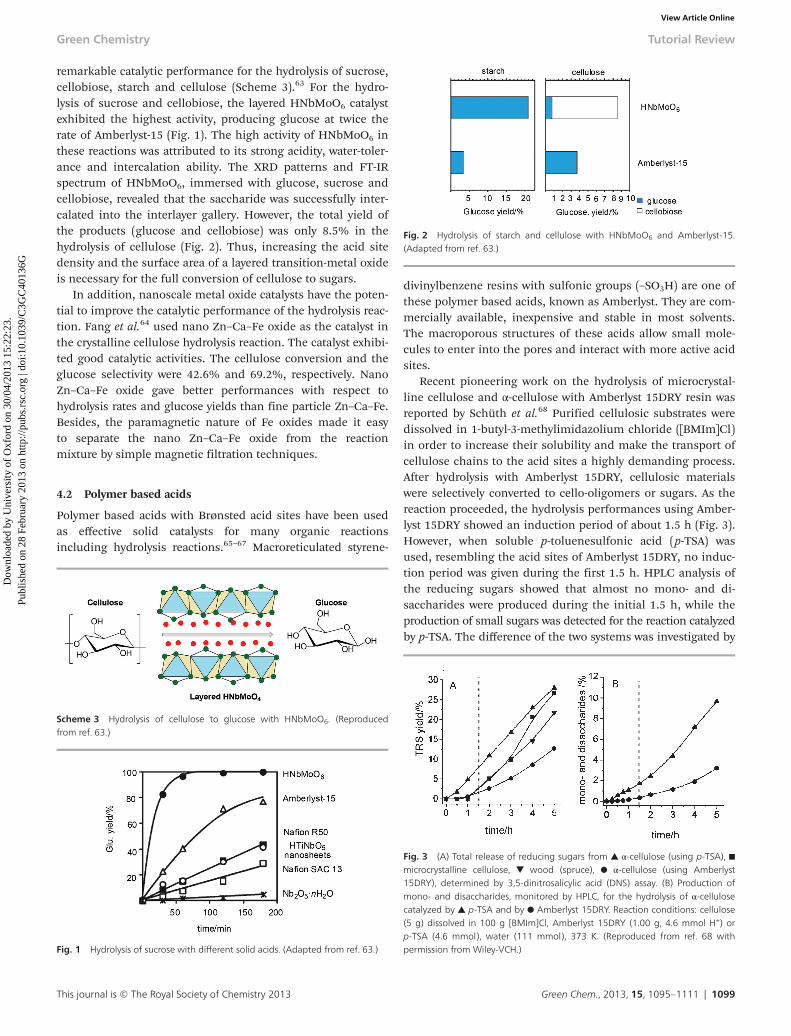
remarkable catalytic performance for the hydrolysis of sucrose,cellobiose, starch and cellulose (Scheme 3).63 For the hydro-lysis of sucrose and cellobiose, the layered HNbMoO6 catalystexhibited the highest activity, producing glucose at twice therate of Amberlyst-15 (Fig. 1). The high activity of HNbMoO6 inthese reactions was attributed to its strong acidity, water-toler-ance and intercalation ability. The XRD patterns and FT-IRspectrum of HNbMoO6, immersed with glucose, sucrose andcellobiose, revealed that the saccharide was successfully inter-calated into the interlayer gallery. However, the total yield ofthe products (glucose and cellobiose) was only 8.5% in thehydrolysis of cellulose (Fig. 2). Thus, increasing the acid sitedensity and the surface area of a layered transition-metal oxideis necessary for the full conversion of cellulose to sugars.
In addition, nanoscale metal oxide catalysts have the poten-tial to improve the catalytic performance of the hydrolysis reac-tion. Fang et al.64 used nano Zn–Ca–Fe oxide as the catalyst inthe crystalline cellulose hydrolysis reaction. The catalyst exhibi-ted good catalytic activities. The cellulose conversion and theglucose selectivity were 42.6% and 69.2%, respectively. NanoZn–Ca–Fe oxide gave better performances with respect tohydrolysis rates and glucose yields than fine particle Zn–Ca–Fe.Besides, the paramagnetic nature of Fe oxides made it easyto separate the nano Zn–Ca–Fe oxide from the reactionmixture by simple magnetic filtration techniques.
4.2 Polymer based acids
Polymer based acids with Brønsted acid sites have been usedas effective solid catalysts for many organic reactionsincluding hydrolysis reactions.65–67 Macroreticulated styrene-
divinylbenzene resins with sulfonic groups (–SO3H) are one ofthese polymer based acids, known as Amberlyst. They are com-mercially available, inexpensive and stable in most solvents.The macroporous structures of these acids allow small mole-cules to enter into the pores and interact with more active acidsites.
Recent pioneering work on the hydrolysis of microcrystal-line cellulose and α-cellulose with Amberlyst 15DRY resin wasreported by Schüth et al.68 Purified cellulosic substrates weredissolved in 1-butyl-3-methylimidazolium chloride ([BMIm]Cl)in order to increase their solubility and make the transport ofcellulose chains to the acid sites a highly demanding process.After hydrolysis with Amberlyst 15DRY, cellulosic materialswere selectively converted to cello-oligomers or sugars. As thereaction proceeded, the hydrolysis performances using Amber-lyst 15DRY showed an induction period of about 1.5 h (Fig. 3).However, when soluble p-toluenesulfonic acid (p-TSA) wasused, resembling the acid sites of Amberlyst 15DRY, no induc-tion period was given during the first 1.5 h. HPLC analysis ofthe reducing sugars showed that almost no mono- and di-saccharides were produced during the initial 1.5 h, while theproduction of small sugars was detected for the reaction catalyzedby p-TSA. The difference of the two systems was investigated by
Scheme 3 Hydrolysis of cellulose to glucose with HNbMoO6. (Reproducedfrom ref. 63.)
Fig. 3 (A) Total release of reducing sugars from ▲ α-cellulose (using p-TSA), ■
microcrystalline cellulose, ▼ wood (spruce), ● α-cellulose (using Amberlyst15DRY), determined by 3,5-dinitrosalicylic acid (DNS) assay. (B) Production ofmono- and disaccharides, monitored by HPLC, for the hydrolysis of α-cellulosecatalyzed by ▲ p-TSA and by ● Amberlyst 15DRY. Reaction conditions: cellulose(5 g) dissolved in 100 g [BMIm]Cl, Amberlyst 15DRY (1.00 g, 4.6 mmol H+) orp-TSA (4.6 mmol), water (111 mmol), 373 K. (Reproduced from ref. 68 withpermission from Wiley-VCH.)Fig. 1 Hydrolysis of sucrose with different solid acids. (Adapted from ref. 63.)
Fig. 2 Hydrolysis of starch and cellulose with HNbMoO6 and Amberlyst-15.(Adapted from ref. 63.)
Green Chemistry Tutorial Review
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 1095–1111 | 1099
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online
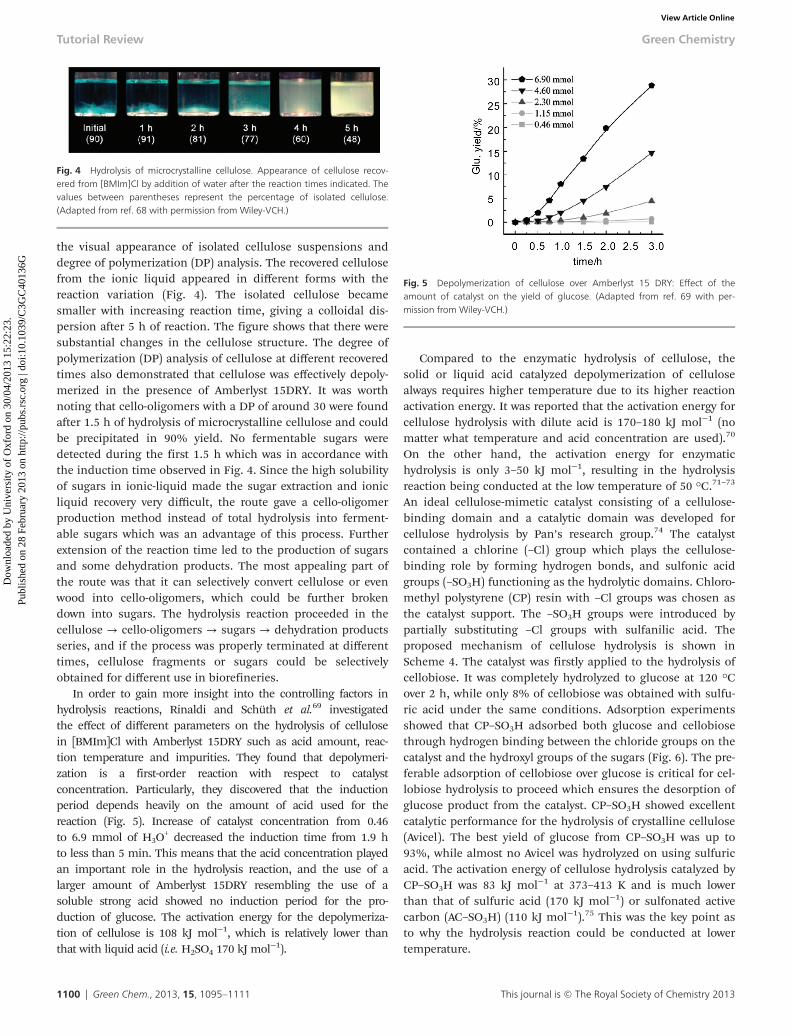
the visual appearance of isolated cellulose suspensions anddegree of polymerization (DP) analysis. The recovered cellulosefrom the ionic liquid appeared in different forms with thereaction variation (Fig. 4). The isolated cellulose becamesmaller with increasing reaction time, giving a colloidal dis-persion after 5 h of reaction. The figure shows that there weresubstantial changes in the cellulose structure. The degree ofpolymerization (DP) analysis of cellulose at different recoveredtimes also demonstrated that cellulose was effectively depoly-merized in the presence of Amberlyst 15DRY. It was worthnoting that cello-oligomers with a DP of around 30 were foundafter 1.5 h of hydrolysis of microcrystalline cellulose and couldbe precipitated in 90% yield. No fermentable sugars weredetected during the first 1.5 h which was in accordance withthe induction time observed in Fig. 4. Since the high solubilityof sugars in ionic-liquid made the sugar extraction and ionicliquid recovery very difficult, the route gave a cello-oligomerproduction method instead of total hydrolysis into ferment-able sugars which was an advantage of this process. Furtherextension of the reaction time led to the production of sugarsand some dehydration products. The most appealing part ofthe route was that it can selectively convert cellulose or evenwood into cello-oligomers, which could be further brokendown into sugars. The hydrolysis reaction proceeded in thecellulose → cello-oligomers → sugars → dehydration productsseries, and if the process was properly terminated at differenttimes, cellulose fragments or sugars could be selectivelyobtained for different use in biorefineries.
In order to gain more insight into the controlling factors inhydrolysis reactions, Rinaldi and Schüth et al.69 investigatedthe effect of different parameters on the hydrolysis of cellulosein [BMIm]Cl with Amberlyst 15DRY such as acid amount, reac-tion temperature and impurities. They found that depolymeri-zation is a first-order reaction with respect to catalystconcentration. Particularly, they discovered that the inductionperiod depends heavily on the amount of acid used for thereaction (Fig. 5). Increase of catalyst concentration from 0.46to 6.9 mmol of H3O
+ decreased the induction time from 1.9 hto less than 5 min. This means that the acid concentration playedan important role in the hydrolysis reaction, and the use of alarger amount of Amberlyst 15DRY resembling the use of asoluble strong acid showed no induction period for the pro-duction of glucose. The activation energy for the depolymeriza-tion of cellulose is 108 kJ mol−1, which is relatively lower thanthat with liquid acid (i.e. H2SO4 170 kJ mol−1).
Compared to the enzymatic hydrolysis of cellulose, thesolid or liquid acid catalyzed depolymerization of cellulosealways requires higher temperature due to its higher reactionactivation energy. It was reported that the activation energy forcellulose hydrolysis with dilute acid is 170–180 kJ mol−1 (nomatter what temperature and acid concentration are used).70
On the other hand, the activation energy for enzymatichydrolysis is only 3–50 kJ mol−1, resulting in the hydrolysisreaction being conducted at the low temperature of 50 °C.71–73
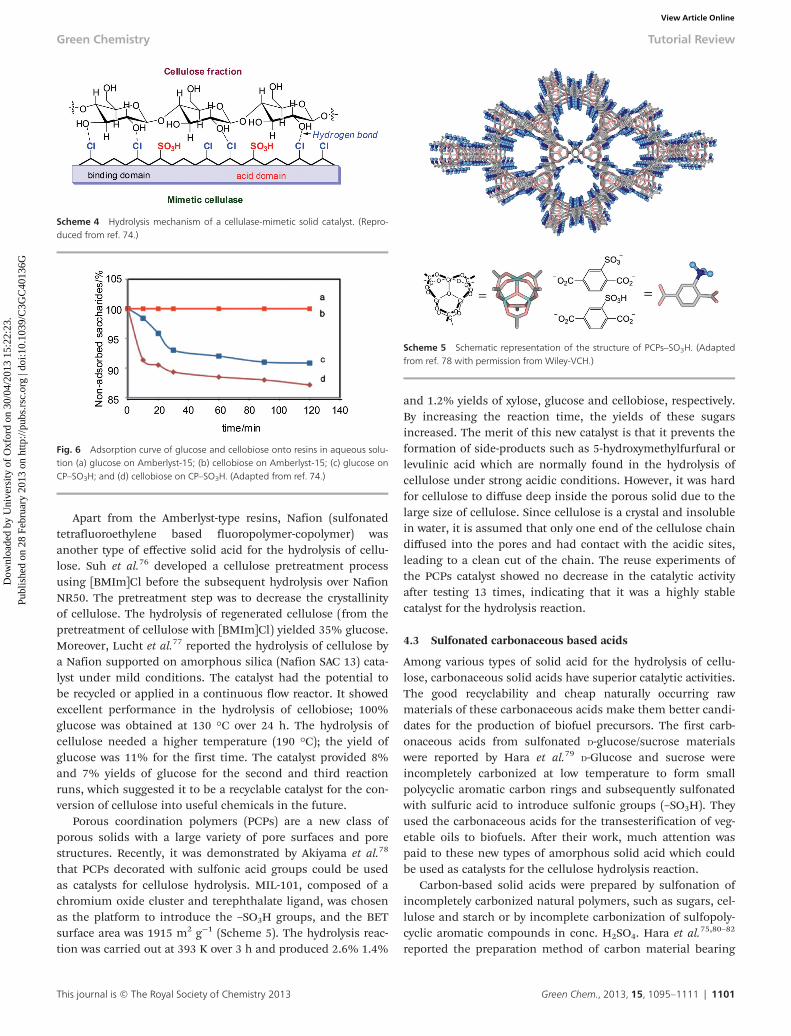
An ideal cellulose-mimetic catalyst consisting of a cellulose-binding domain and a catalytic domain was developed forcellulose hydrolysis by Pan’s research group.74 The catalystcontained a chlorine (–Cl) group which plays the cellulose-binding role by forming hydrogen bonds, and sulfonic acidgroups (–SO3H) functioning as the hydrolytic domains. Chloro-methyl polystyrene (CP) resin with –Cl groups was chosen asthe catalyst support. The –SO3H groups were introduced bypartially substituting –Cl groups with sulfanilic acid. Theproposed mechanism of cellulose hydrolysis is shown inScheme 4. The catalyst was firstly applied to the hydrolysis ofcellobiose. It was completely hydrolyzed to glucose at 120 °Cover 2 h, while only 8% of cellobiose was obtained with sulfu-ric acid under the same conditions. Adsorption experimentsshowed that CP–SO3H adsorbed both glucose and cellobiosethrough hydrogen binding between the chloride groups on thecatalyst and the hydroxyl groups of the sugars (Fig. 6). The pre-ferable adsorption of cellobiose over glucose is critical for cel-lobiose hydrolysis to proceed which ensures the desorption ofglucose product from the catalyst. CP–SO3H showed excellentcatalytic performance for the hydrolysis of crystalline cellulose(Avicel). The best yield of glucose from CP–SO3H was up to93%, while almost no Avicel was hydrolyzed on using sulfuricacid. The activation energy of cellulose hydrolysis catalyzed byCP–SO3H was 83 kJ mol−1 at 373–413 K and is much lowerthan that of sulfuric acid (170 kJ mol−1) or sulfonated activecarbon (AC–SO3H) (110 kJ mol−1).75 This was the key point asto why the hydrolysis reaction could be conducted at lowertemperature.
Fig. 4 Hydrolysis of microcrystalline cellulose. Appearance of cellulose recov-ered from [BMIm]Cl by addition of water after the reaction times indicated. Thevalues between parentheses represent the percentage of isolated cellulose.(Adapted from ref. 68 with permission from Wiley-VCH.)
Fig. 5 Depolymerization of cellulose over Amberlyst 15 DRY: Effect of theamount of catalyst on the yield of glucose. (Adapted from ref. 69 with per-mission from Wiley-VCH.)
Tutorial Review Green Chemistry
1100 | Green Chem., 2013, 15, 1095–1111 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online
Apart from the Amberlyst-type resins, Nafion (sulfonatedtetrafluoroethylene based fluoropolymer-copolymer) wasanother type of effective solid acid for the hydrolysis of cellu-lose. Suh et al.76 developed a cellulose pretreatment processusing [BMIm]Cl before the subsequent hydrolysis over NafionNR50. The pretreatment step was to decrease the crystallinityof cellulose. The hydrolysis of regenerated cellulose (from thepretreatment of cellulose with [BMIm]Cl) yielded 35% glucose.Moreover, Lucht et al.77 reported the hydrolysis of cellulose bya Nafion supported on amorphous silica (Nafion SAC 13) cata-lyst under mild conditions. The catalyst had the potential tobe recycled or applied in a continuous flow reactor. It showedexcellent performance in the hydrolysis of cellobiose; 100%glucose was obtained at 130 °C over 24 h. The hydrolysis ofcellulose needed a higher temperature (190 °C); the yield ofglucose was 11% for the first time. The catalyst provided 8%and 7% yields of glucose for the second and third reactionruns, which suggested it to be a recyclable catalyst for the con-version of cellulose into useful chemicals in the future.
Porous coordination polymers (PCPs) are a new class ofporous solids with a large variety of pore surfaces and porestructures. Recently, it was demonstrated by Akiyama et al.78
that PCPs decorated with sulfonic acid groups could be usedas catalysts for cellulose hydrolysis. MIL-101, composed of achromium oxide cluster and terephthalate ligand, was chosenas the platform to introduce the –SO3H groups, and the BETsurface area was 1915 m2 g−1 (Scheme 5). The hydrolysis reac-tion was carried out at 393 K over 3 h and produced 2.6% 1.4%
and 1.2% yields of xylose, glucose and cellobiose, respectively.By increasing the reaction time, the yields of these sugarsincreased. The merit of this new catalyst is that it prevents theformation of side-products such as 5-hydroxymethylfurfural orlevulinic acid which are normally found in the hydrolysis ofcellulose under strong acidic conditions. However, it was hardfor cellulose to diffuse deep inside the porous solid due to thelarge size of cellulose. Since cellulose is a crystal and insolublein water, it is assumed that only one end of the cellulose chaindiffused into the pores and had contact with the acidic sites,leading to a clean cut of the chain. The reuse experiments ofthe PCPs catalyst showed no decrease in the catalytic activityafter testing 13 times, indicating that it was a highly stablecatalyst for the hydrolysis reaction.
4.3 Sulfonated carbonaceous based acids
Among various types of solid acid for the hydrolysis of cellu-lose, carbonaceous solid acids have superior catalytic activities.The good recyclability and cheap naturally occurring rawmaterials of these carbonaceous acids make them better candi-dates for the production of biofuel precursors. The first carb-onaceous acids from sulfonated D-glucose/sucrose materialswere reported by Hara et al.79 D-Glucose and sucrose wereincompletely carbonized at low temperature to form smallpolycyclic aromatic carbon rings and subsequently sulfonatedwith sulfuric acid to introduce sulfonic groups (–SO3H). Theyused the carbonaceous acids for the transesterification of veg-etable oils to biofuels. After their work, much attention waspaid to these new types of amorphous solid acid which couldbe used as catalysts for the cellulose hydrolysis reaction.
Carbon-based solid acids were prepared by sulfonation ofincompletely carbonized natural polymers, such as sugars, cel-lulose and starch or by incomplete carbonization of sulfopoly-cyclic aromatic compounds in conc. H2SO4. Hara et al.75,80–82
reported the preparation method of carbon material bearing
Scheme 4 Hydrolysis mechanism of a cellulase-mimetic solid catalyst. (Repro-duced from ref. 74.)
Fig. 6 Adsorption curve of glucose and cellobiose onto resins in aqueous solu-tion (a) glucose on Amberlyst-15; (b) cellobiose on Amberlyst-15; (c) glucose onCP–SO3H; and (d) cellobiose on CP–SO3H. (Adapted from ref. 74.)
Scheme 5 Schematic representation of the structure of PCPs–SO3H. (Adaptedfrom ref. 78 with permission from Wiley-VCH.)
Green Chemistry Tutorial Review
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 1095–1111 | 1101
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online

–SO3H groups from microcrystalline cellulose at 723 K overr5 h under N2 flow. The black powder was then boiled in 15 wt%H2SO4 at 353 K under N2 for 10 h; after filtration andwashing with hot water, carbon based acid was obtained.Characterization of this solid acid showed that the carbonmaterial is composed of uniform functionalized graphenesheets, bearing –SO3H, –COOH and phenolic –OH groupswhich are different to conventional solid acids with singlefunctional groups (Scheme 6). The effective surface area of thecarbon material during hydrolysis was about 560 m2 g−1,larger than the BET surface area after hydrolysis (only2 m2 g−1). The H2O vapor adsorption–desorption isothermshowed that a larger amount of water was incorporated intothe bulk of the catalyst. The incorporation ability of hydro-philic molecules made it easy for a cellulose chain in solutionto be in contact with the –SO3H groups in the carbon material,which gave rise to high catalytic performance.
Comparable experiments for the hydrolysis of cellulose withdifferent solid acids revealed that carbon-based acids exhibitremarkable hydrolysis performances (Table 2). After reactingfor 3 h, 68% of cellulose was hydrolyzed to glucose (4% yield)and soluble β-1,4-glucan (64% yield), the liquid acid H2SO4
also has a high activity for the hydrolysis of cellulose (10%glucose and 38% β-1,4-glucan). The enhanced hydrolytic cata-lysis of the amorphous carbon with –SO3H, –OH and –COOHand was further investigated via an adsorption experimentwith cellobiose, a short β-1,4-glucan of cellulose. The results
showed that the strong interactions (hydrogen bonds to oxygenatoms in glycosidic bonds) between the phenolic OH groupsand the glycosidic bonds in β-1,4-glucan led to higher catalyticactivities. Such hydrogen bonds were expected to bind cellu-lose to the surface of the catalysts, making it easier for thehydrolysis to proceed. The activation energy for cellulosehydrolysis with this carbon based acid was 110 kJ mol−1 whichwas lower than that with sulfuric acid (170 kJ mol−1). Theamorphous carbon acid was recovered from the reactionmixture and reused at least 25 times without decrease inactivity. Elemental analysis and ion chromatography revealedthat only 1% –SO3H groups leached into the solution duringthe first reaction and no further leaching was detected in sub-sequent reactions. The excellent performance of the carbonbased catalyst opens the possibility of converting cellulose intovalue-added molecules with cheap and efficient solid acids,which has the potential for use in industrial production in thefuture.
A mild process for the hydrolysis of cellulose to reducingsugars under microwave irradiation was developed by Fu andYin et al.83 They used carbon sulfuric acids (BC–SO3H) derivedfrom bamboo, cotton and starch as catalysts. The introductionof microwave irradiation greatly accelerated the rate of thehydrolysis reaction. Comparison of hydrolysis results betweenthe two heating methods over BC–SO3H revealed that the accel-erating effect of microwave irradiation on the reaction wasevident. The hydrolysis reaction operated at a lower temp-erature of 70–100 °C and the yield of reducing sugars was higherthan that using conventional heating. The yields of hydrolysisproducts were different with the different types of carbonacids. The correlation between the hydrolysis product yieldsand adsorption values confirmed the results from Hara’s workthat the adsorption of the phenolic OH groups of BC–SO3H tothe oxygen atoms in the β-1,4-glycosidic bonds was responsiblefor the high catalytic activities of these carbon-based solidacids when hydrolyzing cellulose to glucose.
Carbonaceous solid acid (CSA) can also be applied to realbiomass substrates under microwave irradiation. The Jiangand Mu research group84 illustrated that lignocellulosicbiomass (e.g. corncob) can be well hydrolyzed by self-derivedCSA under microwave irradiation. The CSA was prepared fromhydrolyzed corncob residue and was used for the hydrolysis of
Scheme 6 Schematic structure of the carbon-based solid acid. (Reproducedfrom ref. 82a.)
Table 2 Hydrolysis of crystalline cellulose by various acid catalysts (ref. 75)a
CatalystFunctionalgroups
Density(mmol g−1)
Max. acidityH0
Surface area(m2 g−1) Yields of hydrolysis products
H2SO4 20.4 −11 — Glucose: 10%, β-1,4-glucan: 38%Niobic acid Acidic OH 0.4 −5.6 90 —H-mordenite Acidic OH 1.4 −5.6 480 —Nafion SO3H 0.9 −11 to −13 <1Amberlyst-15 SO3H 4.8 −2.2 50 —Carbon material (CH0.62O0.54S0.05) SO3H 1.9 −8 to −11 2 Glucose: 4%, β-1,4-glucan: 64%
COOH 0.4 —Phenolic OH 2.0 —
a Conditions: catalyst 0.3 g, cellulose 25 mg, H2O 0.7 g, 3 h.
Tutorial Review Green Chemistry
1102 | Green Chem., 2013, 15, 1095–1111 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online
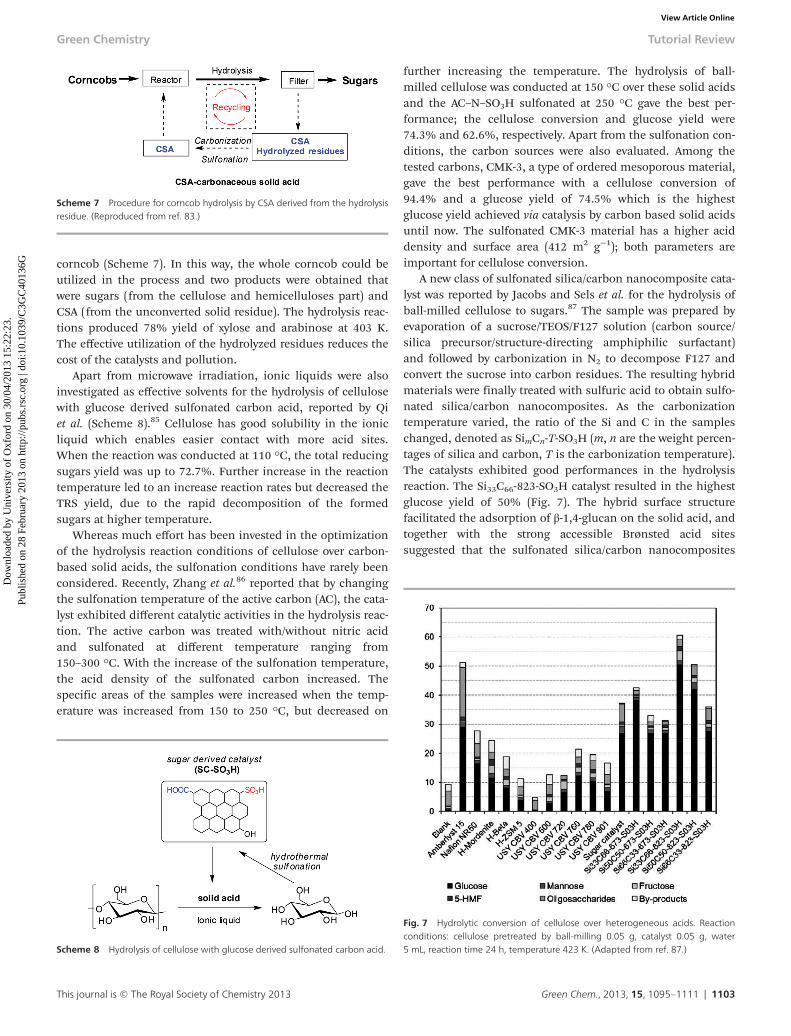
corncob (Scheme 7). In this way, the whole corncob could beutilized in the process and two products were obtained thatwere sugars (from the cellulose and hemicelluloses part) andCSA (from the unconverted solid residue). The hydrolysis reac-tions produced 78% yield of xylose and arabinose at 403 K.The effective utilization of the hydrolyzed residues reduces thecost of the catalysts and pollution.
Apart from microwave irradiation, ionic liquids were alsoinvestigated as effective solvents for the hydrolysis of cellulosewith glucose derived sulfonated carbon acid, reported by Qiet al. (Scheme 8).85 Cellulose has good solubility in the ionicliquid which enables easier contact with more acid sites.When the reaction was conducted at 110 °C, the total reducingsugars yield was up to 72.7%. Further increase in the reactiontemperature led to an increase reaction rates but decreased theTRS yield, due to the rapid decomposition of the formedsugars at higher temperature.
Whereas much effort has been invested in the optimizationof the hydrolysis reaction conditions of cellulose over carbon-based solid acids, the sulfonation conditions have rarely beenconsidered. Recently, Zhang et al.86 reported that by changingthe sulfonation temperature of the active carbon (AC), the cata-lyst exhibited different catalytic activities in the hydrolysis reac-tion. The active carbon was treated with/without nitric acidand sulfonated at different temperature ranging from150–300 °C. With the increase of the sulfonation temperature,the acid density of the sulfonated carbon increased. Thespecific areas of the samples were increased when the temp-erature was increased from 150 to 250 °C, but decreased on
further increasing the temperature. The hydrolysis of ball-milled cellulose was conducted at 150 °C over these solid acidsand the AC–N–SO3H sulfonated at 250 °C gave the best per-formance; the cellulose conversion and glucose yield were74.3% and 62.6%, respectively. Apart from the sulfonation con-ditions, the carbon sources were also evaluated. Among thetested carbons, CMK-3, a type of ordered mesoporous material,gave the best performance with a cellulose conversion of94.4% and a glucose yield of 74.5% which is the highestglucose yield achieved via catalysis by carbon based solid acidsuntil now. The sulfonated CMK-3 material has a higher aciddensity and surface area (412 m2 g−1); both parameters areimportant for cellulose conversion.
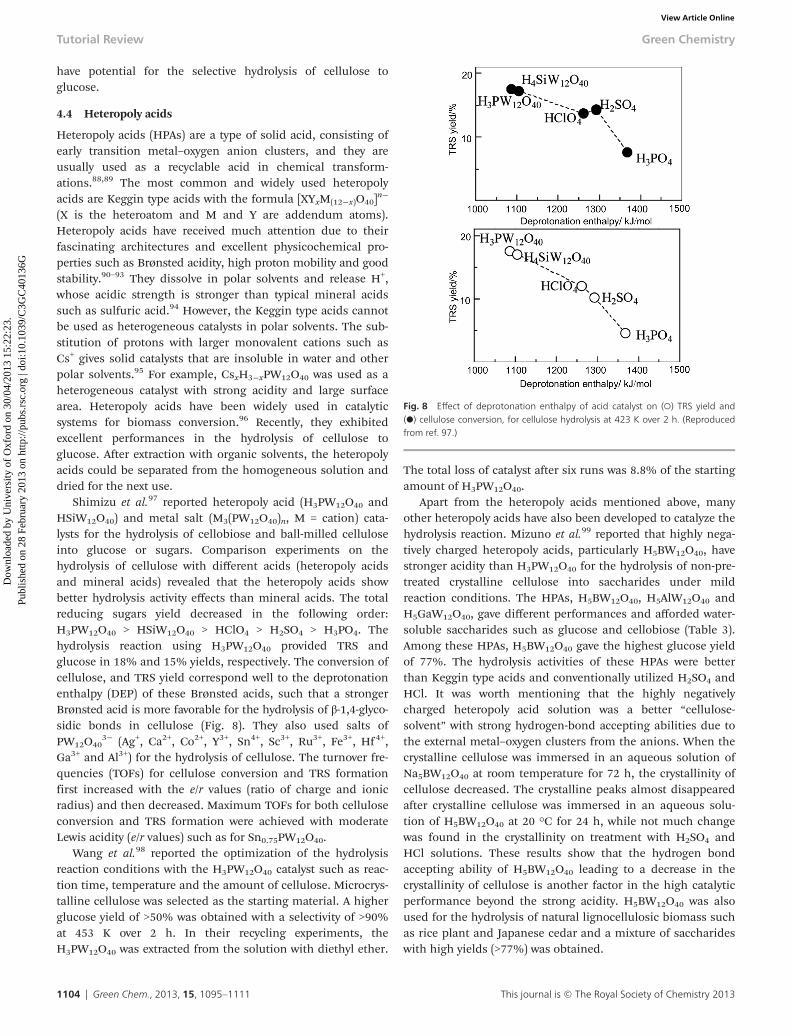
A new class of sulfonated silica/carbon nanocomposite cata-lyst was reported by Jacobs and Sels et al. for the hydrolysis ofball-milled cellulose to sugars.87 The sample was prepared byevaporation of a sucrose/TEOS/F127 solution (carbon source/silica precursor/structure-directing amphiphilic surfactant)and followed by carbonization in N2 to decompose F127 andconvert the sucrose into carbon residues. The resulting hybridmaterials were finally treated with sulfuric acid to obtain sulfo-nated silica/carbon nanocomposites. As the carbonizationtemperature varied, the ratio of the Si and C in the sampleschanged, denoted as SimCn-T-SO3H (m, n are the weight percen-tages of silica and carbon, T is the carbonization temperature).The catalysts exhibited good performances in the hydrolysisreaction. The Si33C66-823-SO3H catalyst resulted in the highestglucose yield of 50% (Fig. 7). The hybrid surface structurefacilitated the adsorption of β-1,4-glucan on the solid acid, andtogether with the strong accessible Brønsted acid sitessuggested that the sulfonated silica/carbon nanocomposites
Scheme 7 Procedure for corncob hydrolysis by CSA derived from the hydrolysisresidue. (Reproduced from ref. 83.)
Scheme 8 Hydrolysis of cellulose with glucose derived sulfonated carbon acid.
Fig. 7 Hydrolytic conversion of cellulose over heterogeneous acids. Reactionconditions: cellulose pretreated by ball-milling 0.05 g, catalyst 0.05 g, water5 mL, reaction time 24 h, temperature 423 K. (Adapted from ref. 87.)
Green Chemistry Tutorial Review
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 1095–1111 | 1103
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online
have potential for the selective hydrolysis of cellulose toglucose.
4.4 Heteropoly acids
Heteropoly acids (HPAs) are a type of solid acid, consisting ofearly transition metal–oxygen anion clusters, and they areusually used as a recyclable acid in chemical transform-ations.88,89 The most common and widely used heteropolyacids are Keggin type acids with the formula [XYxM(12−x)O40]
n−
(X is the heteroatom and M and Y are addendum atoms).Heteropoly acids have received much attention due to theirfascinating architectures and excellent physicochemical pro-perties such as Brønsted acidity, high proton mobility and goodstability.90–93 They dissolve in polar solvents and release H+,whose acidic strength is stronger than typical mineral acidssuch as sulfuric acid.94 However, the Keggin type acids cannotbe used as heterogeneous catalysts in polar solvents. The sub-stitution of protons with larger monovalent cations such asCs+ gives solid catalysts that are insoluble in water and otherpolar solvents.95 For example, CsxH3−xPW12O40 was used as aheterogeneous catalyst with strong acidity and large surfacearea. Heteropoly acids have been widely used in catalyticsystems for biomass conversion.96 Recently, they exhibitedexcellent performances in the hydrolysis of cellulose toglucose. After extraction with organic solvents, the heteropolyacids could be separated from the homogeneous solution anddried for the next use.
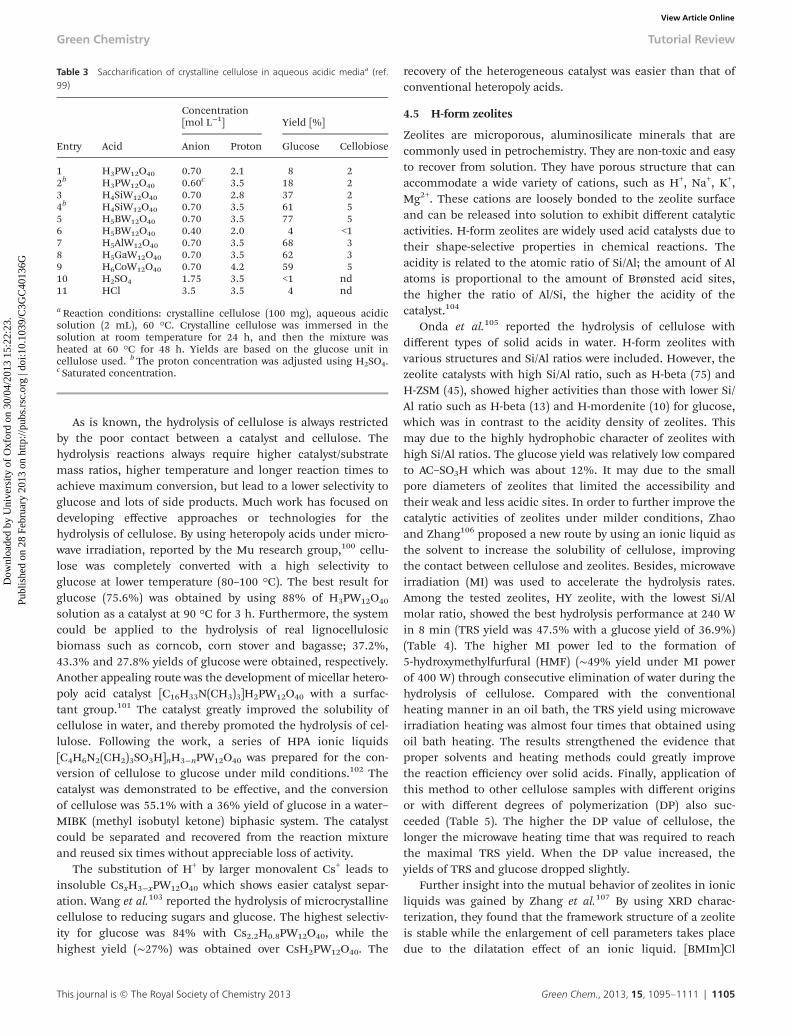
Shimizu et al.97 reported heteropoly acid (H3PW12O40 andHSiW12O40) and metal salt (M3(PW12O40)n, M = cation) cata-lysts for the hydrolysis of cellobiose and ball-milled celluloseinto glucose or sugars. Comparison experiments on thehydrolysis of cellulose with different acids (heteropoly acidsand mineral acids) revealed that the heteropoly acids showbetter hydrolysis activity effects than mineral acids. The totalreducing sugars yield decreased in the following order:H3PW12O40 > HSiW12O40 > HClO4 > H2SO4 > H3PO4. Thehydrolysis reaction using H3PW12O40 provided TRS andglucose in 18% and 15% yields, respectively. The conversion ofcellulose, and TRS yield correspond well to the deprotonationenthalpy (DEP) of these Brønsted acids, such that a strongerBrønsted acid is more favorable for the hydrolysis of β-1,4-glyco-sidic bonds in cellulose (Fig. 8). They also used salts ofPW12O40
3− (Ag+, Ca2+, Co2+, Y3+, Sn4+, Sc3+, Ru3+, Fe3+, Hf4+,Ga3+ and Al3+) for the hydrolysis of cellulose. The turnover fre-quencies (TOFs) for cellulose conversion and TRS formationfirst increased with the e/r values (ratio of charge and ionicradius) and then decreased. Maximum TOFs for both celluloseconversion and TRS formation were achieved with moderateLewis acidity (e/r values) such as for Sn0.75PW12O40.
Wang et al.98 reported the optimization of the hydrolysisreaction conditions with the H3PW12O40 catalyst such as reac-tion time, temperature and the amount of cellulose. Microcrys-talline cellulose was selected as the starting material. A higherglucose yield of >50% was obtained with a selectivity of >90%at 453 K over 2 h. In their recycling experiments, theH3PW12O40 was extracted from the solution with diethyl ether.
The total loss of catalyst after six runs was 8.8% of the startingamount of H3PW12O40.
Apart from the heteropoly acids mentioned above, manyother heteropoly acids have also been developed to catalyze thehydrolysis reaction. Mizuno et al.99 reported that highly nega-tively charged heteropoly acids, particularly H5BW12O40, havestronger acidity than H3PW12O40 for the hydrolysis of non-pre-treated crystalline cellulose into saccharides under mildreaction conditions. The HPAs, H5BW12O40, H5AlW12O40 andH5GaW12O40, gave different performances and afforded water-soluble saccharides such as glucose and cellobiose (Table 3).Among these HPAs, H5BW12O40 gave the highest glucose yieldof 77%. The hydrolysis activities of these HPAs were betterthan Keggin type acids and conventionally utilized H2SO4 andHCl. It was worth mentioning that the highly negativelycharged heteropoly acid solution was a better “cellulose-solvent” with strong hydrogen-bond accepting abilities due tothe external metal–oxygen clusters from the anions. When thecrystalline cellulose was immersed in an aqueous solution ofNa5BW12O40 at room temperature for 72 h, the crystallinity ofcellulose decreased. The crystalline peaks almost disappearedafter crystalline cellulose was immersed in an aqueous solu-tion of H5BW12O40 at 20 °C for 24 h, while not much changewas found in the crystallinity on treatment with H2SO4 andHCl solutions. These results show that the hydrogen bondaccepting ability of H5BW12O40 leading to a decrease in thecrystallinity of cellulose is another factor in the high catalyticperformance beyond the strong acidity. H5BW12O40 was alsoused for the hydrolysis of natural lignocellulosic biomass suchas rice plant and Japanese cedar and a mixture of saccharideswith high yields (>77%) was obtained.
Fig. 8 Effect of deprotonation enthalpy of acid catalyst on (○) TRS yield and(●) cellulose conversion, for cellulose hydrolysis at 423 K over 2 h. (Reproducedfrom ref. 97.)
Tutorial Review Green Chemistry
1104 | Green Chem., 2013, 15, 1095–1111 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online
As is known, the hydrolysis of cellulose is always restrictedby the poor contact between a catalyst and cellulose. Thehydrolysis reactions always require higher catalyst/substratemass ratios, higher temperature and longer reaction times toachieve maximum conversion, but lead to a lower selectivity toglucose and lots of side products. Much work has focused ondeveloping effective approaches or technologies for thehydrolysis of cellulose. By using heteropoly acids under micro-wave irradiation, reported by the Mu research group,100 cellu-lose was completely converted with a high selectivity toglucose at lower temperature (80–100 °C). The best result forglucose (75.6%) was obtained by using 88% of H3PW12O40
solution as a catalyst at 90 °C for 3 h. Furthermore, the systemcould be applied to the hydrolysis of real lignocellulosicbiomass such as corncob, corn stover and bagasse; 37.2%,43.3% and 27.8% yields of glucose were obtained, respectively.Another appealing route was the development of micellar hetero-poly acid catalyst [C16H33N(CH3)3]H2PW12O40 with a surfac-tant group.101 The catalyst greatly improved the solubility ofcellulose in water, and thereby promoted the hydrolysis of cel-lulose. Following the work, a series of HPA ionic liquids[C4H6N2(CH2)3SO3H]nH3−nPW12O40 was prepared for the con-version of cellulose to glucose under mild conditions.102 Thecatalyst was demonstrated to be effective, and the conversionof cellulose was 55.1% with a 36% yield of glucose in a water–MIBK (methyl isobutyl ketone) biphasic system. The catalystcould be separated and recovered from the reaction mixtureand reused six times without appreciable loss of activity.
The substitution of H+ by larger monovalent Cs+ leads toinsoluble CsxH3−xPW12O40 which shows easier catalyst separ-ation. Wang et al.103 reported the hydrolysis of microcrystallinecellulose to reducing sugars and glucose. The highest selectiv-ity for glucose was 84% with Cs2.2H0.8PW12O40, while thehighest yield (∼27%) was obtained over CsH2PW12O40. The
recovery of the heterogeneous catalyst was easier than that ofconventional heteropoly acids.
4.5 H-form zeolites
Zeolites are microporous, aluminosilicate minerals that arecommonly used in petrochemistry. They are non-toxic and easyto recover from solution. They have porous structure that canaccommodate a wide variety of cations, such as H+, Na+, K+,Mg2+. These cations are loosely bonded to the zeolite surfaceand can be released into solution to exhibit different catalyticactivities. H-form zeolites are widely used acid catalysts due totheir shape-selective properties in chemical reactions. Theacidity is related to the atomic ratio of Si/Al; the amount of Alatoms is proportional to the amount of Brønsted acid sites,the higher the ratio of Al/Si, the higher the acidity of thecatalyst.104
Onda et al.105 reported the hydrolysis of cellulose withdifferent types of solid acids in water. H-form zeolites withvarious structures and Si/Al ratios were included. However, thezeolite catalysts with high Si/Al ratio, such as H-beta (75) andH-ZSM (45), showed higher activities than those with lower Si/Al ratio such as H-beta (13) and H-mordenite (10) for glucose,which was in contrast to the acidity density of zeolites. Thismay due to the highly hydrophobic character of zeolites withhigh Si/Al ratios. The glucose yield was relatively low comparedto AC–SO3H which was about 12%. It may due to the smallpore diameters of zeolites that limited the accessibility andtheir weak and less acidic sites. In order to further improve thecatalytic activities of zeolites under milder conditions, Zhaoand Zhang106 proposed a new route by using an ionic liquid asthe solvent to increase the solubility of cellulose, improvingthe contact between cellulose and zeolites. Besides, microwaveirradiation (MI) was used to accelerate the hydrolysis rates.Among the tested zeolites, HY zeolite, with the lowest Si/Almolar ratio, showed the best hydrolysis performance at 240 Win 8 min (TRS yield was 47.5% with a glucose yield of 36.9%)(Table 4). The higher MI power led to the formation of5-hydroxymethylfurfural (HMF) (∼49% yield under MI powerof 400 W) through consecutive elimination of water during thehydrolysis of cellulose. Compared with the conventionalheating manner in an oil bath, the TRS yield using microwaveirradiation heating was almost four times that obtained usingoil bath heating. The results strengthened the evidence thatproper solvents and heating methods could greatly improvethe reaction efficiency over solid acids. Finally, application ofthis method to other cellulose samples with different originsor with different degrees of polymerization (DP) also suc-ceeded (Table 5). The higher the DP value of cellulose, thelonger the microwave heating time that was required to reachthe maximal TRS yield. When the DP value increased, theyields of TRS and glucose dropped slightly.
Further insight into the mutual behavior of zeolites in ionicliquids was gained by Zhang et al.107 By using XRD charac-terization, they found that the framework structure of a zeoliteis stable while the enlargement of cell parameters takes placedue to the dilatation effect of an ionic liquid. [BMIm]Cl
Table 3 Saccharification of crystalline cellulose in aqueous acidic mediaa (ref.99)
Entry Acid
Concentration[mol L−1] Yield [%]
Anion Proton Glucose Cellobiose
1 H3PW12O40 0.70 2.1 8 22b H3PW12O40 0.60c 3.5 18 23 H4SiW12O40 0.70 2.8 37 24b H4SiW12O40 0.70 3.5 61 55 H5BW12O40 0.70 3.5 77 56 H5BW12O40 0.40 2.0 4 <17 H5AlW12O40 0.70 3.5 68 38 H5GaW12O40 0.70 3.5 62 39 H6CoW12O40 0.70 4.2 59 510 H2SO4 1.75 3.5 <1 nd11 HCl 3.5 3.5 4 nd
a Reaction conditions: crystalline cellulose (100 mg), aqueous acidicsolution (2 mL), 60 °C. Crystalline cellulose was immersed in thesolution at room temperature for 24 h, and then the mixture washeated at 60 °C for 48 h. Yields are based on the glucose unit incellulose used. b The proton concentration was adjusted using H2SO4.c Saturated concentration.
Green Chemistry Tutorial Review
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 1095–1111 | 1105
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online

entered into the HY channels and filled the channel space.FT-IR results showed that H+ cations generated in situ from theBrønsted acid sites of the zeolites were the key active speciesfor the hydrolysis of cellulose. According to the charac-terization, the authors proposed a reaction pathway for HY cata-lyzed cellulose hydrolysis in IL (Scheme 9). First, the [BMIM]+
of the IL enters into the channels of zeolite HY and reacts withthe acidic sites to replace the H+. After that, the released H+
moves to the surface of cellulose and hydrolysis occurs.
4.6 Magnetic solid acids
As for the real biomass feedstock and practical process for thehydrolysis of cellulose to glucose, a challenge still exists withrespect to the transformation system. Solid catalysts cannot bedirectly separated when solid residues are formed during thehydrolysis process. Although cellulose can be degraded intosoluble sugars, the lignin components of the actual cellulosicbiomass cannot be converted and humins sometimes form assolid residues, both of which are difficult to separate from therecovered solid catalysts. Additionally, the separation of cata-lysts from the hydrolysis solid residue is important forthe analysis of the recovered catalyst which may be related tothe explanation of the catalytic mechanism. To address theproblem, we developed magnetic sulfonated mesoporous silica
(SBA-15), which can be successfully separated from the reac-tion system with a permanent magnet, in our recentwork.108,109
The magnetic solid acid was prepared by a surfactant-tem-plated sol–gel method similar to reported methods.110,111
Fe3O4 magnetic nanoparticles were dispersed in the blockcopolymer P123 for the co-condensation of tetraethoxysilane(TEOS). After assembly, 3-(mercaptopropyl)trimethoxysilaneMPTMS was added to introduce mercapto groups. H2O2 wasused to oxidize the mercapto groups into sulfonic acid groupsinside the pores of the mesoporous silica. X-Ray photoelectronspectroscopy (XPS) analysis of the sample showed that therewas only one peak (169.01 eV) between 158–177 eV in bindingenergy, demonstrating that the sulfur element in the catalystwas completely in the form of sulfonic acid groups(Scheme 10).
Experiments on the hydrolysis of cellobiose with Fe3O4–
SBA–SO3H showed that the magnetic solid acid gave an evenbetter performance than sulfuric acid. A proposed explanationwas that the channels in the magnetic solid acid contain con-centrated acid sites and the uniform channels allow the reac-tants to easily enter and interact with these acid sites. Furtherhydrolysis of amorphous cellulose (pretreated with 1-butyl-3-methylimidazolium chloride) with Fe3O4–SBA–SO3H gave a50% yield of glucose. The protons diffused from the SBA-15channels on the surface of cellulose undergo hydrolysis reac-tion. When the magnetic solid acid was used for the hydrolysisof microcrystalline cellulose, the yield of glucose decreased to26%. Comparative studies revealed that Fe3O4–SBA–SO3Hexhibited an even better hydrolysis performance than AC–SO3H and Amberlyst-15, presumably due to its higher surface
Table 4 The results of the solid acid-catalyzed hydrolysis of Avicel cellulosea
(ref. 106)
Entry Catalyst Time/min Yield of glucose Yields of TRS
1b HY 600 2.1 7.12c HY 4 20.8 23.43 HY 8 36.9 47.54d HY 15 24.2 34.85 HZSM-5a 9.5 35.2 42.96 HZSM-5b 9.5 33.7 45.37 H-beta 8.5 29.6 41.18 NKC-9 3.3 26.9 38.49e HY 8 4.5 12.710 — 8 7.1 18.8
a Conditions: cellulose 100 mg, [C4mim]Cl 2.0 g, H2O 10 ml, catalyst10 mg, MI at 240 W. bHeated with an oil bath at 100 °C. cWith MI at400 W. dWith MI at 80 W. eHeated with an oil bath at 180 °C.
Table 5 Results of HY zeolite-catalyzed hydrolysis of different cellulosesamples (ref. 106)a
Entry Cellulose type DP Time/minGlucoseyield
TRSyields
1 α-Cellulose 100 7 34.9 46.52 Avicel cellulose 220 8 36.9 47.53 Spruce cellulose 275 9 34.5 44.44 Sigmacell cellulose 450 9.5 32.5 42.45b Cellulose — 1440 12.5 —
a Conditions: cellulose 100 mg in [C4mim]Cl (2.0 g), H2O 10 mg, HYzeolite 10 mg, heated with MI at 240 W. bMilled cellulose 45 mg,H-form zeolite catalyst (HZSM-5 or H-beta) 50 mg, H2O 5 ml, heatedusing an oil bath at 150 °C for 24 h.
Scheme 9 Proposed reaction pathway and recovery of HY. (Adapted from ref.107 with permission from Elsevier). The released H+ can also catalyze the sidereaction, dehydrating glucose into HMF. The recovered catalyst could be reusedby simple calcination.
Tutorial Review Green Chemistry
1106 | Green Chem., 2013, 15, 1095–1111 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online
area. Also, when corncob was used as lignocellulose biomassfor a hydrolysis reaction, the total reducing sugar (TRS) yieldwas 45% (Fig. 9).
The catalyst separation is presented in Scheme 11. Theused Fe3O4–SBA–SO3H can be easily separated from the reac-tion mixture by using a permanent magnet. The recycled cata-lyst was washed with 1 M H2SO4 and water/ethanol, and driedat 80 °C for the next use without deactivation. TEM, XRD andBET analysis of the used catalyst revealed that the mesoporousstructure is retained and the Fe3O4–SBA–SO3H is robust in thehydrothermal conditions.
Another appealing work based on a magnetic solid acid wasreported by Ebitani et al.112 The catalyst was prepared by usingCoFe2O4 as the magnetic core and embedded with silica, fol-lowed by oxidation of thiol groups to –SO3H groups. It was ahighly active catalyst for the hydrolysis of disaccharides andpolysaccharides. The use of nanoparticles was expected toovercome the difficulty of the solid–solid reaction and theywere well dispersed in water solution. By introducing magneticparts in the catalyst, it can be easily separated from solution.The hydrolysis of cellulose was conducted at 423 K in waterand the glucose yield was 7%; the TRS yield was 30% whichwas similar to that using Amberlyst-15. But the TON number
using the MNPs@SiO2–SO3H catalyst was much higher thanthat using Amberlyst-15 (3.8 vs. 0.4) (Table 6).
4.7 Supported metal catalysts
Supported metal catalysts have been widely used in biomassconversion due to their excellent hydrogenation activities.Numerous works have focused on the conversion of celluloseinto sugar alcohols with supported metal catalysts in the pres-ence of acids.113–115 Cellulose was hydrolyzed to sugars overacid and then hydrogenated into the sugar alcohols by sup-ported metal catalysts under H2 pressure. However, thehydrolysis of cellulose to glucose is rarely catalyzed solely bysupported metal catalysts. Recently, Fukuoka et al.116 success-fully converted cellulose to glucose by using a supported Rucatalyst. Various supported materials, including mesoporouscarbon materials (CMKs), carbon black (XC-72), activatedcarbon (AC) and C60 were tested and the CMK supported Rucatalyst (Ru/CMK-3) showed the best performance (the glucoseyield was 34% and the oligosaccharide yield 5%). The loadingamount of Ru on the catalyst had an apparent effect on theproduct distribution. When the Ru loading ranged from 2% to10%, the glucose yield increased from 28% to 34% while theoligosaccharides decreased from 22% to 5%. The yield of totalreducing sugars was ca. 40% regardless of the Ru loading.Besides, CMK-3 itself catalyzed the hydrolysis of cellulose inwater at 503 K and gave a 21% yield of glucose and 22% yieldof oligosaccharides (total 43%). The above results show thatCMK-3 is able to convert cellulose into oligosaccharides andsugars, and Ru plays a very important role in the conversion ofoligosaccharides into glucose. Further experiments on thehydrolysis of cellobiose with CMK-3 and Ru/CMK-3 confirmedthe suspicion. The hydrolysis of cellobiose with Ru/CMK-3
Scheme 10 Preparation of Fe3O4–SBA–SO3H. (Reproduced from ref. 108.)
Fig. 9 Hydrolysis of macrocrystalline cellulose by different solid acids (1.5 gsolid acid catalyst, 1.5 g cellulose, 15 ml H2O, at 150 °C for 3 h). (Reproducedfrom ref. 108.)
Scheme 11 Catalyst separation from the reaction mixture. (Adapted from ref.108.)
Table 6 Hydrolysis of cellulose with MNPs@SiO2–SO3Ha
CatalystAcid amount/mmol g−1
Yield ofTRS
Yield ofglucose TON
MNPs@SiO2–SO3H 0.5 30.2 7.0 3.8Amberlyst-15 4.8 29.3 6.2 0.4
a Conditions: cellulose 0.15 g, catalyst 0.15 g, H2O 1.5 ml, 423 K.
Green Chemistry Tutorial Review
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 1095–1111 | 1107
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online

under the optimized conditions yielded 25% glucose, whilethe CMK-3 support gave a similar performance to the blanktest (6.5% and 7.9% yield of glucose), which meant that thesupport had no activity in the hydrolysis and the Ru speciesplayed the role of hydrolyzing the β-1,4-glycosidic bonds(Scheme 12). Later work showed that the active Ru species wasRuO2·H2O which was proposed to desorb the hydrated water togive a Lewis acid site that can depolymerize cellulose toglucose.117 Another hypothesis was that the Ru species workedas a Brønsted acid by the heterolysis of water molecules on Ruwhich had a low pKa. The new method for the hydrolysis of cel-lulose without acids has a bright future for the development ofnew metal catalysts in the hydrolysis field.
4.8 Others
Recently, many new catalysts have emerged as powerful toolsfor the hydrolysis of cellulose to sugars. Zhang and Fang118
reported the effective use of ferrate CaFe2O4 as a hetero-geneous catalyst for cellulose hydrolysis. In their work, anionic liquid was used for pretreatment to reduce the crystalli-nity. The reaction was carried out at 423 K over 24 h. The bestyield of glucose was achieved with fresh CaFe2O4 catalyst onhydrolyzing the pretreated cellulose; the glucose yield was 37%and the selectivity was 74%. The catalyst could be reusedseveral times without much drop in the activity.
Hydrotalcite [Mg4Al2(OH)12CO3·4H2O], activated by satu-rated Ca(OH)2, was used as an inorganic catalyst for thehydrolysis of cellulose, as reported by Fang et al.119 The reac-tion was carried out at 423 K over 24 h with ball-milled cellu-lose; the conversion and glucose selectivity were 46.6% and85.3%, respectively. The catalyst could be reused 4 times andthe catalytic activity remained. Compared with carbon-basedsolid acids, the activated hydrotalcite catalyst is more stableand can be separated more easily from the reaction mixture.
5. Summary and outlook
The hydrolysis of cellulose to glucose is a critical step for theproduction of cellulosic bioethanol and many other value-added molecules. So far, the hydrolysis process cannot beamplified to industrial scale production due to its loweconomy and efficiency. The catalytic systems mentioned
above were aimed at overcoming these limitations. Solid acidshave many advantages over conventional liquid acids, in separ-ation, recycling and being environmental benign. However, themass transfer limitations between the insoluble polymers andheterogeneous catalysts are bottlenecks for the hydrolysissystem. Conceptually, solid acids having higher specificsurface areas, pore sizes/volume and strong acid sites areprone to exhibit better performances in the reaction. Thenumber of Brønsted acid sites, affinity with the substrates andcatalyst stability are all key factors in the hydrolysis reaction.Among the mentioned solid catalysts, carbon-based solid acidsbearing –SO3H, –COOH and –OH groups and cellulose-mimetic catalysts have strong affinity to the cellulose chainand make the acid sites closer to the β-1,4-glycosidic bonds.Magnetic solid acids are promising materials for the reactiondue to their easier separation and recyclability. Heteropolyacids have been proven to be active catalysts for cellulose con-version, but their water solubility limits their large-scale appli-cation. Incorporation of HPAs into supports or the use ofinsoluble salts of HPAs may solve the problem and requirefurther investigation.
Recent advances in the introduction of ionic liquids, micro-wave irradiation, supercritical water and nanoparticles to thecellulose hydrolysis system may allow the reaction to proceedmore easily under milder conditions. Besides, cellulose pre-treatment can reduce the degree of polymerization of celluloseand increase the surface area which would significantlyimprove the reaction efficiency and reduce the operation cost.All these technologies help to overcome the recalcitrance ofcellulose and depolymerize it to a certain extent. Pretreatmentis also the first step in the conversion of lignocellulosicbiomass to chemicals, thus more convenient and efficient pre-treatment methods, reaction media and advanced heatingmethods should be considered in the hydrolysis of cellulose.
Although cellulose hydrolysis has been developed withsolid acids toward the greener synthesis of sugars, the reactionefficiency and selectivity still need to be improved. The devel-opment of highly acidic catalysts with nanometre size, goodsubstrate affinity and thermal stability is a challenge towardspractical systems. The production of fuels and chemicals frombiomass requires multidisciplinary collaboration of research-ers from enzymatic, homogeneous, and heterogeneous cata-lysis and the bio-/chemical engineering research areas.Economic, environmental, and sustainable aspects should allbe considered when designing new catalytic systems.
We thank the 973 Program (2012CB215305,2012CB215306), NSFC (21172209) and CAS (KJCX2-EW-J02) forfinancial support.
Notes and references
1 W. P. Nel and C. J. Cooper, Energy Policy, 2009, 37, 166.2 S. Shafiee and E. Topal, Energy Policy, 2009, 37, 181.
Scheme 12 Hydrolysis of cellulose by Ru/CMK-3.
Tutorial Review Green Chemistry
1108 | Green Chem., 2013, 15, 1095–1111 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online

3 R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark,J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero,Energy Environ. Sci., 2008, 1, 542.
4 R. Henry, Plant Biotechnol. J., 2010, 8, 288.5 F. Kreith and D. Y. Goswami, in Handbook of Energy
Efficiency and Renewable Energy, CRC Press: Taylor andFrancis Group, Boca Raton, FL, 2007.
6 M. Graziani and P. Fornasiero, in Renewable Resources andRenewable Energy: A Global Challenge, CRC Press: Taylorand Francis Group, Boca Raton, FL, 2007.
7 D. L. Klass, Biomass for Renewable Energy, Fuels, andChemicals, Academic Press, San Diego, 1998.
8 J.-P. Lange, E. Heide, J. Buijtenen and R. Price, Chem-SusChem, 2012, 5, 150.
9 D. Hayes, Catal. Today, 2009, 145, 138.10 J. C. Serrano-Ruiz, R. Luque and A. Sepu lveda-Escribanoa,
Chem. Soc. Rev., 2011, 40, 5266.11 D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem.,
2010, 12, 1493.12 W. Torres, S. S. Pansare and J. G. Goodwin, Catal. Rev. Sci.
Eng., 2007, 49, 407.13 H. de Lasa, E. Salaices, J. Mazumder and R. Lucky, Chem.
Rev., 2011, 111, 5404.14 G. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106,
4044.15 P. Maki-Arvela, B. Holmbom, T. Salmi and D. Murzin,
Catal. Rev. Sci. Eng., 2007, 49, 197.16 C. E. Wyman, B. E. Dale, R. T. Elander, M. Holtzapple,
M. R. Ladisch and Y. Y. Lee, Bioresour. Technol., 2005, 96,1959.
17 Y. H. P. Zhang and L. R. Lynd, Biotechnol. Bioeng., 2004,88, 797.
18 J. A. Geboers, S. Van de Vyver, R. Ooms, B. Op de Beeck,P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714.
19 J. R. Regalbuto, Science, 2009, 325, 822.20 Y. P. Zhang and L. R. Lynd, Biotechnol. Bioeng., 2004, 88,
797.21 P. Engel, R. Mladenov, H. Wulfhorst, G. Jager and
A. C. Spiess, Green Chem., 2010, 12, 1959.22 A. C. Salvador, M. C. Santos and J. A. Saraiva, Green
Chem., 2010, 12, 632.23 J. F. Saeman, Ind. Eng. Chem., 1945, 37, 43.24 R. W. Torget, J. S. Kim and Y. Y. Lee, Ind. Eng. Chem. Res.,
2000, 39, 2817.25 F. Guo, Z. Fang, C. C. Xu and R. L. Smith Jr., Prog. Energy
Combust. Sci., 2012, 38, 672.26 K. D. O. Vigier and F. Jérôme, Top. Curr. Chem., 2010, 295,
63.27 K. Shimizu and A. Satsum, Energy Environ. Sci., 2011, 4,
3140.28 P. L. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969.29 D. Sidiras and E. Koukios, Biomass, 1989, 19, 289.30 T. Tassinari, C. Macy and L. Spano, Biotechnol. Bioeng.,
1980, 22, 1689.31 R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers,
J. Am. Chem. Soc., 2002, 124, 4974.
32 J. Zhang, B. Zhang, J. Zhang, L. Lin, S. Liu and P. Ouyang,Biotechnol. Adv., 2010, 28, 613.
33 T. H. Kim and Y. Y. Lee, Bioresour. Technol., 2005, 96,2007.
34 M. Benoit, A. Rodrigues, Q. Zhang, E. Fourre,K. D. Oliveira Vigier, J. Tatibouet and F. Jerome, Angew.Chem., Int. Ed., 2011, 50, 8964.
35 J. Xue, H. Chen and Z. Kadar, Biomass Bioenergy, 2011, 35,3859.
36 S. Zhu, Y. Wu, Z. Yu, J. Liao and Y. Zhang, ProcessBiochem., 2005, 40, 3082.
37 H. Ma, W. Liu, Y. Wu and Z. Yu, Bioresour. Technol., 2009,100, 1279.
38 A. Payen, C. R. Hebd. Seances Acad. Sci., 1838, 7, 1052.39 A. Brogniart, A. B. Pelonze and R. Dumas, C. R. Chim.,
1839, 8, 51.40 H. Staudinger, Ber. Dtsch. Chem. Ges., 1920, 53, 1073.41 D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew.
Chem., Int. Ed., 2005, 44, 3358.42 M. E. Himmel, S. Y. Ding, D. K. Johnson, W. S. Adney,
M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007,315, 804.
43 A. C. O’Sullivan, Cellulose, 1997, 4, 173.44 M. Jarvis, Nature, 2003, 426, 611.45 Y. Nishiyama, J. Sugiyama, H. Chanzy and P. Langan,
J. Am. Chem. Soc., 2003, 125, 14300.46 M. Wada, Y. Nishiyama, H. Chanzy, T. Forsyth and
P. Langan, Powder Diffr., 2008, 23, 92.47 R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and
A. Rupper, Green Chem., 2010, 12, 972.48 Nanjing Forestry Institute, Plant Hydrolysis Technology,
Agricultural Press, Beijing, 1st edn, 1961.49 W. L. Faith, Ind. Eng. Chem., 1923, 37, 9.50 F. Bergius, Ind. Eng. Chem., 1937, 29, 247.51 P. L. Ragg, P. R. Fields and P. B. Tinker, Philos.
Trans. R. Soc. London, Ser. A, 1987, 321, 537.52 J. F. Saeman, J. L. Bubl and E. E. Harris, Ind. Eng. Chem.,
Anal. Ed., 1945, 17, 35.53 F. Camacho, P. Gonzalez-Tello, E. Jurado and A. Robles,
J. Chem. Technol. Biotechnol., 1996, 67, 350.54 E. E. Harris and E. Beglinger, The Madison Wood-Sugar
Process, Report no. R1617, US Department of Agriculture,Forest Service, Forest Products Laboratory, Madison, WI,1946.
55 D. R. Thompson and H. E. Grethlein, Ind. Eng. Chem.Prod. Res. Dev., 1979, 18, 166.
56 J. F. Harris, A. J. Baker, A.H. Conner, T. W. Jeffries,J. L. Minor, R. C. Pettersen, R. W. Scott, E. L. Springer,T. H. Wegner and J. I. Zerbe, in Two-stage Dilute SulfuricAcid Hydrolysis of Wood: An Investigation of Fundamentals,Gen. Tech. Rep., FPl-45, U. S. Department of Agriculture,Forest Service, Forest Products Laboratory, Madison, WI,1985, p. 73.
57 R. A. Antonoplis, H. W. Blanch, R. P. Freitas,A. F. Sciamanna and C. R. Wilke, Biotechnol. Bioeng., 1983,25, 2757.
Green Chemistry Tutorial Review
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 1095–1111 | 1109
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online

58 P. E. Linnett, J. P. M. Sanders, US Pat., 4677198, 1987.59 R. Erckel, R. Franz, R. Woernle and T. Riehm, US Pat.,
4556431, 1985.60 M. R. Ladisch, Process Biochem., 1979, 14, 21.61 C. Tagusagawa, A. Takagaki, A. Iguchi, K. Takanabe,
J. N. Kondo, K. Ebitani, T. Tatsumi and K. Domen, Chem.Mater., 2010, 22, 2035.
62 C. Tagusagawa, A. Takagaki, A. Iguchi, K. Takanabe,J. N. Kondo, K. Ebitani, S. Hayashi, T. Tatsumi andK. Domen, Angew. Chem., Int. Ed., 2010, 49, 1128.
63 A. Takagaki, C. Tagusagawa and K. Domen, Chem.Commun., 2008, 5363.
64 F. Zhang, X. Deng, Z. Fang, H. Zeng, X. Tian andJ. Kozinski, Petrochem. Technol., 2011, 40, 43.
65 Y. Uozumi and K. Shibatiomi, J. Am. Chem. Soc., 2001,123, 2919.
66 X. Qi, M. Watanabe, T. M. Aida and R. Lee Smith Jr., GreenChem., 2008, 10, 799.
67 M. T. Sanz, R. Murga, S. Beltran and J. L. Cabezas, Ind.Eng. Chem. Res., 2002, 41, 512.
68 R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int.Ed., 2008, 47, 8047.
69 R. Rinaldi, N. Meine, J. vom Stein, R. Palkovits andF. Schüth, ChemSusChem, 2010, 3, 266.
70 H. J. Heeres, B. Girisuta and L. P. B. M. Janssen, Ind. Eng.Chem. Res., 2007, 46, 1696.
71 V. Bravo, M. P. Paez, M. Aoulad and A. Reyes, EnzymeMicrob. Technol., 2000, 26, 614.
72 C. A. Schall, A. P. Dadi and S. Varanasi, Biotechnol.Bioeng., 2006, 95, 904.
73 L. Shuai, Q. Yang, J. Y. Zhu, F. C. Lu, P. J. Weimer,J. Ralph and X. J. Pan, Bioresour. Technol., 2010, 101, 3106.
74 L. Shuai and X. Pan, Energy Environ. Sci., 2012, 5, 6889.75 S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi,
H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008,130, 12787.
76 S. Kim, A. A. Dwiatmoko, J. W. Choi, Y. Suh, D. J. Suh andM. Oh, Bioresour. Technol., 2010, 101, 8273.
77 J. Hegner, K. C. Pereira, B. DeBoef and B. L. Lucht, Tetra-hedron Lett., 2010, 51, 2356.
78 G. Akiyama, R. Matsuda, H. Sato, M. Takata andS. Kitagawa, Adv. Mater., 2011, 23, 3294.
79 M. Toda1, A. Takagaki1, M. Okamura1, J. N. Kondo,S. Hayashi, K. Domen and M. Hara, Nature, 2005, 438,178.
80 M. Kitano, D. Yamaguchi, S. Suganuma, K. Nakajima,H. Kato, S. Hayashi and M. Hara, Langmuir, 2009, 25,5068.
81 K. Fukuhara, K. Nakajima, M. Kitano, H. Kato, S. Hayashiand M. Hara, ChemSusChem, 2011, 4, 778.
82 (a) M. Hara, Energy Environ. Sci., 2010, 3, 601;(b) S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi,H. Kato, S. Hayashi and M. Hara, Solid State Sci., 2010, 12,1029.
83 Y. Wu, Z. Fu, D. Yin, Q. Xu, F. Liu, C. Lu and L. Mao,Green Chem., 2010, 12, 696.
84 Y. Jiang, X. Li, X. Wang, L. Meng, H. Wang, G. Peng,X. Wang and X. Mu, Green Chem., 2012, 14, 2162.
85 H. Guo, X. Qi, L. Li and R. L. Smith Jr., Bioresour. Technol.,2012, 116, 355.
86 J. Pang, A. Wang, M. Zheng and T. Zhang, Chem.Commun., 2010, 46, 6935.
87 S. V. de Vyver, L. Peng, J. Geboers, H. Schepers, F. deClippel, C. J. Gommes, B. Goderis, P. A. Jacobs andB. F. Sels, Green Chem., 2010, 12, 1560.
88 I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171.89 N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199.90 C. L. Hill, J. Mol. Catal. A: Chem., 2007, 262, 2.91 I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 2007, 262, 86.92 I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 2009, 305, 104.93 N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem.
Rev., 2005, 249, 1944.94 J. Macht, M. J. Janik, M. Neurock and E. Iglesia, Angew.
Chem., Int. Ed., 2007, 46, 7864.95 T. Okuhara, Chem. Rev., 2002, 102, 3641.96 W. Deng, Q. Zhang and Y. Wang, Dalton Trans., 2012, 41,
9817.97 K. Shimizu, H. Furukawa, N. Kobayashi, Y. Itayab and
A. Satsuma, Green Chem., 2009, 11, 1627.98 J. Tian, J. Wang, S. Zhao, C. Jiang, X. Zhang and X. Wang,
Cellulose, 2010, 17, 587.99 Y. Ogasawara, S. Itagaki, K. Yamaguchi and N. Mizuno,
ChemSusChem, 2011, 4, 519.100 X. Li, Y. Jiang, L. Wang, L. Meng, W. Wang and X. Mu,
RSC Adv., 2012, 2, 6921.101 M. Chenga, T. Shia, H. Guana, S. Wang, X. Wang and
Z. Jiang, Appl. Catal., B, 2011, 107, 104.102 Z. Sun, M. Cheng, H. Li, T. Shi, M. Yuan, X. Wang and
Z. Jiang, RSC Adv., 2012, 2, 9058.103 J. Tian, C. Fan, M. Cheng and X. Wang, Chem. Eng.
Technol., 2011, 34, 482.104 N. Salman, C. H. Rüscher, J. C. Buhl, W. Lutz, H. Toufar
and M. Stöcker, Microporous Mesoporous Mater., 2006, 90,339.
105 A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008,10, 1033.
106 Z. Zhang and Z. K. Zhao, Carbohydr. Res., 2009, 344, 2069.107 H. Caia, C. Li, A. Wang, G. Xua and T. Zhang, Appl. Catal.,
B, 2012, 123–124, 333.108 D. Lai, L. Deng, Q. Guo and Y. Fu, Energy Environ. Sci.,
2011, 4, 3552.109 D. Lai, L. Deng, J. Li, B. Liao, Q. Guo and Y. Fu, Chem-
SusChem, 2011, 4, 55.110 D. Margolese, J. A. Melero, S. C. Christiansen,
B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12,2448.
111 C. S. Gill, B. A. Price and C. W. Jones, J. Catal., 2007, 251,145.
112 A. Takagaki, M. Nishimura, S. Nishimura and K. Ebitani,Chem. Lett., 2011, 40, 1195.
113 H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci.Technol., 2012, 2, 869.
Tutorial Review Green Chemistry
1110 | Green Chem., 2013, 15, 1095–1111 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online

114 R. Palkovits, K. Tajvidi, A. M. Ruppertc andJ. Procelewska, Chem. Commun., 2011, 47, 576.
115 C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007,46, 7636.
116 H. Kobayashi, T. Komanoya, K. Hara and A. Fukuoka,ChemSusChem, 2010, 3, 440.
117 T. Komanoya, H. Kobayashi, K. Hara, W. J. Chun andA. Fukuoka, Appl. Catal., A, 2011, 407, 188.
118 F. Zhang and Z. Fang, Bioresour. Technol., 2012, 124,440.
119 Z. Fang, F. Zhang, H. Zeng and F. Guo, Bioresour. Technol.,2011, 102, 8017.
Green Chemistry Tutorial Review
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 1095–1111 | 1111
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 3
0/04
/201
3 15
:22:
23.
Publ
ishe
d on
28
Febr
uary
201
3 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
4013
6GView Article Online








