





AAC Accepts, published online ahead of print on 14 July...
35
Avibactam and class C β-lactamases: mechanism of inhibition, conservation of binding pocket 1 and implications for resistance. 2 3 S. D. Lahiri 1δ , M. Johnstone 1 , P.L. Ross 2 , R. McLaughlin 1 , N. B. Olivier 2 and R. A. Alm 1 δ 4 5 1 Infection Innovative Medicines, AstraZeneca R&D Boston, MA, USA 6 2 Discovery Sciences Innovative Medicines, AstraZeneca R&D Boston, MA, USA 7 8 δ To whom correspondence should be addressed. E-mail: [email protected], [email protected]. 9 10 Abstract 11 Avibactam is a novel non-β-lactam β-lactamase inhibitor that inhibits a wide range of β-lactamases. These include 12 class A, class C and some class D enzymes, which erode the activity of β-lactam drugs in multi-drug-resistant 13 pathogens like Pseudomonas aeruginosa and Enterobacteriaceae. Avibactam is currently in clinical development in 14 combination with the β-lactam antibiotics ceftazidime, ceftaroline fosamil and aztreonam. Avibactam has the potential 15 to be the first β-lactamase inhibitor that could provide activity against class C-mediated resistance, which represents 16 a growing concern in both hospital- and community-acquired infections. Avibactam has an unusual mechanism of 17 action: it is a covalent inhibitor that acts via ring-opening but, in contrast to other currently used β-lactamase 18 inhibitors, this reaction is reversible. Here we present a high resolution structure of avibactam bound to a class C β- 19 lactamase, AmpC, from P. aeruginosa that provided insight into the mechanism of both acylation and recyclization in 20 this enzyme class and highlighted the differences observed between class A and class C inhibition. Furthermore, 21 variants resistant to avibactam were isolated that identified the residues that are important for inhibition. Finally, the 22 structural information was used to predict effective inhibition by sequence analysis and functional studies of class C 23 β-lactamases from a large and diverse set of contemporary clinical isolates (P. aeruginosa and several 24 Enterobacteriaceae spp.) obtained from recent infections to understand any pre-existing variability in the binding 25 pocket that could affect inhibition by avibactam. 26 27 AAC Accepts, published online ahead of print on 14 July 2014 Antimicrob. Agents Chemother. doi:10.1128/AAC.03057-14 Copyright © 2014, American Society for Microbiology. All Rights Reserved. on June 26, 2018 by guest http://aac.asm.org/ Downloaded from
-
Upload
truongthuy -
Category
Documents
-
view
215 -
download
0
Transcript of AAC Accepts, published online ahead of print on 14 July...

Avibactam and class C β-lactamases: mechanism of inhibition, conservation of binding pocket 1
and implications for resistance. 2
3
S. D. Lahiri1δ, M. Johnstone1, P.L. Ross2, R. McLaughlin1, N. B. Olivier2 and R. A. Alm1 δ 4
5
1Infection Innovative Medicines, AstraZeneca R&D Boston, MA, USA 6
2Discovery Sciences Innovative Medicines, AstraZeneca R&D Boston, MA, USA 7
8
δ To whom correspondence should be addressed. E-mail: [email protected], [email protected]. 9
10
Abstract 11
Avibactam is a novel non-β-lactam β-lactamase inhibitor that inhibits a wide range of β-lactamases. These include 12
class A, class C and some class D enzymes, which erode the activity of β-lactam drugs in multi-drug-resistant 13
pathogens like Pseudomonas aeruginosa and Enterobacteriaceae. Avibactam is currently in clinical development in 14
combination with the β-lactam antibiotics ceftazidime, ceftaroline fosamil and aztreonam. Avibactam has the potential 15
to be the first β-lactamase inhibitor that could provide activity against class C-mediated resistance, which represents 16
a growing concern in both hospital- and community-acquired infections. Avibactam has an unusual mechanism of 17
action: it is a covalent inhibitor that acts via ring-opening but, in contrast to other currently used β-lactamase 18
inhibitors, this reaction is reversible. Here we present a high resolution structure of avibactam bound to a class C β-19
lactamase, AmpC, from P. aeruginosa that provided insight into the mechanism of both acylation and recyclization in 20
this enzyme class and highlighted the differences observed between class A and class C inhibition. Furthermore, 21
variants resistant to avibactam were isolated that identified the residues that are important for inhibition. Finally, the 22
structural information was used to predict effective inhibition by sequence analysis and functional studies of class C 23
β-lactamases from a large and diverse set of contemporary clinical isolates (P. aeruginosa and several 24
Enterobacteriaceae spp.) obtained from recent infections to understand any pre-existing variability in the binding 25
pocket that could affect inhibition by avibactam. 26
27
AAC Accepts, published online ahead of print on 14 July 2014Antimicrob. Agents Chemother. doi:10.1128/AAC.03057-14Copyright © 2014, American Society for Microbiology. All Rights Reserved.
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

Introduction 28
The continual emergence of multi-drug resistance in Gram-negative bacteria has eliminated many former treatment 29
options. The β-lactam drug class, once the foundation of treatment regimens for many hospital- and community-30
acquired infections, is rapidly becoming obsolete due to the proliferation of β-lactamases (1-3). Even the 31
carbapenems, which for many years represented the last line of defense, are being eroded by the emerging 32
pandemic of carbapenemases such as metallo-β-lactamase containing pathogens (4, 5). The currently available β-33
lactamase inhibitors, such as clavulanic acid and sulbactam, are effective inhibitors of many of the class A β-34
lactamases, but are incapable of inhibiting any other classes, including class C (6). Chromosomally encoded class C 35
β-lactamases are found in many clinically important pathogens, such as P. aeruginosa and many Enterobacteriaceae 36
(7, 8). In many cases, the expression of this enzyme is inducible; however, they can get derepressed, and the 37
subsequent overexpression results in resistance to many β-lactam drugs. Furthermore, there is a myriad of class C 38
enzymes that are encoded on transferable plasmids that enable horizontal transfer of class C mediated β-lactam 39
resistance between bacterial species. 40
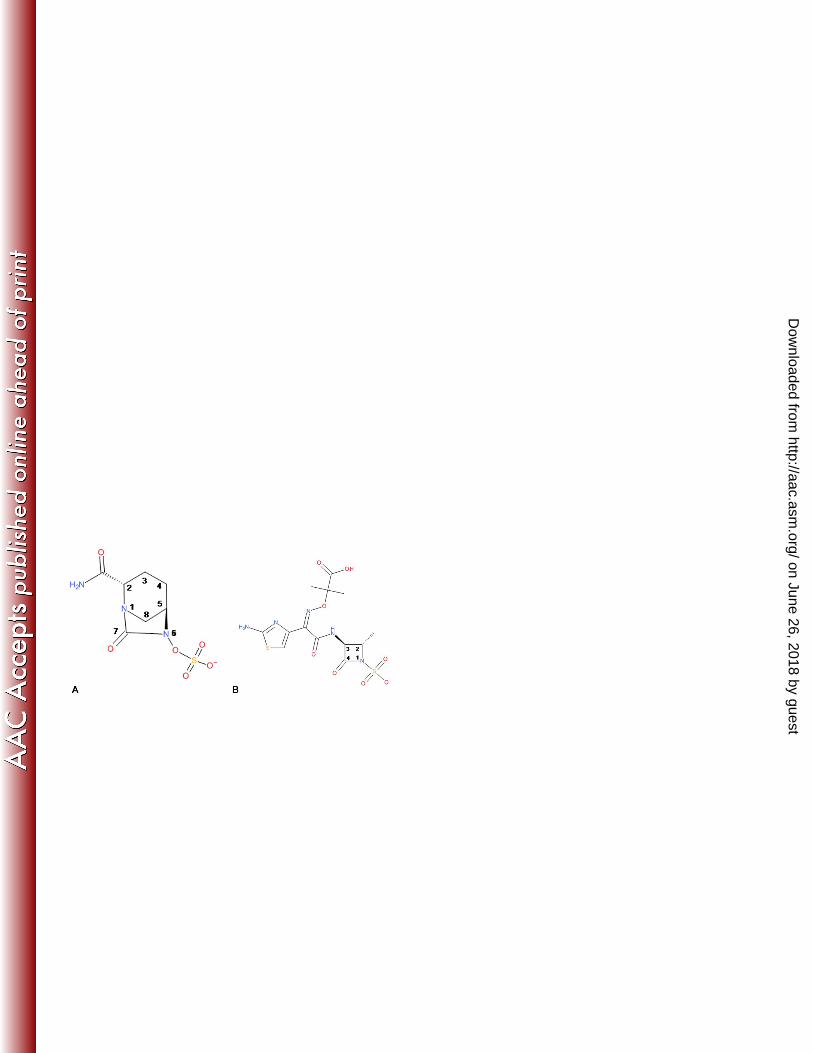
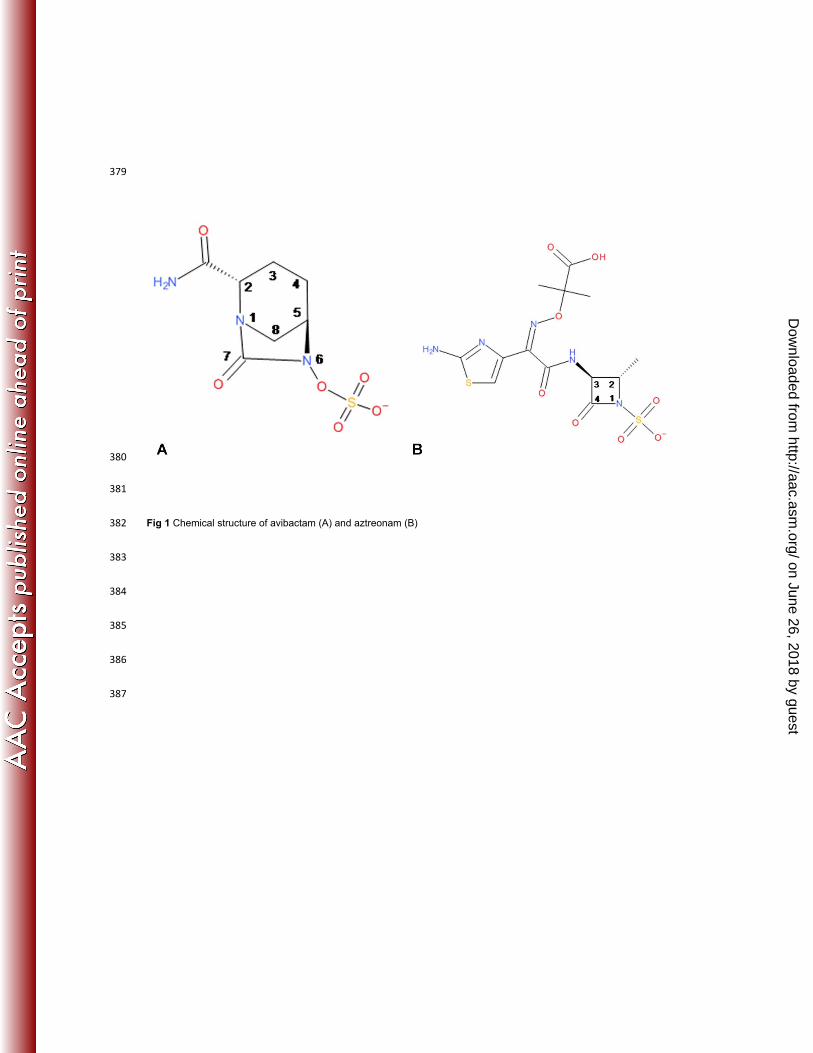
Avibactam (Fig 1A) is a novel non-β-lactam β-lactamase inhibitor that inhibits both class A and class C (and some 41
class D) enzymes, thus providing protection from a diverse range of β-lactamase-mediated resistance mechanism (9-42
11). It is currently in clinical development in combination with the cephalosporins ceftazidime and ceftaroline fosamil, 43
as well as with the monobactam aztreonam (Fig 1B), as alternative therapeutic options for the treatment of infections 44
caused by multi-drug resistant P. aeruginosa and Enterobacteriaceae (12-17). Avibactam is structurally distinct 45
from the clinically used β-lactamase inhibitors in that it does not contain a β-lactam core. In addition, it has a unusual 46
mechanism of inhibition. While the covalent inhibition proceeds in a similar fashion via the opening of the avibactam 47
ring, the reaction is reversible, whereby deacylation results in regeneration of the intact compound as opposed to 48
hydrolysis and turnover (9). This mechanistic difference from clinically used inhibitors contributes towards making 49
avibactam highly effective in providing protection to the β-lactam partner against hydrolysis by chromosomal and 50
plasmidic β-lactamases. 51
We have recently described the mechanism of covalent inhibition of class A enzymes by avibactam as well as a 52
medium resolution structural view of a class C co-complex to rationalize the broad spectrum activity (18). However, 53
the mechanism of inhibition of class C β-lactamases, which is a differentiating attribute of avibactam, could not be 54
confirmed. We now report high resolution P. aeruginosa AmpC structures in complex with avibactam in both the 55
ring-open and ring-closed form, as well as in complex with the monobactam β-lactam, aztreonam. The subsequent 56
analyses have enabled an understanding of the reversible deacylation in class C enzymes as well as a rationale for 57
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

the more rapid recyclization observed with this class C enzyme in comparison to class A β-lactamases. In addition, 58
we also evaluated the conservation of the avibactam binding pocket to assess the risk of any pre-existing pool of 59
resistant class C enzymes by sequencing the chromosomal ampC gene from over 500 diverse clinical isolates. 60
61
Materials and Methods 62
Crystallization, Data Collection, Structure Determination, and Refinement. Crystals of AmpC were obtained as 63
described previously (18). Crystals were soaked with a final concentration of 10mM of avibactam or aztreonam for 15 64
minutes followed by flash freezing in liquid nitrogen. Data were collected at Advanced Photon Source in Chicago at 65
IMCA beamline BM16. Data processing and refinement statistics are provided in Supplementary Table S1. 66
Genome sequencing Total genomic DNA was prepared using the Genomic DNA Purification kit in the Maxwell 16 67
instrument (Promega, Madison, WI). DNA libraries were prepared using the Nextera library construction protocol 68
(Illumina, San Diego, CA) and sequenced on a MiSeq Sequencer (Illumina, San Diego, CA). 69
Data analysis Whole genome data were analyzed using the CLC Bio Workbench software v. 5.5.1 (CLC Bio, 70
Cambridge, MA). Multiple sequence alignments were generated using the Clustal X program. Multi-locus sequence 71
typing (MLST) was performed by comparing the assembled genome sequences of each strain to the database of 72
known P. aeruginosa MLST alleles available at www.pubmlst.org. ConSurf, a bioinformatics tool 73
(www.consurf.tau.ac.il), was used to map the multiple sequence alignment to the AmpC:avibactam structure to 74
visualize the 3-dimensional sequence diversity. Pymol (www.pymol.org) was used to display the diversity, and also to 75
model the residue substitutions identified in the resistant variants. 76
Resistance Selection and Antimicrobial Susceptibility Testing. Resistant variants were selected by plating a 77
bacterial suspension on agar plates containing two-fold increasing concentrations of aztreonam with avibactam held 78
constant at 4ug/mL. The minimum inhibitory concentration (MIC) values were determined by the broth microdilution 79
method following the guidelines established by the Clinical Laboratory Standards Institute. 80
Expression Analysis. RNA from mid-logarithmically grown strains was prepared using the Maxwell 16 LEV 81
simplyRNA purification kit from Promega (Fitchburg, WI). A total of 5 ng RNA was used in a RT-PCR assay using the 82
Qiagen Quantitect SYBR Green RT-PCR kit (Germantown, MD) using a BioRad CFX96™ instrument. The 83
oligonucleotides used to detect the expression of the chromosomal blaampC allele in C. freundii were 5’-84
CGAGGGGAAACCTTATTA-3’ and 5’- TGTATAGGTGGCTAAGTG-3’ and the control oligonucleotides to detect 85
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

expression of the rpsL ribosomal gene were 5’- TAAAAAACCGAACTCCGCA-3’ and 5’- 86
GTCACTTCAAAACCGTTAG-3’. The level of rpsL expression was used for normalization. 87
88
Results 89
High resolution structure of avibactam bound to P. aeruginosa AmpC 90
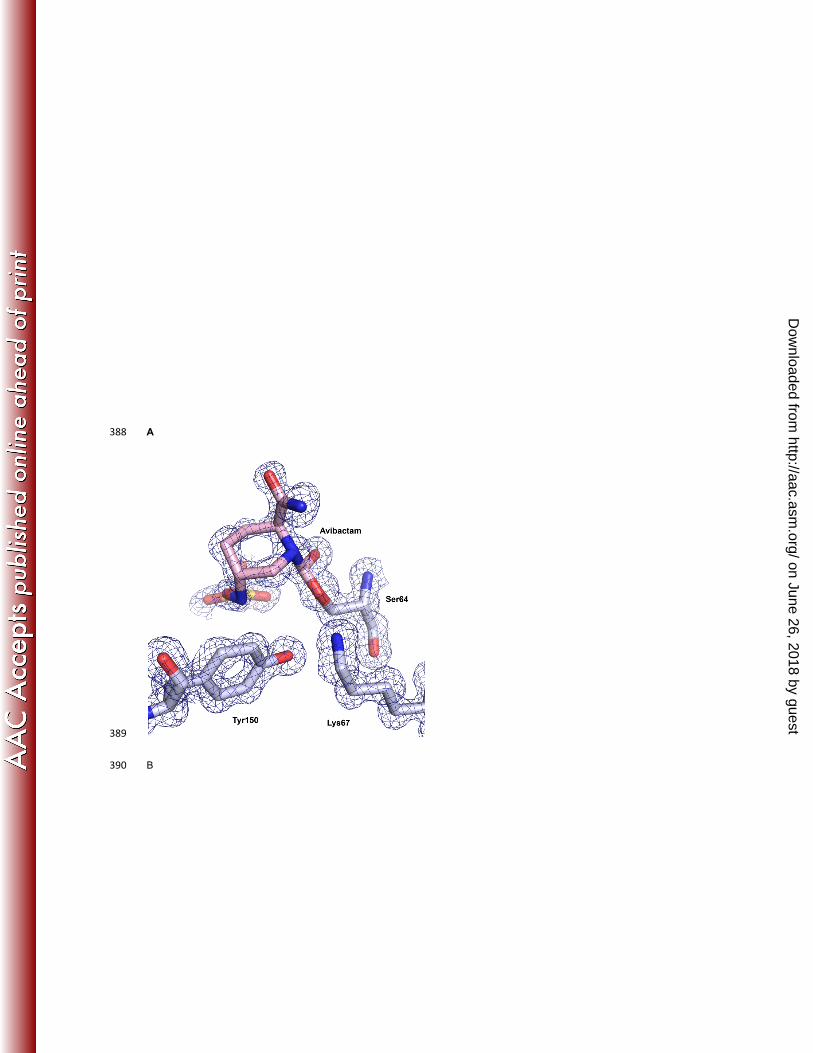
Crystals of avibactam covalently bound to P. aeruginosa AmpC were obtained that diffracted to 1.1 Å. The higher 91
resolution provided improved understanding of avibactam binding, in particular the piperidine ring conformation, the 92
position of the carboxamide and sulfate groups, and most importantly, the accurate positions of the residues involved 93
in catalysis (Fig 2A-C). The high resolution allowed observation of an intramolecular connectivity between the amide 94
group and the N1 nitrogen of the piperidine ring (Fig 2A), suggestive of a short strong hydrogen bond (<2.5 Å) 95
between these two moieties. This interaction is novel to the class C binding mode of avibactam, as the bound 96
structure of a class A β-lactamase at a comparable resolution (18) showed an increased distance between these 97
atoms with no connectivity of electron density. Further, there was additional density observed between the cleaved 98
C7-N6 bonds of the pyrazolidine ring (Fig 2A). The most plausible explanation for this additional density is that a 99
small subpopulation of ring-closed or intermediate state of the inhibitor exists simultaneously in this position. 100
However, refinement in the presence of a closed form of avibactam could not convincingly distinguish the two forms 101
of avibactam and hence the model was refined with the open covalently linked inhibitor (Fig 2B). 102
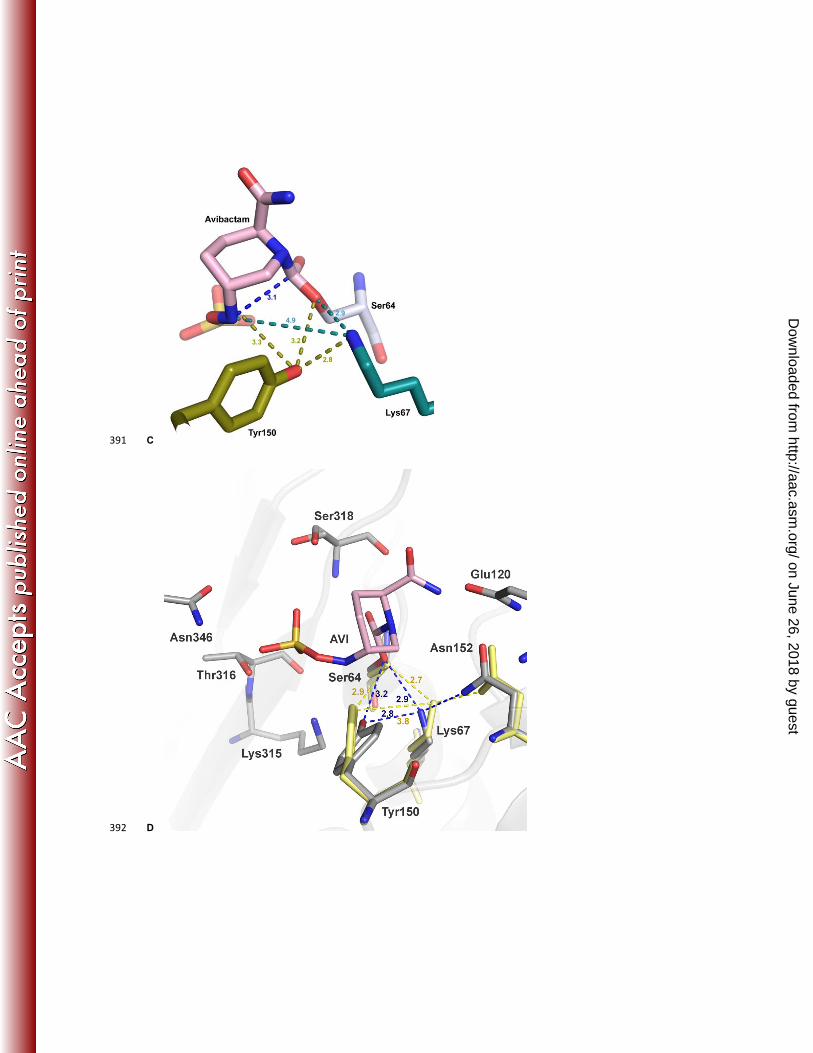
The refined structure showed that eight residues provided three key contributions to the binding interactions with 103
avibactam (Fig 2C). The carboxamide group of avibactam interacted with the side chains of Asn152 and Gln120, the 104
sulfate moiety was positioned by Thr316, Lys315 and Asn346, whereas Tyr150 and Lys67 were positioned to 105
participate in catalytic roles to enable formation of the covalent bond with Ser64. Specifically, the Oη of Tyr150 was 106
equidistant, 3.3Å and 3.2Å respectively, from N6 of avibactam and Oγ of Ser64. The Nζ of Lys67 was 2.9Å from the 107
Oγ of Ser64 and 4.9 Å from N6 of avibactam. Comparison of the apo structure at a similar resolution showed a 108
minimal shift in the binding-pocket residues upon acylation with all residues superimposing exactly except Tyr150, 109
Lys67, and Asn152 (Fig 2D). In the avibactam bound form, the side chain of Tyr150 moves closer to Lys67 by 1.0 Å, 110
indicative of the formation of a new hydrogen bond upon acylation. In addition, a new hydrogen bond is also formed 111
between the carboxamide group of avibactam and Asn152 side chain. Asn152 maintains its hydrogen bond to Lys67 112
upon acylation, that results in a shift in the position of these residues. 113
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

114
Comparison to a substrate binding mode 115
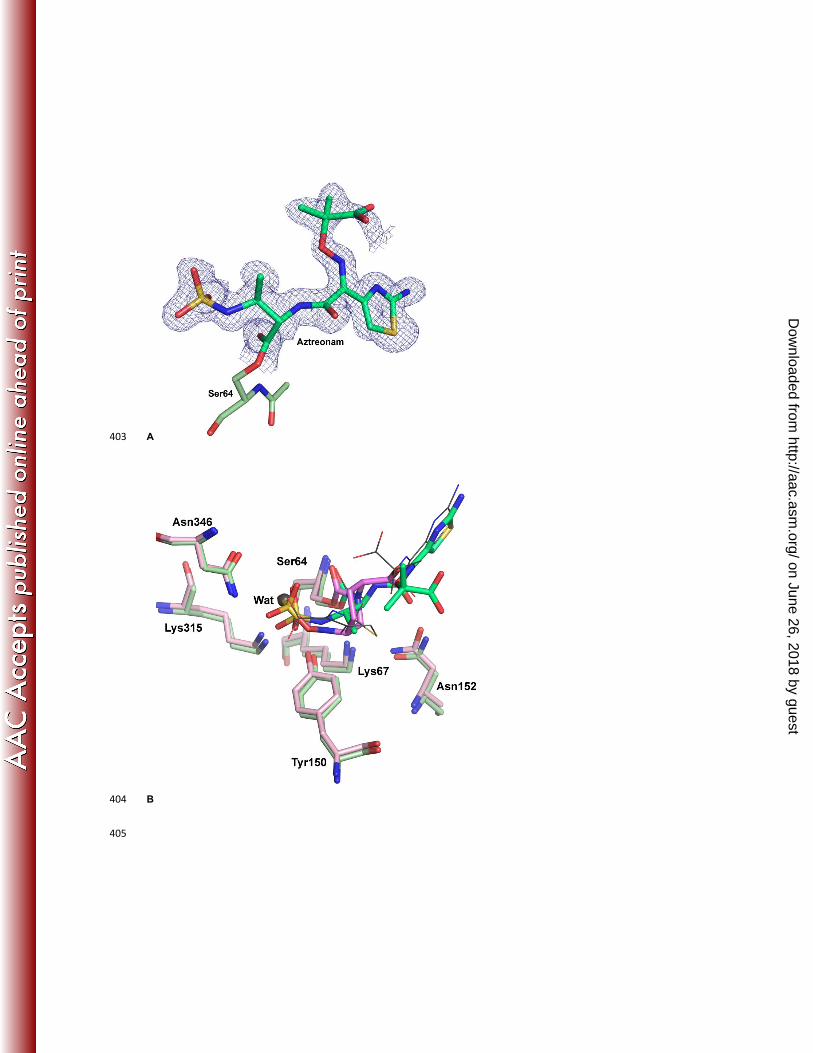
To understand the differences in interactions between a β-lactam drug and avibactam, the structure of aztreonam 116
bound to P. aeruginosa AmpC was solved to 1.3 Å (Fig 3A). Aztreonam contains a sulphonyl group in place of the 117
carboxylate group seen in other β-lactam antibiotics, which makes it structurally closer to avibactam than other β-118
lactams. In addition, whereas aztreonam is hydrolyzed by class C β-lactamases resulting in decreased susceptibility, 119
the rate of hydrolysis is slower than that observed for other β-lactams (6), making it feasible to capture the acyl-120
enzyme complex crystallographically. The structure showed that the position of the acylated aztreonam molecule in 121
P. aeruginosa AmpC was very similar to that previously observed in a Citrobacter freundii class C enzyme, with the 122
exception of the position of the oxyimino group (19). There was a significant similarity in the binding mode and 123
interacting residues between aztreonam and avibactam (Fig 3B). The sulfonyl group of aztreonam and the sulfate 124
group of avibactam were similarly located and interacted with Lys315, Thr316 and Asn346. Additionally, both groups 125
displace the deacylating water molecule (WAT) previously observed in crystal structures (PDB code 1IEL) (20). The 126
position of the N1 of aztreonam upon ring cleavage was in a similar location to N6 of avibactam, although the C3-C4 127
bond of the aztreonam β-lactam ring underwent a rotation of approximately 70⁰ upon ring opening, which displaced 128
the trajectory for recyclization. The side chains of Tyr150 and Lys67 were in identical positions in the two binding 129
modes, as were the interactions of Asn152 with both Lys67 and the ligands. However, in the case of aztreonam, 130
Asn152 formed a hydrogen bond with the carbonyl oxygen, which is opposite in polarity to the amide group of 131
avibactam. Overall, the hydrogen bonding patterns and the key interactions in the acylated forms were very similar 132
between the substrate (aztreonam) and the inhibitor (avibactam) despite the differences in the compound structures. 133
The major differences lay in the overall size and the limited rotational freedom of avibactam compared to aztreonam. 134
135
Conservation of Class C β-lactamase enzymes 136
Given the emerging resistance to β-lactams in P. aeruginosa and evidence that small sequence differences can affect 137
the substrate spectrum (7), there was a need to understand whether avibactam could be expected to inhibit all of the 138
AmpC enzyme variants in this species. There were 36 unique AmpC sequences from P. aeruginosa, representing 67 139
different isolates, available in the public domain (7, 21). An additional 464 isolates, obtained from 25 different 140
countries and isolated from multiple clinical indications from 2008 to 2012 were sequenced. Multi locus sequence 141
typing (MLST) analysis demonstrated a high level of diversity among the isolates. The 531 P. aeruginosa AmpC 142
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

proteins were clustered into groups representing 72 unique sequences. The relative population within each cluster 143
varied, with the largest cluster representing 109 (20.5%) of the isolates examined, whereas 35 clusters were 144
represented by a single isolate (Supplementary section, Fig S1 and Table S2). Although amino acid variations were 145
observed in over 22% of the residues, the eight residues directly involved in avibactam binding were highly 146
conserved, with six of them being conserved in all proteins and the other two (Ser64 and Asn346) having one and 147
two variants, respectively (Table 1). Of these, the isolate containing a Ser64Leu substitution was expected to be non-148
functional because Ser64 is critical for covalent catalysis (22). The changes Asn346Thr and Asn346Ile were both to an 149
amino acids of a similar size, of which, the Asn346Ile substitution also changed the property of the substitution from a 150
polar to a hydrophobic group. 151
Given that an important attribute of avibactam is the inhibition of Class C β-lactamases, the conservation analyses 152
were extended to other Class C enzymes in clinically relevant bacterial pathogens, including the chromosomal AmpC 153
proteins of Escherichia coli, Enterobacter cloacae, Enterobacter aerogenes, and Citrobacter spp. and plasmidic 154
enzymes that can carried by multiple species (Table 1). In the case of E. cloacae, where derepession of the 155
chromosomal blaampC gene is a common resistance mechanism, 79 recent clinical isolates were sequenced, and their 156
chromosomal AmpC proteins, along with 19 sequences from the public domain, were clustered into 57 unique AmpC 157
sequences with almost 40% of the amino acid residues varying among these sequences (Supplementary section, Fig 158
S1 and Table S3). Similarly, unique chromosomal AmpC proteins from E. coli, E. aerogenes and Citrobacter spp. 159
along with 127 plasmidic class C β-lactamase enzymes from public and internal data were also included in the 160
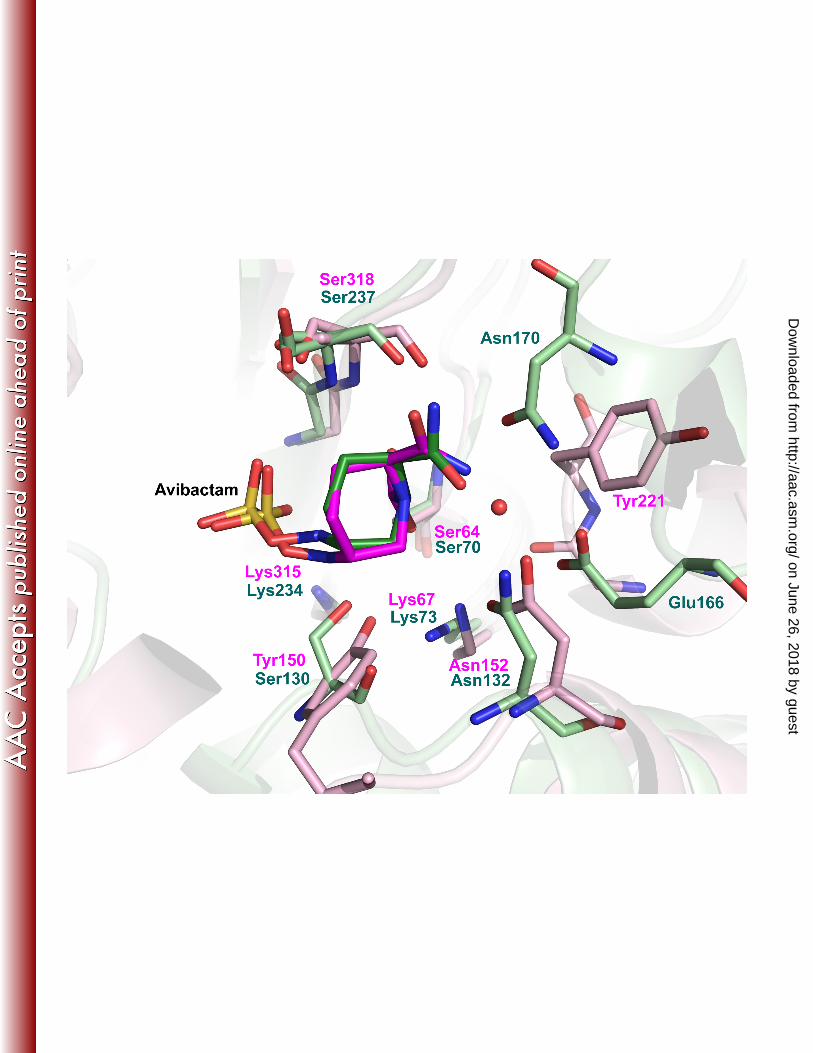
conservation analysis. Despite the dramatic sequence variability observed between these class C enzymes (Fig 4A), 161
Lys67, Tyr150, Asn152 and Lys315 were completely conserved (Fig 4B). A small number of isolates contained 162
variations at Ser64 (n=1), Gln120 (n=2) and Thr316 (n=1). Asn346 was the most variable of the binding pocket 163
residues: three variants among the chromosomal enzymes and 20 plasmidic class C enzymes carried the Asn346Ile 164
substitution (Table 1). This modification had been previously attributed to an extended-spectrum cephalosporinase 165
activity (23, 24). 166
Microbiological susceptibility tests were performed in P. aeruginosa and E. cloacae isolates where the chromosomal 167
blaampC was derepressed and contained no other β-lactamases to enable clear interpretation of the ability of 168
avibactam to inhibit these chromosomal AmpC variants. The susceptibility data confirmed that the restoration of β-169
lactam activity by 4 μg/mL avibactam was not affected by variations in these enzymes and restored the ceftazidime 170
and aztreonam MIC values to susceptible range as defined by CLSI (representative MIC values shown in Table 2). 171
The only variations in the 8 key avibactam binding residues (Table 1) that could not be verified were the Gln120Lys, 172
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

which was not available for testing and the chromosomal Thr316Met which was not derepressed in the isolate. Taken 173
together, these data suggest that there is not a significant pool of pre-existing class C-based resistance to avibactam 174
in clinical isolates of Gram-negative pathogens. 175
Resistance to avibactam in combination with a β-lactam 176
Given the conservation of the avibactam binding-pocket residues and the importance of these residues in β-lactam 177
recognition and catalysis (25-29), we investigated whether any variations in these could compromise the inhibition 178
potency of avibactam while still allowing hydrolysis of the β-lactam drug and thus result in resistance. Spontaneous 179
resistance frequency experiments were carried out in several isolates carrying class C β-lactamases, with initial focus 180
on Enterobacteriaceae spp. where aztreonam in combination with avibactam was more potent than in P. aeruginosa. 181
Resistant variants from a C. freundii and an E. coli isolate that carried multiple β-lactamase enzymes were obtained 182
at low frequencies of 6 x 10-10 and 6 x 10-9, respectively. These mutants had an 8- to 64-fold increase in the 183
aztreonam-avibactam MIC compared with the parental isolate (Table 3). Whole genome sequence analyses of the 184
parent and the daughter strains identified the mutations responsible for the loss of susceptibility. The variation in the 185
resistant C. freundii strains were either a Asn346Tyr or a Tyr150Ser change in the chromosomal AmpC protein, which 186
was confirmed by RT-PCR analysis to be stably derepressed in both the parent and daughter strains to equivalent 187
levels (Table 3 and supplementary Fig S2). The variation in the E. coli mutant was a Tyr150Cys substitution in the 188
plasmid-encoded class C enzyme (CMY-6). 189
Structural models of the AmpC mutations indicated that the Asn346Tyr substitution would result in a steric clash with 190
the sulfate group of avibactam, thus influencing the binding affinity of the inhibitor (Fig 5A). Whereas a similar impact 191
to the sulphonyl of aztreonam is expected, the slightly greater distance between this group and the side chain, as well 192
as the greater flexibility of aztreonam, could still accommodate binding and hydrolysis. While the Tyr150Ser 193
substitution maintains a similar polarity, the reduced size of the Ser residue increases the binding pocket volume 194
(Fig5B). The rigid binding mode of avibactam and the possible role of Tyr150 in the mechanism of inhibition 195
suggested that this change displaced a critical base from the vicinity of acylation. The hydrolysis of aztreonam by a 196
AmpCTyr150Ser variant is not unprecedented, as kinetic studies in laboratory generated mutants have shown that an 197
enzyme carrying this substitution can hydrolyze the substrate, albeit with reduced efficiency, likely by positioning a 198
water molecule in the mutant (30). To further explore this hypothesis, hydrolysis of aztreonam by the cell extract was 199
monitored using LC/MS in the presence and absence of avibactam. The rate of aztreonam hydrolysis was impaired in 200
the Tyr150Ser mutant compared to the parent. The addition of avibactam protected the hydrolysis of aztreonam by 36-201
fold in the parent strain; however, this protection was significantly reduced in the mutant strain, where only a 2-fold 202
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

protection was obtained after 22 h incubation (supplementary Fig S3). A similar rationale is also applicable for the 203
Tyr150Cys E. coli resistant variant due to the similarity in size of the substitutions and their ability to position a water 204
molecule for hydrolysis. 205
206
Discussion 207
Inhibition of class C β-lactamases is one of the most significant and unique attributes of avibactam (31, 32). The 208
structures presented here provide a molecular rationale for the stability of avibactam from hydrolysis by class C 209
enzymes and a hypothesis of the mechanism of acylation and recyclization as well as for a faster rate of recyclization 210
(9, 33) in this class compared to class A enzymes. 211
The lack of hydrolysis can be explained by comparing the structure described here with the well defined mechanism 212
of decylation in class C enzymes (20, 34). In its covalent binding mode, the sulfate group of avibactam has 213
completely displaced the deacylating water molecule (PDB code 1IEL) and in its position is the N6 atom of the 214
scissile bond which remains in an optimal trajectory for the reverse covalent attack on the Ser64 carbamoyl linkage. 215
This, along with a stable carbamoyl covalent linkage of avibactam, explains why intramolecular recyclization of 216
avibactam is preferred over water-mediated hydrolysis. A similar hindrance is also provided by the sulfonate group of 217
aztreonam resulting in its weaker hydrolysis compared to other β-lactams, but the flexibility and shorter length of its 218
sulfonate arm prevents complete protection. In contrast, the limited rotational freedom and the additional length of 219
the sulfate group in avibactam effectively prevent hydrolysis (Fig 3B). This mechanism of protection from hydrolysis is 220
distinct from the class A β-lactamase mechanism, where the hydrolytic water, while still observed in the binding 221
mode, was rendered ineffective by the changed protonation state of the base Glu166 (18). 222
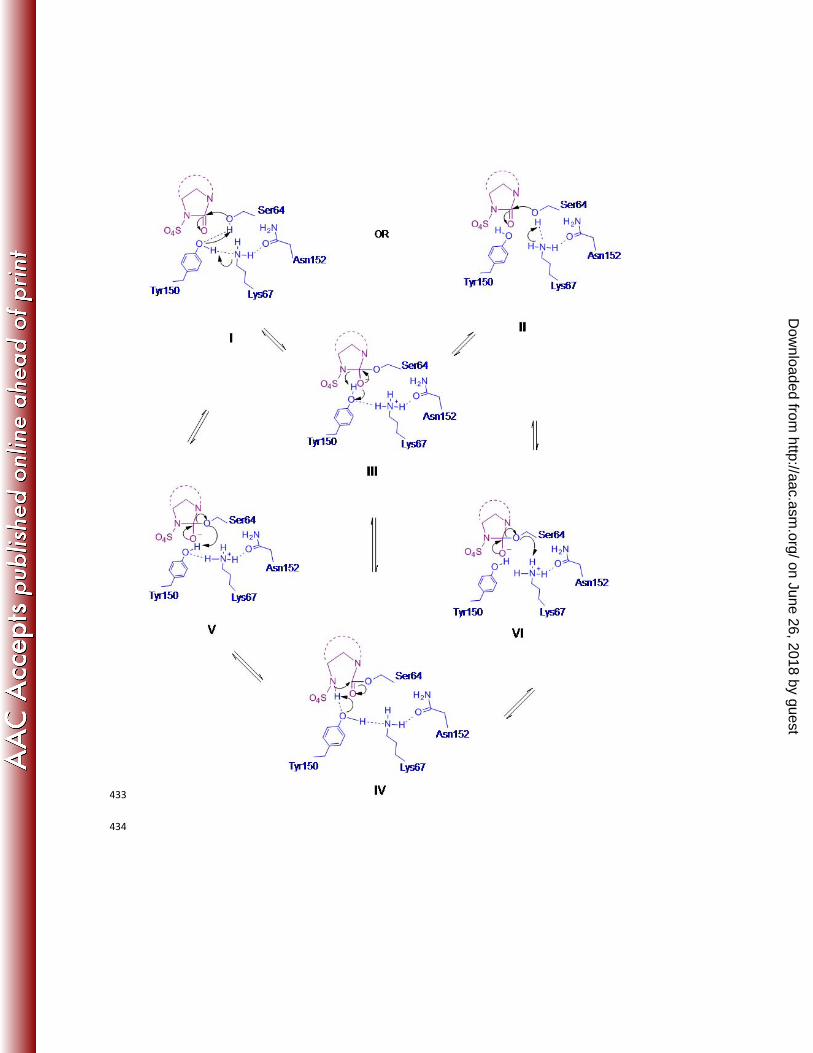
Based on the observed structure, a mechanism of reversible acylation and recyclization of avibactam could be 223
proposed for class C enzymes (Fig 6). The first step for acylation would require the deprotonation of Ser64 and a 224
subsequent protonation of the cleaved N6 nitrogen of avibactam. Both Tyr150 and Lys67 are within hydrogen-225
bonding distances to Ser64, suggesting that either or both of these residues could participate in proton abstraction. 226
However, there is only one residue, Tyr150, within proximity to N6 of avibactam to act as a general base for ring 227
opening. Similarly, in the reverse recyclization reaction, Tyr150 is the only general base in the vicinity that could 228
abstract a proton from N6, while OγSer64 could receive a proton from either Tyr150 or Lys67. Taken together, this 229
suggested that inhibition by avibactam could either proceed via a single base mechanism (Fig 6, I-III-IV-V), where 230
Tyr150 functions as the sole catalytic residue acting as both the general base and the general acid to shuttle the 231
proton from OγSer64 to N6 of avibactam. Alternatively, a conjugated acid-base mechanism can be envisioned, 232
where Lys67 functions as the general base and Tyr150 as the general acid during acylation, and vice versa during 233
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

recyclization (Fig 6, II-III-IV-VI). Both mechanisms would result in a phenolated Tyr150 in the acylated form (Fig 6, 234
IV), which is supported by the structural data where the distance between Tyr150 and Lys67 changes from 3.8Å to 235
2.8Å when covalently linked to avibactam. While this structure cannot distinguish between the single base and the 236
conjugated acid-base mechanisms, insight to the preferred path of electron transfer can be obtained from the 237
resistant variants. The Tyr150Ser and Tyr150Cys substitutions, which increase the distance between the general base 238
and the OγSer64 (Fig 5B) cause weaker but not complete loss of inhibition by avibactam. This suggests that another 239
residue is involved in acylation, thus favoring the conjugated acid-base mechanism (Fig 6, II-III-IV-VI). Further 240
structural and kinetic experiments are needed to better understand the mechanistic complexities. 241
Lastly, the faster recyclization rate of avibactam in class C enzymes compared to A β-lactamases can be explained 242
by the difference in location of the catalytic residues involved in acylation and deacylation (Figure 7). In contrast to 243
class A enzymes (CTX-M-15) where Glu166 and Ser130, the general bases for acylation and recyclization, 244
respectively, were located on the opposite faces of the carbonyl plane (18), the equivalent residues in class C 245
enzymes are located on the same side. The opposite side in the class C binding pocket, equivalent to the Glu166-246
face in class A enzymes, is less polar and formed by the hydrophobic side chain of Tyr122. The lack of polarity and 247
hydrogen-bonding residues in this region likely facilitates the intramolecular hydrogen bond observed between the 248
avibactam carboxamide group and the N1 nitrogen of the piperidine ring (Fig 2A). This strong intramolecular 249
hydrogen bond helps to delocalize electrons from the carbonyl plane, thus making it more favorable for nucleophilic 250
attack by N6. The closed loop of hydrogen bonds between Tyr150-Lys67-Asn152-carboxamide NH2-piperidine N1 in 251
the AmpC binding mode links the two cleaved ends of the scissile bond, which is able to quickly responsive to 252
alterations in electronics between the ring-closed and ring-open forms. 253
Concluding remarks. Drug resistance can occur through subtle changes that maintain substrate recognition and 254
catalysis while no longer permitting optimal inhibitor functionality. Isolates that are resistant to class A β-lactamase 255
inhibitors in current clinical use have emerged that carry β-lactamases with minor modifications, often at positions 256
where the binding mode or the mechanism of the inhibitor differs from that of the substrate, defined as substrate 257
envelope theory (35). To date, the evolutionary pressure on class C β-lactamases has been to adapt to the 258
increasing size of later generations of β-lactam drugs (36). However, they have remained naïve to any selection 259
pressure that would require the enzyme to compromise catalysis in order to avoid inhibition. This is evident by the 260
extremely well conserved binding pocket near the catalytic core among a wide range of chromosomal and plasmidic 261
class C β-lactamases. Avibactam has preserved many of the key features of β-lactam recognition and acylation to 262
efficiently exploit the residues that are critical for β-lactam catalysis. The interactions of avibactam are limited to this 263
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

conserved element, which suggests a very high probability of inhibiting all class C enzymes. In vitro selection 264
experiments hint that the lack of rotational freedom of avibactam could limit its capacity to inhibit certain variants, but 265
the ability of avibactam to mimic the key interaction of a β-lactam substrate combined with its tight binding is likely to 266
bestow a high “genetic barrier” to the development of resistance in the clinic. 267
Acknowledgments All authors are employees of AstraZeneca R&D Boston. The authors acknowledge Jim 268
Whiteaker, Kathy MacCormack and Veronica Kos from Infection, AstraZeneca R&D Boston, for their contributions in 269
whole genome sequencing. We acknowledge Joe Patel from Discovery Sciences, AstraZeneca R&D Boston, for his 270
help during crystallographic data collection. Lastly, we are thankful to Wright Nichols, Patricia Bradford, Dave 271
Ehmann and Thomas Durand-Reville from Infection, AstraZeneca R&D Boston, for careful reading and constructive 272
feedback during the preparation of this manuscript. 273
274
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

REFERENCES 275
1. Bush, K. 2010. The coming of age of antibiotics: discovery and therapeutic value. Ann. N. Y. Acad. Sci. 1213:1-4. 276
doi: 10.1111/j.1749-6632.2010.05872.x; 10.1111/j.1749-6632.2010.05872.x. 277
2. Bassetti M, Merelli M, Temperoni C, Astilean A. 2013. New antibiotics for bad bugs: where are we? Ann. Clin. 278
Microbiol. Antimicrob. 12:22. doi: 10.1186/1476-0711-12-22; 10.1186/1476-0711-12-22. 279
3. Carlet J. 2013. Multi-drug resistant bacteria and antibiotics. Rev. Infirm. 192:17-19. 280
4. Lascols C, Hackel M, Marshall SH, Hujer AM, Bouchillon S, Badal R, Hoban D, Bonomo RA. 2011. Increasing 281
prevalence and dissemination of NDM-1 metallo-beta-lactamase in India: data from the SMART study (2009). J. 282
Antimicrob. Chemother. 66:1992-1997. doi: 10.1093/jac/dkr240. 283
5. Patel G, Bonomo RA. 2013. "Stormy waters ahead": global emergence of carbapenemases. Front. Microbiol. 284
4:48. doi: 10.3389/fmicb.2013.00048; 10.3389/fmicb.2013.00048. 285
6. Jacoby GA. 2009. AmpC beta -lactamases. Clin. Microbiol. Rev. 22:161-182. 286
7. Rodriguez-Martinez JM, Poirel L, Nordmann P. 2009. Extended-spectrum cephalosporinases in Pseudomonas 287
aeruginosa. Antimicrob. Agents Chemother. 53:1766-1771. doi: 10.1128/AAC.01410-08. 288
8. Mammeri H, Nordmann P, Berkani A, Eb F. 2008. Contribution of extended-spectrum AmpC (ESAC) beta-289
lactamases to carbapenem resistance in Escherichia coli. FEMS Microbiol. Lett. 282:238-240. doi: 10.1111/j.1574-290
6968.2008.01126.x. 291
9. Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, 292
reversible, non-beta-lactam beta-lactamase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 109:11663-11668. doi: 293
10.1073/pnas.1205073109. 294
10. Cattoir V. 2011. NXL-104, a novel beta -lactamase inhibitor with broad-spectrum activity. J. Anti-Infect. 13:20-24. 295
11. Drawz SM, Papp-Wallace KM, Bonomo RA. 2014. New beta-lactamase inhibitors: A therapeutic renaissance in 296
an "MDR world"! Antimicrob. Agents Chemother. 58:1835-1846. doi: 10.1128/AAC.00826-13. 297
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

12. Levasseur P,Girard AM, Claudon M, Goossens H, Black MT, Coleman K, Miossec C. 2012. In vitro 298
antibacterial activity of the ceftazidime-avibactam (NXL104) combination against Pseudomonas aeruginosa clinical 299
isolates. Antimicrob. Agents Chemother. 56:1606-1608. doi: 10.1128/AAC.06064-11. 300
13. Shlaes DM. 2013. New beta-lactam-beta-lactamase inhibitor combinations in clinical development. Ann. N. Y. 301
Acad. Sci. 1277:105-114. doi: 10.1111/nyas.12010; 10.1111/nyas.12010. 302
14. Flamm RK, Stone GG, Sader HS, Jones RN, Nichols WW. 2013. Avibactam reverts the ceftazidime MIC of 303
European Gram-negative bacterial clinical isolates to the epidemiological cut-off value. J. Chemother. . doi: 304
10.1179/1973947813Y.0000000145. 305
15. Crandon JL, Nicolau DP. 2013. Human simulated studies of aztreonam and aztreonam-avibactam to evaluate 306
activity against challenging Gram-negative organisms, including metallo-beta-lactamase producers. Antimicrob. 307
Agents Chemother. 57:3299-3306. doi: 10.1128/AAC.01989-12; 10.1128/AAC.01989-12. 308
16. Karlowsky JA, Adam HJ, Baxter MR, Lagace-Wiens PR, Walkty AJ, Hoban DJ, Zhanel GG. 2013. In vitro 309
activity of ceftaroline-avibactam against Gram-negative and Gram-positive pathogens isolated from patients in 310
Canadian hospitals from 2010 to 2012: Results from the CANWARD surveillance study. Antimicrob. Agents 311
Chemother. 57:5600-5611. doi: 10.1128/AAC.01485-13; 10.1128/AAC.01485-13. 312
17. Goldstein EJ, Citron DM, Merriam CV, Tyrrell KL. 2013. Comparative in vitro activity of ceftaroline, ceftaroline-313
avibactam, and other antimicrobial agents against aerobic and anaerobic bacteria cultured from infected diabetic foot 314
wounds. Diagn. Microbiol. Infect. Dis. 76:347-351. doi: 10.1016/j.diagmicrobio.2013.03.019; 315
10.1016/j.diagmicrobio.2013.03.019. 316
18. Lahiri SD, Mangani S, Durand-Reville T, Benvenuti M, De Luca F, Sanyal G, Docquier JD. 2013. Structural 317
insight into potent broad-spectrum inhibition with reversible recyclization mechanism: Avibactam in complex with 318
CTX-M-15 and Pseudomonas aeruginosa AmpC beta-lactamases. Antimicrob. Agents Chemother. 57:2496-2505. 319
doi: 10.1128/AAC.02247-12; 10.1128/AAC.02247-12. 320
19. Oefner C, D'Arcy A, Daly JJ, Gubernator K, Charnas RL, Heinze I, Hubschwerlen C, Winkler FK. 1990. 321
Refined crystal structure of beta-lactamase from Citrobacter freundii indicates a mechanism for beta-lactam 322
hydrolysis. Nature 343:284-288. doi: 10.1038/343284a0. 323
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

20. Powers RA, Caselli E, Focia PJ, Prati F, Shoichet BK. 2001. Structures of ceftazidime and its transition-state 324
analogue in complex with AmpC beta-lactamase: implications for resistance mutations and inhibitor design. Biochem. 325
40:9207-9214. 326
21. Jacoby GA. 2006. Extended-spectrum beta -lactamase controversies. Chinese J. Infect. Chemother. 6:361-370. 327
22. Beadle BM, Trehan I, Focia PJ, Shoichet BK. 2002. Structural milestones in the reaction pathway of an amide 328
hydrolase: substrate, acyl, and product complexes of cephalothin with AmpC beta-lactamase. Structure 10:413-424. 329
23. Lobkovsky E, Billings EM, Moews PC, Rahil J, Pratt RF, Knox JR. 1994. Crystallographic structure of a 330
phosphonate derivative of the Enterobacter cloacae P99 cephalosporinase: mechanistic interpretation of a beta-331
lactamase transition-state analog. Biochem. 33:6762-6772. 332
24. Dahyot S, Broutin I, de Champs C, Guillon H, Mammeri H. 2013. Contribution of asparagine 346 residue to the 333
carbapenemase activity of CMY-2 beta-lactamase. FEMS Microbiol. Lett. 345:147-153. doi: 10.1111/1574-334
6968.12199; 10.1111/1574-6968.12199. 335
25. Hata M, Fujii Y, Tanaka Y, Ishikawa H, Ishii M, Neya S, Tsuda M, Hoshino T. 2006. Substrate deacylation 336
mechanisms of serine-beta-lactamases. Biol. Pharm. Bull. 29:2151-2159. 337
26. Chen Y, McReynolds A, Shoichet BK. 2009. Re-examining the role of Lys67 in class C beta-lactamase 338
catalysis. Protein Sci. 18:662-669. doi: 10.1002/pro.60. 339
27. Lietz EJ, Truher H, Kahn D, Hokenson MJ, Fink AL. 2000. Lysine-73 is involved in the acylation and 340
deacylation of beta-lactamase. Biochem. 39:4971-4981. 341
28. Nukaga M, Kumar S, Nukaga K, Pratt RF, Knox JR. 2004. Hydrolysis of third-generation cephalosporins by 342
class C beta-lactamases. Structures of a transition state analog of cefotoxamine in wild-type and extended spectrum 343
enzymes. J. Biol. Chem. 279:9344-9352. 344
29. Dubus A, Wilkin JM, Raquet X, Normark S, Frere JM. 1994. Catalytic mechanism of active-site serine beta-345
lactamases: role of the conserved hydroxy group of the Lys-Thr(Ser)-Gly triad. Biochem. J. 301 ( Pt 2):485-494. 346
30. Dubus A, Normark S, Kania M, Page MG. 1994. The role of tyrosine 150 in catalysis of beta-lactam hydrolysis 347
by AmpC beta-lactamase from Escherichia coli investigated by site-directed mutagenesis. Biochem. 33:8577-8586. 348
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

31. Coleman K. 2011. Diazabicyclooctanes (DBOs): a potent new class of non-beta -lactam beta -lactamase 349
inhibitors. Curr. Opin. Microbiol. 14:550-555. 350
32. Stachyra T, Pechereau MC, Bruneau JM, Claudon M, Frere JM, Miossec C, Coleman K, Black MT. 2010. 351
Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non-beta-lactam beta-lactamase 352
inhibitor. Antimicrob. Agents Chemother. 54:5132-5138. doi: 10.1128/AAC.00568-10. 353
33. Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Reville TF, Lahiri S, Thresher J, Livchak S, Gao N, 354
Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against Class A, C, and D beta-lactamases. 355
J. Biol. Chem. 288:27960-27971. doi: 10.1074/jbc.M113.485979; 10.1074/jbc.M113.485979. 356
34. Chen Y, Minasov G, Roth TA, Prati F, Shoichet BK. 2006. The deacylation mechanism of AmpC beta-357
lactamase at ultrahigh resolution. J. Am. Chem. Soc. 128:2970-2976. doi: 10.1021/ja056806m. 358
35. Ali A, Bandaranayake RM, Cai Y, King NM, Kolli M, Mittal S, Murzycki JF, Nalam MN, Nalivaika EA, Ozen A, 359
Prabu-Jeyabalan MM, Thayer K, Schiffer CA. 2010. Molecular basis for drug resistance in HIV-1 protease. Viruses 360
2:2509-2535. doi: 10.3390/v2112509; 10.3390/v2112509. 361
36. Bush K, Fisher JF. 2011. Epidemiological expansion, structural studies, and clinical challenges of new beta-362
lactamases from Gram-negative bacteria. Annu. Rev. Microbiol. 65:455-478. doi: 10.1146/annurev-micro-090110-363
102911. 364
365
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
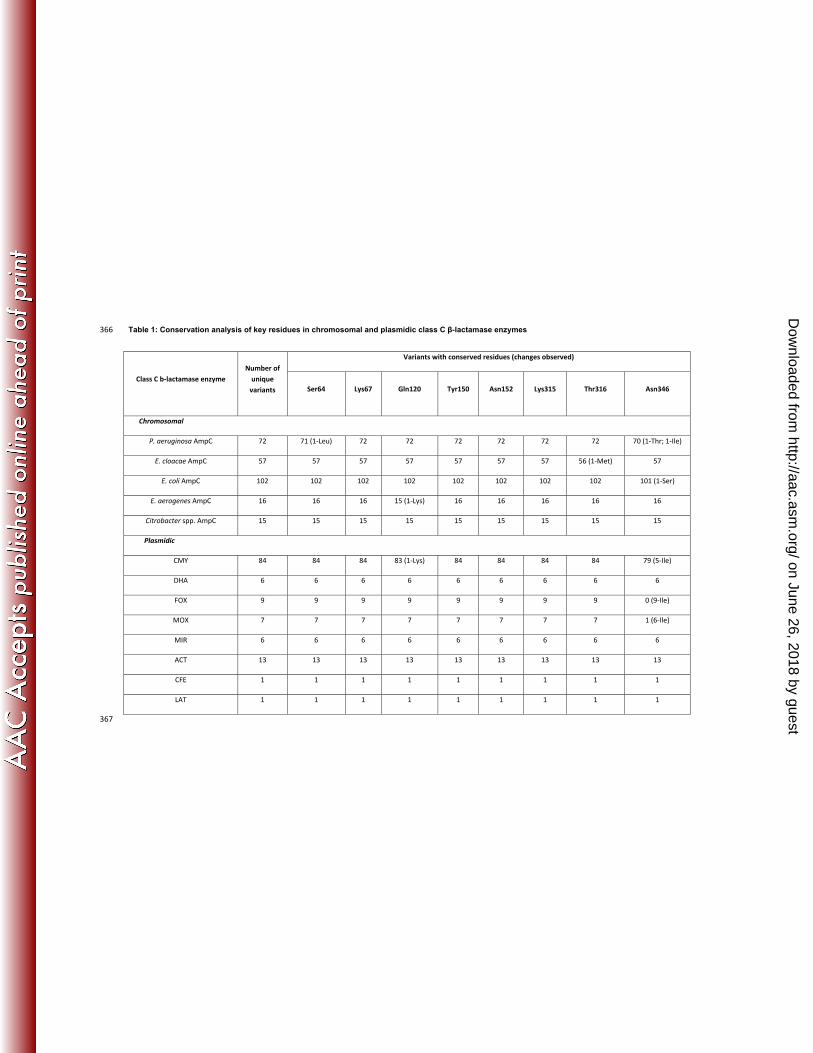
Table 1: Conservation analysis of key residues in chromosomal and plasmidic class C β-lactamase enzymes 366
Class C b-lactamase enzyme Number of
unique variants
Variants with conserved residues (changes observed)
Ser64 Lys67 Gln120 Tyr150 Asn152 Lys315 Thr316 Asn346
Chromosomal
P. aeruginosa AmpC 72 71 (1-Leu) 72 72 72 72 72 72 70 (1-Thr; 1-Ile)
E. cloacae AmpC 57 57 57 57 57 57 57 56 (1-Met) 57
E. coli AmpC 102 102 102 102 102 102 102 102 101 (1-Ser)
E. aerogenes AmpC 16 16 16 15 (1-Lys) 16 16 16 16 16
Citrobacter spp. AmpC 15 15 15 15 15 15 15 15 15
Plasmidic
CMY 84 84 84 83 (1-Lys) 84 84 84 84 79 (5-Ile)
DHA 6 6 6 6 6 6 6 6 6
FOX 9 9 9 9 9 9 9 9 0 (9-Ile)
MOX 7 7 7 7 7 7 7 7 1 (6-Ile)
MIR 6 6 6 6 6 6 6 6 6
ACT 13 13 13 13 13 13 13 13 13
CFE 1 1 1 1 1 1 1 1 1
LAT 1 1 1 1 1 1 1 1 1
367
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
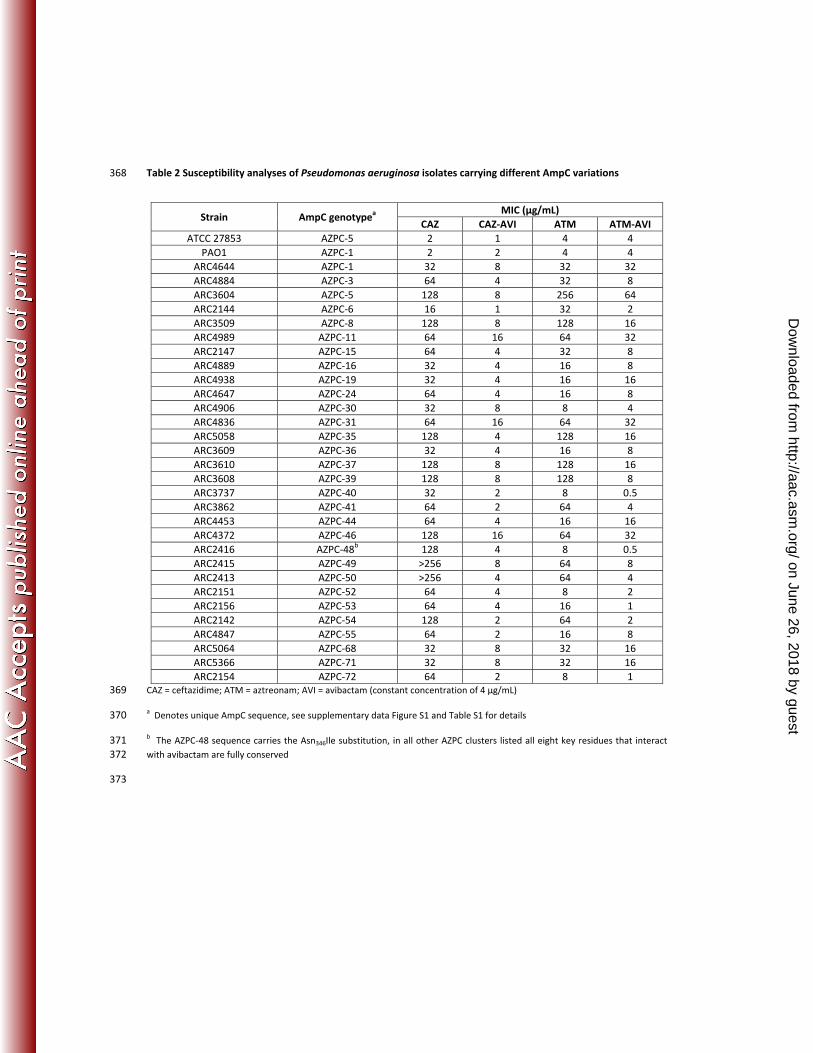
Table 2 Susceptibility analyses of Pseudomonas aeruginosa isolates carrying different AmpC variations 368
CAZ = ceftazidime; ATM = aztreonam; AVI = avibactam (constant concentration of 4 µg/mL) 369
a Denotes unique AmpC sequence, see supplementary data Figure S1 and Table S1 for details 370
b The AZPC-48 sequence carries the Asn346Ile substitution, in all other AZPC clusters listed all eight key residues that interact 371 with avibactam are fully conserved 372
373
Strain AmpC genotypea MIC (µg/mL)
CAZ CAZ-AVI ATM ATM-AVIATCC 27853 AZPC-5 2 1 4 4
PAO1 AZPC-1 2 2 4 4ARC4644 AZPC-1 32 8 32 32ARC4884 AZPC-3 64 4 32 8ARC3604 AZPC-5 128 8 256 64ARC2144 AZPC-6 16 1 32 2ARC3509 AZPC-8 128 8 128 16ARC4989 AZPC-11 64 16 64 32ARC2147 AZPC-15 64 4 32 8ARC4889 AZPC-16 32 4 16 8ARC4938 AZPC-19 32 4 16 16ARC4647 AZPC-24 64 4 16 8ARC4906 AZPC-30 32 8 8 4ARC4836 AZPC-31 64 16 64 32ARC5058 AZPC-35 128 4 128 16ARC3609 AZPC-36 32 4 16 8ARC3610 AZPC-37 128 8 128 16ARC3608 AZPC-39 128 8 128 8ARC3737 AZPC-40 32 2 8 0.5ARC3862 AZPC-41 64 2 64 4ARC4453 AZPC-44 64 4 16 16ARC4372 AZPC-46 128 16 64 32ARC2416 AZPC-48b 128 4 8 0.5ARC2415 AZPC-49 >256 8 64 8ARC2413 AZPC-50 >256 4 64 4ARC2151 AZPC-52 64 4 8 2ARC2156 AZPC-53 64 4 16 1ARC2142 AZPC-54 128 2 64 2ARC4847 AZPC-55 64 2 16 8ARC5064 AZPC-68 32 8 32 16ARC5366 AZPC-71 32 8 32 16ARC2154 AZPC-72 64 2 8 1
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
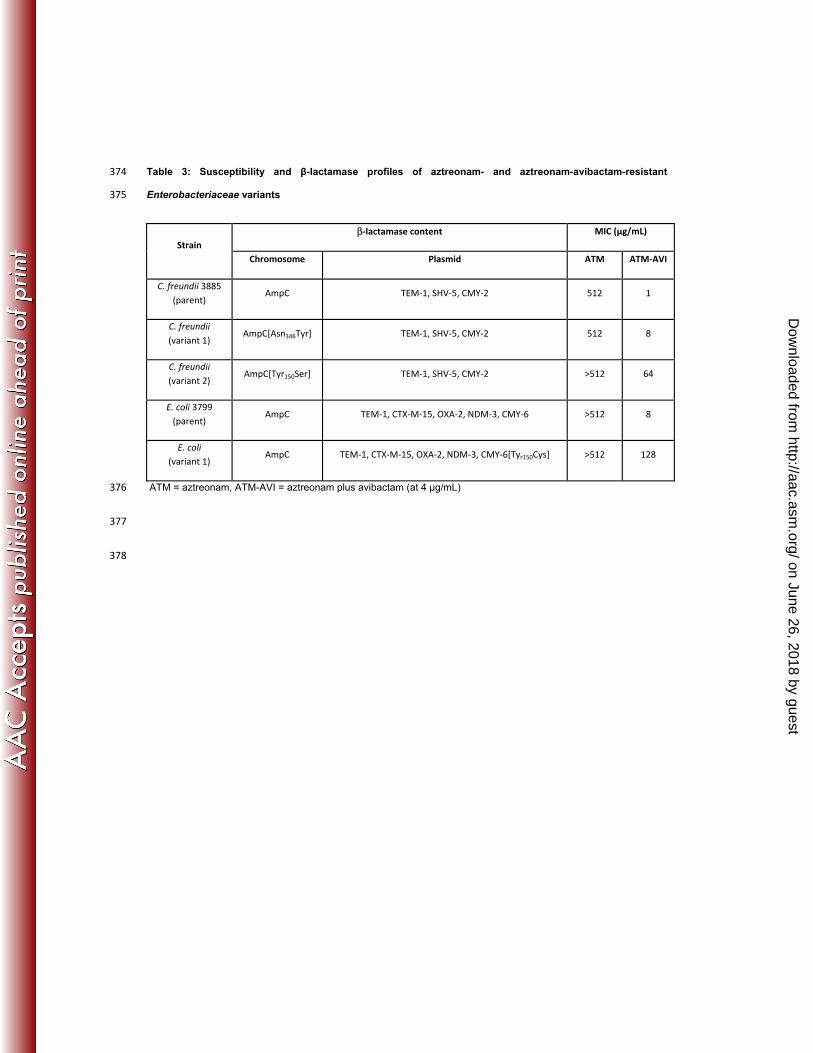
Table 3: Susceptibility and β-lactamase profiles of aztreonam- and aztreonam-avibactam-resistant 374
Enterobacteriaceae variants 375
Strain β-lactamase content MIC (µg/mL)
Chromosome Plasmid ATM ATM-AVI
C. freundii 3885 (parent)
AmpC TEM-1, SHV-5, CMY-2 512 1
C. freundii (variant 1)
AmpC[Asn346Tyr] TEM-1, SHV-5, CMY-2 512 8
C. freundii (variant 2)
AmpC[Tyr150Ser] TEM-1, SHV-5, CMY-2 >512 64
E. coli 3799 (parent)
AmpC TEM-1, CTX-M-15, OXA-2, NDM-3, CMY-6 >512 8
E. coli (variant 1)
AmpC TEM-1, CTX-M-15, OXA-2, NDM-3, CMY-6[Tyr150Cys] >512 128
ATM = aztreonam, ATM-AVI = aztreonam plus avibactam (at 4 μg/mL) 376
377
378 on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
379
380
381
Fig 1 Chemical structure of avibactam (A) and aztreonam (B) 382
383
384
385
386
387
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

Fig 2: Avibactam binding properties in AmpC Avibactam binding pocket in AmpC. (A) The unbiased Fo-Fc 393
electron density (2.9-σ cutoff, green mesh) of avibactam (pink) prior to its addition into the refinement cycle. The 394
connectivity of electron density between carboxamide and pyperidine ring is highlighted in blue arrow while the 395
additional density between observed between the cleaved C7-N6 bonds of the pyrazolidine ring is shown in green 396
arrow (B) Avibactam (pink sticks) and the neighboring residues (gray stick) depicted along with the final refined 2Fo-397
Fc map (1.5-σ cutoff, blue mesh). (C) Distances of Tyr150 (green) and Lys67 (blue) from avibactam (pink) and Ser64 398
(grey). (D) Overlay of avibactam binding pocket (grey sticks) over apo pocket (yellow sticks). Only residues with 399
changed position in apo structure has been shown. Avibactam is depicted in pink sticks. Distances between residues 400
in avibactam structure are depicted in blue lines and labels, and those of apo in yellow. 401
402
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

Fig 3: AmpC crystal structure with aztreonam Aztreonam bound to AmpC (A) The unbiased Fo-Fc electron 406
density (2.9-σ cutoff, blue mesh) of aztreonam (dark green stick) covalently bound to AmpC active site Ser64 (light 407
green stick). (B) Overlay of aztreonam binding pocket (green) on avibactam binding pocket (pink). The ceftazidime 408
and deacylating water molecule (WAT) from the ceftazidime-AmpC crystal structure. 409
410
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

411
412
A413
414
Fig 4: Conservation Conservation of avibactam binding pocket has been mapped on the AmpC crystal structure. (A) 415
Sequence conservation of Class C residues is depicted as a surface map where cyan represents variable regions 416
while purple represents conserved residues. Avibactam is depicted in white stick. (B) Residues in the binding pocket 417
of avibactam (white stick) interacting via their side chains are depicted in stick and color-coded based on 418
conservation. 419
420
421
422
423
424
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

A 425
426
B 427
Fig 5: Resistant mutants. Structural model of resistant variants using crystal structures of avibactam (in pink stick) 428
and aztreonam (in green stick). (A) Asn346 in the avibactam crystal structure is depicted in light pink while modeled 429
Tyr346 change has been depicted in white stick. (B) Tyr150 in aztreonam-AmpC structure is depicted in light green, 430
and Tyr150 in avibactam-AmpC is shown in light pink. The modeled Ser150 change has been depicted in grey stick 431
and the resulting difference in pocket volume due to this change has been depicted in a surface view.. 432
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

Fig 6: Potential mechanism scheme Scheme of proposed mechanism: acylations can proceed via a single base 435
mechanism (I-III-IV-V) or a two base mechanism (II-III-IV-VI) where either Tyr150 alone or Tyr150 and Lys67 436
together, respectively, can participate as the general acids-general bases to acylate followed by deacylate via 437
recyclization. 438
439
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

440
441
Fig 7: Class A vs. Class C Comparison of Class A and Class C binding pocket. Avibactam bound to AmpC (green) 442
has been overlaid on avibactam bound to CTX-M15 (pink) to show differences in residues interacting with the 443
inhibitor. The residues have been labeled in their respective colors. The water molecule observed at the deacylating 444
position in CTXM-15 is shown as a red sphere. 445
446
on June 26, 2018 by guesthttp://aac.asm
.org/D
ownloaded from


![d arXiv:1507.01979v2 [cond-mat.str-el] 9 Nov 2015 · High-resolution synchrotron x-ray powder diffraction pat-terns were collected at the 11-BM-B beamline using an x-ray energy of](https://static.fdocument.org/doc/165x107/5f457820cc53536c49307d57/d-arxiv150701979v2-cond-matstr-el-9-nov-2015-high-resolution-synchrotron-x-ray.jpg)







![Development of Testes and Expression of β-catenin in ... · low semen quality [5]. ... GAPDH, sense: 5’-TGG AGT CTA CTG GCG TCT TC-3’, anti-sense: 5’-ITC ACA CCC ATC ACA AAC](https://static.fdocument.org/doc/165x107/5d2cd5bd88c993136e8b4e7d/development-of-testes-and-expression-of-catenin-in-low-semen-quality.jpg)







