
8 Flüssigkeiten, Lösungen - uni-heidelberg.de · ist, dann nennt man ε die...
26
8. Flüssigkeiten, Lösungen 1 8 Flüssigkeiten, Lösungen ..................................................................................... 3 8.1 Dipol-Dipol-Wechselwirkungen .................................................................. 3 8.1.1 Polarisierung, Partialladung................................................................... 3 8.1.2 Dipolmoment ......................................................................................... 4 8.1.3 Dielektrizitätskonstante ......................................................................... 5 8.2 Induziertes Dipolmoment ............................................................................. 7 8.3 Temporäres Dipolmoment ........................................................................... 7 8.3.1 Reihenfolge der Kräfte .......................................................................... 7 8.4 Wasserstoffbrückenbindung......................................................................... 7 8.4.1 Wasserstoff-Brücken-Bindung und Struktur ......................................... 8 8.4.2 Wasserstoff-Brücken im genetischen Code......................................... 10 8.5 Die flüssige Phase ...................................................................................... 10 8.5.1 Dampfdruck und Molekülgröße .......................................................... 11 8.5.2 Dampfdruck und Wasserstoff-Brücken-Bindung ................................ 11 8.5.3 Siedepunkt ........................................................................................... 11 8.5.4 Verdampfung ....................................................................................... 12 8.6 Prinzip des kleinsten Zwanges ................................................................... 12 8.6.1 Chemische Gleichgewichte ................................................................. 13 8.6.2 Exotherme Reaktion ............................................................................ 14 8.6.3 Endotherme Reaktion .......................................................................... 14 8.7 Phasengleichgewichte ................................................................................ 15 8.7.1 Zustandsdiagramm von Wasser ........................................................... 15
Transcript of 8 Flüssigkeiten, Lösungen - uni-heidelberg.de · ist, dann nennt man ε die...

8. Flüssigkeiten, Lösungen
1
8 Flüssigkeiten, Lösungen .....................................................................................3
8.1 Dipol-Dipol-Wechselwirkungen..................................................................3
8.1.1 Polarisierung, Partialladung...................................................................3
8.1.2 Dipolmoment .........................................................................................4
8.1.3 Dielektrizitätskonstante .........................................................................5
8.2 Induziertes Dipolmoment.............................................................................7
8.3 Temporäres Dipolmoment ...........................................................................7
8.3.1 Reihenfolge der Kräfte ..........................................................................7
8.4 Wasserstoffbrückenbindung.........................................................................7
8.4.1 Wasserstoff-Brücken-Bindung und Struktur.........................................8
8.4.2 Wasserstoff-Brücken im genetischen Code.........................................10
8.5 Die flüssige Phase ......................................................................................10
8.5.1 Dampfdruck und Molekülgröße ..........................................................11
8.5.2 Dampfdruck und Wasserstoff-Brücken-Bindung................................11
8.5.3 Siedepunkt ...........................................................................................11
8.5.4 Verdampfung .......................................................................................12
8.6 Prinzip des kleinsten Zwanges...................................................................12
8.6.1 Chemische Gleichgewichte .................................................................13
8.6.2 Exotherme Reaktion ............................................................................14
8.6.3 Endotherme Reaktion ..........................................................................14
8.7 Phasengleichgewichte ................................................................................15
8.7.1 Zustandsdiagramm von Wasser...........................................................15

8. Flüssigkeiten, Lösungen
2
8.7.2 Zustandsdiagramme anderer Stoffe.....................................................16
8.7.3 Gefrierpunkts-Erniedrigung, Siedepunkts-Erhöhung..........................17
8.7.4 Bestimmung von Molmassen und Teilchenzahlen..............................18
8.7.5 Destillation...........................................................................................18
8.7.6 Azeotrop ..............................................................................................19
8.7.7 Osmose.................................................................................................20
8.8 Solvolyse ....................................................................................................20
8.9 Elektrolytische Leitfähigkeit......................................................................21
8.9.1 Elektrolyse ...........................................................................................21
8.9.2 Leitfähigkeit und Ionenart ...................................................................22
8.9.3 Leitfähigkeit durch Protonen ...............................................................22
8.9.4 Leitfähigkeit durch Hydroxid-Ionen....................................................23
8.9.5 Leitfähigkeit in Salzschmelzen............................................................23
8.9.6 Ein wenig „Stoffchemie“ zum Schluss................................................23
8.10 Index.........................................................................................................25

8. Flüssigkeiten, Lösungen
3
8 Flüssigkeiten, Lösungen
Gase können zu Flüssigkeiten kondensiert werden, weil Atome und Moleküle aufeinander anziehende Kräfte ausüben. Das ideale Gasgesetz beschreibt das Verhalten von Teilchen unter der Voraussetzung, dass sie keine Kräfte aufeinander ausüben. Es gilt daher immer nur angenähert und sicher nicht mehr dann, wenn die durch die Anziehung zwischen den Teilchen bewirkte Absenkung der Energie des Systems die kinetische Energie der Teilchen überkompensiert. (Siehe Kapitel 7, ideale und reale Gase etc.). Die Existenz des flüssigen Zustandes ist ebenso wie die Existenz des festen Zustandes auf das Vorhandensein von Anziehungskräften zurückzuführen. Diejenigen Kräfte, welche Atome durch chemische Bindungen zusammenhalten, wurden bereits besprochen. Diejenigen Anziehungskräfte, welche dazu führen, dass Atome oder Moleküle mit abgeschlossenen Elektronenkonfigurationen (z.B. He, H2, C6H6...) einander wechselseitig anziehen, nennt man „sekundäre Wechselwirkungen“. Es gibt verschiedene Ursachen für das Zustandekommen dieser Kräfte, die sich jeweils in einfachen Modellen beschreiben lassen. Diese Modelle werden nachfolgend vorgestellt.
8.1 Dipol-Dipol-Wechselwirkungen
8.1.1 Polarisierung, Partialladung Verschiedene Arten von Atomen ziehen Elektronen unterschiedlich stark an. Die Gründe hierfür wurden eingehend diskutiert. Als qualitatives Maß, für die Tendenz verschiedener Atomarten, Elektronen anzuziehen, wurde die Skala der Elektronegativität eingeführt. Wenn zwei Atome unterschiedlicher Elektronegativität in einem Molekül aneinander gebunden sind, dann werden die Elektronen, welche die Bindung vermitteln, von den beiden Atomen unterschiedlich stark angezogen. Partialladung In einem Molekül wie Br-Cl (einer „Interhalogenverbindung“) werden die Bindungselektronen mehr zum Chlor als zum Brom hingezogen, denn Chlor ist elektronegativer als Brom (warum?). Damit entsteht eine unsymmetrische Ladungsverteilung: Auf der einen Seite (am Brom) fehlt Elektronendichte, was zu einer „positiven Partialladung“, δ +, führt. Auf der anderen Seite (Chlor) ist mehr Elektronendichte vorhanden, als durch die Kernladung des Atoms ausgeglichen werden kann; das Atom hat eine „negative Partialladung“, δ -. Polarisierung Man sagt auch: „das Atom ist positiv polarisiert“, wenn man ihm die Partialladung δ + zuschreibt. Wenn dem Atom eine negative Partialladung δ - zugeschrieben wird, sagt man entsprechend „ das Atom ist negativ polarisiert“.

8. Flüssigkeiten, Lösungen
4
Symbole für Ladungen Die verschiedenen Symbole, mit denen Ladung angedeutet wird, darf man nicht verwechseln: Ladung von Ionen: z.B. Cl-, SO4
2-, Na+ etc. Diese Ladung „tritt nach außen auf“, sie ist die Ladung der Ionen. Man kann sie messen, sie ist wirklich vorhanden. Formale Ladung: Diese ergibt sich nach den Regeln für das Schreiben von Valenzstrichformeln für jedes Atom in einer Verbindung. In den meisten Fällen ist die formale Ladung der Atome in einer Verbindung gleich Null, sie wird dann nicht angeschrieben. Die Formale Ladung entspricht einer symbolischen Festlegung, sie hat keine „reale Bedeutung“, d.h. sie ist nicht messbar! Die symbolische Festlegung ist aber so beschaffen, dass in jedem Fall gilt: Die Summe der formalen Ladungen ergibt in jedem Fall die nach außen erscheinende Ladung. Partialladung: Mit ihr wird angedeutet, welches von zwei aneinander gebundenen Atomen den „Streit“ um die bindende Elektronendichte gewinnt. Das Atom, das gewinnt, ist dasjenige, das elektronegativer ist. Ihm wird eine Partialladung von δ - zugeschrieben. Dem jeweils anderen Atom wird entsprechend die Partialladung δ + zugeschrieben. Das Symbol δ bedeutet, dass die Ladung klein ist und jedenfalls viel kleiner ist als eine Elementarladung. Es sagt nichts über den Betrag der Partialladung, der klein aber von Fall zu Fall verschieden ist. Die Tatsache, dass die Schwerpunkte positiver und negativer Ladung nicht zusammenfallen, kann durch Messung ermittelt werden.
8.1.2 Dipolmoment In einem zweiatomigen Molekül, in dem die Bindungspartner verschiedene Elektronegativitäten aufweisen, fallen die Schwerpunkte positiver und negativer Ladung nicht zusammen, den Atomen kann eine Partialladung zugeschrieben werden. Damit verhalten sich
solche Moleküle wie elektrische Dipole, sie haben ein „Dipolmoment“. Das elektrische Dipolmoment, µµµµ, ist definiert als das Produkt von Ladung und Abstand. Seine Einheit ist folglich im SI-System C ⋅⋅⋅⋅ m. Die in der Chemie gebräuchliche Einheit ist das „Debye“ (Benannt nach P. Debye, Holland, 1884-1966, Zürich, Göttingen, Leipzig, Berlin, New York).

8. Flüssigkeiten, Lösungen
5
Ein Dipol, in dem zwei Elementarladungen mit entgegengesetztem Vorzeichen 1 Angstroem voneinander entfernt sind, hat ein Dipolmoment von 4.8 Debye (abgekürzt 4.8 D).
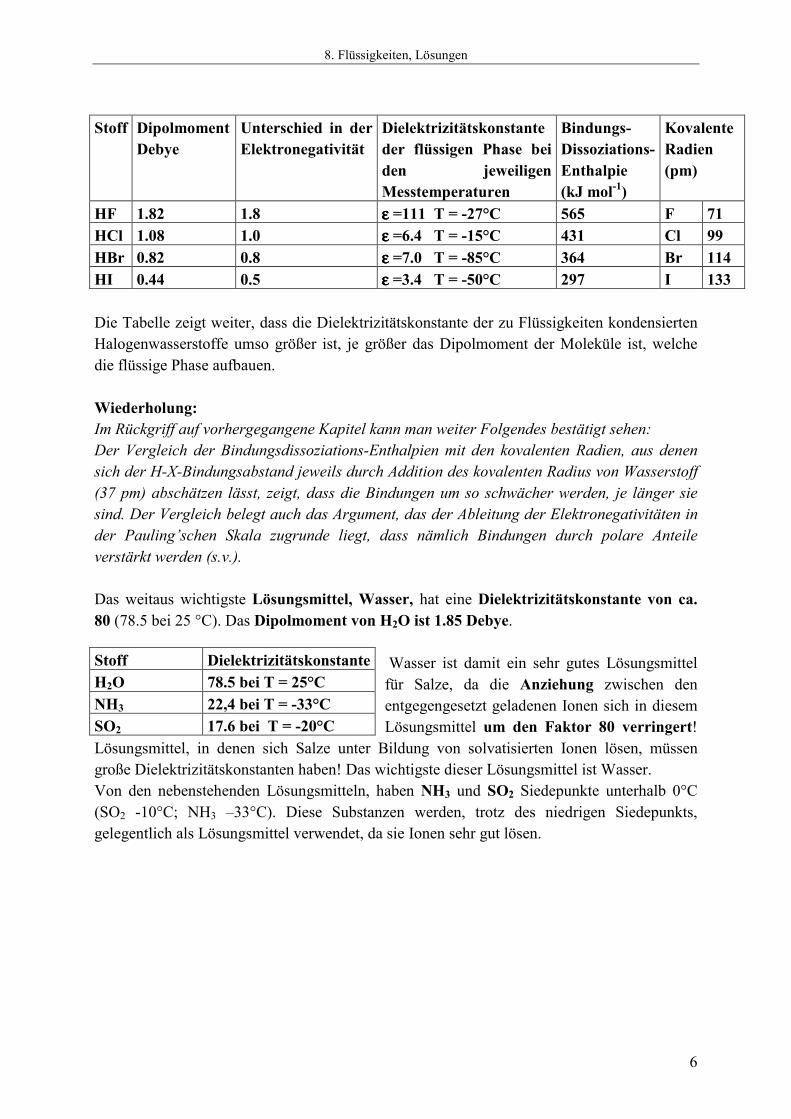
8.1.3 Dielektrizitätskonstante Dipole üben aufeinander Kräfte aus, die durch Coulomb-Wechselwirkung zustande kommen. In einem elektrischen Feld orientieren sich die Dipole in einer Vorzugsrichtung, wobei die im Diagramm symbolisierte vollständige Ordnung der Orientierung wegen der thermischen Unordnung nicht erreicht werden kann. Damit die Dipole sich orientieren, muß Arbeit geleistet werden. Bei gegebener Potentialdifferenz zwischen den Platten (Querschnitt im Bild) der Apparatur, fließt so lange ein Strom, bis das Gleichgewicht zwischen der orientierenden Wirkung des Feldes und dem Bestreben der Dipole nach Unordnung eingestellt ist. Die dabei in der Potentialdifferenz U transportierte Ladungsmenge Q ergibt mit E = U ⋅ Q die Arbeit, die bei diesem Prozess geleistet wurde, oder, anders gesagt, die Energie, die in der Apparatur durch die Orientierung gespeichert wurde. Eine solche Apparatur nennt man einen „Kondensator“. Die Energie, die sich in ihm bei gegebener Spannung speichern lässt, ist seiner „Kapazität“ proportional. Die Einheit der Kapazität ist 1 Faraday = 1C/V. Ein Kondensator hat diese Kapazität, wenn bei ein Volt Potentialdifferenz die Ladung von 1 Coulomb in ihm gespeichert werden kann. Die Ladung, die sich in einem Kondensator speichern lässt, ist bei gegebener Konstruktion des Kondensators und bei gegebener Potentialdifferenz, der „Dielektrizitätskonstanten“ ε ε ε ε des Stoffes proportional, der sich zwischen den Kondensatorplatten befindet. Wenn der Kondensator die Kapazität x Farad hat, wenn der Raum zwischen den Platten leer ist (Vakuum) und εεεε ⋅ x Farad, wenn der Raum zwischen den Platten mit einem Stoff A gefüllt ist, dann nennt man εεεε die Dielektrizitätskonstante des Stoffes A. Die Dielektrizitätskonstante ist eine wichtige Stoffeigenschaft. Das rührt daher, dass die Coulomb-Wechselwirkung zwischen zwei geladenen Teilchen in einem Medium mit der Dielektrizitätskonstanten ε 1/εεεε mal kleiner ist, als im Vakuum. Stoffe, die ein „permanentes Dipolmoment“ haben, haben hohe Dielektrizitätskonstanten. Die nachfolgende Tabelle zeigt am Beispiel der Halogenwasserstoff-Verbindungen, dass die Dipolmomente umso größer sind, je größer die Differenz der Elektronegativität der Bindungspartner ist. Man erkennt an diesen Daten zugleich, dass die Halogenwasserstoff-Verbindungen nicht ionisch gebaut sein können. In HCl findet man einen Bindungsabstand von 127 pm. Bei vollständiger Ladungstrennung würde sich bei diesem Abstand ein Dipolmoment von 1.27 ⋅ 4.8 = 6.08 Debye ergeben. Der beobachtete Wert ist mit 1.08 D wesentlich kleiner. Dies zeigt, dass die Bindung zwar polarisiert ist, dass aber die Ladungstrennung wesentlich kleiner ist, als für ein Ionenpaar H+ X- bei dem jeweiligen Abstand zu erwarten wäre.
8. Flüssigkeiten, Lösungen
6
Stoff Dipolmoment
Debye Unterschied in der Elektronegativität
Dielektrizitätskonstante der flüssigen Phase bei den jeweiligen Messtemperaturen
Bindungs-Dissoziations-Enthalpie (kJ mol-1)
Kovalente Radien (pm)
HF 1.82 1.8 εεεε =111 T = -27°C 565 F 71 HCl 1.08 1.0 εεεε =6.4 T = -15°C 431 Cl 99 HBr 0.82 0.8 εεεε =7.0 T = -85°C 364 Br 114 HI 0.44 0.5 εεεε =3.4 T = -50°C 297 I 133 Die Tabelle zeigt weiter, dass die Dielektrizitätskonstante der zu Flüssigkeiten kondensierten Halogenwasserstoffe umso größer ist, je größer das Dipolmoment der Moleküle ist, welche die flüssige Phase aufbauen. Wiederholung: Im Rückgriff auf vorhergegangene Kapitel kann man weiter Folgendes bestätigt sehen: Der Vergleich der Bindungsdissoziations-Enthalpien mit den kovalenten Radien, aus denen sich der H-X-Bindungsabstand jeweils durch Addition des kovalenten Radius von Wasserstoff (37 pm) abschätzen lässt, zeigt, dass die Bindungen um so schwächer werden, je länger sie sind. Der Vergleich belegt auch das Argument, das der Ableitung der Elektronegativitäten in der Pauling’schen Skala zugrunde liegt, dass nämlich Bindungen durch polare Anteile verstärkt werden (s.v.). Das weitaus wichtigste Lösungsmittel, Wasser, hat eine Dielektrizitätskonstante von ca. 80 (78.5 bei 25 °C). Das Dipolmoment von H2O ist 1.85 Debye.
Wasser ist damit ein sehr gutes Lösungsmittel für Salze, da die Anziehung zwischen den entgegengesetzt geladenen Ionen sich in diesem Lösungsmittel um den Faktor 80 verringert!
Lösungsmittel, in denen sich Salze unter Bildung von solvatisierten Ionen lösen, müssen große Dielektrizitätskonstanten haben! Das wichtigste dieser Lösungsmittel ist Wasser. Von den nebenstehenden Lösungsmitteln, haben NH3 und SO2 Siedepunkte unterhalb 0°C (SO2 -10°C; NH3 –33°C). Diese Substanzen werden, trotz des niedrigen Siedepunkts, gelegentlich als Lösungsmittel verwendet, da sie Ionen sehr gut lösen.
Stoff Dielektrizitätskonstante H2O 78.5 bei T = 25°C NH3 22,4 bei T = -33°C SO2 17.6 bei T = -20°C

8. Flüssigkeiten, Lösungen
7
8.2 Induziertes Dipolmoment Moleküle mit “permanentem Dipolmoment“, in denen die Schwerpunkte der positiven und der negativen Ladung nicht zusammenfallen, ziehen einander an. Zugleich wirken Sie auf die
Elektronendichteverteilung in Molekülen, die selbst kein Dipolmoment haben, denn sie bauen ein lokales elektrisches Feld auf, in dem die Umgebung polarisiert wird. In Molekülen, die selbst kein permanentes Dipolmoment besitzen, wird dadurch ein temporäres Dipolmoment induziert. Daher ziehen sich auch Dipol-Moleküle und unpolare Moleküle an.
8.3 Temporäres Dipolmoment Die Elektronendichteverteilung ist nicht statisch, sondern unterliegt kleinen temporären Schwankungen. Auch in Molekülen, die kein permanentes Dipolmoment aufweisen, entstehen so, temporär, polare Ladungsdichteverteilungen. Diese schwachen temporären Dipole induzieren in den Nachbarmolekülen wiederum Dipole, so dass auch zwischen völlig unpolaren Teilchen dennoch anziehende Kräfte vorhanden sind.
8.3.1 Reihenfolge der Kräfte Die Kräfte, die durch unsymmetrische Ladungsverteilung zustande kommen werden in der folgenden Reihenfolge schwächer: Dipol/Dipol > Dipol/Induzierter Dipol > Temporärer Dipol/Induzierter temporärer Dipol Da bei allen Stoffen wenigstens die letztgenannte Wechselwirkung vorhanden ist, bilden alle Stoffe kondensierte Phasen (s.v.).
8.4 Wasserstoffbrückenbindung Moleküle, die positiv polarisierten Wasserstoff enthalten, üben eine besondere Art von anziehender Wechselwirkung aufeinander aus. In solchen Verbindungen tritt der positiv polarisierte Wasserstoff eines Moleküls mit freien Elektronenpaaren in seiner Umgebung in Wechselwirkung. Diese Wechselwirkung ist dann besonders stark, wenn die freien Elektronenpaare zu elektronegativen Atomen (F, O, N) gehören.

8. Flüssigkeiten, Lösungen
8
Der Grund hierfür liegt darin, dass die Wechselwirkung zwischen zwei Orbitalen - jeweils gleiche Überlappung vorausgesetzt - am stärksten ist, wenn beide Orbitale die gleiche Energie haben. Je größer der energetische Abstand zwischen den Orbitalen ist, die eine Wechselwirkung eingehen sollen, desto schwächer ist diese Wechselwirkung. Das Valenzorbital des Wasserstoffs ist das 1s-Orbital , das bei der niedrigen Energie von -13.6 eV liegt. Nur elektronegative
Atome, wie F, O und N haben Valenzorbitale bei ähnlich niedrigen Energien, so dass nur deren „freie Elektronenpaare“ (im MO-Bild: die Orbitale, welchen die Elektronendichte der freien Elektronenpaare zuzuordnen ist) mit dem Wasserstoff-Valenzorbital in stärkere Wechselwirkung treten können.
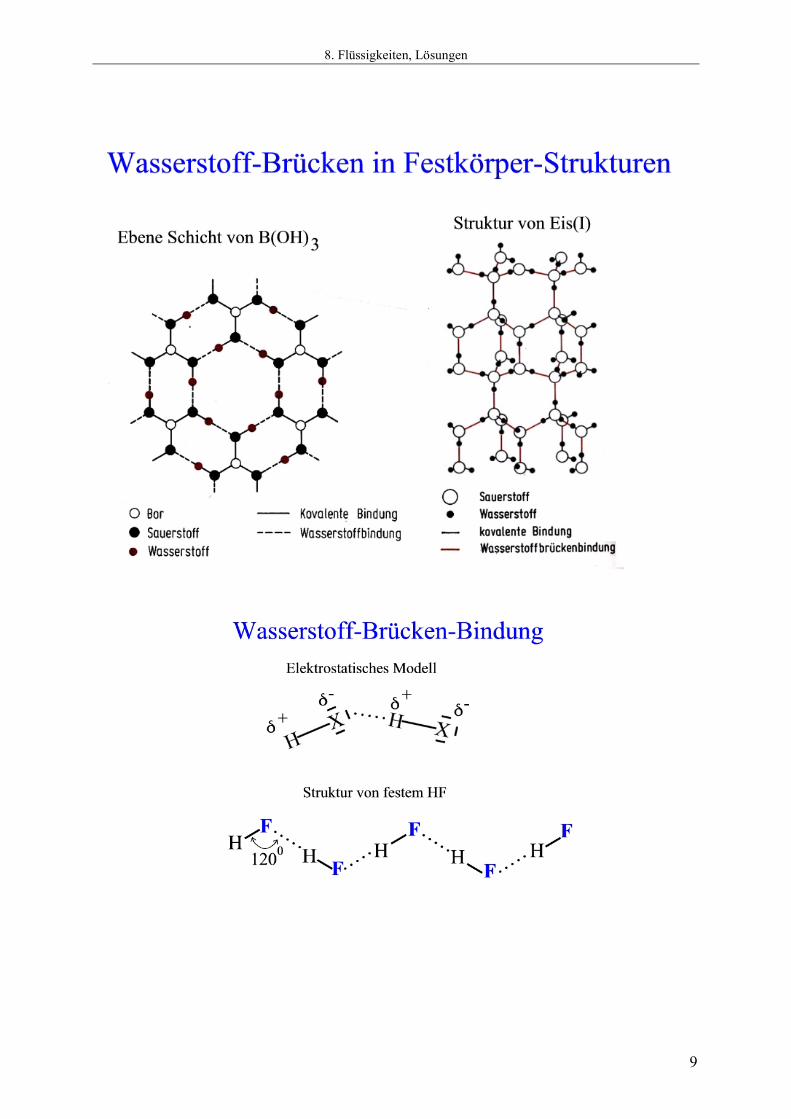
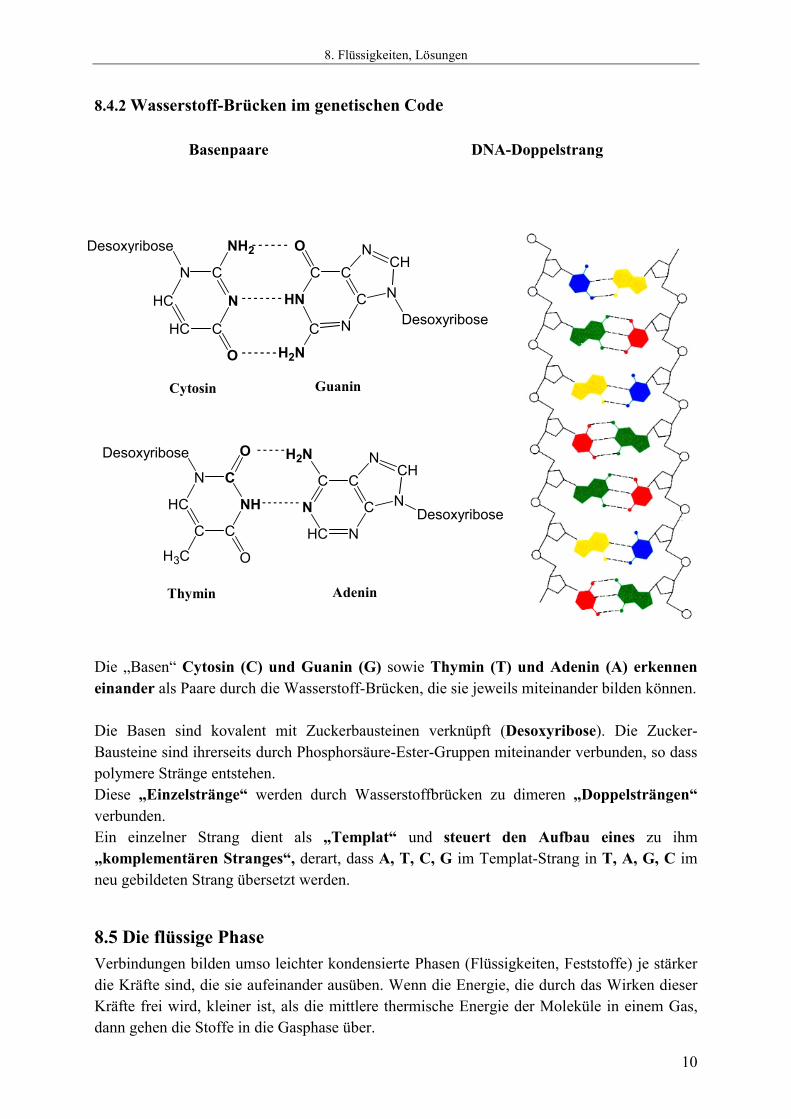
8.4.1 Wasserstoff-Brücken-Bindung und Struktur Die nachfolgenden Diagramme zeigen die Bedeutung von Wasserstoffbrücken für die Wechselwirkung zwischen Molekülen. Dort, wo Wasserstoffbrückenbindungen aufgebaut werden können, bestimmen sie meist die Struktur (Vgl. HF, H2O, B(OH)3, Basenpaare). Die Bindungsenergie von Wasserstoffbrückenbindungen ist deutlich kleiner, als diejenige normaler chemischer Bindungen und beträgt selten mehr als 40 kJ mol-1 (Im Gegensatz zu normalen chemischen Bindungen, die schon als Einfachbindungen Bindungsdissoziations-Enthalpien von um die 400 kJ mol-1 aufweisen!). Dennoch verursachen Wasserstoffbrückenbindungen die stärksten Kräfte zwischen Molekülen mit abgeschlossenen Valenzschalen (D.h. Molekülen, die keine weiteren chemischen Bindungen mehr eingehen müssen um der Edelgasregel zu genügen). Die Erkennung in biologischen Systemen macht von Wasserstoffbrückenbindungen hervorragenden Gebrauch. Die Bindungen können ohne großen Energieaufwand gelöst werden und sind doch in der Summe hinreichend stabil, um die Konformation großer Moleküle (DNA = Desoxyribonucleinsäure = Desoxy RiboNucleic Acid, Proteine) bei den Temperaturen, bei denen sich Leben abspielt, ausreichend stabil zu halten. Das System der H-Brücken in solchen Verbindungen kann man sich vorstellen, wie die einzelnen Glieder eines Reißverschlusses. Auch wenn ein einzelnes Glied nicht sehr stabil ist, so ist das Gesamtsystem der in einer Kette geordneten Glieder von hoher Stabilität. Dennoch kann man es leicht öffnen und schließen.
8. Flüssigkeiten, Lösungen
9
8. Flüssigkeiten, Lösungen
10
8.4.2 Wasserstoff-Brücken im genetischen Code
Basenpaare DNA-Doppelstrang
Die „Basen“ Cytosin (C) und Guanin (G) sowie Thymin (T) und Adenin (A) erkennen einander als Paare durch die Wasserstoff-Brücken, die sie jeweils miteinander bilden können. Die Basen sind kovalent mit Zuckerbausteinen verknüpft (Desoxyribose). Die Zucker-Bausteine sind ihrerseits durch Phosphorsäure-Ester-Gruppen miteinander verbunden, so dass polymere Stränge entstehen. Diese „Einzelstränge“ werden durch Wasserstoffbrücken zu dimeren „Doppelsträngen“ verbunden. Ein einzelner Strang dient als „Templat“ und steuert den Aufbau eines zu ihm „komplementären Stranges“, derart, dass A, T, C, G im Templat-Strang in T, A, G, C im neu gebildeten Strang übersetzt werden.
8.5 Die flüssige Phase Verbindungen bilden umso leichter kondensierte Phasen (Flüssigkeiten, Feststoffe) je stärker die Kräfte sind, die sie aufeinander ausüben. Wenn die Energie, die durch das Wirken dieser Kräfte frei wird, kleiner ist, als die mittlere thermische Energie der Moleküle in einem Gas, dann gehen die Stoffe in die Gasphase über.
HC
N
N
C
CC
H2N
N
CHN
CN
O
NH
C
O
C
HC
CHN
NC
NCH2N
HN
C
O
C
HC C
N
CN
HC
NH2Desoxyribose
O
Desoxyribose
H3C
Desoxyribose
Desoxyribose
Thymin Adenin
GuaninCytosin

8. Flüssigkeiten, Lösungen
11
8.5.1 Dampfdruck und Molekülgröße Daraus folgt die Regel, dass größere Moleküle - das sind zugleich Moleküle mit größerer Molmasse - nur bei hoher Temperatur in die Gasphase übergehen, d.h. schwerer verdampfen als kleine Moleküle. Bei Molekülen, die untereinander keine Wasserstoffbrücken bilden können, ist diese Regel gut erfüllt.
8.5.2 Dampfdruck und Wasserstoff-Brücken-Bindung Bei Molekülen, welche untereinander H-Brücken aufbauen können, beobachtet man dagegen jeweils niedrigere Dampfdrücke über der kondensierten Phase als bei etwa gleich großen Molekülen die keine H-Brücken ausbilden können.
8.5.3 Siedepunkt Die Temperatur, bei welcher der Dampfdruck über einer Flüssigkeit 1 atm (ca. 1 bar, s.v.) erreicht, nennt man die Siedetemperatur oder auch den „Siedepunkt“ des Stoffes, aus dem die Flüssigkeit besteht. Stoff HF HCl HBr HI Siedepunkt (°C) -20 -85 -67 -35 Stoff H2O H2S H2Se H2Te Siedepunkt (°C) 100 -61 -41 -2 Stoff NH3 PH3 AsH3 SbH3 Siedepunkt (°C) -33 -88 -62 -17 Die starken Wasserstoff-Brücken in HF, H2O und NH3 führen dazu, dass diese Stoffe wesentlich höhere Siedetemperaturen haben, als nach ihrer Molmasse zu erwarten wäre. Ab den jeweils schwereren Homologen, HCl, H2S, PH3 steigen die Siedetemperaturen mit zunehmender Molmasse an, wie dies für Verbindungen in Abwesenheit starker H-Brückenbindung erwartet wird und normal ist.

8. Flüssigkeiten, Lösungen
12
8.5.4 Verdampfung Flüssige Phase und Gasphase stehen in einem dynamischen Gleichgewicht miteinander. Wenn je Zeiteinheit ebenso viele Teilchen die Flüssigkeit verlassen, wie Teilchen aus der Gasphase in die Flüssigkeit übergehen, dann ist das System im Gleichgewicht. (Vergleiche Kap 7, chemisches Gleichgewicht).
Störung des Gleichgewichts Wenn das Gleichgewicht „gestört“ wird, so stellt es sich von neuem ein. Druckänderung Eine Störung des Gleichgewichts wäre es etwa, wenn die Teilchen aus der Gasphase durch „Anlegen eines Vakuums“ entfernt würden. Das Gleichgewicht, das einem bestimmten Partialdruck der Teilchen in der Gasphase entspricht, würde sich neu einstellen, indem Teilchen aus der flüssigen Phase in die Gasphase übergingen, solange, bis der dem Gleichgewicht entsprechende Partialdruck wieder erreicht wäre. Die Flüssigkeit würde zum Teil „verdampfen“. Temperaturänderung Erhöhung der Temperatur: Bei Erhöhung der Temperatur hat ein größerer Anteil der Teilchen in der flüssigen Phase ausreichend kinetische Energie (Ekin = ½ mv2 = 3/2 RT, s.v.), um die Anziehungskräfte zu überwinden, die es in der flüssigen Phase festhalten wollen. Die Flüssigkeit verdampft so lange, bis der der jeweiligen Temperatur entsprechende Gleichgewichts-Partialdruck wieder erreicht ist. Erniedrigung der Temperatur: Beim Absenken der Temperatur wird der Anteil derjenigen Teilchen in der Gasphase höher, die eine ausreichend kleine kinetische Energie haben, um unter Energiefreisetzung in der flüssigen Phase gebunden zu werden. Die Gasphase „kondensiert“ so lange, bis der der jeweiligen Temperatur entsprechende Gleichgewichts-Partialdruck wieder erreicht ist.
8.6 Prinzip des kleinsten Zwanges Am Beispiel von Verdampfung und Kondensation lässt sich ein allgemeines Prinzip illustrieren, das für alle System gilt: Das sogenannte „Prinzip des kleinsten Zwanges“, das erstmals von Le Châtelier formuliert wurde (H. L. Le Châtelier, 1850-1936, Paris, und dessen Mitarbeiter K. F. Braun 1887, Formulierung des Prinzips). Dieses Prinzip besagt:

8. Flüssigkeiten, Lösungen
13
Ein im Gleichgewicht befindliches System reagiert auf Störungen stets so, dass es der Ursache der Störung entgegenwirkt. Wenn der Druck erhöht wird, tritt der Prozess ein, der den Druck erniedrigt. Wenn der Druck erniedrigt wird, tritt der Prozess ein, der den Druck erhöht. Auf das oben angeführte Beispiel bezogen heißt das: Wenn der Druck über der Flüssigkeit erniedrigt wird (s.o.), so gehen Teilchen in die Gasphase über und erhöhen den Druck wieder. Wenn die Temperatur des Systems erhöht wird, dann tritt der Prozess ein, der Wärme verbraucht. Verdampfung erfordert Wärme, denn die kinetische Energie der Teilchen in der flüssigen Phase muß erhöht werden, damit mehr Teilchen in die Gasphase gehen. Die Wärmeenergie, die notwendig ist, um ein Mol eines Stoffes zu verdampfen, nennt man seine Verdampfungsenthalpie. Dass Verdampfung Wärme verbraucht, ist aus dem täglichen Leben offensichtlich: Man muß heizen, damit das Kaffeewasser kocht! Man schwitzt, wenn es heiß ist. Der biologisch systemerhaltende Sinn des Schwitzens ist die Regulation der Körpertemperatur: durch die Verdampfung des Schweißes entsteht Abkühlung. Wenn die Temperatur des Systems erniedrigt wird, dann tritt der Prozess ein, der Wärme liefert. Die Kondensation des Gases zur Flüssigkeit liefert Energie, da die Teilchen in der flüssigen Phase sich anziehen. Im täglichen Leben: Das kalte Bier wird warm, wenn am Bierglas Wasserdampf aus der Luft sich zu Tropfen niederschlägt etc.
8.6.1 Chemische Gleichgewichte Auch für chemische Gleichgewichte gilt dieses Prinzip durchwegs. Das Massenwirkungsgesetz entspricht einer quantitativen Fassung dieses Prinzips bezüglich der Stoffkonzentrationen. Im Bezug auf die Reaktionsenthalpie lässt sich aus der van’t Hoff’schen Gleichung eine quantitative Beziehung ableiten:
1. G∆ = -RT ln K; � ln K = - G∆ /RT
2. G∆ = H∆ - T S∆

8. Flüssigkeiten, Lösungen
14
Aus 1. und 2. folgt: d(ln K)/dT = d(- G∆ /RT) / dT � d(ln K)/dT = d(-( H∆ - T S∆ )/RT) / dT d(ln K)/dT = d(- H∆ /RT) / dT + d( S∆ /R) / dT (Wegen d(1/x)/dx = d(x-1) / dx = - x-2 und d( S∆ /R) / dT = 0, weil unabhängig von T).
8.6.2 Exotherme Reaktion Für eine exotherme Reaktion ist H∆ < 0. Damit ist d(ln K)/dT < 0 und das Gleichgewicht verschiebt sich mit steigender Temperatur nach links (d.h. der Logarithmus der Gleichgewichtskonstanten - und damit natürlich auch die Gleichgewichtskonstante selbst - wird kleiner). Es tritt vermehrt der Prozess ein, der Wärme verbraucht, nämlich die endotherme Rückreaktion.
8.6.3 Endotherme Reaktion Bei einer endothermen Reaktion verschiebt sich dagegen das Gleichgewicht mit steigender Temperatur nach rechts. Auch hier tritt der Prozess vermehrt ein, der Wärme verbraucht. In diesem Fall ist das die endotherme Hinreaktion. Im anderen Fall war es die endotherme Rückreaktion.
d(ln K)/dT = H∆ /(RT2)

8. Flüssigkeiten, Lösungen
15
8.7 Phasengleichgewichte Die Phase, in der ein Stoff vorliegt (Gasförmig, Flüssig, Fest) hängt von der Temperatur und
vom Druck ab. Wenn zwei Phasen im Gleichgewicht sein sollen, dann müssen je Zeiteinheit gleich viele Teilchen aus der einen Phase in die andere übergehen, wie aus der anderen Phase in die eine zurückkehren. Wenn eine der Größen, Druck oder Temperatur, festgelegt wird, bei der sich das Gleichgewicht zwischen den Phasen einstellen
soll, dann ist die andere dadurch bereits ebenfalls festgelegt, denn bei einer bestimmten Temperatur kann das Gleichgewicht auch nur bei einem ganz bestimmten Druck erreicht werden. Die Punkte, an denen das der Fall ist, lassen sich in ein Druck/Temperatur-Diagramm als Kurven einzeichnen.
8.7.1 Zustandsdiagramm von Wasser Als ein Beispiel ist das „Zustandsdiagramm“ von Wasser gezeigt. Bei „Atmosphärendruck“ (1.013 bar) sind feste Phase (Eis) und flüssige Phase (Wasser) bei 0°C im Gleichgewicht. Bei höherer Temperatur tritt der Prozess vermehrt ein, der Wärme erfordert. Das ist in diesem Fall das Schmelzen, da das Schmelzen einer Substanz immer ein endothermer Vorgang ist, der „Schmelzwärme“ verbraucht. Die Wärmeenergie, die einem Stoff zugeführt werden muß, damit er schmilzt, nennt man die „Schmelzenthalpie“. Soll bei Atmosphärendruck flüssiges Wasser mit Wasser in der Gasphase im Gleichgewicht sein, dann muß der Partialdruck des Wassers in der Gasphase dem Atmosphärendruck entsprechen. Das ist bei 100°C der Fall (Die Celsius-Skala ist auf den Gefrierpunkt und den Siedepunkt von Wasser geeicht!). Wenn die Temperatur erniedrigt wird, tritt Kondensation ein, dann ist flüssiges Wasser die stabile Phase. Wird die Temperatur erhöht, so verdampft das Wasser, gasförmiges Wasser ist dann die stabile Phase.

8. Flüssigkeiten, Lösungen
16
Nur bei dem Druck und der Temperatur, die jeweils durch die Kurven angegeben sind, sind zwei Phasen miteinander im Gleichgewicht, so dass beide nebeneinander existieren und zeitlich unverändert erhalten bleiben. Der Übergang von der festen Phase in die Gasphase wird durch die „Sublimationskurve“ gekennzeichnet. Nur bei Bedingungen (Druck, Temperatur), die der Lage der Punkte auf der Kurve entsprechen, sind beide Phasen im Gleichgewicht. Wenn der Partialdruck an Wasser in der Luft klein ist, dann sublimieren Eiskristalle, d.h. ohne den Umweg über die flüssige Phase geht die feste Phase in die Gasphase über. Skifahrer kennen das: Auch ohne dass es warm wird und ohne dass der Schnee schmilzt, wird er im Lauf der Zeit weniger, weil er nämlich sublimiert, falls nur die Luft trocken genug ist. Wenn zwischen den drei Phasen, Fest, Flüssig, Gasförmig Gleichgewicht herrschen soll, wenn also alle drei Phasen nebeneinander unbegrenzt lange existieren sollen, dann gibt es nur eine einzige Temperatur und einen einzigen Druck, bei dem dies möglich ist (Tripelpunkt). Druckabhängigkeit des Schmelzpunktes von Wasser Wasser ist insofern ein ungewöhnlicher Stoff, als sich sein Schmelzpunkt mit steigendem Druck zu tieferen Temperaturen verschiebt (siehe Diagramm). Das hat damit zu tun, dass flüssiges Wasser eine höhere Dichte hat als festes Wasser (Eis). (Tägliches Leben: Eis schwimmt auf Wasser; Schlittschuhlaufen funktioniert, weil unter dem Druck der Kufe das Eis schmilzt und flüssiges Wasser als Schmiermittel die Gleitreibung herabsetzt). Eis hat deswegen eine niedrigere Dichte als Wasser, weil der Aufbau eines Kristalls, in dem jeder Sauerstoff in einem Netzwerk von Wasserstoff-Brücken von vier Wasserstoffen (zwei kovalent gebunden und zwei über Wasserstoffbrücken, s.v.) regelmäßig umgeben ist, mehr Platz erfordert, als die regellose Anordnung der Moleküle in der Flüssigkeit. Trotz der Zunahme der Entropie bei der Bildung einer so regelmäßigen Anordnung, wird die Struktur von Eis aufgebaut, da die Energie der in der regelmäßigen Anordnung möglichen maximalen Zahl von Wasserstoffbrückenbindungen den Entropieverlust aufwiegt.
8.7.2 Zustandsdiagramme anderer Stoffe Die Zustandsdiagramme anderer Stoffe sehen im Prinzip ähnlich aus, wie das Zustandsdiagramm des Wassers. Die Schmelzkurve verläuft allerdings in der Regel mit
positiver Steigung und nicht wie bei Wasser mit negativer. D.h. normalerweise steigt der Schmelzpunkt mit steigendem Druck, entsprechend dem normalen Verhalten von Stoffen, dass nämlich die Dichte des Festkörpers größer ist als die der Flüssigkeit. Als Beispiel ist das Phasendiagramm von

8. Flüssigkeiten, Lösungen
17
Kohlendioxid schematisch gezeigt. Der CO2 - Dampfdruck über „Trockeneis“ (festes CO2) erreicht bei –78°C „Atmosphärendruck“ (1.013 bar), so dass festes CO2 als Kühlmittel verwendet werden kann und verwendet wird. Es wird nicht wärmer als –78°C, da es - nach dem Prinzip des kleinsten Zwanges - sich selbst durch Sublimation abkühlt, wenn man ihm Wärme zuführt.
8.7.3 Gefrierpunkts-Erniedrigung, Siedepunkts-Erhöhung Wenn in einem Lösungsmittel eine Substanz X gelöst wird, die keinen eigenen Dampfdruck hat (d.h. ihr eigener Dampfdruck ist unmessbar klein!), dann können nicht alle Teilchen aus der Flüssigkeit in die Gasphase übergehen. Im reinen Lösungsmittel ist der Molenbruch an Lösungsmittel notwendig gleich eins. Wenn auf nl Mole Lösungsmittel nx Mole Substanz gelöst sind, dann ist der Molenbruch des Lösungsmittels in einer solchen Lösung kleiner als eins: xLösungsmittel = nl /(nl + nx). (xa = Molenbruch des Stoffes A).
Nur der dem Molenbruch des Lösungsmittels entsprechende Anteil an Teilchen kann in die Gasphase übergehen, da die anderen Teilchen in der Lösung so fest gehalten werden, dass ihre kinetische Energie nicht ausreicht, um in die Gasphase zu gelangen. Daraus folgt, dass je Zeiteinheit weniger Teilchen in die Gasphase übertreten, als dies beim reinen Lösungsmittel der Fall wäre. Das bedeutet, dass der Dampfdruck über der Lösung kleiner ist, als der
Dampfdruck über dem reinen Lösungsmittel (bei sonst gleichen Bedingungen, d.h. T gleich!). Aus dieser Überlegung folgt - in Ansicht des nebenstehenden Diagramms - auch, dass der Siedepunkt einer Lösung gegenüber dem Siedepunkt des reinen Lösungsmittels erhöht wird. Ebenso folgt daraus, dass der Gefrierpunkt der Lösung erniedrigt wird, d.h. dass er bei tieferer Temperatur liegt, als der Gefrierpunkt des reinen Lösungsmittels. Beide Aussagen - Siedepunktserhöhung, Gefrierpunktserniedrigung - lassen sich in einer Aussage zusammenfassen: Der Existenzbereich der flüssigen Phase wird erweitert! Der Grund hierfür ist, dass die Entropie der Mischung höher ist (In der Mischung ist mehr Unordnung möglich!) als die des reinen Lösungsmittels!

8. Flüssigkeiten, Lösungen
18
8.7.4 Bestimmung von Molmassen und Teilchenzahlen Damit hat man eine Methode, um festzustellen, wie viele Teilchen einer im Lösungsmittel gelösten Substanz in der Lösung enthalten sind. Sofern die gelösten Substanzen keinen eigenen Dampfdruck haben, verhalten sie sich alle - unabhängig von ihrer chemischen Zusammensetzung - in diesem Bezug gleich. Wenn man in einem Lösungsmittel Lösungen bekannter Konzentration eines nicht flüchtigen Stoffes ansetzt und die Gefrierpunktserniedrigung bzw. Siedepunktserhöhung dieser Lösungen misst, so kann man Eichkurven aufstellen, aus denen man ablesen kann, welche Temperaturänderung (Gefrierpunkt, Siedepunkt) jeweils welcher Konzentration entspricht. Damit kann man durch Temperaturmessung die Konzentration des gelösten Stoffes - oder genauer gesagt, die Teilchenzahl - ermitteln und damit Molmassen von Stoffen bestimmen. In dem kleinen Temperaturintervall, das von der Siedepunktserhöhung bzw. der Gefrierpunktserniedrigung überstrichen wird, haben die Eichkurven eine sehr einfache Form: Sie sind linear und haben die Form: T∆ = a ⋅ c, wobei a meist als „molale Siedepunktserhöhung“ bzw. „molale Gefrierpunktserniedrigung“ in °C ⋅ kg ⋅ mol-1 angegeben wird, also als die Temperaturänderung (von Siedepunkt oder Gefrierpunkt), die eintritt, wenn man ein Mol Substanz in 1 kg Lösungsmittel löst. (Dass die Kurven linear sind, ist nicht verwunderlich, da jede Kurve abschnittweise durch eine Gerade wiedergegeben werden kann, wenn der Abschnitt nur klein genug ist). Die Technik des Messens der Siedetemperatur zur Bestimmung der Teilchenzahl nennt man Ebullioskopie; die der Messung des Gefrierpunkts zum gleichen Zweck heißt Kryoskopie.
8.7.5 Destillation Wenn zwei flüssige Stoffe A und B gemischt werden, dann setzt sich die Gasphase über dieser Mischung aus den Molekülen beider Stoffarten zusammen. Wenn der Dampfdruck des einen reinen Stoffes A bei gegebener Temperatur PA ist und der Dampfdruck des anderen reinen Stoffes B PB genannt wird, dann ist der Gesamtdampfdruck über der Mischung: P = xa ⋅ PA + xb ⋅ PB xa, xb : Molenbruch von A bzw. B in der Mischung Raoult’sches Gesetz (F.-M. Raoult, 1830-1901, Grenoble) Der Dampfdruck, der durch A verursacht wird, ist xa ⋅ PA, der Dampfdruck von B ist xb ⋅ PB. Wenn die Dampfdrücke der reinen Stoff A und B verschieden sind, dann ist im Gleichgewicht die Zusammensetzung des Dampfes anders als die Zusammensetzung der Lösung:

8. Flüssigkeiten, Lösungen
19
Der Stoff mit dem höheren Dampfdruck ist in der Gasphase mit höherer Konzentration vorhanden als seinem Molenbruch xa in der Lösung entspricht. Wenn dieser Dampf kondensiert wird, erhält man eine Flüssigkeit, die an dem Stoff angereichert ist, der den höheren Dampfdruck hat. Darauf beruht das Prinzip der Reinigung von Stoffen durch Destillation. Die für den Gesamt-Dampfdruck angegebene Gleichung P = xa ⋅ PA + xb ⋅ PB gilt nur dann genau, wenn die Teilchen A und B keine anderen Kräfte aufeinander ausüben als die Teilchen des Stoffes A unter sich und B unter sich (Ideale Lösungen).
8.7.6 Azeotrop Wenn Lösungen sich nicht ideal verhalten, dann kann es dazu kommen, dass bei einem bestimmten Molenbruch der Stoffe in der flüssigen Phase, d.h. bei einem bestimmten Konzentrationsverhältnis der Stoffe in der Flüssigkeit, der Dampf über der Flüssigkeit die gleiche Zusammensetzung hat, wie die Lösung. Dann kann ein solches Gemisch durch Destillation nicht weiter aufgetrennt werden. Man nennt solche Gemische „Azeotrope“. Beispiele hierfür ist HCl/H2O. Aus einer Lösung, die man durch Einleiten von HCl in Wasser bis zur Sättigung erhält (= konzentrierte Salzsäure, 42%ig, d.h. 42 % Massenanteil an HCl) destilliert beim Erwärmen solange vorwiegend HCl ab, bis die Lösung nur mehr ca. 20% Massenanteil an HCl (ca. 5.5 mol l-1, Dichte = 1.1 kg l-1) enthält. Bei dieser Zusammensetzung der Lösung hat der Dampf die gleiche molare Zusammensetzung wie die Flüssigkeit, es destilliert das Azeotrop. Man kann Salzsäure also nicht durch Abdestillieren von Wasser konzentrieren!). Wenn man ein Gemisch von Ethanol/Wasser destilliert, dann destilliert zunächst eine Mischung aus ca. 96% Ethanol und 4% Wasser als Azeotrop ab, da diese Mischung den höchsten Dampfdruck unter allen möglichen Mischungszusammensetzungen hat. Danach destilliert Wasser ab, das einen höheren Siedepunkt (100°C) hat als C2H5OH (78.5°C).

8. Flüssigkeiten, Lösungen
20
8.7.7 Osmose Die Teilchen in einer verdünnten Lösung verhalten sich wie die Teilchen in einem idealen Gas.
Der osmotische Druck π ist das Äquivalent zum Druck P bei Gasen. Die ideale Gasgleichung findet ihre direkte Übersetzung in π ⋅ V = n ⋅ R ⋅ T, woraus, mit c = n / V die Beziehung π = c ⋅ R ⋅ T folgt.
8.8 Solvolyse Wenn man ein Salz in einem Lösungsmittel löst, das Ionen stabilisieren kann, dann gehen die Bestandteile des Salzes - Anionen und Kationen - getrennt in Lösung. So löst sich NaCl in Wasser unter Bildung von aquatisierten Ionen Na+
aq und Cl-aq.
Der Nachweis dafür kann dadurch erbracht werden, dass man die Teilchenzahl bestimmt (z.B. durch Gefrierpunktserniedrigung oder Siedepunktserhöhung, s.o.) die aus einer Stoffmenge von n Mol NaCl in der Lösung entsteht. Man stellt fest, dass bei NaCl zwei Mol Teilchen je Mol NaCl in der Lösung vorhanden sind.

8. Flüssigkeiten, Lösungen
21
Wenn man Na2SO4-Lösungen in dieser Weise untersucht, findet man, dass je Mol Na2SO4 drei Mol Teilchen gebildet werden etc. Der Ionenverband des Festkörpers wird durch das Lösungsmittel aufgelöst = solvolysiert, da die Lösungsmittelmoleküle mit den Ionen in anziehende Wechselwirkungen treten (s.v.).
8.9 Elektrolytische Leitfähigkeit Lösungen, die Ionen enthalten, leiten den elektrischen Strom. Die Anionen werden, als
negativ geladenen Teilchen, zur Anode, dem positiv geladenen Pol, hingezogen. Die Kationen wandern, als positiv geladene Teilchen, zur Kathode, dem negativen Pol der Spannungsquelle. Solche Lösungen nennt man auch „Elektrolyt-Lösungen“. An der Kathode und an der Anode treten Chemische Umsetzungen ein. Die Elementarschritte dieser Umsetzungen bestehen darin, dass an der Kathode von den Kationen Elektronen aufgenommen werden, während an der Anode die Anionen Elektronen an die Anode abgeben. An der Kathode findet der
Reduktionsprozess statt. An der Anode findet der Oxidationsprozess statt.
8.9.1 Elektrolyse Wenn man HCl in Wasser einbringt, dann bilden sich - in verdünnten Lösungen praktisch quantitativ - aquatisierte Protonen, H+
aq, und aquatisierte Chlorid-Ionen, Cl-aq.
(In anderen Worten: Das Gleichgewicht: HCl + x H2O → H+aq + Cl-
aq liegt „rechts“.) Die Protonen wandern zur Kathode und werden dort reduziert. H+ + e- → ½ H2 Kathodenreaktion = Reduktion Die Anionen wandern zur Anode und werden dort oxidiert: Cl- → ½ Cl2 + e- Anodenreaktion = Oxidation Den Vorgang der Umsetzung eines Stoffes durch die Zuführung oder das Abziehen von Elektronen an negativ bzw. positiv geladenen Elektroden, nennt man „Elektrolyse“.

8. Flüssigkeiten, Lösungen
22
8.9.2 Leitfähigkeit und Ionenart Die Leitfähigkeit wässriger Elektrolyt-Lösungen hängt, bei gleicher Konstruktion des Messgerätes und gleicher Teilchenkonzentration von der Beweglichkeit der Ionen in der Lösung ab, da die Ionen ja durch die Lösung auf die jeweils vorzeichengemäße Elektrode zuwandern müssen. Die Beweglichkeit wird behindert durch die Kräfte, die zwischen Lösungsmittel und Ion wirken, so dass die Ionen Lösungsmittelmoleküle im Schlepp mit durch die Lösung ziehen müssen. Verantwortlich für das Maß an Leitfähigkeit ist daher nicht der Ionenradius (mit dem man z.B. Abstände zwischen Ionen in Kristallgittern abschätzen kann, s.v.), sondern der effektive Radius, den das Ion durch die Solvatation mit Wassermolekülen, die sogenannte Hydratation erhält. Wenn man unter sonst gleich bleibenden Bedingungen die Leitfähigkeit von wässrigen Lösungen misst, welche die Salze LiCl bzw. NaCl in jeweils gleicher Konzentration enthalten, stellt man fest, dass die LiCl-Lösung schlechter leitet als die NaCl-Lösung. Die Anionen sind jeweils Chlorid-Ionen, so dass deren Beitrag zur Leitfähigkeit in beiden Experimenten gleich ist. Der Unterschied muß dann daher rühren, dass die kleinen Li-Ionen stärker von Wasser solvatisiert werden als die größeren Na-Ionen. Mit ihrer Solvathülle sind daher die Li-Ionen größer als die Na-Ionen, so dass sie weniger beweglich sind. Ionen mit kleinem Ionenradius polarisieren die Umgebung also stärker als solche mit größerem Radius.
8.9.3 Leitfähigkeit durch Protonen Das oben verwendete Argument würde zu der Erwartung führen, dass Protonen besonders schlecht leiten sollten, da sie ja die kleinsten einfach positiv geladenen Kationen sind und damit noch weniger beweglich sein sollten als Li-Ionen. Das Gegenteil wird beobachtet! Die Erklärung der besonders guten Leitfähigkeit von Lösungen, die Protonen enthalten, liegt darin, dass nicht die Protonen selbst durch die wässrige Lösung wandern müssen, sondern dass die Eigenschaft eines Wasserstoff-Zentrums, Proton zu sein, weiter gegeben wird. Dieses Beispiel eines „kooperativen Phänomens“ wird wie nebenstehend im Diagramm gezeigt gedeutet.
Das Proton ist an ein H2O-Molekül fest gebunden und bildet mit diesem ein H3O+-Ion. Das H3O+-Ion ist über Wasserstoff-brücken mit anderen H2O-Molekülen verbunden. Indem aus einer Brückenbindung eine kovalente Bindung entsteht und zugleich damit aus einer kovalenten Bindung eine H
OH
HO
H
H
O H
HOH
H O
H
H+
+_

8. Flüssigkeiten, Lösungen
23
Brückenbindung gebildet wird, wandert die positive Ladung durch die Kette von H2O-Molekülen. Im Diagramm ist das dadurch angedeutet, dass die freien Elektronenpaare durch Pfeile bezeichnet sind, die jeweils, von rechts nach links gelesen, als nächste die Bindung mit einem Proton eingehen. Weitergegeben wird die Eigenschaft der positiven Ladung durch sehr kleine Bewegungen auf jeweils kleinem Raum, nämlich dem Sprung eines Protons aus der Brückenbindung in eine kovalente Bindung.
8.9.4 Leitfähigkeit durch Hydroxid-Ionen Für Hydroxid-Ionen, OH-, lässt sich ein analoges Bild entwickeln, das die hohe Leitfähigkeit von Hydroxid-haltigen Lösungen erklärt.
8.9.5 Leitfähigkeit in Salzschmelzen Geschmolzene Salze sind elektrolytisch leitfähig. Die Anionen und Kationen, die im Salzkristall einen hochgeordneten Verband bilden, verhalten sich auch in der weniger geordneten Schmelze immer noch als Kationen und Anionen. Salzschmelzen bilden bei vielen technisch wichtigen Reaktionen das Lösungsmittel. Da Salze eine hohe Gitterenergie aufweisen, haben sie meist auch sehr hohe Schmelzpunkte, was das großtechnische Arbeiten in Salzschmelzen erschwert. Der Gefrierpunkt eines Lösungsmittels kann jedoch herabgesetzt werden, indem man dem Lösungsmittel andere Stoffe beimischt (siehe oben!). Dieses Prinzip macht man sich bei praktisch allen technisch angewendeten Salzschmelzen zu nutze, indem man nicht den reinen Stoff schmilzt sondern ihm einen, unter den angewendeten chemischen Bedingungen „inerten“ anderen Stoff zusetzt, der ausschließlich dazu dient, die Schmelztemperatur herabzusetzen. Beispiele hierfür sind: Al2O3 (Korund)/Na3AlF6 (Kryolith) bei der Schmelzfluß-Elektrolyse zur Gewinnung von Aluminium aus Al2O3. BeO/BeF2/BaF2 oder auch BeCl2/NaCl für die elektrolytische Gewinnung von Beryllium-Metall. MgCl2/CaF2/NaCl bei der Elektrolyse von MgCl2 zu metallischem Mg.
8.9.6 Ein wenig „Stoffchemie“ zum Schluss CaF2 (Struktur s.v.) nennt man auch „Flussspat“, weil er bei vielen metallurgischen Prozessen als „Flussmittel“ zugesetzt wird, um die Schmelztemperatur zu erniedrigen.

8. Flüssigkeiten, Lösungen
24
MgCl2 erhält man aus MgO durch Erhitzen mit Kohle im Chlorstrom (Gleichung?). MgO erhält man aus MgCO3, das beim Erhitzen unter Abgabe von CO2 zerfällt. Nach der gleichen Methode stellt man aus CaCO3 (Kalkstein) „gebrannten Kalk“, CaO, her. MgO (Kochsalzstruktur wie auch Ca , SrO und BaO) hat einen Schmelzpunkt von 2642°C, so dass man für die Elektrolyse besser MgCl2 (Schmelzpunkt: 708°C) verwendet, wobei man den Schmelzpunkt durch die aufgeführten Zusätze noch weiter erniedrigt. CaO, „gebrannter Kalk“, reagiert mit Wasser zu Ca(OH)2 = „gelöschter Kalk“. Gelöschter Kalk ist die billigste von den technisch einfach zugänglichen Verbindungen, die in Wasser OH--Ionen freisetzen. Solche Verbindungen nennt man in einer Einengung des Lewis-Säure-Base-Begriffs „Basen“. Basen werden für viele technisch wichtige Reaktionen gebraucht. Eine kulturhistorisch sehr alte und unverändert technisch bedeutsame Anwendung von Ca(OH)2 beruht auf seiner Reaktion mit dem CO2 der Luft bei seiner Verwendung als „Kalkmörtel“: Es bildet sich dabei zunächst Calciumhydrogencarbonat, Ca(HCO3)2, das unter Abgabe von H2O und CO2 schließlich in CaCO3 (Kalkstein) übergeht. Das Kristallgefüge des bei diesem Prozess entstehenden Kalksteins ist sehr fest und verklebt gut mit allen möglichen Arten von Steinen.

8. Flüssigkeiten, Lösungen
25
8.10 Index
A Adenin .................................................... 10 Anionen .................................................. 21 Anode...................................................... 21 aquatisierten Ionen.................................. 20
B Beispiele zur Leitfähigkeit von
Salzschmelzen .................................... 23 Bindungsdissoziations-Enthalpien............ 6 Bindungsenergie von
Wasserstoffbrückenbindungen ............. 8 Braun ...................................................... 12
C Cytosin.................................................... 10
D Debye........................................................ 4 Desoxyribonucleinsäure ........................... 8 Desoxyribose .......................................... 10 Dielektrizitätskonstante ............................ 5 Doppelstränge......................................... 10 Druckabhängigkeit des Schmelzpunktes
von Wasser ........................................ 16 Druckänderung ..................................... 12
E Ebullioskopie ......................................... 18 Einzelstränge .......................................... 10 Elektrolyse .............................................. 21 Elektrolyt-Lösungen ............................... 21
F Faraday ..................................................... 5 Flussmittel .............................................. 23 Flussspat ................................................. 23 Formale Ladung ..................................... 4 freie Elektronenpaare................................ 8
G gebrannter Kalk ...................................... 24 gelöschter Kalk ....................................... 24 Gewinnung von Aluminium ................... 23 Gewinnung von Beryllium-Metall.......... 23
Gleichgewicht .........................................12 Guanin.....................................................10
H Hydratation..............................................22
I ideale Gasgesetz........................................3 Ideale Lösungen ......................................19 inert .........................................................23 Interhalogenverbindung ............................3
K Kalkmörtel ..............................................24 Kalkstein .................................................24 Kapazität ...................................................5 Kathode ...................................................21 Kationen ..................................................21 komplementärer Strang ...........................10 Kondensator ..............................................5 Korund ....................................................23 Kryolith ...................................................23 Kryoskopie ..............................................18
L Ladung von Ionen ...................................4 Le Châtelier.............................................12
M Massenwirkungsgesetz............................13 molale Gefrierpunktserniedrigung......18 molale Siedepunktserhöhung ...............18 Molenbruch .............................................17
N negative Partialladung...............................3
O osmotische Druck....................................20 Oxidation.................................................21
P Partialladung ...........................................3 Pauling’schen Skala ..................................6 permanentes Dipolmoment .......................7

8. Flüssigkeiten, Lösungen
26
Polarisierung ........................................... 3 positive Partialladung ............................... 3
R Raoult...................................................... 18 Raoult’sches............................................ 18 Reduktion................................................ 21
S Schmelzenthalpie.................................... 15 Schmelzfluß-Elektrolyse ........................ 23 Schmelzwärme........................................ 15 sekundäre Wechselwirkungen .................. 3 Solvathülle .............................................. 22
Störung des Gleichgewichts..................12 Sublimationskurve ..................................16 Symbole für Ladungen ...........................4
T Temperaturänderung ...........................12 Templat ...................................................10 Thymin ....................................................10 Tripelpunkt..............................................16 Trockeneis...............................................17
V van’t Hoff’sche Gleichung......................13 Verdampfungsenthalpie ..........................13


















