
5.62 Physical Chemistry II Spring 2008 For information … Spring 2008 Lecture #29 Page 5 The number...
20
MIT OpenCourseWare http://ocw.mit.edu 5.62 Physical Chemistry II Spring 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
Transcript of 5.62 Physical Chemistry II Spring 2008 For information … Spring 2008 Lecture #29 Page 5 The number...

MIT OpenCourseWare http://ocw.mit.edu
5.62 Physical Chemistry IISpring 2008
For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.

5.62 Spring 2008 Lecture #29 Page 1
Kinetic Theory of Gases: Effusion and Collisions
EFFUSION Consider the process by which molecules escape through a hole in a vesseland into a vacuum
size “d”
slit
oven walls at
definite T
We assume that: (1) d is so small that thepressure in the vessel is unchanged; (2) theeffusion does not perturb the velocity of thegas in the vessel; (3) there are no collisionswhen the molecules pass through the slit.
Molecules that would have been incident on the portion of the wall where the hole is,
now pass through the hole. This creates a flux of particles defined as the number of
particles per unit area per unit time that leave the vessel.
dA
vdt
!
Consider a square hole of area dA. A particle that is a distance vdt from the hole
!moves with speed v and at angle from the surface normal toward the hole.
Draw a parallelepiped around the hole with length equal to vdt, at angle θ from the
normal. All molecules within this volume moving toward the hole (i.e., with the correct
!," angle) with speed v will pass through the hole in time interval dt.
ρ = density of gasVolume of parallelepiped = v cos ! dA dt (Note that, at grazing angles, θ ~ π/2, the volume is small)
# of molecules crossing through dA in dt = ρvcos θdAdt (number density times volume)
#molecules
dAdt= ρvcos ! =FLUX.
We must integrate this expression over the distribution of velocities of the gas to obtainthe average flux J. The Maxwell-Boltzmann distribution (from Lecture #28) is
revised 4/22/08 5:38 PM
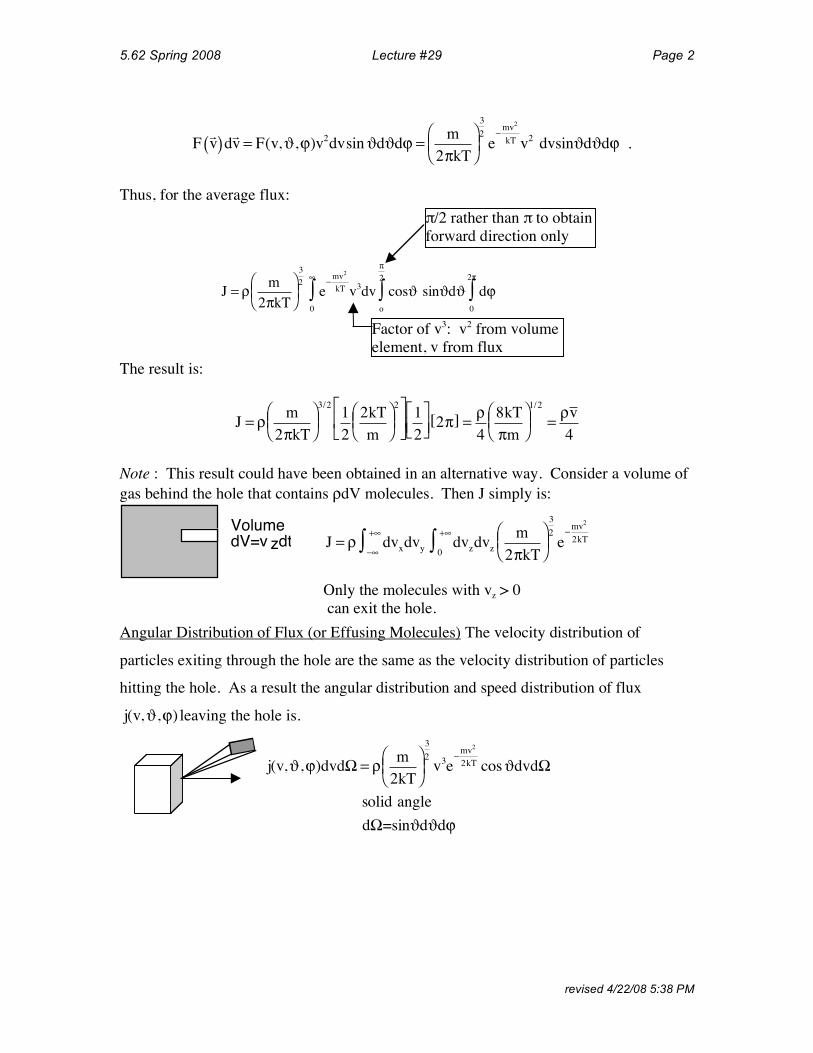
5.62 Spring 2008 Lecture #29 Page 2
F!v( )d!v = F(v,!,")v
2dvsin!d!d" =
m
2#kT
$%&
'()
3
2
e*
mv2
kT v2 dvsin!d!d" .
Thus, for the average flux:
The result is:
J = ! m
2"kT
#$%
&'(
3
2
e)
mv2
kT v3dv cos* sin*d*
o
"2
+0
,
+ d- 0
2"
+
!/2 rather than ! to obtain
forward direction only
Factor of v3: v2 from volume
element, v from flux
J = ! m
2"kT#$%
&'(3/2
1
2
2kT
m
#$%
&'(2)
*+
,
-. 12
)*+
,-.2"[ ] =
!4
8kT
"m#$%
&'(1/2
=!v4
Note : This result could have been obtained in an alternative way. Consider a volume of gas behind the hole that contains ρdV molecules. Then J simply is:
particles exiting through the hole are the same as the velocity distribution of particles
hitting the hole. As a result the angular distribution and speed distribution of flux
Volume
dV=v zdt
Only the molecules with vz > 0
can exit the hole.
J = ! dvx"#
+#$ dvy dvz0
+#$ dvz
m
2%kT
&'(
)*+
3
2
e"mv
2
2kT
Angular Distribution of Flux (or Effusing Molecules) The velocity distribution of
j(v,!,") leaving the hole is.
j(v,!,")dvd# = $ m
2kT
%&'
()*
3
2
v3e+
mv2
2kT cos!dvd#
solid angle
d#=sin!d!d"
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 3
The angular distribution of the flux is
cos !
jangle(")d" = dv0
#
$ j v,!,%( ) =&v
4'cos!d"
0 < ! <'2
, 0 < % < 2'
Effusion is an important mechanism for creating molecular beams that have practical use
(for example in molecular beam epitaxy used in the manufacture of electronic devices)
and for studying collisions and gas phase chemical reactions.
One important application is time of flight verification of the Maxwell Boltzmann
distribution (1955!). The experiment is described in the figure below. Puffs of gas are
released by opening the shutter. The gas spreads as a result of the spread in speed.
{velocity distributions can also be inferred from Doppler shift measurements on spectral
lines}.
q(t) is the number of molecules that
hits the detector between t and t + dt
time
L = vt
gas
sourceshutter
detector
t1 t2 t3
There is a relation between the distribution of arrival times q(t) and the distribution ofspeeds h(v):
. q(t)dt = h(v)dv
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 4
Dynamics determines the exact relation between distance, speed, and time L=vt. Using the Dirac delta function to set this relation gives:
dvh(v)! v" Lt
#$%
&'(
0
)
* = hL
t
#$%
&'( .
Thus measurement of q(t) allows us to infer the functional form of h according to:
hL
t
!"#
$%& = q(t)
dt
dv= q(t)
t2
L.
The flux is obtained by multiplying the amplitude in the arrival time distribution at thecorresponding time t with a factor t2/L. The flux of molecules with large v arrive at the detector early and have not spread out in arrival times as much as the later arriving ones.
Molecular Collisions
Goal: To calculate collision frequency between pairs of molecules in a gas. We begin by defining terms.
Z = average number of collisions of a single particle per unit time.
Collision event occurs when centers of 2 molecules approach within distance d.
Distance d is the hard sphere diameter
Collision cross section ! "d2 =area of circle of
radius d (surrounding particle 1), if the center ofparticle 2 enters (or touches) this circle, a collision occurs.
A collision occurs when the relative velocity permits an encounter between two particles. The relative velocity is defined as:
!vr=!v1!!v2
.
In a time dt, a collision volume dV is swept out by a particle. This volume is:
dV = !d
2 !v
rdt;
!v
r=!v
r"!v
r[ ]1
2 .
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 5
The number of encounters in time dt at relative velocity vr is ρdV. The number of encounters per unit time is
# of encounters/time=!dV
dt= "d
2!!v
r .
The collision frequency is the average of this quantity:
Z=!d
2"!v
r
Since !v
r = v
r, the average of the relative velocity, is
vr = d
!v1 d
!v2!! F(!v1)F(
!v2 )vr
In this expression, we have assumed that the velocity distributions for the two molecules
are independent of each other; this is true. But, at high density, intermolecular
interactions will influence the spatial arrangement of the colliding pairs and this effect is
not taken into account in the above expression. Thus, the average we are calculating is
only valid for dilute gas conditions.
The two distribution functions have the form:
F(!v1) =
m1
2!kT
"#$
%&'
3
2
exp (m1v1
2
2kT
"
#$
%
&' and F(
!v2 ) =
m2
2!kT
"#$
%&'
3
2
exp (m2v2
2
2kT
"
#$
%
&' .
Thus the integral to be done is:
vr =m1
2!kT
"#$
%&'
3
2 m2
2!kT
"#$
%&'
3
2
d!v1 d
!v2(( exp ) m1v1
2+m2v2
2
2kT
"
#$
%
&'
*
+,
-
./ vr
In order to do the integrals we must change variables from
!U
!v
1 and
!v
2to relative and
center of mass velocity :
!v
r=!v
1!!v
2 and M
!U=m
1
!v
1+m
2
!v
2 where M=m
1+m
2
Solving for the particle velocities gives:
!v
1=!U +
m2
M
!v
r and
!v
2=!U !
m1
M
!v
r .
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 6
A bit of algebra shows that the kinetic energy term is:
m1v1
2+m
2v2
2= (m
1+m
2)U
2+m2m1
m1+m
2
vr
2=MU
2+µv
r
2 .
Here the reduced mass µ has been defined by:
1
µ=1
m1
+1
m2
=m1m2
m1+m
2
.
To proceed we need the Jacobian of the transformation for the change in variables:
d!v1d!v2=
!!v1
!!U
"#$
%&'
!!v2
!!U
"#$
%&'
!!v1
!!vr
"
#$
%
&'
!!v2
!!vr
"
#$
%
&'
d!Ud!vr=
1 1
m2
M1 (m1
M1d!Ud!vr= d!Ud!vr
The Jacobian of the transformation is happily equal to unity.
The transformed integral becomes:
vr =m1
2!kT
"#$
%&'
3
2 m2
2!kT
"#$
%&'
3
2
d!U d
!vr(( exp ) MU2
+µvr
2
2kT
"
#$
%
&'
*
+,
-
./ vr
vr =m1
2!kT
"#$
%&'
3
2 m2
2!kT
"#$
%&'
3
2
d!U exp ) MU2
2kT
"
#$
%
&'
*
+,
-
./ d!vr(( exp ) µvr
2
2kT
"
#$
%
&'
*
+,
-
./ vr .
We have no interest in the center of mass variables and can immediately integrate over these variables to obtain:
vr =m1
2!kT
"#$
%&'
3
2 m2
2!kT
"#$
%&'
3
2 2!kT
M
"#$
%&'
3
2
d!vr( exp ) µvr
2
2kT
"
#$
%
&'
*
+,
-
./ vr
vr =µ
2!kT
"#$
%&'
3
2
d!vr( exp ) µvr
2
2kT
"
#$
%
&'
*
+,
-
./ vr .
Note that the Maxwell-Boltzmann distribution for the relative velocity is of the same
form as the Maxwell-Boltzmann distribution for a single particle with replacement of the
particle mass by the reduced mass for the colliding pair.
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 7
In spherical coordinates the average is:
vr = 4! µ
2!kT
"#$
%&'
3
2
dvr( vr
2 exp ) µvr
2
2kT
"
#$
%
&'
*
+,
-
./ vr ,
and we obtain the final result:
vr =8kT
!µ
"
#$
%
&'
1
2
the average relative speed.
For like particles we have µ =m
2so v
r=
16kT
!m
"#$
%&'
1
2
= 2 v . The average relative speed is
times (i.e. slightly larger than) the average speed of one of the particles. vr
v and 2v
One certainly expects that 2
should be between .
particle average collision frequency, but divided by a factor of 2 to avoid double
counting. Thus the average total collision frequency per unit volume is:
The average collision frequency for one particle is
Z=!d2"v
r = 2!d
2"v
in a gas of like particles. The total collision frequency ZTOT will be N times the one
ZTOT
V=
2
2!2"d2v .
Since collision theory is based on collisions, the average total collision frequency per unitvolume is a key quantity.
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 8
Macroscopic Reaction Rates
From Microscopic Properties
Part I.Reaction rates and their corresponding rate constants are macroscopic properties
determined by the microscopic properties of the individual particles that undergo reaction. Goal is to develop a theory to calculate rate constants from the molecular level properties of the reacting particles.
COLLISION THEORY
Consider A + B → C
Upper limit to reaction rate d[C]/dt is the total collision frequency.
d[C]dt = k [A] [B]
VZ
= π d AB
2 8kT
!µ
"
#$
%
&'1/2
ρA ρB
Can we describe the rate constant simply as a hard sphere collision cross section times the average speed?
k = πd AB
2 8kT
!µ
"
#$
%
&'1/2 = πdAB
2 –c –c = –c rel
But !dAB2
� ≡collision (as opposed to reactive) cross section! Not every collision results in a reaction. Many details are missing, e.g. do the colliding molecules have enough energy to react?
1. We want an expression for
… reaction cross section σR
Expect σR to have an energy dependence. Simplest assumption:
σR = 0 if E < Eo and σR = !dAB2
if E ≥ E0
where E ≡ relative energy between colliding particles E0 ≡ critical energy needed to get over barrier.
revised 4/22/08 5:38 PM
5.62 Spring 2008 Lecture #29 Page 9
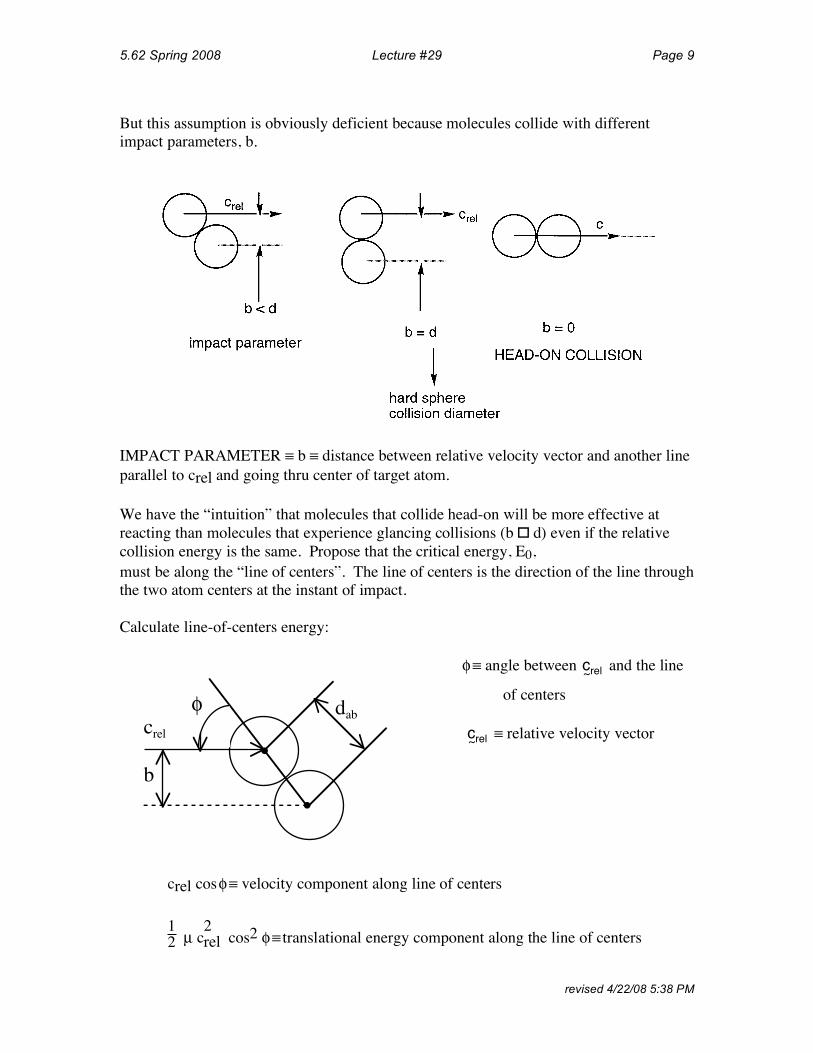
But this assumption is obviously deficient because molecules collide with different impact parameters, b.
IMPACT PARAMETER ≡ b ≡ distance between relative velocity vector and another line parallel to crel and going thru center of target atom.
We have the “intuition” that molecules that collide head-on will be more effective at reacting than molecules that experience glancing collisions (b � d) even if the relative collision energy is the same. Propose that the critical energy, E0, must be along the “line of centers”. The line of centers is the direction of the line through the two atom centers at the instant of impact.
Calculate line-of-centers energy:
φ ≡ angle between and the line
•
•
φ dab
b
crel
c~rel
of centers
≡ relative velocity vector c~rel
crel cosφ≡ velocity component along line of centers
1 2 2 µ crel cos2 φ≡ translational energy component along the line of centers
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 10
1
2µc
rel
2cos
2! =
1
2µc
rel
21" sin
2!( ) # E
0
Since sin φ = b/dAB and 21
µ c rel 2
= Erel ≡ E
Then the requirement for reaction is that the line of centers energy E 1!b2
dAB
2
"
# $
%
& ' ≥ E0
This function relates the component of the relative translational energy that provides thecritical energy along the line of centers to the impact parameter of the collision.
For each E, there is some b, call it b0, such that the line of centers energy is sufficient for reaction:
E 1!b0
2
dAB
2
"
# $
%
& ' = E0
b0 ≡ the largest impact parameter the collision with Erel can have and still have at least E0 along line of centers
b 0
2 = d AB
21!E0
E
"
#
$
%
For each E, all collisions with b ≤ b0 are effective because for these impact parameters the line of centers energy is > E0
So σR(E) = π b The reactive cross-section is a function of the relative translational energy because it includes all values of b ≤ b0and b0 is explicitly dependent on E.
0
2
σR = π b0
2 = πdAB
21!E0
E
"
#
$
% for E > E0
σR = 0 for E ≤ E0
Calculate k by averaging over relative velocities (energies).2.
–k = σR c = πd AB
21!
E0
E
"
#
$
% c
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 11
But, we no longer want to multiply by the average relative velocity because σR is now dependent on the relative energy (velocity). Want to multiply σR by the relative velocity of the specific collision and then want to average over all possible relative energies.
–k = σR c = ∫ σR c f(c) dc
= πd AB
21!E0
E
"
#
$
% 4π
µ
2!kT"#$
%&'3/2
⌡ c3 e–µc2/2kT dc⌠
Change variables to E instead of c because σR(E)
E = 21
µc2 dE = µcdc
k = πd AB
2 ! 1"E0
E
#
$
%
& 4π
µ
2!kT"#$
%&'3/2 dE
µ2E
µ! " #
$ % & 1/ 2
2E
µ! " #
$ % & e –E/kT 2E
µ
!
"#
$
%&1/2
= 8!µ2
µ
2!kT"#$
%&'
3/2 πd AB2 (E!Eo)E0
"
# e –E/kT dE
lower limit is E0 because σR = 0 if E < Eo
dELet x = E !E
0
kTdx = kT
then e–E/kT = e –E0/kT e–x
AB (kT) 2 e–E0/kT k = 8!µ2
µ
2!kT"#$
%&'
3/2 πd
2
x
0
!
" e#xdx
1
! " # $ #
k =8kT
!µ
"
# $
%
& '
1/ 2
relativespeed
! " # $ #
!dAB
2
hard sphere
cross section
% e(E0 / kT
! " $ COLLISION THEORYRATE CONSTANT
fraction of collisions havingnecessary component of energyalong line of centers E0
Collision theory result looks very much like empirical Arrhenius expression (k = A e–Ea/kT) except that the collision theory preexponential factor has a T1/2 dependence
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 12
k = BTm e–E0/kT where m =1/2
Most experimental measurements of k do not show a T dependent pre–exponential factor.This is because measurements of k are made over too small a temperature range to see the weak T1/2 dependence of the pre–exponential factor in the presence of the much stronger T dependence of the exponential term. This pre-exponential T1/2 dependence would show up as non-linearity in plot of lnk vs. 1/T.
RELATIONSHIP BETWEEN E0 (critical line of centers energy) and Ea, EMPIRICAL ARRHENIUS ENERGY
k = A e–Ea/kT
Ead ln k dT = d(lnAEa / kT)
dT= kT2
kCT = 8kT
!m"#$
%&'
1/2 2πdAB e–E0/kT
d ln kCT dT =
d (ln(8k / !µ)1/2!dAB2
+1
2lnT"E0 / kT)
#
$%&
'(
dT
1 = 2T + E0
kT2
Now we require that
d ln k d ln kCT dT = dT
Thus kT
Ea = 2 + E0
Ea must be larger than E0. WHY?
FURTHER ANALYZE COLLISION THEORY RATE CONSTANT:
kCT ∝ ⌡⌠ ⎝⎛1 –
EE
o⎠⎞ E e–E/kT dE
energy dependence of cross section, σ
R relative energies
Ea
kT2=1
2T+E0
kT2
distribution of
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 13
CALCULATE VALUES FOR
kCT = 8kT
!µ
"
#$
%
&'
1/2 2 8kT
!µ
!
"#
$
%&
1/2 2 e1/2 e–Ea/kTπdAB e–E0/kT = πdAB
Get E0 from experimental Ea since E0 = Ea – kT/2
2Get πdAB from transport data
COMPARE TO EXPERIMENT
ln kCT ln k REACTION CALCULATED OBSERVED
CH3 + CH3 → C2H6 10.6 10.5
CF3 + CF3→� C2F6 10.3 10.4
H + CCl4 → HCl + CCl3 0.83 –4.4
Cl + H2 → HCl + H –1.4 –4.7
Conclusions: works well for simple recombination steps where the Ea is small or non-existent and where there is little steric hindrance.
does not work well for reactions with steric hindrance
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 14
— E0 dependence may not be isotropic
— ignores effects of reactant vibrational and rotational energy inovercoming barrier.
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 15
Part II.
A "good" theory must take into account the internal degrees of freedom of thereactants and their angle of approach. An approach known as transition state theory =activated complex theory = absolute rate theory does so in an approximate way.
POTENTIAL ENERGY SURFACE
A correct theory must consider the internal structure of molecules and the forcesacting on atoms in the molecules because bonds are being broken and formed during areaction. During a reactive collision, the force on an atom depends on bothintramolecular forces (forces between atoms in a molecule) and intermolecular forces(forces between molecules). Must treat the two colliding reactants as a single quantum mechanical system. This system exists only during collision process.
The system's potential energy is calculated the same way the potential energy fornuclear vibrational motion is calculated. Within the Born-Oppenheimer approximation, solve
Hel Ψel = Eel Ψel at fixed nuclear configuration.
The resulting Eel is the potential energy at that nuclear configuration. Systematically vary the nuclear configuration (grid of points) to get potential energy as a function of nuclearcoordinates.
Problem: too many nuclear coordinates! Can't plot potential energy as afunction of more than 2 coordinates. A plot of potential energy versus 2 coordinates is a 3-D plot where the potential energy is aSURFACE. Potential energy for more than 2 coordinates is stillcalled a surface even though there are more than 2 coordinates.
revised 4/22/08 5:38 PM
5.62 Spring 2008 Lecture #29 Page 16
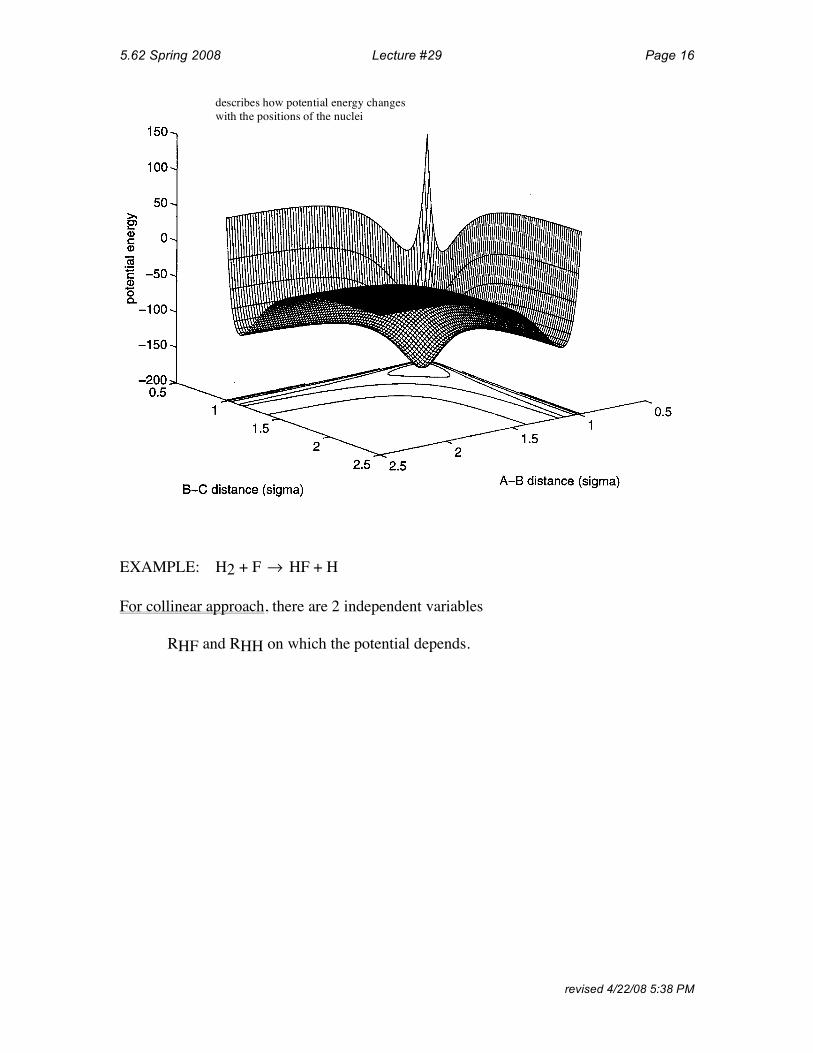
describes how potential energy changes with the positions of the nuclei
EXAMPLE: H2 + F
For collinear approach, there are 2 independent variables
! HF + H
RHF and RHH on which the potential depends.
revised 4/22/08 5:38 PM
5.62 Spring 2008 Lecture #29 Page 17
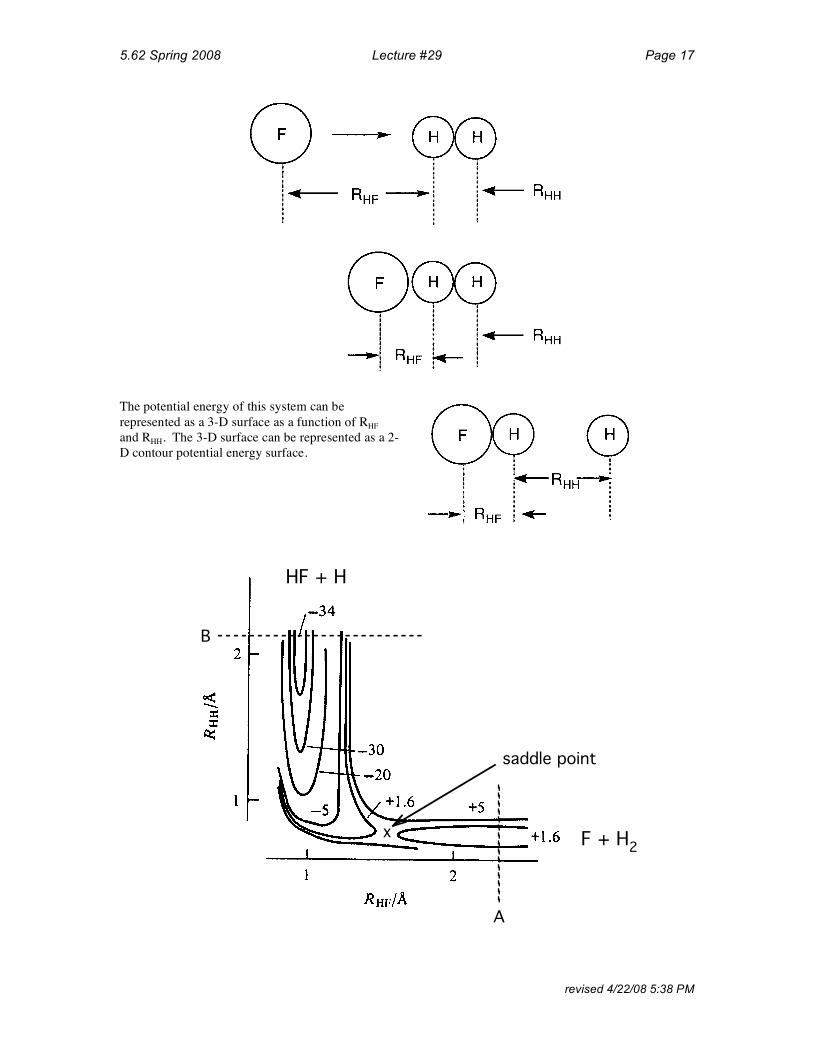
The potential energy of this system can berepresented as a 3-D surface as a function of RHF and RHH. The 3-D surface can be represented as a 2-D contour potential energy surface.
HF + H
B
F + H2 x
saddle point
A
revised 4/22/08 5:38 PM
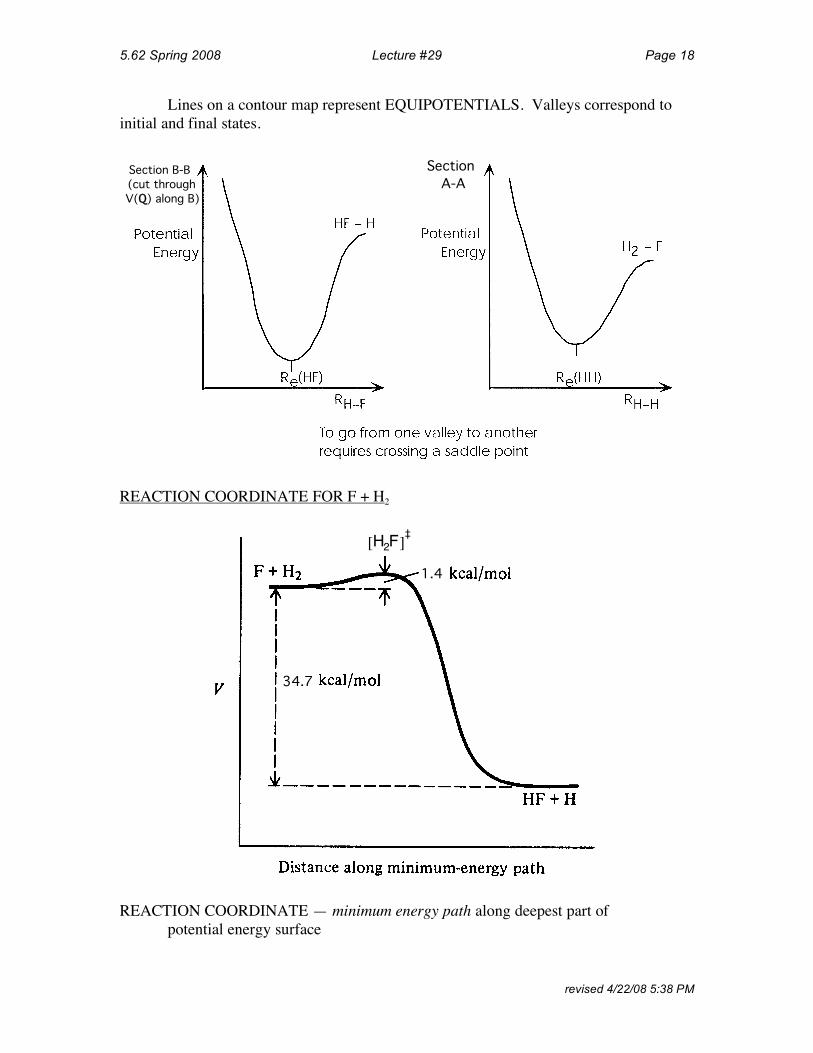
5.62 Spring 2008 Lecture #29 Page 18
Lines on a contour map represent EQUIPOTENTIALS. Valleys correspond to initial and final states.
Section A-A
Section B-B (cut through V(Q) along B)
REACTION COORDINATE FOR F + H2
1.4
H2F[ ]‡
34.7
REACTION COORDINATE — minimum energy path along deepest part of potential energy surface
revised 4/22/08 5:38 PM

5.62 Spring 2008 Lecture #29 Page 19
— note reaction coordinate corresponds to an antisymmetric H2F vibration
TRANSITION STATE — transitory [H2F]‡ complex with a definite structure — dissociates within one half antisymmetric vibration.
TRANSITION STATE THEORY
An approach to calculating a rate constant by reducing the dynamics of thereaction to an equilibrium between the reactants and the transition state along the reaction coordinate.
A + B ↔ [AB]‡ → PRODUCTS
Uses statistical mechanics to treat the equilibrium. The reaction coordinate is the 1-D antisymmetric vibration of the transition state.
revised 4/22/08 5:38 PM


















