
SEPT8 modulates β-amyloidogenic processing of APP by ... · β(Aβ)peptides isthe cleavage of...
15
RESEARCH ARTICLE SEPT8 modulates β-amyloidogenic processing of APP by affecting the sorting and accumulation of BACE1 Kaisa M. A. Kurkinen 1, *, Mikael Marttinen 1, *, Laura Turner 2 , Teemu Natunen 1 , Petra Ma ̈ kinen 1 , Fanni Haapalinna 3 , Timo Saraja ̈ rvi 1 , Sami Gabbouj 1 , Mitja Kurki 4 , Jussi Paananen 4 , Anne M. Koivisto 3 , Tuomas Rauramaa 5 , Ville Leinonen 4 , Heikki Tanila 6 , Hilkka Soininen 3 , Fiona R. Lucas 2 , Annakaisa Haapasalo 3,6, ‡ and Mikko Hiltunen 1,3, ‡ ABSTRACT Dysfunction and loss of synapses are early pathogenic events in Alzheimer’s disease. A central step in the generation of toxic amyloid- β (Aβ) peptides is the cleavage of amyloid precursor protein (APP) by β-site APP-cleaving enzyme (BACE1). Here, we have elucidated whether downregulation of septin (SEPT) protein family members, which are implicated in synaptic plasticity and vesicular trafficking, affects APP processing and Aβ generation. SEPT8 was found to reduce soluble APPβ and Aβ levels in neuronal cells through a post- translational mechanism leading to decreased levels of BACE1 protein. In the human temporal cortex, we identified alterations in the expression of specific SEPT8 transcript variants in a manner that correlated with Alzheimer’s-disease-related neurofibrillary pathology. These changes were associated with altered β-secretase activity. We also discovered that the overexpression of a specific Alzheimer’s- disease-associated SEPT8 transcript variant increased the levels of BACE1 and Aβ peptides in neuronal cells. These changes were related to an increased half-life of BACE1 and the localization of BACE1 in recycling endosomes. These data suggest that SEPT8 modulates β-amyloidogenic processing of APP through a mechanism affecting the intracellular sorting and accumulation of BACE1. KEY WORDS: Alzheimer’s disease, Amyloid precursor protein, Amyloid-β, BACE1, SEPT8 INTRODUCTION Alzheimer’s disease is the most common neurodegenerative disorder in the world, and affects up to 50% of individuals above the age of 85. Owing to the global increase in the aging population, early diagnosis and treatment of Alzheimer’s disease are becoming increasingly important from both a human and socioeconomic perspective. Alzheimer’s disease is clinically associated with a global cognitive decline, and a progressive loss of memory and reasoning. At autopsy, a large number of neuritic plaques and neurofibrillary tangles (NFTs) in the neocortex of the brain are detected in Alzheimer’s disease patients (Cummings, 2004; Mandelkow and Mandelkow, 1998). These consist of amyloid-β (Aβ) peptide and hyperphosphorylated tau protein, respectively. It is well established that the increased β-amyloidogenic processing of amyloid precursor protein (APP), leading to the augmented production of Aβ is a key feature underlying the molecular pathogenesis of Alzheimer’s disease (Neve et al., 2000). The cleavage of APP by β-site APP-cleaving enzyme 1 (BACE1) leads to the formation of the N-terminal soluble APPβ (sAPPβ) and a membrane-bound APP C-terminal fragment, called C99, which is subsequently cleaved by γ-secretase producing Aβ. By contrast, the majority of APP is cleaved by α-secretases, which leads to the release of the soluble ectodomain portion of APP (sAPPα) and prevents Aβ formation. Although the role of sAPPα is still under debate, recent findings have suggested that the overexpression of sAPPα in the brain of Alzheimer’s disease model mice overexpressing human APP with the Swedish mutation and presenilin-1 with deletion of exon 9 (APP/PS1ΔE9) mitigates synaptic and cognitive defects caused by pre-existing pathology (Fol et al., 2016). In neurons, BACE1 shows presynaptic localization and it is largely conveyed in the neuronal recycling endosomes (Cole and Vassar, 2007; Das et al., 2013). BACE1 and APP are segregated in neurons during resting state conditions, whereas, upon activity-dependent induction, these proteins converge into acidic microdomains through an endocytosis-dependent pathway (Das et al., 2013). This is important because BACE1 is optimally active in the acidic intracellular compartments, such as early and late endosomes (Kang et al., 2012). Based on the amyloid cascade hypothesis, increased soluble Aβ peptide levels augment synaptic dysfunction, Ca 2+ dyshomeostasis, inflammation and oxidative stress, as well as tau hyperphosphorylation and the formation of NFTs in specific brain regions in Alzheimer’s disease patients (DeKosky and Scheff, 1990; Hu et al., 2014; Kuperstein et al., 2010). Thus, if started early enough, lowering Aβ levels could be sufficient to slow down the disease process in Alzheimer’s disease. This idea is further supported by the genetic finding indicating that a naturally occurring A673T rare variant in APP protects from Alzheimer’s-disease- and age-related cognitive decline by reducing BACE1-mediated cleavage of APP and/or producing less of the aggregation-prone Aβ40 containing the A→T variation at position 2 (Di Fede et al., 2009; Jonsson et al., 2012; Benilova et al., 2014; Das et al., 2016). Soluble Aβ42 dimers extracted from the Alzheimer’s disease brain have been shown to specifically increase the phosphorylation of tau and subsequently promote neurodegeneration, supporting the intimate link between Aβ and tau in Alzheimer’s disease pathogenesis (Shankar et al., 2008). This observation also concurs with the notion that synaptic dysfunction is the best correlate of the cognitive decline in Alzheimer’s disease, Received 23 December 2015; Accepted 11 April 2016 1 Institute of Biomedicine, School of Medicine, University of Eastern Finland, 70211 Kuopio, Finland. 2 Eisai Ltd., Bernard Katz Building, University College London, London WC1E 6BT, UK. 3 Institute of Clinical Medicine – Neurology, School of Medicine, University of Eastern Finland and Department of Neurology, Kuopio University Hospital, 70211 Kuopio, Finland. 4 Institute of Clinical Medicine – Neurosurgery, School of Medicine, University of Eastern Finland and Neurosurgery of NeuroCenter, Kuopio University Hospital, 70211 Kuopio, Finland. 5 Institute of Clinical Medicine – Pathology, School of Medicine, University of Eastern Finland and Department of Pathology, Kuopio University Hospital, 70211 Kuopio, Finland. 6 Department of Neurobiology, A.I. Virtanen, Institute for Molecular Sciences, School of Medicine, University of Eastern Finland, 70211 Kuopio, Finland. *These authors contributed equally to this work ‡ Author for correspondence ([email protected]; [email protected]) M.H., 0000-0003-3566-4096 2224 © 2016. Published by The Company of Biologists Ltd | Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215 Journal of Cell Science
Transcript of SEPT8 modulates β-amyloidogenic processing of APP by ... · β(Aβ)peptides isthe cleavage of...

RESEARCH ARTICLE
SEPT8 modulates β-amyloidogenic processing of APP byaffecting the sorting and accumulation of BACE1KaisaM. A. Kurkinen1,*, Mikael Marttinen1,*, Laura Turner2, TeemuNatunen1, Petra Makinen1, Fanni Haapalinna3,Timo Sarajarvi1, Sami Gabbouj1, Mitja Kurki4, Jussi Paananen4, Anne M. Koivisto3, Tuomas Rauramaa5,Ville Leinonen4, Heikki Tanila6, Hilkka Soininen3, Fiona R. Lucas2, Annakaisa Haapasalo3,6,‡ andMikko Hiltunen1,3,‡
ABSTRACTDysfunction and loss of synapses are early pathogenic events inAlzheimer’s disease. A central step in the generation of toxic amyloid-β (Aβ) peptides is the cleavage of amyloid precursor protein (APP) byβ-site APP-cleaving enzyme (BACE1). Here, we have elucidatedwhether downregulation of septin (SEPT) protein family members,which are implicated in synaptic plasticity and vesicular trafficking,affects APP processing and Aβ generation. SEPT8 was found toreduce soluble APPβ and Aβ levels in neuronal cells through a post-translational mechanism leading to decreased levels of BACE1protein. In the human temporal cortex, we identified alterations in theexpression of specific SEPT8 transcript variants in a manner thatcorrelated with Alzheimer’s-disease-related neurofibrillary pathology.These changes were associated with altered β-secretase activity. Wealso discovered that the overexpression of a specific Alzheimer’s-disease-associated SEPT8 transcript variant increased the levels ofBACE1 and Aβ peptides in neuronal cells. These changes wererelated to an increased half-life of BACE1 and the localization ofBACE1 in recycling endosomes. These data suggest that SEPT8modulates β-amyloidogenic processing of APP through amechanismaffecting the intracellular sorting and accumulation of BACE1.
KEY WORDS: Alzheimer’s disease, Amyloid precursor protein,Amyloid-β, BACE1, SEPT8
INTRODUCTIONAlzheimer’s disease is the most common neurodegenerative disorderin theworld, and affects up to 50% of individuals above the age of 85.Owing to the global increase in the aging population, early diagnosisand treatment of Alzheimer’s disease are becoming increasinglyimportant from both a human and socioeconomic perspective.Alzheimer’s disease is clinically associated with a global cognitivedecline, and a progressive loss of memory and reasoning. At autopsy,
a large number of neuritic plaques and neurofibrillary tangles (NFTs)in the neocortex of the brain are detected in Alzheimer’s diseasepatients (Cummings, 2004; Mandelkow and Mandelkow, 1998).These consist of amyloid-β (Aβ) peptide and hyperphosphorylated tauprotein, respectively. It is well established that the increasedβ-amyloidogenic processing of amyloid precursor protein (APP),leading to the augmented production ofAβ is a key feature underlyingthe molecular pathogenesis of Alzheimer’s disease (Neve et al.,2000). The cleavage of APP by β-site APP-cleaving enzyme 1(BACE1) leads to the formation of the N-terminal soluble APPβ(sAPPβ) and a membrane-bound APP C-terminal fragment, calledC99, which is subsequently cleaved by γ-secretase producing Aβ. Bycontrast, the majority of APP is cleaved by α-secretases, which leadsto the release of the soluble ectodomain portion of APP (sAPPα) andprevents Aβ formation. Although the role of sAPPα is still underdebate, recent findings have suggested that the overexpression ofsAPPα in the brain ofAlzheimer’s diseasemodelmice overexpressinghumanAPPwith the Swedishmutation and presenilin-1with deletionof exon 9 (APP/PS1ΔE9) mitigates synaptic and cognitive defectscaused by pre-existing pathology (Fol et al., 2016).
In neurons, BACE1 shows presynaptic localization and it is largelyconveyed in the neuronal recycling endosomes (Cole and Vassar,2007; Das et al., 2013). BACE1 and APP are segregated in neuronsduring resting state conditions, whereas, upon activity-dependentinduction, these proteins converge into acidic microdomainsthrough an endocytosis-dependent pathway (Das et al., 2013). Thisis important because BACE1 is optimally active in the acidicintracellular compartments, such as early and late endosomes (Kanget al., 2012). Based on the amyloid cascade hypothesis, increasedsoluble Aβ peptide levels augment synaptic dysfunction, Ca2+
dyshomeostasis, inflammation and oxidative stress, as well as tauhyperphosphorylation and the formation of NFTs in specific brainregions in Alzheimer’s disease patients (DeKosky and Scheff, 1990;Hu et al., 2014; Kuperstein et al., 2010). Thus, if started early enough,lowering Aβ levels could be sufficient to slow down the diseaseprocess in Alzheimer’s disease. This idea is further supported by thegenetic finding indicating that a naturally occurring A673T rarevariant in APP protects from Alzheimer’s-disease- and age-relatedcognitive decline by reducing BACE1-mediated cleavage of APPand/or producing less of the aggregation-prone Aβ40 containing theA→T variation at position 2 (Di Fede et al., 2009; Jonsson et al., 2012;Benilova et al., 2014;Das et al., 2016). SolubleAβ42dimers extractedfrom the Alzheimer’s disease brain have been shown to specificallyincrease the phosphorylation of tau and subsequently promoteneurodegeneration, supporting the intimate link between Aβ and tauin Alzheimer’s disease pathogenesis (Shankar et al., 2008). Thisobservation also concurs with the notion that synaptic dysfunction isthe best correlate of the cognitive decline in Alzheimer’s disease,Received 23 December 2015; Accepted 11 April 2016
1Institute of Biomedicine, School of Medicine, University of Eastern Finland, 70211Kuopio, Finland. 2Eisai Ltd., Bernard Katz Building, University College London,London WC1E 6BT, UK. 3Institute of Clinical Medicine – Neurology, School ofMedicine, University of Eastern Finland and Department of Neurology, KuopioUniversity Hospital, 70211 Kuopio, Finland. 4Institute of Clinical Medicine –
Neurosurgery, School of Medicine, University of Eastern Finland and Neurosurgeryof NeuroCenter, Kuopio University Hospital, 70211 Kuopio, Finland. 5Institute ofClinical Medicine – Pathology, School of Medicine, University of Eastern Finlandand Department of Pathology, Kuopio University Hospital, 70211 Kuopio, Finland.6Department of Neurobiology, A.I. Virtanen, Institute for Molecular Sciences, Schoolof Medicine, University of Eastern Finland, 70211 Kuopio, Finland.*These authors contributed equally to this work
‡Author for correspondence ([email protected]; [email protected])
M.H., 0000-0003-3566-4096
2224
© 2016. Published by The Company of Biologists Ltd | Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience
emphasizing the importance of understanding the early mechanismsleading to synaptotoxicity (Coleman and Yao, 2003). Collectively, inorder to identify factors that could be utilized as novel drug targets or
predictive biomarkers in Alzheimer’s disease, it is necessary to focuson the cellular processes involved in the early steps of Alzheimer’sdisease pathogenesis.
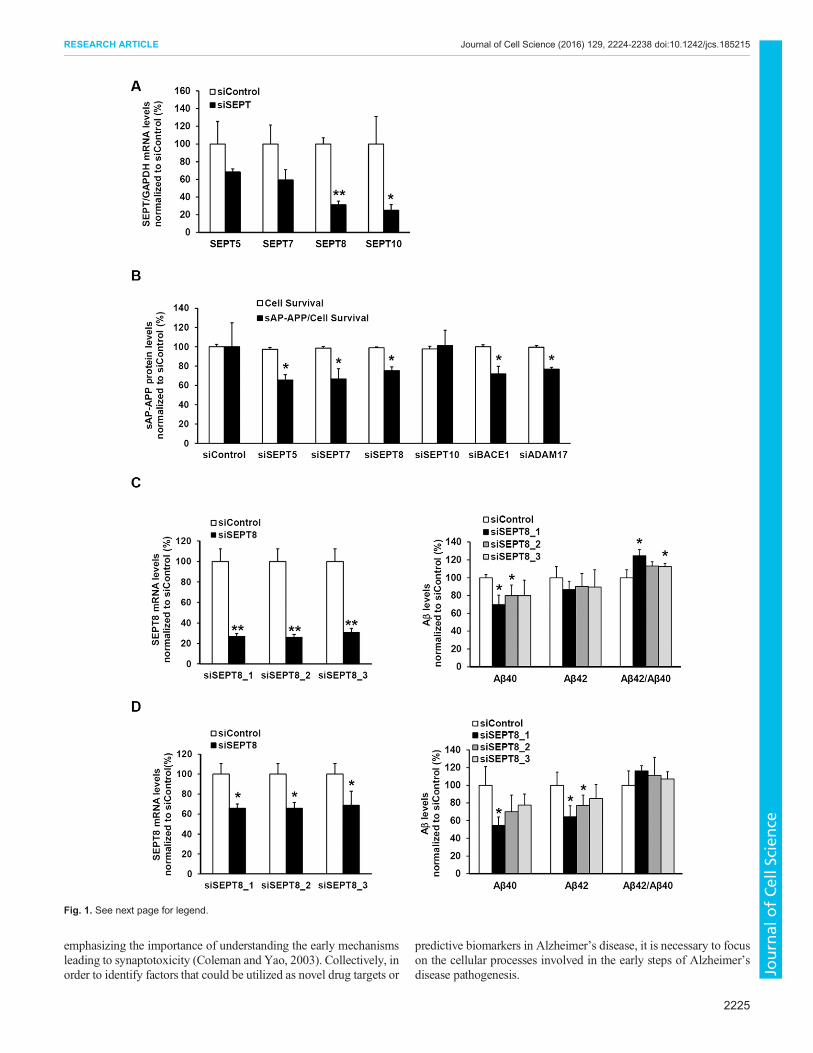
Fig. 1. See next page for legend.
2225
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience

Septins (denoted SEPT) are a conserved family of GTP-bindingproteins, which are highly expressed in the brain (Marttinen et al.,2015). The septin protein family is composed of 13 genes, whichare divided into four subgroups based on their sequence homologyand domain composition: the SEPT2 group (SEPT1, SEPT2,SEPT4 and SEPT5), SEPT3 group (SEPT3, SEPT9 and SEPT12),SEPT6 group (SEPT6, SEPT8, SEPT10, SEPT11 and SEPT14)and SEPT7 group (SEPT7 only). Septins are implicated in theregulation of several cellular processes, including cytokinesis,membrane remodeling and compartmentalization, cytoskeletonrearrangement, apoptosis and vesicle trafficking (Hall and Russell,2012). SEPT8 and SEPT5 have been ascribed a key role in theregulation of presynaptic functions, such as neurotransmitterrelease (Ito et al., 2009) and vesicle trafficking (Amin et al., 2008).In addition to these physiological functions, septins have beenlinked to different neurodegenerative diseases and psychiatricdisorders; for example, Alzheimer’s disease, Parkinson’s disease,Huntington’s disease, frontotemporal dementia, Down syndromeand schizophrenia (Barr et al., 2004; Kinoshita et al., 1998;Munoz-Soriano and Paricio, 2007).Here, we have investigated the involvement of septin family
members SEPT5, SEPT7, SEPT8 and SEPT10 on Alzheimer’sdisease molecular pathogenesis. We show that SEPT8 affects APPprocessing and Aβ peptide production through a molecularmechanism that modulates the intracellular sorting and stabilityof BACE1 in neuronal cells. We also observed an imbalancedSEPT8 transcript variant expression profile that is already presentat the early stages of the Alzheimer’s-disease-relatedneurofibrillary pathology in the human temporal cortex. Thesefindings suggest that SEPT8 is centrally linked to cellularprocesses relevant to Alzheimer’s disease both in vitro and in thehuman brain.
RESULTSRNAi-based screening of septin protein family members innon-neuronal and neuronal cells overexpressing APPidentifies SEPT8 as a target reducing soluble APP and AβlevelsGiven that Aβ-mediated synaptic dysfunction is one of the earliestfeatures in Alzheimer’s disease, and that septin protein family
members are known to play a role in the regulation of presynapticactivity and function (Amin et al., 2008; Ito et al., 2009), westudied the effects of downregulation of septin family members(SEPT5, SEPT7, SEPT8 and SEPT10) on APP processing byassessing the levels of total soluble APP and Aβ in humanHEK293 cells overexpressing alkaline-phosphatase-conjugatedAPP (HEK293-AP–APP) (Lichtenthaler et al., 2003). Validatedsmall interfering RNAs (siRNAs) for BACE1 and ADAM17 wereused as positive controls in the screening of total soluble AP–APP(sAP–APP) and Aβ levels. Downregulation of SEPT5, SEPT7 andSEPT8 (Fig. 1A) significantly decreased sAP–APP levels in thecell culture medium when normalized to the cell survival(Fig. 1B). In addition, a trend towards decreased Aβ40 andAβ42 levels was observed in culture medium samples from cells inwhich SEPT5 and SEPT8 was downregulated (Fig. S1A).Conversely, a trend towards increased Aβ40 and Aβ42 levelswas observed in cells with SEPT7 and SEPT10 downregulation(Fig. S1A). To further elucidate the SEPT8 findings, two othersiRNAs targeting different parts of SEPT8 mRNA sequence werealso applied to the HEK293-AP–APP cells to exclude potentialoff-target effects (Fig. 1C). Decreased Aβ40 and Aβ42 levels wereobserved with all three SEPT8 siRNAs when normalized to totalAPP levels in the cell lysates (Fig. 1C). Total APP-normalized Aβlevels were assessed in order to account for the potential changesin the total APP levels due to the knockdown of SEPT8(Fig. S1B). The same siRNAs were used to downregulateSEPT8 in human SH-SY5Y neuroblastoma cells overexpressingthe APP751 isoform (SH-SY5Y-APP751) (Sarajarvi et al., 2011).Although the downregulation of SEPT8 mRNA levels was not aspronounced in SH-SY5Y-APP751 cells as in HEK293-AP–APPcells, all three SEPT8 siRNAs resulted in a 20–40% decrease inboth Aβ40 and Aβ42 levels (Fig. 1D) when normalized to totalAPP levels in the cell lysates (Fig. S1C). Collectively, these datasuggest that the downregulation of SEPT8 in non-neuronal andneuronal cells affects APP processing by lowering total solubleAPP and Aβ levels.
Downregulation of SEPT8 decreases endogenous BACE1protein levels in SH-SY5Y neuroblastoma cells and primaryhippocampal neuronsThe observation that the downregulation of SEPT8 decreased thelevels of both secreted Aβ and total soluble APP led us to assessthe effects of SEPT8 downregulation on APP processing in thetotal protein lysates of SH-SY5Y-APP751 cells. An average 60%reduction in the γ-tubulin-normalized SEPT8 protein levels(Fig. 2A) did not significantly affect the total protein levels ofAPP (APPtot). However, a trend towards reduced levels of APPC99, but not APP C83, the C-terminal fragment produced in thenon-amyloidogenic APP processing pathway, was observed(Fig. 2A). Furthermore, coinciding with the decreased Aβ levels(Fig. 2B; Fig. S2A), protein levels of endogenous BACE1 werereduced (∼25%) upon the downregulation of SEPT8 (Fig. 2A).Similarly, lentiviral siRNA-mediated downregulation of SEPT8 inprimary hippocampal neuronal cultures prepared from E18 mouseembryos resulted in significantly decreased (∼30%) endogenousBACE1 protein levels (Fig. 2C). Consistent with this finding, thelevels of Aβ40 and Aβ42 were significantly reduced in the culturemedium of hippocampal neurons (Fig. 2D). The mRNA levels ofBACE1 in the primary hippocampal neurons were unchanged[control siRNA (siControl) 100.0±50.0%, siRNA against SEPT8(siSEPT8) 100.8±24.6%, n=4, mean±s.d., independent sample t-test, not significant], suggesting that the decrease in BACE1
Fig. 1. RNAi screening of septin family member proteins in HEK293-AP–APP cells identifies SEPT8 as a target affecting APP processing andAβ40 and Aβ42 levels. (A) qRT-PCR was conducted to assess theknockdown effect of 5 mM siRNAs for each target after 72 h of transfection inHEK293-AP–APP cells. GAPDH-normalized mRNA levels of SEPT5 (n=4),SEPT7 (n=4), SEPT8 (siSEPT8_2, n=3) and SEPT10 (n=4) in siSEPT- andsiControl-transfected cells are presented. (B) The levels of total solublealkaline-phosphatase-linked APP (sAP–APP) in each target siRNA-transfected cell line was normalized to the cell survival (white bars) in eachsample and is shown as the percentage of the level compared to that withsiControl. Cell survival was not affected due to the downregulation of septins.siControl, siSEPT5, siSEPT7, siSEPT10, siBACE1 and siADAM17 (n=4), andsiSEPT8 (siSEPT8_2, n=3). (C) Three different SEPT8 siRNAs significantlydecreased the mRNA levels of SEPT8 in HEK293-AP–APP cells (left). Thelevels of Aβ40 and Aβ42 in the cell culture medium are normalized to APP totalprotein levels in the respective cell lysates and shown as a percentage of thelevel compared to that with siControl (right). n=4. (D) Three different SEPT8siRNAs significantly decreased SEPT8 mRNA levels in SH-SY5Y cells stablyoverexpressing the human APP751 isoform (SH-SY5Y-APP751) (left). Thelevels of Aβ40 and Aβ42 in the cell culture medium are normalized to the totalprotein levels of APP in the respective cell lysates and shown as a percentageof the level compared to that with siControl (right). n=4. In A–D, n values arebiological replicates and results are mean±s.d. *P<0.05, **P<0.01(independent sample t-test).
2226
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience
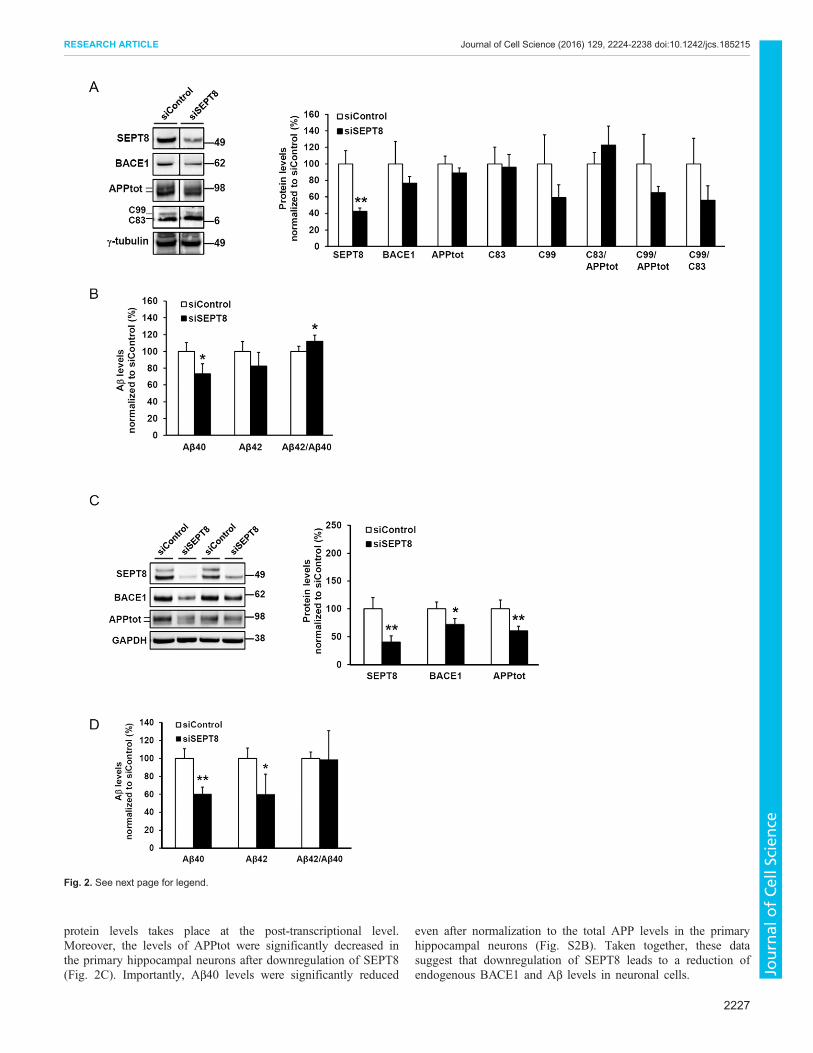
protein levels takes place at the post-transcriptional level.Moreover, the levels of APPtot were significantly decreased inthe primary hippocampal neurons after downregulation of SEPT8(Fig. 2C). Importantly, Aβ40 levels were significantly reduced
even after normalization to the total APP levels in the primaryhippocampal neurons (Fig. S2B). Taken together, these datasuggest that downregulation of SEPT8 leads to a reduction ofendogenous BACE1 and Aβ levels in neuronal cells.
Fig. 2. See next page for legend.
2227
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience

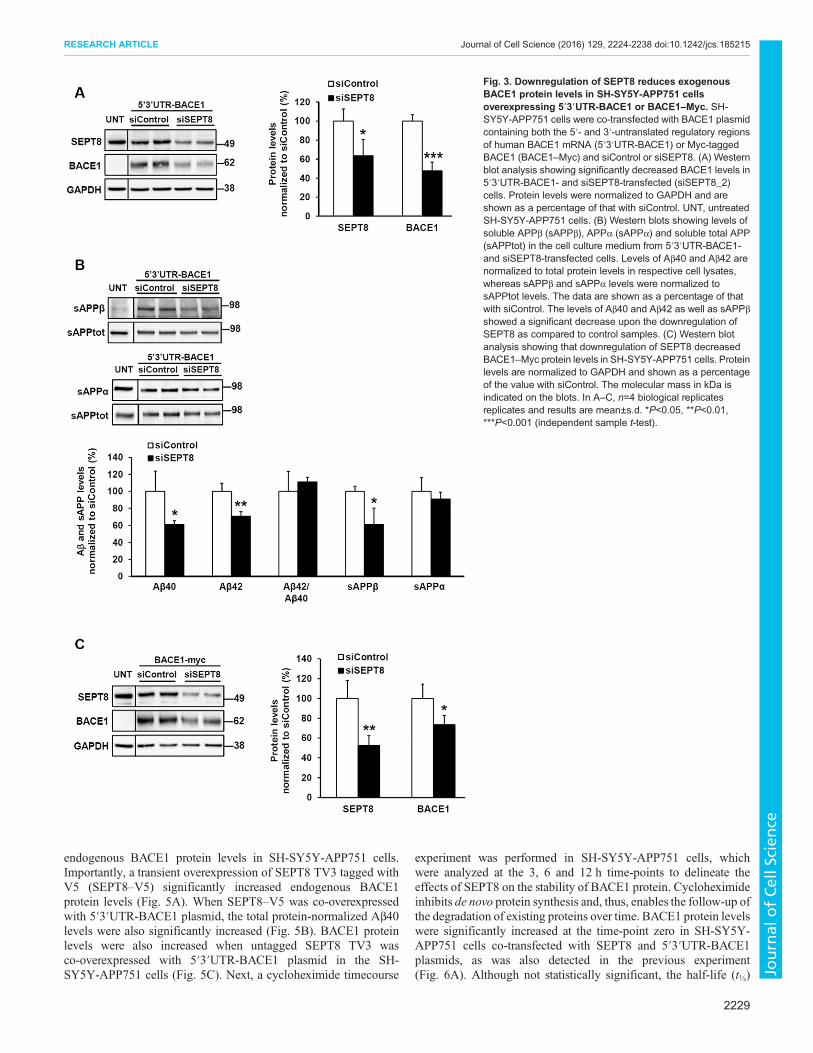
Downregulation of SEPT8 decreases BACE1 protein levelsthrough a post-translational mechanismAs several lines of evidence suggested that the downregulation ofSEPT8 decreases BACE1 protein levels and consequentlyBACE1-mediated APP processing, we next wanted to assesswhether the observed reduction in BACE1 protein levels isregulated at the post-transcriptional or post-translational level. Todo this, we utilized two different BACE1 cDNA plasmids resultingin either high or moderate expression levels of BACE1. A Myc-tagged BACE1 cDNA construct under the cytomegalovirus(CMV) promoter and without 5′ and 3′ untranslated regions(UTRs) of endogenous BACE1 (BACE1–Myc) yields highBACE1 protein levels. Transfection of cells with a cDNAconstruct encompassing the endogenous 5′ and 3′ UTRs ofBACE1 (5′3′UTR-BACE1) results in a moderate overexpressionof BACE1 protein. SH-SY5Y-APP751 cells were co-transfectedeither with control or SEPT8 siRNAs together with 5′3′UTR-BACE1 (Fig. 3A,B) or BACE1–Myc plasmids (Fig. 3C). Thedownregulation of SEPT8 significantly decreased BACE1 levels incells upon expression of either 5′3′UTR-BACE1 or BACE1–Myc.As the decrease was observed in cells transfected with BACE1–Myc, which has no regulatory UTRs, the observed reduction inBACE1 protein levels is likely due to post-translationalmechanisms. In line with the previous findings described above,the downregulation of SEPT8 reduced BACE1-mediated APPprocessing by significantly decreasing Aβ40, Aβ42 and sAPPβ,but not sAPPα levels (Fig. 3B).To further elucidate the post-translational mechanism
underlying reduced BACE1 protein levels after the knockdownof SEPT8, we next assessed whether the trafficking of BACE1and/or APP onto the cell surface was affected by potentiallyaltered intracellular trafficking within the secretory or endocyticrecycling pathways. Western blot analysis performed after cellsurface biotinylation in SH-SY5Y-APP751 cells co-transfectedwith the 5′3′UTR-BACE1 plasmid and control siRNA or siSEPT8(Fig. S3A) demonstrated a moderate, but statistically insignificant,increase in the BACE1 protein levels at the cell surface whennormalized to total levels of BACE1 in the cell lysates (Fig. S3B).The levels of mature APP at the cell surface were not altered afternormalization to the total APP levels in the cell lysates (Fig. S3B).In summary, these results suggest that the knockdown of SEPT8decreases BACE1 levels through a post-translational mechanism,
which does not involve prominent changes in the cell surfacelevels of BACE1.
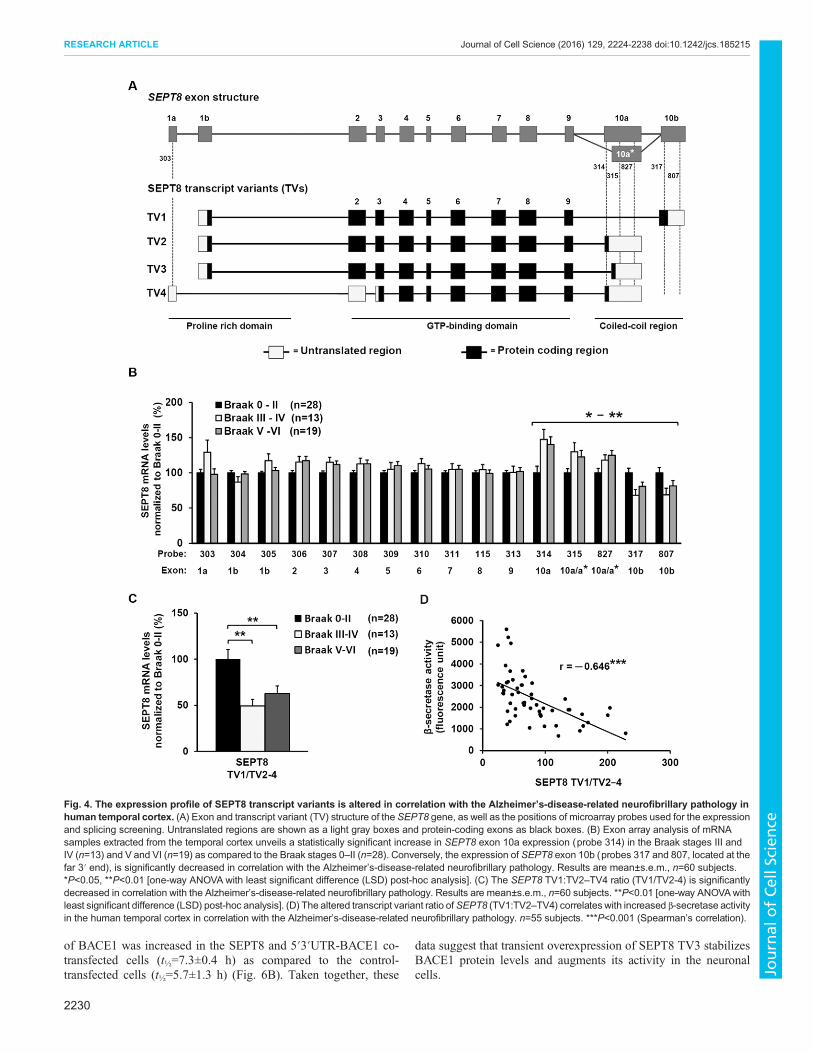
SEPT8 transcript variantmRNAs are differentially expressedin the human temporal cortex in a manner that is correlatedwith Alzheimer’s-disease-related neurofibrillary pathologyNext, we wanted to address the relationship between the expressionof SEPT8 and the Alzheimer’s-disease-associated neurofibrillarypathology in the human temporal cortex. The human SEPT8 geneencodes four different transcript variants (denoted TV) (Fig. 4A):SEPT8 TV1 [483 amino acids (aa), referred to as the ‘canonical’sequence, 56 kDa], SEPT8 TV2 (429 aa, differs from the canonicalsequence as follows: aa 430–483 missing, 50 kDa), SEPT8 TV3(442 aa, differs from the canonical sequence as follows: 429–442,NRSDIGAHQPGMSL→KASGWSSIYSVTIP, aa 443–483missing, 52 kDa), and SEPT8 TV4 (369 aa, differs from thecanonical sequence as follows: aa 1–60 missing, aa 430–483missing, 44 kDa). To determine the expression status of SEPT8transcript variants in our well-established human brain samplecohort (Martiskainen et al., 2015), we applied a microarray-basedapproach in which probes were designed to capture all the exons inthe SEPT8 mRNA (Fig. 4B). This cohort consists of RNA samplesfrom the post mortem temporal cortex of 60 subjects with varyingdegrees of Alzheimer’s-disease-related neurofibrillary pathology(phosphorylated tau protein staining with AT8 antibody) (Table 1).The subjects were grouped based on Braak staging into threesubgroups: Braak stages 0–II, Braak stages III or IV, and Braakstages V or VI (Braak et al., 2006; Martiskainen et al., 2015). Wefound that the expression profile at the 3′ end of SEPT8 wassignificantly different between the Braak groups in the temporalcortex (Fig. 4B). More specifically, probes 317 and 807 targeted tothe exon 10b (TV1 variant) showed decreased binding, whereasprobes 314, 315 and 827 targeted to exon 10a and 10a* (TV2–TV4variants) showed increased binding. This alteration in theexpression of these variants led to a significantly reduced ratio ofTV1 versus TV2–TV4 variants (TV1:TV2–TV4) according to theAlzheimer’s-disease-related neurofibrillary pathology (Fig. 4C).
Based on the results from our in vitro cell culture findings relatingto SEPT8 and BACE1, we next investigated whether the mRNAprofiles of the different SEPT8 transcript variants showed arelationship with β-secretase activity, which was measuredpreviously from the same tissue samples (Laitera et al., 2014;Martiskainen et al., 2015). We observed a significant negativecorrelation between exon 10b (probe 807) and β-secretase activity(r=−0.46, P<0.001, Spearman correlation, n=55) in the temporalcortex. The opposite effect was detected with exon 10a (probe 314)and β-secretase activity (r=0.62, P<10−6, Spearman correlation,n=55). Consequently, the TV1:TV2–TV4 ratio showed significantnegative correlation with β-secretase activity in the temporal cortex(Fig. 4D). Taken together, these data suggest an imbalancedtranscript variant expression of SEPT8 in the early phase ofAlzheimer’s disease pathogenesis (Braak stages 0–II versus III orIV) and that the increased mRNA levels of the SEPT8 TV2–TV4variants coincide with increased β-secretase activity in humantemporal cortex.
Overexpression of TV3 variant of SEPT8 stabilizes BACE1protein in SH-SY5Y neuroblastoma cellsGiven the data that the levels of SEPT8 TV2, TV3 and TV4are increased in Alzheimer’s disease brain and that the increasecorrelated with altered BACE1 activity, next we assessed whethertransient overexpression of the TV3 variant of SEPT8 affects
Fig. 2. Downregulation of SEPT8 leads to decreased levels ofendogenous BACE1 in neuroblastoma cells and cultured mouse primaryhippocampal neurons. (A) Western blot analysis to elucidate the effects onAPP processing in the SH-SY5Y-APP751 neuroblastoma cells transfectedwith SEPT8 (siSEPT8_2) or control siRNAs. On average, a 60% decrease inγ-tubulin-normalized SEPT8 protein levels was observed. Levels of APP C83and C99 are normalized to both γ-tubulin and total APP levels. All sampleswere run on the same gel. (B) Downregulation of SEPT8 led to a reduction inthe Aβ levels. Aβ40 and Aβ42 levels in the cell culture medium are normalizedto total protein levels in the respective cell lysates. (C) Downregulation ofSEPT8 protein levels using shRNA lentiviral particles (20 MOI) in mouseprimary hippocampal neurons cultured for 20 days in vitro (DIV 20). Westernblot analysis shows a significant downregulation of mouse SEPT8 protein14 days after transduction. Downregulation of SEPT8 significantly decreasedendogenously expressed BACE1 protein levels inmouse primary hippocampalneurons. Protein levels are normalized to GAPDH. Themolecular mass in kDais indicated on the blots. (D) Aβ40 and Aβ42 levels were significantlydecreased in mouse primary hippocampal neurons after siSEPT8 lentiviraltransduction. Aβ levels in cell culture medium are normalized to total proteinlevels in the respective cell lysates. In A–D, n=4 biological replicates andresults are mean±s.d. *P<0.05, **P<0.01 (independent sample t-test).
2228
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience
endogenous BACE1 protein levels in SH-SY5Y-APP751 cells.Importantly, a transient overexpression of SEPT8 TV3 tagged withV5 (SEPT8–V5) significantly increased endogenous BACE1protein levels (Fig. 5A). When SEPT8–V5 was co-overexpressedwith 5′3′UTR-BACE1 plasmid, the total protein-normalized Aβ40levels were also significantly increased (Fig. 5B). BACE1 proteinlevels were also increased when untagged SEPT8 TV3 wasco-overexpressed with 5′3′UTR-BACE1 plasmid in the SH-SY5Y-APP751 cells (Fig. 5C). Next, a cycloheximide timecourse
experiment was performed in SH-SY5Y-APP751 cells, whichwere analyzed at the 3, 6 and 12 h time-points to delineate theeffects of SEPT8 on the stability of BACE1 protein. Cycloheximideinhibits de novo protein synthesis and, thus, enables the follow-up ofthe degradation of existing proteins over time. BACE1 protein levelswere significantly increased at the time-point zero in SH-SY5Y-APP751 cells co-transfected with SEPT8 and 5′3′UTR-BACE1plasmids, as was also detected in the previous experiment(Fig. 6A). Although not statistically significant, the half-life (t½)
Fig. 3. Downregulation of SEPT8 reduces exogenousBACE1 protein levels in SH-SY5Y-APP751 cellsoverexpressing 5′3′UTR-BACE1 or BACE1–Myc. SH-SY5Y-APP751 cells were co-transfected with BACE1 plasmidcontaining both the 5′- and 3′-untranslated regulatory regionsof human BACE1 mRNA (5′3′UTR-BACE1) or Myc-taggedBACE1 (BACE1–Myc) and siControl or siSEPT8. (A) Westernblot analysis showing significantly decreased BACE1 levels in5′3′UTR-BACE1- and siSEPT8-transfected (siSEPT8_2)cells. Protein levels were normalized to GAPDH and areshown as a percentage of that with siControl. UNT, untreatedSH-SY5Y-APP751 cells. (B) Western blots showing levels ofsoluble APPβ (sAPPβ), APPα (sAPPα) and soluble total APP(sAPPtot) in the cell culture medium from 5′3′UTR-BACE1-and siSEPT8-transfected cells. Levels of Aβ40 and Aβ42 arenormalized to total protein levels in respective cell lysates,whereas sAPPβ and sAPPα levels were normalized tosAPPtot levels. The data are shown as a percentage of thatwith siControl. The levels of Aβ40 and Aβ42 as well as sAPPβshowed a significant decrease upon the downregulation ofSEPT8 as compared to control samples. (C) Western blotanalysis showing that downregulation of SEPT8 decreasedBACE1–Myc protein levels in SH-SY5Y-APP751 cells. Proteinlevels are normalized to GAPDH and shown as a percentageof the value with siControl. The molecular mass in kDa isindicated on the blots. In A–C, n=4 biological replicatesreplicates and results are mean±s.d. *P<0.05, **P<0.01,***P<0.001 (independent sample t-test).
2229
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience
of BACE1 was increased in the SEPT8 and 5′3′UTR-BACE1 co-transfected cells (t½=7.3±0.4 h) as compared to the control-transfected cells (t½=5.7±1.3 h) (Fig. 6B). Taken together, these
data suggest that transient overexpression of SEPT8 TV3 stabilizesBACE1 protein levels and augments its activity in the neuronalcells.
Fig. 4. The expression profile of SEPT8 transcript variants is altered in correlation with the Alzheimer’s-disease-related neurofibrillary pathology inhuman temporal cortex. (A) Exon and transcript variant (TV) structure of the SEPT8 gene, as well as the positions of microarray probes used for the expressionand splicing screening. Untranslated regions are shown as a light gray boxes and protein-coding exons as black boxes. (B) Exon array analysis of mRNAsamples extracted from the temporal cortex unveils a statistically significant increase in SEPT8 exon 10a expression (probe 314) in the Braak stages III andIV (n=13) and V and VI (n=19) as compared to the Braak stages 0–II (n=28). Conversely, the expression of SEPT8 exon 10b (probes 317 and 807, located at thefar 3′ end), is significantly decreased in correlation with the Alzheimer’s-disease-related neurofibrillary pathology. Results are mean±s.e.m., n=60 subjects.*P<0.05, **P<0.01 [one-way ANOVA with least significant difference (LSD) post-hoc analysis]. (C) The SEPT8 TV1:TV2–TV4 ratio (TV1/TV2-4) is significantlydecreased in correlation with the Alzheimer’s-disease-related neurofibrillary pathology. Results are mean±s.e.m., n=60 subjects. **P<0.01 [one-way ANOVAwithleast significant difference (LSD) post-hoc analysis]. (D) The altered transcript variant ratio ofSEPT8 (TV1:TV2–TV4) correlates with increased β-secretase activityin the human temporal cortex in correlation with the Alzheimer’s-disease-related neurofibrillary pathology. n=55 subjects. ***P<0.001 (Spearman’s correlation).
2230
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience
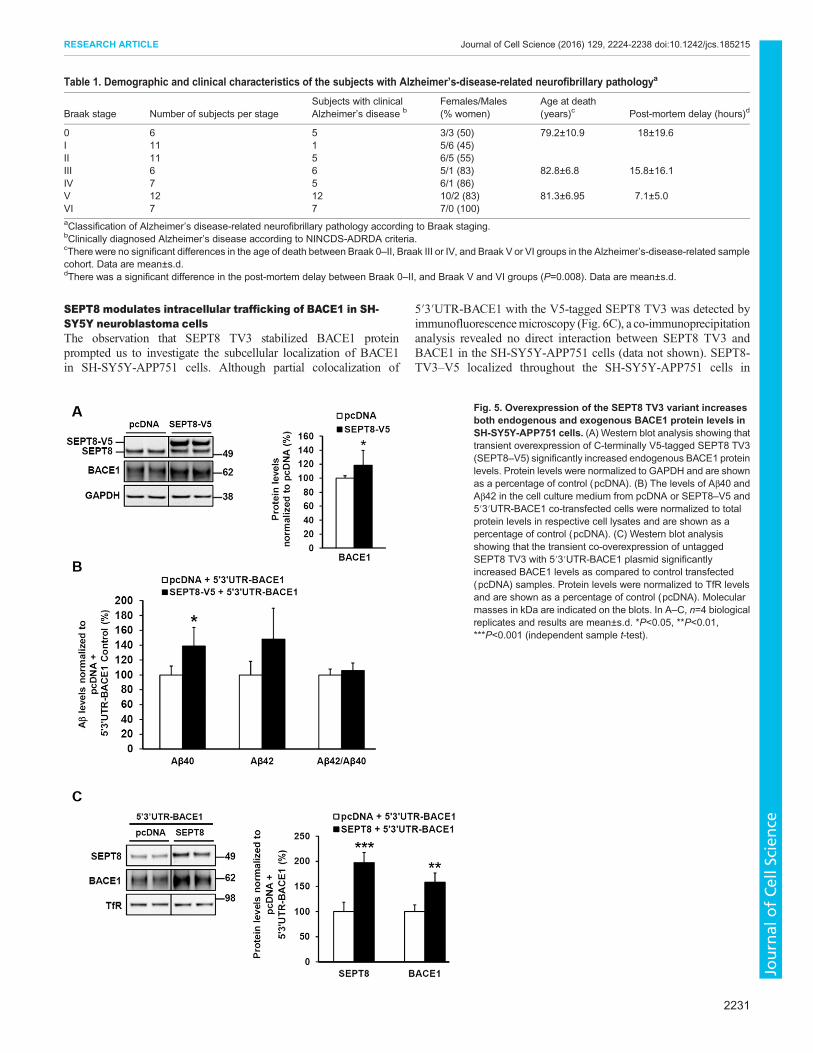
SEPT8 modulates intracellular trafficking of BACE1 in SH-SY5Y neuroblastoma cellsThe observation that SEPT8 TV3 stabilized BACE1 proteinprompted us to investigate the subcellular localization of BACE1in SH-SY5Y-APP751 cells. Although partial colocalization of
5′3′UTR-BACE1 with the V5-tagged SEPT8 TV3 was detected byimmunofluorescencemicroscopy (Fig. 6C), a co-immunoprecipitationanalysis revealed no direct interaction between SEPT8 TV3 andBACE1 in the SH-SY5Y-APP751 cells (data not shown). SEPT8-TV3–V5 localized throughout the SH-SY5Y-APP751 cells in
Fig. 5. Overexpression of the SEPT8 TV3 variant increasesboth endogenous and exogenous BACE1 protein levels inSH-SY5Y-APP751 cells. (A) Western blot analysis showing thattransient overexpression of C-terminally V5-tagged SEPT8 TV3(SEPT8–V5) significantly increased endogenous BACE1 proteinlevels. Protein levels were normalized to GAPDH and are shownas a percentage of control (pcDNA). (B) The levels of Aβ40 andAβ42 in the cell culture medium from pcDNA or SEPT8–V5 and5′3′UTR-BACE1 co-transfected cells were normalized to totalprotein levels in respective cell lysates and are shown as apercentage of control (pcDNA). (C) Western blot analysisshowing that the transient co-overexpression of untaggedSEPT8 TV3 with 5′3′UTR-BACE1 plasmid significantlyincreased BACE1 levels as compared to control transfected(pcDNA) samples. Protein levels were normalized to TfR levelsand are shown as a percentage of control (pcDNA). Molecularmasses in kDa are indicated on the blots. In A–C, n=4 biologicalreplicates and results are mean±s.d. *P<0.05, **P<0.01,***P<0.001 (independent sample t-test).
Table 1. Demographic and clinical characteristics of the subjects with Alzheimer’s-disease-related neurofibrillary pathologya
Braak stage Number of subjects per stageSubjects with clinicalAlzheimer’s disease b
Females/Males(% women)
Age at death(years)c Post-mortem delay (hours)d
0 6 5 3/3 (50) 79.2±10.9 18±19.6I 11 1 5/6 (45)II 11 5 6/5 (55)III 6 6 5/1 (83) 82.8±6.8 15.8±16.1IV 7 5 6/1 (86)V 12 12 10/2 (83) 81.3±6.95 7.1±5.0VI 7 7 7/0 (100)aClassification of Alzheimer’s disease-related neurofibrillary pathology according to Braak staging.bClinically diagnosed Alzheimer’s disease according to NINCDS-ADRDA criteria.cThere were no significant differences in the age of death between Braak 0–II, Braak III or IV, and Braak V or VI groups in the Alzheimer’s-disease-related samplecohort. Data are mean±s.d.dThere was a significant difference in the post-mortem delay between Braak 0–II, and Braak V and VI groups (P=0.008). Data are mean±s.d.
2231
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience
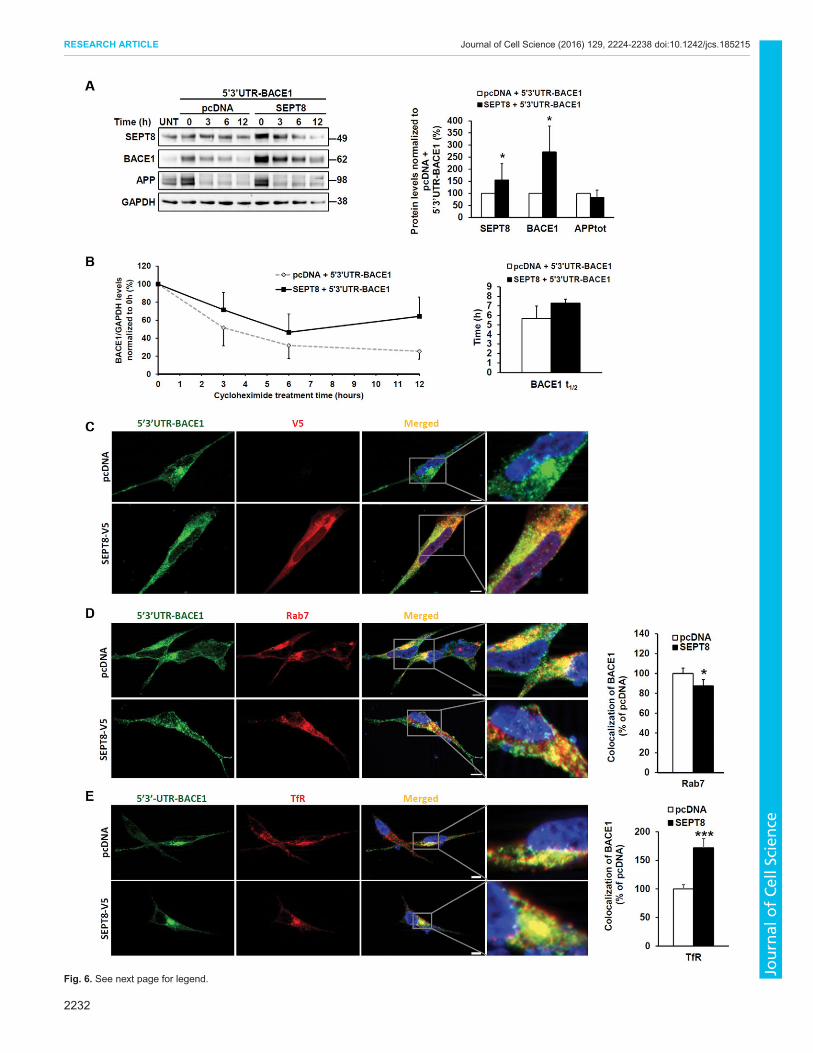
Fig. 6. See next page for legend.
2232
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience

different intracellular compartments, except in the nucleus (Fig. 6C).To validate that the experimental settings in the quantitativeimmunofluorescence microscopy allowed us to detect differencesin the colocalization of two different proteins, we firstdownregulated the well-known BACE1-interacting and sortingprotein Golgi-localized γ-ear-containing ADP-ribosylation factor-binding protein 3 (GGA3) (Tesco et al., 2007) by using siRNA in theSH-SY5Y-APP751 cells transiently overexpressing 5′3′UTR-BACE1 (Fig. S4A). We detected a significantly decreasedcolocalization of BACE1 with GGA3 in these SH-SY5Y-APP751cells. The GGA3 protein levels were decreased by ∼40% and thecolocalization of GGA3 with BACE1 was similarly decreased by∼40% (Fig. S4B). Co-immunostaining of BACE1 and markersspecific for different subcellular compartments was next used toexamine BACE1 subcellular localization. BACE1 localization wasfound to be significantly decreased in the Rab7-positive late-endosomal or lysosomal compartments in cells overexpressingSEPT8–V5 and 5′3′UTR-BACE1 (Fig. 6D). In line with this,colocalization of BACE1with transferrin receptor (TfR; amarker forrecycling endosomes) (Fig. 6E) was significantly increased in cellsco-overexpressing SEPT8–V5 and BACE1 (Das and Pellett, 2011).These findings led us to further study the protein levels of well-
established factors known to regulate endosomal and intracellulartrafficking of BACE1, such as ADP-ribosylation factor 6 (Arf6),GGA1 and GGA3 (He et al., 2005; Sannerud et al., 2011; Tescoet al., 2007). However, no significant changes in the protein levelsof Arf6, GGA1 or GGA3were observed in SH-SY5Y-APP751 cellsco-overexpressing SEPT8 and 5′3′UTR-BACE1 as compared tocells co-transfected with control plasmid and 5′3′UTR-BACE1(data not shown). In addition, overexpression of SEPT8 in SH-SY5Y-APP751 cells did not affect the Lys48- or Lys63-linkedubiquitylation of BACE1 (data not shown). Collectively,cycloheximide timecourse and co-immunostaining data suggestthat the overexpression of SEPT8 TV3 promotes the sorting andaccumulation of BACE1 to the recycling or early endosomalcompartments in SH-SY5Y-APP751 neuroblastoma cells.
DISCUSSIONSeptins are a conserved family of GTP-binding proteins, which arehighly expressed in the brain and known to take part in the
formation, growth and stability of axons and dendrites, synapticplasticity and vesicular trafficking (Marttinen et al., 2015). Beyondthese physiological functions, septins have been linked to differentneurodegenerative and psychiatric disorders, such as Alzheimer’sdisease, Parkinson’s disease, Huntington’s disease, frontotemporaldementia, Down syndrome and schizophrenia (Ageta-Ishihara et al.,2013; Barr et al., 2004; Dong et al., 2003; Gozal et al., 2011; Iharaet al., 2007; Kinoshita et al., 1998; Pissuti Damalio et al., 2012;Zhang et al., 2000).
Based on our initial RNAi screening of SEPT5, SEPT7, SEPT8and SEPT10, we found that SEPT8 in particular affected the β-amyloidogenic processing of APP. More specifically, we found thatthe downregulation of SEPT8 decreased the levels of sAPPβ andAβ in conjunction with reduced protein levels of BACE1 through apotential molecular mechanism involving regulation of sorting andaccumulation of BACE1 protein in neuroblastoma cells. Consistentwith this, endogenous BACE1 protein, but not mRNA levels, aswell as Aβ levels, were significantly downregulated in culturedprimary hippocampal neurons upon lentivirus-mediateddownregulation of SEPT8. However, it should be emphasizedthat the total APP levels were also significantly decreased in thehippocampal neurons, possibly also contributing to the reduced Aβlevels. Although the total APP-normalized Aβ40 levels were stillsignificantly reduced in the primary hippocampal neurons, wecannot completely rule out the possibility that some othermechanism beyond BACE1 might underlie the altered APP- andAβ-related changes. In general, the effects of SEPT8downregulation on the endogenous levels of BACE1 were moresubstantial in the primary hippocampal neurons as compared toneuroblastoma cells. In contrast, overexpression of theAlzheimer’s-disease-associated SEPT8 transcript variant 3(SEPT8 TV3) resulted in an opposite effect to that ofdownregulation, indicated by augmented levels of Aβ andBACE1 protein in neuroblastoma cells. These changes coincidedwith a moderately increased half-life and the alteration of thesubcellular localization of BACE1 from late-endosomal orlysosomal compartments to early and/or recycling endosomes.Previously, a similar change of BACE1 trafficking from late-endosomal or lysosomal compartments to early and recyclingendosomes has been shown to coincide with the stabilization ofBACE1 after depletion of the BACE1-sorting protein GGA3 (Tescoet al., 2007). Thus, our biochemical and immunohistochemicalfindings together suggest that overexpression of SEPT8 TV3promotes sorting and accumulation of BACE1 to the recycling orearly endosomal compartments, which are optimal sites for itsenzymatic activity (Tesco et al., 2007). These changes were notassociated with altered protein levels of factors affecting theendosomal or intracellular trafficking of BACE1, such as Arf6,GGA1 or GGA3, or with alterations in the Lys48- or Lys63-linkedubiquitylation known to regulate the lysosomal and proteosomaltrafficking of BACE1 (Kang et al., 2012; Walker et al., 2012).Importantly, our results are in line with recent studies that haveshown that BACE1 mainly accumulates in the TfR-positiveendosomes and is degraded in the lysosomes (Kandalepas et al.,2013). Collectively, these results suggest SEPT8 is a potential newsorting protein for BACE1 and thus underline its potential role inthe early pathogenesis of Alzheimer’s disease (Fig. 7). However, itshould be noted that the other identified septin family membersbeyond SEPT8 are also potential targets that might affect APPprocessing and the generation of Aβ. It is important to performsimilar detailed molecular characterizations of these septin familymembers as with SEPT8 in the future.
Fig. 6. Overexpression of SEPT8 TV3 stabilizes BACE1 protein levels byincreasing the half-life of BACE1 and enhances the sorting of BACE1 tothe recycling and early endosomal compartments in SH-SY5Y-APP751cells. (A) Western blot analysis of SH-SY5Y-APP751 cells co-transfected withuntagged SEPT8 TV3 and 5′3′UTR-BACE1 and treated with 30 µg/mlcycloheximide for 0, 3, 6 and 12 h. The levels of BACE1 were alreadysignificantly increased at the 0 h time-point in SEPT8-TV3-overexpressingcells as compared to control cells (pcDNA). Total APP (APPtot) was used as acontrol. UNT, untreated SH-SY5Y-APP751 cells. Molecular masses in kDa areindicated on the blots. (B) Quantification of BACE1 protein levels at differenttime-points after cycloheximide treatment suggested that the overexpression ofSEPT8 TV3 reduced the degradation of BACE1. BACE1 protein levels arenormalized to the BACE1:GAPDH ratio at the 0 h time-point. In A and B, n=4biological replicates and results are mean±s.d. (C) BACE1 (green) displayspartial colocalization (yellow) with SEPT8-TV3–V5 (red). SEPT8-TV3–V5localizes in different cellular compartments throughout the cell, except in thenucleus. (D) Overexpression of SEPT8-TV3–V5 (n=69 individual cells)decreases BACE1 (green) colocalization with Rab7 (red), a marker of lateendosomes and lysosomes, as compared to control cells (pcDNA, n=45individual cells). Results are mean±s.e.m. (E) Overexpression of SEPT8-TV3–V5 (n=13 individual cells) significantly increases BACE1 (green) colocalizationwith the recycling and early endosomal marker TfR (red) as compared tocontrol cells (pcDNA, n=21 individual cells). Results are mean±s.e.m. Scalebars: 5 μm. *P<0.05, ***P<0.001 (Mann–Whitney U-test).
2233
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience

As further support for the suggested role of SEPT8 in the earlystages of Alzheimer’s-disease-associated pathophysiology, weidentified transcript-variant-specific alterations in the expressionprofile of SEPT8 mRNA that could be related to the Alzheimer’s-disease-related neurofibrillary pathology in the human temporalcortex. In these assessments, we used a well-established brain tissuesample set, which has been previously used to assess the expressionand splicing status of Alzheimer’s-disease-associated risk genes aswell as α-, β- and γ-secretase activity in the human temporal cortex(Laitera et al., 2014; Martiskainen et al., 2015). The analysis ofSEPT8-exon-specific microarray probes revealed that although totalSEPT8 mRNA levels were unaltered, the levels of TV1 weresignificantly reduced and those of TV2–TV4 were increasedaccording to the Alzheimer’s-disease-related neurofibrillarypathology. This indicates that the ratio of SEPT8 TV1:TV2–TV4mRNAs is significantly reduced even by the early stages of theAlzheimer’s-disease-related neurofibrillary pathology (Braak stageIII). This is a central discovery given that the transcription of theSEPT8 gene is controlled by multiple promoters (Hall et al., 2005),and it underscores the possibility that the expression of differenttranscript variants of SEPT8 might be adapted upon varyingphysiological and pathophysiological circumstances (Fig. 7). Thereduced transcript variant ratio (TV1 versus TV2–TV4) negativelycorrelated with increased activity of BACE1 in the temporal cortex.This observation is in line with the in vitro cell-based data showingthat the overexpression of the SEPT8 TV3 variant increased BACE1levels and the production of Aβ, whereas the opposite effect wasevident upon downregulation of SEPT8, particularly in mouseprimary hippocampal neurons. Nevertheless, it is important to notethat the observed effects of SEPT8 TV3 on BACE1 might differfrom those of SEPT8 TV2 and TV4 due to structural differences.Thus, the effects of the SEPT8 TV2 and TV4 variants should also beelucidated in subsequent studies. The structural differences at theprotein level between SEPT8 transcript variants are primarilylocated at the α-helical coiled-coil domain at the C-terminus, whichis a site that enables isoform-specific contacts with other interactionpartners (Hall et al., 2005; Souza and Barbosa, 2010). Structuralheterogeneity at the C-terminus raises the question of whetherBACE1 is able to specifically interact directly or through otherscaffold partner(s) with certain SEPT8 transcript variants, such as
TV3, which could consequently facilitate the observed sorting ofBACE1 to the recycling or early endosomal compartments.Although immunofluorescence microscopy in the neuroblastomacells suggested that BACE1 and SEPT8 are closely located, a directinteraction between the two proteins was not observed in the co-immunoprecipitation analysis. According to the STRING database(http://string-db.org/), which allows the assessment of the knownand predicted protein–protein interactions, SEPT8 interacts withother septin protein family members, such as SEPT5. Apart fromthis, interaction of SEPT8 with well-established synaptic proteins(CDK5, PARK2 and STX4) linked to neurodegenerative diseaseswas suggested, indicating that SEPT8 is a central component in theinteractome regulating presynaptic functions.
One of the earliest hallmarks in Alzheimer’s disease is thedysfunction and loss of synapses in the hippocampus and associationcortices, but the detailed molecular mechanisms underlying theseprocesses still remain partially elusive (DeKosky and Scheff, 1990;Scheff et al., 2006). Importantly, the degree of synaptic loss shows aclose correlation with the degree of dementia (Coleman and Yao,2003). It has been previously shown that the protein levels ofdifferent septins are altered in the temporal neocortex of Alzheimer’sdisease patients as compared to non-Alzheimer’s disease subjects(Hanai et al., 2004; Musunuri et al., 2014), suggesting that septinscould represent early markers linked to synaptic dysfunction andsynaptotoxicity. Consistent with this idea, presynaptically enrichedSEPT8 controls the formation of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex andthe subsequent docking of synaptic vesicles to the presynapticmembrane (Ito et al., 2009). Based on this, it is conceivable thatSEPT8 could play a central role at the synaptic level in the earlystages of Alzheimer’s-disease-related synaptotoxicity. It has beenrecently shown that protein affinity purification combined with massspectrometry can be utilized for the identification of fragments of thepresynaptic SNARE complex component synaptosomal-associatedprotein 25 (SNAP25) in the cerebrospinal fluid (Brinkmalm et al.,2014). Indeed, significantly increased levels of SNAP25 fragmentshave been observed in the cerebrospinal fluid of Alzheimer’s diseasepatients even by the very early stages of the disease. This pinpointsSNAP25 as a potential biomarker for synaptic integrity even in theearly stages of Alzheimer’s disease and further suggests that
Fig. 7. The potential mechanism of theeffects of SEPT8 in the sorting andaccumulation of BACE1. In non-neuronaland neuronal cells, downregulation of SEPT8levels results in the decreased levels ofBACE1 and subsequently diminishedproduction of sAPPβ and Aβ. In contrast,increased expression of the SEPT8 TV3variant facilitates sorting of BACE1 to earlyendocytic compartments, where BACE1protein is stabilized leading to increasedBACE1 levels and activity, and in theincreased generation of Aβ. Similarly, inAlzheimer’s disease brain, an imbalance inSEPT8 transcript variant expression [reducedTV1:TV2–TV4 variant ratio (TV1/TV2-4)],which occurs during the progression of thebrain pathology, might lead to increasedBACE1 levels and activity, and consequentlyincreased Aβ generation. Full details are in theDiscussion. EE, early endosome; RE,recycling endosome; L, lysosome; PM,plasma membrane.
2234
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience

presynaptic markers beyond SNAP25 might be plausible tools forthe early diagnosis and the assessment of disease progression.Therefore, it would be important to evaluate in the futurewhether theimbalanced transcript variant expression of SEPT8 in thecerebrospinal fluid could serve as a potential biomarker in subjectswith prodromal Alzheimer’s disease. Finally, in order to validate thebiomarker potential and specificity of the SEPT8-related transcriptvariant changes, the comprehensive elucidation of any role forSEPT8 in other neurodegenerative disorders beyond Alzheimer’sdisease is essential.
MATERIALS AND METHODSsiRNA oligonucleotidesThe following siRNAs (Life Technologies) were used to knockdownthe target sequences: human SEPT8 siRNA_1 ID s230020, sense5′-AGAUUAACGCAGUCAUGAATT-3′ and antisense 5′-UUCAUGA-CUGCGUUAAUCUCT-3′; human SEPT8 siRNA_2 ID s230021, sense5′-AGGUGAACAUUAUUCCCAUTT-3′ and antisense 5′-AUGGGAA-UAAUGUUCACCUTG-3′; human SEPT8 siRNA_3 ID s230022,sense 5′-GGAUCCACGUUUGCCUCUATT-3′ and antisense5′-UAGAGGCAAACGUGGAUCCTT-3′; human SEPT5 siRNA ID s22-4294s, sense 5′-CGAGUGCACUGCUGCCUAUTT-3′ and antisense 5′-AUAGGCAGCAGUGCACUCGGT-3′; human SEPT7 siRNA ID s2741,sense 5′-GAAGGGAGCAUGUAGCUAATT-3′ and antisense 5′-UUAG-CUACAUGCUCCCUUCTT-3′; human SEPT10 siRNA ID s455065′-CCAUGAUUCUCGCAUCCAUTT-3′ and antisense 5′-AUGGAUG-CGAGAAUCAUGGTA-3′; ON-TARGETplus Human GGA3 siRNA 5′-GAGAACAAGAGGCGGACUU-3′ (J-012881-09-005, Dharmacon); hu-man BACE1 siRNA ID s24219 (Ambion Life Technologies); and humanADAM17 siRNA ID s13718 (Ambion Life Technologies). All target siR-NAs were compared to siControl Silencer® Select Negative Control #1siRNA (catalog #4390843, Ambion, Life Technologies).
cDNA constructsThe following plasmids were used to overexpress target proteins: SEPT8transcript variant 3 (TV3) in pCMV6-XL vector (Origene), and a plasmid inwhich SEPT8 TV3 cDNA was C-terminally tagged with V5 (SEPT8-TV3-V5) and subcloned into the pcDNA3.1 vector. BACE1 was overexpressedfrom a plasmid with a C-terminally Myc-tagged BACE1 protein under theCMV promoter, and a 5′3′UTR-BACE1 plasmid containing 5′- and 3′-untranslated regulatory regions. pcDNA3.1 served as a control.
Cell culture, transfections and measurement of cell survivalHuman embryonic kidney cells stably overexpressing alkaline-phosphatase-linked APP (HEK293-AP–APP) were cultured in Dulbecco’s modifiedEagle’s medium (DMEM) containing 10% FBS, 2 mM L-glutamine,100 U/ml penicillin and 100 mg/ml streptomycin and supplemented with50 mg/ml hygromycin B and 0.3 µg/ml puromycin at +37°C in a 5% CO2
cell culture incubator (Lichtenthaler et al., 2003; Viswanathan et al., 2011).A human neuroblastoma SH-SY5Y cell line stably overexpressing thehuman APP751 isoform (SH-SY5Y-APP751) was cultured as previouslydescribed (Sarajarvi et al., 2009; Viswanathan et al., 2011, 2013). Cells weretransfected with 5 nM of each target-specific siRNA, using Lipofectamine(Thermo Fisher Scientific) transfection reagent according to themanufacturer’s protocol. Cell survival levels were determined withpropidium iodide and digitonin from HEK293-AP–APP cells plated on a48-well plate and treated as described previously (Loikkanen et al., 1998;Vepsalainen et al., 2013).
Primary hippocampal cell culturesPrimary hippocampal neuronal cultures were prepared from embryonic day18 JAXC57BL/6J mouse brains. All animal experiments were performedaccording to approved guidelines. Briefly, hippocampi were dissected inice-cold HC-buffer containing 1 mg/ml glucose and 10 mM PBS. Tissuewas pelleted by centrifugation at 800 g for 5 min. During the centrifugationpre-warm 5 ml papain-dissociation buffer [in mg: 10 DL-Cysteine-HCl, 10BSA, 250 glucose and 50 ml PBS (pH 7.4) containing 250 µl papain
(10 mg/ml, Sigma) and 5 µl DNAse I (10 mg/ml, Sigma)]. The hippocampiwere dissociated in 5 ml papain-dissociation solution at +37°C for 5 min.Papain-treated tissue was pelleted by centrifugation at 800 g for 5 min. Aftercareful removal of papain-dissociation solution, 2 ml of trituration medium[in ml: 9.8 Ca2+- and Mg2+-free HBSS, 0.1 100 mM sodium pyruvate, 0.11 M HEPES (pH 7.2) and 10 µl DNase I (10 mg/ml)] was added on thehippocampal tissue. The tissue was triturated using a 1-ml pipet by pipettingup and down 15 times. The indissoluble tissue was left to sink to the bottomof the tube, and the supernatant containing the suspension of singular cellswas moved into a clean tube. Then, 2 ml of trituration medium was added ontop of the tissue pellet and trituration was repeated. Cells were harvested bycentrifugation at 800 g for 5 min and resuspended in 10 ml of feedingmedium containing Neurobasal medium supplemented with 1× B27,0.5 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin.Cells were plated on 12-well-plates at a density of 300,000 cells per well,and incubated at +37°C in 5% CO2 cell culture incubator. One-half of thefeeding medium was changed after 5 days in vitro.
Lentiviral transduction shRNA particlesFor validation of the knockdown efficiency of mouse SEPT8 siRNA inmouse primary hippocampal neurons, short hairpin RNA (shRNA) targetingthe open reading frame of mouse SEPT8 mRNA from Sigma (MissionshRNA self-inactivating replication incompetent clone), TRCN0000112992sequence 5′-CCGGCGCCGAGATTAATGCAGTCATCTCGAGATG-ACTGCATTAATCTCGGCGTTTTTG-3′, was used. MISSION® lenti-viral control short hairpin transduction particles, clone ID SHC002H,non-mammalian short hairpin targeting 5′-CCGGCAACAAGATGAAG-AGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT-3′,was used as a control. The bold letters represent the target sequence of thehairpin structure. The lentiviral particles were introduced to the hippocampalneuron cultures at 20 multiplicity of infection (MOI) at 5 days in vitro (DIV).The samples were analyzed at 20 days after transduction.
RNA extraction and cDNA synthesisTotal RNAwas extracted from siRNA-transfected HEK293-AP-APP or SH-SY5Y-APP751 cells or lentivirus-transduced mouse primary hippocampalneurons using TRI Reagent® (Molecular Research Center Inc.) according tothe manufacturer’s protocol. Equal amounts (500 ng) of total RNA sampleswere subjected to cDNA synthesis utilizing SuperScript III ReverseTranscriptase (Invitrogen).
Real-time quantitative PCR analysisTarget-specific primers for human SEPT8 (5′-GAATCACTGCGACTTC-GTGA-3′ and 5′-CTTCCTCTTGGCCTCGTATG-3′), SEPT5 (5′-ACAA-GCAGTACGTGGGCTTC-3′ and 5′-TCAGCACTGAGCAGCTTCC-3′),SEPT7 (5′-TGGGAAAGTCGACATTAATCAAC-3′ and 5′-CCACTGC-ATCTCCAAATCCT-3′), SEPT10 (5′-CCGTTTGCTGTTGTGGGAAG-3′ and 5′-GCCCATTTCCTCCAGTTTGC-3′), BACE1 (5′-ATGGGTGA-GGTTACCAACCA-3′ and 5′-GACAACGTAGAAGCCCTCCA-3′), GA-PDH (5-GGTCTCCTCTGACTTCAACA-3′ and 5′-GTGAGGGTCTCT-CTCTTCCT-3′), and for mouse SEPT8 (5′-ACTCCCTGAAGTCCCTG-GAT-3′ and 5′ATCTGGACTCCGTTGCTGAC-3′), mouse BACE1 (5′-CAACCAGACCGAGGCACT-3′ and 5′-ATGCTCTTGTCGTAGTTGT-ACTCC-3′) and mouse GAPDH (5′-AACTTTGGCATTGTGGAAGG-3′and 5′-ACACATTGGGGGTAGGAACA-3′) were designed to amplify theregion flanking at least two different exons of the target gene. The cDNAsamples were amplified using SYBR Green Master Mix (AppliedBiosystems) or FastStart Universal SYBR Green Master (ROX, Roche) ina real-time quantitative PCR (qRT-PCR) machine (7500 Fast Real TimePCR System, Applied Biosystems). The standard curve method andcomparative Ct-method were used for analyzing mRNA levels, and eachsample was normalized to the GAPDH level from the same sample.
Soluble APP and Aβ measurementsSoluble sAPPα and sAPPβ were detected using a mouse monoclonalantibody (1:1000; 6E10, Biosite) and a specific in-house rat monoclonalantibody (BAWT; 1:10) generated against amino acids 11–16 of Aβ(a kind gift from Prof. Stefan Lichtenthaler, DZNE, Munich, Germany),
2235
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience

respectively. Total sAPP (sAPPtot=sAPPα+sAPPβ) levels were detectedusing a mouse monoclonal antibody recognizing the N-terminus of APP(1:1000; MAB348, clone 22C11; Millipore). Aβ40 and Aβ42 levels weremeasured from HEK293-AP-APP, SH-SY5Y-APP751, and wild-typemouse primary hippocampal neuron cell culture medium using the humanor rat Aβ40 (294-64701) ELISA Kit (Wako) and the Aβ42 (High-Sensitive;292-64501) ELISA Kit (Wako) according to manufacturer’s protocol asdescribed previously (Natunen et al., 2013b).
Western blot analysisTotal proteins were extracted from HEK293-AP–APP, SH-SY5Y-APP751and mouse primary hippocampal neurons using transmembrane proteinextraction reagent (TPER) buffer (Pierce) containing diluted protease andphosphatase inhibitors (1:100; Thermo Scientific). Protein concentrationswere measured by using a bicinchoninic acid (BCA) assay (Pierce). A totalof 10–50 µg of total protein lysates were separated using 4–12% Bis-Trispolyacrylamide gel electrophoresis (PAGE; Invitrogen) and subsequentlytransferred to Hybond-P polyvinylidene fluoride (PVDF) membrane (GEHealthcare). Unspecific antibody binding was prevented by incubatingPVDF membrane in blocking solution containing 5% non-fat milk in 1×Tris-buffered saline containing 0.05% Tween-20 (TBST) for 1 h at roomtemperature. Primary antibodies were diluted in appropriate dilution in 1×TBST and incubated overnight at +4°C. SEPT8 was recognized by anti-SEPT8 polyclonal antibody (1:2000, 11769-1-AP; ProteinTech Group),BACE1 protein was detected by rabbit monoclonal anti-BACE1 D10E5antibody (1:1000, #5606; Cell Signaling Technology). The mousemonoclonal antibody against the N-terminal of APP (1:1000, MAB348,clone 22C11; Millipore) recognized total sAPP (sAPPtot). The solubleAPPα (sAPPα) N terminus was recognized by a mouse monoclonalantibody (1:1000, 6E10; Biosite). Soluble APPβ (sAPPβ) was detected by aspecific rat monoclonal antibody (BAWT, see above). Rabbit polyclonalantibody recognizing the APP C-terminus (1:2000, A8717; Sigma)identified immature APP (APPim), mature APP (APPm) and APP C-terminal fragments (CTFs; C83 and C99). Anti-Golgi-associated, γ-adaptinear containing, ARF binding protein 3 (GGA3) was detected using a mousemonoclonal antibody (1:2500, 612310; BD Transduction Laboratories).Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 6C5) wasrecognized by a mouse monoclonal antibody (1:15,000, ab8245; Abcam).Anti-γ-tubulin rabbit polyclonal antibody (1:1000, ab11317; Abcam) andanti-transferrin receptor mouse monoclonal antibody (1:1000, 13-6800;Zymed) were used for immunoblotting. Secondary anti-mouse-IgG or anti-rabbit-IgG antibodies, conjugated to horseradish peroxidase (HRP), werediluted in 1× TBST. Enhanced chemiluminescence (ECL) substrate (GEHealthcare) was evenly applied on the top of the membrane andsubsequently protein bands were detected with an ImageQuant RT ECLcamera (GE Healthcare) and intensities quantified using Quantity One (Bio-Rad) software.
Cycloheximide timecourseSH-SY5Y-APP751 cells were reverse-transfected using Lipofectaminereagent (Invitrogen) with control plasmid and 5′3′UTR-BACE1, or SEPT8and 5′3′UTR-BACE1. Transfected cells were split into four wells in foursix-well-plates and were assessed at the 0, 3, 6, 12 h time-points for thecycloheximide timecourse. Cycloheximide was added to transfected cells at30 µg/ml. A total of 30 µg protein from each time-point was used for westernblotting. The BACE1 levels at each time-point were normalized to theBACE1 level at 0 h and blotted against the appropriate cycloheximidetreatment to obtain the half-life of BACE1. The half-life of BACE1 wascalculated by plotting intensity values (normalized to the BACE1:GAPDHvalue at 0 h) with respect to chase time. Polynomial fitting was subsequentlyexecuted to obtain half-life calculations.
Biotinylation of cell surface proteinsSH-SY5Y-APP751 cells were rinsed twice with PBS supplemented with0.01 mM CaCl2 and 1 mM MgCl2 (PBS-Ca-Mg), and pre-incubated withfresh PBS-Ca-Mg for 15 min at 4°C. Sulfo-NHS-LC-Biotin (EZ Link™,Pierce) in PBS-Ca-Mg was added to the cells and incubated on a shakerfor 30 min at 4°C. Excess biotin was quenched by incubating cells in
PBS-Ca-Mg supplemented with 0.1 mM glycine for 20 min. Cells werewashed twice with PBS-Ca-Mg. The cells were scraped in T-PERcontaining protease inhibitors and centrifuged at 10,000 g for 10 min at4°C. Protein concentration was measured from the supernatant and 450 µgof proteins were mixed with binding buffer (PBS and 1%Nonident P40) andincubated with agarose beads cross-linked with streptavidin (Pierce)overnight. The samples were centrifuged at 5000 g for 1 min to obtain apellet containing the biotinylated protein fraction and supernatantcontaining the unbiotinylated protein fraction for western blotting.
Immunofluorescence microscopy and quantitative proteincolocalization analysisCells were washed twice in PBS, fixed in 4% paraformaldehyde (PFA) inPBS at room temperature for 15 min, and permeabilized in PBS containing0.1% Triton X-100 for 10 min. Unspecific antibody binding was preventedby incubation in blocking solution containing 1.5% (w/v) goat IgG (Zymed)in PBS at room temperature for 30 min. Cells were incubated with thefollowing primary antibodies for 1.5 h at room temperature: anti-V5 mousemonoclonal antibody (R960-25, Life Technologies, 1:200; recognizing theV5 tag in SEPT8), rabbit anti-BACE1 (D10E5, 5606, Cell Signaling,1:250), mouse anti-Rab7 (B-3, sc-376362, Santa Cruz Biotechnology, 1:50;a marker for late endosomes and lysosomes), anti-TfR monoclonal mouseantibody (13-6800, Zymed, 1:200; a marker of early and recyclingendosomes), and mouse monoclonal anti-GGA3 antibody (612310; BDTransduction Laboratories, 1:50). This was followed by a 1-h incubation atroom temperature with secondary Alexa-Fluor-488-conjugated goat anti-rabbit-IgG and Alexa-Fluor-594-conjugated goat anti-mouse-IgG (both at1:500 dilution; Molecular Probes) antibodies. Cell imaging was performedusing a Zeiss Axio Imager microscope connected to ApoTome.2 at 63×magnification and images were prepared using the Zeiss ZEN 2012program. Colocalization of 5′3′UTR-BACE1 and subcellular markerproteins in pcDNA- (control) or SEPT8-V5-transfected cells wasquantified by assessing the number of colocalizing pixels using ZeissZEN Module Colocalization analysis hardware according to instructions onthe Zeiss web page (http://www.ecu.edu/csdhs/bsomresearchgradstudies/CoreImagingCenter/upload/Colocalization_AIM_ZEN.pdf). Briefly, aregion of interest (ROI) was manually drawn to include each individualanalyzed cell. Every pixel within the ROI was plotted in a scatter diagrambased on its intensity level from each channel. From the scatter diagram, thenumber of colocalizing pixels was quantified in the ZEN ModuleColocalization analysis tool. The values from individual cells wereaveraged and the data are shown as mean±s.e.m. number of colocalizingpixels. Numbers of analyzed cells for each transfection and staining areshown in the figure legends.
Microarray-based global exon expression and splicing screeningof human SEPT8 gene in human brainNeuropathological sample cohortHuman post-mortem brain samples from the temporal lobe were obtainedfromKuopio University Hospital, Kuopio, Finland. The study was approvedby the Ethical Committee of the Kuopio University Hospital. Informedconsent was obtained for all tissue donors and all clinical investigations wereconducted according to the principles expressed in the Declaration ofHelsinki. All subjects (n=60) were investigated at the memory clinic andlater autopsied and evaluated for Alzheimer’s disease-related neurofibrillarypathology (18 males and 42 females; mean±s.d. age 81±9.0 years).Altogether, 41 subjects out of 60 were diagnosed as having clinicalAlzheimer’s disease according to the National Institute of Neurological andCommunicative Disorders and Stroke and the Alzheimer’s disease andRelated Disorders Association (NINCDS-ADRDA) criteria (Table 1)(McKhann et al., 1984). Neurofibrillary pathology of human post-mortembrain temporal cortex samples were assessed with immunostaining ofparaffin sections with AT8 antibody, which detects hyperphosphorylatedtau (Braak and Braak, 1991). The brain samples were subdivided into threegroups based on the degree of the Alzheimer’s-disease-relatedneurofibrillary pathology [Braak stages 0–II (n=28), Braak stages III orIV (n=13), and Braak stages V or VI (n=19)], in which increasedimmunostaining of hyperphosphorylated tau by AT8 antibody denotes more
2236
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience
http://www.ecu.edu/csdhs/bsomresearchgradstudies/CoreImagingCenter/upload/Colocalization_AIM_ZEN.pdf

severe Alzheimer’s disease pathology according to Braak staging (Table 1)(Braak et al., 2006; Natunen et al., 2013a).
The variation of SEPT8 gene exon expression in the inferior temporalcortex was investigated using microarray-based global expression andsplicing screening (Affymetrix Exon Array; Agilent). Briefly, 100 ng oftotal RNA was amplified and labeled using a Low Input Quick Amp WTLabeling kit (5190-2943, Agilent). Samples were processed with the RNASpike-in kit (5188-5282, Agilent). Concentration of RNA, and amplifiedand Cy3-labeled cDNA were measured and quality confirmed beforehybridization of 600 ng of the Cy3-labeled samples onto the Agilent 8×60Kcustom exon chip overnight at +65°C using the Gene ExpressionHybridization Kit (5188-5242, Agilent). Next, samples were washedusing the Gene Expression Wash Pack (5188-5327, Agilent) and the chipswere scanned using an Agilent Technologies Scanner, model G2565CA.Numeric data was produced by Feature Extraction software (version 10.7.3,Agilent).
Statistical analysesIBM SPSS version 21 was used to analyze the data. Statistical comparisonsof results obtained in experiments were performed by independent samplet-test or Mann–Whitney U-test for two-group comparisons. A comparisonof three or more groups was performed using one-way ANOVA followedby a Fisher’s least significant difference (LSD) post-hoc test. Results areexpressed as mean±s.d. when the number of biological replicates wasn≤6 and otherwise as mean±s.e.m. P<0.05 was considered statisticallysignificant.
Competing interestsThe authors declare no competing or financial interests.
Author contributionsK.M.A.K. and M.M. carried out the study designing and performed the majority of thelaboratory experiments, participated in the data analysis and interpretation of dataand wrote the manuscript. L.T. and F.R.L. analyzed data. T.N., P.M., F.H., T.S. andS.G. participated in the biochemical experiments and data analysis. M.M., M.H.,M.K. and J.P. performed bioinformatics data analysis. A.M.K., T.R., V.L. andH.S. collected and analyzed human brain samples. A.H. and M.H. contributedequally to the work, conceived of the study, participated in study design and analysisof the data, and wrote the manuscript. All authors read and approved the finalmanuscript.
FundingThis study was funded by the Suomen Akatemia (Academy of Finland); KuopionYliopistollinen Sairaala (Kuopio University Hospital) [VTR grant V16001]; SigridJuseliuksen Saatio (Sigrid Juselius Foundation); the Ita-Suomen Yliopisto (StrategicFunding of the University of Eastern Finland) [UEF-Brain]; VPH Dementia ResearchEnabled by IT VPH-DARE@IT of the Seventh Framework Programme [grantnumber 601055]; and the EADB project in the JPND-CO-FUND program of theEuropean Commission [grant number 301220].
Supplementary informationSupplementary information available online athttp://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.185215/-/DC1
ReferencesAgeta-Ishihara, N., Yamakado, H., Morita, T., Hattori, S., Takao, K., Miyakawa,T., Takahashi, R. and Kinoshita, M. (2013). Chronic overload of SEPT4, a parkinsubstrate that aggregates in Parkinson’s disease, causes behavioral alterationsbut not neurodegeneration in mice. Mol. Brain 6, 35.
Amin, N. D., Zheng, Y.-L., Kesavapany, S., Kanungo, J., Guszczynski, T., Sihag,R. K., Rudrabhatla, P., Albers, W., Grant, P. and Pant, H. C. (2008). Cyclin-dependent kinase 5 phosphorylation of human septin SEPT5 (hCDCrel-1)modulates exocytosis. J. Neurosci. 28, 3631-3643.
Barr, A. M., Young, C. E., Sawada, K., Trimble, W. S., Phillips, A. G. and Honer,W. G. (2004). Abnormalities of presynaptic protein CDCrel-1 in striatum of ratsreared in social isolation: relevance to neural connectivity in schizophrenia.Eur. J. Neurosci. 20, 303-307.
Benilova, I., Gallardo, R., Ungureanu, A.-A., Castillo Cano, V., Snellinx, A.,Ramakers, M., Bartic, C., Rousseau, F., Schymkowitz, J. and De Strooper, B.(2014). The Alzheimer disease protective mutation A2T modulates kinetic andthermodynamic properties of amyloid-beta (abeta) aggregation. J. Biol. Chem.289, 30977-30989.
Braak, H. and Braak, E. (1991). Neuropathological stageing of alzheimer-relatedchanges. Acta Neuropathol. 82, 239-259.
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H. andDel Tredici, K. (2006).Staging of alzheimer disease-associated neurofibrillary pathology using paraffinsections and immunocytochemistry. Acta Neuropathol. 112, 389-404.
Brinkmalm, A., Brinkmalm, G., Honer, W. G., Frolich, L., Hausner, L., Minthon,L., Hansson, O., Wallin, A., Zetterberg, H., Blennow, K. et al. (2014). SNAP-25is a promising novel cerebrospinal fluid biomarker for synapse degeneration inAlzheimer’s disease. Mol. Neurodegener. 9, 53.
Cole, S. L. and Vassar, R. (2007). The Alzheimer’s disease beta-secretaseenzyme, BACE1. Mol. Neurodegener 2, 22.
Coleman, P. D. and Yao, P. J. (2003). Synaptic slaughter in Alzheimer’s disease.Neurobiol. Aging 24, 1023-1027.
Cummings, J. L. (2004). Alzheimer’s disease. N. Engl. J. Med. 351, 56-67.Das, S. and Pellett, P. E. (2011). Spatial relationships between markers for
secretory and endosomal machinery in human cytomegalovirus-infected cellsversus those in uninfected cells. J. Virol. 85, 5864-5879.
Das, U., Scott, D. A., Ganguly, A., Koo, E. H., Tang, Y. and Roy, S. (2013).Activity-induced convergence of APP and BACE-1 in acidic microdomains via anendocytosis-dependent pathway. Neuron 79, 447-460.
Das, U., Wang, L., Ganguly, A., Saikia, J. M., Wagner, S. L., Koo, E. H. and Roy,S. (2016). Visualizing APP and BACE-1 approximation in neurons yields insightinto the amyloidogenic pathway. Nat. Neurosci. 19, 55-64.
DeKosky, S. T. and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies inAlzheimer’s disease: correlation with cognitive severity.Ann. Neurol. 27, 457-464.
Di Fede, G., Catania, M., Morbin, M., Rossi, G., Suardi, S., Mazzoleni, G., Merlin,M., Giovagnoli, A. R., Prioni, S., Erbetta, A. et al. (2009). A recessivemutation inthe APP gene with dominant-negative effect on amyloidogenesis. Science 323,1473-1477.
Dong, Z., Ferger, B., Paterna, J.-C., Vogel, D., Furler, S., Osinde, M., Feldon, J.and Bueler, H. (2003). Dopamine-dependent neurodegeneration in rats inducedby viral vector-mediated overexpression of the parkin target protein, CDCrel-1.Proc. Natl. Acad. Sci. USA 100, 12438-12443.
Fol, R., Braudeau, J., Ludewig, S., Abel, T., Weyer, S. W., Roederer, J.-P., Brod,F., Audrain, M., Bemelmans, A.-P., Buchholz, C. J. et al. (2016). Viral genetransfer of APPsalpha rescues synaptic failure in an Alzheimer’s disease mousemodel. Acta Neuropathol. 131, 247-266.
Gozal, Y. M., Seyfried, N. T., Gearing, M., Glass, J. D., Heilman, C. J., Wuu, J.,Duong, D. M., Cheng, D., Xia, Q., Rees, H. D. et al. (2011). Aberrant septin 11 isassociated with sporadic frontotemporal lobar degeneration. Mol. Neurodegener.6, 82.
Hall, P. A. and Russell, S. E. H. (2012). Mammalian septins: dynamic heteromerswith roles in cellular morphogenesis and compartmentalization. J. Pathol. 226,287-299.
Hall, P. A., Jung, K., Hillan, K. J. and Russell, S. E. H. (2005). Expression profilingthe human septin gene family. J. Pathol. 206, 269-278.
Hanai, N., Nagata, K.-I., Kawajiri, A., Shiromizu, T., Saitoh, N., Hasegawa, Y.,Murakami, S. and Inagaki, M. (2004). Biochemical and cell biologicalcharacterization of a mammalian septin, Sept11. FEBS Lett. 568, 83-88.
He, X., Li, F., Chang, W.-P. and Tang, J. (2005). GGA proteins mediate therecycling pathway of memapsin 2 (BACE). J. Biol. Chem. 280, 11696-11703.
Hu, X., Li, X., Zhao, M., Gottesdiener, A., Luo, W. and Paul, S. (2014). Taupathogenesis is promoted by Abeta1-42 but not Abeta1-40. Mol. Neurodegener.9, 52.
Ihara, M., Yamasaki, N., Hagiwara, A., Tanigaki, A., Kitano, A., Hikawa, R.,Tomimoto, H., Noda, M., Takanashi, M., Mori, H. et al. (2007). Sept4, acomponent of presynaptic scaffold and lewy bodies, is required for thesuppression of alpha-synuclein neurotoxicity. Neuron 53, 519-533.
Ito, H., Atsuzawa, K., Morishita, R., Usuda, N., Sudo, K., Iwamoto, I., Mizutani,K., Katoh-Semba, R., Nozawa, Y., Asano, T. et al. (2009). Sept8 controls thebinding of vesicle-associated membrane protein 2 to synaptophysin.J. Neurochem. 108, 867-880.
Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson,S., Stefansson, H., Sulem, P., Gudbjartsson, D., Maloney, J. et al. (2012). Amutation in APP protects against Alzheimer’s disease and age-related cognitivedecline. Nature 488, 96-99.
Kandalepas, P. C., Sadleir, K. R., Eimer, W. A., Zhao, J., Nicholson, D. A. andVassar, R. (2013). The Alzheimer’s beta-secretase BACE1 localizes to normalpresynaptic terminals and to dystrophic presynaptic terminals surroundingamyloid plaques. Acta Neuropathol. 126, 329-352.
Kang, E. L., Biscaro, B., Piazza, F. and Tesco, G. (2012). BACE1 proteinendocytosis and trafficking are differentially regulated by ubiquitination at lysine501 and the di-leucine motif in the carboxyl terminus. J. Biol. Chem. 287,42867-42880.
Kinoshita, A., Kinoshita, M., Akiyama, H., Tomimoto, H., Akiguchi, I., Kumar, S.,Noda, M. and Kimura, J. (1998). Identification of septins in neurofibrillary tanglesin Alzheimer’s disease. Am. J. Pathol. 153, 1551-1560.
Kuperstein, I., Broersen, K., Benilova, I., Rozenski, J., Jonckheere, W.,Debulpaep, M., Vandersteen, A., Segers-Nolten, I., Van Der Werf, K.,Subramaniam, V. et al. (2010). Neurotoxicity of Alzheimer’s disease abeta
2237
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience

peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J.29, 3408-3420.
Laitera, T., Sarajarvi, T., Haapasalo, A., Puli, L., Kauppinen, T., Makinen, P.,Rauramaa, T., Tanila, H., Jaaskelainen, J. E., Alafuzoff, I. et al. (2014).Increased gamma-secretase activity in idiopathic normal pressure hydrocephaluspatients with beta-amyloid pathology. PLoS ONE 9, e93717.
Lichtenthaler, S. F., Dominguez, D.-I., Westmeyer, G. G., Reiss, K., Haass, C.,Saftig, P., De Strooper, B. and Seed, B. (2003). The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1.J. Biol. Chem. 278, 48713-48719.
Loikkanen, J. J., Naarala, J. and Savolainen, K. M. (1998). Modification ofglutamate-induced oxidative stress by lead: the role of extracellular calcium. FreeRadic. Biol. Med. 24, 377-384.
Mandelkow, E.-M. and Mandelkow, E. (1998). Tau in Alzheimer’s disease. TrendsCell Biol. 8, 425-427.
Martiskainen, H., Helisalmi, S., Viswanathan, J., Kurki, M., Hall, A., Herukka,S. K., Sarajarvi, T., Natunen, T., Kurkinen, K. M., Huovinen, J. et al. (2015).Effects of Alzheimer’s disease-associated risk loci on cerebrospinal fluidbiomarkers and disease progression: a polygenic risk score approach.J. Alzheimers Dis. 43, 565-573.
Marttinen, M., Kurkinen, K. M. A., Soininen, H., Haapasalo, A. and Hiltunen, M.(2015). Synaptic dysfunction and septin protein family members inneurodegenerative diseases. Mol. Neurodegener. 10, 16.
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D. and Stadlan,E. M. (1984). Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of department of health and humanservices task force on Alzheimer’s disease. Neurology 34, 939-944.
Munoz-Soriano, V. and Paricio, N. (2007). Overexpression of septin 4, thedrosophila homologue of human CDCrel-1, is toxic for dopaminergic neurons.Eur. J. Neurosci. 26, 3150-3158.
Musunuri, S., Wetterhall, M., Ingelsson, M., Lannfelt, L., Artemenko, K.,Bergquist, J., Kultima, K. and Shevchenko, G. (2014). Quantification of thebrain proteome in Alzheimer’s disease using multiplexed mass spectrometry.J. Proteome Res. 13, 2056-2068.
Natunen, T., Martiskainen, H., Sarajarvi, T., Helisalmi, S., Pursiheimo, J.-P.,Viswanathan, J., Laitinen, M., Makinen, P., Kauppinen, T., Rauramaa, T. et al.(2013a). Effects of NR1H3 genetic variation on the expression of liver X receptoralpha and the progression of Alzheimer’s disease. PLoS ONE 8, e80700.
Natunen, T., Parrado, A. R., Helisalmi, S., Pursiheimo, J. P., Sarajarvi, T.,Makinen, P., Kurkinen, K. M., Mullin, K., Alafuzoff, I., Haapasalo, A. et al.(2013b). Elucidation of the BACE1 regulating factor GGA3 in Alzheimer’s disease.J. Alzheimers Dis. 37, 217-232.
Neve, R. L., McPhie, D. L. and Chen, Y. (2000). Alzheimer’s disease: a dysfunctionof the amyloid precursor protein(1). Brain Res. 886, 54-66.
Pissuti Damalio, J. C., Garcia, W., Alves Macêdo, J. N., de Almeida Marques, I.,Andreu, J. M., Giraldo, R., Garratt, R. C. and Ulian Araujo, A. P. (2012). Selfassembly of human septin 2 into amyloid filaments. Biochimie 94, 628-636.
Sannerud, R., Declerck, I., Peric, A., Raemaekers, T., Menendez, G., Zhou, L.,Veerle, B., Coen, K., Munck, S., De Strooper, B. et al. (2011). ADP ribosylationfactor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediatingthe endosomal sorting of BACE1. Proc. Natl. Acad. Sci. USA 108, E559-E568.
Sarajarvi, T., Haapasalo, A., Viswanathan, J., Makinen, P., Laitinen, M.,Soininen, H. and Hiltunen, M. (2009). Down-regulation of seladin-1 increasesBACE1 levels and activity through enhanced GGA3 depletion during apoptosis.J. Biol. Chem. 284, 34433-34443.
Sarajarvi, T., Tuusa, J. T., Haapasalo, A., Lackman, J. J., Sormunen, R.,Helisalmi, S., Roehr, J. T., Parrado, A. R., Makinen, P., Bertram, L. et al.(2011). Cysteine 27 variant of the delta-opioid receptor affects amyloid precursorprotein processing through altered endocytic trafficking. Mol. Cell. Biol. 31,2326-2340.
Scheff, S. W., Price, D. A., Schmitt, F. A. and Mufson, E. J. (2006). Hippocampalsynaptic loss in early Alzheimer’s disease and mild cognitive impairment.Neurobiol. Aging 27, 1372-1384.
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith,I., Brett, F. M., Farrell, M. A., Rowan, M. J., Lemere, C. A. et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synapticplasticity and memory. Nat. Med. 14, 837-842.
Souza, T. A. C. B. and Barbosa, J. A. R. G. (2010). Cloning, overexpression,purification and preliminary characterization of human septin 8. Protein J. 29,328-335.
Tesco, G., Koh, Y. H., Kang, E. L., Cameron, A. N., Das, S., Sena-Esteves, M.,Hiltunen, M., Yang, S.-H., Zhong, Z., Shen, Y. et al. (2007). Depletion of GGA3stabilizes BACE and enhances beta-secretase activity. Neuron 54, 721-737.
Vepsalainen, S., Koivisto, H., Pekkarinen, E., Makinen, P., Dobson, G.,McDougall, G. J., Stewart, D., Haapasalo, A., Karjalainen, R. O., Tanila, H.et al. (2013). Anthocyanin-enriched bilberry and blackcurrant extracts modulateamyloid precursor protein processing and alleviate behavioral abnormalities in theAPP/PS1 mouse model of Alzheimer’s disease. J. Nutr. Biochem. 24, 360-370.
Viswanathan, J., Haapasalo, A., Bottcher, C., Miettinen, R., Kurkinen, K. M. A.,Lu, A., Thomas, A., Maynard, C. J., Romano, D., Hyman, B. T. et al. (2011).Alzheimer’s disease-associated ubiquilin-1 regulates presenilin-1 accumulationand aggresome formation. Traffic 12, 330-348.
Viswanathan, J., Haapasalo, A., Kurkinen, K. M. A., Natunen, T., Makinen, P.,Bertram, L., Soininen, H., Tanzi, R. E. and Hiltunen, M. (2013). Ubiquilin-1modulates gamma-secretase-mediated epsilon-site cleavage in neuronal cells.Biochemistry 52, 3899-3912.
Walker, K. R., Kang, E. L., Whalen, M. J., Shen, Y. and Tesco, G. (2012).Depletion of GGA1 and GGA3 mediates postinjury elevation of BACE1.J. Neurosci. 32, 10423-10437.
Zhang, Y., Gao, J., Chung, K. K. K., Huang, H., Dawson, V. L. and Dawson, T. M.(2000). Parkin functions as an E2-dependent ubiquitin- protein ligase andpromotes the degradation of the synaptic vesicle-associated protein, CDCrel-1.Proc. Natl. Acad. Sci. USA 97, 13354-13359.
2238
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 2224-2238 doi:10.1242/jcs.185215
Journal
ofCe
llScience


















