Report complete vFS18 - chem.uzh.ch · to α-substituted derivatives of the type 2. Depending on...
7
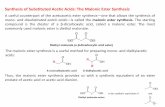
Report N° 3, p. 1 Laboratory Report CHE212, Project 23 by Peter Muster bench no. 43 supervised by Stefan Bienz February 21 th , 2018 Stereoselective α -Bromination of an Evans Amide Summary The Evans amide of a β-silylated propionic acid derivative was brominated in α-position to the carboxyl group by conversion to its enol boronate, followed by reaction with NBS as the bromine source. The reaction delivered 80% of a 4:1 mixture of two diastereoisomers, which was readily separated by chromatography. Starting with (R)-configured Evans auxiliary, the reaction delivered as the major isomer the product with (S)-configuration at the newly formed stereogenic center. 1. Introduction Organic molecules of the same constitution but differing in the three-dimensional arrangement of groups and/or substituents are called stereoisomers. Depending upon the number of stereogenic units contained in such molecules, the number of stereoisomers can be rather high (2 n with n = number of stereogenic units). Since stereoisomers are distinct compounds, possessing indivi- dual properties, it is important to have synthetic control over the spatial arrangement of groups within molecules. Such control is given, e.g., with stereoselective reactions. A very famous moiety to effect stereoselectivity is the Evans auxiliary A (Scheme 1). [1] It is a carbamate group that can be attached to carboxylic acids. The respective imides of the type 1 can be used as starting materials for a number of reactions in which new stereogenic units are formed in a controlled manner. For instance, enolization of Evans amide 1 delivers an enolate species that reacts with electrophiles E + to α-substituted derivatives of the type 2. Depending on the exact reaction conditions, either diastereoisomer 2a or 2b is formed predominantly. [1,2] The two diastereoisomers can usually be separated without special effort.
Transcript of Report complete vFS18 - chem.uzh.ch · to α-substituted derivatives of the type 2. Depending on...

Report N° 3, p. 1
Laboratory Report
CHE212, Project 23
by Peter Muster bench no. 43
supervised by Stefan Bienz
February 21th, 2018
Stereoselective α-Bromination of an Evans Amide
Summary
The Evans amide of a β-silylated propionic acid derivative was brominated in α-position to the carboxyl group by conversion to its enol boronate, followed by reaction with NBS as the bromine source. The reaction delivered 80% of a 4:1 mixture of two diastereoisomers, which was readily separated by chromatography. Starting with (R)-configured Evans auxiliary, the reaction delivered as the major isomer the product with (S)-configuration at the newly formed stereogenic center.
1. Introduction
Organic molecules of the same constitution but differing in the three-dimensional arrangement of groups and/or substituents are called stereoisomers. Depending upon the number of stereogenic units contained in such molecules, the number of stereoisomers can be rather high (2n with n = number of stereogenic units). Since stereoisomers are distinct compounds, possessing indivi-dual properties, it is important to have synthetic control over the spatial arrangement of groups within molecules. Such control is given, e.g., with stereoselective reactions. A very famous moiety to effect stereoselectivity is the Evans auxiliary A (Scheme 1).[1] It is a carbamate group that can be attached to carboxylic acids. The respective imides of the type 1 can be used as starting materials for a number of reactions in which new stereogenic units are formed in a controlled manner. For instance, enolization of Evans amide 1 delivers an enolate species that reacts with electrophiles E+ to α-substituted derivatives of the type 2. Depending on the exact reaction conditions, either diastereoisomer 2a or 2b is formed predominantly.[1,2] The two diastereoisomers can usually be separated without special effort.

Report N° 3, p. 2
Scheme 1. General scheme for the stereoselective α-derivatization of carboxylic acids via the Evans amide.
Since the Evans auxiliary is synthesized in enantiomerically pure form from phenylalanine, enantio-merically enriched products can be accessed. Removal of the auxiliary from 2a and 2b delivers products 3a and 3b, which are mirror images of each other (enantiomers).
We report below upon the α-bromination of Evans substrate 4 (Scheme 2), which was intended to be used as an intermediate for the synthesis of optically active silicon-containing amino acids.
2. Results and Discussion
Starting material 4 was obtained by a procedure reported by Smith et al.[3] and was directly treated with Bu2BOTf (dibutylboryl trifluoromethane sulfonate) in CH2Cl2 at −78 °C to effect formation of boron enolate 5 (Scheme 2). According to literature, this reaction should be at least 99% stereo-selective for the Z(O)-enolate, for which the boron atom is chelated to the oxazolidine carboxyl O-atom as shown. Due to the rather rigid structure of 5, where the top face of the molecule is steri-cally hindered by the benzyl substituent (Bn) of the oxazolidine moiety, electro-
Scheme 2. Reaction scheme for the stereoselective α-bromination of an Evans amide.
O
N O
O
Bn
1
Me
O
N O
O
Bn
Me
E
O
N O
O
Bn
Me
E
O
OHMe
E
O
OHMe
E
2a 3a
2b 3b
A
O
N O
O
Bn
4
SiClMe Me
EtN(i-Pr)2Bu2BOTf,
CH2Cl2, –78 °C
O
N O
O
Bn
5
SiClMe Me
NBS, CH2Cl2, –78 °C
81%
O
N O
O
Bn
6a (16%)
SiClMe MeBu2
B
O
N O
O
Bn
6b (65%)
SiClMe Me
Br
Br

Report N° 3, p. 3
philic attack would be expected to occur preferentially at the bottom face of the enolate. This was in fact the case. Treatment of the enolate 6 with NBS (N-bromosuccinimide) at low temperature delivered two products that were readily separated by column chromatography and turned out to be the bromo derivatives 6a (16%) and 6b (65%), formed in a dr (diasteroisomeric ratio) of 1:4 in favor of the expected major product.
The common structural framework of the two compounds was readily verified with the data of the MS, 1H-, and 13C-NMR spectra. The CI-MS reveal at m/z 439/437/435 the signals of the ionized molecules [M + NH4]
+ with the typical isotopic distributions for BrCl derivatives, the 1H-NMR spec-tra show the expected new signals for the α-positioned protons at δ 5.94 and 5.90 ppm, respec-tively, and the corresponding signals for the C nuclei are found at δ 41.5 and 41.6 ppm in the 13C-NMR spectra. Not surprisingly, however, the analytical data of the two diastereoisomeric products 6a and 6b do not differ characteristically, and their spectra do not reveal any diagnostic informa-tion that would allow the assignment of the relative configurations of the stereogenic centers of the compounds. In particular, no significant differences were found for the coupling constants relating to the magnetic interactions of the HC(2’) with the two diastereotopic H2C(3’) that might have given some information about the preferred conformation of the molecules and finally the relative configurations of the two stereogenic centers. For 6a the respective coupling constants were found to be 9.3 and 6.3 Hz, for 6b 9.9 and 6.0 Hz. Luckily, derivative 6b arose as a solid. Careful crystallization of 6b from hexane delivered single crystals that were amenable for single crystal X-ray analysis. This revealed that the relative configuration of the two stereogenic centers of the sample molecules is (R*,S*). Since the absolute configuration at C(4) is known to be (R), the configuration of the stereogenic center at C(2’) has to be (S). The configuration of 6b is thus (4R,2’S) and the configuration of its diastereomeric relative 6a (4R,2’R).
The selectivity of the reaction was not very high. In fact, we were rather disappointed to observe solely a selectivity of 4:1. Since the major diastereoisomer was easily separated from the minor component by chromatography, however, virtually isomerically pure material, which can be used for subsequent transformations, was attained by the procedure.
Description of the next experiment(s) in a prose-type style as above.
.........
3. Conclusion
We have shown that the enolboronate of the propionic Evans substrate 4 can readily be α-bro-minated, that this reaction, however, proceeds with rather poor stereoselectivity. The major isomer is the expected (4R,2’S)-configured product 6b, which can readily be separated by chromatogra-phy from the undesired (4R,2’R)-configured isomer 6a. Thus, the procedure allows to prepare the

Report N° 3, p. 4
α-bromopropionic acid derivative 6b in enantiomerically and diastereoisomerically pure form, albeit in rather low yield.
Experimental Part
1. General. Unless otherwise stated, all chemicals were of reagent grade and purchased from Sigma–Aldrich. Reactions were carried out under N2 in oven-dried (100 °C) glass equipment and monitored for completion by analyzing a small sample (after suitable workup) by TLC, GC, HPLC, or NMR. Solvents for reactions were of p.a. grade or distilled prior to their use; dry CH2Cl2 was ob-tained by distillation over CaH2. Extracts were neutralized, washed with H2O and brine, and dried over anhydrous MgSO4. Evaporation of the solvents in vacuo was done with the rotary evaporator at the given bath temperature and pressure. pH: Merck indicator paper pH 1–14 (universal indi-cator) or Metrohm 713 pH meter equipped with pH sensitive electrode. Chromatography: Merck silica gel 60 (40–63 µm) with the indicated solvent system. Thin layer chromatography (TLC): Merck TLC plates silica gel 60 on aluminum with the indicated solvent system; the spots were visualized by UV light (254 and 366 nm) and exposure to I2 vapor, stains of KMnO4, p-anisalde-hyde, and/or platiniummolybdate. Specific optical rotation [α]D: Perkin–Elmer Polarimeter 241 MC; measured at 23 °C. M.p.: Büchi 510; heating rate 2 °C min−1; range 2/3 to fully molten. Refractive index nD: refractometer of Zeiss or Weso; measured at 23 °C. UV-Vis spectra: Cary Series spectrophotometer (Agilent technologies); λmax (logε) and λmin (logε) in nm. IR spectra: Spec-trumTwo FT-IR Spectrometer (Perkin–Elmer) equipped with a Specac Golden GateTM ATR (attenua-ted total reflection) accessory; applied as neat samples; 1/λ in cm−1. 1H-NMR spectra in CDCl3; Bruker AV-300 (300 MHz); δ in ppm rel. to CHCl3 (δ 7.24; corresponds to TMS (δ 0.00)), J in Hz. 13C-NMR spectra in CDCl3; Bruker AV-300 (75.5 MHz); δ in ppm rel. to CDCl3 (δ 77.0; corresponds to TMS (δ 0.0)); multiplicities from DEPT-135 and DEPT-90 experiments. Gas chromatogra-phy/electron impact ionization-mass spectrometry (GC/EI-MS): Thermo Scientific ISQ GC/MS equipped with a Trace 1300 GC, split/splitless injector at 250 °C; flow rate 1 mL min−1; Thermo Scientific TG-SQC capillary column, 15 m, 0.25 mm i.d., 0.25 µm film thickness; gradient 20 °C min−1 from 60–300 °C then isothermal for another 3 min, Rt in min; EI at 70 eV; single stage quadrupole mass analyzer, mass range 50–600 amu at 2 scans min−1 in full scan mode; in m/z (rel.%) for molecular ions and characteristic fragments (with interpretation) and for all further signals of �5 rel.%. Elemental analysis: LECO Truespec CHNS(O) microanalyzer. High-resolution electrospray ionization mass spectra (HR-ESI-MS): QExactive (Thermo Fisher Scientific, Bremen, Germany) with a heated ESI source connected to a Dionex Ultimate 3000 UHPLC system. Samples dissolved in MeOH or H2O at ca. 50 µg mL−1; injection of 1 µL on-flow with an auto-sampler (mo-bile phase: MeOH + 0.1% HCOOH or CH3CN/H2O 2:8 + 0.1% HCOOH; flow rate 120 µL min−1); ion source parameters: spray voltage 3.0 kV, capillary temperature 320 °C, sheath gas 5 L min−1, s-lens RF level 55.0; full scan MS in alternating (+)/(−)-ESI mode; mass ranges 80–1’200, 133–2’000, or 200–3’000 amu; resolution (full width half-maximum) 70’000; automatic gain control (AGC) target 3.00·106; maximum allowed ion transfer time (IT) 30 ms; mass calibration <2 ppm

Report N° 3, p. 5
accuracy for m/z 130.06619–1621.96509 in (+)-ESI and for m/z 265.14790–1779.96528 in (−)-ESI with Pierce® ESI calibration solutions (Thermo Fisher Scientific, Rockford, USA); lock mas-ses: ubiquitous erucamide (m/z 338.34174, (+)-ESI) and palmitic acid (m/z 255.23295, (−)-ESI).
Insert here a list of abbreviations, but only if unusual (uncommon or self-defined) abbrevia-tions are used in the document.
2. (4R)-4-Benzyl-3-{(2R)-2-bromo-3-[(chloromethyl)(dimethyl)silyl]propanoyl}-1,3-oxazolidin-2-one and (4R)-4-Benzyl-3-{(2S)-2-bromo-3-[(chloromethyl)(dimethyl)silyl]propanoyl}-1,3-oxazolidin-2-one (6a and 6b) (analogous to[3]).
EtN(i-Pr)2 (500 mg, 3.83 mmol) and a soln. of Bu2BOTf (1M in CH2Cl2, 3.85 mL 3.85 mmol) were added subsequently and dropwise at −78 °C (bath of acetone/dry ice) by syringe to a soln. of 4 ((R)-4-benzyl-3-{3-[(chloromethyl)(dimethyl)silyl]propanoyl}-1,3-oxazolidin-2-one, 930 mg, 2.74 mmol[3]) in CH2Cl2 (10 mL) in a 50 mL two-necked flask equipped with a septum. The color of the mixture turned immediately to yellow. The mixture was kept at −78 °C for 4 h, while the color deepened and became almost brown, before it was transferred with a double-tipped needle (insu-lated to maintain the temp.) to a soln. of NBS (650 mg, 3.64 mmol) in CH2Cl2 (20 mL) at −78 °C. When TLC control showed complete consumption of 4, after 1 h, a sat. aq. NaHCO3 soln. (10 mL) was added to the now reddish soln., and the mixture was allowed to warm to 23 °C. Extraction with CH2Cl2 (3 × 20 mL), drying of the combined extracts with MgSO4, and evaporation of the sol-vent in vacuo (40 °C, 850 mbar) delivered a crude product (125 mg), which was chromatographed (25 g SiO2, AcOEt/hexane 5:95) to afford, after evaporation of the solvent in vacuo (40 °C, 200 mbar) and removal of residual solvent at 23 °C and 1 mbar for 3 h, 6a (183 mg, 0.43 mmol, 16%) and 6b (739 mg, 1.76 mmol, 65%); yield for both products 81%. The spectroscopic data of 6a and 6b (IR, 1H-NMR, 13C-NMR) are in agreement with those reported in literature as far as given there;[3] the signal assignments were performed with correlation data.[4]
Data of 6a ((4R,2'R)-configured, colorless oil, estimated by 1H-NMR to be >95% pure, enantio-meric purity not determined but assumed to be retained from the starting material (>99% ee)).
TLC: Rf = 0.67 (AcOEt/hexane 5:1).
Cl Si NMe Me
O
O
O
Bn
Cl Si NMe Me
O
O
O
Bn
Cl Si NMe Me
O
O
O
Bn
NBSCH2Cl2, –78 °C
81%+
6a (16%) 6b (65%)1 : 4
Br Br
4
C16H21BrClNO3Si(418.79)
C16H22ClNO3Si(339.89)
C16H21BrClNO3Si(418.79)

Report N° 3, p. 6
nD23 = 1.0763 (lit.: 1.0781[3]).
b.p.: 122–124 °C (40 mbar).
[α]D23= −36.7 (c = 2.86, CHCl3, lit.: −35.8, c = 0.5, CHCl3[3]).
UV-Vis (MeOH): λmax 200 (4.50), λmin 239 (0.56), λmax 259 (2.20) (in agreement with[3]).
IR (CHCl3): 3613m, 3082w, 3060w, 3028w, 3001w, 2961m, 2922m, 2869w, 1777s (C=O), 1703s (C=O), 1607m (C=C, arom.), 1583m (C=C, arom.), 1494m, 1482w, 1453m, 1445w, 1384s, 1350s, 1289m, 1261s, 1208s, 1201s, 1176m, 1107m, 1073m, 1045m, 1031m, 1014m, 1002w, 971m, 919w, 874m, 847s, 802w, 761m, 750m, 734m, 707m, 643m, 623m, 602m. 1H-NMR: 7.39–7.20 (m, 5 arom. H); 5.94 (dd, J = 9.6, 6.3, BrHC); 4.71–4.62 (m, HCN); 4.28–4.16 (m, H2CO); 3.41 (dd, J = 13.3, 3.4, 1 H of PhCH2); 2.85, 2.83 (AB, J = 13.4, ClCH2); 2.71 (dd, J = 13.3, 10.3, 1 H of PhCH2); 1.98 (dd, J = 14.5, 9.6, 1 H of SiCH2); 1.66 (dd, J = 14.5, 6.3, 1 H of CH2Si); 0.23, 0.22 (2s, 2 MeSi) (in agreement with[3]). 13C-NMR: 169.6 (s, CON); 152.5 (s, COO); 135.0 (s, arom. C); 129.4, 129.1 (2d, 2×2 arom. C); 127.5 (d, arom. C); 66.4 (t, H2CO); 56.1 (d, HCN) 41.5 (d, BrHC); 37.9 (t, PhCH2); 30.0 (t, ClCH2); 21.3 (t, SiCH2); −3.9, −4.1 (2q, 2 MeSi).
CI-MS (NH3): 439/437/435 (30/100/73, [M + NH4]+), 357 (9), 338 (6, [M − Br]+), 249 (30), 195
(9), 164 (28), 147 (34), 115 (9).
HR-ESI-MS (MeOH): 418.02505 (100, C16H22BrClNO3Si+; [M + H]+; calc. 418.02409; Δ = +2.3 ppm).
Anal. calc. for C16H21BrClNO3Si (418.79): C 45.89, H 5.05, N 3.34; found: C 45.97, H 5.13, N 3.31.
Data of 6b ((4R,2'S)-configured, colorless crystals, estimated by 1H-NMR to be >95% pure, enan-tiomeric purity not determined but assumed to be retained from the starting material (>99% ee)). Crystals suitable for single crystal X-ray analysis were obtained by recrystallization of an aliquote from hexane.
TLC: Rf = 0.63 (AcOEt/hexane 5:1).
M.p. 99.2–100.0 °C (hexane) (lit.: 99 °C[3]).
[α]D20= −39.2 (c = 1.35, CHCl3, lit.: −39.5, c = 0.5, CHCl3[3]).
UV-Vis: λmax 200 (4.50), λmin 239 (0.56), λmax 259 (2.20) (in agreement with[3]).
IR (CHCl3): 3614m, 3087w, 3062w, 3029w, 3001w, 2960m, 2922m, 2869w, 1778 (C=O), 1707s (C=O), 1603m (C=C, arom.), 1584m (C=C, arom.), 1497m, 1482w, 1455m, 1446w, 1387s, 1349s, 1288m, 1262s, 1212s, 1201s, 1176m, 1104m, 1073m, 1047m, 1030m, 1017m, 1002w, 971m, 922w, 877m, 844s, 800w, 759m, 751m, 734m, 707m, 643m, 625m, 601m.

Report N° 3, p. 7
1H-NMR: 7.37–7.22 (m, 5 arom. H); 5.90 (dd, J = 9.8, 6.1, BrHC); 4.76–4.67 (m, HCN); 4.27–4.18 (m, H2CO); 3.32 (dd, J = 13.5, 3.4, 1 H of PhCH2); 2.83 (s, ClCH2); 2.79 (dd, J = 13.5, 9.6, 1 H of PhCH2); 1.97 (dd, J = 14.5, 9.9, 1 H of SiCH2); 1.67 (dd, J = 14.4, 6.0, 1 H of SiCH2); 0.21, 0.20 (2s, 2 MeSi) (in agreement with[3]). 13C-NMR: 169.5 (s, CON); 152.4 (s, COO); 134.8 (s, arom. C); 129.4, 129.0 (2d, 2×2 arom. C); 127.4 (d, arom. C); 66.2 (t, H2CO); 55.2 (d, HCN) 41.6 (d, BrHC); 37.0 (t, PhCH2); 30.1 (t, ClCH2); 21.3 (t, SiCH2); −3.9, −4.2 (2q, 2 MeSi).
CI-MS (NH3): 439/437/435 (29/100/73, [M + NH4]+), 359 (6), 357 (17), 338 (9, [M − Br]+), 249
(55), 195 (12), 164 (25), 115 (8).
HR-ESI-MS (MeOH): 418.02484 (100, C16H22BrClNO3Si+; [M + H]+; calc. 418.02409; Δ = +1.8 ppm).
Anal. calc. for C16H21BrClNO3Si (418.79): C 45.89, H 5.05, N 3.34; found: C 45.51, H 4.98, N 3.25.
For single crystal X-ray analysis, see separate report.
3. Next experiment…….
REFERENCES
[1] D. A. Evans, M. D. Ennis, D. J. Mathre, J. Am. Chem. Soc. 1982, 104, 1737–1739. [2] M. A. Walker, C. H. Heathcock, J. Org. Chem. 1991, 56, 5747–5750. [3] R. J. Smith, S. Bienz, Helv. Chim. Acta 2004, 87, 1681–1696. [4] S. Bienz, L. Bigler, T. Fox, H. Meier, Spektroskopische Methoden in der organischen Chemie /
Hesse–Meier–Zeeh, 9. Ed., Thieme, Stuttgart 2016.