
Production of stable quinine nanodispersions using esterified γ-polyglutamic acid biopolymer
8
Biochemical Engineering Journal 79 (2013) 259–266 Contents lists available at ScienceDirect Biochemical Engineering Journal jou rnal h om epage: www.elsevier.com/locate/bej Regular article Production of stable quinine nanodispersions using esterified -polyglutamic acid biopolymer Christoph Hoennscheidt a , Dirk Kreyenschulte a,b , Argyrios Margaritis b , Rainer Krull a,∗ a Institute of Biochemical Engineering, Technische Universität Braunschweig, Germany b Department of Chemical and Biochemical Engineering, University of Western Ontario, London, Canada a r t i c l e i n f o Article history: Received 4 April 2013 Received in revised form 5 July 2013 Accepted 9 August 2013 Available online 31 August 2013 Keywords: -Polyglutamic acid Biopolymer Surfactant, Dispersion Quinine Solvent evaporation a b s t r a c t Novel methods are needed for the development of nanodispersed drug formulations to enhance bioavail- ability of many hydrophobic pharmaceuticals. The poorly water-soluble quinine is a well-known anti-malaria drug which can be used as a promising model compound for the development of novel nanodispersed formulations. In addition to hydrophobic drug’s own affecting properties, surfactants play an important role for the enhancement of their low bioavailability by preparing stable dispersions. Amphiphilic compounds can efficiently be used to stabilize colloidal fragments by preventing the precipi- tation or crystallization of poorly water-soluble active ingredients during fabrication. A novel biopolymer derivative based on the biotechnologically produced -polyglutamic acid (-PGA) from Bacillus licheni- formis cultivation was developed for encapsulation of the active ingredient. High-molecular -PGA is an anionic polyelectrolyte that was optimized and modified with hydrophobic l-phenylalanine ethyl ester (l-PAE) to form an amphiphilic comb polymer P(-GA-r-l-PAE) with surfactive properties. The approach of the nanodispersion polymer concentration, molecular weight and grafting degree enables the efficient stabilization of the poorly water-soluble model drug. The research presented in this report indicates the potential benefits of hydrophobically modified -PGA and suggests its potential role in forming stable dispersions for future pharmaceutical applications. © 2013 Elsevier B.V. All rights reserved. 1. Introduction Due to significant progress in the field of pharmaceutical nano- technology, the development of enhanced bioavailable drugs in nanodispersion reservoirs and nano-sized drug delivery vehicles is extensively investigated [1]. An important prerequisite for med- ical applications is the biocompatibility of the employed materials [2]. Thus, several biodegradable polymers, such as chitosan, algi- nate and -polyglutamic acid, shortened as -PGA, have been introduced for drug encapsulation and delivery [3–7]. -PGA is an anionic, water soluble polyamide consisting of repeated glutamic Abbreviations: CuSO4, copper(II) sulfate; DMSO, dimethyl sulfoxide; DSC, differential scanning calorimetry; EDC, 1-ethyl-3-(3-dimethylaminopropyl) car- bodiimide; DO, dissolved oxygen; EDTA, ethylendiaminetetraacetic acid; GPC, gel permeation chromatography; l-PAE, l-phenylalanine ethyl ester; OD450, optical density (450 nm); MWCO, molecular weight cut off; P(-GA-r-l-PAE), poly(-glutamic acid-random-l-phenylalanine ethyl ester); SEM, scanning electron microscopy; THF, tetrahydrofuran; -PGA, -polyglutamic acid. ∗ Corresponding author at: Institute of Biochemical Engineering, Technische Uni- versität Braunschweig, Gaußstraße 17, 38106 Braunschweig, Germany. Tel.: +49 0531 391 7653; fax: +49 0531 391 7652. E-mail addresses: [email protected], [email protected] (R. Krull). acid units linked between the -amino and -carboxylic acid func- tional groups [8]. This polyamide can generally be produced by cultivation of various Bacillus species, most commonly Bacillus sub- tilis and Bacillus licheniformis [9,10]. A high molar mass of this type of biopolymer is required to function as a surfactant. -PGA is capable of binding certain metal cations through ionic interactions because it is an anionic polymer. As a component of bacterial capsules, this binding or trapping phenomenon serves to provide essential metal cations for enzymatic reactions or struc- tural components of the cell [11]. As a result, several studies have examined the metal binding characteristics of -PGA [12,13]. These studies indicate the addition of metal ions, such as Cu 2+ , Al 3+ , Cr 3+ or Fe 3+ , induced the flocculation of dissolved -PGA [13]. Manocha and Margaritis [14] investigated whether the metal induced -PGA precipitation could be used to separate -PGA from B. licheniformis cultivation broth. Divalent copper ions exhibited the best proper- ties for the selective precipitation and subsequent resolubilization of -PGA. Due to its highly anionic nature, this polyamide can be used to form polymer–drug complexes with cationic compounds as the anticancer drug doxorubicin [6,15]. Another promising approach is the use of -PGA in combination with other polymeric com- pounds, such as chitosan or polylysine, to form polyionic complexes [16–18]. The successful delivery of insulin was demonstrated 1369-703X/$ – see front matter © 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.bej.2013.08.004
Transcript of Production of stable quinine nanodispersions using esterified γ-polyglutamic acid biopolymer

R
P�
Ca
b
a
ARRAA
K�BSQS
1
tnii[nia
dbgopm
vT
1h
Biochemical Engineering Journal 79 (2013) 259– 266
Contents lists available at ScienceDirect
Biochemical Engineering Journal
jou rna l h om epage: www.elsev ier .com/ locate /be j
egular article
roduction of stable quinine nanodispersions using esterified-polyglutamic acid biopolymer
hristoph Hoennscheidta, Dirk Kreyenschultea,b, Argyrios Margaritisb, Rainer Krull a,∗
Institute of Biochemical Engineering, Technische Universität Braunschweig, GermanyDepartment of Chemical and Biochemical Engineering, University of Western Ontario, London, Canada
r t i c l e i n f o
rticle history:eceived 4 April 2013eceived in revised form 5 July 2013ccepted 9 August 2013vailable online 31 August 2013
eywords:-Polyglutamic acidiopolymerurfactant, Dispersion
a b s t r a c t
Novel methods are needed for the development of nanodispersed drug formulations to enhance bioavail-ability of many hydrophobic pharmaceuticals. The poorly water-soluble quinine is a well-knownanti-malaria drug which can be used as a promising model compound for the development of novelnanodispersed formulations. In addition to hydrophobic drug’s own affecting properties, surfactantsplay an important role for the enhancement of their low bioavailability by preparing stable dispersions.Amphiphilic compounds can efficiently be used to stabilize colloidal fragments by preventing the precipi-tation or crystallization of poorly water-soluble active ingredients during fabrication. A novel biopolymerderivative based on the biotechnologically produced �-polyglutamic acid (�-PGA) from Bacillus licheni-formis cultivation was developed for encapsulation of the active ingredient. High-molecular �-PGA is an
uinineolvent evaporation
anionic polyelectrolyte that was optimized and modified with hydrophobic l-phenylalanine ethyl ester(l-PAE) to form an amphiphilic comb polymer P(�-GA-r-l-PAE) with surfactive properties. The approachof the nanodispersion polymer concentration, molecular weight and grafting degree enables the efficientstabilization of the poorly water-soluble model drug. The research presented in this report indicates thepotential benefits of hydrophobically modified �-PGA and suggests its potential role in forming stabledispersions for future pharmaceutical applications.
. Introduction
Due to significant progress in the field of pharmaceutical nano-echnology, the development of enhanced bioavailable drugs inanodispersion reservoirs and nano-sized drug delivery vehicles
s extensively investigated [1]. An important prerequisite for med-cal applications is the biocompatibility of the employed materials
2]. Thus, several biodegradable polymers, such as chitosan, algi-ate and �-polyglutamic acid, shortened as �-PGA, have beenntroduced for drug encapsulation and delivery [3–7]. �-PGA is annionic, water soluble polyamide consisting of repeated glutamic
Abbreviations: CuSO4, copper(II) sulfate; DMSO, dimethyl sulfoxide; DSC,ifferential scanning calorimetry; EDC, 1-ethyl-3-(3-dimethylaminopropyl) car-odiimide; DO, dissolved oxygen; EDTA, ethylendiaminetetraacetic acid; GPC,el permeation chromatography; l-PAE, l-phenylalanine ethyl ester; OD450,ptical density (450 nm); MWCO, molecular weight cut off; P(�-GA-r-l-PAE),oly(�-glutamic acid-random-l-phenylalanine ethyl ester); SEM, scanning electronicroscopy; THF, tetrahydrofuran; �-PGA, �-polyglutamic acid.∗ Corresponding author at: Institute of Biochemical Engineering, Technische Uni-
ersität Braunschweig, Gaußstraße 17, 38106 Braunschweig, Germany.el.: +49 0531 391 7653; fax: +49 0531 391 7652.
E-mail addresses: [email protected], [email protected] (R. Krull).
369-703X/$ – see front matter © 2013 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.bej.2013.08.004
© 2013 Elsevier B.V. All rights reserved.
acid units linked between the �-amino and �-carboxylic acid func-tional groups [8]. This polyamide can generally be produced bycultivation of various Bacillus species, most commonly Bacillus sub-tilis and Bacillus licheniformis [9,10]. A high molar mass of this typeof biopolymer is required to function as a surfactant.
�-PGA is capable of binding certain metal cations through ionicinteractions because it is an anionic polymer. As a component ofbacterial capsules, this binding or trapping phenomenon serves toprovide essential metal cations for enzymatic reactions or struc-tural components of the cell [11]. As a result, several studies haveexamined the metal binding characteristics of �-PGA [12,13]. Thesestudies indicate the addition of metal ions, such as Cu2+, Al3+, Cr3+
or Fe3+, induced the flocculation of dissolved �-PGA [13]. Manochaand Margaritis [14] investigated whether the metal induced �-PGAprecipitation could be used to separate �-PGA from B. licheniformiscultivation broth. Divalent copper ions exhibited the best proper-ties for the selective precipitation and subsequent resolubilizationof �-PGA.
Due to its highly anionic nature, this polyamide can be usedto form polymer–drug complexes with cationic compounds as the
anticancer drug doxorubicin [6,15]. Another promising approachis the use of �-PGA in combination with other polymeric com-pounds, such as chitosan or polylysine, to form polyionic complexes[16–18]. The successful delivery of insulin was demonstrated
260 C. Hoennscheidt et al. / Biochemical Engineering Journal 79 (2013) 259– 266
Table 1Technical data for the HPLC system.
Mobile phase9 mM sulfuric acid
Flow rate0.6 mL min−1
Column temperature50 ◦C
Injection volume25 �L
Column Aminex HPX-87H ion exclusion column (7.8 mm × 300 mm)
Detectors Waters 2487 Dual � Absorbance Detector at 192 nmWaters 410 Differential Refractometer at 30 ◦C
Pump Waters 1525 Binary HPLC Pump
Autosampler Waters 717plus Autosampler
usetsia�asthcd
edaoast
2
2
CiBsAwelvos3kcr0a1wm5p
w
Degasser
Software
sing nanoparticle assembly by ionic interactions between oppo-itely charged chitosan and �-PGA [5]. In previous studies, Akagit al. [19] developed a new self-assembling drug delivery sys-em utilizing hydrophobically modified �-PGA. The hydrophobicegments consisting of l-phenylalanine ethyl ester (l-PAE) werentroduced into the hydrophilic biopolymer backbone to producen amphiphilic comb polymer P(�-GA-r-l-PAE) [20]. The modified-PGA could subsequently be used to prepare nanoparticles withn inner hydrophobic core surrounded by a hydrophilic polymerhell. The resulting nanoparticles were employed as delivery sys-ems for various proteins and steroid drugs [19,21–23]. Due to theirydrophobic core, disperse systems containing amphiphilic �-PGAan enhance the bioavailability of poorly water-soluble organicrugs.
In the present study, the organic drug quinine was partiallyncapsulated with modified high molecular weight �-PGA pro-uced by B. licheniformis 9945A. Quinine is one of the first knownnti-malarial drugs and is associated with severe side-effects uponverdose [24–26]. Quinine itself is poorly water-soluble, and servess a model substance of this class of hydrophobic drugs. Finally,table dispersions were prepared using the solvent evaporationechnique, and further characterized.
. Materials and methods
.1. Bacterial strain and bioreactor cultivation
B. licheniformis 9945A was obtained from the American Typeulture Collection (ATCC). The batch cultivation was performed
n a 3 L bioreactor (BioFlo 110 Modular Benchtop Fermentor, Newrunswick Scientific Co., Inc.) with a working volume of 1.4 L. Theensors included pH and dissolved oxygen probes (Mettler-Toledo).
resistance temperature detector and a conductive level probeere used. The level probe triggered the addition of antifoam B
mulsion (Sigma–Aldrich) when the culture broth reached a certainevel due to foaming. To ensure sufficient mixing during the culti-ation, a baffle was installed with a dual six-blade Rushton impellerperated at 750 min−1. Aeration was provided by a standard ringparger at 1 L L−1 min−1, and the temperature was controlled at7 ◦C by heating or cooling the surrounding water jacket. pH wasept constant at 6.5 by automatic feeding of 2 M NaOH. The mediumomposition (g L−1) implied: 80 glycerol, 20 l-glutamic acid, 12 cit-ic acid, 7 NH4Cl, 0.5 MgSO4·7H2O, 0.5 K2HPO4, 0.15 CaCl2·2H2O,.1 MnSO4·H2O and 0.04 FeCl3·6H2O. The inoculum for the biore-ctor cultivation was prepared in a 500 mL shaking flask containing00 mL of the production medium. 250 �L of the glycerol stockere added and incubated at 37 ◦C and 200 min−1 for approxi-ately 24 h. When the culture broth reached a biomass of 0.3 g L−1,
0 mL were used as inoculum for bioreactor cultivation. Broth sam-les of about 30–40 mL were taken during cultivation.
To monitor the cell growth of B. licheniformis the dry cell weightas determined as a function of time. Therefore 5 mL of the culture
Waters In-Line Degasser
Waters Breeze Chromatography Software Version 3.20
broth were transferred to 50 mL falcon tubes and diluted withapproximately 30 mL of deionized water. The cultivation brothwas centrifuged at 12,100 × g at room temperature for 10 min, andthe supernatant was discarded. Depending on broth viscosity and�-PGA concentration, this procedure was repeated a maximumof three times to separate the cells from the biopolymer. The cellpellet was dissolved in deionized water and filtered through pre-weighed 0.2 �m cellulose nitrate membrane filters. After washingwith deionized water the filters were dried at 95 ◦C for 6–10 h. Thefilters were cooled in a desiccator and weighed to determine thedry biomass concentration. For each sample, measurements wereperformed as duplicates.
The concentrations of citric acid and glycerol were determinedby high performance liquid chromatography (HPLC). After appro-priate dilution of the samples with filtered deionized water, 950 �Lwas transferred to HPLC vials and analyzed with an HPLC system(Waters) as specified in Table 1. For each sample, the measurementswere performed as duplicates. The l-glutamic acid concentrationwas determined by using a commercially available enzymatic anal-ysis kit (Megazyme l-Glutamic Acid Assay Kit, K-GLUT) utilizing theenzymes glutamate dehydrogenase and diaphorase.
2.2. Separation and purification of �-PGA
Biopolymer purification was achieved by CuSO4 precipitationdeveloped by Manocha and Margaritis [14]. An equal volumeof 1 M divalent copper ions was used to induce the precipi-tation of non-soluble Cu2+/�-PGA complex and cells in culturebroth. The precipitate was solubilized in a 0.5 M EDTA solutionat pH 8 after separation from the supernatant. Subsequently, thecells were removed by centrifugation at 12,100 × g for 20 minto obtain soluble �-PGA in aqueous solution. Copper salts,EDTA and other low-molecular-weight impurities were removedby diafiltration against deionized water using a cross-flow fil-tration membrane with a MWCO of 50 kDa (Polyethersulfonecrossflow cassette, Vivaflow). The purified retentate containing�-PGA was frozen at −20 ◦C and freeze-dried at −50 ◦C (FreezeDryer ALPHA 1–4 LD, Martin Christ) to obtain a colorless pow-der.
SDS gel electrophoresis and gel permeation chromatography(GPC) were used to examine the mean size distribution of the puri-fied biopolymer. Conventional homogeneous 4% Tris–Glycine gelswere used with a HiMarkTM pre-stained high-molecular-weight(HMW) protein standard (Life Technologies) for electrophoresis.The resulting polymer distribution could be stained with a 3% solu-tion of methylene blue [27]. GPC measurements were carried outsimilar to HPLC detections. Samples were diluted in a 100 mM
phosphate buffer (pH = 7.4) followed by separation in a GPC col-umn (Ultrahydrogel linear columns in series (7.8 mm × 300 mm),Waters) under a flow rate of 0.8 mL min−1 at 30 ◦C. Detection wasperformed with a Differential Refractometer (Waters) at 30 ◦C. The
C. Hoennscheidt et al. / Biochemical Engineering Journal 79 (2013) 259– 266 261
F E) yies s the
rd
2
mdbhNEsimeartsD
2
twesSwtwtfrt
ig. 1. The hydrophobic modification of �-PGA with l-phenylalanine ethyl ester (l-PAide chain of �-PGA using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) a
esidual protein impurities were determined using a previouslyescribed Bradford assay [28].
.3. Hydrophobic modification of �-PGA
As depicted in Fig. 1, �-PGA was hydrophobicallyodified with l-PAE by conventional EDC (1-ethyl-3-(3-
imethylaminopropyl)carbodiimide) coupling to generate aiopolymeric surfactant [29]. The purified �-PGA and l-PAEydrochloride (Sigma–Aldrich) were dissolved in cold 50 mMaHCO3 solution. The mixture was cooled in a water/ice bath.DC (Sigma–Aldrich) was dissolved separately in 50 mM NaHCO3olution and added dropwise to the mixture of �-PGA and l-PAEmmediately to prevent further hydrolysis by EDC in aqueous
edia [30,31]. After 24 h, the solution was transferred to cellulosester dialysis tubes with a MWCO of 8–10 kDa (Spectrumlabs)nd dialyzed against 5 L of deionized water to remove undesiredeaction products. Several water exchanges were performed, andhe dialysis was stopped after approximately 3 days. The purifiedolution was frozen at −20 ◦C and freeze-dried at −50 ◦C (Freezeryer ALPHA 1–4 LD, Martin Christ).
.4. Preparation and characterization of quinine dispersion
Quinine nanodispersions were prepared using solvent evapora-ion technique. Tetrahydrofuran (THF) was chosen as a non-polarater miscible solvent [32–34]. Due to its high vapor pressure, THF
nables a polarity shift in a solvent mixture with polar water byubsequent evaporation. A solution of 1 g L−1 quinine (anhydrous,igma–Aldrich) and 1 g L−1 amphiphilic polymer, P(�-GA-r-l-PAE),as made in a THF/H2O (1:1) mixture. During THF evapora-
ion quinine solubility slowly decreased and succeeded nucleationith subsequent colloidal growth of the drug. The purpose of
he amphiphilic polymer was to act as an efficient surfactant byorming stabilized nanoparticles of the drug, Fig. 2, in polar envi-onment. The THF was completely evaporated after approximatelyhree days. A stable aqueous dispersion was obtained consisting of
lds an amphiphilic P(�-GA-r-l-PAE) complex. l-PAE is attached to the �-carboxylatecoupling agent.
a mixture of molecular quinine, primary particles and reversibleagglomerates.
To quantitate the total proportion of dispersed quinine, the sam-ples were first dried. After complete solubilization of quinine inDMSO, the absorption was determined spectroscopically at a wave-length of 332 nm. A calibration curve was used to determine thetotal amount of quinine in dispersion. In addition, to comprehendthe amounts of molecular quinine and its colloidal forms the sam-ples were separated with 0.1 and 0.45 �m PP syringe filters. Thefiltrates were treated as described above to determine the differentportions quantitatively.
In addition to the enhanced bioavailability, there is also inter-est in the stability of the dispersions under cooled conditions toenhance drug half-life. Therefore, differential scanning calorimetry(DSC 1, Mettler Toledo) was used to study enthalpy driven tran-sitions during cooling [35]. Unfortunately, agglomeration of thecolloidal forms followed by sedimentation can be the result duringcooling process due to the loss of structural integrity and smallersurface area [36].
To illustrate the colloidal reservoirs in dispersion, a high-resolution SEM was used to measure freeze-dried samples.
3. Results and discussion
3.1. Microbial �-PGA production
The parameters during B. licheniformis cultivation were definedconstant as described in chapter 2.1. Dry biomass, dissolved oxy-gen (DO) concentration and �-PGA concentration were determinedoffline over time as well as the concentrations of the three main car-bon sources citric acid, glycerol and l-glutamic acid were shown inFig. 3.
Microbial growth began after a lag-phase of approximately6 h with a specific growth rate of � = 0.13 h−1, Fig. 3(A).Growth was temporarily reduced by limited oxygen avail-
ability after 21 h, but resumed between 23 and 55 h with� = 0.32 h−1. A subsequent stationary phase with a dry biomassconcentration of approximately 4 g L−1 was observed until asecond growth phase began at 79 h with � = 0.129 h−1. The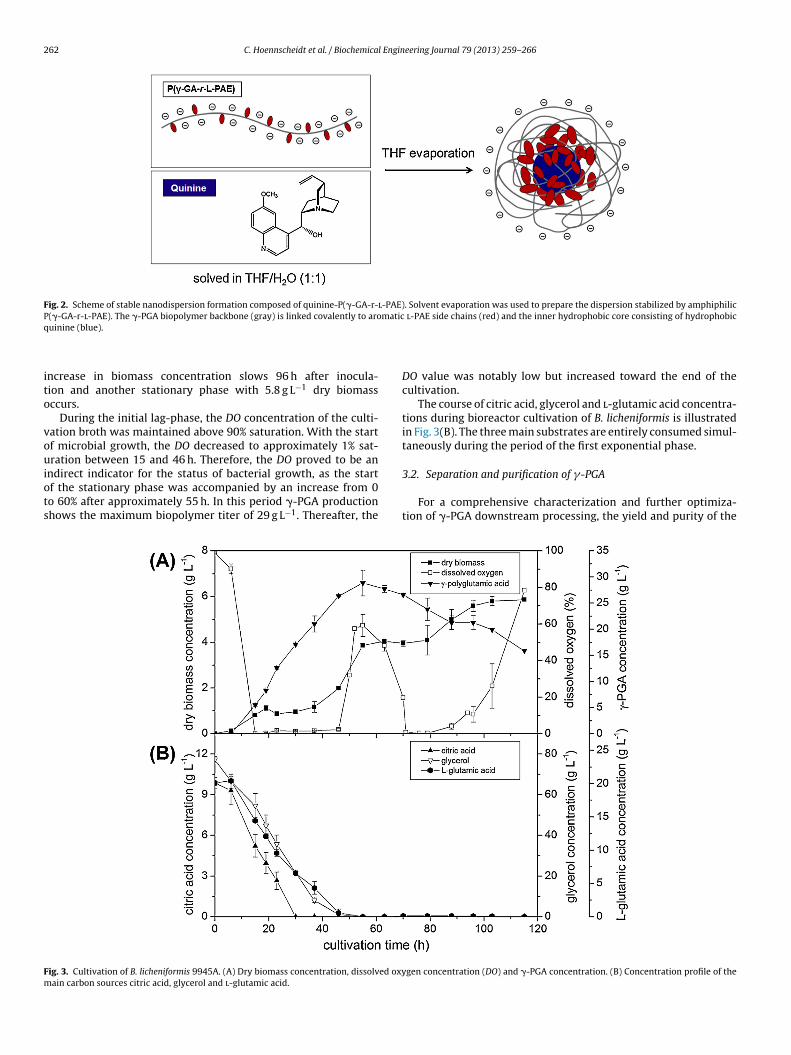
262 C. Hoennscheidt et al. / Biochemical Engineering Journal 79 (2013) 259– 266
Fig. 2. Scheme of stable nanodispersion formation composed of quinine-P(�-GA-r-l-PAE). Solvent evaporation was used to prepare the dispersion stabilized by amphiphilicP(�-GA-r-l-PAE). The �-PGA biopolymer backbone (gray) is linked covalently to aromatic l-PAE side chains (red) and the inner hydrophobic core consisting of hydrophobicq
ito
vouiots
Fm
uinine (blue).
ncrease in biomass concentration slows 96 h after inocula-ion and another stationary phase with 5.8 g L−1 dry biomassccurs.
During the initial lag-phase, the DO concentration of the culti-ation broth was maintained above 90% saturation. With the startf microbial growth, the DO decreased to approximately 1% sat-ration between 15 and 46 h. Therefore, the DO proved to be an
ndirect indicator for the status of bacterial growth, as the startf the stationary phase was accompanied by an increase from 0o 60% after approximately 55 h. In this period �-PGA productionhows the maximum biopolymer titer of 29 g L−1. Thereafter, the
ig. 3. Cultivation of B. licheniformis 9945A. (A) Dry biomass concentration, dissolved oxyain carbon sources citric acid, glycerol and l-glutamic acid.
DO value was notably low but increased toward the end of thecultivation.
The course of citric acid, glycerol and l-glutamic acid concentra-tions during bioreactor cultivation of B. licheniformis is illustratedin Fig. 3(B). The three main substrates are entirely consumed simul-taneously during the period of the first exponential phase.
3.2. Separation and purification of �-PGA
For a comprehensive characterization and further optimiza-tion of �-PGA downstream processing, the yield and purity of the
gen concentration (DO) and �-PGA concentration. (B) Concentration profile of the
C. Hoennscheidt et al. / Biochemical Engin
bt
st�rtt4p
t
Foa
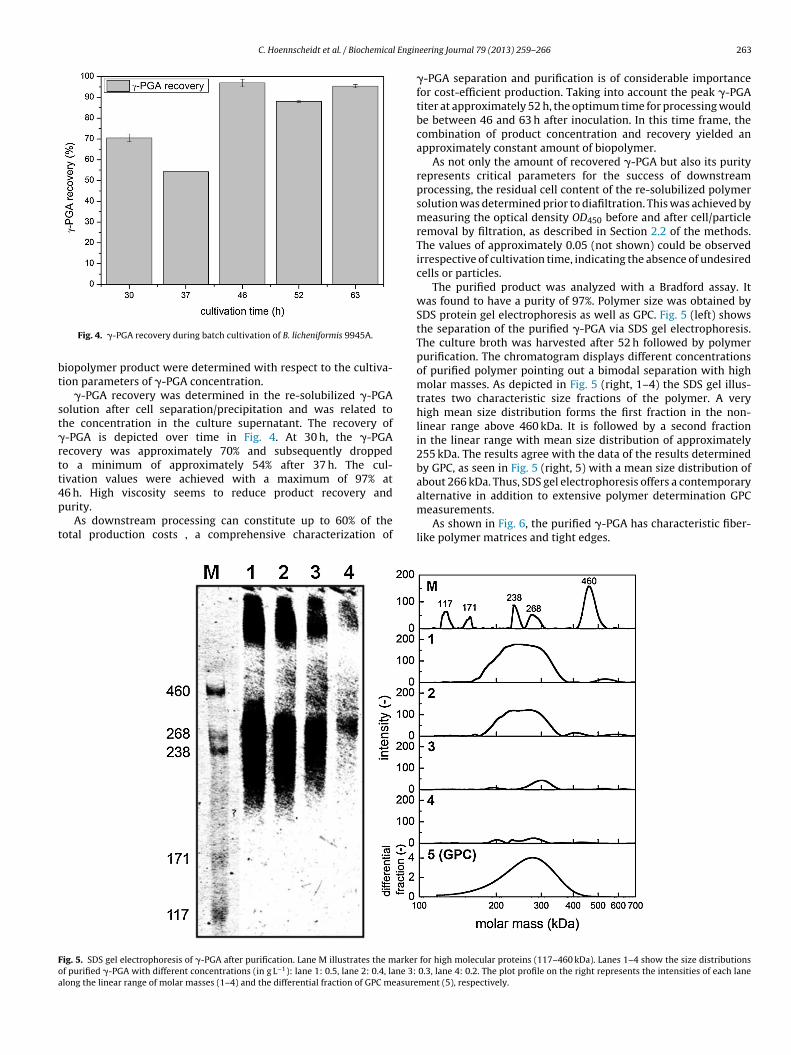
Fig. 4. �-PGA recovery during batch cultivation of B. licheniformis 9945A.
iopolymer product were determined with respect to the cultiva-ion parameters of �-PGA concentration.
�-PGA recovery was determined in the re-solubilized �-PGAolution after cell separation/precipitation and was related tohe concentration in the culture supernatant. The recovery of-PGA is depicted over time in Fig. 4. At 30 h, the �-PGAecovery was approximately 70% and subsequently droppedo a minimum of approximately 54% after 37 h. The cul-ivation values were achieved with a maximum of 97% at
6 h. High viscosity seems to reduce product recovery andurity.As downstream processing can constitute up to 60% of theotal production costs , a comprehensive characterization of
ig. 5. SDS gel electrophoresis of �-PGA after purification. Lane M illustrates the markerf purified �-PGA with different concentrations (in g L−1): lane 1: 0.5, lane 2: 0.4, lane 3:
long the linear range of molar masses (1–4) and the differential fraction of GPC measure
eering Journal 79 (2013) 259– 266 263
�-PGA separation and purification is of considerable importancefor cost-efficient production. Taking into account the peak �-PGAtiter at approximately 52 h, the optimum time for processing wouldbe between 46 and 63 h after inoculation. In this time frame, thecombination of product concentration and recovery yielded anapproximately constant amount of biopolymer.
As not only the amount of recovered �-PGA but also its purityrepresents critical parameters for the success of downstreamprocessing, the residual cell content of the re-solubilized polymersolution was determined prior to diafiltration. This was achieved bymeasuring the optical density OD450 before and after cell/particleremoval by filtration, as described in Section 2.2 of the methods.The values of approximately 0.05 (not shown) could be observedirrespective of cultivation time, indicating the absence of undesiredcells or particles.
The purified product was analyzed with a Bradford assay. Itwas found to have a purity of 97%. Polymer size was obtained bySDS protein gel electrophoresis as well as GPC. Fig. 5 (left) showsthe separation of the purified �-PGA via SDS gel electrophoresis.The culture broth was harvested after 52 h followed by polymerpurification. The chromatogram displays different concentrationsof purified polymer pointing out a bimodal separation with highmolar masses. As depicted in Fig. 5 (right, 1–4) the SDS gel illus-trates two characteristic size fractions of the polymer. A veryhigh mean size distribution forms the first fraction in the non-linear range above 460 kDa. It is followed by a second fractionin the linear range with mean size distribution of approximately255 kDa. The results agree with the data of the results determinedby GPC, as seen in Fig. 5 (right, 5) with a mean size distribution ofabout 266 kDa. Thus, SDS gel electrophoresis offers a contemporaryalternative in addition to extensive polymer determination GPCmeasurements.
As shown in Fig. 6, the purified �-PGA has characteristic fiber-like polymer matrices and tight edges.
for high molecular proteins (117–460 kDa). Lanes 1–4 show the size distributions0.3, lane 4: 0.2. The plot profile on the right represents the intensities of each lanement (5), respectively.
264 C. Hoennscheidt et al. / Biochemical Engineering Journal 79 (2013) 259– 266
Ff
3
dacm
wbgsri[mt3attppfPsw
Fo
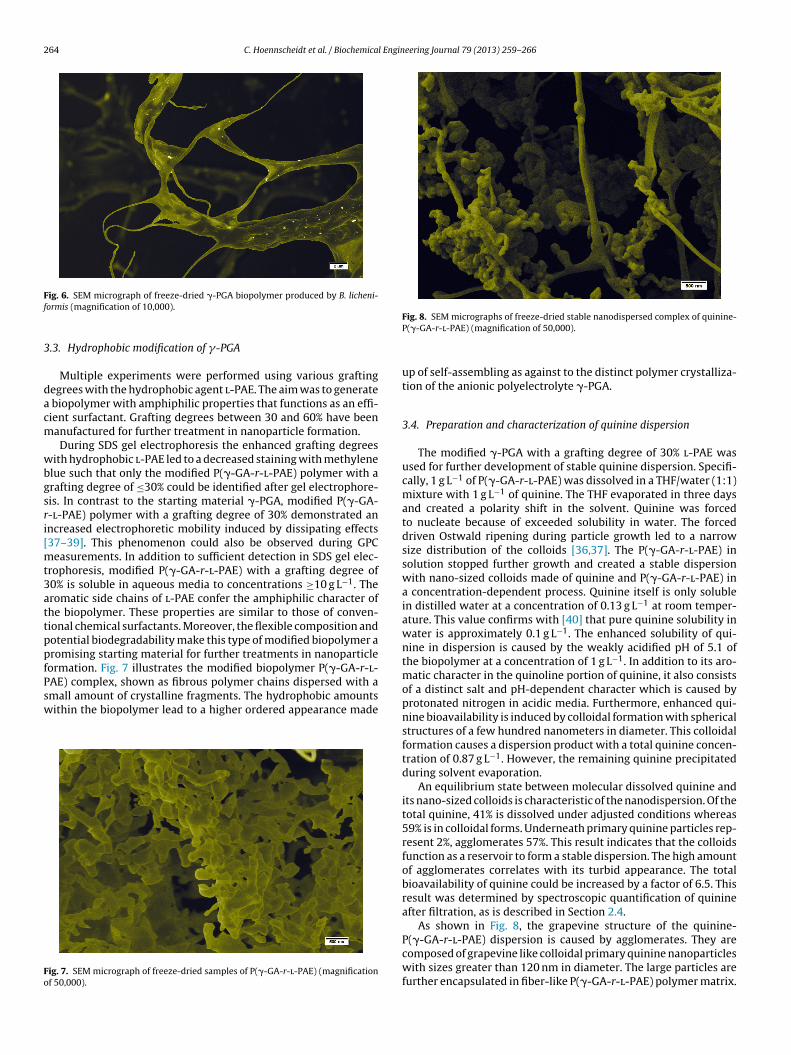
ig. 6. SEM micrograph of freeze-dried �-PGA biopolymer produced by B. licheni-ormis (magnification of 10,000).
.3. Hydrophobic modification of �-PGA
Multiple experiments were performed using various graftingegrees with the hydrophobic agent l-PAE. The aim was to generate
biopolymer with amphiphilic properties that functions as an effi-ient surfactant. Grafting degrees between 30 and 60% have beenanufactured for further treatment in nanoparticle formation.During SDS gel electrophoresis the enhanced grafting degrees
ith hydrophobic l-PAE led to a decreased staining with methylenelue such that only the modified P(�-GA-r-l-PAE) polymer with arafting degree of ≤30% could be identified after gel electrophore-is. In contrast to the starting material �-PGA, modified P(�-GA--l-PAE) polymer with a grafting degree of 30% demonstrated anncreased electrophoretic mobility induced by dissipating effects37–39]. This phenomenon could also be observed during GPC
easurements. In addition to sufficient detection in SDS gel elec-rophoresis, modified P(�-GA-r-l-PAE) with a grafting degree of0% is soluble in aqueous media to concentrations ≥10 g L−1. Theromatic side chains of l-PAE confer the amphiphilic character ofhe biopolymer. These properties are similar to those of conven-ional chemical surfactants. Moreover, the flexible composition andotential biodegradability make this type of modified biopolymer aromising starting material for further treatments in nanoparticleormation. Fig. 7 illustrates the modified biopolymer P(�-GA-r-l-AE) complex, shown as fibrous polymer chains dispersed with a
mall amount of crystalline fragments. The hydrophobic amountsithin the biopolymer lead to a higher ordered appearance madeig. 7. SEM micrograph of freeze-dried samples of P(�-GA-r-l-PAE) (magnificationf 50,000).
Fig. 8. SEM micrographs of freeze-dried stable nanodispersed complex of quinine-P(�-GA-r-l-PAE) (magnification of 50,000).
up of self-assembling as against to the distinct polymer crystalliza-tion of the anionic polyelectrolyte �-PGA.
3.4. Preparation and characterization of quinine dispersion
The modified �-PGA with a grafting degree of 30% l-PAE wasused for further development of stable quinine dispersion. Specifi-cally, 1 g L−1 of P(�-GA-r-l-PAE) was dissolved in a THF/water (1:1)mixture with 1 g L−1 of quinine. The THF evaporated in three daysand created a polarity shift in the solvent. Quinine was forcedto nucleate because of exceeded solubility in water. The forceddriven Ostwald ripening during particle growth led to a narrowsize distribution of the colloids [36,37]. The P(�-GA-r-l-PAE) insolution stopped further growth and created a stable dispersionwith nano-sized colloids made of quinine and P(�-GA-r-l-PAE) ina concentration-dependent process. Quinine itself is only solublein distilled water at a concentration of 0.13 g L−1 at room temper-ature. This value confirms with [40] that pure quinine solubility inwater is approximately 0.1 g L−1. The enhanced solubility of qui-nine in dispersion is caused by the weakly acidified pH of 5.1 ofthe biopolymer at a concentration of 1 g L−1. In addition to its aro-matic character in the quinoline portion of quinine, it also consistsof a distinct salt and pH-dependent character which is caused byprotonated nitrogen in acidic media. Furthermore, enhanced qui-nine bioavailability is induced by colloidal formation with sphericalstructures of a few hundred nanometers in diameter. This colloidalformation causes a dispersion product with a total quinine concen-tration of 0.87 g L−1. However, the remaining quinine precipitatedduring solvent evaporation.
An equilibrium state between molecular dissolved quinine andits nano-sized colloids is characteristic of the nanodispersion. Of thetotal quinine, 41% is dissolved under adjusted conditions whereas59% is in colloidal forms. Underneath primary quinine particles rep-resent 2%, agglomerates 57%. This result indicates that the colloidsfunction as a reservoir to form a stable dispersion. The high amountof agglomerates correlates with its turbid appearance. The totalbioavailability of quinine could be increased by a factor of 6.5. Thisresult was determined by spectroscopic quantification of quinineafter filtration, as is described in Section 2.4.
As shown in Fig. 8, the grapevine structure of the quinine-P(�-GA-r-l-PAE) dispersion is caused by agglomerates. They are
composed of grapevine like colloidal primary quinine nanoparticleswith sizes greater than 120 nm in diameter. The large particles arefurther encapsulated in fiber-like P(�-GA-r-l-PAE) polymer matrix.
l Engin
pcmaiocmSl
4
pP(emtTf�aesw
cptas
cis
patootsdsppmtd
A
tBaFtUs
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[[
[
[
[
[
C. Hoennscheidt et al. / Biochemica
DSC measurements demonstrate the freezing behavior in theresence of disperse material. Whereas water crystallized during aooling rate of 10 K mol−1 at −15.5 ◦C, quinine suspension and poly-er solutions with concentrations of 1 g L−1 crystallized at −19.5
nd −18.2 ◦C, respectively. Similar behavior could be observed dur-ng the cooling of quinine dispersion. A crystallization temperaturef −18.6 ◦C was determined. In comparison to deionized water, therystallization temperature of dispersion is decreased to approxi-ately −3.2 ◦C. No agglomeration or aggregation, as discussed in
ection 2.4, could be observed during cooling process so that aong-term durability of the nanodispersion could be sustained.
. Conclusions
Microbial cultivation processes are a promising method toroduce biodegradable and non-allergenic biopolymers, such as �-GA, that can be used in novel nanosized drug delivery systemsDDS). The main objective is to optimize such bioprocesses and gen-rate polymers in high titer and high grades of purity. Furthermore,odifying these biopolymers to create an amphiphilic surfac-
ant prior to formation of dispersions was the focus of this work.he biotechnological process that generates high-molecular �-PGArom B. licheniformis was used for production and purification. The-PGA was further modified to an amphiphilic component actings a stabilizer with a hydrophobic part built by l-phenylalaninethyl ester, l-PAE. The enhanced bioavailability of the poorly water-oluble model substance quinine by addition of P(�-GA-r-l-PAE)as demonstrated.
The production of �-PGA was achieved with a biopolymer con-entration of approximately 29 g L−1 in the first exponential growthhase. Growth and product formation caused the depletion ofhe main carbon sources. The correct selection of separation timellowed recovery rates ≥ 97% to be achieved. This result demon-trates the efficiency of copper sulfate precipitation.
Hydrophobic modifications of purified �-PGA were achieved byoupling with l-PAE. Modified �-PGA biopolymer with a 30% graft-ng degree of l-PAE was further used for subsequent nanoparticletudies.
A solvent evaporation process was established to prepare dis-ersions with P(�-GA-r-l-PAE) as a surfactant. As quinine servess a model substance, it is expected that the P(�-GA-r-l-PAE) sys-em can be transferred to other hydrophobic drugs. The formationf dispersions was achieved by the evaporation of the non-polar,rganic solvent THF. The THF induced a nucleation process ofhe poorly water-soluble drug. The nanoparticulate colloids wereubsequently encapsulated by the generated biosurfactant. Stableispersion could be achieved and was affected by an equilibriumtate between molecular dissolved quinine and colloidal forms. Theure quinine colloids are composed of a hydrophobic core of theoorly water-soluble drug surrounded by the amphiphilic biopoly-er shell. The research presented in this report strongly indicates
he potential of hydrophobically modified �-PGA to form stableispersions for future pharmaceutical applications.
cknowledgments
The authors kindly thank Prof. Dr. U. Rau and Mr. W. Grassl, Insti-ute for Biochemistry and Biotechnology, Technische Universitätraunschweig, for the implementation of GPC measurements. Theuthors gratefully acknowledge funding by the German Research
oundation (DFG) in the Research Unit 856 mikroPART “Microsys-ems for Particulate Life Science Products” at the Technischeniversität Braunschweig (Prof. Dr. R. Krull), Germany, and theupport of Natural Sciences and Engineering Research Council of
[
[
eering Journal 79 (2013) 259– 266 265
Canada (NSERC) through Discovery Grant No. 4388, awarded toProf. Dr. A. Margaritis.
References
[1] V. Wagner, A. Dullaart, A.-K. Bock, A. Zweck, The emerging nanomedicine land-scape, Nat. Biotechnol. 24 (2006) 1211–1217.
[2] R.H. Müller, K. Mäder, S. Gohla, Solid lipid nanoparticles (SLN) for controlleddrug delivery – a review of the state of the art, Eur. J. Pharm. Biopharm. 50(2000) 161–177.
[3] H. Katas, Z. Hussain, T.C. Ling, Chitosan nanoparticles as a percutaneous drugdelivery system for hydrocortisone, J. Nanomater. (2012) 1–11.
[5] Y.-H. Lin, F.-L. Mi, C.-T. Chen, W.-C. Chang, S.-F. Peng, H.-F. Liang, et al., Prepara-tion and characterization of nanoparticles shelled with chitosan for oral insulindelivery, Biomacromolecules 8 (2007) 146–152.
[6] B. Manocha, A. Margaritis, Controlled release of doxorubicin fromdoxorubicin/�-polyglutamic acid ionic complex, J. Nanomater. 2010 (2010) 9.
[7] H.H. Tønnesen, J. Karlsen, Alginate in drug delivery systems, Drug Dev. Ind.Pharm. 28 (2002) 621–630.
[8] A. Richard, A. Margaritis, Poly(glutamic acid) for biomedical applications, Crit.Rev. Biotechnol. 21 (2001) 219–232.
[9] I. Bajaj, R. Singhal, Poly(glutamic acid) – an emerging biopolymer of commercialinterest, Bioresour. Technol. 102 (2011) 5551–5561.
10] J.M. Buescher, A. Margaritis, Microbial biosynthesis of polyglutamic acidbiopolymer and applications in the biopharmaceutical, biomedical and foodindustries, Crit. Rev. Biotechnol. 27 (2007) 1–19.
11] T.J. Beveridge, R.G. Murray, Uptake and retention of metals by cell walls ofBacillus subtilis, J. Bacteriol. 127 (1976) 1502–1518.
12] T.J. Beveridge, R.G. Murray, Sites of metal deposition in the cell wall of Bacillussubtilis, J. Bacteriol. 141 (1980) 876–887.
13] R.J. McLean, D. Beauchemin, L. Clapham, T.J. Beveridge, Metal-binding charac-teristics of the gamma-glutamyl capsular polymer of Bacillus licheniformis ATCC9945, Appl. Environ. Microbiol. 56 (1990) 3671–3677.
14] B. Manocha, A. Margaritis, A novel method for the selective recovery and purifi-cation of �-polyglutamic acid from Bacillus licheniformis fermentation broth,Biotechnol. Prog. 26 (2010) 734–742.
15] F. Hellmers, P. Ferguson, J. Koropatnick, R. Krull, A. Margaritis, Characterizationand in vitro cytotoxicity of doxorubicin-loaded �-polyglutamic acid-chitosancomposite nanoparticles, Biochem. Eng. J. 75 (2013) 72–78.
16] Z. Keresztessy, M. Bodnár, E. Ber, I. Hajdu, M. Zhang, J.F. Hartmann, et al.,Self-assembling chitosan/poly-�-glutamic acid nanoparticles for targeted drugdelivery, Colloid Polym. Sci. 287 (2009) 759–765.
17] I. Hajdu, M. Bodnár, G. Filipcsei, J.F. Hartmann, L. Daróczi, M. Zrínyi, et al.,Nanoparticles prepared by self-assembly of chitosan and poly-�-glutamic acid,Colloid Polym. Sci. 286 (2007) 343–350.
18] T. Akagi, K. Watanabe, H. Kim, M. Akashi, Stabilization of polyion complexnanoparticles composed of poly(amino acid) using hydrophobic interactions,Langmuir 26 (2010) 2406–2413.
19] T. Akagi, T. Kaneko, T. Kida, M. Akashi, Preparation and characterization ofbiodegradable nanoparticles based on poly(gamma-glutamic acid) with l-phenylalanine as a protein carrier, J. Control. Release 108 (2005) 226–236.
20] D.J. Irvine, A.M. Mayes, L.G. Griffith, Nanoscale clustering of RGD peptides atsurfaces using Comb polymers. 1. Synthesis and characterization of Comb thinfilms, Biomacromolecules 2 (2001) 85–94.
21] T. Akagi, X. Wang, T. Uto, M. Baba, M. Akashi, Protein direct delivery to dendriticcells using nanoparticles based on amphiphilic poly(amino acid) derivatives,Biomaterials 28 (2007) 3427–3436.
22] D. Hudson, A. Margaritis, Biopolymer nanoparticle production for controlledrelease of biopharmaceuticals, Crit. Rev. Biotechnol. (2013) 1–19.
23] M. Ryu, T. Nakazawa, T. Akagi, T. Tanaka, R. Watanabe, M. Yasuda, et al., Suppres-sion of phagocytic cells in retinal disorders using amphiphilic poly(�-glutamicacid) nanoparticles containing dexamethasone, J. Control. Release 151 (2011)65–73.
24] J. Achan, A.O. Talisuna, A. Erhart, A. Yeka, J.K. Tibenderana, F.N. Baliraine, et al.,Quinine, an old anti-malarial drug in a modern world: role in the treatment ofmalaria, Malaria J 10 (2011) 12.
25] A. Jaeger, Quinine and chloroquine, Medicine 35 (2007) 652–653.26] P. Murambiwa, B. Masola, T. Govender, S. Mukaratirwa, C.T. Musabayane, Anti-
malarial drug formulations and novel delivery systems: a review, Acta Trop.118 (2011) 71–79.
27] M. Kambourova, M. Tangney, G. Fergus, Regulation of polyglutamic acid syn-thesis by glutamate in Bacillus licheniformis and Bacillus subtilis regulation ofpolyglutamic acid synthesis by glutamate in Bacillus licheniformis and Bacillussubtilis, Appl. Environ. Microbiol. 67 (2001) 1004–1007.
28] M.M. Bradford, A rapid and sensitive method for the quantitation of micro-gram quantities of protein utilizing the principle of protein-dye binding, Anal.Biochem. 72 (1976) 248–254.
29] A. Williams, I.T. Ibrahim, Carbodiimide chemistry: recent advances, Chem. Rev.81 (1981) 589–636.
30] N. Nakajima, Y. Ikada, Mechanism of amide formation by carbodiimide for
bioconjugation in aqueous media, Bioconjugate Chem. 6 (1995) 123–130.31] N. Wrobel, M. Schinkinger, V.M. Mirsky, A novel ultraviolet assay for testingside reactions of carbodiimides, Anal. Biochem. 305 (2002) 135–138.
32] B.C.J.K. Yu, A.J. Hurd, A. Eisenberg, Syntheses of silica/polystyrene-block-poly(ethylene oxide) films with regular and reverse mesostructures of large

2 l Engin
[
[
[
[
[
[
[
66 C. Hoennscheidt et al. / Biochemica
characteristic length scales by solvent evaporation-induced self-assembly,Langmuir 17 (2001) 7961–7965.
33] H. Yabu, T. Higuchi, M. Shimomura, Unique phase-separation structures ofblock-copolymer nanoparticles, Adv. Mater. 17 (2005) 2062–2065.
34] M. Trotta, F. Debernardi, O. Caputo, Preparation of solid lipid nanoparticles by asolvent emulsification–diffusion technique, Int. J. Pharm. 257 (2003) 153–160.
35] H.W. Sheng, Z.Q. Hu, K. Lu, Melting and freezing behaviors of Pb nanoparticles
embedded in an Al matrix, Nanostruct. Mater. 9 (1997) 661–664.36] Y.-H. Lin, C.-K. Chung, C.-T. Chen, H.-F. Liang, S.-C. Chen, H.-W. Sung, Prepa-ration of nanoparticles composed of chitosan/poly-gamma-glutamic acid andevaluation of their permeability through Caco-2 cells, Biomacromolecules 6(2005) 1104–1112.
[
eering Journal 79 (2013) 259– 266
37] R.D. Groot, T.J. Madden, D.J. Tildesley, On the role of hydrodynamic interac-tions in block copolymer microphase separation, J. Chem. Phys. 110 (1999)9739–9749.
38] S.G. Schulz, H. Kuhn, G. Schmid, C. Mund, J. Venzmer, Phase behavior ofamphiphilic polymers: a dissipative particles dynamics study, Colloid Polym.Sci. 283 (2004) 284–290.
39] H. Guo, K. Kremer, Molecular dynamics simulation of the phase behav-
ior of lamellar amphiphilic model systems, J. Chem. Phys. 119 (2003)9308–9320.40] R. Banerjee, P.M. Bhatt, N.V. Ravindra, G.R. Desiraju, Saccharin salts of activepharmaceutical ingredients, their crystal structures, and increased water solu-bilities, Cryst. Growth Des. 5 (2005) 2299–2309.


















