
PLZF Controls the Development of Fetal-Derived IL-17 Vg6 gd T Cells
10
of February 13, 2018. This information is current as T Cells δ γ + 6 γ V + Fetal-Derived IL-17 PLZF Controls the Development of Ying Lu, Xin Cao, Xianyu Zhang and Damian Kovalovsky http://www.jimmunol.org/content/195/9/4273 doi: 10.4049/jimmunol.1500939 September 2015; 2015; 195:4273-4281; Prepublished online 25 J Immunol Material Supplementary 9.DCSupplemental http://www.jimmunol.org/content/suppl/2015/09/25/jimmunol.150093 average * 4 weeks from acceptance to publication Speedy Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • ? The JI Why References http://www.jimmunol.org/content/195/9/4273.full#ref-list-1 , 21 of which you can access for free at: cites 40 articles This article Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved. 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on February 13, 2018 http://www.jimmunol.org/ Downloaded from by guest on February 13, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of PLZF Controls the Development of Fetal-Derived IL-17 Vg6 gd T Cells

of February 13, 2018.This information is current as
T Cellsδγ +6γV+Fetal-Derived IL-17PLZF Controls the Development of
Ying Lu, Xin Cao, Xianyu Zhang and Damian Kovalovsky
http://www.jimmunol.org/content/195/9/4273doi: 10.4049/jimmunol.1500939September 2015;
2015; 195:4273-4281; Prepublished online 25J Immunol
MaterialSupplementary
9.DCSupplementalhttp://www.jimmunol.org/content/suppl/2015/09/25/jimmunol.150093
average*
4 weeks from acceptance to publicationSpeedy Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
?The JIWhy
Referenceshttp://www.jimmunol.org/content/195/9/4273.full#ref-list-1
, 21 of which you can access for free at: cites 40 articlesThis article
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved.1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
PLZF Controls the Development of Fetal-DerivedIL-17+Vg6+ gd T Cells
Ying Lu,1 Xin Cao,1 Xianyu Zhang, and Damian Kovalovsky
Expression of promyelocytic leukemia zinc finger (PLZF) protein directs the effector differentiation of invariant NKT (iNKT) cells
and IL-4+ gd NKT cells. In this study, we show that PLZF is also required for the development and function of IL-17+ gd T cells.
We observed that PLZF is expressed in fetal-derived invariant Vg5+ and Vg6+ gd T cells, which secrete IFN-g and IL-17,
respectively. PLZF deficiency specifically affected the effector differentiation of Vg6+ cells, leading to reduced numbers of mature
CD272CD44+ phenotype capable of secreting IL-17. Although PLZF was not required for Vg5+ gd T cells to develop, when these
cells were reprogrammed into IL-17–secreting cells in Skint-1 mutant mice, they required PLZF for their effector maturation,
similarly to Vg6+ gd T cells. The impaired effector differentiation of PLZF-deficient Vg6+ gd T cells was not due to increased
apoptosis and it was related to reduced proliferation of immature CD27+CD442 Vg6+ gd T cells, which was required for their
differentiation into mature CD272CD44+ IL-17–secreting cells. Thus, the present study identifies that PLZF function is not
restricted to NKT or IL-4+ T cells, but it also controls the development of IL-17+ gd T cells. The Journal of Immunology,
2015, 195: 4273–4281.
Conventional T cells leave the thymus as naive cells. Onlyafter recognition of their cognate Ag in the periphery dothey differentiate into cytokine-secreting cells belonging
mainly to the Th1, Th2, and Th17 T cell subsets. “Innate-like”T cells acquire cytokine-secreting functions within the thymus andpreferentially migrate to nonlymphoid tissues, providing a firstline of defense against invading pathogens (1, 2)A common feature of different subsets of innate-like T cells within
the adult T cell compartment, including invariant NKT (iNKT) cells,gd NKT cells (Vg1.1+Vd6.3+ gd T cells), and human mucosal-associated invariant T cells, is the expression of the promyelocyticleukemia zinc finger (PLZF) protein (3–7). PLZF is considereda determinant of innate-like differentiation, as it is necessary for theacquisition of effector functions in iNKT cells and gd NKT cells.PLZF-deficient iNKT cells have a naive phenotype, preferentiallysecrete IL-2, and migrate to lymph nodes (3, 4, 6). Conversely,transgenic PLZF expression during T cell development is sufficientto provide some “innate” effector functions to conventional T cells.These are the acquisition of a memory/activated phenotype, theability to co-secrete IL-4 and IFN-g, and preferential migration to theliver microvasculature, unique characteristics of iNKT cells (4, 8, 9).During fetal hematopoiesis gd T cell subsets with innate-like
properties are generated, such as invariant Vg5+ and Vg6+ gd
T cells. Hematopoietic progenitors that enter the thymus at approx-
imately embryonic day 14 (E14) first rearrange the Vd1 and Vg5TCR loci. Successful rearrangement allows the development ofdendritic epidermal T cells, which secrete IFN-g and migrate to theintraepithelial compartment of the skin (10, 11). In contrast, uponunproductive Vg5 rearrangements, cells attempt to rearrange the Vg6loci, leading to the generation of invariant Vg6Vd1 gd T cells, whichsecrete IL-17 and colonize the epithelium of the uterus, tongue, lungs,and peritoneum. IL-17+ gd T cells are broadly identified as CD272
cells in peripheral tissues (12, 13) and preferentially proliferate inresponse to IL-7 signals, allowing their self-renewal in peripheraltissues (14). IL-17+ gd T cells are a major source of innate IL-17 inseveral mucosal infection models such as Mycobacterium tubercu-losis, Escherichia coli, and Listeria monocytogenes infections (15,16). Innate IL-17 and IL-21 secretion by gd T cells in response toIL-1 and IL-23 was shown to mediate autoimmune inflammation inan experimental autoimmune encephalomyelitis model (17).In this study, we have uncovered a novel function of PLZF in the
development of innate-like IL-17+Vg6+ gd T cells. Although PLZFis expressed in both Vg5+ and Vg6+ fetal thymic precursors, it isspecifically required for the development of Vg6+ gd T cells,allowing their normal maturation, expansion, and acquisition of IL-17 secretion after selection. Interestingly, despite PLZF expression inVg5+ gd T cells, PLZF deficiency did not affect their developmentor their ability to colonize the skin. The inability of PLZF-deficientVg6+ gd T cells to expand intrathymically was not due to increasedapoptosis, but to an impairment of Vg6+ gd T cells to proliferateand acquire a mature CD272CD44+ phenotype and function.
Materials and MethodsMaterials and mice
C57BL/6 mice were purchased from The Jackson Laboratory. PLZF-deficient (PLZF knockout [KO]) mice have been described (3). Lck-Bcl2transgenic mice were previously described (18). Rag2/gc-deficient micewere purchased from Taconic. FVB/N mice from Taconic, which presentthe Skin-1 mutation, were crossed with PLZF2/2 mice to generate com-pound PLZF2/2 Skint-1 mutant mice. A similar cross was performedbetween FVB/N mice from The Jackson Laboratory, which does notcontain the Skint-1 mutation, with PLZF2/2 mice to obtain PLZF2/2
Skint-1 wild-type mice. All animals were bred in-house and surgeries were
Experimental Immunology Branch, National Cancer Institute, National Institutes ofHealth, Bethesda, MD 20892
1Y.L. and X.C. contributed equally to this work.
Received for publication April 23, 2015. Accepted for publication August 31, 2015.
This work was supported by National Institutes of Health intramural funds(1ZIABC011429).
Address correspondence and reprint requests to Dr. Damian Kovalovsky, Experi-mental Immunology Branch, National Cancer Institute, National Institutes ofHealth, 9000 Rockville Pike, Building 10/4B17, Bethesda, MD 20892. E-mail:[email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: DN, double-negative; E, embryonic day; ETP,early thymic progenitor; iNKT, invariant NKT; KO, knockout; PLZF, promyelocyticleukemia zinc finger.
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1500939
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

performed under approved animal protocols by the National CancerInstitute Animal Care and Use Committee.
Tissue dissections, Abs, and flow cytometry
Pregnant day 16 mothers were euthanized by bottled CO2, and fetuseswere isolated and placed on ice for 30–60 min. Neonatal day 1 pups weresimilarly sacrificed by hypothermia. Fetuses and neonates were washedin PBS and tail samples were digested for genotyping. Thymuses wereextracted under a magnifier and dispersed under a 70-mm cell strainer(Falcon) to obtain single-cell suspensions. gd T cells from uterus and skinwere isolated by Liberase (Roche) and DNaseΙ (Sigma-Aldrich) digestionof the tissues, washing, and filtration through a 70-mm cell strainer (Fal-con). Immune cells in these cell suspensions were identified by stainingwith an anti-CD45.2 (104)-specific Ab. Ab staining was performed for 30min on ice and FACS analysis was done using a BD LSRFortessa cell an-alyzer (BD Biosciences). The following Ab clones were used: lineage biotinpanel (TER-119, RB6-8C5, RA3-6B2, M1/70, 145-2C11), c-Kit (2B8),CD44 (IM7), CD25 (PC61), TCR gd (GL3), CD3ε (145-2C11), CD27(16.7F9/ LG.7F9), Vg5 (536), CCR6 (140706), CD127 (A7R34), andCD62L (MEL-14). a-Galactosylceramide–CD1d tetramers were obtainedfrom the National Institutes of Health Tetramer Core Facility. Identifica-tion of Vg6+ gd T cells was performed by prestaining with GL3 Ab, fol-lowed by staining with the unconjugated rabbit 17D1 IgM Ab and a labeledsecondary anti-IgM Ab (Jackson ImmunoResearch Laboratories). Whenidentifying Vg6+ gd T cells, a simultaneous staining with Vg5-specific Abwas performed to exclude Vg5+ cells from the analysis.
Intranuclear PLZF staining was performed after surface staining usingthe Foxp3 transcription factor staining buffer set (eBioscience) followedby staining with the anti–PLZF-PE Ab (Mags.21F7) or isotype control(eBioscience). Analysis of IL-17+ gd T cells was performed by sorting gdT cells according to surface markers using the FACSAria cell sorter andstimulation for 4 h with PMA plus ionomycin and brefeldin A, followed bysurface staining, fixation, and permeabilization of cells using an intracel-lular staining kit (BD Biosciences) and staining with the anti–IL-17a Ab(TC11-18H10.1, BioLegend). FACS was performed using FITC, PE,PerCP5.5, PE-Cy7, allophycocyanin, and Pacific Blue dyes. Dead cellswere excluded by propidium iodide staining or Live/Dead fixable deadcell stain kits (Life Technologies), and doublets were excluded based onforward scatter width and size distributions.
BrdU labeling
Analysis of in vivo proliferation was performed by a single i.p. injection ofBrdU (10 mg/ml) in PBS to the mother. Twenty-four hours later, motherswere sacrificed and fetuses dissected to analyze BrdU incorporation inthymic gd T cells by FACS analysis
RT-PCR
Quantitative PCR was analyzed using SYBR Green in a 7900KT Fast real-timePCR system. The following primers were used: Zbtb16 forward, 59-TCTGA-CAAAGATGGGGATGATCC-39, reverse, 59-CAGTATTCCGTGCAGATGGT-ACAC-39; Bgl2 forward, 59-GGTGGGGTCATGTGTGTGG-39, reverse, 59-CGGTTCAGGTACTCAGTCATCC-39; Vg6 forward, 59-GAAGCCCGATG-CATACATAC-39, reverse, 59-GGGAAATGTCTGCATCAAGC-39; Vg5forward, 59-GAGGATCCCGCTTGGAAATGGATGAGA-39, reverse, 59-CTTATGGAGATTTGTTTCAGC-39; Sox13 forward, 59-TACTCACA-GACTGATCCCCAG-39, reverse, 59-AAGTGCAGTTTCACCCATCAC-39; Runx1 forward, 59-TGAACCACTCCACTGCCTTTAA-39, reverse,59-AATGGGTGTCGCTGGGTG-39; Rorc forward, 59-TGCAAGAC-TCATCGACAAGGC-39, reverse, 59-AGCTTTTCCACATGTTGGCTG-39; Hprt forward, 59-TCAGTCAACGGGGGACATAAA-39, reverse, 59-GGGGCTGTACTGCTTAACCAG-39.
Statistical analysis
Plotting of the data and statistical analysis were performed using GraphPadPrism software using an unpaired standard t test to compare two pop-ulations. The significance obtained is represented in the graphs.
ResultsPLZF is expressed in fetal Vg5+ and Vg6+ gd T cells
PLZF is required for the innate-like differentiation of iNKT andVg1.1Vd6.3 gd T cells. As fetal hematopoiesis preferentially givesrise to innate-like subsets, we assessed whether PLZF expression isalso shared with other innate-like T cells of fetal origin. To this end,we analyzed intracellular PLZF levels in different thymic subsets in
E16 and day 1 neonatal thymi. PLZF was present in early thymicprogenitors (ETPs) of fetal origin, increasing at the double-negative(DN)2a stage and then gradually decreasing as cells progressedtoward the DN3 and DN4 stages. Analysis of PLZF mean fluo-rescence intensity levels in fetal and neonatal gd T cells identified∼50% lower levels in day 1 neonatal cells as compared with fetalgd T cells (Fig. 1A). We next analyzed whether the presence ofPLZF protein correlated with increased expression by measuringZbtb16 (PLZF) mRNA transcripts in fetal-derived gdT cells.We included CD4 single-positive thymocytes, iNKT cells, and gdT cells from adult thymus as reference controls for the analysis.Zbtb16 mRNA levels were high in fetal (E16) gdT cells and werereduced 80-fold in neonatal (day 1) gd T cells (Fig. 1B), indicatingthat high PLZF expression is restricted to gdT cell subsets thatdevelop at the earliest fetal stages. The low Zbtb16 mRNA levelsthat we detected in adult gd T cells may correspond to the PLZF+
Vg1.1Vd6.3 gd T cell subset that develops after birth (6). Inter-estingly, Zbtb16 mRNA levels in fetal gd T cells were ∼4-foldhigher than those of adult iNKT cells. Furthermore, the high levelsof PLZF in DN2 thymocytes in the fetal thymus, which are theprecursors of all T cells and have not been committed to the gdT cell lineage, indicated that PLZF expression occurs before gdT cell selection.To investigate the gd T cell subsets that express PLZF in the
fetal (E16) thymus, we set up to identify Vg5+ and Vg6+ gd T cellsubsets by FACS analysis. We used the 17D1 Ab that recognizesthe Vg5Vd1 gd TCR but can also recognize the Vg6Vd1 gd TCRwhen cells are prestained with the anti-gd TCR GL3 Ab clone(19). To verify the specificity of the 17D1 Ab for Vg6+ gd T cells,we first performed a double staining with an anti-Vg5 Ab and17D1. We observed that 17D1+ cells were Vg5+ as well as Vg52
cells, that some Vg5+ cells did not present 17D1 staining, and thatmany gd T cells did not present Vg5 or 17D1 staining (Fig. 1C).Vg5217D12 gd T cells corresponded mostly to the Vg4 gd T cellsubset (Supplemental Fig. 1). To clearly identify which of these gdT cell subpopulations corresponded to the Vg6 and Vg5 subsets,we sorted the different gd T cell subpopulation and analyzed Vg5and Vg6 transcripts by RT-PCR. As expected, Vg5 transcriptswere present only in Vg5+ gd T cells irrespective of staining withthe 17D1 Ab whereas expression of the Vg6 TCR was restricted toVg5217D1+ gd T cells (Fig. 1D), indicating that the combinationof 17D1 and Vg5 staining could be used to specifically identifyVg6+ gd T cells. Sequence analysis of the purified Vg6 RT-PCRproduct further confirmed that it corresponded to the canonicalinvariant Vg6+ gd TCR (data not shown). High Zbtb16 transcriptlevels were found in both Vg5+ and Vg6+ but not in Vg52Vg62
gd T cell subsets in the E16 thymus (Fig. 1D), suggesting thatPLZF expression may play a role in the development of theselineages.To evaluate whether Vg5+ and Vg6+ gd T cells found in adult
mice were exclusively derived from PLZF-expressing cells, weperformed fate mapping using compound PLZF-CRE transgenic 3Rosa-Tdtomato mice. In this strain, expression of CRE is con-trolled by the PLZF promoter, leading to the excision of a stopcassette in the Rosa promoter and to the constitutive expression ofTdtomato. Therefore, even the transient expression of PLZF leadsto the permanent label of cells and their progeny. Using this fate-mapping model we observed that 100% of Vg5+ gd T cells in theadult skin and 100% of Vg6+ gd T cells in the adult uterus werederived from PLZF-expressing cells, whereas ∼30% of gd T cellsfound in the spleen were derived from PLZF-expressing cells(Fig. 1E). This 30% Tdtomato label in adult gd T cells is a con-sequence of PLZF expression in hematopoietic stem cells, whichalso leads to a 30% Tdtomato label in CD42CD82 DN and CD4+
4274 PLZF CONTROLS THE DEVELOPMENT OF IL-17+ gd T CELLS
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
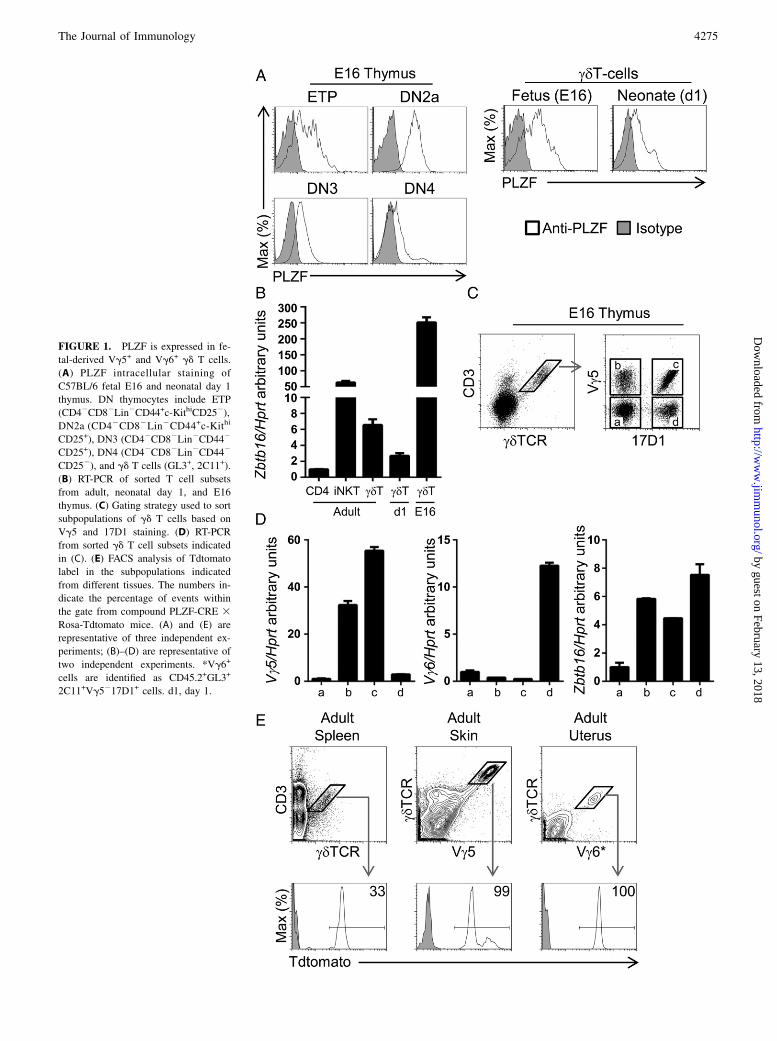
FIGURE 1. PLZF is expressed in fe-
tal-derived Vg5+ and Vg6+ gd T cells.
(A) PLZF intracellular staining of
C57BL/6 fetal E16 and neonatal day 1
thymus. DN thymocytes include ETP
(CD42CD82Lin2CD44+c-KithiCD252),
DN2a (CD42CD82Lin2CD44+c-Kithi
CD25+), DN3 (CD42CD82Lin2CD442
CD25+), DN4 (CD42CD82Lin2CD442
CD252), and gd T cells (GL3+, 2C11+).
(B) RT-PCR of sorted T cell subsets
from adult, neonatal day 1, and E16
thymus. (C) Gating strategy used to sort
subpopulations of gd T cells based on
Vg5 and 17D1 staining. (D) RT-PCR
from sorted gd T cell subsets indicated
in (C). (E) FACS analysis of Tdtomato
label in the subpopulations indicated
from different tissues. The numbers in-
dicate the percentage of events within
the gate from compound PLZF-CRE 3Rosa-Tdtomato mice. (A) and (E) are
representative of three independent ex-
periments; (B)–(D) are representative of
two independent experiments. *Vg6+
cells are identified as CD45.2+GL3+
2C11+Vg5217D1+ cells. d1, day 1.
The Journal of Immunology 4275
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
CD8+ double-positive thymocytes as well as in CD4 and CD8T cells in spleen (20) (Supplemental Fig. 2).Thus, PLZF is expressed in Vg5+ and Vg6+ gd T cells in the
fetal thymus.
PLZF is required for normal development of Vg6+ but not ofVg5+gd T cells
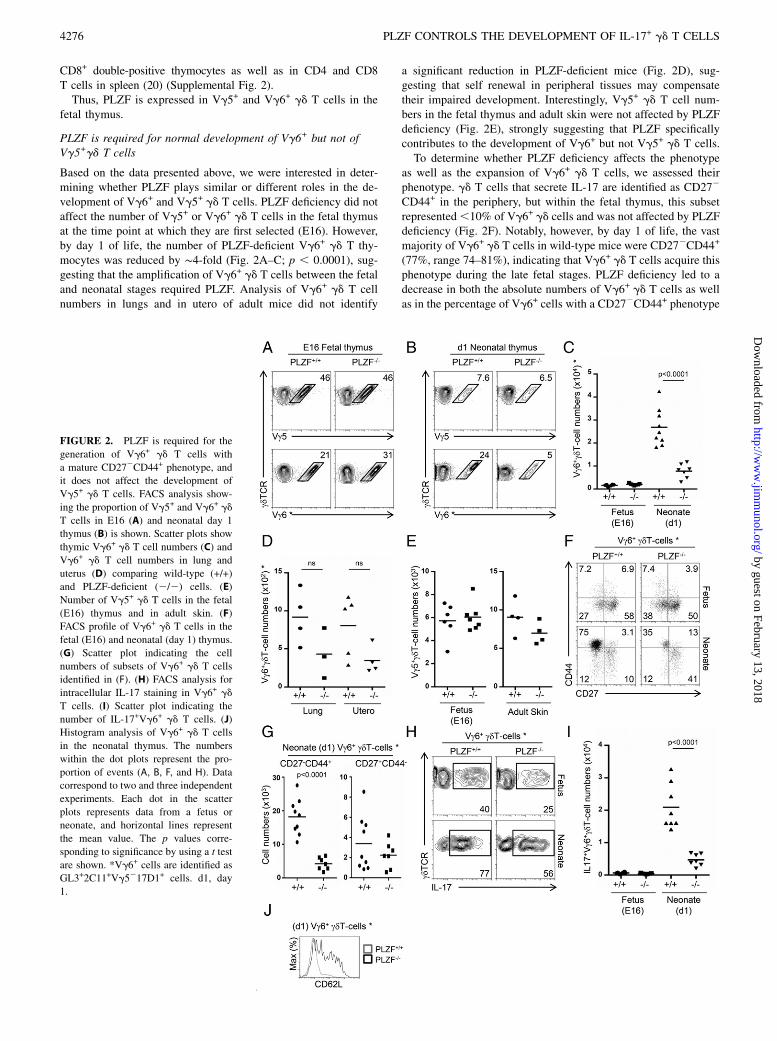
Based on the data presented above, we were interested in deter-mining whether PLZF plays similar or different roles in the de-velopment of Vg6+ and Vg5+ gd T cells. PLZF deficiency did notaffect the number of Vg5+ or Vg6+ gd T cells in the fetal thymusat the time point at which they are first selected (E16). However,by day 1 of life, the number of PLZF-deficient Vg6+ gd T thy-mocytes was reduced by ∼4-fold (Fig. 2A–C; p , 0.0001), sug-gesting that the amplification of Vg6+ gd T cells between the fetaland neonatal stages required PLZF. Analysis of Vg6+ gd T cellnumbers in lungs and in utero of adult mice did not identify
a significant reduction in PLZF-deficient mice (Fig. 2D), sug-gesting that self renewal in peripheral tissues may compensatetheir impaired development. Interestingly, Vg5+ gd T cell num-bers in the fetal thymus and adult skin were not affected by PLZFdeficiency (Fig. 2E), strongly suggesting that PLZF specificallycontributes to the development of Vg6+ but not Vg5+ gd T cells.To determine whether PLZF deficiency affects the phenotype
as well as the expansion of Vg6+ gd T cells, we assessed theirphenotype. gd T cells that secrete IL-17 are identified as CD272
CD44+ in the periphery, but within the fetal thymus, this subsetrepresented,10% of Vg6+ gd cells and was not affected by PLZFdeficiency (Fig. 2F). Notably, however, by day 1 of life, the vastmajority of Vg6+ gd T cells in wild-type mice were CD272CD44+
(77%, range 74–81%), indicating that Vg6+ gd T cells acquire thisphenotype during the late fetal stages. PLZF deficiency led to adecrease in both the absolute numbers of Vg6+ gd T cells as wellas in the percentage of Vg6+ cells with a CD272CD44+ phenotype
FIGURE 2. PLZF is required for the
generation of Vg6+ gd T cells with
a mature CD272CD44+ phenotype, and
it does not affect the development of
Vg5+ gd T cells. FACS analysis show-
ing the proportion of Vg5+ and Vg6+ gd
T cells in E16 (A) and neonatal day 1
thymus (B) is shown. Scatter plots show
thymic Vg6+ gd T cell numbers (C) and
Vg6+ gd T cell numbers in lung and
uterus (D) comparing wild-type (+/+)
and PLZF-deficient (2/2) cells. (E)
Number of Vg5+ gd T cells in the fetal
(E16) thymus and in adult skin. (F)
FACS profile of Vg6+ gd T cells in the
fetal (E16) and neonatal (day 1) thymus.
(G) Scatter plot indicating the cell
numbers of subsets of Vg6+ gd T cells
identified in (F). (H) FACS analysis for
intracellular IL-17 staining in Vg6+ gd
T cells. (I) Scatter plot indicating the
number of IL-17+Vg6+ gd T cells. (J)
Histogram analysis of Vg6+ gd T cells
in the neonatal thymus. The numbers
within the dot plots represent the pro-
portion of events (A, B, F, and H). Data
correspond to two and three independent
experiments. Each dot in the scatter
plots represents data from a fetus or
neonate, and horizontal lines represent
the mean value. The p values corre-
sponding to significance by using a t test
are shown. *Vg6+ cells are identified as
GL3+2C11+Vg5217D1+ cells. d1, day
1.
4276 PLZF CONTROLS THE DEVELOPMENT OF IL-17+ gd T CELLS
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

(50%, range 34–64%) (Fig. 2F). This change in phenotyperesulted in reduced numbers of CD272CD44+ Vg6+ gd T cells inPLZF-deficient mice and similar numbers of CD27+CD442 Vg6+
gd T cells in wild-type and PLZF-deficient mice (Fig. 2G). Theseresults suggest that PLZF specifically regulates the generation ofthe CD272CD44+ subset from immature precursors.
PLZF is required for the IL-17 effector differentiation of Vg6+
gd T cells
Our results indicated that intrathymic Vg6+ gd T cells can besubdivided into different subpopulations based on the expressionof CD27 and CD44. They also showed that the predominant Vg6+
gd T cell subset in the fetal E16 thymus was CD27+CD442
whereas a CD272CD44+ phenotype was predominant in the neo-natal thymus. We therefore assessed whether these phenotypescorresponded to differences in the ability to secrete IL-17 and,furthermore, whether PLZF was required for the acquisition ofthis effector function. Approximately 40% of wild-type Vg6+ gdT cells from the fetal thymus had the capacity to secrete IL-17 andthis proportion increased to ∼80% in the neonatal thymus. Inter-estingly, PLZF deficiency led to a reduced proportion and numbersof IL-17+Vg6+ gd T cells in both the fetal and neonatal thymus(Fig. 2H, 2I), indicating that in the absence of PLZF, the effectormaturation of Vg6+ gd T cells was affected. The impaired ac-quisition of IL-17 secretion in PLZF-deficient Vg6+ gd T cells didnot correlate with increased IFN-g secretion as shown by intra-cellular staining (Supplemental Fig. 3), indicating that PLZF doesnot prevent an alternative differentiation fate into IFN-g–secretingcells. These results show that PLZF is required for the acquisitionof IL-17 effector functions in Vg6+ gd T cells during the fetalperiod. Impaired effector maturation of PLZF-deficient Vg6+ gd
T cells was also evident, as cells failed to downregulate CD62Llevels, a common characteristic of maturation in innate-likeT cells (Fig. 2J).Collectively, these data indicate that PLZF expression is required
for the acquisition of IL-17 secretion in Vg6+ gd T cells, as wellas their maturation into a CD62L2CD272CD44+ phenotype.
PLZF is required for immature CD27+CD442 Vg6+ gd T cellsto develop into mature CD272CD44+ IL-17–secreting cells
To specifically determine whether CD27+CD442 Vg6+ gd T cellsfound in the E16 fetal thymus were the precursors of CD272
CD44+ cells, we sorted CD27+CD442 Vg6+ gd T cell subsetsfrom wild-type and PLZF-deficient E16 thymuses and assessedtheir differentiation potential in vitro in cocultures with the stromacell line OP9-DL1 for 3 d. Wild-type CD27+CD442 Vg6+ gd
T cells gave rise to a majority of CD272CD44+ cells after culture,indicating that CD27+CD442 cells are the precursors of CD272
CD44+ cells. Importantly, CD27+CD442 Vg6+ gd T cells wereunable to generate the CD272CD44+ subset in the absence ofPLZF, revealing a requirement for PLZF at this maturation step(Fig. 3A). Thus, PLZF is required for the maturation of CD27+
CD442 Vg6+ gd T cells to a CD272CD44+ phenotype.We next assessed whether PLZF-deficient immature CD27+
CD442 Vg6+ gd T cells from fetal E16 thymus could acquire IL-17 secretion after in vitro culture. Approximately 20% of sortedimmature CD27+CD442 E16 Vg6+ wild-type gd T cells werecapable of secreting IL-17, and this proportion increased to 50%after a 3-d in vitro coculture on OP9-DL1 stroma, indicating thatmaturation into a CD272CD44+ phenotype correlates with theacquisition of IL-17 secretion. Immature PLZF-deficient CD27+
CD442 Vg6+ gd T cells already presented a reduced IL-17 se-cretion profile and, consistent with their lack of maturation in theOP9-DL1 system, the proportion of IL-17+ cells did not increase
(Fig. 3B). During these in vitro differentiation cocultures we didnot detect a significant increase in apoptosis in either wild-type orPLZF-deficient Vg6+ gd T cells (Fig. 3C), suggesting that PLZF isnot providing a prosurvival role.Interestingly, analysis of proliferation by dilution of cell trace
label identified that PLZF-deficient CD27+CD442 Vg6+ gd Tcells proliferated at a lower rate than did wild-type cells duringthe OP9-DL1 cocultures (Fig. 3D). We also observed that PLZF-deficient E16 Vg6+ gd T cells incorporated lower levels ofBrdU 24 h after BrdU injection into the mother (Fig. 3E). Theseresults indicate that PLZF is required for the proliferation ofimmature Vg6+ gd T cells, opening the possibility that lack ofproliferation may be a causal mechanism impairing the maturationof PLZF-deficient cells.To test whether impaired proliferation was sufficient to prevent
the maturation of Vg6+ gd T cells, we sorted wild-type CD27+
CD442 Vg6+ gd T cells and cocultured them with OP9-DL1stroma in the presence of different concentrations of a cyclin ki-nase 4/6 inhibitor (PD0332991) to inhibit their proliferation. Weobserved that 100 nM treatment led to stronger inhibition ofproliferation than did 50 nM treatment, as observed by dilutionof CellTrace Violet (Fig. 3F). Interestingly, 50 and 100 nMPD0332991 treatment led to ∼20 and 65% inhibition in the gen-eration of mature CD272CD44+Vg6+ gd T cells from immatureCD27+CD442 progenitors, respectively (Fig. 3G), strongly sug-gesting that the inability of PLZF-deficient CD27+CD442 Vg6+
gd T cells to proliferate is causative of their maturation defect. Incorrelation with the requirement of proliferation for cells to ac-quire a mature CD272CD44+ phenotype, inhibition of prolifera-tion during the in vitro cultures also led to a reduction of IL-17secretion in wild-type Vg6+ gd T cells (Fig. 3H). Impaired IL-17effector maturation of PLZF-deficient Vg6+ gd T cells correlatedwith impaired expression of Sox13, Rorc, and Runx1, whichcontrol the IL-17 program in gd T cells and Th17 cells (Fig. 3I).Altogether, these results show that PLZF-deficient CD27+
CD442 Vg6+ gd T cells fail to proliferate and acquire a ma-ture CD272CD44+ phenotype with increased IL-17 secretion.
Transgenic Bcl2 expression does not rescue Vg6+ gd T celldevelopment in PLZF-deficient mice
Although we did not observe increased apoptosis of PLZF-deficientVg6+ gd T cells in vitro, reduced survival of PLZF-deficientVg6+ cells in vivo could still contribute to their impaired devel-opment. To evaluate this hypothesis, we generated compoundPLZF-deficient 3 lck–Bcl-2 transgenic mice because transgenicexpression of Bcl2 has been shown to prevent the apoptosisof lymphocytes to different stimuli (21). Whereas Bcl2 expressionin sorted Vg6+ gd T cells from transgenic neonatal thymus wasincreased by 20-fold (Fig. 4A), there was no rescue of Vg6+ gd
T cells in the Bcl2-transgenic PLZF-deficient neonatal thymus(Fig. 4B, 4C). The reduced proportion of IL-17+Vg6+ gd T cells inPLZF-deficient neonates was also not rescued by transgenic Bcl2expression (Fig. 4D, 4E). These results suggest that increasedapoptosis is not the underlying mechanism responsible for thedevelopmental impairment of PLZF-deficient Vg6+ gd T cells.
PLZF deficiency impairs the IL-17 effector maturation of Skint-1 mutant Vg5+ gd T cells
Skint-1 is an Ig family gene expressed in thymic epithelial cellsand keratinocytes. Interestingly, in Skint-1 mutant mice, Vg5+ gdT cells do not acquire the ability to secrete IFN-g and insteadacquire a IL-17–secreting fate and migrate to the epithelia of theuterus instead of the skin, similar to Vg6+ gd T cells (22). Basedon this, it was postulated that the IL-17 program in gd T cells is
The Journal of Immunology 4277
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
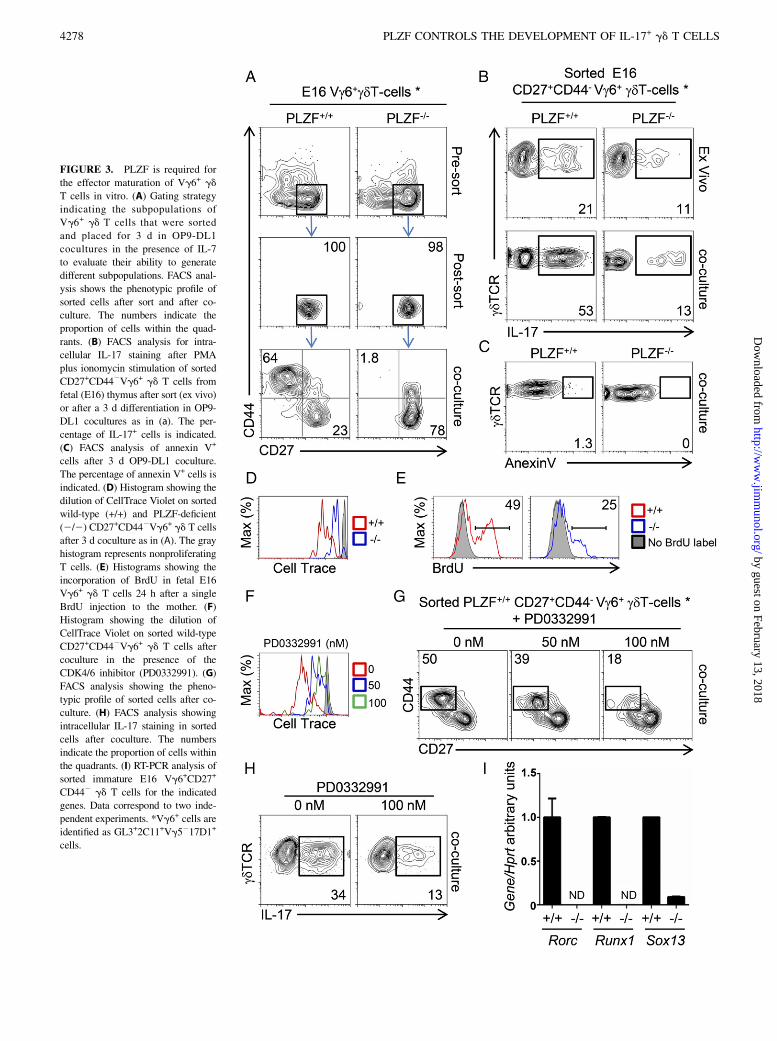
FIGURE 3. PLZF is required for
the effector maturation of Vg6+ gd
T cells in vitro. (A) Gating strategy
indicating the subpopulations of
Vg6+ gd T cells that were sorted
and placed for 3 d in OP9-DL1
cocultures in the presence of IL-7
to evaluate their ability to generate
different subpopulations. FACS anal-
ysis shows the phenotypic profile of
sorted cells after sort and after co-
culture. The numbers indicate the
proportion of cells within the quad-
rants. (B) FACS analysis for intra-
cellular IL-17 staining after PMA
plus ionomycin stimulation of sorted
CD27+CD442Vg6+ gd T cells from
fetal (E16) thymus after sort (ex vivo)
or after a 3 d differentiation in OP9-
DL1 cocultures as in (a). The per-
centage of IL-17+ cells is indicated.
(C) FACS analysis of annexin V+
cells after 3 d OP9-DL1 coculture.
The percentage of annexin V+ cells is
indicated. (D) Histogram showing the
dilution of CellTrace Violet on sorted
wild-type (+/+) and PLZF-deficient
(2/2) CD27+CD442Vg6+ gd T cells
after 3 d coculture as in (A). The gray
histogram represents nonproliferating
T cells. (E) Histograms showing the
incorporation of BrdU in fetal E16
Vg6+ gd T cells 24 h after a single
BrdU injection to the mother. (F)
Histogram showing the dilution of
CellTrace Violet on sorted wild-type
CD27+CD442Vg6+ gd T cells after
coculture in the presence of the
CDK4/6 inhibitor (PD0332991). (G)
FACS analysis showing the pheno-
typic profile of sorted cells after co-
culture. (H) FACS analysis showing
intracellular IL-17 staining in sorted
cells after coculture. The numbers
indicate the proportion of cells within
the quadrants. (I) RT-PCR analysis of
sorted immature E16 Vg6+CD27+
CD442 gd T cells for the indicated
genes. Data correspond to two inde-
pendent experiments. *Vg6+ cells are
identified as GL3+2C11+Vg5217D1+
cells.
4278 PLZF CONTROLS THE DEVELOPMENT OF IL-17+ gd T CELLS
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
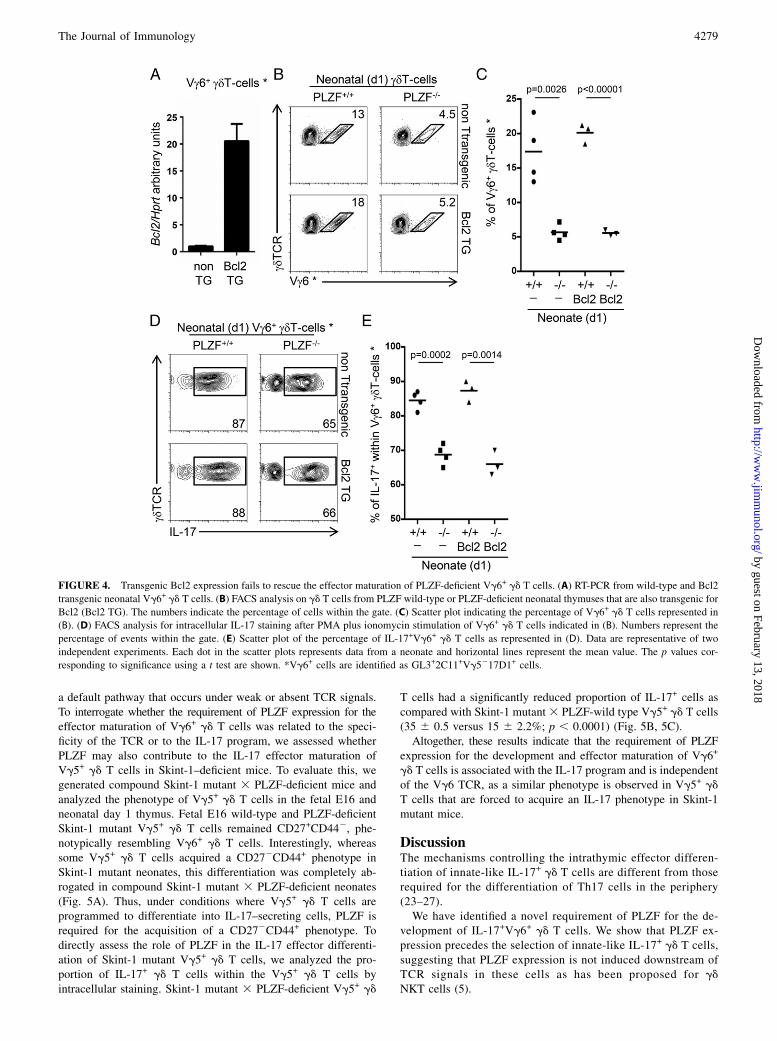
a default pathway that occurs under weak or absent TCR signals.To interrogate whether the requirement of PLZF expression for theeffector maturation of Vg6+ gd T cells was related to the speci-ficity of the TCR or to the IL-17 program, we assessed whetherPLZF may also contribute to the IL-17 effector maturation ofVg5+ gd T cells in Skint-1–deficient mice. To evaluate this, wegenerated compound Skint-1 mutant 3 PLZF-deficient mice andanalyzed the phenotype of Vg5+ gd T cells in the fetal E16 andneonatal day 1 thymus. Fetal E16 wild-type and PLZF-deficientSkint-1 mutant Vg5+ gd T cells remained CD27+CD442, phe-notypically resembling Vg6+ gd T cells. Interestingly, whereassome Vg5+ gd T cells acquired a CD272CD44+ phenotype inSkint-1 mutant neonates, this differentiation was completely ab-rogated in compound Skint-1 mutant 3 PLZF-deficient neonates(Fig. 5A). Thus, under conditions where Vg5+ gd T cells areprogrammed to differentiate into IL-17–secreting cells, PLZF isrequired for the acquisition of a CD272CD44+ phenotype. Todirectly assess the role of PLZF in the IL-17 effector differenti-ation of Skint-1 mutant Vg5+ gd T cells, we analyzed the pro-portion of IL-17+ gd T cells within the Vg5+ gd T cells byintracellular staining. Skint-1 mutant 3 PLZF-deficient Vg5+ gd
T cells had a significantly reduced proportion of IL-17+ cells ascompared with Skint-1 mutant 3 PLZF-wild type Vg5+ gd T cells(35 6 0.5 versus 15 6 2.2%; p , 0.0001) (Fig. 5B, 5C).Altogether, these results indicate that the requirement of PLZF
expression for the development and effector maturation of Vg6+
gd T cells is associated with the IL-17 program and is independentof the Vg6 TCR, as a similar phenotype is observed in Vg5+ gd
T cells that are forced to acquire an IL-17 phenotype in Skint-1mutant mice.
DiscussionThe mechanisms controlling the intrathymic effector differen-tiation of innate-like IL-17+ gd T cells are different from thoserequired for the differentiation of Th17 cells in the periphery(23–27).We have identified a novel requirement of PLZF for the de-
velopment of IL-17+Vg6+ gd T cells. We show that PLZF ex-pression precedes the selection of innate-like IL-17+ gd T cells,suggesting that PLZF expression is not induced downstream ofTCR signals in these cells as has been proposed for gdNKT cells (5).
FIGURE 4. Transgenic Bcl2 expression fails to rescue the effector maturation of PLZF-deficient Vg6+ gd T cells. (A) RT-PCR from wild-type and Bcl2
transgenic neonatal Vg6+ gd T cells. (B) FACS analysis on gd T cells from PLZF wild-type or PLZF-deficient neonatal thymuses that are also transgenic for
Bcl2 (Bcl2 TG). The numbers indicate the percentage of cells within the gate. (C) Scatter plot indicating the percentage of Vg6+ gd T cells represented in
(B). (D) FACS analysis for intracellular IL-17 staining after PMA plus ionomycin stimulation of Vg6+ gd T cells indicated in (B). Numbers represent the
percentage of events within the gate. (E) Scatter plot of the percentage of IL-17+Vg6+ gd T cells as represented in (D). Data are representative of two
independent experiments. Each dot in the scatter plots represents data from a neonate and horizontal lines represent the mean value. The p values cor-
responding to significance using a t test are shown. *Vg6+ cells are identified as GL3+2C11+Vg5217D1+ cells.
The Journal of Immunology 4279
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Although PLZF is not expressed at the earliest stages of T celldevelopment in the adult thymus, we have found that ETPs in thefetal thymus are PLZF-expressing cells. A unique characteristicof fetal hemopoietic stem cells is the expression of Lin28b, whichblocks the production of mature Let-7 microRNAs (28). As PLZFwas recently shown to be regulated by Let-7 microRNAs (29), itis reasonable to speculate that the increase of PLZF levels duringfetal hematopoiesis might be due to increased Lin28b and reducedLet-7 levels in fetal progenitors. We have also observed that PLZFlevels are further increased in DN2 thymocytes in the fetal thy-mus. Interestingly, Zbtb16 (PLZF) expression has been previouslyidentified in DN2 thymocytes of adult mice (30), indicating thatPLZF may be induced as cells develop into DN2a thymocytes. AsIL-7 plays a critical role for the generation of DN2a thymocytes aswell as in the maintenance of IL-17+ gd T cells, it is possible thatPLZF is positively regulated or maintained by IL-7 signals. Al-though, PLZF expression already occurs before gdT cell selectionin the fetal thymus, a role of TCR signals in regulating PLZFexpression cannot be ruled out. It was originally postulated thatthe IL-17 program in fetal Vg6+ gd T cells occurs in the absenceof TCR engagement (22); however, later work identified that Vg6+
gd T cells have received TCR signals during development (31),and therefore it is possible that TCR signals maintain PLZF ex-pression in Vg6+ gd T cells.We also show that PLZF specifically allows the differentiation
of fetal-derived IL-17+ gdT cells, but not IFN-g+ gd T cells, de-spite sharing a common PLZF+ precursor. Therefore, PLZF-driveninnate-like differentiation is not exclusive to NKT cells or IL-4+
T cell subsets (5, 32), but it also controls the differentiation ofinnate-like IL-17+ gd T cells.We have observed that PLZF is required for both the develop-
ment and acquisition of IL-17 secretion in Vg6+ gd T cells as wellas in Vg5+ gd T cells when redirected into a IL-17–secretingphenotype in Skint-1 mutant mice, indicating that the requirementof PLZF expression for the development of IL-17–secreting cellsis independent of the specificity of the TCR. Interestingly, de-velopment of Vg5+ gd T cells was not affected by the absence ofPLZF in Skint-1 wild-type mice when acquiring their IFN-gprogram, suggesting that TCR signals during differentiation maycompensate the requirement of PLZF expression. In fact, theability of transgenic PLZF expression to confer effector functionsto the conventional naive CD4 T cell population in the absence ofTCR stimulation suggests that PLZF and TCR signals may playredundant roles in providing the effector maturation to T cells(8, 33).Lack of surface CD27 identifies IL-17+ gd T cells in peripheral
tissues (12, 13). However, in the fetal thymus, some Vg6+ gd
T cells acquired IL-17 secretion but have not yet downregulatedCD27 levels, indicating that downregulation of CD27 is not re-quired for their effector maturation.We have characterized the different developmental stages of
Vg6+ gd T cells and have shown that CD27+CD442 Vg6+ gdT cells are the precursors of CD272CD44+ Vg6+ gd T cells,which are the major subset present in the neonatal day 1 thymusand secrete IL-17. PLZF was required for this developmentalstep, as the number of mature CD272CD44+Vg6+ gd T cellswas highly reduced in the neonatal thymus, and PLZF-deficientimmature CD27+CD442Vg6+ gd T cells did not proliferate andgenerate mature CD272CD44+Vg6+ gd T cells in vitro. PLZF-mediated proliferation of immature CD27+CD442Vg6+ gd T cellsis required for the effector maturation into a CD272CD44+IL-17+
phenotype, as blocking proliferation in wild-type immatureCD27+CD442Vg6+ gd T cells also prevented their maturation.Impaired maturation of PLZF-deficient Vg6+ gdT cells was alsoevident as inefficient CD62L downregulation, a phenotype asso-ciated with the maturation of innate-like T cells that is disruptedin PLZF-deficient iNKT and gd NKT cells (4, 6). It is not clearhow a defect in proliferation leads to a maturation block in Vg6+
gd T cells. However, previous reports identified a requirement ofproliferation for developmental progression in T cells. The ef-fector maturation of Th cells into IFN-g–, IL-4–, and IL-10–se-creting cells was shown to be dependent on proliferation (34, 35),and, more recently, b-selection–induced proliferation was shownto be required for the generation of double-positive thymocytes(36). Presumably, epigenetic changes during the cell cycle may berequired for developmental progression (37).It is also not clear how PLZF controls the proliferation of im-
mature Vg6+ gd T cells. Although overexpression of PLZF leadsto a block of cell cycle progression in a myelomonocytic cellline (U937T) (38), it was recently described that PLZF expressionmay stimulate proliferation in NIH3T3 cells by repressing ex-pression of Cdkn1a (p21), a negative regulator of cell cycle pro-gression (39); however, we did not find increased Cdkn1a levels inPLZF-deficient Vg6+ gd T cells (data not shown), indicating thatPLZF is required for other mechanisms of cell cycle control inVg6+ gd T cells.Despite the deficient development of Vg6+ gd T cells in the
absence of PLZF, Vg6+ gd T cells were present in the lungs anduterus of adult mice, indicating that PLZF-deficient Vg6+ gdT cells are able to self-renew in the periphery.Although transgenic PLZF expression protects from superantigen-
mediated negative selection in conventional T cells (33), a prosurvival
FIGURE 5. Impaired IL-17 effector maturation of PLZF-deficient Skint-
1 mutant Vg5+ gd T cells. (A) FACS analysis of gated Vg5+ gd T cells
from fetal (E16) and neonatal (day 1) thymus in the indicated strain. The
numbers indicate the percentage of cells within the gate. (B) FACS analysis
for intracellular IL-17 staining after PMA plus ionomycin stimulation of
Vg5+ gd T cells in Skint-1 mutant (E16) thymus. (C) Scatter plot of the
percentage of IL-17+Vg5+ gd T cells as represented in (B). Data are rep-
resentative of two independent experiments. Each dot in the scatter plots
represents data from a fetus and horizontal lines represent the mean value.
The p values corresponding to significance using a t test are shown.
4280 PLZF CONTROLS THE DEVELOPMENT OF IL-17+ gd T CELLS
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

function does not seem to be the main role of PLZF in controllingthe effector differentiation of Vg6+ gd T cells. We did not detectincreased apoptosis of PLZF-deficient Vg6+ gdT cells; also, trans-genic Bcl2 expression, which protects against several apoptoticstimuli in thymocytes, did not rescue the development of PLZF-deficient Vg6+ gd T cells.Interestingly, a recent report identified that PLZF is expressed
in human Th17 cells, binds to the CCR6 promoter, and regulatesthe expression of IL-17–associated genes (40), suggesting thatPLZF may control IL-17 effector maturation in other cell types.Collectively, we have identified that PLZF not only controls
the effector maturation of NKT and IL-4+gdT cells but also thatof innate-like IL-17+ gd T cells.
AcknowledgmentsWe thank Dr. Naomi Taylor and Dr. Avinash Bhandoola for critically
reviewing the manuscript, Dr. Derek Sant’Angelo for providing the
PLZF-CRE 3 Rosa-Tdtomato strain, Dr. Robert Tigelaar for the 17D1
Ab, and Dr. Juan Carlos Zuniga-Pflucker for the OP9-DL1 cell line.
DisclosuresThe authors have no financial conflicts of interest.
References1. Vantourout, P., and A. Hayday. 2013. Six-of-the-best: unique contributions of gd
T cells to immunology. Nat. Rev. Immunol. 13: 88–100.2. Constantinides, M. G., and A. Bendelac. 2013. Transcriptional regulation of the
NKT cell lineage. Curr. Opin. Immunol. 25: 161–167.3. Kovalovsky, D., O. U. Uche, S. Eladad, R. M. Hobbs, W. Yi, E. Alonzo, K. Chua,
M. Eidson, H. J. Kim, J. S. Im, et al. 2008. The BTB-zinc finger transcriptionalregulator PLZF controls the development of invariant natural killer T cell ef-fector functions. Nat. Immunol. 9: 1055–1064.
4. Savage, A. K., M. G. Constantinides, J. Han, D. Picard, E. Martin, B. Li,O. Lantz, and A. Bendelac. 2008. The transcription factor PLZF directs theeffector program of the NKT cell lineage. Immunity 29: 391–403.
5. Kreslavsky, T., A. K. Savage, R. Hobbs, F. Gounari, R. Bronson, P. Pereira,P. P. Pandolfi, A. Bendelac, and H. von Boehmer. 2009. TCR-inducible PLZFtranscription factor required for innate phenotype of a subset of gd T cells withrestricted TCR diversity. Proc. Natl. Acad. Sci. USA 106: 12453–12458.
6. Alonzo, E. S., R. A. Gottschalk, J. Das, T. Egawa, R. M. Hobbs, P. P. Pandolfi,P. Pereira, K. E. Nichols, G. A. Koretzky, M. S. Jordan, and D. B. Sant’Angelo.2010. Development of promyelocytic zinc finger and ThPOK-expressing innategd T cells is controlled by strength of TCR signaling and Id3. J. Immunol. 184:1268–1279.
7. Gerart, S., S. Siberil, E. Martin, C. Lenoir, C. Aguilar, C. Picard, O. Lantz,A. Fischer, and S. Latour. 2013. Human iNKT and MAIT cells exhibit a PLZF-dependent proapoptotic propensity that is counterbalanced by XIAP. Blood 121:614–623.
8. Kovalovsky, D., E. S. Alonzo, O. U. Uche, M. Eidson, K. E. Nichols, andD. B. Sant’Angelo. 2010. PLZF induces the spontaneous acquisition of memory/effector functions in T cells independently of NKT cell-related signals. J.Immunol. 184: 6746–6755.
9. Thomas, S. Y., S. T. Scanlon, K. G. Griewank, M. G. Constantinides,A. K. Savage, K. A. Barr, F. Meng, A. D. Luster, and A. Bendelac. 2011. PLZFinduces an intravascular surveillance program mediated by long-lived LFA-1-ICAM-1 interactions. J. Exp. Med. 208: 1179–1188.
10. Girardi, M., J. Lewis, E. Glusac, R. B. Filler, L. Geng, A. C. Hayday, andR. E. Tigelaar. 2002. Resident skin-specific gammadelta T cells provide local,nonredundant regulation of cutaneous inflammation. J. Exp. Med. 195: 855–867.
11. Jameson, J., K. Ugarte, N. Chen, P. Yachi, E. Fuchs, R. Boismenu, andW. L. Havran. 2002. A role for skin gd T cells in wound repair. Science 296:747–749.
12. Ribot, J. C., A. deBarros, D. J. Pang, J. F. Neves, V. Peperzak, S. J. Roberts,M. Girardi, J. Borst, A. C. Hayday, D. J. Pennington, and B. Silva-Santos. 2009.CD27 is a thymic determinant of the balance between interferon-g- and inter-leukin 17-producing gd T cell subsets. Nat. Immunol. 10: 427–436.
13. Haas, J. D., S. Ravens, S. D€uber, I. Sandrock, L. Oberdorfer, E. Kashani,V. Chennupati, L. Fohse, R. Naumann, S. Weiss, et al. 2012. Development ofinterleukin-17-producing gd T cells is restricted to a functional embryonic wave.Immunity 37: 48–59.
14. Michel, M. L., D. J. Pang, S. F. Haque, A. J. Potocnik, D. J. Pennington, andA. C. Hayday. 2012. Interleukin 7 (IL-7) selectively promotes mouse and humanIL-17-producing gd cells. Proc. Natl. Acad. Sci. USA 109: 17549–17554.
15. Matsuzaki, G., and M. Umemura. 2007. Interleukin-17 as an effector molecule ofinnate and acquired immunity against infections.Microbiol. Immunol. 51: 1139–1147.
16. O’Brien, R. L., and W. K. Born. 2010. gd T cell subsets: a link between TCR andfunction? Semin. Immunol. 22: 193–198.
17. Sutton, C. E., S. J. Lalor, C. M. Sweeney, C. F. Brereton, E. C. Lavelle, andK. H. Mills. 2009. Interleukin-1 and IL-23 induce innate IL-17 production fromgd T cells, amplifying Th17 responses and autoimmunity. Immunity 31: 331–341.
18. Linette, G. P., Y. Li, K. Roth, and S. J. Korsmeyer. 1996. Cross talk between celldeath and cell cycle progression: BCL-2 regulates NFAT-mediated activation.Proc. Natl. Acad. Sci. USA 93: 9545–9552.
19. Roark, C. L., M. K. Aydintug, J. Lewis, X. Yin, M. Lahn, Y. S. Hahn,W. K. Born, R. E. Tigelaar, and R. L. O’Brien. 2004. Subset-specific, uniformactivation among Vg6/Vd1+ gd T cells elicited by inflammation. J. Leukoc. Biol.75: 68–75.
20. Lynch, L., X. Michelet, S. Zhang, P. J. Brennan, A. Moseman, C. Lester,G. Besra, E. E. Vomhof-Dekrey, M. Tighe, H. F. Koay, et al. 2015. RegulatoryiNKT cells lack expression of the transcription factor PLZF and control thehomeostasis of Treg cells and macrophages in adipose tissue. Nat. Immunol. 16:85–95.
21. Chao, D. T., G. P. Linette, L. H. Boise, L. S. White, C. B. Thompson, andS. J. Korsmeyer. 1995. Bcl-XL and Bcl-2 repress a common pathway of celldeath. J. Exp. Med. 182: 821–828.
22. Turchinovich, G., and A. C. Hayday. 2011. Skint-1 identifies a common mo-lecular mechanism for the development of interferon-g-secreting versusinterleukin-17-secreting gd T cells. Immunity 35: 59–68.
23. Rachitskaya, A. V., A. M. Hansen, R. Horai, Z. Li, R. Villasmil, D. Luger,R. B. Nussenblatt, and R. R. Caspi. 2008. Cutting edge: NKT cells constitutivelyexpress IL-23 receptor and RORgt and rapidly produce IL-17 upon receptorligation in an IL-6-independent fashion. J. Immunol. 180: 5167–5171.
24. Lochner, M., L. Peduto, M. Cherrier, S. Sawa, F. Langa, R. Varona,D. Riethmacher, M. Si-Tahar, J. P. Di Santo, and G. Eberl. 2008. In vivo equi-librium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgt+
T cells. J. Exp. Med. 205: 1381–1393.25. Shibata, K., H. Yamada, T. Sato, T. Dejima, M. Nakamura, T. Ikawa, H. Hara,
S. Yamasaki, R. Kageyama, Y. Iwakura, et al. 2011. Notch-Hes1 pathway isrequired for the development of IL-17-producing gd T cells. Blood 118: 586–593.
26. Malhotra, N., K. Narayan, O. H. Cho, K. E. Sylvia, C. Yin, H. Melichar,M. Rashighi, V. Lefebvre, J. E. Harris, L. J. Berg, and J. Kang, ImmunologicalGenome Project Consortium. 2013. A network of high-mobility group boxtranscription factors programs innate interleukin-17 production. Immunity 38:681–693.
27. Serre, K., and B. Silva-Santos. 2013. Molecular mechanisms of differentiation ofmurine pro-inflammatory gd T cell subsets. Front. Immunol. 4: 431.
28. Yuan, J., C. K. Nguyen, X. Liu, C. Kanellopoulou, and S. A. Muljo. 2012.Lin28b reprograms adult bone marrow hematopoietic progenitors to mediatefetal-like lymphopoiesis. Science 335: 1195–1200.
29. Pobezinsky, L. A., R. Etzensperger, S. Jeurling, A. Alag, T. Kadakia,T. M. McCaughtry, M. Y. Kimura, S. O. Sharrow, T. I. Guinter, L. Feigenbaum,and A. Singer. 2015. Let-7 microRNAs target the lineage-specific transcriptionfactor PLZF to regulate terminal NKT cell differentiation and effector function.Nat. Immunol. 16: 517–524.
30. Li, L., M. Leid, and E. V. Rothenberg. 2010. An early T cell lineagecommitment checkpoint dependent on the transcription factor Bcl11b.Science 329: 89–93.
31. Wencker, M., G. Turchinovich, R. Di Marco Barros, L. Deban, A. Jandke,A. Cope, and A. C. Hayday. 2014. Innate-like T cells straddle innate andadaptive immunity by altering antigen-receptor responsiveness. Nat. Immunol.15: 80–87.
32. Lee, Y. J., K. L. Holzapfel, J. Zhu, S. C. Jameson, and K. A. Hogquist.2013. Steady-state production of IL-4 modulates immunity in mousestrains and is determined by lineage diversity of iNKT cells. Nat. Immunol.14: 1146–1154.
33. Savage, A. K., M. G. Constantinides, and A. Bendelac. 2011. Promyelocyticleukemia zinc finger turns on the effector T cell program without requirement foragonist TCR signaling. J. Immunol. 186: 5801–5806.
34. Bird, J. J., D. R. Brown, A. C. Mullen, N. H. Moskowitz, M. A. Mahowald,J. R. Sider, T. F. Gajewski, C. R. Wang, and S. L. Reiner. 1998. Helper T celldifferentiation is controlled by the cell cycle. Immunity 9: 229–237.
35. Richter, A., M. Lohning, and A. Radbruch. 1999. Instruction for cytokine ex-pression in T helper lymphocytes in relation to proliferation and cell cycleprogression. J. Exp. Med. 190: 1439–1450.
36. Kreslavsky, T., M. Gleimer, M. Miyazaki, Y. Choi, E. Gagnon, C. Murre,P. Sicinski, and H. von Boehmer. 2012. b-Selection-induced proliferation isrequired for ab T cell differentiation. Immunity 37: 840–853.
37. Mullen, A. C., A. S. Hutchins, A. V. Villarino, H. W. Lee, F. A. High, N. Cereb,S. Y. Yang, X. Hua, and S. L. Reiner. 2001. Cell cycle controlling the silencingand functioning of mammalian activators. Curr. Biol. 11: 1695–1699.
38. Konrad, T. A., A. Karger, H. Hackl, I. Schwarzinger, I. Herbacek, and R. Wieser.2009. Inducible expression of EVI1 in human myeloid cells causes phenotypesconsistent with its role in myelodysplastic syndromes. J. Leukoc. Biol. 86: 813–822.
39. Choi, W. I., M. Y. Kim, B. N. Jeon, D. I. Koh, C. O. Yun, Y. Li, C. E. Lee, J. Oh,K. Kim, and M. W. Hur. 2014. Role of promyelocytic leukemia zinc finger(PLZF) in cell proliferation and cyclin-dependent kinase inhibitor 1A (p21WAF/CDKN1A) gene repression. J. Biol. Chem. 289: 18625–18640.
40. Singh, S. P., H. H. Zhang, H. Tsang, P. J. Gardina, T. G. Myers, V. Nagarajan,C. H. Lee, and J. M. Farber. 2015. PLZF regulates CCR6 and is critical for theacquisition and maintenance of the Th17 phenotype in human cells. J. Immunol.194: 4350–4361.
The Journal of Immunology 4281
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from


















