
Pharmacological postconditioning with the phytoestrogen genistein
9
Original article Pharmacological postconditioning with the phytoestrogen genistein R. Tissier a,b,c,1 , X. Waintraub a,b,c,1 , N. Couvreur a,b,c , M. Gervais f , P. Bruneval e , C. Mandet e , R. Zini a,b,c , B. Enriquez a,b,c , A. Berdeaux a,b,c,d, ⁎ , B. Ghaleh a,b,c,d a INSERM, U 660, Créteil, F-94010, France b Université Paris XII, Faculté de Médecine, Laboratoire de Pharmacologie, Créteil, F-94000, France c Ecole Nationale Vétérinaire d’Alfort, Maisons-Alfort, F-94700, France d AP-HP, Groupe hospitalier Henri Mondor - Albert Chenevier, Fédération de Cardiologie, Créteil, F-94000, France e INSERM U 652, Institut des Cordeliers, F-75005, Paris, France f CNRS UMR 7054, Créteil, France Received 16 August 2006; received in revised form 19 October 2006; accepted 20 October 2006 Abstract Estrogens are known to activate the phosphatidyl-inosityl 3-kinase (PI3K)/Akt pathway, which is central in the cardioprotection afforded by ischemic postconditioning. Therefore, our goal was to investigate whether a phytoestrogen, genistein, could induce a pharmacological postconditioning and to investigate potential mechanisms. We used low doses of genistein in order to avoid tyrosine kinases inhibition. Thus, pentobarbital-anesthetized rabbits underwent a coronary artery occlusion followed by 4 h of reperfusion. Prior to reperfusion, they randomly received an i.v. injection of either saline (Control), 100 or 1000 μg/kg of genistein (Geni 100 and Geni 1000 , respectively), and 10 or 100 μg/kg of 17β-estradiol (17β 10 and 17β 100 , respectively). Infarct size (IS, % area at risk) was significantly reduced in Gen 100 , Gen 1000 and 17β 100 but not in 17β 10 (6 ± 2, 16 ± 5, 12 ± 3 and 29 ± 7%, respectively) vs. Control (35 ± 4%). A significant decrease in the percentage of TUNEL-positive nuclei within infarcted area was observed in Gen 100 and 17β 100 vs. Controls. The estrogen receptor antagonist fulvestrant (1 mg/kg i.v.) and the PI3K inhibitor wortmaninn (0.6 mg/kg) abolished the cardioprotective effect of genistein. Western blots also demonstrated an increase in Akt posphorylation in Gen 100 . In the same group, in vitro mitochondrial swelling studies demonstrated a significant inhibition of calcium-induced opening of mitochondrial transition pore vs. Controls. In conclusion, genistein exerts pharmacological postconditioning with a similar potency as 17β-estradiol through a pathway involving activation of the estrogen receptor, of PI3K/Akt and mitochondrial preservation. Therefore, genistein should not be only considered as an inhibitor of tyrosine kinase but also as a cardioprotective estrogen. © 2006 Elsevier Inc. All rights reserved. Keywords: Estrogens; Infarction; Mitochondria; Reperfusion; Ischemia; Postconditioning; Phytoestrogens; Estradiol; Genistein; Rabbit; Cardioprotection 1. Introduction Although the cardioprotective effects of hormone replace- ment therapy remain controversial [1–6], the acute pretreatment with 17-β estradiol has been clearly demonstrated to reduce myocardial infarct size in dogs [7–9] and rabbits [10–13], in both males [7–9,12,13] and females [8,10,13]. In humans, a single intracoronary administration of estrogens has also been shown to render the myocardium more resistant to ischemia during balloon angioplasty [14]. This acute beneficial effect of estrogens administered before the onset of ischemia is thought to activate signaling pathways which are also involved in ischemic postconditioning, i.e., mitochondrial ATP-dependent potassium channel (K ATP ) [8,14–16] and phosphatidylinositol 3-kinase (PI3K)/Akt antiapoptotic pathway [16–20]. The administration of 17β-estradiol could indeed induce a pharma- cological postconditioning, reducing infarct size when instituted just before reperfusion in male dogs [7] and cats [21]. Therefore, acute treatment with estrogens could be proposed as a therapeutic strategy in adjunction to early revascularization for the treatment of acute myocardial infarction [14]. Journal of Molecular and Cellular Cardiology 42 (2007) 79 – 87 www.elsevier.com/locate/yjmcc ⁎ Corresponding author. Laboratoire de Pharmacologie, INSERM U660, Faculté de Médecine de Créteil, 8, rue du Général Sarrail, 94000 CRETEIL, France. Tel: +33 1 49 81 36 51; fax: +33 1 48 98 36 61. E-mail address: [email protected] (A. Berdeaux). 1 Both authors contributed equally to this work. 0022-2828/$ - see front matter © 2006 Elsevier Inc. All rights reserved. doi:10.1016/j.yjmcc.2006.10.007
Transcript of Pharmacological postconditioning with the phytoestrogen genistein

Journal of Molecular and Cellular Cardiology 42 (2007) 79–87www.elsevier.com/locate/yjmcc
Original article
Pharmacological postconditioning with the phytoestrogen genistein
R. Tissier a,b,c,1, X. Waintraub a,b,c,1, N. Couvreur a,b,c, M. Gervais f, P. Bruneval e, C. Mandet e,R. Zini a,b,c, B. Enriquez a,b,c, A. Berdeaux a,b,c,d,⁎, B. Ghaleh a,b,c,d
a INSERM, U 660, Créteil, F-94010, Franceb Université Paris XII, Faculté de Médecine, Laboratoire de Pharmacologie, Créteil, F-94000, France
c Ecole Nationale Vétérinaire d’Alfort, Maisons-Alfort, F-94700, Franced AP-HP, Groupe hospitalier Henri Mondor - Albert Chenevier, Fédération de Cardiologie, Créteil, F-94000, France
e INSERM U 652, Institut des Cordeliers, F-75005, Paris, Francef CNRS UMR 7054, Créteil, France
Received 16 August 2006; received in revised form 19 October 2006; accepted 20 October 2006
Abstract
Estrogens are known to activate the phosphatidyl-inosityl 3-kinase (PI3K)/Akt pathway, which is central in the cardioprotection afforded byischemic postconditioning. Therefore, our goal was to investigate whether a phytoestrogen, genistein, could induce a pharmacologicalpostconditioning and to investigate potential mechanisms. We used low doses of genistein in order to avoid tyrosine kinases inhibition. Thus,pentobarbital-anesthetized rabbits underwent a coronary artery occlusion followed by 4 h of reperfusion. Prior to reperfusion, they randomlyreceived an i.v. injection of either saline (Control), 100 or 1000 μg/kg of genistein (Geni100 and Geni1000, respectively), and 10 or 100 μg/kg of17β-estradiol (17β10 and 17β100, respectively). Infarct size (IS, % area at risk) was significantly reduced in Gen100, Gen1000 and 17β100 but not in17β10 (6±2, 16±5, 12±3 and 29±7%, respectively) vs. Control (35±4%). A significant decrease in the percentage of TUNEL-positive nucleiwithin infarcted area was observed in Gen100 and 17β100 vs. Controls. The estrogen receptor antagonist fulvestrant (1 mg/kg i.v.) and the PI3Kinhibitor wortmaninn (0.6 mg/kg) abolished the cardioprotective effect of genistein. Western blots also demonstrated an increase in Aktposphorylation in Gen100. In the same group, in vitro mitochondrial swelling studies demonstrated a significant inhibition of calcium-inducedopening of mitochondrial transition pore vs. Controls. In conclusion, genistein exerts pharmacological postconditioning with a similar potency as17β-estradiol through a pathway involving activation of the estrogen receptor, of PI3K/Akt and mitochondrial preservation. Therefore, genisteinshould not be only considered as an inhibitor of tyrosine kinase but also as a cardioprotective estrogen.© 2006 Elsevier Inc. All rights reserved.
Keywords: Estrogens; Infarction; Mitochondria; Reperfusion; Ischemia; Postconditioning; Phytoestrogens; Estradiol; Genistein; Rabbit; Cardioprotection
1. Introduction
Although the cardioprotective effects of hormone replace-ment therapy remain controversial [1–6], the acute pretreatmentwith 17-β estradiol has been clearly demonstrated to reducemyocardial infarct size in dogs [7–9] and rabbits [10–13], inboth males [7–9,12,13] and females [8,10,13]. In humans, a
⁎ Corresponding author. Laboratoire de Pharmacologie, INSERM U660,Faculté de Médecine de Créteil, 8, rue du Général Sarrail, 94000 CRETEIL,France. Tel: +33 1 49 81 36 51; fax: +33 1 48 98 36 61.
E-mail address: [email protected] (A. Berdeaux).1 Both authors contributed equally to this work.
0022-2828/$ - see front matter © 2006 Elsevier Inc. All rights reserved.doi:10.1016/j.yjmcc.2006.10.007
single intracoronary administration of estrogens has also beenshown to render the myocardium more resistant to ischemiaduring balloon angioplasty [14]. This acute beneficial effect ofestrogens administered before the onset of ischemia is thoughtto activate signaling pathways which are also involved inischemic postconditioning, i.e., mitochondrial ATP-dependentpotassium channel (KATP) [8,14–16] and phosphatidylinositol3-kinase (PI3K)/Akt antiapoptotic pathway [16–20]. Theadministration of 17β-estradiol could indeed induce a pharma-cological postconditioning, reducing infarct size when institutedjust before reperfusion in male dogs [7] and cats [21].Therefore, acute treatment with estrogens could be proposedas a therapeutic strategy in adjunction to early revascularizationfor the treatment of acute myocardial infarction [14].

80 R. Tissier et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 79–87
Interestingly, many authors suggested the use of naturalestrogens derived from plants, i.e., “phytoestrogens”, as atherapeutic alternative to classical estrogens [22–26]. The maincompound of this family is the isoflavone genistein which isable to exert an immediate vasodilation with a similar potencythan 17β-estradiol in human forearm vasculature [26]. Inaddition, a single administration of genistein during a 45 minischemia has been shown to reduce infarct size in ovariecto-mized female rats [27]. This was observed with low (0.1–1 mg/kg i.v.) but not with higher doses of genistein (1.5–5 mg/kg i.v.),as pleiotropic effect such as inhibition of tyrosine kinases mayappear with the later doses [28,29]. The investigation of thecardioprotective effect of genistein is therefore relevant fromdifferent points of view: (1) the pharmacological point of view,in order to propose and understand a cardioprotective strategy,(2) the nutritional point of view, as genistein is contained inmany food products such as soy [22] and (3) the methodologicalpoint of view, as this compound is sometimes only considered asan inhibitor of tyrosine kinases, occulting the potentialestrogenic cardioprotective properties.
Accordingly, the first goal of this study was to compare themagnitude of the cardioprotection afforded by 17β-estradioland low doses of genistein. These compounds were adminis-tered prior to reperfusion to induce pharmacological postcondi-tioning. The second goal was to investigate the mechanism ofthe cardioprotection induced by genistein, i.e., to examinewhether inhibition of apoptosis, activation of the estrogenreceptor and the PI3K/Akt pathway are involved. Finally, asinhibition of the opening of the mitochondrial permeabilitytransition pore is now considered as one end-effector ofischemic postconditioning [30], we also investigated this aspectin the genistein-induced postconditioning. For all theseinvestigations, we used a rabbit model of myocardial infarction[31,32].
2. Methods
The experiments were performed in accordance with theofficial regulations edicted by the French Ministry ofAgriculture.
2.1. Animal surgery
As previously described [32], anesthesia was induced inmale New Zealand rabbits (2–2.5 kg) by zolazepam andtiletamine (20–30 mg/kg i.v. each). Animals were intubated andmechanically ventilated with 100% oxygen. Anesthesia wasmaintained by pentobarbital (20–30 mg/kg, i.v.). A catheter waspositioned in the ear marginal artery of the rabbit for arterialpressure measurement (P23ID strain gauge, Statham Instru-ments, Oxnard, CA, USA). An external electrocardiogram wasalso recorded. A left thoracotomy was performed at the fourthintercostal space under sterile conditions. The pericardium wasopened and a 3/0 Prolene suture was passed beneath a majorbranch of the left coronary artery. The ends of the ligature werepassed through a short segment of propylene tubing to form asnare. A coronary artery occlusion (CAO) was induced by
pulling the snare through the tubing. Ischemia was confirmedby the occurrence of ST segment deviation of the electro-cardiogram. Reperfusion was induced by releasing the snare.The chest was then closed in layers and a small tube was left inthe thorax to evacuate air and fluids after surgery.
2.2. Measurement of risk area and infarct size
After completion of reperfusion, the animals receivedheparin and sodium pentobarbital (60 mg/kg, i.v.). Potassiumchloride was then administered intravenously to induce cardiacarrest and the hearts were excised. The ascending aorta wascannulated and perfused (120 mm Hg) retrogradely with salinefollowed by Evans blue (5%) after ligation of the previouslyoccluded artery. The left ventricle was cut into 6–8 slices whichwere weighed and incubated with 1% triphenyltetrazoliumchloride (Sigma, Poole, U.K.) in a pH 7.4 buffer at 37 °C. Sliceswere fixed in 10% formaldehyde and then photographed with adigital camera mounted on a stereomicroscope. Using acomputerized planimetric program (Scion Image, Scion Cor-poration, Frederick, MD, USA), the area at risk and theinfarcted zones were quantified. The area at risk was identifiedas the non-blue region and was expressed as a percentage of theleft ventricle weight. Infarcted area was identified as the TTC-unstained tissue and was expressed as a percentage of the area atrisk.
2.3. Experimental protocol
The animals were included into 4 different protocols asillustrated in Fig. 1. They were all subjected to a 20 mincoronary artery occlusion.
In Protocol A, we investigated the magnitude of protectionafforded by two different doses of 17β-estradiol (10 and100 μg/kg i.v.) and genistein (100 and 1000 μg/kg i.v.).Accordingly, the rabbits received at random an intravenousinjection of either saline (Control), 10 μg/kg of 17β-Estradiol(17β-E10), 100 μg/kg of 17β-Estradiol (17β-E100), 100 μg/kgof genistein (Geni100) and 1000 μg/kg of genistein (Geni1000).These intravenous injections were performed during the last5 min of ischemia, prior to reperfusion. The doses of 17β-estradiol were chosen as 10 μg/kg i.v. is commonly used in thecardioprotection setting in dogs [8,9] and rabbits [10,12,13] andin order to investigate a higher dose, 100 μg/kg i.v. Moreover,the 10 μg/kg i.v. dose of 17β-Estradiol is known to elicitmaximal blood concentrations averaging those observed inwomen during midcycle [8]. The 100 and 1000 μg/kg i.v. dosesof genistein were demonstrated to be cardioprotective in rats[27]. Following coronary artery occlusion, rabbits underwent a4 h reperfusion.
In Protocol B, our aim was to confirm the cardioprotectionelicited by genistein after a prolonged reperfusion duration.Control and Geni100 groups of rabbits were further investigatedand were therefore subjected to a 72 h reperfusion in order toconfirm the results observed in Protocol A. We chose the100 μg/kg dose of genistein as it was able to induce a strongprotection in Protocol A.

Fig. 1. Experimental protocol. CAO, coronary artery occlusion; CAR, coronaryartery reperfusion; 17β-E10, intravenous administration of 10 μg/kg of 17-βestradiol during the last 5 min of CAO; 17β-E100, intravenous administration of100 μg/kg of 17-β estradiol during the last 5 min of CAO; Geni100, intravenousadministration of 100 μg/kg of genistein during the last 5 min of CAO; Geni1000,intravenous administration of 1000 μg/kg of genistein during the last 5 min ofCAO; Fulv, intravenous administration of fulvestrant 1 h before the onset ofCAO; Geni100+Fulv, administration of Fulv and Geni100; Wort, intravenousadministration of wortmaninn 10 min before the onset of CAO; Geni100+Wort,administration of Wort and Geni100.
81R. Tissier et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 79–87
In Protocol C, in order to determine whether the estrogenreceptors and the PI3K/Akt pathway were involved in theprotective effect of genistein, rabbits were randomly dividedin several groups receiving either the estrogen receptorantagonist fulvestrant (1 mg/kg i.v., Fulv group) or the PI3K
inhibitor wortmaninn (0.6 mg/kg i.v., Wort group) alone orcombined with 100 μg/kg of genistein (Geni100+Fulv andGeni100+Wort, respectively). Fulvestrant and wortmaninn wereadministered intravenously 1 h and 10 min before the onset ofcoronary artery occlusion, respectively. The reperfusion dura-tion reached 4 h before infarct sizing. The dose of fulvestrantwas selected on the basis of preliminary experiments. Weindeed demonstrated that 0.35 mg/kg i.v. of fulvestrant[8,10,13] tended to inhibit the infarct size reduction elicitedby genistein at 0.1 mg/kg i.v. (infarct sizes=35.8±5.8 and26.2±6.5% of the area at risk with fulvestrant alone or incombination with genistein, n=4 and 6, respectively). At ahigher dose of 1 mg/kg i.v., fulvestrant totally abolished theeffect of genistein.
In Protocol D, Control and Geni100 rabbits were sacri-ficed at 10 min of reperfusion in order to perform myo-cardial sampling for Western blots and for mitochondriaextraction.
2.4. Histology, terminal dUTP nick end labeling (TUNEL) andimmunohistochemistry
As previously described [33], we investigated histologicalabnormalities on heart sections stained with hematoxylin–eosin–saffron. Detection of apoptosis using TUNEL techni-que was performed on paraffin tissue sections with aTUNEL ApopTag kit (Q. Biogene, Carlsbad, CA, USA).Hemorrhage was determined blindly and expressed by ascore from 0 (no hemorrhage) to 4 (huge hemorrhage)within the infarcted area. The percentage of TUNEL positivenuclei within the infarcted area was also determined blindly.Anti-CD31 monoclonal antibody (Dako, Trappes, France)was used as an endothelial cell marker in order to identifymicrocirculatory network.
2.5. Western blots
A sample of the ventricular tissue at risk was excised andfrozen in liquid nitrogen before being stored at −80 °C(Protocol D). Tissue lysates were prepared with cold 1%Nonidet P-40 in 50 mM Tris–HCl pH 7.4 containing120 mM NaCl, 1 mM EDTA, 50 mM NaF, 0.1 mMNa3VO4 and protease inhibitor cocktail. Total protein levelswere quantified using the BCA assay. Lysates (100 μg oftotal protein) were then subjected to SDS-PAGE andtransferred to PVDF membranes. The membranes wereprobed with monoclonal phospho-Ser473 specific anti-Akt(Cell Signaling Technology, Beverly, MA, USA), monoclonalphospho specific anti-ERK1/2 (Upstate, Charlottesville, VA)and biotinylated anti-actin (Santa Cruz Biotechnology, SantaCruz, CA, USA) antibodies overnight at 4 °C. After incu-bation with alkaline phosphatase-linked secondary antibodies,immunoreactive proteins were visualized by NBT/BCIP orAttophos AP fluorescent (Promega, Madison, WI, USA)substrates, respectively. Quantitative analysis of phospho-Aktwas performed using QuantityOne software (Bio-Rad, Her-cules, CA, USA).

82 R. Tissier et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 79–87
2.6. Investigation of calcium-induced mitochondrial swelling
In Protocol D, a sample of the ventricular tissue at risk wasexcised and mitochondria were extracted after 10 min ofcoronary artery reperfusion, as previously described [34].Mitochondrial volume changes were estimated by monitoringchanges in light scattering measured at 540 nm. Briefly, assayswere performed in 1 ml of a buffer composed of 50 mMsucrose, 100 mM KCl, 10 mM HEPES and 5 ml KH2PO4 (pH7.4 at 37 °C) containing 0.4 mg of mitochondrial proteins.Pyruvate and malate (10/10 mM) were added and after 1 minof incubation at 37 °C, the swelling was initiated by additionof different concentrations of calcium (25 to 800 μM) inseparate samples. Maximal changes in light scattering weremeasured at steady-state after 10 min. For each experiment,the quality of the preparation was checked by addition ofcyclosporine A (1 μM) in a separate sample of mitochondrialextract.
2.7. Statistical analysis
Data are reported as mean±SEM. Values of left ventricleweights, areas at risk and infarct sizes were analyzed using a oneway ANOVA followed by PLSD Fisher test if necessary.Comparisons of heart rate and mean arterial pressure wereperformed between groups at baseline, at 5 and 19-min CAOand at 5-min CAR using a one-way ANOVA for repeatedmeasures. Histological scores of hemorrhage were comparedbetween groups using a non-parametric Kruskall–Wallis testfollowed if necessary by a Mann–Whitney test. Percentage ofTUNEL positive nuclei within infarction was comparedbetween groups using a one-way ANOVA followed by PLSDFisher test. For the investigation of mitochondrial calcium-induced volume changes, changes in light scattering at 540 nmwere compared between groups using a one-way ANOVA forrepeated measures, according to increasing calcium concentra-tions. Significant differences were determined as p<0.05.
3. Results
Eighty-nine rabbits were included in the present study.In Protocol A, 12, 7, 8, 6 and 6 rabbits were included in
Table 1Hemodynamics in Protocol A
Group n Heart rate (beats/min)
Baseline CAO 5′ CAO 19′
Control 12 260±10 263±9 253±717β-E10 7 259±8 252±8 250±717β-E100 8 234±9 228±10 223±5Geni100 6 242±14 236±11 230±9Geni1000 6 256±9 264±6 250±12
Values are mean±SEM; n, number of rabbits.CAO, coronary artery occlusion; CAR, coronary artery reperfusion; CAO 5′, 5 mintravenous administration of 10 μg/kg of 17β-estradiol during the last 5 min of CAlast 5 min of CAO; Geni100, intravenous administration of 100 μg/kg of genistein duof genistein during the last 5 min of CAO.
the Control, 17β-E10, 17β-E100, Geni100 and Geni1000groups, respectively. In Protocol B, 9 and 8 rabbits wereincluded in Control and Geni100 groups, respectively. InProtocol C, 6, 7, 5 and 5 rabbits were included in Fulv,Geni100+Fulv, Wort and Geni100+Wort groups, respectively.In Protocol D, 5 rabbits were investigated in each of theControl and the Geni100 groups for Western blots analysis,respectively. Out of them, 3 and 5 were also used for themitochondrial experiments in Control and Geni100 groups,respectively.
3.1. Hemodynamics
As shown in Table 1, values of heart rate and mean arterialpressure were not significantly different among groups atbaseline, at 5 and 19-min CAO and at 5-min CAR in ProtocolA. In other protocols, hemodynamic parameters were alsosimilar among groups (data not shown).
3.2. Area at risk and infarct sizes
As shown in Table 2, left ventricle weights and sizes ofthe area at risk were similar among groups in eachprotocol. In Protocol A (Fig. 2), infarct sizes were sig-nificantly reduced by 65%, 83%, and 54% in 17β-E100,Geni100 and Geni1000 groups as compared to Control group(12±3, 6±2, 16±5% vs. 35±4%, respectively, p<0.05).However, it was not significantly modified in the 17β-E10
group (29±7%).As illustrated in Fig. 3, infarct size reduction was indeed
confirmed after a 72 h reperfusion in Geni100 vs. Controlgroups in Protocol B (17±4% vs. 35±3%, respectively,p<0.05). In Protocol C, both fulvestrant and wortmaninnabolished the infarct size reduction elicited by Geni100 withouteffect by their own (Fig. 4).
3.3. Histology, TUNEL and immunohistochemistry
In Protocol A, hearts from Control, 17β-E100 andGeni100 groups were investigated by histology as illustratedin Fig. 5 (n=12, 8 and 6, respectively). In Control group,myocardial infarction consisted in a core of necrosis with
Mean arterial pressure (mm Hg)
CAR 5′ Baseline CAO 5′ CAO 19′ CAR 5′
242±9 79±5 75±5 69±5 69±5247±9 75±6 72±5 77±7 77±7224±8 69±5 65±6 66±6 70±6244±11 79±9 68±5 69±5 69±6255±11 72±4 78±4 78±6 73±8
in of CAO; CAO 19′, 19 min of CAO; CAR 5′, 5 min of CAR; 17β-E10,O; 17β-E100, intravenous administration of 100 μg/kg of 17 estradiol during thering the last 5 min of CAO; Geni1000, intravenous administration of 1000 μg/kg
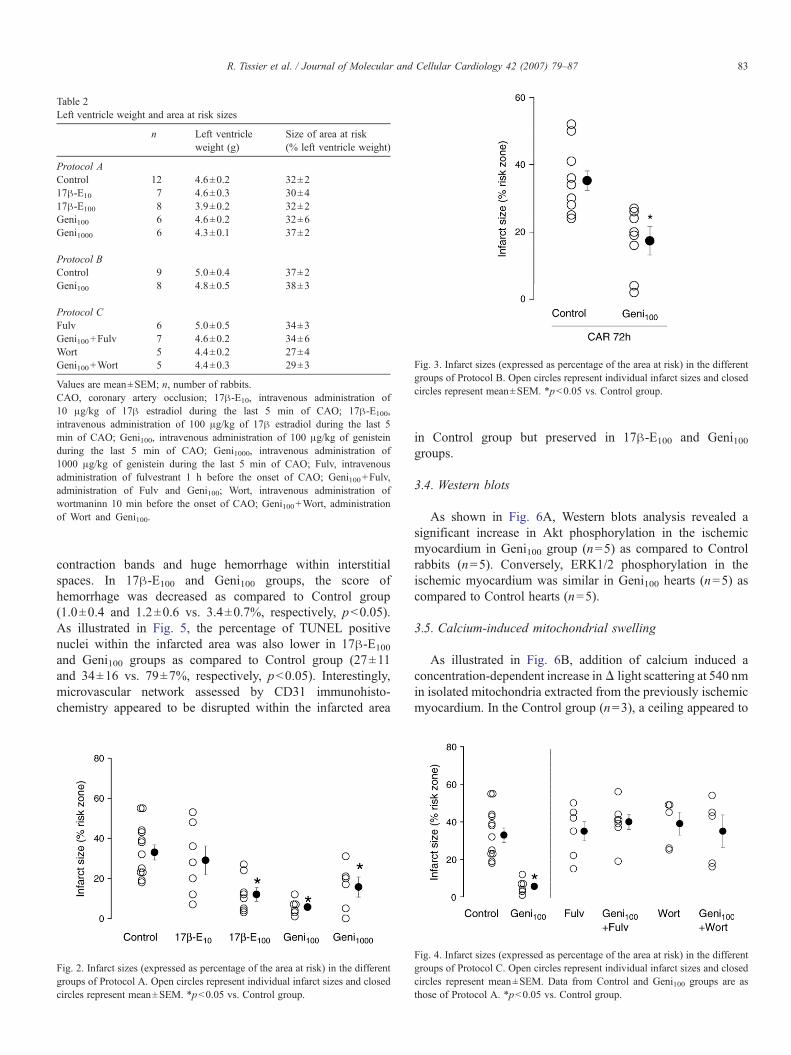
Fig. 3. Infarct sizes (expressed as percentage of the area at risk) in the differentgroups of Protocol B. Open circles represent individual infarct sizes and closedcircles represent mean±SEM. *p<0.05 vs. Control group.
Table 2Left ventricle weight and area at risk sizes
n Left ventricleweight (g)
Size of area at risk(% left ventricle weight)
Protocol AControl 12 4.6±0.2 32±217β-E10 7 4.6±0.3 30±417β-E100 8 3.9±0.2 32±2Geni100 6 4.6±0.2 32±6Geni1000 6 4.3±0.1 37±2
Protocol BControl 9 5.0±0.4 37±2Geni100 8 4.8±0.5 38±3
Protocol CFulv 6 5.0±0.5 34±3Geni100+Fulv 7 4.6±0.2 34±6Wort 5 4.4±0.2 27±4Geni100+Wort 5 4.4±0.3 29±3
Values are mean±SEM; n, number of rabbits.CAO, coronary artery occlusion; 17β-E10, intravenous administration of10 μg/kg of 17β estradiol during the last 5 min of CAO; 17β-E100,intravenous administration of 100 μg/kg of 17β estradiol during the last 5min of CAO; Geni100, intravenous administration of 100 μg/kg of genisteinduring the last 5 min of CAO; Geni1000, intravenous administration of1000 μg/kg of genistein during the last 5 min of CAO; Fulv, intravenousadministration of fulvestrant 1 h before the onset of CAO; Geni100+Fulv,administration of Fulv and Geni100; Wort, intravenous administration ofwortmaninn 10 min before the onset of CAO; Geni100+Wort, administrationof Wort and Geni100.
83R. Tissier et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 79–87
contraction bands and huge hemorrhage within interstitialspaces. In 17β-E100 and Geni100 groups, the score ofhemorrhage was decreased as compared to Control group(1.0±0.4 and 1.2±0.6 vs. 3.4±0.7%, respectively, p<0.05).As illustrated in Fig. 5, the percentage of TUNEL positivenuclei within the infarcted area was also lower in 17β-E100
and Geni100 groups as compared to Control group (27±11and 34±16 vs. 79±7%, respectively, p<0.05). Interestingly,microvascular network assessed by CD31 immunohisto-chemistry appeared to be disrupted within the infarcted area
Fig. 2. Infarct sizes (expressed as percentage of the area at risk) in the differentgroups of Protocol A. Open circles represent individual infarct sizes and closedcircles represent mean±SEM. *p<0.05 vs. Control group.
in Control group but preserved in 17β-E100 and Geni100groups.
3.4. Western blots
As shown in Fig. 6A, Western blots analysis revealed asignificant increase in Akt phosphorylation in the ischemicmyocardium in Geni100 group (n=5) as compared to Controlrabbits (n=5). Conversely, ERK1/2 phosphorylation in theischemic myocardium was similar in Geni100 hearts (n=5) ascompared to Control hearts (n=5).
3.5. Calcium-induced mitochondrial swelling
As illustrated in Fig. 6B, addition of calcium induced aconcentration-dependent increase inΔ light scattering at 540 nmin isolated mitochondria extracted from the previously ischemicmyocardium. In the Control group (n=3), a ceiling appeared to
Fig. 4. Infarct sizes (expressed as percentage of the area at risk) in the differentgroups of Protocol C. Open circles represent individual infarct sizes and closedcircles represent mean±SEM. Data from Control and Geni100 groups are asthose of Protocol A. *p<0.05 vs. Control group.
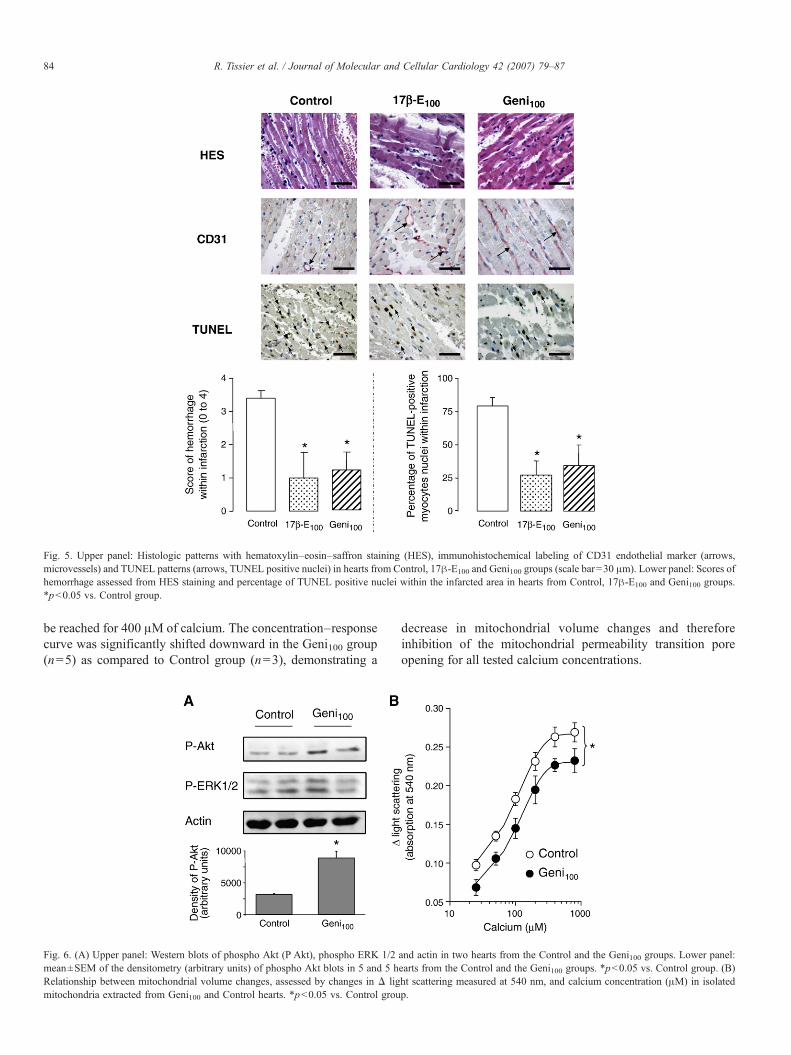
Fig. 5. Upper panel: Histologic patterns with hematoxylin–eosin–saffron staining (HES), immunohistochemical labeling of CD31 endothelial marker (arrows,microvessels) and TUNEL patterns (arrows, TUNEL positive nuclei) in hearts from Control, 17β-E100 and Geni100 groups (scale bar=30 μm). Lower panel: Scores ofhemorrhage assessed from HES staining and percentage of TUNEL positive nuclei within the infarcted area in hearts from Control, 17β-E100 and Geni100 groups.*p<0.05 vs. Control group.
84 R. Tissier et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 79–87
be reached for 400 μM of calcium. The concentration–responsecurve was significantly shifted downward in the Geni100 group(n=5) as compared to Control group (n=3), demonstrating a
Fig. 6. (A) Upper panel: Western blots of phospho Akt (P Akt), phospho ERK 1/2mean±SEM of the densitometry (arbitrary units) of phospho Akt blots in 5 and 5 hRelationship between mitochondrial volume changes, assessed by changes in Δ ligmitochondria extracted from Geni100 and Control hearts. *p<0.05 vs. Control grou
decrease in mitochondrial volume changes and thereforeinhibition of the mitochondrial permeability transition poreopening for all tested calcium concentrations.
and actin in two hearts from the Control and the Geni100 groups. Lower panel:earts from the Control and the Geni100 groups. *p<0.05 vs. Control group. (B)ht scattering measured at 540 nm, and calcium concentration (μM) in isolatedp.

85R. Tissier et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 79–87
4. Discussion
To our knowledge, this is the first report to demonstrate that aphytoestrogen, like genistein, is able to induce a pharmacolo-gical postconditioning through a pathway involving activationof the estrogen receptor and the PI3K/Akt pathway. Thiscardioprotective effect of genistein results from inhibition of theopening of mitochondrial permeability transition pore and thedecrease in the percentage of TUNEL positive nuclei within theinfarcted area. The magnitude of the protection elicited bygenistein at the investigated doses appears to be close to that of17β-estradiol (100 μg/kg i.v.).
We first investigated the cardioprotection induced by twodoses of 17β-estradiol (10 and 100 μg/kg i.v.) and genistein(100 and 1000 μg/kg i.v.) administered prior to reperfusion.Regarding 17β-estradiol, the highest dose was the only one tosignificantly reduce infarct size in our experimental conditions.As the pre-ischemic administration of 10 μg/kg of 17β-estradiolwas previously shown to be cardioprotective in male rabbits[10,12,13], our data suggest that higher doses are required toreach the protective threshold in the postconditioning setting.However, the intravenous administration of 10 μg/kg of 17β-estradiol in male dogs was also shown to reduce infarct sizewhen administered at the end of a 60 min ischemia [7]. We canhypothesize that ischemia severity is greater in rabbits whichare devoided of native collateral circulation and that thethreshold of protection could be greater. The doses of 100 and1000 μg/kg i.v. of genistein were chosen on the basis that theyreduce infarct size in ovariectomized rats subjected to 45 min ofischemia [27]. We did not investigate greater doses as they arewell known to induce tyrosine kinases inhibition [29], whichwas proposed to alter the protection induced through estrogenicactivation [27]. The effect of tyrosine kinase inhibition appears,however, complex in the setting of myocardial infarction as,e.g., it could be deleterious by inducing an inhibition ofpreconditioning [35] or in contrast, protective by the selectiveinhibition of Src tyrosine kinases during or after a prolongedischemia [35,36]. In our report, we observed a significantprotection with both doses of genistein and importantly, themagnitude of infarct size reduction was similar to that observedwith 17β-estradiol. This suggests that genistein could be a goodalternative to 17β-estradiol in the protection against reperfusioninjury. Furthermore, we demonstrated that the protectionafforded by genistein was still observed after prolongedreperfusion, i.e., 72 h.
The protection induced by intravenous administration of100 μg/kg of genistein prior to reperfusion was abolished by theestrogen receptor antagonist fulvestrant, also known as ICI182–780 [37]. This estrogen receptor antagonist was shown toinhibit the protective effect of pre-ischemic administration of17β-estradiol in male rabbits [10]. As fulvestrant is not selectiveof a particular estrogen receptor sub-type [38], we do not knowwhether genistein-induced protection involved the α and/or βsub-types but this phytoestrogen is thought to have a greateraffinity for the β-receptor [39]. Conversely, the pre-ischemiccardioprotective effect of 17β-estradiol is supposed to involvemainly the α-receptor [11]. In this context, it is reasonable to
speculate that 17β-estradiol administered prior to reperfusionalso depends upon estrogen receptor but we have no directevidence to support such mechanism in the present study.
We also demonstrated that genistein-induced postcondition-ing activates the PI3K/Akt pathway, these two kinases beingimplicated in the acute beneficial effect of estrogens adminis-tered prior to ischemia [17]. Therefore, the protection affordedby genistein shares a common “Reperfusion Induced SalvageKinases” (RISK) pathway with ischemic and isofluranepostconditioning [16,19,20,31,40,41]. A direct interactionbetween the estrogen receptor and the p85alpha regulatorysubunit of PI3K was indeed previously demonstrated [42] and itis well known that these kinases mediate the acute beneficialeffect of estrogens administered prior to ischemia [17].Conversely, genistein did not activate ERK 1/2 in our experi-mental conditions, although these kinases could be altered byestrogens in other conditions [43].
We further demonstrated that genistein induced aninhibition in calcium induced-opening of mitochondrialpermeability transition pore and a decrease in the percentageof TUNEL positive nuclei within the infarcted area. Theantiapoptotic effect of the PI3K/Akt pathway is indeed wellknown [44] as well as the relationship between activation ofthe RISK pathway and inhibition of mitochondrial perme-ability transition pore opening [45]. Altogether these findingssuggest that genistein inhibited, at least in part, mitochondrialapoptosis and that it allows infarct size reduction. Thecardiac antiapoptotic actions of chronic treatment withestrogens are moreover well known following coronaryartery ligation in rats [18]. All these data therefore supportthe concept that genistein actually mimic postconditioning byreducing infarct size when administered prior to reperfusionand by activating protective pathways similar to those ofischemic postconditioning [16,19,20,31,40,41]. Whether othermechanisms could be involved was not investigated in thisreport but previous investigations with 17β-estradiol demon-strated that connexin-43 might be also involved in myocytesprotection [7].
Finally, histological analysis demonstrated that hemorrhagewithin the infarcted area was inhibited by the intravenousadministration of both 100 μg/kg of genistein and 17β-estradiol. This was associated with preservation of themicrocirculatory network as compared to Controls. Theseresults suggest that estrogens might also induce a pharmaco-logical postconditioning against microvascular disruption. Thispoint was not investigated in the present report but thevasculoprotective effect of both 17β-estradiol and genistein iswell-known in rabbits [46,47] as well as in humans [26].
In conclusion, we demonstrated for the first time thatgenistein induces a pharmacological postconditioning effectwith a similar potency than 17β-estradiol through directactivation of the estrogen receptor, the PI3K/Akt pathway andthe decrease in both mitochondrial permeability transition poreopening and in cardiomyocyte apoptotic cell death. Thusgenistein should not be only considered as an inhibitor oftyrosine kinases but also as a cardioprotective estrogen at lowdoses.

86 R. Tissier et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 79–87
Acknowledgments
This study was supported by grants from the French “AgenceNationale pour la Recherche” (Ischermdiol project) and“Fondation de l'Avenir” (ET5 #406). Xavier Waintraub wassupported by a grant from the “Fondation pour la RechercheMédicale”. The authors are greatly indebted to Fanny Lidourenand Alain Bizé for their excellent technical support.
References
[1] Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, BlackH, et al. Effects of conjugated equine estrogen in postmenopausal womenwith hysterectomy: the women's health initiative randomized controlledtrial. JAMA 2004;291:1701–12.
[2] Manson JE, Hsia J, Johnson KC, Rossouw JE, Assaf AR, Lasser NL, et al.Estrogen plus progestin and the risk of coronary heart disease. N Engl JMed 2003;349:523–34.
[3] Ouyang P, Michos ED, Karas RH. Hormone replacement therapy and thecardiovascular system lessons learned and unanswered questions. J AmColl Cardiol 2006;47:1741–53.
[4] McNulty PH, Jagasia D, Whiting JM, Caulin Glaser T. Effect of 6 wkestrogen withdrawal or replacement on myocardial ischemic tolerance inrats. Am J Physiol Heart Circ Physiol 2000;278:H1030–4.
[5] van Eickels M, Patten RD, Aronovitz MJ, Alsheikh Ali A, Gostyla K,Celestin F, et al. 17 beta estradiol increases cardiac remodeling andmortality in mice with myocardial infarction. J Am Coll Cardiol2003;41:2084–92.
[6] Zhai P, Eurell TE, Cotthaus RP, Jeffery EH, Bahr JM, Gross DR. Effects ofdietary phytoestrogen on global myocardial ischemia–reperfusion injury inisolated female rat hearts. Am J Physiol Heart Circ Physiol 2001;281:H1223–32.
[7] Lee TM, Lin MS, Chou TF, Tsai CH, Chang NC. Adjunctive 17beta-estradiol administration reduces infarct size by altered expression of caninemyocardial connexin43 protein. Cardiovasc Res 2004;63:109–17.
[8] Lee TM, Su SF, Tsai CC, Lee YT, Tsai CH. Cardioprotective effects of 17beta-estradiol produced by activation of mitochondrial ATP-sensitive K(+)Channels in canine hearts. J Mol Cell Cardiol 2000;32:1147–58.
[9] Tsai CH, Su SF, Chou TF, Lee TM. Differential effects of sarcolemmal andmitochondrial K(ATP) channels activated by 17 beta-estradiol onreperfusion arrhythmias and infarct sizes in canine hearts. J PharmacolExp Ther 2002;301:234–40.
[10] Booth EA, Marchesi M, Kilbourne EJ, Lucchesi BR. 17Beta-estradiol as areceptor-mediated cardioprotective agent. J Pharmacol Exp Ther2003;307:395–401.
[11] Booth EA, Obeid NR, Lucchesi BR. Activation of estrogen receptor-alphaprotects the in vivo rabbit heart from ischemia–reperfusion injury. Am JPhysiol Heart Circ Physiol 2005;289:H2039–47.
[12] Hale SL, Birnbaum Y, Kloner RA. beta-Estradiol, but not alpha-estradiol,reduced myocardial necrosis in rabbits after ischemia and reperfusion. AmHeart J 1996;132:258–62.
[13] Hale SL, Birnbaum Y, Kloner RA. Estradiol, administered acutely, protectsischemic myocardium in both female and male rabbits. J CardiovascPharmacol Ther 1997;2:47–52.
[14] Lee TM, Su SF, Chou TF, Tsai CH. Pharmacologic preconditioning ofestrogen by activation of the myocardial adenosine triphosphate-sensitivepotassium channel in patients undergoing coronary angioplasty. J Am CollCardiol 2002;39:871–7.
[15] Lee TM, Chou TF, Tsai CH. Differential role of K(ATP) channels activatedby conjugated estrogens in the regulation of myocardial and coronaryprotective effects. Circulation 2003;107:49–54.
[16] Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple,brief coronary occlusions during early reperfusion protect rabbit hearts bytargeting cell signaling pathways. J Am Coll Cardiol 2004;44:1103–10.
[17] Patten RD, Karas RH. Estrogen replacement and cardiomyocyteprotection. Trends Cardiovasc Med 2006;16:69–75.
[18] Patten RD, Pourati I, Aronovitz MJ, Baur J, Celestin F, Chen X, et al.17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro viaactivation of phospho-inositide-3 kinase/Akt signaling. Circ Res2004;95:692–9.
[19] Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinasepathway: a common target for both ischemic preconditioning andpostconditioning. Trends Cardiovasc Med 2005;15:69–75.
[20] Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM. Postconditioning: aform of “modified reperfusion” protects the myocardium by activating thephosphatidylinositol 3-kinase-Akt pathway. Circ Res 2004;95:230–2.
[21] Delyani JA, Murohara T, Nossuli TO, Lefer AM. Protection frommyocardial reperfusion injury by acute administration of 17 beta-estradiol.J Mol Cell Cardiol 1996;28:1001–8.
[22] Lissin LW, Cooke JP. Phytoestrogens and cardiovascular health. J Am CollCardiol 2000;35:1403–10.
[23] Dubey RK, Gillespie DG, Imthurn B, Rosselli M, Jackson EK, Keller PJ.Phytoestrogens inhibit growth and MAP kinase activity in human aorticsmooth muscle cells. Hypertension 1999;33:177–82.
[24] Figtree GA, Griffiths H, Lu YQ, Webb CM, MacLeod K, Collins P. Plant-derived estrogens relax coronary arteries in vitro by a calcium antagonisticmechanism. J Am Coll Cardiol 2000;35:1977–85.
[25] Souzeau E, Belanger S, Picard S, Deschepper CF. Dietary isoflavonesduring pregnancy and lactation provide cardioprotection to offspring ratsin adulthood. Am J Physiol Heart Circ Physiol 2005;289:H715–21.
[26] Walker HA, Dean TS, Sanders TA, Jackson G, Ritter JM, Chowienczyk PJ.The phytoestrogen genistein produces acute nitric oxide-dependentdilation of human forearm vasculature with similar potency to 17beta-estradiol. Circulation 2001;103:258–62.
[27] Deodato B, Altavilla D, Squadrito G, Campo GM, Arlotta M, Minutoli L,et al. Cardioprotection by the phytoestrogen genistein in experimentalmyocardial ischaemia–reperfusion injury. Br J Pharmacol 1999;128:1683–90.
[28] Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, et al.Genistein, a specific inhibitor of tyrosine-specific protein kinases. J BiolChem 1987;262:5592–5.
[29] Fryer RM, Schultz JE, Hsu AK, Gross GJ. Pretreatment with tyrosinekinase inhibitors partially attenuates ischemic preconditioning in rat hearts.Am J Physiol 1998;275:H2009–15.
[30] Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M.Postconditioning inhibits mitochondrial permeability transition.Circulation 2005;111:194–7.
[31] Tessier-Vetzel D, Tissier R, Waintraub X, Ghaleh B, Berdeaux A.Isoflurane inhaled at the onset of reperfusion potentiates thecardioprotective effect of ischemic postconditioning through a NO-dependent mechanism. J Cardiovasc Pharmacol 2006;47:487–92.
[32] Tissier R, Aouam K, Berdeaux A, Ghaleh B. Evidence for a ceiling ofcardioprotection with a nitric oxide donor-induced delayed preconditioningin rabbits. J Pharmacol Exp Ther 2003;306:528–31.
[33] Tissier R, Souktani R, Bruneval P, Giudicelli JF, Berdeaux A, Ghaleh B.Adenosine A(1)-receptor induced late preconditioning and myocardialinfarction: reperfusion duration is critical. Am J Physiol Heart Circ Physiol2002;283:H38–43.
[34] Morin C, Zini R, Tillement JP. Anoxia-reoxygenation-induced cytochromec and cardiolipin release from rat brain mitochondria. Biochem BiophysRes Commun 2003;307:477–82.
[35] Hattori R, Otani H, Uchiyama T, Imamura H, Cui J, Maulik N, et al.Src tyrosine kinase is the trigger but not the mediator of ischemicpreconditioning. Am J Physiol Heart Circ Physiol 2001;281:H1066–74.
[36] Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, et al. Srcblockade stabilizes a Flk/cadherin complex, reducing edema and tissueinjury following myocardial infarction. J Clin Invest 2004;113:885–94.
[37] Howell A. Faslodex (ICI 182780). An oestrogen receptor downregulator.Eur J Cancer 2000;36(Suppl 4):S87–8.
[38] Nuttall ME, Pendrak I, Emery JG, Nadeau DP, Fisher PW, Nicholson TA,et al. Antagonism of oestrogen action in human breast and endometrialcells in vitro: potential novel antitumour agents. Cancer ChemotherPharmacol 2001;47:437–43.

87R. Tissier et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 79–87
[39] Lee GS, Choi KC, Kim HJ, Jeung EB. Effect of genistein as a selectiveestrogen receptor beta agonist on the expression of Calbindin-D9k in theuterus of immature rats. Toxicol Sci 2004;82:451–7.
[40] Yang XM, Philipp S, Downey JM, Cohen MV. Postconditioning'sprotection is not dependent on circulating blood factors or cells butinvolves adenosine receptors and requires PI3-kinase and guanylyl cyclaseactivation. Basic Res Cardiol 2005;100:57–63.
[41] Chiari PC, Bienengraeber MW, Pagel PS, Krolikowski JG, Kersten JR,Warltier DC. Isoflurane protects against myocardial infarction during earlyreperfusion by activation of phosphatidylinositol-3-kinase signaltransduction: evidence for anesthetic-induced postconditioning in rabbits.Anesthesiology 2005;102:102–9.
[42] Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK.Interaction of oestrogen receptor with the regulatory subunit ofphosphatidylinositol-3-OH kinase. Nature 2000;407:538–41.
[43] Nuedling S, Kahlert S, Loebbert K, Meyer R, Vetter H, Grohe C.Differential effects of 17beta-estradiol on mitogen-activated protein kinasepathways in rat cardiomyocytes. FEBS Lett 1999;454:271–6.
[44] Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt andapoptosis: size matters. Oncogene 2003;22:8983–98.
[45] Davidson SM, Hausenloy D, Duchen MR, Yellon DM. Signalling via thereperfusion injury signalling kinase (RISK) pathway links closure of themitochondrial permeability transition pore to cardioprotection. Int JBiochem Cell Biol 2006;38:414–9.
[46] Nascimento CA, Kauser K, Rubanyi GM. Effect of 17beta-estradiol inhypercholesterolemic rabbits with severe endothelial dysfunction. Am JPhysiol 1999;276:H1788–94.
[47] Watanabe Y, Littleton-Kearney MT, Traystman RJ, Hurn PD. Estrogenrestores postischemic pial microvascular dilation. Am J Physiol Heart CircPhysiol 2001;281:H155–60.

![Genistein induces apoptosis of colon cancer cells by ...€¦ · pathway [3]. In this study, we demonstrated that GEN can inhibite proliferation and induce apoptosis of colon cancer](https://static.fdocument.org/doc/165x107/6091035508039222da437990/genistein-induces-apoptosis-of-colon-cancer-cells-by-pathway-3-in-this-study.jpg)















![The Effects of Pharmacological Carbonic Anhydrase ...S-nitrosylation targets upon infection with the oomycete Phytophthora infestans [14]. Additionally, it is worth noting that the](https://static.fdocument.org/doc/165x107/60f89da2a24b6b558f15cb7b/the-effects-of-pharmacological-carbonic-anhydrase-s-nitrosylation-targets-upon.jpg)
