
Pancreatic β-Cell–Derived IP-10/CXCL10 Isletokine Mediates ...
11
Pancreatic b-Cell–Derived IP-10/CXCL10 Isletokine Mediates Early Loss of Graft Function in Islet Cell Transplantation Gumpei Yoshimatsu, 1 Faisal Kunnathodi, 1 Prathab Balaji Saravanan, 1 Rauf Shahbazov, 1 Charles Chang, 2 Carly M. Darden, 2 Sandra Zurawski, 3 Gulbahar Boyuk, 4 Mazhar A. Kanak, 5 Marlon F. Levy, 5 Bashoo Naziruddin, 6 and Michael C. Lawrence 1 Diabetes 2017;66:2857–2867 | https://doi.org/10.2337/db17-0578 Pancreatic islets produce and secrete cytokines and che- mokines in response to inflammatory and metabolic stress. The physiological role of these “isletokines” in health and disease is largely unknown. We observed that islets release multiple inflammatory mediators in patients undergoing islet transplants within hours of infusion. The proinflammatory cytokine interferon-g–induced protein 10 (IP-10/CXCL10) was among the highest released, and high levels correlated with poor islet transplant outcomes. Transgenic mouse studies confirmed that donor islet– specific expression of IP-10 contributed to islet inflamma- tion and loss of b-cell function in islet grafts. The effects of islet-derived IP-10 could be blocked by treatment of donor islets and recipient mice with anti–IP-10 neutraliz- ing monoclonal antibody. In vitro studies showed induction of the IP-10 gene was mediated by calcineurin-dependent NFAT signaling in pancreatic b-cells in response to ox- idative or inflammatory stress. Sustained association of NFAT and p300 histone acetyltransferase with the IP-10 gene required p38 and c-Jun N-terminal kinase mitogen- activated protein kinase (MAPK) activity, which differen- tially regulated IP-10 expression and subsequent protein release. Overall, these findings elucidate an NFAT-MAPK signaling paradigm for induction of isletokine expression in b-cells and reveal IP-10 as a primary therapeutic target to prevent b-cell–induced inflammatory loss of graft func- tion after islet cell transplantation. Islet endocrine cells have been shown to produce cytokines under conditions of physical, inflammatory, and metabolic stress (1–6). Expression of interleukin (IL)-1b has been ob- served in b-cells exposed to hyperglycemic conditions and in patients with type 2 diabetes (7). IL-1b signaling further promotes cytokine expression in b-cells (3,5,6,8–10). Acute effects of IL-1b to enhance insulin expression and induce proliferation in b-cells indicate a physiological role of cyto- kines to enable islets to adapt to inflammatory stress and metabolic demand (11–16). However, inappropriate expres- sion of cytokines by islets is associated with endoplasmic reticulum and oxidative stress responses that promote b-cell death and dysfunction (17–20). Overexposure of is- lets to osmotic, metabolic, oxidative, or inflammatory stress induces p38 and c-Jun N-terminal kinase (JNK) mitogen- activated protein kinase (MAPK) signaling (21–24). This leads to downstream effects of nuclear factor-kB to upre- gulate genes that promote apoptosis (17,25). During islet transplantation, islets are exposed to mul- tiple physical and chemical stresses throughout the islet cell isolation and infusion procedures that induce expression of cytokines. These inflammatory mediators are secreted by islets and persist for days within culture. Upon transplantation of islets, an innate immune response is observed that is largely responsible for a loss of up to 50% or more of the initial islet graft mass during the immediate posttransplant period. This 1 Islet Cell Laboratory, Baylor Research Institute, Dallas, TX 2 Institute of Biomedical Studies, Baylor University, Waco, TX 3 Baylor Institute for Immunology Research, Dallas, TX 4 Adacell Medical Research Center, Department of Endocrinology and Metabolism, Diskapi Yildirim Beyazit Training and Research Hospital, Ankara, Turkey 5 Department of Surgery, Virginia Commonwealth University School of Medicine, Richmond, VA 6 Annette C. and Harold C. Simmons Transplant Institute, Baylor University Medical Center, Dallas, TX Corresponding authors: Michael C. Lawrence, [email protected], and Bashoo Naziruddin, [email protected]. Received 16 May 2017 and accepted 22 August 2017. This article contains Supplementary Data online at http://diabetes .diabetesjournals.org/lookup/suppl/doi:10.2337/db17-0578/-/DC1. G.Y. and F.K. contributed equally to this study © 2017 by the American Diabetes Association. Readers may use this article as long as the work is properly cited, the use is educational and not for profit, and the work is not altered. More information is available at http://www.diabetesjournals .org/content/license. Diabetes Volume 66, November 2017 2857 IMMUNOLOGY AND TRANSPLANTATION Downloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022
Transcript of Pancreatic β-Cell–Derived IP-10/CXCL10 Isletokine Mediates ...

Pancreatic b-Cell–Derived IP-10/CXCL10 IsletokineMediates Early Loss of Graft Function in Islet CellTransplantationGumpei Yoshimatsu,1 Faisal Kunnathodi,1 Prathab Balaji Saravanan,1 Rauf Shahbazov,1 Charles Chang,2
Carly M. Darden,2 Sandra Zurawski,3 Gulbahar Boyuk,4 Mazhar A. Kanak,5 Marlon F. Levy,5
Bashoo Naziruddin,6 and Michael C. Lawrence1
Diabetes 2017;66:2857–2867 | https://doi.org/10.2337/db17-0578
Pancreatic islets produce and secrete cytokines and che-mokines in response to inflammatory and metabolicstress. The physiological role of these “isletokines” inhealth and disease is largely unknown. We observed thatislets release multiple inflammatory mediators in patientsundergoing islet transplants within hours of infusion. Theproinflammatory cytokine interferon-g–induced protein10 (IP-10/CXCL10) was among the highest released, andhigh levels correlated with poor islet transplant outcomes.Transgenic mouse studies confirmed that donor islet–specific expression of IP-10 contributed to islet inflamma-tion and loss of b-cell function in islet grafts. The effectsof islet-derived IP-10 could be blocked by treatment ofdonor islets and recipient mice with anti–IP-10 neutraliz-ing monoclonal antibody. In vitro studies showed inductionof the IP-10 gene was mediated by calcineurin-dependentNFAT signaling in pancreatic b-cells in response to ox-idative or inflammatory stress. Sustained association ofNFAT and p300 histone acetyltransferase with the IP-10gene required p38 and c-Jun N-terminal kinase mitogen-activated protein kinase (MAPK) activity, which differen-tially regulated IP-10 expression and subsequent proteinrelease. Overall, these findings elucidate an NFAT-MAPKsignaling paradigm for induction of isletokine expressionin b-cells and reveal IP-10 as a primary therapeutic targetto prevent b-cell–induced inflammatory loss of graft func-tion after islet cell transplantation.
Islet endocrine cells have been shown to produce cytokinesunder conditions of physical, inflammatory, and metabolicstress (1–6). Expression of interleukin (IL)-1b has been ob-served in b-cells exposed to hyperglycemic conditions andin patients with type 2 diabetes (7). IL-1b signaling furtherpromotes cytokine expression in b-cells (3,5,6,8–10). Acuteeffects of IL-1b to enhance insulin expression and induceproliferation in b-cells indicate a physiological role of cyto-kines to enable islets to adapt to inflammatory stress andmetabolic demand (11–16). However, inappropriate expres-sion of cytokines by islets is associated with endoplasmicreticulum and oxidative stress responses that promoteb-cell death and dysfunction (17–20). Overexposure of is-lets to osmotic, metabolic, oxidative, or inflammatory stressinduces p38 and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinase (MAPK) signaling (21–24). Thisleads to downstream effects of nuclear factor-kB to upre-gulate genes that promote apoptosis (17,25).
During islet transplantation, islets are exposed to mul-tiple physical and chemical stresses throughout the islet cellisolation and infusion procedures that induce expression ofcytokines. These inflammatory mediators are secreted by isletsand persist for days within culture. Upon transplantation ofislets, an innate immune response is observed that is largelyresponsible for a loss of up to 50% or more of the initial isletgraft mass during the immediate posttransplant period. This
1Islet Cell Laboratory, Baylor Research Institute, Dallas, TX2Institute of Biomedical Studies, Baylor University, Waco, TX3Baylor Institute for Immunology Research, Dallas, TX4Adacell Medical Research Center, Department of Endocrinology and Metabolism,Diskapi Yildirim Beyazit Training and Research Hospital, Ankara, Turkey5Department of Surgery, Virginia Commonwealth University School of Medicine,Richmond, VA6Annette C. and Harold C. Simmons Transplant Institute, Baylor University MedicalCenter, Dallas, TX
Corresponding authors: Michael C. Lawrence, [email protected],and Bashoo Naziruddin, [email protected].
Received 16 May 2017 and accepted 22 August 2017.
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db17-0578/-/DC1.
G.Y. and F.K. contributed equally to this study
© 2017 by the American Diabetes Association. Readers may use this article aslong as the work is properly cited, the use is educational and not for profit, and thework is not altered. More information is available at http://www.diabetesjournals.org/content/license.
Diabetes Volume 66, November 2017 2857
IMMUNOLOGYAND
TRANSPLANTATIO
ND
ownloaded from
http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022

phenomenon was first described as an instant blood-mediatedinflammatory reaction that is characterized by a heparin-sensitive, platelet-mediated activation of the complement cas-cade (26,27). We hypothesized that release of islet-derivedcytokines by stressed islets contributes to the early inflam-matory loss of islet cell tissue upon transplantation.
In this study, we monitored circulating inflammatorymediators in patients immediately after islet transplantation andidentified interferon-g–induced protein 10 (IP-10/CXCL10)as a major chemokine released immediately upon islet infu-sion that adversely affects transplant outcomes. Protection ofislet grafts with b-cell–specific deletion of IP-10 in an islettransplant model confirmed a role of donor islet–specific IP-10to contribute to early inflammatory loss of islet graft function.Further analysis of IP-10 in b-cells indicated that NFAT andstress-activated MAPK signaling directly induced expressionof the IP-10 gene in response to oxidative or inflammatory stress.Moreover, high glucose stimulated release of IP-10 protein fromstressed b-cells. Finally, a monoclonal antibody (mAb) di-rected toward IP-10 could prevent early loss of islet graftstransplanted in mice. These findings highlight IP-10 as akey isletokine that can be targeted to prevent islet inflam-mation and improve islet cell transplant outcomes.
RESEARCH DESIGN AND METHODS
Cell and Tissue SamplesBlood samples were collected from 34 patients undergoingislet transplants at Baylor University Medical Center at timeintervals after islet infusion in accordance with InstitutionalReview Board–approved protocols. Isolated human isletsfrom multiple donors (.90% purity) were provided by theIntegrated Islet Distribution Program at City of Hope andfrom the cGMP Islet Cell Processing Laboratory at BaylorUniversity Medical Center. C57BL/6 mouse pancreatic isletswere isolated as described below. Isolated islets were cultured1–2 days before use. Isolated islets, FACS-purified b-cells,and the MIN6 b-cell line were cultured in RPMI 1640 orKrebs-Ringer bicarbonate HEPES buffer media at 37°C in5% CO2 humidified air.
Mouse Islet IsolationThe use and care of laboratory animals for all studies wereperformed in accordance with guidelines and protocols approvedby the Institutional Animal Care and Use Committee. Islets wereisolated fromwild-type and IP-10 knockout (IP-102/2) C57BL/69- to 12-week-oldmalemice (The Jackson Laboratory). Pancreatawere removed frommice after type V collagenase (C9263; Sigma-Aldrich) perfusion, digested for 18 min at 37°C, and washedwithHanks’ balanced salt solution. Islets were separated fromdigested pancreas preparations by centrifugation on a 1.077and 1.100 g/mL Ficoll gradient solution. The separated islet tissuelayer was collected and washed with Hanks’ balanced salt solu-tion. Intact islets were hand selected from the purifiedpreparation by pipette and cultured overnight at 37°C.
Reagents and Recombinant DNA ConstructsAntibodies were used as follows: NFATc2, p300, andHDAC3 (Santa Cruz Biotechnology, Inc.), and extracellular
signal–regulated kinase (ERK)1/2, p38, and JNK (Cell Sig-naling Technology). The mouse IP-10 promoter-reportervector containing Gaussia luciferase reporter was obtainedfrom GeneCopoeia. Anti–IP-10 mAb was obtained fromthe Baylor Research Institute Biotechnology Core. Vectorsexpressing green fluorescent protein (GFP) and mouseNFATc2 or sequence-scrambled short hairpin (sh)RNA wereobtained from OriGene. Expression vectors containingdominant negative NFAT PxIxIT motif (dnNFAT) andmutated dnNFAT AxAxAA motif (dnNFATm) were pre-viously described (5,28).
DNA Transfection and Promoter AssaysMIN6 cells were transfected with plasmids and shRNAconstructs by Lipofectamine 2000 24 h before purificationand cell treatments. Cells were harvested with 13 ReporterLysis buffer, and luciferase/Renilla enzyme activity was de-tected by the Dual-Luciferase Reporter Assay System (Promega)using a TD20/20 Luminometer.
Flow CytometryHuman islets andMIN6 cells underwent FACS on aMoFlo high-speed cell sorter. b-Cells were purified based on zinc-sensitivefluorescence of Newport Green 29,79-dichlorofluoresceindiacetate, as previously described (5). Newport Green–stained b-cell fractions were screened for exclusive expres-sion of insulin by immunoassay. MIN6 cells transfectedwith shNFAT-GFP and sh-scrambled-GFP were trypsinizedand purified based on relative GFP fluorescence.
ImmunoassaysProteins from human serum collected from patients un-dergoing islet transplants and supernatant samples fromisolated human islets (300 islet equivalent [IEQ] in 1 mLmedium) were measured by multiplex assays with magneticbeads using the Luminex platform system (EMD Millipore).ELISAs were performed to measure IP-10 and insulin usinga Cytation 5 microplate reader (BioTek).
Immunofluorescent StainingFormalin-fixed and paraffin-embedded sections of isolatedhuman islets were incubated with primary antibody (1:250)in PBS containing 0.1% Triton X-100 and 4% BSA overnightat 4°C. Samples were washed, underwent fluorescein iso-thiocyanate and Cy3 conjugated secondary antibody (1:500),and imaged by an Olympus BX61 TRF Fluorescent Microscope.
Islet TransplantationWild-type C57BL/6, IP-102/2 C57BL/6, and nude mice(Envigo) (9- to 12-week old males) were injected with strep-tozotocin (STZ; 200 mg/kg body weight). Mice with bloodglucose.400 mg/dL for$2 consecutive days were selectedas diabetic recipients for islet transplantation. For mouseislet recipients, 200 islets were infused to C57BL/6 recipi-ents via the portal vein. For human islet recipients, 2,000IEQ were infused to subcapsular kidney sites in nude mice,and kidneys were removed after 6 h for analysis of isletgraft cytokine gene expression.
2858 IP-10 Mediates Loss of Islet Graft Function Diabetes Volume 66, November 2017
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022

Glucose MeasurementsBlood glucose and body weight were monitored twice weeklyfor up to 30 days. After 30 days, mice were fasted for 8 hbefore an intraperitoneal administration of 20% glucosesolution at 2 g glucose/kg body weight. Blood glucose wasmeasured at 30-min intervals for intraperitoneal glucosetolerance test (IPGTT) analyses.
Nuclear Run-on AssayMIN6 cells were treated and lysed in Tris buffer containing0.5% Triton X-100. Nuclei were washed and isolated bycentrifugation on a 0.6 mol/L sucrose cushion at 600g in atable top centrifuge. Intact fractionated nuclei were incu-bated at 30°C for 30 min in Tris buffer containing 2 unitsRNasin recombinant ribonuclease inhibitor (Promega) and30% glycerol in the presence of 200 mmol/L ribonucleosidetriphosphate. RNA was isolated using TRI Reagent andtranscribed to cDNA. Relative changes in gene targets and18S mRNA were quantified by quantitative (q)PCR.
Chromatin Immunoprecipitation AssaysChromatin immunoprecipitation (ChIP) assays were per-formed as previously described (6). Briefly, MIN6 cells werefixed, and chromatin DNA-protein was cross-linked with1% formaldehyde for 10 min, quenched with 125 mmol/Lglycine for 5 min, and washed with PBS. Fixed cells weresonicated with a Bioruptor 200 (Diagenode) and lysed inradioimmunoprecipitation assay buffer containing proteaseinhibitor cocktail. DNA-protein complexes were immunopre-cipitated with the indicated antibodies or IgG isotype con-trols and extensively washed. Cross-links were reversed at65°C for 4 h in Tris buffer containing 5 mol/L NaCl and 0.5mol/L EDTA. DNA was extracted by phenol/CHCl3 and pre-cipitated with ethanol, followed by proteinase K and ribo-nuclease A treatment. Precipitated DNA and 1% controlinputs were analyzed by qPCR.
Real-time qPCRqPCR was performed using the Bio-Rad CFX Connect systemwith RT2 SYBR Green qPCR Mastermix. RT2 qPCR PrimerAssays were used to detect specific cDNA-converted mRNAtranscripts for 18S and target genes (Qiagen). Primer se-quences used to detect input, and immunoprecipitated DNAsequences from 2266 to 2122 of the 59-flank mouse IP-10promoter region were 59-TCTGCAAGGCACTCATCTGAT and59-CGAGGGCATTGCTTGTGTTT.
RESULTS
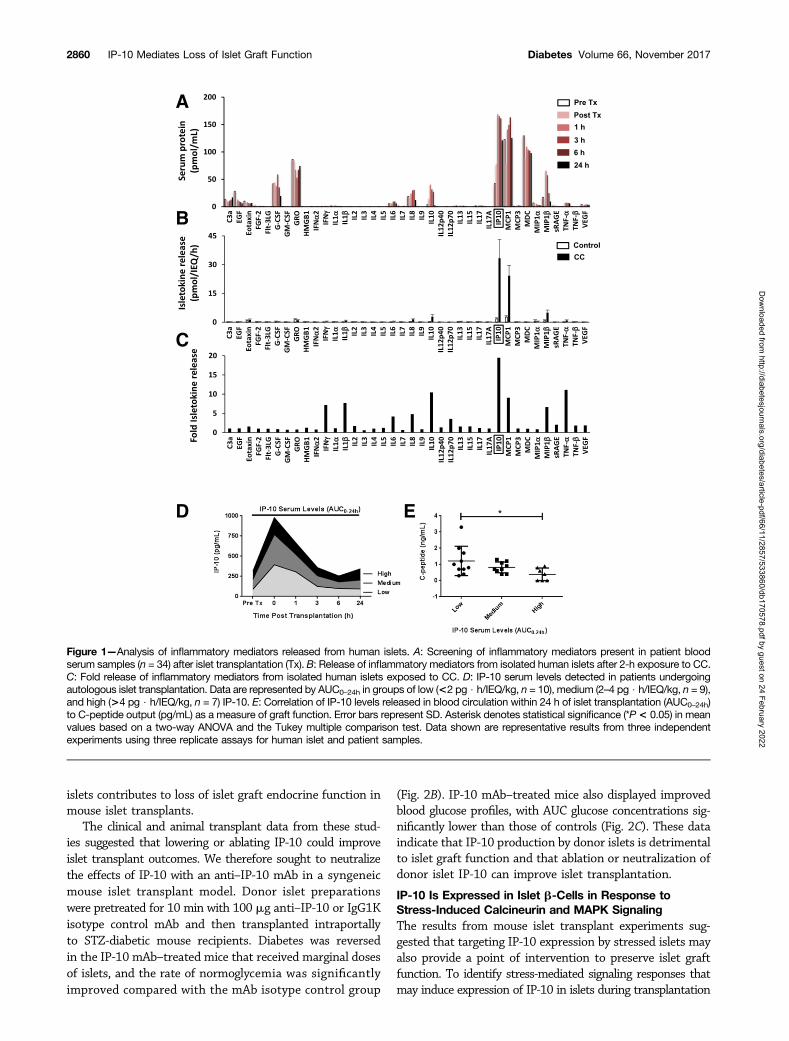
High IP-10 Released From Islets Upon Transplantation IsAssociated With Poor Clinical Transplant OutcomesWe analyzed clinical samples for inflammatory mediatorspresent in the blood plasma circulation immediately afterislet infusion. Samples from islet transplant patients beforeand after islet infusion were analyzed by Luminex multiplexprotein screening to identify early inflammatory mediatorspresent upon infusion into the portal vein. Several isleto-kines were present in the blood circulation within hours of
islet transplantation (Fig. 1A), with particularly high levelsof IP-10, MCP-1, and MDC (.100 pmol/mL). Notably, IP-10showed the largest increase in expression (approximatelyfourfold) in the circulation within 1 h of islet infusion rel-ative to pretransplant control samples.
To identify isletokines directly produced by islets underinflammatory conditions, we exposed isolated human islets(3,000 IEQ) to a proinflammatory cytokine cocktail (CC)containing 5 ng/mL IL-1b, 10 ng/mL tumor necrosis factor-a (TNF-a), and 50 ng/mL interferon-g (IFN-g) for 2 h.Islets were washed extensively, and we analyzed proteinsreleased into the medium 6 h after treatment. The analysisshowed that chemokines IP-10, MCP-1, and MIP-1b hadthe highest rates of release (.5 pmol/IEQ/h) (Fig. 1B). Largeinductions (more than fivefold) were observed for IFN- g,IL-1b, IL-10, IP-10, MCP-1, MIP-1b, and TNF-a (Fig. 1C).Among all cytokines tested, IP-10 had the highest increasein induction and amount of protein released into the circu-lation after islet infusion or into the medium in vitro afterislet exposure to inflammatory stress.
To investigate the role of IP-10 in innate inflammatoryreactions immediately after islet transplantation in isolationfrom adaptive immune responses associated with allotrans-plantation, we analyzed serum samples from autologousislet transplant patients (n = 26) collected at various intervalsimmediately after infusion and correlated them to trans-plant outcomes assessed more than 6 months after transplan-tation. Transplant patients were grouped by IP-10 area underthe curve (AUC) plasma levels over 24 h (AUC0–24h) of low(,2 pg $ h/IEQ/kg, n = 10), medium (2–4 pg $ h/IEQ/kg,n = 9), and high (.4 pg $ h/IEQ/kg, n = 7) (Fig. 1D). Theanalyses showed that patients in the group with higherlevels of IP-10 had significantly reduced islet graft functionas assessed by C-peptide (Fig. 1E). Collectively, these dataindicate that IP-10 plays a role in the inflammatory re-sponse and loss of islet function observed during the im-mediate peritransplant period. The data also suggest thatthat lowering IP-10 released by islets could improve trans-plant outcomes.
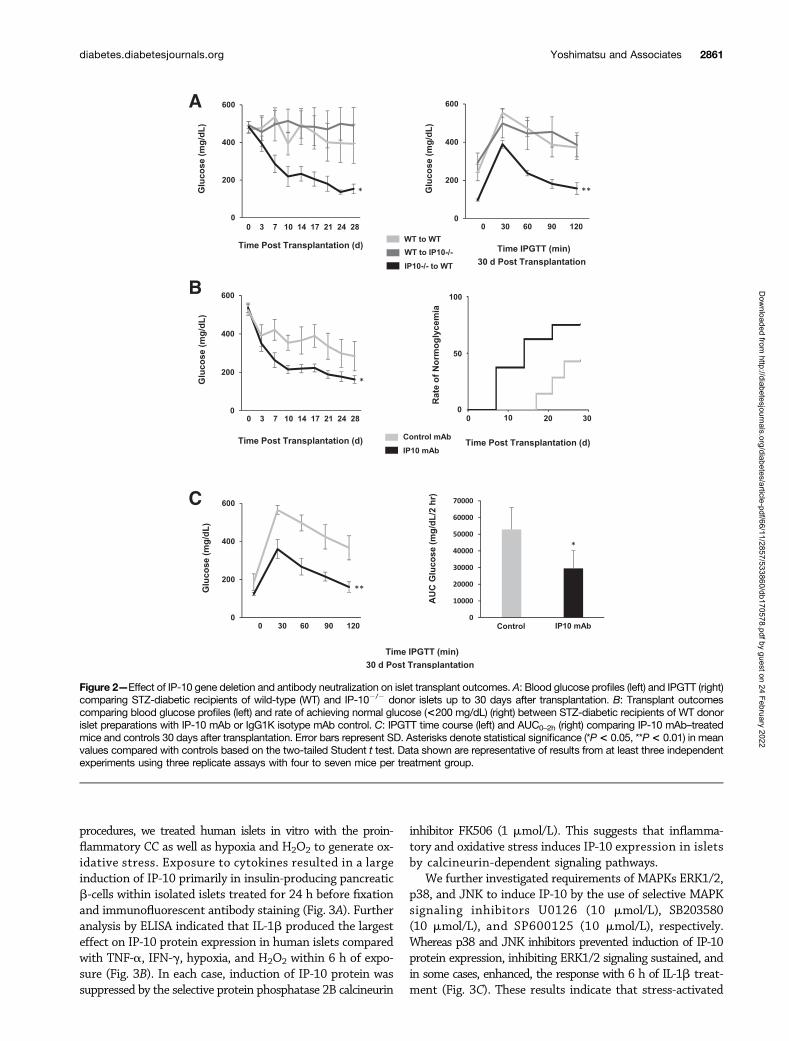
Effects of IP-10 on Islet Transplant Outcomes Is Donor-Islet Specific and Can Be Reversed by Anti–IP-10 mAbTo determine the direct contributions of islet-derived IP-10on transplant outcomes, we used transgenic IP-102/2 miceto compare effects of IP-10 gene deletion in islet donortissue and in islet recipients on graft function. STZ-induceddiabetic mice were recipients of marginal doses of 200 iso-lated mouse donor islets intraportally. Blood glucose wasmonitored in recipients up to 30 days after transplantation,and glucose profiles were assessed by IPGTT (Fig. 2A). Hy-perglycemia was reversed in those receiving IP-102/2 donorislets up to 30 days after transplantation. IPGTTs showedreversal of blood glucose to normal concentrations within2 h of the glucose challenge. In contrast, recipients of wild-type donor islets remained diabetic and exhibited poor glu-cose tolerance 30 days after transplantation. The resultsindicate that IP-10 specifically derived from b-cells of donor
diabetes.diabetesjournals.org Yoshimatsu and Associates 2859
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022
islets contributes to loss of islet graft endocrine function inmouse islet transplants.
The clinical and animal transplant data from these stud-ies suggested that lowering or ablating IP-10 could improveislet transplant outcomes. We therefore sought to neutralizethe effects of IP-10 with an anti–IP-10 mAb in a syngeneicmouse islet transplant model. Donor islet preparationswere pretreated for 10 min with 100 mg anti–IP-10 or IgG1Kisotype control mAb and then transplanted intraportallyto STZ-diabetic mouse recipients. Diabetes was reversedin the IP-10 mAb–treated mice that received marginal dosesof islets, and the rate of normoglycemia was significantlyimproved compared with the mAb isotype control group
(Fig. 2B). IP-10 mAb–treated mice also displayed improvedblood glucose profiles, with AUC glucose concentrations sig-nificantly lower than those of controls (Fig. 2C). These dataindicate that IP-10 production by donor islets is detrimentalto islet graft function and that ablation or neutralization ofdonor islet IP-10 can improve islet transplantation.
IP-10 Is Expressed in Islet b-Cells in Response toStress-Induced Calcineurin and MAPK SignalingThe results from mouse islet transplant experiments sug-gested that targeting IP-10 expression by stressed islets mayalso provide a point of intervention to preserve islet graftfunction. To identify stress-mediated signaling responses thatmay induce expression of IP-10 in islets during transplantation
Figure 1—Analysis of inflammatory mediators released from human islets. A: Screening of inflammatory mediators present in patient bloodserum samples (n = 34) after islet transplantation (Tx). B: Release of inflammatory mediators from isolated human islets after 2-h exposure to CC.C: Fold release of inflammatory mediators from isolated human islets exposed to CC. D: IP-10 serum levels detected in patients undergoingautologous islet transplantation. Data are represented by AUC0–24h in groups of low (<2 pg $ h/IEQ/kg, n = 10), medium (2–4 pg $ h/IEQ/kg, n = 9),and high (>4 pg $ h/IEQ/kg, n = 7) IP-10. E: Correlation of IP-10 levels released in blood circulation within 24 h of islet transplantation (AUC0–24h)to C-peptide output (pg/mL) as a measure of graft function. Error bars represent SD. Asterisk denotes statistical significance (*P< 0.05) in meanvalues based on a two-way ANOVA and the Tukey multiple comparison test. Data shown are representative results from three independentexperiments using three replicate assays for human islet and patient samples.
2860 IP-10 Mediates Loss of Islet Graft Function Diabetes Volume 66, November 2017
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022
procedures, we treated human islets in vitro with the proin-flammatory CC as well as hypoxia and H2O2 to generate ox-idative stress. Exposure to cytokines resulted in a largeinduction of IP-10 primarily in insulin-producing pancreaticb-cells within isolated islets treated for 24 h before fixationand immunofluorescent antibody staining (Fig. 3A). Furtheranalysis by ELISA indicated that IL-1b produced the largesteffect on IP-10 protein expression in human islets comparedwith TNF-a, IFN-g, hypoxia, and H2O2 within 6 h of expo-sure (Fig. 3B). In each case, induction of IP-10 protein wassuppressed by the selective protein phosphatase 2B calcineurin
inhibitor FK506 (1 mmol/L). This suggests that inflamma-tory and oxidative stress induces IP-10 expression in isletsby calcineurin-dependent signaling pathways.
We further investigated requirements of MAPKs ERK1/2,p38, and JNK to induce IP-10 by the use of selective MAPKsignaling inhibitors U0126 (10 mmol/L), SB203580(10 mmol/L), and SP600125 (10 mmol/L), respectively.Whereas p38 and JNK inhibitors prevented induction of IP-10protein expression, inhibiting ERK1/2 signaling sustained, andin some cases, enhanced, the response with 6 h of IL-1b treat-ment (Fig. 3C). These results indicate that stress-activated
Figure 2—Effect of IP-10 gene deletion and antibody neutralization on islet transplant outcomes. A: Blood glucose profiles (left) and IPGTT (right)comparing STZ-diabetic recipients of wild-type (WT) and IP-102/2 donor islets up to 30 days after transplantation. B: Transplant outcomescomparing blood glucose profiles (left) and rate of achieving normal glucose (<200 mg/dL) (right) between STZ-diabetic recipients of WT donorislet preparations with IP-10 mAb or IgG1K isotype mAb control. C: IPGTT time course (left) and AUC0–2h (right) comparing IP-10 mAb–treatedmice and controls 30 days after transplantation. Error bars represent SD. Asterisks denote statistical significance (*P< 0.05, **P< 0.01) in meanvalues compared with controls based on the two-tailed Student t test. Data shown are representative of results from at least three independentexperiments using three replicate assays with four to seven mice per treatment group.
diabetes.diabetesjournals.org Yoshimatsu and Associates 2861
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022
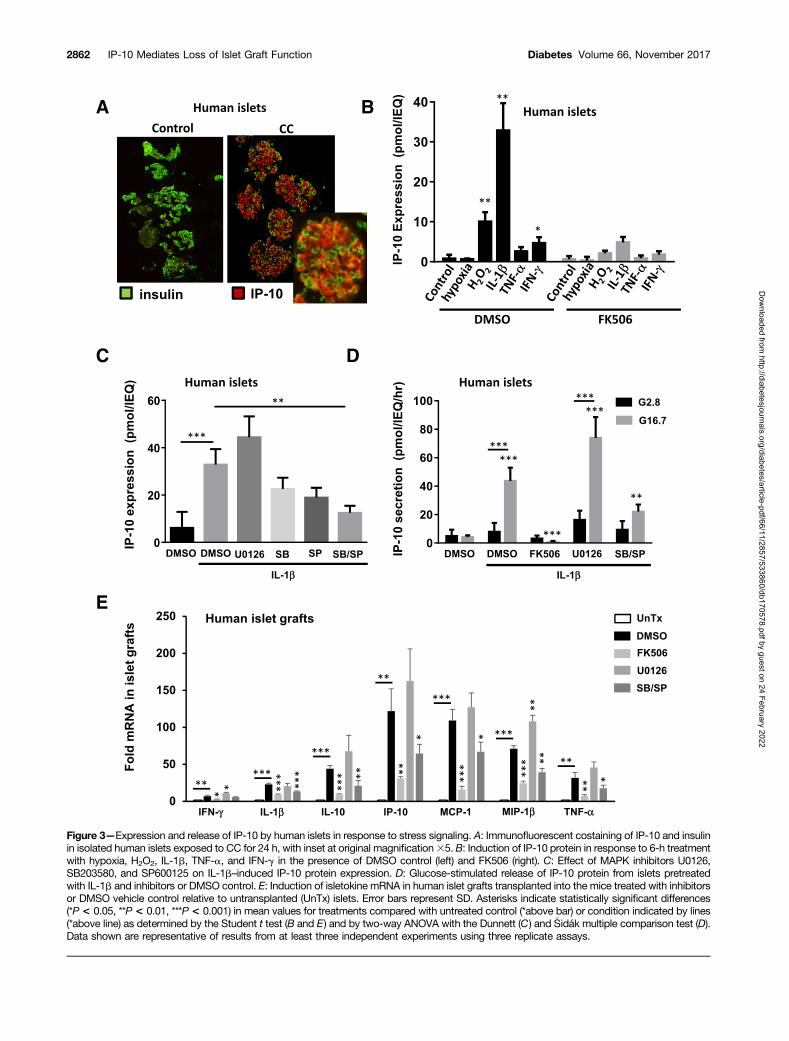
Figure 3—Expression and release of IP-10 by human islets in response to stress signaling. A: Immunofluorescent costaining of IP-10 and insulinin isolated human islets exposed to CC for 24 h, with inset at original magnification35. B: Induction of IP-10 protein in response to 6-h treatmentwith hypoxia, H2O2, IL-1b, TNF-a, and IFN-g in the presence of DMSO control (left) and FK506 (right). C: Effect of MAPK inhibitors U0126,SB203580, and SP600125 on IL-1b–induced IP-10 protein expression. D: Glucose-stimulated release of IP-10 protein from islets pretreatedwith IL-1b and inhibitors or DMSO control. E: Induction of isletokine mRNA in human islet grafts transplanted into the mice treated with inhibitorsor DMSO vehicle control relative to untransplanted (UnTx) islets. Error bars represent SD. Asterisks indicate statistically significant differences(*P< 0.05, **P< 0.01, ***P< 0.001) in mean values for treatments compared with untreated control (*above bar) or condition indicated by lines(*above line) as determined by the Student t test (B and E) and by two-way ANOVA with the Dunnett (C) and �Sidák multiple comparison test (D).Data shown are representative of results from at least three independent experiments using three replicate assays.
2862 IP-10 Mediates Loss of Islet Graft Function Diabetes Volume 66, November 2017
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022

MAPKs p38 and JNK have selective if not reciprocal roles inthe induction of intracellular IP-10 protein expression inhuman islets in response to IL-1b.
We also examined conditions that could influence IP-10release from stressed islets. We observed that human isletstreated with IL-1b for 6 h had significantly increased ratesof IP-10 release (.40 pmol/IEQ/h) when exposed to highglucose (Fig. 3D). The effect of glucose on the release ofIP-10 from islets was likely independent of insulin action,because exogenous insulin up to 100 nmol/L could not di-rectly stimulate IP-10 secretion alone or in combination withglucose (Supplementary Fig. 1). Glucose alone was not suf-ficient to induce IP-10 expression but was required forIP-10 release after 6 h induction with IL-1b. Moreover,glucose-stimulated IP-10 protein release was significantlyreduced in human islets exposed to IL-1b in the presenceof p38 and JNK but not ERK1/2 inhibitor. Notably, glucose-stimulated insulin secretion was not affected by MAPK in-hibitors (Supplementary Fig. 2). Taken together, these dataindicate that calcineurin signaling and MAPKs p38 and JNKare required for IP-10 expression and that glucose can stim-ulate its subsequent release from human islets. Importantly,the results demonstrate that IP-10 signaling pathways can betargeted for suppression without affecting b-cell function.
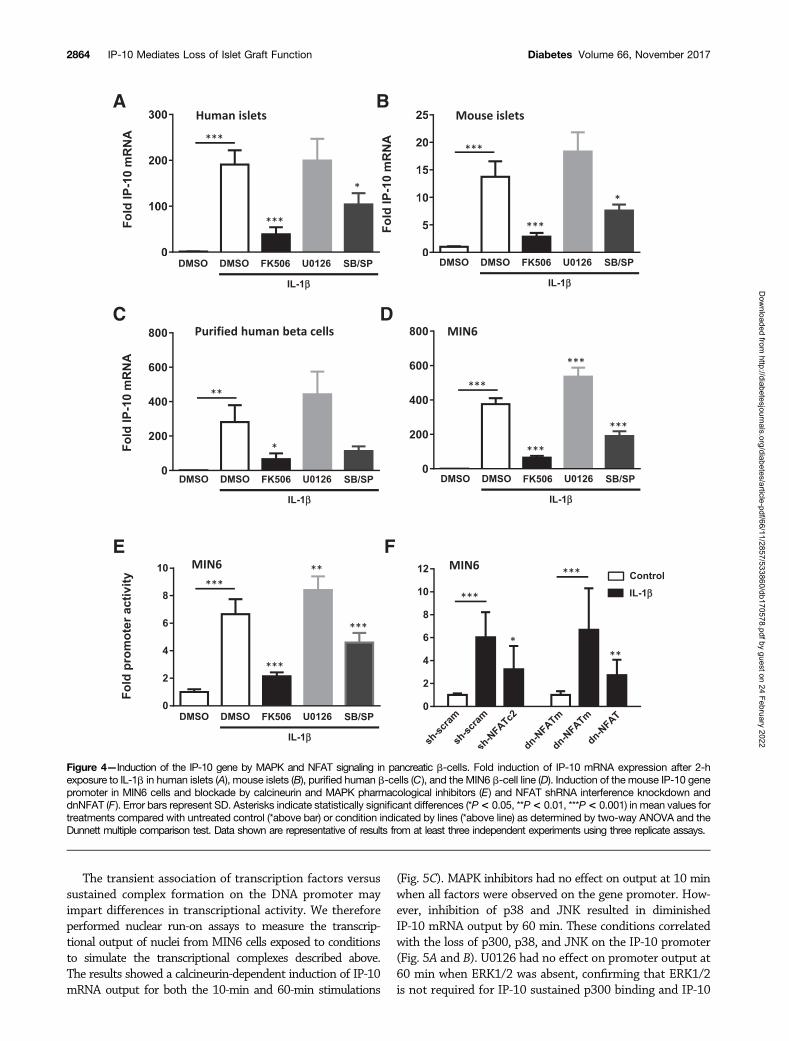
Calcineurin and MAPKs Regulate NFAT-MediatedIsletokine Gene Transcription in Pancreatic b-CellsThe previous experiments demonstrated that human isletscould express IP-10 in response to stress-activated signalingpathways. To determine whether islets expressed isleto-kines during islet transplantation, we transplanted isolatedhuman islets into the kidney capsules of diabetic nude miceand removed the islet grafts by scalpel dissection after 6 hto evaluate the expression of isletokine genes. The geneexpression assays showed a significant induction of mRNAin human islet xenografts for all isletokines tested (Fig. 3E).In each case, isletokine mRNA was blocked or largely re-duced by pretreatment of islets with calcineurin and MAPKp38 and JNK inhibitors. These results indicate that IP-10and other isletokines are induced in human islets upontransplantation at the transcriptional level via a commonmechanism involving calcineurin and MAPK signaling path-ways. Similarly, IP-10 mRNA was induced in isolated humanand mouse islets in vitro within 2 h of exposure to IL-1band suppressed by both calcineurin and MAPK p38 andJNK inhibitors (Fig. 4A and B). These responses werealso observed in FACS-purified human b-cells and MIN6b-cell lines (Fig. 4C and D). The results were further cor-roborated using a different set of highly selective MAPKinhibitors in MIN6 cells (Supplementary Fig. 3). Moreover,calcineurin and MAPKs p38 and JNK were required to in-duce IP-10 gene promoter activity in MIN6 cells as detectedby luciferase promoter-reporter assays (Fig. 4E). These ex-periments collectively demonstrate that IP-10 induction isregulated by calcineurin and MAPKs in pancreatic isletb-cells during islet transplantation and under conditionsof inflammatory stress.
That calcineurin activates downstream target NFAT-family transcription factors to directly regulate cytokinegenes in immune cells has been well established (29–39).Interfering with the ability of calcineurin to bind to anddephosphorylate NFAT-family member substrates preventsNFAT from regulating cytokine gene transcription. Thus,we used an RNA interference knockdown approach to testthe requirement of NFAT in the induction of IP-10 pro-moter activity in MIN6 cells. Overexpression of NFATc2shRNA reduced induction of IP-10 promoter activity inMIN6 cells treated for 6 h with IL-1b compared with scram-bled shRNA controls. In addition, overexpression of adnNFAT PxIxIT motif to interfere with NFAT interactionwith calcineurin suppressed IL-1b–induced IP-10 promoteractivity compared with an alanine-substituted AxAxAAdnNFATm control (Fig. 5F). These results indicated thatNFAT is an essential component for the activation of theIP-10 gene promoter by calcineurin in pancreatic b-cells.
To determine whether NFAT directly interacts with theIP-10 gene in b-cells, we performed ChIP assays to examinethe association of NFATc2 with the 59 flanking IP-10 genepromoter in MIN6 cells. Time course analyses indicatedthat the IP-10 promoter was enriched with NFATc2 up to40-fold within 10 min of IL-1b exposure (Fig. 5A). Theassociation of NFATc2 with the IP-10 promoter was largelysustained for up to 60 min. In contrast, FK506 abolishedNFATc2 binding. MAPK inhibitors had little effect on theacute (10 min) association of NFATc2 with the IP-10 genebut showed a significant reduction in a sustained (60 min)response. Histone acetylase p300 showed a biphasic associ-ation with the IP-10 promoter. However, whereas ERK1/2inhibition had no effect on p300 association, inhibitors ofp38 and JNK prevented a sustained response. The biphasickinetics of p300 were inverse to what was observed withthe HDAC3 association where p38 and JNK inhibitionenhanced the response to IL-1b. This inverse relationshipindicates competitive MAPK signaling requirements forp300 and HDAC3 to regulate the IP-10 promoter. The re-sults also suggest that calcineurin signaling is required forrelative changes of association of p300 and HDAC3 withthe IP-10 promoter to occur.
MAPKs have previously been shown to translocate to thenucleus and associate with DNA-protein complexes withingene promoters (13,40,41). Thus, we also examined effectsof IL-1b to induce MAPK association with the IP-10 pro-moter. p38 and JNK both associated with the IP-10 pro-moter with similar kinetics as observed with p300 (Fig. 5B).This contrasted with ERK1/2, which was transiently enrichedwithin 10 min but sharply subsided by 60 min. In each case,FK506 prevented MAPKs from associating with the IP-10 pro-moter. MAPK inhibitors had little effect on the acute accumu-lation of MAPKs on the promoter but selectively inhibitedlate-phase responses. Together, these results suggest that cal-cineurin signaling is required for NFAT, p300, HDAC3, andMAPK association with the IP-10 promoter and that MAPKactivity is required for sustained formation of the protein-DNA chromatin complex.
diabetes.diabetesjournals.org Yoshimatsu and Associates 2863
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022
The transient association of transcription factors versussustained complex formation on the DNA promoter mayimpart differences in transcriptional activity. We thereforeperformed nuclear run-on assays to measure the transcrip-tional output of nuclei from MIN6 cells exposed to conditionsto simulate the transcriptional complexes described above.The results showed a calcineurin-dependent induction of IP-10mRNA output for both the 10-min and 60-min stimulations
(Fig. 5C). MAPK inhibitors had no effect on output at 10 minwhen all factors were observed on the gene promoter. How-ever, inhibition of p38 and JNK resulted in diminishedIP-10 mRNA output by 60 min. These conditions correlatedwith the loss of p300, p38, and JNK on the IP-10 promoter(Fig. 5A and B). U0126 had no effect on promoter output at60 min when ERK1/2 was absent, confirming that ERK1/2is not required for IP-10 sustained p300 binding and IP-10
Figure 4—Induction of the IP-10 gene by MAPK and NFAT signaling in pancreatic b-cells. Fold induction of IP-10 mRNA expression after 2-hexposure to IL-1b in human islets (A), mouse islets (B), purified human b-cells (C), and the MIN6 b-cell line (D). Induction of the mouse IP-10 genepromoter in MIN6 cells and blockade by calcineurin and MAPK pharmacological inhibitors (E) and NFAT shRNA interference knockdown anddnNFAT (F). Error bars represent SD. Asterisks indicate statistically significant differences (*P< 0.05, **P< 0.01, ***P< 0.001) in mean values fortreatments compared with untreated control (*above bar) or condition indicated by lines (*above line) as determined by two-way ANOVA and theDunnett multiple comparison test. Data shown are representative of results from at least three independent experiments using three replicate assays.
2864 IP-10 Mediates Loss of Islet Graft Function Diabetes Volume 66, November 2017
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022
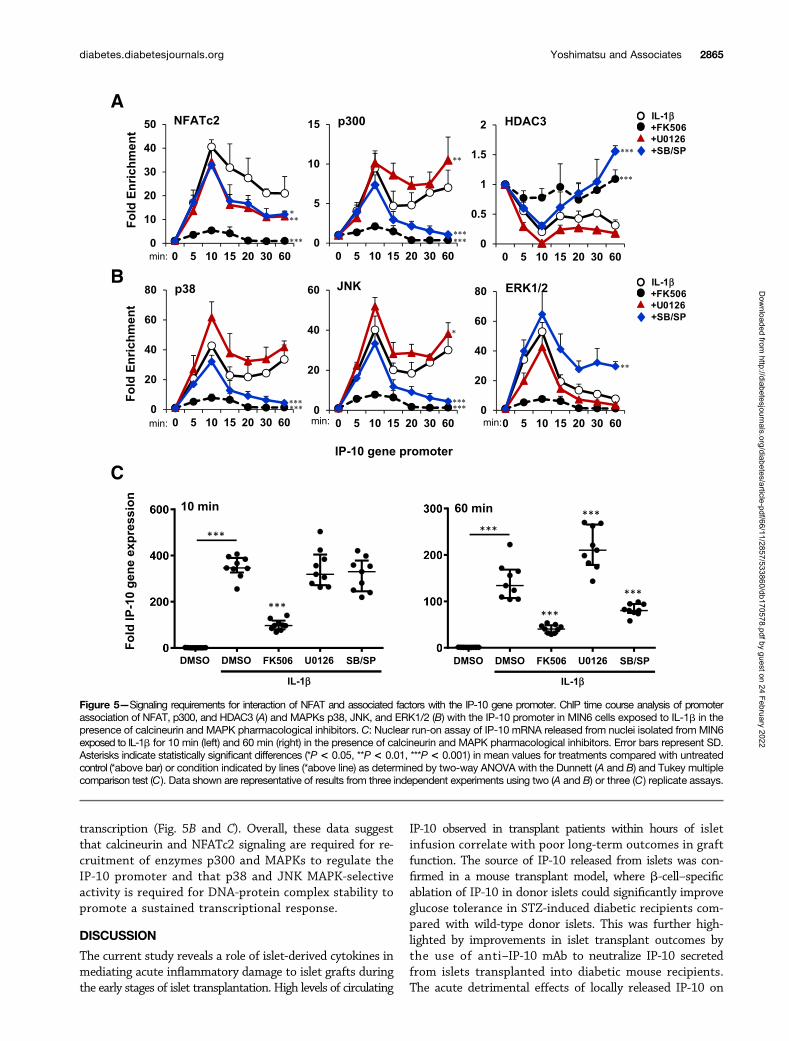
transcription (Fig. 5B and C). Overall, these data suggestthat calcineurin and NFATc2 signaling are required for re-cruitment of enzymes p300 and MAPKs to regulate theIP-10 promoter and that p38 and JNK MAPK-selectiveactivity is required for DNA-protein complex stability topromote a sustained transcriptional response.
DISCUSSION
The current study reveals a role of islet-derived cytokines inmediating acute inflammatory damage to islet grafts duringthe early stages of islet transplantation. High levels of circulating
IP-10 observed in transplant patients within hours of isletinfusion correlate with poor long-term outcomes in graftfunction. The source of IP-10 released from islets was con-firmed in a mouse transplant model, where b-cell–specificablation of IP-10 in donor islets could significantly improveglucose tolerance in STZ-induced diabetic recipients com-pared with wild-type donor islets. This was further high-lighted by improvements in islet transplant outcomes bythe use of anti–IP-10 mAb to neutralize IP-10 secretedfrom islets transplanted into diabetic mouse recipients.The acute detrimental effects of locally released IP-10 on
Figure 5—Signaling requirements for interaction of NFAT and associated factors with the IP-10 gene promoter. ChIP time course analysis of promoterassociation of NFAT, p300, and HDAC3 (A) and MAPKs p38, JNK, and ERK1/2 (B) with the IP-10 promoter in MIN6 cells exposed to IL-1b in thepresence of calcineurin and MAPK pharmacological inhibitors. C: Nuclear run-on assay of IP-10 mRNA released from nuclei isolated from MIN6exposed to IL-1b for 10 min (left) and 60 min (right) in the presence of calcineurin and MAPK pharmacological inhibitors. Error bars represent SD.Asterisks indicate statistically significant differences (*P < 0.05, **P < 0.01, ***P < 0.001) in mean values for treatments compared with untreatedcontrol (*above bar) or condition indicated by lines (*above line) as determined by two-way ANOVA with the Dunnett (A and B) and Tukey multiplecomparison test (C). Data shown are representative of results from three independent experiments using two (A and B) or three (C) replicate assays.
diabetes.diabetesjournals.org Yoshimatsu and Associates 2865
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022

transplanted islets is likely indirect because direct treatmentof isolated islets with concentrations of up to 100 ng/mLrecombinant IP-10 did not affect b-cell function or apopto-sis within 24 h of exposure (Supplementary Fig. 4A and B).
These results provide proof of principle for the potentialuse of anticytokine therapy to protect islets during clinicalislet transplantation procedures. Indeed, promising resultshave been observed with the use of anti-inflammatory re-combinant protein antagonists to neutralize cytokines duringislet transplantation. Our center has successfully used etaner-cept and anakinra to block TNF-a and IL-1b, respectively, toimprove islet transplant outcomes (42). These observationswere corroborated in a controlled mouse transplant studythat demonstrated combined effects of etanercept and ana-kinra to improve islet cell function and engraftment inimmunodeficient mice (43). These results show a combina-torial approach to targeting circulating inflammatory medi-ators, such as IP-10, TNF-a, and IL-1b, could conceivablyhave synergistic effects to improve islet transplant out-comes or reduce IEQ tissue required. The use of neutralizingprotein antagonists to selectively target key isletokines re-leased upon islet infusion to improve transplant outcomeswarrants further exploration for clinical use.
The study also identifies upstream molecular targetsto block isletokine production by islet b-cells. In vitro andin vivo results both indicated that a set of common signal-ing components is activated by oxidative or inflammatorystress, which induces expression of isletokine genes inb-cells. One common signaling component is the calcium-dependent calcineurin/NFAT pathway. The selective calci-neurin inhibitor FK506 suppressed multiple cytokines inhuman islet grafts transplanted in mice. Further analysisshowed that the downstream transcriptional target of cal-cineurin, NFATc2, and histone acetylase p300 were associ-ated with the IP-10 promoter in response to IL-1b.
The other identified common signaling component in-volves stress-activated MAPKs. The activity of chromatin-bound p38 and JNK promoted the association of bothNFATc2 and p300 while opposing the accumulation ofHDAC3. Transcriptional activity of IP-10 in b-cells corre-lated closely with kinetics of promoter-bound NFATc2 andp300, indicating that p38 and JNK were required for geneexpression. ERK1/2, however, had little effect on the IP-10gene promoter, which highlights a kinase-selective roleof stress-activated p38 and JNK in islet inflammation. To-gether, these results indicate that multiple isletokines ex-pressed during islet transplantation can be blocked byperturbing calcineurin/NFATc2 and p38/JNK signaling.
NFAT interacts with basic helix-loop-helix (bZIP) familyproteins to regulate cytokine gene promoters (44–50). ThesebZIP proteins can enhance or repress promoter activity, depend-ing on the cell signaling and gene promoter context. We pre-viously showed that NFAT requires bZIPs c-Jun and ATF2 toactivate the TNF-a promoter in b-cells when exposed to highglucose and IL-1b (5). However, under normal conditions,b-cell–specific bZIP MafA interacts with NFAT to both reg-ulate the insulin gene in response to glucose conditions and
maintain the TNF-a gene in a silenced state (5,6). Furtherstudies are required to determine specific NFATc2-interactingcofactors that bind to and regulate IP-10 and other isletokinepromoters in b-cells during islet cell transplantation.
Lastly, proinflammatory cytokines are systemically elevatedduring the progression of type 1 and type 2 diabetes. Althoughinflammatory cells largely contribute to this inflammatorystate, the role of isletokines in the context of diabetes hasnot been fully explored. Considering that islets can evoke aninflammatory response when stressed and transplanted,b-cells could conceivably contribute to inflammatory re-sponses during a metabolic or inflammatory disease state.Thus, b-cells could play a role in the initiation or exacerba-tion of their own immune destruction by release of isleto-kines when damaged or distressed. In this broader context,targeting isletokines may have the potential to prevent ordelay progression of metabolic diseases propagated by isletinflammation and b-cell–targeted immune destruction.
Acknowledgments. The authors thank Nofit Borenstein-Auerbach andRituparna Bhattacharjee for technical assistance and Monila Khadka for databaseanalysis assistance, all from Baylor Scott & White Research Institute. The authorsacknowledge the editorial assistance of Cindy Orticio, Baylor Scott & White Health.Funding. This work was supported by Roche Diagnostic Corporation, Baylor Scott& White Health, and the Tubitak Graduate Scholarship to G.B. The National Institutesof Health grants U19-AI-057234 funded mAb development and 1UC4-DK-098085funded the Integrated Islet Distribution Program.Duality of Interest. No potential conflicts of interest relevant to this articlewere reported.Author Contributions. G.Y., F.K., P.B.S., R.S., C.C., C.M.D., S.Z., G.B., andM.A.K. researched data. M.F.L. contributed to discussion. B.N. contributed todiscussion and reviewed and edited the manuscript. M.C.L. researched data andwrote the manuscript. M.C.L. is the guarantor of this work and, as such, had fullaccess to all the data in the study and takes responsibility for the integrity of the dataand the accuracy of the data analysis.Prior Presentation. Parts of this study were presented in abstract form at the2015 International Pancreas and Islet Transplant Association, Melbourne, Australia,15–19 November 2015; the 76th Scientific Sessions of the American DiabetesAssociation, New Orleans, LA, 10–14 June 2016; the 2015 American TransplantCongress, Philadelphia, PA, 2–6 May 2015; and at the 2016 American TransplantCongress, Boston, MA, 11–15 June 2016.
References1. Chen MC, Schuit F, Eizirik DL. Identification of IL-1beta-induced messengerRNAs in rat pancreatic beta cells by differential display of messenger RNA. Dia-betologia 1999;42:1199–12032. Cardozo AK, Kruhøffer M, Leeman R, Orntoft T, Eizirik DL. Identification of novelcytokine-induced genes in pancreatic beta-cells by high-density oligonucleotidearrays. Diabetes 2001;50:909–9203. Kutlu B, Cardozo AK, Darville MI, et al. Discovery of gene networks regulatingcytokine-induced dysfunction and apoptosis in insulin-producing INS-1 cells. Di-abetes 2003;52:2701–27194. Jacobsen ML, Rønn SG, Bruun C, et al. IL-1beta-induced chemokine and Fasexpression are inhibited by suppressor of cytokine signalling-3 in insulin-producingcells. Diabetologia 2009;52:281–2885. Lawrence MC, Naziruddin B, Levy MF, Jackson A, McGlynn K. Calcineurin/nuclear factor of activated T cells and MAPK signaling induce TNF-alpha gene ex-pression in pancreatic islet endocrine cells. J Biol Chem 2011;286:1025–10366. Lawrence MC, Borenstein-Auerbach N, McGlynn K, et al. NFAT targets signalingmolecules to gene promoters in pancreatic b-cells. Mol Endocrinol 2015;29:274–288
2866 IP-10 Mediates Loss of Islet Graft Function Diabetes Volume 66, November 2017
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022

7. Maedler K, Sergeev P, Ris F, et al. Glucose-induced beta cell production ofIL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 2002;110:851–8608. Cardozo AK, Proost P, Gysemans C, Chen MC, Mathieu C, Eizirik DL. IL-1betaand IFN-gamma induce the expression of diverse chemokines and IL-15 in humanand rat pancreatic islet cells, and in islets from pre-diabetic NOD mice. Diabetologia2003;46:255–2669. Taylor-Fishwick DA, Weaver JR, Grzesik W, et al. Production and function ofIL-12 in islets and beta cells. Diabetologia 2013;56:126–13510. Lopes M, Kutlu B, Miani M, et al. Temporal profiling of cytokine-induced genesin pancreatic b-cells by meta-analysis and network inference. Genomics 2014;103:264–27511. Comens PG, Wolf BA, Unanue ER, Lacy PE, McDaniel ML. Interleukin 1 is potentmodulator of insulin secretion from isolated rat islets of Langerhans. Diabetes 1987;36:963–97012. Maedler K, Schumann DM, Sauter N, et al. Low concentration of interleukin-1beta induces FLICE-inhibitory protein-mediated beta-cell proliferation in humanpancreatic islets. Diabetes 2006;55:2713–272213. Lawrence MC, Shao C, McGlynn K, Naziruddin B, Levy MF, Cobb MH. Multiplechromatin-bound protein kinases assemble factors that regulate insulin gene tran-scription. Proc Natl Acad Sci U S A 2009;106:22181–2218614. Roep BO, Kleijwegt FS, van Halteren AG, et al. Islet inflammation and CXCL10 inrecent-onset type 1 diabetes. Clin Exp Immunol 2010;159:338–34315. Arous C, Ferreira PG, Dermitzakis ET, Halban PA. Short term exposure of betacells to low concentrations of interleukin-1b improves insulin secretion through focaladhesion and actin remodeling and regulation of gene expression. J Biol Chem 2015;290:6653–666916. Hajmrle C, Smith N, Spigelman AF, et al. Interleukin-1 signaling contributes toacute islet compensation. JCI Insight 2016;1:e8605517. Ortis F, Pirot P, Naamane N, et al. Induction of nuclear factor-kappaB and itsdownstream genes by TNF-alpha and IL-1beta has a pro-apoptotic role in pancreaticbeta cells. Diabetologia 2008;51:1213–122518. Gurzov EN, Ortis F, Cunha DA, et al. Signaling by IL-1beta+IFN-gamma and ERstress converge on DP5/Hrk activation: a novel mechanism for pancreatic beta-cellapoptosis. Cell Death Differ 2009;16:1539–155019. Hasnain SZ, Borg DJ, Harcourt BE, et al. Glycemic control in diabetes is re-stored by therapeutic manipulation of cytokines that regulate beta cell stress. NatMed 2014;20:1417–142620. Gorasia DG, Dudek NL, Veith PD, et al. Pancreatic beta cells are highly sus-ceptible to oxidative and ER stresses during the development of diabetes. J ProteomeRes 2015;14:688–69921. Paraskevas S, Aikin R, Maysinger D, et al. Activation and expression of ERK,JNK, and p38 MAP-kinases in isolated islets of Langerhans: implications for culturedislet survival. FEBS Lett 1999;455:203–20822. Nishiki Y, Adewola A, Hatanaka M, Templin AT, Maier B, Mirmira RG. Trans-lational control of inducible nitric oxide synthase by p38 MAPK in islet b-cells. MolEndocrinol 2013;27:336–34923. Noguchi H, Nakai Y, Ueda M, et al. Activation of c-Jun NH2-terminal kinase(JNK) pathway during islet transplantation and prevention of islet graft loss by in-traportal injection of JNK inhibitor. Diabetologia 2007;50:612–61924. Fukuda K, Tesch GH, Yap FY, et al. MKK3 signalling plays an essential role inleukocyte-mediated pancreatic injury in the multiple low-dose streptozotocin model.Lab Invest 2008;88:398–40725. Abdelli S, Ansite J, Roduit R, et al. Intracellular stress signaling pathwaysactivated during human islet preparation and following acute cytokine exposure.Diabetes 2004;53:2815–282326. Bennet W, Groth CG, Larsson R, Nilsson B, Korsgren O. Isolated human isletstrigger an instant blood mediated inflammatory reaction: implications for intraportalislet transplantation as a treatment for patients with type 1 diabetes. Ups J Med Sci2000;105:125–13327. Ozmen L, Ekdahl KN, Elgue G, Larsson R, Korsgren O, Nilsson B. Inhibition ofthrombin abrogates the instant blood-mediated inflammatory reaction triggered by
isolated human islets: possible application of the thrombin inhibitor melagatran inclinical islet transplantation. Diabetes 2002;51:1779–178428. Chow CW, Rincón M, Davis RJ. Requirement for transcription factor NFAT ininterleukin-2 expression. Mol Cell Biol 1999;19:2300–230729. Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification ofa putative regulator of early T cell activation genes. Science 1988;241:202–20530. Emmel EA, Verweij CL, Durand DB, Higgins KM, Lacy E, Crabtree GR. Cyclo-sporin A specifically inhibits function of nuclear proteins involved in T cell activation.Science 1989;246:1617–162031. Bierer BE, Mattila PS, Standaert RF, et al. Two distinct signal transmissionpathways in T lymphocytes are inhibited by complexes formed between an immunophilinand either FK506 or rapamycin. Proc Natl Acad Sci U S A 1990;87:9231–923532. Flanagan WM, Corthésy B, Bram RJ, Crabtree GR. Nuclear association of a T-celltranscription factor blocked by FK-506 and cyclosporin A. Nature 1991;352:803–80733. Clipstone NA, Crabtree GR. Identification of calcineurin as a key signallingenzyme in T-lymphocyte activation. Nature 1992;357:695–69734. Cockerill PN, Shannon MF, Bert AG, Ryan GR, Vadas MA. The granulocyte-macrophage colony-stimulating factor/interleukin 3 locus is regulated by an induciblecyclosporin A-sensitive enhancer. Proc Natl Acad Sci U S A 1993;90:2466–247035. Park J, Yaseen NR, Hogan PG, Rao A, Sharma S. Phosphorylation of thetranscription factor NFATp inhibits its DNA binding activity in cyclosporin A-treatedhuman B and T cells. J Biol Chem 1995;270:20653–2065936. Wesselborg S, Fruman DA, Sagoo JK, Bierer BE, Burakoff SJ. Identification ofa physical interaction between calcineurin and nuclear factor of activated T cells(NFATp). J Biol Chem 1996;271:1274–127737. Loh C, Shaw KT, Carew J, et al. Calcineurin binds the transcription factorNFAT1 and reversibly regulates its activity. J Biol Chem 1996;271:10884–1089138. Luo C, Burgeon E, Carew JA, et al. Recombinant NFAT1 (NFATp) is regulated bycalcineurin in T cells and mediates transcription of several cytokine genes. Mol CellBiol 1996;16:3955–396639. Okamura H, Aramburu J, García-Rodríguez C, et al. Concerted dephosphorylationof the transcription factor NFAT1 induces a conformational switch that regulatestranscriptional activity. Mol Cell 2000;6:539–55040. Lawrence MC, McGlynn K, Shao C, et al. Chromatin-bound mitogen-activatedprotein kinases transmit dynamic signals in transcription complexes in beta-cells.Proc Natl Acad Sci U S A 2008;105:13315–1332041. Nelson JD, LeBoeuf RC, Bomsztyk K. Direct recruitment of insulin receptor andERK signaling cascade to insulin-inducible gene loci. Diabetes 2011;60:127–13742. Matsumoto S, Takita M, Chaussabel D, et al. Improving efficacy of clinical islettransplantation with iodixanol-based islet purification, thymoglobulin induction, andblockage of IL-1b and TNF-a. Cell Transplant 2011;20:1641–164743. McCall M, Pawlick R, Kin T, Shapiro AM. Anakinra potentiates the protectiveeffects of etanercept in transplantation of marginal mass human islets in immuno-deficient mice. Am J Transplant 2012;12:322–32944. Jain J, McCaffrey PG, Miner Z, et al. The T-cell transcription factor NFATp is asubstrate for calcineurin and interacts with Fos and Jun. Nature 1993;365:352–35545. Northrop JP, Ullman KS, Crabtree GR. Characterization of the nuclear andcytoplasmic components of the lymphoid-specific nuclear factor of activated T cells(NF-AT) complex. J Biol Chem 1993;268:2917–292346. Tsai EY, Jain J, Pesavento PA, Rao A, Goldfeld AE. Tumor necrosis factor alphagene regulation in activated T cells involves ATF-2/Jun and NFATp. Mol Cell Biol1996;16:459–46747. Sun LJ, Peterson BR, Verdine GL. Dual role of the nuclear factor of activatedT cells insert region in DNA recognition and cooperative contacts to activator protein1. Proc Natl Acad Sci U S A 1997;94:4919–492448. Yang TT, Chow CW. Transcription cooperation by NFAT.C/EBP composite en-hancer complex. J Biol Chem 2003;278:15874–1588549. Falvo JV, Lin CH, Tsytsykova AV, et al. A dimer-specific function of the tran-scription factor NFATp. Proc Natl Acad Sci U S A 2008;105:19637–1964250. Walters RD, Drullinger LF, Kugel JF, Goodrich JA. NFATc2 recruits cJun ho-modimers to an NFAT site to synergistically activate interleukin-2 transcription. MolImmunol 2013;56:48–56
diabetes.diabetesjournals.org Yoshimatsu and Associates 2867
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/66/11/2857/533860/db170578.pdf by guest on 24 February 2022










![Computational Modeling of Glucose Toxicity in Pancreatic Β-cells [Update]](https://static.fdocument.org/doc/165x107/577cb4f61a28aba7118cd93d/computational-modeling-of-glucose-toxicity-in-pancreatic-cells-update.jpg)







