
On functional and structural heterogeneity of VIM-type metallo-β
10
Journal of Antimicrobial Chemotherapy (2003) 51, 257–266 DOI: 10.1093/jac/dkg067 Advance Access publication 6 January 2003 257 ................................................................................................................................................................................................................................................................... © 2003 The British Society for Antimicrobial Chemotherapy On functional and structural heterogeneity of VIM-type metallo-β -lactamases Jean-Denis Docquier 1 , Josette Lamotte-Brasseur 2 , Moreno Galleni 2 , Gianfranco Amicosante 3 , Jean-Marie Frère 2 and Gian Maria Rossolini 1 * 1 Dipartimento di Biologia Molecolare, Sezione di Microbiologia, Università di Siena, Policlinico ‘Le Scotte’, Viale Bracci, I-53100 Siena; 3 Dipartimento di Scienze e Tecnologie Biomediche – Cattedra di Biochimica Clinica, Università di L’Aquila, Via Vetoio, Loc. Coppito, I-67100 L’Aquila, Italy; 2 Laboratoire d’Enzymologie & Centre d’Ingénierie des Protéines, Institut de Chimie, Université de Liège, Bat. B6 Allée de la Chimie, Sart Tilman, B-4000 Liège, Belgium Received 21 January 2002; returned 5 July 2002; revised 1 August 2002; accepted 28 October 2002 The VIM metallo-β-lactamases are emerging resistance determinants, encoded by mobile genetic elements, that have recently been detected in multidrug-resistant nosocomial isolates of Pseudomonas aeruginosa and other Gram-negative pathogens. In this work a T7-based expression system for overproduction of the VIM-2 enzyme by Escherichia coli was developed, which yielded ∼80 mg of protein per litre of culture. The enzyme was mostly released into the medium, from which it was recovered at >99% purity by an initial ammonium sulphate pre- cipitation followed by two chromatography steps, with almost 80% efficiency. Determination of kinetic parameters of VIM-2 under the same experimental conditions previously used for VIM-1 (the first VIM-type enzyme detected in clinical isolates, which is 93% identical to VIM-2) revealed significant differences in K m values and/or turnover rates with several substrates, including penicillins, cephalosporins and carbapenems. Compared with VIM-1, VIM-2 is more susceptible to inactivation by chelators, indicating that the zinc ions of the latter are probably more loosely bound. These data indicated that at least some of the amino acid differences between the two proteins have functional significance. Molecular modelling of the two enzymes identified some amino acid substitutions, including those at positions 223, 224 and 228 (in the BBL numbering), that could be relevant to the changes in catalytic behaviour. Introduction Acquired metallo-β-lactamases (molecular class B, group 3 of the functional classification) 1 are emerging resistance determinants in several bacterial species of clinical relevance, including members of the family Enterobacteriaceae, Pseudo- monas spp. and other non-fastidious Gram-negative non- fermenters. 2,3 The ability to hydrolyse most β-lactam compounds efficiently, including carbapenems, the lack of clinically useful inhibitors and the mobile nature of their genetic determinants identify these enzymes as major threats to efficacy of antimicrobial chemotherapy. 2,3 In the chal- lenging quest for new drugs and inhibitors, a detailed under- standing of their biochemical properties and structure is desirable. 4–7 During the last decade, two types of acquired metallo-β- lactamases have been detected in Gram-negative pathogens, namely the IMP- and VIM-type enzymes. The IMP-type enzymes were detected in the early 1990s 8,9 and have been the subject of several biochemical and structural investiga- tions. 7,9–15 The VIM-type enzymes were identified more recently, 16,17 and considerably less is known about their func- tional and structural properties. 17,18 The VIM-type enzymes belong to subclass B1 of molecu- lar class B and include at least three variants: VIM-1, VIM-2 and VIM-3. 16,17,19 Among them, VIM-2 is apparently the most .................................................................................................................................................................................................................................................................. *Corresponding author. Tel: +39-0577-233455; Fax: +39-0577-233325; E-mail: [email protected] Downloaded from https://academic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019
Transcript of On functional and structural heterogeneity of VIM-type metallo-β

Journal of Antimicrobial Chemotherapy (2003) 51, 257–266DOI: 10.1093/jac/dkg067Advance Access publication 6 January 2003
257. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
© 2003 The British Society for Antimicrobial Chemotherapy
On functional and structural heterogeneity of VIM-type metallo-β-lactamases
Jean-Denis Docquier1, Josette Lamotte-Brasseur2, Moreno Galleni2, Gianfranco Amicosante3,
Jean-Marie Frère2 and Gian Maria Rossolini1*
1Dipartimento di Biologia Molecolare, Sezione di Microbiologia, Università di Siena, Policlinico ‘Le Scotte’, Viale Bracci, I-53100 Siena; 3Dipartimento di Scienze e Tecnologie Biomediche – Cattedra di Biochimica Clinica, Università di L’Aquila, Via Vetoio, Loc. Coppito, I-67100 L’Aquila, Italy; 2Laboratoire d’Enzymologie & Centre
d’Ingénierie des Protéines, Institut de Chimie, Université de Liège, Bat. B6 Allée de la Chimie, Sart Tilman, B-4000 Liège, Belgium
Received 21 January 2002; returned 5 July 2002; revised 1 August 2002; accepted 28 October 2002
The VIM metallo-β-lactamases are emerging resistance determinants, encoded by mobilegenetic elements, that have recently been detected in multidrug-resistant nosocomial isolatesof Pseudomonas aeruginosa and other Gram-negative pathogens. In this work a T7-basedexpression system for overproduction of the VIM-2 enzyme by Escherichia coli was developed,which yielded ∼80 mg of protein per litre of culture. The enzyme was mostly released into themedium, from which it was recovered at >99% purity by an initial ammonium sulphate pre-cipitation followed by two chromatography steps, with almost 80% efficiency. Determination ofkinetic parameters of VIM-2 under the same experimental conditions previously used for VIM-1(the first VIM-type enzyme detected in clinical isolates, which is 93% identical to VIM-2) revealedsignificant differences in Km values and/or turnover rates with several substrates, includingpenicillins, cephalosporins and carbapenems. Compared with VIM-1, VIM-2 is more susceptibleto inactivation by chelators, indicating that the zinc ions of the latter are probably more looselybound. These data indicated that at least some of the amino acid differences between the twoproteins have functional significance. Molecular modelling of the two enzymes identified someamino acid substitutions, including those at positions 223, 224 and 228 (in the BBL numbering),that could be relevant to the changes in catalytic behaviour.
Introduction
Acquired metallo-β-lactamases (molecular class B, group 3of the functional classification)1 are emerging resistancedeterminants in several bacterial species of clinical relevance,including members of the family Enterobacteriaceae, Pseudo-monas spp. and other non-fastidious Gram-negative non-fermenters.2,3 The ability to hydrolyse most β-lactamcompounds efficiently, including carbapenems, the lack ofclinically useful inhibitors and the mobile nature of theirgenetic determinants identify these enzymes as major threatsto efficacy of antimicrobial chemotherapy.2,3 In the chal-lenging quest for new drugs and inhibitors, a detailed under-
standing of their biochemical properties and structure isdesirable.4–7
During the last decade, two types of acquired metallo-β-lactamases have been detected in Gram-negative pathogens,namely the IMP- and VIM-type enzymes. The IMP-typeenzymes were detected in the early 1990s8,9 and have beenthe subject of several biochemical and structural investiga-tions.7,9–15 The VIM-type enzymes were identified morerecently,16,17 and considerably less is known about their func-tional and structural properties.17,18
The VIM-type enzymes belong to subclass B1 of molecu-lar class B and include at least three variants: VIM-1, VIM-2and VIM-3.16,17,19 Among them, VIM-2 is apparently the most
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
*Corresponding author. Tel: +39-0577-233455; Fax: +39-0577-233325; E-mail: [email protected]
Dow
nloaded from https://academ
ic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019

J.-D. Docquier et al.
258
widespread (it has been detected in clinical isolates fromvarious European countries and the Far East),17,19–21 whereasVIM-1 and VIM-3 producers encountered so far were isolatedin Italy16 and Taiwan,19 respectively. The amino acid sequencesof VIM-1 and VIM-2 diverge by 7%16,17 whereas VIM-3 dif-fers from VIM-2 by only two residues.19 Notwithstandingtheir close structural similarity, notable differences have beenreported for the kinetic parameters of VIM-1 and VIM-2 withsome substrates,17,18 suggesting that the few structural differ-ences between these two natural variants have a functionalsignificance. However, in those studies, the experimentalconditions used for kinetic measurements were not thesame.17,18
In this work an expression system for high-level produc-tion of the VIM-2 enzyme in Escherichia coli was developed,and biophysical and biochemical characterizations of thepurified VIM-2 enzyme were carried out under conditionsidentical to those previously adopted for VIM-1.18 Significantdifferences in the behaviour of the two enzymes with varioussubstrates were confirmed, and molecular modelling wasused to investigate the potential correlations between struc-tural differences and the different enzyme kinetics.
Materials and methods
Bacterial strains and culture media
E. coli XL-1 blue (Stratagene Inc., La Jolla, CA, USA) wasroutinely used for molecular cloning and plasmid propaga-tion. E. coli BL21(DE3) (Novagen Inc., Madison, WI, USA)was used for metallo-β-lactamase gene expression. Bacteriawere grown in Luria–Bertani (LB) medium,22 or in BufferedSuper Broth medium (BSB: yeast extract, 20 g/L; tryptone,35 g/L; NaCl, 5 g/L; buffered with 50 mM sodium phosphatebuffer pH 7.0) supplemented with the appropriate antibiotic.Media components were from Difco Laboratories (Detroit,MI, USA).
Construction of the expression system for overproduction of VIM-2 in E. coli
The blaVIM-2 gene was amplified by PCR with primers VIM-2-Fwd (5′-GGAATTCCATATGTTCAAACTTTTGAGT-AAG), which added EcoRI (bold) and NdeI (underlined)restriction sites at the 5′-end of the gene, and VIM-2-Rev(5′-CCGGATCCTGCTACTCAACGACTG), which added aBamHI restriction site (bold) after the blaVIM-2 stop codon.Amplification was carried out in 50 µL using 50 pmol of eachprimer and the Expand PCR system (Roche Biochemicals,Mannheim, Germany), under the conditions recommendedby the manufacturer, and the following cycling parameters:initial denaturation at 94°C for 3 min; denaturation at 94°Cfor 1 min, annealing at 52°C for 1 min, extension at 72°C for1 min, repeated for 30 cycles; final extension step at 72°C for
10 min. Plasmid pVRP193 (50 ng)21 was used as template.The resulting 0.8 kb amplicon was digested with EcoRI andBamHI and cloned into the plasmid vector pBC-SK (Strata-gene) to give recombinant plasmid pJDD-V2. After con-firmatory sequencing, to rule out the presence of unwantedmutations introduced by PCR, the 0.8 kb NdeI–BamHIfragment of pJDD-V2, containing the blaVIM-2 gene, wassubcloned into the T7-based expression vector pET-9a(Novagen) to obtain plasmid pET-9/VIM-2.
Expression experiments
E. coli BL21(DE3)(pET-9/VIM-2) was grown aerobically at37°C in 100 mL of BSB medium containing kanamycin,50 mg/L. When the OD at 600 nm reached a value of ∼0.8, theculture was split into two and to one subculture isopropyl-β-D-thiogalactopyranoside (IPTG) (final concentration 1 mM)was added. β-Lactamase activity was monitored spectrophoto-metrically, using 100 µM imipenem as substrate in 10 mMHEPES buffer (pH 7.5) (HB), at 30°C, both in cell extractsand in culture supernatants from samples obtained at differenttimes. Cell extracts were prepared by centrifuging the culture,resuspending the cells in the same volume of HB, and dis-rupting them by sonication [five cycles, 20 s for each cycle, at45 W, using a B. Braun Labsonic L sonicator (Melsungen,Germany)]. The amount of enzyme was calculated on the basisof the following kinetic parameters for imipenem: kcat = 34/sand Km = 9 µM.
Purification of VIM-2
The VIM-2 enzyme produced by E. coli BL21(DE3)(pET-9/VIM-2) was purified from the culture supernatants of 200 mLstationary phase cultures grown aerobically at 37°C in BSBmedium. Cells were removed by centrifugation (10 000g, for30 min at 4°C) and solid ammonium sulphate was added to thesupernatant to achieve a 50% saturation. After 1 h of gentlestirring at 4°C, the sample was centrifuged (13 000g, for 1 h at4°C). Solid ammonium sulphate was added to the clarifiedsupernatant to 80% saturation, and the precipitate, collectedas described above, was solubilized in 20 mM triethanol-amine (pH 7.2) (1/20 of the original volume) and loaded (flowrate 2 mL/min) onto an HR column (5 × 1.6 cm) packed with10 mL of Source 15Q gel (Amersham–Pharmacia Biotech,Uppsala, Sweden), previously equilibrated with the samebuffer. Elution was performed with the same buffer using alinear NaCl gradient (0–1 M in 200 mL), at a flow rate of2 mL/min. Fractions containing β-lactamase activity werepooled and concentrated ∼10-fold using a Centriplus YM10system (Millipore, Bedford, MA, USA). The concentratedsample was then injected onto a Superdex 75 HR 10/30column (Amersham–Pharmacia Biotech) equilibrated withHB containing 50 µM ZnCl2 and 0.2 M NaCl, and proteinswere eluted in the same buffer at a flow rate of 0.8 mL/min.
Dow
nloaded from https://academ
ic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019

Biochemistry of VIM metallo-β-lactamases
259
Fractions containing β-lactamase activity were pooled andstored at –20°C. During the purification procedure thepresence of β-lactamase activity was monitored as describedabove. Protein concentration in solution was determined withthe Bio-Rad Protein assay (Bio-Rad, Richmond, CA, USA),using bovine serum albumin as the standard. The molarextinction coefficient at 280 nm of the purified enzyme wasdetermined as the average of values obtained by the colori-metric protein concentration determination and by theoreticalcalculation.23
Protein electrophoretic techniques
SDS–PAGE was performed according to Laemmli,24 usingfinal acrylamide concentrations of 12% and 5% (w/v) for theseparating and the stacking gels, respectively. After electro-phoresis the protein bands were stained with CoomassieBrilliant Blue R-250. Analytical isoelectric focusing (IEF) ofthe purified protein and detection of enzyme activity wereperformed as described previously.16
Gel-permeation chromatography
Gel-permeation chromatography to determine the molecularmass of the native VIM-2 enzyme was carried out on a Super-dex 75 HR 10/30 column (Amersham–Pharmacia Biotech)equilibrated with HB containing 0.15 M NaCl, to preventunwanted protein–column matrix interactions. The purifiedenzyme (100 µL, at a concentration of 0.5 mg/mL) was elutedin the same buffer at a flow rate 0.8 mL/min. The low-rangegel filtration calibration kit (Amersham–Pharmacia Biotech)was used for column calibration. Apparent partition coeffi-cients (Kav) were calculated as described previously.25
N terminus sequencing and electrospray mass spectrometry
The amino-terminal sequence of the purified VIM-2 proteinwas determined using a gas-phase sequencer (Procise-492,Applied Biosystems, Foster City, CA, USA), after redissolv-ing the protein (50 pmol) in 0.1% (v/v) trifluoroacetic acid inwater and loading the sample onto a PVDF membrane (Milli-pore Corp.). Electrospray mass spectrometry was carried outusing a PE-Sciex API III triple quadrupole mass spectrometerequipped with an ion-spray source (Perkin-Elmer, Rahway,NJ, USA). The sample (150 pmoles) was redissolved in 1%(v/v) potassium formate/70% (v/v) acetonitrile in water andinjected into the source of the mass spectrometer at a flow rateof 20 µL/min. Source and cone voltages were 5.5 kV and60 V, respectively. The source temperature was kept at 50°C.Twenty-four scans covering 800–1800 atomic mass unitswere accumulated and data were analysed with the softwaredelivered with the instrument.
Determination of kinetic parameters
Substrate hydrolysis by the purified enzymes was monitoredby following the absorbance variation, at 30°C, using a Cary100 UV-Vis spectrophotometer (Varian Instruments, WalnutCreek, CA, USA), in a total reaction volume of 500 µL. Thesources, wavelengths, changes in extinction coefficients andreaction buffer (HB containing 50 µM ZnCl2) used in thespectrophotometric assays were the same as described previ-ously.11,18 The final enzyme concentrations ranged from 4.2 to84 nM. Enzyme dilutions were prepared in the reaction buffersupplemented with 20 mg/L BSA, and were discarded aftereach working session. The steady-state kinetic parameters(Km and kcat) were determined under initial-rate conditionsusing the Hanes–Woolf plot26 and, when standard deviationvalues exceeded 5%, they were also verified by the analysis ofthe complete hydrolysis time-courses as described by DeMeester et al.27 Km values lower than 10 µM were measured asinhibition constants (Kis) with a competitive model, using100 µM nitrocefin as the reporter substrate, as described pre-viously.18 Purified VIM-1 enzyme for kinetic measurementswas prepared as described previously.18
Inactivation by chelating agents
Inactivation time-courses were monitored by following thehydrolysis of 150 µM imipenem in the presence of differentconcentrations of EDTA, dipicolinic acid or 1,10-o-phenan-throline. Reactions were carried out at 30°C in HB in a finalreaction volume of 500 µL. Enzyme dilutions were preparedin the same buffer supplemented with 20 mg/L BSA, and thefinal enzyme concentration was 2.1 nM. Pseudo-first-orderinactivation rate constants (ki) were determined and theinactivation efficiencies (k+2/K) were calculated according tothe proposed model.28
Molecular modelling
VIM-1 and VIM-2 structural models were based on the avail-able X-ray diffraction three-dimensional structure of CcrAfrom Bacteroides fragilis.5,29 Molecular models were built byknowledge-based modelling using the HOMOLOGY moduleof the Insight II software (Molecular Simulations, San Diego,CA, USA) running on a Silicon Graphics Indy workstation(Silicon Graphics Inc., Mountain View, CA, USA). Histidinespresent at the active site were taken as neutral; for the others,the tautomeric form of the imidazole ring was chosen accord-ing to the X-ray structure, after geometric analysis of thepotential hydrogen bonds. The zinc-coordinated cysteine wasin the thiolate form. Other titratable sites were assigned theirstandard protonation states at pH 7. Hydrogen atoms wereadded to the structure using the PROTONATE module ofthe AMBER version 4.1 software.30 Atomic charges of the
Dow
nloaded from https://academ
ic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019
J.-D. Docquier et al.
260
AMBER version 4.1 all-atom library were generally used inthe calculations, except for the residues of the active site.Atomic point charges for the zinc ions, the zinc coordinatingresidues and active-site water molecules were those proposedby Banci et al.31 Docking experiments were carried out withthe Insight II software using the benzylpenicillin structureoptimized by AM1 semi-empirical methods. The correspond-ing complex structures were then optimized by molecularmechanics methods using AMBER version 4.1, first bysteepest descent energy minimization of atoms with a force>500 kcal/mol/Å and then by conjugate gradient energy mini-mization until the rms gradient was <0.1 kcal/mol. Graphicswere realized with MOLMOL molecular graphics software.32
The BBL numbering scheme is used throughout this paper.33
Results and discussion
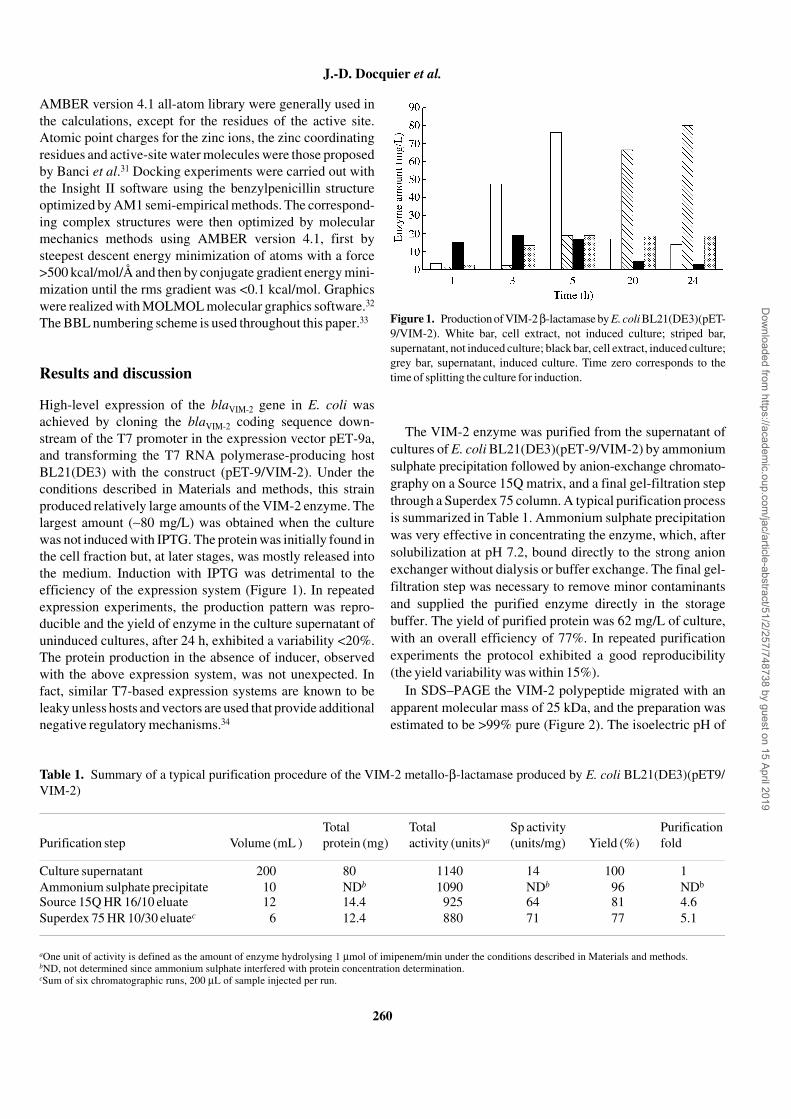
High-level expression of the blaVIM-2 gene in E. coli wasachieved by cloning the blaVIM-2 coding sequence down-stream of the T7 promoter in the expression vector pET-9a,and transforming the T7 RNA polymerase-producing hostBL21(DE3) with the construct (pET-9/VIM-2). Under theconditions described in Materials and methods, this strainproduced relatively large amounts of the VIM-2 enzyme. Thelargest amount (∼80 mg/L) was obtained when the culturewas not induced with IPTG. The protein was initially found inthe cell fraction but, at later stages, was mostly released intothe medium. Induction with IPTG was detrimental to theefficiency of the expression system (Figure 1). In repeatedexpression experiments, the production pattern was repro-ducible and the yield of enzyme in the culture supernatant ofuninduced cultures, after 24 h, exhibited a variability <20%.The protein production in the absence of inducer, observedwith the above expression system, was not unexpected. Infact, similar T7-based expression systems are known to beleaky unless hosts and vectors are used that provide additionalnegative regulatory mechanisms.34
The VIM-2 enzyme was purified from the supernatant ofcultures of E. coli BL21(DE3)(pET-9/VIM-2) by ammoniumsulphate precipitation followed by anion-exchange chromato-graphy on a Source 15Q matrix, and a final gel-filtration stepthrough a Superdex 75 column. A typical purification processis summarized in Table 1. Ammonium sulphate precipitationwas very effective in concentrating the enzyme, which, aftersolubilization at pH 7.2, bound directly to the strong anionexchanger without dialysis or buffer exchange. The final gel-filtration step was necessary to remove minor contaminantsand supplied the purified enzyme directly in the storagebuffer. The yield of purified protein was 62 mg/L of culture,with an overall efficiency of 77%. In repeated purificationexperiments the protocol exhibited a good reproducibility(the yield variability was within 15%).
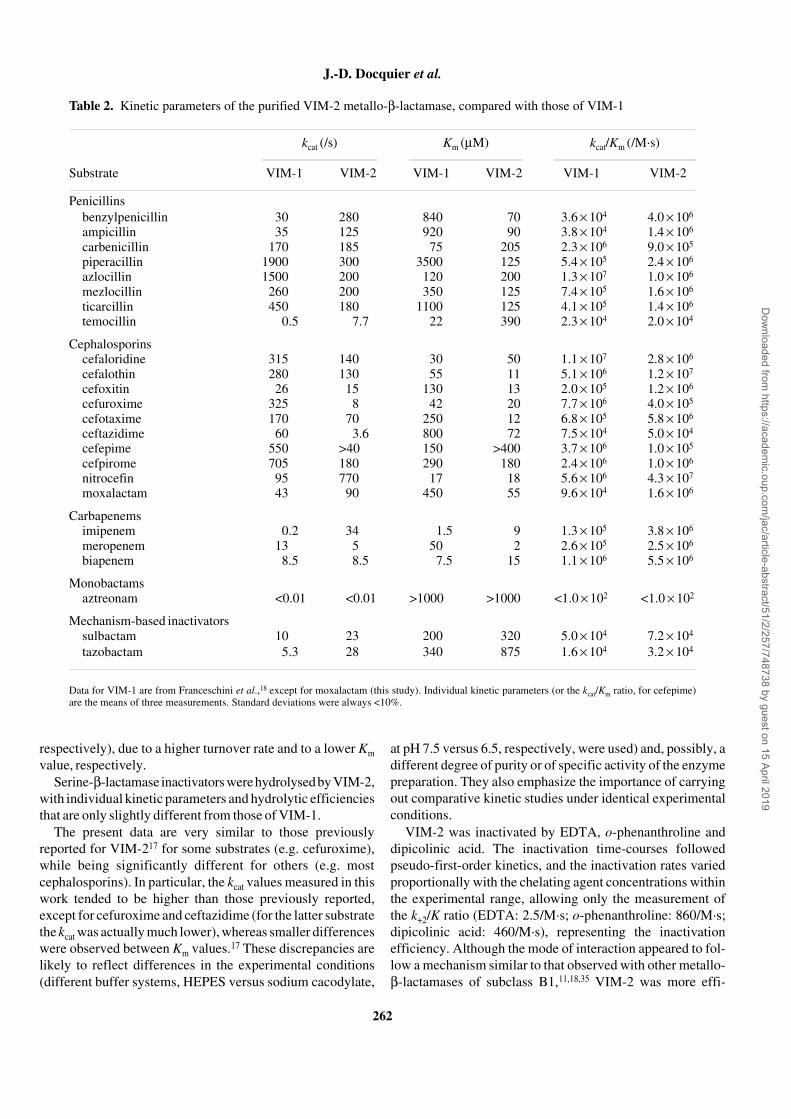
In SDS–PAGE the VIM-2 polypeptide migrated with anapparent molecular mass of 25 kDa, and the preparation wasestimated to be >99% pure (Figure 2). The isoelectric pH of
Figure 1. Production of VIM-2 β-lactamase by E. coli BL21(DE3)(pET-9/VIM-2). White bar, cell extract, not induced culture; striped bar,supernatant, not induced culture; black bar, cell extract, induced culture;grey bar, supernatant, induced culture. Time zero corresponds to thetime of splitting the culture for induction.
Table 1. Summary of a typical purification procedure of the VIM-2 metallo-β-lactamase produced by E. coli BL21(DE3)(pET9/VIM-2)
aOne unit of activity is defined as the amount of enzyme hydrolysing 1 µmol of imipenem/min under the conditions described in Materials and methods.bND, not determined since ammonium sulphate interfered with protein concentration determination.cSum of six chromatographic runs, 200 µL of sample injected per run.
Purification step Volume (mL )Total protein (mg)
Total activity (units)a
Sp activity (units/mg) Yield (%)
Purification fold
Culture supernatant 200 80 1140 14 100 1Ammonium sulphate precipitate 10 NDb 1090 NDb 96 NDb
Source 15Q HR 16/10 eluate 12 14.4 925 64 81 4.6Superdex 75 HR 10/30 eluatec 6 12.4 880 71 77 5.1
Dow
nloaded from https://academ
ic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019

Biochemistry of VIM metallo-β-lactamases
261
purified VIM-2, measured by analytical IEF, was 5.15 (datanot shown), in good agreement with the theoretical value(4.9). The molecular mass of the purified enzyme, estimatedby gel-filtration, was 25 000 Da, indicating that the nativeenzyme is monomeric. This value is somewhat smaller thanthat previously reported (29.7 kDa),17 but is consistent withresults of SDS–PAGE and mass spectrometry (see above andbelow). The molar extinction coefficient for pure VIM-2 wascalculated as 28 500/M·cm. The amino-terminal sequence ofthe purified protein was determined as NH2-VDSSG, indicat-ing that the mature enzyme is generated by the cleavage of a26-amino-acid signal peptide and not of a 20-amino-acidsignal peptide, as originally predicted.17 The cleavage posi-tion of the signal peptide was identical to that in VIM-1,although the sequences of the two pro-enzymes are differentaround that position.16–18 Electrospray mass spectrometryyielded a value of 25 515 ± 1 Da, in excellent agreementwith the calculated molecular mass of the mature protein(25 515.4 Da).
The kinetic parameters of VIM-2 were determined forseveral substrates under the same conditions as previouslyused for VIM-1.18 Moreover, the kinetic parameters of VIM-1were determined for moxalactam and redetermined for somesubstrates (benzylpenicillin, ampicillin, meropenem). In thelatter cases, the values obtained were always found to bewithin the confidence limits of those previously reported.18
Under the experimental conditions employed, VIM-2hydrolysed all the tested compounds except aztreonam. Theindividual kinetic parameters (Km and kcat) of VIM-2 withseveral substrates, and a comparison with those of VIM-1, arereported in Table 2.
With penicillins, the individual kinetic parameters of VIM-2were quite homogeneous and the hydrolytic efficiencies werehigh (kcat/Km ratios were in the range 0.9–4 × 106/M·s), exceptfor temocillin, which was hydrolysed 50- to 100-fold lessefficiently. This lower efficiency was mostly due to a lowerturnover rate. Compared with those of VIM-1, the kinetic
parameters of VIM-2 were similar for some substrates (car-benicillin, mezlocillin) but remarkably different for others. Insome cases these differences resulted in notable differences inhydrolytic efficiency as well (e.g. benzylpenicillin andampicillin, which were better substrates for VIM-2 than forVIM-1 due to both lower Km values and higher turnover rates,and azlocillin, which was a better substrate for VIM-1, mostlydue to a higher turnover rate). In other cases the differencesin individual kinetic parameters balanced out, resulting insimilar hydrolytic efficiencies (e.g. piperacillin, ticarcillinand temocillin). Comparison of the kinetic parameters ofticarcillin and temocillin revealed that the presence of a6-α-methoxy group was detrimental to the activity of bothenzymes. Interestingly, however, this effect was muchstronger for VIM-2, and the responses of individual kineticparameters were remarkably different with the two enzymes(e.g. with VIM-1 the Km was strongly decreased whereas withVIM-2 it was increased by the presence of the 6-α-methoxygroup).
Hydrolytic efficiencies of VIM-2 with cephalosporinswere highly variable, with values of kcat/Km ratios rangingfrom 5 × 104 to 1 × 107/M·s. Except for cefepime, the Kmvalues were quite low and usually lower than those of peni-cillins, whereas the turnover rates exhibited an overall highervariability, which often directly influenced the hydrolyticefficiencies. With cefepime, only a pseudo-first-order rateconstant was measurable, since the initial velocities remainedproportional to substrate concentration up to a value of 400 µM.Compared with VIM-1, both the kcat and Km values of VIM-2tended to be lower (with some exceptions for cefepime,nitrocefin and moxalactam). With some substrates (e.g.cefaloridine, cefalothin, ceftazidime and cefpirome) thedifferences in individual kinetic parameters again balancedout, resulting in similar hydrolytic efficiencies for the twoenzymes, whereas with other substrates (e.g. cefoxitin, cefur-oxime, cefotaxime and moxalactam) the kcat/Km ratios werealso different. The largest difference in hydrolytic efficien-cies (almost 40-fold higher for VIM-1) was observed withcefepime, a finding that was consistent with in vitro suscep-tibility data.16,17
VIM-2 exhibited high hydrolytic efficiencies with all carba-penems (kcat/Km ratios were in the range 2.5–5.5 × 106/M·s),resulting from a combination of very low Km values andrelatively low turnover rates. Overall this behaviour wassimilar to that observed for VIM-1, and differentiates theVIM-type enzymes from all other zinc-β-lactamases thatachieve similar hydrolytic efficiencies toward carbapenemsas a result of higher turnover rates combined with higher Kmvalues.18 Notwithstanding this common trend, differences inindividual kinetic constants between VIM-2 and VIM-1 werealso apparent with carbapenems, resulting in different hydro-lytic efficiencies. In particular, imipenem and meropenemwere hydrolysed more efficiently by VIM-2 (30- and 10-fold,
Figure 2. SDS–PAGE analysis of the purified VIM-2 protein (10 µg).Protein size standards are reported in kDa on the right.
Dow
nloaded from https://academ
ic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019
J.-D. Docquier et al.
262
respectively), due to a higher turnover rate and to a lower Kmvalue, respectively.
Serine-β-lactamase inactivators were hydrolysed by VIM-2,with individual kinetic parameters and hydrolytic efficienciesthat are only slightly different from those of VIM-1.
The present data are very similar to those previouslyreported for VIM-217 for some substrates (e.g. cefuroxime),while being significantly different for others (e.g. mostcephalosporins). In particular, the kcat values measured in thiswork tended to be higher than those previously reported,except for cefuroxime and ceftazidime (for the latter substratethe kcat was actually much lower), whereas smaller differenceswere observed between Km values.17 These discrepancies arelikely to reflect differences in the experimental conditions(different buffer systems, HEPES versus sodium cacodylate,
at pH 7.5 versus 6.5, respectively, were used) and, possibly, adifferent degree of purity or of specific activity of the enzymepreparation. They also emphasize the importance of carryingout comparative kinetic studies under identical experimentalconditions.
VIM-2 was inactivated by EDTA, o-phenanthroline anddipicolinic acid. The inactivation time-courses followedpseudo-first-order kinetics, and the inactivation rates variedproportionally with the chelating agent concentrations withinthe experimental range, allowing only the measurement ofthe k+2/K ratio (EDTA: 2.5/M·s; o-phenanthroline: 860/M·s;dipicolinic acid: 460/M·s), representing the inactivationefficiency. Although the mode of interaction appeared to fol-low a mechanism similar to that observed with other metallo-β-lactamases of subclass B1,11,18,35 VIM-2 was more effi-
Table 2. Kinetic parameters of the purified VIM-2 metallo-β-lactamase, compared with those of VIM-1
Data for VIM-1 are from Franceschini et al.,18 except for moxalactam (this study). Individual kinetic parameters (or the kcat/Km ratio, for cefepime)are the means of three measurements. Standard deviations were always <10%.
kcat (/s) Km (µM) kcat/Km (/M·s)
Substrate VIM-1 VIM-2 VIM-1 VIM-2 VIM-1 VIM-2
Penicillinsbenzylpenicillin 30 280 840 70 3.6 × 104 4.0 × 106
ampicillin 35 125 920 90 3.8 × 104 1.4 × 106
carbenicillin 170 185 75 205 2.3 × 106 9.0 × 105
piperacillin 1900 300 3500 125 5.4 × 105 2.4 × 106
azlocillin 1500 200 120 200 1.3 × 107 1.0 × 106
mezlocillin 260 200 350 125 7.4 × 105 1.6 × 106
ticarcillin 450 180 1100 125 4.1 × 105 1.4 × 106
temocillin 0.5 7.7 22 390 2.3 × 104 2.0 × 104
Cephalosporinscefaloridine 315 140 30 50 1.1 × 107 2.8 × 106
cefalothin 280 130 55 11 5.1 × 106 1.2 × 107
cefoxitin 26 15 130 13 2.0 × 105 1.2 × 106
cefuroxime 325 8 42 20 7.7 × 106 4.0 × 105
cefotaxime 170 70 250 12 6.8 × 105 5.8 × 106
ceftazidime 60 3.6 800 72 7.5 × 104 5.0 × 104
cefepime 550 >40 150 >400 3.7 × 106 1.0 × 105
cefpirome 705 180 290 180 2.4 × 106 1.0 × 106
nitrocefin 95 770 17 18 5.6 × 106 4.3 × 107
moxalactam 43 90 450 55 9.6 × 104 1.6 × 106
Carbapenemsimipenem 0.2 34 1.5 9 1.3 × 105 3.8 × 106
meropenem 13 5 50 2 2.6 × 105 2.5 × 106
biapenem 8.5 8.5 7.5 15 1.1 × 106 5.5 × 106
Monobactamsaztreonam <0.01 <0.01 >1000 >1000 <1.0 × 102 <1.0 × 102
Mechanism-based inactivatorssulbactam 10 23 200 320 5.0 × 104 7.2 × 104
tazobactam 5.3 28 340 875 1.6 × 104 3.2 × 104
Dow
nloaded from https://academ
ic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019

Biochemistry of VIM metallo-β-lactamases
263
ciently inactivated than VIM-118 by all three chelators (thek+2/K ratios were 4- to 20-fold higher for o-phenanthrolineand dipicolinic acid, respectively), indicating that zinc ionsare probably more tightly bound in the latter enzyme.
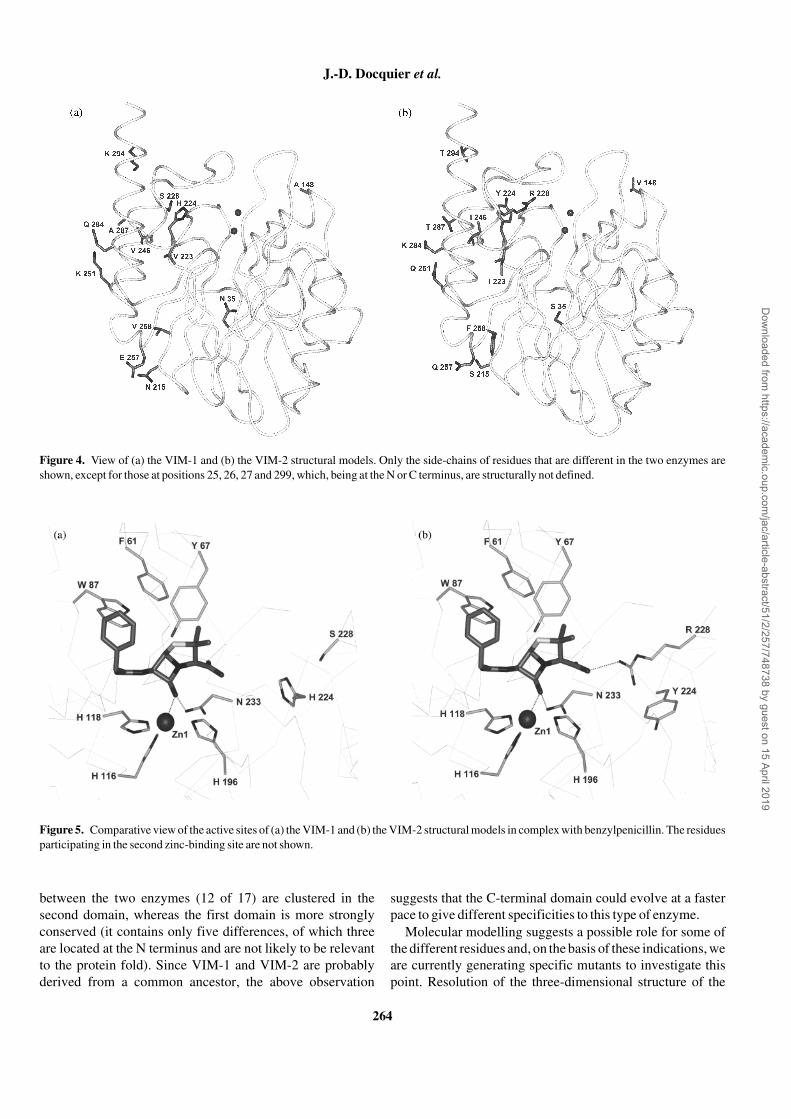
At the sequence level VIM-1 and VIM-2 are 93% identicaland can be aligned without the introduction of major gaps withall the subclass B1 β-lactamases,33 including those (Bc-II,CcrA and IMP-1) for which three-dimensional structures areavailable.4,5,7,29 Of the latter enzymes, CcrA was chosen as thebasis for construction of VIM-1 and VIM-2 structural models,since although Bc-II exhibits a higher similarity, the CcrAstructure has better defined coordinates for the loop betweenstrands 3 and 4.29 Compared with CcrA, the differences interms of deletions/insertions for VIM-1 and VIM-2 consist of:(i) the lack of one residue (W64) in the L1 loop betweenstrands 3 and 4; and (ii) the insertion of two residues(L172 and E173) in the L2 loop, between helix 3 and strand 8(Figure 3).
Of the 17 residues differentiating VIM-2 from VIM-1,most (including those at positions 25, 26, 27, 35, 148, 215,
246, 251, 257, 258, 284, 287, 294 and 299) are located at theprotein surface and are distant from the active site, while three(at positions 223, 224 and 228) are located in the neighbour-hood of the active site (in loop L3) and could influencesubstrate binding (Figure 4).
Docking experiments with benzylpenicillin support thisview and revealed some interesting structural features thatcould explain the different affinities of VIM-1 and VIM-2 forthis substrate. In both VIM-1 and VIM-2, the phenyl ringof benzylpenicillin could interact with the aromatic bulkyside-chains of F61, Y67 and W87 (located in loop L1) by meansof aromatic–aromatic interactions, whereas the carbonyloxygen of the β-lactam fits in the oxyanion hole formed by thezinc atom of the first binding site (Zn1) and the side-chain ofN233, which is conserved in most metallo-β-lactamases.33,36
A unique feature of VIM-type enzymes is represented by thevariability encountered at position 224. In other enzymes ofsubclass B1 a conserved lysine residue, whose side-chainloosely interacts with the β-lactam carboxylate,37 is foundhere. This could differentially affect the orientation andstabilization of the substrate in the active site. At position 224,VIM-1 has a histidine whose side-chain is shorter and prob-ably neutral in this environment, and would not be expected tocreate an interaction with the substrate, whereas VIM-2 has atyrosine whose slightly longer side-chain might positivelyaffect stabilization with some substrates, including thosecarrying a positively charged C3 group, such as ceftazidime,for which VIM-2 exhibits a higher affinity than VIM-1. More-over, stabilization of the substrate in the active site could beinfluenced in different ways by the different residues atpositions 228. At this position, VIM-2 has an arginine, whoseside-chain protrudes into the active site so that the positivelycharged guanidium group could interact directly with thebenzylpenicillin carboxylate (O–NH2 distance: 3.09 Å),whereas VIM-1 has a serine, whose side-chain is definitelytoo short to create any interaction with the substrate (Figure 5).The supplementary interaction between R228 and thesubstrate in VIM-2 could explain the higher affinity forbenzylpenicillin observed with the latter enzyme comparedwith VIM-1.
Conclusions
Comparative biochemical analysis carried out under the sameexperimental conditions allowed us to confirm that there arenotable differences between VIM-1 and VIM-2 in their inter-actions with several β-lactam substrates and also with metalchelators. This means that at least some of the amino aciddifferences between the two proteins must have functionalsignificance. For this reason, the two natural VIM variantspresent a relevant model for understanding the roles ofspecific residues in the mechanism of zinc-β-lactamases. Itshould be noted that although the two α/β domains constitut-ing the protein are similar in size, most of the differences
Figure 3. Structural alignment of CcrA, VIM-1 and VIM-2 amino acidsequences, generated with the help of the HOMOLOGY module of theInsight II software. Identical residues in the three sequences are shadedin grey. Subsitutions between VIM-1 and VIM-2 are shaded in black.Relevant positions are numbered according to the BBL scheme.33
Positions of the zinc–ligand residues (grey and italicized) are indicated.Secondary structure elements (S, strands; H, helices; L, loops) areindicated below the sequences.
Dow
nloaded from https://academ
ic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019
J.-D. Docquier et al.
264
between the two enzymes (12 of 17) are clustered in thesecond domain, whereas the first domain is more stronglyconserved (it contains only five differences, of which threeare located at the N terminus and are not likely to be relevantto the protein fold). Since VIM-1 and VIM-2 are probablyderived from a common ancestor, the above observation
suggests that the C-terminal domain could evolve at a fasterpace to give different specificities to this type of enzyme.
Molecular modelling suggests a possible role for some ofthe different residues and, on the basis of these indications, weare currently generating specific mutants to investigate thispoint. Resolution of the three-dimensional structure of the
Figure 4. View of (a) the VIM-1 and (b) the VIM-2 structural models. Only the side-chains of residues that are different in the two enzymes areshown, except for those at positions 25, 26, 27 and 299, which, being at the N or C terminus, are structurally not defined.
Figure 5. Comparative view of the active sites of (a) the VIM-1 and (b) the VIM-2 structural models in complex with benzylpenicillin. The residuesparticipating in the second zinc-binding site are not shown.
Dow
nloaded from https://academ
ic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019

Biochemistry of VIM metallo-β-lactamases
265
VIM enzymes is also underway, and will provide an essentialcontribution to the understanding of the mechanistic pro-perties of these clinically relevant enzymes. The functionaldiversity encountered among metallo-β-lactamases producedby opportunistic pathogens could be critical to the develop-ment of effective ‘broad-spectrum’ inactivators of theseenzymes.
Acknowledgements
Thanks are due to Isabel Garcia-Saez for determination of themass spectra, and to Nicola Franceschini for providing uswith some purified VIM-1 enzyme. This work was supportedby the European research network on metallo-β-lactamaseswithin the Training and Mobility of Researchers (TMR)Programme (Contract No. FMRX-CT98-0232), by grant no.20011068755-003 from the Italian ‘M.I.U.R.’ (PRIN 2001)and by a grant from the Belgian federal government (PAIP5/33).
References
1. Bush, K., Jacoby, G. A. & Medeiros, A. A. (1995). A functionalclassification scheme for β-lactamases and its correlation withmolecular structure. Antimicrobial Agents and Chemotherapy 39,1211–33.
2. Bush, K. (1999). β-Lactamases of increasing clinical importance.Current Pharmaceutical Design 11, 839–45.
3. Livermore, D. M. & Woodford, N. (2000). Carbapenemases: aproblem in waiting? Current Opinion in Microbiology 5, 489–95.
4. Carfi, A., Pares, S., Duée, E., Galleni, M., Duez, C., Frère, J. M.et al. (1995). The 3-D structure of a zinc metallo-β-lactamase fromBacillus cereus reveals a new type of protein fold. EMBO Journal14, 4914–21.
5. Concha, N. O., Rasmussen, B. A., Bush, K. & Herzberg, O.(1996). Crystal structure of the wide-spectrum binuclear zincβ-lactamase from Bacteroides fragilis. Structure 4, 823–36.
6. Ullah, J. H., Walsh, T. R., Taylor, I. A., Emery, D. C., Verma,C. S., Gamblin, S. J. et al. (1998). The crystal structure of the L1metallo-β-lactamase from Stenotrophomonas maltophilia at 1.7 Åresolution. Journal of Mololecular Biology 284, 125–36.
7. Concha, N. O., Janson, C. A., Rowling, P., Pearson, S.,Cheever, C. A., Clarke, B. P. et al. (2000). Crystal structure of theIMP-1 metallo β-lactamase from Pseudomonas aeruginosa and itscomplex with a mercaptocarboxylate inhibitor: binding determinantsof a potent, broad-spectrum inhibitor. Biochemistry 39, 4288–98.
8. Watanabe, M., Iyobe, S., Inoue, M. & Mitsuhashi, S. (1991).Transferable imipenem resistance in Pseudomonas aeruginosa.Antimicrobial Agents and Chemotherapy 35, 147–51.
9. Osano, E., Arakawa, Y., Wacharotayankun, R., Ohta, M., Horii,T., Ito, H. et al. (1994). Molecular characterization of an entero-bacterial metallo-β-lactamase found in a clinical isolate of Serratiamarcescens that shows imipenem resistance. Antimicrobial Agentsand Chemotherapy 38, 71–8.
10. Muramo, K., Takeda, A., Nakamura, Y. & Nakaya, K. (1995).Purification and characterization of metallo-β-lactamase fromSerratia marcescens. Microbiology and Immunology 39, 27–33.
11. Laraki, N., Franceschini, N., Rossolini, G. M., Santucci, P.,Meunier, C., de Pauw, E. et al. (1999). Biochemical characterizationof the Pseudomonas aeruginosa 101/1477 metallo-β-lactamaseIMP-1 produced by Escherichia coli. Antimicrobial Agents andChemotherapy 43, 902–6.
12. Prosperi-Meys, C., Llabres, G., de Seny, D., Paul Soto, R.,Hernandez Valladares, M., Laraki, N. et al. (1999). Interactionbetween class B β-lactamases and suicide substrates of active-siteserine β-lactamases. FEBS Letters 443, 109–11.
13. Haruta, S. , Yamaguchi, H., Yamamoto, E. T., Eriguchi, Y., Nuk-aga, M., O’Hara, K. et al. (2000). Functional analysis of the activesite of a metallo-β-lactamase proliferating in Japan. AntimicrobialAgents and Chemotherapy 44, 2304–9.
14. Riccio, M. L., Franceschini, N., Boschi, L., Caravelli, B.,Cornaglia, G., Fontana, R. et al. (2000). Characterization of themetallo-β-lactamase determinant of Acinetobacter baumanniiAC-54/97 reveals the existence of blaIMP allelic variants carried bygene cassettes of different phylogeny. Antimicrobial Agents andChemotherapy 44, 1229–35.
15. Haruta, S., Yamamoto, E. T., Eriguchi, Y. & Sawai, T. (2001).Characterization of the active-site residues asparagine 167 andlysine 161 of the IMP-1 metallo β-lactamase. FEMS MicrobiologyLetters 197, 85–9.
16. Lauretti, L., Riccio, M. L., Mazzariol, A., Cornaglia, G., Amico-sante, G. Fontana, R. et al. (1999). Cloning and characterization ofblaVIM, a new integron-borne metallo-β-lactamase gene from aPseudomonas aeruginosa clinical isolate. Antimicrobial Agents andChemotherapy 43, 1584–90.
17. Poirel, L., Naas, T., Nicholas, D., Collet, L., Bellais, S., Cavallo,J. D. et al. (2000). Characterization of VIM-2, a carbapenem-hydrolyzing metallo-β-lactamase and its plasmid- and integron-borne gene from a Pseudomonas aeruginosa clinical isolate inFrance. Antimicrobial Agents and Chemotherapy 44, 891–7.
18. Franceschini, N., Caravelli, B., Docquier, J. D., Galleni, M.,Frère, J. M., Amicosante, G. et al. (2000). Purification andbiochemical characterization of the VIM-1 metallo-β-lactamase.Antimicrobial Agents and Chemotherapy 44, 3003–7.
19. Yan, J. J., Hsueh, P. R., Ko, W. C., Luh, K. T., Tsai, S. H., Wu,H. M. et al. (2001). Metallo-β-lactamases in clinical Pseudomonasisolates in Taiwan and identification of VIM-3, a novel variant ofthe VIM-2 enzyme. Antimicrobial Agents and Chemotherapy 45,2224–8.
20. Mavroidi, A., Tsakris, A., Tzelepi, E., Pournaras, S., Loukova,V. & Tzouvelekis, L. S. (2000) Carbapenem-hydrolysing VIM-2metallo-β-lactamase in Pseudomonas aeruginosa from Greece.Journal of Antimicrobial Chemotherapy 46, 1041–2.
21. Pallecchi, L., Riccio, M. L., Docquier, J. D., Fontana, R. & Ros-solini, G. M. (2001). Molecular heterogeneity of blaVIM-2-containingintegrons from Pseudomonas aeruginosa plasmids encoding theVIM-2 metallo-β-lactamase. FEMS Microbiology Letters 195,145–50.
Dow
nloaded from https://academ
ic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019

J.-D. Docquier et al.
266
22. Sambrook, J., Fritsch, E. F. & Maniatis, T. (2001). MolecularCloning: A Laboratory Manual, 3rd edn. Cold Spring Harbor Labora-tory Press, Cold Spring Harbor, NY, USA.
23. Pace, C. N., Vajdos, F., Fee, L., Grimsley, G. & Gray, T. (1995).How to measure and predict the molar absorption coefficient of aprotein. Protein Science 4, 2411–23.
24. Laemmli, U. K. (1970). Cleavage of structural proteins duringthe assembly of the head of bacteriophage T4. Nature 227, 680–5.
25. Hagel, J. (1989). Gel filtration. In Principles, High ResolutionMethods and Applications (Janson, J. C. & Rydén, L., Eds),pp. 63–106. VCH Publishers Inc., New York, NY, USA.
26. Segel, I. H. (1975). Rapid equilibrium partial and mixed-typeinhibition. In Enzyme Kinetics, Behavior and Analysis of RapidEquilibrium and Steady-State Enzyme Systems, pp. 210–2. JohnWiley & Sons, New York, NY, USA.
27. De Meester, F., Joris, B., Reckinger, G., Bellefroid-Bourguignon, C., Frère, J. M. & Waley, S. G. (1987). Automatedanalysis of enzyme inactivation phenomena. Application toβ-lactamases and DD-peptidases. Biochemical Pharmacology 36,2393–403.
28. Hernandez Valladeres, M., Felici, A., Weber, G., Adolph, H. W.,Zeppezauer, M., Rossolini, G. M. et al. (1997). Zn(II) dependence ofthe Aeromonas hydrophila AE036 metallo-β-lactamase activity andstability. Biochemistry 36, 11534–41.
29. Carfi, A., Duée, E., Paul-Soto, R., Galleni, M., Frère, J. M. &Dideberg, O. (1998). X-ray structure of the ZnII β-lactamase fromBacteroides fragilis in an orthorhombic crystal form. Acta Crystallo-graphica Section D Biological Crystallography 54, 45–57.
30. Pearlman, D., Case, D. A., Caldwell, J. A., Ross, W. S., Cheath-man, T. E., Ferguson, D. M. et al. (1995). AMBER 4.1. University ofCalifornia, San Francisco, CA, USA.
31. Banci, L., Schröder, S. & Kollman, P. A. (1992). Moleculardynamics characterization of the active cavity of carboxypeptidaseA and some of its inhibitor adducts. Proteins 13, 288–305.
32. Koradi, R., Billeter, M. & Wüthrich, K. (1996). MOLMOL: aprogram for display and analysis of macromolecular structures.Journal of Molecular Graphics 14, 51–5.
33. Galleni, M., Lamotte-Brasseur, J., Rossolini, G. M., Spencer, J.,Dideberg, O. & Frère, J. M. (2001). Standard numbering scheme forclass B β-lactamases. Antimicrobial Agents and Chemotherapy 45,660–3.
34. Studier, F. W., Rosenberg, A. H., Dunn, J. J. & Dubendorff,J. W. (1990). Use of T7 RNA polymerase to direct expression ofcloned genes. Methods in Enzymology 185, 60–89.
35. Rossolini, G. M., Franceschini, N., Riccio, M. L., Mercuri, P. S.,Perilli, M., Galleni, M. et al. (1998). Characterization and sequenceof the Chryseobacterium (Flavobacterium) meningosepticumcarbapenemase: a new molecular class B β-lactamase showing abroad substrate profile. Biochemical Journal 332, 145–52.
36. Wang, Z., Fast, W., Valentine, A. M. & Benkovic, S. J. (1999).Metallo-β-lactamase: structure and mechanism. Current Opinion inChemical Biology 3, 614–22.
37. Prosperi-Meys, C., Wouters, J., Galleni, M. & Lamotte-Brasseur, J. (2001). Substrate binding and catalytic mechanism ofclass B β-lactamases: a molecular modelling study. Cellular andMolecular Life Sciences 58, 2136–43.
Dow
nloaded from https://academ
ic.oup.com/jac/article-abstract/51/2/257/748738 by guest on 15 April 2019



![9. Heterogeneity: Latent Class Modelspeople.stern.nyu.edu › wgreene › DiscreteChoice › 2014 › DC2014-9-LCModels.pdf[Topic 9-Latent Class Models] 3/66 Latent Classes • A population](https://static.fdocument.org/doc/165x107/5f03e2617e708231d40b3e43/9-heterogeneity-latent-class-a-wgreene-a-discretechoice-a-2014-a-dc2014-9-lcmodelspdf.jpg)


![Metallo-β-Lactamases: A Major Threat to Human Healthfile.scirp.org/pdf/AJMB_2014071615272629.pdf · tobacter baumannii [9]-[11]is facile transfer of genetic inform). ... ased on](https://static.fdocument.org/doc/165x107/5b7a3ec67f8b9ae7368b5881/metallo-lactamases-a-major-threat-to-human-tobacter-baumannii-9-11is.jpg)

![kp∂n A^vIm¿ - SYS State Committee — SUNNI …sysstatecommittee.com/afkar/2011/01-17.pdf12 Xplv^-Øp¬ apPm-ln-Zo≥ tIcf Ncn-{X-Øns‚ I\epw ImXepw alvaqZv ]\-ßm-ßc 18 hmb-\-](https://static.fdocument.org/doc/165x107/5acb9c507f8b9a27628b809a/kpn-avim-sys-state-committee-sunni-xplv-p-appm-ln-zo-ticf-ncn-x-ns.jpg)









