
Inmunogenética en medicina clínica - Acta Médica Colombiana · Defectos enzimáticos no...
39
25 Actualizaciones Inmunogenética en medicina clínica Parte III. Asociación HLA-Enfermedad Gloria de Egea, Eduardo Egea, Marcela Salazar, Antonio Iglesias, Juan Yunis, Scarlet Lechín, Iván Yunis, Edmond J. Yunis Debido a la importancia que tiene el Complejo Mayor de Histocompatibilidad (CMH) en el con- trol genético de la respuesta inmunitaria y por ende en la homeostasis de la inmunorregulación, una vez aclarados los mecanismos genéticos subya- centes a la expresión de las moléculas de clase I y II, la investigación se dirigió al estudio de la aso- ciación sistema HLA-enfermedad. Cualquier alteración, bien sea por exceso o por defecto, en la expresión de las moléculas de clase I y II tiene importantes consecuencias en el tipo de respuesta inmune producida por el individuo y en la asociación de dicha respuesta con determinadas enfermedades. Para analizar los grados de asocia- ción entre determinados marcadores genéticos y la susceptibilidad a presentar una determinada enfer- medad, se han utilizado los siguientes métodos epidemiológicos: a) Estudio de la probabilidad o riesgo de padecer la enfermedad portando un alelo específico en un sistema polimórfíco. b) Determi- nación del riesgo relativo, para lo cual se calcula el riesgo de padecer una enfe r medad en una pobla- Drs. Gloria de Egea, Eduardo Egea Bermejo, Antonio Iglesias : División of Inmunogenetics, Dana Farber Cancer Institute, Department of Pathology Har- vard Medical School: Facultad de Medicina Universidad del Norte, U. Libre, Barranquilla, Colombia; Dra. Marcela Salazar Vallejo: Department of Patholo- gy Harvard Medical School; Drs. Juan Yunis, Iván Yunis: División of Inmuno- genetics, Dana Farber Cancer Institute, Department of Pathology Harvard Medical School; Dr. Scarlet Lechin: División of Inmunogenetics, Dana Farber Cancer Institute, Boston, Department of Pathology Harvard Medical School, Sección de Psicofarmacología y Medicina Psicosomática, Instituto Medicina Experimental, Universidad Central de Venezuela, Caracas; Dr. Edmond J. Yunis: División of Inmunogenetics, Dana Farber Cancer Institute, Department of Pa- thology Harvard Medical School, Chief Division of Inmunogenetics, Dana Farber Cancer Institute, Professor of Pathology, Harvard Medical School. Solicitud de separatas al Dr. Iglesias. ción de sujetos que expresan un marcador en rela- ción con el riesgo en una población control, c) Comparación de los marcadores de un individuo que tiene un haplotipo determinado con los marca- dores de los mismos haplotipos de sujetos norma- les. d) Estudio de los haplotipos extendidos de indi- viduos con una determinada enfermedad y los de su familia y compración con los haplotipos exten- didos de familias sanas. Los primeros estudios acerca de la asociación HLA-enfermedad fueron llevados a cabo por Lilly en ratones en 1964 (1), con la demostración del papel del CMH en un tipo de leucemia inducida por virus. Tres años más tarde Amiel (2) analizó la asociación del CMH con la enfermedad de Hodg- kin. En 1973, fue informada la asociación entre espondilitis anquilosante y el antígeno HLA-B27 (3) y desde entonces se ha podido demostrar la asociación de un gran número de patologías en el ser humano con el CMH. A pesar de que existe un número de enfermedades en las cuales se encuen- tran alelos específicos de varios loci del CMH, és- tos se expresan también en individuos normales. A partir del V Taller Internacional de Histocompati- bilidad, reunido en 1972 (4), se pudo demostrar que la mayoría de los antígenos del sistema HLA varían considerablemente en frecuencia de acuer- do a las diferentes áreas geográficas, como conse- cuencia de variaciones alélicas a nivel de los isoti- pos del CMH. Por ello, es importante que cada país tenga cuidadosamente establecida la antigenicidad en su población de control con el fin de poderla comparar con la de la enfermedad a estudiar. Acta Médica Colombiana Vol. 15 N°1 - Enero-Febrero - 1990
Transcript of Inmunogenética en medicina clínica - Acta Médica Colombiana · Defectos enzimáticos no...

25
Actualizaciones
Inmunogenética en medicina clínica Parte III. Asociación HLA-Enfermedad
Gloria de Egea, Eduardo Egea, Marcela Salazar, Antonio Iglesias, Juan Yunis, Scarlet Lechín, Iván Yunis, Edmond J. Yunis
Debido a la importancia que tiene el Complejo Mayor de Histocompatibilidad (CMH) en el con-trol genético de la respuesta inmunitaria y por ende en la homeostasis de la inmunorregulación, una vez aclarados los mecanismos genéticos subya-centes a la expresión de las moléculas de clase I y II, la investigación se dirigió al estudio de la aso-ciación sistema HLA-enfermedad.
Cualquier alteración, bien sea por exceso o por defecto, en la expresión de las moléculas de clase I y II tiene importantes consecuencias en el tipo de respuesta inmune producida por el individuo y en la asociación de dicha respuesta con determinadas enfermedades. Para analizar los grados de asocia-ción entre determinados marcadores genéticos y la susceptibilidad a presentar una determinada enfer-medad, se han utilizado los siguientes métodos epidemiológicos: a) Estudio de la probabilidad o riesgo de padecer la enfermedad portando un alelo específico en un sistema polimórfíco. b) Determi-nación del riesgo relativo, para lo cual se calcula el riesgo de padecer una enfermedad en una pobla-
Drs. Gloria de Egea, Eduardo Egea Bermejo, Antonio Iglesias : División of Inmunogenetics, Dana Farber Cancer Institute, Department of Pathology Har-vard Medical School: Facultad de Medicina Universidad del Norte, U. Libre, Barranquilla, Colombia; Dra. Marcela Salazar Vallejo: Department of Patholo-gy Harvard Medical School; Drs. Juan Yunis, Iván Yunis: División of Inmuno-genetics, Dana Farber Cancer Institute, Department of Pathology Harvard Medical School; Dr. Scarlet Lechin: División of Inmunogenetics, Dana Farber Cancer Institute, Boston, Department of Pathology Harvard Medical School, Sección de Psicofarmacología y Medicina Psicosomática, Instituto Medicina Experimental, Universidad Central de Venezuela, Caracas; Dr. Edmond J. Yunis: División of Inmunogenetics, Dana Farber Cancer Institute, Department of Pa-thology Harvard Medical School, Chief Division of Inmunogenetics, Dana Farber Cancer Institute, Professor of Pathology, Harvard Medical School.
Solicitud de separatas al Dr. Iglesias.
ción de sujetos que expresan un marcador en rela-ción con el riesgo en una población control, c) Comparación de los marcadores de un individuo que tiene un haplotipo determinado con los marca-dores de los mismos haplotipos de sujetos norma-les. d) Estudio de los haplotipos extendidos de indi-viduos con una determinada enfermedad y los de su familia y compración con los haplotipos exten-didos de familias sanas.
Los primeros estudios acerca de la asociación HLA-enfermedad fueron llevados a cabo por Lilly en ratones en 1964 (1), con la demostración del papel del CMH en un tipo de leucemia inducida por virus. Tres años más tarde Amiel (2) analizó la asociación del CMH con la enfermedad de Hodg-kin. En 1973, fue informada la asociación entre espondilitis anquilosante y el antígeno HLA-B27 (3) y desde entonces se ha podido demostrar la asociación de un gran número de patologías en el ser humano con el CMH. A pesar de que existe un número de enfermedades en las cuales se encuen-tran alelos específicos de varios loci del CMH, és-tos se expresan también en individuos normales. A partir del V Taller Internacional de Histocompati-bilidad, reunido en 1972 (4), se pudo demostrar que la mayoría de los antígenos del sistema HLA varían considerablemente en frecuencia de acuer-do a las diferentes áreas geográficas, como conse-cuencia de variaciones alélicas a nivel de los isoti-pos del CMH. Por ello, es importante que cada país tenga cuidadosamente establecida la antigenicidad en su población de control con el fin de poderla comparar con la de la enfermedad a estudiar.
Acta Médica Colombiana Vol. 15 N°1 - Enero-Febrero - 1990
Administrador
Line
Administrador
Line

26 G. de Egea y cols.
Entre los mecanismos propuestos para explicar la asociación entre HLA y enfermedad se encuen-tran los siguientes:
1. Alteración de los antígenos propios de cla-se I. Desde 1971 Jerne (5) y en 1978 Benacerraf observaron que las células T periféricas tienen poca reactividad para antígenos autólogos del CMH, y en cambio responden bien a antígenos alogénicos. Zinkernagel y Doherty en 1974 (6) demostraron la restricción genética de dicha respuesta; con estos antecedentes Geczy en 1981 (7) pudo identificar en cultivos celulares de Klebsiella pneumoniae (cepa K43) un factor que podía modificar un com-ponente celular del B27 o cerca de él, y descubrió que esta modificación podía inducir a las células efectoras para que lesionaran tejidos específicos como la sinovial (produciendo sinovitis), con cier-ta predilección por el tejido conectivo del esquele-to axial. Esta alteración en la inmunorregulación desembocaría en una serie de eventos responsa-bles de la aparición de espondilitis y uveitis. Pos-teriormente fue posible establecer que algunos microorganismos comparten ciertas estructuras con antígenos de productos génicos del HLA y con base en ellos Ogasawara y Yu (8) pudieron de-mostrar el posible mimetismo celular entre la pa-red de la membrana celular de la Klebsiella y el antígeno B27 a partir de una sesuencia de seis aminoácidos. La asociación con el B27 entre los pacientes con espondilitis anquilosante con com-promiso axial y uveitis anterior es del 90% en la población caucásica, comparándola con una pre-sencia de B27 en sólo 5 a 10% de los controles sanos (9, 10). La asociación del B27 con espondi-litis anquilosante en cuatro grupos raciales (cau-cásicos, negros, japoneses y amerindios) sugiere que los genes ligados al B27 están comprometidos en la patogénesis de la enfermedad. En otras artri-tis reactivas producidas por otros microorganis-mos como Shigella, Campylobacter, Yersinia y Salmonella no se ha podido evidenciar alteración o mimetismo molecular entre la estructura de su membrana y los genes del alelo B27. Por lo tanto la susceptibilidad de los individuos a espondilitis-uveítis, parece que se hereda en forma dominante (7, 10).
2. Disminución o ausencia de genes supreso-res dominantes y su asociación con el HLA-D/ DR. En estudios realizados en ratones y cobayos, se encontró evidencia de que la ausencia o dismi-nución de ciertos genes supresores se asocia a al-gunas enfermedades relacionadas con el HLA-D/ DR, como se aprecia en las cepas de ratones NZB, MRL/Cpr, BXSB con problemas autoinmunes. En el humano, un número de enfermedades autoin-munes, o probablemente de origen autoinmune muestran una fuerte asociación con el HLA-D/Dr, como ocurre con la diabetes mellitus juvenil (tipo I), el lupus eritematoso sistémico (LES), la enfer-medad de Graves, la miastenia gravis, el síndrome de Sjögren primario, la hepatitis crónica activa, la enfermedad de Addison idiopática, la enfermedad celíaca, la dermatitis herpetiforme y su asociación con el Dw3 y el DR3 en caucásicos; la artritis reu-matoide y la diabetes mellitus con el DR4, la es-clerosis múltiple y la lepra tuberculoide con el DR2. En los japoneses el haplotipo HLA-Bw52, Dwl2 y el DR2 se asocian a una hiperrespuesta a ciertos antígenos solubles y no solubles como el toxoide tetánico, el polen del cedro y el antígeno de la pa-red del estreptococo; y los haplotipos Bw54, DYT y DR4 se asocian a una alta respuesta a los antíge-nos mencionados anteriormente (9-13).
En estudios de función celular, se ha documen-tado una alteración de la función supresora en muchas de las enfermedades autoinmunes y en la hiporrespuesta a ciertos antígenos, lo que sugiere un defecto en las células T supresoras, debido a la ausencia de genes supresores dominantes asocia-da al HLA (13).
3. Los antígenos del CMH como receptores. Se ha planteado la hipótesis de que los antígenos del HLA pudieran ser receptores para ciertos pató-genos. Al respecto está demostrado que el antíge-no Duffy de los eritrocitos actúa como receptor para la malaria; en cambio aquellos individuos que carecen del antígeno Duffy tienen resistencia a la malaria. La susceptibilidad a la malaria, depen-diente del antígeno Duffy, es dominante (14). He-lelnius y col en 1978 demostraron que los antíge-nos HLA-A y HLA-B pudieran ser los receptores para el virus de Semliki Forest.

Inmunogenética en medicina clínica 27
4. Mimetismo molecular. En los últimos años, se ha postulado la posibilidad de que los productos génicos del HLA tengan una estructura parecida a la de ciertos determinantes antigénicos de algunos microorganismos, especialmente bacterias Gram (-), lo que explicaría la reactividad cruzada entre el microorganismo patógeno y un antígeno del sis-tema HLA. Este mecanismo se invoca actualmen-te para explicar la relación entre HLA-B27 y la espondiloartropatía seronegativa; la enfermedad de Behcet y el HLA-B5; la tiroiditis aguda de De Quervain y el HLA-Bw35.
Esta predisposición para desarrollar artritis re-activa no es habitual en las enfermedades reumáti-cas, lo que permite plantear la posibilidad de cier-tas precondiciones para que se establezca la artri-tis. Esta predisposición está relacionada con algu-nos mecanismos inmunes y con ciertos serotipos de patógenos entéricos como las bacterias Gram (-), tales como: Yersinia, Salmonella, Shigella, Klebsiella y Campylobacter. Esta artritis reactiva podría ser el producto de la interrelación entre el microorganismo y el CMH, posiblemente a través de la hipótesis del mimetismo molecular (15, 16). Llama la atención que la mayoría de las artritis reactivas postdisentéricas están relacionadas con el HLA-B27, HLA-B7 y HLA-Bw60, es decir, la susceptibilidad se encuentra ligada al loci B del CMH (17).
5. Genes ligados al HLA. Se han logrado docu-mentar algunos defectos de genes ligados al HLA y relacionados con el sistema del complemento, en el cual el defecto puede deberse a mutaciones que ocurren a nivel de haplotipos extendidos, ex-presándose a nivel de sexo masculino preferen-cialmente. Al analizar, estudios de desequilibrio de ligamiento, estudiando segmentos cromosómi-cos se ha podido documentar la asociación entre deficiencia del C2 con haplotipos extendidos del HLA-A25, HLA-B18, HLA-DR2, BFS, C4AB2. Por lo tanto se intuyó que la deficiencia del C2 asociada con los haplotipos antes mencionados fuese producto de la mutación del gen de C2 en forma reciente a nivel de haplotipos extendidos; por esto parece que existe una relación muy estre-cha entre una alteración génica y la ontogenia de
los haplotipos extendidos y se acuñó por primera vez el término complotipo (18,19) como haploti-pos del complemento (19).
6. HLA y alelos anormales en la diferencia-ción de genes humanos. De Wolf y col (20) estu-diaron en 1979 la región H2 en el murino y obser-varon que la diferenciación en el ratón está con-trolada genéticamente. Por ello se planteó la posi-bilidad de que en ciertas asociaciones del HLA se encuentre un desequilibrio de unión con ciertos alelos de genes de diferenciación en el humano y la expresión fenotípica pudiera ser anormal. Por ejemplo, en la asociación del teratocarcinoma tes-ticular en el humano y el Dw27.
7. Defectos enzimáticos no inmunológicos relacionados con el HLA. Está plenamente docu-mentado que los genes de la 21 α y β hidroxilasa se encuentran localizados en el HLA y por lo tanto la deficiencia de la 21 α hidroxilasa en la vía de la esteroidogénesis aunque, sin ninguna regulación inmunológica, está relacionada con el HLA (21).
8. Defecto en la expresión bioquímica de al-gunas proteínas. Se ha logrado observar en algu-nas enfermedades que existe un defecto en la pro-ducción de determinadas proteínas que posible-mente está codificado por la región del HLA. Esto ocurre en la hemocromatosis idiopática y su aso-ciación con el HLA-A3. La expresión bioquímica parcial se observa en los portadores heterocigotes de dicho alelo, pero la acumulación excesiva de hierro se observa especialmente en los homocigo-tes (22, 23).
9. Alteración de la expresión de los antígenos de clase I y II inducidos por microorganismos y citoquinas. Se ha podido observar que la expre-sión de los antígenos de clase I y II puede ser in-ducida por virus o citoquinas. Uno de los principa-les mediadores para la expresión de genes del CMH en forma aberrante es el γ-interferón. Lo ; virus son los microorganismos mejor estudiados, ya que incrementan la producción de interferón creando una expresión aberrante de genes del CMH. Cam-bell y col han demostrado que el 7-interferón in-crementa la expresión de antígenos de clase I del CMH en células 6 del páncreas y el mismo grupo ha demostrado que la infección por reovirus incre-
Acta Med Colomb Vol. 15 N°1 ~ 1990

28 G. de Egea y cols.
menta la expresión de proteínas clase I del CMH sobre células B humanas y en células de ratas de la cepa RINm5F (24-26).
El grupo de Botazzo y Feldmann en Inglaterra es el que más ha estudiado la posibilidad del papel de la expresión del HLA-DR aberrante, que fun-cionaría como autoantígeno con pérdida de la to-lerancia inmunológica y activación de las células T, las cuales no reconocerían autoantígenos espe-cíficos asociados con los antígenos de clase II. Este grupo ha propuesto como uno de los mecanismo de inducción de enfermedades endocrinas de tipo autoinmune (diabetes, tiroiditis) la expresión del HLA-DR aberrante.
10. Asociación con inmunodeficiencia del complemento. La deficiencia de C2 es la más fre-cuente de las deficiencias del complemento en la población caucásica (un individuo homozigote por cada 10.000 donadores en un estudio y 1.2% de heterozigotes en otro). Primero se documentó esta inmunodeficiencia en la población normal y pos-teriormente en el LES, en el síndrome de Henoch-Schonlein, en la polimiositis y las vasculitis. La inmunodeficiencia se debe a la expresión de un alelo null en el loci estructural. Siempre se asocia al haplotipo HLA-A25, HLA-B18 y al HLA-DR2. Se ha descrito deficiencia de C4 homozigota y he-terozigota asociada a enfermedades inmunológi-cas especialmente de tipo autoinmune, como LES, dermatomiositis y vasculitis.
Marcadores de enfermedad Desde hace tiempo se conoce que un gran nú-
mero de enfermedades tienen asociación con el CMH, por ejemplo, los pacientes muestran dife-rente distribución de los marcadores del CMH, particularmente en el caso de los antígenos del HLA, al compararlos con los diferentes controles étnicos (27). En el caso de los pacientes con dia-betes mellitus tipo I hay un aumento en la frecuen-cia de los antígenos HLA B15 (w62), B18, DR3, DR4 (28), así como del alelo del complemento BF*F1 (29), y una disminución de las frecuencias de B7 y DR2. Con el estudio de un gran número de individuos diabéticos se determinó que la distri-bución del alelo BF*F1 era consistente con una
herencia simple recesiva de este marcador y un aumento de susceptibilidad a la diabetes tipo I (30). Analizando marcadores de CMH entre los diabéti-cos y sus familias, fue aparente que la mayoría de los pacientes presentaban incremento en los si-guientes haplotipos: [HLA-B8, F1C30, DR3]; [HLA-B 18, F1C30, DR3]; [HLA-B15(w62), SC33, DR4] y HLA-B16(38), SC21, DR4] (31).
En los pacientes con enteropatía sensible al glu-ten (32) dos haplotipos extendidos se encontraban en un 60% de ellos: [HLA-B8, SCO1 , DR3] y [HLA-B44, FC31, DR7],
Hay dos excepciones al fenómeno aparente de que los haplotipos extendidos, más que los alelos individuales de CMH son los marcadores de sus-ceptibilidad a las enfermedades asociadas al CMH; en el caso de la espondilitis anquilosante, el HLA-B27 está fuertemente aumentado en pacientes de una gran variedad de grupos étnicos, lo cual sugie-re que este alelo definido serológicamente es por sí mismo un alelo de susceptibilidad. La segunda excepción es la esclerosis múltiple, en la cual HLA-DR2 o un subtipo de DR2 (33), más que un haplo-tipo extendido es el marcador en pacientes caucá-sicos, especialmente en aquellos de origen nórdi-co europeo.
Aunque la mayoría de las enfermedades asocia-das al CMH tienen aspectos inmunológicos, y qui-zá etiologías diferentes, hay dos entidades que no funcionan de esta manera, tal es el caso de la hemo-cromatosis (34) y la hiperplasia adrenal congénita por deficiencia de 21 a hidroxilasa (35). En esta última patología, la razón es evidente, hay dos genes para el citocromo p450 y las 21-hidroxila-sas adrenales α y β, cada uno situado a 3' inmedia-tamente al lado de los genes para C4A y C4B (36, 37). Unicamente se expresa 21-OHβ y los defec-tos o deleciones de este gen se asocian con hiper-plasia adrenal congénita (38, 39). Este último es un trastorno recesivo de penetrancia completa. En los pacientes caucásicos en el área de Boston, con la forma perdedora de sal de la enfermedad, apro-ximadamente un 20% de los haplotipos del CMH corresponden al haplotipo extendido [HLA-Bw47, FC91, DR7] (40), el cual contiene la deleción de 21-OHβ (36, 37). El haplotipo extendido [HLA-

Inmunogenética en medicina clínica 29
B14, Sc2 (1,2) DR1] es común entre los pacientes con inicio tardío de la enfermedad, especialmente entre aquellos de ancestro del sur de Europa (41); hay que anotar que en los pacientes procedentes de Venezuela con esta enfermedad no se encuen-tra ninguno de los haplotipos extendidos caracte-rísticos de la población europea (42); esto ilustra claramente la especificidad étnica de los marcado-res, tanto en salud como en enfermedad.
La ausencia de [HLA-B8, SCO1, DR3] entre los pacientes con hiperplasia adrenal congénita, se debe a la presencia de un gen normal para la 21-OHβ en su haplotipo extendido.
Los mecanismos para las asociaciones entre HLA y enfermedad son desconocidos para un gran número de entidades. Aunque se ha tratado de res-ponsabilizar de estas asociaciones a anormalida-des en los genes de la respuesta inmune (43), el hecho de que los haplotipos extendidos, más que los alelos individuales sean frecuentemente los marcadores, requiere que tengamos cuidado ya que cualquier alelo dentro del grupo del haplotipo ex-tendido puede ser el responsable.
Los complotipos en la medicina clínica Además de los casos en que se sospecha una
deficiencia hereditaria de una de las proteínas del complemento, la tipificación genética del comple-mento tiene gran aplicación en los trasplantes de tejidos, especialmente en el trasplante de médula ósea (44). En aquellos pacientes que requieren trasplante de médula ósea, la identificación del donante solamente mediante tipificación de HLA puede ser difícil para los pacientes, los cuales pue-den presentar reacciones de autoanticuerpos, leu-copenia, etc. Ya que los complotipos se encuen-tran codificados por genes cercanos a la región HLA-D, y el poliformismo del complotipo es ex-tenso, la determinación de los complotipos nos da una información valiosa en la identificación de los donantes.
La identificación in útero de los fetos afectados por enfermedades asociadas al CMH aún no es posible, con una excepción: en una familia con un hijo que padezca deficiencia de 21 α OH, es posi-ble algunas veces, mediante examen del DNA de
las células amnióticas fetales, determinar si el feto también se encuentra afectado (45).
La deficiencia hereditaria de C2 es el resultado de la herencia de ambos C6p's del alelo nulo C2*Q0. Este alelo se encuentra con mayor fre-cuencia en relación con el haplotipo extendido [HLA-(A25), B18, S042, DR2] (46, 48). Ya que esta deficiencia ha sido informada únicamente en europeos caucásicos y que casi todos los haploti-pos de los pacientes homozigotos para la deficien-cia tienen elementos de este haplotipo extendido, se puede postular que estos haplotipos descienden de una mutación simple en el loci C2 en el haploti-po del CMH de un paciente caucásico. Las varia-ciones en la composición alélica específica de C2*Q0 unida a los haplotipos puede explicarse por el cruce al azar entre el haplotipo ancestral y los haplotipos caucásicos. Con estudios de fre-cuencia de los diferentes haplotipos podemos con-cluir que la mutación que llevó a esta deficiencia ocurrió probablemente entre 600 y 1.300 años atrás.
De acuerdo con los mecanismos patogénicos expuestos anteriormente, asociados a diferentes enfermedades, planteamos la siguiente clasifica-ción:
1. Alteración aberrante de los antígenos de cla-se I y II inducidos por un agente infeccioso. Ej.: Klebsiella pneumoniae cepa K43 capaz de modi-ficar el componente celular del B27 produciendo la espondilitis anquilosante a través de la teoría del mimetismo molecular; endocrinopatías auto-inmunes (tiroiditis, diabetes juvenil).
2. Inmunodeficiencia de los componentes del complemento como C2 y C4. Ej.: LES, síndrome de Henoch-Schonlein, polimiositis, vasculitis.
3. Asociada con haplotipos extendidos. Ej.: Diabetes mellitus tipo I, asociada a ciertos haplo-tipos extendidos [HLA-B8, SC01, DR3]; [HLA-B18,F1C30,DR3], [HLA-B15 (w62),SC33,DR4] y [HLA-B16 (38), SC21, DR4], Enteropatía por gluten [HLA-B8, SC01, DR3], [HLA-B44, FC31, DR7],
4. Herencia poligénica. Además de genes rece-sivos o dominantes existe una relación con los an-tígenos de clase I y II, lógicamente asociada a un factor ambiental. Ej.: LES, esclerosis múltiple,
Acta Med Colomb Vol. 15 N°1 ~ 1990
30 G. de Egea y cols.
diabetes mellitus juvenil, enfermedad de Graves, enfermedad celiaca, etc.
5. Deficiencia de la 21-hidroxilasa. Un ejemplo es la deficiencia de la 21-hidroxilasa asociada a hiperplasia adrenal congénita; esta es una enzima importante que participa en el metabolismo del cortisol. La deficiencia de dicha enzima produce
virilización y pérdida de sal; además esta entidad se ha asociado a un haplotipo extendido el [B w47, FC031, DR7].
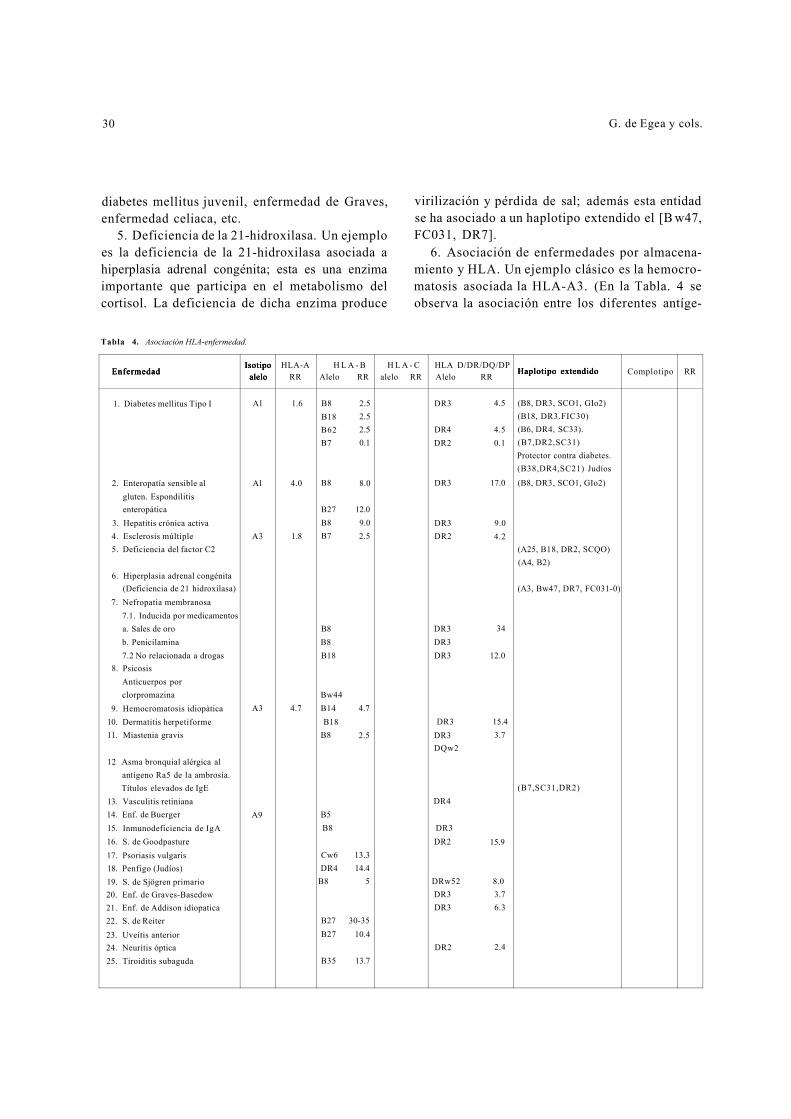
6. Asociación de enfermedades por almacena-miento y HLA. Un ejemplo clásico es la hemocro-matosis asociada la HLA-A3. (En la Tabla. 4 se observa la asociación entre los diferentes antíge-
Tabla 4. Asociación HLA-enfermedad.
EEEnnnfffeeerrrmmmeeedddaaaddd IIIsssoootttiiipppooo
aaallleeelllooo HLA-A H L A - B H L A - C HLA D/DR/DQ/DP
HHHaaapppllloootttiiipppooo eeexxxttteeennndddiiidddooo RR Alelo RR alelo RR Alelo RR Complotipo RR
1. Diabetes mellitus Tipo I Al 1.6 B8 B18 B62 B7
2.5 2.5 2.5 0.1
DR3
DR4 DR2
4.5
4.5 0.1
(B8, DR3, SCO1, GIo2) (B18, DR3.FIC30) (B6, DR4, SC33). (B7,DR2,SC31) Protector contra diabetes. (B38,DR4,SC21) Judíos
2. Enteropatía sensible al Al 4.0 B8 8.0 DR3 17.0 (B8, DR3, SCO1, GIo2) gluten. Espondilitis enteropática B27 12.0
3. Hepatitis crónica activa B8 9.0 DR3 9.0 4. Esclerosis múltiple A3 1.8 B7 2.5 DR2 4.2 5. Deficiencia del factor C2 (A25, B18, DR2, SCQO)
(A4, B2) 6. Hiperplasia adrenal congénita
(Deficiencia de 21 hidroxilasa) (A3, Bw47, DR7, FC031-0) 7. Nefropatia membranosa
7.1. Inducida por medicamentos a. Sales de oro B8 DR3 34 b. Penicilamina B8 DR3 7.2 No relacionada a drogas B18 DR3 12.0
8. Psicosis Anticuerpos por clorpromazina Bw44
9. Hemocromatosis idiopàtica A3 4.7 B14 4.7 10. Dermatitis herpetiforme B18 DR3 15.4 11. Miastenia gravis B8 2.5 DR3
DQw2 3.7
12 Asma bronquial alérgica al antígeno Ra5 de la ambrosía. Títulos elevados de IgE (B7,SC31,DR2)
13. Vasculitis retiniana DR4 14. Enf. de Buerger A9 B5 15. Inmunodeficiencia de IgA B8 DR3 16. S. de Goodpasture DR2 15.9 17. Psoriasis vulgaris Cw6 13.3 18. Penfigo (Judíos) DR4 14.4 19. S. de Sjögren primario B8 5 DRw52 8.0 20. Enf. de Graves-Basedow DR3 3.7 21. Enf. de Addison idiopatica DR3 6.3 22. S. de Reiter B27 30-35
23. Uveítis anterior B27 10.4 24. Neuritis óptica DR2 2.4 25. Tiroiditis subaguda B35 13.7

Inmunogenética en medicina clínica 31
nos clase I, II y III y haplotipos extendidos con las diferentes enfermedades. Se hace énfasis en que la mayoría de estos haplotipos se encuentran defini-dos en diversos estudios.
En un intento de estudiar e identificar marcado-res del CMH en la respuesta inmune específica contra el virus de hepatitis B (HVB), se realizó la
Tabla 4. (Continuación)
tipificación de los antígenos de las clases I, II y III en personas que recibieron la vacuna de la hepati-tis B. Los resultados mostraron que las personas que no respondieron a la vacunación tenían un incremento en la frecuencia de dos haplotipos ex-tendidos: HLA-B8, DR3, SC01 y HLA-B44, DR7, FC31, lo que sugiere que los genes que controlan
Enfermedad Isotipo
alelo HLA-A
RR HLA
Alelo - B RR
H L A - C alelo RR
HLA D/DR/DQ/DP Alelo RR
Haplotipo extendido Complotipo RR
26. Enfermedades de Behcet B5 6.3 DR2 (Uveítis en tunecinos)
B5 (Turcos) 10 DR4-DR7
(Otrasexpresiones en tunecinos)
Bw51 (Turcos) 15 DR4-DR7-DQw3 Bw51 (Inglés) 2
27. Enfermedad de Buerger A9 B5 28. Enfermedades autoinmunes en
chinos de Hong Kong (A2,Bw46,DRw9) 29. Polimiositis juvenil DR3 19
(Latinoamericanos) DR3 4 (Caucásicos) DR3 13 (Negros) (DRw6 (Negros) DR2/DQwl
30. Polimiositis del adulto B8 5 DR3 7 a. Asociada al anticuerpo Jo-1 DR3/DR6 1 86
DRw52 31. Lupus eritematoso generalizado DR2 3 (DR2, DOwl, C4AQO) (C4AQ0) 6
DR3 3 (DR3, Dqw2, C4AQO) (C4BQ0) a. Anti-Ro (SS-A) B8 6 DR2/DR3 17 (C2Q0) a 1. Títulos altos de anti-Ro DQwl/DQw2 b. Blancos y negros americanos (C4AQ0) 3 c. Homozigotos (C4AQ0) 17 d. Inducido por hidralazina DR4 6 e. Inducido por procainamida DR6 6 f. Madres de niños con lupus neonatal DR3 8 g. S. de Sjögren secund. a lupus DR2 2
32. Esclerosis sistémica progresiva B8 DR5 3 (B8.BFS) (C4BQ0) DR3 2 (C4AQ0) 9
33. Arteritis de células gigantes DR4 3 (C4AQ0, C4B1)
34. Artritis psoriásica B13 4 Cw6 9 B17 6 Bw38 9 Bw39 5
Acta Med Colomb Vol. 15 N°1 ~ 1990
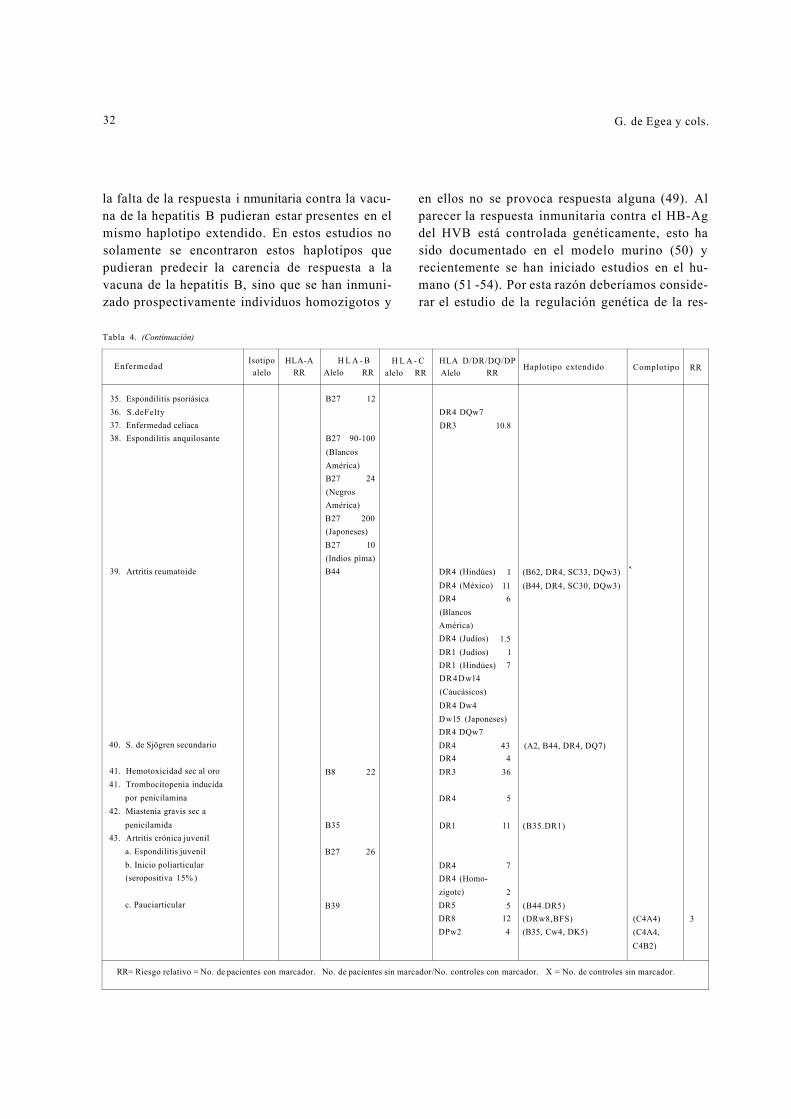
32 G. de Egea y cols.
la falta de la respuesta i nmunitaria contra la vacu-na de la hepatitis B pudieran estar presentes en el mismo haplotipo extendido. En estos estudios no solamente se encontraron estos haplotipos que pudieran predecir la carencia de respuesta a la vacuna de la hepatitis B, sino que se han inmuni-zado prospectivamente individuos homozigotos y
en ellos no se provoca respuesta alguna (49). Al parecer la respuesta inmunitaria contra el HB-Ag del HVB está controlada genéticamente, esto ha sido documentado en el modelo murino (50) y recientemente se han iniciado estudios en el hu-mano (51 -54). Por esta razón deberíamos conside-rar el estudio de la regulación genética de la res-
Tabla 4. (Continuación)
Enfermedad Isotipo
alelo HLA-A
RR H L A - B
Alelo RR H L A - C
alelo RR HLA D/DR/DQ/DP Alelo RR
Haplotipo extendido Complotipo RR
35. Espondilitis psoriásica B27 12 36. S.deFelty DR4 DQw7 37. Enfermedad celiaca DR3 10.8 38. Espondilitis anquilosante B27 90-100
(Blancos América) B27 24 (Negros América) B27 200 (Japoneses) B27 10 (Indios pima)
39. Artritis reumatoide B44 DR4 (Hindúes) 1 (B62, DR4, SC33, DQw3) «
DR4 (México) 11 (B44, DR4, SC30, DQw3) DR4 6 (Blancos América) DR4 (Judíos) 1.5 DR1 (Judíos) 1 DR1 (Hindúes) 7 DR4Dwl4 (Caucásicos) DR4 Dw4 Dwl5 (Japoneses) DR4 DQw7
40. S. de Sjögren secundario DR4 43 (A2, B44, DR4, DQ7) DR4 4
41. Hemotoxicidad sec al oro B8 22 DR3 36 41. Trombocitopenia inducida
por penicilamina DR4 5 42. Miastenia gravis sec a
penicilamida B35 DR1 11 (B35.DR1) 43. Artritis crónica juvenil
a. Espondilitis juvenil B27 26 b. Inicio poliarticular DR4 7 (seropositiva 15% ) DR4 (Homo-
zigote) 2 c. Pauciarticular B39 DR5 5 (B44.DR5)
DR8 12 (DRw8,BFS) (C4A4) 3 DPw2 4 (B35, Cw4, DK5) (C4A4,
C4B2)
RR= Riesgo relativo = No. de pacientes con marcador. No. de pacientes sin marcador/No. controles con marcador. X = No. de controles sin marcador.

Inmunogenética en medicina clínica 33
puesta inmunitaria a la vacuna de la hepatitis B y el estudio de la respuesta en individuos receptores a la vacuna HBV para justificar o descartar con bases moleculares, biológicas e inmunológicas, la implementación de un programa de inmunización contra la HVB en nuestro país.
Cayzer (55), de la Universidad de Brisbane, Australia, ha encontrado una relación dosis-efecto con el sistema HLA en la respuesta a la vacuna de la hepatitis B en el hombre, todos sus pacientes que respondieron tuvieron uno o más de los si-guientes alelis: B7, B8 o B44, Al , A2, A3, mien-tras que en los que no respondieron se encontró la presencia de DR4 y DR7. En las mismas condicio-nes Sasazuki del Departamento de Genética de la Universidad de Kyushu, en el Japón, al estudiar la relación falta de respuesta a la vacuna de la hepati-tis B y haplotipos, encontró que todos los indivi-duos positivos para HLA-Bw54, DR4, DR53, Dqw4, no respondieron al HB-Ag. Ninguno de los linfocitos de los pacientes que no respondieron mostró un buen manejo de la respuesta in vitro al HB-Ag; sin embargo, si se hacía depleción de las células T-CD8 de las muestras de sangre periféri-ca de los pacientes que no respondieron, se obser-vaba un aumento en la producción de anticuerpos (Tabla 5).
Los estudios de regulación genética de la res-puesta humoral HB-Ag en el modelo murino (50, 51, 56), indican que esta respuesta está regulada por genes localizados en el CMH en el ratón. Los mecanismos celulares responsables de la falta de respuesta al HB s-Ag o a la vacuna de la hepatitis B aún no se han delineado muy bien.
Por lo anterior, decimos que la respuesta inmu-
Tabla 5. Genes de inmunosupresión (Is) en el CMH asociados a la falta de respuesta a la vacuna del HBV.
1. HLA-Bw54, DR4 DRw53,DQw2 (Dominante) Japoneses*
2. HLA-B8, SC031, DR3a, DRw52a, DQw2a (Recesivo) HLA-B44, FC31.DR7, DRw53, DQw2 (Caucásicos)
Mediado por células T supresoras *Abstract: Histocompatibility and Immunogenetics Conference. New York, November 18,1987.
ne a un antígeno, en particular el HBs-Ag, fallaría si el macrófago no procesa el antígeno, o lo pre-senta en forma inadecuada a las células T. Por otro lado, las células B pueden perder los receptores necesarios para la interacción con las células T o pudiera ser que estas deficiencias se deban a la ausencia de una clase de clono de células T, el cual potencialmente reaccionaría con un epítope y éste podría ser tan similar a la región variable de los genes de la clase II, que haría que las células T lo reconocieran como propio.
En este orden de ideas se haría un estudio de familias en poblaciones de áreas endémicas con la finalidad de encontrar haplotipos extendidos, aso-ciado a la falta de respuesta a la vacuna, para lo cual se compararía la frecuencia de estos haploti-pos extendidos en los pacientes que responden y en los que no responden y se determinaría la rela-ción con la vacuna. Todo lo anterior, aunado a ensayos de inmunorregulación en la misma mues-tra, daría lugar a resultados que podrán aplicarse a los programas de salud en nuestro país.
Un segundo aspecto toca la asociación de las enfermedades atópicas y los haplotipos extendi-dos. Repasemos brevemente la genética y las bases inmunológicas de la respuesta atópica. La IgE, una glicoproteína con funciones de anticuerpo, tiene dos cadenas pesadas y dos livianas; es la inmuno-globulina con menor concentración en el suero (entre 0.10 y 0.40 u/ml). La propiedad biológica más destacada de esta molécula es la de sensibili-zar tejidos homólogos (57) a través de la capaci-dad de unirse a la membrana de basófilos y células cebadas. Los mastocitos son la mayor fuente de histamina y otros mediadores preformados que participan en los estadios iniciales de los procesos alérgicos.
La reacción alérgica por excelencia se lleva a cabo cuando un alergeno interactúa con dos molé-culas de IgE sobre la membrana del linfocito, de-sencadenando toda una secuencia de eventos bio-químicos y enzimáticos que determinan a la postre los cambios fisiológicos que expresa el cuadro clí-nico. Los eventos anteriormente descritos se resu-men en la Figura 7 (58, 59).
Las enfermedades atópicas, entre ellas el asma
Acta Med Colomb Vol. 15 N°1 ~ 1990

34 G. de Egea y cols.
bronquial alérgica, son el resultado de la interac-ción de múltiples factores, tales como la funciona-lidad del sistema inmunitario, la físiopatología del árbol traqueobronquial y los mecanismos genéti-cos y endocrinos comprometidos en ellas. El con-trol genético del asma es complejo; podemos decir que existen evidencias para considerar la implica-ción de varios genes asociados al CMH. El fenoti-po de la enfermedad también puede ser regulado por niveles de IgE determinados genéticamente; esto puede ser la resultante de un balance entre los genes de la respuesta inmune (Ir) y los genes de inmunosupresión (Is) (localizados en el cromoso-na 6) y los genes del IgE total (localizados en cro-mosomas diferentes al 6).
Los primeros estudios del carácter hereditario del asma fueron realizados por Cooke y Vandzer Veer en 1916 (52-54). Ellos y otros, esclarecieron su predisposición genética (60-64). Recientemen-te se ha venido estudiando la relación de marcado-res genéticos con los posibles mecanismos de he-rencia en las enfermedades atópicas. Al respecto, los investigadores han enfocado sus esfuerzos hacia el sistema HLA; sin embargo, los resultados en la asociación CMH y enfermedad hasta el momento son polémicos (62-65) y en su mayoría fueron es-tudios de población. En contraste, los estudios
familiares (66) mostraron segregación entre un haplotipo del HLA con la sensibilización al Ag RaE de la ambrosía; otros estudios también han aportado los mismos resultados (67-69). Por otro lado la respuesta IgE específica al Ag Ra5 de la ambrosía se encuentra altamente asociado con el DR2 y Dw2 (70, 71).
Los genes Ir e Is que controlan la respuesta IgE específica al Ra5 de la ambrosía, se hallan con frecuencia en el haplotipo HLA-B27, SC31, DR2, DQwl y probablemente se hereden de manera recesiva, produciendo el asma bronquial. Pero la respuesta baja al mismo antígeno se asocia con el haplotipo HLA-B8, SC01, DR3, DRw52, DQw2, y se hereda en forma dominante para producir ri-nitis. (Tabla 6) (72). No sabemos cuál molécula del CMH, clase II, por ejemplo, es la responsable de estas respuestas. Su identificación indicaría que algún subtipo aún desconocido se encontraría en desequilibrio de enlace con estos haplotipos. Cuan-do se describan los haplotipos extendidos que contengan esta clase de genes en las poblaciones colombianas, también será necesario identificarlos subtipos que explicarán las bases moleculares de las respuestas inmunitarias en Colombia.
Interrelación entre los diferentes antígenos y las moléculas de clase I y II en la expresión de la enfermedad
Plantear que la enfermedad es producto de dos categorías amplias: comportamental, en la cual la enfermedad es una falla primaria del organismo como un todo; mecanicista, en la cual la enferme-dad es resultado de una falla específica y una falla ambiental, sin profundizar en los nuevos avances
Tabla 6. Genes Ir e Is asociados al CMH (alergia a la ambrosía "Ragweed").
1. HLA-B27, SC31, DR2, DQwl (Recesivo) Producción de IgE específica contra Ra5 asociada a asma bronquial*.
2. HLA-B8, SC01, DR3, DRw52, DQw2 (Dominante) Baja respuesta de la IgE específica al Ra5 asociada con rinitis alérgjca al Ra5 (IgE) (rinitis alérgica).
* Este mecanismo puede ser ocasionado por la ausencia de células T supresoras. Estos hallazgos requieren confirmación en otros laboratorios de investigación.

Inmunogenética en medicina clínica 35
científicos es tener un concepto muy ortodoxo sobre la enfermedad. Para que la enfermedad se exprese existen una serie de factores de riesgo que nos explican una predisposición, mas no la causa de la misma. Esta predisposición es la que tiene dife-rentes expresiones y una de ellas es la herencia. El polimorfismo genético, las uniones entre los dife-rentes antígenos con las moléculas de clase I y II, la interacción trimolecular entre isotipos del CMH, péptidos o antígenos y el receptor de las células T que producirían implicaciones a nivel de la inmu-norregulación normal, traduciría en la era moder-na de la introspección el concepto moderno de enfermedad, que no es otra cosa sino los múltiples factores interrelacionados que al acoplarse inade-cuadamente producen la enfermedad, pero si los factores antes mencionados realizan el acopla-miento adecuado, se constituye la homeostasis normal. Por ello no estamos de acuerdo con la teo-ría hiperbólica expresada en 1898 por von Behr-ing (73), la cual decía que todas las enfermedades eran producto de diferentes microorganismos, ni con la expresada por Angelí, editor del New En-gland Journal en 1985, quien pensaba que la en-fermedad es producto de la psique (74). Pero sí estamos de acuerdo parcialmente con Sagan (75), quien postula que la longevidad en la última cen-turia debe ser atribuida a la modernización que incluye cambios en la organización social, el con-trol ambiental, control sanitario y dietas, más que al desarrollo de la investigación médica. A través de estos capítulos hemos analizado las diferentes interrelaciones entre las moléculas de clase I y II, los diferentes antígenos implicados en su patogé-nesis y la expresión multifactorial de las enferme-dades sin suscribirnos a meras especulaciones sino con hechos establecidos a nivel experimental.
En la última década el avance de la biomedicina ha sido explosivo debido principalmente al enten-dimiento y conocimiento de nuevas tecnologías como la biología molecular, y su aplicación a los diversos campos de la medicina, como en la inmu-nología, la endocrinología, la inmunogenética y la reumatología; de esta manera se ha facilitado un diagnóstico precoz de las enfermedades, y una comprensión de su fisiopatología lo cual lleva por
supuesto al empleo de un tratamiento más efec-tivo.
Sin embargo a pesar del avance tecnológico en muchas áreas aún desconocemos el antígeno, el papel que éste desempeña en la inmunorregula-ción, su relación inmunogénica con el huésped y la capacidad de éste para reconocer y procesar dicho antígeno. Si el reconocimiento y procesamiento del antígeno es el adecuado, el resultado que pro-duce la inmunorregulación normal es la salud; pero si éste es inadecuado sobreviene la enfermedad.
De acuerdo con la evolución de la medicina, a principios de este siglo era imposible plantear el título de este capítulo, ya que se desconocía el agente causal de la enfermedad y en ocasiones se acuñaban términos que hacían imposible en esa época diferenciar las enfermedades. Por ejemplo, la palabra reumatismo, se refería a múltiples en-fermedades, como: artritis reumatoide, LES, gota, osteoartritis, etc., entidades en las cuales existen antígenos diferentes y fisiopatologías diferentes. En algunas podemos encontrar una base autoin-mune, en otras un defecto enzim§tico o en otras el mimetismo molecular hacia diferentes antígenos como mecanismo causal de la enfermedad por la pérdida de la tolerancia inmunológica. Otro ejem-plo lo encontramos en el caso de las enfermedades infecciosas, en las cuales desde los trabajos de Koch, Pasteur, Vidal, quienes desarrollaron la bacteriología y establecieron un axioma clásico con respecto a la etiología: "una bacteria es igual a una enfermedad infecciosa". Posteriormente con el desarrollo de la dinámica farmacológica apare-ce la era de la antibioticoterapia, logrando contro-lar gran parte de las enfermedades infecciosas, fallando en el caso de los virus.
De esta manera Jenner en 1724 (76) desarrolló el proceso de la vacunación y creó la vacuna como un mecanismo de tipo inmunológico que inducía al huésped a producir anticuerpos contra determi-nados antígenos virales como la viruela; poste-riormente se desarrollaron las vacunas jontra sa-rampión, parotiditis, etc. Muchos virus también evolucionaron, y hoy en día los retrovirus (tipo I, II y III) han burlado el sistema inmunológico, siendo agentes causales de enfermedades tales
Acta Med Colomb Vol. 15 N°1 ~ 1990

36 G. de Egea y cols.
como el SIDA, los linfomas y las leucemias de células T, y la mielopatía espástica del Pacífico colombiano; a pesar de muchos esfuerzos, aún no se ha logrado la vacuna contra dichos agentes.
Igual ocurre con ciertos agentes parasitarios, en los cuales el repertorio antigénico es variable, y los esfuerzos para lograr una vacuna sintética con-tra dichos antígenos (malaria, leishmania, tripano-soma) apenas se están iniciando. Otras áreas en cuales el papel del antígeno no está definido son el cáncer y las enfermedades autoinmunes. A finales de la década de los 80 y comienzos del 90 debe-mos plantearnos una pregunta importante: ¿cuál es el antígeno que induce la expresión de genes que van a iniciar una respuesta celular y humoral con subsecuente elaboración de una variedad de mediadores y efectores de una respuesta; enzimas, inmunoglobulinas, proteínas del complemento, factores de crecimiento celular que contribuyen a la pérdida de la inmunorregulación, y de esta ma-nera inducen a una proliferación celular ocasio-nando las diversas manifestaciones en los tejidos y órganos, produciendo las diferentes enfermeda-des de acuerdo al antígeno causal?
De acuerdo con las características inmunoge-néticas del huésped y su interacción con el antíge-no causal, la expresión de la enfermedad podría ser secundaria a: 1. Un agente infeccioso. 2. Una estimulación antigénica persistente, (detritus de pared celular bacteriana). 3. Mimetismo molecu-lar. 4. Defecto enzimático. 5. Autoinmunidad. 6. Proliferación celular anormal (neoplasias).
Por lo tanto los esfuerzos de la investigación biomédica deben dirigirse a la identificación del antígeno causal, el cual produce epifenómenos como la autoinmunidad, antígenos aberrantes de clase I y II con pérdida de la tolerancia inmunoló-gica, inflamación aguda y crónica, proliferación celular, trastornos de la inmunorregulación, ela-boración de citoquinas, etc.
Otro de los problemas difíciles en la investiga-ción biomédica ha sido el relacionado con la de-tección de los antígenos implicados en la autoin-munidad, en la cual diferentes antígenos celulares hacen parte de las 10.000 o más proteínas que se encuentran en la célula humana y solamente 30 ó
40 son blancos comunes de la autoinmunidad. Pero con las técnicas de cultivo celular, hibridomas, anticuerpos monoclonales, inmunofluorescencia y clonación celular se han logrado caracterizar va-rios antígenos especialmente en pacientes con enfermedades endocrinas, infecciosas y reumáti-cas, que podrían facilitar el tratamiento futuro con vacunación, o a través de anticuerpos antiidiotípi-cos. En este capítulo sólo analizaremos los antíge-nos que induzcan trastornos en la inmunorregula-ción de un huésped susceptible inmunogenética-mente y que esté bien definida su asociación con los isotipos de las moléculas de clase I y II del CMH; por lo tanto los antígenos de tipo infeccioso o tumoral no son parte del objetivo de este capítu-lo; para ello queremos analizar además algunos mecanismos inmunológicos inducidos por el antí-geno y su interrelación con el HLA para producir la enfermedad.
Mimetismo molecular Se ha sugerido que las infecciones virales pue-
den producir una enfermedad autoinmune, para ello se planteó la posibilidad de que un determinante antigénico sobre un virus simule determinantes antigénicos a nivel de los tejidos del huésped, y que éste produzca anticuerpos contra los determi-nantes virales, y de esta manera dichos anticuer-pos reaccionarán contra las células, tejidos o es-tructuras celulares del huésped. Esta teoría ha sido denominada del mimetismo molecular, debido a que el huésped es incapaz de diferenciar lo propio de lo no-propio.
El mimetismo molecular se define como un mecanismo biológico en el cual se comparten di-ferentes epítopes de microorganismos de diferen-tes especies, con epítopes de proteínas normales del huésped, induciéndose a través de un microor-ganismo invasor una respuesta inmune de reac-ción cruzada contra diferentes tejidos ocasionan-do la autorreactividad, autoinmunidad o la infla-mación. El epítope o el sitio de la célula normal que reacciona con el anticuerpo producido por el microorganismo no se conoce.
El mimetismo molecular se ha estudiado prin-cipalmente en virus DNA y RNA. Muchos virus

Inmunogenética en medicina clínica 37
comparten sitios antigénicos con componentes celulares normales del huésped; estas característi-cas comunes han sido demostradas por compara-ción directa de secuencias de aminoácidos linea-les, conformando una estructura homóloga con el huésped; casi siempre la secuencia de aminoáci-dos que produce la reacción cruzada se limita a unos pocos aminoácidos (77,78). Oldstone y Fuji-nami (79) observaron que sólo seis aminoácidos fueron suficientes para producir reactividad cru-zada, ya que los anticuerpos monoclonales utiliza-dos reconocen sólo unos pocos aminoácidos. El hecho de encontrar anticuerpos monoclonales que reaccionan con constituyentes del huésped y del virus sugiere que el virus tiene el potencial de dis-parar una respuesta inmune y ocasionar una enfer-medad, y que una vez iniciado el proceso no nece-sariamente se va a encontrar el virus. También se ha demostrado que además de los anticuerpos, los linfocitos y las linfotoxinas generadas contra el virus pueden reaccionar contra proteínas propias y así de esta manera no es necesaria la presencia de una partícula viral.
Lane y Hoeffler (80) utilizando anticuerpos monoclonales, mostraron que los antígenos T del virus 40 del simio, y las proteínas normales de células del huésped tienen sitios antigénicos co-munes. Oldstone y col (77) encontraron que la fosfoproteína P3 del virus del sarampión, la pro-teína de 140 kilodaltons del virus del herpes, y la hemaglutinina del virus de la vacinia reaccionan con diferentes epítopes sobre filamentos interme-dios o intracitoplásmicos; este mismo grupo ha demostrado que péptidos virales con secuencias de aminoácidos homólogos a proteínas propias del huésped pueden inducir anticuerpos que reaccio-nan en forma cruzada y células linfoides que pro-liferan en respuesta a las proteínas propias, de esta manera se genera una respuesta inflamatoria in vivo en el sitio de la localización de la proteína propia. Para demostrar este fenómeno, Oldstone y col (79) analizaron la homología en la secuencia de la pro-teína básica de la mielina (MBP) y la polimerasa del virus de la hepatitis B (HBVP). Al practicar un análisis computarizado se demostró una secuencia de aminoácidos similar entre estas dos proteínas, y
Al inmunizar siete conejos con HB VP utilizan-do ocho aminoácidos, cinco de ellos desarrollaron niveles de anticueipos contra la MBP; estos títulos de anticuerpos oscilaron entre 5.5 y 8.7. Cuando tales anticuerpos se probaron contra el péptido de HBVP los títulos oscilaron entre 10.4 y 14.5.
Además se pudo observar la generación de an-ticuerpos y la presencia de linfocitos que reaccio-naban en forma cruzada con los péptidos virales y MBP, con producción de infiltrados a nivel del SNC de los conejos. El sitio encefalitogénico in-ducido por la MBP es un determinante de las célu-las del huésped, que a la vez ha sido mapeado en varias especies de animales; este es un ejemplo clásico de autorreactividad y autoinmunidad en enfermedades asociadas a la desmielinización (77-83).
El mecanismo patogénico posible es la genera-ción de linfocitos efectores que reaccionen en for-ma cruzada, o de anticuerpos que reconocen de-terminantes antigénicos específicos sobre células o tejidos blanco. Aun cuando los glicopéptidos de virus participan en estos eventos, las proteínas vi-rales se encuentran dentro de las células y no se expresan a nivel de la membrana celular, pudién-dose inducir la generación de linfocitos T citotóxi-cos durante las infecciones experimentales y natu-rales, y a la vez este tipo de daño inmunopatológi-co no requiere la presencia del microorganismo causal. En ocasiones el microorganismo podría ser depurado, pero los componentes del ataque inmu-ne continúan lesionando los elementos propios, que
Acta Med Colomb Vol. 15 N°1 ~ 1990
se encontró una similitud de las proteínas antes mencionadas con las nucleoproteínas y hemaglu-tininas del virus de la influenza, la cápside del vi-rus del polioma, la proteína central del adenovi-rus, la poliproteína de Rouss del sarcoma de las aves, el virus de la leucemia de Abelson y la pro-teína EC-LF2 del virus de Epstein-Barr. Ejemplo:

38 G. de Egea y cols.
a la vez liberarían los antígenos propios o neoantí-genos, continuándose de esta manera el daño tisu-lar. Un ejemplo clínico clásico lo observamos en las encefalopatías virales en humanos, en las cua-les el agente induce el daño inmunológico, pero rara vez se aisla el agente causal.
En estudios recientes realizados en pacientes con sarampión, quienes han padecido encefalitis viral o panencefalitis esclerosante subaguda, se han obtenido células mononucleares de sangre perifé-rica y del líquido cefalorraquídeo, que proliferan cuando se cultivan con la proteína básica de la mielina. Este trabajo in vitro corrobora otro punto para explicar los mecanismos patogénicos induci-dos por la proteína básica de la mielina. Finalmen-te aplicando la teoría de la red de idiotipo-anti-idiotipo se especula que la autoinmunidad ocurre cuando existen determinantes virales que tienen una configuración molecular en espejo con la del huésped pudiendo de esta forma inducir la enfer-medad (77-83).
1. Producción de anticuerpos contra el HLA-B27. La espondilitis anquilosante y el síndrome de Reiter son dos enfermedades reumáticas de etiolo-gía desconocida, que tienen en común el hecho de tener una susceptibilidad genética relacionada con el HLA-B 27. A pesar de ello existen casos de gemelos idénticos que difieren en la susceptibili-dad de la espondilitis anquilosante. Existe otro hecho y es que el síndrome de Reiter se encuentra precedido por un discreto episodio de alguna in-fección de tipo gastrointestinal o genital; entre los organismos que más se le asocian encontramos: Salmonella, Shigella, Yersinia en el Reiter y la Klebsiellapneumoniae en la espondilitis (84-86).
Se ha observado que existe una alta incidencia de Klebsiella en la flora intestinal de individuos con espondilitis y que esto coincide con la exacer-bación de la enfermedad; también se ha encontra-do un incremento de los niveles de IgA contra Klebsiella (87).
Mientras que 80% de los casos con síndrome de Reiter que tienen compromiso axial son B27+, solamente el 7% de los controles sanos tienen el B27+ por lo tanto los individuos B27+ tiene un alto riesgo de contraer la enfermedad. Posiblemente
genes localizados dentro del complejo del HLA, codifican para una respuesta inmune que produce susceptibilidad parala enfermedad (88).
Después de analizar la secuencia proteica del Database (Dayhoff Databank) se notaron varias secuencias que comparten el HLA-B27 y la Kleb-siella pneumoniae", entre estas secuencias se en-contró una mejor homología entre el HLA-B27, en los residuos 72-79 y. los residuos 188-193 de la Klebsiella. Mediante la técnica de ELISA se de-mostró que siete de 24 sueros, 29% de los pacien-tes con espondilitis anquilosante, presentaban anti-cuerpos que se unían a péptidos sintéticos que re-presentan los residuos 69-84 del HLA-B27, y con-tienen los residuos homólogos de la Klebsiella (89). En contraste, ninguno de los 22 individuos HLA-B27 sanos ten²an el mismo anticuerpo (p < 0,01); 34 de los individuos HLA-B27 con síndrome de Reiter tenían el anticuerpo que se une al péptido sintético de los residuos 69-84. Dicho anticuerpo está dirigido contra la región hipervariable del HLA-B27 que comparte regiones homologas con la Klebsiella pneumoniae (90). Estos resultados sugieren que una respuesta inmune dirigida ini-cialmente durante la infección contra Klebsiella, también reacciona contra la secuencia homóloga del HLA-B27, produciendo de esta forma una res-puesta contra determinantes del huésped (91), como se puede apreciar en la Tabla 7. A pesar de que la teoría del mimetismo molecular explica en algo la autoinmunidad al compartir algunos aminoácidos homólogos entre el huésped y el microorganismo y el hecho de que esta autoinmunidad pueda per-petuarse sin la presencia del germen causal, debi-do a la reactividad cruzada que esta hace con el huésped, deja muchos interrogantes tales como: 1. En la mayoría de los grupos étnicos estudiados los individuos con espondilitis anquilosante son B27+ (más del 85% de los individuos estudiados), pero la enfermedad sólo ocurre en menos del 50% de los individuos B27+. ¿Qué pasa con el resto de estos individuos? 2. La distribución limitada de la población HLA B27 + (blancos, 8%; negros, 1%). 3. La discordancia de la enfermedad en gemelos idénticos. 4. La relación de agentes infecciosos que afectan la flora intestinal como las bacterias

Inmunogenética en medicina clínica 39
Klebsiella pneumoniae, Escherichia coli, Shige-lla, Salmonella con espondilitis anquilosante, sín-drome de Reiter y artritis reactiva.
Todas estas posibilidades antes mencionadas suponen la posibilidad de que exista un factor que interactúe con las células B27+ in vivo y desenca-dene la expresión de la espondilitis anquilosante (90,91).
2. Enfermedades virales. Estudios previos sugieren la posibilidad de que las infecciones vira-les podrían producir una enfermedad autoinmune. Se ha observado que algunos determinantes anti-génicos de un virus pueden simular los determi-nantes antigénicos del huésped. En estos casos el anticuerpo se produce contra los determinantes virales y puede reaccionar contra las células del huésped. Srinivasappa y col produjeron anticuer-pos monoclonales dirigidos contra Coxackie B, dengue, virus de la encefalitis japonesa, coriome-ningitis linfocítica, sarampión, rabia, estomatitis vesicular, citomegalovirus, herpes simple y vacci-nia. Los hibridomas se generaron después de in-munizar ratones Balb/c con dichos virus, y los esplenocitos de ratón fueron fusionados con célu-las de mieloma múltiple; cada uno de los hibrido-mas fue clonado, y la actividad viral se determinó por un tamizado de los sobrenadantes de los hibri-domas utilizando para ello varias técnicas tales como: neutralización viral, radioinmunoensayo, ELISA, hemaglutinación, inmunoíluorescencia e inmunoprecipitación. La actividad anti-tisular se obtuvo por un tamizado de los hibridomas clona-dos por IFI con tejidos normales como cerebro, pituitaria, glándulas salivares, tiroides, tráquea, timo, corazón, glándula adrenal, riñon, páncreas, testículo, ovario, estómago e intestino. De los 635 anticuerpos monoclonales del screening 21 fueron
positivos contra 10 ó más tejidos. Varios de estos anticuerpos reaccionaron contra múltiples órga-nos y reconocieron a la vez más antígenos en otros órganos.
Este trabajo aporta la posibilidad de que exista un anticuerpo contra un agente infeccioso y que dicho anticuerpo reaccione contra tejidos norma-les del huésped; a pesar de ser una teoría nueva, aún no existe una prueba definitiva (92).
3. Filamentos intermedios. Al utilizar anti-cuerpos monoclonales contra la fosfoproteína del virus del sarampión y la proteína del virus del her-pes simple tipo I se ha observado que reaccionan en forma cruzada contra una proteína de los fila-mentos intermedios, como es la vimentina, la cual tiene un peso molecular de 52.000, mientras que el peso molecular de la fosfoproteína es de 70.000 y la proteína del herpes es de 146.000; tanto la fos-foproteína del virus del sarampión como la proteí-na del virus del herpes, sólo reaccionan con los diferentes determinantes antigénicos del filamen-to intermedio, la proteína del virus del sarampión no reacciona contra la del herpes (77).
Los anticuerpos contra el citoesqueleto como en el caso de la vimentina, desmina y citoquerati-na, podrían jugar un papel en la patogénesis de las enfermedades autoinmunes; sin embargo algunas evidencias indirectas no indican que esté relacio-nada con los virus (93,94). Estos anticuerpos diri-gidos contra el esqueleto cada vez se estudian más en enfermedades como el LES.
4. Enfermedades desmielinizantes. Existen algunas evidencias que sugieren que la etiología de la esclerosis múltiple y otras enfermedades desmielinizantes tienen el antecedente de una in-fección viral asociada a factores autoinmunes. Un modelo experimental para explicar este fenómeno
Tabla 7. Homología y reacción cruzada entre HLA y klebsiella pneumoniae nitrogenasa.
Péptido Origen Localización del No. de residuos
Secuencia de aminoácidos Anticuerpo Reactividad
1. 31 HLAB27.1 6 9 - 8 4 AKAQTDREDLRTLLRY ++++ 0.940
2. 45 Klebsiella nitrogenasa 185 - 196+ NSRQTDREDELIGGC + + 0.259
3. 43 HLAB27.1 6 7 - 8 4 CKAKAQTDREDLRTLLRY ++++ 1.200
4. 46 HLAB27.1 68 - 83+ KAKAQTDREDLRTURGGC +++ 0.603
Acta Med Colomb Vol. 15 N°1 ~ 1990

40 G. de Egea y cols.
ha sido el desarrollo con la enfermedad alérgica experimental (EAE), la cual se puede producir por inmunización en animales de laboratorio con teji-dos del sitema nervioso central, o con proteina básica de la mielina (MBP). Fujinami y Oldstone (77-79) han informado que la MBP y la polimera-sa del virus de la hepatitis B producen lesiones de EAE en los conejos. Una de las proteínas más abundantes del sistema nervioso central es la pro-teolípido de mielina, la cual se encuentra embe-bida en la membrana de la mielina; su secuencia de aminoácidos ha sido analizada y muestra fuer-tes evidencias que sugieren similitud entre seg-mentos de proteínas-proteolípidos de los virus que infectan a los humanos. Así de esta forma los virus podrían inducir: a) fenómenos autoinmunes por el hecho de compartir determinantes antigénicos con moléculas de las membranas celulares normales de los tejidos y la inducción de anticuerpos contra estos tejidos normales (mimetismo molecular), b) la alteración de la inmunorregulación y estimula-ción de células T citotóxicas, c) liberación de antí-genos secuestrados, d) formación de neoantígenos y e) susceptibilidad genética.
5. Carditis reumática y miocardiopatía por Tripanosoma cruzi. Una de las mejores eviden-cias para explicar el mimetismo molecular fue realizada por van de Rijn y col (95) al absorber suero de pacientes con el antígeno adecuado. Esta evidencia se obtuvo en sueros de pacientes con carditis reumática, al observar que el anticuerpo contra el estreptococo beta hemolítico reaccionó con corazón normal y con corazón de pacientes con enfermedad de Chagas. Los anticuerpos con-tra el Tripanosoma cruzi reaccionan contra cora-zón normal y nervios. Khoury y col (96), utilizan-do anticuerpos monoclonales contra estreptoco-cos y Tripanosoma cruzi, Cunnigham y col (97) y Woody y col (98) han probado que éstos reaccio-nan en forma cruzada contra los tejidos normales.
Autoinmunidad órgano-específica Casi siempre esta patología se encuentra aso-
ciada a la autoinmunidad endocrina, y clínicamen-te se encuentra relacionada con la afección de una o varias glándulas debido a la presencia de un
autoanticuerpo dirigido contra la membrana celu-lar de los diferentes tejidos glandulares o compo-nentes citoplásmicos de las células. La respuesta innunológica dirigida contra el determinante anti-génico o epítope del tejido blanco es única, órga-no-específica e incluso célulo-específica.
Casi simultáneamente con el reconocimiento de los anticuerpos anti-nucleares al comienzo de la década de los 50, Roitt y col (99) en 1956 demos-traron la base autoinmune de la tiroiditis de Hashi-moto y posteriormente se pudo demostrar este concepto en las gastritis atrófica, anemia pernicio-sa, enfermedad de Addison y síndrome de Schmidt (100, 101). 18 años después de esta descripción, Botazzo y col (102) demostraron por vez primera la presencia de anticuerpos contra la célula de los islotes (ICA) y posteriormente en 1975 (103) se describieron los anticuerpos contra la hipófisis. Hasta el momento se han descrito anticuerpos contra casi todas las glándulas endocrinas excepto la pineal. La presencia de estos anticuerpos produ-ce en la glándula : síntomas clínicos o subclínicos, infiltración linfo-monocitaria en la glándula co-rrespondiente y alteración de la secreción hormo-nal (101).
Las endocrinopatías autoinmunes pueden ocu-rrir solas o en combinación. Se clasifican en dos tipos: tipo I caracterizado por hipoadrenalismo, hipoparatiroidismo idiop§tico y candidiasis muco-cutánea crónica. Tipo II o síndrome de Schmidt que se caracteriza por hipoadrenalismo primario idiop§tico, diabetes mellitus tipo I, e hipogonadis-mo primario idiop§tico (104). En ocasiones en el tipo II se incluye la enfermedad de Graves y la tiroiditis atrofica. Este tipo de patología se asocia conelHLA-B8 y DR3 en el caucásico (105). Otras manifestaciones autoinmunes se asocian a las endocrinopatías inmunes tales como vitíligo, mias-tenia gravis, enfermedad cel²aca, anemia pernicio-sa y serositis.
La autoinmunidad órgano-específica puede comprometer órganos blancos a través de varios mecanismos:
a) Un proceso destructivo lento. En este tipo de patología el parénquima es invadido por linfocitos

Inmunogenética en medicina clínica 41
activados y citotóxicos y anticuerpos dirigidos contra determinantes antigénicos de la membrana celular. Como resultado de este tipo de agresión se produce pérdida de los tejidos normales y en oca-siones se reemplaza por tejido conectivo; como consecuencia de ello se produce la pérdida de la función fisiológica. Como ejemplos encontramos anticuerpos contra los enterocitos (EC-Ab) en la enfermedad de Crohn; auto-anticuerpos contra las células de la mucosa, colitis ulcerativa; en la dia-rrea idiop§tica, la tiroiditis de Hashimoto, mixe-dema primario, gastritis fúndica (tipo A), gastritis antral (tipo B), enfermedad de Addison, insufi-ciencia gonadal, hipoparatiroidismo idiop§tico, diabetes mellitus autoinmune, vitíligo, alopecia areata y alopecia universal (100-104).
b) Por estimulación de receptores. En este tipo de patología se observa la presencia de anticuer-pos que reaccionan contra los receptores hormo-nales sobre la superficie celular, mimetizando al-gunos de los efectos de las hormonas tróficas. Como resultado final de este tipo de mecanismo estos anticuerpos tienen la capacidad de producir: 1. Hormonas T3 y T4 con hipertiroidismo en la enfermedad de Graves. 2. Estimulación del creci-miento. Ejemplo: bocio o hiperplasia (99). 3. Pro-ducción de péptidos específicos y mediadores quí-micos (106).
Adams, en 1956 (107), demostró en el suero de un paciente con hipertiroidismo el "long acting thyroid stimulator (LATS)" de la enfermedad de Graves. 30 años después se ha podido documentar un anticuerpo que estimula la glándula tiroides (TSAb), que se encuentra dirigido contra el recep-tor de TSH (TSH-R), que a la vez mimetiza la ac-ción de la TSH, estimulando la producción de T3 y T4. Doniach en 1976 (108) y Drexhage en 1980 (109) han demostrado la presencia de inmunoglo-bulinas que producen el crecimiento de los órga-nos y por ende la formación del bocio.
A nivel del tracto gastrointestinal se han produ-cido auto-anticuerpos que inducen hipersecreción de gastrina y a la vez hipertrofia de las células parietales.
c) Producción de auto-anticuerpos bloqueado-res. En este tipo de patología se ha observado la
presencia de anticuerpos que compiten con los receptores hormonales u otros mediadores. Esto se explica a través de la imagen en espejo de la teoría de la red idiotipo-anti-idiotipo. Un ejemplo clásico de este mecanismo es el de mixedema pri-mario en todas las edades, la tiroiditis atrófica y la posibilidad de anticuerpos que atraviesan la pla-centa y son responsables de algunos de los casos de cretinismo (101, 110-114). Entre otras enfer-medades en las cuales se han descrito anticuerpos antibloqueadores, tenemos: diabetes mellitus aso-ciada a acantosis nigricans, anticuerpos contra los receptores de la paratohormona en algunos casos de insuficiencia renal, anticuerpos contra los re-ceptores ɓ2 adrenérgicos en asma; o los receptores de la acetilcolina en el caso de la miastenia gravis. Anticuerpos contra los receptores de la gastrina (gastritis fúndica), contra los receptores de las gonadotrofinas (insuficiencia gonadal).
Uno de los mecanismos implicados en la pato-génesis de estas enfermedades es la presencia de anticuerpos anti-receptores, debido a los anticuer-pos anti-idiotipo relacionado con la presencia de anticuerpos antirreceptor hormonal. Es decir, el idiotipo del anticuerpo antirreceptor hormonal pudiera ser el sustrato para la generación de anti-cuerpos anti-idiotipos que tienen la misma imagen del anticuerpo antirreceptor (101,110-114). En la Figura 8 se explica la teoría de la imagen interna de la red idiotipo-anti-idiotipo y las diferentes en-fermedades relacionadas con los anticuerpos anti-rreceptores.
Alteración aberrante de los antígenos de clase I y II
Estudios realizados por Pujol-Borrell y col en 1983, al utilizar preparados de células tiroideas humanas estimuladas con fitohemaglutinina y otras lectinas, mostraron la propiedad de las células epi-teliales o tirocitos de expresar antígenos de clase II, ya que normalmente estas células son HLA cla-se II negativas.
Simultáneamente Hanafusa y col (116) en 1983, en pacientes con tiroiditis de Hashimoto y enfer-medad de Graves, encontraron un incremento en los antígenos de clase I. Estudios realizados por
Acta Med Colomb Vol. 15 N°1 ~ 1990

42 G. de Egea y cols.
Botazzo y Feldman demostraron que el interfón y (IFN) a la dosis de menos de 1 μ/ml es efectiva en inducir la expresión de antígenos de clase I y II en los tirocitos, sin embargo, ni la IL-2 ni el interferon a ó (3 incrementan la expresión de dichos antíge-nos (101, 117). Todd y col (118), utilizando dosis bajas 5 a 10 Õ/ml de ɔ-IFN inducen la expresión de DR pero no de las subregiones DP o DQ. Pero si los tirocitos se cultivan con 500 Õ/ml de ɔ-IFN se expresan todas las subregiones, es decir, DR, DP y DQ. Utilizando además ɔ-IFN asociado al factor de necrosis tumoral alfa o a la linfotoxina hay ex-presión de los antígenos de clase II, pero el factor de necrosis tumoral alfa o beta por sí solo induce la expresión de los antígenos de clase II.
Por lo anteriormente analizado, se plantea la posibilidad de que una inapropiada expresión de las moléculas de clase II por las células epiteliales, sea capaz por sí misma de expresar sus moléculas de superficie y de esta manera simular ser célula presentadora de antígeno, lo que provocaría la producción de células T reactivas, causando el efecto inflamatorio.
Botazzo, Londei y Feldman (101, 115-121) describen tres tipos de enfermedades en las cuales existe una expresión aberrante de los antígenos de clase I y II. Un primer grupo conformado por en-docrinopatías autoinmunes que afectan la glándu-la tiroides y el páncreas (tiroiditis autoinmune y
diabetes mellitus insulino dependiente). Un se-gundo grupo lo conforman las enfermedades in-flamatorias del intestino y un tercer grupo en el cual existen anticuerpos órgano-específico contra las mitocondrias en la cirrosis biliar primaria y contra las ribonucleoproteínas Ro/SS-A y La/SS-B en el síndrome de Sjögren primario. En estas enfermedades es posible que las células ductales epiteliales expresen antígenos mitocondriales en la superficie celular.
Finalmente la expresión aberrante de las molé-culas de clase II por los órganos blancos de los antígenos de clase I y II y la complejidad de la regulación epitelial de la expresión de los antíge-nos de clase I y II, explican la heterogeneidad del curso clínico de estas enfermedades. Es posible que exista un factor iniciador que no conocemos, causal de la expresión aberrante de dichos antíge-nos y del trastorno de la inmunorregulación.
Anticuerpos antinucleares y antígenos nucleares extraíbles
El objetivo de involucrar algunos antígenos como los filamentos intermedios, los anticuerpos antinucleares, los antígenos nucleares extraíbles y la cardiolipina dentro de esta revisión, tiene una connotación más que histórica pues existe una re-lación estrecha entre estos dos antígenos y las moléculas de clase I y II y los serotipos del com-plemento (122).
¿Qué son los anticuerpos antinucleares (ANA)? Es pregunta habitual de los estudiantes, médicos y residentes cuando están frente a una enfermedad autoinmune. Desde hace 40 años, uno de los obje-tivos de la investigación ha sido determinar la es-pecificidad de estos anticuerpos para delinear un grupo o subgrupo de enfermedades, y así de esta manera se desarrollaron una variedad de pruebas serológicas para la detección de estos anticuerpos.
¿Cuál es el papel de este antígeno y su interac-ción con los antígenos de clase I y II? Precisamen-te este antígeno o antígenos, son el blanco de los anticuerpos y de esta manera se forman los com-plejos inmunes circulantes que tienen un papel patogénico en la expresión de enfermedades como lupus, enfermedad mixta, etc., en un individuo

Inmunogenética en medicina clínica 43
genéticamente susceptible. Así de esta forma surge la célula LE, siendo la
primera prueba serológica para la detección del lupus (123). Su origen se remonta a 1945, cuando Hargraves de la Mayo Clinic, visitó a Scheicher y Sharp del Minneapolis General Hospital y les en-señó una técnica para encontrar células de la mé-dula ósea; y en 1948 con Morton, un residente de medicina, publicó la descripción de la célula LE utilizando para ello la técnica de la concentración e incubación y no el examen directo. Haserick (124) descubrió que el suero de pacientes con lupus pro-ducía el fenómeno LE cuando se incubaba con médula ósea normal; posteriormente se emplea-ron polimorfonucleares. En un artículo clásico, Coons y Kaplan (125) en 1950, demostraron que el antígeno podía ser detectado por anticuerpos fluorescentes. En 1957 Friou (126) y col y Barda-wil y col (127) escribieron un artículo demostran-do la aplicación clínica de los anticuerpos fluores-centes o del ANA (FANA). En 1961 Beck (128) describe los diferentes patrones de inmunofluo-rescencia. Estos estudios de los anticuerpos anti-nucleares marcaron un hito histórico en el desa-rrollo y conocimiento de las enfermedades auto-inmunes.
Otro tipo de antígeno fue reconocido en 1959 por Deicher, Holman y Kunke (129), utilizando técnicas de inmunodifusión, detectaron los antíge-nos nucleares extraíbles o ENA. Posteriormente se emplearon métodos bioquímicos como protea-sas y nucleasas para digerir antígenos; esto se rea-lizó como un medio para determinar si los antíge-nos de interés reaccionaban con proteínas y ácidos nucleicos, por ello se logró demostrar que la ma-yoría de los anticuerpos.de interés reaccionaban contra proteínas, y así se demostró que la ribonu-cleoproteína o RNP era un antígeno sensible a la ribonucleasa, mientras que el antígeno Sm (antí-geno Smith de un individuo con lupus) no era sen-sible (130).
A pesar de los grandes avances de la inmunolo-gía poco se sabía acerca de la función de estos antígenos que reaccionaban con los anticuerpos; sólo hasta 1979 Lemer y Steitz (131) utilizaron suero de pacientes que inmunoprecipitaba con
antígenos radiomarcados. En este procedimiento el RNA se marcó con P32 y la proteína con sulfuro de metionina 35; se logró documentar que la ma-yor parte de los ENA está conformada por RNA y ribonucleoproteína. La subdividieron en dos gru-pos: Snurps = (Small nuclear ribonucleoproteins) y el otro grupo Scyrps = (Small cytoplasmic ribo-nucleoproteins).
Usualmente el anticuerpo reacciona contra el determinante antigénico sobre el componente pro-teico de esas partículas o sus complejos. Por ejem-plo, el Snurp es la partícula que porta el determi-nante RNP, y está compuesto de una sola pieza de RNA llamada Ul , la cual se encuentra rodeada de nueve proteínas denominadas 68K, A, B, B', C, D, E, F, G. Generalmente los anticuerpos anti-RNP reaccionan con la proteína 68K, A y C del Snurp (131-134).
El antígeno Sm se encuentra usualmente sobre la proteína B y B' de una serie de Snurps. Los Snurps que llevan el antígeno Sm contienen los RNA lla-mados Ul , U2, U4, U5 y U6. Parece que la partí-cula a la cual se une el anticuerpo anti-RNP es un subgrupo de las partículas a las cuales se une el anti-Sm (135). Otra serie de antígenos ENA son el Ro y La que antiguamente se conocían como Ro/ SS-A y La/SS-B que se encuentran presentes en un complejo de RNA y proteína. Posiblemente las partículas Ro y La están comprometidas en el pro-cesamiento del RNA del núcleo al citoplasma. La nucleoproteína La es una fosfoproteína que se asocia con la transcripción de la RNA III polime-rasa. Se ha demostrado que algunos autoantígenos SJT, SS-B y Ha, son autoantígenos determinantes que se encuentran en la porción proteica de la molécula, son macromoléculas similares al antí-geno La (136, 137).
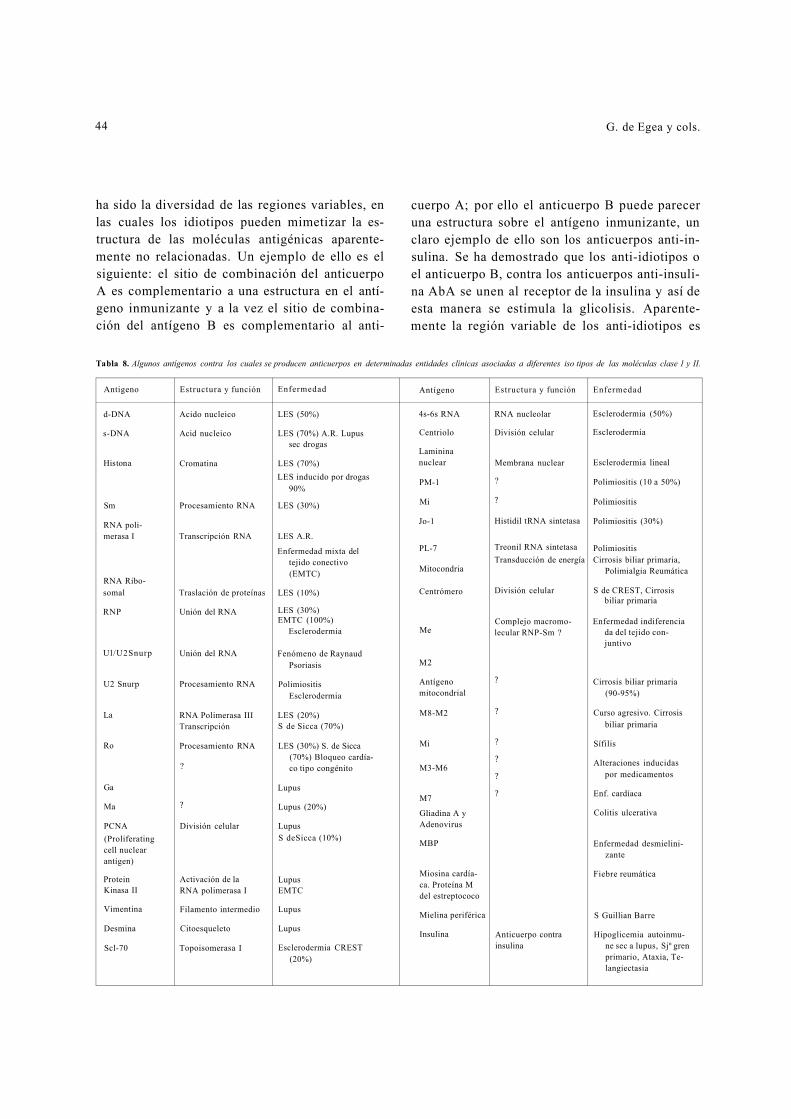
A continuación elaboramos una lista de algu-nos antígenos que se encuentran definidos y que hacen parte de algunas entidades clínicas y que se asocian a diferentes isotipos de las moléculas de clase I y II (Tabla 8).
Red de idiotipos anti-idiotipos De acuerdo con la teoría de Jerne, uno de los
problemas en el proceso de la inmunorregulación
Acta Med Colomb Vol. 15 N°1 ~ 1990
44 G. de Egea y cols.
ha sido la diversidad de las regiones variables, en las cuales los idiotipos pueden mimetizar la es-tructura de las moléculas antigénicas aparente-mente no relacionadas. Un ejemplo de ello es el siguiente: el sitio de combinación del anticuerpo A es complementario a una estructura en el antí-geno inmunizante y a la vez el sitio de combina-ción del antígeno B es complementario al anti-
cuerpo A; por ello el anticuerpo B puede parecer una estructura sobre el antígeno inmunizante, un claro ejemplo de ello son los anticuerpos anti-in-sulina. Se ha demostrado que los anti-idiotipos o el anticuerpo B, contra los anticuerpos anti-insuli-na AbA se unen al receptor de la insulina y así de esta manera se estimula la glicolisis. Aparente-mente la región variable de los anti-idiotipos es
Tabla 8. Algunos antígenos contra los cuales se producen anticuerpos en determinadas entidades clínicas asociadas a diferentes iso tipos de las moléculas clase 1 y II.
Antigeno Estructura y función Enfermedad Antígeno Estructura y función Enfermedad
d-DNA Acido nucleico LES (50%) 4s-6s RNA RNA nucleolar Esclerodermia (50%)
s-DNA Acid nucleico LES (70%) A.R. Lupus Centriolo División celular Esclerodermia
Histona Cromatina
sec drogas
LES (70%) LES inducido por drogas
90%
Laminina nuclear
PM-1
Membrana nuclear ?
Esclerodermia lineal
Polimiositis (10 a 50%)
Sm
RNA poli-merasa I
RNA Ribo-somal
RNP
Ul/U2Snurp
Procesamiento RNA
Transcripción RNA
Traslación de proteínas
Unión del RNA
Unión del RNA
LES (30%)
LES A.R.
Enfermedad mixta del tejido conectivo (EMTC)
LES (10%)
LES (30%) EMTC (100%)
Esclerodermia
Fenómeno de Raynaud Psoriasis
Mi
Jo-1
PL-7
Mitocondria
Centrómero
Me
M2
?
Histidil tRNA sintetasa
Treonil RNA sintetasa Transducción de energía
División celular
Complejo macromo-lecular RNP-Sm ?
Polimiositis
Polimiositis (30%)
Polimiositis Cirrosis biliar primaria,
Polimialgia Reumática
S de CREST, Cirrosis biliar primaria
Enfermedad indiferencia da del tejido con-juntivo
U2 Snurp Procesamiento RNA Polimiositis Esclerodermia
Antígeno mitocondrial
? Cirrosis biliar primaria (90-95%)
La RNA Polimerasa III Transcripción
LES (20%) S de Sicca (70%)
M8-M2 ? Curso agresivo. Cirrosis biliar primaria
Ro
Ga
Ma
PCNA (Proliferating cell nuclear antigen)
Protein Kinasa II
Vimentina
Desmina
Scl-70
Procesamiento RNA
?
?
División celular
Activación de la RNA polimerasa I
Filamento intermedio
Citoesqueleto
Topoisomerasa I
LES (30%) S. de Sicca (70%) Bloqueo cardía-co tipo congénito
Lupus
Lupus (20%)
Lupus S deSicca (10%)
Lupus EMTC
Lupus
Lupus
Esclerodermia CREST (20%)
Mi
M3-M6
M7
Gliadina A y Adenovirus
MBP
Miosina cardía-ca. Proteína M del estreptococo
Mielina periférica
Insulina
?
?
?
?
Anticuerpo contra insulina
Sífilis
Alteraciones inducidas por medicamentos
Enf. cardíaca
Colitis ulcerativa
Enfermedad desmielini-zante
Fiebre reumática
S Guillian Barre
Hipoglicemia autoinmu-ne sec a lupus, Sjºgren primario, Ataxia, Te-langiectasia

Inmunogenética en medicina clínica 45
parecida a algún segmento o polipéptido de la molécula de insulina y la región variable del anti-cuerpo B es considerada como una manifestación de una imagen interna del antígeno externo (139-141) (Figura 9).
Vacunas anti-idiotípicas El principio del mimetismo idiotípico pudiera
utilizarse como un método de inmunización, ya que puede simular un antígeno microbiano. Un anti-idiotipo podría ser utilizado como vacuna, que tendría la ventaja importante de no contener un agente infeccioso; además de la facilidad de pro-ducir grandes cantidades a través de los anticuer-pos monoclonales anti-idiotípicos. Estos anticuer-pos pueden inducir una respuesta efectiva de célu-las T, más que un virus inactivo. Finalmente, los anti-idiotipos pueden mimetizar determinantes antigénicos selectivos que pueden ser construidos por ingeniería genética (138, 142). Una de las va-cunas anti-idiotípicas desarrollada ha sido contra el antígeno de superficie del virus de la hepatitis B (142).
Los anti-idiotipos se pueden utilizar como in-munoterapia de los cánceres de células B, cuyas inmunoglobulinas a nivel de la superficie celular expresarían idiotipos privados. Un anti-idiotipo puede unirse específicamente al idiotipo y causar inhibición del crecimiento o lisis del tumor sin afectar los tejidos normales. En los cánceres de células B humanas, los anticuerpos anti-idiotípi-cos han sido utilizados en leucemia linfocítica aguda y crónica y en linfomas (142).
Las cardiolipinas Ciertos eventos trombóticos son más comunes,
en un subgrupo de pacientes con lupus, que tienen VDRL falso positivo (143). Desde 1952, varios grupos de investigadores han descrito casos de lupus con anticoagulantes circulantes de tipo ad-quirido. Este anticoagulante lúpico se demostró, era una inmunoglobulina que prolonga el tiempo parcial de tromboplastina y menos frecuentemen-te el tiempo de protrombina, por interferir con el complejo activador de la protrombina. Este anti-cuerpo se encontró en los pacientes con lupus que
tenían VDRL + llamaba la atención que tenían paradójicamente una tendencia a la trombosis y no a la hemorragia (144-146). La presencia de estos anticuerpos anticardiolipinas predispone a las pacientes a presentar abortos espontáneos recu-rrentes, muerte fetal in útero, trombocitopenia, síndrome de Evans, síndrome de Snedon, hiper-tensión pulmonar y enfermedad neurológica aso-ciada al lupus.
En 1983, Harris y col purificaron la cardiolipi-na y desarrollaron un radioinmunoensayo de fase sólida para la detección de anticuerpos anti-car-diolipina que es 200 a 400 veces más sensible que el VDRL. Inoue y col (150) notaron ciertas simili-tudes estéricas entre la cardiolipina y el sostén de la unión azúcar-fosfato del DNA por lo cual pen-saron que los sueros de los pacientes tenían una reacción cruzada con los anticuerpos anti-nuclea-res, los cuales son responsables del VDRL falso positivo. La presencia de este anticuerpo en los pacientes con lupus se hereda en un subgrupo de pacientes que constituyen el denominado síndro-me de anticardiolipina.
CONCLUSIONES En este último capítulo hemos analizado los
mecanismos asociados a diferentes moléculas de clase I, II y III, haplotipos extendidos en los cuales intervienen distintos mecanismos asociados y a la vez participan antígenos en la expresión de la en-fermedad.
Entre los mecanismos analizados que expresan antígenos o epitopes, que a la vez son los blancos
Acta Med Colomb Vol. 15 N°1 - 1990

46 G. de Egea y cols.
de anticuerpos naturales son el mimetismo mole-cular, la autoinmunidad órgano-específica y los diferentes mecanismos asociados; la alteración aberrante de los antígenos de clase I y II, el ANA, el ENA, la red de idiotipos-anti-idiotipos y las car-diolipinas como causales de enfermedad. A través del análisis de los diferentes mecanismos implica-dos podemos observar que algunos factores del huésped determinan: 1. El tipo de enfermedad y la respuesta clínica y el tiempo en el cual un indivi-duo pudiera ser susceptible por un antígeno deter-minado genéticamente. 2. Ciertos epítopes de las moléculas del HLA participan en la predisposi-ción a una enfermedad determinada. 3. El proce-samiento de los diferentes antígenos por las célu-las presentadoras de antígenos y la subsecuente interacción entre los pequeños péptidos, las molé-culas de clase I y II y el receptor de las células T, son etapas diferentes pero algunas enfermedades podrían estar relacionadas con alguna alteración en algunas de las etapas. 4. Es posible que el antí-geno desencadenante de una enfermedad, esté re-lacionado con haplotipos determinados que pue-den variar en algunos aminoácidos de acuerdo al grupo étnico estudiado y tener la enfermedad un comportamiento diferente. 5. Es fundamental para las futuras investigaciones, tratar de identificar el antígeno blanco y los otros genes comprometidos en la susceptibilidad para entender mejor la pato-génesis de las enfermedades antes mencionadas. 6.- La susceptibilidad de la mayoría de las enfer-medades reumáticas tiene una fuerte influencia de los genotipos del HLA y el tipo de herencia usual-mente es poligénica. 7. Con las tecnologías del DNA recombinante y su aplicabilidad en la com-prensión de estas enfermedades, va a producirse una ayuda valiosa en el diagnóstico y tratamiento de ellas. 8. El entendimiento de las enfermedades asociadas al HLA se ha retardado por la carencia de modelos en animales pero la tecnología trans-génica disponible es una oportunidad para alcan-zar un mejor conocimiento de las enfermedades, y de esta manera se pensara en el futuro en la posibi-lidad de curación de muchas entidades crónicas para las cuales sólo disponemos de tratamiento paliativo en la actualidad.
N u e v a n o m e n c l a t u r a d e l o s a n t í g e n o s de los leucocitos y nuevos alelos de los genes del CMH adoptados en el X Taller Internacio-nal de Histocompatibilidad
El comité de nomenclatura del último taller de histocompatibilidad revisó y adicionó nuevas es-pecificidades, identificadas por pruebas serológi-cas y técnicas celulares. También denominó nue-vos genes evidenciados por las novedosas técni-cas de biología molecular. Para ello se basaron en las reglas establecidas en previos talleres (151-156).
Sólo se consideraron cambios en los nombres de los productos de los genes de los antígenos de clase I y II.
Los datos provenientes del análisis molecular han logrado definir e identificar el orden de los genes en la región HLA. Se concluyó que estos nuevos alelos que se expresan en la región HLA-D se les pudiera designar en forma oficial sin deslin-darse de las reglas ya establecidas y revisadas en los capítulos anteriores. Todos los genes en esta región son denominados con la letraD como pre-fijo y les agregan las letras R, P, Q para designar la subregión (subregión DR, DP y DQ). Se agregan las letras A o B para nominar la cadena α ó β, o la secuencia relacionada en el caso de los llamados pseudogenes. A lo anterior se le debe agregar un número cuando hay más de un gen α ó β. De esta manera el gen de la cadena α en la región DQ se denominó HLA-DQA1. Las dos nuevas subregio-nes se denominaron DO (inicialmente conocida como DO) y DN (anteriormente llamada DZ o DO). La lista completa de los nuevos genes con su de-signación previa se halla en la Tabla 9. El orden de los genes se muestra en una forma esquemática en la Figura 10.
Se debe destacar que los datos de la secuencia nucleotídica han definido una amplia variedad de alelos. Muchas veces estos alelos corresponden a una sola especificidad definida previamente por técnicas serológicas o celulares. Esto ha permitido una nueva definición del alelo que se considera hoy en día como la secuencia nucleotídica del gen incluyendo los intrones. Sin embargo, en la prác-tica es más importante el producto génico y su función. De esta forma podemos considerar que

Inmunogenética en medicina clínica 47
Tabla 9. Nuevas denominaciones para los genes de la región HLA una definición más adecuada para un alelo ser²a la secuencia aminopeptídica de la proteína codifica-da por un gen determinado.
A cada alelo así definido se le dio un número, el cual se colocó después de un asterisco a continua-ción de la denominación del gen. Cada alelo se identificó con cuatro dígitos de acuerdo con las siguientes reglas: los primeros dos dígitos descri-ben la especificidad más cercana y los otros dos identifican al alelo. De esta forma se intenta tanto como sea posible mantener la relación entre el ale-lo y la especificidad serológica. Un ejemplo de esto se relaciona con la especificidad del B27 para correlacionar los hallazgos de isoelectroenfoque de los clonos de linfocitos T y de la secuencia pep-tídica: HLA-B*2701. Esta misma denominación se aceptó para los antígenos de clase I. En las ta-blas 10 y 11 se muestran los nuevos nombres para algunos alelos de los loci HLA-A, HLA-B y HLA-D. Las especificidades o los productos génicos obtenidos a través de las metodologías del "poli-morfismo de la longitud de los segmentos de res-tricción" (RFLP), y del isoelectroenfoque en una o en dos dimensiones se consideraron de mucha ayuda para definir fenotipos y genotipos tanto de las especificidades conocidas como de las nuevas, pero se concluyó que ellas no contribuían mucho a la designación formal. Esto podrá ser de mucho valor en el futuro cuando se comparen haplotipos relacionados y agrupados en patrones definidos por el polimorfismo de la longitud de los segmen-
Nombre Denominación anterior
Características moleculares
HLA-E 6.2 K E, "6.2" Fragmento de restricción produ-cido por la enzima Hind III asociado con clase I
HLA-DRA DR alfa Cadena alfa del DR
HLA-DRB1 DRbetal.DRIB DR betal - DR1, DR3, DR4, DR5, etc.
HLA-DRB2 DR beta11, DR2B Seudogen con secuencia pareci-da al DRbeta
HLA-DRB 3 DR betaIII, DR3B DRbeta3 del DRw52 y Dw24, Dw25, Dw26
HLA-DRB4 DRbetalV, DR4B DRbeta4 del DRw53
HLA-DQA1 DQalfal .DQlA Cadena DQ alfa
HLA-DQB1 DQbetal, DQ1B Cadena DQ beta
HLA-DQA2 DXalfa, DQ2a Secuencia relacionada con la cadena alfa del DQ
HLA-DQB2 DXbeta, DQ2B Secuencia relacionada con la cadena beta del DQ.
HLA-DOB DObeta Cadena DO beta
HLA-DNA DZalfa Cadena DN alfa
HLA-DPA1 DPalfal .DPlA Cadena DP alfa
HLA-DPB1 DPbetal.DPlB Cadena DP beta
HLA-DPA2 DPalfa2,DP2A Cadena DP alfa
HLA-DPB2 DPbeta2,DP2B Cadena DP beta
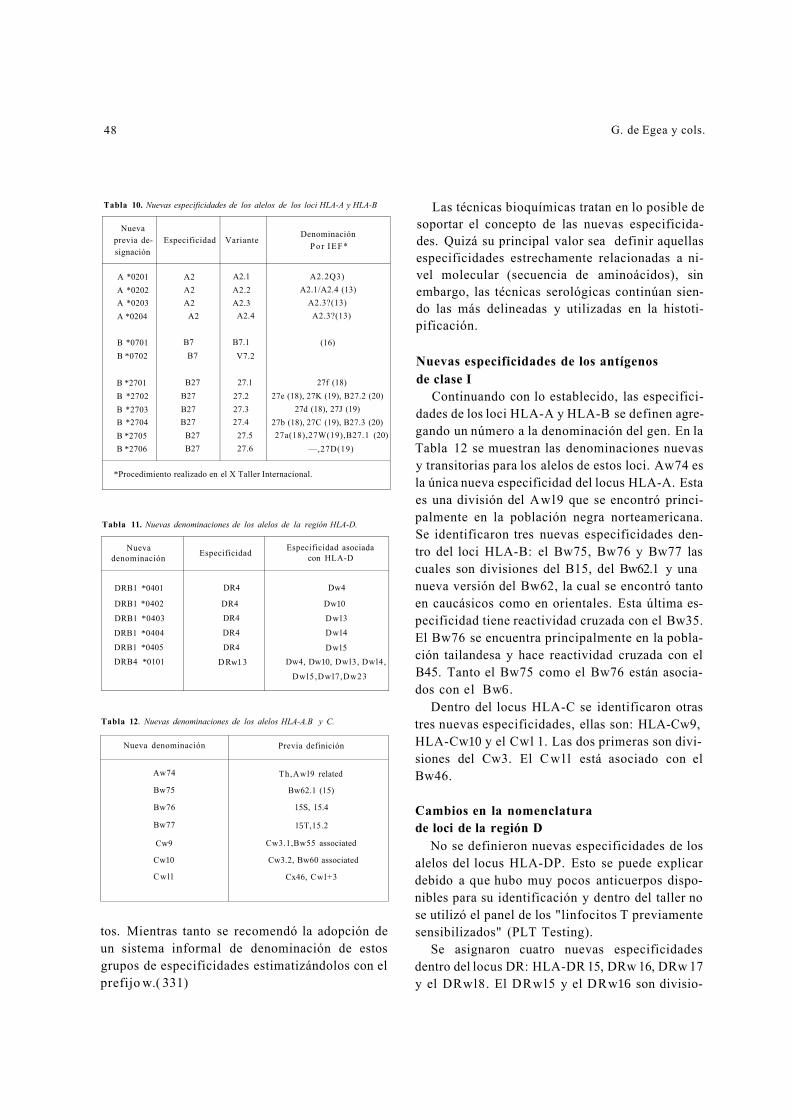
48 G. de Egea y cols.
Tabla 10. Nuevas especificidades de los alelos de los loci HLA-A y HLA-B
Nueva previa de-signación
Especificidad Variante Denominación
Po r IEF*
A *0201 A2 A2.1 A2.2Q3) A *0202 A2 A2.2 A2.1/A2.4 (13) A *0203 A2 A2.3 A2.3?(13) A *0204 A2 A2.4 A2.3?(13)
B *0701 B7 B7.1 (16) B *0702 B7 V7.2
B *2701 B27 27.1 27f (18) B *2702 B27 27.2 27e (18), 27K (19), B27.2 (20) B *2703 B27 27.3 27d (18), 27J (19) B *2704 B27 27.4 27b (18), 27C (19), B27.3 (20) B *2705 B27 27.5 27a(18),27W(19),B27.1 (20) B *2706 B27 27.6 —,27D(19)
*Procedimiento realizado en el X Taller Internacional.
Tabla 11. Nuevas denominaciones de los alelos de la región HLA-D.
Nueva denominación Especificidad Especificidad asociada
con HLA-D
DRB1 *0401 DR4 Dw4
DRB1 *0402 DR4 Dw10
DRB1 *0403 DR4 Dwl3
DRB1 *0404 DR4 Dwl4
DRB1 *0405 DR4 Dwl5 DRB4 *0101 D Rw13 Dw4, Dw10, Dwl3, Dwl4,
Dwl5,Dwl7,Dw23
Tabla 12. Nuevas denominaciones de los alelos HLA-A.B y C.
Nueva denominación Previa definición
Aw74 Th,Awl9 related
Bw75 Bw62.1 (15)
Bw76 15S, 15.4
Bw77 15T,15.2
Cw9 Cw3.1,Bw55 associated
Cw10 Cw3.2, Bw60 associated
Cwl l Cx46, Cwl+3
tos. Mientras tanto se recomendó la adopción de un sistema informal de denominación de estos grupos de especificidades estimatizándolos con el prefijo w.( 331)
Las técnicas bioquímicas tratan en lo posible de soportar el concepto de las nuevas especificida-des. Quizá su principal valor sea definir aquellas especificidades estrechamente relacionadas a ni-vel molecular (secuencia de aminoácidos), sin embargo, las técnicas serológicas continúan sien-do las más delineadas y utilizadas en la histoti-pificación.
Nuevas especificidades de los antígenos de clase I
Continuando con lo establecido, las especifici-dades de los loci HLA-A y HLA-B se definen agre-gando un número a la denominación del gen. En la Tabla 12 se muestran las denominaciones nuevas y transitorias para los alelos de estos loci. Aw74 es la única nueva especificidad del locus HLA-A. Esta es una división del Awl9 que se encontró princi-palmente en la población negra norteamericana. Se identificaron tres nuevas especificidades den-tro del loci HLA-B: el Bw75, Bw76 y Bw77 las cuales son divisiones del B15, del Bw62.1 y una nueva versión del Bw62, la cual se encontró tanto en caucásicos como en orientales. Esta última es-pecificidad tiene reactividad cruzada con el Bw35. El Bw76 se encuentra principalmente en la pobla-ción tailandesa y hace reactividad cruzada con el B45. Tanto el Bw75 como el Bw76 están asocia-dos con el Bw6 .
Dentro del locus HLA-C se identificaron otras tres nuevas especificidades, ellas son: HLA-Cw9, HLA-Cw10 y el Cwl 1. Las dos primeras son divi-siones del Cw3. El C w l l está asociado con el Bw46.
Cambios en la nomenclatura de loci de la región D
No se definieron nuevas especificidades de los alelos del locus HLA-DP. Esto se puede explicar debido a que hubo muy pocos anticuerpos dispo-nibles para su identificación y dentro del taller no se utilizó el panel de los "linfocitos T previamente sensibilizados" (PLT Testing).
Se asignaron cuatro nuevas especificidades dentro del locus DR: HLA-DR 15, DRw 16, DRw 17 y el DRwl8. El DRwl5 y el DRw16 son divisio-

Inmunogenética en medicina clínica 49
nes del HLA-DR2. El DRwl7 incluye la mayor parte de las especificidades originalmente defini-das dentro del DR3 las cuales en general se en-cuentran asociadas con el DQw2. El DR18 es una pequeña especificidad del DR3 que se encuentra en la población blanca.
A los alelos del locus DQ se le asignaron seis nuevas y transitorias especificidades. Es de anotar que la mayoría de estas especificidades se identifi-caron utilizando anticuerpos monoclonales. Ellas se muestran en la Tabla 13.
Nuevas especificidades del loci HLA-D definidas por técnicas celulares
En adición a las técnicas que emplean las célu-las homozigotes para tipificar los alelos de este loci, recientemente se han utilizado otras metodo-logías tales como el clonaje de los linfocitos T ci-totóxicos (PTLc y CTLc). Estas nuevas designa-ciones se muestran también en la Tabla 13, al igual que una lista completa de las especificidades reco-nocidas hasta el momento.
La tecnología recombinante del ácido deoxirribunucleico; su aplicación en inmuno-genética
En el período comprendido entre 1967 y 1972, tres descubrimientos hicieron posible un enorme avance en la investigación de los desórdenes clíni-cos, y por consiguiente en su tratamiento. El pri-mero de ellos fue el descubrimiento de la endonu-cleasa de restricción que es una enzima bacteriana que produce, por medio de la digestión enzim§ti-ca, pequeños fragmentos de ácido deoxirribonu-cleico (ADN). El segundo fue el descubrimiento de las ligasas, enzimas que tienen la facultad de unir segmentos de ADN y, el tercero, fue la cons-trucción de una molécula de ADN mediante la unión de un segmento de ADN humano al ADN de un plásmido para formar "un organismo" capaz de reproducirse en bacterias. Estos tres pilares del desarrollo tecnológico hicieron posible el clonaje del ADN humano y establecieron las bases para el desarrollo de la metodología del aislamiento y la ampliación en el estudio de los genes.
Desde comienzos de la época de los 70 se pro-
Tabla 13. Lista completa de todas las especificidades reconocidas hasta la fecha del sistema HLA.
A B C D DR DQ DP
Al B5 Cwl Dwl DRl DQwl DPwl
A2 B7 Cw2 Dw2 DR2 DQw2 DPw2
A3 B8 Cw3 Dw3 DR3 DQw3 DPw3
A9 B12 Cw4 Dw4 DR4 DQw4 DPw4
A10 B13 Cw5 Dw5 DR5 DQw5 (wl) DPw5
A 11 B14 Cw6 Dw6 DRw6 DQw6 (wl) DPw6
Awl9 B15 Cw7 Dw7 DR7 DQw7(w3)
A23(9) B16 Cw8 Dw8 DRw8 DQw8 (3)
A24 (9) B17 Cw9 (w3) Dw9 DR9 DQw9 (w3)
A25(10) B18 Cwl0(w3) Dw10 DRw10
A26 (10) B21 Cwl l D w l l (w7) DRwl 1 (5)
A28 Bw22 Dwl2 DRwl2
A29 (wl9) B27 Dwl 3 DRwl3 (w6)
A30(wl9) B35 Dwl4 DRwl4 (w6)
A31(wl9) B37 D w l 5 DRwl 5 (2)
A32 (wl9) B38 (16) Dwl6 DRwl 6 (2)
Aw33 (wl9) B39 (16) Dwl7 (w7) DRwl 7 (3)
Aw34 (10) B40 Dwl8(w6) DRwl 8 (3)
Aw36 Bw41 Dwl9(w6)
Aw43 Bw42 Dw20 DRw52
Aw66 (10) B44 (12) Dw21
Aw68 (28) B45 (12) Dw22 DRw53
Aw69 (28) Bw46 Dw23
Aw74 (w29) Bw47 Dw24
Bw48 Dw25
B49 (21) Dw26
Bw50 (21)
B51 (5)
Bw52 (5)
Bw53
Bw54 (w22)
Bw55 (w22)
Bw56 (w22)
Bw57 (17)
Bw58 (17)
Bw59
Bw60 (w40)
Bw61 (w40)
Bw62 (15)
Bw63 (15)
Bw64 (14)
Bw65 (14)
Bw67
Bw70
Bw71 (w70)
Bw72(w70)
Bw73
Bw75 (15)
Bw76(15)
Bw77 (15)
Bw4
Bw6
dujo el acelerado desarrollo del estudio del geno-ma humano, tanto en su estructura como en su función. Inicialmente, la secuencia nucleotídica del material genómico se pudo estudiar a través de
Acta Med Colomb Vol. 15 N°1 ~ 1990

50 G. de Egea y cols.
métodos indirectos, tales como la secuencia de aminoácidos dentro de una proteína o la secuencia nucleotídica del ácido ribonucleico. Hoy la situa-ción ha cambiado completamente; ahora es posi-ble el estudio específico del ADN humano me-diante técnicas moleculares haciendo posible el entendimiento de los complejos mecanismos por medio de los cuales el genoma humano controla el comportamiento biológico y funcional del hom-bre (158).
La tecnología del ADN recombinante com-prende una mezcla de técnicas, algunas novedosas y otras en desarrollo desde mucho tiempo atrás. Quizás las más importantes son: la fragmentación del ADN con enzimas de restricción, la hibridiza-ción del material nuclear en estudio, utilizando para ello secuencias nucleotídicas, conocidas las cua-les se han marcado con material radiactivo (sonda de ADN) que hacen posible la identificación de las secuencias específicas del ADN o del ARN y el clonaje del ADN. En esta última,un fragmento específico del ADN se integra a un material genó-mico en replicación acelerada, Ej. un plásmido o un virus. De tal manera, este último al inoculársele a una bacteria o a una levadura se reproduce y se amplifica. Finalmente el estudio de la secuencia de los nucleótidos en un fragmento clonado de ADN ha permitido el estudio estructural del geno-ma; así, de esta manera, se logró extrapolar el re-conocimiento estructural del genoma al diagnósti-co-de algunas enfermedades (159). De todas las técnicas anteriormente descritas quizás la manipu-lación y el análisis del ADN por las técnicas deno-minadas "blotting" son las que tienen más aplica-ción en el estudio de la estructura y función del sistema HLA. Las más usadas son las siguientes: el uso de las enzimas de restricción, la separación de los segmentos de ADN por métodos electrofo-réticos, la transferencia de estos fragmentos a fil-tros de nitrocelulosa o membranas de nylon, la hibridización de estos fragmentos utilizando son-das de ADN marcadas con material radiactivo, el aislamiento y clonaje de los fragmentos útiles para el estudio, la localización de un fragmento especí-fico en una posición singular dentro de un cromo-soma dado.
Enzimas de restricción. Las endonucleasas de restrición son enzimas, producidas por bacterias, cuya función biológica es proteger a estos micro-organismos de la invasión por ADN extraño (160). Estas enzimas son capaces de cortar al ADN en sitios específicos de aproximadamente cuatro a seis nucleótidos de longitud al reconocer una secuen-cia específica, produciendo pequeños fragmentos de ADN denominados fragmentos de restricción. Muchas de estas enzimas han sido purificadas de diferentes especies bacterianas y en la actualidad existen más de 100 de ellas en el comercio. Com-parando el tamaño de los fragmentos del ADN que se producen sobre una región genética, en particu-lar después del uso de una combinación de dife-rentes endonucleasas de restricción se puede con-formar un mapa de restricción genético para mos-trar la localización de los posibles alelos en un gen determinado. Los patrones de fragmentos gene-rados por las enzimas de restricción serán idénti-cos en todos los alelos de un individuo y aun entre diferentes individuos; lo anterior indjca una ho-mología en la secuencia de las bases del ADN, sin embargo, este mapa de restricción puede variar si la secuencia es ligeramente distinta como es el caso de los individuos con distintos alelos, mutaciones, delecciones, inversiones e inserciones (161). Las variaciones de los patrones que se generan refle-jan el polimorfismo de una región genómica dada. El estudio por medio del cual se consiguen estas diferencias en el genoma humano se ha denomina-do "estudio del polimorfismo genético basado en la longitud de los fragmentos de restricción". PLFR (traducido de las siglas en inglés, RFLP) (161).
Hibridización de los ácidos nucleicos con sondas de ADN complementarias. Se define como el proceso en el cual un segmento genómico se introduce en una secuencia de nucleótidos es-pecífica. Por medio de este procedimiento se pue-de identificar y aislar un gen específico. Se debe recordar que una cadena de ADN o una molécula de RNA siempre se aparea con una segunda cade-na que posee una secuencia complementaria con la primera; este apareamiento ocurre aun en el evento que las cadenas no sean completamente

Inmunogenética en medicina clínica 51
homólogas y sólo se requiere que un 70% de sus bases sean complementarias. Una sonda de ADN se obtiene cuando la enzima DNA sintetasa-trans-criptasa reversa del ARN sintetiza una secuencia de ADN complementaria (cADN), de tal manera que la secuencia nucleotídica del cADN es com-plementaria a la secuencia nucleotídica del ARN, por ejemplo una secuencia CCG (citosina citosina guanina) en el ARN se complementara con una secuencia (sintetizada por la enzima) GGC (gua-nina guanina citosina) en la cADN (162). Para evidenciar la hibridización se le debe adicionar un marcador a la sonda de ADN, la mayoría de las veces fósforo radiactivo (32P). La hibridización se lleva a cabo sobre el ADN celular después de que en éste se han separado sus dos bandas por medio de algún procedimiento; un ejemplo es el uso de altas temperaturas permitiendo así el aco-plamiento del cADN a la secuencia complementa-ria específica en el ADN. El procedimiento más utilizado en la hibridización del ADN es el South-ern Blot (163).
Las técnicas del polimorfismo de la longitud de los segmentos de restricción y Southern Blot se han aplicado en una forma amplia al estudio de los antígenos de histocompatibilidad y al análisis de los genes del complejo mayor de histocompatibi-lidad (MHC). Básicamente esta técnica consiste en: extracción y purificación del ADN provenien-tes de las muestras a estudiar, la digestión enzimà-tica del ADN utilizando endonucleasas de restric-ción, la fragmentación y separación de los frag-mentos por medio de la electroforesis en geles de agarosa, la transferencia de estos fragmentos a un soporte sólido (las más utilizadas son las membra-nas de nylon y nitrocelulosa), la hibridización de la membrana utilizando cADN marcadas y, final-mente, la visualización de los fragmentos a los cuales se le ha unido el cADN por medio de la autorradiografía (si la sonda ha sido marcada con P32), o por un sistema de detección colorimétrica como cuando se utiliza biotina-avidina, este últi-mo es el denominado protocolo de hibridización con materiales no radiactivos. Cuando la técnica que se emplea es la autorradiografía, el análisis de los resultados se evidenciará con base en un pa-
trón de bandas, las cuales aparecerán en la película de rayos X. La Figura 11 muestra en forma esque-mática el procedimiento.
El polimorfismo de la longitud de los fragmen-tos de restricción se hereda en forma mendeliana, de tal manera que sirve no solamente para distin-guir a dos individuos genéticamente diferentes sino también para determinar la verdadera relación biológica entre dos o más individuos.
En los últimos cinco años, debido a la gran can-tidad de sondas que se han obtenido, alrededor de 1.000 (164) se ha podido identificar un alto grado de polimorfismo en loci de los genes.
El uso de las sondas de ADN en las pruebas de paternidad responsable. Este procedimiento se puede llevar a cabo a partir de 10cc de sangre periférica de cada uno de los miembros a estudiar.
Acta Med Colomb Vol. 15 N°1 ~ 1990

52 G. de Egea y cols.
Figura 12A. Prueba de paternidad negativa mediante la aplicación de los patrones de bandas de restricción. Aquí se observa que el patrón de las bandas de los segmentos de restricción del ADN del niño es totalmente diferente a los del supuesto padre.
Para llevar a cabo esta prueba se requiere analizar muestras de ADN provenientes de la madre, el niño y del supuesto padre. También es necesario incluir un control positivo que debe ser proveniente de un individuo de quien se conoce el polimorfismo de la longitud de sus fragmentos de restricción; es necesario también incluir marcadores de los cua-les se conocen sus pesos moleculares y sirven como guía para determinar el peso molecular de cada uno de los fragmentos a estudiar. También debe incluirse una mezcla de ADN proveniente del niño y del padre para determinar si algunos de los frag-mentos que parecen ser de tamaño similar son en realidad los mismos alelos.
Los procedimientos estandarizados permiten tener un alto control en el análisis de los estudios del ADN (164, 165). En forma muy generalizada se puede decir que la prueba resultaría negativa si el patrón de restricción del niño revela una banda, la cual no se detecta en ninguno de los dos padres (Figura 12A). La prueba se considera positiva cuando varias o todas las bandas del patrón de res-tricción del niño son iguales a la de los padres (Figura 12B). El cálculo de las probabilidades de las diferentes combinaciones a través del análisis estadístico, así como también el índice de paterni-dad se obtienen aplicando una serie de fórmulas establecidas y definidas por el comité de nomen-clatura del taller internacional en el mapeo del genoma humano (164).
Polimorfismo molecular de los genes del complejo mayor de histocompatibilidad. El CMH es el sistema más polimórfico del ser huma-no. Esta característica es uno de los aspectos más sobresalientes de los genes que integran este siste-ma. El CMH se encuentra ubicado dentro de una región del brazo corto del cromosoma 6 la cual tiene una amplitud de sólo 2.5 centimorgans (cM). Este complejo se caracteriza por el marcado dese-quilibrio de enlace genético que existe entre' los loci de los genes de clase I y II. Entre los antígenos de clase I, el locus HLA-A consta de 24 alelos; el locus HLA-B (el más polimórfico de todos) repre-senta un total de 50 alelos, y el locus HLA-C sólo comprende 11 alelos. En el grupo de los antígenos
Figura 12B. Prueba de paternidad positiva mediante el uso de la metodología del PLSR. En el ejemplo esquematizado arriba se puede observar claramente que tanto la madre como el supuesto padre biológico comparten el mismo patrón de las bandas análogas a los alelos compartidos. En un caso como éste, los investigadores podrían asegurar con un alto grado de certeza que el alega-do padre es en realidad el verdadero padre biológico.
de clase II, el más polimórfico de todos ellos lo constituye el grupo de los alelos del locus DR, dentro del cual se han identificado 20 alelos. El locus DQ posee 10 alelos y el locus DP sólo con seis alelos descritos hasta la fecha. Este polimor-fismo también se extiende para algunas proteínas del sistema del complemento cuyos genes que las codifican se encuentran ubicados dentro del siste-ma del CMH. Los genes de estos antígenos, deno-minados antígenos de clase III, son Bf, C4a, C4b y C2 (166, 167).
Con el advenimiento de las técnicas de biología molecular hemos podido aprender y comprender en parte la complejidad de los genes que confor-man el CMH. Hoy conocemos por lo menos el

Inmunogenética en medicina clínica 53
número de genes que comprenden cada clase, la reorganización de sus exones y entrones y en la mayoría de las veces su secuencia nucleotídica (168, 169). Existen actualmente 24 genes dentro de la clase I; en cada región existe un limitado número de genes funcionales y otro tanto de genes no funcionales, la mayoría de ellos son los análo-gos de los genes murinos Qa y Tla. Para las molé-culas de clase II existen por lo menos siete genes que codifican la síntesis de la cadena (3 y por lo menos uno más para las cadenas Ŭ.
La complejidad de la región HLA-D es cada día mayor, aunque está dividida en tres grupos princi-pales de genes (subregiones, DP, DQ, DR), cada una de ellas está compuesta por múltiples genes funcionales de las cadenas Ŭ y ɓ (diversos loci). Cuando estos loci no son funcionales se les deno-minan pseudogenes, tal es el caso del gen DRB2 del loci DR. Además de las regiones anteriormen-te mencionadas se han identificado otras tres sub-regiones: DN, DO, DX. A los genes ubicados en estas zonas no se les ha podido demostrar una fun-ción biológica determinada y se piensa que ello se ha originado a través de duplicaciones genéticas dentro de la región HLA-D.
Normalmente en cada subregión existe un apa-reamiento de las cadenas Ŭ y ɓ pertenecientes a las mismas, sin embargo, puede resultar la formación de moléculas híbridas con un mayor nivel de com-plejidad por ejemplo cuando se aparean la cadena Ŭ del DP con la cadena ɓ del DQ (160).
El análisis del material genómico en la región del CMH, utilizando la metodología del PLFR está enfocada hacia el análisis genético de la familia de estos genes y a la determinación de haplotipos, a partir de la información de los fenotipos.
El uso de las cADN (sondas de amplia y corta longitud) permiten generar los patrones de frag-mentos, los cuales conllevan a interpretar la loca-lización y la secuencia nucleotídica de los posi-bles alelos cuando se correlacionan con la longi-tud de los fragmentos resultantes. En caso de los antígenos de clase I, el número de bandas que se pueden detectar varía de 15 a 25 de acuerdo con las enzimas y sondas utilizadas (170-175). Este dato es compatible con el número de genes que se
estima para el número de antígenos de clase 1(176). El análisis de la distribución de estos patrones de restricción entre los haplotipos muestra que existe un grupo de ellos, de tal manera que algunos frag-mentos tienden a aparecer en los mismos haploti-pos. El hecho de que estos grupos se encuentren dentro de haplotipos, los cuales previamente se han clasificado con base en especificidades defi-nidas serológicamete, ha permitido estudiar la re-lación entre el polimorfismo definido a un nivel genómico definido por técnicas serológicas.
El análisis de la variabilidad de los antígenos de clase II se llevó acabo inicialmente mediante prue-bas celulares como el cultivo mixto de linfocitos. Posteriormente, mediante el uso de las técnicas serológicas y bioquímicas, a través de la utiliza-ción de antisueros obtenidos de sueros de indivi-duos sensibilizados (aloanticuerpos), con el uso de los anticuerpos monoclonales y las electrofore-sis de proteínas en dos dimensiones ha sido posi-ble documentar en una forma más extensa el poli-formismo existente en los antígenos de clase II.
Desde el punto de vista molecular existen algu-nas observaciones claras en la relación funcional y estructural de algunos de los genes de la región HLA-D.
El gen DRB1 en asociación con el gen Ŭ codifi-ca las proteínas para los antígenos DR 1 Ŭ DRW 15, mientras que el gen DRB3 y el gen DR Ŭ codifica la síntesis de la familia de las moléculas del DRW52. Por otro lado el DRB4 en asocio con la cadena DRŬ codifica la síntesis de la familia de las moléculas del DRW52. Por otro lado el DRB4 en asocio con la cadena D RŬ codifica la síntesis del antígeno DRW53. El análisis de la secuencia de los ácidos nucleicos y de los aminoácidos tanto de los genes como de los productos génicos del DRB ha mostrado que el polimorfismo reside bá-sicamente a nivel de la cadena DRB 1, lo cual per-mite la producción de múltiples determinantes y por consiguiente las diferentes especificidades funcionales para los antígenos DR.
Las variaciones al azar en una estructura gené-tica básica estarían explicadas por el fenómeno de la mutación genética, bien sea múltiple o única o bien por las conversiones genéticas debidas a las
Acta Med Colomb Vol. 15 N°1 ~ 1990

54 G. de Egea y cols.
inserciones de secuencias de ácidos nucleicos de loci vecinos.
La selección natural de los genes polimórficos sobre los genes monomórficos. En esta última teo-ría están implicados los microorganismos patóge-nos. Es posible que estos dos tipos de mecanismos participen en la producción del polimorfismo. Una imbricación importante que resulta de las dos hi-pótesis anteriormente expuestas, lo constituiría el grupo de sujetos en los cuales las moléculas de clase I y II no interactuarían adecuadamente con el receptor de las células T (TcR), produciría una respuesta inmunitaria inadecuada en contra de ciertos microorganismos patógenos. Esta carencia de los conocimientos de los determinantes antigé-nicos representaría lo que se conoce como "punto ciego" dentro del repertorio de las células T. Apa-rentemente el polimorfismo del CMH es una com-pensación de los puntos ciegos de las células T y aquellos individuos que son portadores de los puntos ciegos tienden a desaparecer.
Como se mencionó anteriormente, los alelos de las subregiones DR, DP y DQ se heredan en blo-que. Esto debido a la corta distancia que existe entre ellos lo cual hace casi improbable la existen-cia de recombinación genética (cross over).
Polimorfismo genético y asociación con enfermedades. En los estudios de la asociación del sistema HLA con las enfermedades, el fenó-meno de desequilibrio de enlace genético en el complejo mayor de histocompatibilidad, la mayo-ría de las veces ha dificultado el reconocimiento del posible locus de susceptibilidad para una en-fermedad dada; un ejemplo lo constituye la diabe-tes mellitus insulino dependiente, la cual se ha aso-ciado con el antígeno HLA-DR3 cuando éste for-ma parte del haplotipo B8 DR3 o del B18 DR3 (177). Debe destacarse que en esta entidad el alelo del "C4 null" se encuentra asociado a ambos ha-plotipos y debe considerarse como un posible fac-tor genético de contribución en la patogénesis de esta entidad (178).
Como otro ejemplo podemos citar el caso del DR4 que generalmente se asocia con el DQw7. Esto podría causar dificultad en el análisis de la
importancia de los alelos DR y DQ en la suscepti-bilidad a las enfermedades. Singal y col muestran un incremento del DR4, DQw7 (anteriormente DQw3) en pacientes con artritis reumatoidea, es-pecialmente en el caso de la enfermedad grave, pero otros estudios no demuestran tal incremento en grupos de pacientes similares. Nepom y col plantean la posibilidad de que existan diferencias estructurales entre las cadenas (3 del DR4 que pue-dan influenciar la interacción entre el CMH y el receptor de células T, produciendo un incremento en la habilidad de tales células para reconocer y responder a antígenos extraños. A través del aná-lisis computarizado en tres dimensiones en mode-los de la molécula de clase II identifican una posi-ble hélice alfa en la tercera región hipervariable de la cadena DRB que podría estar involucrada en el reconocimiento por las células T. Esto podría ex-plicar el porqué un grupo de determinantes antigé-nicos relacionados en diferentes subtipos del DR4 (especificidades celulares) pueden conferir la sus-ceptibilidad a la enfermedad.
Las moléculas de clase II poseen pequeñas re-giones de hipervariabilidad alélica, especialmente a nivel de los dominios A1 y B1, que varían entre los diferentes individuos, pero estas regiones son idénticas en las diferentes células de un individuo determinado. El sitio donde se une al antígeno se encuentra localizado a nivel de la región hiperva-riable.
Como se mencionó anteriormente, la mayoría de los estudios en población con los diferentes grupos étnicos han confirmado una fuerte asocia-ción entre el HLA-DR4 y artritis reumatoide en 70-75% de los casos. En otros pequeños grupos raciales se encuentra una asociación entre AR y el haplotipo HLA-DR 1. Tratando de definir el epíto-pe responsable de la susceptibilidad a la artritis reumatoide, utilizando para ello una serie de téc-nicas, tales como MLR, serología, RFLP, endonu-cleasas de restricción y clonos de células T, Du-quesnoy y col a través de un aloantisuero lograron definir un epítope en pacientes con AR con HLA-DR4, pero también lo encontraron en pacientes con HLA-DR1. Otro grupo de investigadores lo-gró definir un epítope, utilizando para ello el anti-

Inmunogenética en medicina clínica 55
cuerpo monoclonal (abl09db) en 93% de los pa-cientes con AR.
Con la ayuda de la biología molecular Grege-sen y col han logrado evidenciar que a nivel de la tercera región hipervariable del primer dominio de los subtipos DR4 (Dw4, Dwl0, Dwl3, Dw 14 y Dwl5) y del DR1 comparten una secuencia de aminoácidos similares, especialmente los subtipos Dw4 y Dw 14 y el tipo DR 1. Llama la atención que al compartir estos epítopes se ha observado una mayor susceptibilidad a la artritis reumatoide, pero el subtipo DwlO no parece asociarse a la artritis reumatoide. La secuencia de aminoácidos del sub-tipo Dwl4 y del tipo DR 1 es similar y sólo difieren con el subtipo Dw4 a nivel de un solo aminoácido en la posición 71. En cambio estos dos subtipos (Dw4 y Dwl4) y el tipo DR1 difieren en tres ami-noácidos con el subtipo DwlO (Tabla 13).
Se sugiere que el aminoácido en la posición 57 del DQB1 a nivel de la primera región hipervaria-ble del primer dominio puede ser importante en la susceptibilidad para evolucionar hacia formas se-veras de AR; este aminoácido es el ácido aspártico cargado negativamente en la posición 57.
La genética molecular y la tecnología del trasplante. Después del descubrimiento del siste-ma HLA a mediados de la década del 60 y del éxito del trasplante renal entre gemelos idénticos, comenzaron los programas nacionales e interna-cionales de intercambio, con el fin de encontrar el apareamiento de individuos comprometidos en un trasplante. Para esa época ya se conocía que el apareamiento de los locis A y B no tenían mayor importancia en la supervivencia del trasplante renal del donante cadavérico y que el locus más impor-
Tabla 14. Seciemcoas a.ompácodpsDW uDRl
66 67 68 69 70 71 72 73 74
Dw4 Asp Leu Leu Gli Gli Lys Lys Ala Ala
Dw10 Ile - - - Asp Gli - - -Dwl4 - - - - - Arg - - -
Dwl5 - - - - - Arg - - -
DR1 - - - - - Arg - - -
tante era un nuevo locus DR, el cual había sido descrito en trabajos que utilizaron como diseño experimental el cultivo mixto de linfocitos. Poste-riormente, en el desarrollo del taller internacional de Histocompatibilidad, celebrado en 1975 en Aarhus, utilizando una nueva técnica descrita por Dupont y otros autores (179-181) mediante el uso de las células homozigotes (Homozigous typing cells: HTCs) se definieron unas series de alelos pertenecientes al nuevo locus, denominado HLA-D. También quedó claro que estos alelos se encon-traban en fuerte desequilibrio de enlace genético con los genes del locus HLA-B (181). En 1977, un nuevo taller internacional llevado a cabo en la ciu-dad de Oxford, mostró los resultados provenientes de diferentes laboratorios, los cuales, utilizando anticuerpos provenientes de sueros de multíparas, encontraron una serie de patrones de reactividad serológica, los cuales se correspondían con los tipos de HLA-D al utilizar el panel de las células HTCs (182). Estos nuevos determinantes identificados serológicamente se denominaron HLA-DR para resaltar su relación con los fenotipos de los genes HLA-D, estos anticuerpos que definían las especi-ficidades Dwl, Dw2, Dw3 se denominaron DR1, DR2 y DR3 respectivamente. Después de todo este avance también se esclareció que la región DR tie-ne muy poco desequilibrio de enlace genético con la región DP por un lado y por la región DQ del otro lado.
En las siguientes líneas trataremos de presentar algunos conceptos claros acerca del uso de las sondas ADN en la región HLA-D y sus aportes para un mejor desarrollo de las pruebas de tipifi-cación, después que diferentes grupos en el nove-no taller internacional encontraron (183) este nue-vo soporte en las técnicas de histotipificación, otros grupos de investigadores (184, 185) esclarecieron que las cADN entre individuos no relacionados genéticamente pero con homogocidad idéntica entre el HLA-DW/DR mostraban un patrón de restricción idéntico (185). Estos hallazgos indican que existe una secuencia de nucleótidos específi-cos que se conserva a nivel de estos alelos y que sólo las bandas que son DR1, 2 ó 4, tienen bandas de especificidades que no tienen otros haplotipos
Acta Med Colomb Vol. 15 N°1 ~ 1990

56 G. de Egea y cols.
(186, 187). Además se sabe hoy que el DR3, 5 y DRW6 se pueden agrupar de acuerdo con sus pa-trones de restricción. Es necesario anotar que to-dos estos resultados se han obtenido utilizando la cADN del DR (3 y en la mayoría de los experimen-tos, el material genómico fue digerido utilizando las enzimas de restricción BamHl y Pvull (186-188). De manera análoga también se ha encontra-do polimorfismo de los alelos del DQ en grupos de individuos genéticamente diferentes pero con fe-notipos idénticos, los cuales a menudo muestran un mismo patrón de restricción al usar las sondas de la cadena (3 del DQ (188-189). Poco se conoce acerca de la importancia de la diferencia de los patrones de restricción del DQ en aquellos indivi-duos idénticos al locis DR que han sido sometidos a trasplantes renales, igual se puede decir en rela-ción con el locis DR.
Se ha especulado que la incompatibilidad DP debe ser importante en el trasplante de médula ósea; este concepto ha emergido de la alta proporción de pacientes que desarrollan la enfermedad injerto vs. huésped. A pesar de que estos individuos se selec-cionan con un alto apareamiento en los loci HLA-A, B, C y DR entre individuos genéticamente di-ferentes, los cuales tienen una baja o nula respues-ta en el cultivo mixto de linfocitos, pero con una fuerte estimulación de PLT (190).
Existen varios puntos que deben clarificarse antes de presentar conclusiones acerca del papel de la biología molecular, en el estudio de los antí-genos de clase II y su aplicación en la clínica del trasplante: en primera instancia debe tenerse en cuenta la distribución tisular de los antígenos DR, DP y DQ. Estos antígenos no se expresan en igua-les cantidades ni tampoco de la misma forma en los diferentes tipos de células de los mismos y di-ferentes individuos (191). Por otro lado, la distri-bución del DQ varía en las diferentes subpobla-ciones de monocitos (192). Otros autores también han encontrado diferencias en la expresión de los distintos antígenos de clase II sobre la superficie de los linfocitos B a través de las diferentes fases de su diferenciación (192). Casi nada se conoce acerca de la expresión de los antígenos DP sobre la superficie de las células que pudieran ser de im-
portancia en el trasplante de riñón. Por otro lado, los niveles de la variabilidad de los genes de la cadena Ŭ y ɓ del DQ demostrado por los estudios de la técnica del ADN recombinante han excedido las especificidades que se pueden reconocer a tra-vés de las técnicas celulares y serológicas que se conocen hasta el momento.
Los estudios que han utilizado técnicas bioquí-micas han confirmado lo anterior (193). Para las moléculas DP la situación no es diferente, muy poco se conoce en relación con la expresión a los determinantes aloantigénicos en las moléculas DP ya que éstas no se pueden determinar con antisue-ros; esta dificultad puede ser debido a la densidad con que se expresan estos antígenos DP en los lin-focitos de sangre periférica.
Posiblemente la continuidad de los estudios en el campo del análisis molecular de los genes del MHC permitan esclarecer la verdadera importan-cia de estas nuevas especificidades y su aplicación en la escogencia del donante perfecto.
La técnica de la reacción en cadena de la po-limerasa del ADN. Este nuevo método ha revolu-cionado la tecnología del ADN recombinante, permitiendo que los estudios se puedan hacer en una forma más rápida y más sensible (194). Este método permite amplificar material genómico de tal manera que intenta reproducir la replicación natural del ADN de una manera similar a como se da in vivo. En él, la secuencia del ADN en pocas horas se puede amplificar un millón de veces. La metodología se basa en una técnica que se divide en tres etapas, todas conducidas en una forma re-petitiva bajo diferentes condiciones controladas de temperatura. Inicialmente, la muestra de ADN en estudio se somete a altas temperaturas con la fina-lidad de desnaturalizarla y así poder mantener las dos hélices disociadas. A continuación se utilizan dos oligonucleótidos como moldes, se producirá la síntesis de dos nuevas moléculas, las cuales contienen cada una de ellas una banda del ADN nativo; la secuenci a del nucleótido es determinada por la secuencia del ADN. En el tercer paso de la técnica denominada proceso de extensión, la poli-merasa del ADN reproducirá grandes cantidades

Inmunogenética en medicina clínica 57
del segmento del genoma inicialmente reproduci-do. Actualmente la polimerasa que se utiliza es derivada del Thermus acuaticus (188), la cual es altamente estable a temperaturas altas. En la técni-ca, cada ciclo comprende tres pasos: desnaturali-zación; apareamiento y extensión. La Figura 13 intenta explicar en forma esquemática el procedi-miento; en ella se observa claramente como el seg-mento genómico que se quiere amplificar después de tres ciclos produce cantidades acumuladas re-velantes (189, 190).
La reacción en cadena de la polimerasa (RCP) tiene varias aplicaciones en medicina clínica. Una de ellas es su aplicabilidad en las enfermedades genéticas (191-192), tales como anemia de células falciformes, la fenil cetonuria, etc. Otra ventaja de la técnica es que ella no utiliza ningún material radiactivo, haciéndola más accesible a los países del tercer mundo. Si se logra obtener suficiente material genético después de amplificarlo éste se pudiera colorear fácilmente después de colorear el gel con bromuro de ethidium. Esta técnica es tan sensible,que se pueden utilizar simples hebras de cabello (189), o la obtención del ADN por medio de un frotis de la mucosa de los carrillos. En el campo de diagnóstico de las enfermedades infec-ciosas la técnica ya se ha aplicado a la detección del virus del SIDA (191) y al papilovavirus huma-no (192). Es de esperarse que por su rapidez y su bajo costo y poco riesgo laboral, este método se pueda aplicar a una gran variedad de ensayos diagnósticos en la clínica.
Es de lógica considerar que la inmunogenética y la genética clínica han comenzado a correr por un nuevo sendero, el de la biología molecular. Uno de los sistemas que más se ha beneficiado es el sistema HLA, en el cual actualmente ha sido posi-ble detectar, alrededor de unos 200 polimorfismos. Con el descubrimiento de nuevos marcadores de polimorfismo tal vez se dé un gran avance en el campo de la asociación del CMH con otras enfer-medades. Es posible también que una lista de nue-vas entidades clínicas, asociadas con el sistema HLA, se agregue a las ya conocidas, bien sea con la susceptibilidad o resistencia a ellas. Este tipo de tecnología permitirá un nuevo avance en la pre-
dicción y prevención de las enfermedades, dando así la oportunidad de una mejor aplicabilidad a los conceptos de la medicina preventiva. Sin embar-go, la máxima aplicación de esta tecnología, du-rante la segunda mitad de este decenio, lo consti-tuye la oportunidad de entender los mecanismos fisiopatológicos de diversas enfermedades al identificar y caracterizar, en forma exacta, el gen o los genes responsables de la misma.
GLOSARIO Alelo. La forma alternante de un gen que se expresa
en el mismo locus. Alogénico. Individuos que pertenecen a una mis-
ma especie pero son genéticamente distintos. Centimorgan. Unidad de medida que define la
distancia entre dos genes. Se puede decir que un centimorgan equivale a 1% de recombinación.
Codominante. El estado en el cual cada gen ex-presa su efecto en el heterozigoto.
Centrómero. Sitio en donde el cromosoma se une
Acta Med Colomb Vol. 15 N°1 ~ 1990

58 G. de Egeaycols.
al huso durante la división celular. Clon. Familia de células provenientes de una mis-
ma célula ancestral. También se refiere este tér-mino a un grupo de moléculas idénticas genéti-camente.
Clonaje. Metodología del ADN recombinante por medio de la cual se hace posible clonar genes.
Deleción. Pérdida de un segmento genómico en un cromosoma dado.
Desequilibrio de enlace genético. Estado en el cual dos o más genes están presentes en el mis-mo haplotipo con una frecuencia mayor de la esperada.
Dominios. Secuencia polipeptídica de la molécu-la de una inmunoglobulina, la cual se encuentra fisicoquímicamente definida por puentes de disulfuros, estableciéndose asas que en la ma-yoría de las veces cumplen funciones biológi-cas.
Endonucleasas de restricción. Enzima capaz de degradar el ADN extraño a través del reconoci-miento específico de secuencias nucleotídicas en el interior de la molécula. Esta enzima res-peta el ADN del hospedero a causa de la med-iación del mismo.
Entrecruzamiento. Intercambio de segmentos entre cromosomas homólogos.
Fenotipos. Los rasgos característicos externos producidos por los genes.
Gen. Factor unitario de la herencia. Genotipo. Constitución genética de un individuo
u organismo. Genoma. El total del material genético de un indi-
viduo u organismo. Genes de respuesta inmunitaria. Conjuntos de
genes pertenecientes al CMH, los cuales con-trolan la respuesta inmunitaria a antígenos es-pecíficos (genes Ir).
Haplotipo. Es el conjunto de genes localizados en el CMH estrechamente ligados entre sí.
Heterozigoto. Tenencia de diferentes alelos en un locus determinado a nivel de cromosomas ho-mólogos.
Homozigoto. Tenencia de un mismo alelo en cro-mosomas homólogos.
Hibridización. Es el proceso mediante el cual dos
bandas separadas de ADN interactúan para for-mar una molécula de doble hélice. Esta interac-ción se basa en la formación de puentes de hi-drógeno entre las bases complementarias de cada una de las bandas.
Ligasa. Enzima que cataliza la formación de los puentes de fosfodiesterol en el sitio de escisión de una molécula de ADN nativo.
Locus. Sitio que ocupa un gen a lo largo de un cro-mosoma determinado.
Mutación. Un cambio en el material genético de un individuo, el cual se transmite en forma he-reditaria.
Nucleótido. Es la unidad básica de los ácidos nu-cleicos. Bioquímicamente corresponde a un nucleósido que es fosforilado en uno de los gru-pos hidroxilos de las pentosas.
Plásmido. Se trata de una estructura de ADN cir-cular extracromosómica, la cual se replica autó-nomamente dentro de la célula bacteriana y no es esencial para el mantenimiento de la célula.
Polimorfismo. Existencia de dos o más fenotipos distintos en una población determinada.
Polimerasa. Enzima catalizadora del ensamblaje de los rribonucleótidos o de los deoxiribonu-cleótidos al interior del ADN o del ARN res-pectivamente.
Recombinación. Es el fenómeno mediante el cual un entrecruzamiento permite que el gameto contenga los genes tanto de origen materno como paterno.
Riesgo relativo. Es el término que asocia a un marcador genético con el hecho biológico de presentar una enfermedad dada.
Southern Blot. Es la metodología que permite hacer la transferencia de los fragmentos de ADN, obtenidos mediante electroforesis del gel a cier-tos tipos diferentes de soportes. Los fragmentos que se transfieren posteriormente se detectan mediante la utilización de la hibridización con sondas de ADN o ARN.
Sonda. Moléculas de ADN o de ARN las cuales se encuentran "marcadas" (acopladas) radiactivas o inmunológicamente y que se usan en los ex-perimentos de hibridización para detectar la presencia de secuencias complementarias.

Inmunogenética en medicina clínica 59
Transcriptasa reversa. Una enzima de ciertos virus de ARN la cual cataliza la polimerización del ADN a partir de un templete de ARN. Tam-bién se le conoce como ADN polimerasa, ARN dependiente.
ABSTRACT Recent research on the association between dis-
ease and HLA system is discussed in this review paper. In addition, the conclusions of the "Nomen-clature Committee" of the 10th International His-tocompatibility meeting, as well as the significance of Recombinant DNA technology are carefully analized. Practical clinical uses of all this complex knowledge are minutely illustrated.
REFERENCIAS 1: Lilly F. The inheritance to the gross leukemia virus in mice. Genetics
1966; 53: 529-535.
2. Amiel JL . Study of the leucocyte phenotypes in the Hodg Kins's disease. Histocompatibility testing 1967. Copenhagen: Munksgaard; 1970: 79-81.
3. Brewerton D, Caffrey M, Hart F. Ankylosing spondylitis and HLA-B27. Lancet 1973; 2: 904-906.
4. Bodmer JG, Rocques P, et al. Joint report of the fifth international histocompatibility workshop. In: Dausset I, Colombani J, eds. Histo-compatibility Testing 1972. Copenhagen: Munksgaard; 1973: 619.
5. Jerne NK.The somatic generation of immune recognition. Eur J Immunol 1971; 1:1-8.
6. Zinkernagel RM, Doherty PC. Restriction of in vitro T cell mediated cytotoxicity in lymphocyte chorionomeningitis within a syngenic or semiallogenic system. Nature 1974; 248:701 -702.
7. Geczy AF, Alexander K, Bashir HV, Edmonds JP, Upfold L, Sulli-van J. HLA-B27, Klebsiella and ankylosing spondylitis: Biological and chemical studies. In: Moller G, ed. Imnmnol Rev Copenhagen: Munksgaard, 1981; 70: 23-50.
8. Ogasawara M, Kono DM, Yu DTY. Mimicry of human histocompati-bility HLA-B27 antigens by klebsiella pneumoniae. Infect lmmunol 1986; 51:901-908.
9. Svejgaard A, Platz P, Ryder LP. HLA and disease 1982, a survey. In: Moller G, ed. Immunol Rev. Copenhagen: Munksgaard, 1983; 70:193-218.
10. Weiss EH, Kvon W, Dorner C, Lang M, Riethmuller G. Organization, sequence and expression of the HLA-B27 gene: A molecular approach to analyze HLA and disease associations. Immunobiology 1985; 170:367-380.
11. Teresaki PL. Histocompatibility testing 1980. Los Angeles: UCLA Tis-sue typing laboratory; 1980.
12 Thomson G, Bodmer W. HLA Haplotype associations with disease. Tissue Antigens 1979; 13:91-103.
12. Alper CA. Exdxtended MHC haplotypes and disease markers. lSl Atlas of Science 1988:79-83.
13. Babbitt BP, Allen PM, Matsneda G, Haber E, Unanne ER. Binding of immunogenic peptides to I a histocompatibility molecules. Nature 1985; 317:359-362.
Sasazuki T, Nishimura Y, M u t o M, Ohta N. HLA linked genes con-trolling immune response and disease susceptibility. In: Moller G ed. Immunol Rev Copenhagen: Munksgaard; 1981;70:51-75.
14. Miller LH, Mason SJ, Dvorak JA, McG innis MH, Rotchman IK. Erythrocyte receptors for Plasmodium knowlwesi malaria. Duffy group determinants. Science 1975; 189:561-564.
15. Inman RD. The interplay or microbes and MHC in the pathogenesis of the Reiter's syndrome. Clin Exp Rheumatol 1986; 4:75-82.
16. Inman RD, Chiu B, Johnston MEA, Falk J. Molecular mimicry in Reiters syndrome: cytotoxicity and ELISA studies of HLA- microbial relationships. Immunology 1986; 58:501-506.
17. Inman RD, Johnston MEA, Hodge M, Flak J, Helewa A. Post dysen-teric reactive arthritis. A clinical and immunogenetic study following an outbreak of salmonellosis. Arthritis Reum 1988;31:1377-1383.
18. Alper CA, Awdeh ZL, Raum D, Yunis EJ. Extended major histocom-patibility complex haplotypes in man: Role of alleles analogous to murine. Clin Immunol Immunopath 1982; 24:276-282
19. Alper CA, Raum D, Karp S, Awdeh ZL, Yunis EJ. Serum comple-ment "supergenes" of the major histocompatibility complex in man (Complotypes). Vox Sang 1983; 45:62-67.
20. De Wolf WC, Lange PH, Einarson ME, Yunis EJ. HLA and testicular cancer. Nature 1979; 277:216-219.
21. Dupont B, Pollack MS, Levine LS, O'Neil G J, Hawkins BR,New MI. Joint report congenital adrenal hyperplasia. In: Teresaki PI ed. Histo-compatibility testing 1980. Los Angeles: UCLA Tissue typing labora-tory; 1980.
22. Cartwright GE, Edwards CQ, Kravit K, Skolnick M, Amos DB, Johnson A, Buskaajar L. Hereditary hemochromatosis. Phenotypic expression of the disease. N Engl J Med 1979; 301:175-182.
23. Amos DB, Johnson AH, Cartwright G, Edwards C, Scholnick M. HLA and b cell antigens in hemochromatosis. Tissue Antigens 1977; 10:206-211.
24. Miller JFPA. Histocompatibility genes and diabetes. Clin Immunol News 1988; 9:149-151.
25. Campbell IL, Wong GHW, Schrader JW, Harrison LC. Interferon gamma enhances the expression of the major histocompatibility class I antigens on mouse pancreatic beta cells. Diabetes 1985; 34:1205-1209.
26. Campbell IL, Harrison RG, Ashcroft RG, Jack I. Retrovirus infection enhances expression of class I MHC protein on human b cell nad rat RINm5F cell. Diabetes 1988; 37:362-365.
27. Ryder LP, Svejgaard A. Genetic of HLA disease association. Ann Rev Genet 1981; 15:169-187.
28. Svejgaard A, Platz P, Ryder LP. Insulin dependent diabetes mellitus. In: Teresaki PI ed. Histocompatibility Testing 1980. Copenhagen: Munksgaard; 1980:638-656.
29. Raum D, Alper CA, Stein R, Gabbay KH. Genetic marker for insulin-dependent diabetes mellitus. Lancet 1979; i:1208-1210.
30. Raum D, Awdeh ZL, Alper CA, BF. Types and the mode of inheritance of insulin dependent diabetes mellitus (IDDM). Immunogenetics 1981; 12:59-74.
31. Raum D, Awdeh ZL, Yunis EJ, Alper CA, Gabbay KH. Extended major histocompatibility complex haplotypes in type I diabetes mellitus. J Clin Invest 1984; 74:449-454.
32. Alper CA, Fleischnick E, Awdeh ZL, Katz AJ, Yunis EJ. Extended major histocompatibility complex haplotypes in patients with gluten-sensitive enteropathy. J Clin Invest 1987; 79:251-256.
33. Van der Berg-Loonen EM, Lucas KD. LD 7A typing in 46 patients with multiple sclerosis. In: Teresaki PI ed. Histocompatibility testing 1975. Copenhagen: Munksgaard; 1975:773-777.
34. Simon M, Pawlotsky Y, Bourel M, Fauchet R, Genentet B. Hemo-chromatose idiopathique: maladie associe a l'antigene tissulaire HL-A3? Nouv Presse Med 1975; 4:1432.
35. Dupont B, Oberfeld SE, Smithwick EM, Lee TD, Levine LS. Close genetic linkage between HLA and congenital adrenal hyperplasia (21-hidroxylase deficiency). Lancet 1977; ii: 1309-1312.
36. Carrol MC, Campbell RD, Porter RP. Mapping of steroid 21 -hydrox-ylase genes adjacent to complement component C4 genes in HLA, the major histocompatibility complex in man. P roc Natl Acad Sci USA 1985;
Acta Med Colomb Vol. 15 N°1 ~ 1990

60 G. de Egea y cols.
82:521-525. 37. White PC, Grpssberg D, Oufer BJ, Chaplin DD, New MI, Dupont B,
Strominger JL. Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci USA 1985; 82:1089-1093.
38. White PC, New MI, Dupont B. HLA- linked congenital adrenal hyper-plasia results from a defective gene encoding a cytochrome p-450 spe-cific for steroid 21-hydroxylation. Proc Natl Acad Sci USA 1984; 81:7505- 7509.
39. Carrol MC, Palsdottir A, Belt KT, Porter RR. Deletion of comple-ment C4 and steroid 21-hydroxylase gene in the HLA class III region. EMBO 1985; 4:2547-2552.
40. Fleischnick E, Awdeh ZL, Raum D, et al. Extended MHC haplotypes in 21-hydroxylase-deficiency congenital adrenal hyperplasia: shared genotypes in unrelated patients. Lancet 1983; i:l52-l56.
41. Laron Z, Pollack MS, Zamir R, et al. Late onset 21-hydroxylase defi-ciency and HLA in the Ashkenazi population: a new allele at the 21-hydroxylase loci. Human Immunol 1980; 1:55-56.
42. Layrisse Z, White C, Gunczler P, et a l. Sharing of MHC haplotypes among apparently unrelated patients with congenital adrenal hyperplasia due to 21-hydroxilase deficiency. Immuno getics 1987; 25:99-103.
43. McDevitt HO, Bodmer WF: HLA immune response genes and disease. Lancet 1974; i:l269-1275.
44. Alper CA, Awdeh ZL, Rappeport J, Raum DD, Yunis EJ. Complotypes in bone marrow transplantation. Transpl Proc 1985; 17:440-441.
45. Whitehead AS, Woods DE, Fleischnick E, et al. DNA polymorphism of the C4 genes: a new marker for analysis of the major histocompatibil-ity complex. N Engl J Med 1984; 310:88-91.
46. Fu SM, Stern R, Kunkel HG, et al. Mixed lymphocyte culture determi-nants and C2 deficiency: LD-7a associated with C2 deficiency in four families. J Exp Med 1975; 142:495-506.
47. Awdeh ZL, Raum DD, Glass D, et al. Complement human histocom-patibility antigen haplotypes in C2 deficiency. J Clin lnvest 1981; 67:581 -583.
48. Fu SM, Kunkel HG, Brusman HP, Allen FH Jr, Fotino M. Evidence for linkage between HLA histocompatibility genes and those involved in the synthesis of the second component of complement. J Exp Med 1974; 140:1108-1110.
49. Craven DE, Kunches LM, Dienstag JL, et al. Non-responsiveness to hepatitis B veccine in health care workers; results of re-vaccination and genetic typing. Ann Int Med 1986; 105:356.
50. Vento S. Antigen specific cell function in autoimmune chronic active hepatitis. Lancet 1984:1200-1204.
51. Walker ME, Szmuness W, Stevens CE, Rubinstein P. Gentetics of anti-HBs responsiveness: I HLA-DR27 and non-responsiveness to hepa-titis vaccination. Transfusion 1981; 21:601.
52. Usonis V, Kuhnl P, Rede HD, Doerr HW. Humoral immune response after hepatitis B vaccination: Kinetics of anti-HBs antibodies and de-mostration of HLA antigens. Zentra Bacteriol Microbiol Hyg 1986; 262:377-384.
53. Penichi G, Cappellacis Mole A, Morelli M, Lulli P, Pescini A. HLA and hyporesponsibiliiy to anti-HBV vaccination (genetic study of non-responder sublects to anti-hepatitis B viral Vaccine). Boll Inst Seroter Milan 1986; 65:459-463.
54. Gorzynski TJ, Davis CS. Immune response gene-associated antigens la/DR structure and function in immunologically related diseases. Mayo Clin Proc 1983; 58:457-466.
55. Cayzer T. Proceedings of the International symposium on viral Hepati-tis and liver disease. Barbican Center 1987; 26-28.
56. Milich D, Leroux-Roels GG, Chisari FV. Genetic regulation of the response after hepatitis B vaccination: Kinetics of anti-HBs antibodies and demostration of HLA. Zentra Bacteriol Microbiol Hyg 1986; 262:377-384.
57. Cars RM, Anderson RR. The disappearance rate of skin-sensitivity antibody. J Allergy 1986; 42:29.
58. Bennich H, Johansson SGO. Structure and function of human Immunoglobulin E. Adv Immunol 1971; 13:1.
59. Katz DH, Marcelleti JF. Regulation of the IgE antibody system in humans and experimetal animals. Yamura Tada eds. Progress in Immunology. V Intern Congress of Immunology 1983:465-482.
60. Smith JM. Epidemiology and natural history of asthma, allergic rhinitis and atopic dermatitis In: Middleton E, Reed C, Ellis E, eds, Allergy Principals and Practice. St Louis: Mosby 1983:771.
61. Blumenthal M, Mendell N, Yunis EJ. Immunogenetics of atopic diseases. J All Clin Immunol 1980; 65:403-405.
62. Vaughn W, Black JH. Heredity in practice of Allergy. 3th Ed St. Louis: Mosby 1954:74.
63. Deweck A, Blumenthal M, Yunis EJ, Jeannet M.HLA and allergy. In: Dausset J, Svejgaard A eds. HLA and disease. Baltimore: Williams and Wilkins Company; 1977.
64. Blumenthal M, Namboodiri K, Mendell N, Gleich G, Elston RC, Yunis EJ. Genetic transmission of serum IgE levels. J Med Genet 1981; 10:219-228.
65. Hopp R, Coleman R, Bewerta A, Townley R. Methacholine inhalation challenge as a potential genetic marker. American Academy of Pediat-rics section of Allergy and Immunology 1984.
66. Levine E, Stember R, Fontino M. Ragweed, Hay fever, genetic control and linkage to HLA-A haplotypes. Science 1972; 178:1201.
67. Blumental M, Amos D, Noreen H, Mendell N, Yunis EJ. Genetic mapping of Ir loci in man. Linkage to second loci of HLA. Science 1974; 184:1301.
68. Mendell NR, Blumenthal M, Amos DB, Yunis EJ, Elston RC. Rag-weed sensitivity. Segregation analysis and linkage to HLA-B. Cytogenetics 1978;22:330-334.
69. Blumenthal MN, Yunis EJ, Mendell NR, Amos DB. HLA and Rag-weed allergy. Monographs in Allergy; 11:83-88.
70. Marsh D, Hsu SH, Roebber M. HLA-Dw2, A genetic marker for hu-man immune responses to short ragweed allergen Ra5, I: response result-ing primarily from natural antigenic exposure. J Exp Med 1982; 155:1439-1451.
71. Marsh D, Meyers D, Freidhoff L. HLA-D w2 A genetic mark er for human response to short ragweed allergen Ra5, II: response after rag-weed immunotherapy. J Exp Med 1982; 155:1452-1463.
72. Scheleimer RP, McGlashan DW, Peters SP, Naderia R, Proud D, Adkinson NF, Lichenstentein LM: Inflammatory mediators and mecha-nism of release form purified human basophils and mast cells. J Allergy Clin Immunol 1984; 4:473-479.
73. Jones K, Moon G. Health disease and Society. London: Routledge and Kegan Paul Ltd; 1987.
74. Angell M. Disease as a reflection of the psyche. N Engl J Med 1985; 312:1570-1572.
75. Sagan LA. True causes of sickness and well-being. The health of nations. New York: Basic Books; 1987.
76. Jenner E. An inquiry into the causes and effects of the variolae vaccinal a disease discovered in some of the western countries of England, Par-ticulary Gloucestershire and known by the name of the cow-Pox, Lon-don. Sampson Low; 1978.
77. Fujinami RS, Oldstone MBA, Wroblewska Z, Frankel ME, Koprow-sky H. Molecular mimicry in virus infection: Cross-reaction of measles virus phosphoprotein or of herpes simplex virus protein with human in-termediate filaments. Proc natl Acad Sci USA 1983; 80:2346-2350.
78. Ahmed R, Oldstone MBA. Mechanism and biological implications of virus-induced polyclonal B-cell activation. In: AL Notkins and MBA Oldstone eds. Concepts in viral pathogenesis. New York: Springer Ver-lag; 231-238.
79. Fujinami RS, Oldstone MBA. Aminoacid homology between the encephalitogenic site of myelin basic protein and virus: Mechanism for autoimmunity. Science 1985; 230:1043-1045.
80. Lane DP, Hoeffler WK. SV40 large T shares an antigenic determinant with a cellular protein of molecular weight 68000. Nature 1980; 288:167-

Inmunogenética en medicina clínica 61
170.
81. Dales S, Fujinami RS, Oldstone MBA. Infection with vacinia favors the selection of hybridomas synthetisizing autoantibodies against inter-mediate filaments, one of the cross-reacting with the virus hemagglutinin. J Immunol 1983; 131:1546-1553.
82. Buchmeir MJ, Lewicki HA, Tomori O, Oldstone MBA. Monoclonal antibodies to lymphocytic choriomeningitis and pichinde viruses: Gen-eration, characterization, and cross-reactivity with other arenav Virology 1981; 113:73-85.
83. Shaw S Y, Laursen RA, Lees MB. Analogous amino acid sequences in myelin proteolipid and viral proteins. FEBS letters 1986; 207:266-270.
84. Eastmond CJ, Woodrow JC. Discordance for ankylosing spondylitis in monocytic twins. Ann Rheum Dis 1977; 36:360-365.
85. Keat A. Is spondylitis caused by klebsiella? Immunol To day, 7:144-145. 86. Rosembaum JT. Why HLA-B27: An analysis based on two animal
models. Ann Intern Med 1981; 94:261 -263. 88. Schlosstein L. Teresaki PI, Bluestone R, Pearson CM. High associa-
tion of an HLA antigen w27 with ankylosing spondylitis. N Engl J Med 1973;288:704-709.
89. Schwimmbeck PL, Yu DTY, Oldstone MBA. Auto antibodies to HLA -B27 in the sera of HLA-B27 patients with ankylosing spondylitis and Reiter's syndorme. J Exp Med 1987; 166:173-181.
90. Sullivan J, Upfold L, Geczy AF, Baschir HV, Edmonds J. Immuno-chemical characterization of klebsiella antigens which specifically mod-ify an HLA-B27 associated cell-surface component. Hum Immunol 1982; 5:295-307.
91. Sullivan JS, Prendergast JK, Geczy AF, Edmonds JP et al. Cross-reacting bacterial detenninants in ankylosing spondilitis. Am J Med 1988; 85 (Suppl 6A):54-55.
92. Srinivasappa J, Saegusa J, Prabhakar BS et al. Molecular mimicry: Frecuency of reactivity of monoclonal antiviral antibodies with normal tissues. J Virology 1986; 57:397-401.
93. Senecal JL, Rothfield HF, Oliver JM. Immunoglobulin M autoanti-body to vimentin intermediate filaments. J Clin lnvest 1982; 69:716-721.
94. Kurki P, Helve T, Virtanene I. Antibodies to cytoplasmic intermediate filaments in rheumatic diseases. J Rheumatol 1983; 10:558-562.
95. Van de Risn, Zabriskie JB, McCarthy. Group A streptococcal anti-gens cross-reactive with myocardium: Purification of heart reactive anti-body and isolation and characterization of the streptococcal antigen. J Exp Med 1977; 146:579-599.
96. Khoury EL, Ritacco V, Cossio PM, Laguens RP, Szarfman A, Diez C, Avana RM. Circulating antibodies to peripheral nerve in american trypanosomiasis. Chagas disease. Clin Exp Immunol 1979; 36:8-15.
97. Cunninghan MW, Krisher K, Graves DC. Murine monoclonal anti-bodies reactive with human heart and group A streptococcal membrane antigens. Infect lmmunol 1974; 46:34-41.
98. Wood JN, Hudson L, Jessell TM, Yamamato M. A monoclonal anti-body defining antigenic determinants on sub-populations of mammalian neurones and trypanosoma cruzi parasites. Nature 1982; 296:34-38.
99. Roit IM, Doniach D, Campbell PN, Vaughan-Hudson R. Autoanti-bodies in Hashimoto's disease (Lymphadenoid gitre). Lancet 1956; 2:820-823.
100. Doniach D, Roitt IM. Human organ specific autoimmunityL: Personal memories. Autoimmunity 1988; 1:11-13.
101. Botazo GF, Todd I, Mirakian R, Belfiore A, Pujol-Borrell R. Organ-specific autoimmunity: A 1986 Overview. Immunological reviews 1986; 94:137-168.
102. Botazzo GF, Florin-Christensen A, Doniach D. Islet cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiences. Lancet 1974;2:1279-1283.
103. BotazzoGF,Pouplard A, Florin-Christensen A,Doniach D. Autoanti-bodies to prolactin-secreting cells of human pituitary. Lancet 1975; 2:97-101.
104. Eisenbarth GS. Immunoendocrinopathy syndromes. In: Wilson JD, Foster DW eds. Williams Texbook of endocrinology. 7th Ed. Philadel-
phia: WB Saunders; 1985:1290-1300. 105. Farid NR, Bear JC. The human major histocompatibility complex and
endocrine disease. Endocrine Reviews 1981; 2:50-86. 106. Loveridge N, Bitensky L, Chayen J, et al. Inhibition of parietal cell
function by human gamma-globulin containing gastric parietal cell antibodies. Clin Exp Immunol 1980; 41:264-270.
107. Adams DD. The clinical status of patients whose scrheve given the ab-normal response when assagent for thyrotropin. Pore univ otago Med Sch 1956; 34:29-35.
108. Doniach D. Clinical observations and hypothesis related to TSH recep-tors. In: Muhlen A Schleusener eds. Biochemical basis of thyroid stimu-lation and thyroid hormone action. Sttugart: Georgethiene; 34-36.
109. Drexhage HA, Botazzo GF, Doniach D, Bitensky L, Chayen J. Evi-dence for thyroid growth stimulating immunoglobulins in some goitrous thyroid diseases. Lancet 1980; 2:287-292.
110. Drexhage HA, Botazzo GF, Bitensky L, Chayen J, Doniach D. Thy-roid growth-blocking antibodies in primary myxedema. Nature 1981; 289:594-596.
111. Kahn CR, Kasuga M, King GL, Grunfeld C. Autoantibodies to insulin receptors in man: Immunological determinants and mechanism of ac-tion. In: Evered D, ed. Receptors, autoantibodies and disease. CIBA symposium 90, London, Pitman; 91-105.
112. Konishi J, Lida Y, Kasagi K, Eudo K, Misaki T, Torizuka K. Thyro-tropin receptor blocking antibodies. In: Labrie F, Proul eds Endocrinol-ogy. Amsterdam: Excerpta Medica; 559-562.
113. Compston A, Vincent A. Multiple sclaerosis and Myastenia Gravis. Clin Immunol Allergy 1985; 5:569-584.
114. Dawkins RL, Garlepp MJ. Autoimmune diseases of muscle: Myas-tenia Gravis and Myositis. In; Rose NR, Mackay IR eds. The autoim-mune disease; 591-615.
115. Pujol-Borrel R, Hanafusa T, Chiovato L. Botazzo GF. Lecitin-In-duced expression of DR antigen on human cultured follicular thyroid cells. Nature 1983:304.
116. Hanafusa T, Pujol-Borrel R, Chiovato L, Russell RCG, Doniach D, Botazzo GF. Aberrant expression of HLA-DR antigen on thyrocytes of Graves' disease: Relevance to thyroid autoimmunity. Lancet 1983; 2:1111-1115.
117. Botazzo GF, Pujol-Borrel R, Hanafusa T, Feldman M. Role of aber-rant HLA-DR expression and antigen presentation in induction of endo-crine autoimmunity. Lancet 1983; 1115-1119.
118. Tood I, Pujol-Borrel R, Hammond LJ, Botazzo GF. Interferon gamma induces HLA-DR expression by thyroid epithelium. Clin Exp Immunol 1985;61:265-270.
119. Londei M,Lamb JR, Botazzo GF, Feldman M. Epithelial cell express-ing aberrant MHC class I determinants can present antigen to cloned human T cells. Nature 1984; 312:639-641.
120. Londei M, Botazzo GF, Meldnann M. Human T cell clones from au-toimmune thy roid glands: specific recognition of autologous thyroid cells. Science 1985; 228:85-89.
121. Grubeck-Loebenstein B, Londei M,Greenall C, Pirich K, Kassal H, Waldhausl W, Feldman M. Pathogenic relevance of HLSA class II expressing thyroid follicular cells in nontoxic gotierand in Graves' disease. J Clin lnvest 1988; 81:1608-1614.
122. Granados J, Alarcón-Segovia D, Oria CV, et al. Anticardiolipin au-toantibodies (ACLA) in families of patients with systemic lupus erythe-matosus (SUE), relationship to complement serotypes (Abstr) Arthritis Rheum 1987; Supp 30:522.
123. Hargraves MM, Richmond H, Morton R. Presentation of two bone marrow elements: the "tart" and the "LE" cell. Mayo Clin Proc 1948; 23:25-28.
124. Haserick JR, Bortz DW. Normal bone marrow inclusion phenomena induced by lupus Erythematosus plasma. J Invest Dermatol 1949; 13:47-49.
125. Coons AH, Kaplan MH. Localization of antigen in human cells, I. Im-provements in a method for detection of antigen by means of a fluores-
Acta Med Colomb Vol. 15 N°1 ~ 1990

62 G. de Egea y cols.
cent antibody. J Exp Med1950; 91:1-13. 126. Frion GJ. Clinical application of lupus serum nucleoprotein reaction
using fluorescent antibody technique. J Clin Invest 1957; 36:890-896. 127. Bardawill WA, Toy BL, Galins BA, et al. Disseminated lupus erythe-
matosus, scleroderma, and dermatomyositis as manifestations of sensiti-zation to DNA protein, I, an immunohistochemical approach. Am J Pathol 1958;34:607-629.
128. Beck JS. Variation in the morphological patterns of "autoimmune" nu-clear fluorescence. Lancet 1961; 1:1203-1205.
129. Deicher HRG, Holman HR, Kunkel HG. The precipitin reaction be-tween DNA nad serum factor in systemic lupus erythematosus. J Exp Med 1959; 109:97-114.
130. Sharp GC, Irvin, WS, Tan EM, et al. Mixed Connective tissue disease: An apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen. Am J Med 1972; 52:148-159.
131. Lerner MR, Steitzz JA. Antibodies to small nuclear RNAs complexed with proteins are produced by patients with systemic lupus erythematosus. Natl Acad Sci USA 1979; 76:5495-5499.
132. Lerner MR, Boyle JA, Moun t SM, et al. Are RNPs involves in splicing? Nature 1980; 283:220-224.
133. Petterson I, Hinterberger M, Mimori T, et al. The structure of ma-malian small nuclear ribonucleoprotein. J Biol Chem 1984; 259:5907-5914.
134. Yang VW, Lerner MR, Steitz. J A, et al. A small nuclear ribonucleopro-tein is required for splicing of adenoviral early RNA sequences. Proc Natl Acad Sci USA 1981; 78:1371-1375.
135. Fischer DE, Reeves WH, Conner GE, et al. Pulse labeling of small nuclear ribonucleoproteins in vivo reveals distintc patterns of antigen recognition by human autoimmune antibodies. Proc Natl Acad Sci USA 1984.
136. Mathews MB, Francoeur AM. The antigen recognizes and binds to the 3'oligouidylate tail of a small RNA. Mol Cell Biol 1984; 4:1134-1140.
137. Whittingham S, Naselli G, McNeilage LJ, Coppel RS, Sturgess AD. Serological diagnosis of primary Sjögrens syndrome by means of hu-man recombinant La (SS-B) as nuclear antigen. Lancet 1987; 2:1-3.
138. Jerne NK.The natural-selection theory of antibody formation. Proc Natl Acad Sci USA 1955; 41:849-857.
139. Flier JS, Kahn CR, Roth J. Receptors, antireceptor antibodies and mechanisms of insulin resistance. N Eng J Med 1979; 300:413-419.
140. Benson EA, HoP, WangC, Wu PC, Fredlung PN, Yueng RT.Insulin autoimmunity as a cause of hypoglycemia. Arch Inter Med 1984; 144:2351-2354.
141. Moller DE, Ratner RE, Borenstein DG, Taylor SI. Autoantibodies to the insulin receptor as a cause of autoimmune hypoglycemia in systemic lupus erythematosus. Am J Med 1988; 84:334-338.
142. Burdette S, Schwartz RS. Network idiotype and anti-idiotype. N Engl J Med 1987; 317:219-224.
143. Conley CL, Hartman RC. A hemorrhagic disorder caused by circulat-ing anticoagulant in patients with disseminated lupus erythematosus. J Clin lnvest 1952; 31:621-622.
144. Frick PG. Acquired circulating anticoagulants in systemic "Collagen disease": Autoimmune thromboplastin deficiency. Blood 1955; 10:691-706.
145. Schleider MA, Nachman RL, Jaffe EA, et al. A clinical study of the lupus anticoagulant. Blood 1976; 48:499-509.
146. Boxer M, Ellman L, Carvallo A. The lupus anticoagulant. Arthritis Rheum 1976; 19:1244-1248.
147. Harris EN, Gharavi AE, Boey ML, et al. Anticardiolipin antibodies: Detection by radioimmunoassay and association with thrombosis in sys-temic lupus erythematosus. Lancet 1983; 2:1211-1214.
148. Hughes GRv. Thrombosis, abortion, cerebral disease and the lupus anticoagulant. Br Med J 1983; 1088-1089.
149. Harris EN, Gharavi AE, Hughes GRV. Anti-phospholipid antibodies.
Clin Rheum Dis 1985; 11:591-610. 150. Inove K, Nojima S, Tomizawa T. The specificity of cardiolipin in the
serologic reaction. J Biochem 1965; 57:824-826. 151. Bulletin of the World Health Organization, 1968:39:483.
152. Terasaki PI ed. Histocompatibility Testing, 1970 Copenhagen: Munksgaard; 1970:49.
153. Bulletin of the World Health Organization 1972; 47:659.
154. Bulletin of the World Health Organization, 52. 155. Bulletin of the World Health Organization, 1978; 461. 156. Albert ED, et al. Histocompatibility Testing 1984. Heidelberg, Sprin-
ger Verlaag. 157. Shows TB, AlperC A, Boostsma D, et al. International System for Human
Gene nomenclature. Gene 1979; 25:96-116.
158. Albert Bruce, et al. Molecular Biology of the cell. New York. Garland publishig Inc., 1983.
159. Watkins PC. Restriction Fragment Length Plymorphism (RFLP): Ap-plications in human chromosome mapping and genetic disease research. Biotechniques 1988; 6:310-320.
160. Nathans D, Smith HO. Restriction endonucleases in the analysis of DNA molecules. Ann Rev Biochem 1975; 44:273-292.
161. Botstein D, et al. Construction of a genetic linkage map in man using restriction fragment lenght polymorphism. Am J Hum Genet 1980; 32:314,
162. Simon M.Establishment of an approved clinical laboratory based on the analysis of restriction fragment length polymorphisms. In Nucleic acis probes in the diagnosis of human genetic diseases. AM Wiley, ed New York: Liss Inc.; 1988:195-216.
163. Southern E.Detection of specific sequences among DNA fragment sepa-rated by gel electrophoresis. J Mol Biol 1975; 98:503-512.
164. Frezal J, Klinger HP, eds. Human Gene Mapping 9: The Paris confer-ence 1987. Cytogenetics and Cell Genetics 1978; 44:1.
165. Baird M, Wexier K, Clyne M, Meade E, Ratzlaff L, Smalls G, Benn P, Glassberg J, Blazs I. The application of DNA-print for the estimation of paternity. Advances in forensic haemogenetics. New York: Springer-Verlag; 1987.
166. Bodmer WF. The HLA system, 1984. Histocompatibility testing 1984. Albert ED, ed. Berlin: Heiberg Springer-Verlag 1984:11-28.
167. The nomenclature commnittee on leukocyte antigens. Nomenclature for factors of the HLA systems, 1987. Immuno gentiics. Springer-Verlag 1988:391-398.
168. High KA,BenzEJ, Jr.The ABC's of molecular genetics:A haemotolo-gist's introduction. In: Hoffbrand A V ed. Recen t Adv Haematol. 1985; 4:25-61.
169. Weatherall DJ ed. The new genetics and clinical practice. New York: Oxford University Press; 1982.
170. Parham P. Function and polymorphism of human leukocyte antigen A-B-C molecules. Am Journal Med 1988; 85 (Suppl 6a.):2-5.
171. Strachan T, Sodoyer R, Damotte M, Jorgan BR. Complete Sequence of functional class I HLA gene, HLA-A3: implications for the of HLA genes. EMBO 1984; 3:887-894.
172. Sodoyer R, Damotte M, Delovich TL, Trucy J, Jordan BR, Strachan T. Complete nucleotide sequence of a gene encoding a functional human histocompatibility antigen (HLA-Cw3). EMBO J 1984; 3:879-885.
173. Gustafsson K, Wiman K, Emmoth E, et al. Mutations and selection in the generation of class II histocompatibility antigen polymorphism. EMBO 1984;3:1655-1662.
174. Kappes DJ, Arnot D, Okada K, Strminger J. Structure and polymor-phism of HLA class II SB lightchain genes. EMBO J 1984; 3:2985-2993.
175. Sood Ak, Pereira D, Weissman S. Isolation and partial nucleotide of a cDNA clone for human histocompatibility antigen HLA-B by use of deoxynucleotide primer. Porc Natl Acad Sci USA 1981; 78:616-620.
176. Cohen D, et al. DNA polymorphism of HLA class I and class /I regions. Immunol reviews 1985; 85:87-105.
177. Svejgaard A, Platz P, Ryder LP. Insulin-dependent diabetes mellitus.

Inmunogenética en medicina clínica 63
In: Histocompatibility testing Terasochi Ped: UCLA Tissue typing labo-ratory. Los Angeles 1980:638.
178. Awdeh ZL, Raum D, Yunis EJ, Alper CA. Extended HLA comple-ment allele haplotypes: Evidence for T/t like complex in man. Proc Natl Acad Sci USA 1983; 80:259.
179. Jorgensen R, Lamm LU, Kissmeyer-Nielsen F. Mixed lymphocyte culture with inbred individuals. An approach to MLC typing. Tissue Antigen 1973; 3:323.
180. Dupont B, Jersild C, Hansen GS, Nielsen LS, Thomsen M, Svejgaard A. Typing of MLC determinants by means of LD-homozigous and LD-heterozigous reference cells. Transplant Proc, 5:1543.
181. Mempel W, Grosse-Wilde H, Barumann P, et al. Population genetics of the MLC response: typing for MLC determinants using homozygous and heterozygous reference cells. Transplant Proc 1973; 5:1529.
182. Nuñez G, Giles RC, Ball EJ, Hurley CK, Capra JD, Stansky P. Ex-pression of HLA-DR, MB, MT and SB antigens on human mononuclear cells: Identification of two phenotypically distinct monocyte populations. J Immunol 1984; 133:1300.
183. Albert ED, Baur MP, Mayr WR eds. Histocompatibility testing 1984. Berlin: Springer, 1984.
184. Wake C, Long E, Mach B. Allelic polymorphism and complexity of the genes for the HLA-DR beta gene. Direct analysis by DNA-DNA hybridization. Nature 1982; 300:372.
185. Andersson M, Bohme J, Andersson G, Moler E, Thorsby E, Rask L, Peterson PA. Genomic hybridization with class II transplantation
186. Carlsson B, Bohme J, Lundgren G, Moller E, Petersen PA, Rask L, Ringden O, Wallin J. HLA class II genes estudied with genomic hy-bridization in kidney and bone marrow transplantation donor-recipient pairs .Transplant Proc 1985; 17:952.
187. Wallin J. Bohme J, Carlsson B, Moller E, Peterson PA, Rask L. HLA class II polymorphism: restriction fragment patterns correlated to Ninth Workshop serology and function. In: Albert ED, Baur MP, Mayr WD eds. Histocompatibility testing 1984. Berlin: Springer; 572-576.
188. Moller E, Carlsson B, Wallin J. Implications of structural class II gene
polymorphism for the concept of serologic specificities. Immunol Rev 1985; 85:107.
189. Bohme J, Anderson M, Hammerling U, et al.HLA-DRb genes vary in number between different HLA-DR specificities, whereas the number of DQb genes is constant .J Immunol 1985; 135:2149.
190. Shaw S, Johnson AH, Shearer GM. Evidence for a new segregant se-ries of B-cell antigens that are encoded in the HLA-D region and that stimulate secondary allogeneic proliferative and cytotoxic responses. J Exp Med 1980; 152:565.
191. Nuñez G, Ball E, Stastny P. Antigen presentation by adherent cells from human peripheral blood. Correlation between T-cell activation and ex-pression of HLA-DQ and DR antigens. Human Immunol 1987; 19:29-39.
192. Heyningen van V, Guy K, Newman R, Steele CM. Human MHC class II molecules as a differentiation markers. Immunogenetics 1982; 16:459.
193. Giles RC, Capra DC. Structure, function, and genetics of human class II molecules. Adv Immunol 1985; 37:1.
194. Dilella A, Huang W-M, Wou SLC. Screening for phenylketonuria mutations by DNA amplification with the polymerase chain reaction. Lancet 1988; ii:497-99.
188. Kogan SC, Doherty ABM, Gitscher J. An improved method for prena-tal diagnosis of genetic diseases by analysis of amplified DNA sequences. NEngl J Med 1987; 317:985-990.
189. Higuchi R, von Beraldingen CH, Sensabaugh GF, Erlich HA. DNA typing from single hairs. Nature 1988; 332:543-46.
190. Nicholas L, Philp S, Robert W. Simple, non-invasive method to obtain DNA for gene analysis.Lancet 1988; ii:1356-58.
191. Ou CZ, Mitchell SW, Krebs J, et al. DNA amplification for direct detection of human immunodeficiency virus-1 (HIV-1) in DNA of pe-ripheral mononuclear cells. Science 1987; 239:295-97.
192. Shibata DK, Amheim N, Martin WJ. Detection of human papiloma virus in paraffin-embedded tissue using the polymerase chain reaction. J Exp Med 1988; 167:225-30.
Acta Med Colomb Vol. 15 N°1 ~ 1990






