Reactions of α -Hydrogens : Aldol and Claisen Condensation Reactions

651.06
139
Pericyclic Reactions
-describes a family of reactions, all concerted, which proceed through a transition state
involving the cyclic movement of electrons
3 types:
electrocyclic reaction cycloaddition
( 1 ring 0 ring ) ( 2 mol. 1 ring )
conversion of σ−>π bond conversion of π −> 2 σ bonds
sigmatropic rearrangement
( acyclic acyclic )
no change in bonding
Electrocyclic Reactions
-formation of single bond between ends of a linear π system or the reverse process
ex:
175o
one interesting feature is its stereospecificity:

651.06
140
175o
cis-3,4-dimethylcyclobutene (E,Z)-2,4-hexadiene
175o
trans (E, E)
Winter, TL 1965, 1207
What accounts for this specificity?
-Woodward & Hoffman proposed the “conservation of orbital symmetry”
see: Hoffman & Woodward, Accts. Chem. Res. 1968, 1, 17
c2
!'
!
symmetry elements present in s.m. & pdt
H
H
H
H

651.06
141
!*
"*
"
!
"4*
"3*
"2
"1
Now consider the likely transition states for this reaction:
(the π orbitals of c1 & c4 must rotate to form σ orbital)
vs.
conrotatory disrotatory
note that not all symmetry elements are maintained in the two transition states:
conrotatory has only a c2 axis
disrotatory has only the σ plane
Now we classify orbitals on the basis of these symmetry elements:

651.06
142
!*
"*
"
!
"4*
"3*
"2
"1
A
S
A
S
S
A
S
A
c2 c2
conrotatory process
(note that orbitals of like symmetry are correlated & that correlation lines of like
symmetry do not cross)
Note that bonding orbitals in s.m. correlate with bonding orbitals in pdt
⇒ symmetry allowed
Now, let’s see the disrotatory process:
!*
"*
"
!
"4*
"3*
"2
"1
A
A
S
S
A
S
A
S
! !
disrotatory process

651.06
143
Note that, due to symmetry considerations, π2 correlates with π3*, meaning that the
product is in an excited (higher energy) state
⇒ THIS IS THERMALLY FORBIDDEN
Thus, electrocyclic ring opening of cyclobutene is predicted to be conrotatory
Another formulation used to explain these process was created by Dewar & Zimmerman:
Dewar, M.J.S. Angew. Chem. Int. Ed. Engl. 1971, 10, 761
Zimmerman, H.E. JACS 1966, 1564, 1566
Zimmerman, H.E. Accts. Chem. Res. 1971, 4, 272
This approach uses interaction diagrams
consider s.m. as a series of basis (p) orbitals:
look at both transition states and connect lobes which were bonded:
vs.
conrotatory disrotatory
Note that in the conrotatory diagram, the interaction path must travel across the plane of
the ring, while disrotatory path is on the same side. This can be seen more clearly by
ignoring the molecular framework altogether & simply tracing a path between orbitals:

651.06
144
conrotatory vs. disrotatory
These two situations are referred to as Huckel (disrotatory) & anti-Huckel or Mobius
(conrotatory)
Now, let’s phase the orbitals:
Möbius Hückel
(1phase inversion) (0 phase inversions)
No matter what the phasing, the Möbius has one inversion
For Hückel transition states, we know how to define aromaticity:
4n+2 electrons ⇒ aromatic
4n electrons ⇒ anti-aromatic
What about Möbius transition states?
The Hückel 4n+2 rule is reversed:
4n+2 electrons ⇒ anti-aromatic
4n electrons ⇒ aromatic
Heilbronner, TL 1964, 1923

651.06
145
Conclusion:
4n+2 systems: Hückel thermally allowed, Möbius thermally forbidden
4n systems: Möbius thermally allowed, Hückel thermally forbidden
Caveats:
1) these rules apply to thermal, ground state reactions
2) these rules only consider electronics; steric strain can change things
651.06
146
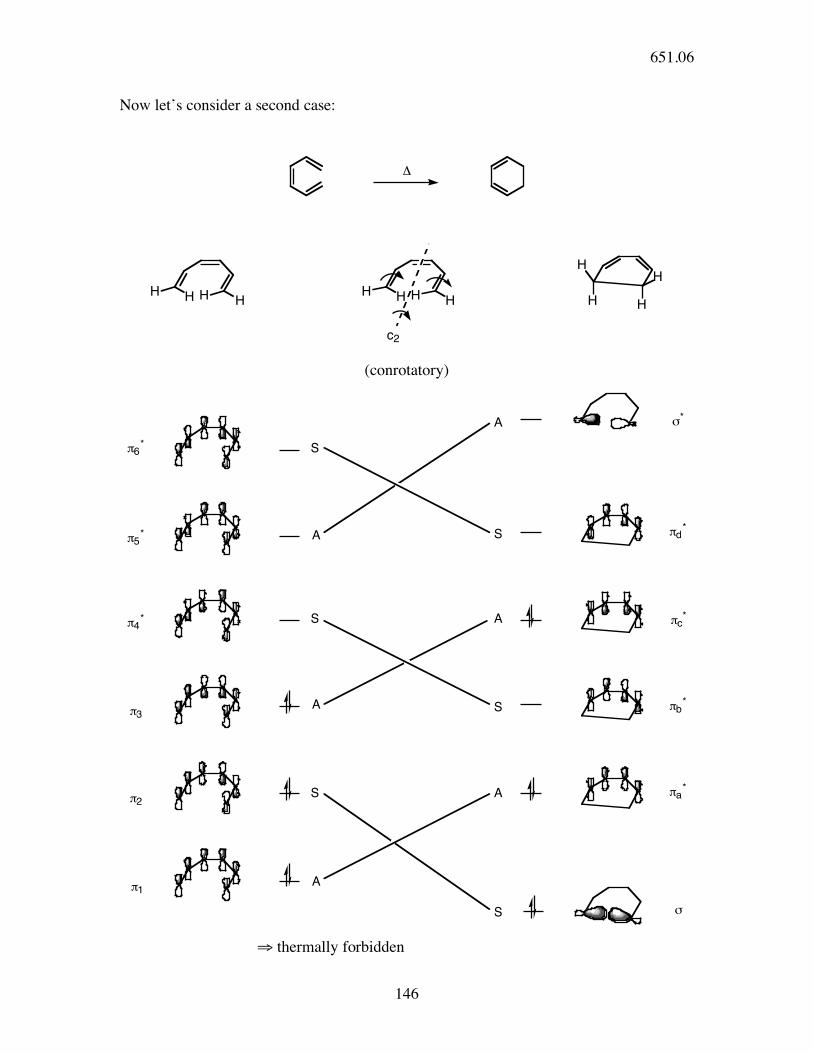
Now let’s consider a second case:
!
HH H
HH
H HH
H
H
H
H
c2 (conrotatory)
!6*
!5*
!4*
!3
!2
!1
S
A
S
A
S
A
S
A
S
A
S
A "*
!d*
!c*
!b*
!a*
"
⇒ thermally forbidden
651.06
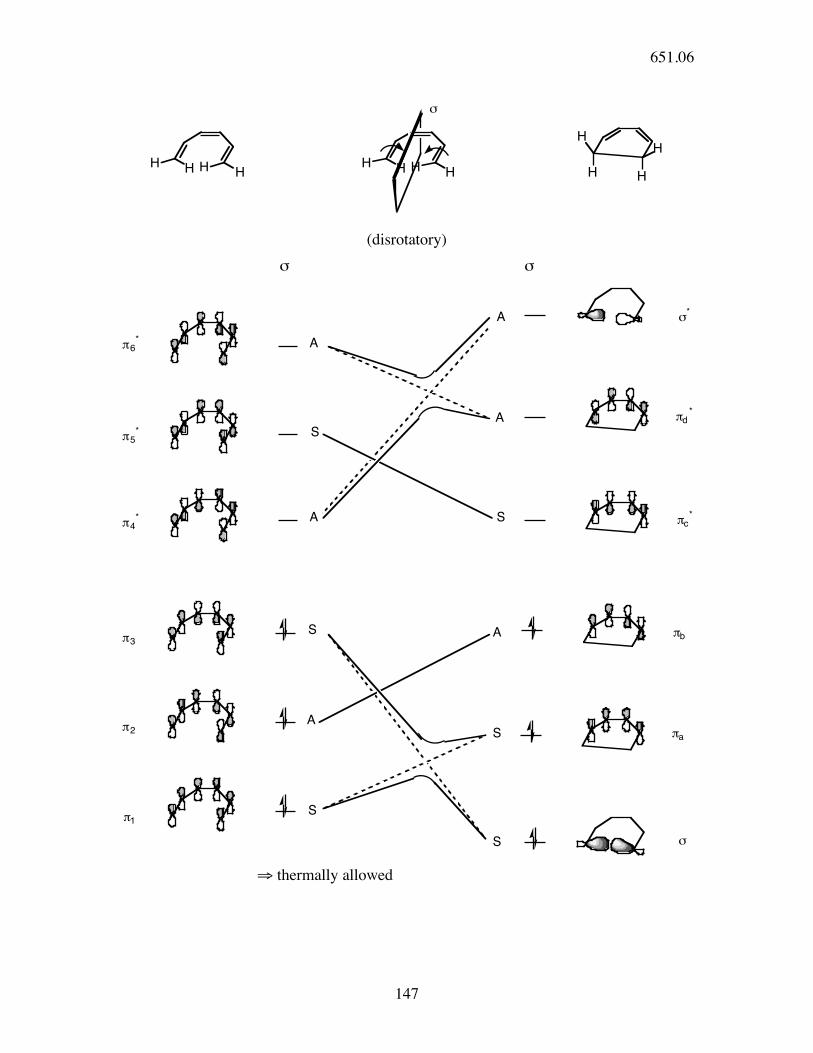
147
HH H
HH
H HH
H
H
H
H
!
(disrotatory)
σ σ
!6*
!5*
!4*
!3
!2
!1
"*
!d*
!c*
!b
!a
"
A
S
A
S
A
S
S
S
A
S
A
A
⇒ thermally allowed
651.06
148
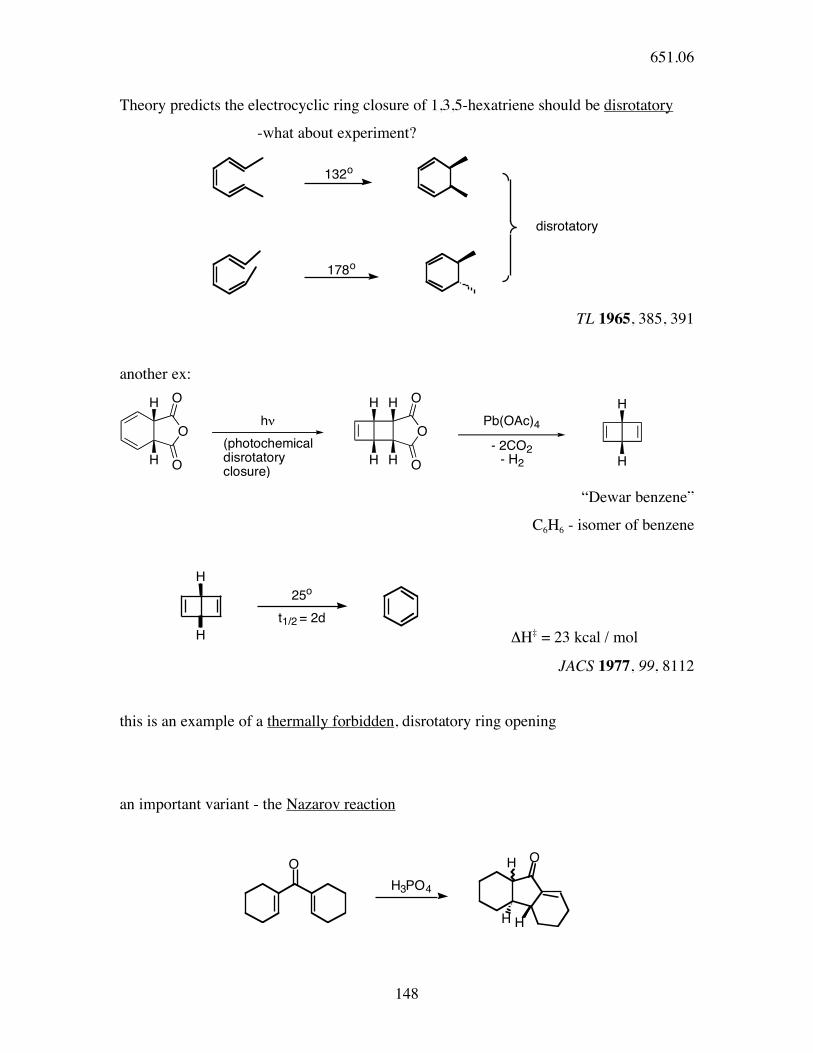
Theory predicts the electrocyclic ring closure of 1,3,5-hexatriene should be disrotatory
-what about experiment?
132o
178o
disrotatory
TL 1965, 385, 391
another ex:
O O
O
O
O
O
H
H H
H
H
H
h!
(photochemicaldisrotatoryclosure)
- 2CO2- H2
Pb(OAc)4
H
H
“Dewar benzene”
C6H6 - isomer of benzene
H
H
25o
t1/2 = 2d
ΔH‡ = 23 kcal / mol
JACS 1977, 99, 8112
this is an example of a thermally forbidden, disrotatory ring opening
an important variant - the Nazarov reaction
OO
H
H H
H3PO4
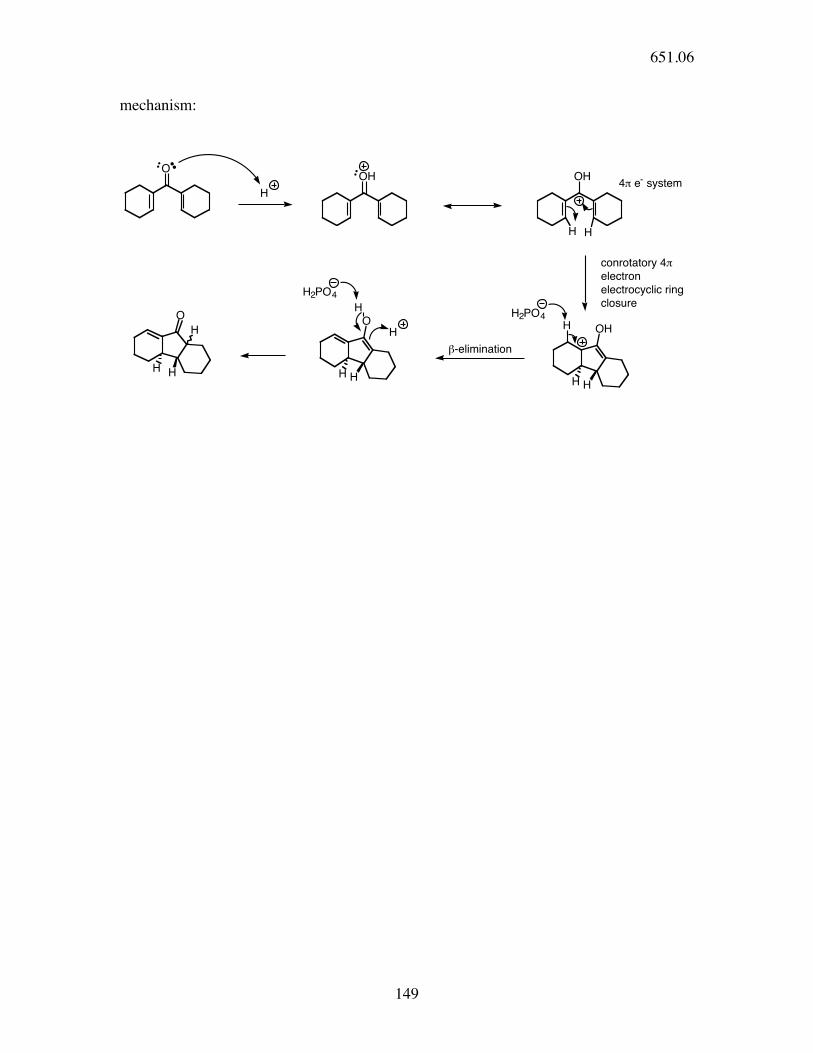
651.06
149
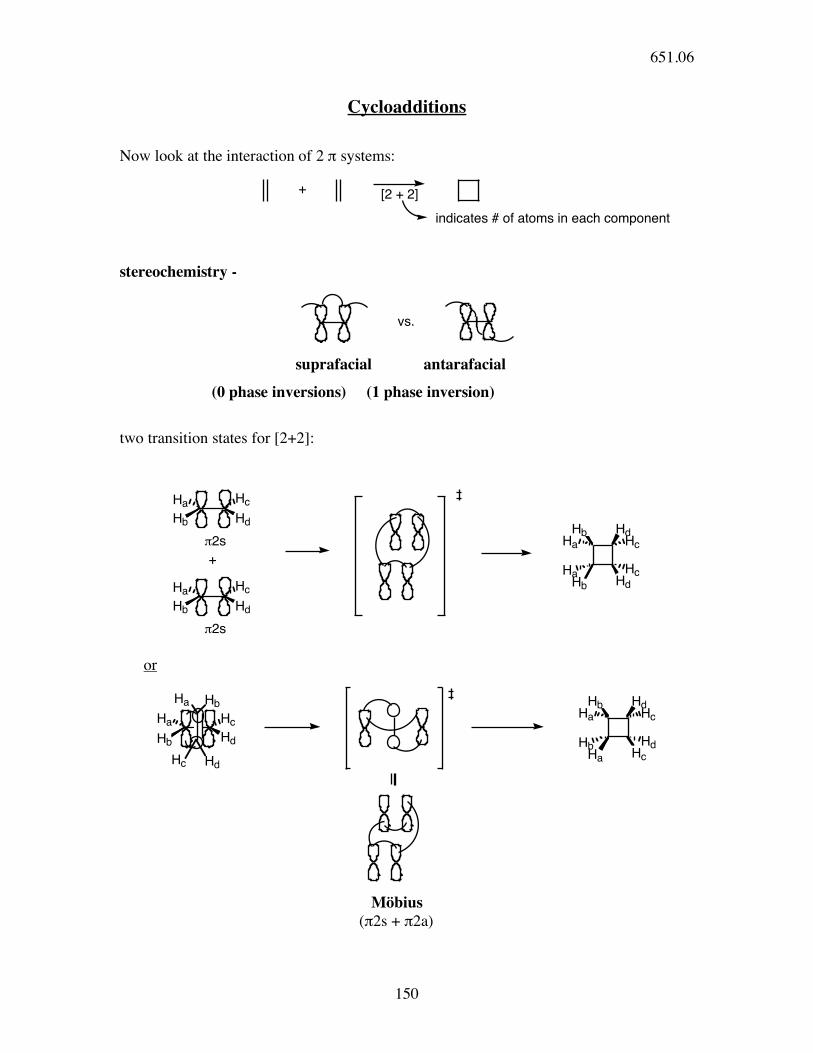
mechanism:
O
H
OH OH
HH
OH
H H
O
H H
O
H
4! e- system
conrotatory 4!
electron
electrocyclic ring
closure
HH2PO4
"-elimination
H
H2PO4
H
H H
651.06
150
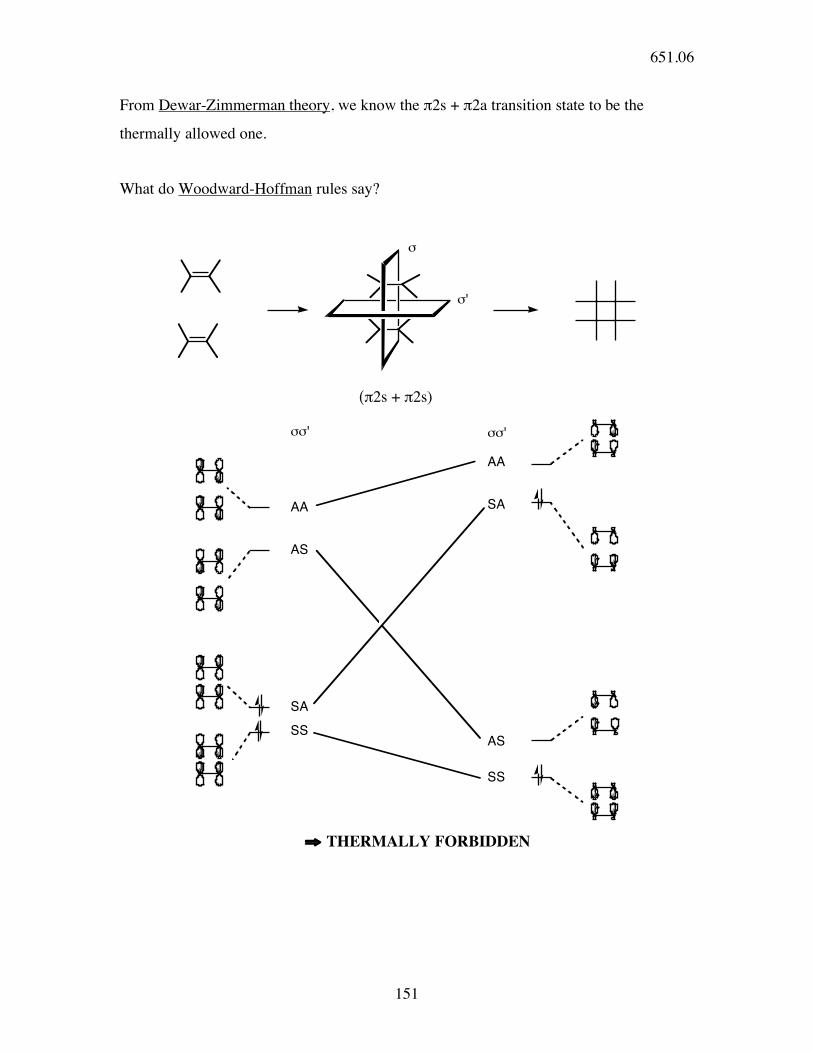
Cycloadditions
Now look at the interaction of 2 π systems:
[2 + 2]+
indicates # of atoms in each component
stereochemistry -
vs.
suprafacial antarafacial
(0 phase inversions) (1 phase inversion)
two transition states for [2+2]:
Hb Hd
Hb Hd
Ha Hc
HcHa
!2s
!2s
+
Hb
Hb
Hd
Hd
Hc
Hc
Ha
Ha
or
Hb Hd
HcHa
Ha Hb
Hc Hd
Hb
Ha
Hd
Hc
Hc
Hd
Ha
Hb
Möbius (π2s + π2a)
651.06
151
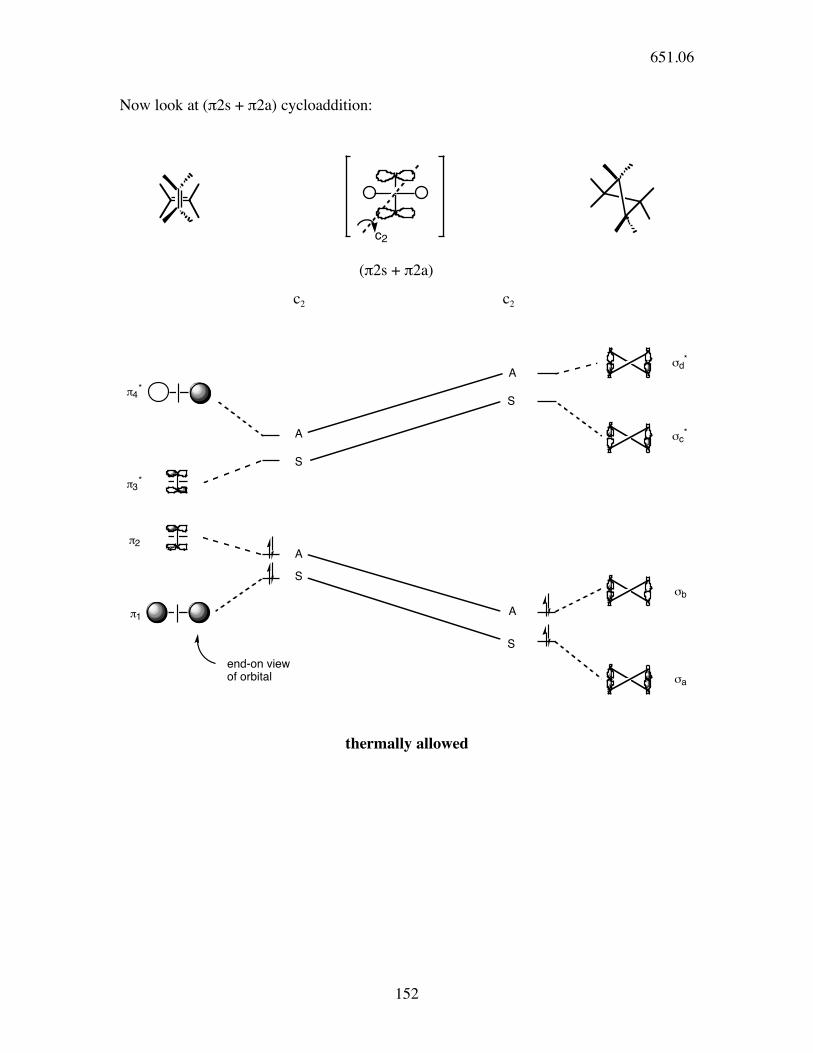
From Dewar-Zimmerman theory, we know the π2s + π2a transition state to be the
thermally allowed one.
What do Woodward-Hoffman rules say?
!'
!
(π2s + π2s)
!!' !!'
AA
AS
SA
SS
SS
AS
SA
AA
⇒ THERMALLY FORBIDDEN
651.06
152
Now look at (π2s + π2a) cycloaddition:
c2 (π2s + π2a)
c2 c2
A
S
A
S
A
S
A
S
!4*
!3*
!2
!1
end-on view
of orbital
"d*
"c*
"b
"a
thermally allowed
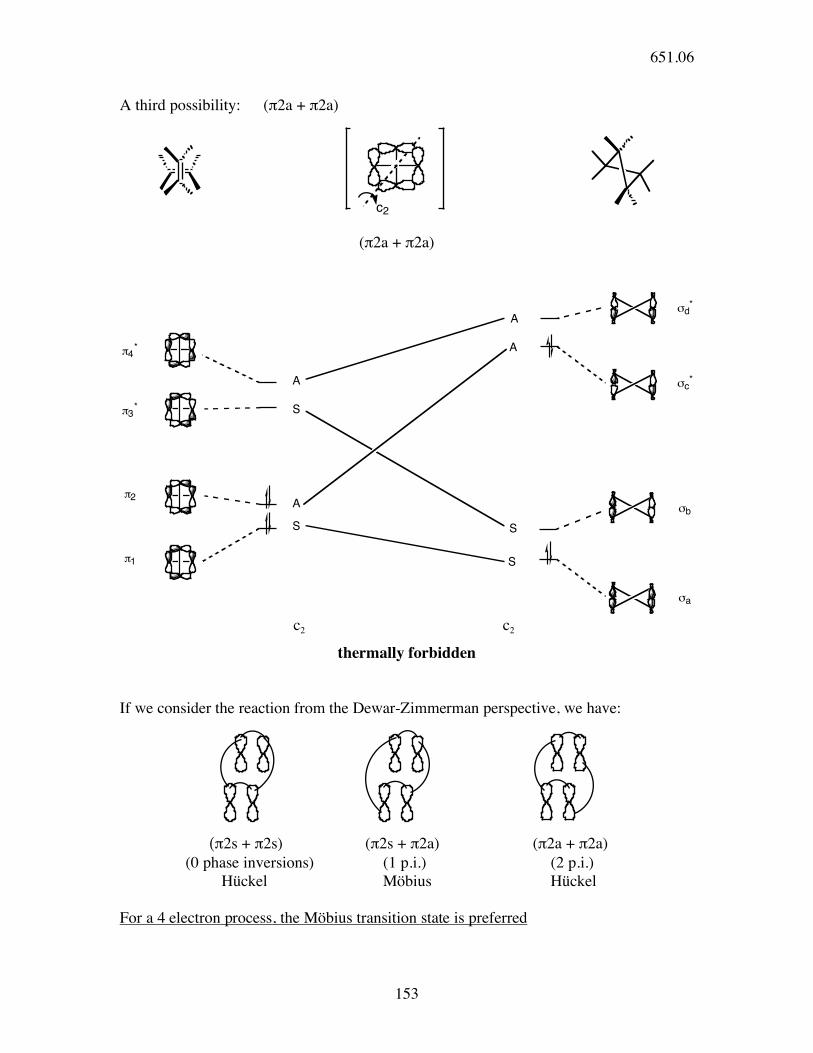
651.06
153
A third possibility: (π2a + π2a)
c2 (π2a + π2a)
c2 c2
thermally forbidden
If we consider the reaction from the Dewar-Zimmerman perspective, we have:
(π2s + π2s) (π2s + π2a) (π2a + π2a) (0 phase inversions) (1 p.i.) (2 p.i.) Hückel Möbius Hückel
For a 4 electron process, the Möbius transition state is preferred
A
S
A
S
A
A
S
S
!4*
!3*
!2
!1
"d*
"c*
"b
"a
651.06
154
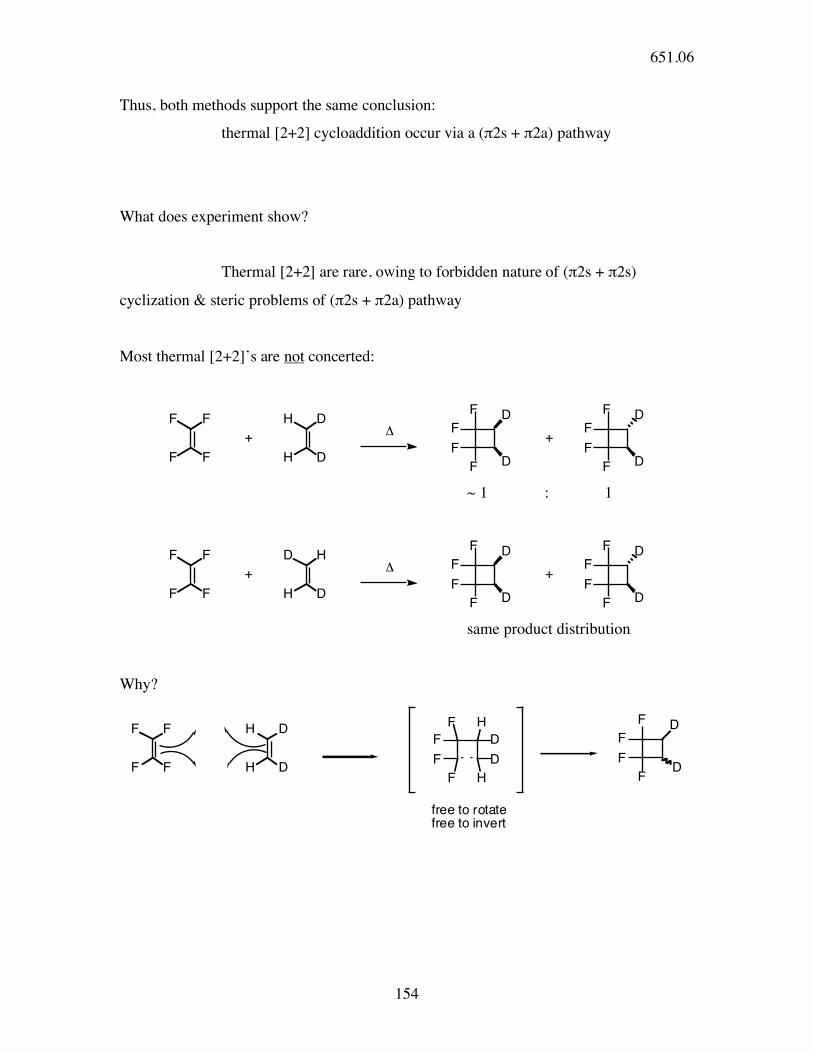
Thus, both methods support the same conclusion:
thermal [2+2] cycloaddition occur via a (π2s + π2a) pathway
What does experiment show?
Thermal [2+2] are rare, owing to forbidden nature of (π2s + π2s)
cyclization & steric problems of (π2s + π2a) pathway
Most thermal [2+2]’s are not concerted:
F F
F F
H D
H D
F
F
F
F+
!+
F
F
F
F
D
D D
D
~ 1 : 1
F F
F F
D H
H D
F
F
F
F+
!+
F
F
F
F
D
D D
D
same product distribution
Why?
F F
F F
H D
H D
F
F
F
F
D
D
F
F
F
F
H
D
D
H
free to rotate
free to invert
651.06
155
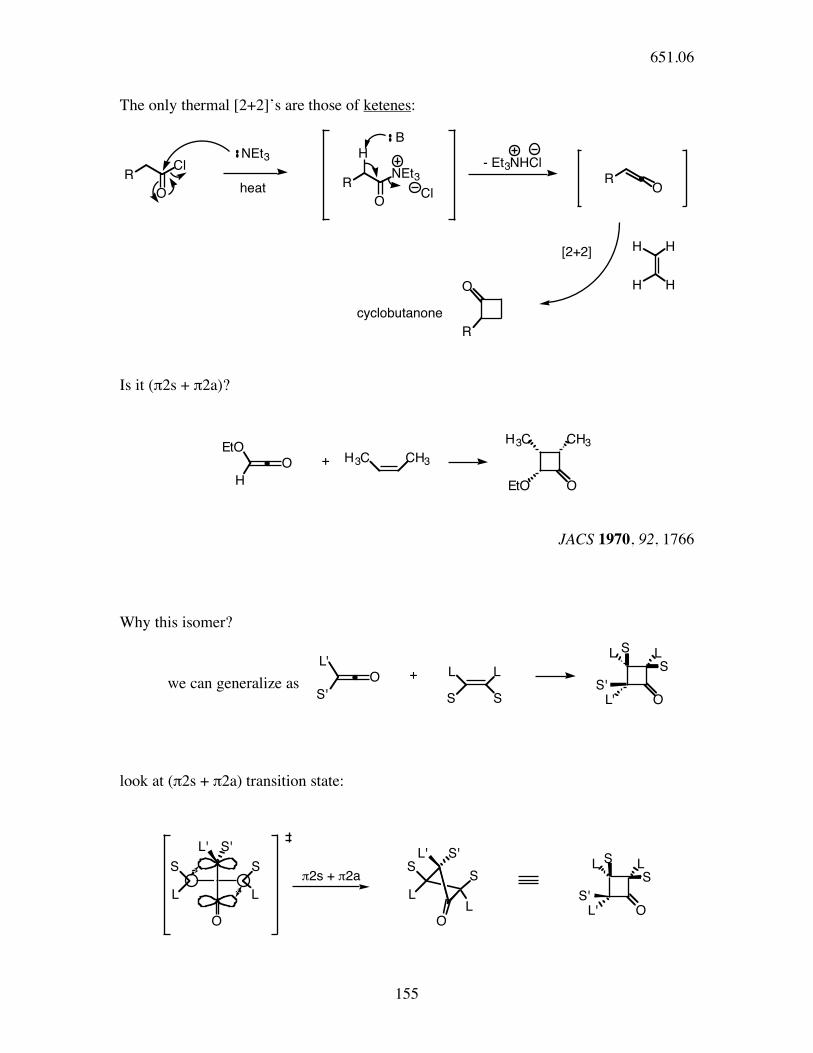
The only thermal [2+2]’s are those of ketenes:
RCl
OR
NEt3
O
H
RO
HH
H HO
R
NEt3
heat Cl
- Et3NHCl
[2+2]
B
cyclobutanone
Is it (π2s + π2a)?
O
EtO
H
CH3H3C
H3C CH3
EtO O
+
JACS 1970, 92, 1766
Why this isomer?
we can generalize as
O
L'
S'
LL
L L
L' O
+
S
S
S'
S S
look at (π2s + π2a) transition state:
S
L
S
L
L' S'
S
L
S
L
L' S'
O
!2s + !2a
O
L L
L' O
S
S
S'
651.06
156
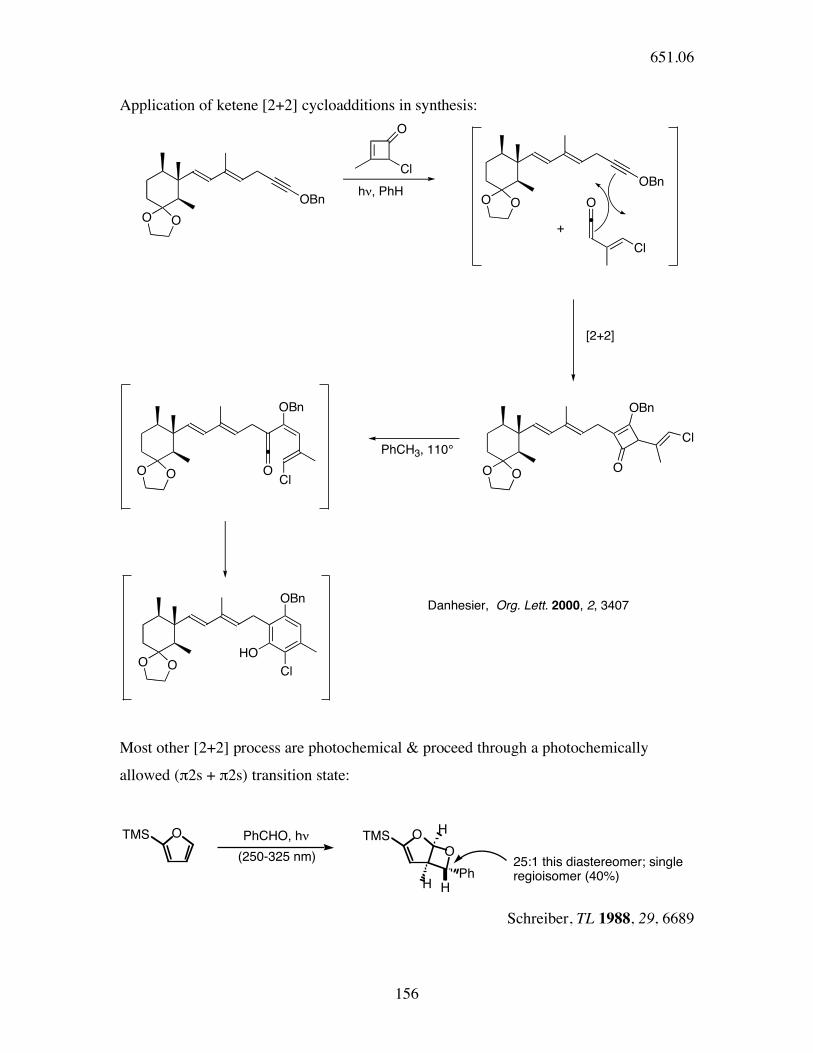
Application of ketene [2+2] cycloadditions in synthesis:
O O
OBn
Cl
O
h!, PhHO O
OBn
+
Cl
O
[2+2]
O O
OBn
O
ClPhCH3, 110°
O O OCl
OBn
O OCl
OBn
HO
Danhesier, Org. Lett. 2000, 2, 3407
Most other [2+2] process are photochemical & proceed through a photochemically
allowed (π2s + π2s) transition state:
OTMS PhCHO, h!
(250-325 nm)
OTMS
O
H
PhHH
25:1 this diastereomer; single regioisomer (40%)
Schreiber, TL 1988, 29, 6689
651.06
157
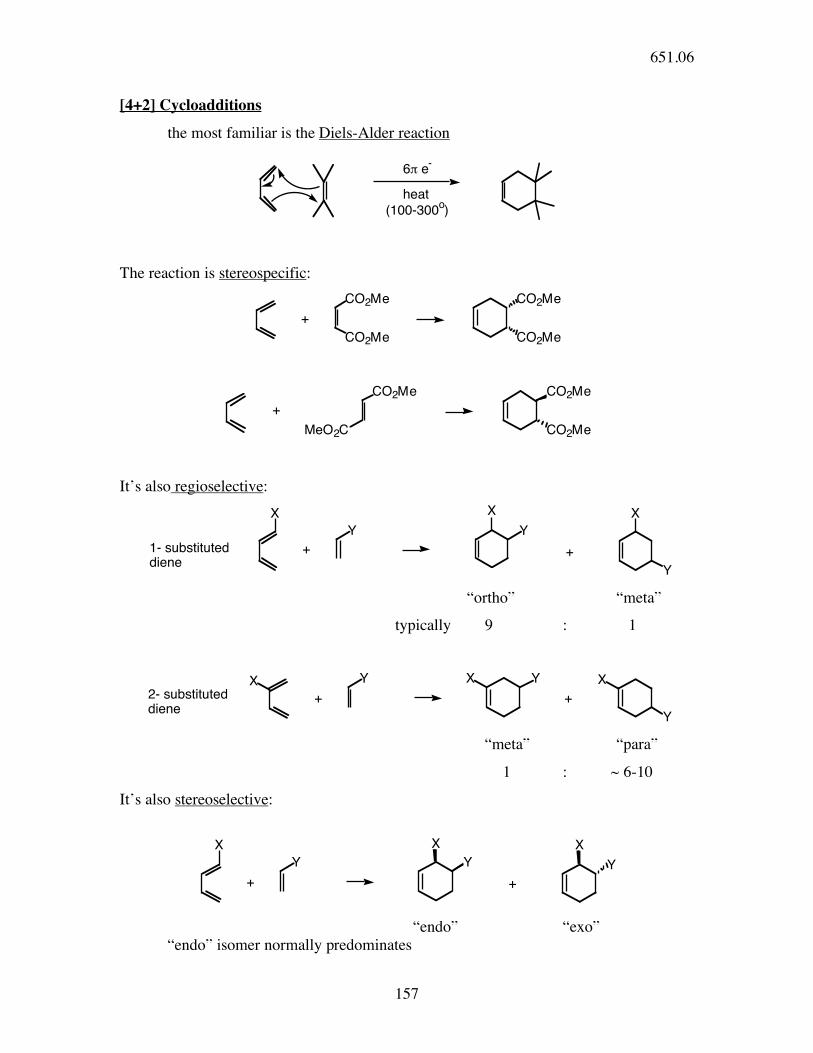
[4+2] Cycloadditions
the most familiar is the Diels-Alder reaction
6! e-
heat
(100-300o)
The reaction is stereospecific: CO2Me
CO2Me
CO2Me
CO2Me
+
CO2Me CO2Me
CO2Me
+
MeO2C
It’s also regioselective:
Y Y
+
X X
1- substituted
diene+
X
Y “ortho” “meta”
typically 9 : 1
Y Y
+2- substituted
diene+
Y
X XX
“meta” “para”
1 : ~ 6-10
It’s also stereoselective:
Y Y
+
X X
+
X
Y
“endo” “exo” “endo” isomer normally predominates
651.06
158
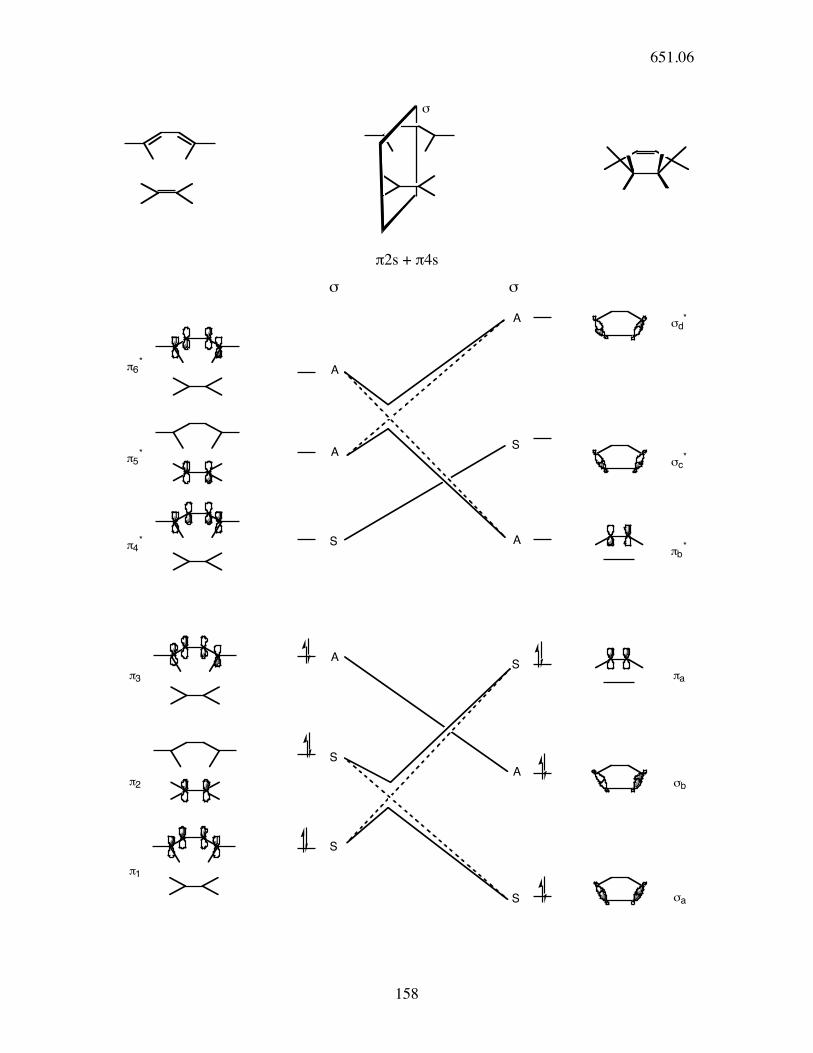
!
π2s + π4s
σ σ
!6*
!5*
!4*
!3
!2
!1
"d*
"c*
!b*
!a
"b
"a
S
S
S
A
AS
S A
A
A
A
S
651.06
159
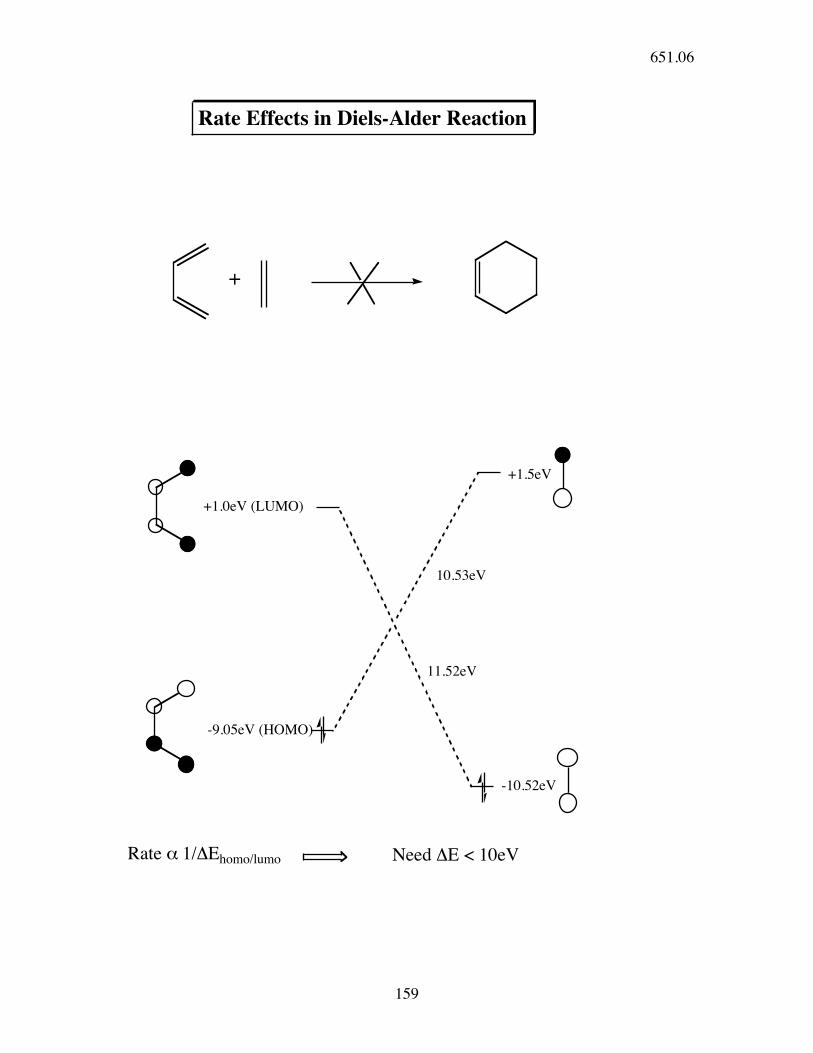
+
+1.0eV (LUMO)
-9.05eV (HOMO)
+1.5eV
-10.52eV
Rate Effects in Diels-Alder Reaction
10.53eV
Rate ! 1/"Ehomo/lumo Need "E < 10eV
11.52eV
651.06
160
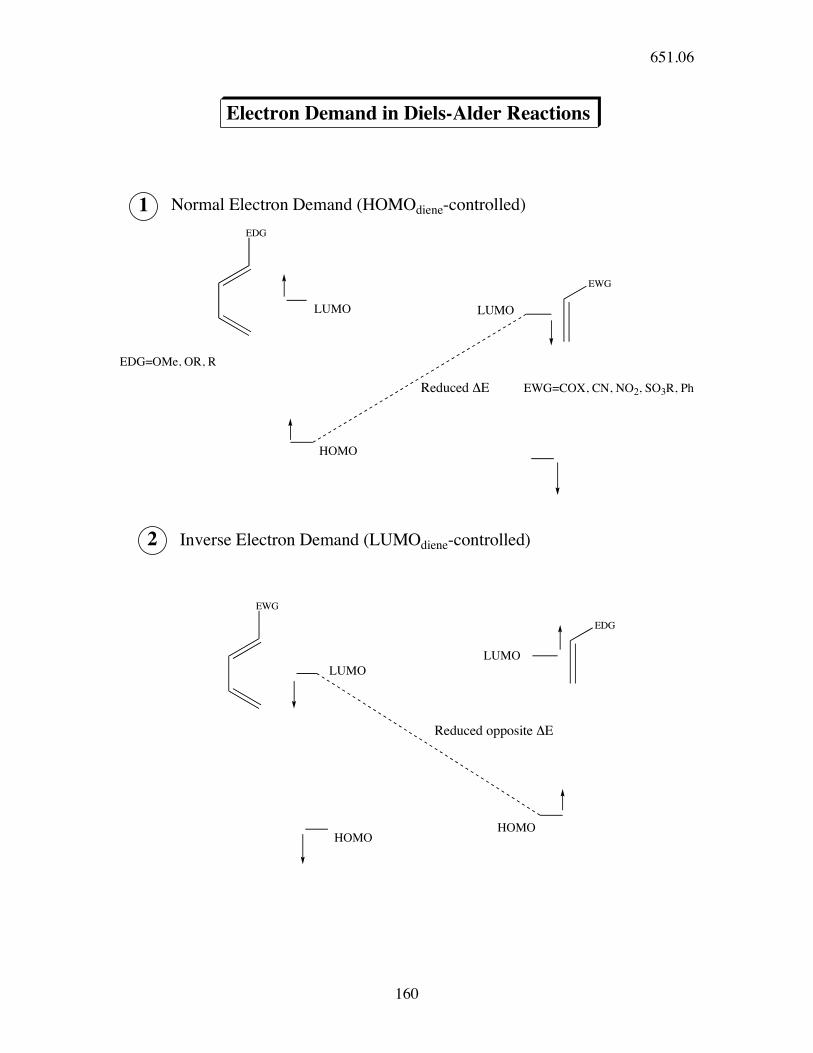
Electron Demand in Diels-Alder Reactions
EDG
EWG
HOMO
Reduced !E
1
LUMO
2
EWG
Normal Electron Demand (HOMOdiene-controlled)
EDG
Inverse Electron Demand (LUMOdiene-controlled)
LUMO
HOMO
LUMO
LUMO
HOMO
Reduced opposite !E
EDG=OMe, OR, R
EWG=COX, CN, NO2, SO3R, Ph
651.06
161
O
O
O
MeO Cl
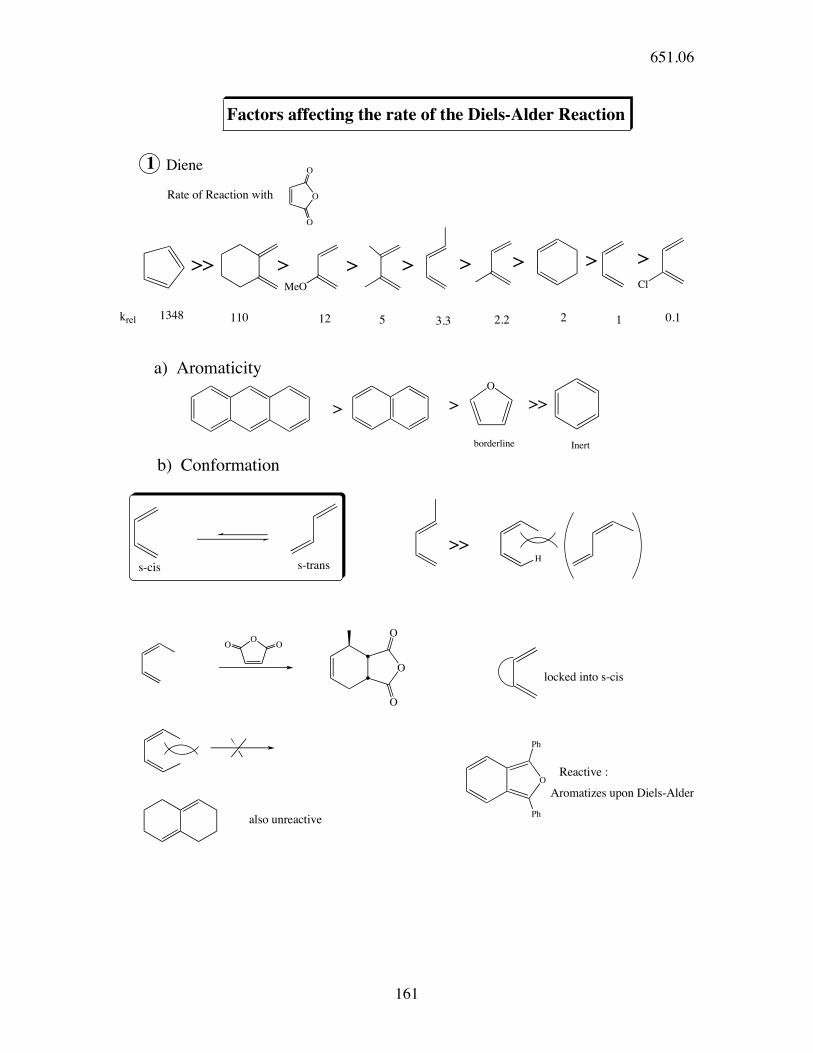
Factors affecting the rate of the Diels-Alder Reaction
1 Diene
Rate of Reaction with
>> > > > > > > >
O
110 2.2 2 1
OO O
>
a) Aromaticity
>>
Inert
O
O
O
0.1
borderline
locked into s-cis
b) Conformation
krel 1348 12 5 3.3
O
s-cis
Ph
Ph
s-trans
>>
>
Reactive :
H
Aromatizes upon Diels-Alder
also unreactive
651.06
162
CNNC
CNNC
COCl SO2Ph NO2 COCH3CO2Me CN
W
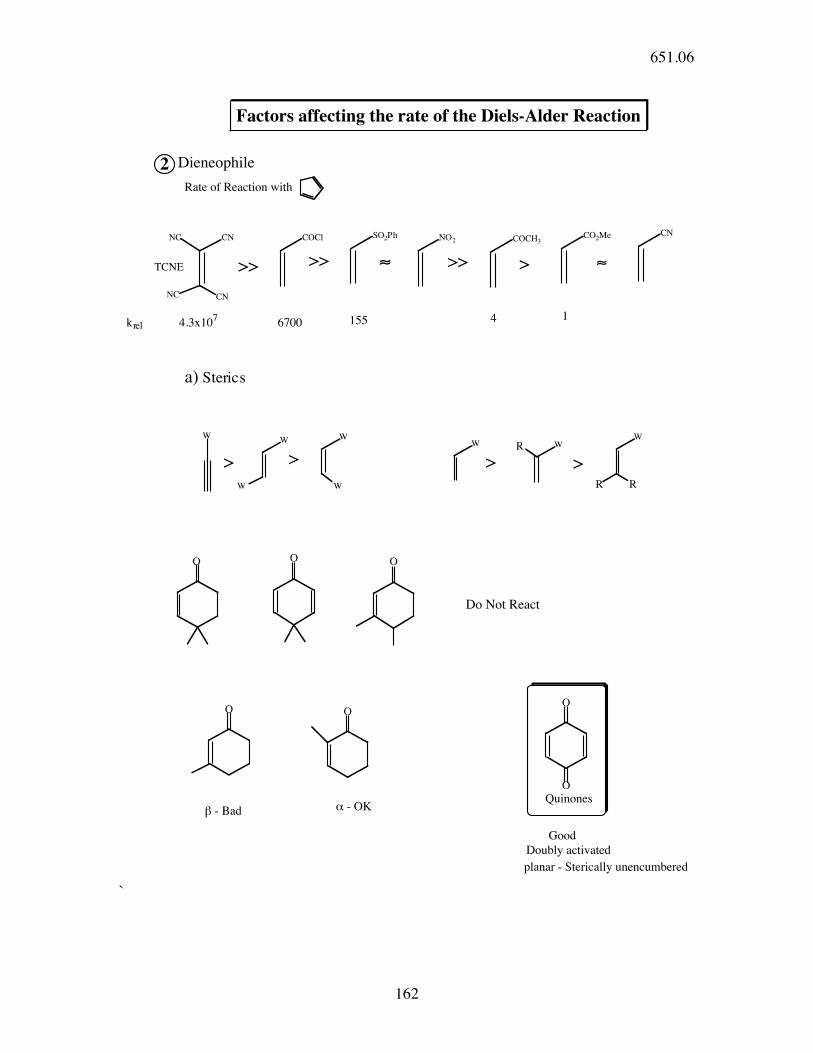
Factors affecting the rate of the Diels-Alder Reaction
W
W
2 Dieneophile
Rate of Reaction with
W
krel
W
4.3x107
W
6700
W
155
W
4
R
R R
1
>> >> >> >
O O O
a) Sterics
>
O
>
O
> >
Do Not React
O
! - Bad
O
" - OK
Good
Doubly activated
planar - Sterically unencumbered
Quinones
#TCNE #
`
651.06
163
O
CN
O
CN
O
CN
O
CN
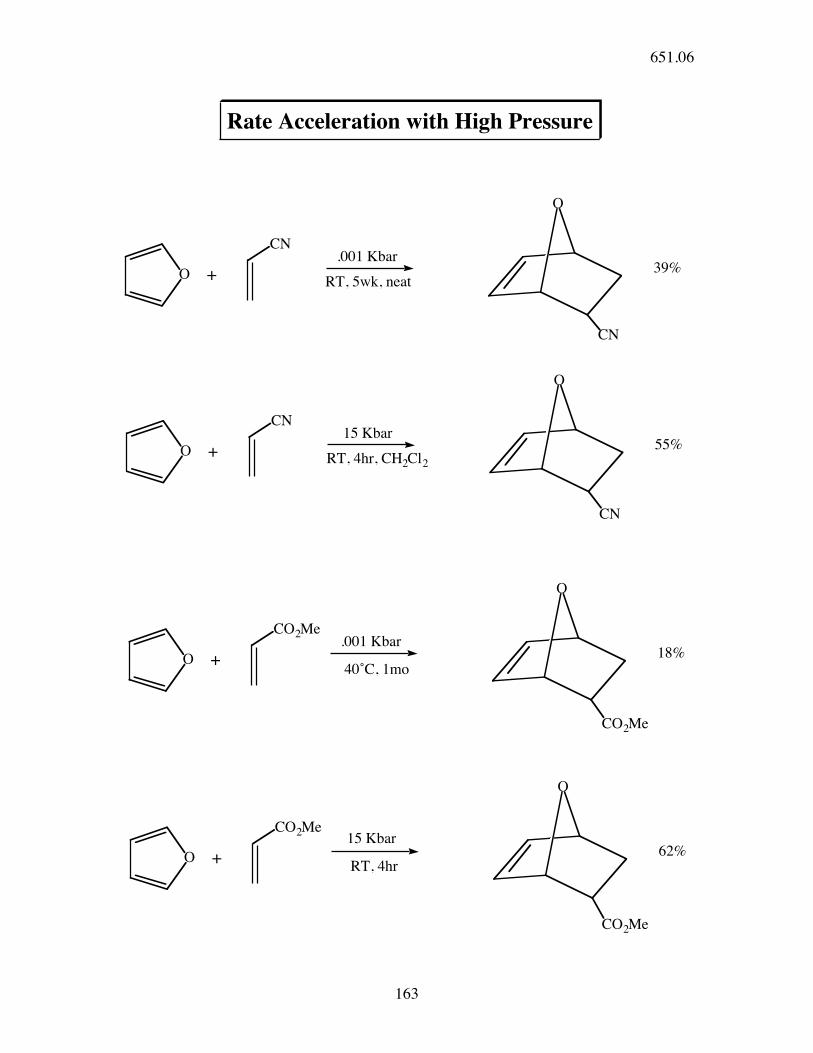
Rate Acceleration with High Pressure
O
+
CO2Me
.001 Kbar
+
15 Kbar
RT, 4hr, CH2Cl2
O
RT, 5wk, neat
CO2Me
39%
55%
O +
CO2Me
.001 Kbar
+
15 Kbar
RT, 4hr
O
40˚C, 1mo
CO2Me
18%
62%
651.06
164
O
O
O
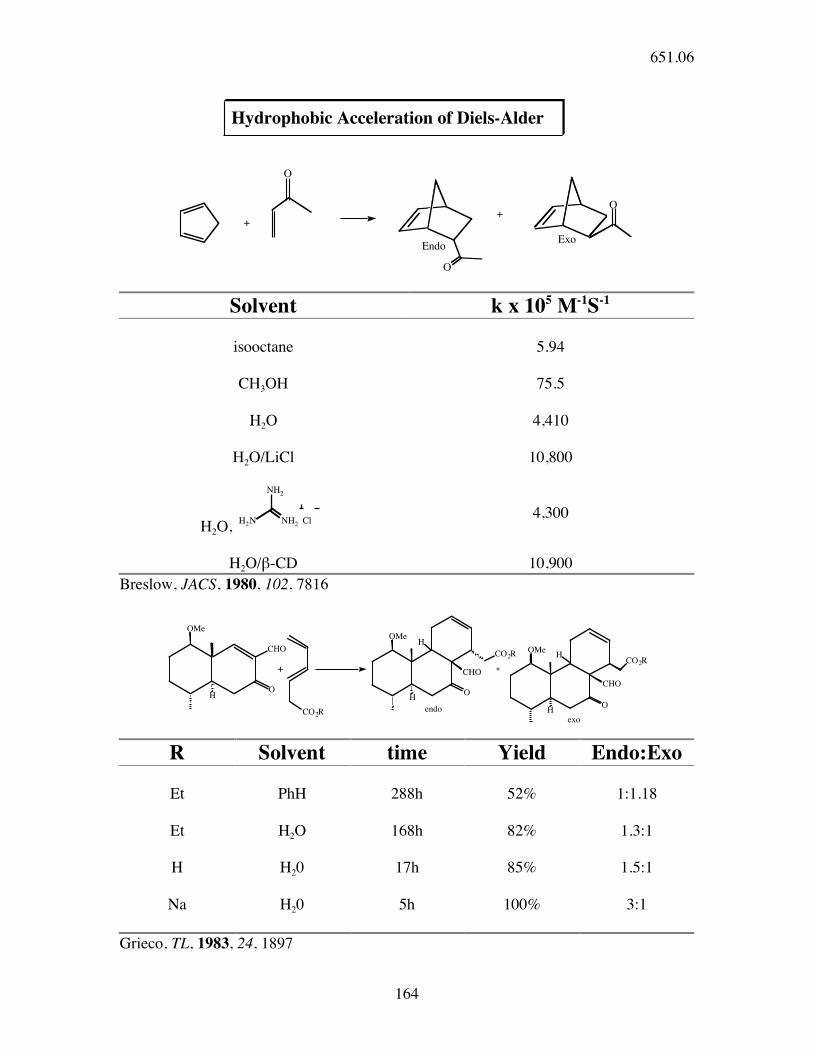
Hydrophobic Acceleration of Diels-Alder
++
EndoExo
Solvent k x 105 M-1S-1
isooctane 5.94
CH3OH 75.5
H2O 4,410
H2O/LiCl 10,800
H2O,
NH2
H2N NH
2Cl
4,300
H2O/β-CD 10,900
Breslow, JACS, 1980, 102, 7816
OMe
OH
CO2R
OMe
OH
OMe
OH
CHO
H
H
CHO
CO2R
+ +
endo
exo
CO2R
CHO
R Solvent time Yield Endo:Exo
Et PhH 288h 52% 1:1.18
Et H2O 168h 82% 1.3:1
H H20 17h 85% 1.5:1
Na H20 5h 100% 3:1
Grieco, TL, 1983, 24, 1897
651.06
165
E
AlCl3
(HOMO) (HOMO)
OMe
!E = 2.2eV
Examples
++
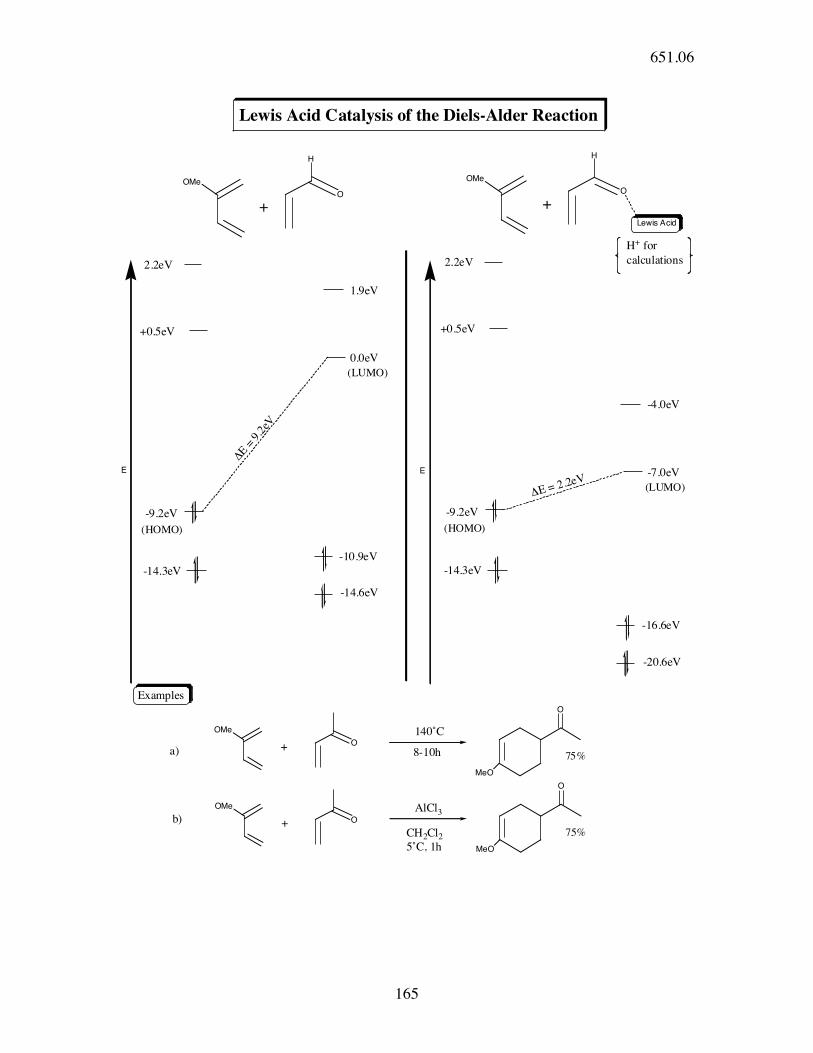
Lewis Acid Catalysis of the Diels-Alder Reaction
2.2eV
-14.3eV
1.9eV
O
H
-14.6eV
OMe
O
H
Lewis Acid
+0.5eV
!E =
9.2
eV
-16.6eV
2.2eV
-14.3eV
-4.0eV
-20.6eV
OMe
O
-9.2eV-9.2eV
-7.0eV
+0.5eV
O
0.0eV
140˚C
OMe
-10.9eV
O
8-10h
+
75%
CH2Cl25˚C, 1h
O
+
75%
a)
b)
(LUMO)
(LUMO)
H+ for
calculations
MeO
MeO
E
651.06
166
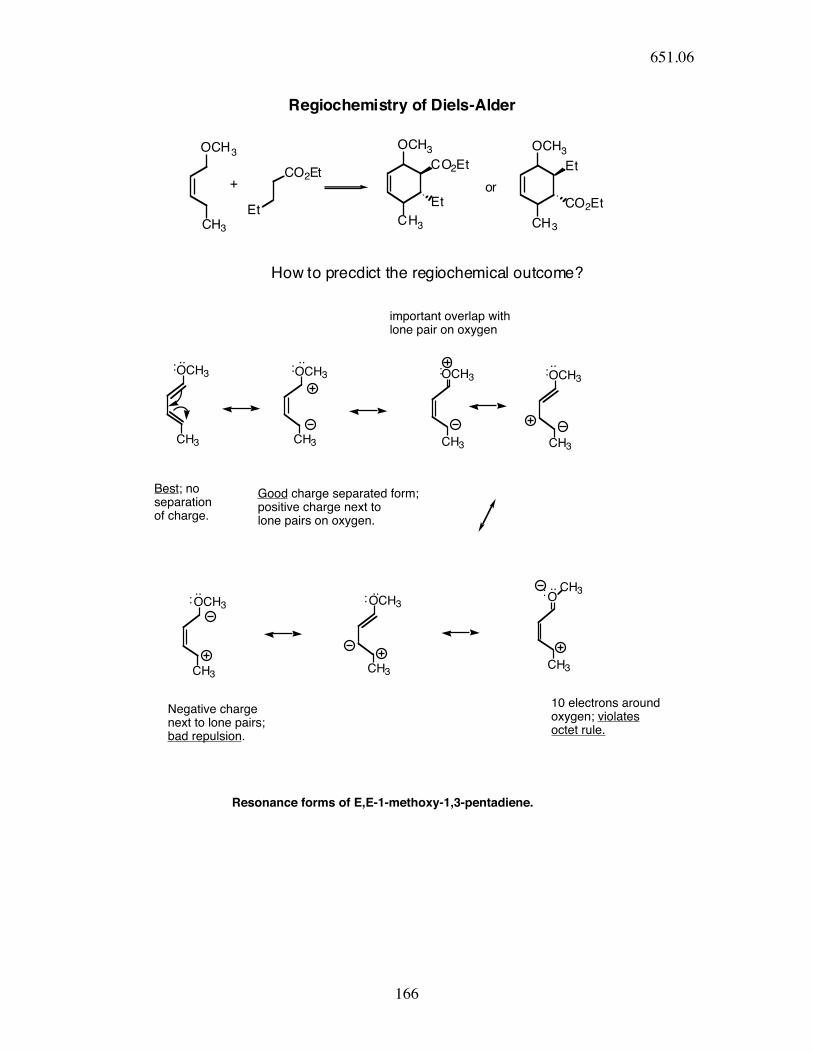
Regiochemistry of Diels-Alder
OCH3
CH3
+CO2Et
Et
OCH3
CH3
CO2Et
Et
OCH3
CH3
Et
CO2Et
or
How to precdict the regiochemical outcome?
OCH3
CH3
OCH3
CH3
OCH3
CH3
OCH3
CH3
O
CH3
OCH3
CH3
OCH3
CH3
CH3..
..: : :
::
:
:
Best; no separationof charge.
Good charge separated form; positive charge next tolone pairs on oxygen.
important overlap with lone pair on oxygen
Negative chargenext to lone pairs;bad repulsion.
10 electrons aroundoxygen; violates octet rule.
Resonance forms of E,E-1-methoxy-1,3-pentadiene.
.. ..
....
651.06
167
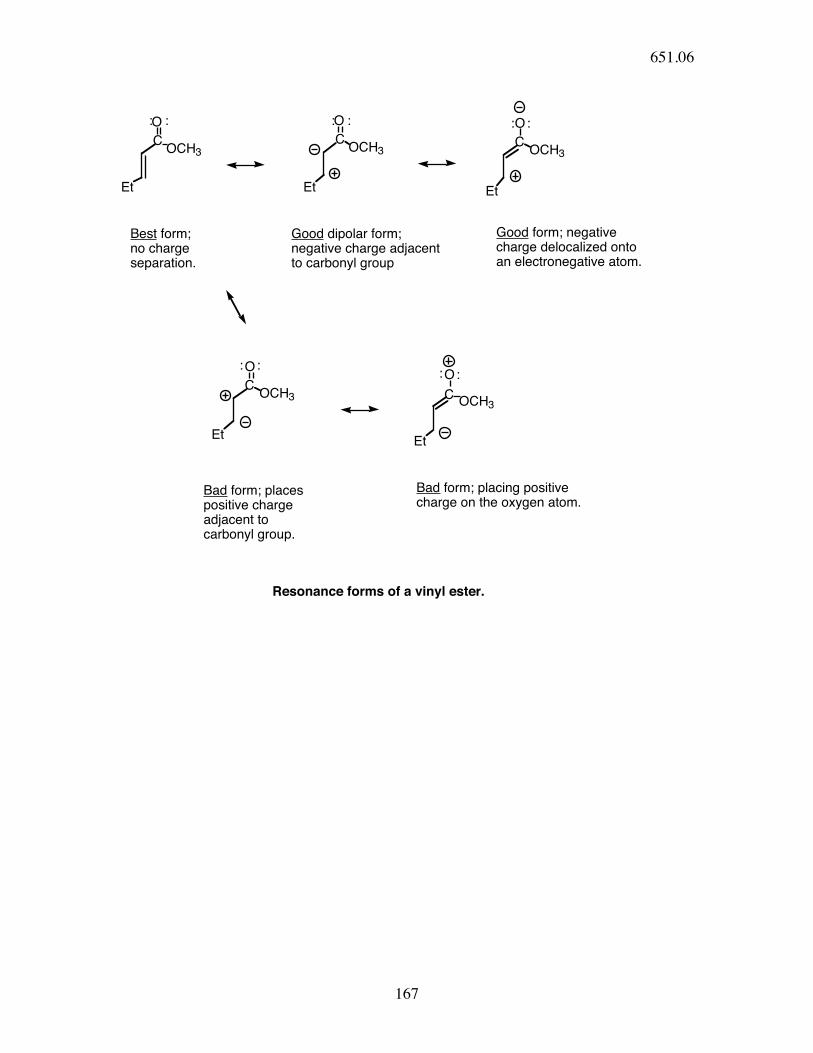
C
O
OCH3C
O
OCH3
Et Et
C
O
OCH3
Et
C
O
OCH3
Et
C
O
OCH3
Et
Good form; negativecharge delocalized ontoan electronegative atom.
: : : : : :
: :: :
Best form;no charge separation.
Good dipolar form;negative charge adjacentto carbonyl group
Bad form; placespositive chargeadjacent tocarbonyl group.
Bad form; placing positivecharge on the oxygen atom.
Resonance forms of a vinyl ester.
651.06
168
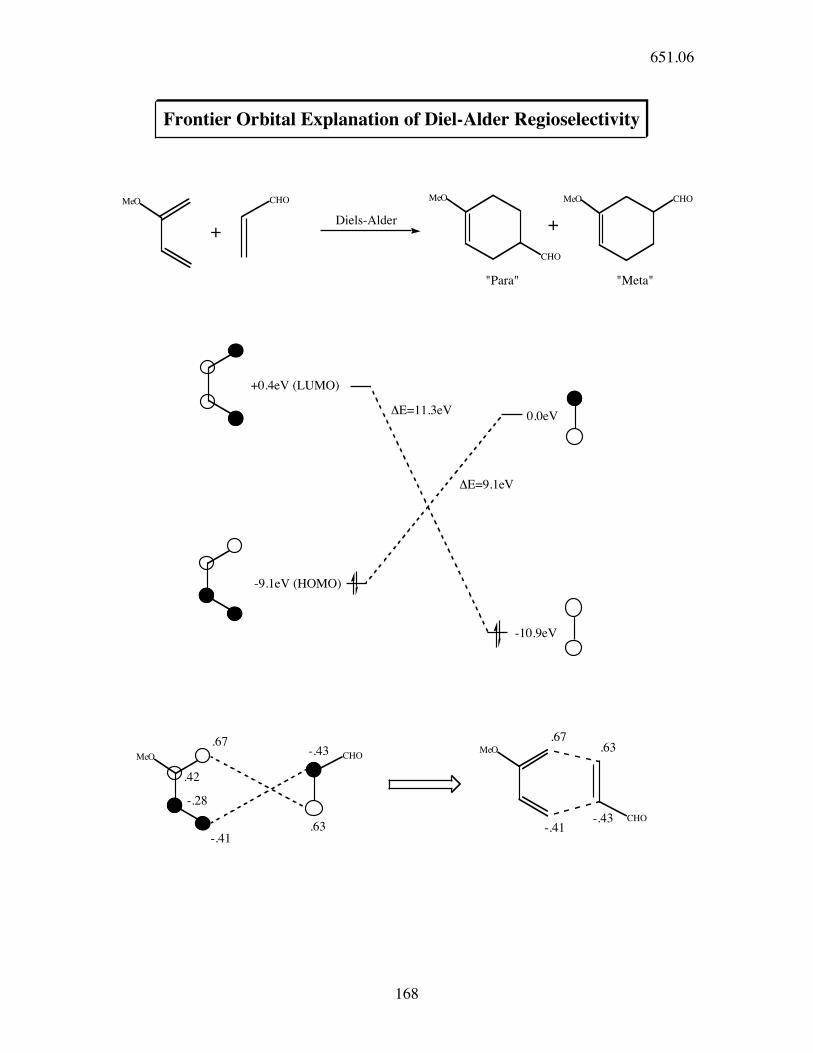
MeO CHO MeO CHOMeO
CHO
+Diels-Alder +
"Para" "Meta"
+0.4eV (LUMO)
MeO
-9.1eV (HOMO)
CHO
0.0eV
-10.9eV
.67
.42
-.28
-.41
MeO-.43
CHO.63
.67
-.41
.63
-.43
Frontier Orbital Explanation of Diel-Alder Regioselectivity
!E=11.3eV
!E=9.1eV
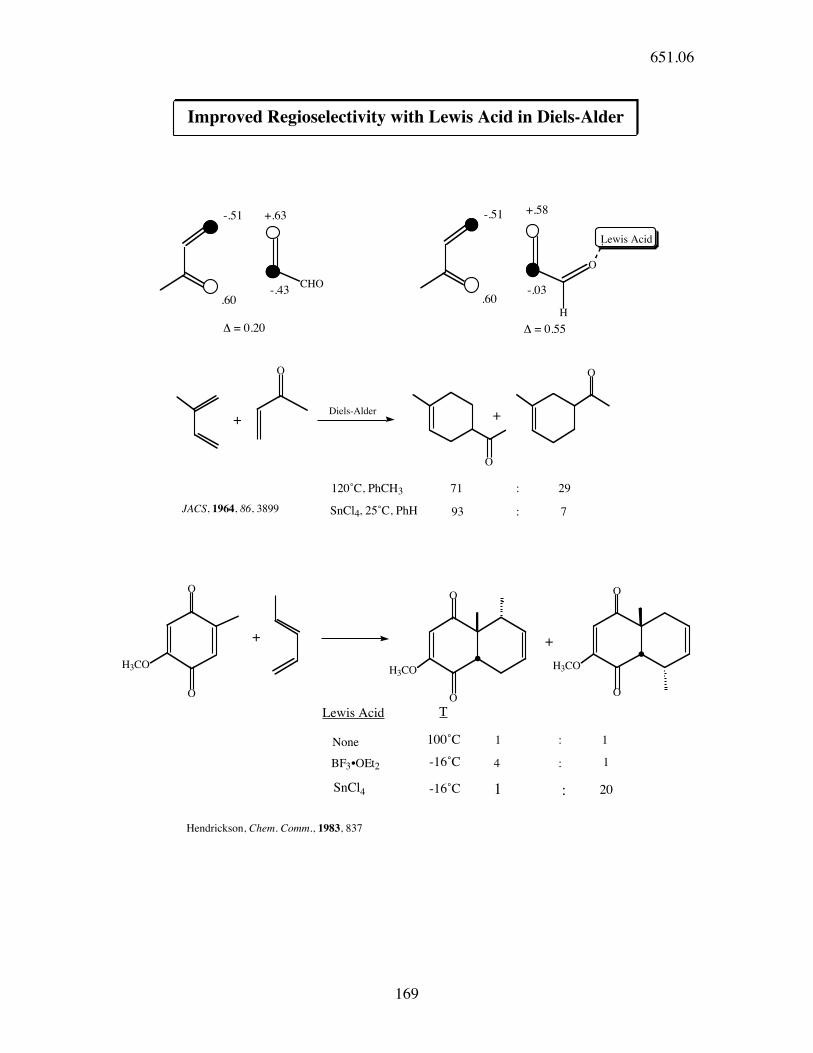
651.06
169
-.51
CHO
-.51
.60-.43
+.63
O
Lewis Acid
93
O
O
O
+
H
.60
71
Diels-Alder
:
SnCl4, 25˚C, PhH
+
29
O
O
H3CO
! = 0.20
-.03
+.58
O
O
H3CO
: 7JACS, 1964, 86, 3899
O
O
H3CO
+
Improved Regioselectivity with Lewis Acid in Diels-Alder
120˚C, PhCH3
+
! = 0.55
None 1 :
BF3•OEt2
1
4 : 1
100˚C
-16˚C
SnCl4 -16˚C 1 : 20
Hendrickson, Chem. Comm., 1983, 837
Lewis Acid T
651.06
170
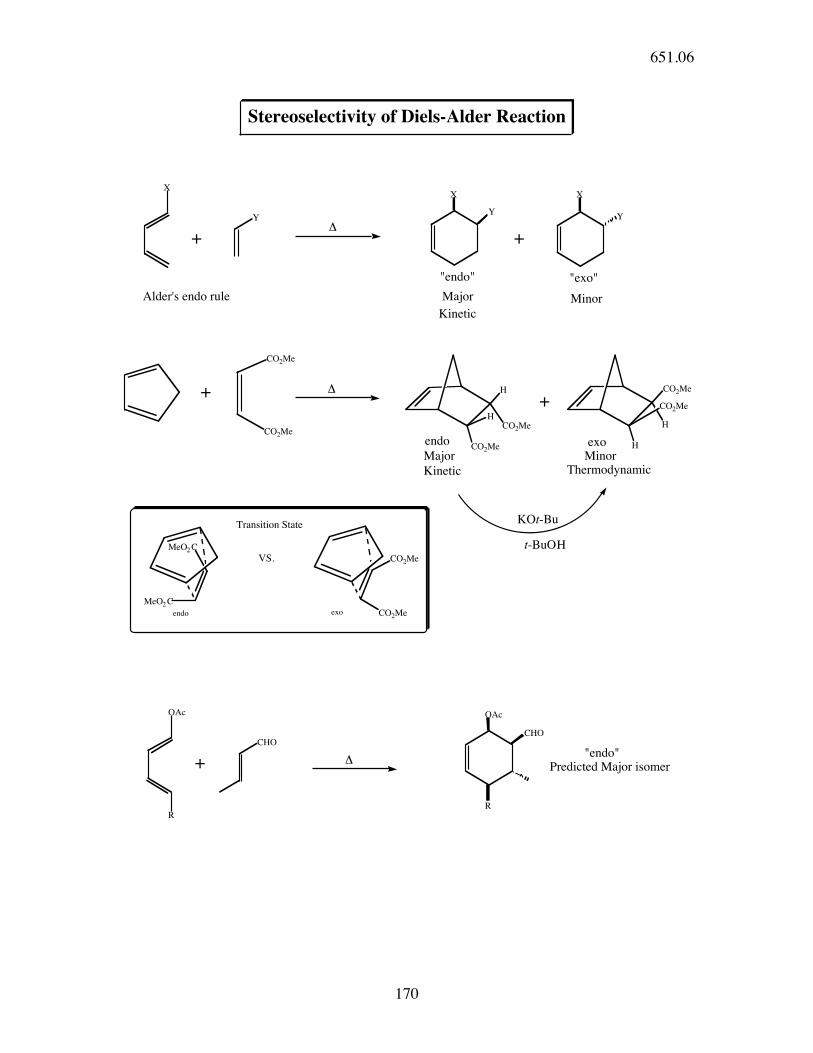
Y
XX
Y
X
Y
CO2Me
CO2Me
H
CO2MeH
CO2Me
CO2Me
H
CO2Me
H
Stereoselectivity of Diels-Alder Reaction
++!
"endo" "exo"
Major Minor
+ !
MeO2 C
MeO2 C
+
KOt-Bu
CO2Me
CO2Me
t-BuOH
endo
CHO
OAc
+ !"endo"
Predicted Major isomer
R
Minor
VS.
Major
OAc
CHO
R
Thermodynamic
Kinetic
exoendo
Kinetic
exo
Transition State
Alder's endo rule
651.06
171
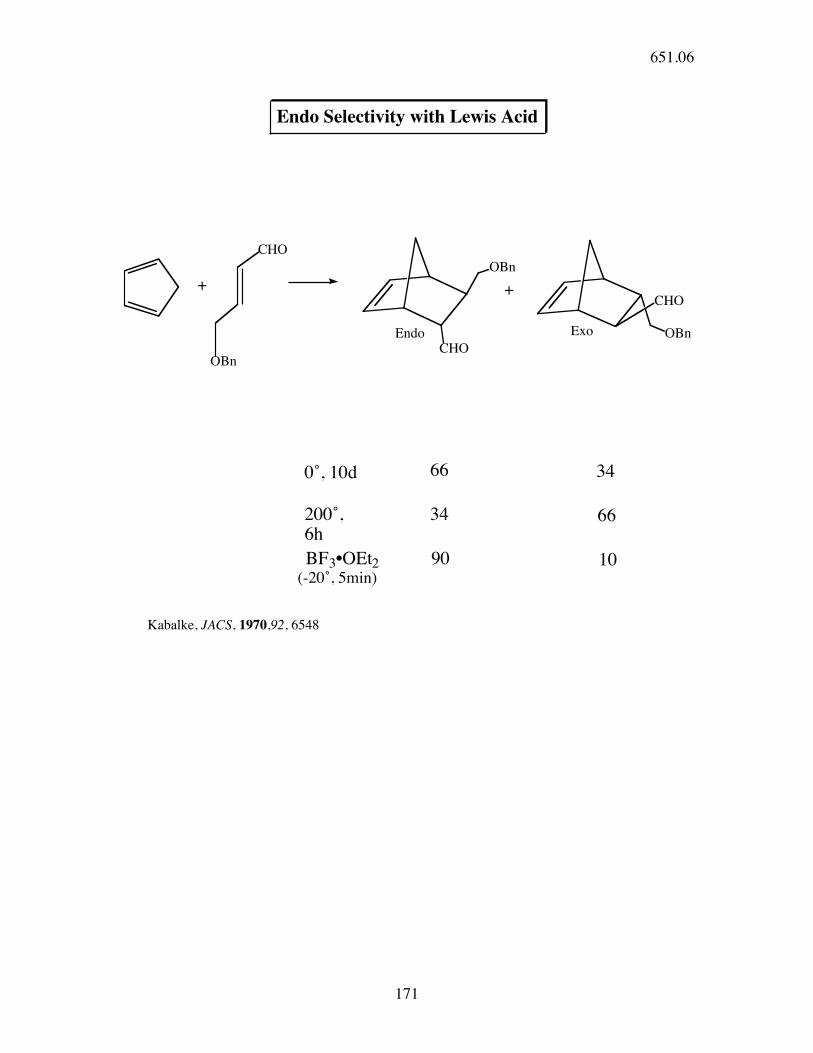
OBn
CHOOBn
CHO
Endo Selectivity with Lewis Acid
+ +
Endo Exo
0˚, 10d 66 34
200˚, 6h
34 66
BF3•OEt2(-20˚, 5min)
90 10
CHO
OBn
Kabalke, JACS, 1970,92, 6548
651.06
172
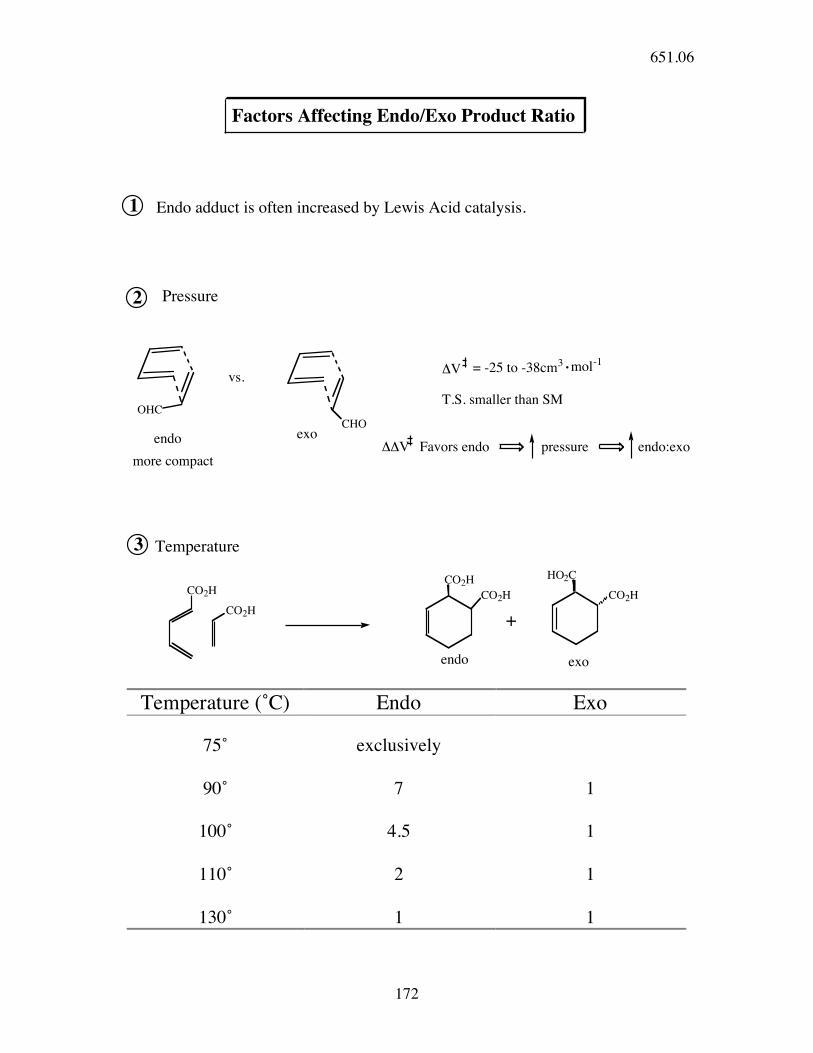
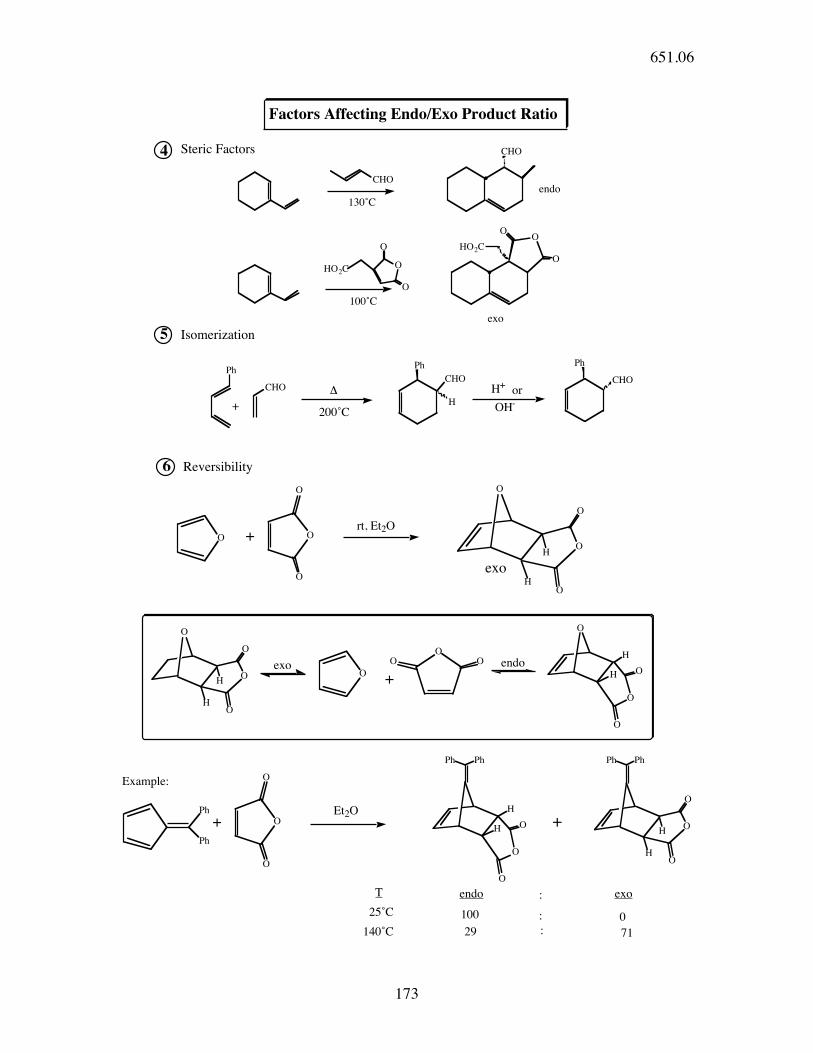
Factors Affecting Endo/Exo Product Ratio
1 Endo adduct is often increased by Lewis Acid catalysis.
2 Pressure
OHC
vs.
CHO
endo
more compact
exo
!V = -25 to -38cm3 mol-1
T.S. smaller than SM
3
!!V Favors endo pressure endo:exo
Temperature
CO2H
CO2H+
CO2H HO2C
CO2H CO2H
endo exo
Temperature (˚C) Endo Exo
75˚ exclusively
90˚ 7 1
100˚ 4.5 1
110˚ 2 1
130˚ 1 1
651.06
173
Ph
CHO
Ph Ph
CHO CHO
CHO
CHO
OHO2C
O
O
O
O
O
HO2C
O O
O
O
O
O
O
O
H
H
O
OO O
O
O
O
O
H
H
O
O
O
O
H
H
5
Steric Factors
O
O
O
6
100˚C
+
Isomerization
Reversibility
130˚C
Ph
Ph
4
O
O
O
H
H
rt, Et2O
exo endo
+
Factors Affecting Endo/Exo Product Ratio
+ O
O
O
H
H
!
exo
+
H+or
OH-
endo
Et2O+
Example:
endo exoT
25˚C 100
:
: 0
140˚C
Ph Ph Ph Ph
29 : 71
H200˚C
exo
651.06
174
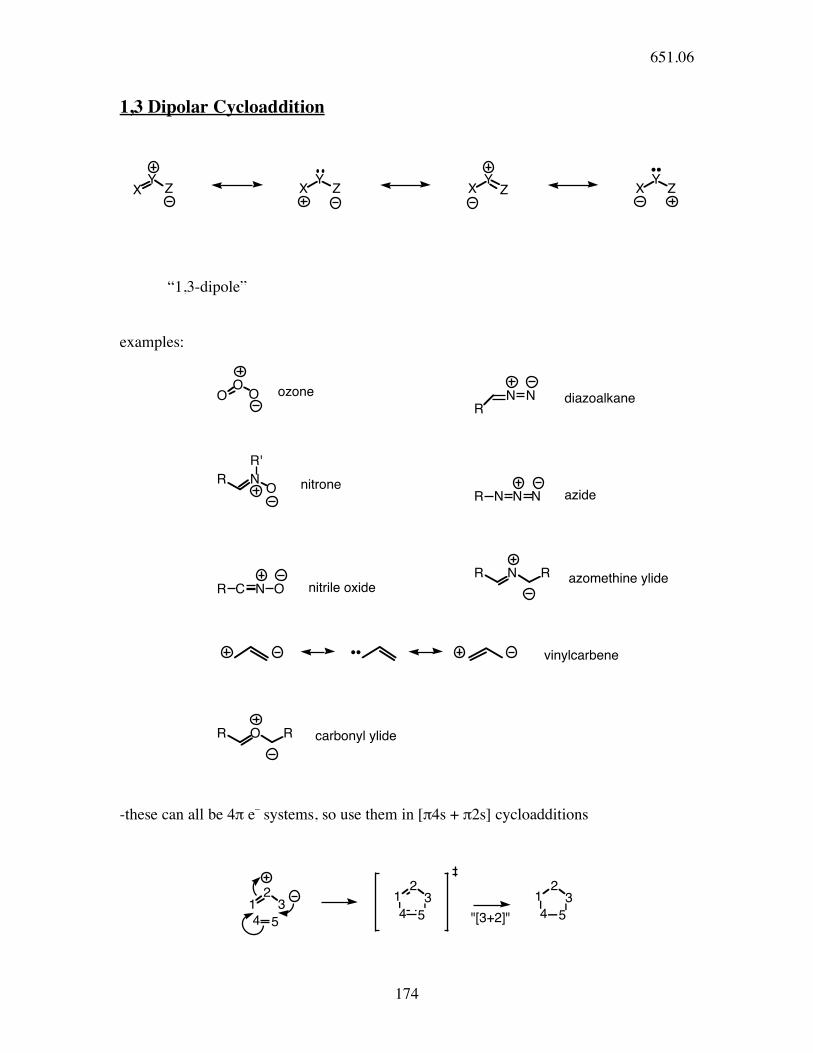
1,3 Dipolar Cycloaddition
YX Z
YX Z
YX Z
YX Z
“1,3-dipole”
examples:
O
O O ozone
N N
Rdiazoalkane
NO
Rnitrone
R'
R N N N azide
R C N O nitrile oxide
NR R azomethine ylide
vinylcarbene
OR R carbonyl ylide
-these can all be 4π e_ systems, so use them in [π4s + π2s] cycloadditions
1
4 5
32 1
4 5
32
1
4 5
32
"[3+2]"

651.06
175
5-membered ring (heterocyclic) products are the usual signatures of [3+2] cycloadditions.
familiar examples:
O
OO
O3
[3+2] retro - [3+2]
[3+2]
mol-ozonide
O
O
OO O
O
OO
O
O
O
more synthetically important examples:
nitrile oxides
R NO2RCNO
R R'
O OH
R R'
O O
R'
R' C CH
N O
N O
R
R'
R'
R
isoxazoline
isoxazole
Zno
HCl
1O2
[4+2]
PhNCO
50o
651.06
176
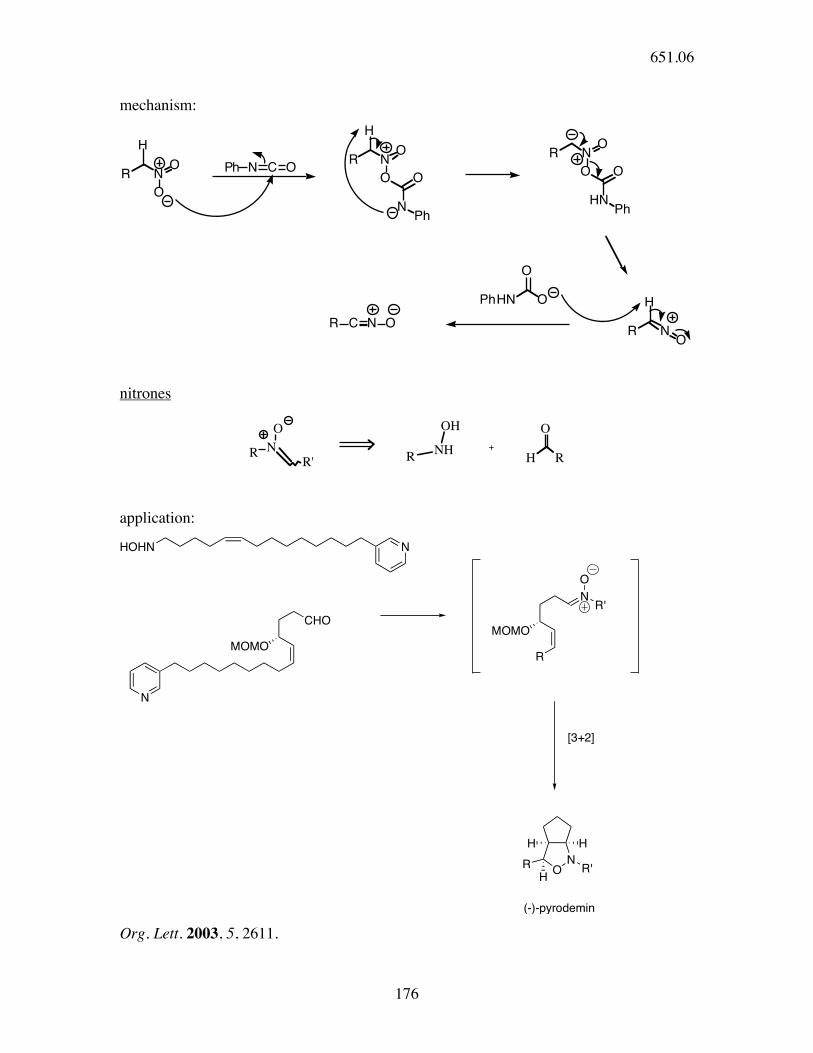
mechanism:
R NO
O
Ph N C OR N
O
O O
NPh
R NO
O O
HNPh
H
R NO
HPhHN O
O
R C N O
H
nitrones
N
O
RR'
RNH
OH O
H R
+
application:
N
MOMO
CHO
HOHN N
R
MOMO
NR'
O
[3+2]
ONR'
H H
HR
(-)-pyrodemin Org. Lett. 2003, 5, 2611.
651.06
177
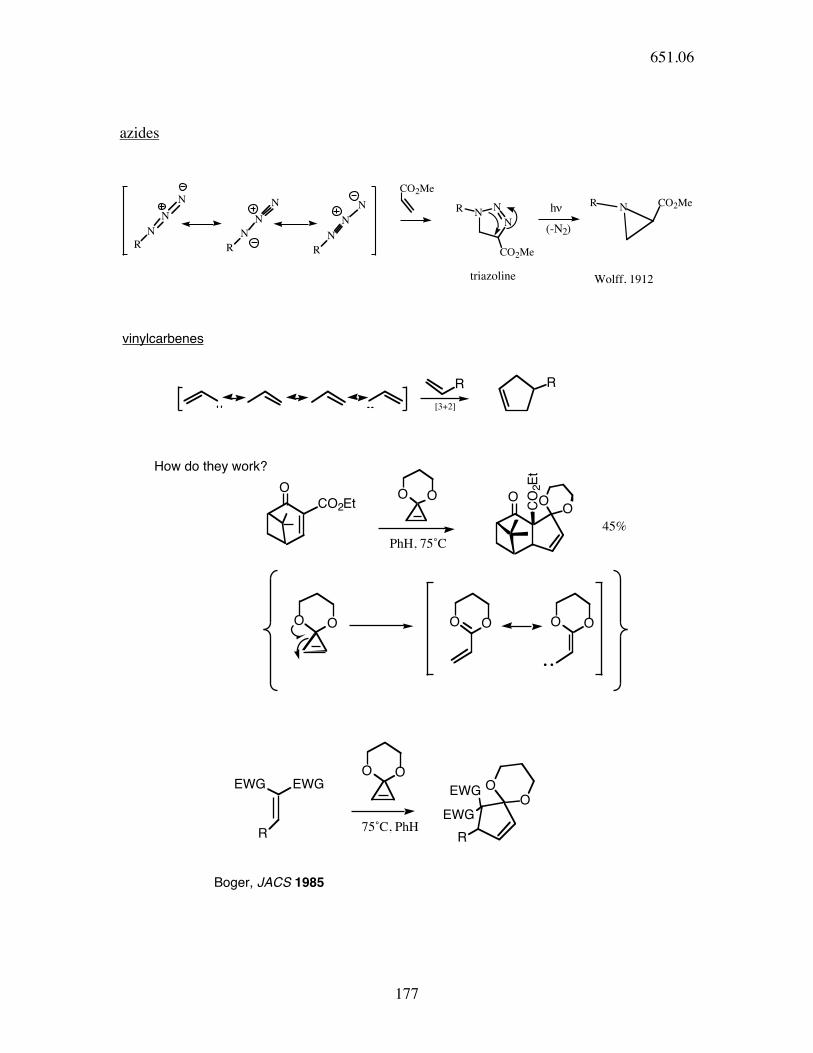
azides
N
N
N
R
N
N
N
R
N
N
N
R
CO2Me
NN
NR
CO2Me
NR CO2Meh!
(-N2)
triazoline Wolff, 1912
R R
O
CO2EtOO O
CO
2E
t
OO
OO OO OO
EWG
R
EWGOO
OOEWG
EWG
R
How do they work?
75˚C, PhH
PhH, 75˚C
45%
vinylcarbenes
[3+2]
Boger, JACS 1985
651.06
178
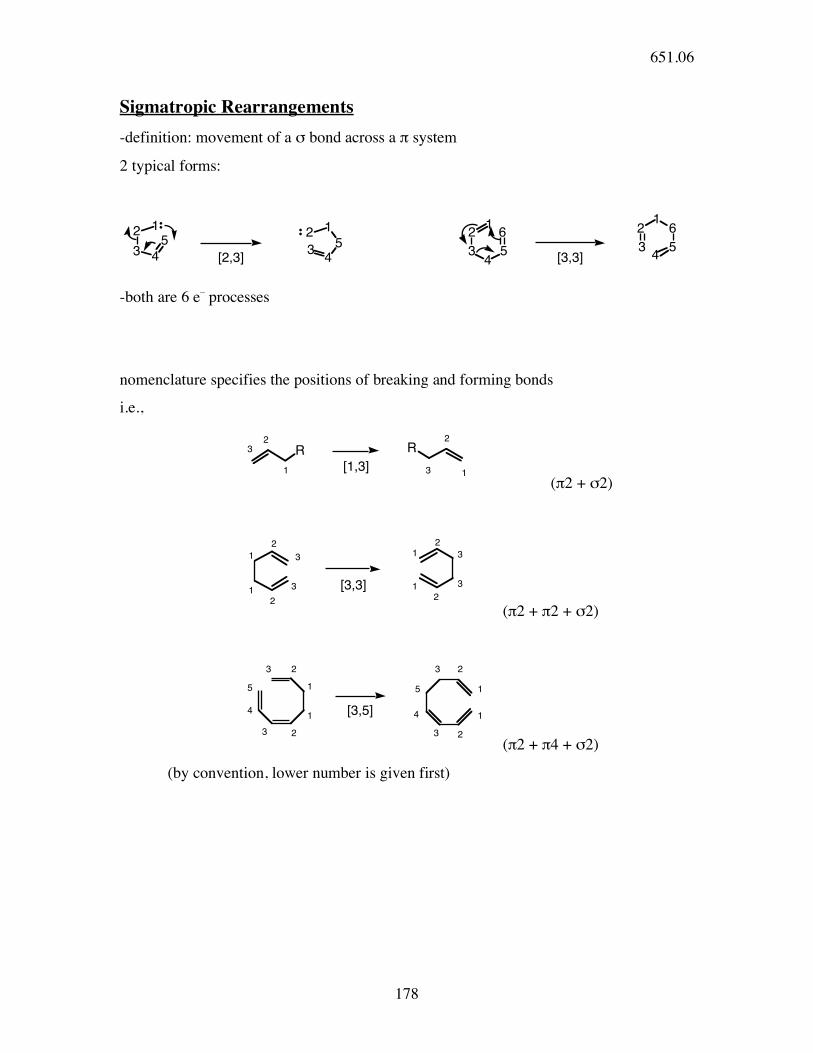
Sigmatropic Rearrangements
-definition: movement of a σ bond across a π system
2 typical forms:
2
3 4
51
2
345
1
[2,3]
2
345
61 2
345
61
[3,3]
-both are 6 e_ processes
nomenclature specifies the positions of breaking and forming bonds
i.e.,
R R3
11
2 2
3[1,3]
(π2 + σ2)
3
2
1
3
21
3
21
3
21[3,3]
(π2 + π2 + σ2)
1
23
5
4
3
1
2
1
23
5
4
3
1
2
[3,5]
(π2 + π4 + σ2)
(by convention, lower number is given first)
651.06
179
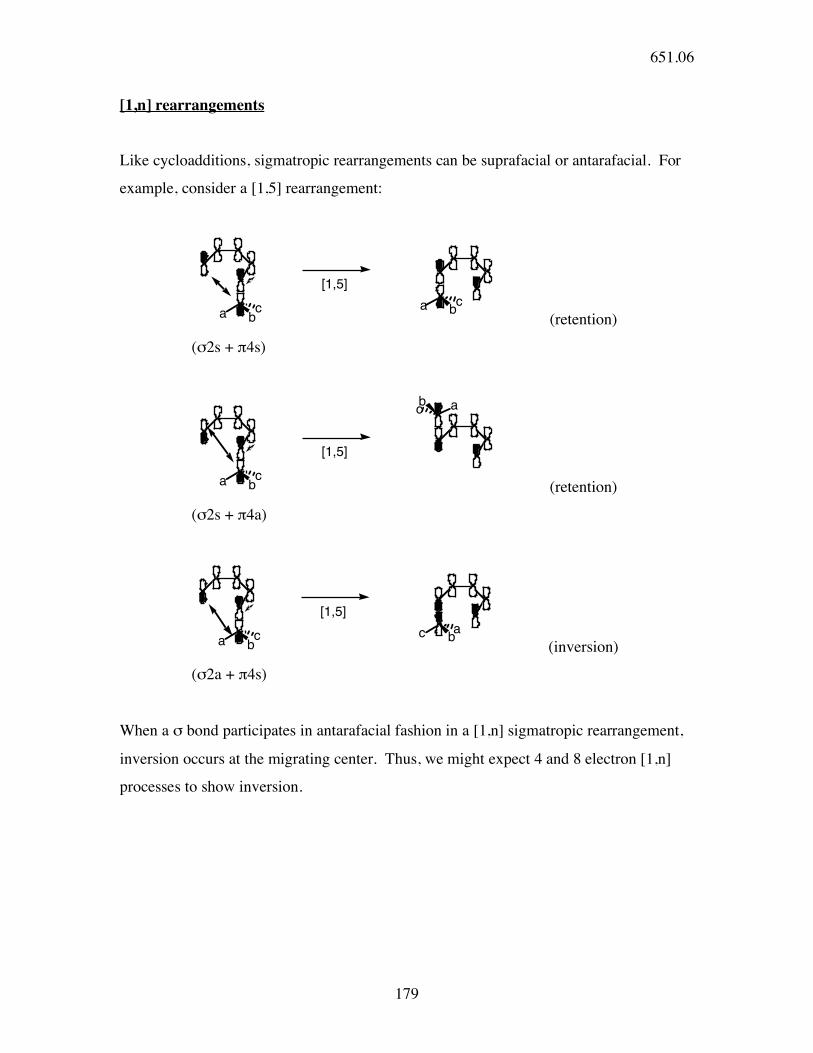
[1,n] rearrangements
Like cycloadditions, sigmatropic rearrangements can be suprafacial or antarafacial. For
example, consider a [1,5] rearrangement:
a bc
[1,5]
a bc
(retention)
(σ2s + π4s)
a bc
[1,5]
abc
(retention)
(σ2s + π4a)
a bc
[1,5]
c ba
(inversion)
(σ2a + π4s)
When a σ bond participates in antarafacial fashion in a [1,n] sigmatropic rearrangement,
inversion occurs at the migrating center. Thus, we might expect 4 and 8 electron [1,n]
processes to show inversion.
651.06
180
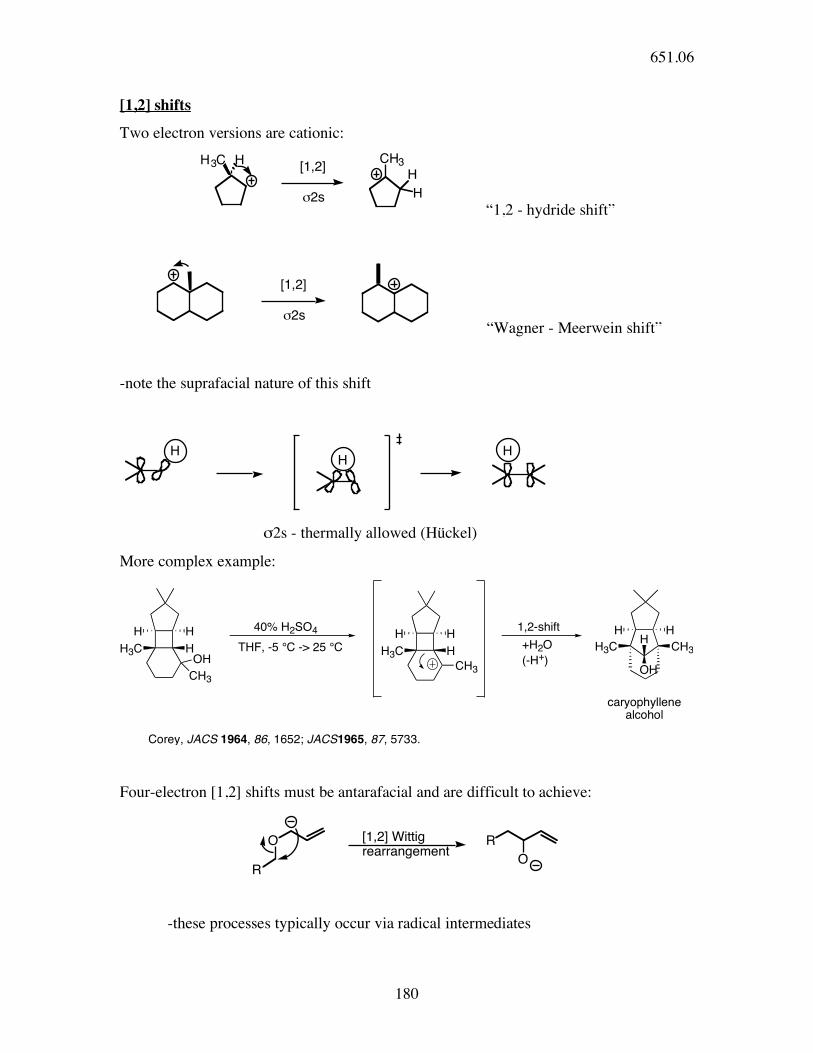
[1,2] shifts
Two electron versions are cationic: H3C H
[1,2]
!2s
CH3H
H
“1,2 - hydride shift”
[1,2]
!2s “Wagner - Meerwein shift”
-note the suprafacial nature of this shift
HH
H
σ2s - thermally allowed (Hückel)
More complex example:
H3C H
H H
CH3
OH
40% H2SO4
THF, -5 °C -> 25 °C H3C H
H H
CH3
1,2-shiftH
OH
H3C CH3
H H
+H2O
(-H+)
caryophyllenealcohol
Corey, JACS 1964, 86, 1652; JACS1965, 87, 5733.
Four-electron [1,2] shifts must be antarafacial and are difficult to achieve:
O
R
R
O
[1,2] Wittig rearrangement
-these processes typically occur via radical intermediates
651.06
181
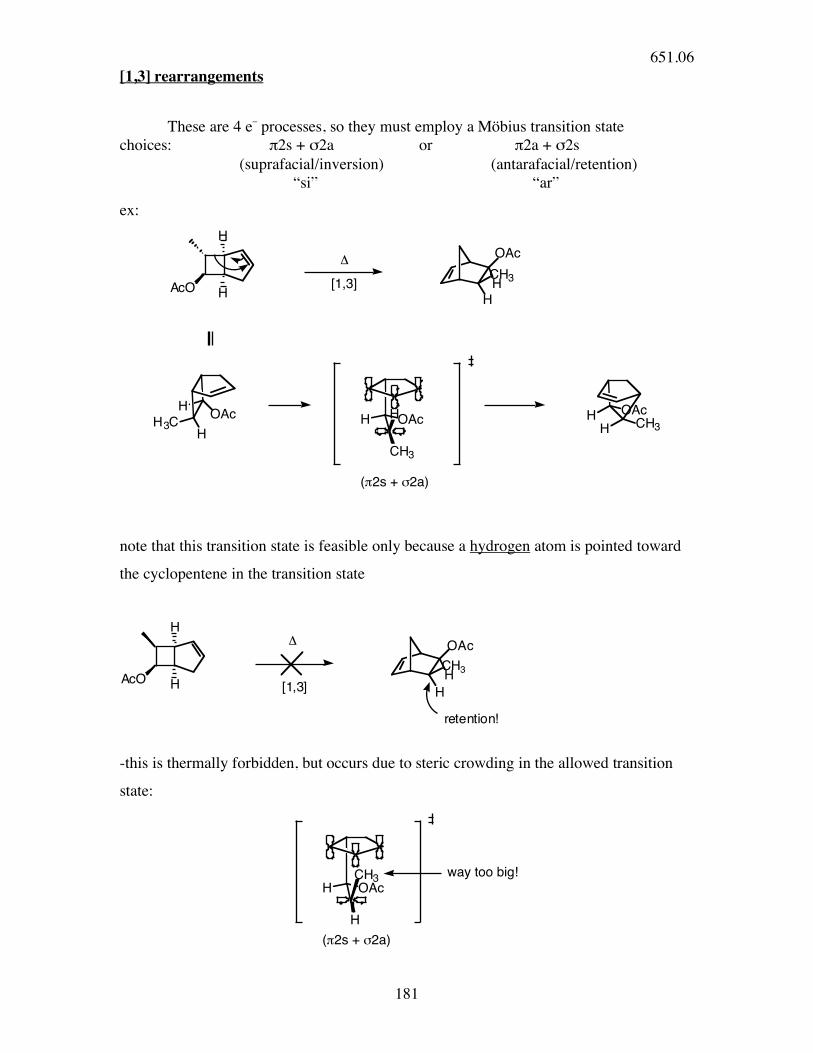
[1,3] rearrangements
These are 4 e_ processes, so they must employ a Möbius transition state choices: π2s + σ2a or π2a + σ2s (suprafacial/inversion) (antarafacial/retention) “si” “ar”
ex:
H
AcO
H
H
OAc
H
HCH3
!
[1,3]
OAc
HH3C H OAc
CH3
H OAcCH3H
H
("2s + #2a)
note that this transition state is feasible only because a hydrogen atom is pointed toward
the cyclopentene in the transition state
!
[1,3]AcO
H
H
OAc
H
HCH3
retention!
-this is thermally forbidden, but occurs due to steric crowding in the allowed transition
state:
H OAc
H
CH3
(!2s + "2a)
way too big!
651.06
182
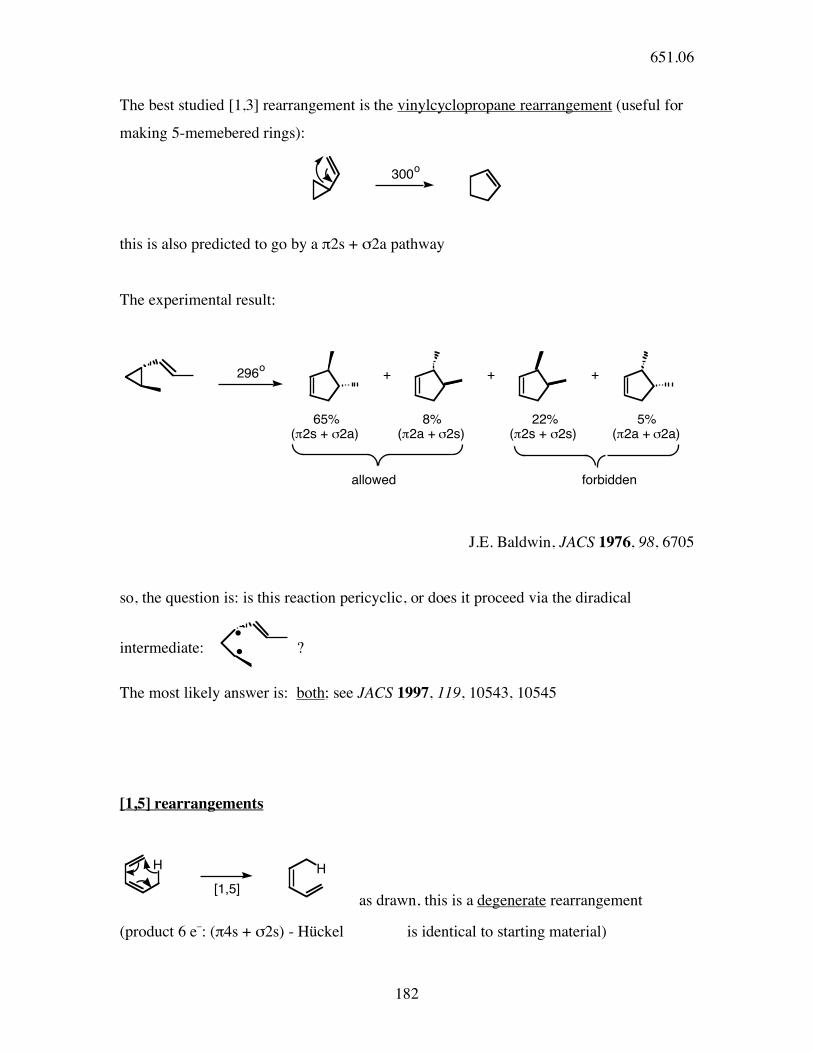
The best studied [1,3] rearrangement is the vinylcyclopropane rearrangement (useful for
making 5-memebered rings):
300o
this is also predicted to go by a π2s + σ2a pathway
The experimental result:
296o
+ + +
65% 8% 22% 5%(!2s + "2a) (!2a + "2s) (!2s + "2s) (!2a + "2a)
allowed forbidden
J.E. Baldwin, JACS 1976, 98, 6705
so, the question is: is this reaction pericyclic, or does it proceed via the diradical
intermediate:
?
The most likely answer is: both; see JACS 1997, 119, 10543, 10545
[1,5] rearrangements
H H
[1,5] as drawn, this is a degenerate rearrangement
(product 6 e_: (π4s + σ2s) - Hückel is identical to starting material)
651.06
183
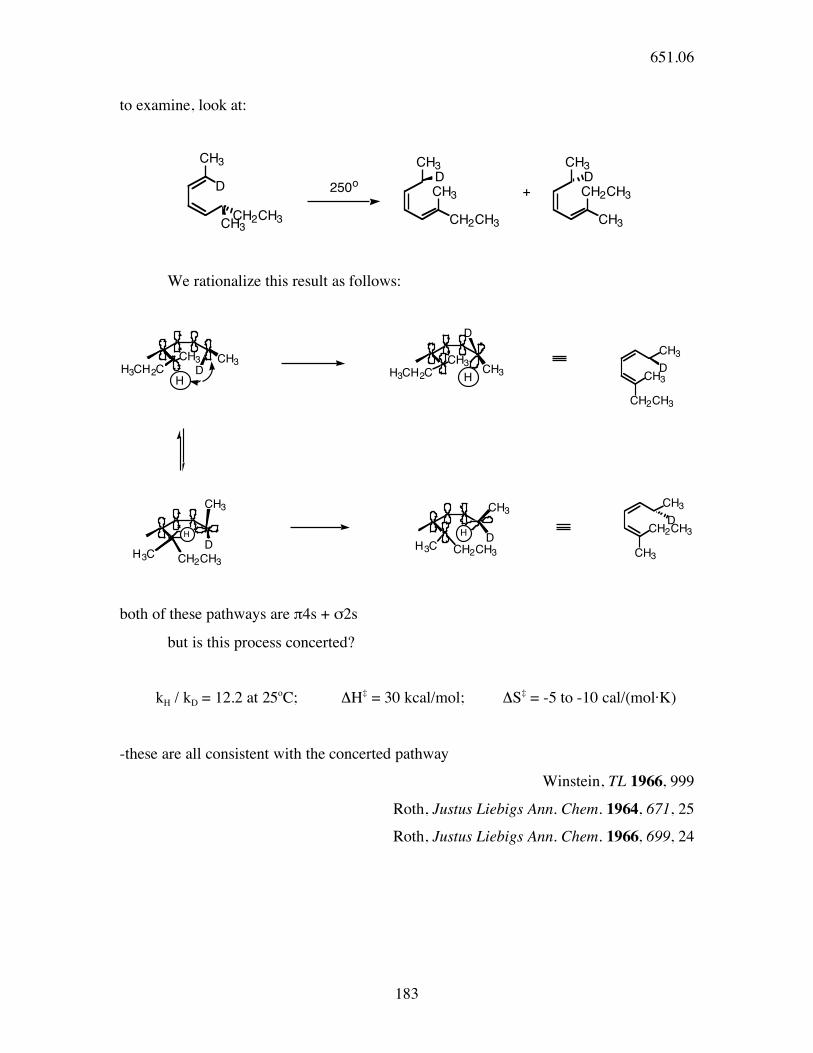
to examine, look at:
DCH3 CH2CH3
CH3 CH3 CH3
CH3CH2CH3CH2CH3
D
CH3
D
+250o
We rationalize this result as follows:
D
CH3H3CH2C
CH3
H
D
CH3H3CH2C
CH3
H CH3D
CH3
CH2CH3
D
CH3
CH2CH3H3C
HD
CH3
CH2CH3H3C
H CH2CH3D
CH3
CH3
both of these pathways are π4s + σ2s
but is this process concerted?
kH / kD = 12.2 at 25oC; ΔH‡ = 30 kcal/mol; ΔS‡ = -5 to -10 cal/(mol·K)
-these are all consistent with the concerted pathway
Winstein, TL 1966, 999
Roth, Justus Liebigs Ann. Chem. 1964, 671, 25
Roth, Justus Liebigs Ann. Chem. 1966, 699, 24
651.06
184
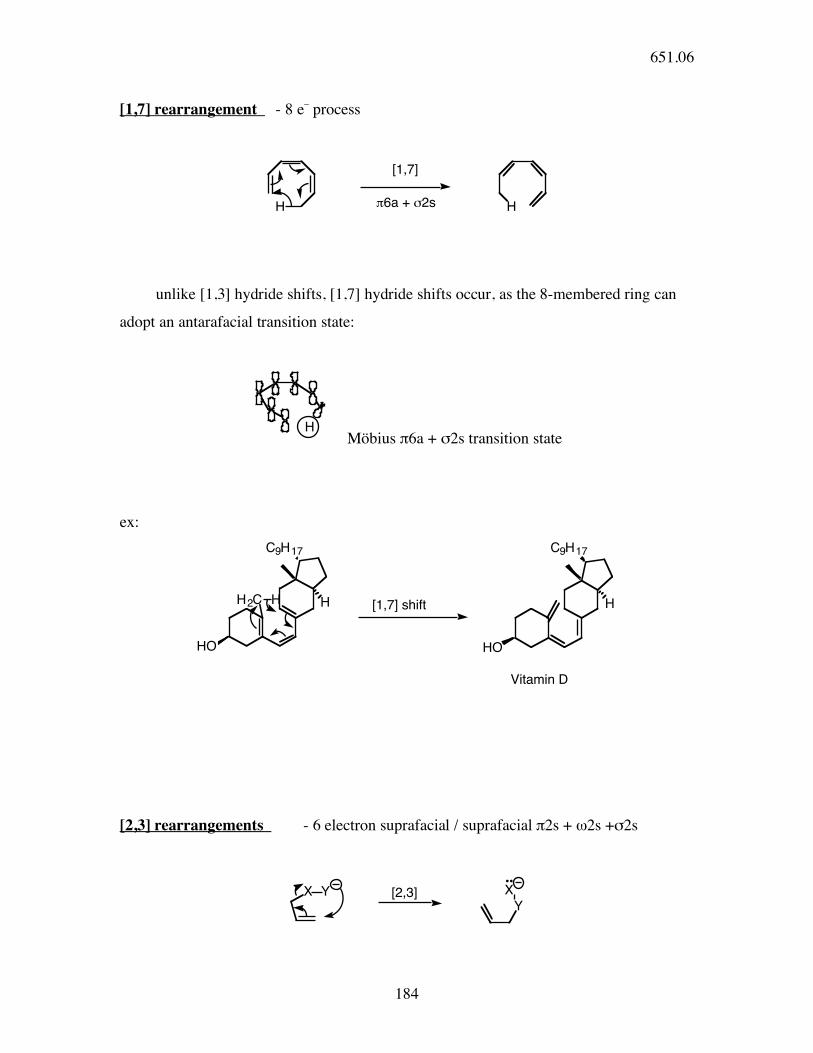
[1,7] rearrangement - 8 e_ process
H H
[1,7]
!6a + "2s
unlike [1,3] hydride shifts, [1,7] hydride shifts occur, as the 8-membered ring can
adopt an antarafacial transition state:
H
Möbius π6a + σ2s transition state
ex:
HO
C9H17
H2C H H
HO
C9H17
H[1,7] shift
Vitamin D
[2,3] rearrangements - 6 electron suprafacial / suprafacial π2s + ω2s +σ2s
X YY
X[2,3]
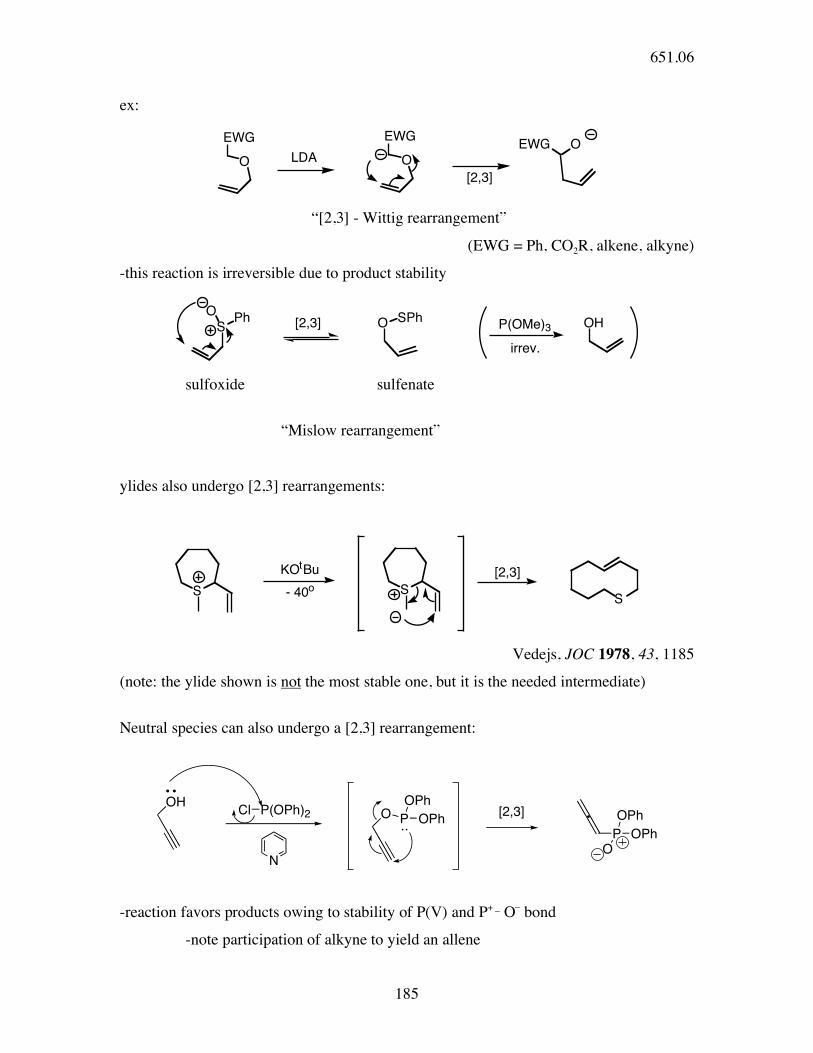
651.06
185
ex:
EWG
O
EWG
OLDA
[2,3]
EWG O
“[2,3] - Wittig rearrangement”
(EWG = Ph, CO2R, alkene, alkyne)
-this reaction is irreversible due to product stability
O
SPh SPhO OHP(OMe)3
irrev.
[2,3]
sulfoxide sulfenate
“Mislow rearrangement”
ylides also undergo [2,3] rearrangements:
SS
S
KOtBu
- 40o
[2,3]
Vedejs, JOC 1978, 43, 1185
(note: the ylide shown is not the most stable one, but it is the needed intermediate)
Neutral species can also undergo a [2,3] rearrangement:
OHCl P(OPh)2 O P
OPh
OPhP
O
OPh
OPh
[2,3]
N -reaction favors products owing to stability of P(V) and P+ _ O_ bond
-note participation of alkyne to yield an allene
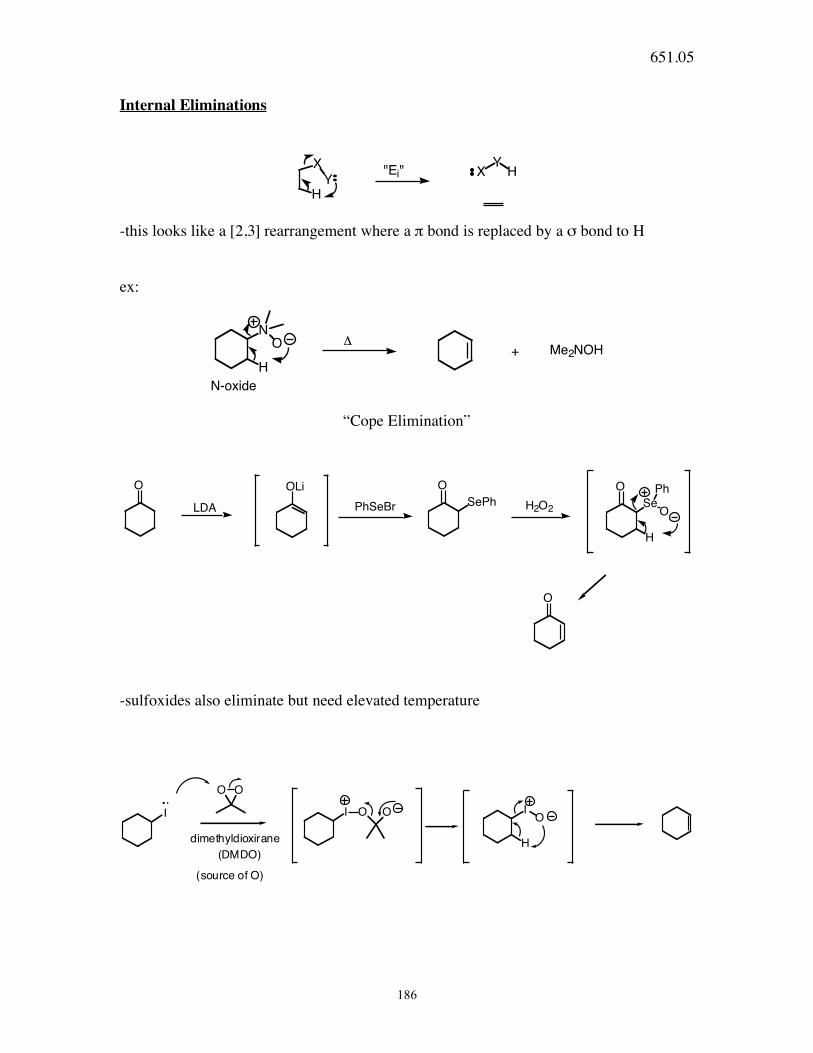
651.05
186
Internal Eliminations
H
Y
X YX H"Ei"
-this looks like a [2,3] rearrangement where a π bond is replaced by a σ bond to H
ex:
N
O
H
+ Me2NOH!
N-oxide
“Cope Elimination”
O OLi O
SePh
O
Se
Ph
O
H
O
LDA PhSeBr H2O2
-sulfoxides also eliminate but need elevated temperature
I
O O
I O O IO
Hdimethyldioxirane
(source of O)
(DMDO)
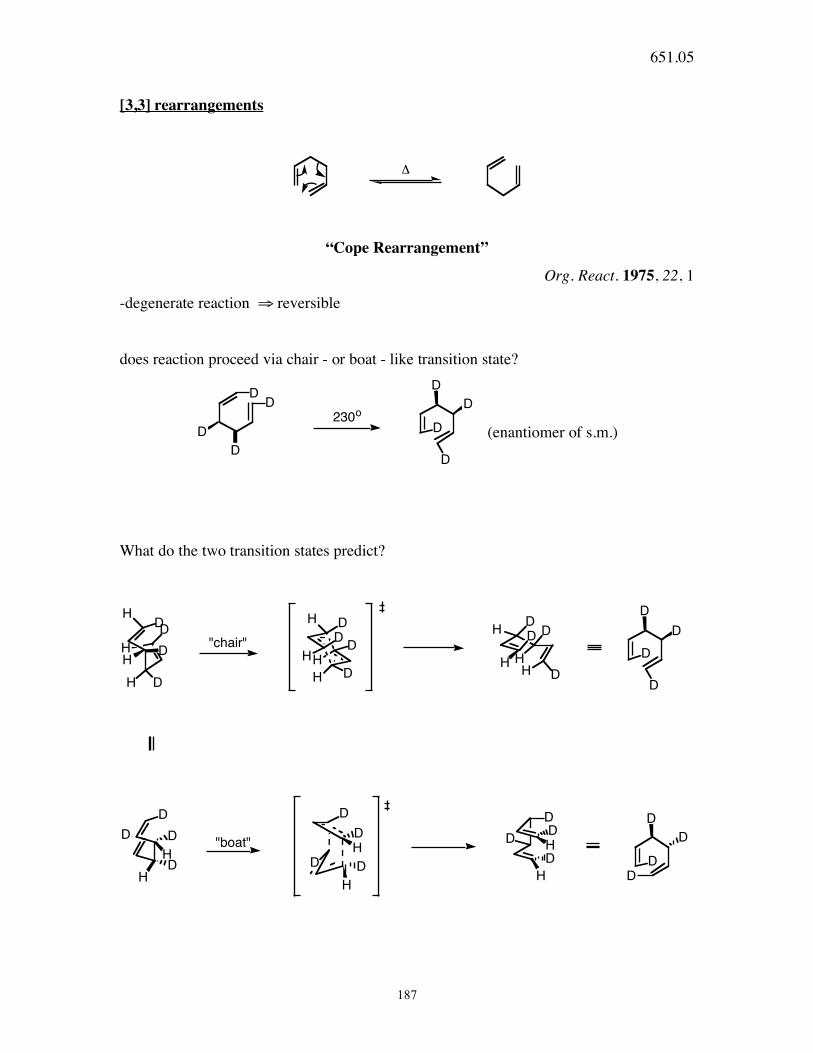
651.05
187
[3,3] rearrangements
!
“Cope Rearrangement”
Org. React. 1975, 22, 1
-degenerate reaction ⇒ reversible
does reaction proceed via chair - or boat - like transition state?
DD
D
D
D
D
D
D
230o
(enantiomer of s.m.)
What do the two transition states predict?
HH
HDD
DH
DH
DH
H
H
DD
D DH
DH D
H
D
HD
D
D
D"chair"
D
H
H
D
D
D D
D
H
H
D
D
D
DD
D
H
H
D
D
D
D
"boat"
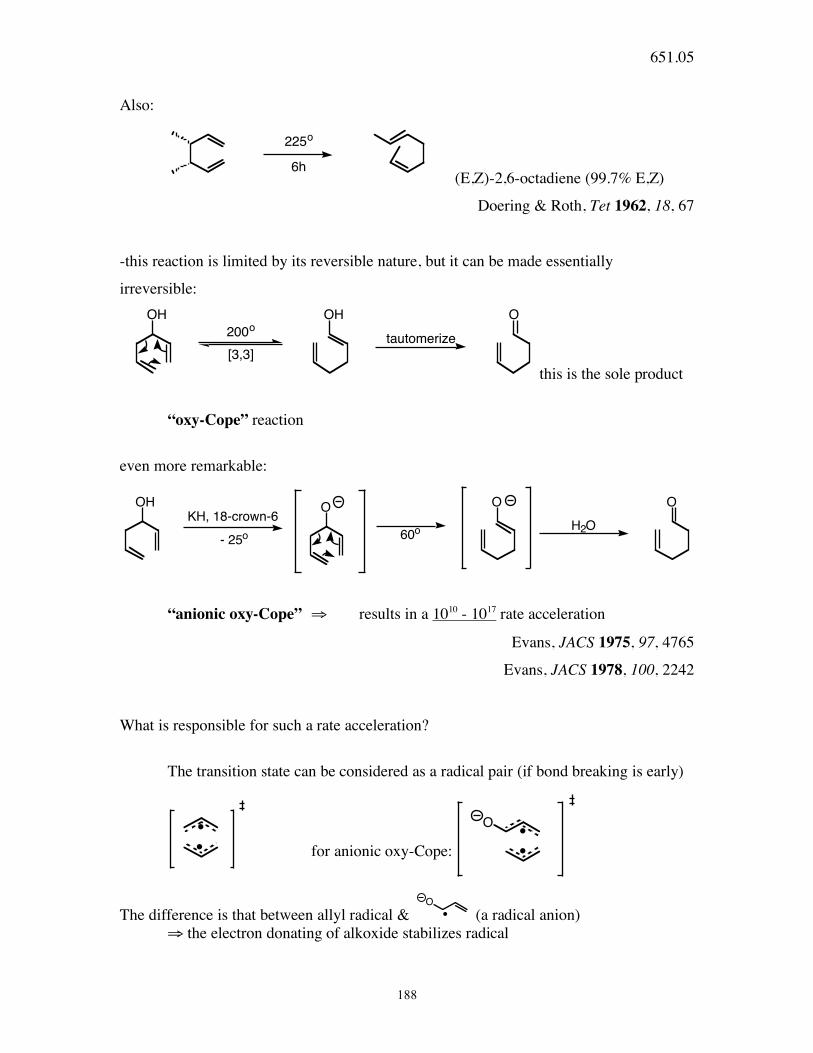
651.05
188
Also:
225o
6h (E,Z)-2,6-octadiene (99.7% E,Z)
Doering & Roth, Tet 1962, 18, 67
-this reaction is limited by its reversible nature, but it can be made essentially
irreversible: OH OH O
tautomerize200o
[3,3]
this is the sole product
“oxy-Cope” reaction
even more remarkable:
O O O
H2O60o
OHKH, 18-crown-6
- 25o
“anionic oxy-Cope” ⇒ results in a 1010 - 1017 rate acceleration
Evans, JACS 1975, 97, 4765
Evans, JACS 1978, 100, 2242
What is responsible for such a rate acceleration?
The transition state can be considered as a radical pair (if bond breaking is early)
for anionic oxy-Cope:
O
The difference is that between allyl radical & O
(a radical anion) ⇒ the electron donating of alkoxide stabilizes radical
651.05
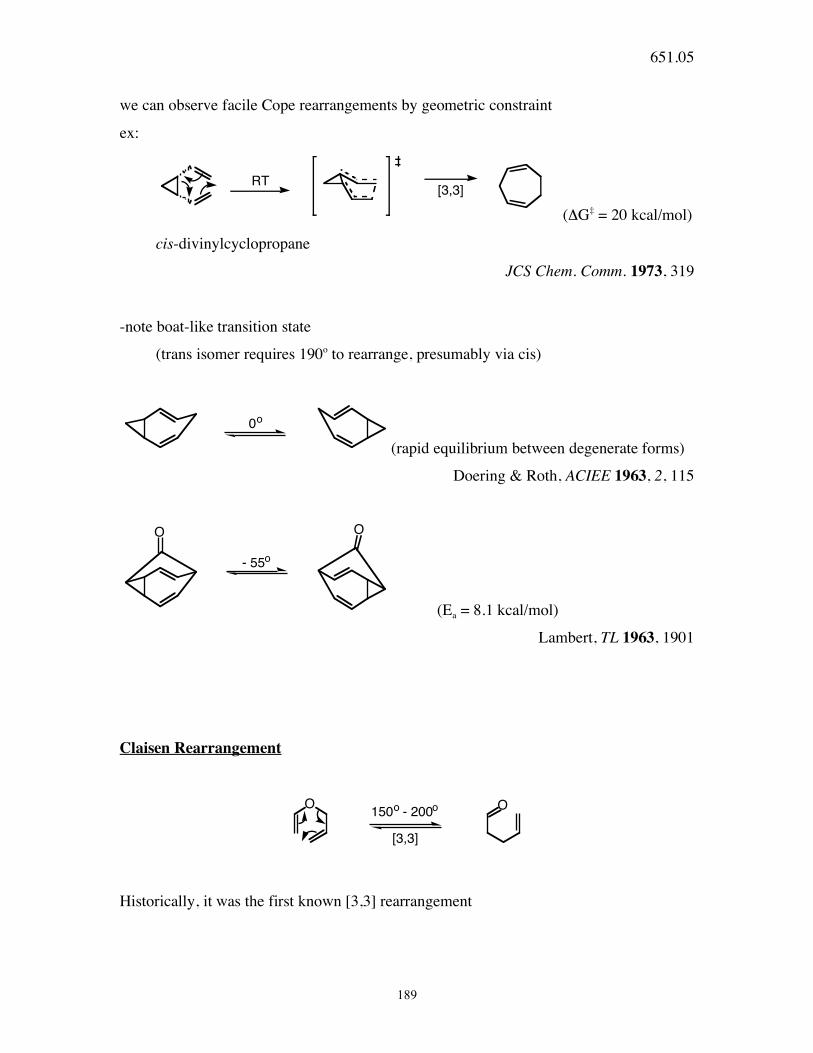
189
we can observe facile Cope rearrangements by geometric constraint
ex:
RT[3,3]
(ΔG‡ = 20 kcal/mol)
cis-divinylcyclopropane
JCS Chem. Comm. 1973, 319
-note boat-like transition state
(trans isomer requires 190o to rearrange, presumably via cis)
0o
(rapid equilibrium between degenerate forms)
Doering & Roth, ACIEE 1963, 2, 115
- 55o
O O
(Ea = 8.1 kcal/mol)
Lambert, TL 1963, 1901
Claisen Rearrangement
O O150o - 200o
[3,3]
Historically, it was the first known [3,3] rearrangement
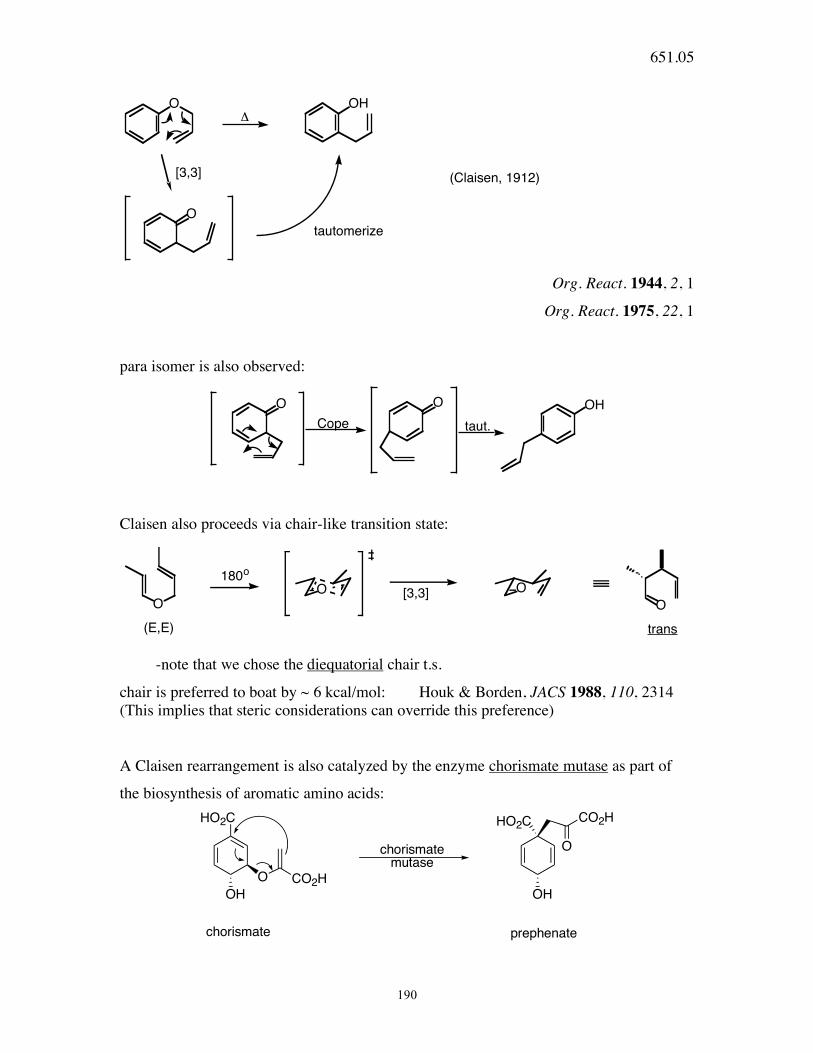
651.05
190
O
O
OH!
[3,3]
tautomerize
(Claisen, 1912)
Org. React. 1944, 2, 1
Org. React. 1975, 22, 1
para isomer is also observed:
O O OH
Cope taut.
Claisen also proceeds via chair-like transition state:
O O
O O180o
[3,3]
trans(E,E)
-note that we chose the diequatorial chair t.s.
chair is preferred to boat by ~ 6 kcal/mol: Houk & Borden, JACS 1988, 110, 2314 (This implies that steric considerations can override this preference)
A Claisen rearrangement is also catalyzed by the enzyme chorismate mutase as part of
the biosynthesis of aromatic amino acids: HO2C
O
OHCO2H
chorismatemutase
HO2C
O
CO2H
OH
chorismate prephenate
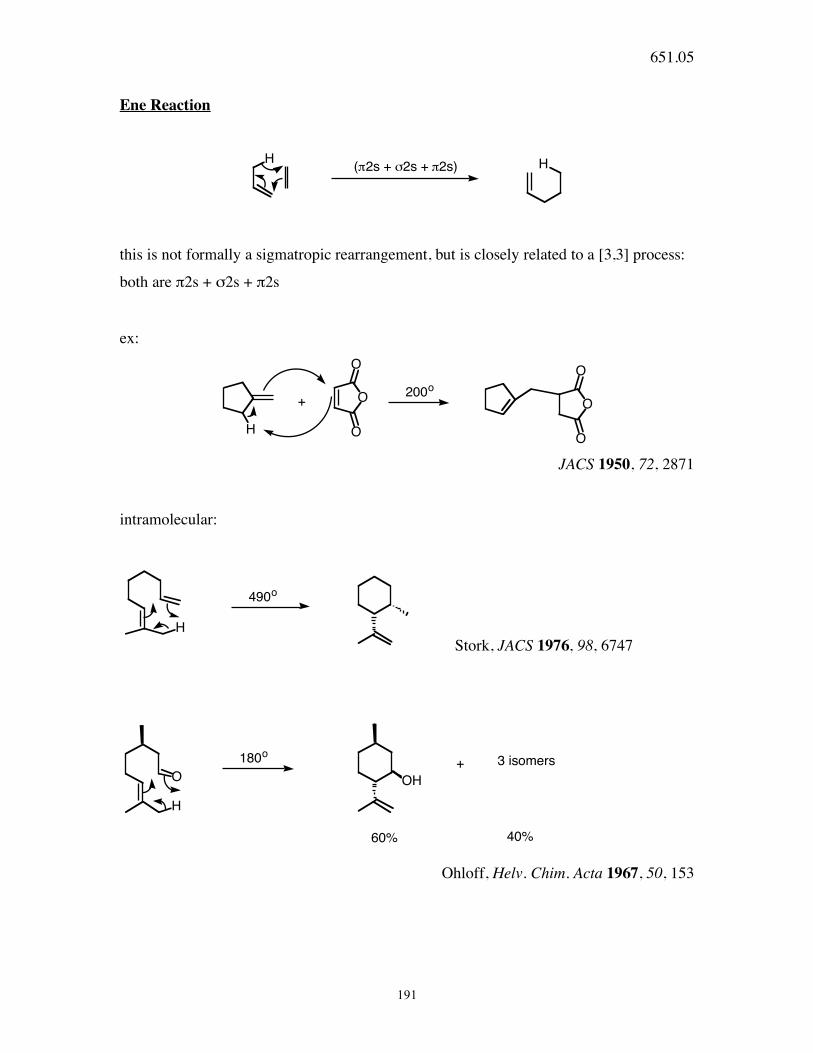
651.05
191
Ene Reaction
H H(!2s + "2s + !2s)
this is not formally a sigmatropic rearrangement, but is closely related to a [3,3] process:
both are π2s + σ2s + π2s
ex:
OO
H
O
O
O
O
200o
+
JACS 1950, 72, 2871
intramolecular:
H
490o
Stork, JACS 1976, 98, 6747
O
H
OH
180o
+ 3 isomers
60% 40%
Ohloff, Helv. Chim. Acta 1967, 50, 153
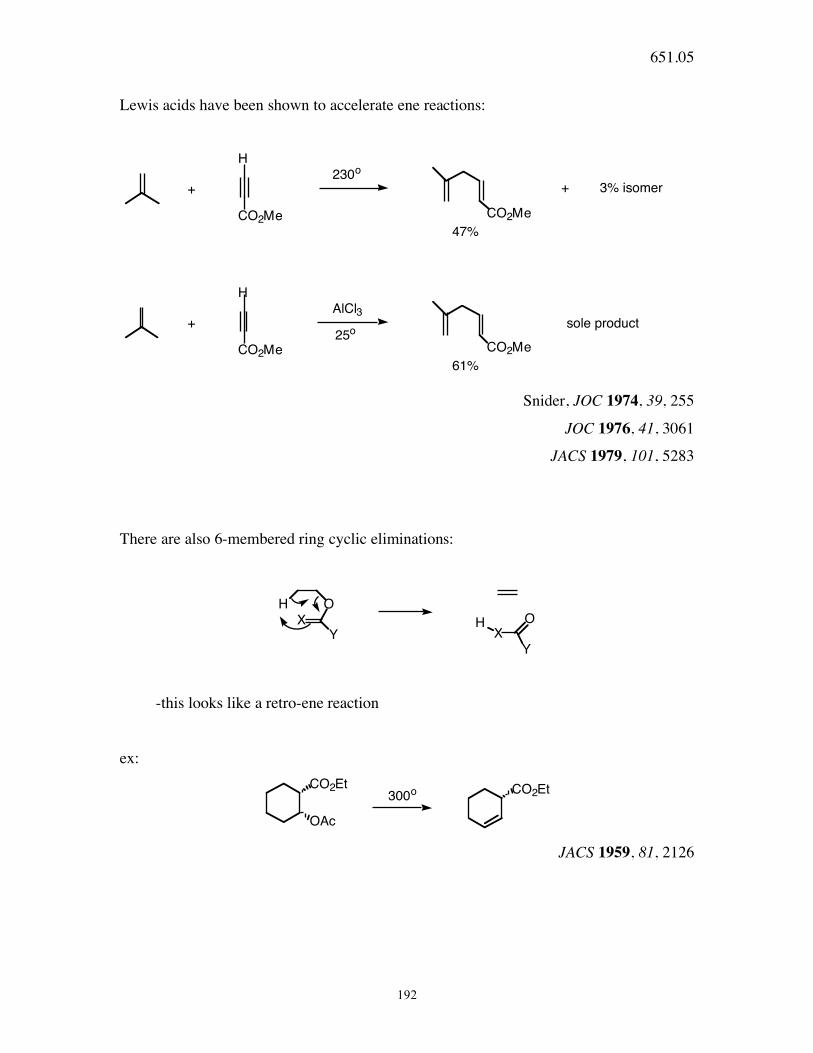
651.05
192
Lewis acids have been shown to accelerate ene reactions:
H
CO2Me CO2Me
+
230o
+ 3% isomer
47%
H
CO2Me CO2Me
+AlCl3
sole product
61%
25o
Snider, JOC 1974, 39, 255
JOC 1976, 41, 3061
JACS 1979, 101, 5283
There are also 6-membered ring cyclic eliminations:
H O
X
Y X
O
Y
H
-this looks like a retro-ene reaction
ex: CO2Et
OAc
CO2Et300o
JACS 1959, 81, 2126
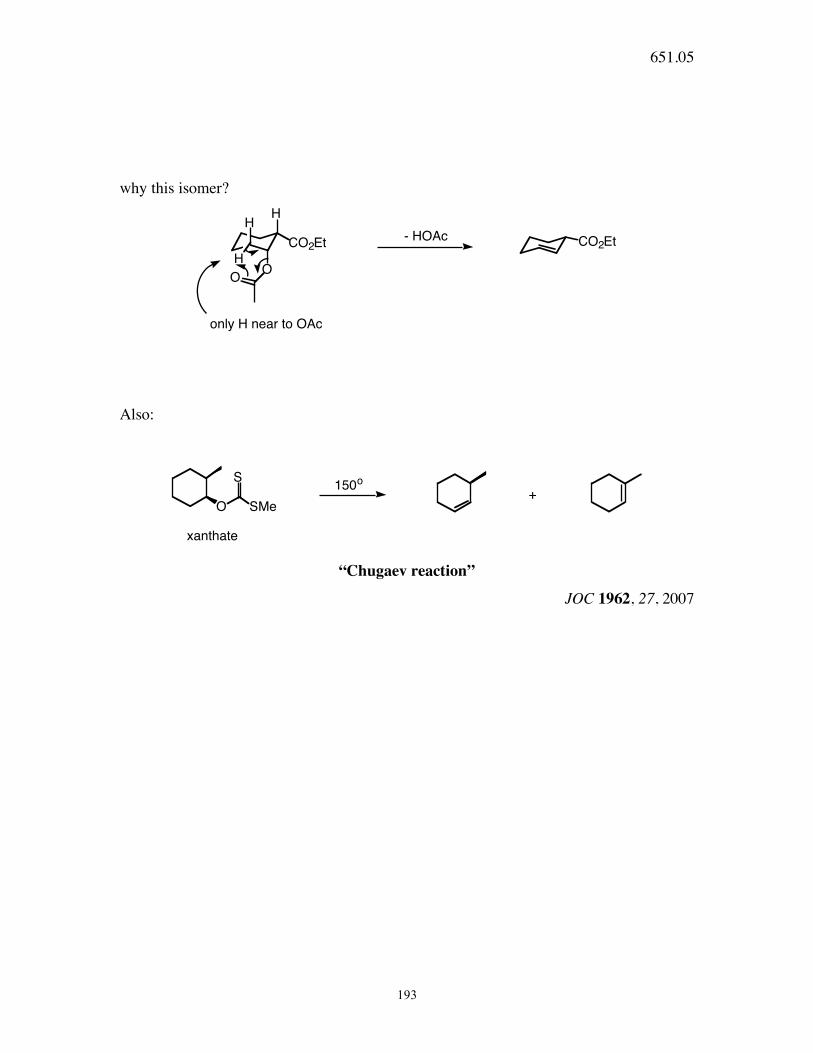
651.05
193
why this isomer? H
H
HO
O
CO2EtCO2Et- HOAc
only H near to OAc
Also:
O SMe
S150
o
+
xanthate
“Chugaev reaction”
JOC 1962, 27, 2007