
and tumorigenesis in multiple cancer types RSPO signaling...
38
1 Therapeutic targeting of tumor-derived R-spondin attenuates β-catenin signaling and tumorigenesis in multiple cancer types Running title: RSPO signaling in cancer Cecile Chartier 1,5 , Janak Raval 1 , Fumiko Axelrod 1 , Chris Bond 1,2 , Jennifer Cain 1 , Cristina Dee-Hoskins 1 , Shirley Ma 1 , Marcus M. Fischer 1 , Jalpa Shah 1,3 , Jie Wei 1,4 , May Ji 1 , Andrew Lam 1 , Michelle Stroud 1 , Wan-Ching Yen 1 , Pete Yeung 1 , Belinda Cancilla 1 , Gilbert O’Young 1 , Min Wang 1 , Ann M. Kapoun 1 , John Lewicki 1 , Timothy Hoey 1 , and Austin Gurney 1 1 OncoMed Pharmaceuticals Inc., Redwood City, CA 2 Current affiliation: Center for Drug Research and Development, Vancouver, BC 3 Current affiliation: ZS Associates, San Mateo, CA 4 Current affiliation: Rinat Laboratories, Pfizer Inc., South San Francisco, CA 5 Correspondence: Dr. Cecile Chartier, OncoMed Pharmaceuticals, 800 Chesapeake Drive, Redwood City, CA 94063 Telephone: 650-995-8200 Fax: 650-298-8600 E-mail: [email protected] Financial support: Research funding was provided by OncoMed Pharmaceuticals. Key words: RSPO, beta-catenin, cancer, antibody therapeutics, cancer stem cells Conflict of interest statement: The authors have no conflicts of interest to disclose. Research. on January 20, 2019. © 2015 American Association for Cancer cancerres.aacrjournals.org Downloaded from Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561
Transcript of and tumorigenesis in multiple cancer types RSPO signaling...

1
Therapeutic targeting of tumor-derived R-spondin attenuates β-catenin signaling
and tumorigenesis in multiple cancer types
Running title: RSPO signaling in cancer
Cecile Chartier1,5, Janak Raval1, Fumiko Axelrod1, Chris Bond1,2, Jennifer Cain1,
Cristina Dee-Hoskins1, Shirley Ma1, Marcus M. Fischer1, Jalpa Shah1,3, Jie Wei1,4, May
Ji1, Andrew Lam1, Michelle Stroud1, Wan-Ching Yen1, Pete Yeung1, Belinda Cancilla1,
Gilbert O’Young1, Min Wang1, Ann M. Kapoun1, John Lewicki1, Timothy Hoey1, and
Austin Gurney1
1OncoMed Pharmaceuticals Inc., Redwood City, CA
2Current affiliation: Center for Drug Research and Development, Vancouver, BC
3Current affiliation: ZS Associates, San Mateo, CA
4Current affiliation: Rinat Laboratories, Pfizer Inc., South San Francisco, CA
5Correspondence: Dr. Cecile Chartier, OncoMed Pharmaceuticals, 800 Chesapeake
Drive, Redwood City, CA 94063
Telephone: 650-995-8200
Fax: 650-298-8600
E-mail: [email protected]
Financial support: Research funding was provided by OncoMed Pharmaceuticals.
Key words: RSPO, beta-catenin, cancer, antibody therapeutics, cancer stem cells
Conflict of interest statement: The authors have no conflicts of interest to disclose.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

2
ABSTRACT
Deregulation of the β-catenin signaling has long been associated with cancer.
Intracellular components of this pathway, including axin, APC, and β-catenin, are
frequently mutated in a range of human tumors, but the contribution of specific
extracellular ligands that promote cancer development through this signaling
axis remains unclear. We conducted a reporter-based screen in a panel of human
tumors to identify secreted factors that stimulate β-catenin signaling. Through
this screen and further molecular characterization, we found that R-spondin
(RSPO) proteins collaborate with Wnt proteins to activate β-catenin. RSPO family
members were expressed in several human tumors representing multiple
malignancies, including ovarian, pancreatic, colon, breast, and lung cancer. We
generated specific monoclonal antibody antagonists of RSPO family members
and found that anti-RSPO treatment markedly inhibited tumor growth in human
patient-derived tumor xenograft models, either as single agents or in combination
with chemotherapy. Furthermore, blocking RSPO signaling reduced the
tumorigenicity of cancer cells based on serial transplantation studies. Moreover,
gene expression analyses revealed that anti-RSPO treatment in responsive
tumors strongly inhibited β-catenin target genes known to be associated with
cancer and normal stem cells. Collectively, our results suggest that the RSPO
family is an important stimulator of β-catenin activity in many human tumors, and
highlight a new effective approach for therapeutically modulating this
fundamental signaling axis.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

3
INTRODUCTION
Beta-catenin signaling is essential to developmental processes and is de-regulated in a
wide range of diseases including cancer.(1-3) WNT ligands are able to activate beta-
catenin-dependent signaling by binding to frizzled (FZD) receptor and LRP5/6 co-
receptor complex, inducing the release of unphosphorylated beta-catenin from a
cytosolic destruction complex comprised of axin, APC, CKI, and GSK3 and its
translocation to the nucleus where it associates with the TCF/LEF family of transcription
factors and induces canonical gene transcription. Intracellular components of the
signaling pathway such as APC, axin, and beta-catenin itself are mutated in a number
of human cancers. However, no WNT ligand overexpression has yet been directly
associated to the disease in humans.(3)
The RSPO family of 4 proteins represents another group of secreted factors that
enhance beta-catenin signaling. RSPO1-4 have a similar structure and harbor two furin
domains, which are necessary and sufficient for beta-catenin activation, and one
thrombospondin type I domain, which promotes non-canonical Wnt signaling.(4,5)
Three classes of transmembrane proteins have been discovered that interact with
RSPO: syndecans, Leu-rich repeat-containing G protein-coupled receptors (LGRs),
and E3 ubiquitin ligases.(5)-(10)) Of note, one of these receptors, LGR5, is a robust
marker of stem cells (SCs).(11)-(18)) A mechanism of action for RSPO has been
elucidated; RSPOs protect FZD receptors from internalization and degradation and
recruit IQGAP1 into the Wnt signaling complex, leading to both an increase in beta-
catenin and Rho GTPase signaling activities.(9),(19) The LGR receptors involved in this
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

4
process are members of the superfamily of seven-transmembrane receptors,
suggesting the potential for additional signaling mechanisms elicited by RPSO.
Like WNTs, RSPOs have important roles in development and act as powerful SC
growth factors.(20) RSPO1 is used as an indispensable agent for in vitro propagation of
intestinal SCs and its activity cannot be replaced by exogenous WNT protein.(21) In vivo,
RSPO1 controls the phenotypic sex of humans and mice, RSPO2 is necessary for
development of numerous mouse organs, RSPO3 is involved in angiogenesis during
placenta development in mice, and RSPO4 is the gene disrupted in humans with
congenital anonychia.(22) In most cases, RSPOs function by enhancing Wnt/beta-
catenin signaling. However, RSPO2 or 3 were also shown to signal through cascades
that are separate from the canonical pathway.(5),(8),(19),(23)-(25)
Several lines of evidence suggest a role for RSPOs in cancer development. The
identification of Rspo2 and Rspo3 as sites of integration for MMTV-induced mammary
tumors in mice supported a role as breast cancer oncogenes.(26)-(28)) Ectopic
expression of Rspo2 was shown to increase tumorigenic and invasive properties of
mouse breast cell lines.(25) Genomic rearrangements and transcriptional activation in
human tumors that result in elevated RSPO expression have been identified,
suggesting RSPO may be functionally important for tumor development.(29)-(32))
However, no direct demonstration of a functional role for RSPO in cancer development
and maintenance has been reported to date.
Here, we report an analysis of patient-derived xenograft (PDX) tumor samples for
RSPO expression and activity. Functional RSPO activity was found to be produced by
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

5
human tumors of multiple types. The importance of RSPO in driving the growth of over-
expressing tumors was tested in vivo with anti-RSPO mAbs directed against various
members of the family. Efficacy was observed in PDX models established with
minimally passaged human tumors of multiple types. Gene expression analyses
revealed that RSPO promoted expression of genes associated with Wnt signaling
pathway and normal and cancer SCs (CSCs). These data provide the first
demonstration that RSPO family members play a major role in the growth of human
cancer and validate RSPO1-3 as therapeutic targets.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

6
MATERIALS AND METHODS
Xenograft models and tissue processing. Non-obese diabetic/severe combined
immunodeficient (NOD/SCID) mice were purchased from Harlan Laboratories and
maintained under specific pathogen-free conditions and provided with sterile food and
water ad libitum. Animals were housed in a U.S. Department of Agriculture-registered
facility in accordance with NIH guidelines for the care and use of laboratory animals.
The mice were allowed to acclimate for several days before the studies.
The establishment of OMP PDX models was described previously.(33) Patient-derived
tumors were passaged in mice without any intervening cell culture. Tumor cells were
stored in liquid nitrogen. Experiments for obtaining fresh tumor cells or testing antibody
therapeutic efficacy were initiated from frozen cell stocks. CR3150, CR1560, CR2506,
and CR2513 models were established at Crown BioScience (Beijing, China).
For in vitro assays and in vivo tumorigenicity assessments, single cell suspensions were
obtained from freshly dissociated tumors according to a previously described
procedure.(34) Cells were then incubated with biotinylated antibodies (α-mouse CD45-
biotin 1:200 dilution and α-mouse H2Kd-biotin 1:100 dilution, BioLegend, San Diego,
CA) on ice for 30 min followed by addition of 50 ul MagnaBind streptavadin-labeled
magnetic beads (Thermo Scientific, Waltham, MA) per 10E6 cells/ml. Mouse cells were
removed on a magnetic stand.
Cell lines. The HEK-293 STF cell line was a kind gift from Dr. Jeremy Nathans received
in 2005. It contains the TOP-FLASH reporter system comprised of 8 copies of a TCF
binding site (AGATCAAAGG) upstream of a minimal promoter and firefly luciferase.
Cells were cultured in DMEM, 10% FBS, 500 ug/ml Geneticin (Life Technologies,
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

7
Carlsbad, CA) and incubated at 37oC, 5% CO2. HEK-293T (CRL-3216TM) and WNT3A-
L cells (CRL-2647TM) were obtained in 2005 from ATCC (Manassas, VA) and cultured
according to the ATCC guidelines.
STF reporter assay. Tumor cells were cultured in DMEM, 10% FBS (Life Technology,
Carlsbad, CA) for 24 hours at 37oC and 5% CO2. One volume of conditioned medium
and cells was added on top of a monolayer of HEK-293 STF cells grown in 1 volume of
DMEM, 10% FBS. When used, 5 ng/ml of recombinant human RSPO (R&D systems,
Minneapolis, MN) were added to the reporter cells. WNT3A-conditioned medium
prepared from WNT3A-L cells was added to the tumor cell conditioned medium or
purified RSPO at a final concentration of 25% v/v. The activity of purified soluble
receptors (10 ug/ml) and monoclonal antibodies (40 ug/ml) was compared to the control
immunoadhesin JAG1-Fc and control antibody LZ1. Reporter cells were incubated for
16 hours prior to luciferase activity measurement using Steady-Glo® according to the
manufacturer instructions (Promega, Madison, WI).
Real-time RT-PCR. For RSPO gene expression analyses, RNA was extracted from
xenograft tumors using an RNeasy Fibrous Tissue mini kit (Qiagen, Germantown, MD)
according to the manufacturer’s instructions. Thirty ng RNA were submitted to a one-
step RT-PCR reaction using Applied Biosystems 7900HT instrument and buffer system
according to the manufacturer’s instructions (Life Technologies, Carlsbad, CA). GUSB
was used as endogenous control against which delta Cts were calculated and the
lowest RSPO expressing sample as baseline for the delta-delta Ct calculations.
Relative expressions were determined by raising 2 to the power of the negative value of
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

8
delta-delta Ct for each sample. All gene expression assays were purchased from ABI
and verified for species specificity prior to screening the OMP PDXs.
- GUSB: Hs00939627_m1
- RSPO1: Hs01045335_mH
- RSPO2: Hs00379983_m1
- RSPO3: Hs00262176_m1
- RSPO4: Hs01382765_m1
For select target gene expression analyses, frozen tumor tissues were pulverized in
liquid nitrogen with a mortar and pestle. RNA was extracted as above. All RNAs were
verified to contain intact ribosomal bands by Bioanalyzer (Agilent, Mountain View, CA).
cDNA template was generated from 50 ng RNA using Cells Direct one-step qRT-PCR
kit (Life Technologies, Carlsbad, CA). PCR was performed on ABI 7900HT (Life
Technologies, Carlsbad, CA) using the relative standard curve method. Gene
expression normalization was performed with GAPDH. Statistical calculations were
performed using t-test with Excel software (Microsoft, Redmond, WA). Primer (forward
and reverse) and probe sequences were as follows. TDGF1: 5’-
CCAGAGTGCTGAAGGAATGGA-3’, 5’-CCTCTCTCTTCTATTTGCTTCCTCTT-3’, and
5’-CCTACCCAGTCTCCCTGCACACACG-3’; ASCL2: 5’-CCCCTCCCCACAGCTTCT-
3’, 5’-AAATGGATTCTCTGTGCCCTTAGA-3’, and 5’-
CACCAACACTTGGAGATTTTTCCGGAGG-3’; CLDN2: 5’-
CCTAAGTCCCCAACCCTCAAC-3’, 5’-GGGCAAAGGGATCCTCTGA-3’, and 5’-
TGAAACCCCATTCCCTTAAGCCAGGA-3’; PTPRO: 5’-
GAACTCTGTTGCTGTCTGAGCAA-3’, 5’-GCTCTCACATGTATGCAGGTCAA-3’, and
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

9
5’-CGTGGTGCCTAGACTTTGCATTCCTTG-3’; RPRD1A: 5’-
CACCTGAGGCAGATCATGCA-3’, 5’-ACGGCACCATGCTTTTCTCT-3’, and 5’-
CTTTGAGTGCAGTTTGGTCTGACCCCTC-3’; LGR5: 5’-
TGGACTCAAGAGACTCAGTAACGTATTAT-3’, 5’-
TAAGCAGAGAAGTAATGTTCCTAACATCA-3’, and 5’-
ATTTAGCTTGGTTTTAGCTGTGTTCTCTCTGGATAACC-3’; ZNRF3: 5’-
CCATGTCTTATGTTGAGAGTGTGACA-3’, 5’-TGCAAATATGTAAAATCTGTGTGCAA-
3’, and 5’-TGGAATAATCATTGAAAATGACTAACACAAGACCCTGTAA-3’; AXIN2: 5’-
CCTTTCCTCCACACACCTTCA-3’, 5’-TGTATAGTACAAGTAACAATGGCAAACAG-3’,
and 5’-ATGTACAGATTAACTTAACACAAAAACCCGAACATCAAA-3’; GAPDH: 5’-
CCACCCATGGCAAATTCC-3’, 5’-TGGGATTTCCATTGATGACAAG-3’, and 5’-
TGGCACCGTCAAGGCTGAGAACG-3’.
Antibodies. Anti-RSPO antibodies were generated by immunizing mice with purified
recombinant human RSPO1, RSPO2, or RSPO3 (R&D systems, Minneapolis, MN)
followed by hybridoma generation and characterization. The antibodies LZ1, an anti-
lysozyme monoclonal generated by panning a phage display library obtained from
MorphoSys (Martinsried/Planegg, Germany) and 1B711, a murine monoclonal directed
against dinitrophenol (anti-hapten) and obtained from the ATCC (Manassas, VA) were
used as negative controls in the in vitro and in vivo experiments, respectively.(35) Anti-
RPSO3 monoclonal antibody #2 was isolated by a mammalian cell antibody display
technique, MAbTrap, in which antibody sequences derived from the immunized mice
were transiently expressed and then retained on the cell surface by interaction with a
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

10
membrane-anchored CH2-CH3 antibody fragment. FACS was used to isolate cells
expressing the antibody of interest and corresponding cDNA isolated.
The binding of anti-RSPO antibodies to RSPO was assessed by flow cytometry. First,
HEK-293T cells were transiently transfected with cDNA expression vectors for RSPO
extracellular domain fused to human CD4 transmembrane domain fused to GFP using
Fugene 6 (Promega, Madison, WI). Transfected cells were then incubated with 20 ug/ml
antibody and washed with PBS. Antibody binding was detected with a PE-conjugated
anti-mouse Fc antibody and light signals measured for PE and GFP, using a Canto II
instrument (BD Biosciences, San Jose, CA).
To assess ability of anti-RSPO antibodies to block interaction with LGR, binding studies
were conducted. RSPO proteins were expressed as Fc-fusion proteins containing
RSPO N-terminal furin domains and human IgG1 Fc. HEK-293T cells were transiently
transfected with cDNA expression vector encoding the N-terminal extracellular domain
of human LGR fused to human CD4 transmembrane domain fused to GFP. After 24
hours cells were incubated with RSPO-Fc fusion proteins and anti-RSPO antibodies at
10 ug/ml. RSPO binding was detected by flow cytometry with a PE-conjugated anti-
human Fc antibody and light signals measured for PE and GFP.
In vivo efficacy experiments. About 50,000 freshly thawed tumor cells re-suspended
in 100 ul 50% matrigel in FACS buffer (OMP PDXs) or tumor fragments (Crown
BioScience PDXs) were injected sub-cutaneously into the left flank region of 6- to 8-
week old NOD/SCID mice with a 25-gauge needle. Tumor-bearing animals were
randomized and treatment started when the mean tumor volumes reached 100 to 150
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

11
mm3. Chemotherapeutic agents were given once weekly at various doses ranging from
10-100 mg/kg as specified in the figure legends. Antibodies were dosed once weekly or
bi-weekly at a dose ranging from 10-25 mg/kg. All agents were administered
intraperitoneally.
For the tumorigenicity study, single tumor cell suspensions prepared as described from
control and treated tumors were counted and diluted to 50 cells in 100 ul 50% matrigel
in FACS buffer. Ten NOD/SCID mice were injected subcutaneously per group. Tumor
growth was monitored for 59 days without any further treatment.
Microarray analysis. RNA was isolated and microarray analyses were conducted as
detailed in Supplementary Data. Data are available through GEO database, accession
GSE73906.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

12
RESULTS
Identification of RSPO as a tumor-derived beta-catenin stimulating activity
In order to identify possible beta-catenin stimulating activities produced by human tumor
cells, a functional screen was developed. Tumor cells freshly obtained from minimally
passaged PDXs (referred to as OMP-xxxx tumors hereafter) were co-cultured with a
HEK-293 cell-based reporter cell line containing a TCF-luciferase reporter (STF). This
reporter cell line responds to WNT and RSPO signals through beta-catenin-driven
transcription. Forty eight OMP tumors (Table S1) were screened in this assay, of which
3 (6.25%) induced reporter activity more than 2 fold relative to control (Fig 1A and Table
S2). OMP-LU2, OMP-LU25, and OMP-OV38 induced luciferase activity 6.7, 4.7, and
3.1 fold over medium alone, respectively. In this survey, WNT3A was also tested, alone
as a positive control and added to the co-culture assay. Interestingly, the 3 tumors that
were positive for beta-catenin signaling activity displayed further elevated activation
upon addition of WNT3A (Fig 1A). OMP-LU2, OMP-LU25, and OMP-OV38 boosted
WNT3A activity 4.3, 9.6, and 1.6 fold over WNT3A alone. Such potentiation of WNT
has been noted as a hallmark of RSPO activity, suggesting that RSPO ligands might be
responsible for the activation of the beta-catenin reporter in the assay.(4) Of note,
additional OMP tumors were shown to score in the assay only in the presence of
WNT3A, possibly reflecting lower levels of the beta-catenin activating factor(s). OMP-
B37, OMP-B39, and OMP-LU102 increased WNT3A activity 2.7, 9, and 1.8 fold,
respectively (Fig 1A and Table S2). To examine the tumor-derived activity further,
FZD8-Fc and LGR5-Fc decoy receptors were tested in the assay. FZD8-Fc acts as an
inhibitor of Wnt signaling by binding WNT ligands, whereas LGR5-Fc binds to RSPO
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

13
and antagonizes RSPO potentiation of Wnt signaling (Fig 1B and C). In conditions in
which FZD8-Fc abolished WNT3A-induced reporter activity, it partially inhibited the
activity induced by the tumor cells alone and dramatically decreased the activity induced
by the tumor cells in presence of exogenously added WNT3A. Conversely, while LGR5-
Fc remained mostly ineffective at inhibiting WNT3A alone, it efficiently decreased the
activity of the tumor cells alone and supplemented with WNT3A. LGR5-Fc reduced the
activity of the co-cultures containing added WNT3A to a level comparable to that
observed with WNT3A alone (Fig 1D). These results suggest that the activation of the
beta-catenin reporter observed in the tumor cell culture assay reflects the presence of
RSPO activity potentiating low levels of endogenous WNT ligands. The modest
inhibition of the tumor-induced activation by FZD8-Fc suggests the presence of a
distinct (WNT or non-WNT) ligand not bound by FZD8-Fc.
Interestingly, the tumors previously found to exhibit beta-catenin activity possessed
markedly elevated levels of particular RSPO family members. Generally only a single
RPSO family member was highly expressed within a tumor as determined by
quantitative real time polymerase chain reaction (qPCR) (Fig 2 and Table S2). Thus,
OMP-LU2, OMP-LU25 and OMP-OV38 expressed high levels of RSPO2 or RSPO3.
Additional RSPO mRNA-positive tumors, e.g. OMP-OV19 and OMP-PN7 (Fig1A and
Table S2), had not been identified by the in vitro functional assay, maybe due to the low
sensitivity of the assay. It has been reported that RSPO2 and RSPO3 genes are targets
of rearrangements in colon cancer and that resulting fusions increase gene
expression.(29,31) Based on these reports, we performed a PCR-based screen and
also mRNAseq analysis. These analyses did not find fusions in either RSPO2 or
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

14
RSPO3 in our tumor bank, including RSPO2-high OMP-C28, suggesting that other
mechanisms are involved in the over-expression of RSPO in these tumors. RSPO3-high
human CRC tumors possessing the reported PTPRK-RSPO3 gene fusion were
identified at an external service provider (Fig S1).
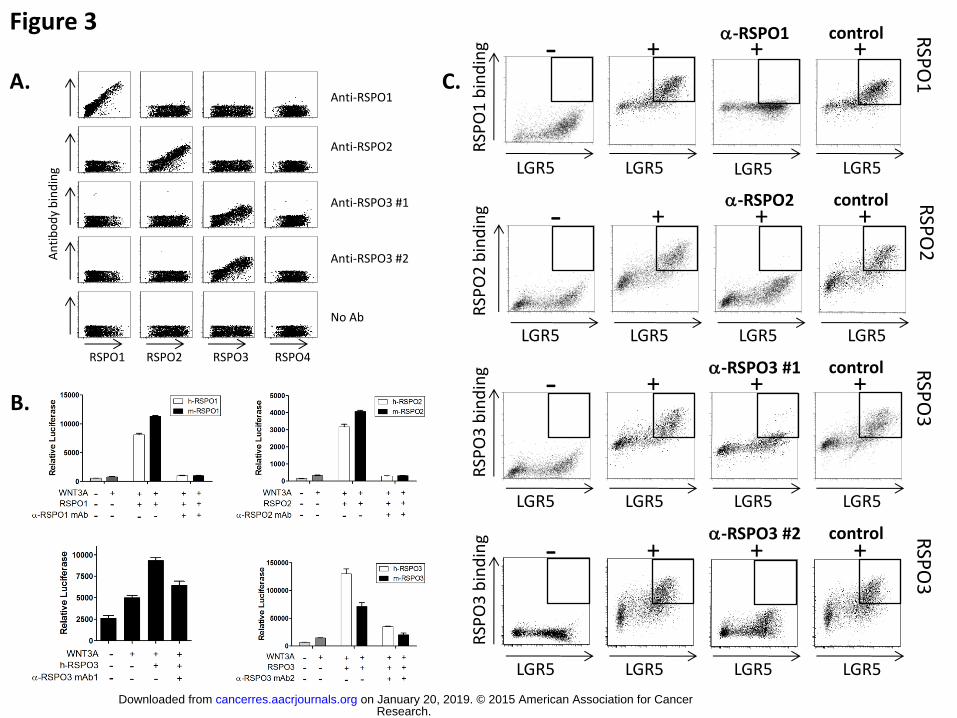
In order to investigate the functional consequence of inhibiting the activity of specific
RSPOs, monoclonal antibodies were generated that bind RSPO1, RSPO2, or RSPO3
(Fig 3A) and block the ability of RSPO to potentiate WNT stimulation of the beta-catenin
luciferase reporter through blocking binding to LGR (Fig 3B and C). All antibodies were
shown to be specific for their targeted RSPO and not to cross-react with the other
RSPOs. Anti-RSPO antibodies were tested for impact on the tumor-induced beta-
catenin activity observed in the luciferase reporter screen. Antibody to RSPO2 blocked
the activity produced by OMP-LU2 tumor cells and antibody to RSPO3 blocked the
activity observed in response to OMP-LU25, OMP-LU102, and OMP-OV38 tumor cells
(Fig 4). In each case, the antibodies inhibited the reporter activity back to control levels.
None of the 3 antibodies affected the reporter activity when non-beta-catenin inducing
tumor cells such as OMP-PN25 and OMP-M6 were used in the assay (Fig S2). Overall,
the results parallel the relative RSPO gene expression pattern (Fig 2, Table S2),
strongly implicating the RSPO proteins as the inducers of the observed beta-catenin
signaling activity produced by these human tumor cells. In particular, the above data
revealed RSPO2 or RSPO3 as the principal factors responsible for the beta-catenin
activity derived from several of our patient-derived tumors, making potential therapeutic
targets of RSPO2 and RSPO3.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

15
RSPO antagonists inhibit the growth of human tumor xenografts
The impact of inhibiting RSPO signaling on tumor growth was assessed using PDXs in
mice. Tumor-bearing mice were treated with antibodies to individual RSPO family
members. Anti-RSPO treatment produced significant inhibition of tumor growth in
several types of human tumors (Fig 5A). Growth of OMP-OV19, an ovarian tumor noted
to express RSPO1, was inhibited by antibody to RSPO1. OMP-C28 and OMP-PN7, a
colon and a pancreatic tumor, respectively, noted to express RSPO2, were inhibited by
antibody to RSPO2. Anti-RSPO3 displayed significant anti-tumor activity in several
RSPO3-high tumors including non-small cell lung cancer (NSCLC) models OMP-LU25
and OMP-LU102, colorectal cancer (CRC) models CR3150, CR2513, and CR2506, and
ovarian cancer OMP-OV38. Colon tumor CR3150 is noteworthy as a tumor possessing
a RSPO3 chromosomal translocation identical to those previously reported.(29) Thus,
anti-RSPO treatments showed single agent therapeutic activity in most RSPO-high
models. Combination with chemotherapy, including taxane treatment in NSCLC and
ovarian models, gemcitabine treatment in pancreatic tumor, and irinotecan in CRC
models, resulted in further significant inhibition of tumor growth beyond the impact of
chemotherapy alone (Fig 5A). The mechanism of combination activity with paclitaxel
was investigated in OMP-OV38. Anti-RSPO3 treatment combined with paclitaxel was
found to increase mitotic cell death (Fig S3).
Not all RSPO-high tumors were sensitive to RSPO inhibition. For example, the NSCLC
OMP-LU2, despite high levels of RPSO2 mRNA, and CRC CR1560, despite high levels
of RSPO3 mRNA, were not significantly inhibited by anti-RSPO2 and anti-RSPO3,
respectively, whether alone or combined with chemotherapy (Fig 5A). Analysis of LGR4,
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

16
5, and 6 gene expressions revealed no correlation between receptor expression and
resistance to antibody treatment (Fig S4). Tumors that did not express RSPO were
unresponsive to antibody treatment (Fig S5).
The responsive NSCLC model OMP-LU25 was used to assess the impact of RSPO3
inhibition on CSCs, also known as tumor initiating cells. For this, tumors previously
treated with antibody and/or chemotherapy were harvested and processed to a single
cell suspension and 50 cells were injected into recipient mice for a second (treatment
free) round of propagation. The ability of this low cell number to re-grow a tumor
provided a direct measure of their CSC content. In such a setting, anti-RSPO3
significantly reduced the tumorigenicity of OMP-LU25 tumor cells both as a single agent
and in combination with paclitaxel (Fig 5B). While all 10 control mice and 8 out of 9
paclitaxel group mice developed tumors, the anti-RSPO3 and combination-treated
tumor cells engrafted poorly (7:10 and 6:10, respectively) and grew slowly. Similar
reductions in tumorigenicity after anti-RSPO treatment were observed in other tumors
including recently obtained pancreatic and lung tumors that were not included in our
original panel (Fig S6). In addition, levels of CD44, a well-known colon CSC marker,
were significantly reduced in anti-RSPO3-treated tumors (Fig S7).(33) These results
show that RSPO3 is important to maintain CSCs in multiple tumor types.
RSPO blockade inhibits stem cell signaling pathways in tumors
RSPO1 functions as a potent regulator of SC growth in colon.(21) The expression of a
panel of genes associated with SC and/or RSPO signaling was thus monitored in anti-
RSPO3-treated CRC xenografts. Control and antibody-treated tumor RNAs from the in
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

17
vivo efficacy experiments conducted with CR3150, CR2506, CR2513, and CR1560
were tested for the expression of ASCL2, AXIN2, CLDN2, LGR5, PTPRO, RPRD1A,
TDGF1, and ZNRF3 genes, using qPCR. Strikingly, 7 of the 8 genes were down-
regulated in response to anti-RSPO3 in all 3 sensitive CRC models (Fig 6). The down-
regulation was highly significant in at least 2 of the models for these genes. For
example, ASCL2 expression could no longer be detected in any of the anti-RSPO3-
treated CR2506 and CR2513 samples, LGR5 was down-regulated 7.24 and 5.75 fold in
treated CR2506 and CR3150 tumors, respectively, and TDGF1 mRNA levels were
reduced in all 3 models, up to 234 fold. All 7 down-regulated genes are reported
canonical Wnt signaling target genes. (36,37) Several of these genes, ASCL2, LGR5,
and TDGF1, have also been directly associated with CSCs in various tumor types,
supporting a role for the RSPO pathway in CSC biology and consistent with the in vivo
tumorigenicity results presented herein.(38-40) Down-regulation of Wnt and SC genes
by anti-RSPO treatment could be involved in the sensitization of tumor cells to
chemotherapeutic agents.(41,42) Two of the 8 studied genes, AXIN2 and ZNRF3, were
down-regulated by anti-RSPO3 in the non-responding CR1560 model. The 2 genes are
the only Wnt target genes of the panel that were highly expressed in this model,
possibly reflecting a lower state of Wnt signaling activation. Thus, isolated Wnt targets
could be regulated by anti-RSPO3 therapy and not be associated with growth inhibition
of non-RSPO/Wnt-driven tumors.
To further study the mechanism of action of RSPO3, microarray analysis was performed
on anti-RSPO3-treated colon tumors. Findings were consistent with the qPCR results
(Fig. 6) and also revealed new insights into the RSPO3 mechanism of action (Table
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

18
S3). In the 3 responsive models, modulation of Wnt signaling was confirmed by the
down-regulation of numerous Wnt genes that included CLDN2, EPHB2, CD44, LRP4,
ASCL2, AXIN2, LGR5, PTPRO, TDGF1, and ZNRF3. In addition, effects on Notch
signaling were revealed by the down-regulation of genes such as NOTCH1 and PSEN1.
Both Wnt and Notch pathways are associated with CSCs (43-45). Consistently,
numerous SC genes were inhibited by anti-RSPO3 in the 3 responders. Interestingly,
differentiation genes including BAMBI, CADM1, POU2F2, and RGS2 were up-regulated.
Most of the genes described above were not significantly modulated by anti-RSPO3 in
the non-responsive CR1560. Together, these results show that RSPO3 blockade
inhibited CSC signaling pathways in CRC, and suggest that the tumor cells may adopt a
more differentiated cell fate. Microarray data for anti-RSPO3 plus irinotecan-treated
CR2506 further support this mechanism by showing that most Wnt and SC genes
targeted by anti-RSPO3 remained modulated upon addition of chemotherapeutics, with
some of them responding to a greater extent to the combination than to anti-RSPO3
alone (Table S3). Overall, the gene expression data support the inhibition of self-
renewal pathways as the main mechanism of anti-RSPO3’s anti-tumor activity.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

19
DISCUSSION
Since the initial discovery of WNT1 as a gene product capable of promoting breast
cancer thirty years ago, there has been a recognition that this signaling pathway may
contribute to human cancer.(46) This recognition has been greatly strengthened by the
understanding that intracellular components of the signaling cascade, APC, axin, and
beta-catenin, are mutated in approximately 90% of human colon tumors.(47) Mutations
in Wnt pathway components have also now been observed in a broad range of human
tumors.(3) Recently, therapeutic agents have been developed that target the Wnt
pathway, and these agents show efficacy in a range of preclinical tumor models.(43,48)
OMP-18R5 (vantictumab), an antibody that functions by blocking WNT binding to five of
the ten human FZD receptors, demonstrates activity in inhibiting the growth of a range
of human tumor xenograft models, indicating that extracellular Wnt pathway signals play
an important role in the growth of many human tumors. Despite this progress, a major
unanswered question remains the specific contribution of various members of the large
family of WNT ligands in human tumors, and other signaling proteins that might
contribute to beta-catenin activation, particularly in tumors that do not possess known
mutations impacting intracellular signaling components. For instance, despite the
demonstration that murine WNT1 can drive the formation of breast tumors when over-
expressed due to retroviral MMTV integration, there has not been clear evidence that
WNT1 is frequently over-expressed in human tumors. To address this question of
which factor might be responsible for driving beta-catenin signaling in human tumors,
we surveyed a series of human tumors for production of beta-catenin signaling activity
using a functional reporter assay. This effort has identified RSPO family members as
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

20
important stimulators of beta-catenin signaling activity in a number of human tumors.
Patient-derived lung and ovarian tumor cells were shown to induce beta-catenin
activation in an in vitro cell-based assay and the activity was directly dependent on
RSPO2 or RSPO3 expression as demonstrated using specific blocking anti-RSPO
antibodies. Interestingly, while RSPO1- or RSPO4-high tumors were identified, none of
them displayed any detectable activity in the STF co-culture assay. This could reflect
the assay detection limit or/and the induction of differential beta-catenin activation levels
by different RSPO proteins.
Subsequently, the growth of several of the RSPO-producing tumors was shown to be
driven by the RSPO activity when implanted in NOD/SCID mice. Using anti-RSPO
antibodies alone and in combination with standard of care chemotherapeutic agents, we
were able to significantly decrease the growth rate of RSPO3-positive lung and ovarian
tumors that were identified in our cell-based screen. In addition, RSPO expressing
tumors with no detectable ex vivo beta-catenin activating properties, were also shown to
respond to antibody-mediated anti-RSPO treatment. Anti-RSPO1 was shown to inhibit
the in vivo growth of an RSPO1-high ovarian tumor and anti-RSPO2 that of RSPO2-
high colon and pancreas tumors. This suggests that the reporter assay used to monitor
tumor cell-derived RSPO activity is not sensitive enough to allow for detecting low levels
of functional RSPO or that a beta-catenin-independent function of RSPO is at play in
these models. Our results also suggest that gene expression analysis may be sufficient
to identify tumors that respond to anti-RSPO therapy. Accordingly, three xenograft
models in which no RSPO gene expression was detected failed to respond to antibody-
mediated RSPO inhibition. Testing additional models will be required to further defining
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

21
minimal RSPO gene expression levels associated with anti-RSPO treatment sensitivity.
Finally, the identification of an anti-RSPO2 antibody-resistant lung tumor and an anti-
RSPO3-resistant colon tumor despite respective high RSPO2 and RSPO3 expression
indicates that not all instances of high RSPO expression may reflect tumor dependence
upon this signaling axis. Of note, RSPO4 was not tested as a therapeutic target in this
work. However, its relative prevalence across the OMP tumor bank supports evaluating
RSPO4 in future studies. Overall, the comprehensive set of in vivo efficacy data
presented here demonstrates the therapeutic value of RSPO blockade to treat RSPO-
expressing tumors.
In anti-RSPO3-sensitive CRC models, the significant down-regulation of target genes
associated with both normal stem cells and CSCs supports a mechanism of action by
which the antibody would inhibit tumor growth by decreasing CSC frequency, a most
desirable outcome for long-term cancer therapies. CSCs are defined as the most
tumorigenic subset of malignant cells that supports tumor growth by conferring stem cell
properties, including self-renewal and multipotency to tumor cells.(49) The major impact
of RSPO on stem cells is thought to be due to RSPO potentiation of the Wnt/beta-
catenin signaling and thus, one hypothesis is that RSPO may have limited signaling
activity in absence of WNT ligands.(4,50) In this model, tumors may achieve enhanced
beta-catenin signaling by elevated RSPO which functions to potentiate low levels of
WNT that are present in the tumor microenvironment, rather than by substantial up-
regulation of the expression of WNT family members. Thus, it appears that, in many
tumors, a synergistic potentiation of beta-catenin function is mediated by tumor-derived
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

22
RSPO and blocking RSPO represents a most efficient therapeutic approach in these
tumors.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

23
ACKNOWLEDGEMENTS
We thank many people at OncoMed Pharmaceuticals for their contributions to this work,
including Peter Stathis, Ian Scott, Esohe Idusogie, Jim Evans, Xiaomei Song, Diane
Pardi and Michael Mulkerrin. We thank Zhun Wang, Jie Cai and Henry Li from Crown
Bioscience for their assistance. Research funding was provided by OncoMed
Pharmaceuticals.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

24
REFERENCES
1. Wang J, Sinha T, Wynshaw-Boris A. Wnt signaling in mammalian development: lessons from mouse genetics. Cold Spring Harbor perspectives in biology 2012;4(5).
2. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell 2012;149(6):1192-205. 3. Polakis P. Wnt signaling in cancer. Cold Spring Harbor perspectives in biology 2012;4(5). 4. Kazanskaya O, Glinka A, del Barco Barrantes I, Stannek P, Niehrs C, Wu W. R-Spondin2 is a
secreted activator of Wnt/beta-catenin signaling and is required for Xenopus myogenesis. Developmental cell 2004;7(4):525-34.
5. Ohkawara B, Glinka A, Niehrs C. Rspo3 binds syndecan 4 and induces Wnt/PCP signaling via clathrin-mediated endocytosis to promote morphogenesis. Developmental cell 2011;20(3):303-14.
6. Carmon KS, Gong X, Lin Q, Thomas A, Liu Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proceedings of the National Academy of Sciences of the United States of America 2011;108(28):11452-7.
7. de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011;476(7360):293-7.
8. Glinka A, Dolde C, Kirsch N, Huang YL, Kazanskaya O, Ingelfinger D, et al. LGR4 and LGR5 are R-spondin receptors mediating Wnt/beta-catenin and Wnt/PCP signalling. EMBO reports 2011;12(10):1055-61.
9. Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012;485(7397):195-200.
10. Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012;488(7413):665-9.
11. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449(7165):1003-7.
12. Jaks V, Barker N, Kasper M, van Es JH, Snippert HJ, Clevers H, et al. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nature genetics 2008;40(11):1291-9.
13. Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell stem cell 2010;6(1):25-36.
14. Brzeszczynska J, Ramaesh K, Dhillon B, Ross JA. Molecular profile of organ culture-stored corneal epithelium: LGR5 is a potential new phenotypic marker of residual human corneal limbal epithelial stem cells. International journal of molecular medicine 2012;29(5):871-6.
15. de Visser KE, Ciampricotti M, Michalak EM, Tan DW, Speksnijder EN, Hau CS, et al. Developmental stage-specific contribution of LGR5(+) cells to basal and luminal epithelial lineages in the postnatal mammary gland. The Journal of pathology 2012;228(3):300-9.
16. Plaks V, Brenot A, Lawson DA, Linnemann JR, Van Kappel EC, Wong KC, et al. Lgr5-expressing cells are sufficient and necessary for postnatal mammary gland organogenesis. Cell reports 2013;3(1):70-8.
17. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013;494(7436):247-50.
18. Yee KK, Li Y, Redding KM, Iwatsuki K, Margolskee RF, Jiang P. Lgr5-EGFP marks taste bud stem/progenitor cells in posterior tongue. Stem cells (Dayton, Ohio) 2013;31(5):992-1000.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

25
19. Carmon KS, Gong X, Yi J, Thomas A, Liu Q. RSPO-LGR4 functions via IQGAP1 to potentiate Wnt signaling. Proceedings of the National Academy of Sciences of the United States of America 2014;111(13):E1221-9.
20. Schuijers J, Clevers H. Adult mammalian stem cells: the role of Wnt, Lgr5 and R-spondins. The EMBO journal 2012;31(12):2685-96.
21. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009;459(7244):262-5.
22. Jin YR, Yoon JK. The R-spondin family of proteins: emerging regulators of WNT signaling. The international journal of biochemistry & cell biology 2012;44(12):2278-87.
23. Friedman MS, Oyserman SM, Hankenson KD. Wnt11 promotes osteoblast maturation and mineralization through R-spondin 2. The Journal of biological chemistry 2009;284(21):14117-25.
24. Mulvaney JF, Yatteau A, Sun WW, Jacques B, Takubo K, Suda T, et al. Secreted factor R-Spondin 2 is involved in refinement of patterning of the mammalian cochlea. Developmental dynamics : an official publication of the American Association of Anatomists 2013;242(2):179-88.
25. Klauzinska M, Baljinnyam B, Raafat A, Rodriguez-Canales J, Strizzi L, Greer YE, et al. Rspo2/Int7 regulates invasiveness and tumorigenic properties of mammary epithelial cells. Journal of cellular physiology 2012;227(5):1960-71.
26. Lowther W, Wiley K, Smith GH, Callahan R. A new common integration site, Int7, for the mouse mammary tumor virus in mouse mammary tumors identifies a gene whose product has furin-like and thrombospondin-like sequences. Journal of virology 2005;79(15):10093-6.
27. Theodorou V, Kimm MA, Boer M, Wessels L, Theelen W, Jonkers J, et al. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nature genetics 2007;39(6):759-69.
28. Callahan R, Mudunur U, Bargo S, Raafat A, McCurdy D, Boulanger C, et al. Genes affected by mouse mammary tumor virus (MMTV) proviral insertions in mouse mammary tumors are deregulated or mutated in primary human mammary tumors. Oncotarget 2012;3(11):1320-34.
29. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature 2012;488(7413):660-4.
30. Watson AL, Rahrmann EP, Moriarity BS, Choi K, Conboy CB, Greeley AD, et al. Canonical Wnt/beta-catenin signaling drives human schwann cell transformation, progression, and tumor maintenance. Cancer discovery 2013;3(6):674-89.
31. Shinmura K, Kahyo T, Kato H, Igarashi H, Matsuura S, Nakamura S, et al. RSPO fusion transcripts in colorectal cancer in Japanese population. Molecular biology reports 2014;41(8):5375-84.
32. Gong X, Yi J, Carmon KS, Crumbley CA, Xiong W, Thomas A, et al. Aberrant RSPO3-LGR4 signaling in Keap1-deficient lung adenocarcinomas promotes tumor aggressiveness. Oncogene 2015;34(36):4692-701.
33. Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, et al. Phenotypic characterization of human colorectal cancer stem cells. Proceedings of the National Academy of Sciences of the United States of America 2007;104(24):10158-63.
34. Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PloS one 2008;3(6):e2428.
35. Rothe C, Urlinger S, Lohning C, Prassler J, Stark Y, Jager U, et al. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. Journal of molecular biology 2008;376(4):1182-200.
36. Van der Flier LG, Sabates-Bellver J, Oving I, Haegebarth A, De Palo M, Anti M, et al. The Intestinal Wnt/TCF Signature. Gastroenterology 2007;132(2):628-32.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

26
37. Kim M, Kim H, Jho EH. Identification of ptpro as a novel target gene of Wnt signaling and its potential role as a receptor for Wnt. FEBS letters 2010;584(18):3923-8.
38. Zhu R, Yang Y, Tian Y, Bai J, Zhang X, Li X, et al. Ascl2 knockdown results in tumor growth arrest by miRNA-302b-related inhibition of colon cancer progenitor cells. PloS one 2012;7(2):e32170.
39. Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457(7229):608-11.
40. da Silva-Diz V, Sole-Sanchez S, Valdes-Gutierrez A, Urpi M, Riba-Artes D, Penin RM, et al. Progeny of Lgr5-expressing hair follicle stem cell contributes to papillomavirus-induced tumor development in epidermis. Oncogene 2013;32(32):3732-43.
41. Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2012;18(3):869-81.
42. Liu YS, Hsu HC, Tseng KC, Chen HC, Chen SJ. Lgr5 promotes cancer stemness and confers chemoresistance through ABCB1 in colorectal cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2013;67(8):791-9.
43. Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proceedings of the National Academy of Sciences of the United States of America 2012;109(29):11717-22.
44. Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell stem cell 2009;5(2):168-77.
45. Yen WC, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21(9):2084-95.
46. Nusse R. Wnt signaling. Cold Spring Harbor perspectives in biology 2012;4(5). 47. Schepers A, Clevers H. Wnt signaling, stem cells, and cancer of the gastrointestinal tract. Cold
Spring Harbor perspectives in biology 2012;4(4):a007989. 48. Baarsma HA, Konigshoff M, Gosens R. The WNT signaling pathway from ligand secretion to gene
transcription: molecular mechanisms and pharmacological targets. Pharmacology & therapeutics 2013;138(1):66-83.
49. Clevers H. The cancer stem cell: premises, promises and challenges. Nature medicine 2011;17(3):313-9.
50. Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cellular signalling 2014;26(3):570-9.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

27
FIGURE LEGENDS
Figure 1. Detection of OMP tumor sample-derived beta-catenin activation
The ability of human tumor-derived soluble factors to activate beta-catenin was
assessed in the STF assay. A. Fold inductions are shown for a subset of the 48 OMP
tumor samples tested. They represent the ratio of tumor (red bars), WNT3A (green
bars), and tumor + WNT3A (purple bars)-induced luciferase activity relative to the
medium alone. B. FZD8.Fc inhibitory activity was demonstrated in the STF reporter
assay. STF cells were exposed to WNT3A or non-WNT3A (solid triangle) conditioned
medium supplemented with the indicated concentrations of FZD8.Fc (hollow dots).
Mouse JAG1-Fc (solid dots) was used as negative control. Results are displayed as
luciferase activity levels (RLU, Relative Light Units) as a function of the test agent’s
concentration (g/ml). C. Serial dilutions of LGR5.Fc were used to inhibit the induction
of luciferase by RSPO1 (squares), RSPO2 (circles), RSPO3 (triangles), or RSPO4
(diamonds) added to WNT3A-conditioned medium. Results are shown as the
measured RLUs as a function of the decoy protein concentration. D. Both FZD8.Fc and
LGR5.Fc inhibited the luciferase reporter activity (RLUs) of STF cells stimulated with
tumor cell cultures supplemented (purple bars) or not (red bars) with WNT3A. WNT3A
activity was only inhibited by FZD8.Fc (green bars).
Figure 2. RSPO gene expression profile across multiple different tumor types
RSPO1, 2, 3, and 4 gene expression in breast, colon, lung, melanoma, ovary and
pancreas tumor models was assessed by real time RT-PCR and expressed as a
relative quantity using the lowest positive sample as baseline and GUSB as
endogenous reference. Each purple bar represents one patient-derived tumor sample.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

28
Figure 3. Characterization of anti-RSPO1, RSPO2, and RSPO3 monoclonal antibodies
RSPO binding properties of the anti-RSPO monoclonal antibodies were assessed by
flow cytometry. The ability of anti-RSPO antibodies to inhibit RSPO signaling function
was monitored in the cell-based beta-catenin reporter assay. A. RSPO1, 2, 3, or 4 was
displayed at the surface of HEK-293 cells by means of a transiently transfected RSPO1,
2, 3, or4.CD4TM.GFP fusion cDNA expression vector that tethers RSPO to the cell
surface via a transmembrane anchor. Binding of the RSPO moieties by the anti-
RSPO1, anti-RSPO2, and two anti-RSPO3 monoclonal antibodies was detected using a
PE-conjugated secondary antibody. The PE (y axis) and GFP/FITC (x axis) signals are
plotted for each individual cell analyzed. The relative increase of both signals translates
specific antibody binding to the antigen. B. Anti-RSPO antibodies’ inhibitory activity was
confirmed in the STF reporter assay. STF cells were exposed to WNT3A conditioned
medium supplemented with RSPO1, RSPO2, or RSPO3. Anti-RSPO antibodies were
added. Results are displayed as luciferase activity levels (RLU, Relative Light Units). C.
LGR5 was displayed at the surface of HEK-293T cells by means of a transiently
transfected LGR5.CD4TM.GFP fusion cDNA expression vector that expresses the N-
terminal extracellular domain of LGR5 tethered to the cell surface via a transmembrane
anchor. Ability of anti-RSPO monoclonal antibody to block the binding of the indicated
RSPO was assessed by incubating cells with RSPO-Fc fusion protein and monoclonal
antibody as indicated and followed by detection of bound RSPO protein with PE-
conjugated anti-human Fc secondary antibody. Specific RSPO-LGR5 binding is
indicated by detection of cells within the inset box of the flow cytometry plot.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

29
Figure 4. Inhibition of tumor-derived beta-catenin activity by the anti-RSPO antibodies
The use of specific anti-RSPO antibodies in the tumor-STF cell co-culture reporter
assay identified RSPO ligands as the tumor-derived factors responsible for inducing
beta-catenin transcriptional activity. OMP-LU2, -LU25, -LU102, and -OV38 cells were
tested for their ability to induce luciferase activity of a TCF-responding element in
absence (1st series) and presence (2nd series) of WNT3A when combined with a control
mAb (red bars), anti-RSPO1 (tan bars), anti-RSPO2 (purple bars), or anti-RSPO3 mAb1
(blue bars) antibody. Effect of the anti-RSPO antibodies could not be assessed (ND) on
OV38 in absence of WNT3A. Relative luciferase activity (RLU) was plotted for each
condition next to a no tumor cell control (black bars).
Figure 5. Inhibition of RSPOhigh tumor growth in vivo using specific anti-RSPO
antibodies
Ovarian (OMP-OV19 and –OV38), pancreatic (OMP-PN7), lung (OMP-LU2, –LU25, and
–LU102), and colorectal (OMP-C28, CR3150, CR2513, CR2506, and CR1560)
xenograft models were treated with anti-RSPO1 (green symbols), anti-RSPO2 (blue
symbols), or anti-RSPO3 mAb2 (red symbols) antibody alone (up-pointing triangles) or
in combination with chemo (down-pointing triangles) in order to assess the role of
RSPO in driving the in vivo growth of these models. Anti-RSPO1 was used at 10 mg/kg
once weekly; anti-RSPO2 was used at 10 (OMP-PN7) or 15 (OMP-LU2) mg/kg once
weekly or 25 mg/kg every other week (OMP-C28); anti-RSPO3 mAb2 was used at 25
mg/kg once weekly. The chemo agents and regimens were as follows: 15 mg/kg Taxol
once weekly for OMP-OV19, -OV38, and –LU25, 10 mg/kg Taxol once weekly for OMP-
LU2, 100 mg/kg gemcitabine once weekly for OMP-PN7, and 10 mg/kg irinotecan once
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

30
weekly for OMP-C28, CR3150, CR2513, CR2506, and CR1560. A. Tumor volumes
were measured weekly, averaged for each treatment group (n=10) and graphed as a
function of time to obtain growth curves relative to the control antibody (open black
circles) and chemo (black circles) groups. Tumor models were deemed responsive
when p-values calculated using a 2-way ANOVA analysis and Bonferroni post-tests
were <0.05 (*), <0.01 (**), or <0.001 (***) at termination. Antibody alone was compared
to control antibody and chemo combination was compared to control antibody + chemo
agent as depicted by black vertical lines on the graphs. B. The tumorigenicity of anti-
RSPO3-treated OMP-LU25 tumors was assessed upon passaging low cell doses in
naïve NOD/SCID mice. All 10 individual tumor volumes were plotted for each group
after 100% take rate was observed in the control group.
Figure 6. Down-regulation of select Wnt target genes in anti-RSPO3-treated CRC
xenografts
ASCL2, AXIN2, CLDN2, LGR5, PTPRO, RPRD1A, TDGF1, and ZNRF3 genes were
analyzed for expression levels in CRC xenografts CR3150, CR2506, CR2513, and
CR1560 treated with control antibody (small plaid pattern bars) or anti-RSPO3 (large
plaid pattern bars). Only high quality RNA samples were included in the analysis,
resulting in the following sample sizes: n=15 for control antibody in CR3150 and
CR2506, n=14 for control antibody in CR2513 and CR1560, n=3 for anti-RSPO3 in
CR3150, n=10 for anti-RSPO3 in CR2506, n=8 for anti-RSPO3 in CR2513, and n=9 for
anti-RSPO3 in CR1560. Results were expressed as relative quantity over GAPDH
endogenous control gene. P values were calculated using a t test with Welch’s
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

31
correction and anti-RSPO3-induced decrease considered significant when below 0.05
(*), 0.01 (*), or 0.001 (***).
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561
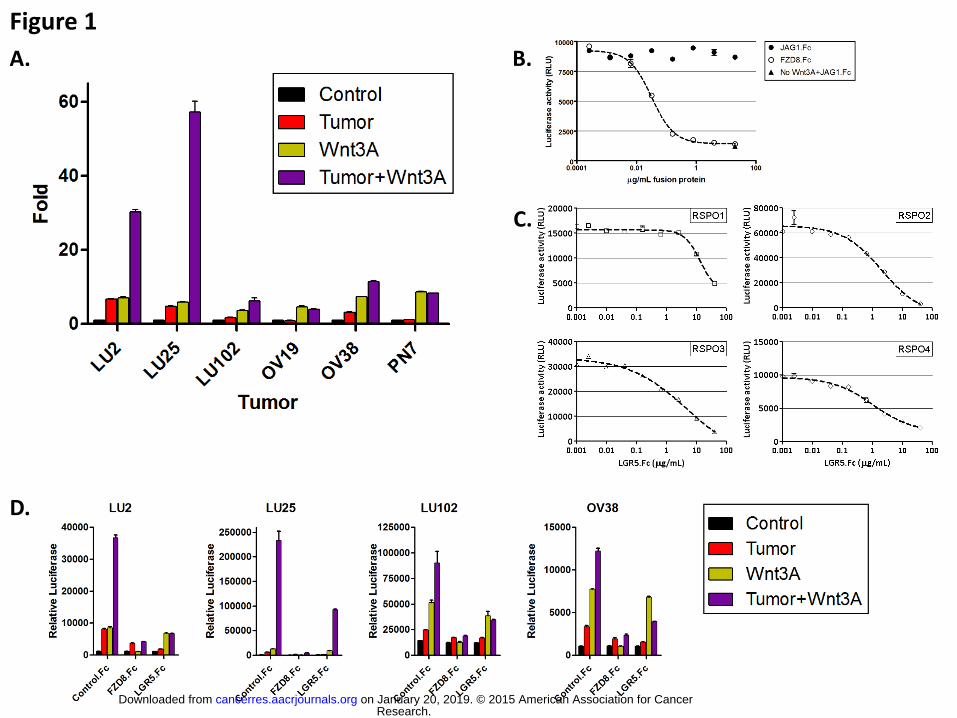
Figure 1
A. B.
D.
C.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561
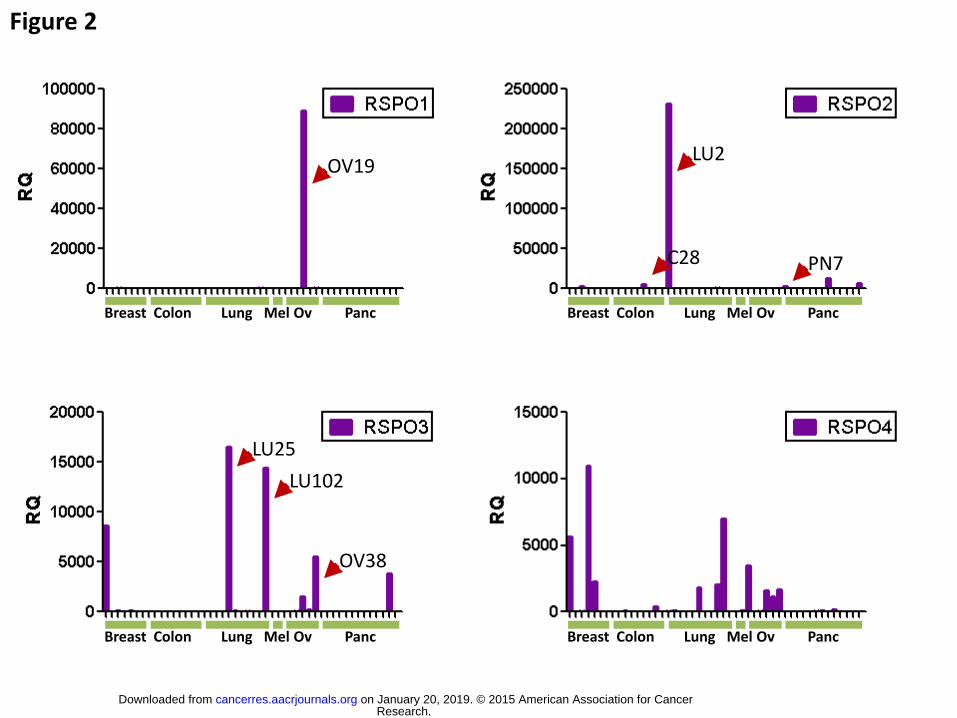
Figure 2
Breast Colon Lung Mel Ov Panc
OV19 LU2
LU25
LU102
OV38
Breast Colon Lung Mel Ov Panc
Breast Colon Lung Mel Ov Panc Breast Colon Lung Mel Ov Panc
PN7 C28
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561
An
tib
od
y b
ind
ing
Anti-RSPO1
Anti-RSPO2
Anti-RSPO3 #1
Anti-RSPO3 #2
No Ab
RSPO1 RSPO2 RSPO3 RSPO4
Figure 3
A.
B.
C.
LGR5 LGR5 LGR5 LGR5
RSP
O1
bin
din
g
a-RSPO1 control RSP
O1
- + + +
a-RSPO2 control
LGR5 LGR5 LGR5 LGR5
RSP
O2
bin
din
g
RSP
O2
- + + +
RSP
O3
bin
din
g a-RSPO3 #2 control RSP
O3
- + + +
LGR5 LGR5 LGR5 LGR5
RSP
O3
bin
din
g a-RSPO3 #1 control RSP
O3
- + + +
LGR5 LGR5 LGR5 LGR5
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561
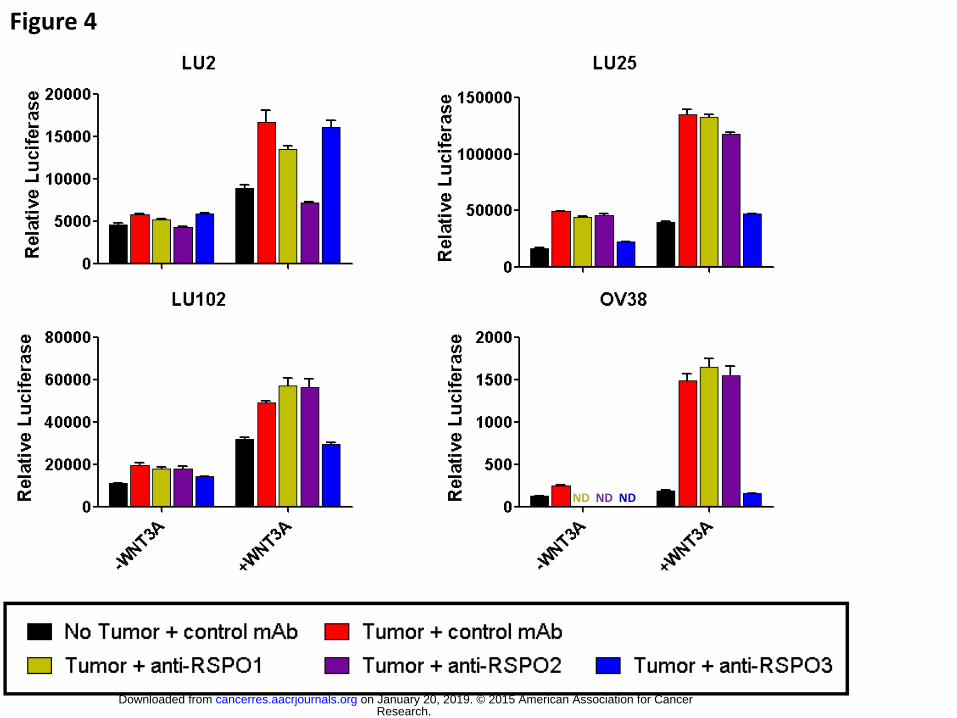
Figure 4
ND ND ND
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561
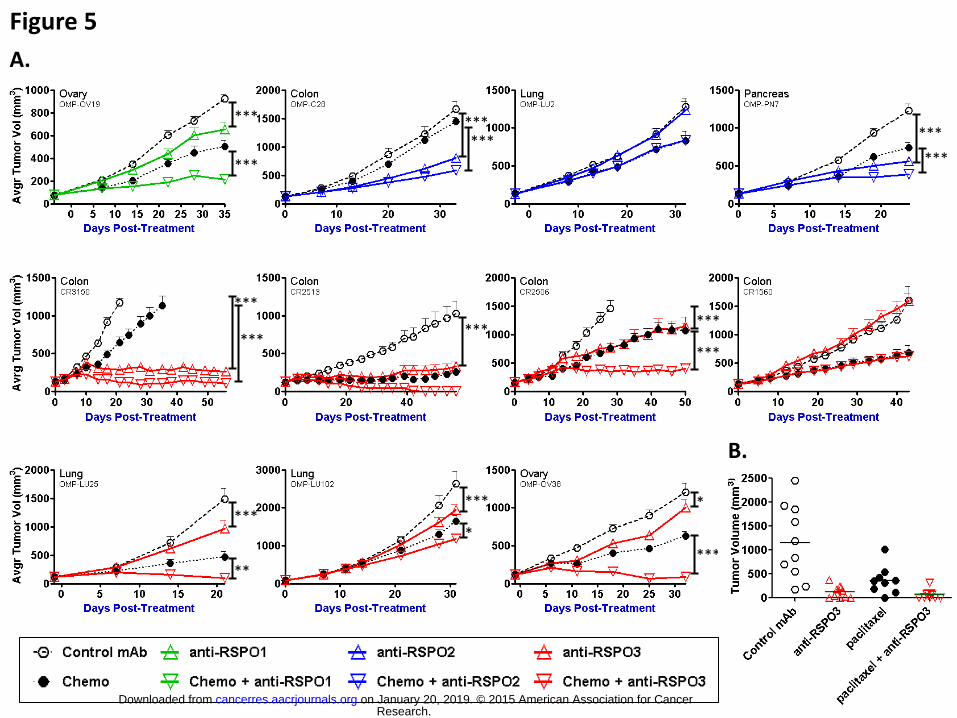
Figure 5
***
***
*** *** ***
***
***
**
***
*
*
***
***
*** ***
***
***
A.
B.
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561
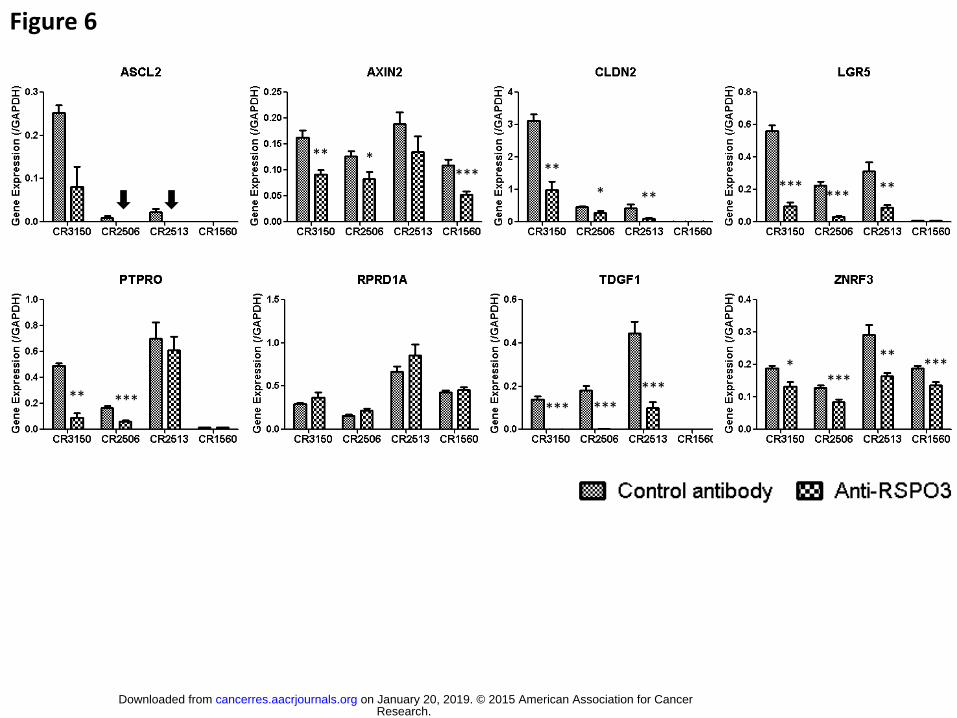
Figure 6
** * *** **
* ** ***
*** **
** *** *** *** ***
* ***
** ***
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561

Published OnlineFirst December 30, 2015.Cancer Res Cecile Chartier, Janak Raval, Fumiko Axelrod, et al. -catenin signaling and tumorigenesis in multiple cancer types
βTherapeutic targeting of tumor-derived R-spondin attenuates
Updated version
10.1158/0008-5472.CAN-15-0561doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2015/12/30/0008-5472.CAN-15-0561.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/early/2015/12/30/0008-5472.CAN-15-0561To request permission to re-use all or part of this article, use this link
Research. on January 20, 2019. © 2015 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on December 30, 2015; DOI: 10.1158/0008-5472.CAN-15-0561


















