
α,ω-Diferrocenyl Cumulene Molecular Wires Studied by Density Functional Theory
11
r,ω-Diferrocenyl Cumulene Molecular Wires Studied by Density Functional Theory Benno Bildstein, ² Olga Loza, ‡ and Yuri Chizhov* ,‡ Institute of General, Inorganic and Theoretical Chemistry, University of Innsbruck, Innrain 52a, 6020 Innsbruck, Austria, and Faculty of Physics, Photonics Department, St. Petersburg State University, 198904 St. Petersburg, Russia Received October 13, 2003 Density functional theory calculations for neutral and oxidized diferrocenyldiphenyl- cumulenes, [Fc(Ph)C n (Ph)Fc] with n ) 2, 4, 6, 8, were performed at the B3LYP/6-31G level without any restrictions in symmetry. Geometrically, of the four possible diasteromeric structures of the neutral cumulenes, no preferential conformation was revealed, and the calculated structures reproduce the experimental X-ray structural data quite well. The spin density of all mono- and dications is located on the iron atoms, but not on the cumulene chain. Calculations of the ionization potentials for monocations show a breakdown of Koopmans’ theorem. A calibration procedure was proposed for evaluation of vertical ionization potentials, resulting in values with experimental accuracy. UV-visible absorptions are assigned in terms of their band maximums, transition energies, and bandwidths on the basis of deconvoluted experimental spectra and on the basis of comparison with structurally related compounds. Calculations of the characteristic high-frequency symmetric vibrations of the cumulene chain show a very good match with experimental Raman bands. Redox potentials in solution are calculated by a simplified model on the basis of empirical findings; the results reproduce the experimental half-wave potentials quite satisfactorily and allow an estimate of an electrochemical decay slope and an effective conjugation limit in these cumulene molecular wires. The structural, chemical, optical, electrochemical, and molecular orbital properties of 2-31 make ferrocenyl cumulenes very promising molecular objects with great potential in molecular-scale electronics in comparison with pure organic molecular wires. 1. Introduction In a recent paper 1 we reported on the synthesis, properties, and electrochemistry of a new family of ferrocenyl cumulenes. These organometallic molecules have been designed as one-dimensional unsaturated systemssthe cumulene subunitswith end-capping fer- rocenyl substituents as electrochemical probes for study- ing charge transmittal through the cumulene molecular wire. This is a research contribution in the field of molecular electronics with the principal question “How good is a cumulene as a conducting molecular wire?”. In this contribution, a full account of the theoretical description of such ferrocenyl cumulenes is given, including their geometry, electronic structure, optical properties, and electrochemical features. Only cumu- lenes with an even number of carbons (1,C 2 ; 2,C 4 ; 3, C 6 ; 4,C 8 ) are the subject of this paper (Chart 1), because electrochemical measurements 1 have shown that odd cumulenes C 3 ,C 5 , and C 7 are electronically decoupled and therefore of no interest as potential one-dimensional conductors. 2. Results and Discussion The geometries of of [2n +1]cumulenes Fc(Ph)Cd C(Ph)Fc (1), Fc(Ph)CdCdCdC(Ph)Fc (2), Fc(Ph)CdCd CdCdCdC(Ph)Fc (3), and Fc(Ph)CdCdCdCdCdCd CdC(Ph)Fc (4) have been fully optimized at the B3LYP DFT level, where B3LYP stands for Becke’s three- parameter nonlocal exchange functional 2 with the non- * To whom correspondence should be addressed. E-mail: chizhov@ photonics.phys.spbu.ru. ² University of Innsbruck. ‡ St. Petersburg State University. (1) Skibar, W.; Kopacka, H.; Wurst, K.; Salzmann. C.; Ongania, K.- H.; Fabrizi de Biani, F.; Zanello, P.; Bildstein, B. Organometallics 2004, 23, 1024. (2) (a) Becke, A. D. J. Chem. Phys. 1992, 96, 2155. (b) Becke, A. D. J. Chem. Phys. 1992, 97, 9173. (c) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. Chart 1. Overview of Cumulenes Studied by DFT Calculations a a Abbreviations: Fc ) ferrocenyl, Ph ) phenyl. Only the trans stereoisomers are shown. 1825 Organometallics 2004, 23, 1825-1835 10.1021/om034234d CCC: $27.50 © 2004 American Chemical Society Publication on Web 03/05/2004
Transcript of α,ω-Diferrocenyl Cumulene Molecular Wires Studied by Density Functional Theory

r,ω-Diferrocenyl Cumulene Molecular Wires Studied byDensity Functional Theory
Benno Bildstein,† Olga Loza,‡ and Yuri Chizhov*,‡
Institute of General, Inorganic and Theoretical Chemistry, University of Innsbruck,Innrain 52a, 6020 Innsbruck, Austria, and Faculty of Physics, Photonics Department,
St. Petersburg State University, 198904 St. Petersburg, Russia
Received October 13, 2003
Density functional theory calculations for neutral and oxidized diferrocenyldiphenyl-cumulenes, [Fc(Ph)Cn(Ph)Fc] with n ) 2, 4, 6, 8, were performed at the B3LYP/6-31G levelwithout any restrictions in symmetry. Geometrically, of the four possible diasteromericstructures of the neutral cumulenes, no preferential conformation was revealed, and thecalculated structures reproduce the experimental X-ray structural data quite well. The spindensity of all mono- and dications is located on the iron atoms, but not on the cumulenechain. Calculations of the ionization potentials for monocations show a breakdown ofKoopmans’ theorem. A calibration procedure was proposed for evaluation of vertical ionizationpotentials, resulting in values with experimental accuracy. UV-visible absorptions areassigned in terms of their band maximums, transition energies, and bandwidths on the basisof deconvoluted experimental spectra and on the basis of comparison with structurally relatedcompounds. Calculations of the characteristic high-frequency symmetric vibrations of thecumulene chain show a very good match with experimental Raman bands. Redox potentialsin solution are calculated by a simplified model on the basis of empirical findings; the resultsreproduce the experimental half-wave potentials quite satisfactorily and allow an estimateof an electrochemical decay slope and an effective conjugation limit in these cumulenemolecular wires. The structural, chemical, optical, electrochemical, and molecular orbitalproperties of 2-31 make ferrocenyl cumulenes very promising molecular objects with greatpotential in molecular-scale electronics in comparison with pure organic molecular wires.
1. Introduction
In a recent paper1 we reported on the synthesis,properties, and electrochemistry of a new family offerrocenyl cumulenes. These organometallic moleculeshave been designed as one-dimensional unsaturatedsystemssthe cumulene subunitswith end-capping fer-rocenyl substituents as electrochemical probes for study-ing charge transmittal through the cumulene molecularwire. This is a research contribution in the field ofmolecular electronics with the principal question “Howgood is a cumulene as a conducting molecular wire?”.In this contribution, a full account of the theoreticaldescription of such ferrocenyl cumulenes is given,including their geometry, electronic structure, opticalproperties, and electrochemical features. Only cumu-lenes with an even number of carbons (1, C2; 2, C4; 3,C6; 4, C8) are the subject of this paper (Chart 1), becauseelectrochemical measurements1 have shown that oddcumulenes C3, C5, and C7 are electronically decoupledand therefore of no interest as potential one-dimensionalconductors.
2. Results and Discussion
The geometries of of [2n +1]cumulenes Fc(Ph)CdC(Ph)Fc (1), Fc(Ph)CdCdCdC(Ph)Fc (2), Fc(Ph)CdCdCdCdCdC(Ph)Fc (3), and Fc(Ph)CdCdCdCdCdCdCdC(Ph)Fc (4) have been fully optimized at the B3LYPDFT level, where B3LYP stands for Becke’s three-parameter nonlocal exchange functional2 with the non-
* To whom correspondence should be addressed. E-mail: [email protected].
† University of Innsbruck.‡ St. Petersburg State University.(1) Skibar, W.; Kopacka, H.; Wurst, K.; Salzmann. C.; Ongania, K.-
H.; Fabrizi de Biani, F.; Zanello, P.; Bildstein, B. Organometallics 2004,23, 1024.
(2) (a) Becke, A. D. J. Chem. Phys. 1992, 96, 2155. (b) Becke, A. D.J. Chem. Phys. 1992, 97, 9173. (c) Becke, A. D. J. Chem. Phys. 1993,98, 5648.
Chart 1. Overview of Cumulenes Studied by DFTCalculationsa
a Abbreviations: Fc ) ferrocenyl, Ph ) phenyl. Only thetrans stereoisomers are shown.
1825Organometallics 2004, 23, 1825-1835
10.1021/om034234d CCC: $27.50 © 2004 American Chemical SocietyPublication on Web 03/05/2004

local correlation functional of Lee, Yang, and Parr.3 Bothspin-restricted DFT for neutral complexes and spin-unrestricted DFT for cations was used with the stan-dard 6-31G basis set for C and H and the Hay-Wadtpseudo-potential for Fe.4 This choice is a reasonablecompromise between the accuracy of the calculationsand an acceptable computational expenditure. Thegeometry of mono- and dioxidized cumulenes was cal-culated without any restriction in symmetry (broken-symmetry approach). The total spin (S2) values showsome spin contamination (SC) for doublet and tripletion states. The observed SC is completely characteristicand acceptable for such large systems. No attempts weremade to annihilate the SC, since the latter did notexceed a 12% contribution of higher multiplets and hasnegligible influence on the geometry and Kohn-Shamenergies as well as on other properties.5 All data for thedications are given for the ground electronic tripletstates (high-spin states HS).
2.1. Geometry. [2n+1]Cumulenes 1-4 may exist infour different syn/anti/cis/trans diastereoisomeric forms,A-D, as has been discussed in more detail in our recentexperimental paper.1 Hence, the first question to answeris “Which isomer is the most stable, and how are therelative stabilities affected by an increase of the cumu-lene chain length?” For the olefin Fc(Ph)CdC(Ph)Fc (1)the cis/syn/anti isomers B-D are nonexistent due tosteric hindrance; therefore, this stereochemical problemwas only addressed for Fc(Ph)CdCdCdC(Ph)Fc (2) andFc(Ph)CdCdCdCdCdC(Ph)Fc (3), respectively (Table1).
The differences in the total energies of the fourisomers do not exceed 0.87 kcal/mol, which is theaverage thermal energy at room temperature, except inthe case of the anti-trans isomer of 2 (which is also theonly existing isomer of olefin 1). Therefore, all cumuleneA-D isomers of systems with more than four cumulatedcarbons are energetically more or less equivalent, andthere is no prevailing geometry in solution or in the gasphase; thus, we chose the anti/trans conformation(minimal steric hindrance) for all further calculations.Table 2 summarizes pertinent interatomic distances andtilt angles of substituents for the neutral and oxidizedferrocenyl cumulenes; experimental X-ray data1 (whereavailable) are cited in parentheses for comparison. TheDFT-optimized molecular structures are shown in Fig-ure 1. The structure of olefin 1 differs notably from thegeometries of the longer cumulenes 2-4. Due to stericcrowding in 1, the phenyl rings are oriented almostorthogonally (78°) in relation to the π-plane of theolefinic moiety, whereas the tilt angles of the Cp planesare approximately 22° according to the computationalstructure optimization. The phenyl rings have a con-
siderable decrease in tilt angle for the longer cumulenes,and the ferrocenyl groups are bent out of the cumuleneπ-plane by only 12-16°. In addition, the computedstructure of 1 is in rather poor agreement with theexperimental X-ray single-crystal structure,1 but for thelonger cumulenesswhere steric crowding is negligiblesthe DFT-computed structures comply satisfactorily withreality, evidenced by the good match of experimental1
and theoretical data for cumulene 2.Besides these conformational features, the structure
of the cumulene sp carbon chain itself is of primeinterest. The calculations predict a distinct alternationof adjacent CdC bonds; the C(1)-C(2) bond lengths areamazingly constant in the undistorted cumulenes 2-4and are equal to 135 pm, very close to the double-bondlength in ethylene (132 pm). The bond distances of thefollowing C(2)-C(3) bonds are also approximately con-stant with 126 pm, but their value is closer to the CtCtriple-bond length in acetylene (121.2 pm). The averageC-C bond length tends to decrease with increasingcumulene chain length and is equal to 137.0, 132.0,130.8, and 130.2 pm for 1-4, correspondingly. Thus, theaverage C-C bond length is closer to a CdC double-bond distance, as expected. We also note that the C-Cbond alternation in 1-4 is enhanced in comparison withunsubstituted cumulenes H2C2n+1H2, because the π-con-jugated ferrocenyl and phenyl substituents intensify theC-C bond alternation and decrease the average C-Cbond distance.
(3) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.(4) (a) Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 270. (b)
Wadt, W. R.; Hay, P. J. J. Chem. Phys. 1985, 82, 284. (c) Hay, P. J.;Wadt, W. R. J. Chem. Phys. 1985, 82, 299.
(5) Graefenstein, J.; Kraka, E.; Filatov, M.; Cremer, D. Int. J. Mol.Sci. 2002, 3, 360.
Table 1. Relative Total Energies (kcal/mol) forA-D Isomers of 2 and 3
A anti/trans B syn/trans C anti/cis D syn/cis
2 +1.178 0.000 +0.084 +0.0693 +0.207 0.000 +0.138 +0.277
Table 2. Selected Interatomic Distances (pm) andTilt Angles (deg) of Substituents for the Neutral
and Oxidized Ferrocenyl Cumulenes 1-41b 2b 3b 4b
C(1)-C(2) 137.1 (129)c 135.1 (134)c 135.2 135.2139.1 137.1 137.8 137.8137.5 135.8 136.0 136.6
C(2)-C(3) 126.1 (125)c 126.4 126.6124.8 124.7 124.8125.7 125.9 125.7
C(3)-C(4) 135.1 (138)c 130.8 130.4137.4 132.7 132.4135.9 131.3 131.4
C(4)-C(5) 126.9125.1126.0
C(R)-C(ω)d 137.1 (129)c 396.1 (397)c 653.9 911.2139.1 399.1 657.7 915.2137.5 397.3 654.8 913.3
Fe(1)-Fe(2)e 753.5 918.2 1141.7 1370.0753.4 926.5 1125.0 1329.1778.4 951.6 1167.4 1424.0
Fe-Cav(Cp)f 210.9 (203.8)c 211.1 (204.4)c 211.2 211.2216.9; 211.7 218.9; 212.3 213.7 213.6216.8 217.5 217.4 217.7
tilt angle Cp vs 22.0 (6.1)c 12.6 (22.3)c 16.1 16.6cumulene π-plane 21.3; 3.5 10.7; 8.1 10.9 11.4
26.8 13.5 15.0 16.5
tilt angle Ph vs 78.3 (85.0)c 36.7 (27.2)c 29.1 29.4cumulene π-plane 85.0; 72,3 42.5; 37.7 34.0 32.9
77.2 39.7 34.0 29.9a Experimental X-ray data are cited in parentheses. b For each
bond length and tilt angle, the first entry is for the neutral speciesand the second and third entries are for the mono- and dications,respectively. HS denotes the high-spin state of dication. c Experi-mental values.1 d C(R)-C(ω): cumulene chain length. Fe(1)-Fe(2). e Distance between iron atoms. Fe-Cav(Cp). f Average Fe-Cbond length in Fc groups.
1826 Organometallics, Vol. 23, No. 8, 2004 Bildstein et al.

In the mono- and dioxidized cumulenium compounds1+/2+-4+/2+ the cumulene chain remains largely unaf-fected; only a slight enhancement of the C-C bondalternation is observed for monocations, but almost nochange occurs in the case of dicationic cumulenes. Thecumulene chain length C(R)-C(ω) remains constantwithin limits of 1% during oxidation; only the inter-atomic Fe-Fe distances change to a greater extent, due
to conformational changes of differently oriented ferro-cenyl groups on sequential oxidation.
However, oxidation affects the geometry of the ferro-cenyl groups. The average Fe-C(Cp) distances of thedications are increased by 6.2 pm in each ferrocenylgroup and do not depend substantially on the length ofthe cumulene chain. For comparison, this increase ofthe Fe-C(Cp) distance is approximately 4.7 pm forsimple ferrocenium monocations.
Especially noteworthy is that calculations predictasymmetrical geometries for short-length species 1+ and2+. The asymmetry touches only Fc units and is evidentas an increase in Fe(1)-Cav(Cp) distances only in oneferrocenyl group and in different tilt angles of ferrocenylas well as phenyl rings versus the cumulene π-plane ofmonocations 1+ and 2+. This is clear evidence forremoval of an electron from only one Fc group, whilethe second Fc group remains unoxidized (vide infra, part2.2.2). Monocations 3+ and 4+ have a symmetric struc-ture in terms of both bond distances and tilt angles,shown by an increase of 2.4 pm for the Fe(1)-Cav(Cp)distances of both Fc groups simultaneously.
In summary, calculations predict that in single anddouble oxidation only Fe atoms are involved, while thecumulene chains remain unaffected. This structuralstability of the cumulene chain meets the principle ofminimal structural distortion during electron transfer;therefore, one might expect that ferrocenyl cumulenemolecular wires can sustain a large current density.
2.2. Electronic Structure. Table 3 summarizes theKohn-Sham energies, symmetry, and character ofmolecular orbitals for the four [2n+1]cumulenes 1-4.Figure 2 gives the MO level diagram, important frontierMO’s are shown in Figure 3, and total spin densitiesfor mono- and dications of extended molecular wires areoutlined in Figure 4. Table 4 gives the adiabatic andvertical ionization potentials as well as the dipolemoments and total spin densities for the singly anddoubly oxidized cumulenes 1-4. The ionization poten-tials were calculated by the ∆SCF method. The verticalionization potentials (IPv) have been calculated for theoptimized geometries of the neutral compounds, fol-lowed by a single-point calculation of the energy of theradical cation. The adiabatic ionization potentials (IPa)
Figure 1. Molecular structures of ferrocenyl cumulenesaccording to DFT calculations at the B3LYP/6-31G level.
Figure 2. Energy MO diagram for 1-4 according to DFTcalculations in the B3LYP/6-31G approximation.
R,ω-Diferrocenyl Cumulene Molecular Wires Organometallics, Vol. 23, No. 8, 2004 1827

have been calculated for the optimized geometries of theneutral and ionized complexes.
2.2.1. Molecular Orbitals. In principle, the valenceMO’s are formed by MO interactions of the cumulenechain, ferrocenyl groups, and phenyl substituents. How-ever, only the π-levels of the cumulene bridge interactstrongly with the HOMO’s of the ferrocenyl moieties,forming the upper valence bands of 1-4, while theπ-MO’s of the phenyl substituents are considerablylower in energy and therefore contribute insignificantly.All MO’s are classified by ag and au types of the Ci
symmetry group, allowing only interactions betweenfragment MO’s of matching symmetry.
It is evident from Table 3 and Figure 3 that π-HOMO1’s of 2-4 are localized predominantly on thecumulene bridge. The 3d Fe atomic orbital contributionsare not very high and decrease from 27 to 18%, whilethe π-MO cumulene character builds up more rapidlywith increasing bridge length. In the case of 1, HOMO1has predominantly 3d Fe character; actually, it is the1au 3d(x2-y2) Fe MO which is destabilized by theethylene π-system. The ethylene π-MO is lower in
Table 3. Molecular Orbital Kohn-Sham Energies EKS (eV) for 1-4: Symmetry Types and MO Characterwith 3d Fe and [n]cumulene Chain Contributions
MO 1 2 3 4 character
LUMO2 1au -0.251 1ag -0.376 1au -0.734 1ag -1.500 π*(Ph), “π2*”
LUMO1 1ag -0.882 (42% 3d Fe,20% π- cumulene)
1au -1.965 (17% 3d Fe,44% π-cumulene)
1ag -2.395 (12% 3d Fe,55% π-cumulene)
1au -2.668 (11% 3d Fe,61% π-cumulene)
π1*-[2n + 1] cum
HOMO1 1au -5.060 (68% 3d Fe,9% π-cumulene)
1ag -4.852 (27% 3d Fe,36% π-cumulene)
1au 4.768 (20% 3d Fe,47% π-cumulene)
1ag -4.722 (18% 3d Fe,53% π-cumulene)
π1-[2n + 1] cum
HOMO2 1ag -5.239 1au -5.465 1ag -5.508 1au -5.544 3d-(x2 - y2) FeHOMO3 2au -5.285 2ag -5.471 2au -5.519 2ag -5.566 3d-(xy) FeHOMO4 2ag -5.306 2au -5.475 2ag -5.521 2au -5.572 3d+(xy) FeHOMO5 3au -5.662 3ag -5.557 3au -5.574 3ag -5.611 3d+(x2 - y2) Fe
HOMO6-9 4au -6.669 3ag -6.230 4au -5.875 "π1"-[2n + 1] cum5ag -6.656 4ag -6.401 π2-[2n + 1] cum
3ag -6.380 3au -6.546 4ag -6.611 5ag -6.672 3d-(z2) Fe4au -6.419 4ag -6.566 4au -6.623 5au -6.676 3d+(z2) Fe
Figure 3. Frontier molecular orbitals and MO energies for 1-4 according to DFT calculations at the B3LYP/6-31G level.
1828 Organometallics, Vol. 23, No. 8, 2004 Bildstein et al.

energy than the HOMO’s of the substituents: in ourcase the 3d Fe HOMO of the ferrocenyl groups. There-fore, the interaction of 1au 3d(x2-y2)Fe and 3au π-eth-ylene MO’s leads to an antibonding HOMO1 localizedprimarily on the Fe atoms. The next three HOMO’s of1 and four HOMO’s of 2-4 are almost completelynonbonding MO’s with contributions of 75-80% on theFe atoms. They form very narrow zones of 0.09-0.06eV, ruling out any significant electron coupling betweenthe 3d(xy) and 3d(x2-y2) Fe atomic orbitals of differentferrocenyl groups. The centers of gravity of these zonesequal -5.277, -5.492, -5.530, and -5.573 eV for 1-4,respectively. The gradual decrease of the MO energieswith increasing chain length is rather unusual and is anoteworthy feature of the electronic structure of thesecumulenes, suggesting stabilization of ferrocenyl MOenergies due to the negative inductive effect of thecumulene chain. Interestingly, the increasing redoxpotentials of 1-4 correlate well with this stabilizationof the 3d(xy) and 3d(x2-y2) Fe MO’s (vide infra).
The next two totally nonbonding MO’s are practicallydegenerate, with close to 90% contribution of the 3d(z2)Fe AO. Thus, any electron coupling between 3d(z2) FeAO’s of cumulene end-capping ferrocenyl groups iscompletely absent. The energies of these MO’s arestabilized by approximately the same value as the 3d-(xy) and 3d(x2-y2) Fe MO’s, caused by the inductive
effect of the cumulene chain. According to calculations,one 3ag MO of 3 and the two 4au and 4ag MO’s of 4 arelocated between the 3d(xy) and 3d(x2-y2) Fe MO zoneand 3d(z2) Fe levels. The 3ag MO of 3 and the 4au MOof 4 are π-type MO’s, but they are oriented in thecumulene plane. Such MO’s are usually denoted aspseudo-π-MO’s or “π”-MO’s. The 4ag MO of 4 is the π2-MO of the [7]cumulene chain. All other MO’s are locatedbelow -6.44 eV and are localized on the ferrocenyl andphenyl substituents, respectively.
The lowest unoccupied molecular orbitals are pre-dominantly antibonding π*-orbitals. The π*-cumulenecontributions increase from 20 to 61%, while the 3d Fecontributions decrease from 42 to 11% with increasingcumulene length. The next LUMO2 has a differentcharacter for compounds 1-4. In 1 and 2 it is localizedon the phenyl rings, while in 3 and 4 the LUMO2’s areantibonding “π*”-MO’s of the cumulene chain. As canbe seen from Figure 2, the frontier MO’s show differentslopes: LUMO1 changes more strongly than HOMO1,and therefore, the bathochromic shift of the first UV-vis absorption corresponds mainly to a decrease of theLUMO1 energies.
2.2.2. Total Spin Densities, Dipole Moments andIonization Potentials. The ∆SCF method in DFTcalculations commonly understate IP’s to some extent,but in general trends are reproduced correctly. As can
Figure 4. Isosurfaces of total spin density for 3+, 4+ and 32+, 42+ according to DFT calculations in the B3LYP/6-31Gapproximation (TSD level: 0.005 e/a0
3, a0 ) 0.53 Å, Bohr radius).
Table 4. Adiabatic and Vertical Ionization Potentials of 1-4 (eV), Dipole Moments (D), and Total SpinDensities (e) for Single and Double Ionization According to DFT Calculations at the B3LYP/6-31G Level
1 2 3 4
IPa+ 5.560 5.528 5.512 5.418
IPv+ 5.801 5.755 5.684 5.566
correctedIPv
+6.70 6.92 6.91 6.86
IPa2+ 13.723 13.628 13.386 13.191
IPv2+ 13.938 13.916 13.698 13.437
dipole 0.003 0.004 0.087 0.029momenta 8.663 7.878 0.198 0.037
0.045 0.070 0.075 0.751
spin density 0.00, 0.00 0.00, 0.00 0.00, 0.00 0.00, 0.00Fe(1), Fe(2)a (0.991, 0.009) (0.995, 0.005) 0.51, 0.49 0.400, 0.403
1.006, 0.994 1.017, 0.983 0.910, 0.917 0.850, 0.948a The first entry is for the neutral species, and the second and third entries are for the mono- and dications, respectively.
R,ω-Diferrocenyl Cumulene Molecular Wires Organometallics, Vol. 23, No. 8, 2004 1829

be seen from Table 4, the first adiabatic and verticalionization potentials change very insignificantly, whileIP2+ values decrease approximately by 0.1 eV with eachchain lengthening by a CdC unit. The corrected IPv
+
will be obtained in section 2.2.3.One of the main goals of our DFT calculations is to
specify the charge distribution in the 1+-4+ mono-cations in order to determine the extent of electroniccoupling between the end-capping ferrocenyl groupsalong the cumulene sp-carbon wire. As one can see fromTable 4, the dipole moments show symmetrical chargedistribution for all di- and monocations, except for theshort monocations 1+ and 2+, for which an asymmetricalgeometry was obtained. Hence, two important pointsdraw attention and need to be discussed. First, theunpaired electrons are localized totally on the Fe atomsof both mono- and dications. The small contribution ofspin density on the cumulene chain (about 20%) wasfound only for cations 4+ and 42+ (Table 4, Figure 4).These findings are in contrast with the shape ofHOMO1, indicating that the order of ion states and theMO order are different (vide infra, section 2.2.3).
Second, calculation predicts the slightly asymmetricalgeometry, high spin asymmetry, and consequently, largedipole moments for the 1+ and 2+ monocations, incontrast to monocations 3+ and 4+, where symmetricalgeometry, spin density, and very low dipole momentsare found.
The same asymmetry solution was reported by Bohm7
for unrestricted INDO calculations of biferrocenyleneand biferrocene cations, compounds similar to 1-4. Thegood agreement between calculated and measured IP’swas treated as clear evidence of an oxidation processwith a final localized cation state. A rather sophisticatedexplanation of this phenomena was reported for bi-nuclear transition-metal compounds.8
A simpler model based on results of Snyder9 andBroer10 can be used here: the total energy lowering fora cation with broken symmetry is due to the electronrelaxation energy. This effect is essentially an atomiceffect, and it reaches its maximum value only if a holeis created on one site. If an electron hole is delocalizedover N atomic centers only about 1/N of the atomicrelaxation energy is obtained. The relaxation energy ofthe cation is defined as the difference IPa - IPv and canbe easily obtained from Table 4. The calculated relax-ation energies for monocations 1+-4+ are equal to-0.24, -0.23, -0.17, and -0.15 eV, correspondingly.The reduced relaxation energies for 3+ and 4+ indicatea symmetric charge distribution in ions due to someadmixture of the π-HOMO of the cumulene chain, whilethe slightly higher relaxation energies for 1+ and 2+ giveevidence for an additional energy gain of -0.08 eV dueto hole localization on one site. Thus, the calculationsat the given level of theory strongly support Snyder’sapproach9 and show that the asymmetrical ion geometrycan be stable because of a small energy gain caused by
strong charge localization. A more general conclusionfollows from the above considerations: the necessarycondition for broken symmetry solution is the totalabsence of π- or σ-MO bridge contribution in the groundelectronic state of the mixed-valence binuclear complex.At the same time we assume that much more theoreticaland experimental work should be done in order to proveunambiguously the reality of broken symmetry geom-etries predicted at the given UDFT level.
Coming back to the mainstream of this work, oneshould note that the longer monocation complexes 3+
and 4+ belong to Robin and Day10 class III withdominant half-to-half spin localization on metal centers,as in the case of dications 12+-42+.
2.2.3. Ionization Potentials and Kohn-ShamEnergies. The comparison of the total spin densitydistribution with the shape of the MO’s gives evidencethat the first IP’s of 1-4 are connected with electronremoval from HOMO2 and not from HOMO1, indicatingthat the order of ion states and the MO order aredifferent. Usually such a violation of MO sequence isregarded in HF theory as breakdown of Koopmans’theorem approximation12a widely used for IP evaluation.As shown quite recently, there is an analogue ofKoopmans’ theorem for Kohn-Sham energies inDFT:12b,c
where Dk is the energy correction term traditionallydenoted as Koopmans’ defect. The Dk value depends onmodel exchange-correlation functional and can reachapproximately 2-3 eV. As was reported for Fc, vinyl-Fc, styrenyl-Fc, and phenyl-Fc, the Dk values stronglydepend on the electron density localization.13 It wasfound that at the B3LYP/6-31G level calculated Dkvalues are equal to 1.46 ( 0.10 eV for the 3d(x2-y2),3d(xy) Fe MO, 0.60 ( 0.11 eV for the 3d(z2)Fe MO, and2.14 ( 0.13 eV for all π-MO’s. These findings are ofessential importance, since they allow a calibration ofKohn-Sham energies via eq 1 and allow thereby anexact evaluation of IPv
+ for substituted ferrocenes,including 1-4. Applying this procedure to HOMO1 andHOMO2 shows that the corrected IPv
+ value of 1 isdefinitely connected with HOMO2, while in the case of2-4 electron removal from HOMO2 or HOMO1 isequally possible, due to uncertainities of the Koopmans’defect. The corrected IPv
+ are presented in Table 4. Theproposed calibration procedure gives ionization poten-tials for 1-4 practically with experimental accuracy;therefore we will use only these corrected IPv
+ valuesin the following. As a general remark it should be notedhere that the direct use of the Kohn-Sham energies asIP’s without additional analysis can lead to erroneousconclusions.
2.3. Optical Properties. 2.3.1. UV-Vis Spectros-copy. From the experimentally obtained UV-vis spec-
(6) Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.;Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S. J.; Windus, T. L.; Dupluis, M.; Montgomery, J. A. J. Comput.Chem. 1993, 14, 1347; GAMESS Version 2001.
(7) Bohm, M. C.; Gleiter, R.; Delgado-Pena, F. D.; Covan, D. O. J.Chem. Phys. 1983, 79, 1164.
(8) Bohm, M. C. Theor. Chim. Acta 1982, 60, 455.(9) Snyder, L. C. J. Chem. Phys. 1971, 55, 95.(10) Broer, R. Ph.D. Thesis, Rijksuniversiteit Groningen, 1981.
(11) Robin, M. B.; Day, P. Adv. Inorg. Chem. Radiochem. 1967, 10,247.
(12) (a) Koopmans, T. Physica (Amsterdam) 1933, 1, 104. (b) Chong,D. P.; Gritsenko, O. V.; Baerends, E. J. J. Chem. Phys. 2002, 116, 1760.(c) Gritsenko, O. V.; Baerends, E. J. J. Chem. Phys. 2002, 117, 9154.
(13) (a) Loza, O. A.; Shakleina, I. V.; Chizhov, Yu. V. On the problemof the physical interpretation of the Kohn-Sham orbital energies. Bookof Abstracts, 5th Session of the V. A. Fock School on Quantum andComputational Chemistry, Novgorod the Great, Russia, 2002; p 58.
εks ) -IP + Dk (1)
1830 Organometallics, Vol. 23, No. 8, 2004 Bildstein et al.
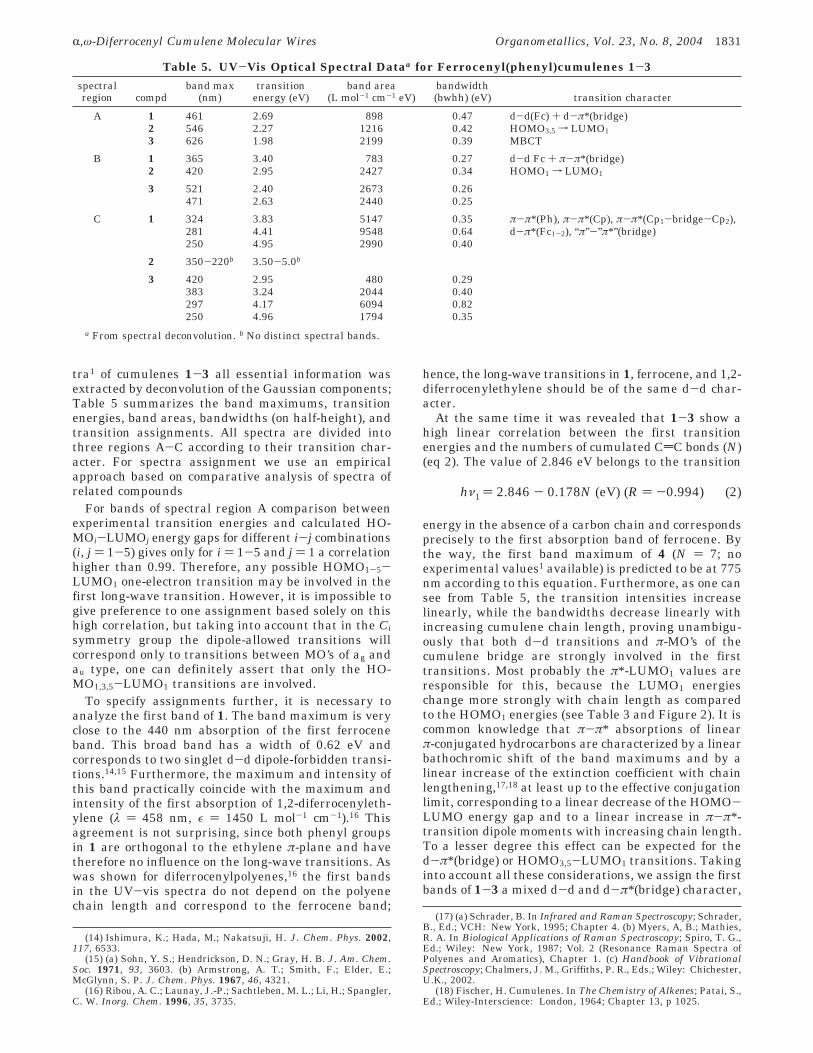
tra1 of cumulenes 1-3 all essential information wasextracted by deconvolution of the Gaussian components;Table 5 summarizes the band maximums, transitionenergies, band areas, bandwidths (on half-height), andtransition assignments. All spectra are divided intothree regions A-C according to their transition char-acter. For spectra assignment we use an empiricalapproach based on comparative analysis of spectra ofrelated compounds
For bands of spectral region A comparison betweenexperimental transition energies and calculated HO-MOi-LUMOj energy gaps for different i-j combinations(i, j ) 1-5) gives only for i ) 1-5 and j ) 1 a correlationhigher than 0.99. Therefore, any possible HOMO1-5-LUMO1 one-electron transition may be involved in thefirst long-wave transition. However, it is impossible togive preference to one assignment based solely on thishigh correlation, but taking into account that in the Cisymmetry group the dipole-allowed transitions willcorrespond only to transitions between MO’s of ag andau type, one can definitely assert that only the HO-MO1,3,5-LUMO1 transitions are involved.
To specify assignments further, it is necessary toanalyze the first band of 1. The band maximum is veryclose to the 440 nm absorption of the first ferroceneband. This broad band has a width of 0.62 eV andcorresponds to two singlet d-d dipole-forbidden transi-tions.14,15 Furthermore, the maximum and intensity ofthis band practically coincide with the maximum andintensity of the first absorption of 1,2-diferrocenyleth-ylene (λ ) 458 nm, ε ) 1450 L mol-1 cm-1).16 Thisagreement is not surprising, since both phenyl groupsin 1 are orthogonal to the ethylene π-plane and havetherefore no influence on the long-wave transitions. Aswas shown for diferrocenylpolyenes,16 the first bandsin the UV-vis spectra do not depend on the polyenechain length and correspond to the ferrocene band;
hence, the long-wave transitions in 1, ferrocene, and 1,2-diferrocenylethylene should be of the same d-d char-acter.
At the same time it was revealed that 1-3 show ahigh linear correlation between the first transitionenergies and the numbers of cumulated CdC bonds (N)(eq 2). The value of 2.846 eV belongs to the transition
energy in the absence of a carbon chain and correspondsprecisely to the first absorption band of ferrocene. Bythe way, the first band maximum of 4 (N ) 7; noexperimental values1 available) is predicted to be at 775nm according to this equation. Furthermore, as one cansee from Table 5, the transition intensities increaselinearly, while the bandwidths decrease linearly withincreasing cumulene chain length, proving unambigu-ously that both d-d transitions and π-MO’s of thecumulene bridge are strongly involved in the firsttransitions. Most probably the π*-LUMO1 values areresponsible for this, because the LUMO1 energieschange more strongly with chain length as comparedto the HOMO1 energies (see Table 3 and Figure 2). It iscommon knowledge that π-π* absorptions of linearπ-conjugated hydrocarbons are characterized by a linearbathochromic shift of the band maximums and by alinear increase of the extinction coefficient with chainlengthening,17,18 at least up to the effective conjugationlimit, corresponding to a linear decrease of the HOMO-LUMO energy gap and to a linear increase in π-π*-transition dipole moments with increasing chain length.To a lesser degree this effect can be expected for thed-π*(bridge) or HOMO3,5-LUMO1 transitions. Takinginto account all these considerations, we assign the firstbands of 1-3 a mixed d-d and d-π*(bridge) character,
(14) Ishimura, K.; Hada, M.; Nakatsuji, H. J. Chem. Phys. 2002,117, 6533.
(15) (a) Sohn, Y. S.; Hendrickson, D. N.; Gray, H. B. J. Am. Chem.Soc. 1971, 93, 3603. (b) Armstrong, A. T.; Smith, F.; Elder, E.;McGlynn, S. P. J. Chem. Phys. 1967, 46, 4321.
(16) Ribou, A. C.; Launay, J.-P.; Sachtleben, M. L.; Li, H.; Spangler,C. W. Inorg. Chem. 1996, 35, 3735.
(17) (a) Schrader, B. In Infrared and Raman Spectroscopy; Schrader,B., Ed.; VCH: New York, 1995; Chapter 4. (b) Myers, A, B.; Mathies,R. A. In Biological Applications of Raman Spectroscopy; Spiro, T. G.,Ed.; Wiley: New York, 1987; Vol. 2 (Resonance Raman Spectra ofPolyenes and Aromatics), Chapter 1. (c) Handbook of VibrationalSpectroscopy; Chalmers, J. M., Griffiths, P. R., Eds.; Wiley: Chichester,U.K., 2002.
(18) Fischer, H. Cumulenes. In The Chemistry of Alkenes; Patai, S.,Ed.; Wiley-Interscience: London, 1964; Chapter 13, p 1025.
Table 5. UV-Vis Optical Spectral Dataa for Ferrocenyl(phenyl)cumulenes 1-3spectralregion compd
band max(nm)
transitionenergy (eV)
band area(L mol-1 cm-1 eV)
bandwidth(bwhh) (eV) transition character
A 1 461 2.69 898 0.47 d-d(Fc) + d-π*(bridge)2 546 2.27 1216 0.42 HOMO3,5 f LUMO1
3 626 1.98 2199 0.39 MBCT
B 1 365 3.40 783 0.27 d-d Fc + π-π*(bridge)2 420 2.95 2427 0.34 HOMO1 f LUMO1
3 521 2.40 2673 0.26471 2.63 2440 0.25
C 1 324 3.83 5147 0.35 π-π*(Ph), π-π*(Cp), π-π*(Cp1-bridge-Cp2),281 4.41 9548 0.64 d-π*(Fc1-2), “π”-”π*”(bridge)250 4.95 2990 0.40
2 350-220b 3.50-5.0b
3 420 2.95 480 0.29383 3.24 2044 0.40297 4.17 6094 0.82250 4.96 1794 0.35
a From spectral deconvolution. b No distinct spectral bands.
hν1 ) 2.846 - 0.178N (eV) (R ) -0.994) (2)
R,ω-Diferrocenyl Cumulene Molecular Wires Organometallics, Vol. 23, No. 8, 2004 1831
with the latter contribution designated as a MBCT(metal to bridge charge transfer) transition.
The same approach can be used for examination ofthe transitions in the B spectral region. It should benoted that for 3 we placed two closely spaced absorp-tions into one spectral band. A linear correlation be-tween the second transition energies and the numbersof CdC bonds (N) is also observed in this case (eq 3).
The value of 3.618 eV is close to the second d-d excitedstate of ferrocene at 3.8 eV, and the transition intensi-ties increase linearly with chain length, just as in thecase of the A bands considered above. However, thebandwidths are quite narrow and nearly constant,similar to the case for π-π* bands of simple non-ferrocenyl cumulenes.18 Therefore, these B bands areassigned as transitions of mixed d-d and π-π*(bridge)character, with a dominant contribution from the latter.
The unambiguous assignment of the C spectral regionis virtually impossible, due to strongly overlapped π-π*transitions of the Ph and Cp rings, π-π* transitions ofthe (Cp1-bridge-Cp2) moiety, d-π* MLCT transitions,and “π”-”π*”(bridge) transitions.
We assume that the intense absorption connectedwith the first long-wave transition might be used inmolecular photonics, since the metal to bridge chargetransfer will favor one-electron transition along thecumulene chain, suggesting gating by visible light as apossible application.
2.3.2. Raman Spectroscopy. We performed DFTcalculations of the vibration frequencies for 1-3 as wellas for related “model” cumulenes, including unsubsti-tuted cumulenes H2CnH2, mono- and diferrocenyl cu-mulenes H2Cn(H)Fc and Fc(H)Cn(H)Fc, and mono- anddiphenylcumulenes H2Cn(H)Ph and Ph(H)Cn(H)Ph. Theresults are in complete agreement with the character-istic high-frequency symmetric vibrations of the cumu-lene chain (Table 6). The vibration frequencies calcu-lated by all ab initio methods are somewhat higher thanthe experimental values; therefore, scaling coefficientsare widely used for correction. In our case a scalingcoefficient of 0.93 gives a good agreement with anaccuracy of 4% between calculated and experimentalvalues. This satisfactory agreement clearly indicatesthat the cumulene sp-carbon chains in 1-3 are linear.
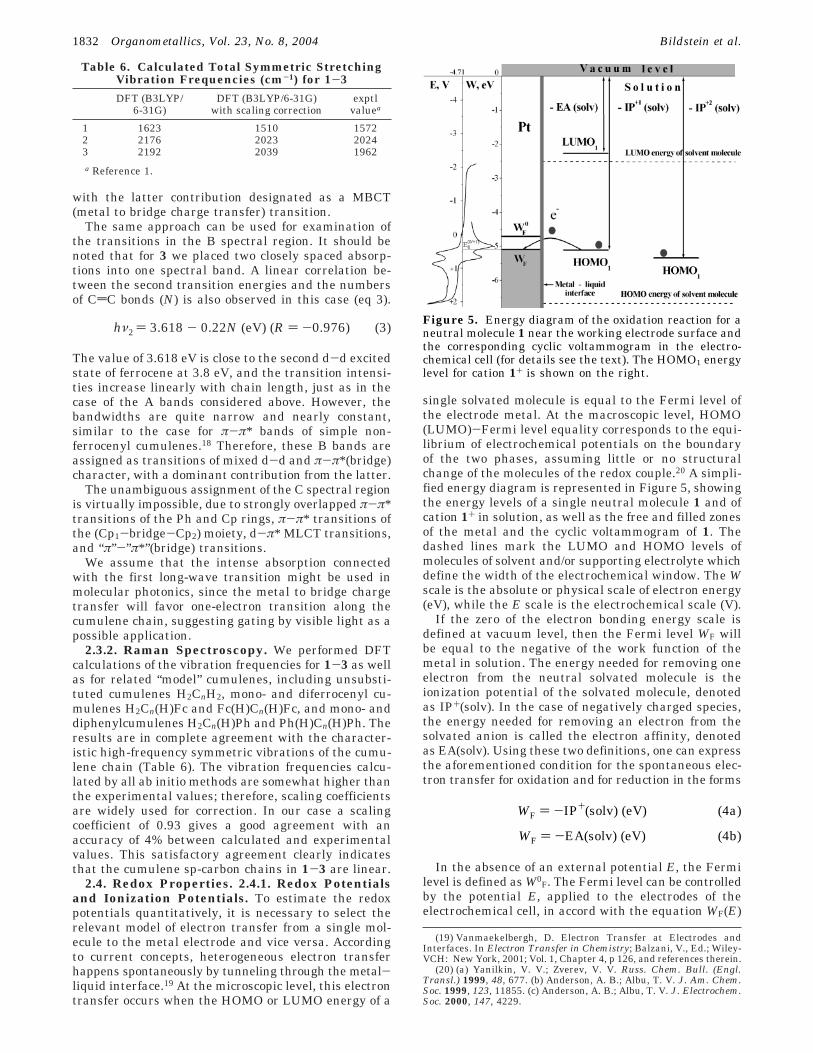
2.4. Redox Properties. 2.4.1. Redox Potentialsand Ionization Potentials. To estimate the redoxpotentials quantitatively, it is necessary to select therelevant model of electron transfer from a single mol-ecule to the metal electrode and vice versa. Accordingto current concepts, heterogeneous electron transferhappens spontaneously by tunneling through the metal-liquid interface.19 At the microscopic level, this electrontransfer occurs when the HOMO or LUMO energy of a
single solvated molecule is equal to the Fermi level ofthe electrode metal. At the macroscopic level, HOMO(LUMO)-Fermi level equality corresponds to the equi-librium of electrochemical potentials on the boundaryof the two phases, assuming little or no structuralchange of the molecules of the redox couple.20 A simpli-fied energy diagram is represented in Figure 5, showingthe energy levels of a single neutral molecule 1 and ofcation 1+ in solution, as well as the free and filled zonesof the metal and the cyclic voltammogram of 1. Thedashed lines mark the LUMO and HOMO levels ofmolecules of solvent and/or supporting electrolyte whichdefine the width of the electrochemical window. The Wscale is the absolute or physical scale of electron energy(eV), while the E scale is the electrochemical scale (V).
If the zero of the electron bonding energy scale isdefined at vacuum level, then the Fermi level WF willbe equal to the negative of the work function of themetal in solution. The energy needed for removing oneelectron from the neutral solvated molecule is theionization potential of the solvated molecule, denotedas IP+(solv). In the case of negatively charged species,the energy needed for removing an electron from thesolvated anion is called the electron affinity, denotedas EA(solv). Using these two definitions, one can expressthe aforementioned condition for the spontaneous elec-tron transfer for oxidation and for reduction in the forms
In the absence of an external potential E, the Fermilevel is defined as W0
F. The Fermi level can be controlledby the potential E, applied to the electrodes of theelectrochemical cell, in accord with the equation WF(E)
(19) Vanmaekelbergh, D. Electron Transfer at Electrodes andInterfaces. In Electron Transfer in Chemistry; Balzani, V., Ed.; Wiley-VCH: New York, 2001; Vol. 1, Chapter 4, p 126, and references therein.
(20) (a) Yanilkin, V. V.; Zverev, V. V. Russ. Chem. Bull. (Engl.Transl.) 1999, 48, 677. (b) Anderson, A. B.; Albu, T. V. J. Am. Chem.Soc. 1999, 123, 11855. (c) Anderson, A. B.; Albu, T. V. J. Electrochem.Soc. 2000, 147, 4229.
Table 6. Calculated Total Symmetric StretchingVibration Frequencies (cm-1) for 1-3DFT (B3LYP/
6-31G)DFT (B3LYP/6-31G)
with scaling correctionexptl
valuea
1 1623 1510 15722 2176 2023 20243 2192 2039 1962a Reference 1.
hν2 ) 3.618 - 0.22N (eV) (R ) -0.976) (3)Figure 5. Energy diagram of the oxidation reaction for aneutral molecule 1 near the working electrode surface andthe corresponding cyclic voltammogram in the electro-chemical cell (for details see the text). The HOMO1 energylevel for cation 1+ is shown on the right.
WF ) -IP+(solv) (eV) (4a)
WF ) -EA(solv) (eV) (4b)
1832 Organometallics, Vol. 23, No. 8, 2004 Bildstein et al.

) WF0 - eE (e is the electron charge). Since E is
measured versus the reference electrode, the standardpotential of the reference electrode ERE has to be addedto WF
0 with a reverse sign. Finally, the relation betweenthe Fermi level of the working electrode and the appliedpotential is given by
Since WF(E) depends on the applied potential, WF(E)can be matched with -IP+(solv) at formal potential E0according to eqs 4a and 5:
This equation includes some experimental quantitiessuch as the work function of the electrode metal, usuallyPt, and the standard potential of the reference electrode.A direct measurement of IP+(solv) values is ratherdifficult, while quantum-chemical calculations are oftennot precise enough. However, these values are easilydetermined from linear correlations between the formaloxidation potentials and experimental gas-phase IP’s.
2.4.2. E0-IP Correlations and Ionization Poten-tials of Solvated Molecules. Since 1980 a number oflinear correlations21-23 between E0 and experimentalgas-phase IP’s were established for structurally relatedorganic and organometallic compounds, but up to nowtheir physical meaning has not been recognized. Onlysome selected data are given below:
The finding of an essentially linear relationship withvery similar parameters cannot be accidental. To revealthe underlying physical reasons, we revised all relevantavailable data concerning ferrocene derivatives and bis-(benzene)chromium (BBCr) derivatives.22-27 As a result,a representative data set covering a wide range ofpotentials from -0.98 to +1.3 V and IP’s from 5.01 to7.40 eV were obtained. After all electrochemical data
were reduced to the same experimental conditions(reference electrode SCE, solvent dichloromethane, sup-porting electrolyte [Bu4N][B(C6F5)4]) with correspondingcorrections,28 a generalized linear regression was ob-tained for these 28 sandwich Fe and Cr complexes
with E0(Fc0/+) ) 0.46 V and E0(BBCr0/+) ) -0.66 V asreference points.
Now if we compare eqs 6 and 7, the physical meaningof regression (7) becomes clear: the first free term ofeq 7 is equal to the Fermi level of the working Ptelectrode in dichloromethane shifted on reference (SCE)electrode potential, while the second term is the ioniza-tion potential of the solvated molecule IP+(solv). It hasbeen shown earlier29 that the absolute potential of thestandard hydrogen electrode (SHE) is equal to thenegative of the Fermi level of the Pt electrode insolution. Reported values29 include 4.44 ( 0.02 V(aqueous solutions), 4.60 ( 0.10 V (acetonitrile solu-tions), and 4.29 ( 0.07 V (formamide solutions), respec-tively. Although the absolute potential of the SHE fordichloromethane was not reported, a value of 4.44 Vseems very reasonable. Taking into account that thestandard potential of SCE is equal to 0.24 V, the finalvalue for WF
0 - eERE is -4.68 eV, very close to the freeterm of eq 7. Therefore, we conclude that 4.71 V is theabsolute potential of the standard hydrogen electrodein the SCE-dichloromethane-[Bu4N][B(C6F5)4] elec-trochemical system.
Due to solvation effects, IP+(solv) energies are alwayslower in value than gas-phase IP+, and according to eq7 the IP’s of solvated molecules are three-fourths of theIP’s of free molecules. They can now be easily evaluatedif the IP values are known. The gradient in regression(7) is an averaged quantity which characterizes theextent of lowering of gas-phase IP’s at solvation, de-pending on molecule and solvent properties.
Table 7 summarizes the relevant experimental1 andcalculated data. The redox potentials E0
0/+ were calcu-lated according to regression (7) with corrected IPv
+
values from Table 4. We regard the agreement with
(21) (a) Howell, J. O.; Goncalves, J. M.; Amatore, C.; Klasinc, L.;Wightman, R. M.; Koshi, J. K. J. Am. Chem. Soc. 1984, 106, 3968. (b)Li, T. T.-T.; Brubaker, C. H. J. Organomet. Chem. 1981, 216, 223. (c)Gassman, P. G.; Yamaguchi, R. J. Am. Chem. Soc. 1979, 101, 1308.
(22) Yur’eva, L. P.; Peregudova, S. M.; Kravtsov, D. N.; Vasilkov,A. Yu.; Nekrasov, L. N.; Asfandiarov, N. L.; Timoshenko, M. M.;Chizhov, Yu. V. J. Organomet. Chem. 1987, 336, 371.
(23) Al-Saeed, A. M.; Seddon, E. A.; Seddon, K. R.; Shimran, A. A.;Tompkins, S.; Grossel, M. C.; Knychala, J. P. J. Organomet. Chem.1988, 347, C25.
(24) Gritzner, G.; Kuta, J. Pure Appl. Chem. 1984, 56, 461.(25) Bailey, S. I.; Leung, W.-P.; Ritchie, I. M. Electrochim. Acta 1985,
30, 861.(26) (a) Cabelli, D. E.; Cowley, A. H.; Lagowski, J. J. Inorg. Chim.
Acta 1982, 57, 195. (b) Timoshenko, M. M.; Yur’eva, L. P.; Zaitseva,N. N.; Uralets, I. A.; Kravtsov, D. N.; Chizhov, Yu. V. J. Organomet.Chem. 1989, 361, 79.
(27) (a) Vondrak, T. J. Organomet. Chem. 1984, 275, 93. (b) Vondrak,T. J. Organomet. Chem. 1986, 306, 89. (c) Khvostenko, V. I.; Asfan-diarov, N. L. J. Electron Spectrosc. Relat. Phenom. 1993, 63, 419.
(28) Geiger, W. E. Redox Reactions of Symmetrical Metal SandwichCompounds. J. Organomet. Chem. Libr. 1990, 22, 142.
(29) Trasatti, S. Pure Appl. Chem. 1986, 58, 956.
WF(E) ) WF0 - eERE - eE (eV) (5)
eE0 ) WF0 - eERE + IP+(solv) (eV) (6)
Eox1/2 ) -4.88 + 0.78(IP)
(cyclic saturated hydrocarbons)21c
Eox1/2 ) -4.08 + 0.62(IPv) (R ) 0.991; for 20 bis
(benzene)chromium derivatives)22
Eo ) -4.163 + 0.681(IPv) (R )
0.946; for 20 ferrocene derivatives)23
Table 7. Experimental and Calculated RedoxPotentials, CV Wave Splitting, and Metal-Metal
Distancesa for 1-4E0
0/+ E0+/2+ dE0
exptlb calcd exptlb calcd exptl calcdcL(Fe1-Fe2)
calcd
1 0.37 0.32 0.68 0.73 0.31 0.36 (0.41) 7.542 0.40 0.48 0.78 0.72 0.38 0.32 (0.24) 9.183 0.43 0.47 0.54 0.63 0.11 0.20 (0.16) 11.424 0.44 0.53 - 0.06d (0.09) 13.70
a Redox potentials are given in V, and L(Fe1-Fe2) is the distancebetween Fe atoms in Å. b All experimental redox potentials1 arecorrected for 0.04 V to fit E0(Fc0/+) ) 0.46 V. c Difference betweencalculated E0
+/2+ and experimental E00/+; values in parentheses
are differences between calculated E0+/2+ and E0
0/+ values. d Ex-trapolated from linear regression dE0 ) 0.75 - 0.049[L(Fe1-Fe2)]based on mean dE0 values.
E00/+ )
-4.71((0.14) + 0.75((0.02)(IPv) (R ) 0.989) (7)
R,ω-Diferrocenyl Cumulene Molecular Wires Organometallics, Vol. 23, No. 8, 2004 1833

experimental redox potentials as very satisfactory,taking into account the complexity of the problem.
The dication redox potentials E0+/2+ should obey a
similar equation, since the tunneling mechanism ofheterogeneous electron transfer does not depend on thecharge of the reacting species. However, the dicationgradient value should be much smaller, because solva-tion energy increases strongly for dications. We findsatisfactory results at a dication gradient equal to 0.39,which is nearly half of the monocation gradient (eq 8).As expected, electrochemical oxidation of monocations1-4 occurs more easily with bridge lengthening accord-ing to our calculations.
In studies of mixed-valence compounds the dE0 )E0
+/2+ - E00/+ splitting is of special interest, since it
gives additional information on the distance dependenceof electron coupling, allowing an estimation of bothdecay slope and the effective conjugation length orconjugation limit, respectively.30a The calculated wavesplittings in Table 7 concur favorably with the experi-mental data.1 We include calculated distances betweenremote Fe atoms in Table 7 in order to show moredistinctly the distance dependence of the redox wavesplitting. The average calculated dE0 values agree withthe exponential decay regression dE0 ) -0.05 + 1.76exp(- 0.18[L(Fe1-Fe2)]) (R ) -0.990) with a decay slopeequal to 0.18 A-1 and an effective conjugation limit (dE0) 0) at a distance Leff ) 16.0 ( 0.9 Å. The obtained“electrochemical” decay slope is in reasonable agreementwith “spectral” decay slopes reviewed recently for sixseries of binuclear mixed-valence metal complexes.30b
2.4.3. Redox Potentials and Electron Affinitiesof Solvated Molecules. During the reduction of aneutral molecule in solution, an additional electronoccupies the LUMO via tunneling through the metal-liquid interface and generates the anion, surrounded byits solvation shell. Just as in the case of cations (eq 6),the redox potential E0
-/0 is connected by eqs 9 and 10with the electron affinity of the solvated molecule, EA-(solv).
Equation 10 shows that there is a simple relationbetween redox potentials and electron affinity of thesolvated molecule. Experimental gas-phase EA’s of morecomplex molecules are very rarely available, while first-principles calculations are successful only with verylarge basis sets. Therefore, the proposed approach seemsto be useful for an approximate evaluation of electronaffinities of larger molecules.
Table 8 summarizes all information relating to thereduction of 1-3. There is an amazingly good agreementbetween the difference of oxidation and reductionpotentials ∆E0 (V), which can be called the “electro-chemical gap”, and the first transition energies hν1,often called the “optical gap”. The latter values are
higher than the former ones by only 0.20 eV. To the bestof our knowledge, this is the first such correspondencereported for organometallics, and only recently haveoptical/electrochemical gap correlations been found fornanoparticles.31,32 Furthermore, there is an excellentlinear correlation between ∆E0 values and the LUMO1f HOMO3,5 energy gap, proving that the unpairedelectron populates the LUMO1 at reduction of 1-3 andthat it is delocalized on the cumulene bridge.
2.5. Summary. Both the spin-restricted DFT/B3LYPmethod for neutral complexes and the spin-unrestrictedDFT/B3LYP method for cations was used with thestandard 6-31G basis set for C and H and the Hay-Wadtpseudopotential for Fe. The geometry of mono- anddioxidized diferrocenyldiphenylcumulenes 1-4 was cal-culated with full optimization without any restrictionin symmetry.
The computational conformation analysis did notreveal any preferable conformation among four possiblediasteromeric structures; therefore, all calculations for1-4 were performed in the trans-anti conformationonly. The DFT calculation reproduces the experimentalstructural data very well. The calculations show astrong asymmetric charge distribution for 1+ and 2+
with corresponding large dipole moments and strongasymmetry of total spin densities. In contrast, 3 and 4monocations have a symmetric geometry and chargedistribution with equal spin density of the unpairedelectron on each Fe atom. No apparent spin density wasfound on the cumulene chain for any of the mono- anddications, and the two unpaired electrons are distributedsymmetrically between remote Fe atoms for all dications12+-42+. The adiabatic and vertical IP’s were calculatedby the ∆SCF method for both cations and dications. Ouranalysis showed unambiguously that the breakdown ofthe Koopmans theorem for Kohn-Sham MO’s occurs for1-4. A calibration procedure for the Kohn-Sham ener-gies was applied, affording the vertical IP’s with ex-perimental accuracy. From the experimentally obtainedUV-vis spectra of cumulenes 1-3 all essential informa-tion was extracted by deconvolution on the Gaussiancomponents: the band maximums, transition energies,band areas, and bandwidths. The assignments werebased on a detailed comparative analysis of the spectraof related compounds. The calculated frequencies ofhigh-frequency symmetric vibrations of the cumulenechain for 1-4 agree very well with the experimentalRaman bands.
A simplified model based on empirical findings wasproposed for modeling of redox potentials. The results
(30) (a) Ward, M. D. Chem. Soc. Rev. 1995, 24, 121. (b) Launay, J.-P. Chem. Soc. Rev. 2001, 30, 386.
(31) (a) Chen, S.; Ingrma, R. S.; Murrey, R. W. Science 1998, 280,2098. (b) Templeton, A. C.; Wuelfing, W. P.; Murrey, R. W. Acc. Chem.Res. 2000, 33, 27.
(32) Haram, S. K.; Quinn, B. M.; Bard, A. J. J. Am. Chem. Soc. 2001,123, 8860.
E0+/2+ ) -4.71 + 0.39(IPv
2+) (8)
eE0-/0 ) WF
0 - eERE + EA(solv) (eV) (9)
eE0-/0 ) -4.71 + EA(solv) (eV) (10)
Table 8. Experimental Redox Potentials E0-/0 (V),
Electron Affinities of Solvated Molecules EA(solv)(eV), Electrochemical Gap ∆E0 (V), and Optical
Gap hν1 (eV) for 1-3E0
-/0 EA(solv)b ∆E0 hν1
1a g-2.3 e+2.41 g2.63 2.692 -1.70 +3.01 2.06 2.273 -1.35 +3.36 1.74 1.98
a Lower limit value of E0-/0. b Calculated by eq 10.
1834 Organometallics, Vol. 23, No. 8, 2004 Bildstein et al.

obtained for E00/+ and E0
+/2+ reproduce the experimentalvalues very satisfactorily, taking into account thecomplexity of the problem. An essentially exponentialdecay of calculated CV wave splitting with increasingFe1-Fe2 distance was found, indicating an electrochemi-cal decay slope of 0.18 A-1 and an effective conjugationlimit of 16.0 Å in these diferrocenylcumulene com-pounds. A surprisingly good agreement between theelectrochemical and optical gap was obtained, indicatinga very close relationship between electrochemical andoptical properties of 1-3.
The structural, chemical, optical, electrochemical, andmolecular orbital properties of 1-4 make ferrocenylcumulenes very promising molecular objects with greatpotential in molecular scale electronics, in comparisonwith purely organic molecular wires.33
First, the electron transfer involves only iron centersand not the cumulene bridge, which remains un-changed, stable, and constant in length, meeting theprinciple of minimal structural distortion during elec-tron transfer. This structural stability and rigid molec-ular architecture of the cumulene chain is a keyadvantage in comparison with less rigid but more
common polyene or polyphenyl chains, implying thatsuch rigid molecular wires can sustain a large currentdensity.
Second, due to the rich synthetic chemistry of theferrocenyl moiety it is not a principal problem to anchorit to a metal or semiconductor junction, e.g. by a thiol/gold interface.34 In addition, donor/acceptor substituentson the ferrocenyl termini may tailor the ionizationpotentials IP1 of ferrocenyl cumulenes, thereby makingelectron-transfer control more flexible.
Third, the intense absorption in the visible spectralregion may open new possibilities in molecular photo-nics applications. The very close relationship betweenthe electrochemical and the optical gaps of 1-3 mayprove useful for gating by visible light.
Fourth, the localized high spin properties of dications12+-42+ may find interesting applications in the devel-opment of new magneto-sensitive molecular devices.
Acknowledgment. B.B. thanks the Austrian Sci-ence Fund FWF (Grant No. P13073-PHY) for supportof this research. Y.C. thanks the Ministry of Educationof the Russian Federation (Grant No. E02-3-358). Y.C.is grateful to V. Phillipov, M.Sc., for help in visualizationsoftware development. The Center of High-PerformanceCalculation of St. Petersburg State University is grate-fully acknowledged for unlimited computational time.
Supporting Information Available: Text giving fullcomputational procedure details and all calculated geometries.This material is available free of charge via the Internet athttp://pubs.acs.org.
OM034234D
(33) (a) Molecular Electronics: Science and Technology; Aviram, A.,Ed.; Confer. Proc. No. 262; American Institute of Physics: New York,1992. (b) Miller, J. S. Adv. Mater. 1990, 2, 378, 495, 601. (c) Molecularand Biomolecular Electronics, Birge, R. R., Ed.; Adv. Chem. Ser. 240;American Chemical Society: Washington, DC, 1991. (d) Nanostructuresand Mesoscopic Systems, Kirk, W. P., Reed, M. A., Eds.; Academic:New York, 1992. (e) Samanta, M. P.; Tian, W.; Datta, S.; Henderson,J. I.; Kubiak, C. P. Phys. Rev. B 1996, 53, 7626.
(34) (a) Bain, C. D.; Troughton, E. B.; Tao, Y.-T.; Evall, J.; White-sides, G. M.; Nuzzo, R. G. J. Am. Chem. Soc. 1989, 111, 321. (b) Sellers,H.; Ulman, A.; Shnidman, Y.; Eilers, J. E. J. Am. Chem. Soc. 1993,115, 9389.
R,ω-Diferrocenyl Cumulene Molecular Wires Organometallics, Vol. 23, No. 8, 2004 1835


















