
Rights / License: Research Collection In Copyright - …36082/... · Es entsteht dabei...
67
Research Collection Doctoral Thesis Die Umsetzung von 3β-Acetoxy-{Delta}-20 -18,20-cyclo-5α- pregnen mit Blei(IV)-acetat Author(s): Schorta, Roman Publication Date: 1964 Permanent Link: https://doi.org/10.3929/ethz-a-000293982 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection . For more information please consult the Terms of use . ETH Library
Transcript of Rights / License: Research Collection In Copyright - …36082/... · Es entsteht dabei...
Research Collection
Doctoral Thesis
Die Umsetzung von 3β-Acetoxy-{Delta}-20 -18,20-cyclo-5α-pregnen mit Blei(IV)-acetat
Author(s): Schorta, Roman
Publication Date: 1964
Permanent Link: https://doi.org/10.3929/ethz-a-000293982
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 3523
I. Die Umsetzung von 3/?-Acetoxy- a20
-18,20-cyclo-5<X-pregnen mit Blei(IV)-acetat
II. Zur Photochemie des Testosterons
Von der
EIDGENÖSSISCHEN TECHNISCHEN
HOCHSCHULE IN ZÜRICH
zur Erlangung
der Würde eines Doktors der Naturwissenschaften
genehmigte
PROMOTIONSARBEIT
vorgelegt von
ROMAN SCHORTA
dipl. Naturwissenschafter ETH
von Zernez |Kt. Graubünden)
Referent: Herr Prof. Dr. O. Jeger
Korreferent: Herr Prof. Dr. A. Eschenmoser
Juris-Verlag Zürich
1964

Leer - Vide - Empty
i

Meinen lieben Eltern und meiner lieben Frau
in Dankbarkeit gewidmet
Leer - Vide - Empty

Meinem sehr verehrten Lehrer,
Herrn Prof. Dr. O. Jeger ,
unter dessen Leitung die vorliegende Arbeit ausgeführt wurde, möchte ich für seine
Unterstützung, sowie für das mir stets entgegengebrachte Wohlwollen aufs herzlichste
danken.
Besonders herzlich danken möchte ich aber auch
Herrn Dr. K.Schaffner,
für die vielen wertvollen Ratschläge, die unermüdliche Hilfe und das stete Interesse,
die er mir jederzeit zuteil werden liess.

Der Marc-Birkigt-Stiftung danke ich für die mir gewährten Stipendien.

- 7 -
Inhaltsverzeichnis
20I. Die Umsetzung von 3ß -Acetoxy- A -18,20-cyclo-5oL-pregnen mit
Blei(IV)-acetat
THEORETISCHER TEIL 9
Einleitung 9
Reaktionen von Blei(IV) -acetat mit Olefinen 11
Eigene Arbeiten 21
201) Behandlung von 3(3-Acetoxy-A -18,20-cyclo-5oi/-pregnen
mit Blei(IV)-acetat 21
2) Verknüpfung von Produkt B mit Produkt A 22
3) Struktur des Produktes B 24
4) Struktur des Produktes A 25
Diskussion 27
Zur Stereochemie des Produktes B 30
EXPERIMENTELLER TEIL 32
Literatur 41
H. Zur Photochemie des Testosterons
THEORETISCHER TEIL 43
Zur UV.-Bestrahlung von cO, (i -ungesättigten
Carbonylverbindungen 43
1) Inter- und intramolekulare Cycloadditionen"
44

- 8 -
2) Verschiebung der oü, (3 -Doppelbindung in die ß, Y -Stellung 52
3) Reaktionen unter Teilnahme des Lösungsmittels 52
4) Umlagerungsreaktionen in Abhängigkeit des Lösungsmittels 53
Eigene Arbeiten 55
1) Bestrahlung von O-Acetyl-testosteron in Hexan 55
2) Bestrahlung von O-Acetyl-testosteron in Aethanol 56
3) Destrahlung von O-Acetyl-testosteron in Diäthyläther 57
EXPERIMENTELLER TEIL 59
Literatur 63
ZUSAMMENFASSUNG 65
- 9 -
20I. Die Umsetzung von 3 ß - Acetoxy - A - 1 8
,20 -cyclo-5oc-pregnen
mit Blei(IV)-acetat
THEORETISCHER TEIL
Einleitung
Das Nebennierenrinden-Hormon Aldosteron ist aus natürlichen Quellen nur in
sehr geringen Mengen erhältlich. Für seine künstliche Herstellung schienen nebst
dem wenig ergiebigen totalsynthetischen Zugang vor allem partialsynthetische Metho¬
den wünschenswert.
Ein solcher Zugang stellte aber die chemisch attraktive Aufgabe, neue Metho¬
den zur selektiven Einführung der das Aldosteron auszeichnenden Sauerstoffunktion
an der angulären Methylgruppe 18 zu suchen. Diese erstmals im Laboratorium von
J e g e r gelöste Problemstellung veranlasste in der Folge zahlreiche Arbeitsgrup¬
pen zur Ausarbeitung verschiedener Methoden, die selektive, intramolekulare Sub¬
stitutionen an nicht aktivierten Alkanstellen gestatten.
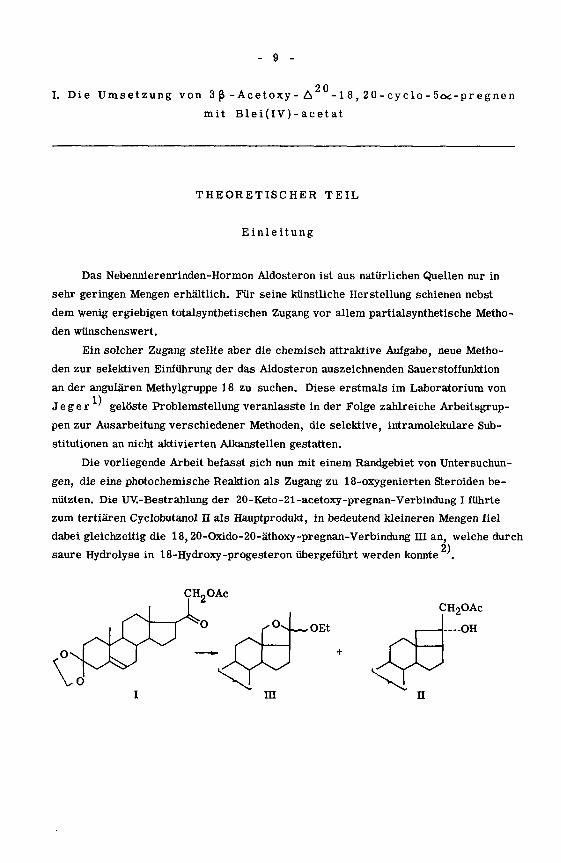
Die vorliegende Arbeit befasst sich nun mit einem Randgebiet von Untersuchun¬
gen, die eine photochemische Reaktion als Zugang zu 18-oxygenierten Steroiden be¬
nützten. Die UV.-Bestrahlung der 20-Keto-21-acetoxy-pregnan-Verbindung I führte
zum tertiären Cyclobutanol II als Hauptprodukt, in bedeutend kleineren Mengen fiel
dabei gleichzeitig die 18,20-Oxido-20-äthoxy-pregnan-Verbindung HI an, welche durch
2)saure Hydrolyse in 18-Hydroxy-progesteron übergeführt werden konnte
.
CHgOAc
OEt
- 10 -
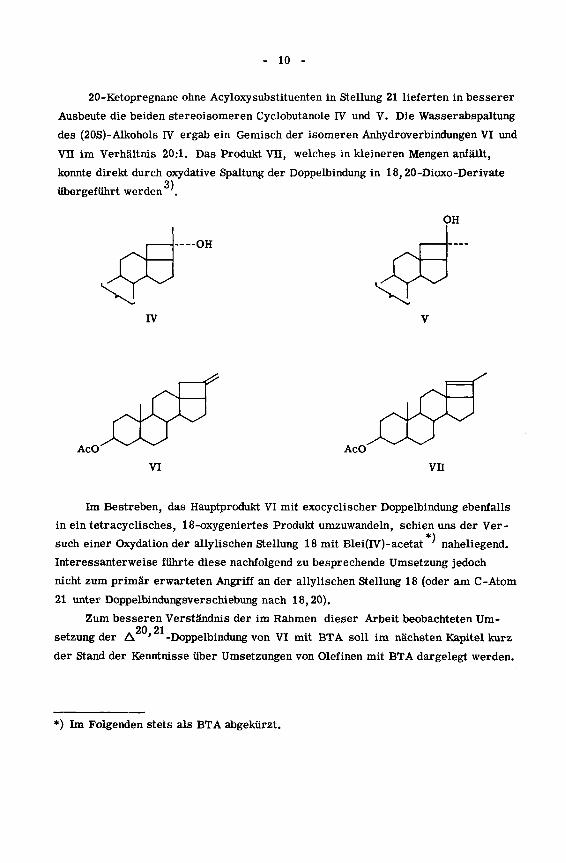
20-Ketopregnane ohne Acyloxysubstituenten in Stellung 21 lieferten in besserer
Ausbeute die beiden stereoisomeren Cyclobutanole IV und V. Die Wasserabspaltung
des (20S)-Alkohols IV ergab ein Gemisch der isomeren Anhydroverbindungen VI und
VII im Verhältnis 20:1. Das Produkt VU, welches in kleineren Mengen anfällt,
konnte direkt durch oxydative Spaltung der Doppelbindung in 18,20-Dioxo-Derivate3)
übergeführt werden.
OH
--OH
IV
AcO AcO
VI vn
Im Bestreben, das Hauptprodukt VI mit exocyclischer Doppelbindung ebenfalls
in ein tetracyclisches, 18-oxygeniertes Produkt umzuwandeln, schien uns der Ver-
*)such einer Oxydation der allylischen Stellung 18 mit Blei(IV)-acetat naheliegend.
Interessanterweise führte diese nachfolgend zu besprechende Umsetzung jedoch
nicht zum primär erwarteten Angriff an der allylischen Stellung 18 (oder am C-Atom
21 unter Doppelbindungsverschiebung nach 18,20).
Zum besseren Verständnis der im Rahmen dieser Arbeit beobachteten Um-
20 21Setzung der A ' -Doppelbindung von VI mit BTA soll im nächsten Kapitel kurz
der Stand der Kenntnisse über Umsetzungen von Olefinen mit BTA dargelegt werden.
*) Im Folgenden stets als BTA abgekürzt.

- 11 -
Reaktionen von Blei(IV) - acetat mit Olefinen
Die nachfolgend besprochenen Beispiele zeigen eine ausserordentliche Vielfalt
von Reaktionsmöglichkeiten der Olefine mit BTA. Bis heute sind jedoch nur spärli¬
che Angaben über den genauen Verlauf der verschiedenen Reaktionen bekannt.
Während der Oxydation wird vierwertiges Blei auf die zweiwertige Oxydationsstufe
reduziert. Dabei entsteht Bleidiacetat unter Ausstossung zweier Acetoxygruppen.
Dieser Austritt lässt sich auf drei verschiedene Arten formulieren:
(1) Es entstehen zwei Acetoxyradikale,
(2) es wird je ein Acetoxy-Kätion und Acetoxy-Anion gebildet;
(3) schliesslich kann das Blei zwei Elektronen aufnehmen und dadurch zwei Acetat-
Anionen abgeben.
Durch Wahl des Lösungsmittels und der Reaktionsbedingungen können an dem¬
selben Substrat verschiedene Resultate erhalten werden. Dabei lassen sich als haupt¬
sächlichste Reaktionstypen Substitutionen und Additionen feststellen. Die meisten
BTA-Oxydationen wurden bis anhin in Benzol und in Eisessig ausgeführt. In aproti-
schen Lösungsmitteln wie Benzol sind aus Stabilitätsgründen radikalische Ueber-
gangszustände vorzuziehen, während in Eisessig heterolytische Reaktionsmechanismen
vorherrschen dürften. Dass jedoch auch in Benzol wahrscheinlich ionische Umsetzun¬
gen stattfinden können, zeigt das Beispiel von Norbornen (siehe unten), bei welchem
sowohl die Oxydation mit BTA als auch die Umsetzung mit Chlor und Brom'
zu
analogen Produkten führt. Es ist also anzunehmen, dass bei den erwähnten Reaktionen
analog gebaute, ionische Zwischenstufen durchlaufen werden. Auch beim Angriff an
einer Doppelbindung in Eisessig dürften ionische Zwischenstufen vorherrschen.
Criegee zum Beispiel nimmt an, dass primär die Fragmente (AcO)„Pb und AcO"
an die Doppelbindung angelagert werden. Die daraus resultierende bleiorganische
Verbindung zerfällt nach dieser Auffassung in Folge der hohen Elektronenaffinität
des Bleis in ein Carboniumion und in ein (AcOLPb" -Anion. Das letztere liefert Blei¬
diacetat und ein weiteres Acetoxy-Anion. Das Kation kann sich je nach Struktur der
18)Substrate stabilisieren. Mo s her hingegen zieht für die Spaltung von intermediär
entstehenden Bleialkoxylatverbindungen in Analogie zum eingehend untersuchten
Zerfall von Chromsäureestern eine heterolytische Spaltung vor. Als Argument gegen
einen homolytischen Zerfall führt er aber lediglich das Fehlen von Dimeren an, die

- 12 -
bei einem radikalischen Mechanismus entstehen sollten. Die Lösung dieses Problems
dürfte wohl oft darin liegen, dass von Fall zu Fall schwer abschätzbare Stabilitäts¬
faktoren die Mechanismusart beeinflussen können.
4)1923 hatte Dimroth das BTA in die Praxis des organischen Chemikers
eingeführt, indem er beobachtete, dass bei der Behandlung von 4-Propenylanisol
(Vin) mit BTA in guter Ausbeute zwei Acetatreste an die Doppelbindung addiert wer¬
den.
MeO—V \—CH=CH-CH„ •» MeO—^ \-CH— CH-CH0
\=/ 3 \=/ i I 3
OAc OAc
vrn De
5)In der Folge wurden zahlreiche analoge Anisol-Derivate untersucht. C r i e g e e
stellte fest, dass die Substitution des aromatischen Ringes einen wesentlichen Einfluss
auf den Gang der Reaktion ausübt. So ergibt die Oxydation von p-Methoxystyrol (X)
ein Diacetat XI, dessen Entstehung durch die Annahme einer 1,2-Verschiebung des
Phenylkernes verständlich wird.
tf~\ /TA /OAcMeO—</ x>—CH=CH„ - MeO—V \)—CH„-CHC
*=*2 \=/ *
"OAc
X XI
Währenddessen zeigt das unsubstituierte Styrol (Xu) einen gänzlich andersarti¬
gen Reaktionsverlauf. Er kommt im Endeffekt einer Addition von Methylacetat gleich.
f\-c
w -CH=CH2 - <^J—ÇH-CH2-CH3OAc
xn xm
Im allgemeinen reagieren aber aliphatische oder cyclische Olefine nicht in der
von Dimroth beschriebenen Form. Dazu bedarf es meist besonderer Reaktions-
bedingungen (z.B. Temperaturerhöhung usw.), wobei die Acetataddition stark durch
Substitutionen in Allylstellung konkurrenziert wird. Zudem sind diese Reaktionstypen
auch oft mit molekularen Umlagerungen gekoppelt.
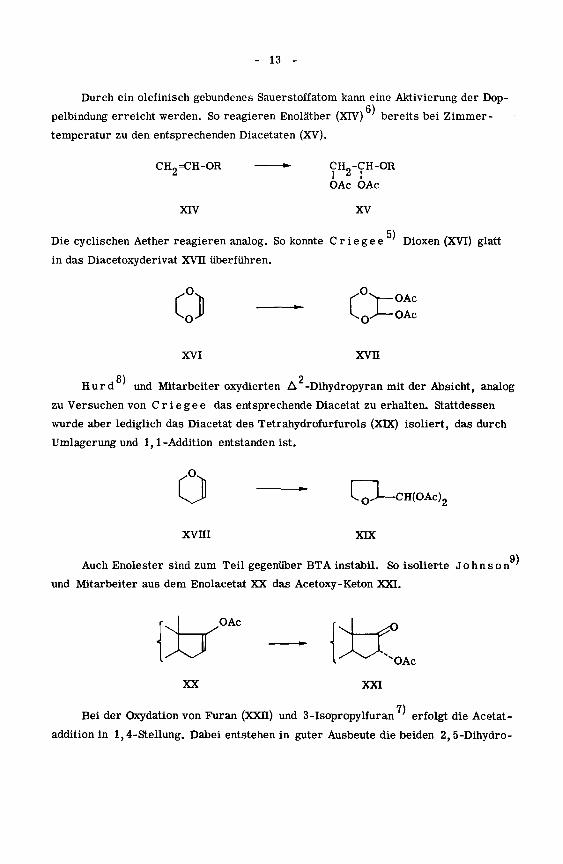
- 13 -
Durch ein olefinisch gebundenes Sauerstoffatom kann eine Aktivierung der Dop-
pelbindung erreicht werden. So reagieren Enoläther (XIV) bereits bei Zimmer¬
temperatur zu den entsprechenden Diacetaten (XV).
CH„=CH-OR •- CH0-CH-OR
OAc OAc
XIV XV
5)Die cyclischen Aether reagieren analog. So konnte C r i e g e e Dioxen (XVI) glatt
in das Diacetoxyderivat XVII überführen.
Ü — CcOAc
^ ^0^-OAc
xvi xvn
Hurd und Mitarbeiter oxydierten A -Dihydropyran mit der Absicht, analog
zu Versuchen von Crie gee das entsprechende Diacetat zu erhalten. Stattdessen
wurde aber lediglich das Diacetat des Tetrahydrofurfurols (XDC) isoliert, das durch
Umlagerung und 1,1-Addition entstanden ist.
011
- I—I
—CH(OAc)2
xvni XDC
9)
Auch Enolester sind zum Teil gegenüber BTA instabil. So isolierte Johnson '
und Mitarbeiter aus dem Enolacetat XX das Acetoxy-Keton XXI.
OAc
\XJXX
7)
Bei der Oxydation von Furan (XXII) und 3-Isopropylfuran'
erfolgt die Acetat-
addition in 1,4-Stellung. Dabei entstehen in guter Ausbeute die beiden 2,5-Dihydro-
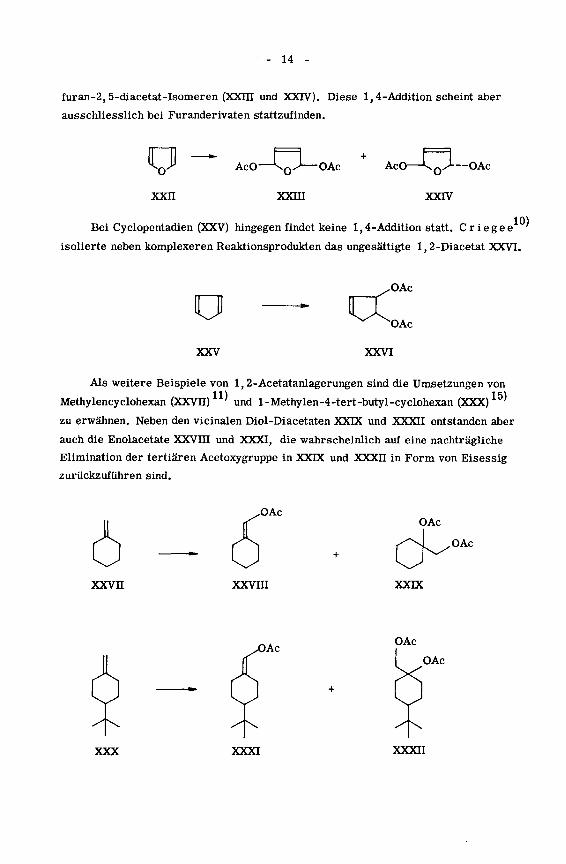
- 14 -
furan-2, 5-diacetat-Isomeren (XXin und XXIV). Diese 1,4-Addition scheint aber
ausschliesslich bei Furanderivaten stattzufinden.
o
xxn
AcO
xxni
OAc AcO
xxrv
OAc
Bei Cyclopentadien (XXV) hingegen findet keine 1,4-Addition statt. Criegee
isolierte neben komplexeren Reaktionsprodukten das ungesättigte 1,2-Diacetat XXVI
10)
o
XXV
CC,OAc
OAc
XXVI
Als weitere Beispiele von 1,2-Acetatanlagerungen sind die Umsetzungen von
Methylencyelohexan (XXVII) und l-Methylen-4-tert-butyl-cyclohexan (XXX)
zu erwähnen. Neben den vicinalen Diol-Diacetaten XXDC und XXXII entstanden aber
auch die Enolacetate XXVin und XXXI, die wahrscheinlich auf eine nachträgliche
Elimination der tertiären Acetoxygruppe in XXDC und XXXII in Form von Eisessig
zurückzuführen sind.
„OAcOAc
„OAc
xxvn xxvm XXDC
OAc
OAc
XXX XXXI XXXII
- 15 -
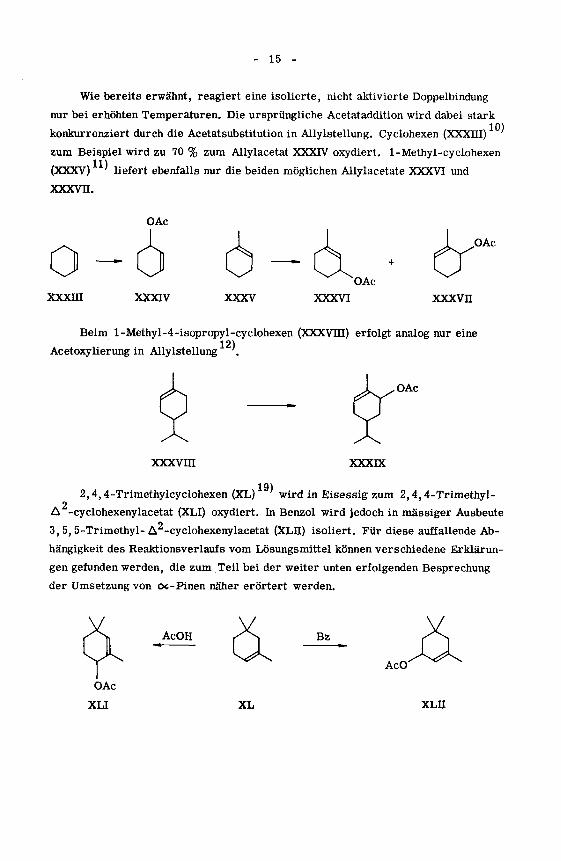
Wie bereits erwähnt, reagiert eine isolierte, nicht aktivierte Doppelbindung
nur bei erhöhten Temperaturen. Die ursprüngliche Acetataddition wird dabei stark
konkurrenziert durch die Acetatsubstitution in Allylstellung. Cyclohexen (XXXIII)
zum Beispiel wird zu 70 % zum Allylacetat XXXIV oxydiert. 1-Methyl-cyclohexen
(XXXV) ' liefert ebenfalls nur die beiden möglichen Allylacetate XXXVI und
xxxvn.
OAc
oxxxni
OAc
XXXIV XXXV XXXVI
&xxxvn
Beim l-Methyl-4-isopropyl-cyclohexen (XXXVTII) erfolgt analog nur eine
12)Acetoxylierung in Allylstellung '.
OAc
xxxvm XXXIX
19)2,4,4-Trimethylcyclohexen (XL) wird in Eisessig zum 2,4,4-Trimethyl-
o
A -cyclohexenylacetat (XLI) oxydiert. In Benzol wird jedoch in massiger Ausbeute
3, 5, 5-Trimethyl-A -cyclohexenylacetat (XLII) isoliert. Für diese auffallende Ab¬
hängigkeit des Reaktionsverlaufs vom Lösungsmittel können verschiedene Erklärun¬
gen gefunden werden, die zum Teil bei der weiter unten erfolgenden Besprechung
der Umsetzung von oc-Pinen näher erörtert werden.
AcOH Bz
AcO
XL XLII
- 16 -
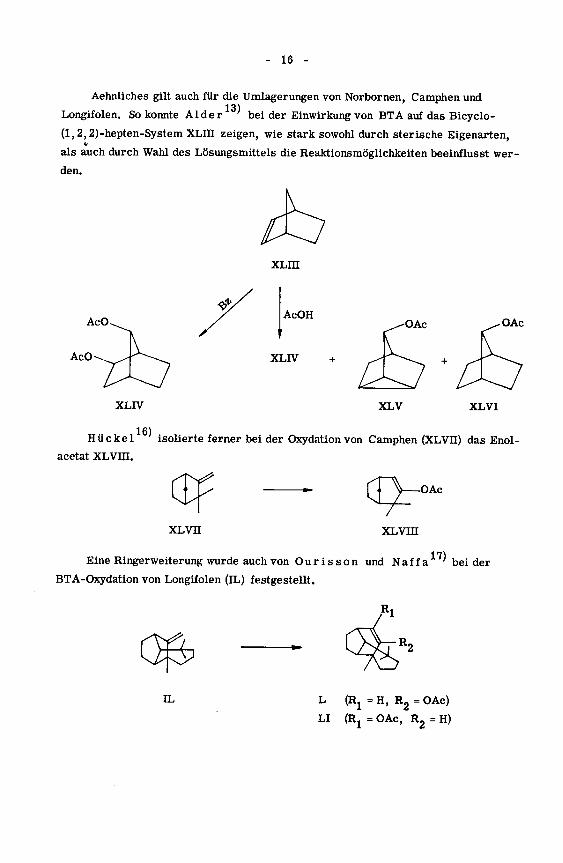
Aehnliches gilt auch für die Umlagerungen von Norbornen, Camphen und
13)Longifolen. So konnte Aider bei der Einwirkung von BTA auf das Bicyclo-
(l,2,2)-hepten-System XLHI zeigen, wie stark sowohl durch sterische Eigenarten,
als auch durch Wahl des Lösungsmittels die Reaktionsmöglichkeiten beeinflusst wer¬
den.
XLHI
AcO
AcO
AcOH
"
XLIV
OAc OAc
XLIV XLV XLVI
,16),.Hü ekel isolierte ferner bei der Oxydation von Camphen (XLVU) das Enol-
acetat XLVm.
XLvn
T
xLvra
OAc
,17)Eine Ringerweiterung wurde auch von Our is son und Naff a 'beider
BTA-Oxydation von Longifolen (IL) festgestellt.
if,
IL L (Rj = H, R2 = OAc)
LI (Rx = OAc, R2 = H)
- 17 -
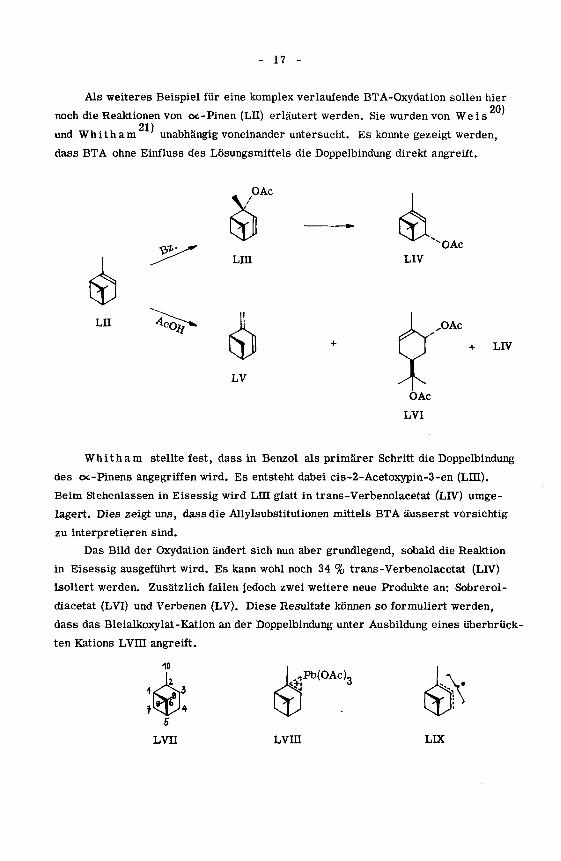
Als weiteres Beispiel für eine komplex verlaufende BTA-Oxydation sollen hier
20)noch die Reaktionen von oc-Pinen (Lu) erläutert werden. Sie wurden von Weis
21)und Whitham unabhängig voneinander untersucht. Es konnte gezeigt werden,
dass BTA ohne Einfluss des Lösungsmittels die Doppelbindung direkt angreift.
OAc
"OAc
LIU LIV
LII
LV
Whitham stellte fest, dass in Benzol als primärer Schritt die Doppelbindung
des cx-Pinens angegriffen wird. Es entsteht dabei cis-2-Acetoxypin-3-en (LUI).
Beim Stehenlassen in Eisessig wird LUI glatt in trans-Verbenolacetat (LIV) umge¬
lagert. Dies zeigt uns, dass die Allylsubstitutionen mittels BTA äusserst vorsichtig
zu interpretieren sind.
Das Bild der Oxydation ändert sich nun aber grundlegend, sobald die Reaktion
in Eisessig ausgeführt wird. Es kann wohl noch 34 % trans-Verbenolacetat (LIV)
isoliert werden. Zusätzlich fallen jedoch zwei weitere neue Produkte an: Sobrerol-
diacetat (LVI) und Verbenen (LV). Diese Resultate können so formuliert werden,
dass das Bleialkoxylat-Kation an der Doppelbindung unter Ausbildung eines überbrück¬
ten Kations LVHI angreift.
Pb(OAc),
Lvm LDC
- 18 -
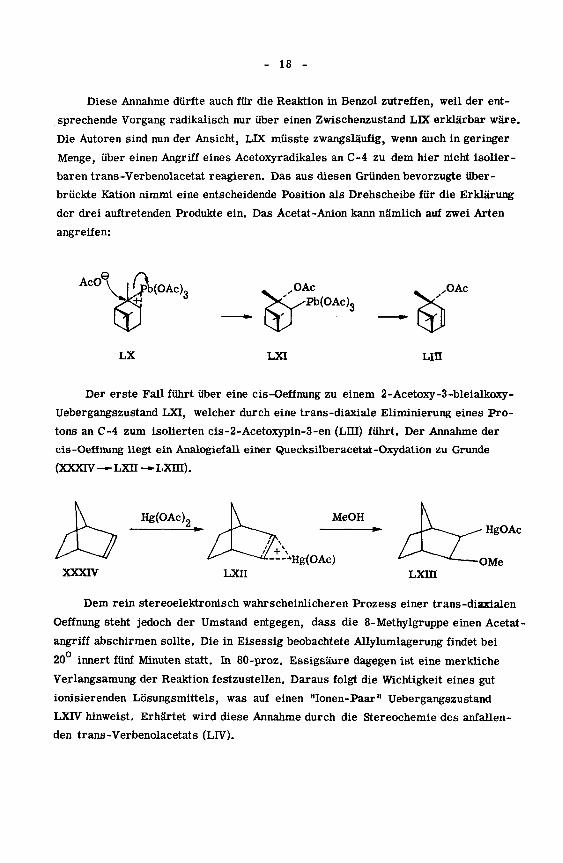
Diese Annahme dürfte auch für die Reaktion in Benzol zutreffen, weil der ent¬
sprechende Vorgang radikalisch nur über einen Zwischenzustand LDC erklärbar wäre.
Die Autoren sind nun der Ansicht, LDC müsste zwangsläufig, wenn auch in geringer
Menge, über einen Angriff eines Acetoxyradikales an C-4 zu dem hier nicht isolier¬
baren trans-Verbenolacetat reagieren. Das aus diesen Gründen bevorzugte über¬
brückte Kation nimmt eine entscheidende Position als Drehscheibe für die Erklärung
der drei auftretenden Produkte ein. Das Acetat-Anion kann nämlich auf zwei Arten
angreifen:
AcO W?b(OAc)3 . ,OAc „OAc
LX LXI Lin
Der erste Fall führt über eine cis-Oeffnung zu einem 2-Acetoxy-3-bleialkoxy-
Uebergangszustand LXI, welcher durch eine trans-diaxiale Eliminierung eines Pro¬
tons an C-4 zum isolierten cis-2-Acetoxypin-3-en (LUI) führt. Der Annahme der
cis-Oeffnung liegt ein Analogiefall einer Quecksilberacetat-Oxydation zu Grunde
(xxxiv—Lxn—Lxm).
Hg(OAc)2 MeOH
''
—^Hg(OAc)XXXIV LXII
— ^A-^^-HgOAc
Lxin
-OMe
Dem rein stereoelektronisch wahrscheinlicheren Prozess einer trans -diaxialen
Oeffnung steht jedoch der Umstand entgegen, dass die 8-Methylgruppe einen Acetat-
angriff abschirmen sollte. Die in Eisessig beobachtete Allylumlagerung findet bei
20 innert fünf Minuten statt. In 80-proz. Essigsäure dagegen ist eine merkliche
Verlangsamung der Reaktion festzustellen. Daraus folgt die Wichtigkeit eines gut
ionisierenden Lösungsmittels, was auf einen "Ionen-Paar" Uebergangszustand
LiXTV hinweist. Erhärtet wird diese Annahme durch die Stereochemie des anfallen¬
den trans-Verbenolacetats (LIV).
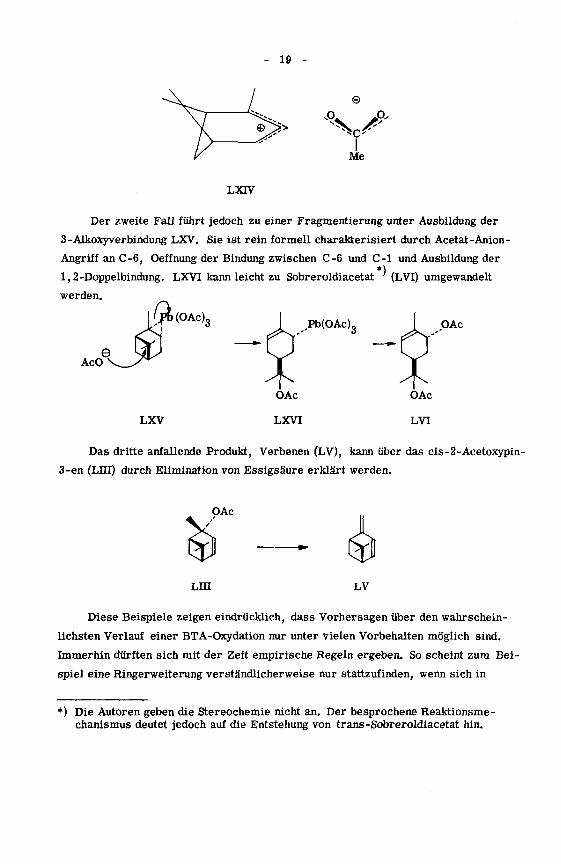
19 -
e
IMe
LXIV
Der zweite Fall führt jedoch zu einer Fragmentierung unter Ausbildung der
3-Alkoxyverbindung LXV. Sie ist rein formell charakterisiert durch Acetat-Anion-
Angriff an C-6, Oeffnung der Bindung zwischen C-6 und C-1 und Ausbildung der
1,2-Doppelbindung. LXVI kann leicht zu Sobreroldiacetat (LVI) umgewandelt
werden.
|$(OAc)3 | pb(OAc)3 i PAc
eAcO
LXV
Das dritte anfallende Produkt, Verbenen (LV), kann über das cis-2-Acetoxypin-
3-en (Lin) durch Elimination von Essigsäure erklärt werden.
OAc
LIU LV
Diese Beispiele zeigen eindrücklich, dass Vorhersagen über den wahrschein¬
lichsten Verlauf einer BTA-Oxydation nur unter vielen Vorbehalten möglich sind.
Immerhin dürften sich mit der Zeit empirische Regeln ergeben. So scheint zum Bei¬
spiel eine Ringerweiterung verständlicherweise nur stattzufinden, wenn sich in
*) Die Autoren geben die Stereochemie nicht an. Der besprochene Reaktionsme¬
chanismus deutet jedoch auf die Entstehung von trans-Sobreroldiacetat hin.

- 20 -
oU -Stellung einer exocyclischen Doppelbindung ein quaternäres C-Atom befindet
(vgl. Camphen (XLVII) und Longifolen (IL) ).
Zusammenfassend kann gesagt werden, dass das Lösungsmittel mit Sicherheit
die Vielfalt der entstehenden Produkte einer Olefin-BTA-Oxydation beeinflusst.
Der ursprünglich zu Grunde liegende Mechanismus des BTA-Angriffs an der Dop¬
pelbindung jedoch scheint kaum wesentlich davon abhängig zu sein.
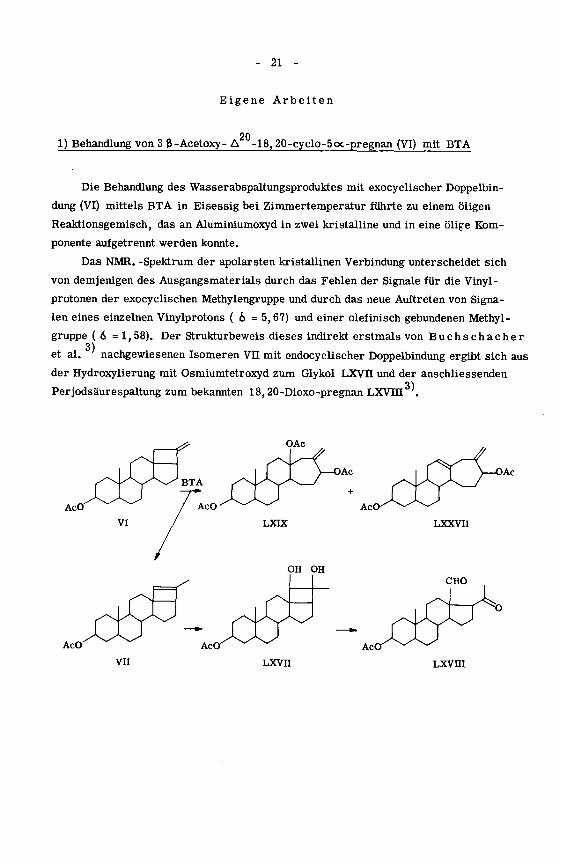
21
Eigene Arbeiten
201) Behandlung von 3 ß-Acetoxy- A -18,20-cyelo-5cx-pregnan (VI) mit BTA
Die Behandlung des Wasserabspaltungsproduktes mit exocyclischer Doppelbin¬
dung (VI) mittels BTA in Eisessig bei Zimmertemperatur führte zu einem öligen
Reaktionsgemisch, das an Aluminiumoxyd in zwei kristalline und in eine ölige Kom¬
ponente aufgetrennt werden konnte.
Das NMR. -Spektrum der apolarsten kristallinen Verbindung unterscheidet sich
von demjenigen des Ausgangsmaterials durch das Fehlen der Signale für die Vinyl -
protonen der exocyclischen Methylengruppe und durch das neue Auftreten von Signa¬
len eines einzelnen Vinylprotons ( o = 5,67) und einer olefinisch gebundenen Methyl¬
gruppe ( 6 = 1,58). Der Strukturbeweis dieses indirekt erstmals von Buchschacher
3)et al. nachgewiesenen Isomeren Vu mit endocyclischer Doppelbindung ergibt sich aus
der Hydroxylierung mit Osmiumtetroxyd zum Glykol LXVII und der anschliessenden3)
Perjodsäurespaltung zum bekannten 18,20-Dioxo-pregnan LXVHI '.
AcO'
AcO
vn LXVII Lxvm

- 22 -
Die zweite, im Folgenden als Produkt A bezeichnete Verbindung ist tetra-
cyclisch und weist auf Grund ihrer IR. -, NMR. - (siehe unten) und UV. -Spektren
(keine intensive Absorption oberhalb 220 mu) die folgenden Teilstrukturen auf:
1 -^C-CHg, 2 ^CH-OAc, >C=CH-, >C=CH2.Die dritte, im folgenden als Produkt B bezeichnete Verbindung, unterscheidet
sich von Produkt A durch den Mehrgehalt von 1 Mol Essigsäure und besitzt auf
Grund der spektroskopischen Daten die Teilstrukturen: 1 ^C-CH„, 2 ^CH-OAc,
1 ^C-OAc, ^C=CH„. Produkt B konnte im Gegensatz zu den ersten beiden Ver¬
bindungen nicht kristallin, wohl aber als einheitliches Oel erhalten werden.
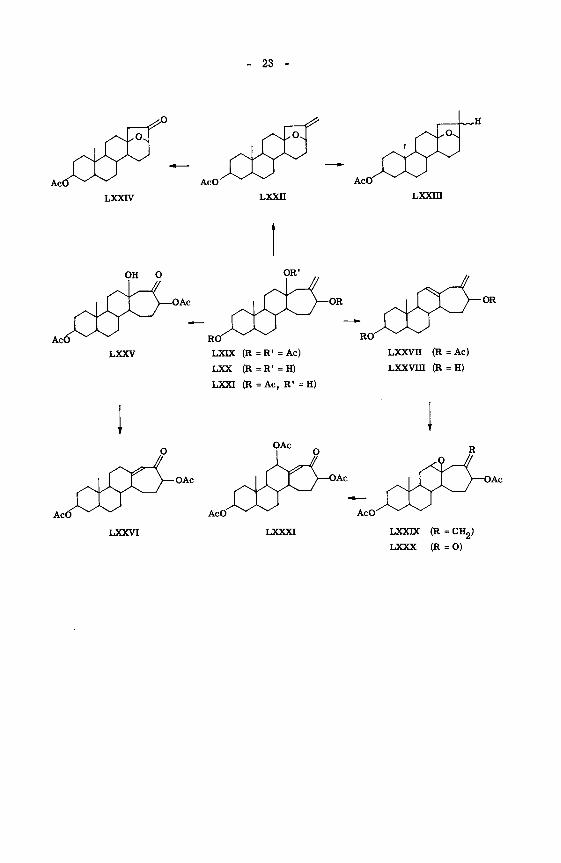
2) Verknüpfung von Produkt B (LXDC) mit Produkt A (LXXVH)
Die Reduktion des öligen Produktes B (LXDC) mittels Lithiumaluminiumhydrid
führte in guter Ausbeute zum kristallinen Triol LXX, das bei Zimmertemperatur
über Nacht in Pyridin/Acetanhydrid-l:l-Gemisch acetyliert wurde. Dass es sich
bei der nicht veresterbaren Hydroxylgruppe des resultierenden Trioldiacetats LXXI
( \> = 3540, 1732, 1245) erwartungsgemäss um die tertiär gebundene Sauer-
stoffunktion handelt, ist aus den NMR. -Spektrum der Verbindung ersichtlich (kein
Signal im Bereich von 6 = 3,0 bis 4,0 für eine >CH-OH Gruppierung).
Die Abspaltung der tertiären Hydroxylgruppe des Trioldiacetates LXXI mittels
Phosphoroxychlorid in Pyridin ergab in 35-proz. Ausbeute Produkt A (LXXVH).
Die dadurch aufgezeigte strukturelle Beziehung der beiden Verbindungen A und B
dürfte darauf hinweisen, dass sie durch denselben Fragmentierungstypus aus VI
mittels BTA entstanden sind.
- 23 -
AcO
<^
Lxxn
AcO
Lxxm
OAc
OAc
LXDC (R =R' =Ac)
LXX (R = R' = H)
LXXI (R = Ac, R' = H)
OAc
LXXVH (R = Ac)
LXXVin (R = H)
AcO AcO'
LXXXI LXXDC (R = CH2)LXXX (R = O)

- 24 -
3) Struktur des Produktes B (LXDC)
Das Trisdeacetyl-Derivat (LXX) schien von Anfang an günstig, um die relative
Lage der neu eingeführten Acetatgruppen in Produkt B (LXDC) zu bestimmen. Dass
es sich nicht um ein cx-Glykol handelt, beweist die Rückgewinnung von Ausgangs¬
material nach der Behandlung von LXX mit Perjodsäure. Lässt man dagegen einige
Tropfen konz. Schwefelsäure in Aceton auf das Triol LXX einwirken, kann eine auf¬
schlussreiche intramolekulare Aetherbildung unter Verlust eines Moleküls Wasser
beobachtet werden. Das reacetylierte Produkt LXXII, C23H„.03, ist auf Grund
des IR.-Spektrums ( v =3060, 1730, 1665, 1245, 885) weiterhin ungesättigt.
Im NMR. -Spektrum sind die zwei Vinylprotonen der exocyclischen Methylengruppe
als breites Band (6 = 4,84) sichtbar. Dass der Aethersauerstoff eine tertiäre und
eine sekundäre Haftstelle aufweist, zeigt ein undeutlich strukturiertes Signal für ein
einziges Proton bei 6 = 4,48 neben dem CH-3 Proton bei 6 = 4,64. Der chemische
Nachweis der exocyclischen Doppelbindung von LXXII liess sich wie folgt erbringen:
Die katalytische Hydrierung ergab ein gesättigtes Dihydro-Derivat LXXIQ, das auf
Grund des NMR. -Spektrums eine neue, sekundär gebundene Methylgruppe
( 6 = 1,04/d, J = 7 Hz) aufweist. Der Abbau des ungesättigten Aethers LXXH
mittels Osmiumtetroxyd und Natriummetaperjodat führte schliesslich zu einem Ke-
-1 *)
ton LXXTV, dessen IR. -Spektrum ( v = 1760 cm ) ' das Vorhandensein eines' r
max
5-Ringketones anzeigt.
*) Die für ein 5-Ringketon relativ hochliegende IR. -Absorptionsbande dürfte auf das
Vorhandensein eines Heteroatoms im Bicyclosystem von LXXTV zurückzuführen
sein.
Vergleiche:
24) 25)
Djerassi und Ourisson;
H3C\.0^/-CH3h3c-;QS:h3
1742 cm"1 1754 cm"

- 25 -
Ozonisation des ungesättigten Trioldiacetats LXXI ergab ein gesättigtes Nor-
keton LXXV und damit den chemischen Beweis, dass auch im Produkt B eine exo-
cyclische Methylengruppe vorliegt. Wasserelimination aus dem Produkt LXXV
durch Behandlung mit Natriumacetat in Eisessig führte in 35-proz. Ausbeute zu
einem ex, ß-ungesättigten Diacetoxyketon (LXXVI, Ä =246/14*400), das nach
NMR. -Spektrum lediglich ein Vinylproton aufweist (6=5,79/s). Vorgängige Ver¬
suche, diese Elimination mittels Phosphoroxychlorid oder starken organischen Ba¬
sen (Natrium-methylat, Kalium-tert. -butylat) zu erzwingen, waren erfolglos ge¬
blieben. Für die Festlegung der Position der sekundären Acetatgruppe, die bei der
Oxydation mit BTA neu eingeführt worden ist, in der ex-Stellung zur exocyclischen
Doppelbindung von LXCC (C-17) bzw. zur Carbonylgruppe von LXXV können zwei
Argumente angeführt werden: Einmal die Reaktionsfolge LXXI — LXXH - LXXTV
und schliesslich die signifikante Verschiebung nach tiefer Feldstärke des NMR. -
Signals von jenem Proton (vergleiche LXDC: 6 = 5,50 ; LXXI: 6 = 5,36), das am
gleichen C-Atom haftet wie die zur Diskussion stehende Acetatgruppe. In diesem Zu¬
sammenhang sind die vergeblichen Versuche bemerkenswert, in welchen die
<x-Acetoxygruppe von LXXV einer reduktiven Entfernung mittels Zink-Eisessig,
-Acetanhydrid oder -Aethanol widerstand.
Wenn man nun von der plausiblen Annahme ausgeht, dass der Ring C des Produk¬
tes B noch unverändert 6-gliedrig ist und gleichzeitig den induktiven Effekt einer
oc-Acetoxygruppe auf die Carbonylfrequenz eines Keton-Carbonyls (Verschiebung
nach höherer Wellenlänge) berücksichtigt, ist die Bande des ungesättigten Ketons
LXXVI bei 1690 cm" zusammen mit der Reaktionsfolge LXXI -- LXXH — LXXIV
für das Vorhandensein eines 7-gliedrigen Ringes D im Produkt B beweisend.
4) Struktur des Produktes A (LXXVII)
Produkt A (LXXVII) unterscheidet sich von Produkt B (LXIX) im Minderge-
halt einer Acetoxygruppe, besitzt jedoch zusätzlich eine Doppelbindung. Da das
NMR. -Spektrum auf eine trisubstituierte Doppelbindung hinweist (ein Vinylproton
bei 6 = 5,42/breites Signal), kommen für diese nur zwei Positionen in Betracht:
12 13 13 18A ' oder A '
. Gegen eine 13,18-Lage spricht die Tatsache, dass kein
Absorptionsmaximum im UV. -Gebiet oberhalb von 220 mu festgestellt werden

- 26 -
konnte. Dass es sich tatsächlich um eine 12,13-ungesättigte Verbindung handelt,
konnte in folgender Weise gezeigt werden:
Epoxydierung des Produktes A (LXXVII) mit einem Mol Benzopersäure führte zu
einem Monoepoxyd LXXDC. Das NMR. -Spektrum dieser Verbindung zeigt bei
6 = 4, 98 ein breites Signal von zwei Protonen, das auf die Erhaltung der exocycli-
schen Doppelbindung hinweist. Damit in Uebereinstimmung ergab die Ozonisierung
ein Ketoepoxyd LXXX. Durch Chromatographie an Kieselgel wurde das Epoxyd zu
einer hydroxylhaltigen Verbindung geöffnet, die direkt acetyliert wurde. Das resul¬
tierende Produkt LXXXI stellt ein oc, ß -ungesättigtes Triacetoxyketon dar
(X = 239/11'200). Im NMR.-Spektrum können die Methylprotonen der dreiIUoX
Acetoxygruppen ( 6 = 1,98; 2,07; 2,11), die Protonen an C-3, C-12 und C-17
( 6 = 4,57/1H; 5,26/2 H) und das Vinylproton an C-18 ( 6 = 5,90/s) lokalisiert
werden.
Die Ursachen für die Bevorzugung der 12,13-Lage für die trisubstituierte
Doppelbindung von LXXVII können verschiedener Natur sein. Für eine sterische
Kontrolle spricht der Umstand, dass die Wasserabspaltung am Trioldiacetat LXX
nur zum Produkt LXXVII führte, obwohl durch die Ausbildung einer 13,18-Doppel-
bindung ein konjugiertes Diensystem entstehen würde. Andererseits dürfte der Reak¬
tionsmechanismus dieser Fragmentierung die Frage aufwerfen, ob an C-13 inter¬
mediär überhaupt ein Carboniumion ausgebildet wird. In der folgenden Diskussion
soll auf diese Probleme näher eingegangen werden.

- 27 -
Diskus sion
Die beschriebene BTA-Oxydation zeigt zwei grundlegend verschiedene Reak¬
tionen: Eine Isomerisierung (VI—Vu) und eine Fragmentierung (VI — LXDC,
LXXVH).
Die Isomerisierung der Doppelbindung von der exocyclischen Anordnung in VI
zur endocyclischen Lage von VU stellt eine Ergänzung zur Wasserabspaltung aus
dem tertiären (20S)-Alkohol IV dar, die bekanntlich zu einem 20:1-Gemisch der
20,21- und 18,20-Doppelbindungsisomeren führt. Die Verschiebung der Doppelbin¬
dung in VI in die endocyclische Lage in VII verbessert den Zugang zu 18-oxygenier-
ten Steroiden aus dem (20S)-Alkohol IV, indem nun beide Anhydroprodukte dieser
Verbindung zu- diesem Zweck eingesetzt werden können. Damit war eine Lösung
der eingangs gestellten Problemstellung in allerdings wenig befriedigender Weise
(Ausbeute des Uebergangs IV -*> VE : 23 %'. ) gefunden worden.
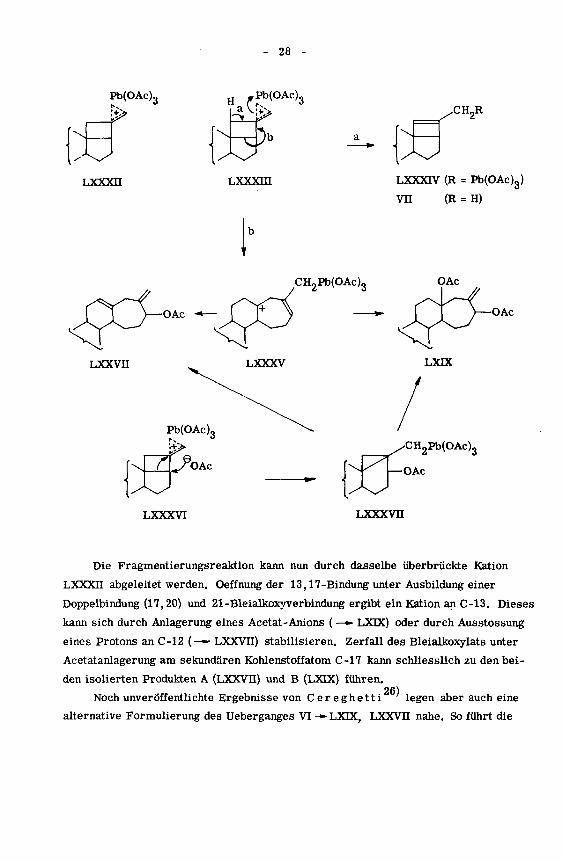
Die Frage nach dem Mechanismus der Umwandlungen, welche das Methylen-
cyclobutan-Derivat VI bei der Umsetzung mit BTA in Eisessig erfährt, kann wohl
am besten in Anlehnung an die auf Seite 18 erwähnten mechanistischen Betrachtun¬
gen bezüglich der oc-Pinen-Oxydation diskutiert werden. Die Ausbildung eines von
15)
jenen Autoren '
postulierten überbrückten Kations dürfte im Falle von VI zu der
sterischen Anordnung LXXXII führen. So erfolgt auch die Benzopersäure-Oxydation
der gleichen Verbindung unter Angriff des Reagens an der exo-Seite des Bicyclo-
[3,2,0]heptan-Systems .Elimination eines Protons an C-18 ergibt eine unge¬
sättigte 21-Bleialkoxyverbindung (LXXXIV), die nun z.B. homolytisch zerfallen
und unter Anlagerung eines WasserStoffradikals zum isolierten Isomerisierungs-
produkt Vu führen kann.
24)Gil-Av und Herling isomerisierten das unsubstituierte Methylen-
cyclobutan mittels eines Natrium/Aluminiumoxyd-Katalysators zum Methylcyclo-
buten. Bei dieser Reaktion stellte sich ein Gleichgewicht ein, das stark zu Gunsten
der endocyclischen Form verschoben war (85 % : 15 %). Diese katalytische Isomeri¬
sierung konnte jedoch nicht auf das hier vorliegende Steroidbeispiel angewandt wer¬
den. Diesbezügliche Versuche führten nur zu Ausgangsmaterial.
- 28 -
Pb(OAc),
...ô
LXXXII
H rPb(OAc)3
o»
LXXXIII
CH2R
LXXXIV (R = Pb(OAc)g)VII (R = H)
Lxxvn
CH2Pb(OAc)3
LXXXV
OAc
LXDC
OAc
Pb(OAc),
Z*/0AcCH2Pb(OAc)3
—OAc
LXXXVI LXXXVH
Die Fragmentierungsreaktion kann nun durch dasselbe überbrückte Kation
LXXXII abgeleitet werden. Oeffnung der 13,17-Bindung unter Ausbildung einer
Doppelbindung (17,20) und 21 -Bleialkoxyverbindung ergibt ein Kation an C-13. Dieses
kann sich durch Anlagerung eines Acetat -Anions ( —» LXDC) oder durch Ausstossung
eines Protons an C-12 {—— LXXVU) stabilisieren. Zerfall des Bleialkoxylats unter
Acetatanlagerung am sekundären Kohlenstoffatom C-17 kann schliesslich zu den bei¬
den isolierten Produkten A (LXXVU) und B (LXIX) führen.
Noch unveröffentlichte Ergebnisse von Cereghetti legen aber auch eine
alternative Formulierung des Ueberganges VI -*• LXDC, LXXVU nahe. So führt die
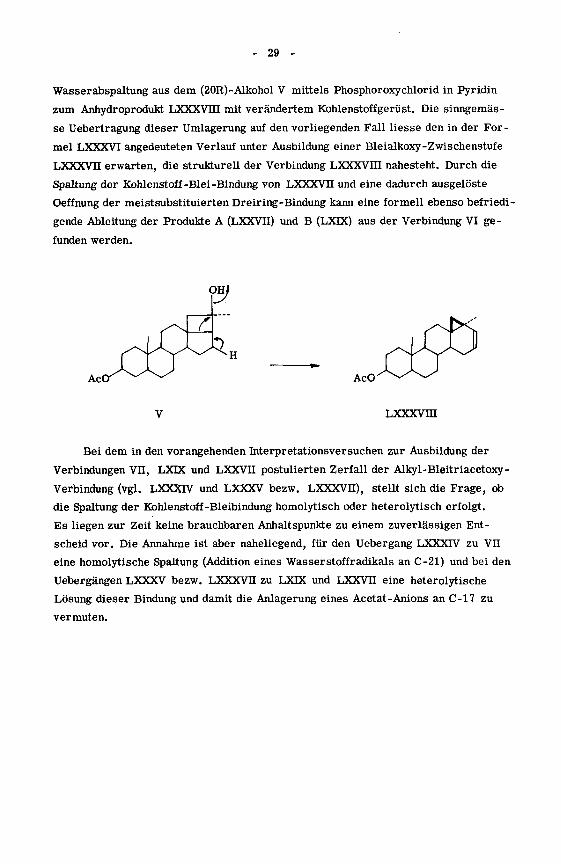
- 29 -
Wasserabspaltung aus dem (20R)-Alkohol V mittels Phosphoroxychlorid in Pyridin
zum Anhydroprodukt LXXXVHI mit verändertem Kohlenstoffgerüst. Die sinngemäs¬
se Uebertragung dieser Umlagerung auf den vorliegenden Fall Hesse den in der For¬
mel LXXXVI angedeuteten Verlauf unter Ausbildung einer Bleialkoxy-Zwischenstufe
LXXXVn erwarten, die strukturell der Verbindung LXXXVHI nahesteht. Durch die
Spaltung der Kohlenstoff-Blei-Bindung von LXXXVII und eine dadurch ausgelöste
Oeffnung der meistsubstituierten Dreiring-Bindung kann eine formell ebenso befriedi¬
gende Ableitung der Produkte A (LXXVII) und B (LXTX) aus der Verbindung VI ge¬
funden werden.
AcO
Lxxxvm
Bei dem in den vorangehenden Interpretationsversuchen zur Ausbildung der
Verbindungen Vu, LXDC und LXXVÜ postulierten Zerfall der Alkyl-Bleitriacetoxy-
Verbindung (vgl. LXXXTV und LXXXV bezw. LXXXVH), stellt sich die Frage, ob
die Spaltung der Kohlenstoff-Bleibindung homolytisch oder heterolytisch erfolgt.
Es liegen zur Zeit keine brauchbaren Anhaltspunkte zu einem zuverlässigen Ent¬
scheid vor. Die Annahme ist aber naheliegend, für den Uebergang LXXXTV zu VII
eine homolytische Spaltung (Addition eines Wasserstoffradikals an C-21) und bei den
Uebergängen LXXXV bezw. LXXXVII zu LXTX und LXXVII eine heterolytische
Lösung dieser Bindung und damit die Anlagerung eines Acetat-Anions an C-17 zu
vermuten.

- 30 -
Zur Stereochemie des Produktes B (LXDC)
Die räumliche Anordnung der neu eingeführten Acetoxysubstituenten an C-13
und C-17 im Produkt B lässt sich nicht mit Sicherheit auf Grund der mechanisti¬
schen Betrachtungen, wie sie auf Seite 28 angeführt wurden, ableiten. Wohl müsste
unter der Annahme eines synchronen Reaktionsablaufes bei der Schrittfolge
LXXXIII —LXXXV—LXDC ein 13ß, 17£-Acetoxy-Derivat (Konfigurationsumkeh-
rung an C-13) und bei der Variante LXXXVI — LXXXVTI -* LXDC, LXXVH ein
13 ß,17ß-Diacetoxy-Derivat (Konfigurationsumkehrung an C-13 und C-17) entste¬
hen. Eine solche Annahme ist aber keinesfalls bewiesen, d. h. eine thermodyna-
mischkontrollierte Einführungder Acetoxysubstituenten ist in jedem Fall ebenso gut
möglich. Um dennoch wenigstens Anhaltspunkte für die Konfiguration an C-13 des
*)Produktes B (LXDC) zu erhalten, wurde das Circulardichrogramm vom Triol-
*)diacetat-keton LXXV gemessen. Das ER. -Spektrum von LXXV zeigt in starker
Verdünnung (Tetrachlorkohlenstoff) eine Hydroxylabsorption bei 3572 cm",was auf
eine interne Wasserstoffbrücke hinweist. Die Differenz zur Wellenlänge einer freien
tertiären Hydroxylgruppe (3617 cm" ) von etwa 45 cm" ergibt nach der Formel
29) o
von Kuhn eine Wasserstoffbrücke von 2,1 A. Das Hydroxyketon LXXV weist
nun einen stark negativen Cottoneffekt von A£ = - 3,0 mit einem Maximum bei
293 nui auf. An Siebenringen wurden bis anhin nur selten Cottoneffekte vermessen.
28)Bei A-homo-Steroiden z.B. war die Interpretation der Messungen durch die
Vielfalt von möglichen Konformationen erschwert. Im vorliegenden Fall lassen je¬
doch nur zwei 13 ß-Hydroxy-Konformationen einen negativen Cottoneffekt erwarten
unter der Voraussetzung, dass eine Wasserstoffbrücke zwischen der tertiären Hydro¬
xylgruppe und dem Ketonsauerstoff vorliegt. Es ist hier aber zu betonen, dass die
Möglichkeit einer Brückenbildung mit dem Carbonylsauerstoff der Acetatgruppen
nicht ausgeschlossen werden kann und daher der Wert der nachfolgenden Diskussion
erheblich eingeschränkt wird. Bei der ersten, in Frage kommenden Konformation (a)
misst die Wasserstoffbrücke 2,5 Ä, beim zweiten Typus (b) ergibt sich eine Bindungs¬
länge von 2,1 À (Dreidingmodelle). Bei einer 13<x-Hydroxykonformation ist nur eine
Brücke von ca. 2 A möglich, wobei jedoch nach der Octantenregel ein positiver
Cottoneffekt zu erwarten wäre.
Herrn Dr. G.Snatzke, Universität Bonn, danke ich bestens für die Aufnahmedes ER. -Spektrums sowie für die Circulardichroismus-Messungen, die er mir
freundlicherweise auch interpretierte.
- 31 -
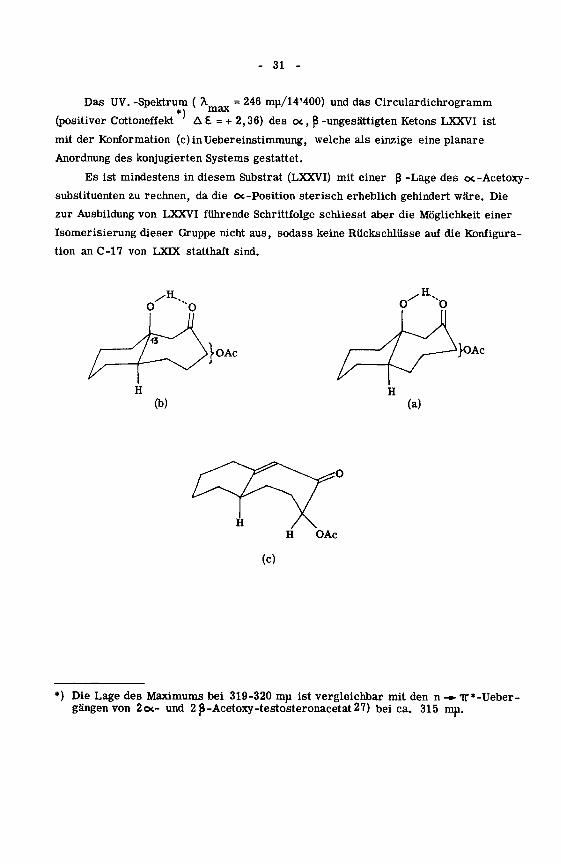
Das UV. -Spektrum ( A = 246 mu/14'400) und das Circulardichrogramm^\ max
(positiver Cottoneffekt'A £ = + 2,36) des oc, ß -ungesättigten Ketons LXXVI ist
mit der Konformation (c)inUebereinstimmung, welche als einzige eine planare
Anordnung des konjugierten Systems gestattet.
Es ist mindestens in diesem Substrat (LXXVI) mit einer ß -Lage des oc-Acetoxy-
substituenten zu rechnen, da die oc-Position sterisch erheblich gehindert wäre. Die
zur Ausbildung von LXXVI führende Schrittfolge schliesst aber die Möglichkeit einer
Isomerisierung dieser Gruppe nicht aus, sodass keine Rückschlüsse auf die Konfigura¬
tion an C-17 von LXDC statthaft sind.
OAc
H
OAc
(c)
*) Die Lage des Maximums bei 319-320 mu ist vergleichbar mit den n-»- ir*-Ueber-
gängenvon 2oc- und 2 ß-Acetoxy-testosteronacetat 27) bei ca. 315 mp.

- 32 -
EXPERIMENTELLER TEIL
Die Smp. sind nicht korrigiert und wurden in einer im Hochvakuum zuge¬
schmolzenen Kapillare im Oelbad bestimmt.
Die spezifischen Drehungen wurden in Chloroformlösung in einem Rohr von
0, 5 dm Länge gemessen.
Die Analysenpräparate wurden entweder im Hochvakuum sublimiert oder je
nach Smp. bei Zimmertemperatur oder in der Wärme ca. 48 Std. im Hochvakuum
getrocknet.
Uebliche Aufarbeitungbedeutet: Aufnahme des Reaktionsgemisches in Aether, Wa¬
schen der organischen Phase mit Wasser bis zum Neutralpunkt und Eindampfen der
über wasserfreiem Natriumsulfat getrockneten Aetherlösung im Rotationsverdampfer.
Bei der Dünnschichtchromatographie diente Kieselgel G, Merck als Träger;
der Nachweis der Substanzflecken erfolgte durch Besprühen mit 50-proz. Schwefel¬
säure.
Die Aufnahme der UV. -Spektren erfolgte in Aethanol-, die der IR. -Spektren
wenn nichts anderes vermerkt wurde, in Chloroformlösung.
Die NMR.-Spektren wurden in 5-8-proz. CDC1,-Lösung bei 60 Megahertz
aufgenommen. Die Lage der Signale ist in 6 -Werten angegeben, bezogen auf internes
Tetramethylsilan (6 =0). Die Signale werden wie folgt charakterisiert:
s (Singlett), d (Dublett), t (Triplett), q (Quadruple«;), b (breites, undeutlich struk¬
turiertes Signal).
***
Die Analysen wurden im mikroanalytischen Laboratorium der ETH (Leitung
W. Manser) ausgeführt. Frau A. von Wartburg und Herr Chr. Chylewsky
besorgten die Aufnahme der NMR.-Spektren, die Herren R. Dohner und
F. Müller die Aufnahme der IR. -Spektren. Für diese wertvolle Mitarbeit danke
ich allen Beteiligten bestens.
Herrn P. Meier,
der an unserem Laboratorium seine Laboranten-Lehrjahre
absolviert, möchte ich an dieser Stelle für seine experimentelle Mithilfe herzlich
danken.

- 33 -
Umsetzung von 3ß-Acetoxy- A
mit BTA
201 8
,20 - cy clo - 5oc-pr egnen (VI)
7,5 g exocyclisches Wasserabspaltungsprodukt (VI) wurden in 200 ml Eisessig
gelöst, mit 12 g Blei(IV)-acetat (10 % AcOH angefeuchtet) versetzt und bei Zim¬
mertemperatur 20 Stunden gerührt. Das gelbe Reaktionsgemisch wurde am Rotations¬
verdampfer bei 30 auf ca. 40 ml eingeengt, in Aether aufgenommen und neutralge¬
waschen (Wasser, KJ, Thiosulfat, Bicarbonat, Wasser). Aus der organischen Phase
resultierten ca. 10 g Oel, welche an 300 g neutralem A1„0„ (Akt. n) chromatogra-
phiert wurden.
Lösungsmittel Eluat, mg Zusammensetzung
Petrolaether-Benzol 4:1
Petrolaether-Benzol 1:1
Benzol
1843
1405
4091
Produkt Vu krist.
Produkt A (LXXVH)
Produkt B (LXDC) ölig
18Produkt VU: 3ß-Acetoxy-A -18, 20-cyclo-5<x-pregnen.
Smp. 102-103 (viermal aus Methylenchlorid/Methanol umkrist.). [ot] _ = -13
(c=0,8). IR.-Spektrum (CS0): V„
=3020, 1735, 1640, 1245 cm"1.
NMR.-Spektrum (CC14): 6 = 0, 80/s (3) CHg-19; 1, 58/t/j=l, 5 Hz (3) CHg-21;1,96/s (3) 3-OCOCH3; 2,48/b (1) CH-17; 4, 59/b (1) CH-3; 5,67/t/j=l, 5 Hz (1)
CH-18.
C23H24°2 Ber.
Gef.
C 80,65C 80,58
H
H
10,01 %9,87 %
Produkt A: Doppelt-ungesättigtes Diacetat LXXVII.
Smp. 125-126 (dreimal aus Methylenchlorid/Methanol umkrist. ).
[oc]D = - 60° (c = 1,05). IR.-Spektrum (CS2): vmax
= 1735, 1645, 1245, 910,
895 cm"1. NMR.-Spektrum (CDC13/CC14- 8 %): 6 = 0,77/s (3) CHg-19; 1,98/s (3)
3-OCOCH3; 2,04/s(3) 17-OCOCH3; 2,63/d, 3,02/d (J = je 14 Hz) (2) CH2-18;4,70/b (1) CH-3; 4,86/d/j = 2 Hz (2) CH2-21; ca. 5,42/b (1+1) CH-12 und CH-17.
C25H36°4 Ber.
Gef.
C 74,96C 75,09
H 9,06 %H 9,18 %

- 34 -
Verseifung von LXXVII:
65 mg Produkt LXXVII wurden in 10 ml 5-proz. methanolischer KOH-Lösung
über Nacht bei Zimmertemperatur stehengelassen. Die übliche Aufarbeitung ergab
51 mg kristallinen Dialkohol LXXVIII welcher dreimal aus Aceton/Petroläther um-
kristallisiert wurde.
Smp. 169-170°; [oc] = - 74° (c = 0, 75)
Et. -Spektrum (Nujol): 3430, 3280, 1645, 890 cm".
C91H,„0„ Ber. C 79,70 H 10,19 %£1 JZ Z
Gef. C 79,75 H 10,18 %
Produkt B: Ungesättigtes Triacetat LXIX.
Farbloses Oel; [oc]D = - 13,2° (c = 0,65)
IR.-Spektrum: v =1722, 1650, 1255, 960, 920 cm"1.r
max' ' ' '
NMR.-Spektrum (CDCl3/40 mg/0,45 ml): 6 = 0,77/s (3) CHj-19; 2,03/s(3+3)
3- + 13-OCOCH3; 2,07/s (3) 17-OCOCH3; 2,48/d + 3,08/d (1 + 1) CH2-18;4,68/b (1) CH-3; 4,98/b + 5,06/b (1 + 1) CH2~21; 5, 50/b (1) CH-17.
Osmium(VIII)-oxyd-Oxydation von Produkt VII
714 mg Produkt VII wurden in 10 ml Pyridin gelöst, mit 536 mg Osmium-
tetroxyd versetzt und 3 Tage bei Zimmertemperatur im Dunkeln stehengelassen.
Nach halbstündigem Einleiten von Schwefelwasserstoff wurde über Cellit filtriert,
in Aether aufgenommen, mit 2n-HCl, Bicarbonat und Wasser neutralgewaschen und
anschliessend eingedampft. Es resultierten 610 mg grünes Oel, welches an neutra¬
lem A1„0„, Aktivität HI, chromatographiert wurde. Die Benzol-Aether-l:l-Eluate
enthielten 409 mg Glykol LXVII, welches zweimal aus Methylenchlorid/Hexan um¬
kristallisiert wurde.
Smp. 199-200° ; [oc] = - 3,9° (c = 0, 5).
m.-Spektrum (Nujol) : 3540, 3320, 1730, 1715, 1250 cm"1.
C„H„fiO. Ber. C 73,36 H 9,64 %M
Gef. C 73,42 H 9,67 %

- 35 -
Glykolspaltung mit Perjodsäure
305 mg Glykol LXVII wurden in 40 ml Methanol und 10 ml Pyridin gelöst und
mit einer Lösung von 2 g Perjodsäure in 5 ml Wasser versetzt. Nach 45-minütigem
Rühren bei Zimmertemperatur wurde das Reaktionsgemisch in Aether aufgenommen
und neutralgewaschen. Es resultierten 297 mg Kristalle, die dreimal aus Methylen¬
chlorid/Hexan umkristallisiert wurden. Die Tollensprobe war positiv. Das Präparat3)
ergab bei der Mischprobe mit dem von Cereghetti etal. erhaltenen Aldehyd
keine Depression. Es liegt 3ß-Acetoxy-18,20-dioxo-5oc-pregnan (LXVHI) vor.
Smp. 116°; [oc]D = 23,8° (c = 0,48)
IR.-Spektrum (Nujol): 2660, 1725, 1705, 1255 cm"1
(CHC13): 2740, 1725 - 1705, 1260 cm"1
NMR. -Spektrum (CDClg/40 mg/0,4 ml): 6 = 0, 75/s (3) CHg-19; 2,02/s (3)
3-OCOCH3; 2,13/s (3) 21-CH3; 4,68/m (1) CH-3; 9,83/s(l) CHO-18.
CMHSd°4 Ber- C 73.76 H 9,15 %iö ö* *
Gef. C 73,33 H 9,08 %
Reduktion des Produktes B (LXIX)
1,2 g öliges Produkt LXDC wurden in 150 ml Aether gelöst, mit 1,2 g LiAlH.
versetzt und 2 Std. zum Sieden erhitzt. Die übliche Aufarbeitung ergab 1,076 g unge¬
sättigtes Triol LXX, welches zur Analyse dreimal aus Wasser/Methanol umkristalli-
siert wurde.
Smp. 192-193°, [oe] = -19° (c = 0, 5) in Nujol.
IR. -Spektrum (Nujol): 3330, 3250, 1650 cm"1.
C91H„ O Ber. C 75,40 H 10,25 %*
Gef. C 75,60 H 10,26 %
Ungesättigter Aether LXXII
150 mg Triol LXX wurden in 15 ml Aceton suspendiert, mit 8 Tropfen konzen¬
trierter Schwefelsäure versetzt und 31/2 Std. bei Zimmertemperatur gerührt. Das
gelbe Reaktionsgemisch wurde anschliessend in 50 ml ges. Bicarbonatlösung einge¬
rührt und 20 Minuten stehengelassen. Die Aufarbeitung ergab ein gelbes Oel, welches
roh in Acetanhydrid/Pyridin-(1:1)- Gemisch acetyliert wurde. Es resultierten

- 36 -
161 mg Oel, welche an 3,5 g neutralem A1„0 (Aktivität U) chromatographiert
wurden. Es konnten 98 mg Kristalle eluiert werden, die aus Methylenchlorid/Methanol
zur Analyse dreimal umkristallisiert wurden.
Smp. 176-177°; [oe] D= - 19, 5° (c = 0,3).
IR.-Spektrum (CC14): 3060, 1730, 1665, 1245, 885.
NMR. -Spektrum (CDClg/CCl4 - 5 %) : 6 = 0,85/s (3) CHg-19; 1,99/s (3)
3-OCOCH3; 2,24/b (2)CH2-18; 4,48/b (1) CH-17; 4,64/b (1) CH-3; 4, 84/b (2)
CH2-21.
C„H„.0 Ber. C 77,05 H 9,56 %z ö °
Gef. C 77,38 H 9,42 %
Hydrierung des ungesättigten Aethers LXXII
77 mg ungesättigtes Produkt LXXII wurden in 20 ml Benzol gelöst, mit 100 mg
5-proz. Pd/Kohle versetzt und 36 Stunden hydriert. Die Wasserstoffaufnahme betrug
4,2 ml (0,9 Mol). Das anfallende Rohprodukt wurde an neutralem AlqO. (Aktivität II)
chromatographiert. Es resultierten 50 mg Hydrierungsprodukt LXXHI, welches
nach zweimaliger Kristallisation aus Methylenchlorid/Methanol bei 181-182
schmolz.
[oc]D = -l,7° <c=0,S).
IR.-Spektrum (CC14): 1735, 1250, 1030 cm"1.
NMR. -Spektrum (CDCl3-8, 5 %) : 6=0,85/s (3) CHg-19; 1,04/d (J = 7 Hz) (3)
CH3-21; 1,96/s (3)3-OCOCH3; 3,89/b (1) CH-17; 4,62/b (1) CH-3.
C„H„.0„ Ber. C 76,62 H 10,07 %£ä J4 ä
Gef. C 76,53 H 10,07 %
Oxydation von LXXII mit Osmium(VIII)-oxyd und Natrium-
metaper jodat
157 mg ungesättigter Aether LXXII wurden in 5 ml Dioxan gelöst und mit 2 ml
Wasser versetzt. Unter Rühren wurden 20 mg Osmiumtetroxyd zugegeben. Nach
10 Minuten verfärbte sich das Reaktionsgemisch braun. Anschliessend wurden innert
20 Minuten 700 mg Natriummetaperjodat zugegeben und weitere 3 Stunden bei Zim¬
mertemperatur gerührt. Das Reaktionsgemisch wurde in Aether aufgenommen und
wie üblich aufgearbeitet. Es resultierten 105 mg Oel, welches nach Filtration an

- 37 -
neutralem A1„0„ (Akt. in) kristallisierte. Nach dreimaligem Umkristallisieren aus
Methylenchlorid/Petroläther schmolz das Aether-Keton LXXIV bei 178-179°.
[oc]D = -20° (c=0,3).
IR.-Spektrum (CC14): 1760, 1735, 1250 cm.
NMR. -Spektrum (CDC13/CC14 - 7, 5%): è = 0, 87/s (3) CHg-19; 2,01/s (3)
3-OCOCH3; 2,21/s (2) CH2-18; 3,98/b (1) CH-17; 4,64/b (1) CH-3.
C,„H„„0 Ber. C 73,30 H 8,95 %^ ä *
Gef. C 73,36 H 8,96 %
Ungesättigtes Trioldiacetat LXXI
309 mg Triol LXX wurden über Nacht bei Zimmertemperatur in Acetan-
hydrid/Pyridingemisch (1 : 1) acetyliert. Es resultierten 392 mg Rohprodukt,
welches an 9 g neutralem A1„0„ (Akt. H) chromatographiert wurde. 350 ml Petrol-
äther/Benzol-l:l-Gemisch eluierten 204 mg Kristalle, die zweimal aus Methylen¬
chlorid/Petroläther umkristallisiert wurden.
Smp. 156-157° ; [oc] = - 8° (c = 0,7).
IR.-Spektrum (CC14): 3540, 1732, 1635, 1245 cm"1.
NMR. -Spektrum (CDC13/CC14 - 8%): 6 = 0,81/s (3) CHg-19 ; 1,97/s (3) 3-OCOCH3;2,07/s (3) 17-OCOCH3; 2,24/s (2) CH2-18; 4, 59/b (1) CH-3; 5,00/b + 5,14/b (1 + 1)
CH2-21; 5,42/b(l) CH-17.
C„-H„fiOt. Ber. C 71,74 H 9,15 %"a J °
Gef. C 71,67 H 9,24 %
Ozonisierung des ungesättigten Trioldiacetats LXXI
185 mg Trioldiacetat wurden in 25 ml Methylenchlorid gelöst, auf -70° abge¬
kühlt und während einer halben Stunde mit einem trockenen Ozonstrom behandelt.
Nach dem Auftauen wurde auf 5 ml eingeengt, mit 5 ml Wasser versetzt und 2 Std.
auf dem Wasserbad stehengelassen. Das erkaltete Reaktionsgemisch wurde in Aether
aufgenommen und neutralgewaschen. Es resultierten 151 mg Trioldiacetat-Keton
LXXV, das dreimal aus Methylenchlorid/Hexan umkristallisiert wurde.
Smp. 168-169°; [txJD=101° (c=0,58)
IR.-Spektrum: 3580, 1740-1715, 1250 cm"1.

- 38 -
NMR. -Spektrum (CDClg-8 %): 6=0, 79/s (3) CHg-19; 2,02/s (3) 3-OCOCH3;2,14/s (3) 17-OCOCH3; 2,33/d + 2,92/d (J = 12 Hz) (1 + 1) CH2-18; 4,63/b (1)
CH-3; 5,36/b (1) CH-17.
C„.H„R0R Ber. C 68,54 H 8,63 %^ ö0 °
Gef. C 68,38 H 8,61 %
Wasserabspaltung am Produkt LXXV
600 mg (3-Hydroxy-Keton LXXV wurden in 50 ml Eisessig gelöst, mit
600 mg krist. Natriumacetat versetzt und 2 Stunden am Rückfluss gekocht. Dann wur¬
de zur Trockene eingedampft, in Aether aufgenommen und neutralgewaschen. Es re¬
sultierten 578 mg gelbes Oel, das an 25 g Kieselgel chromatographiert wurde. Es
konnten 228 mg oc, ß -ungesättigtes Keton LXXVI eluiert werden (dreimal umkristal¬
lisiert aus Methylenchlorid/Methanol).
Smp. 174-175°; [oc] ß= + 40° (c = 0, 7)
UV. -Spektrum: 246 mu (14'400)
IR.-Spektrum: 1725, 1690, 1655, 1250 cm"1.
NMR.-Spektrum (CDCl3-8%): 6 = 0,80/s (3) CHg-19; 2,03/s (3) 3-OCOCH3;2,13/s (3) 17-OCOCH3; 4,65/b (1) CH-3; 5,31/b (1) CH-17; 5,79/s (1) CH-18.
C94HS4°* Ber- c 71>61 H 8>51 %Z4 ö °
Gef. C 71,59 H 8,52 %
Wasserabspaltung am ungesättigten Trioldiacetat LXXI
192 mg Produkt LXXI wurden in 5 ml Pyridin gelöst, mit 2 g Phosphoroxy-
chlorid versetzt und 2 Stunden bei 90 stehen gelassen. Nach dem Erkalten wurde
auf Eis gegossen, in Aether aufgenommen und wie üblich neutralgewaschen. Es re¬
sultierten 132 mg rotes Oel, welches an A120„ (Aktivität n) chromatographiert
wurde. Die Benzoleluate lieferten 72 mg Kristalle mit dem Schmelzpunkt 122-124
(zweimal umkristallisiert aus Methylenchlorid/Methanol).
Nach IR. -, NMR. -Spektrum und Mischschmelzpunkt ist das vorliegende Was¬
serabspaltungsprodukt mit Produkt A (LXXVH) identisch.

- 39 -
Selektive Epoxydierung des Produktes A (LXXVII)
200 mg Produkt LXXVII wurden in 50 ml Chloroform gelöst, auf 0° abgekühlt
und mit 70 mg Benzopersäure versetzt. Das Reaktionsgemisch liess man an¬
schliessend 16 Stunden im Eisbad stehen. Bei der üblichen Aufarbeitung resultierten
195 mg Monoepoxyd LXXIX, das dreimal aus Methylenchlorid/Hexan umkristallisiert
wurde.
Smp. 173-174°; [oc] = - 15, 5° (c = 0, 8).
IR.-Spektrum: 1725, 1648, 1250, 910 cm"1.
NMR. -Spektrum (CDClg): 4 = 0,74/s (3) CHg-19; 2,00/s (3) 3-OCOCH3;2,07/s (3) 17-OCOCH3; 2,05/d + 2,67/d (J = 14 Hz) (1 + 1) CH2-18; 3,10/b (1)
CH-12; 4,65/b (1) CH-3; 5,06/b + 4,98/b (1 + 1) CH2-21; 5,37/b (1) CH-17.
C-.H-.O. Ber. C 72,08 H 8,71 %àx> â0 °
Gef. C 72,03 H 8,78 %
Ozonisation des ungesättigten Epoxyds LXXIX
496 mg Epoxyd LXXIX wurden in 40 ml Essigester gelöst, auf -70° abgekühlt
und während 20 Minuten Ozon eingeleitet. Nach dem Auftauen wurde mit 25 ml Was¬
ser versetzt und 2 Stunden am Rückfluss gekocht. Das Reaktionsgemisch wurde aus-
geäthert und neutralgewaschen. Es resultierten 456 mg kristallines Ketoepoxyd
LXXX, das zur Analyse fünfmal aus Methylenchlorid/Methanol umkristallisiert wurde.
Smp. 188-189°; [oe] = - 40° (c = 0,5)
IR.-Spektrum: 1740, 1725, 1250 cm"1.
NMR. -Spektrum (CDCl3/37 mg) : i = 0,73/s (3) CHg-19; 1,98/s (3) 3-OCOCH3;2,12/s (3) 17-OCOCH3; 3,15/b (1) CH-12; 3,15/d (1) (J = 12 Hz) CH(H)-18;
4,62/b (l)CH-3; 5,12/b (1) CH-17.
C94H-j-i0fi Ber- C 68>87 H 8,19 %^ d4 °
Gef. C 68,55 H 8,31 %
Epoxydöffnung am Produkt LXXX
420 mg Ketoepoxyd LXXX wurden an 20 g Kieselgel (unter 0,08 mm) chro-
matographiert. Die Benzol/Essigester-2:l-Eluate ergaben 203 mg kristallines Aus¬
gangsmaterial. Mittels Essigester konnten weitere 182 mg eines farblosen Oels
eluiert werden, dessen IR. -Spektrum eine charakteristische Hydroxylbande aufwies.

- 40 -
Das Rohprodukt wurde über Nacht in 30 ml Pyridin/Acetanhydrid-l:l-Gemisch ace-
tyliert. Nach Filtration an Alox IV resultierten 156 mg Kristalle, die nach zwei¬
maligem Umkristallisieren aus Methylenchlorid/Hexan bei 179-180 schmolzen.
[oc]D=+54°(c=0,5)UV. -Spektrum: X
mgx= 239 mu (11 '200).
IR.-Spektrum: 1730, 1675, 1635, 1245 cm"1.
NMR.-Spektrum (CDClg/41 mg/0, 5 ml): 4 =0,73/s (3) CHg-19; 1,98/s (3)
3-OCOCH3; 2,07/s (3) 12-OCOCH3; 2,11/s (3) 17-OCOCH3; 4, 57/b (1) CH-3;
5,26/b (1 + 1) CH-12 und CH-17; 5,90/s (1) CH-18.
C„fiH„R07 Ber. C 67,80 H 7,88 %M ö0 '
Gef. C 67,67 H 7,82 %

- 41 -
Literaturverzeichnis
1) G.Cainelli, M. Lj. Mihailovic, D.Arigoni, O.Jeger,Helv. 42, 1126 (1959); vgl. aber auch:
E.J.Corey, W.R.Hertler, J. Amer.chem.Soc. 80, 2903 (1958), (U, 5209
F.Greuter, J. Kalvoda, O.Jeger, Proc. ehem. Soc. 1958, 349 (1959)P. Buchschacher, J. Kalvoda, D.Arigoni, O.Jeger, J.Amer.
ehem.Soc. 80, 2905 (1958).
2)H.Wehrli, M. Cereghetti, K.Schaffner, O.Jeger, Helv. 43,367 (1960).
3) P. Buchschacher, M. Cereghetti, H.Wehrli, K.Schaffner,O.Jeger, Helv. 42, 2122 (1959).M. Cereghetti, H.Wehrli, K.Schaffner, O.Jeger, Helv. 43,354 (1960); vgl. aber auch:
N.C.Yang, D.D. H.Yang, J. Amer. ehem. Soc. 80, 2913 (1958).M.Barnard, N.C.Yang, Proc.ehem.Soc. 1958,~~302.
4) W.Dimroth, R.Schweizer, Chem. Ber. 56, 1375 (1923).
5)R.Criegee, P.Dimroth, K.Noll, R.Simon, C.Weis, Chem.
Ber. 90, 1070 (1957).
6) M. Levas, Ann.Chim. 127, 697 (1952).
7) N.Elming, N. Clauson-Kaas, Acta chem.Scand. 6, 535(1952).
8) C.D.Hurd, O.E.Edwards, J.org.Chem. 19, 1319(1954).
9) W.S.Johnson, B . Gastambide, R.Pappo, J.Amer. chem. Soc. 79,
1991 (1957).
10) R.Criegee, Liebigs Ann.Chem. 481, 263 (1930).
11) M. Mousseron, R. Jacquier, Bull. Soc. chim. France 1952, 467.
12) A.Kergomard, Ann.Chim. 8, 153 (1953).
13) K. Aider, F.H.Flock, H.Wirtz, Chem. Ber. 9J_, 609(1958).
14) S.Winstein, M.Shatavsky, C.Norton, R. B. Woodward,J.Amer.chem.Soc. 77, 4183 (1956).
15) B. Cross, C.H.Whitham, J. chem. Soc. 1960, 3895.
16) W.Hückel, Chem.Ber. 80, 41 (1947).
17) P.Naffa, G.Ourisson, Bull. Soc. chim. France 1954, 1115.
18) W.A.Mo s her, J. org. Chem. 26, 1044 (1961).
19) J.Alkonyi, Chem.Ber. 96, 1873 (1963).
20) C.Weis, Diss. Karlsruhe 1953.
21) C.H.Whitham, J.chem.Soc. 1961, 2232.
22) L.Schmerling, J.P.Luvisi, R.W.Welch, J. Amer. chem. Soc. 78,2819 (1956).

- 42 -
23) E.Gil-Av, J. Her ling, Tetr.Let. 1961, 27.
24) CDjerassi, E.Wilfrid, L.Vises, A.J.Lemin, «Lorg.Chem. 18
1449 (1953).
25) G.Ourisson, C.Sandris, Bull.Soc.chim.France, 1956, 958.
26) M. Cereghetti, Diss. ETH, 1962.
27) F. Sondheimer, S.Kaufmann, J.Romo, H.Martinez, J.Amer.
chem. Soc. 75, 4712 (1953).
28) G.Snatzke, B.Zeeh, E. Müller, Tetr.Let. 1963, 1425.
29) L.P.Kuhn, J.Amer.chem.Soc. 74, 2492(1952).

- 43 -
II. Zur Photochemie des Testosterons
THEORETISCHER TEIL
Zur UV.-Bestrahlung von oc, ß-ungesättigten
Carbonylverbindungen
Die Untersuchungen lichtkatalysierter Umwandlungen von Naturstoffen erfuhren
in den letzten Jahren eine bedeutende Intensivierung.
Als eines der dabei erschlossenen Teilgebiete zeichnet sich die Photochemie
der gekreuzt konjugierten Dienone durch bemerkenswert vielseitige und komplexe
Folgen von lichtinduzierten Umlagerungsreaktionen aus. Diese schon sehr eingehend
untersuchten Umlagerungen erinnern in hohem Masse an ähnliche Reaktionsabläufe,
die bei Dunkelreaktionen mit kationischen Zwischenstufen allgemein bekannt sind
(z.B. 1,2-Alkylwanderungen nach Wagner-Meerwein, Dienon-Phenolumlage-
rungen).
Bei der Aufnahme der nachfolgend zu besprechenden photochemischen Unter¬
suchung des einfach oc, ß -ungesättigten Cyclohexenons Testosteron war ein ähnliches
Verhalten dieses Chromophoren unter UV. -Einwirkung noch unbekannt. In der Tat
liessen sich zu jenem Zeitpunkt im wesentlichen drei grundsätzlich verschiedene
Photoreaktionstypen unterscheiden, die an verschiedenen oc, ß- sowie oc, ß , t, 6 -
ungesättigten Carbonylverbindungen beschrieben worden sind:
- Inter- und intramolekulare Cycloadditionen,
- Verschiebung der oc, ß -Doppelbindung in die ß ,V- -Stellung,
- Reaktionen unter Teilnahme des Lösungsmittels.

- 44 -
1) Inter- und intramolekulare Cycloadditionen
Als weitaus häufigster Reaktionstypus wurde die sogenannte Cycloaddition be¬
schrieben, die bei der Mehrzahl der Fälle einen Zusammenschluss von zwei Doppel¬
bindungen zu einem Cyclobutansystem einschliessen. Die zur Auslösung einer sol¬
chen Photoreaktion erforderliche elektronische Anregung der Doppelbindung kann
prinzipiell auf zwei Arten gewährleistet werden: Einmal durch Uebertragung der An¬
regungsenergie mittels eines Sensibilisators, sowie dadurch, dass eine der beiden
Doppelbindungen Teil eines konjugierten Systems (z.B. ot, ß-ungesättigte Carbonyl-
verbindung) ist, welches in dem in der Praxis verwendeten UV. -Bereich über
200 mu absorbiert. Beim zweiten Doppelbindungspartner kann es sich um eine zweite
derartige Gruppe oder auch um eine isolierte Doppelbindung handeln - eine Tatsache,
die zeigt, dass nur eine der Doppelbindungen elektronisch angeregt werden muss und
die zweite Doppelbindung lediglich als "Akzeptor" der angeregten T-Elektronen des
5b)Partners fungiert . Beteiligen sich zwei ungesättigte Moleküle am beschriebenen
Vorgang, so werden Addukte bezw. Dimerisierungsprodukte erhalten, während eine
günstige stereochemische Anordnung von zwei Doppelbindungssystemen im gleichen
Molekül zu intramolekularen Ringschlussreaktionen führen kann.
Bereits 1867 beobachtete Fritzsche,
das Anthracen (I) unter dem Ein-
2)fluss von Sonnenlicht zu Dianthracen (HI) dimerisiert. Schönberg nimmt als
Ursache dieser Dimerisierung ein durch Licht verursachtes Gleichgewicht
(I = n) an. Die Diradikalform n kann nun unter Ausbildung eines Achtringes di-
merisieren.
I n m
Im Gegensatz dazu dimerisieren die oc, ß-ungesättigten Carbonylverbindungen
meist unter Ausbildung von Cyclobutan-Derivaten. Treib s' liess Sonnenlicht
auf 3,5-Dimethylcyclohexenon (IV) in verdünnter, äthanolischer Lösung einwirken.
Dabei entstand u. a. ein Photodimeres, dem die allgemeine Struktur V zukommt.
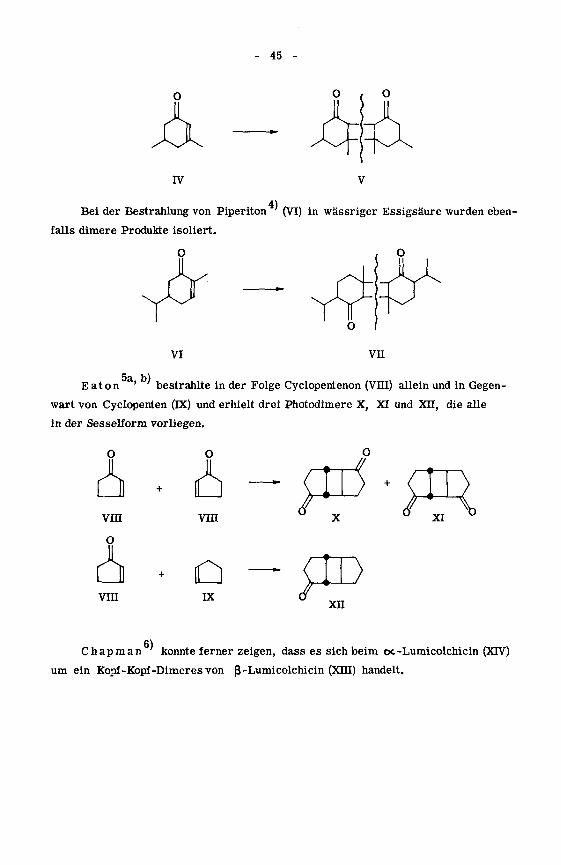
45 -
O
IV
4)Bei der Bestrahlung von Piperiton (VI) in wässriger Essigsäure wurden eben¬
falls dimere Produkte isoliert.
O
O (
VI vn
5a b)Eaton
' bestrahlte in der Folge Cyclopentenon (VHI) allein und in Gegen¬
wart von Cyclopenten (DC) und erhielt drei Photodimere X, XI und Xu, die alle
in der Sesselform vorliegen.
vin
O
vra
vra
ÛDC
/
O
4—L
XI
xn
Chapman konnte ferner zeigen, dass es sich beim oc-Lumicolchicin (XTV)
um ein Kopf-Kopf-Dimeres von ß -Lumicolchicin (XHI) handelt.
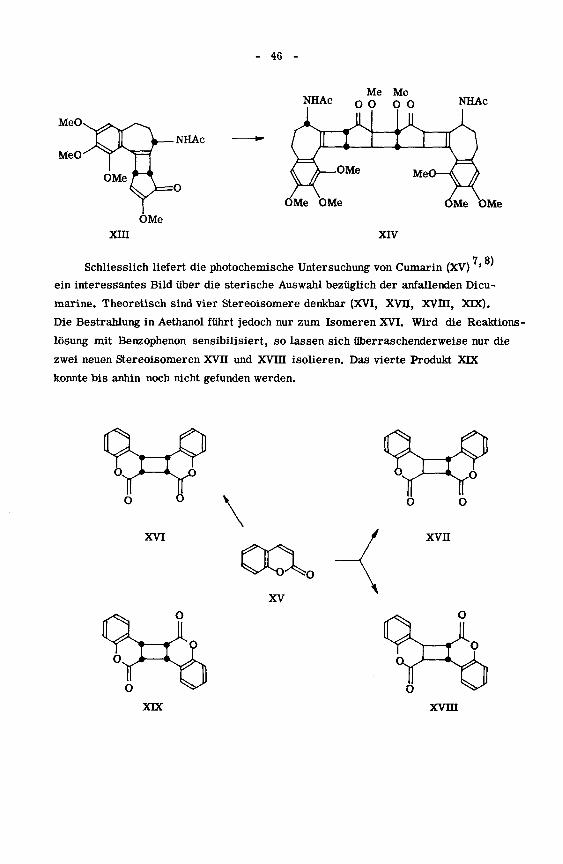
- 46 -
Me MeNHAc
0 0 0 0 NHAc
NHAc
OMe OMe
OMe
xra XIV
OMe
,7,8)
Schliesslich liefert die photochemische Untersuchung von Cumarin (XV)
ein interessantes Bild über die sterische Auswahl bezüglich der anfallenden Dicu-
marine. Theoretisch sind vier Stereoisomere denkbar (XVI, XVII, XVHI, XDC).
Die Bestrahlung in Aethanol führt jedoch nur zum Isomeren XVI. Wird die Reaktions -
lösung mit Benzophenon sensibilisiert, so lassen sich überraschenderweise nur die
zwei neuen Stereoisomeren XVII und XVin isolieren. Das vierte Produkt XDC
konnte bis anhin noch nicht gefunden werden.
O oo o
XVI
oa0XV
xvn
XDC xvm
- 47 -
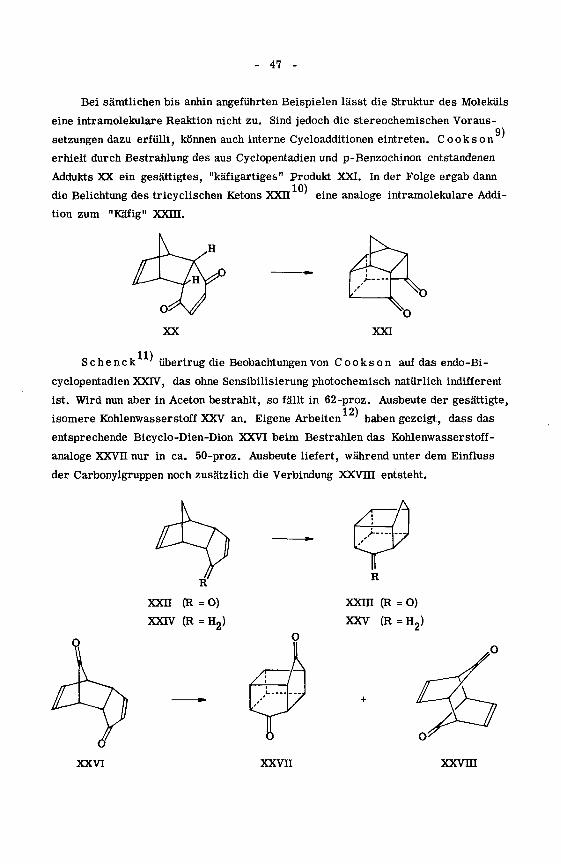
9)
Bei sämtlichen bis anhin angeführten Beispielen lässt die Struktur des Moleküls
eine intramolekulare Reaktion nicht zu. Sind jedoch die stereochemischen Voraus¬
setzungen dazu erfüllt, können auch interne Cycloadditionen eintreten. Cooks on"
erhielt durch Bestrahlung des aus Cyclopentadien und p-Benzochinon entstandenen
Addukts XX ein gesättigtes, "käfigartiges" Produkt XXI. In der Folge ergab dann
die Belichtung des tricyclischen Ketons XXII eine analoge intramolekulare Addi¬
tion zum "Käfig" XXm.
XXI
S c h e n c k übertrug die Beobachtungen von C o o k s o n auf das endo-Bi-
cyclopentadien XXIV, das ohne Sensibilisierung photochemisch natürlich indifferent
ist. Wird nun aber in Aceton bestrahlt, so fällt in 62-proz. Ausbeute der gesättigte,12)
isomere Kohlenwasserstoff XXV an. Eigene Arbeiten;haben gezeigt, dass das
entsprechende Bicyclo-Dien-Dion XXVI beim Bestrahlen das Kohlenwasserstoff¬
analoge XXVn nur in ca. 50-proz. Ausbeute liefert, während unter dem Einfluss
der Carbonylgruppen noch zusätzlich die Verbindung XXVHI entsteht.
XXH (R =0)
XXIV (R = H2)
xxm (R =0)
XXV (R = H2)
,o
XXVI XXVII xxvni
- 48 -
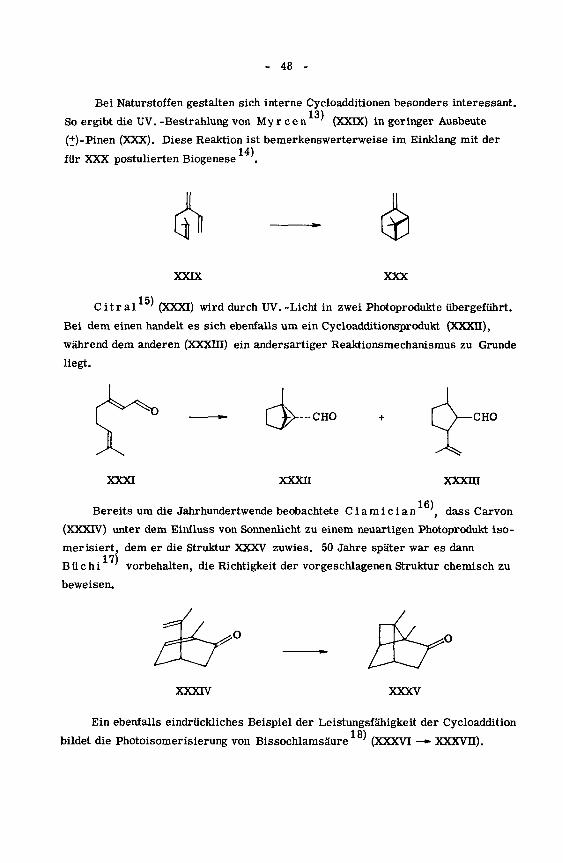
Bei Naturstoffen gestalten sich interne Cycloadditionen besonders interessant.13)
So ergibt die UV. -Bestrahlung von My r c e n' (XXIX) in geringer Ausbeute
(+)-Pinen (XXX). Diese Reaktion ist bemerkenswerterweise im Einklang mit der
14)für XXX postulierten Biogenese .
XXIX XXX
15)Citral ' (XXXI) wird durch UV. -Licht in zwei Photoprodukte übergeführt.
Bei dem einen handelt es sich ebenfalls um ein Cycloadditionsprodukt (XXXII),
während dem anderen (XXXin) ein andersartiger Reaktionsmechanismus zu Grunde
liegt.
-CHO CHO
XXXI xxxn xxxm
.16)Bereits um die Jahrhundertwende beobachtete Ciamician "', dass Carvon
(XXXTV) unter dem Einfluss von Sonnenlicht zu einem neuartigen Photoprodukt iso-
merisiert, dem er die Struktur XXXV zuwies. 50 Jahre später war es dann
17)Büchi vorbehalten, die Richtigkeit der vorgeschlagenen Struktur chemisch zu
beweisen.
-/O Éy
XXXIV XXXV
Ein ebenfalls eindrückliches Beispiel der Leistungsfähigkeit der Cycloaddition
bildet die Photoisomerisierung von Bissochlamsäure (XXXVI —» XXXVII).
- 49 -
Vv°
o^o^o
XXXVI xxxvn
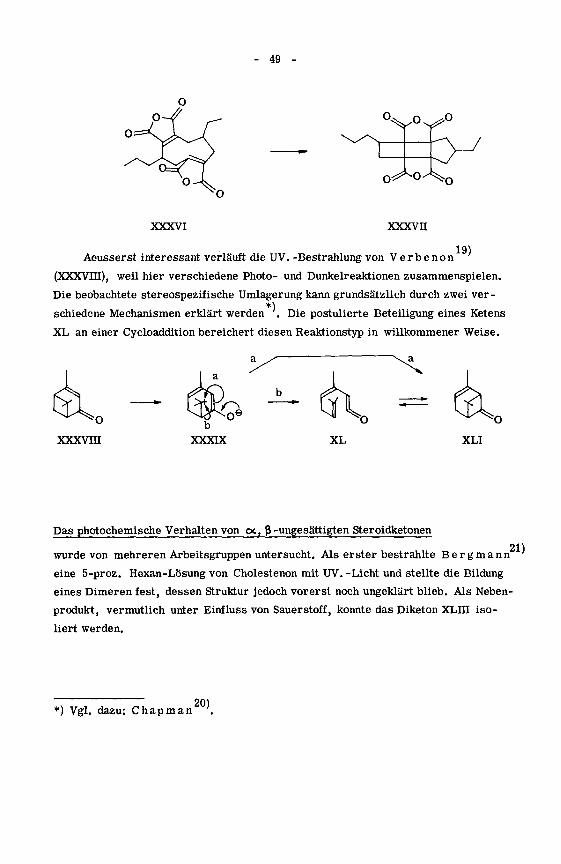
19)Aeusserst interessant verläuft die UV. -Bestrahlung von Verbenon
(XXXVm), weil hier verschiedene Photo- und Dunkelreaktionen zusammenspielen.
Die beobachtete stereospezifische Umlagerung kann grundsätzlich durch zwei ver-
*)
schiedene Mechanismen erklärt werden '. Die postulierte Beteiligung eines Ketens
XL an einer Cycloaddition bereichert diesen Reaktionstyp in willkommener Weise.
XLI
Das photochemische Verhalten von <x, ß-ungesättigten Steroidketonen
wurde von mehreren Arbeitsgruppen untersucht. Als erster bestrahlte Bergmann'
eine 5-proz. Hexan-Lösung von Cholestenon mit UV. -Licht und stellte die Bildung
eines Dimeren fest, dessen Struktur jedoch vorerst noch ungeklärt blieb. Als Neben
produkt, vermutlich unter Einfluss von Sauerstoff, konnte das Diketon XLIQ iso¬
liert werden.
21)
*) Vgl. dazu: Chapman
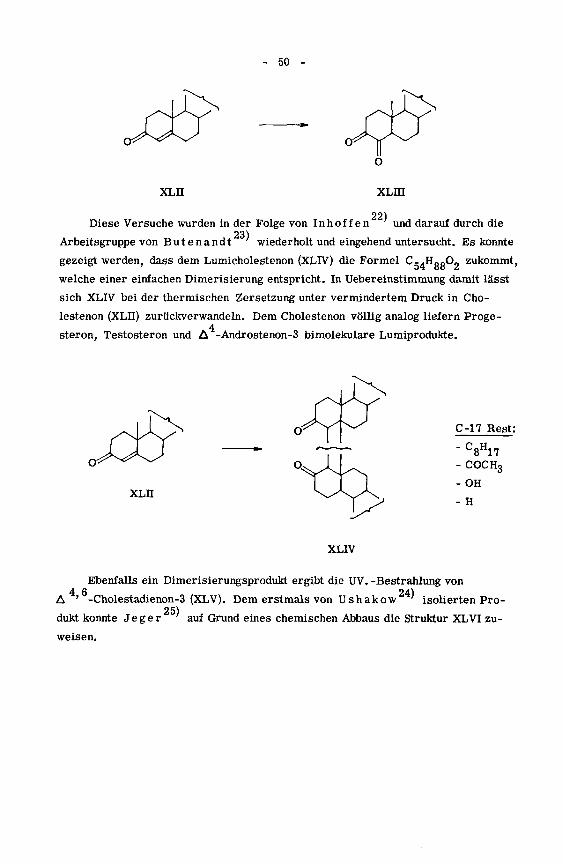
- 50
XLII xLm
22)Diese Versuche wurden in der Folge von Inhoffen und darauf durch die
23)Arbeitsgruppe von Butenandt wiederholt und eingehend untersucht. Es konnte
gezeigt werden, dass dem Lumicholestenon (XLIV) die Formel Cg4HggO„ zukommt,
welche einer einfachen Dimerisierung entspricht. In Uebereinstimmung damit lässt
sich XLIV bei der thermischen Zersetzung unter vermindertem Druck in Cho¬
lestenon (XLII) zuriickverwandeln. Dem Cholestenon völlig analog liefern Proge-4
steron, Testosteron und A -Androstenon-3 bimolekulare Lumiprodukte.
oJ^J^JXLII
C-17 Rest:
' C8H17- COCH3-OH
- H
XLIV
Ebenfalls ein Dimerisierungsprodukt ergibt die UV. -Bestrahlung von4.6
„L_,__i_J_. „ _„, . TT 24)A *'
"-Cholestadienon-3 (XLV). Dem erstmals von Ushakow
25)dukt konnte J e g e r
weisen.
isolierten Pro¬
auf Grund eines chemischen Abbaus die Struktur XLVI zu-
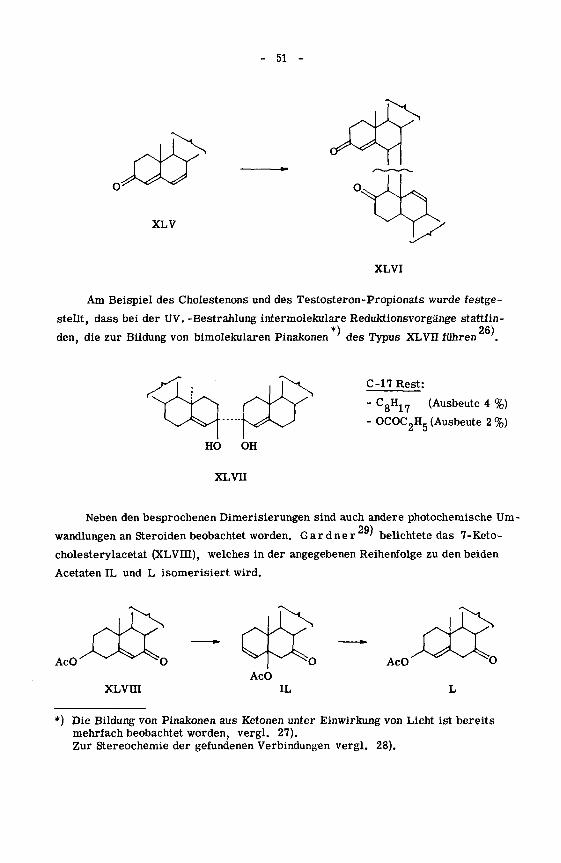
51
O
XLV
XLVI
Am Beispiel des Cholestenons und des Testosteron-Propionats wurde festge¬
stellt, dass bei der UV. -Bestrahlung intermolekulare Reduktionsvorgänge stattfin-
den, die zur Bildung von bimolekularen Pinakonen des Typus XLVu führen '.
C-17 Rest:
- CgH17 (Ausbeute 4 %)
- OCOC2H5 (Ausbeute 2 %)
Neben den besprochenen Dimerisierungen sind auch andere photochemische Um¬
wandlungen an Steroiden beobachtet worden. Gard ne r' belichtete das 7-Keto-
cholesterylacetat (XLVIII), welches in der angegebenen Reihenfolge zu den beiden
Acetaten IL und L isomerisiert wird.
AcO AcO
AcO
XLVIII IL
*) Die Bildung von Pinakonen aus Ketonen unter Einwirkung von Licht ist bereits
mehrfach beobachtet worden, vergl. 27).Zur Stereochemie der gefundenen Verbindungen vergl. 28).
- 52 -
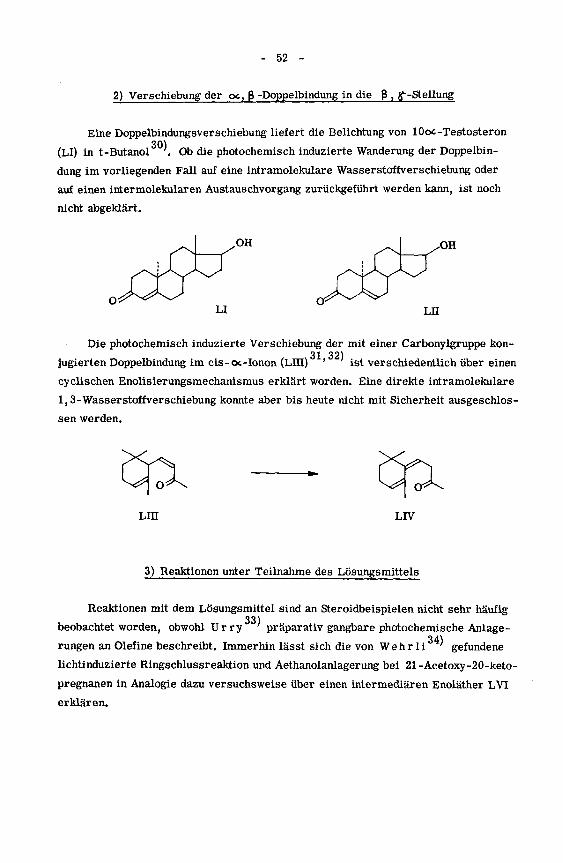
2) Verschiebung der oc, ß -Doppelbindung in die ß, ^-Stellung
Eine Doppelbindungsverschiebung liefert die Belichtung von lOoc -Testosteron
30)(LI) in t-Butanol . Ob die photochemisch induzierte Wanderung der Doppelbin¬
dung im vorliegenden Fall auf eine intramolekulare Wasserstoffverschiebung oder
auf einen intermolekularen Austauschvorgang zurückgeführt werden kann, ist noch
nicht abgeklärt.
OH OH
Die photochemisch induzierte Verschiebung der mit einer Carbonylgruppe kon-
31 32}jugierten Doppelbindung im cis-ot-Ionon (LUI) ' ist verschiedentlich über einen
cyclischen Enolisierungsmechanismus erklärt worden. Eine direkte intramolekulare
1,3-WasserStoffverschiebung konnte aber bis heute nicht mit Sicherheit ausgeschlos¬
sen werden.
O-
Lin
O'
LIV
3) Reaktionen unter Teilnahme des Lösungsmittels
Reaktionen mit dem Lösungsmittel sind an Steroidbeispielen nicht sehr häufig33)
beobachtet worden, obwohl Urry präparativ gangbare photochemische Anlage¬
rungen an Olefine beschreibt. Immerhin lässt sich die von Wehr li'
gefundene
lichtinduzierte Ringschlussreaktion und Aethanolanlagerung bei 21-Acetoxy-20-keto-
pregnanen in Analogie dazu versuchsweise über einen intermediären Enoläther LVI
erklären.
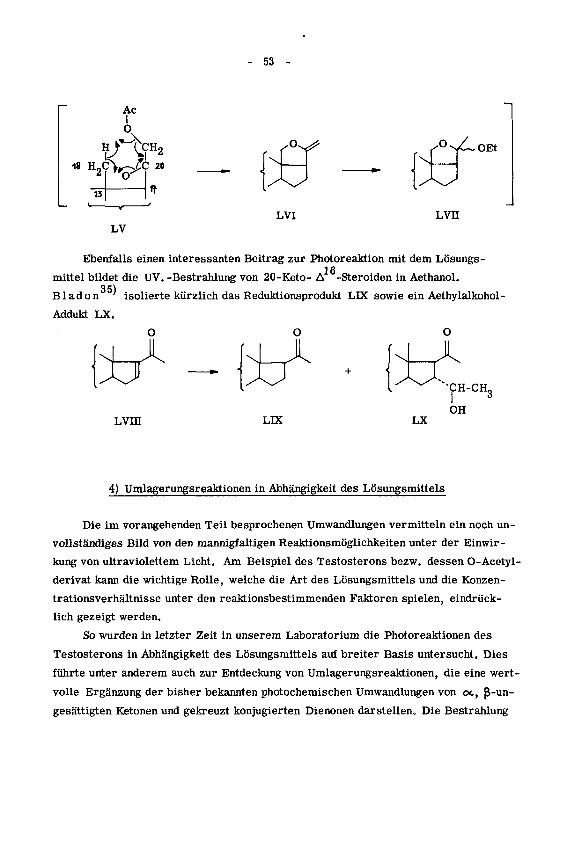
- 53 -
AcI
V %18 HgÇV-Zç 20
13
LV
-O^ 'OvA OEt
LVI Lvn
Ebenfalls einen interessanten Beitrag zur Photoreaktion mit dem Lösungs-1 R
mittel bildet die UV. -Bestrahlung von 20-Keto- A -Steroiden in Aethanol.35)
B lad on' isolierte kürzlich das Reduktionsprodukt LIX sowie ein Aethylalkohol-
Addukt LX.
O
XJX
Lvra
o
X
LLX
O
X
SCH-CH„
LX
OH
4) Umlagerungsreaktionen in Abhängigkeit des Lösungsmittels
Die im vorangehenden Teil besprochenen Umwandlungen vermitteln ein noch un¬
vollständiges Bild von den mannigfaltigen Reaktionsmöglichkeiten unter der Einwir¬
kung von ultraviolettem Licht. Am Beispiel des Testosterons bezw. dessen O-Acetyl -
dérivât kann die wichtige Rolle, welche die Art des Lösungsmittels und die Konzen-
trationsverhältnisse unter den reaktionsbestimmenden Faktoren spielen, eindrück¬
lich gezeigt werden.
So wurden in letzter Zeit in unserem Laboratorium die Photoreaktionen des
Testosterons in Abhängigkeit des Lösungsmittels auf breiter Basis untersucht. Dies
führte unter anderem auch zur Entdeckung von Umlagerungsreaktionen, die eine wert¬
volle Ergänzung der bisher bekannten photochemischen Umwandlungen von oc, ß-un-
gesättigten Ketonen und gekreuzt konjugierten Dienonen darstellen. Die Bestrahlung
- 54 -
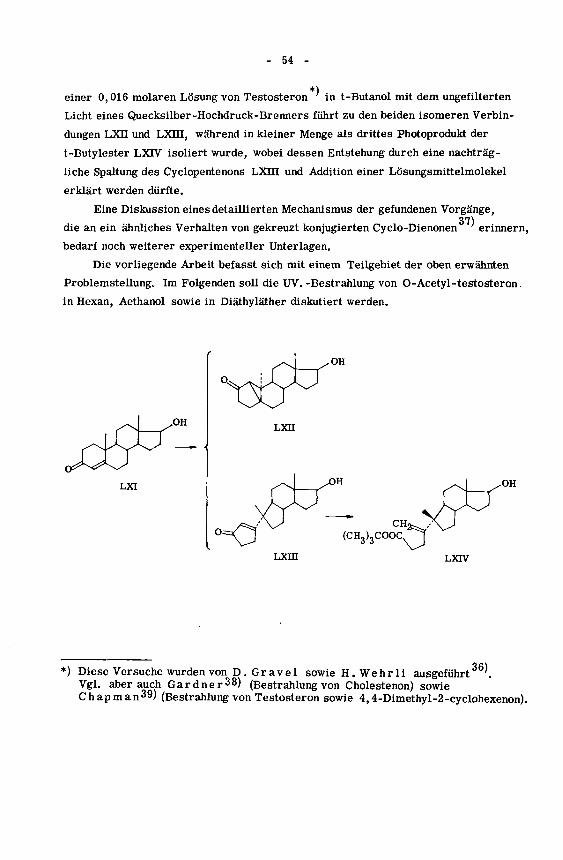
*)
einer 0,016 molaren Lösung von Testosteron'in t-Butanol mit dem ungefilterten
Licht eines Quecksilber-Hochdruck-Brenners führt zu den beiden isomeren Verbin¬
dungen LXII und LXni, während in kleiner Menge als drittes Photoprodukt der
t-Butylester LXTV isoliert wurde, wobei dessen Entstehung durch eine nachträg¬
liche Spaltung des Cyclopentenons LXHI und Addition einer Lösungsmittelmolekel
erklärt werden dürfte.
Eine Diskussion eines detaillierten Mechanismus der gefundenen Vorgänge,37)
die an ein ähnliches Verhalten von gekreuzt konjugierten Cyclo-Dienonen erinnern,
bedarf noch weiterer experimenteller Unterlagen.
Die vorliegende Arbeit befasst sich mit einem Teilgebiet der oben erwähnten
Problemstellung. Im Folgenden soll die UV. -Bestrahlung von O-Acetyl-testosteron
in Hexan, Aethanol sowie in Diäthyläther diskutiert werden.
LXI
W^OH
LXII
-o
.OH
(CH,),COOC.^
.OH
•^'y-
Lxin LXIV
*) Diese Versuche wurden von D. Gravel sowie H. Wehr li ausgeführt '.
Vgl. aber auch Gardner38) (Bestrahlung von Cholestenon) sowie
Chapman39) (Bestrahlung von Testosteron sowie 4,4-Dimethyl-2-cyclohexenon).
55 -
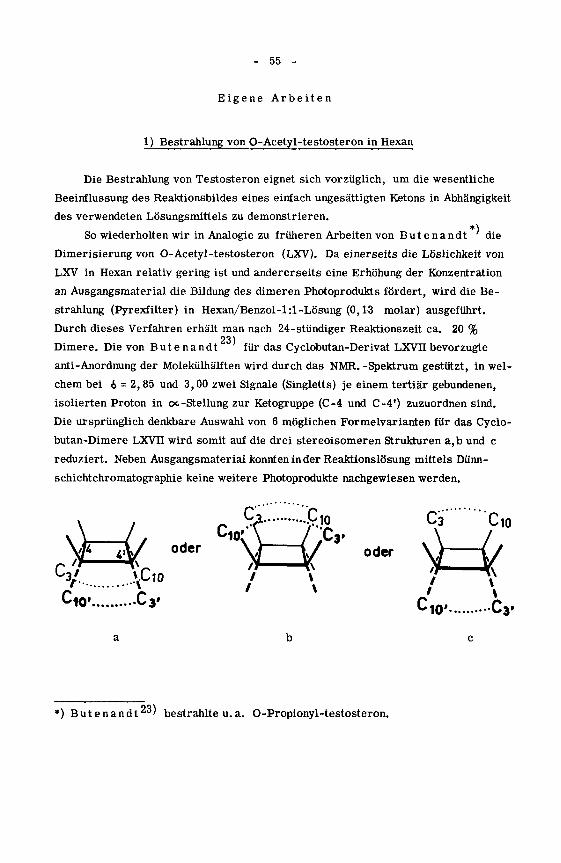
Eigene Arbeiten
1) Bestrahlung von O-Acetyl-testosteron in Hexan
Die Bestrahlung von Testosteron eignet sich vorzüglich, um die wesentliche
Beeinflussung des Reaktionsbildes eines einfach ungesättigten Ketons in Abhängigkeit
des verwendeten Lösungsmittels zu demonstrieren.
So wiederholten wir in Analogie zu früheren Arbeiten von Butenandt die
Dimerisierung von O-Acetyl-testosteron (LXV). Da einerseits die Löslichkeit von
LXV in Hexan relativ gering ist und andererseits eine Erhöhung der Konzentration
an Ausgangsmaterial die Bildung des dimeren Photoprodukts fördert, wird die Be¬
strahlung (Pyrexfilter) in Hexan/Benzol-l:l-Lösung (0,13 molar) ausgeführt.
Durch dieses Verfahren erhält man nach 24-stündiger Reaktionszeit ca. 20 %23)
Dimere. Die von Butenandt für das Cyclobutan-Derivat LXVII bevorzugte
anti-Anordnung der Molekülhälften wird durch das NMR. -Spektrum gestützt, in wel¬
chem bei 6 - 2, 85 und 3,00 zwei Signale (Singletts) je einem tertiär gebundenen,
isolierten Proton in oc-Stellung zur Ketogruppe (C-4 und C-4') zuzuordnen sind.
Die ursprünglich denkbare Auswahl von 6 möglichen Formelvarianten für das Cyclo-
butan-Dimere LXVTf wird somit auf die drei stereoisomeren Strukturen a,b und c
reduziert. Neben Ausgangsmaterial konnten in der Reaktionslösung mittels Dünn¬
schichtchromatographie keine weitere Photoprodukte nachgewiesen werden.
C3 Cio
C3/ )fu>Cio* C3' :10' C3»
*) Butenandt23' bestrahlte u. a. O-Propionyl-testosteron.
- 56 -
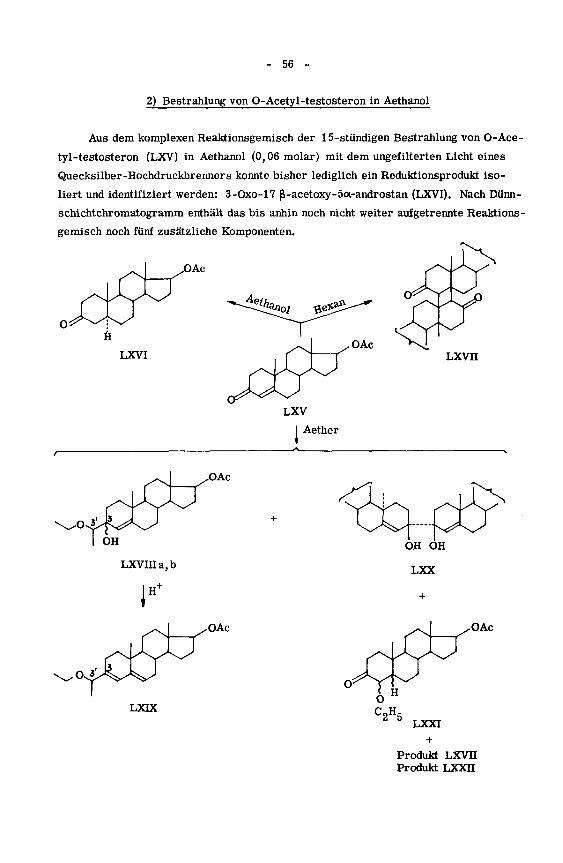
2) Bestrahlung von O-Acetyl-testosteron in Aethanol
Aus dem komplexen Reaktionsgemisch der 15-stündigen Bestrahlung von O-Ace¬
tyl-testosteron (LXV) in Aethanol (0,06 molar) mit dem ungefilterten Licht eines
Quecksilber-Hochdruckbrenners konnte bisher lediglich ein Reduktionsprodukt iso¬
liert und identifiziert werden: 3-Oxo-17 ß-acetoxy-5ot-androstan (LXVI). Nach Dünn-
schichtchromatogramm enthält das bis anhin noch nicht weiter aufgetrennte Reaktions¬
gemisch noch fünf zusätzliche Komponenten.
.OAc
LXVI
X^-O
OAc
LXVIIIa.b
1"
OH OH
LXX
OAc
LXIX
OAc
LXXI
Produkt LXVII
Produkt LXXH

- 57 -
3) Bestrahlung von O-Acetyl-testosteron in Diäthyläther
Die Bestrahlung von Testosteronacetat (LXV) in Diäthyläther mit einem Queck-*)
silber-Brenner (sowohl Hochdruck als auch Niederdruck ) ergab nach 24 Stunden
ein komplexes Reaktionsgemisch, von welchem bisher 6 Photoprodukte abgetrennt
und teilweise aufgeklärt werden konnten:
a) 2 % Cyclobutanderivat LXVII, vgl. Abschnitt 1).
b) 30 % Pinakon LXX. Sowohl das IR.-Spektrum ( v =3610, 3540, 1724,m IU3X
1670, 1255 cm) als auch das NMR. -Spektrum (6 = 0, 81/s (6) CHg-18,18' ;
l,04/s(6)CHg-19,19* ; 2,03/s (6) 17,17'-OCOCH3 ; ca. 4,6/b (2) CH-17,17';
5,33/s (2) CH-4, 4') deuten auf eine Pinakonstruktur, was durch Spaltung mittels
Perjodsäure zu O-Acetyl-testosteron chemisch bestätigt werden konnte.
c) Je 15 % von zwei Verbindungen mit der Bruttoformel C2J.H40O..Beide Verbindungen weisen praktisch ein deckungsgleiches IR. -Spektrum auf
(vgl. z.B. LXVma: v =3570, 1725, 1660, 1260 cm"1) sowie ein ebenfalls
sehr ähnliches NMR.-Spektrum. Ein 1:1-Gemisch von LXVIIIa und LXVmb in
Dioxan mit einer Spur Schwefelsäure ergibt 80 % eines doppelt ungesättigten An-
hydroprodukts LXK (NMR. -Spektrum: 6 = 0,84/s und 0,93/s je eine anguläre Me¬
thylgruppe; 1,16/t (3) (J = 7Hz) CH3-CH2 ; 1,25/d (3) (J = 6 Hz) CHg-CH ;
ca. 3,3/m (2) -CHg-O- ; 3,80/q (1) r:CH-0- ; ca. 5,4/b (1) und 5,86/s (1) je
ein Vinylproton). Das Kernresonanz- sowie das UV. -Spektrum (X = 240 mu/
22*200) stehen in bester Uebereinstimmung mit der vorgeschlagenen Struktur LXDC.
Diese Resultate erlauben die Folgerung, dass es sich bei den Verbindungen LXVIIIa
und LXVHIb um Diastereomere handelt. Die äusserst leicht zugängliche Wasser¬
abspaltung sowie die NMR. -Daten (insbesondere die Signale bei 4 = 1,18/t (3)
(J = 7 Hz) CH3-CH2- ; 1,23/d (3) (J = 6 Hz) CH3-CHC5 ca. 3,5/m (3)
-CH,-0-CHC sowie 5,14/s (1) (trisubst. DB)) stimmen mit der Struktur LXVHIa, b
überein. Für den tertiären Charakter der Hydroxylgruppe sprechen zusätzlich ein
experimenteller Befund (weder acetylier- noch oxydierbar), sowie ein spektrosko¬
pisches Resultat (im NMR. -Spektrum keine zusätzlichen Signale bei 6=3,0 bis
4,0). Die Stereochemie an den beiden neu eingeführten, asymmetrischen Kohlen¬
stoffatomen C-3 und C-3' ist noch unbekannt.
*) 90-proz. Emission bei 2537 Ä.

- 58 -
d) 5 % Photoprodukt LXXI. In dieser Verbindung scheint die ursprüngliche Car-
bonylgruppierung noch vorhanden zu sein. Das IR.-Spektrum ( v = 1725-
1700, 1260 cm" ) weist auf ein sechsgliedriges Ringketon hin. Das NMR. -
Spektrum deutet auf eine Aethoxylgruppe hin ( 4 = 1,16/t (3); ca. 3,3/m (3)).
In Anlehnung zu den beobachteten Reduktionen dürfte es sich beim gesättigten
Produkt LXXI um eine 3-Oxo-4-äthoxy-Verbindung handeln.
e) 5 % Photoprodukt LXXH. Eine befriedigende C,H-Analyse liegt nicht vor. Die
bis anhin erhaltenen Werte kommen der Bruttoformel C-JH.-O. am nächsten.
Auch das ER.-Spektrum ( v =3630, 1720, 1260 cm-1) weist eine gewisse
Aehnlichkeit mit den Verbindungen LXVHIa und LXVHIb auf. Im NMR. -Spektrum
dagegen ist die eindeutige Interpretation der auftretenden Signale nicht möglich.
Am Beispiel des O-Acetyl-testosterons ist somit die wichtige Rolle deutlich
ersichtlich, welche die Art des Lösungsmittels (Isomerisierung, Reduktion und Addi¬
tion von Lösungsmittelmolekülen) und die Könzentrationsverhältnisse (Dimerisierung)
unter den reaktionsbestimmenden Faktoren spielen. Dabei ist die Tatsache bemerkens¬
wert, dass in Diäthyläther die Isomerisierungsreaktionen durch die beobachtete Lö¬
sungsmitteladdition und Pinakoribildung vollständig unterdrückt werden.

- 59 -
EXPERIMENTELLER TEIL
(Vergleiche Seite 32)
O-Acetyl-testosteron Dimeres
5 g Testosteronacetat (LXV) wurden in 60 ml Benzol gelöst, mit weiteren
60 ml Hexan versetzt und während 20 Stunden mit einem Quecksilber-Hochdruck¬
brenner (Pyrexfilter) belichtet. Das anfallende Dimere (LXVn) begann nach 10 Stun¬
den in feinen Kristallen auszufallen. Filtration der Reaktionslösung lieferte ca.
1 g Kristalle mit Smp. 325 Zersetzung), umkristallisierbar aus Aethanol.
MG.-Bestimmung: Gef. : 651,9 (Ber. 660).
UV. -Spektrum: Kein Absorptionsmaximum, [ot] _ = + 40° (c = 0, 5)
ER.-Spektrum: v =1725, 1680, 1260 cm-1.max ' '
NMR.-Spektrum: £=0,77/s(6) CHg-18; 0,91/s (6) CH3-19; 2,04/s (6)
17-OCOCH3; 2,27/m (4) CH2-2; 2, 85/s (1) und 3,00/s (1) CH-4,4' ; 4,60/m (2)
CH-17.
C49H«n°ß Ber- c 76>32 H 9,15 %*z bu b
Gef. C 76,46 H 9,14 %
Bestrahlung von Testosteronacetat in Aethanol
900 mg Testosteronacetat (LXV) wurden in 450 ml Aethanol gelöst und 15 Stun¬
den mit einem Quecksilber-Hochdruckbrenner bestrahlt. Nach dem Eindampfen des
Reaktionsgemisches resultierten 980 mg farbloses Oel, das nach Dünnschichtchro¬
matographie neben Ausgangsmaterial noch 5 weitere Komponenten enthielt. Chroma¬
tographie an 60-fâcher Menge A120„ der Aktivität n lieferte mit Petroläther/Benzol
1:1 -Gemisch als Eluierungsmittel 172 mg Kristalle vom Smp. 154.
IR.-Spektrum: Vmax= 1735, 1705, 1255 cm"
.Die Mischprobe mit 3-Qxo-17ß -
acetoxy-androstan (LXVI) ergab keine Depression. Auch waren die IR. -Absorptions¬
spektren deckungsgleich.
C-.H^O, Ber. C 75,86 H 9,70 %Z1 äi ô
Get. C 75,63 H 9,70 %
Die Benzoleluate enthielten 102 mg Ausgangsmaterial, während die restlichen
Eluate noch nicht in einheitliche Fraktionen aufgetrennt werden konnten.

- 60 -
Bestrahlung von Testosteronacetat in Diäthyläther
1 g Testosteronacetat (LXV) wurde in 160 ml Diäthyläther gelöst und unter
Stickstoff während 24 Stunden mit einem Hanau-Niederdruckbrenner belichtet. Das
Belichtungsgemisch wurde vorsichtig eingedampft (Peroxydbildung ?) und direkt auf
einer Kieselgelsäule chromatographiert. Dadurch konnte eine gute Vortrennung in
eine polare und in eine apolare Fraktion erzielt werden. Die letztere konnte durch
mehrmaliges Chromatographieren an Kieselgel in 4 Komponenten (LXVHIa,b;
LXXI, LXXII) aufgetrennt werden, während das gleiche Verfahren bei der polaren
Fraktion zu den Photoprodukten LXVII und LXX führte.
Die physikalischen Daten der isolierten Verbindungen:
Produkt LXVma: (ca. 15 %)
Smp. 115. [o^Jtj = + 45 (c = 0,5). UV.-Spektrum: Keine Absorption.
IR. -Spektrum: v =3570, 1725, 1660, 1260 cm"1.r
max' ' '
NMR.-Spektrum: 6=0,81/s(3); 1,05/s (3); 1,18/t (3) J = 7 Hz; 1,23/d (3) J = 6Hz;
2,03/s(3); ca. 3,5/m(3); ca. 4,6/b(l); 5,14/s (1).
C„,H O Ber. C 74,21 H 9,97 %0
Gef. C 73,97 H 10,00 %
Produkt LXVHTb: (ca. 15 %)
Oelig. [ocJD= + 30 (c = 0, 5). UV.-Spektrum: Keine Absorption.
ER.-Spektrum: vmax=3580> 1725> 1660> 1260 cm"1.
NMR.-Spektrum: 6=0,82/s(3); l,07/s(3); 1,18/t (3) J = 7 Hz; l,26/d(3)
J=6Hz; 2,05/s(3); ca. 3,5/m(3); ca. 4,6/b(l); 5,30/s (1).
C H O Ber. C 74,21 H 9,97 %"° w *
Gef. C 74,16 H 9,94 %
Produkt LXXI: (ca. 5 %)
Oelig; UV. -Spektrum: Keine Absorption.
IR.-Spektrum: v =1725-1700, 1260 cm"1.K
max'
NMR.-Spektrum: Signale bei 6 = 0,82/s(3); 1,16/t (3); 1,18/s (3); 2,03/s (3);
ca. 3,3/m (3); ca. 4,6/b (1).
Produkt LXXH: (ca. 5 %)
Smp. 127.UV. -Spektrum: Keine Absorption.

- 61 -
m.-Spektrum: v =3630, 1720, 1260 cm". NMR.-Spektrum: 4 = 0,82/s(3);
ca. 1,1/m (6 ?); 2, 05/s (3); ca. 4,6/m (1); ca. 5,3/m (1).
C,H-Bestimmung: Gef. C 74,92 H 9,85 %.
Produkt LXVn: (ca. 2 %)
Smp. 325° (Zersetzung). Nach Mischschmelzpunkt und den restlichen spektrosko¬
pischen Daten mit dem aus der Hexan-Bestrahlung erhaltenen Testosteronacetat-
Dimeren identisch.
Produkt LXX: (ca. 30 %)
Smp. 230°. [oc] D= + 63°. UV. -Spektrum: Keine Absorption.
MG.-Bestimmung: Gef. 635,8 (Ber. 660).
IR.-Spektrum: V =3620-3540, 1725, 1675, 1255 cm"1.r
max' ' '
NMR.-Spektrum: 6 = 0,81/s (6) CHg-18; 1,04/s (6) CHg-19; 2,03/s (6)
17-OCOCH3; 4,59/m (2) CH-17; 5,33/s (2) CH-4.
Bei den restlichen 28 % des rohen Bestrahlungsgemisches handelt es sich um
ca. 10% Ausgangsmaterial sowie um ein z.T. verharztes, uneinheitliches Oel.
Behandlung des Pinakons LXX mit Perjodsäure
100 mg Pinakon LXX wurden in 4 ml Pyridin gelöst, mit 8 ml Methanol ver¬
dünnt und mit 500 mg Perjodsäure in 1, 5 ml Wasser versetzt. Nach einstündigem
Stehen wurde die Reaktionslösung in Aether aufgenommen und neutralgewaschen.
Daraus resultierten 92 mg eines vorerst öligenRohproduktes, das nach Filtration über
Alox II n kristallisierte. Nach Mischschmelzpunkt, IR. - und UV. -Spektrum handelt
es sich um Testosteronacetat (LXV).
Wasserabspaltung am Produkt LXVIII
214 mg 1:1-Gemisch des an C-3 epimeren Photoprodukts LXVIII wurden in
5 ml Methylenchlorid gelöst, mit 5 ml Dioxan verdünnt und mit einem Tropfen
2n-Schwefelsäure versetzt. Nach 14-stündigem Stehen bei Zimmertemperatur wurde
das Reaktionsgemisch in Aether aufgenommen und neutralgewaschen. Es resultierten
171 mg Kristalle, die nach Dünnschichtchromatographie leicht verunreinigt waren.
Nach zweimaligem Umkristallisieren aus Methylenchlorid/Hexan lag der Schmelz¬
punkt bei 150-152°.

- 62 -
UV. -Spektrum: A,
=240 mu/22'200.
m. -Spektrum: V = 1725, 1655, 1260 cm".
NMR.-Spektrum: 6=0,84/s(3); 0,93/s (3); 1,16/t J = 7 Hz (3); 1,25/d J = 6 Hz
(3); 2,07/8 0); ca. 3,3/m (2); 3,8/q(l); ca. 4,7/b(l); ca. 5,4/b (1);
5,36/s(l).
C„-H„ftO, Ber. C 77,67 H 9,91 %Z0 ä0 ä
Get. C 76,97 H 8,89 %

- 63 -
Literaturverzeichnis
1) J.Fritzsche, J.prakt.Chem. 101, 337(1867).
2) A. Schönberg, Trans. Farad. Soc. 3_2, 517 (1936).
3) W.Treibs, J.prakt.Chem. 138, 299 (1933).
4) W.Treibs, Chem.Ber. 63, 2738 (1930).
5a) P. E. Eaton, J. Amer.ehem.Soc. 84, 2344(1962).
5b) P. E. Eaton, J.Amer.ehem.Soc. 84, 2454 (1962).
6) O.L.Chapman, R.W.King, J.Amer.ehem.Soc. 85, 806 (1963).
7) R.Anet, Chem.and Ind. 1960, 897.
8) G.O.Schenck, I.v.Wilucki, C.H.Krauch, Chem.Ber. 95, 1409
(1962).~~
9) R. C. Cookson, E.Crundwell, J.Hudec, Chem. and Ind. 1958,1003.
10) R. C. Cookson, J.Hudec, R.O.Williams, Tetr. Let. 1960,Nr. 22, 29.
11) G.O.Schenck, R.Steinmetz, Chem.Ber. 96, 520(1963).
12) R.Schorta, unveröffentlichte Versuche.
13) K.J.Crowley, Proc.chem.Soc. 1962, 245.
14) L.Ruzicka, Experientia 9, 357(1953).
15) R. C. Cookson, J.Hudec, S.A.Knight, B.R
. D. Whitear,
Tetrahedron 19, 1995 (1963).
16) G.Ciamician, P.Silber, Chem.Ber. 41^, 1928(1909).
17) G.Büchi, I.M.Goldman, J.Amer.chem.Soc. 79, 4741(1957).
18) D. H.R.Barton, Pure and Appl. Chem. 6, 663 (1963).
19) J.J.Hurst, G.H.Whitham, J. chem. Soc. 1960, 2864.
20) O.L.Chapman in W.A.Noyes, G.S.Hammond, J.N.Pitts:
Advances in Photochemistry, Vol. I. London-New York: Interscience Publ. 1963.
21) E.Bergmann, Y.Hirshberg, Nature 142, 1037(1938).
22) H. H. Inhoffen, Huang-Minlon, Naturw. 27, 167(1939).
23) A.Butenandt, A. Wolff, Chem.Ber. 72, 1121 (1939); A.Butenandt,L. Karlson-Poschmann, G.Failer, U.Schiedt, E.Bickert,Liebigs Ann. Chem. 575, 123 (1952).
24) M. I. Ushakow, N. F. Kosheleva, J.gen.Chem.(U.S.S.R.) 14, 1138
(1944); Chem.Abstr. 40, 4071 (1946).
25) H.Throndsen, G.Cainelli, D.Arigoni, O.Jeger, Helv. 45,2342 (1962).

- 64 -
26) A.Butenandt, L. Poschmann, Chem.Ber. 73, 893 (1940).
27) G.Ciamician, P. Silber, Chem.Ber. 33, 2912; 36, 1517; 44, 1280.
28) P.Bladon, J. W. Cor nf orth, R.H.Jaeger, J. chem.Soc. 1958,863.
29) P.D.Gardner, H.F.Hamil, J. Amer. ehem. Soc. 83, 3531(1961).
30) H.Wehrli, R.Wenger, K.Schaffner, O.Jeger, Helv. 46, 678
(1963)."~
31) M. Mousseron-Canet, M. Mousseron, P.Legendre, Bull.Soc.
chim. France 1961, 1509.
32) P. de Mayo, J. B. Stother s, R.W.Sip, Can. J. ehem. 39, 2135
(1961).—
33) W. H.Urry, F.W.Stacey, O.O
.Juveland
,C
. H. Me . Donnall,J. Amer. chem.Soc. 75, 250(1953); W.H.Urry, F.W.Stacey,E.S.Huyser, ibid. 76, 450(1954).
34) H.Wehrli, M. C er eghetti, K.Schaffner, O.Jeger, Helv. 43,367 (1960).
~~
35) I.A.Williams, P.Bladon, Tetr.Let. 1964, 257.
36) B.Nann, D.Gravel, R.Schorta, H.Wehrli, K.Schaffner,O.Jeger, Helv. 46, 2473 (1963).
37)H.Dutler, C.Ganter, H.Ryf, E .C
.Ut zinger , K.Weinberg,
K.Schaffner, D.Arigoni, O.Jeger, Helv. 45, 2346(1962);C.Ganter, E . C. Ut zinger, K.Schaffner, D.Arigoni, O.Jeger,Helv. 45, 2403 (1962).
38) W.W.Kwie, B.A.Shoulders, P.D.Gardner, J.Amer.chem.Soc.
84, 2268 (1962).
39) O. L. Chapman, T.A.Rettig, A. A. Griswold, A.I.Dutton,P.Fitton, Tetr.Let. 1963, 2049.

- 65 -
ZUSAMMENFASSUNG
I. Teil
20Die Umsetzung von 3 |i-Acetoxy-18,20-cyclo- A -5(x-pregnen mit Blei(IV)-
acetat führte zu zwei grundsätzlich verschiedenen Reaktionen:
- Isomerisierung der exocyclischen Doppelbindung in die endocyclische Lage.
- Fragmentierung der 13,17-Bindung unter Ausbildung von D-bishomo-Steroiden.
II. Teil
Als Teil eines grösseren Programms photochemischer Untersuchungen von
alicyclischen <x, ß -ungesättigten Ketonen wurde O-Acetyl-testosteron in Hexan,
Aethanol und Diäthyläther bestrahlt. Die Natur des Lösungsmittels spielt bei den
zu beobachtenden Photoreaktionen eine ausschlaggebende Rolle. Dimerisierung,
Pinakonbildung, Reaktion zwischen Steroid- und Lösungsmittelmolekül können teil¬
weise oder ganz an die Stelle von stereospezifischen, molekularen Umlagerungen
treten.

Lebenslauf
Am 22. Juni 1936 wurde ich in Chur geboren. Nach dem Besuch der Primar¬
schule trat ich 1948 ins Gymnasium der Bündner-Kantonsschule ein, wo ich 1955
die Maturitätsprüfung (Typus A) ablegte. Im Herbst 1956 begann ich mein Studium
an der Abteilung für Naturwissenschaften der Eidgenössischen Technischen Hoch¬
schule in Zürich und bestand im Frühling 1961 die Diplomprüfung. Seither arbeitete
ich am organisch-chemischen Laboratorium der Eidgenössischen Technischen Hoch¬
schule unter der Leitung von Herrn Prof. Dr. O. Jeger an der vorliegenden Pro¬
motionsarbeit.
Zürich, im Februar 1964 Roman Schorta





![p-adische Galoisdarstellungen und ’; -Moduln · Ziel der Arbeit ist es, onFtaines teilweise skizzenhaften Beweis ([Fon90, Abschnitte A1 und A3]) genau auszuführen, dabei einige](https://static.fdocument.org/doc/165x107/5e147d02a6eb11536a3de613/p-adische-galoisdarstellungen-und-a-moduln-ziel-der-arbeit-ist-es-onftaines.jpg)












