







γλώσσες
Σελίδες
Νομικός


Orden de corto alcance en aleaciones binariasVidrios y líquidosEstructuras supramoleculares
Cristales líquidos
Polímeros

Orden de corto alcance en aleaciones binarias
A
Bb
A
Aa
C
Cró
C
Cr
−−=
−−=
11ηη
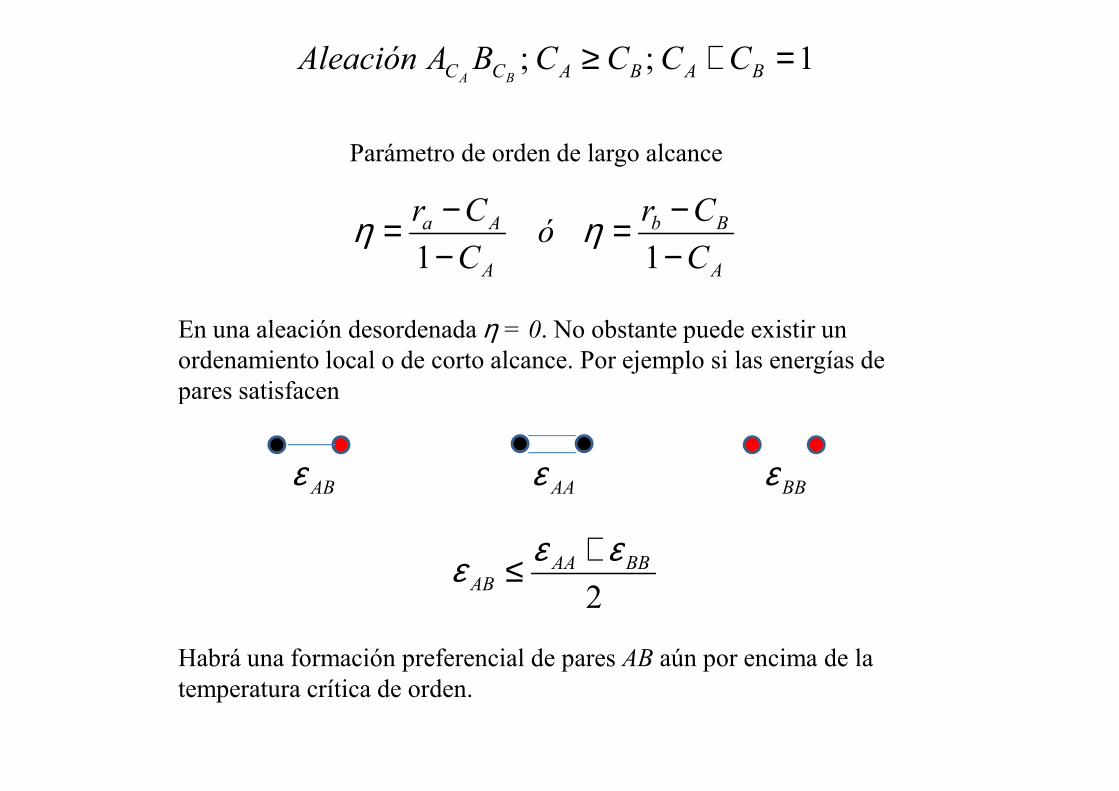
1;; =+≥ BABACC CCCCBAAleaciónBA
Parámetro de orden de largo alcance
En una aleación desordenada η = 0. No obstante puede existir un ordenamiento local o de corto alcance. Por ejemplo si las energías de pares satisfacen pares satisfacen
ABε BBεAAε
2BBAA
AB
εεε +≤
Habrá una formación preferencial de pares AB aún por encima de la temperatura crítica de orden.
AABAA z��� =+2
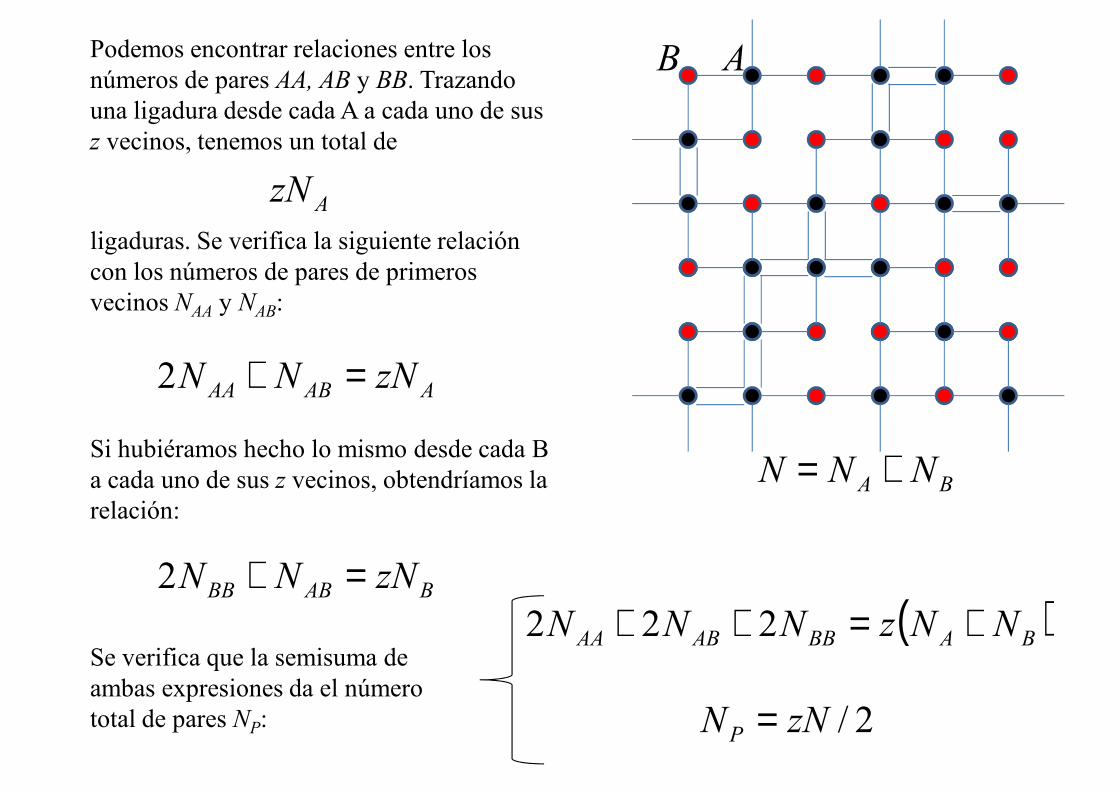
ABPodemos encontrar relaciones entre los números de pares AA, AB y BB. Trazando una ligadura desde cada A a cada uno de sus z vecinos, tenemos un total de
Az�
ligaduras. Se verifica la siguiente relación con los números de pares de primeros vecinos �AA y �AB:
AABAA z��� =+2
Si hubiéramos hecho lo mismo desde cada B a cada uno de sus z vecinos, obtendríamos la relación:
BABBB z��� =+2
Se verifica que la semisuma de ambas expresiones da el número total de pares �P:
( )BABBABAA ��z��� +=++ 222
2/z��P =
BA ��� +=

la semidiferencia es:
( ) 2/BABBAA ��z�� −=−2/z���� BBABAA =++
o:
En el límite de baja concentración de B
0≈BB� ( ) ( ) 2/2/ BABAAA CCz���z� −=−=
Reemplazando en la expresión de la semisuma
( ) 2/1 BAAB CCz�� +−=
o:
BAB z�C� =
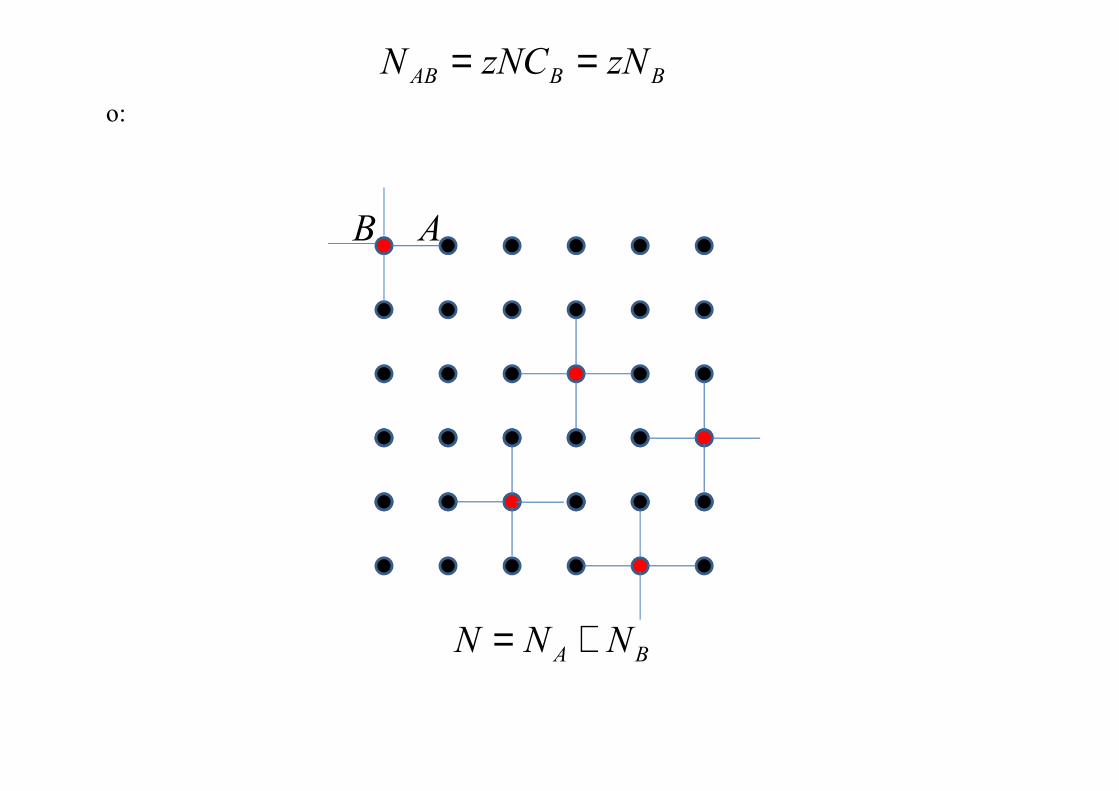
o:
BBAB z�z�C� ==
AB
BA ��� +=

Definimos las probabilidades de pares AA, AB, BB
BAjiz����P ijPijij ,,;/2/ ===
y el parámetro de orden de corto alcance como
( )BAABBAAB
AB CCPPPP −=
−=Γ 2/42
4 2
En el caso desordenado (distribución aleatoria) PAB = 2CACB
0=ΓAB
también llamada función de correlación AB.

Podemos estimar PAB usando las energías de interacción de a pares
ABε BBεAAε
AB
BB
AA
eCCP
eCP
eCP
BAAB
BBB
AAA
βε
βε
βε
−
−
−
∝
∝
∝
2
;
;2
2
kT/1=β
eCCP BAAB ∝ 2
consideramos el caso εAA = εBB y cambiamos el cero de la energía:
ABε
AAε
0
0
ε
ε−βε
βε
βε
eCCP
eCP
eCP
BAAB
BBB
AAA
2
;
;2
2
∝
∝
∝−
−
BBε

El factor de proporcionalidad en las expresiones de las probabilidades es la inversa de la suma de probabilidades (a su vez proporcional a la Función de Partición):
βεβεβε eCCeCeCTZ BABA 2)( 22 ++= −−
ZeCCP
ZeCP
BAAB
AAA
/2
;/2
βε
βε
=
= −
La función de correlación nos queda:
eeCC βεβε
si
2/1== BA CC
βεβε
βε
−+=
ee
ePAB
( ) 2/)( βεβε eeTZ += −
−=
−=Γ 144
Z
eCCCC
Z
eCCBABA
BAAB
βεβε
≈Γ≈
⇒>>1
11
AB
ABPsi βε

En ausencia de interacción (ε = 0)
4
1;
2
1 === BBAAAB PPP
En correspondencia con una aleación desordenada.
Si CA >> CB,
βεβε eCCeCTZ BAA 2)( 2 +≈ − eCCeCTZ BAA 2)( +≈
βεβε
βε
eCCeC
eCCP
BAA
BAAB 2
22 +
= −
Para βε << 1
( ) ( )( ) BABCABABAB CPCCeCCPA
221/2/21,1
2 ≈ →+≈≈ <<≈ βεβε βε

Podemos obtener el número de pares AB:
BBBPABAB z�z�Cz�
C�P� ==≈=2
2
Como ya habíamos deducido para esta situación.
Extensión de la correlación a vecinos más alejados
La función de correlación AB puede extenderse a pares de átomos separados cualquier distancia RAB
( )BA
ab
AB
ab
AB ααα −=Γ 4
obviamente,
BBAA CC == αα ;
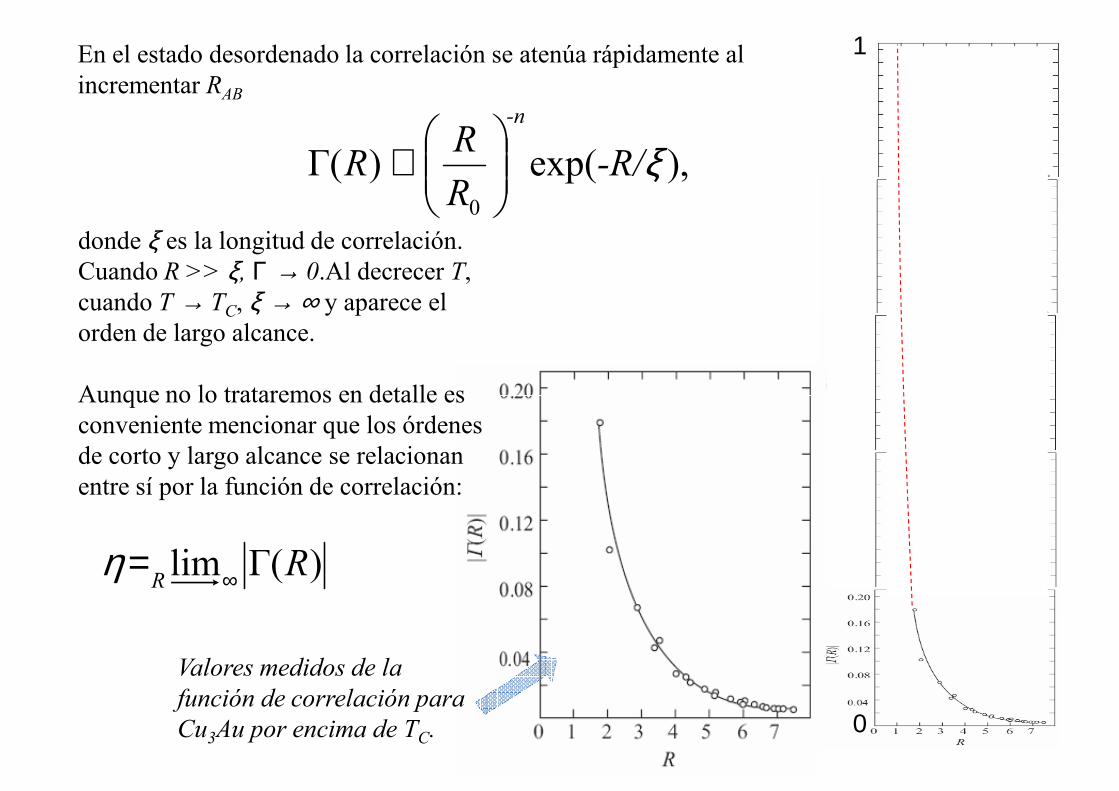
En el estado desordenado la correlación se atenúa rápidamente al incrementar RAB
),exp()Γ(0
ξ-R/R
RR
-n
∝
donde ξ es la longitud de correlación. Cuando R >> ξ, Γ → 0.Al decrecer T, cuando T → TC, ξ → ∞ y aparece el orden de largo alcance.
Aunque no lo trataremos en detalle es
1
)Γ(lim RR ∞=η
Valores medidos de la
función de correlación para
Cu3Au por encima de TC.
Aunque no lo trataremos en detalle es conveniente mencionar que los órdenes de corto y largo alcance se relacionan entre sí por la función de correlación:
0

Vidrios y líquidos
Fusión de un cristal
Debajo de TF las vibraciones térmicas causan un movimiento irregular de los átomos, que esencialmente no se alejan de sus posiciones asignadas en la red.
Encima de TF los átomos se “delocalizan” y ya no permanecen en la vecindad de un punto específico, aunque puede manifestarse un orden de corto alcance.
Simulaciones computacionales: 32 objetos con interacciones
Sólido cristalino a alta temperatura líquido
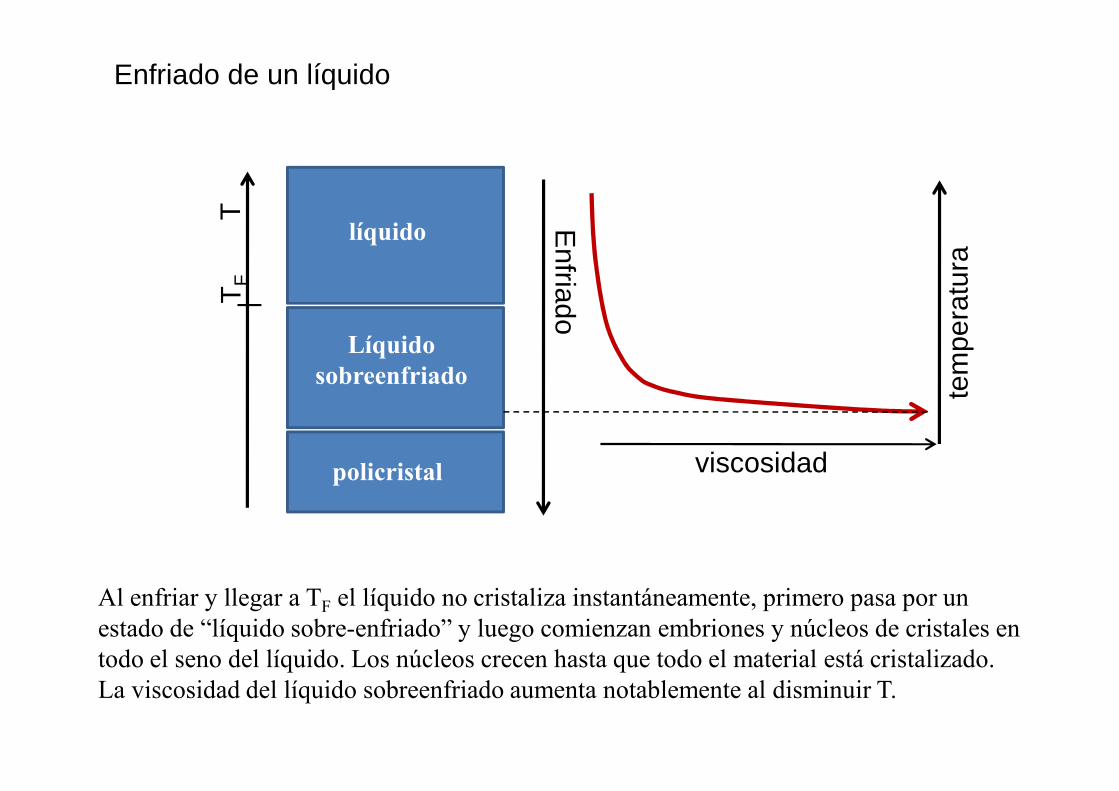
Enfriado de un líquido
líquido
Líquido sobreenfriado
Enfriado
TT
F
tem
pera
tura
policristal viscosidad
Al enfriar y llegar a TF el líquido no cristaliza instantáneamente, primero pasa por un estado de “líquido sobre-enfriado” y luego comienzan embriones y núcleos de cristales en todo el seno del líquido. Los núcleos crecen hasta que todo el material está cristalizado. La viscosidad del líquido sobreenfriado aumenta notablemente al disminuir T.
tem
pera
tura

Si el líquido se enfría muy rápido no alcanza a cristalizar porque el aumento de la viscosidad impide que los átomos alcancen las posiciones de mínima energía del arreglo ordenado: solidifica en forma de un vidrio.
líquido
Líquido sobreenfriado
TT
F
Enfriado
tem
pera
tura
vidrio
Vidrios comunes
Óxidos basados en SiO2
Estructuras cristalinas complejas dT/dt ∼∼∼∼ 10-4 – 10-1 K/s
Baja tasa de enfriado
Metales y aleacionesEstructuras cristalinas
simples dT/dt ∼∼∼∼ 106 K/s
Alta tasa de enfriado
viscosidad
Condición suficiente
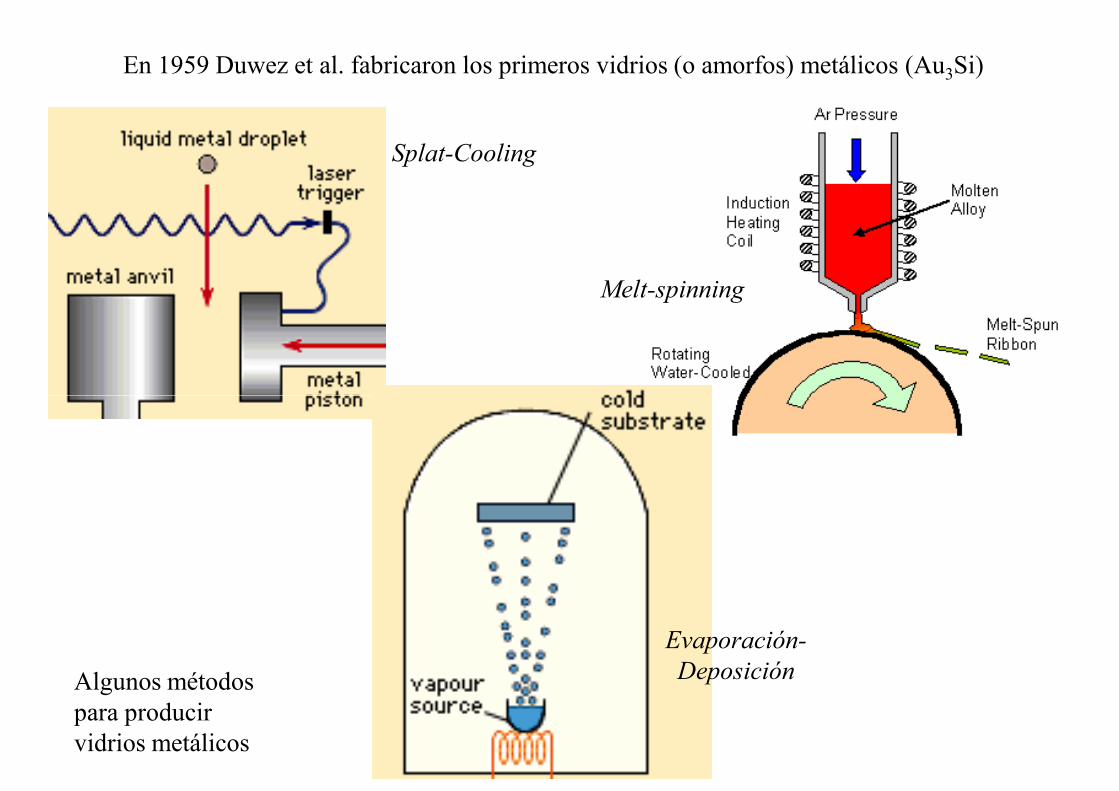
En 1959 Duwez et al. fabricaron los primeros vidrios (o amorfos) metálicos (Au3Si)
Splat-Cooling
Melt-spinning
Evaporación-
DeposiciónAlgunos métodos para producir vidrios metálicos

Fabricación de un microhilo amorfo (vidrio) de aleación de base Fe
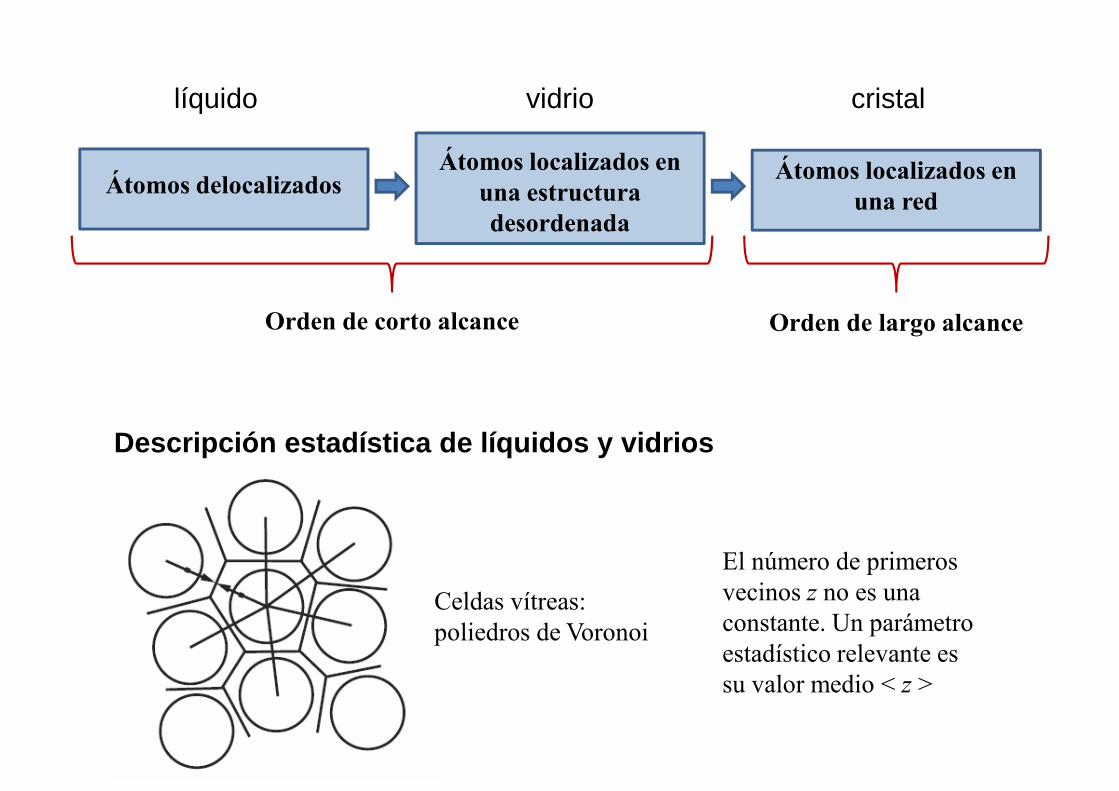
Átomos delocalizadosÁtomos localizados en
una estructura desordenada
Átomos localizados en una red
líquido vidrio cristal
Orden de corto alcance Orden de largo alcance
Descripción estadística de líquidos y vidrios
Celdas vítreas: poliedros de Voronoi
El número de primeros vecinos z no es una constante. Un parámetro estadístico relevante es su valor medio < z >

Función de distribución atómica
…
Definimos las densidades de probabilidad de 1, 2, 3…, s átomos en las posiciones r1, r2, r3..., rs, por
La probabilidad de encontrar un átomo en el volumen dr1 ubicado en la vecindad de r1, otro en el volumen dr2 ubicado en la vecindad de r2, etc., es:
La densidad de probabilidad normalizada es
está asociada a la función de correlación ).Γ(R
Veremos que para obtener esta última hay que hacer una integración de la primera en cáscara esférica de ancho atómico de radio |r2 – r1| centrada en r1.
densidad media
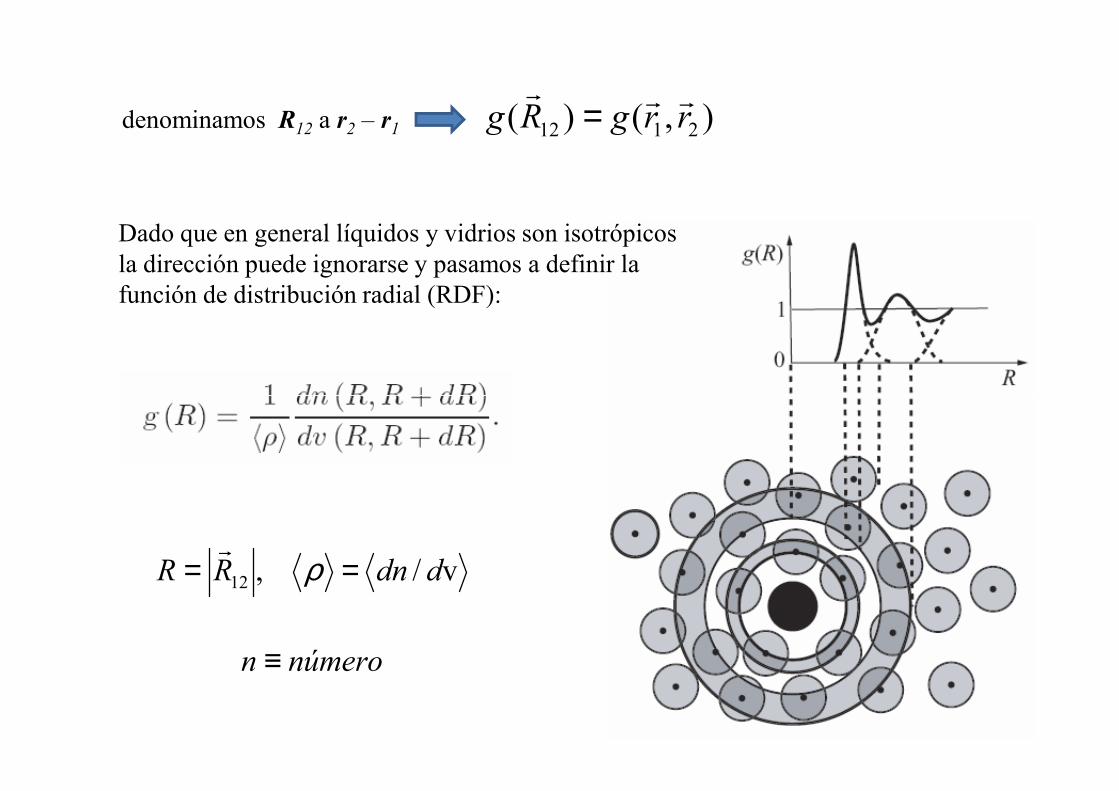
denominamos R12 a r2 – r1 ),()( 2112 rrgRgrrr
=
Dado que en general líquidos y vidrios son isotrópicos la dirección puede ignorarse y pasamos a definir la función de distribución radial (RDF):
v/,12 ddnRR == ρr
númeron ≡
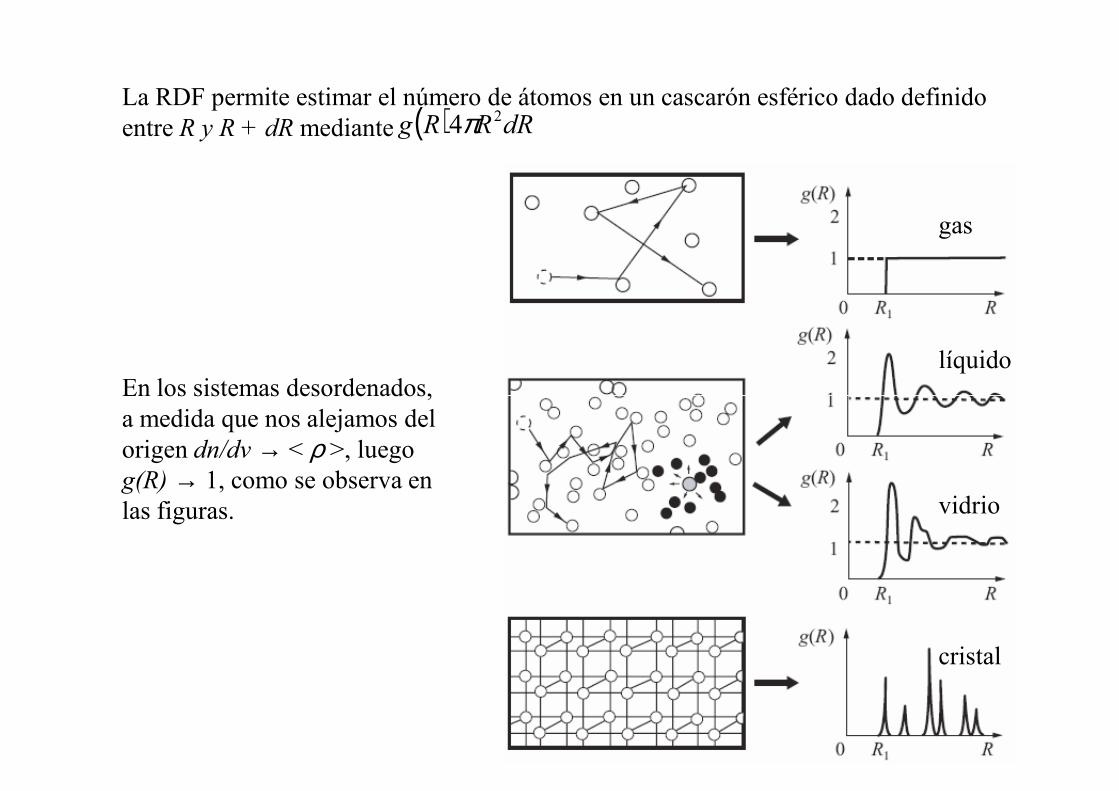
La RDF permite estimar el número de átomos en un cascarón esférico dado definido entre R y R + dR mediante ( ) dRRRg 24π
En los sistemas desordenados,
gas
líquidoEn los sistemas desordenados, a medida que nos alejamos del origen dn/dv → < ρ >, luego g(R) → 1, como se observa en las figuras. vidrio
cristal
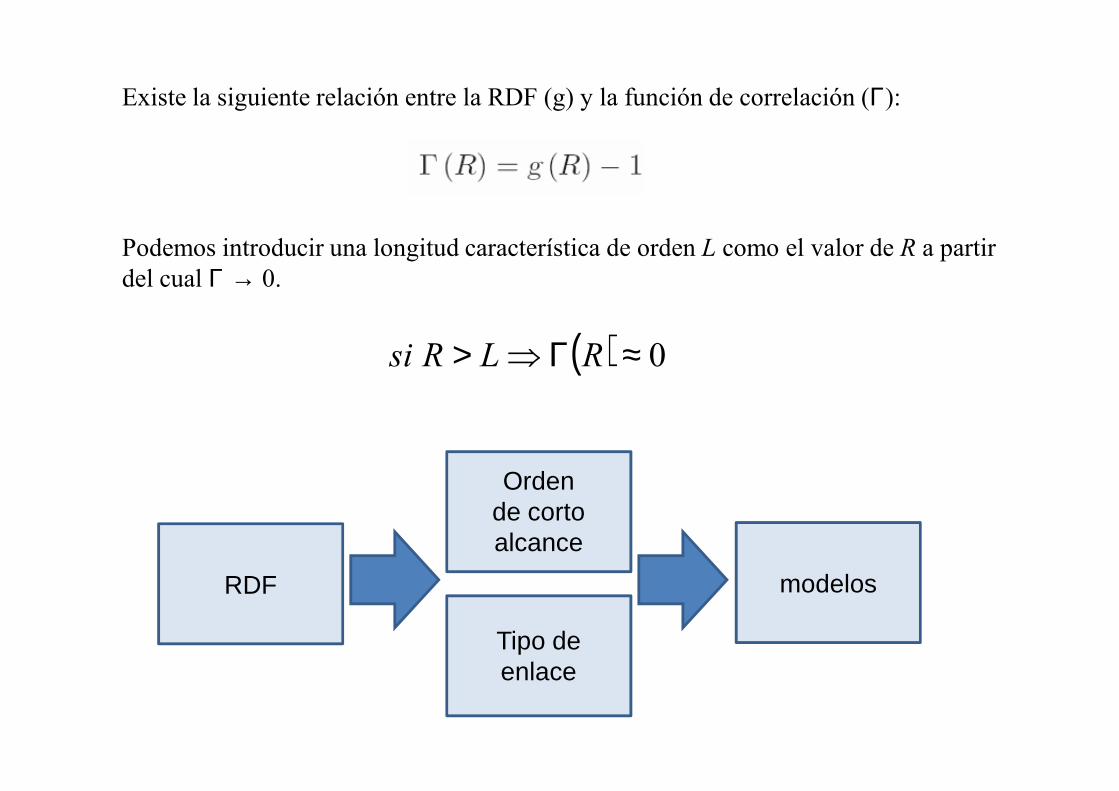
Existe la siguiente relación entre la RDF (g) y la función de correlación (Γ):
Podemos introducir una longitud característica de orden L como el valor de R a partir del cual Γ → 0.
( ) 0≈Γ⇒> RLRsi
RDF
Orden de corto alcance
Tipo de enlace
modelos

Modelos para líquidos y vidrios
Modelo de empaquetamiento denso aleatorio de esferas (J.D. Bernal 1959)
Estudio empírico con lentejas o bolitas idénticas de dulce o plastilina, o esferas para rodamientos, colocadas en contenedores sin paredes planas (de vidrio y de goma).
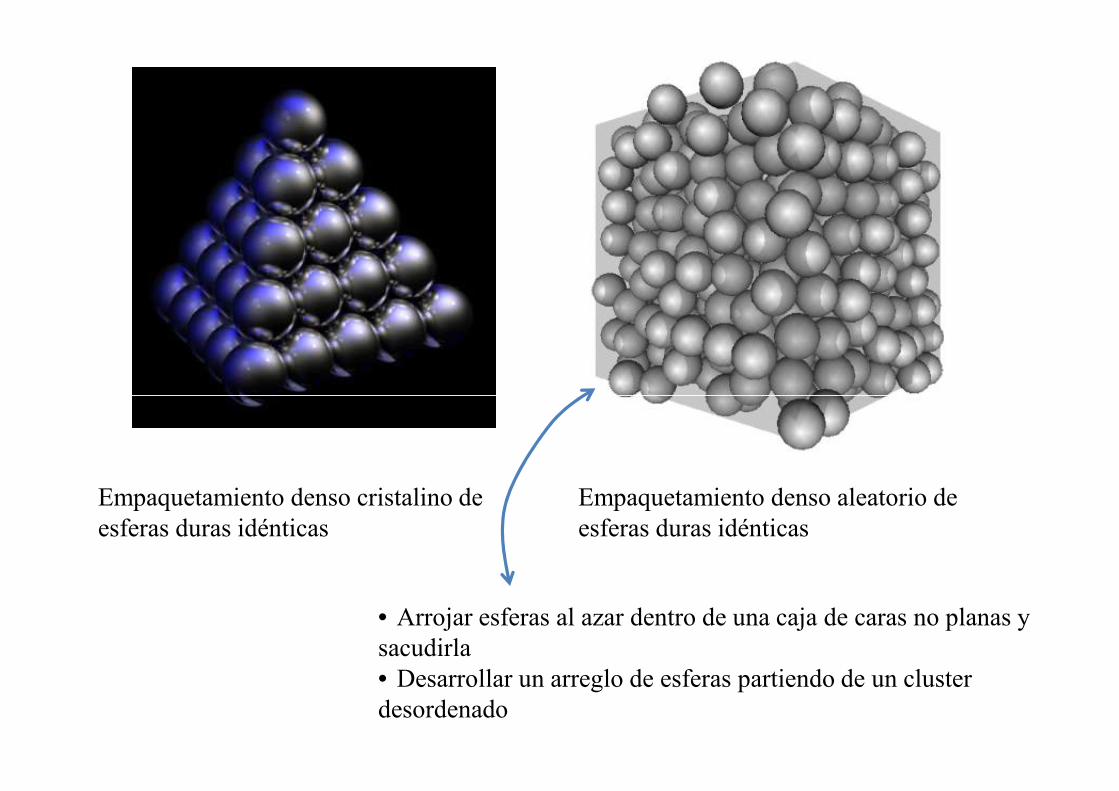
• Arrojar esferas al azar dentro de una caja de caras no planas y sacudirla• Desarrollar un arreglo de esferas partiendo de un cluster desordenado
Empaquetamiento denso cristalino de esferas duras idénticas
Empaquetamiento denso aleatorio de esferas duras idénticas
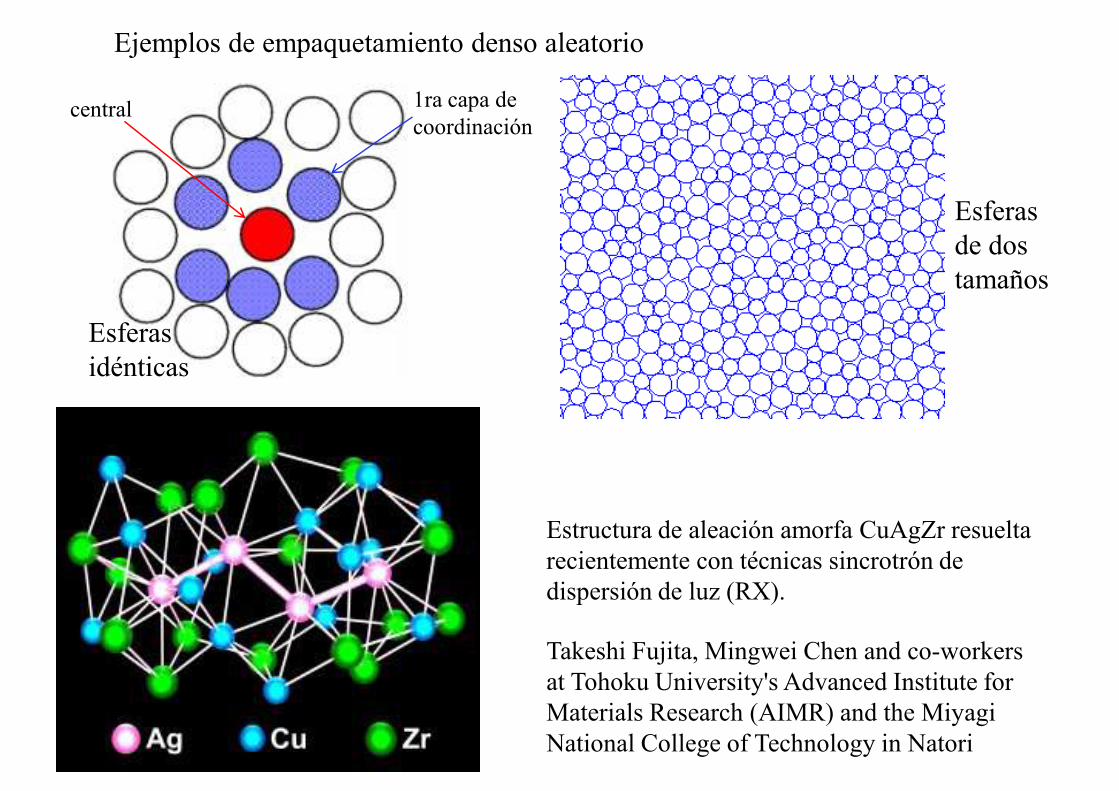
Ejemplos de empaquetamiento denso aleatorio
Esferas idénticas
Esferas de dos tamaños
central 1ra capa de coordinación
Takeshi Fujita, Mingwei Chen and co-workers at Tohoku University's Advanced Institute for Materials Research (AIMR) and the Miyagi National College of Technology in Natori
Estructura de aleación amorfa CuAgZr resueltarecientemente con técnicas sincrotrón de dispersión de luz (RX).
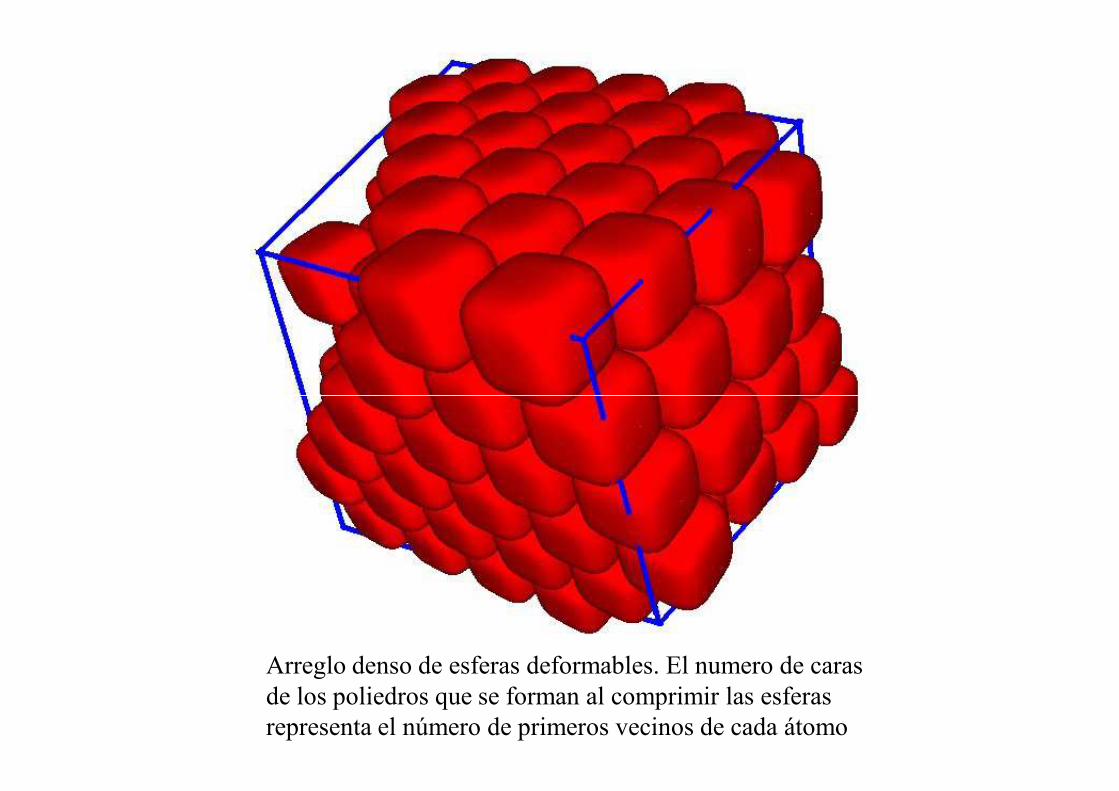
Arreglo denso de esferas deformables. El numero de caras de los poliedros que se forman al comprimir las esferas representa el número de primeros vecinos de cada átomo
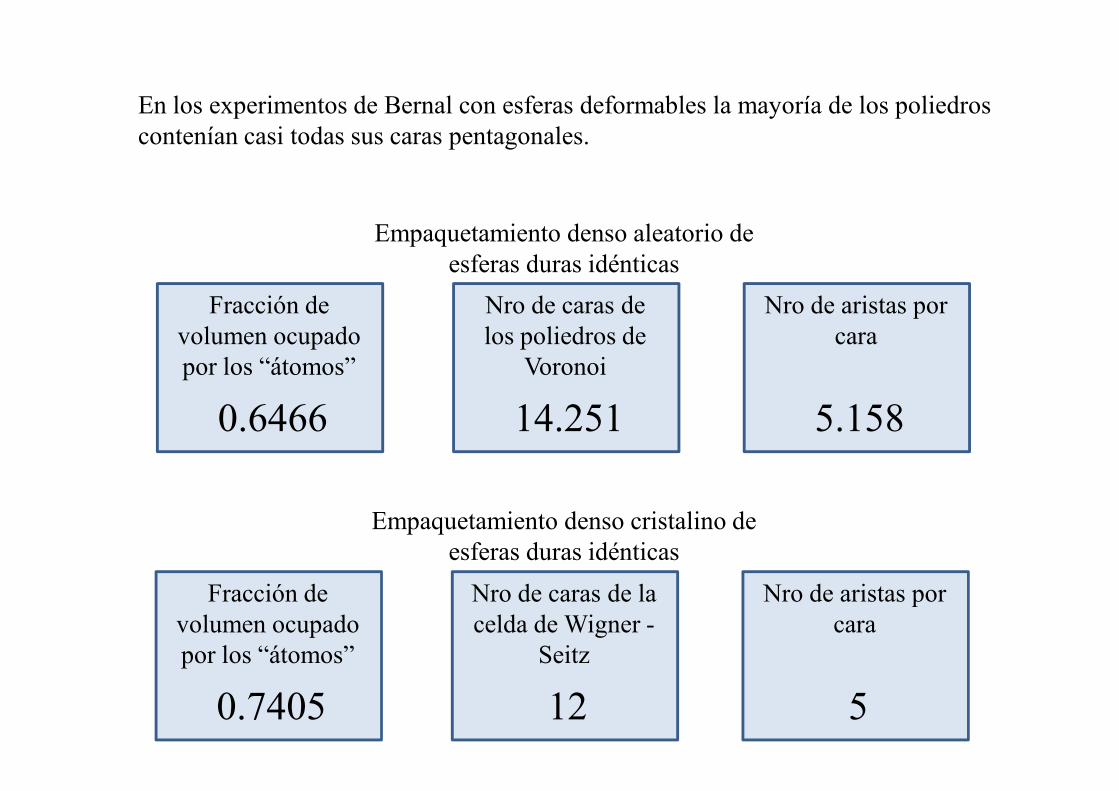
En los experimentos de Bernal con esferas deformables la mayoría de los poliedros contenían casi todas sus caras pentagonales.
Fracción de volumen ocupado por los “átomos”
0.6466
Nro de caras de los poliedros de
Voronoi
14.251
Nro de aristas por cara
5.158
Empaquetamiento denso aleatorio de esferas duras idénticas
0.6466 14.251 5.158
Empaquetamiento denso cristalino de esferas duras idénticas
Fracción de volumen ocupado por los “átomos”
0.7405
Nro de caras de la celda de Wigner -
Seitz
12
Nro de aristas por cara
5
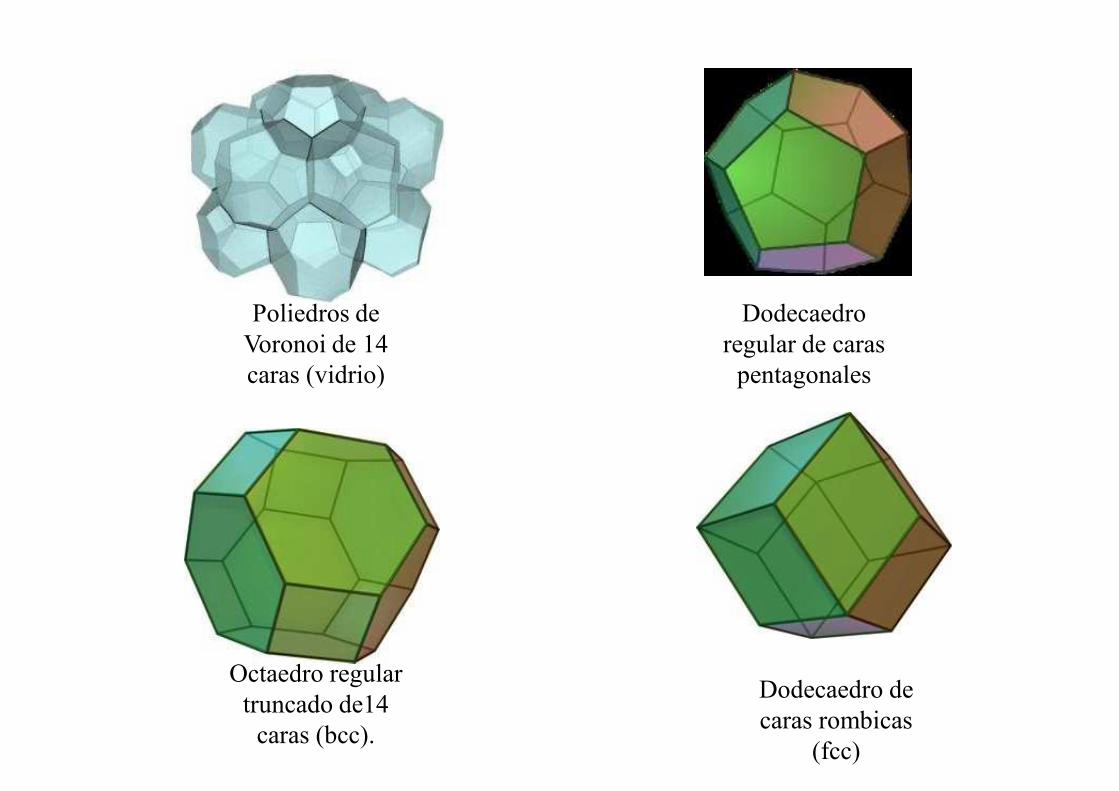
Poliedros de Voronoi de 14 caras (vidrio)
Dodecaedro regular de caras
pentagonales
Octaedro regular truncado de14
caras (bcc).
Dodecaedro de caras rombicas
(fcc)
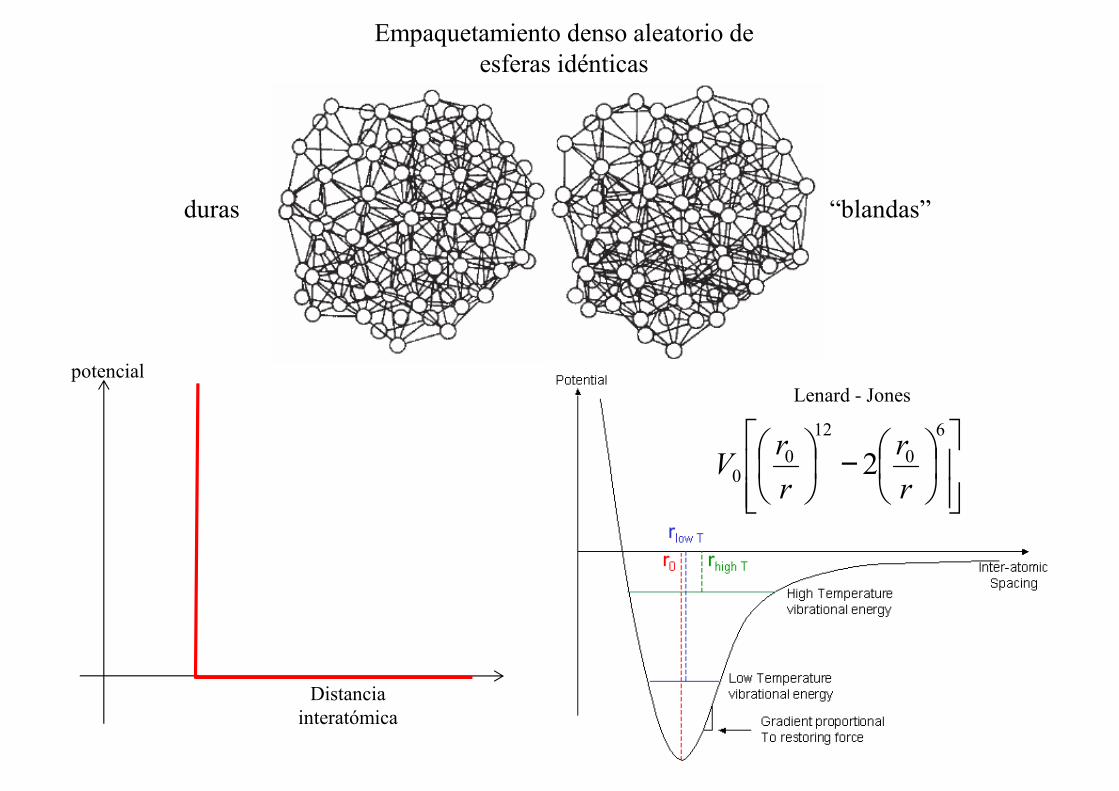
Empaquetamiento denso aleatorio de esferas idénticas
duras “blandas”
potencialLenard - Jones
Distancia interatómica
Lenard - Jones
−
6
0
12
00 2
r
r
r
rV
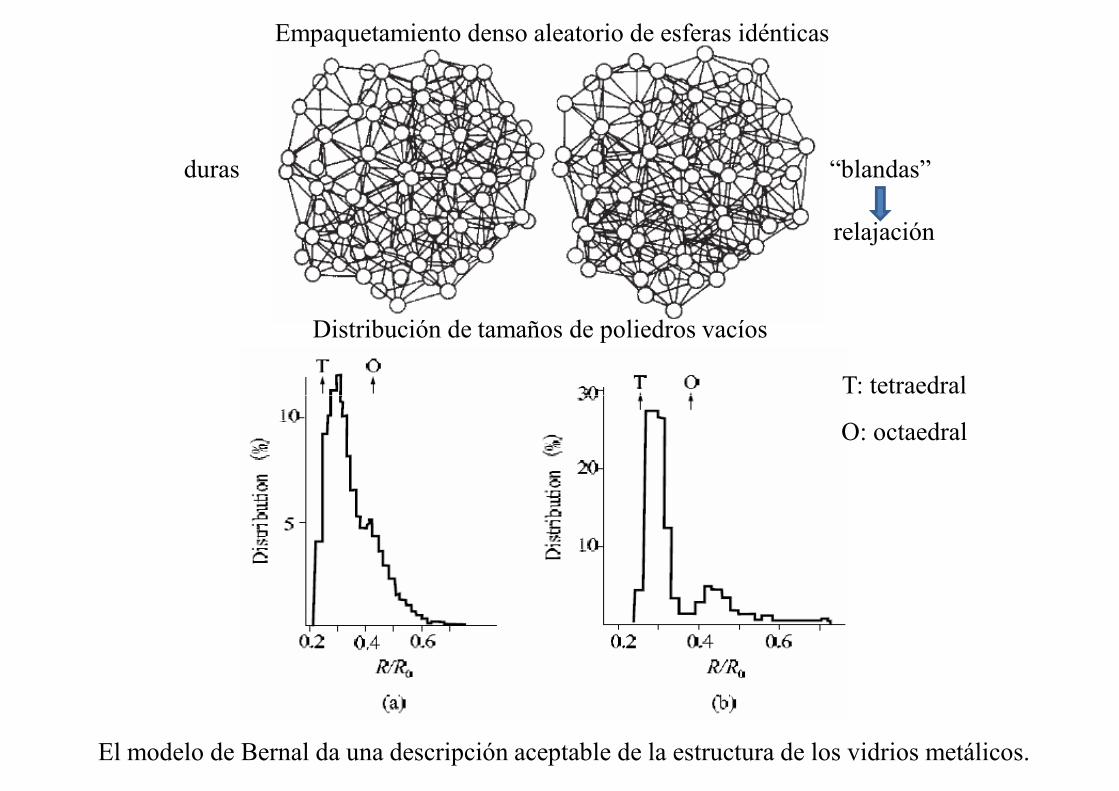
Empaquetamiento denso aleatorio de esferas idénticas
duras “blandas”
Distribución de tamaños de poliedros vacíos
T: tetraedral
relajación
T: tetraedral
O: octaedral
El modelo de Bernal da una descripción aceptable de la estructura de los vidrios metálicos.
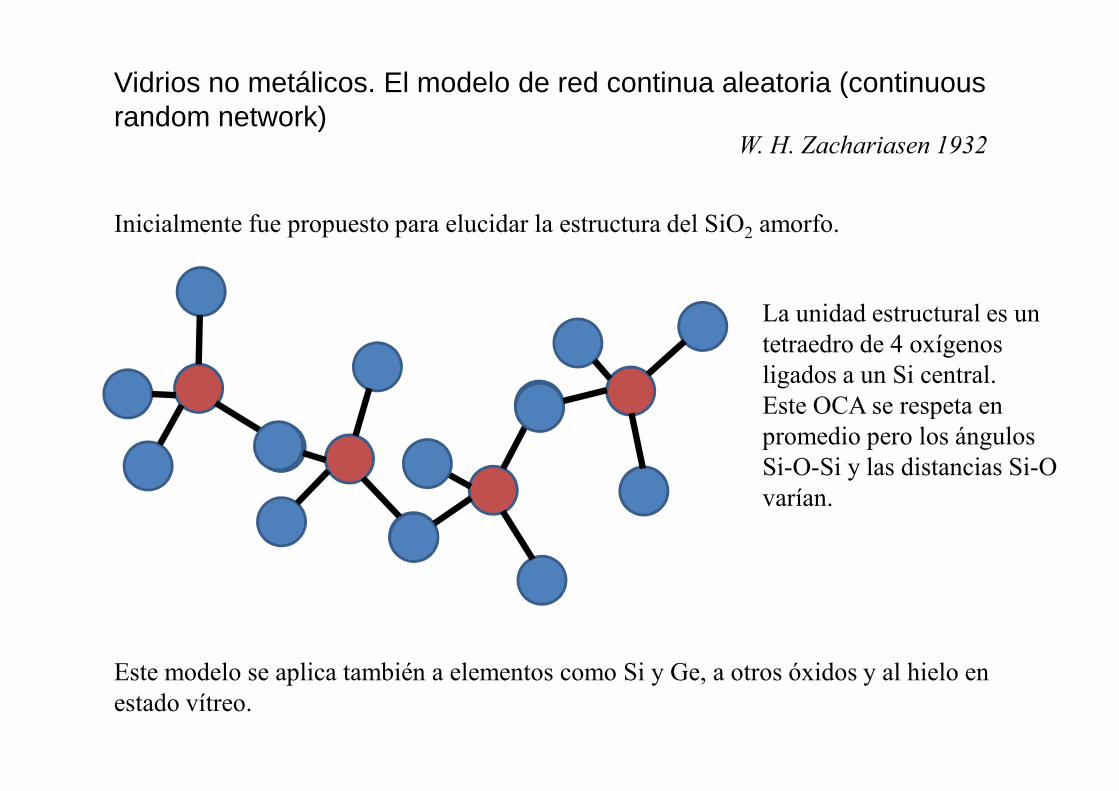
Vidrios no metálicos. El modelo de red continua aleatoria (continuous random network)
W. H. Zachariasen 1932
Inicialmente fue propuesto para elucidar la estructura del SiO2 amorfo.
La unidad estructural es un tetraedro de 4 oxígenos ligados a un Si central. Este OCA se respeta en Este OCA se respeta en promedio pero los ángulos Si-O-Si y las distancias Si-O varían.
Este modelo se aplica también a elementos como Si y Ge, a otros óxidos y al hielo en estado vítreo.
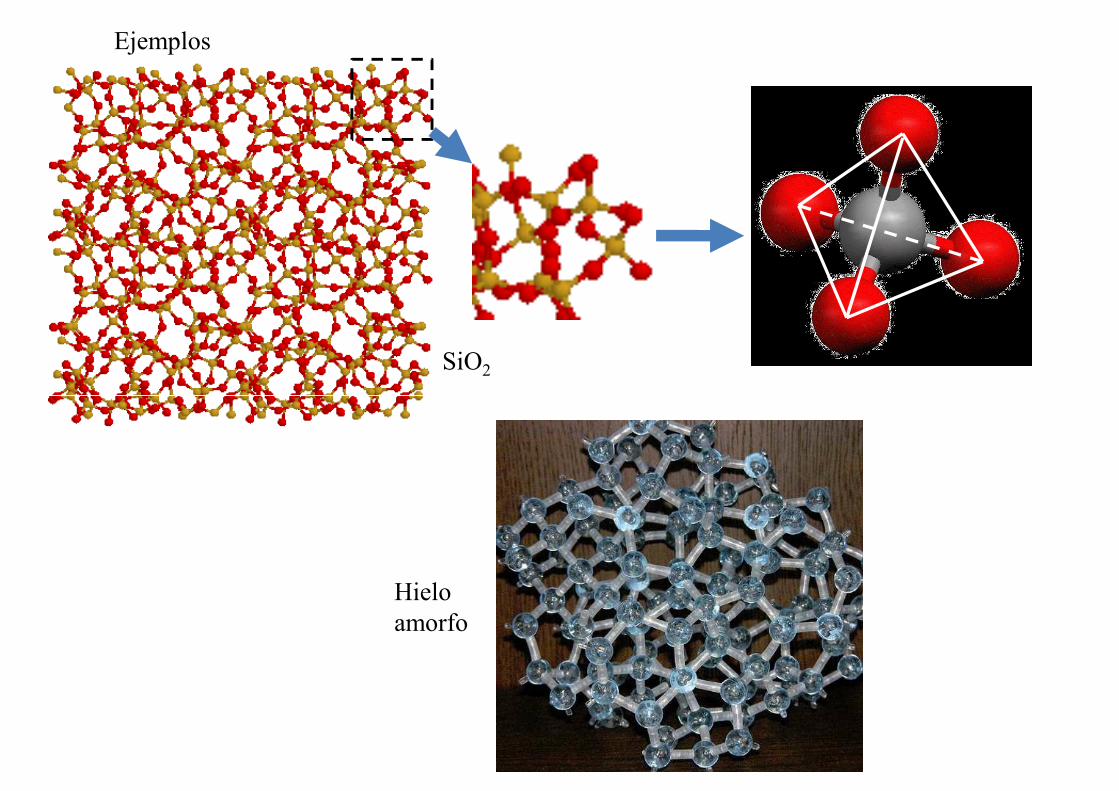
Ejemplos
SiO2
Hielo amorfo
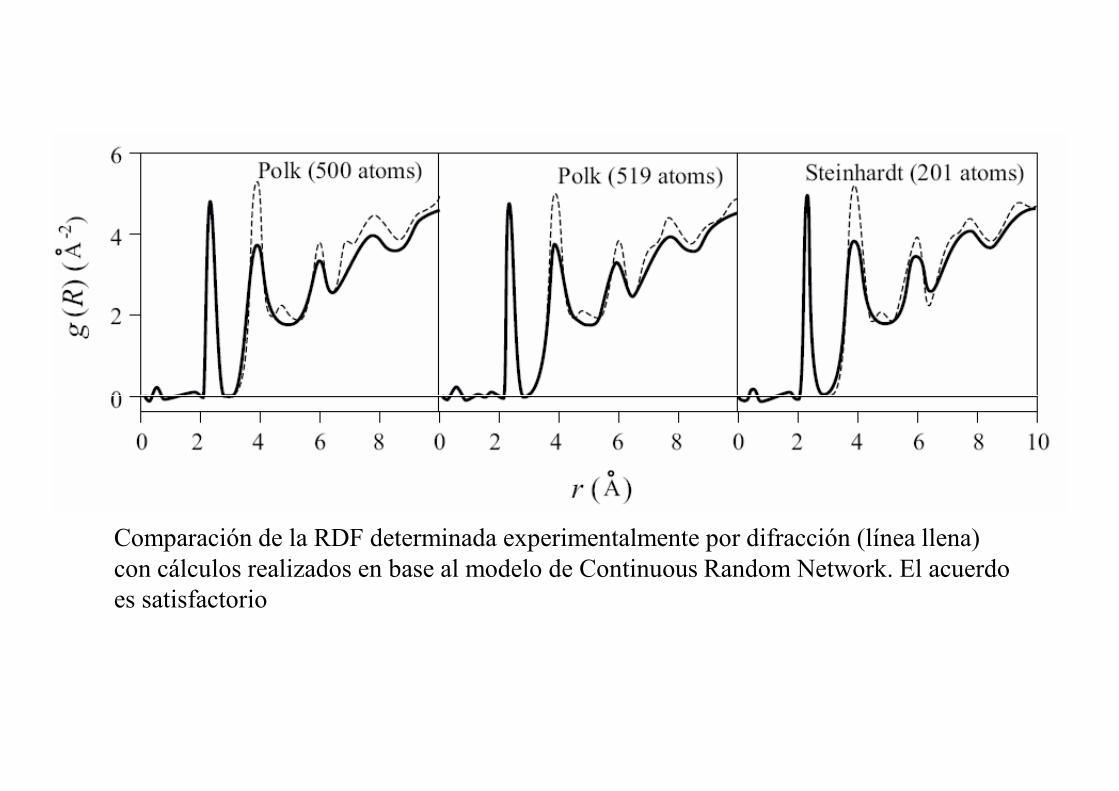
Comparación de la RDF determinada experimentalmente por difracción (línea llena) con cálculos realizados en base al modelo de Continuous Random Network. El acuerdo es satisfactorio
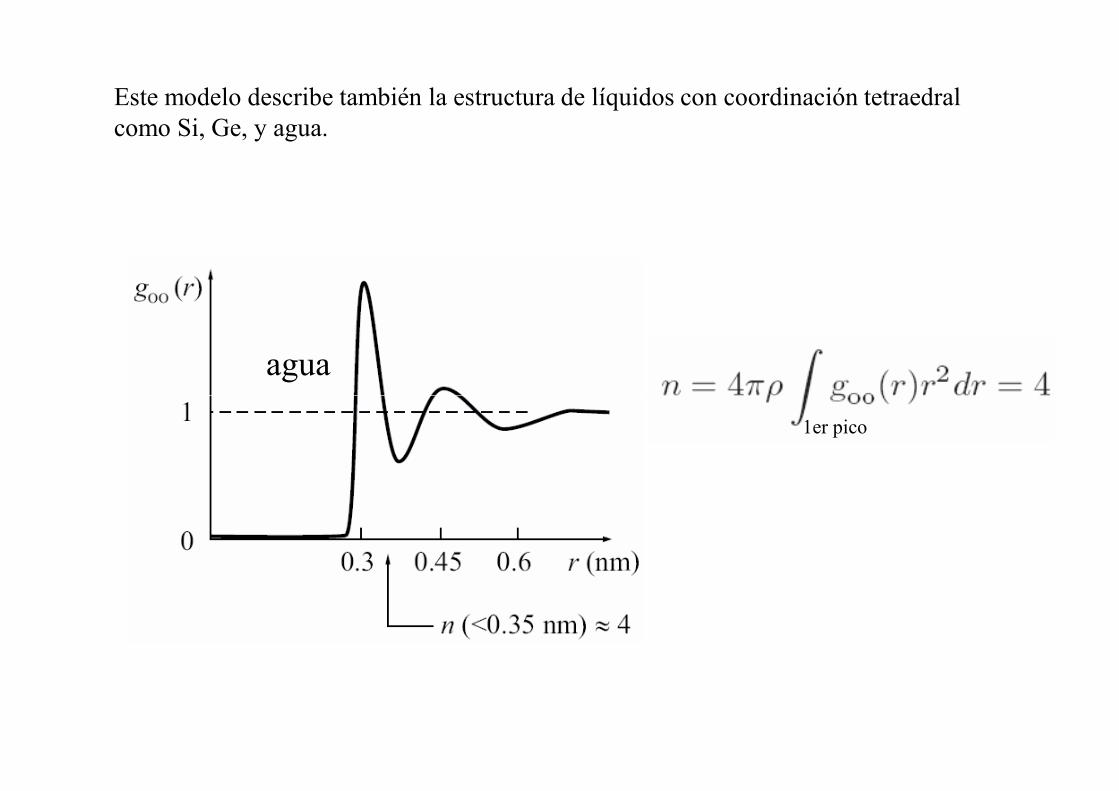
Este modelo describe también la estructura de líquidos con coordinación tetraedral como Si, Ge, y agua.
agua
1er pico
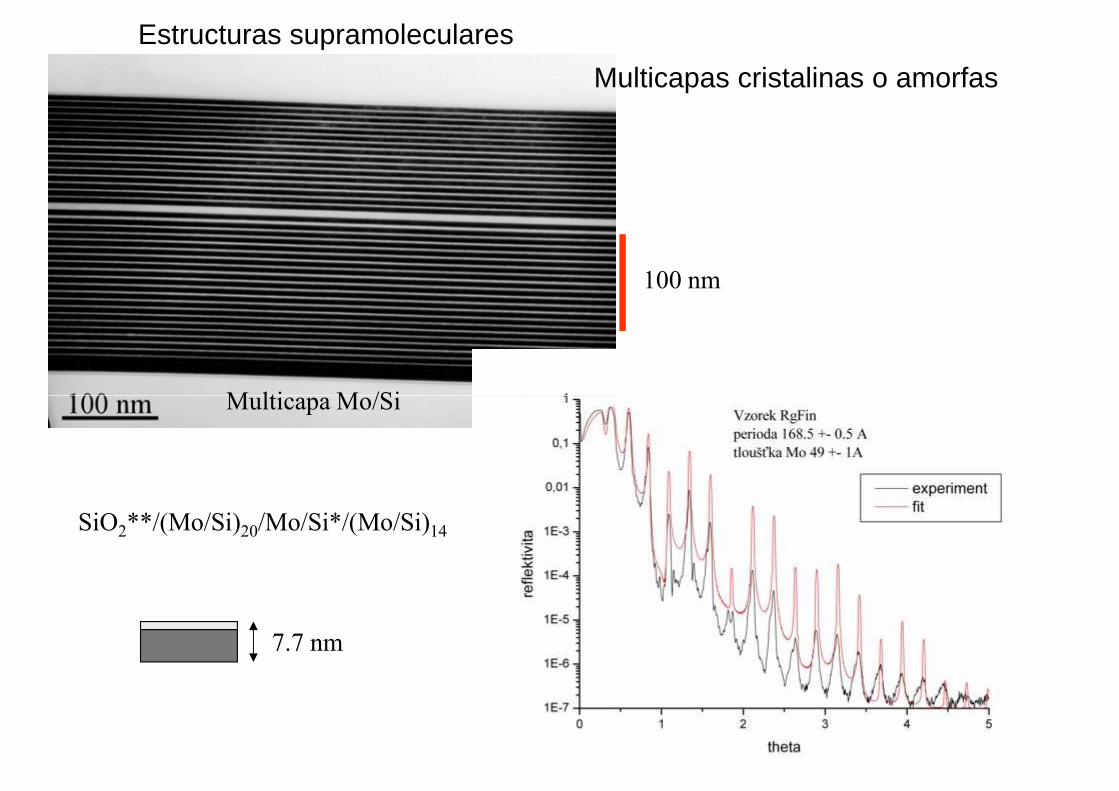
Multicapa Mo/Si
Estructuras supramoleculares
Multicapas cristalinas o amorfas
100 nm
Multicapa Mo/Si
SiO2**/(Mo/Si)20/Mo/Si*/(Mo/Si)14
7.7 nm
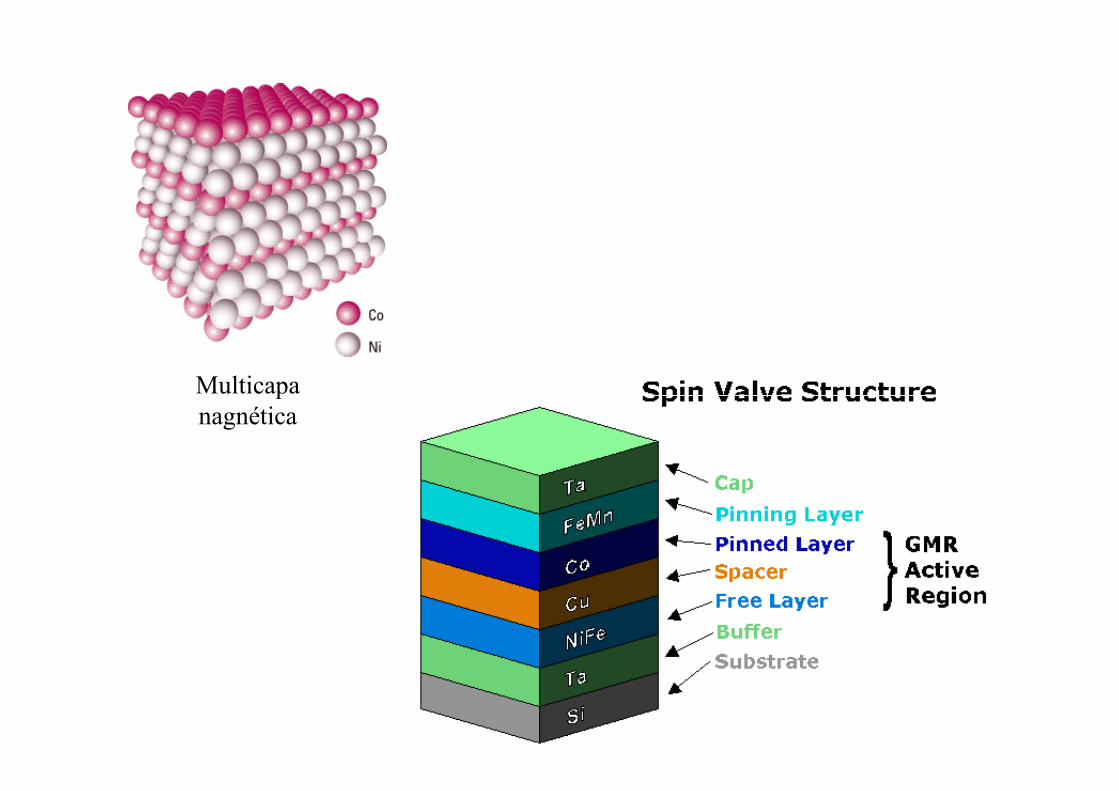
Multicapa Multicapa nagnética

Magnetoresistencia gigante
Fe/CrFe/Cr
Peter Grünberg, Nobel Prize in Physics 2007
Albert Fert, Nobel Prize in Physics 2007
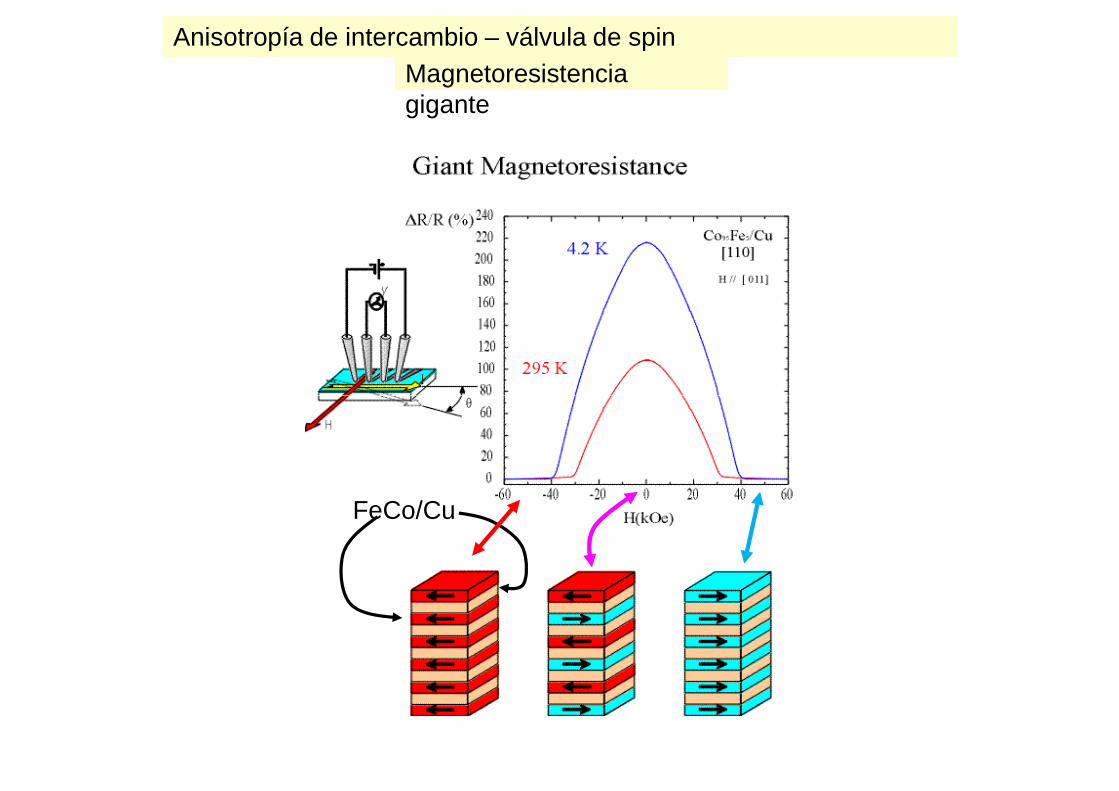
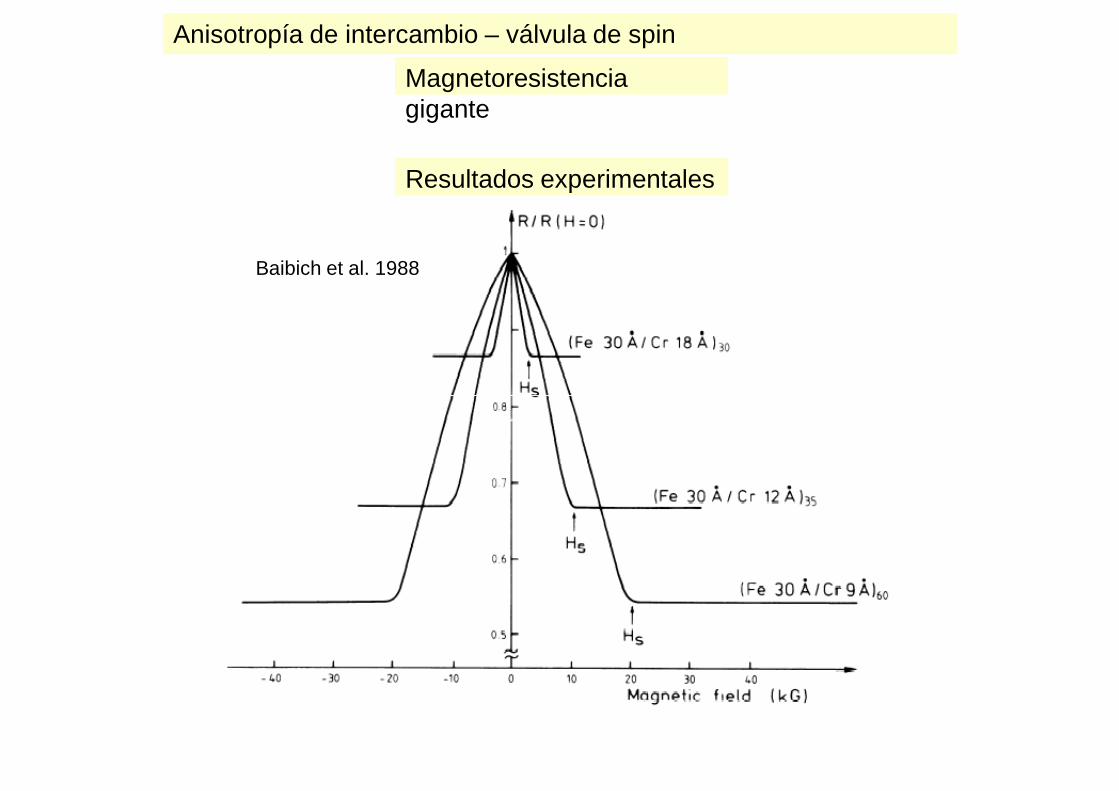
Anisotropía de intercambio – válvula de spinMagnetoresistencia gigante
FeCo/Cu
Anisotropía de intercambio – válvula de spin
Magnetoresistencia gigante
Baibich et al. 1988
Resultados experimentales
Anisotropía de intercambio – válvula de spin
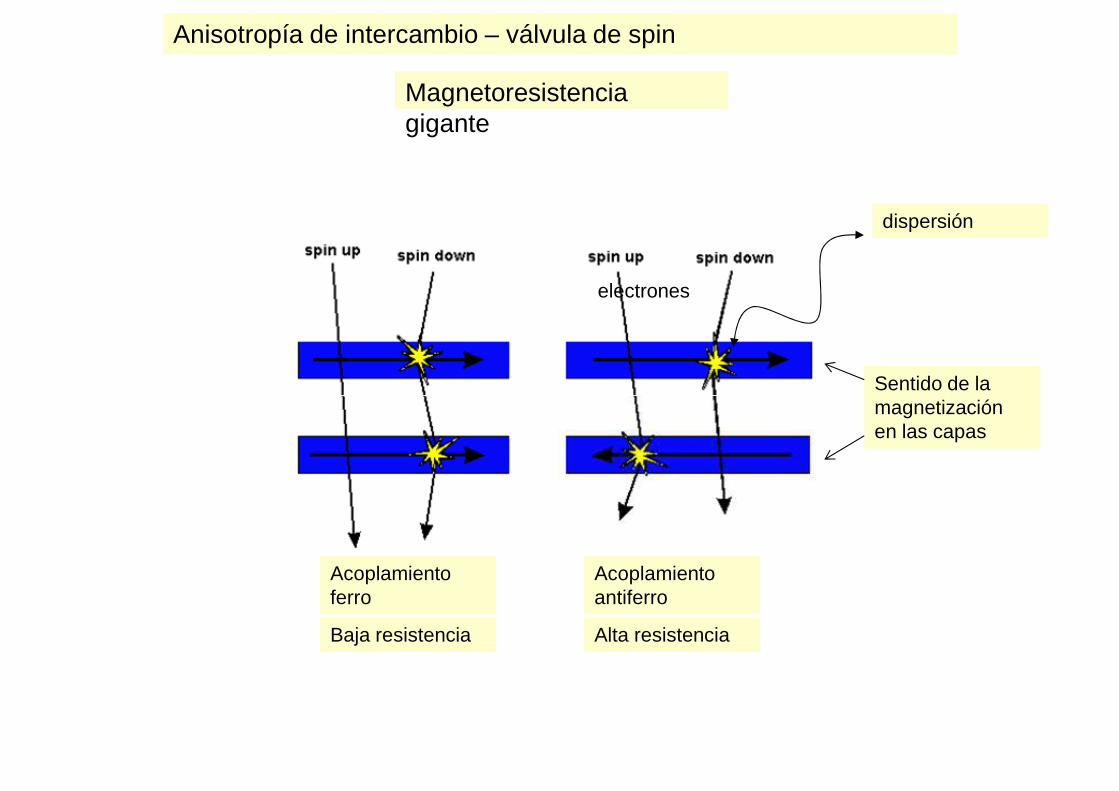
Magnetoresistencia gigante
Sentido de la
dispersión
electrones
magnetización en las capas
Acoplamiento ferro
Acoplamiento antiferro
Alta resistenciaBaja resistencia
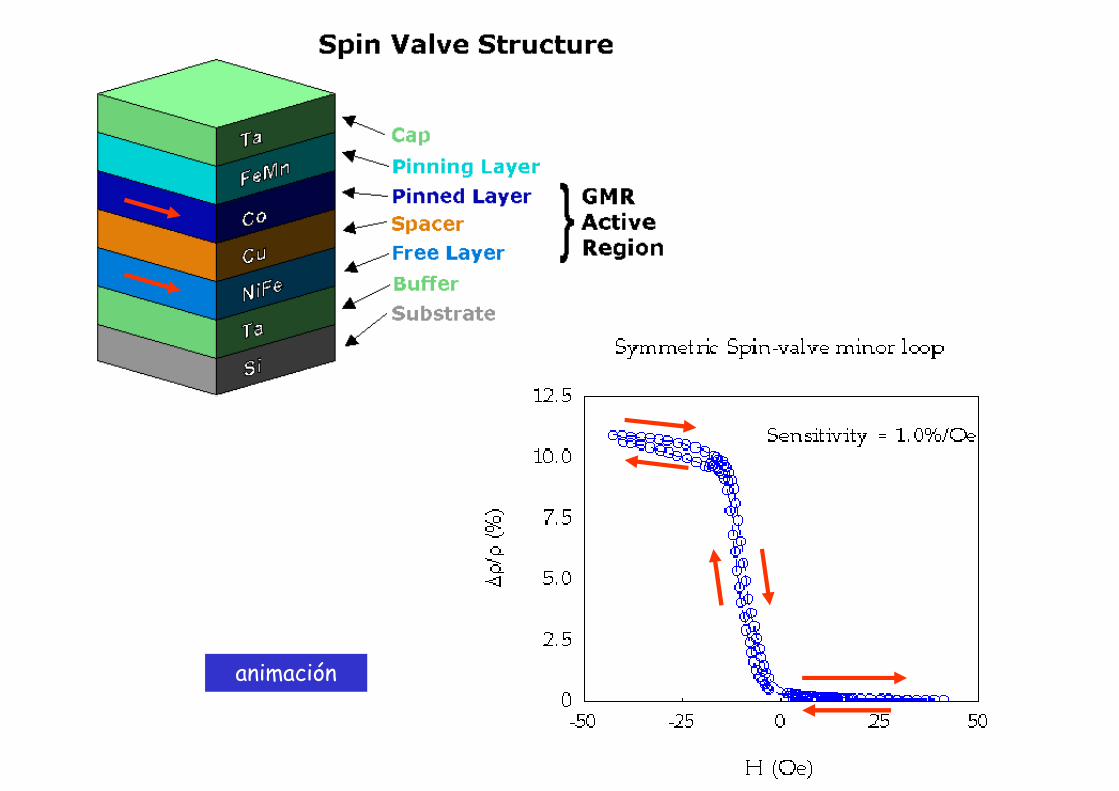
animación
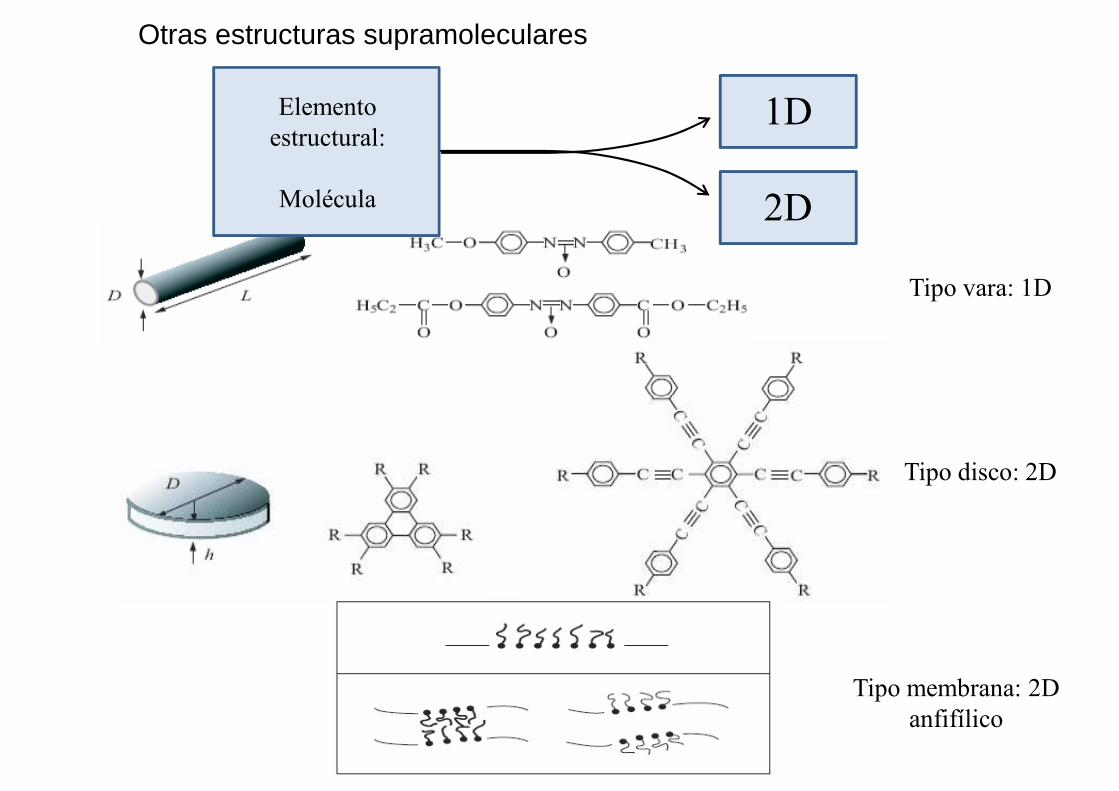
Otras estructuras supramoleculares
Tipo vara: 1D
Elemento estructural:
Molécula
1D
2D
Tipo disco: 2D
Tipo membrana: 2Danfifílico
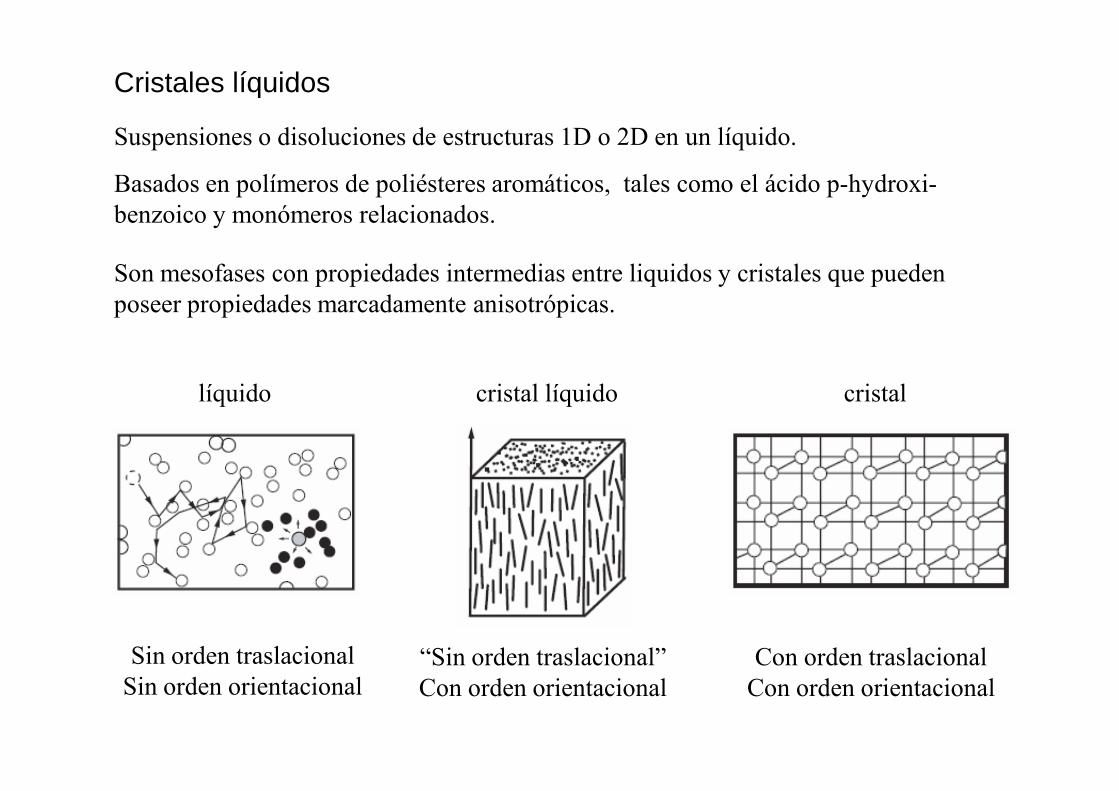
Cristales líquidos
Suspensiones o disoluciones de estructuras 1D o 2D en un líquido.
Son mesofases con propiedades intermedias entre liquidos y cristales que pueden poseer propiedades marcadamente anisotrópicas.
líquido cristal líquido cristal
Basados en polímeros de poliésteres aromáticos, tales como el ácido p-hydroxi-benzoico y monómeros relacionados.
líquido cristal líquido cristal
Sin orden traslacionalSin orden orientacional
“Sin orden traslacional”Con orden orientacional
Con orden traslacionalCon orden orientacional
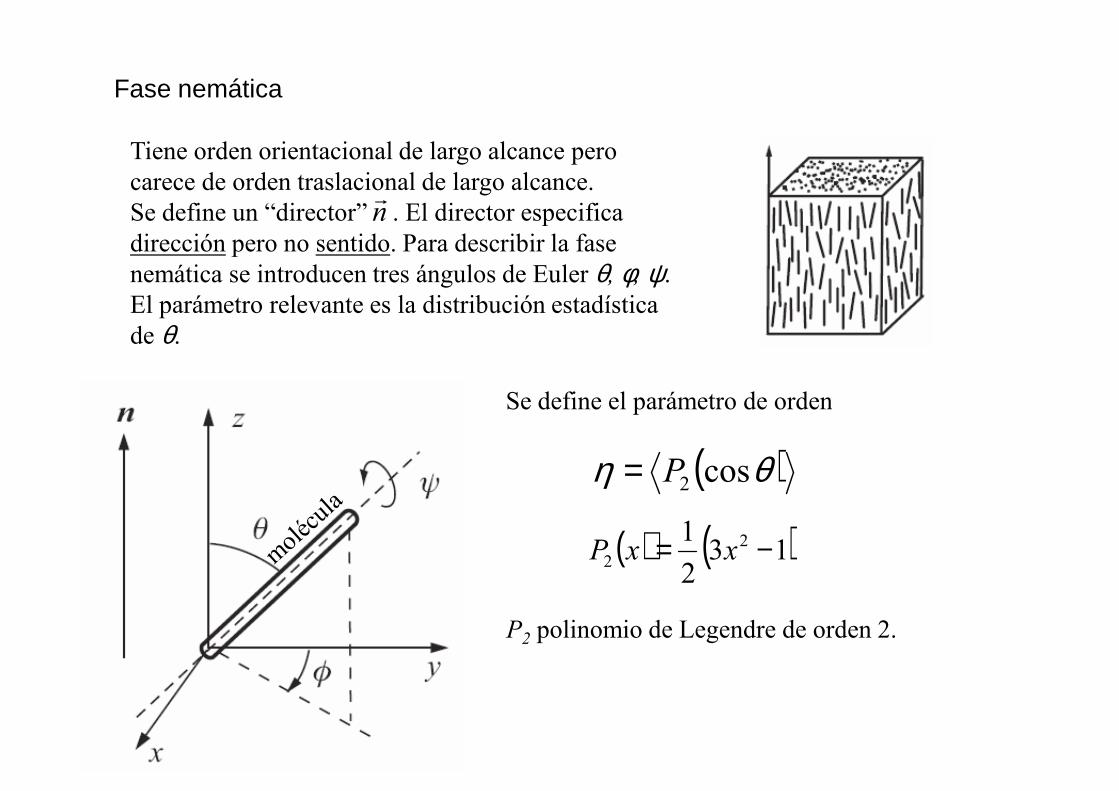
Fase nemática
Tiene orden orientacional de largo alcance pero carece de orden traslacional de largo alcance.Se define un “director” . El director especifica dirección pero no sentido. Para describir la fase nemática se introducen tres ángulos de Euler θ, φ, ψ. El parámetro relevante es la distribución estadística de θ.
nr
Se define el parámetro de orden
P2 polinomio de Legendre de orden 2.
Se define el parámetro de orden
( )θη cos2P=
( ) ( )132
1 22 −= xxP
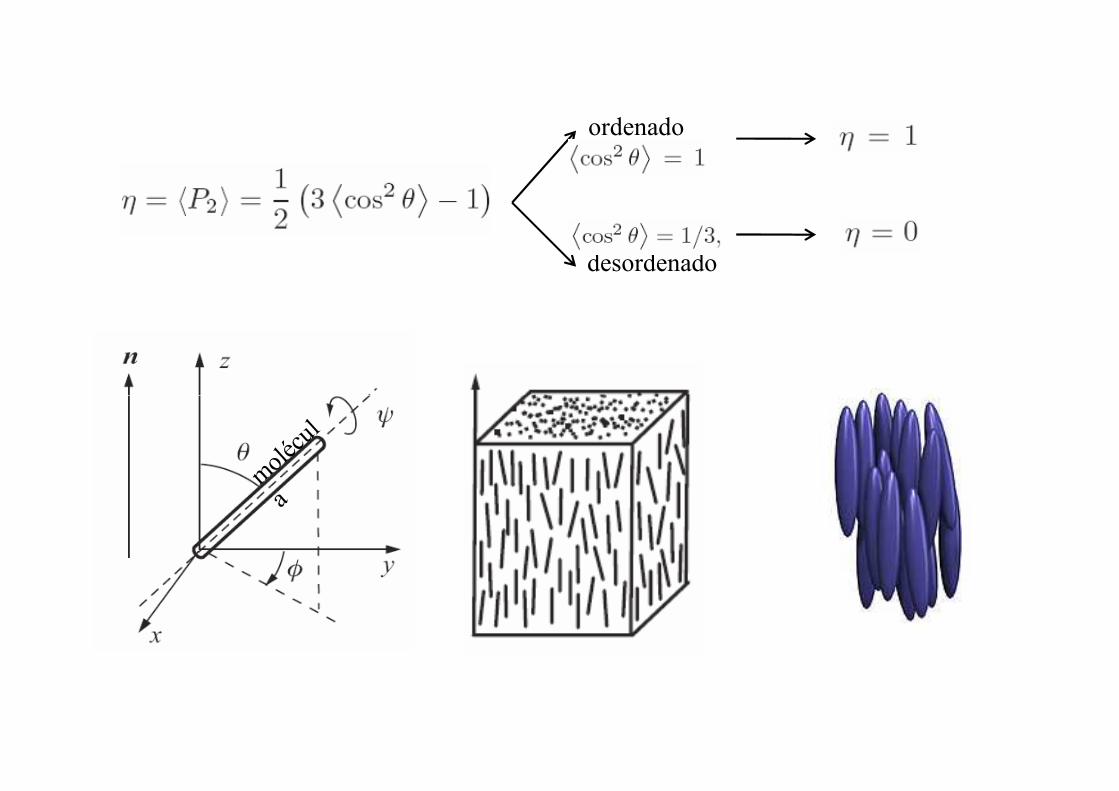
ordenado
desordenado

Fase colestérica o nemática quiral
Tiene orden orientacional de largo alcance pero carece de orden traslacional de largo alcance dentro de cada capa. Sin embargo hay orden traslacional de largo alcance de capas. Las moléculas son largas y usualmente planas, arregladas en capas laminares a causa de la interacción entre las bases en sus extremos. Las moléculas están orientadas en el plano de la capa. La orientación del director rota con un paso L en función de la posición de la capa sobre un eje normal a la misma, de acuerdo con las expresiones:
Nemática quiral
L
las expresiones:
La fase nemática se recupera cuando L = ∞ (o qC = 0). Como
( )o
3000 Α≈= TLL
Se observa difracción de Bragg con luz visible y el material cambia de color con la temperatura.
Parámetros de orden: , ó L,η ., Cqη
Con materiales de este tipo se desarrollan prototipos de e-paper.

E-books
Fujitsu

Kindle Amazon

Fases esméctica y columnar
En la fase esméctica moléculas tipo “vara” se ordenan en capas periódicas paralelas regularmente espaciadas. Dentro de las capas las moléculas se alinean en la dirección de . Ésta puede ser normal a la capa (Fase esméctica A) u oblicua (Fase esméctica C), las posiciones de los centros de masa permanecen desordenadas. En esta fase hay simetría traslacional de las capas. Las moléculas sólo pueden moverse dentro de la capa. Las capas pueden deslizar una respecto de otra.
nr
zn,r
El orden se describe a través de tres parámetros:
z es la coordenada vertical* de los centros de masa moleculares.
esméctica
* Normal a las capas.
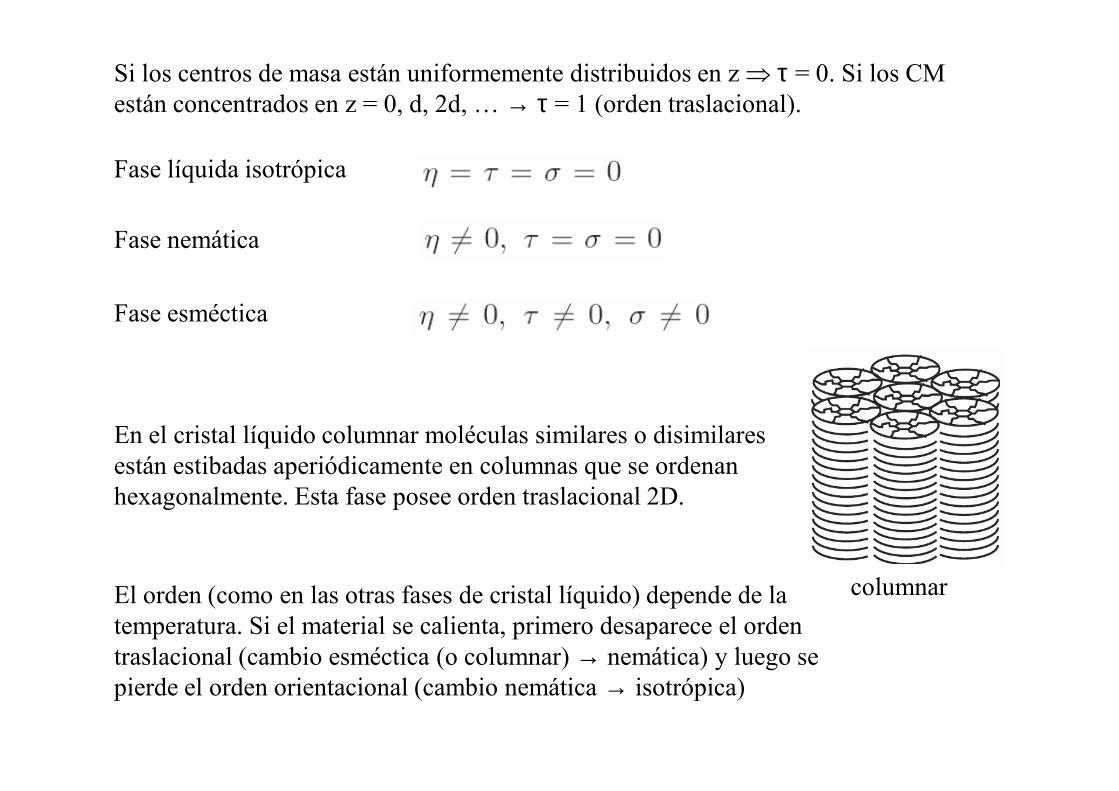
Fase líquida isotrópica
Fase nemática
Fase esméctica
Si los centros de masa están uniformemente distribuidos en z ⇒ τ = 0. Si los CM están concentrados en z = 0, d, 2d, … → τ = 1 (orden traslacional).
En el cristal líquido columnar moléculas similares o disimilares están estibadas aperiódicamente en columnas que se ordenan hexagonalmente. Esta fase posee orden traslacional 2D.
El orden (como en las otras fases de cristal líquido) depende de la temperatura. Si el material se calienta, primero desaparece el orden traslacional (cambio esméctica (o columnar) → nemática) y luego se pierde el orden orientacional (cambio nemática → isotrópica)
columnar
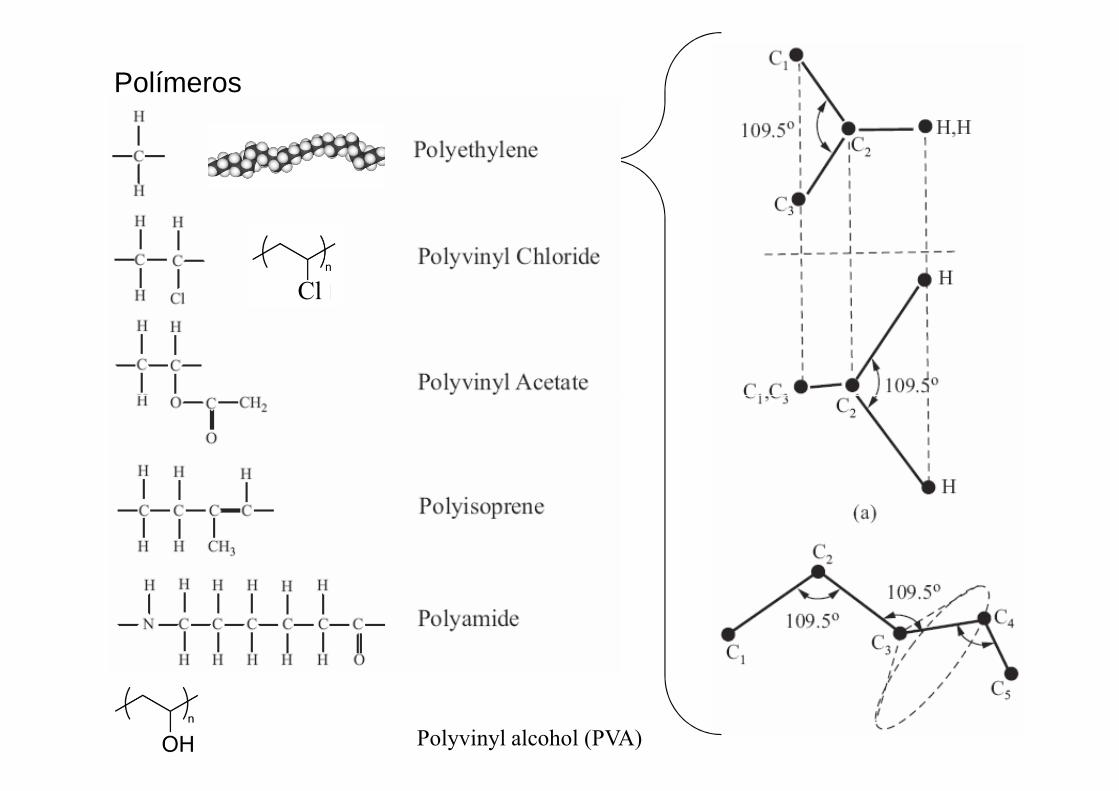
Polímeros
Cl
Polyvinyl alcohol (PVA)
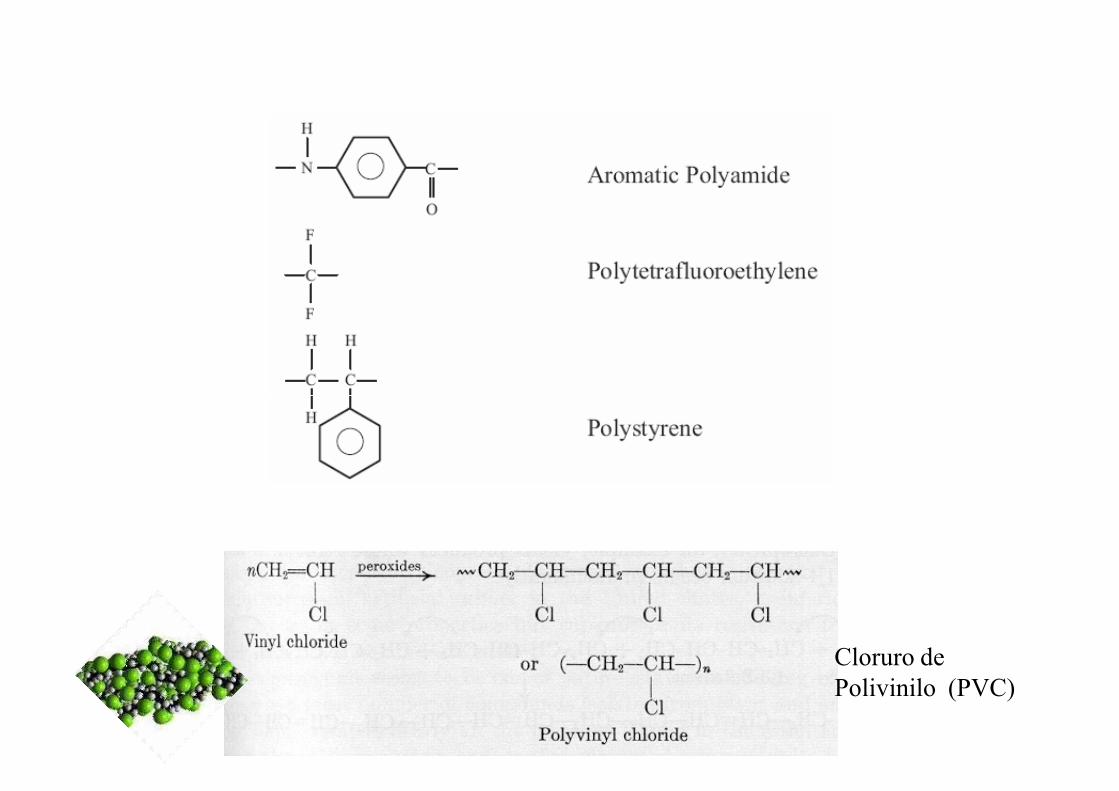
Cloruro de Polivinilo (PVC)
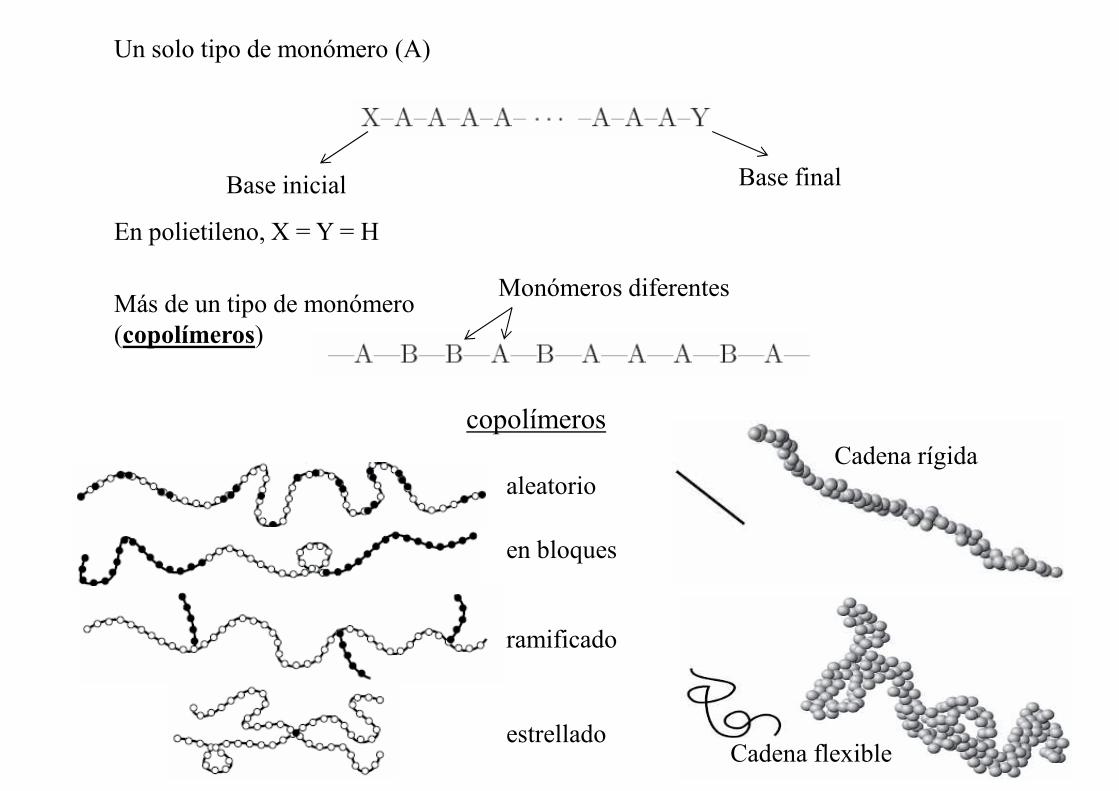
Un solo tipo de monómero (A)
Base inicial Base final
En polietileno, X = Y = H
Más de un tipo de monómero(copolímeros)
Monómeros diferentes
aleatorio
en bloques
ramificado
estrellado
copolímerosCadena rígida
Cadena flexible
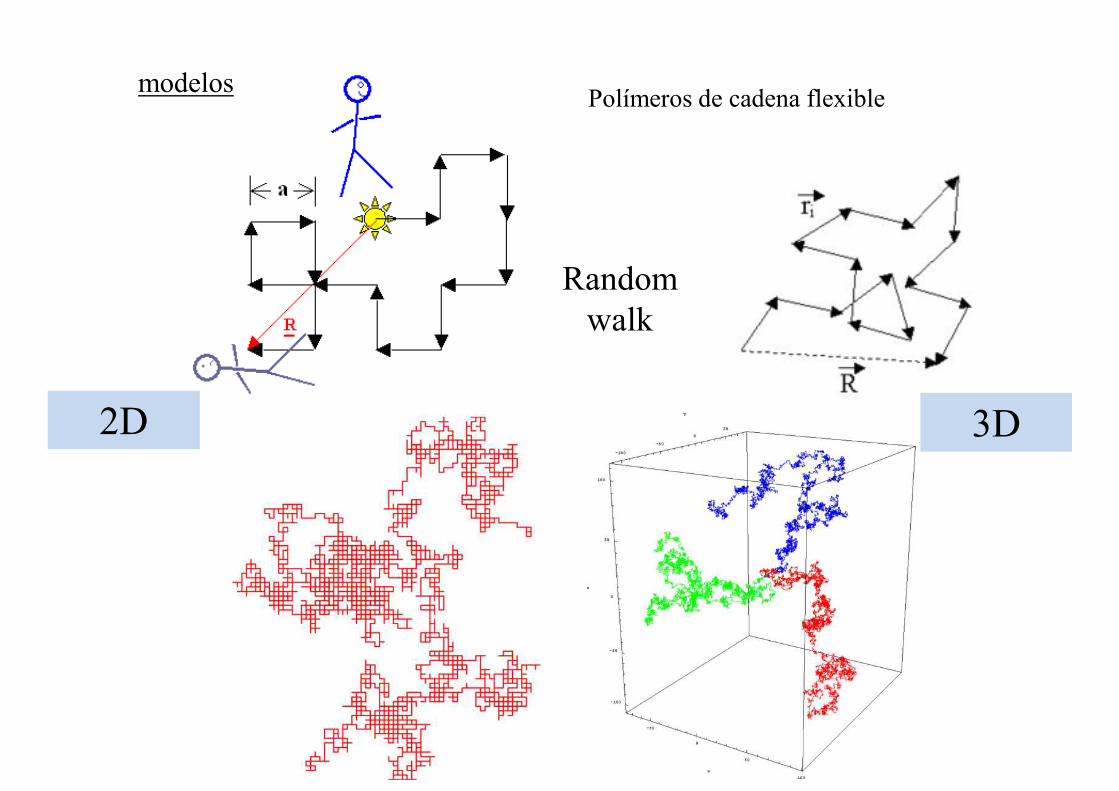
modelos
Random walk
Polímeros de cadena flexible
2D 3D
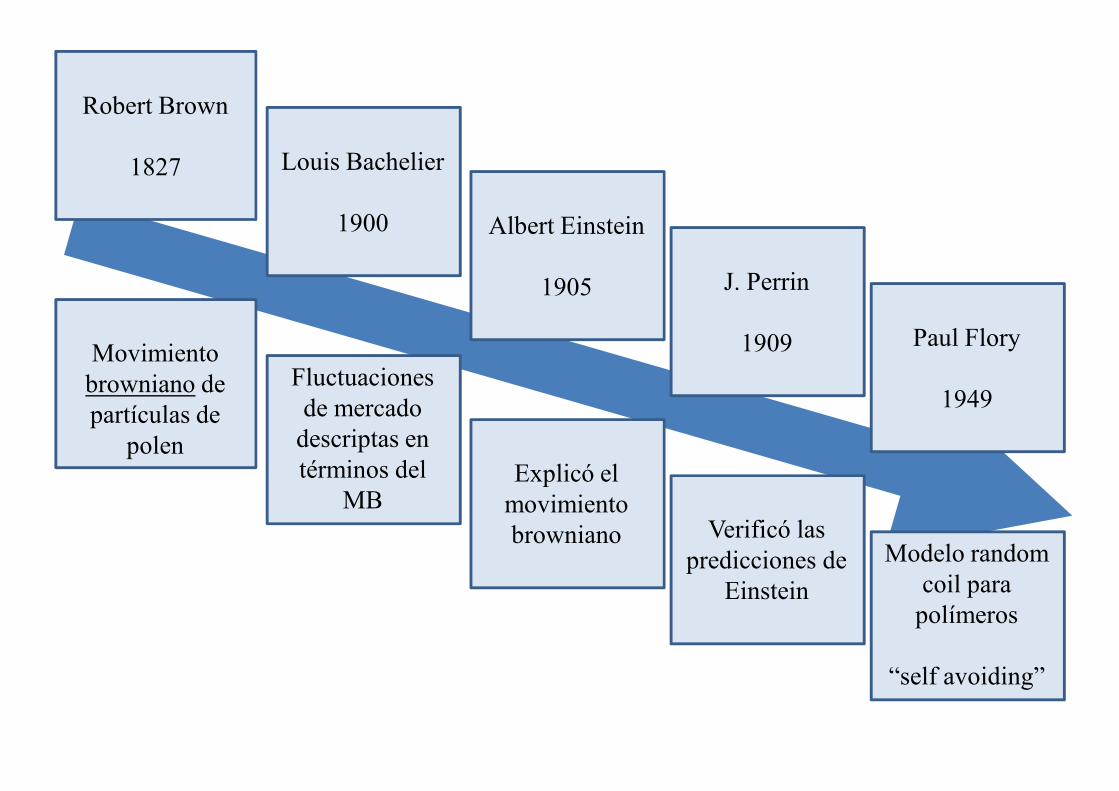
Albert Einstein
1905
Paul Flory
1949
J. Perrin
1909
Robert Brown
1827
Movimiento browniano de
Louis Bachelier
1900
Fluctuaciones de mercado
Explicó el movimiento browniano
1949
Modelo random coil para
polímeros
“self avoiding”
Verificó las predicciones de
Einstein
browniano de partículas de
polen
de mercado descriptas en términos del
MB
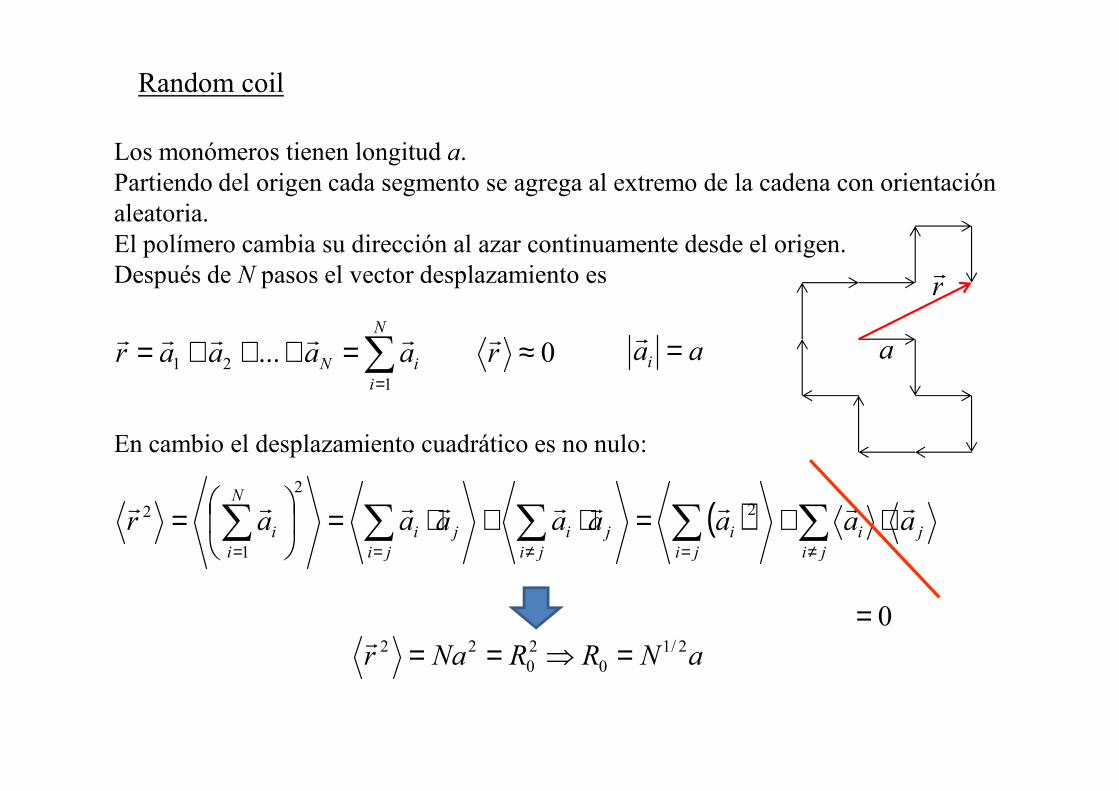
Random coil
Los monómeros tienen longitud a. Partiendo del origen cada segmento se agrega al extremo de la cadena con orientación aleatoria. El polímero cambia su dirección al azar continuamente desde el origen. Después de � pasos el vector desplazamiento es
0...1
21 ≈=+++= ∑=
raaaar�
i
i�
rrrrrra
rr
aai =r
( ) ∑∑∑∑∑≠=≠==
⋅+=⋅+⋅=
=ji
ji
ji
i
ji
ji
ji
ji
�
i
i aaaaaaaarrrrrrrrrr 2
2
1
2
a�RR�ar 2/10
20
22 =⇒==r
En cambio el desplazamiento cuadrático es no nulo:
0=
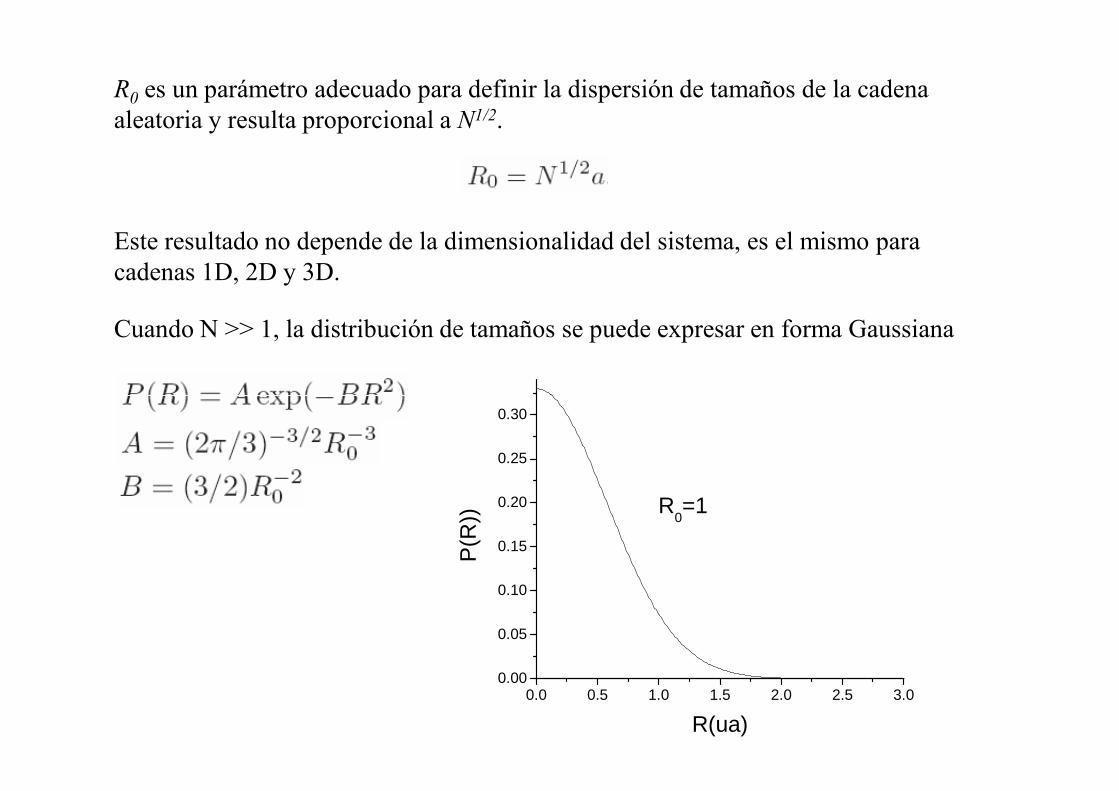
R0 es un parámetro adecuado para definir la dispersión de tamaños de la cadena aleatoria y resulta proporcional a �1/2.
Este resultado no depende de la dimensionalidad del sistema, es el mismo para cadenas 1D, 2D y 3D.
Cuando N >> 1, la distribución de tamaños se puede expresar en forma Gaussiana
0.0 0.5 1.0 1.5 2.0 2.5 3.00.00
0.05
0.10
0.15
0.20
0.25
0.30
P(R
))
R(ua)
R0=1

El modelo random coil es el más simple, pero los resultados presentan múltiples cruces que en la práctica representarían uniones adicionales. Éstas no son practicables a causa de las interacciones estéricas repulsivas. A causa de ello se introduce el modelo “self-avoiding walk”. Se impone la condición de que la cadena no puede cruzarse a sí misma.
Cruce evitado
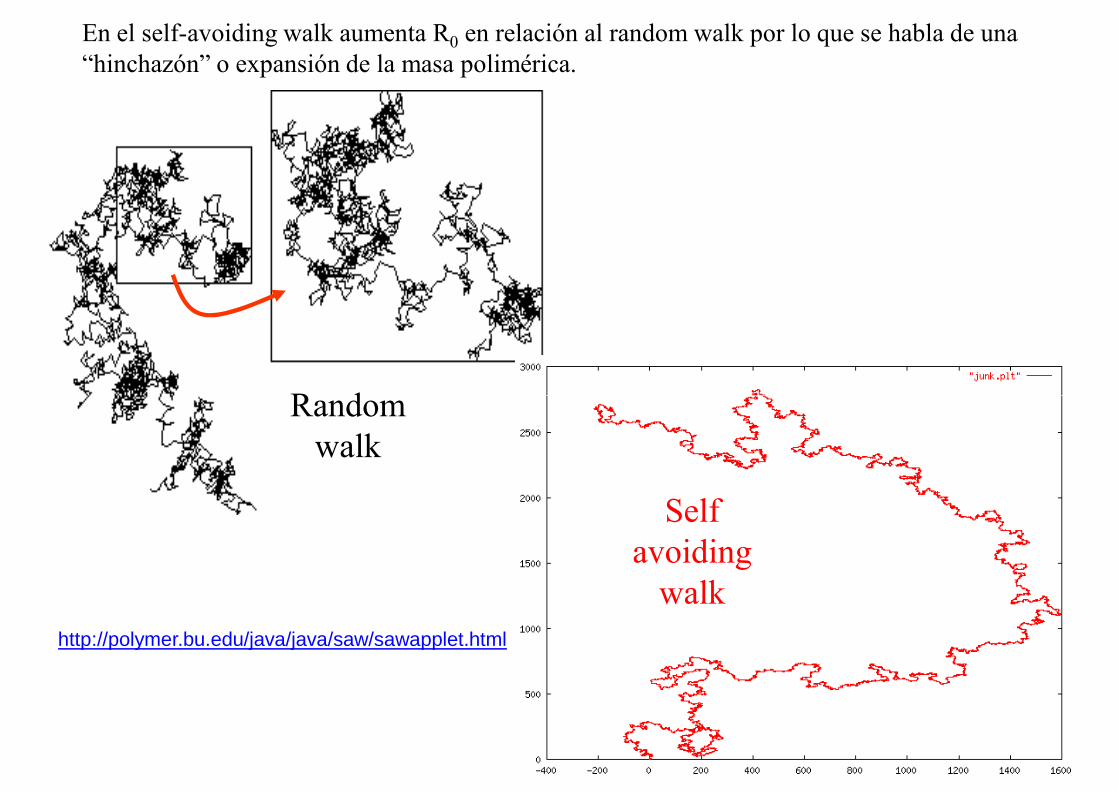
Random
En el self-avoiding walk aumenta R0 en relación al random walk por lo que se habla de una “hinchazón” o expansión de la masa polimérica.
Random walk
Self avoiding
walkhttp://polymer.bu.edu/java/java/saw/sawapplet.html

El self-avoiding walk es un problema matemático difícil que aún no tiene solución analítica. A partir de aproximaciones o simulaciones se encuentra que
Donde ν depende de la dimensíonalidad de la cadena
33
24
3
11
==
==
==
d
d
d
ν
ν
ν
35
3 == dν
Para una cadena unidimensional , es decir crece siempre en mismo sentido porque de lo contrario se intersectaría a sí misma.
�aR =0
Como νSAW > νRW (para d = 1 – 3) las cadenas SAW resultan de tamaño mayor.
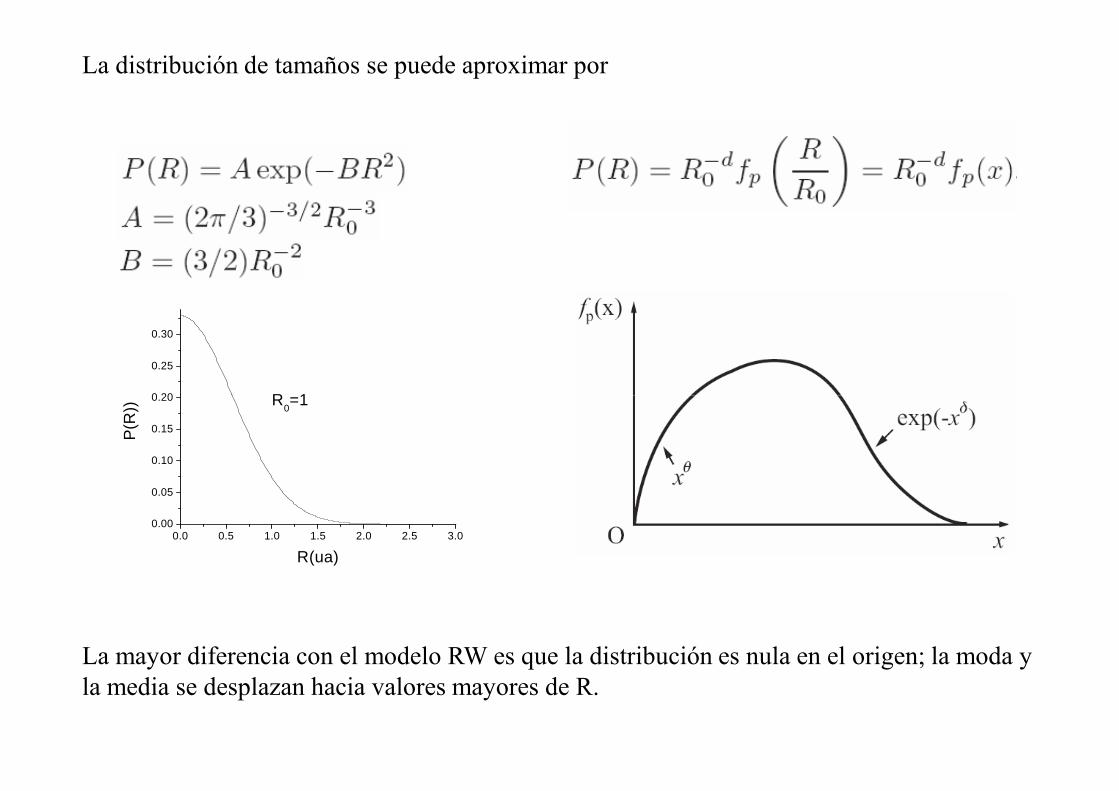
La distribución de tamaños se puede aproximar por
0.20
0.25
0.30
R =1
La mayor diferencia con el modelo RW es que la distribución es nula en el origen; la moda y la media se desplazan hacia valores mayores de R.
0.0 0.5 1.0 1.5 2.0 2.5 3.00.00
0.05
0.10
0.15
0.20
P(R
))
R(ua)
R0=1

Polímeros parcialmente ordenados
Un fenómeno que conduce a la cristalización de polímeros es el plegamiento de la cadena molecular (molecular chain folding), equematizado en la figura para una película con un espesor de varios micrones.
Película de polímero cristalizado

En la escala macroscópica zonas cristalinas y amorfas se presentan mezcladas, prevaleciendo el amorfo (al menos 60 % en volumen)
La cristalización se favorece por la aplicación de esfuerzos lineales o por la La cristalización se favorece por la aplicación de esfuerzos lineales o por la precipitación desde una fase de cristal líquido. Por ejemplo la solución de moléculas de poliaramida en ácido sulfúrico concentrado presenta orden orientacional. Las fibras precipitadas desde esta solución tienen una tenacidad muy elevada, superior a la de cuerdas de acero. Ello se debe a que los enlaces C-C de las macromoléculas (que son muy fuertes) están orientados.
aceropolímero KK 8≈
poliaramida
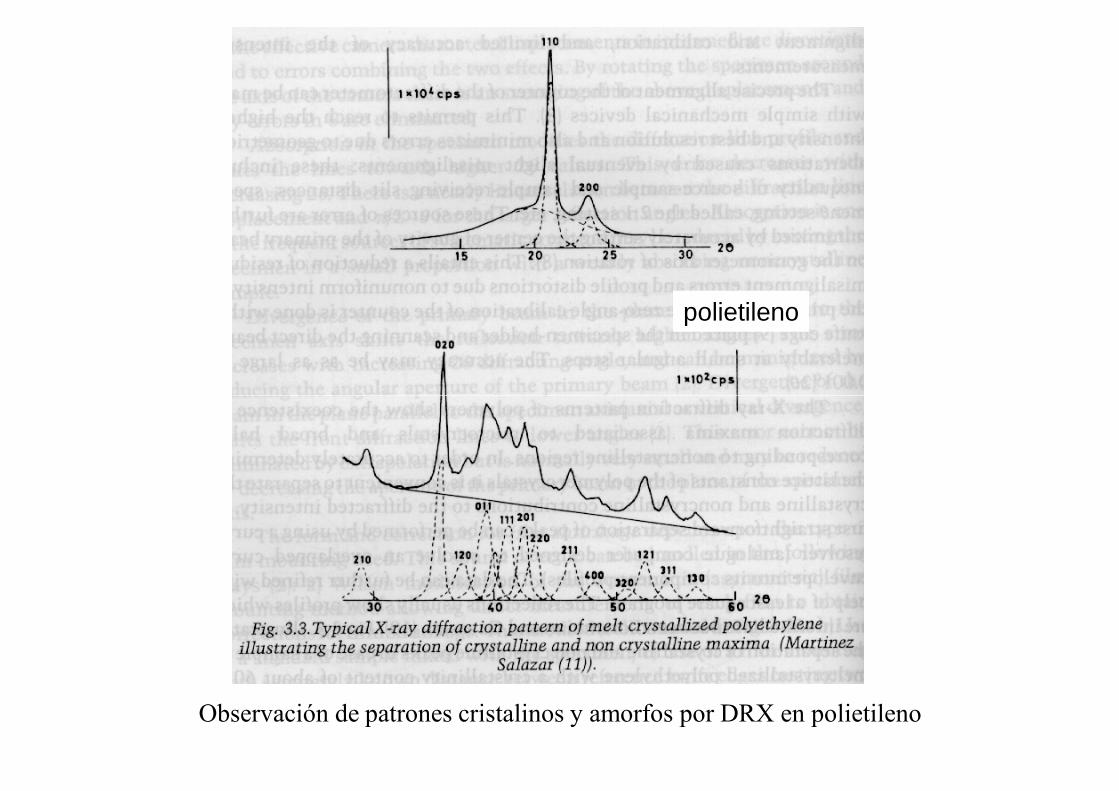
polietileno
Observación de patrones cristalinos y amorfos por DRX en polietileno
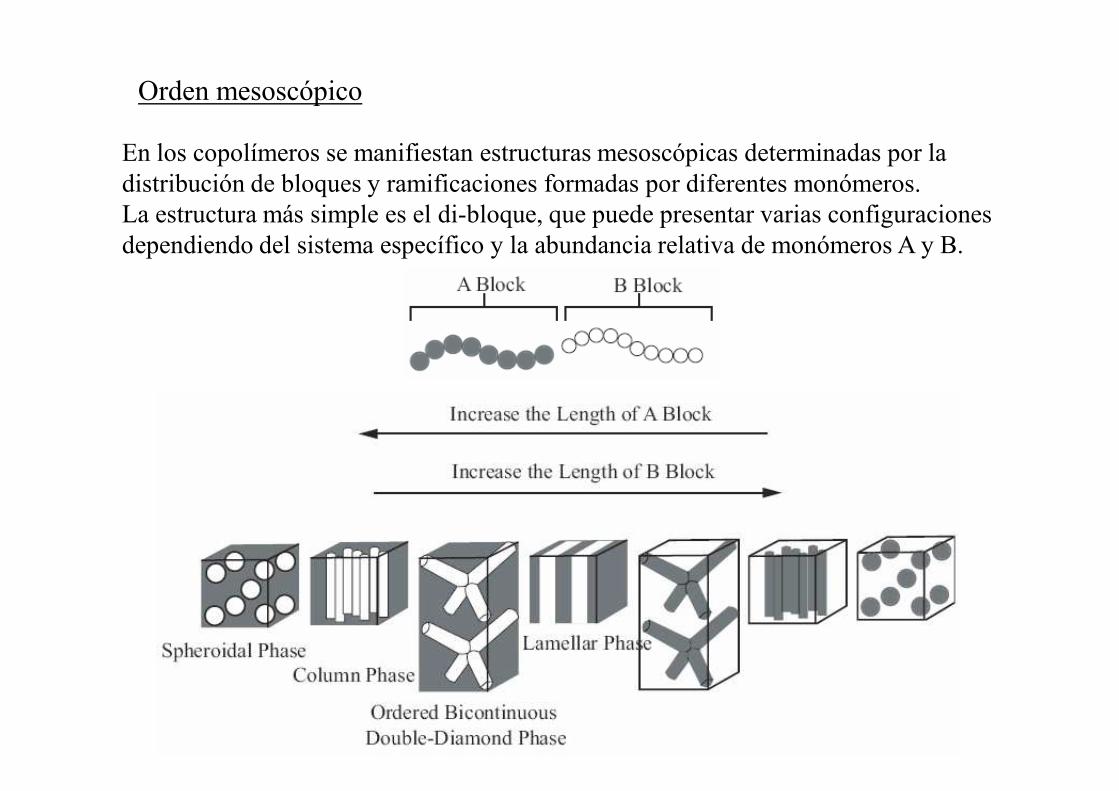
Orden mesoscópico
En los copolímeros se manifiestan estructuras mesoscópicas determinadas por la distribución de bloques y ramificaciones formadas por diferentes monómeros.La estructura más simple es el di-bloque, que puede presentar varias configuraciones dependiendo del sistema específico y la abundancia relativa de monómeros A y B.
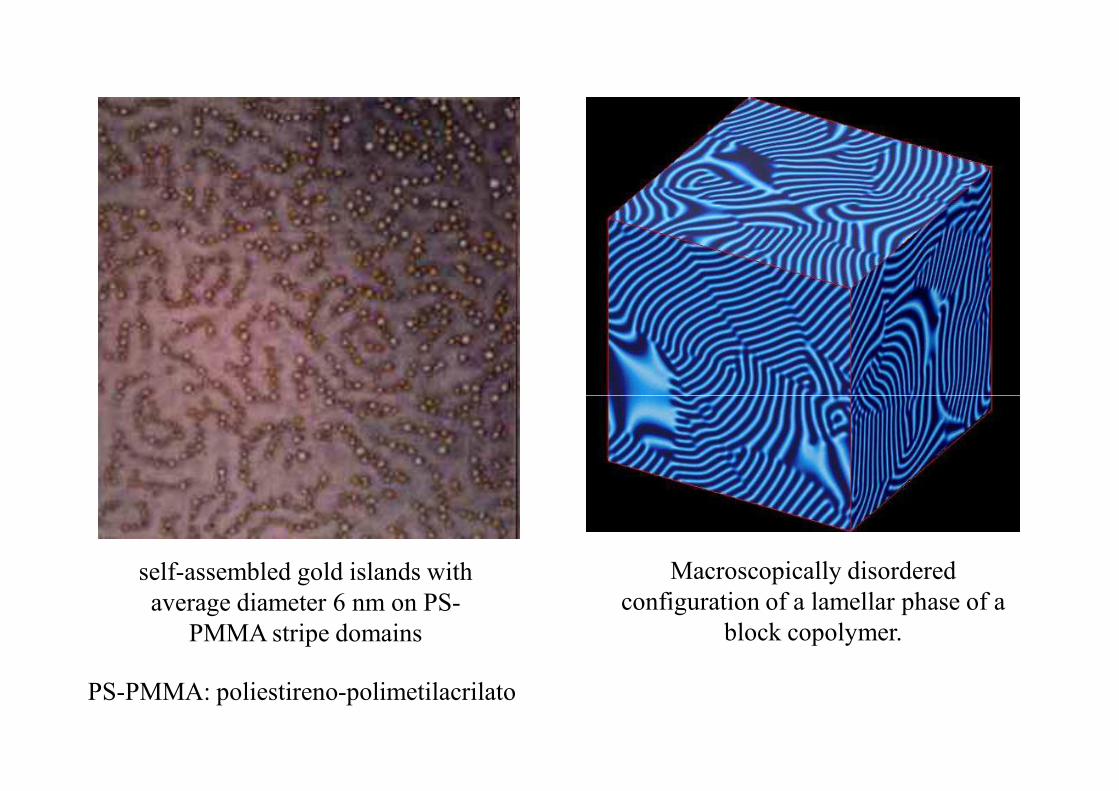
self-assembled gold islands with average diameter 6 nm on PS-
PMMA stripe domains
Macroscopically disordered configuration of a lamellar phase of a
block copolymer.
PS-PMMA: poliestireno-polimetilacrilato
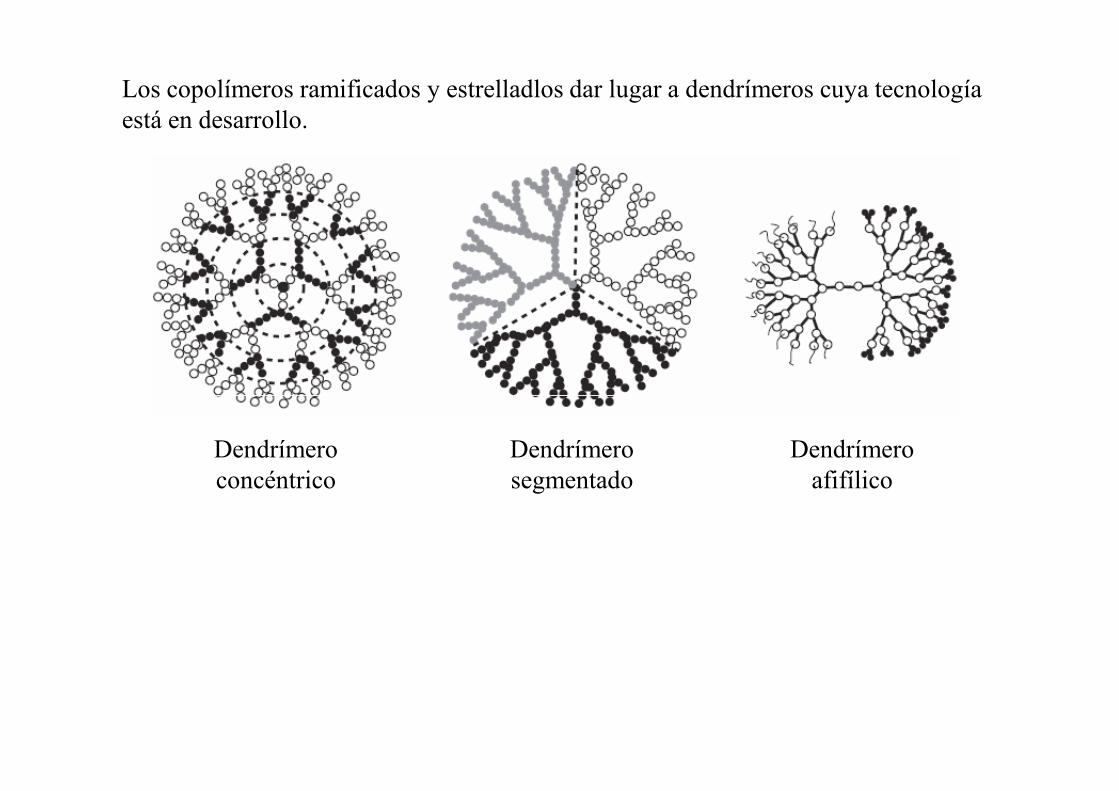
Los copolímeros ramificados y estrelladlos dar lugar a dendrímeros cuya tecnología está en desarrollo.
Dendrímero concéntrico
Dendrímero segmentado
Dendrímero afifílico
Top Related