







γλώσσες
Σελίδες
Νομικός


Functional Expression and Characterization of Histamine-gated Chloride Channels Arunesh Saras Ph.D. Dissertation To be presented by permission of the department of Cellphysiology of the Ruhr-University, Bochum
& International Graduate School of Neuroscience
2005

Contents
i
Contents Chapter 1: Introduction
1.1 GABA-receptors and the GABAergic system
1.1.1 The inhibitory γ-aminobutyric acid system 1
- a general overview
1.1.2 GABA-receptors: GABAB and GABAC 1
1.1.3 General properties of GABAA receptors 2
1.1.4 Types of heteromultimeric GABAA receptors 4
and their location and properties
1.1.5 Homomultimeric GABAA receptors 5
1.1.6 Trafficking of GABA-receptors and interacting proteins 6
1.1.7 Potentiation and modulation of GABAA receptors 8
1.1.7.1 Modulation of GABAA receptors by Propofol 8
1.1.7.2 Modulation of GABAA receptors by further 13
chemicals
1.1.8 Function of distinct GABAA subunits in vivo 14
investigated by knockout mice
1.2 Histamine-receptors and the histaminergic system 17
1.2.1 Histamine in the nervous system 17
1.2.2 Metabotropic histamine receptors 18
1.2.3 Interaction of histamine antagonists with GABAA receptors 22
1.2.4 Ionotropic histamine receptors and direct modulatory 22
action of histamine to ion channels
1.2.5 Histamine functions and knockout mice 24
1.2.6 Diseases where histamine is involved 25
1.2.7 Aims of the work 27

Contents
ii
Chapter 2: Materials 28
2.1 Chemicals and enzymes 28
2.2 Drugs used for pharmacological characterizations 29
2.3 Primers 31
2.4 Standards for DNA 32
2.5 Consumption materials 32
2.6 Kits 33
2.7 RNase free materials and chemicals 33
2.8 Frequently used buffer 34
2.9 Bacterial strains 37
2.10 Plasmid vectors 37
2.11 Softwares 38
Chapter 3: Methods 39
3.1 Characterizing, isolating and concentrating nucleic acids 39
3.1.1 Determination of concentrations of nucleic acids 39
3.1.2 Gel electrophoresis 39
3.1.3 Phenol: chloroform extraction of nucleic acids 39
3.1.4 Ethanol precipitation of nucleic acids 40
3.1.5 QIAquick PCR-Purification Kit 41
3.1.6 RNA Extraction 42
3.1.7 Quick preparation of plasmid DNA 42
3.1.8 Maxi preparation of plasmid DNA using the 43
QIAGEN Plasmid Maxi Kit
3.2 PCR Methods 43
3.2.1 Reverse transcription 43

Contents
iii
3.2.3 PCR-based generation of chimeric cDNAs and 45 site-directed mutagenesis
3.2.4 Phosphorylation op PCR primers 46
3.3 Cloning of DNA 46
3.3.1 Restriction 46
3.3.2 Dephosphorylation 46
3.3.3 Polishing of DNA using T4 DNA polymerase and T4 PNK 47
3.3.4 Fill in reaction 47
3.3.5 Ligation of DNA 48
3.3.6 Culturing of bacteria 48
3.3.7 Transformation of plasmid DNA 48
3.3.8 Sequencing of DNA 48
3.4 RNA techniques 48
3.4.1 In vitro transcription 49
3.5 Functional expression of LGICs in Xenopus laevis 50
3.5.1 Surgery 50
3.5.2 Oocyte preparation and injection of cRNA 50
3.5.3 Electrophysiological recording using two-electrode 51
voltage clamp
3.6 Functional Expression of LGICs in HEK 293 cells 52
3.6.1 Culture of HEK 293 cells and transfection 52
3.6.2 Patch clamp investigation of GABA receptors 53
expressed in HEK 293 cells
Chapter 4: Results I 55
4.1 Bioinformatical search for histamine-gated channels 55
4.2 Construction of expression vectors for GABAA receptors 56
4.3 Establishing functional expression GABAA receptors in 61
Xenopus oocytes
4.4 Direct effects of histamine on heteromultimeric GABAA receptors 65
4.5 Modulation of heteromultimeric GABAA receptors by histamine 65

Contents
iv
4.5.1 Potentiation of α1β1 GABAA receptors by histamine 65
4.6.1 Potentiation of α1β1 GABAA receptors by histidine 69
4.6.2 Characterization of histidine potentiation 70
4.6.3 Dependence of average histamine and histidine 72
potentiation on GABA concentration
4.6.4 Histidine does not alter the I/V curve and retains 73
selectivity for the permeability of the channel
4.7 Potentiation of GABAA receptors in HEK 293 cells 74
4.8 Dependence of histamine potentiation on GABAA receptor 76
subunit combinations
4.9 Homomultimeric channels of β1 subunit and the effect of 81
histamine and histidine
4.9.1 Homomultimeric channels of β3 subunit and effect of histamine 84
4.10 Molecular cloning of ρ1 subunit of GABAC receptors 84
4.10.1 GABAC receptors: No potentiation by histamine and histidine 86
4.11 Possible mechanisms of the histamine action 87
4.11.1 Histamine binding site is different from pentobarbital 87
binding-site on β3 subunit of GABAA receptors
4.11.2 Histamine binding site is similar to propofol binding- 88
site on β3 subunit of GABAA receptors –
Experiments on homomultimeric β3 subunit
4.11.3 Histamine binding site is similar to propofol binding 90
Site on β3 subunit of GABAA receptors
- Experiments on heteromultimeric α1β1 receptors
4.11.4 Effect of histamine on EC50 of GABA on GABAA receptors 91
4.11.5 Molecular cloning of point mutation in β1 subunits 96
4.11.6 β1(M286W) mutation completely abolishes potentitaion 96
mediated by histamine
4.11.7 Histamine and propofol have similar binding sites 98
4.11.8 β1 (M286W) mutation completely abolishes potentitaion 101
mediated by histidine
4.11.9 Sequence alignment with GABAC receptors depicts that 101
histamine has similar binding site to propofol

Contents
v
4.11.10 β1 (M286W) mutation does not interfere with the 102
potentiaion mediated by other modulators
Chapter 5: Results II 106
5. Characterization of homomultimeric β3 channels 106
5.1 Homomultimeric β3 receptors behave like histamine-gated ion channels 106
5.2 Histamine gated homomultimeric β3 receptors behave like typical 106
ligand-gated chloride channels
5.3 Relative comparison of various agonist of histamine with GABA 110
- relative agonists efficacy compared to GABA
5.4 Pharmacological characterization of histamine-gated homomultimeric 112
β3 receptors
5.4.1 Thioperamide acts as a competitive blocker for 113
histamine-evoked current
5.5 Comparison of L- and D-histidine action on β3 homomultimeric receptors 116
5.6 Inhibition of Propofol-induced current by thioperamide on 118
homomultimeric β3 receptors
Chapter 6: Results III 121
6. Characterization of homomulitmeric β2 channels 121
6.1 Homomultimeric β2 receptors behave like histamine-gated ion channels 121
6.2 Histamine gated homomultimeric β2 receptors behave like typical ligand- 122
gated chloride channels.
6.3 Relative comparison of various agonists of histamine with GABA 122
6.4 Pharmacological characterization of histamine-gated homomultimeric 122
β2 receptors
6.5 Relative comparison of homomultimeric β2 and β3 homomultimeric 126
receptors

Contents
vi
Chapter 7: Results IV 132
7. Characterization of homomultimeric ε subunit 132
7.1 Characterization of ε subunit containing receptors 132
7.2 ε contianing receptors behave like histamine-gated ion channels 133
7.3 Pharmacology of ε subunit 135
7.4 Expression of α1β1ε, β1ε and ε in HEK 293 cells 136
7.5 Molecular cloning of GFP-tagged subunits of GABAA receptors 136
7.5.1 Molecular cloning of GFP-tagged α1 and β1 subunits of 137
GABAA receptors
7.5.2 Molecular cloning of GFP-tagged ε subunit of 137
GABAA receptor
7.6 Expression of α1, β1 and ε in HEK 293 cells 142
Chapter 8: Results V 145
8.1 Properties of α1β2γ2 receptors and direct activation by histamine 145
8.2 Homomultimeric γ2 receptors: Activation by histamine and its 147
metabolites
Chapter 9: Discussion 150
Chapter 10: Summary 167
Chapter 11: References 169
Appendix: 189
Appendix I 189
Appendix II 190
Acknowledgements 191
Curriculum Vitae 192

Chapter 1 Introduction
1
1. INTRODUCTION
1.1: GABA-receptors and the GABAergic system
1.1.1: The inhibitory γ-aminobutyric acid system - a general overview
Gamma–aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the
mammalian central nervous system. It regulates many physiological functions and
emotional and cognitive behaviors through neurosynaptic contacts widespread in the
brain (Costa, 1982). In the mammalian brain the GABA is synthesized primarily from
glutamate in a reaction which is catalyzed by two glutamic acid decarboxylase (GAD)
enzymes, GAD65 and GAD67 (Bloom and Iversen, 1971). In the synaptic vesicle GABA
is loaded by a vesicular neurotransporter (VGAT) (Fon and Edwards, 2001) and it is
liberated into nerve terminal by calcium dependent exocytosis. However, no-vesicular
GABA secretion is being described and might play a role during development (Attwell et
al., 1993; Taylor and Gordon-Weeks, 1991). The effect of GABA can be mediated by
ionotropic or metabotropic receptors, which are localized post - or presynaptically. The
termination of GABA activation can happen either by its reuptake into the nerve
terminals or surrounding glial cells by a class of plasma membrane GABA transporters
(GATs) (Cherubini and Conti, 2001). Thereafter, GABA is metabolized by a
transamination reaction that is catalyzed by GABA transaminase (GABA-T). GABA acts
on 3 types of receptors which are phylogenitically conserved across different species:
GABAA, GABAB and GABAC receptors (Friedl et al., 1988).
1.1.2: GABA-receptors: GABAB and GABAC
GABAB receptors are bicuculline insensitive, chloride independent, metabotropic
receptors (Hill and Bowery, 1981; Bowery et al., 1980; Nicoll, 1988) and belong to the
superfamily of G-protein coupled receptors. GABAB receptors were shown to mediate

Chapter 1 Introduction
2
presynaptic inhibition on some nerve endings and postsynaptic inhibition on some cell
bodies or dendrites. GABAB receptors exist as GABAB1a / GABAB2 and GABAB1b /
GABAB2 and are associated with G–proteins. They have seven transmembrane domains.
GABAB receptors are localized both pre- and postsynaptically and they use different
mechanisms at these locations to regulate cell excitability. Presynaptic inhibition occurs
through a GABAB receptor mediated reduction in calcium current at the nerve terminal
and a subsequent reduction in transmitter release, whereas postsynaptic inhibition occurs
by GABAB receptor mediated activation of potassium currents that hyperpolarize the
neuron (Connors et al., 1988).
Like GABAA receptors, GABAC receptors are ligand-gated ion channel receptors (Sigel,
1995; Johnston, 1996; Enz and Cutting, 1998). This receptor is a chloride-selective ion
channel, but is insensitive to the GABAA receptor antagonist bicuculline (Bormann and
Feigenspan, 1995). GABAC receptors are believed to be homo - or heteropentameric
proteins that are composed of a single or multiple ρ subunits. They are also different from
GABAB receptors being insensitive to baclofen but are responsive to cis-4-aminocrotonic
acid, a structural analogue of GABA. GABAC receptors can be considered as
pharmacological variants of GABAA receptors (Mehta and Ticku, 1999; Bormann, 2000).
1.1.3: General properties of GABAA receptors
The GABAA receptors are members of the ligand-gated ion channel superfamily, which
also includes nicotinic acetylcholine, glycine and serotonin (5-HT3) receptors. GABAA
receptors are the primary mediators of GABA-induced rapid inhibitory neurotransmission
(Sieghart, 1995) and are believed to be heteropentameric proteins that are constructed
from subunits derived from several related genes or gene families (Macdonald and Olsen,
1994). At present, six α subunits, three β subunits, three γ subunits, one δ subunit, one ε
subunit, one π subunit and one θ subunit have been identified in mammals (Macdonald
and Olsen, 1994; Schofield et al., 1987; Mehta and Ticku, 1999). All the subunits are
related to each other and have molecular weights of about 50 kD. These various subunits
provide enormous subunit combinations but only certain subunit combinations are

Chapter 1 Introduction
3
preferred (McKernan and Whiting, 1996). Native receptors contain at least one α, one β
and one γ subunit. The δ, ε, π and θ subunits able to substitute for the γ–subunit
(McKernan and Whiting, 1996). GABAA receptors are integral membrane proteins,
which are formed by assembly of five homologous subunits around a central ion channel
(Chang et al., 1996). Each subunit has a large extracellular N-terminal domain and a C-
terminal domain containing four transmembrane segments, designated M1-M4, and
connected by relatively short loops. The extracellular N-terminal domains are believed to
form the agonist binding sites, whereas the transmembrane domains form the channel;
with the five M2 domains being the primary lining of the ion-conducting pore of the
receptor (Xu and Akabas, 1996). The M2 domain is thought to be a key channel-lining
component, which determines channel properties such as conductance, rectification, and
desensitization. Determined by the pore-forming M2-region, GABAA receptors carry
primarily chloride ions, however other anions, such as bicarbonate (HCO3-) can also
permeate the channel pore, although less efficiently (Kaila, 1994; Bormann et al., 1987;
Moss and Smart 2001).
Fig. 1.1: Structure of GABAA receptors. Proposed structure of a ligand gated ion channel. A receptor subunit contains four hydrophobic transmembrane (TM) domains. TM2 is believed to form the lining of the ion channel. The large amino terminal domain is located extracellularly and believed to incorporate neurotransmitter and some modulators binding sites. The intracellular domain in between TM3 and TM4 comprises ~ 10 % of the mass of each subunit. This domain is the most divergent part of individual receptor subunits and contains numerous consensus sites for the action of both serine/threonine and lysine protein kinases. Adapted from (Moss and Smart, 2001).

Chapter 1 Introduction
4
1.1.4: Types of heteromultimeric GABAA receptors and their location and
properties
Although molecular biology revealed seven types of homologous GABAA subunit types
by now, the subunit composition and the arrangement of subunits within a functional
GABAA receptor in the brain remains unknown in detail. Of the many subunit
combinations that are theoretically possible, only a few dozen have been shown to exist,
reflecting the differential distribution of subunit types among brain regions (Wisden et
al., 1992; Fritschy and Mohler, 1995; Pirker et al., 2000). The most abundantly
expressed receptor subtype in the brain is formed from α1, β2 and γ2 subunits (Sieghart
and Sperk, 2002; McKernan and Whiting, 1996; Whiting, 2003). The likely
stoichiometry is two α, two β and one γ subunit (Tretter et al., 1997; Farrar et al., 1999),
with the subunits arranged around the ion channel pore in the sequence γ-β-α-β-α,
(Baumann et al., 2002). Other common assemblies also contain α, β and γ2 subunits (for
example, α2β3γ2, α3β3γ2, α4βxγ2, α5β3γ2 and α6βxγ2), whereas receptors in which the γ2
subunit is replaced by γ1, γ3, or δ are less abundant. Further variability arises from the fact
that individual pentamers might contain two different α or two different β subunit
isoforms (Sieghart and Sperk, 2002). In some cases, the γ subunit can be replaced by α, ε,
α or π subunit, and the π and θ subunits might also be capable of co-assembling with α, β
and γ subunits to form receptors that contain subunits from four families (Neelands and
Macdonald, 1999; Bonnert et al., 1999). This molecular heterogeneity has important
functional consequences for GABAA receptor subtypes: subunit composition dictates not
only the properties of the receptors, but also their cell surface distribution and dynamic
regulation (Luscher and Keller, 2004; Sieghart and Sperk, 2002; Hevers and Luddens,
1998).
A combination of several methods allowed more precise subcellular localization of
GABAA receptors, and enrichment of the α1, α2, α3, α6, β2, β3 and γ2 subunits within the
postsynaptic membrane of GABAergic synapses. Each of these receptor subunits was
also found in extrasynaptic plasma membranes, and no GABAA receptor subunit type has

Chapter 1 Introduction
5
yet been found to have an exclusively synaptic location. Even in the case of α1β2γ2
GABAA receptors, which are highly enriched in synapses, more receptors are found
outside than inside synaptic junctions. Some GABAA receptors do not seem to
accumulate at synaptic junctions; for example, the δ subunit was shown to be present
exclusively in the extra - synaptic somatic and dendritic membranes of cerebellar granule
cells (Nusser et al., 1995) and at extra-synaptic and peri-synaptic locations in
hippocampal dentate gyrus granule cells (Wei et al., 2003). The lack of a γ subunit is
probably responsible for δ subunit failure to be incorporated at the synapse and δ subunit
containing receptors seems to be purely extra - synaptic. In general, receptors containing
a γ2 subunit in association with α1, α2, α3 subunits are the predominant receptor subtypes
that mediate synaptic inhibition and receptors that contain α4 or α6 subunits in
combination with δ subunits are predominantly or exclusively extra synaptic.
A vital property of a ligand-gated ion channel is its sensitivity to endogenous agonists.
For recombinant receptors that contain α, β and γ subunit, sensitivity to GABA is most
strongly affected by the type of α subunit that is present, with α3 subunits conferring the
highest and α6 subunits the lowest EC50 values (Knoflach et al., 1996; Fisher and
Macdonald, 1997; Bohme et al., 2004; 2004; Minier and Sigel, 2004). The absolute
EC50 values for specific subunit combinations reported by different groups is
considerably variable, but, in studies in which α subunits have been compared, the rank
order was shown to be α6 < α1 < α2 < α4 < α5 < α3, Bohme et al., 2004). Replacing the
γ2 subunit in α4β3γ2 assemblies with a δ subunit decreases the EC50 for GABA (Brown et
al. 2002). Overall α6β3γ2 or α6β3δ combinations have the lowest EC50s for GABA (~0.3–
0.7 µM), whereas for α1β3γ2 or α2β3γ2 subtypes they are an order of magnitude higher
(~6–14 µM).
1.1.5: Homomultimeric GABAA receptors
It is reported that some GABAA receptor subunits indeed form homomultimeric channels.
Among these subunits, the β subunit is thought to be a key component to assemble

Chapter 1 Introduction
6
heteromultimeric functional ion channels, to play a central role in determining the
subcellular locations of GABAA receptors (Connolly et al., 1996) and to bear binding
sites for agonists (Sigel et al., 1990; Amin and Weiss, 1993) and some clinically
important drugs such as general anesthetics (Cestari et al., 1996; Hill-Venning et al.,
1997). The β subunits are found to be capable of forming homomultimeric functional
channels when expressed in Xenopus oocytes or mammalian cells (Sigel et al., 1989;
Krishek et al., 1996). Channels composed of β1 subunits are constitutively active and
show spontaneous currents whereas GABAA receptors that contain β3 subunits are
inactive in the absence of GABA but they also form homomulitmeric channels, in which
the GABA current can be potentiated by pentobarbital and propofol. It was shown
(Martinez-Torres and Miledi, 2004) that the human γ2 subunit could also form
homomultimeric channels with an EC50 of 300 µM. The γ2 receptors were blocked by
bicuculline and were potentiated by pentobarbital and flunitrazepam. The other possible
homomultimeric receptors are suspected to be retained in the endoplasmic reticulum by
interactions with the Ig-binding protein BiP or calnexin and are then rapidly degraded
(Bollan et al., 2003; Gorrie et al., 1997).
1.1.6: Trafficking of GABA-receptors and interacting proteins
It is documented that GABAA receptors can be inserted and removed rapidly at synapses
(Kittler and Moss, 2003). This process is important in the synaptic inhibition and causes
the enhancement in the amplitude of miniature postsynaptic currents (mIPSC) (Wan et
al., 1997). Insulin induces the rapid insertion of GABAA receptors in to the synaptic
membrane by phosphorylating β subunits through Phosphoinositide-3 Kinase (PI3K)
(Wang et al., 2003). Conversely, removal of the receptors occurs by the activity of brain
derived neurotrophic factors (NFS) leading to suppression of mIPSC (Jovanovic et al.,
2004). Like glutamate receptors, there are both relatively immobile and highly mobile
GABAA receptors on the surface of neurons (Velazquez et al., 1989), with certain
subunits (for example, α1 and α6) being responsible for anchoring at the surface (Peran et
al., 2004). Several proteins have been identified that bind directly to GABAA to regulate
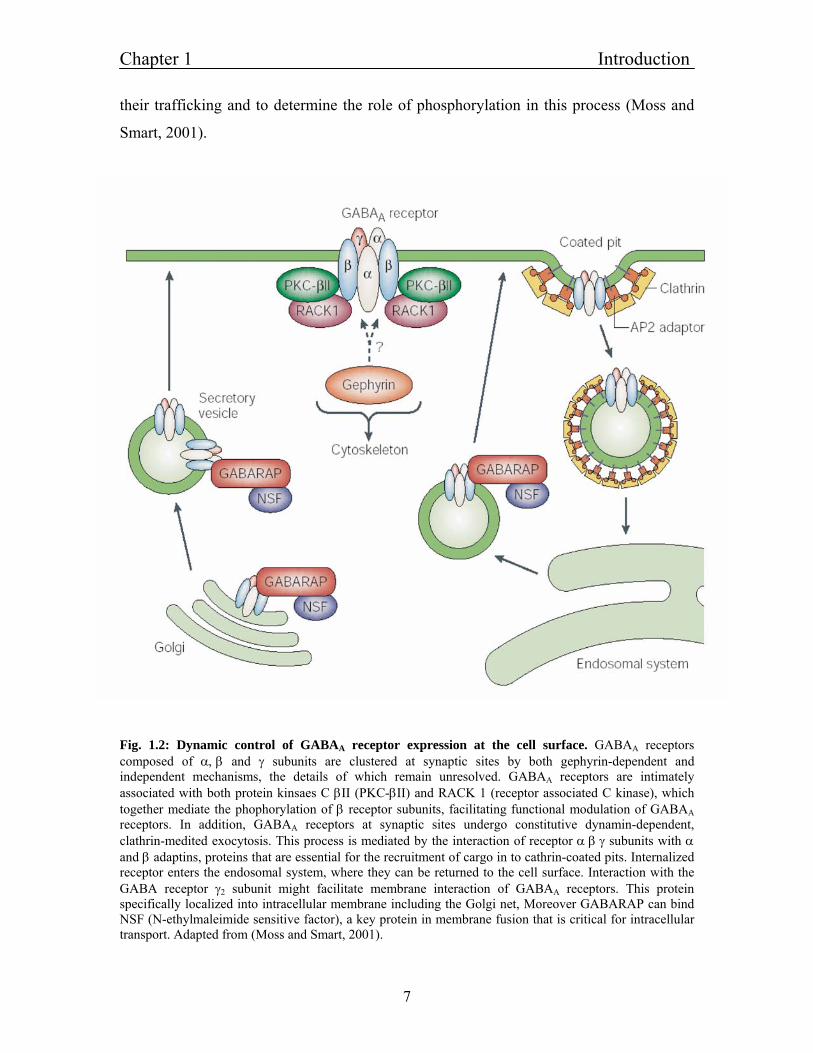
Chapter 1 Introduction
7
their trafficking and to determine the role of phosphorylation in this process (Moss and
Smart, 2001).
Fig. 1.2: Dynamic control of GABAA receptor expression at the cell surface. GABAA receptors composed of α, β and γ subunits are clustered at synaptic sites by both gephyrin-dependent and independent mechanisms, the details of which remain unresolved. GABAA receptors are intimately associated with both protein kinsaes C βII (PKC-βII) and RACK 1 (receptor associated C kinase), which together mediate the phophorylation of β receptor subunits, facilitating functional modulation of GABAA receptors. In addition, GABAA receptors at synaptic sites undergo constitutive dynamin-dependent, clathrin-medited exocytosis. This process is mediated by the interaction of receptor α β γ subunits with α and β adaptins, proteins that are essential for the recruitment of cargo in to cathrin-coated pits. Internalized receptor enters the endosomal system, where they can be returned to the cell surface. Interaction with the GABA receptor γ2 subunit might facilitate membrane interaction of GABAA receptors. This protein specifically localized into intracellular membrane including the Golgi net, Moreover GABARAP can bind NSF (N-ethylmaleimide sensitive factor), a key protein in membrane fusion that is critical for intracellular transport. Adapted from (Moss and Smart, 2001).

Chapter 1 Introduction
8
Direct binding partners include GABA-receptor-associated protein (GABARAP) (Wang
and Olsen, 2000; Wang et al., 1999), receptor for activated C-kinase (RACK1) (Brandon
et al., 1999), Src and AP2 (Mochly-Rosen and Gordon, 1998; Chang et al., 1998;
Yarwood et al., 1999). The GABA receptors are retained in the Golgi complex by
GABARAP protein (Wang et al., 1999) and their exit from this compartment could
involve interactions between GABARAP and N-ethyl maleimide sensitive factor (NSF)
and / or catalytically inactive phospholipase C (PLC)-related protein. Once inserted at
synapses, GABAA receptors are stabilized by their interaction with gephyrin and other
clustering molecules (Kneussel et al., 2001). Endocytosis of GABAA receptors might also
involve an interaction with (ubiquitin related protein), Plic-1, which could protect them
from degradation (Bedford et al., 2001).
1.1.7: Potentiation and modulation of GABAA receptors
The GABAA receptors are modulated by various chemical agents like benzodiazepines
(Sigel, 2002; Boileau and Czajkowski, 1999), neurosteroids (Rick et al., 1998),
barbiturates (Olsen et al., 1986), anesthetics (Krasowski et al., 1998) and alcohol (Mihic
et al., 1997). In total, GABAA receptors incorporate more than ten distinct binding sites
which have made this receptor a well recognized target for drug development (Korpi,
1994). In this study, it turned out that the modulatory site for propofol was the most
important one; therefore it is described in greater detail.
1.1.7.1: Modulation of GABAA receptors by Propofol
Propofol belongs to the class of general anesthetics enhancing GABAA-receptor function.
General anesthetic administration induces a state characterized by loss of consciousness,
amnesia, analgesia and immobility (Yamakura et al., 2001). At the level of ion channels,
intravenous anesthetic effects on GABAA receptors are concentration dependent. At low
concentrations, GABA-active anesthetic potentiate submaximal GABA-induced currents.
At higher concentration, they directly open channels in the absence of GABA
Chapter 1 Introduction
9
(Yamakura et al., 2001; Belleli et al., 1999). At even higher concentrations, some
anesthetics inhibit currents.
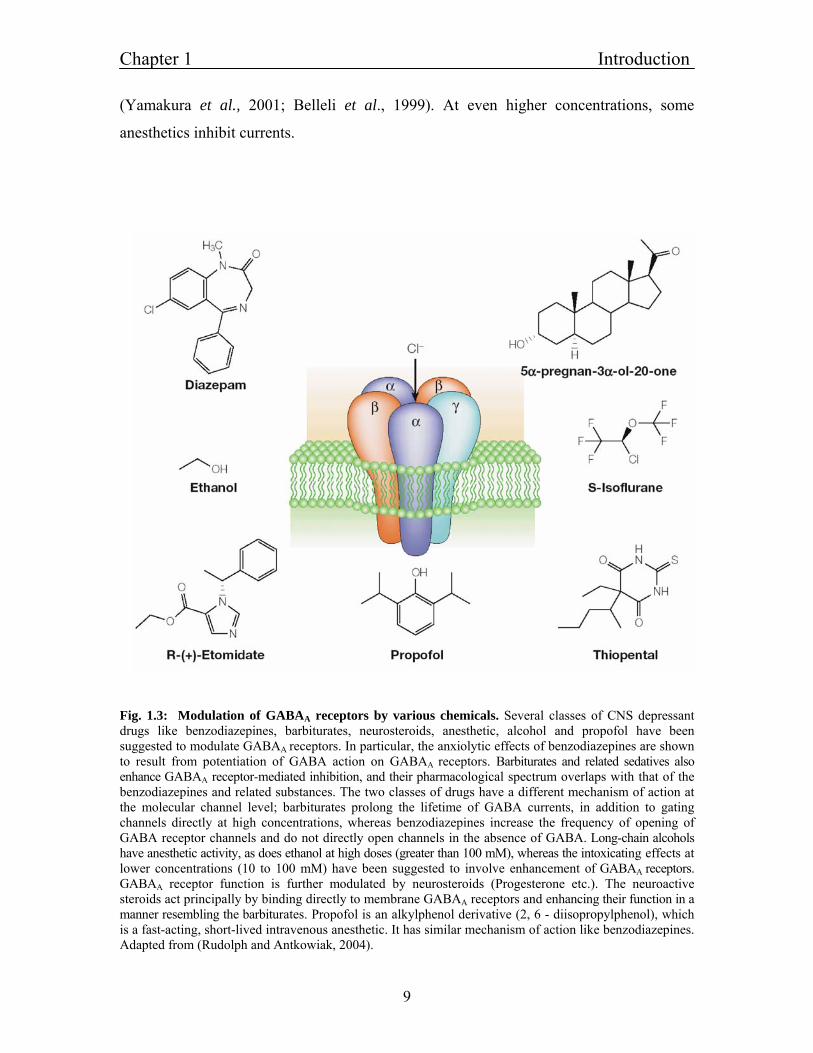
Fig. 1.3: Modulation of GABAA receptors by various chemicals. Several classes of CNS depressant drugs like benzodiazepines, barbiturates, neurosteroids, anesthetic, alcohol and propofol have been suggested to modulate GABAA receptors. In particular, the anxiolytic effects of benzodiazepines are shown to result from potentiation of GABA action on GABAA receptors. Barbiturates and related sedatives also enhance GABAA receptor-mediated inhibition, and their pharmacological spectrum overlaps with that of the benzodiazepines and related substances. The two classes of drugs have a different mechanism of action at the molecular channel level; barbiturates prolong the lifetime of GABA currents, in addition to gating channels directly at high concentrations, whereas benzodiazepines increase the frequency of opening of GABA receptor channels and do not directly open channels in the absence of GABA. Long-chain alcohols have anesthetic activity, as does ethanol at high doses (greater than 100 mM), whereas the intoxicating effects at lower concentrations (10 to 100 mM) have been suggested to involve enhancement of GABAA receptors. GABAA receptor function is further modulated by neurosteroids (Progesterone etc.). The neuroactive steroids act principally by binding directly to membrane GABAA receptors and enhancing their function in a manner resembling the barbiturates. Propofol is an alkylphenol derivative (2, 6 - diisopropylphenol), which is a fast-acting, short-lived intravenous anesthetic. It has similar mechanism of action like benzodiazepines. Adapted from (Rudolph and Antkowiak, 2004).

Chapter 1 Introduction
10
Propofol is an alkylphenol derivative (2, 6 - diisopropylphenol), which is a fast-acting,
short-lived intravenous anesthetic. The behavioral actions of propofol cover a large
concentration range. High concentration produces sleep, sedation, hypnosis and
immobility, whereas mild sedation and impairment of memory occurs at lower
concentration (around 3 % of those needed to induce immobility) (Veselis et al., 2002;
Smith et al., 1994). At sedative concentration, propofol reduces neuronal activity
prominently in cortical networks. At higher, hypnotic concentration, subcortical
structures, including the thalamus, midbrain reticular formation and possibly the
hypothalamus, are also affected (Rudolph and Antkowiak, 2004). Interestingly, there is a
linear relationship between the regional benzodiazepine binding site densities, consistent
with a similar mechanism of action of propofol and benzodiazpines (Alkire and Haier,
2001).
During propofol-induced hypnosis, global cerebral blood flow and glucose metabolism
seem to be significantly decreased, and some brain areas show a markedly higher degree
of depression than others. These regions are localized in diverse cortical areas, and also in
the thalamus and midbrain (Fiset et al., 1999; Alkire, 1998). Electroencephalography
(EEG) (Alkire, 1998) has provided evidence that thalamic structures are inhibited at
hypnotic propofol concentrations. In an elegant approach (Hofbauer et al., 2004) showed
that at mildly sedating concentration, human subjects ratings of thermal pains were
increased, and there was a corresponding increase in evoked activity in the thalamus and
somatosensory cortex. When subjects lost consciousness, noxious stimuli evoked
thalamic responses were abolished. Bonhomme used a similar experimental design,
tactile stimuli were applied during sedative and hypnotic propofol
concentration (Bonhomme et al., 2001). With hypnotic concentrations of propofol,
thalamic and cortical responses ceased. Magoun and Moruzzi found that several nuclei in
the midbrain reticular formation are involved in arousal, wakefulness and sleep and these
structures are plausible targets for general anesthetic to produce some of their sedative
and hypnotic effects (Moruzzi and Magon, 1949). General anesthesia and sleep share
some common features like depression of sensory input and motor output and similar
EEG patterns. Moreover, similar to sleep a recovery process takes place in anesthesia
Chapter 1 Introduction
11
(Tung et al., 2004). So, hypothalamic networks that are involved in sleep regulation
might have a key role in mediating anesthetic-induced hypnosis. The hypnotic effects of
several anesthetic applied to TM nucleus-hypothalamic region involved in regulation of
sleep and wakefulness, are consistent with such a mechanism (Nelson et al., 2002).
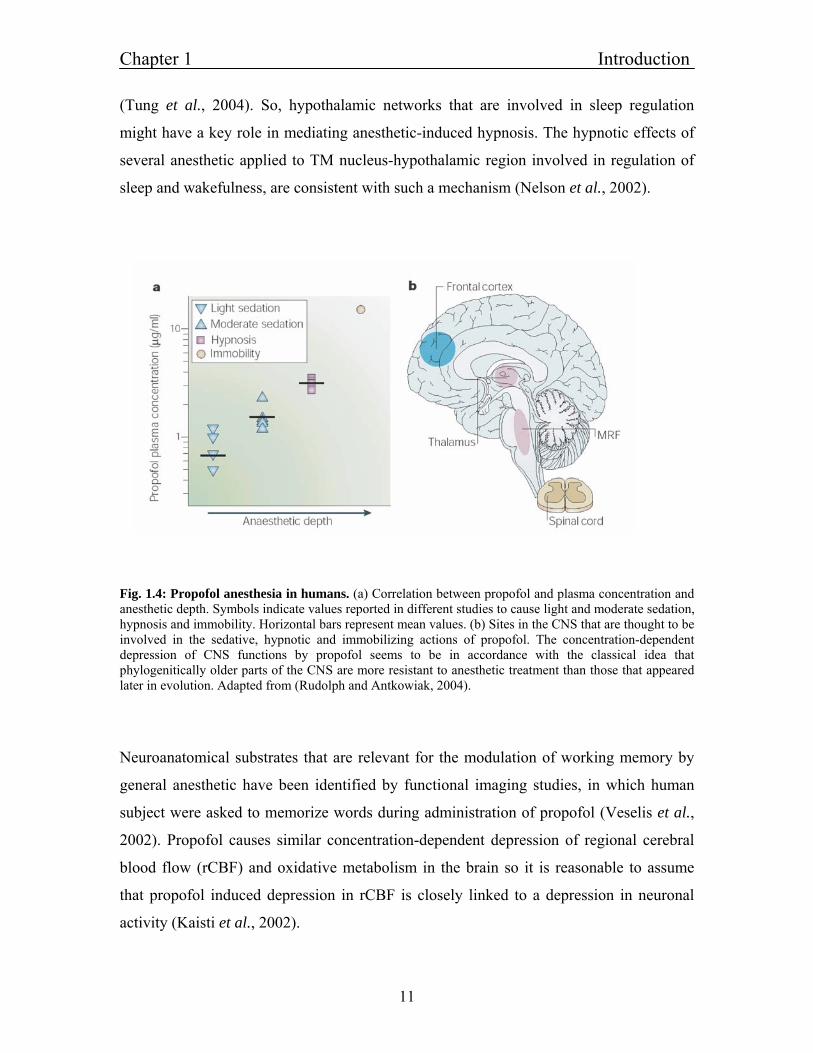
Fig. 1.4: Propofol anesthesia in humans. (a) Correlation between propofol and plasma concentration and anesthetic depth. Symbols indicate values reported in different studies to cause light and moderate sedation, hypnosis and immobility. Horizontal bars represent mean values. (b) Sites in the CNS that are thought to be involved in the sedative, hypnotic and immobilizing actions of propofol. The concentration-dependent depression of CNS functions by propofol seems to be in accordance with the classical idea that phylogenitically older parts of the CNS are more resistant to anesthetic treatment than those that appeared later in evolution. Adapted from (Rudolph and Antkowiak, 2004).
Neuroanatomical substrates that are relevant for the modulation of working memory by
general anesthetic have been identified by functional imaging studies, in which human
subject were asked to memorize words during administration of propofol (Veselis et al.,
2002). Propofol causes similar concentration-dependent depression of regional cerebral
blood flow (rCBF) and oxidative metabolism in the brain so it is reasonable to assume
that propofol induced depression in rCBF is closely linked to a depression in neuronal
activity (Kaisti et al., 2002).

Chapter 1 Introduction
12
The GABAA receptors have attracted considerable attention as a target for anesthetic
agents. Using knock-in point mutations in mice, (Jurd et al., 2003) have provided
definitive evidence that specific GABAA receptors are involved in the actions of propofol.
Whereas sites on both α and β subunits are crucial for volatile anesthetic action (Mihic et
al., 1997) for example α1-S270, α1-A291, β2-N265 and β2-M286, only the sites on β
subunits have been found to be relevant for the actions of the intravenous anesthetic
propofol (Krasowski et al., 1998). Two groups have recently reported the introduction of
point mutation in to β subunits on the GABAA receptors. Jurd showed the generation and
analysis of β3 (N265M) mice (Jurd et al., 2003). This point mutation abolished the
modulatory and direct effect of etomidate and propofol in vitro, and subsequently
reduced the modulatory actions of enflurane, whereas the modulatory actions of
neuroactive steroid alphaxalone was preserved (Siegwart et al., 2002). The duration of
the loss of the righting reflex in response to etomidate and propofol was reduced in β3
(N265M) mice compared with wild-type mice, indicating that the hypnotic activity is
mediated in part by GABAA receptors that contain the β3 subunit and in part by other
targets, possibly GABAA receptors that contain the β2 subunit. A point mutation in β1
subunit (M286W) abolished potentiation of GABA by propofol but did not alter direct
activation of the receptor by higher concentrations of propofol (Krasowski et al., 1998).
This point mutation in M3 of the β1 subunit (M286W) eliminated GABA potentiation by
1 µM propofol. In fact, submaximal GABA currents at the α2β1 (M286W) mutant
receptor were not enhanced by propofol at concentration up to 10 µM. Cysteine
substituted for these residues was used to determine whether propofol could protect it by
sulfhydryl – reactive reagents p-chloromercuribenzensulfonate (pCMBS) (Bali and
Akabas, 2004). The pCMBS reaction rate with an engineered Cys depends on two major
factors: first accessibility of the Cys to bulk solution and second reactivity of the Cys
with sulfhydrylreagents. Accessibilty depends on stearic and electrostatic factors in the
access pathway from bulk solution to the site of Cys. Their results showed that propofol
protected the substituted Cys at β2 M286W by a stearic effect caused by the local
presence of propofol and hence they concluded that this residue lies near the propofol-
binding site. The other β2 subunit residue, β2N265C was not protected by propofol.
Chapter 1 Introduction
13
Methionine oocupies a volume 43 Aº greater than asparagine and is more hydrophobic.
Thus, stearic bulk at position 265 can alter propofol binding, perhaps by inducing a
conformational change at the propofol binding site equivalent 100 Aº away.
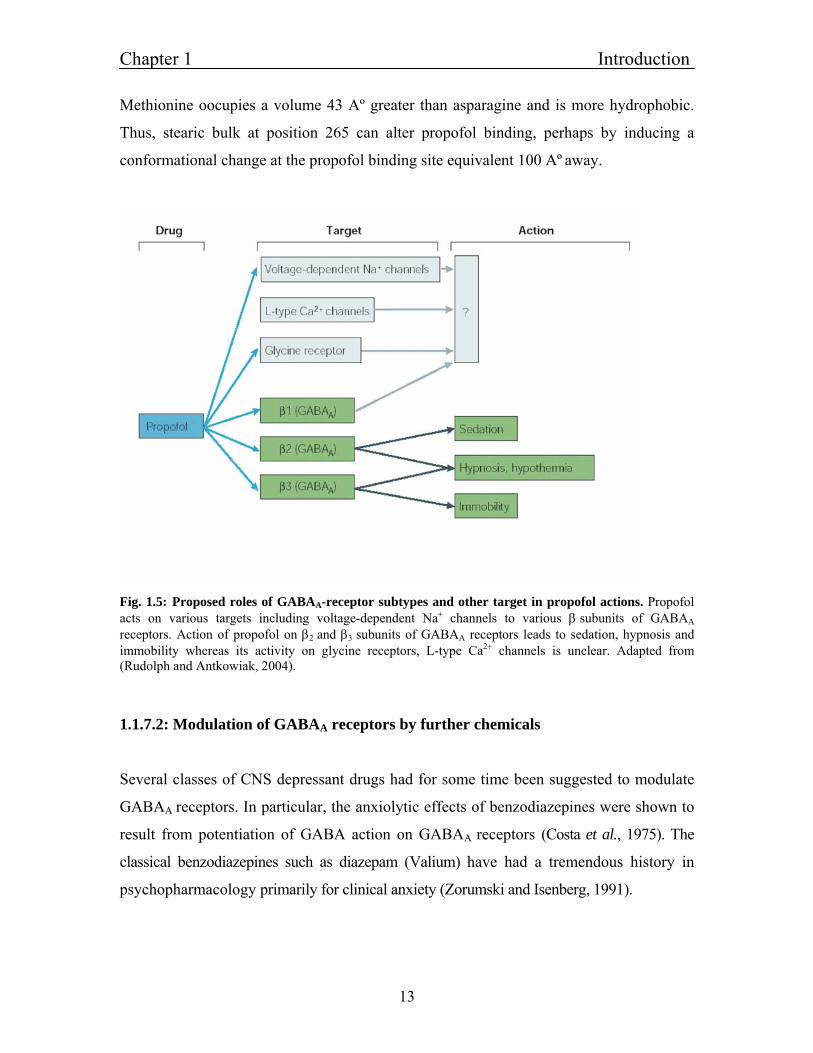
Fig. 1.5: Proposed roles of GABAA-receptor subtypes and other target in propofol actions. Propofol acts on various targets including voltage-dependent Na+ channels to various β subunits of GABAA
receptors. Action of propofol on β2 and β3 subunits of GABAA receptors leads to sedation, hypnosis and immobility whereas its activity on glycine receptors, L-type Ca2+ channels is unclear. Adapted from (Rudolph and Antkowiak, 2004).
1.1.7.2: Modulation of GABAA receptors by further chemicals
Several classes of CNS depressant drugs had for some time been suggested to modulate
GABAA receptors. In particular, the anxiolytic effects of benzodiazepines were shown to
result from potentiation of GABA action on GABAA receptors (Costa et al., 1975). The
classical benzodiazepines such as diazepam (Valium) have had a tremendous history in
psychopharmacology primarily for clinical anxiety (Zorumski and Isenberg, 1991).

Chapter 1 Introduction
14
Other uses of benzodiazepines include sedation, muscle relaxation, and a significant
utilization for treatment of panic (Biggio et al., 1995). Barbiturates and related sedatives also
enhance GABAA receptor-mediated inhibition, and their pharmacological spectrum
overlaps with that of the benzodiazepines and related substances. The selective actions of
benzodiazepines not shown by barbiturates or vice versa arise from heterogeneity in
GABA receptor sensitivity to the drugs, and corresponding heterogeneity in brain regions
and functions. Some GABA-receptors are insensitive to benzodiazepines but not to
barbiturates. In addition, the two classes of drugs have a different mechanism of action at
the molecular channel level; barbiturates prolong the lifetime of GABA currents, in
addition to gating channels directly at high concentrations, whereas benzodiazepines
increase the frequency of opening of GABA receptor channels and do not directly open
channels in the absence of GABA (Study and Barker, 1981). Alcohols are CNS
depressants known to enhance GABAA receptor currents with a pharmacological spectrum
of action overlapping those of the benzodiazepines and barbiturates. Long-chain alcohols
have anesthetic activity, as does ethanol at high doses (greater than 100 mM), whereas the
intoxicating effects at lower concentrations (10 to 100 mM) have been suggested to
involve enhancement of GABAA receptors (Suzdak et al., 1986). GABAA receptor function
is further modulated by neurosteroids. The neurosteroids are endogenous steroid hormone
metabolites that have direct and rapid actions on cells not involving steroid hormone
receptors or regulation of gene expression. Progesterone was shown to produce rapid
sedative activity. Progesterone has anxiolytic and anticonvulsant activity; discontinuation
after long-term administration leads to withdrawal signs that are clearly CNS mediated.
The neuroactive steroids act principally by binding directly to membrane GABAA
receptors and enhancing their function in a manner resembling the barbiturates (Lambert et
al., 1995).
1.1.8: Function of distinct GABAA subunits in vivo investigated by knockout
mice
Gene targeting and transgenic mice have demonstrated several important roles for GABA in
the CNS. Knockouts of both GAD67 and GABAA receptor subunit β3 lead to early

Chapter 1 Introduction
15
neonatal lethality (Asada et al., 1997). GAD65 knockout mice show increased anxiety,
increased sensitivity to benzodiazepines (Kash et al., 1999). Epilepsy results from knockout
of GAD65, GABA β3, and GABA receptor δ subunit. Mice targeted for this subunit have
a phenotype remarkably similar to Angelman syndrome, especially the epilepsy, but also
including the cognitive, motor and sleep impairment (DeLorey et al., 1998). The γ2 subunit
knockout mice show early neonatal lethality (Gunther et al., 1995), without cleft palate,
involving impaired clustering of GABAA receptors at synapses (Essrich et al., 1998).
Because GABA receptors are important drug targets, some GABA receptors subunit
knockout mice have impaired sensitivity to drugs, such as decreased response to
benzodiazepines in γ2 homozygous knockouts. Increased response to benzodiazepines is
seen in γ2 heterozygous knockouts or in γ2L null mutants (Quinlan et al., 2000). Reduced
sensitivity to anesthetics was seen in β3 but not α6 knockouts, and reduced sensitivity to
neuroactive steroids is observed in the δ subunit knockout (Mihalek et al., 1999).
Gene targeting in mice also has been employed to ‘‘knock in’’ a mutation of the α1 subunit
H101N, which prevents benzodiazepine binding to GABA receptors containing this subunit
(McKernan et al., 2000). The resulting animals have greatly impaired sensitivity to the
sedative but not the anxiolytic actions of the benzodiazepines, whereas anticonvulsant
activity is partially reduced. This finding indicates that the subtypes of GABA receptors
containing the α1 subunit and the brain circuits in which they function are the substrates
for benzodiazepine-stimulated sedation.
α1 subunit-containing GABAA receptors in forebrain contribute to the effect of inhaled
anesthetics on conditioned fear. Knockout mice were 75 to 145 % less sensitive to the
amnestic effects of the inhaled anesthetic isoflurane. These results indicate that α1-
containing GABAA receptors in the hippocampus, amygdala, and / or cortex influence the
amnestic effects of inhaled anesthetics (Sonner et al., 2005). Also α1 knockout mice
show impaired dendritic spine maturation. There was a concomitant decreased density of
mature mushroom-shaped spines, which became more pronounced in adults. In contrast,
dendritic arborization was not altered in these mice (Heinen et al., 2003). α5 knockout

Chapter 1 Introduction
16
mice showed enhanced learning and memory and altered GABAergic synaptic
transmission (Collinson et al., 2002). In the CA1 region of hippocampal brain slices from
α5 knockout mice, the amplitude of the IPSCs was decreased, and paired-pulse
facilitation of field EPSP (fEPSP) amplitudes was enhanced indicating α5 containing
GABAA receptors play a key role in cognitive processes by controlling a component of
synaptic transmission in the CA1 region of the hippocampus.
Requirement of α5 GABAA receptors for the development of tolerance to the sedative
action of diazepam in knock-in mice, in which the α1, α2, α3, or α5 GABAA receptors had
been rendered insensitive to diazepam by histidine-arginine point (van Rijnsoever et al.,
2004). A reduction in α5 subunit-containing gamma-aminobutyric acid GABAA receptors
has been reported to enhance some forms of learning in mutant mouse models (Yee et al.,
2004). Moreover, the largely extrasynaptic α5 GABAA receptors in hippocampal
pyramidal cells are implicated as control elements of the temporal association of threat
cues in trace fear conditioning (Crestani et al., 2002).
Wild type, α2 (H101R), and α3 (H126R) mice showed a robust diminution of the motor-
depressant drug action. In contrast, α5 (H105R) mice failed to display any sedative
tolerance. α1 (H101R) mice showed no alteration of motor activity with chronic
diazepam treatment. Thus, the chronic activation of α5 GABAA receptors is crucial for the
normal development of sedative tolerance to diazepam, which manifests itself in
conjunction with α1 GABAA receptors. To identify the molecular and neuronal target
mediating the anxiolytic action of benzodiazepines, (Low et al., 2000) generated and
analyzed two mouse lines in which the α2 or α3 GABAA receptors, respectively, were
rendered insensitive to diazepam by a knock-in point mutation. The anxiolytic action of
diazepam was absent in mice with the α2 (H101R) point mutation but present in mice
with the α3 (H126R) point mutation. These findings indicate that the anxiolytic effect of
benzodiazepine drugs is mediated by α2 GABAA receptors, which are largely expressed
in the limbic system, but not by α3 GABAA receptors, which predominate in the reticular
activating system. In another study it was shown that by introducing a histidine-to-

Chapter 1 Introduction
17
arginine point mutation at position 101 of the murine alpha1-subunit gene, that α1-type
GABAA receptors, are rendered insensitive to allosteric modulation by benzodiazepine-
site ligands, whilst regulation by the physiological neurotransmitter gamma-aminobutyric
acid is preserved (Rudolph et al., 1999). Alpha1(H101R) mice failed to show the
sedative, amnesic and partly the anticonvulsant action of diazepam. In contrast, the
anxiolytic-like, myorelaxant, motor-impairing and ethanol-potentiating effects were fully
retained, and are attributed to the nonmutated GABAA receptors found in the limbic
system (α2, α5), in monoaminergic neurons (α3) and in motoneurons (α2, α5).
1.2: Histamine-receptors and the histaminergic system
1.2.1: Histamine in the nervous system
Histamine is one of the aminergic neurotransmitters, playing an important role in the
regulation of several physiological processes. Histamine is synthesized and transported to
brains of almost all animal species. The content of histamine varies between species,
being higher in lower vertebrates and to be a lower level in mammals (Reite, 1972;
Almeida and Beaven, 1981). Histamine containing nerve cells in the brain are found
exclusively in the tubomamillary nucleus of the hypothalamus (TM nucleus) and they
project throughout the brain and to all fields of hippocampus (Schwartz et al., 1991). In
all mammals, the cerebral cortex, amygdala, substantia niagra and striatum receive
moderate or dense histaminergic innervations. The density of projections in the
hippocampus and thalamus varies, and the retina and spinal cord also receive
histaminergic fibers from the TM nucleus. Also, afferent projections to TM neurons are
wide spread and come from prominent sources like infralimbic cortex, lateral septum and
preoptic nucleus (Ericson et al., 1991). The brain stem innervations in to TM nucleus
include, the adrenergic cell group C1-C3, from noradrenergic groups A1-A3, and from
serotonergic group B5-B9 also, only few fibers from locus coeruleus and the
dopaminergic groups of substantia nigra and ventral tegmentum innervates TM nucleus.
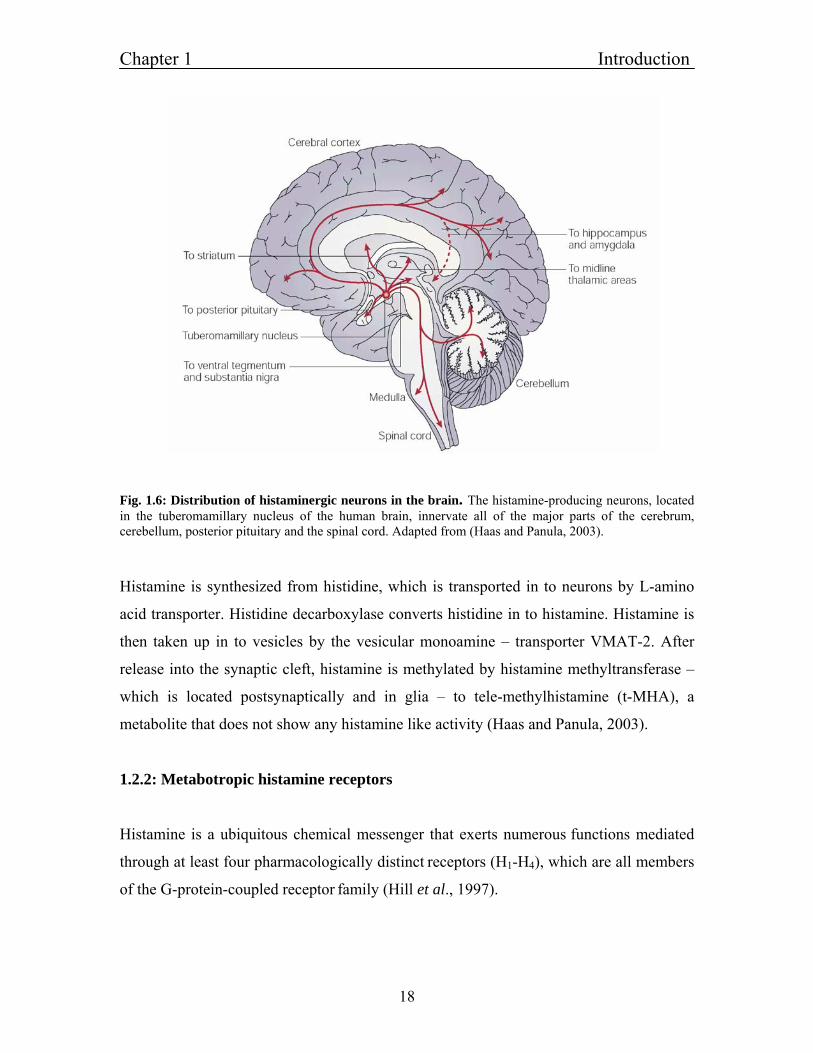
Chapter 1 Introduction
18
Fig. 1.6: Distribution of histaminergic neurons in the brain. The histamine-producing neurons, located in the tuberomamillary nucleus of the human brain, innervate all of the major parts of the cerebrum, cerebellum, posterior pituitary and the spinal cord. Adapted from (Haas and Panula, 2003).
Histamine is synthesized from histidine, which is transported in to neurons by L-amino
acid transporter. Histidine decarboxylase converts histidine in to histamine. Histamine is
then taken up in to vesicles by the vesicular monoamine – transporter VMAT-2. After
release into the synaptic cleft, histamine is methylated by histamine methyltransferase –
which is located postsynaptically and in glia – to tele-methylhistamine (t-MHA), a
metabolite that does not show any histamine like activity (Haas and Panula, 2003).
1.2.2: Metabotropic histamine receptors
Histamine is a ubiquitous chemical messenger that exerts numerous functions mediated
through at least four pharmacologically distinct receptors (H1-H4), which are all members
of the G-protein-coupled receptor family (Hill et al., 1997).

Chapter 1 Introduction
19
Fig. 1.7: Metabolism of Histamine in the neurons The L-amino-acid transporter brings histidine into neurons where histamine is synthesized by the specific enzyme histidien decarboxylase. Histamine is then taken up into vesicles by the vesicular monoamine-transporter VMAT-2. After release, histamine is methylated by histamine-methyltransferase which is located postsynaptically and in glia to tele-methylhistamine, a metabolite that does not show any histamine-like activity. Adapted from (Haas and Panula 2003).
The H1 receptor is expressed in the brain, endothelial cells, and smooth muscle cells. The
most characteristic roles for H1 receptor activation are smooth muscle contraction and
increases in vascular permeability (Ash and Schild, 1966). The H1 receptor is a 486-491
amino acid protein encoded by an intronless gene (Yamashita et al., 1991). H1 receptors
mediate excitatory actions on whole brain activity. At the cellular level excitation is
achieved by the activation of the Gq/11 heterotrimeric G-protein and its downstream
effector phospholipase C (PLC). Stimulation of the Gq/11 - PLC pathway by the H1-
receptor results in the synthesis of inositol-1, 4,5-trisphosphate and 1,2-diacylglycerol,
which in turn stimulate an increase in intracellular Ca2+ and the activation of protein
kinase C (PKC). H1 receptor activation can lead to activation of several other signaling
pathways like stimulation of nitric oxide synthase activity (via a Ca2+/calmodulin-
dependent pathway) and subsequent activation of soluble guanylyl cyclase in a variety of
different cell types (Leurs et al., 1991; Casale et al., 1985; Duncan et al., 1980).

Chapter 1 Introduction
20
The H2 receptor was first cloned from dog and later found in several species. H2 receptor
is an intronless gene and protein consists of 358-359 amino acids. The H2 receptor has
been demonstrated to function as a key modulator for gastric acid secretion, and H2
receptor antagonists are widely used for the treatment of gastrointestinal ulcers (Soll and
Walsh, 1979). The direct action on neuronal membranes is usually excitatory or
potentiates excitation. The H2 receptors signals through Gs-G-proteins, adenylyl cyclase
and PKA, which phosphorylates proteins and activates the transcription factor cyclic-
AMP response element binding protein.
The H3 receptor was first characterized as an auto-receptor - regulating histamine
synthesis and release from rat cerebral cortex, striatum, and hippocampus (Arrang et al.,
1983, 1985). H3-receptor-mediated inhibition of histamine release has also been observed
in human cerebral cortex (Arrang et al., 1988). H3 receptor is located presynaptically on
histaminergic neurons. By alternative splicing several isoform of H3 receptors, consisting
of 326-445 amino acids, are derived from a single gene. H3 also provides negative
feedback to the release of other transmitter such as glutamate, acetylcholine and
noradrenaline. H3 receptors are coupled to Gi\o and high voltage activated Ca2+ channels.
The H3 receptors are coupled negatively by cAMP and activates the mitogen activated
protein kinase pathways (Drutel et al., 2001).
The H4 receptor is detected predominantly in the periphery, for example in bone marrow
and leucocytes. The amino acid sequence of the H4 receptor has a 35 % amino acid
homology with the H3 receptor and a much lower homology to H1 and H2 receptors. Very
little is known about the actual biological function of H4 receptor. The H4 receptor can
mediate chemotaxis and calcium influx in mast calls and eosinophils (O'Reilly et al.,
2002; Hofstra et al., 2003).
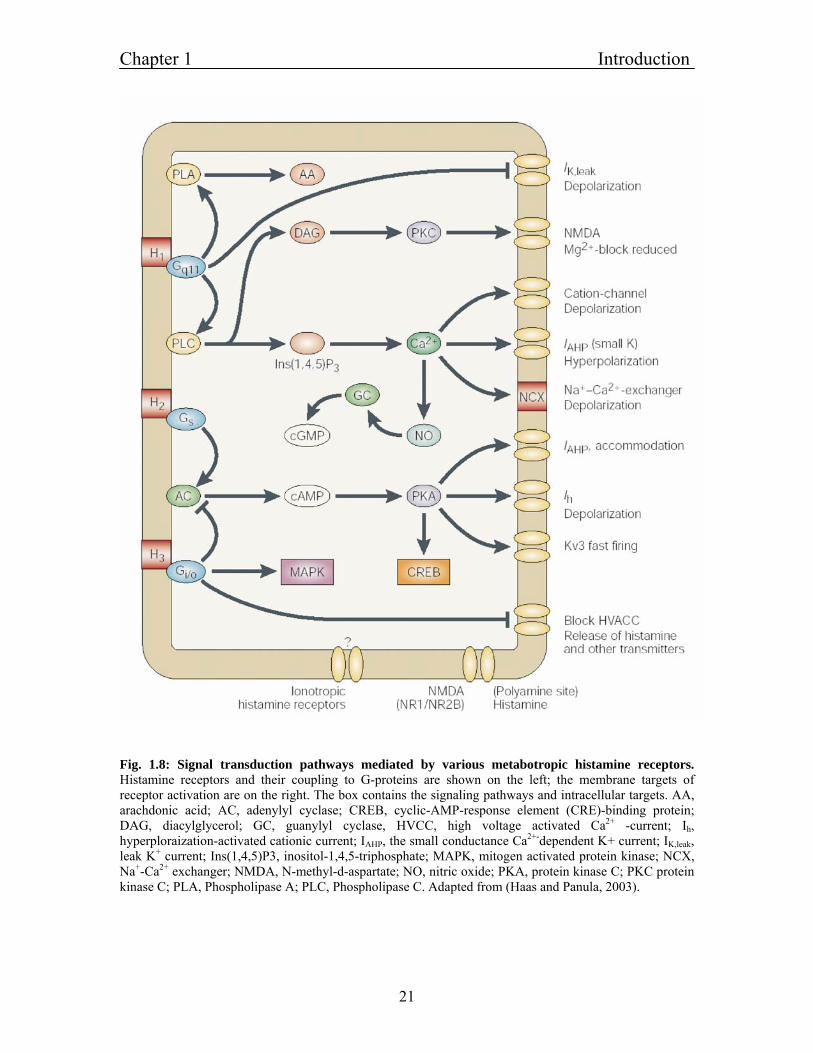
Chapter 1 Introduction
21
Fig. 1.8: Signal transduction pathways mediated by various metabotropic histamine receptors. Histamine receptors and their coupling to G-proteins are shown on the left; the membrane targets of receptor activation are on the right. The box contains the signaling pathways and intracellular targets. AA, arachdonic acid; AC, adenylyl cyclase; CREB, cyclic-AMP-response element (CRE)-binding protein; DAG, diacylglycerol; GC, guanylyl cyclase, HVCC, high voltage activated Ca2+ -current; Ih, hyperploraization-activated cationic current; IAHP, the small conductance Ca2+-dependent K+ current; IK,leak, leak K+ current; Ins(1,4,5)P3, inositol-1,4,5-triphosphate; MAPK, mitogen activated protein kinase; NCX, Na+-Ca2+ exchanger; NMDA, N-methyl-d-aspartate; NO, nitric oxide; PKA, protein kinase C; PKC protein kinase C; PLA, Phospholipase A; PLC, Phospholipase C. Adapted from (Haas and Panula, 2003).

Chapter 1 Introduction
22
1.2.3: Interaction of histamine antagonists with GABAA receptors
In one report the interaction of histamine H2 receptor antagonists with GABA and
benzodiazepine binding sites in the CNS was analyzed (Lakoski et al., 1983). The
histamine H2-receptor antagonist cimetidine potently inhibited [H3] muscimol and
enhanced [H3] flunitrazepam binding in membranes prepared from several brain regions
in the rat, including the dorsal raphe nucleus. As further examined in cortical membranes,
this effect on both GABA and benzodiazepine binding sites was specific for imidazole-
derived H2 receptor antagonists (potency: cimetidine greater than metiamide greater than
tiotidine) and not observed with either several H1 receptor antagonists or histamine. Their
data indicate a striking similarity between the actions of cimetidine (and other imidazole-
derived H2 receptor antagonists) and GABA on binding parameters at the GABA receptor
complex.
In one report the in vitro antagonism of benzodizepines binding to cerebral receptors by
H1 and H2 histamine antagonists was checked (Speeg et al., 1981). They investigated
about the depressant action of histamine antagonists in CNS. It was demonstrated that
cimetidine and pyrilamine are competitive antagonists of 3H-benzodiazepine binding to
human cerebral receptor in vitro. Therefore, the interaction of antihistamine with CNS
receptors other than histamine receptor may explain, at least in part, the side effect of
sedation.
1.2.4: Ionotropic histamine receptors and direct modulatory actions of histamine to
ion channels
In another study the effect of histamine H2-receptor antagonists on the GABA-responses
of the intestine was investigated. GABA and the GABAA agonist muscimol were applied
to isolated ileal guinea pig preparations in the absence and presence of two H2 receptor
antagonists, famotidine and cimetidine. Both GABA and muscimol produced a
concentration-dependent contractile effect on the guinea pig ileum. Famotidine and
cimetidine modified this contractile effect, either by enhancing or by inhibiting it. The

Chapter 1 Introduction
23
differing results depended not only on the antagonist concentration, but also on the
concentration of GABA or muscimol. In conclusion, the interaction of H2 receptor
antagonists with GABA receptors is not limited to the central nervous system, but it also
extends to the peripheral nervous system. The receptor interaction mainly involves
GABAA receptors and depends on both the specific H2 antagonist and the concentration
used (Koutsoviti-Papadopoulou et al., 2003).
To test the hypothesis that cimetidine-like drugs produce CNS effect like seizure and
analgesia effects via inhibition of GABAA receptors, the actions of these drugs were
studied. The H2 antagonists famotidine and tiotidine produced competitive and reversible
inhibition of GABA-evoked currents in HEK 293 cells transfected. In contrast, the H2
antagonist ranitidine and the cimetidine congener improgan had very weak (if any)
effects. Authors concluded that cimetidine-like drugs do not appear to produce seizures
or analgesia by directly inhibiting GABAA receptors (Cannon et al., 2004).
In contrast to the multiple genes for metabotropic histamine receptors, no genes for
ionotropic histamine receptors have been identified in mammals up to now. There are few
hints from electrophysiological experiments that in mammals such direct activated
channels may also exist and that histamine mediate fast synaptic inhibition of rat
supraoptic oxytocin neurons via chloride conductance activation. Up to now, the ion
channels mediating this action were not identified (Hatton and Yang, 2001). At N-
methyl-D-aspartate (NMDA) receptors, histamine enhances the glutamate-evoked current
by direct binding to the channel protein itself. Histamine causes a direct facilitation of the
NMDA-receptor through its polyamine modulatory sites. When applied to cultured
hippocampal neurons, histamine selectively increased by up to tenfold the amplitude of
the component of synaptic transmission that was mediated by NMDA-receptor (Bekkers,
1993). By selectively enhancing the NMDA component of neurotransmission, histamine
should enhance process in which NMDA currents participate, such as triggering of Long-
term potentiation. Conversely, pathological conditions that deplete histamine in the brain
might lead to a reduced ability to trigger Long-term potentiation and so to memory loss.

Chapter 1 Introduction
24
In insects, histamine-acivated chloride channels were known for a long time. Native
ionotropic histamine receptors of invertebrates have been characterized in vivo,
particularly in the large monopolar neurons of the visual system of Drosophila (Hardie,
1989), the heart ganglion (Hashemzadeh-Gargari and Freschi, 1992), and the olfactory
receptor neurons of lobster (McClintock and Ache, 1989), where they mediate the pre-
synaptic inhibition of ORNs (Wachowiak et al., 2002). The ionotropic histamine
receptors mediate rapid neurotransmission in the visual system of invertebrates (Burg et
al., 1993; Hardie, 1989). Recently, genes for histamine-gated ion channels were
identified (Zheng et al., 2002; Gisselmann et al., 2002). Two histamine receptor subunits
have been so far cloned from Drosophila: HisCl-α1 (alias hisCl2, ort, hclA and Dm-
HACL1) and HisCl–α2 (alias hisCl1, ort, hclB and Dm-HACL2) (Gengs et al., 2002;
Zheng et al., 2002; Gisselmann et al., 2002). Both form homomultimeric chloride
channels when expressed in Xenopus oocytes, where HisCl-α2 is about an order of
magnitude more sensitive than HisCl-α (Zheng et al., 2002; Gisselmann et al., 2002).
1.2.5: Histamine functions and knockout mice
The histaminergic neurons are involved in many functions such as memory, sleep, and
alertness and feeding. Histaminergic neurons send widespread projections to most
cerebral regions, including those known to be important in sleep-wake control, such as
the cortex, thalamus, and posterior and preoptic / anterior hypothalamus, and to the
forebrain and brainstem aminergic and cholinergic structures (Inagaki et al., 1988; Panula
et al., 1989). In these target areas, histamine modulates neuronal activity-excitability via
H1, H2, and H3 receptors. Moreover, histaminergic neurons firing rate varies across the
sleep-wake cycle, being highest during waking and lowest during rapid-eye movement
sleep.
Administration of various substances impairing histaminergic transmission increases slow
wave sleep, whereas enhancement of transmission promotes wakefulness (Monti et al.,
1991). Muscimol-induced inactivation of the posterior hypothalamus containing
histaminergic cells results in hypersomnia in both normal and experimentally induced

Chapter 1 Introduction
25
insomniac cats (Lin, 2000). Finally, inhibition of histamine synthesis in the same area
increases slow wave sleep, whereas inhibition of histamine degradation elicits long-
lasting arousal (Lin et al., 1986, 1988). In histidine decarboxylase knockout mice,
disruption of histamine synthesis causes permanent changes in the cortical EEG and
sleep-wake cycle and that, at moments when high vigilance is required (lights off,
environmental change etc.), mice lacking brain histamine are unable to remain awake
(Parmentier et al., 2002). Neuronal histamine has been shown to suppress food intake
through activation of histamine H1 receptors in the ventromedial hypothalamus or
inhibition of the H3 receptor in the paraventricular nucleus (Sakata et al., 1988; Ookuma
et al., 1989) each of which is involved in satiety regulation.
Leptin, an ob gene product (Zhang et al., 1994) has been demonstrated to promote
histamine turnover by affecting the post - transcriptional process of histidine
decarboxylase formation or histamine release per se (Yoshimatsu et al., 1999). In
addition, concentration or turnover rate of hypothalamic histamine was lowered in leptin-
deficient ob/ob and leptin receptor-mutated db/db mice, but it was increased in diet-
induced obese animals (Yoshimatsu et al., 1999). In H1 receptor knockout mice it has
been shown that H1 - receptor is a key receptor for downstream signaling of leptin in the
brain that contributes to regulation of feeding, fat deposition, and UCP mRNA expression
(Masaki et al., 2001). Histamine also alters thermoregulation; hypothalamic
histaminergic neurons are activated not only peripherally by high ambient temperature,
but also centrally by Interleukin L-1beta as endogenous pyrogen (Kang et al., 1994). H3
receptor knockout mice display reduced locomotion and body temperature (Toyota et al.,
2002). Histamine neurons stimulate the sympathetic nervous system to increase lipolysis
in the adipose tissue (Bugajski and Janusz, 1981) an effect that depends more on H1
receptor than H2. Also, Stimulation of supraoptic nucleus by histamine causes synthesis
and release of vasopressin which in turn induces antidiuresis (Haas et al., 1975;
Armstrong and Sladek, 1985; Tuomisto et al., 1980).

Chapter 1 Introduction
26
1.2.6: Diseases where histamine is involved
Histamine is assumed to be involved in neurodegenerative disorders like in Alzheimer.
Numerous neurofibrillary tangles were found in the Alzheimer hypothalami, concentrated
in the tuberomammillary area. Most of them were of globular type and extracellular, and
only a minority were histamine immunoreactive. They may represent remnants of
degenerated TM (Nakamura, 1993). Decrease in brain histamine as well as histidine may
contribute to the cognitive decline in Alzheimer's disease directly or through the
cholinergic system (Schneider et al., 1997). The TM neurons seem morphologically
normal in patients with Parkinson disease though, the central histaminergic system
appears to be activated in Parkinson disease, and since the histaminergic innervation is
increased in the substantia nigra. Also, modulation of the histamine H3 receptor occurs in
Parkinson disease at the level of the mRNA expression in the striatum and receptor
density in the substantia nigra. Marked increase occurs in histamine H3 receptors in the
striatum and substantia nigra by tonic dopaminergic inputs (Ryu et al., 1994).
There is growing evidence to suggest the involvement of histaminergic pathways in the
pathophysiology of schizophrenia. In agreement, decreased H1 receptor-mediated
response to histamine is consistently observed among schizophrenic patients (Rauscher et
al., 1980; Nakai et al., 1991). Levels of t-MHA, the major histamine metabolite in brain
(Schwartz et al., 1971) are significantly enhanced in the cerebrospinal fluid of
schizophrenic patients (Prell et al., 1995). Finally, a polymorphism within the H2 receptor
gene was recently reported to be associated with schizophrenia (Orange et al., 1996).
Many patients diagnosed as schizophrenic have either a chronic excess or deficiency of
blood histamine. Nutritional treatment correcting these imbalances has led to great
improvement or recovery for most such patients. Histamine is used to promote alpha
wave activity in the brain, which enables an individual to handle anxiety and stress easier
(McLeod et al., 1998). If the person is deficient in histidine, it leads to a lack of histamine
and creates unbalances in calming alpha-rhythms in the brain allowing the excitatory beta
waves (responsible for the brain activity that leads to anger and tension to promote)
(McLeod et al., 1998).

Chapter 1 Introduction
27
1.2.7: Aims of the work
In Insects, the existence of histamine-gated chloride channels is long known. The
possible occurrence of such channels in vertebrates has been long postulated but no gene
was identified until now. Such channels have a fair chance to belong to the gene-family
of ligand- gated channels.
There were some indications that a so far undiscovered correlation between histamine
and GABA on the level of receptors exist. In insects, GABA and histamine gate the same
channel. In mammals, GABAA receptors are co-localized in close proximity to
histaminergic neurons, but specific interrelationship between GABA and histamine has
not been investigated yet. Therefore the aims of my work were to identify possible
candidates with bioinformatical means for histamine-gated or modulated channels in
vertebrates and to check especially members of the class of ligand-gated ion channels for
possible genes with similarity to insect histamine-gated channels.
The cDNA of found candidates should be cloned and functionally expressed in Xenopus
oocytes. The action of histamine should then be characterized by a two-electrode voltage
clamp measurements.

Chapter 2 Materials
28
Chapter 2
2: Materials
2.1: Chemicals and enzymes
Agarose LE, analytical grade, Biozym
Albumine, bovine, Fraction V, Sigma,
Alkaline Phosphatase, Shrimp, Roche
Ampicillin Disodium Salt, Sigma
ATP, Disodium Salt, Sigma
Collagenase, Worthington Biochemical Corporation
DMSO, Sigma
dNTPs, Invitrogen
DTT, Invitrogen
Diethyl Pyrocarbonate, Sigma
EDTA Disodium Salt, Sigma
Ethidium Bromide solution, Sigma
Fetal Bovine Serum, Invitrogen
Formamide, Sigma
Goat serum, Gibco, Sigma
Herring Sperm DNA, Roche
Levamisole, Sigma
Proteinase K, Roche
Restriction enzymes from: MBI Fermentas, Roche
RNase A, pancreatic, Roche
RNase H, Roche
RNasin, MBI Fermentas
RNase-free DNaseI, Roche, Biozym

Chapter 2 Materials
29
SUPERSCRIPT III RNase H- Reverse Transcriptase, Invitrogen
T4 DNA ligase, MBI Fermentas
T4 DNA polymerase, MBI Fermentas
T4 polynucleotide Kinase, MBI Fermentas
Taq DNA Polymerase, Invitrogen
All other standard chemicals were from Sigma, Fluka, Aldrich Baker, Gerbu, Merck,
Pharmacia, Promega, Riedel de Haen, Roth and Serva and used typically in p.a. quality.
2.2: Drugs used for pharmacological characterizations
Stocks solutions of drugs used for pharmacological characterizations were prepared as
indicated in the following list. If Xenopus Ringer was used as the solvent, care was taken
to check and if necessary adjust the pH to 7.4 after solving of the drugs as especially
histamine acidifies the agonist solutions.
Neurotransmitters:
Acetylcholine 100 mM Xenopus Ringer Sigma
ATP 100 mM Xenopus Ringer Sigma
Dopamine 100 mM Xenopus Ringer Sigma
GABA 1 M Xenopus Ringer Sigma
Glycine 1 M Xenopus Ringer Sigma
Glutamate 1 M Xenopus Ringer Sigma
Histamine 1 M Xenopus Ringer Sigma
Octopamine 100 mM Xenopus Ringer Sigma
Serotonin 10 mM Xenopus Ringer Sigma
GABA-receptor related drugs:
Bemegride 300 mM DMSO Acros
Chapter 2 Materials
30
Diazepam 100 mM DMSO Roche
Pentobarbital 10 mM DMSO Sigma
Propofol 50 mM DMSO Tocris
Flunitrazepam 1 mM Xenopus Ringer Ratiopharm
Histamine-receptor related drugs:
Doxylamine 10 mM DMSO RBI
DM235 100 mM DMSO Sigma
Cimetidine 10 mM Xenopus Ringer Sigma
Famotidine 10 mM Xenopus Ringer Wallgreen's
HTMT 30 mM DMSO Tocris
Histidine 100 mM Xenopus Ringer Sigma
Pyrilamine 100 mM DMSO RBI
R-alpha-Methylhistamine 100 mM Xenopus Ringer Tocris
tele-Methylhistamine 30 mM Xenopus Ringer RBI
Thioperamide 30 mM Xenopus Ringer Tocris
Others:
Harmane 100 mM DMSO Tocris
PTX 3 mM Xenopus Ringer Sigma
Chapter 2 Materials
31
2.3: Primers
All oligonucleotides (primers) were purchased from Invitrogen or MWG Biotech and solved in H2O to a concentration of 100 pmol / µl. Primers S.No. Subunit Primer Sequence
1 θ hGABA-th Not1-Stop GAC TGC GGC CGC TTA ATC GAT ATA CAT ATG GTA TAC CCA
2 θ r GABA-th –fw GCC ATC CAC ATT ACT GAC GAG CTA CAC 3 θ r GABA-th.5-new GCC GAA TTC GCC ATG GGC ATC CGA GGC ATC CGA
GGC ATG CTG 4 θ Gaba-t-Eco.ATG GAC GAA TTC CAC CAT GCT GCG AGC CGC TGA GCT
CCT 5 θ m GABA–th-3 GCA TGG GCC CTA ATA GAC ATG GTA TAA CCA 6 θ r GABA–th-rev TAT CAG GCC ATC CTG CAC ATG TGC TAC 7 θ Gaba-t-Cla1-sp-Xho1 CTC GAG CTA ATC GAT ATA CAT ATG GTA TAA CCA
GTA 8 θ mGABA-th-fw GCA TGC GGC CGC CAT CCA CAT TAC TGA TGG GCT G 9 θ r GABA –th-1000-fw GGC TCG AGC TCC TGG ATA TCA TTT TGG ATG 10 θ mGABA –th-rev GCC ATG TGA ACA CCA AGG ATC CTA GAC 11 θ rGAB-th-3-new ATC CTC GAG CCT GCT GCT GTG GTG ATA CTC 12
θ θ
mGABA-th-5 rGABA–th-3-new
GCA TGC GGC CGC CAT GGG CAT CCG AGG TAT GCT G ATC CTC GAG CCT GCT GCT GTG GTG ATA CTC
13 ε rGe-EV-r ATC ATA CTC TTG GGT CCT CTT AGA ATT CC 14 ε rGAe-up GCT GAG ATG TTG CCT AAA GTT CTC C 15 ε rG e–EV ATA ACC ATA CCC AAC CAG ATG GC 16 ε Mus-rat-gaba-e-up ACC ATG GTG CCT AAA GTT CTC CTG ATG 17 ε Mus-rat-gaba-e-down CCA GCT GGA GCC TAC AGG TTA AGG 18 ε r GA e-down TCC TGG GGA ACT GAG GTG ATT GC 19 β1 H-B1-rev GTG TAC ATA GTA AAG CCA ATA AAC 20 β1 r GABA b1-down GAG TCT AA CCG AAC CAT GAG AC 21 β1 H-B1-fw TGG ACA GTA CAA AAT CGA GAG AG 22 β1 r-B1 –fw TGG ACA GTA CAA AAT CGA GAG AGT TTG 23 β1 r-B1-rev GTG TAC ATA GTA AAG CCA ATA AAC GA 24 β2 RR-GABA – B2-up GCC ACC ATG TGG GGC TTT GCG GGA GGA AGG 25 β2 RR-GABA- B2-Do ATC AAG TGT TAA CAT AGT ACA GCC AG 26 β3 r-GABA-B3-Del Stop GTT AAC ATA GTA CAG CCA GTA AAC TAA 27 γ2 r-GABA –g2-atg-BamH1 CCG GAT CCA CCA TGA GTT CGC CAA ATA CAT GG 28 α1 r-GA-a1-4 CAA GCC CGT GAT GAA GAA AAG TCG Stop codon:
29 β3 H-GABA –b3 Not1-Stop –EV
GAC TGC GGC CGC TAG ATA TCG TTA ACA TAG TAC AGC CAG TA
30 γ2 H-GABA g2-Not 1-Stop-EV GAC TGC GGC CGT TAG ATA TCC AGA TAA AGA TAG GAG ACC CA
31 α1 GABA a1-Not1-Stop-Ev GAC TGC GGC CGC TAG ATA TCT TGA TGG GGT GTG GGG
32
γ2 H-GABAA g2- Not 1-Stop – EV
GAC TGC GGC CGC TAG ATA TCC AGA TAA AGA TAG GAG ACC CA
33 γ2 rGABA–g2-Stop-Xho1 GCC TCG AGT CAC AGA TAA AGA TAG GAG AC 34 ε rGABA-e-Not1-Stop–EV GAC TGC GGC CGC TAG ATA TCC AGG TTA AGG CAA
ATC ACC CAG TA

Chapter 2 Materials
32
35 ε HGABA-e-rev-O CAA GTT AAG GCA AAC AAG CCA GTA GAG CAC 36 ε HGABA–e-fw-O TTG TCG AAA GTT CTT CCA GTC TTC CTA GGC 37 ε RGABA-e-fw-O TTG CCT AA GTT CTC CTG ATG CTC CTC 38 ε RGABA-e-rev-O CAG GTT AAG GCA AAT CAC CCA GTA GAC 39 ε Rat-ep-start-H3-r CTG GAA GCT TCT TTC CAC TAG GCT GAG GCT GAG
GCC CAA AG 40 GFP 1\2 EcoRV-GFP-5` ATC GTG AGC AAG GGC GAG GAG CTG TTC ACC 41 GFP GFP down w\o stop ATC CTT GTA CAG CTC GTC CAT GCC
2.4: Standards for DNA
Following pre-made DNA size standards were used:
Gene Ruler 100 bp DNA Ladder, MBI Fermentas
Gene Ruler 1 Kbp DNA Ladder, MBI Fermentas 250 bp DNA Leiter, Diagonal
2.5: Consumption materials
Borosilicate glass capillaries (GC150TF-10) for electrophysiological studies were from
Clark Electronical Instruments. Pipette tips were from Sarstaed. Other plastic ware like
15 ml and 50 ml polypropylene tubes, 50 ml plastic tubes, 0.5-1.5 ml reaction tubes as
well as PCR-tubes, syringes and Petri dishes were bought from Sarstaed, Eppendorf or
Biozym. Cell culture dishes were from Nunc.
Distilled water was prepared in a Quarz-double distilling unit and autoclaved at 121°C
and 20 PSI for 20 minutes. Plastic ware that had to be sterile was either autoclaved under
the same conditions for 15 minutes or was used from unopened bags.

Chapter 2 Materials
33
2.6: Kits
RNA isolation:
TRIzol Reagent, Invitrogen
DNA cleanup and isolation:
QIAquick PCR Purification Kit, Qiagen,
QIAquick Nucleotide Removal Kit, Qiagen,
QIAquick Gel Extraction Kit, QiagenPlasmid DNA isolation
Qiagen plasmid maxi Kits, Qiagen
Others:
Amplicap T7 or T3 Kit, Biozyme
First strand synthesis: SuperScript III, Invitrogen
2.7: RNase free materials and chemicals
When working with RNA only RNase free solutions and vessels are used, to avoid
degradation of RNA.
For the production of RNase free water (Sambrook et al., 1989), ddH2O water was
combined with DEPC to a final concentration of 0.1 %, incubated for 30 minutes at room
temperature and then autoclaved for 15 minutes at 20 PSI and 121° C to expel DEPC.
DEPC destroys RNases, but can also kill RNA. However, when it comes in contact with
moisture, it hydrolyzes to form ethanol and carbon dioxide and is therefore rendered
harmless through appropriate treatment.
Glassware was baked over night at 180° C in an oven and solutions were made with
DEPC-water in those RNase free vessels.

Chapter 2 Materials
34
2.8: Frequently used buffers
Agar-plates: 15 g/l in LB-/NZ-Medium,
autoclaved
Alkaline Phosphatase-buffer, 10 x: 500 mM Tris-HCl; pH 8.5
50 mM MgCl2
Ampicillin stock: 10 mg/ml in H2O
Barth´s solution: 88 mM NaCl
1 mM KCl
0.82 mM MgSO4
0.33 mM Ca(NO3)2
0.41 mM CaCl2
2.4 mM NaHCO3
5 mM Tris-HCl, pH 7.4
Ethidiumbromide stock: 10 mM in H2O
HBS, 10 x: 1.4 M NaCl
0.25 M HEPES
14 mM Na2HPO4
LB (Luria-Bertani) medium; pH 7.4: 10 g/l Tryptone
5 g/l yeast extract
10 g/l NaCl
autoclaved
Ligation buffer, 5 x: 250 mM Tris-HCl; pH 7.6
50 mM MgCl2
5 mM ATP
5 mM DTT
Lysis buffer: 10 mM Tris-HCl; pH 7.9
1 mM EDTA
15 % Sucrose
2 mg/ml Lysozyme
0.2 mg/ml RNase

Chapter 2 Materials
35
0.1 mg/ml BSA
M10-Medium: 500 ml MEM
50 ml FBS
5 ml L-Glutamine, 200 mM
5 ml Pen/Strep-Soln.
ND96 99.6 mM NaCl
2 mM KCl
1 mM MgCl2
5 mM HEPES, pH 7.5
Pen/Strep solution: 10,000 U Penicilline
10 mg Streptomycine
in 1 ml 150 mM NaCl soln.
Probe buffer, 5 x: 20 % Ficoll 400, (w/v)
100 mM EDTA
0,025 % Bromephenolblue
0,025 % Xylenecyanole, (w/v)
PNK buffer: 250 mM Tris-HCl; pH 7.6
50 mM MgCl2
25 mM DTT
0,5 mM Spermidine
0,5 mM EDTA
Buffer P1: 50 mM Tris-HCl; pH 7.9
10 mM EDTA
100 µg/ml RNase A
Buffer P2: 200 mM NaOH
1 % SDS
Buffer P3: 3 M Potassium acetate; pH 7.4
Buffer QBT: 750 mM NaCl
50 mM MOPS; pH 7.0
15 % Ethanol (v/v)
0.15 % Triton X-100, (v/v)

Chapter 2 Materials
36
Buffer QC: 1 M NaCl
0.05 M MOPS; pH 7.0
15 % Ethanol
Buffer QF: 1.25 M NaCl
0.05 M Tris-HCl; pH 8.5
15 % Ethanol (v/v)
Reverse Transcriptase buffer, 5x 250 mM Tris acetate, pH 8.4
375 mM Potassium acetate
40 mM Magnesium acetate
1.-Strand buffer, 5 x: 250 mM Tris-HCl, pH 8.3
375 mM KCl
15 mM MgCl2
Taq-DNA-Polymerase buffer, 10 x: 200 mM Tris-HCl; pH 8.4
500 mM KCl
TBE: 90 mM Tris-HCl; pH 8.3
90 mM Boric acid
2 mM EDTA
TE, 1 x: 10 mM Tris-HCl; pH 7.9
1 mM EDTA
Transcription buffer: 400 mM Tris-HCl; pH 8.0
60 mM MgCl2
100 mM Dithiothreitol
20 mM Spermidine
Xenopus Ringer's soln. 115 mM NaCl
2.5 mM KCl
1.8 mM CaCl2
10 mM Hepes, pH 7.2

Chapter 2 Materials
37
2.9: Bacterial strains
XL1-Blue (Stratagene) recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1
lac [F´proAB lacIqZ∆M15 Tn10 (Tetr)]c
2.10: Plasmid vectors
General cloning and expression vectors: pSGEM: Oocyte expression vector derived from pGEMHE vector (3022 bp); (Liman et
al., 1992) Kindly provided by Dr. Michael Hollmann, Bochum.
pBluescript II KS (+) (Stratagene)
pRC/CMV (Invitrogen)
pCDNA3 (Invitrogen)
GABA-receptor expression vectors:
Following vectors originate from the German Genome Resource Center Berlin (RZPD):
GABAA-receptors:
Human delta subunit in pCMV-SPORT6 clone IRATp670E0653D6
Human alpha 1 subunit in pBluescriptR clone IRAKp961A1533Q
Human alpha 3 subunit in pBluescriptR clone IRAKp961F0547Q
Human alpha 5 subunit in pT3T7 clone IRATp970H0853D6
Human alpha 6 subunit in pSPORT6-sfi clone DKFZp686D23263Q
Human beta 3 subunit in pCMV-SPORT6 clone IRAKp961K0613Q
Human gamma 1 subunit in pBluesciptR clone IRAKp961J0448Q
Human gamma 2L subunit in pCMV-SPORT6 clone IRAKp961L0931Q

Chapter 2 Materials
38
GABAC-receptors:
Human rho1 subunit in pT3T7-PacI clone IMAGp998P2111525Q
The plasmid pCDNA3.1-GABA-myc-theta containing the human GABAA-theta subunit
cDNA was a kindly gift of P. Wingrove, MSD. The plasmid pCDNA-GABA-β2
containing the rat GABAA-β2 subunit cDNA was kindly provided by R. Ruprecht,
München.
The plasmids:
pCDNA3-rGAα1 containing the rat GABA(A) alpha1-subunit cDNA in pCDNA3
pSGEM-rGAα1 containing the rat GABA(A) alpha1-subunit cDNA in pSGEM
pCDNA3-rGAβ1 containing the rat GABA(A) beta1-subunit cDNA in pCDNA3
pSGEM-rGAβ1 containing the rat GABA(A) beta1-subunit cDNA in pSGEM
pCDNA3-rGA ε containing the rat GABA(A) epsilon-subunit cDNA in pCDNA3
pSGEM-rGA ε containing the rat GABA(A) epsilon-subunit cDNA in pSGEM
were from the Lehrstuhl für Zellphysiologie plasmid collection.
2.11: Software
General sequence analysis was done with the DNASTAR program package. Alignments
of DNA sequences were done using the program Megalign. DNA sequences can be
translated into amino acid sequences with Mapdraw, which also can find restriction sites
for restriction enzymes. Protein as well as DNA Sequence comparisons to known and
published sequences were performed in the Internet (NCBI USA) with BLAST (Basic
Local Alignment Search Tool) Search, according to (Altschul et al., 1990).

Chapter 3 Methods
39
Chapter 3
3: Methods
3.1: Characterizing, isolating and concentrating nucleic acids
3.1.1: Determination of concentrations of nucleic acids
The quantification of the amount of nucleic acids in solutions was measured by
adsorption of light with a wavelength of 260 nm in a Thermo Helios Gamma photometer,
Thermo Biotech. One optical density (OD) is equivalent to a concentration of 50 µg / ml
for double stranded DNA molecules, to 40 µg / ml for RNA molecules and to 33 µg / ml
for oligonucleotides.
3.1.2: Gel electrophoresis
Following amplification of DNA by PCR (3.2.2) or linearization by restriction reactions
(3.3.1) there is need to determine, whether the appropriate product has been produced.
This is typically accomplished by agarose gel electrophoresis.
Samples were mixed with loading buffer to a final concentration of 1 x and loaded in the
wells of the gel, which was then run in a gel chamber in 1 x TBE buffer at a voltage of
approximately 10 V / cm.
The gel-running buffer was supplied with EtBr, an intercalating dye, to a final
concentration of 40 ng / ml high enough to make DNA visible under the UV-light at 302
nm, which was done with a ChemiImager 4400 (Biozym). To measure the size of a
fragment, the 1 KB DNA marker ladder was used (gene ruler, MBI-Fermentas).

Chapter 3 Methods
40
3.1.3: Phenol:chloroform extraction of nucleic acids
Phenol:chloroform extraction is used to remove protein and other contaminants from
nucleic acids and is often required to remove enzymes after an enzymatic reaction.
To one volume of DNA or RNA one volume 50:50 phenol:chloroform was added. After a
30 seconds vortexing step, the solution turned milky white. The upper aqueous phase and
the lower organic phase were separated by centrifugation for 5 minutes at 13,000 rpm in a
microcentrifuge. The upper phase was carefully removed, mixed with one volume of
chloroform and again vortexed for 30 seconds. After another centrifugation step the
supernatant was transferred to a clean tube and purification proceeded with ethanol
precipitation (3.1.4).
3.1.4: Ethanol precipitation of nucleic acids
Ethanol precipitation is a standard method used to concentrate nucleic acids. The method
relies on the tendency of nucleic acids to precipitate in a solution of salt and ethanol,
particularly at colder temperatures. Sodium acetate is the most commonly used salt, but
ammonium acetate is often used when co-precipitation of single nucleotides is to be
avoided.
To 1 volume of DNA or RNA either 0.1 volumes of 3 M sodium acetate, pH 5.5 and 2
volumes of 100 % ethanol were added. Tubes were placed at -20° C for at least 30
minutes. After a brief thaw samples were spun in a microfuge for 15 minutes at 13,000
rpm. The supernatant was carefully drawn off and discarded. After adding 70 % ethanol,
the reaction tubes were spun again at the same conditions. The pellet was dried briefly in
a SpeedVac Concentrator (SAVANT) or air-dried. The nucleic acids were then dissolved
in a desired volume of water or TE buffer.

Chapter 3 Methods
41
3.1.5: QIAquick Agarose Gel Extraction Kit
DNA can be extracted from agarose gels using the QIAquick method. The DNA bands
were excised from the agarose gel and distributed in centrifuge tubes. Three volumes of
Buffer QG, a chaotropic salt, were added to the gel slice containing DNA. The tubes were
incubated at 50° C until the agarose had completely melted. The molten gel was
centrifuged through a QIAquick column (1 min, 13.000 rpm). The DNA is absorbed to
the DNA-binding resin under these conditions of high ionic strength and low pH.
Subsequently, the column was washed with 650 µl PE buffer. At the wash it was
necessary to remove all traces of PE buffer. Therefore the tubes were spun again to get
rid of all the liquid. To elute the DNA, 50 µl of EB buffer were added and the samples
were incubated for 1 minute at room temperature. The tubes were centrifuged for 30
seconds and the eluate containing the pure DNA was collected into a clean tube. All
centrifugation steps were performed at 13,000 rpm in a microfuge, which corresponds to
approximately 10,000 g.
3.1.6: QIAquick PCR-Purification Kit
This protocol was designed for clean-up PCR-generated DNA fragments or the
purification of templates for in-vitro transcription.
To the foregoing reaction 3 volumes of buffer PB were added per one volume of the
reaction sample and the mixed solution was layered onto a QIAquick column. A flow-
through was produced by centrifugation, which was discarded afterwards. The column
was washed with 750 µl of buffer PE and drained by another centrifugation step. To elute
the DNA 50 µl of buffer EB were applied, let stood for 1 minute and then centrifuged.
All centrifugation steps were performed at 13000 rpm in a microfuge, which corresponds
to approximately 10000 g.

Chapter 3 Methods
42
3.1.7: RNA Extraction
RNA was isolated from mouse or rat brain tissues using Trizol (Invitrogen). 100 mg
tissue that RNA was to be isolated from was placed in glass homogenizer. Ten volumes
of Trizol reagent (1 ml) were added, and the tissues were thoroughly homogenized. The
homogenate was incubated at room temperature for 5 min before 200 µl chloroform was
added per 1 ml of homogenized tissue. The samples were shaken by hand for 15 sec and
incubated at room temperature for 3 min. The samples were centrifuged for 10 min at
13000 rpm. The top layer of the three distinct resultant layers was carefully transferred to
a new sterile tube where 500 µl isopropanol was added per ml of sample solution. This
was incubated at room temperature for 10 min and centrifuged for 10 min at 13000 rpm.
Following this centrifugation, the isopropanol was carefully removed, leaving the RNA
pellet intact. One milliliter of 70 % ethanol was then added to the tube and the samples
were centrifuged for 10 min. The ethanol was carefully removed and the RNA pellet was
allowed to air dry. Once dry, the pellet was resuspended in 50 µl DEPC treated water and
then stored at -70° C.
3.1.8: Quick preparation of plasmid DNA
For a quick and convenient preparation of plasmid DNA of multiple cultures for clone
characterization for cloning purpose the Easy-prep method was used (Berghammer and
Auer, 1993). 1.5 ml of an over night culture was centrifuged at 13,000 rpm in a table top
centrifuge, the supernatant discarded and the pellet resuspended in 50 µl lysis buffer. The
tubes were incubated for 1 min at 95° C and subsequently incubated on ice for one more
minute. The tubes were centrifuged for 20 min at 13,0000 rpm and 20 µl of the
supernatant was transferred into a fresh tube. 4 µl of the DNA solution was typically used
for analytical digestions with restriction enzymes (3.3.1). The DNA was stored at -20° C.

Chapter 3 Methods
43
3.1.9: Maxi preparation of plasmid DNA using the QIAGEN Plasmid Maxi
Kit
The QIAGEN plasmid purification is based on a modified alkaline lysis procedure,
followed by binding of plasmid DNA to QIAGEN Anion-Exchange Resin under
appropriate low salt and pH conditions. RNA, proteins, dyes and low molecular weight
impurities are removed by a medium salt wash. Plasmid DNA is eluted in a high-salt
buffer, and then concentrated and desalted by isopropanol precipitation.
Up to 500 µg of high-copy plasmid DNA from 250 ml LB cultures of E. coli can be
recovered. Bacterial cultures were spun down and the pellet resuspended in 10 ml of
buffer P1. After addition of the same amount of buffer P2, the solution was gently, but
thoroughly mixed, by inverting the tube 4-6 times. The same procedure was carried out
with 10 ml prechilled buffer P3 and the mixture incubated on ice for 5 minutes. While
centrifuging the solution at 10,000 g for 10 minutes, the necessary amount of QIAGEN-
tip 500 filter tips was equilibrated with 10 ml of buffer QBT each and the column was
allowed to empty by gravity flow. The supernatant from the centrifugation step was then
applied to the column. The QIAGEN-tip 500 was washed 2 times with 10 ml buffer QC
and the DNA eluted into a clean tube by applying 8 ml of buffer QF. The DNA was
precipitated with 0.7 volumes of room temperature isopropanol and immediately
centrifuged at 10000 g for 30 minutes in a microfuge and the supernatant carefully
decanted. The DNA was washed in 5 ml 70 % ethanol, air-dried for 5 minutes and
redissolved in 500 µl TE buffer.
3.2: PCR Methods
3.2.1: Reverse transcription The synthesis of first strand cDNA was accomplished using SUPERSCRIP III RNase H-
Reverse Transcriptase (RT), which is a DNA polymerase that synthesizes a
complementary DNA strand from single-stranded RNA. 2 µg of mouse or rat brain RNA

Chapter 3 Methods
44
(3.1.6), 100 ng of oligo dT(17-22) primer and RNase free water were mixed and heated
for 5 minutes at 70° C (total volume 10 µl). Immediately after the samples had been
chilled on ice, they were quickspun. 4 ml 5x Reverse Transcriptase buffer (Invitrogen),
10 U RNasin, 4 µl 25 mM MgCl2, 2 µl 0.1 M DTT and 1 µl 10 mM dNTPs were added at
room temperature (water up to 19 µl). The reaction was started by adding 100 U
SUPERSCRIPT III RNase H- Reverse Transcriptase. After 1-½ hours incubation at 42°
C, the reaction was stopped by adding 1 µl 0.5M EDTA, pH 8.0. The tubes were boiled
for 2 minutes at 95° C to stop the reaction.
3.2.2: Polymerase Chain Reaction
PCR (Mullis and Faloona, 1987) is used to amplify a region of DNA. The reaction
generally requires design of exact oligonucleotide primers (at least 17 bp; preferably > 20
bp) at either end of the region of interest, though there are several ways around this
necessity. Through a series of cycles of denaturation of the DNA template, annealing of
the primers and extension of DNA by a DNA polymerase, the region of DNA between
the primers is exponentially amplified. The Taq DNA polymerase is isolated from
Thermus aquaticus and catalyzes the polymerization of nucleotides into duplex DNA in
the 5’→3’ direction in the presence of magnesium. Taq exhibits furthermore a 5’→3’
exonuclease activity. Reactions contained 1x PCR buffer, 1.5 mM MgCl2, 200 µM
dNTPs, 2.5 U Taq Polymerase, combined with 0.125 U Pfu and 20-50 pmol upward
sense and downward antisense primer each. The entire reaction volume was 50-100 µl
and the fluid was overlaid with a drop of mineral oil to prevent condensation.
The polymerase chain reaction was carried out in a thermal cycler according to the
following amplification conditions:
Step 1: Denaturation 94° C, 1 minute
Step 2: hold at 80° C, enzyme added (hot-start)
Step 3: Denaturation 94° C, 1 minute

Chapter 3 Methods
45
Step 4: Annealing 50-65° C, 1 minute
Step 5: Extension 72° C, 1-3 minutes
Step 6: Final extension 72° C, 10 minutes
After 25-40 cycles were performed, temperature was held at 12° C.
The melting temperature TM of a primer molecule can be calculated in accordance to the
following formula:
TM = 2° C x number of nucleotides (A + T) + 4° C x number of nucleotides (G + C)
The annealing temperature was experimental adjusted between 50-65° C in different
applications.
3.2.3: PCR-based generation of chimeric cDNAs and site-directed mutagenesis
In order to generate chimeric cDNA, for example constructs composed of the 5'-end of
the GABACrho1 subunit and the 3' end of the GABAAβ1 subunit, the 'ligate & PCR'
method was used. The corresponding 3'- and 5'-fragments that need to be fused together
were independently amplified by PCR (3.2.2). Primers located at the site of later fusion
were phosphorylated prior to PCR (3.2.4). In this case the primer located at the 3'-end of
the 5'-fragment and the primer located at the 5'-end of the 3'-fragment. After PCR and
gel-purification of both products (3.1.2 and 3.1.5), about 100 ng of each fragment were
combined and ligated (3.3.5). 1 µl of this ligation mix was used as a template for PCR
with primers located at the 5'-end of the 5'-fragment and the 3'-end of the 3'-fragment.
These are the primers flanking the desired chimeric cDNA, in our example the GABAC ρ1
subunit 'ATG' forward primer and the GABAAβ1 subunit 'Stop' reverse primer. The PCR
products generated in this manner were gel-purified (3.1.2) and the product of the right
size isolated from the gel (3.1.5) and cloned blunt end (3.3.3) or after digestion with
suitable restriction enzymes (3.3.1) into the pSGEM vector. Site-directed mutagenesis
was performed essentially by the same method. In this case, the desired mutation was
incorporated in on of the PCR primers.

Chapter 3 Methods
46
3.2.4:Phosphorylation of PCR primers
For the PCR-mediated generation of chimeric cDNAs (3.2.3), 5'-phosphorylated PCR
primers were needed. In order to phosporylate the oligonucleotide, a 20 µl reaction mix
was set up containing 1 nmol of the corresponding oligonucleotide, 1 mM ATP, 1x PNK
buffer and 5 U of PNK enzyme. After 30 min of incubation at 37° C, the reaction was
terninated by heat (20 min 75° C). 1 µl of this reaction mix containing 50 nmol of
phosphorylated primer was directly used for PCR (3.2.2) without further purification.
3.3: Cloning of DNA
3.3.1: Restriction
Restriction enzymes recognize short, specific (often palindromic) DNA sequences. They
cleave double-stranded DNA at specific sites within or adjacent to their recognition
sequences. Each restriction enzyme has specific requirements to achieve optimal activity.
Conditions such as temperature, pH, enzyme cofactors; salt composition and ionic
strength affect enzyme activity and stability. An analytical scale restriction enzyme digest
is usually performed in a volume of 20 µl on 0.1-0.5 µg of substrate DNA (preparative
scale, 5-10 µg DNA, 20 - 50 µl), using a 2- to 10-fold excess of enzyme over DNA. For
calculation of the amount of enzyme it was assumed that app. 1 U of enzyme cuts 1 µg
DNA in a volume of 50 µl at 37° C in one hour. DNA to be restricted was incubated with
the appropriate restriction enzyme. For all restrictions 10x buffer was used.
3.3.2: Dephosphorylation
Alkaline phosphatases from both bacterial and animal sources are widely used in
molecular biology for the dephosphorylation of 5'-phosphorylated ends of DNA.
Dephosphorylation of a restriction enzyme digested plasmid (5-20 pmoles of 5' ends, 0.1-
0.5 U / pmoles 5' ends) reduces re-ligation and transformation to < 0.5 % of control
(undigested vector). Shrimp Alkaline Phosphatase is a high specific activity, heat-labile
⋅

Chapter 3 Methods
47
alkaline phosphatase for molecular biology applications isolated from Arctic shrimp.
Vectors were linearized with the appropriate restriction enzymes (3.3.1). The phosphate
rest at that 5' end of the vector was cleaved off by adding 10x SAP buffer (Roche) and 1-
5 U enzyme (SAP or CIAP) for 1 µg DNA. Incubation was done for 1 hour at 37° C.
1/20th volume of 500 mM EDTA was added and the tubes were heated for 10 minutes to
70° C to terminate the reaction, followed by purification by gel electrophoresis (3.1.2 and
3.1.5).
3.3.3: Polishing of DNA using T4 DNA polymerase and T4 PNK
To clone PCR products into a blunt end vector it is necessary to polish the DNA
molecules and to ensure the presence of a phosphate rest at their end, which can be
performed using T4 DNA polymerase and T4 PNK. T4 DNA Polymerase catalyzes the
synthesis of DNA in the 5'→3' direction. Its applications are 3' overhang removal and 5'
overhang fill-in to form blunt ends. T4 Polynucleotide Kinase catalyzes the transfer and
exchange of Pi from ATP to the 5' hydroxyl terminus of polynucleotides (double- and
single-stranded DNA). PCR samples were incubated typically in a 1x medium salt
restriction buffer (Orange buffer, MBI-Fermentas) with 1 U T4 DNA polymerase, 5 U T4
PNK, ATP and dNTPs at final concentrations of 1000 µM and 500 µM, respectively. The
samples were let stood at room temperature for 1 hour. To stop the reaction 1 µl 0.5 M
EDTA was added and the tubes were heated for 10 minutes at 70º C.
3.3.4: Fill in reaction
In some cases a filling step using the DNA Polymerase I Large (Klenow) fragment was
used to fill 5' protruding ends hence rendering blunt ends. The fill in protocol was
performed according to the product instructions. In brief 1 - 10 µg of the digested DNA
were mixed with 1 µl Klenow (5 U), 10x buffer, 40 µM of each dNTP in a final volume
of 20 µl. The reaction was incubated at room temperature for 10 minutes and stopped by
heating the mixture at 75° C for 10 minutes.

Chapter 3 Methods
48
3.3.5: Ligation of DNA
The enzyme, which is used in this application, T4 DNA ligase, catalyzes the formation of
phosphodiester bonds in the presence of ATP between double-stranded DNAs with 3'
hydroxyl and 5' phosphate termini. For the ligation reaction 100-200 ng vector was used
and 2-15 µl purified fragment (3.1.5, typically a 3 fold molar excess) was added. Both
DNA molecules were incubated in a 20 µl volume containing 2 µl 5x ligation buffer and
1 µl ligase (5 U) over night at 4-20° C.
3.3.6: Culturing of bacteria
Bacteria were grown on LB Agar plates containing 100 µg Ampicillin per ml. The
antibiotic was added, after the Agar was autoclaved at 121° C for 20 minutes. When
inoculating liquid cultures, LB contained 100 µg Ampicillin per ml.
3.3.7: Transformation of plasmid DNA
Following ligation (3.3.5), 50 µl aliquots of competent XL1-Blue bacteria were combined
with 4-20 µl of the ligation reaction in prechilled 1.5 ml plastic tubes and incubated on
ice for 30 minutes. Bacteria were heat shocked in a 42° C water bath for 45-50 seconds
and instantly placed back on ice for 2 minutes. This procedure caused the cells to become
permeable to the plasmid. Seven volumes of LB medium were added to the tube and
bacteria were incubated one hour in a 37° C incubator. The transformation reaction was
plated and the bacteria then incubated over night at 37° C. Until further use plates were
stored upside down at 4° C.
3.3.8: Sequencing of DNA
Sequencing of DNA was done using the sequencing service of the Lehrstuhl of
molekulare Neurobiochemie, Bochum. Sequencing reactions were done by the Sanger
method using fluorescine-labelled primers or oligonucleotides.

Chapter 3 Methods
49
3.4: RNA techniques
3.4.1: In vitro transcription
For the generation of templates for run-off transcripts, plasmids were linearized using
restriction enzymes (3.3.1) cutting downstream of the cDNA insert (for pSGEM-
constructs, PacI was used always) and purified using the QIAquick PCR purification kit
(3.1.6). Capped complementary RNA (cRNA) was transcribed in vitro using the
Amplicap-T7 kit (Biozym) according to the manufacturer's protocol. The resulting cRNA
was purified by ammonium acetate purification. The integrity and the quantity of the
cRNAs were investigated by agarose gel electrophoresis and visualization under UV light
(3.1.2).
Fig. 3.1: Steps of in vitro cRNA transcription.The common RNA polymerases used for in vitro transcription are SP6, T7 and T3 polymerases. Depending on the orientation of the DNA equence relative to the promoter, the template may be designed to generate a sense or antisense strand RNA (adapted from Swanson and Folander, 1992).

Chapter 3 Methods
50
3.5: Functional expression of LGICs in Xenopus laevis
Xenopus laevis is one species among 14 in the Xenopus genus. They are clawed aquatic
frogs, which are found in veldt ponds and lakes in arid and semi-arid regions across
southern Africa. Xenopus laevis were purchased from Horst Kehler GmbH, Hamburg or
Nasco (USA). The frogs were housed in the animal facility of the Lehrstuhl für
Zellphysiologie in tanks containing fresh water under standard keeping conditions at
20° C.
3.5.1: Surgery
The animal was anesthetized with a 1 % tricaine solution. The surgery was performed on
ice under semi-sterile conditions. First, the animal’s skin was rinsed with sterile Barth’s
solution. A celiotomy, 0.5 to 1 cm long and approximately 2 cm above to and parallel
with the crease formed by the hind limb of the frog, gave access to the pleuroperitonial
cavity. The oocytes were carefully pulled out of the abdomen and placed into calcium-
free ND-96. Sutures of the abdominal wall and skin were performed using medical silk.
The frog was monitored in a recovery bath for 2 to 4 hours, before it was taken back to
the animal facility.
3.5.2: Oocyte preparation and injection of cRNA
The oocyte sacks were opened and all lobules cut into pieces of approximately 0.5 cm3
for better separation. Several stages of washing were carried out in a 50 ml plastic
Falcone tube, which was placed on a vertical rotator. After rotating for 15 min, the
calcium-free ND-96 solution was poured off and replaced by 30 ml fresh calcium-free
ND-96. This procedure was repeated five times.
In order to defolliculate oocytes, the oocytes were subjected to gently shaking for 2 h in
calcium-free ND-96 containing 2 mg / ml collagenase type Ia. Afterwards, the oocytes
were rinsed with several changes (3-4) of calcium-free ND-96 and rotated for 15 min. As

Chapter 3 Methods
51
a final step, the oocytes were washed with normal (Ca-containing) ND-96 solution.
The oocytes were transferred to a petri dish containing Barth's medium and selected for
size, stage and damage. The oocytes were stored in 24-well dishes in Barth's media at a
temperature of 15-18° C. Injection of cRNA (3.4.1) was performed with an electronically
controlled air pressure injector using glass pipettes. Glass capillaries were pulled using a
Kopf vertical puller. The tip was manually broken under the microscope (diameter of
about 10-20 µm) and loaded with cRNA by suction (usually 1-2 µl). Oocytes were then
placed into a 35 mm petri dish with a polypropylene mesh glued to the bottom to fix the
oocytes and injected with a given volume of cRNA. 50 nl RNA solution (app. 1 µg / µl)
was injected into an individual oocyte judged by the number of oocytes injected with 2 µl
of the RNA solution. After injection oocytes were incubated at 15-18° C in Barth´s
solution supplemented with 100 U ml-1 penicillin, 50 µg ml-1 streptomycin. Oocytes were
tested for functional expression of LGICs typically after 2–7 days.
3.5.3: Electrophysiological recording using two-electrode voltage clamp
Fig. 3.2: The two-electrode voltage clamp set-up.The Xenopus laevis oocyte is impaled with two electrodes. One electrode measures the voltage and the other injects currents.

Chapter 3 Methods
52
Two-electrode voltage-clamp recording was used to obtain current responses to applied
substances. Agonists and antagonists were diluted to the concentrations indicated with
Xenopus Ringer's solution and were applied by means of a multibarrel, single-tip
superfusion device or, for screening of multiple different solutions to one individual
oocyte by manual application of 100 µl solution by an automatic pipette. Electrodes were
pulled from borosilicate glass using a Kopf vertical pipette puller and were backfilled
with 3 M KCl. Both current and voltage electrode had resistances of 0.2-1 MΩ.
Membrane potential was controlled and current signals were recorded with a two-
electrode voltage-clamp amplifier (TURBO TEC-03, npi) and pCLAMP software (Axon
Instruments). For measuring, one oocyte was placed into the perfusion chamber and
superfused with isotonic Xenopus Ringer at a flow rate of 1-2 ml min-1. For voltage
clamp experiments, membrane potential was typically clamped at -50 mV. In case of very
high or low evoked current amplitude, the holding potential was adjusted in a range from
-30 up to -100 mV.
3.6: Functional expression of LGICs in HEK 293 cells
3.6.1: Culture of HEK 293 cells and transfection
Human embryonic kidney cell line HEK 293 were cultured in M10 medium at 37° C and
5 % CO2 in a cell culture incubator (Heareus) under standard growing conditions. One or
two days prior transfection, cells were seeded at an amount of 2-10x 104 cells into a 3.5
cm dish containing 2 ml M10 medium. Transfections were carried out using Calcium
phosphate precipitation. Cell culture medium was exchanged to 1 ml fresh medium
before starting the transfection. The transfection mix was prepared as follows: a total of
18 µg expression plasmids and 2 µg pIRES-eGFP (Clontech) was solved in 225 µl ddH20
and mixed with 25 µl 2.5 M CaCl2. The solution was carefully overlaid with drop-wise
addition of 250 µl 2x HBS, mixed by up and down pipetting and incubated for 15 min.
100 µl of the transfection mix was used for each dish. After 4-6 h, dished were washed
with PBS twice and fresh M10 medium was added. Electrophysiological experiments

Chapter 3 Methods
53
were performed 1-3 day after transfection. Efficiency of transfection, typically 10-50 %,
was checked by GFP-fluorescence detection of the GFP coded by the pIRES-eGFP.
3.6.2: Patch clamp investigation of GABA receptors expressed in HEK293 cells
Patch clamp technique is an important method for studying electrophysiological
properties of biological membranes, which was developed by Neher and Sakmann. It
allows the recording of macroscopic whole-cell currents flowing across biomembranes
through ion channels. One can study membrane-contained ion channels, receptor
activated channels and second-messenger activated channels. The electrical circuit used
in the patch clamp technique is shown in the following figure.
Fig. 3.3: Schematic representation of patch clamp circuit. The diagram illustrates the circuits in the patch clamp technique. The patch pipette is in direct contact with the cellular membrane to form a seal. The current flowing through the ion channels on the membrane enters the patch pipette through an electrode. With pipette potential of Vp, the current through it (Ip) is then fed to a current-voltage converter (head stage amplifier), which has a feedback resistor (R).
Mammalian culture cells such as HEK 293 are frequently employed for studying
recombinant expression of individual receptors. A vast amount of publications exist
where HEK 293 cells have been used for investigation of GABAA receptors. Cultured
mammalian cells have the advantage of providing an environment that is similar to the

Chapter 3 Methods
54
milieu of the natural mammalian cells. To a great degree, channel permeability, post-
translational processing, signaling and coupling to other cellular factors in these cells are
similar to the processes in most mammalian cells. Post-translational processing like
protein glycosylation varies in other expression systems like Xenopus oocytes. This could
probably interfere with the proper assembly of subunits.
Transfected HEK 293 cells (3.6.1) transiently expressing green fluorescent protein and
the recombinant GABA channels were recorded in the whole-cell voltage-clamp
configuration (Hamill et al., 1981) under visual control using an inverted microscope
(Zeiss, Jena, Germany). The cells were kept in an external solution containing: 138 mM
NaCl, 5 mM KCl, 0.5 mM CaCl2, 1.5 mM MgCl2, 10 mM Glucose and 10 mM HEPES.
pH was adjusted to 7.4 with NaOH. Patch electrodes were pulled from borosilicate glass
(Clark Electromedical Instruments) using a horizontal pipette puller (DMZ Universal
Puller, Zeitz-Instruments) to yield pipettes with resistances of 3 to 6 MΩ. Pipettes were
filled with a solution containing: 140 mM KCl, 0.1 mM CaCl2, 1 mM MgCl2, 5 mM
EGTA, 10 mM HEPES adjusted to 7.3 with KOH. After getting the whole cell mode, the
cell was lifted and it was positioned in front of a perfusion system consisting of a theta
tube flow pipe. Voltage protocols were delivered and current signals were recorded with
an EPC-7 amplifier using the pCLAMP software on a 486-IBM compatible PC running
under DOS. Typical holding potential was -60 mV. Data were collected and analyzed
with pCLAMP 7 software (Axon Instruments) in combination with SigmaPlot 7.0 (SPSS,
Inc).

Chapter 4 Results I
55
Chapter 4 4.1: Bioinformatical search for histamine-gated channels
For histamine, one of the aminergic neurotransmitters, molecular biology techniques lead
to the identification of various metabotropic histamine receptors (H1 - H4), which are all
members of the G-protein-coupled receptor family (Hill et al., 1997). There are some
indications from electrophysiological studies, that there might be an additional histamine-
gated ion channel in vertebrates (Hatton and Yang, 2001) but no genes were identified so
far. Histamine was pointed out to be the major neurotransmitter in the invertebrate retina
(Hardie, 1989) but only recently the genes HisCl-α1 and HisCl-α2 for histamine-gated ion
channels were identified (Zheng et al., 2002; Gisselmann et al., 2002) in Drosophila
melanogaster. The discovery that invertebrate histamine-gated chloride channels belong
to the super family of ligand-gated ion channels gave raise to speculations about the
nature of potential histamine-gated channels in vertebrates. Identification of histamine-
gated ion channels in vertebrate, may lead to several physiological and pharmacological
important development in histamine research field.
We started using bioinformatics approaches, to find potential candidates of histamine-
gated ion channel genes or cDNAs in vertebrates. We looked for the sequence homology
for HisCl-α1 and HisCl-α2 in various vertebrates genomes (e.g. Zebra fish, rat, mouse,
human etc). For this, we scanned genomes with the complete coding regions of HisCl- α1
and HisCl-α2 receptor subunits as well as separate scanning for all of the predicted
features characteristics of ligand-gated ion channel subunits: signal peptide, two cysteines
separated by 13 amino acids (cysteine-bridge) and four putative transmembrane (TM1 to
TM4) domains. All these parameters did not show any sequence homology for any
unknown ligand-gated ion channel. However, it exhibited some similarities with known
GABAA receptors subunits as well as some extent to the glycine receptors. We also

Chapter 4 Results I
56
looked for any sequence homology in the EST database, even for incomplete vertebrate
genomes (e.g. Chimpanze and Xenopus laevis).
4.2: Construction of expression vectors for GABAA receptors
As our genome-scanning results showed that GABAA receptors have some similarities
with histamine-gated ion channels; we decided to investigate the action of histamine to
the class of ionotropic GABAA receptors. It was planned that the electrophysiological
characterization to be performed in Xenopus oocytes with two electrode-voltage clamping
(3.5.3). To generate the corresponding cRNA, it was necessary to clone the cDNA for the
GABA receptors to be investigated into the pSGEM vector as this vector allows to in
vitro transcribe cRNA suitable for an effective high level expression. Different subunits
of GABAA receptors from rat (α1, β1, θ, ε), human (α3, α5, β3) and human ρ1 GABAC
were cloned into pSGEM. The constructs pSGEM-mouse γ2 as well as pSGEM-rat β2 and
pSGEM-δ were already available from other projects of our department.
For cloning of pSGEM-α1, -α5, -β1, -β3 and -ε the open reading frame of the cDNA insert
present in various plasmids used as template (see table 1) was amplified by PCR (3.2.2)
using the primers indicated in table 1. The PCR products were gel purified (3.1.2), made
blunt-end and phosphorylated for cloning into EcoRV/SmaI cut pSGEM (3.3.1) or were
cut with suitable restriction sites incorporated into the PCR primers (2.3) and cloned into
the corresponding polylinker sites of the dephosphorylated pSGEM vector (3.3.2). After
ligation (3.3.5) and transformation into E. coli XL1-blue (3.3.7), plasmids isolated from
several colonies (3.1.8) were analyzed by restriction mapping (3.3.1) and from clones
with the correct insert, large amounts of plasmid was prepared by the maxi prep method
(3.1.9). Fig. 4.1 shows the schematic representations of the cloning of α1, β1 and α5
subunits of GABAA receptors. The identity and integrity of the cDNA insert was
confirmed by sequencing (3.3.8) typically using primers flanking the polylinker site (T7
and pSGEM-down or SP6). The final plasmids were named according to table 1.

Chapter 4 Results I
57
Table 1: List of plasmids containing GABAA receptors cDNAs used and templates for PCR as well
For construction of an expression vector for the rat θ subunit, no cloned cDNA was
available. Therefore it was decided to amplify the cDNA by RT-PCR. As a template for
this PCR, rat cDNA was used generated by reverse transcription (3.2.1) from total RNA
isolated from rat brain (3.2.2). As it turned out that it was impossible to amplify the
complete open reading frame of the theta subunit for cloning in one piece, the open
reading frame was amplified in two pieces using the primer pairs theta-ATG, theta-5`up
for the 5'-fragment and theta-stop, theta-3`down for the 3'-fragment (3.2.2). Cloning of
the complete open reading frame was achieved by first cloning of 5' fragment into the
NotI and EcoRI site of pSGEM (3.3.1 - 3.3.7). Into the resulting plasmid, the 3'-fragment
was inserted between the EcoRV and ApaI sites. The final plasmid pSGEMrat-θ
contained the complete open reading frame cloned into the NotI and ApaI sites of the
pSGEM polylinker. The singularly site EcoRV in the theta cDNA was used for fusing
together both fragments. The final plasmid was verified by sequencing (3.3.8) with
primers flanking the polylinker (T7, pSGEM-down) and primer theta-seq-1 sequencing
the fusion site of the two fragments.
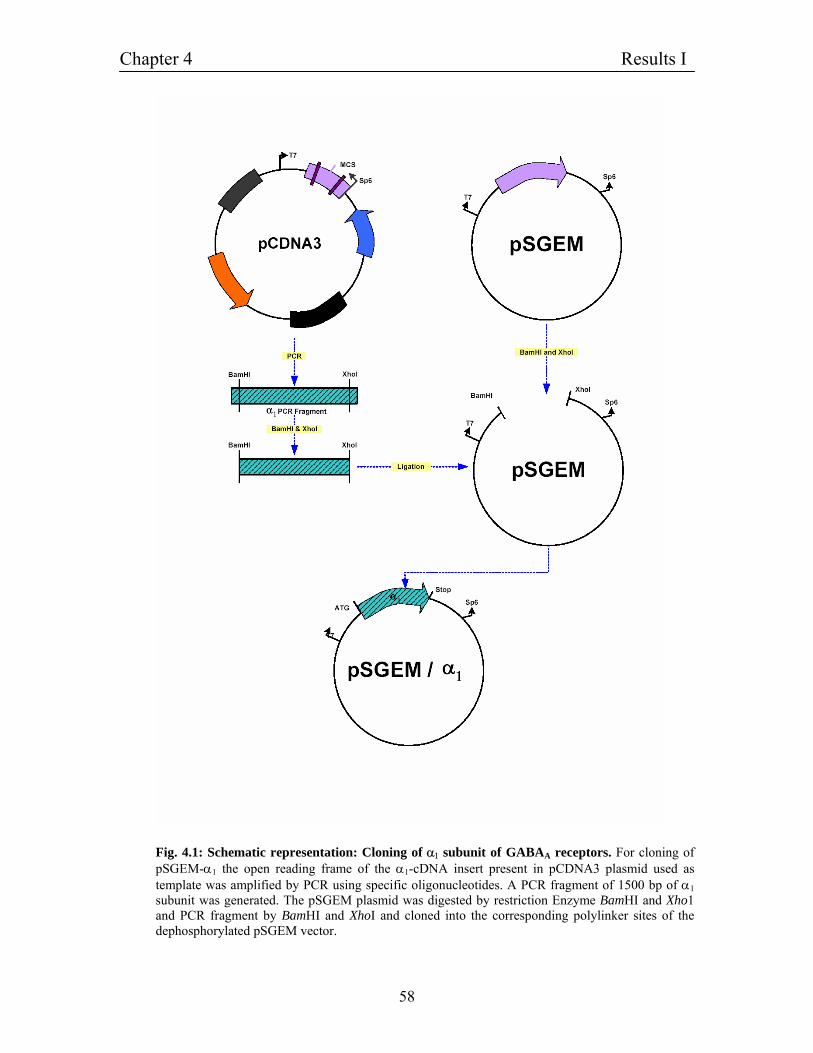
Chapter 4 Results I
58
Fig. 4.1: Schematic representation: Cloning of α1 subunit of GABAA receptors. For cloning of pSGEM-α1 the open reading frame of the α1-cDNA insert present in pCDNA3 plasmid used as template was amplified by PCR using specific oligonucleotides. A PCR fragment of 1500 bp of α1 subunit was generated. The pSGEM plasmid was digested by restriction Enzyme BamHI and Xho1 and PCR fragment by BamHI and XhoI and cloned into the corresponding polylinker sites of the dephosphorylated pSGEM vector.
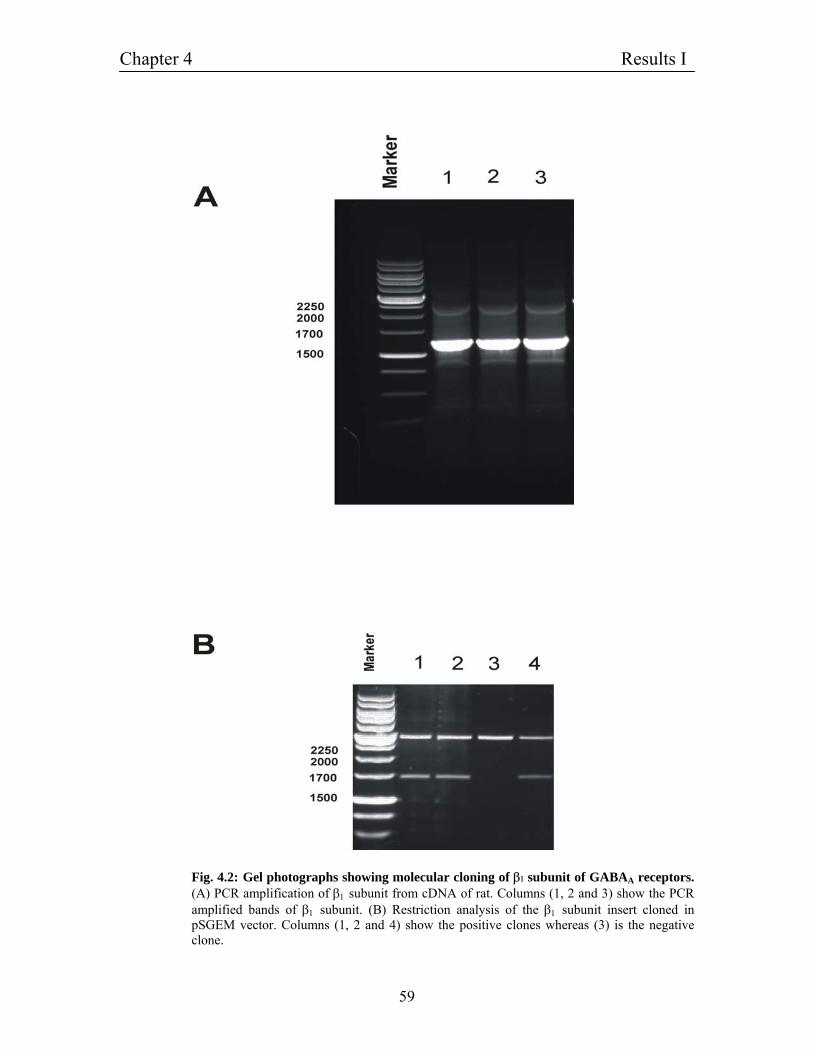
Chapter 4 Results I
59
Fig. 4.2: Gel photographs showing molecular cloning of β1 subunit of GABAA receptors. (A) PCR amplification of β1 subunit from cDNA of rat. Columns (1, 2 and 3) show the PCR amplified bands of β1 subunit. (B) Restriction analysis of the β1 subunit insert cloned in pSGEM vector. Columns (1, 2 and 4) show the positive clones whereas (3) is the negative clone.
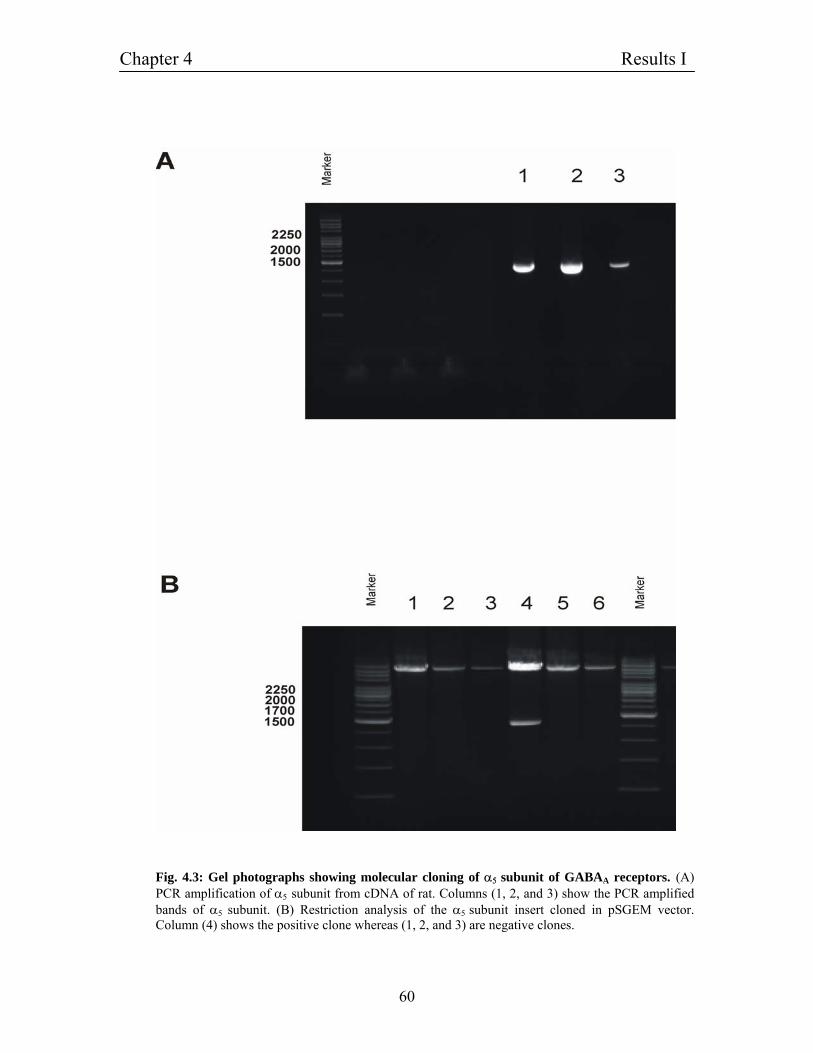
Chapter 4 Results I
60
Fig. 4.3: Gel photographs showing molecular cloning of α5 subunit of GABAA receptors. (A) PCR amplification of α5 subunit from cDNA of rat. Columns (1, 2, and 3) show the PCR amplified bands of α5 subunit. (B) Restriction analysis of the α5 subunit insert cloned in pSGEM vector. Column (4) shows the positive clone whereas (1, 2, and 3) are negative clones.

Chapter 4 Results I
61
4.3: Establishing functional expression GABAA receptors in Xenopus oocytes
In the first electrophysiological part of this work it had to be shown that GABAA
receptors could be successfully expressed in Xenopus oocytes using our expression
vectors. Further it was intended to test, whether the basic properties of the expressed
GABA receptors, like the affinity for GABA match the data known from literature.
To functionally characterize the GABAA subunits, various combinations of their cRNA
generated by in vitro transcription (3.4.1) were injected into Xenopus oocytes (3.5.2) and
measured using two-electrode voltage clamp technique (3.5.3). Application of GABA to
oocytes injected with cRNA of various subunit combinations elicited inward membrane
currents (typically measured at -60 mV), which are due to the opening of membrane
channels permeable mainly to chloride ions. Dose-response curves were generated by
application of various concentrations of GABA.
Of all the β subunit-containing GABAA receptors in the brain, only 19-25 % contains the
β3 subunit, with 55-60 % containing the β2 subunit and 16-18 % containing the β1 subunit
(Benke et al., 1994). We checked the effect of various β subunits for GABA affinity
when combined with the α1 subunit. Fig. 4.5A shows the dose-response curves for α1β1,
α1β2 and α1β3. GABA activated these heteromultimeric receptors with an EC50 of 8.14 ±
0.03 µM, (n = 9), 3.9 ± 0.08 µM, (n = 5) and 1.03 ± 0.15 µM, (n = 5) respectively.
Incorporation of β3 subunit in to the combination leads to a higher reduction in the EC50
of GABA (Fig. 4.4A).
There are 6 different subtypes of α subunit of GABAA receptors. α and β are described
as having binding site for GABA and in conjunction with γ2 subunit α forms binding site
for benzodiazepines (Minier and Siegel, 2004; Bohme et al., 2004; Gunther et al., 1995;
Walters et al., 2000). Fig. 4.4C shows dose-response curve and bar-diagram for α3β1 and
α5β1. GABA evoked EC50 for these receptors are 1.28 ± 0.07 µM, (n = 3) and 3.7µM ±
0.11, (n = 5); respectively. α3β1 seems to be most sensitive GABA combination when
keeping β1 as a constant subunit.
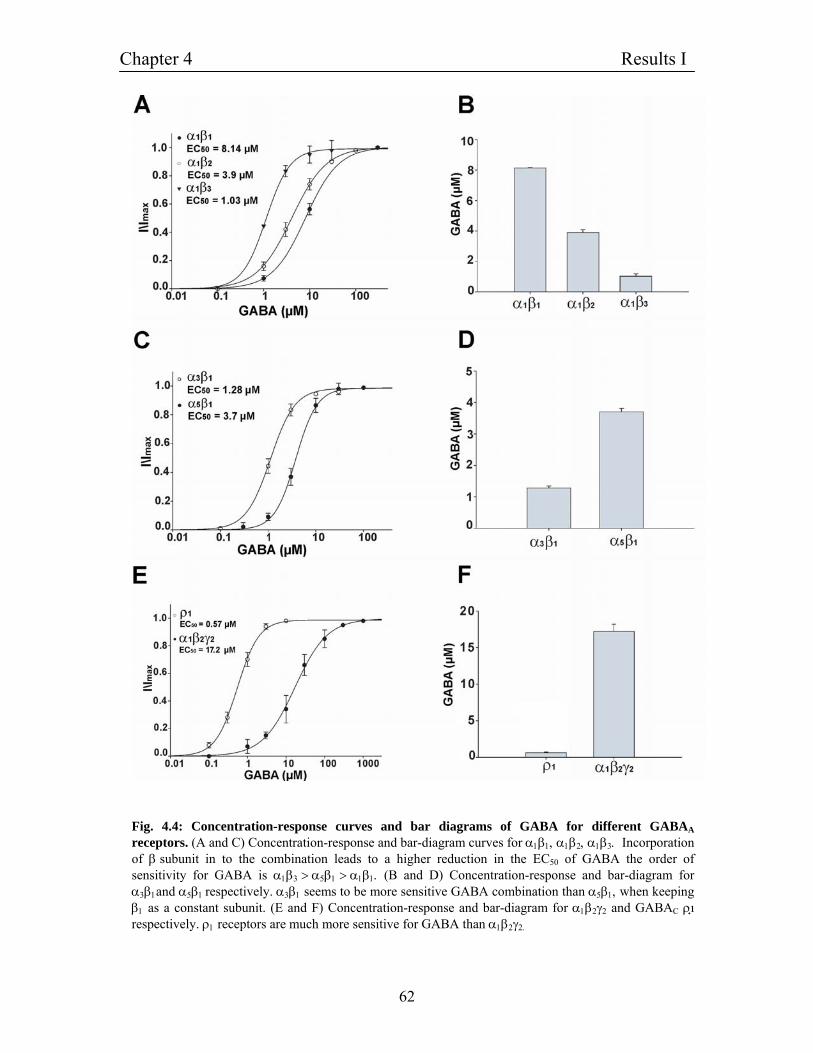
Chapter 4 Results I
62
Fig. 4.4: Concentration-response curves and bar diagrams of GABA for different GABAA receptors. (A and C) Concentration-response and bar-diagram curves for α1β1, α1β2, α1β3. Incorporation of β subunit in to the combination leads to a higher reduction in the EC50 of GABA the order of sensitivity for GABA is α1β3 > α5β1 > α1β1. (B and D) Concentration-response and bar-diagram for α3β1and α5β1 respectively. α3β1 seems to be more sensitive GABA combination than α5β1, when keeping β1 as a constant subunit. (E and F) Concentration-response and bar-diagram for α1β2γ2 and GABAC ρ respectively. ρ1 receptors are much more sensitive for GABA than α1β2γ2.

Chapter 4 Results I
63
The major receptor subtype of the GABAA receptor in the brain most probably consists of
α1, β2 and γ2 subunits. The most likely stoichiometry is two α subunits, two β subunits
and one γ subunit. α1β2γ2 is the most prominent GABAA receptor combination in the
CNS, contributing 70 % of the total GABAA receptors (Mehta and Ticku 1999). The ρ1 is
the GABAC receptors and is insensitive for commonly known modulators like
neurosteroids and barbiturates. Fig. 4.4E shows the dose response curve for α1β2γ2 and ρ1
where GABA activates these receptors with an EC50 of 17.2 ± 0.1 µM, (n = 5) and 0.57 ±
0.01 µM, (n = 5), respectively.
Table 2 shows the EC50 of various GABAA receptor subunit combinations, checked in
this study. α1β3 is the most sensitive subunit combination we studied. The order of
sensitivity for GABA is α1β3 > α3β1 > α1β2 > α5β1 > α1β1> α1β2γ2.
S.No. Subunit Combinations
EC50 (µM)
1. α1β3 1.03 ± 0.15 2. α3β1 1.28 ± 0.07 3. α1β2 3.19 ± 0.08 4. α5β1 3.70 ± 0.11 5. α1β1 8.14 ± 0.03 6. α1β2γ2 17.2 ± 0.10
Table 2: EC50 of various tested GABAA receptor subunit combinations.
For the combination α1β1, an even more detailed characterization was performed. The
reversal potential -25 mV of the GABA evoked current determined by I/V curve fits the
fact that GABA evoked a chloride current (data not shown). Also the basic
pharmacological properties of GABAA receptors like block by PTX and bicuculline were
experimentally confirmed (data not shown). To characterize the potentiation properties of
α1β1, we co-applied 2µM GABA with various concentrations of pentobarbital and
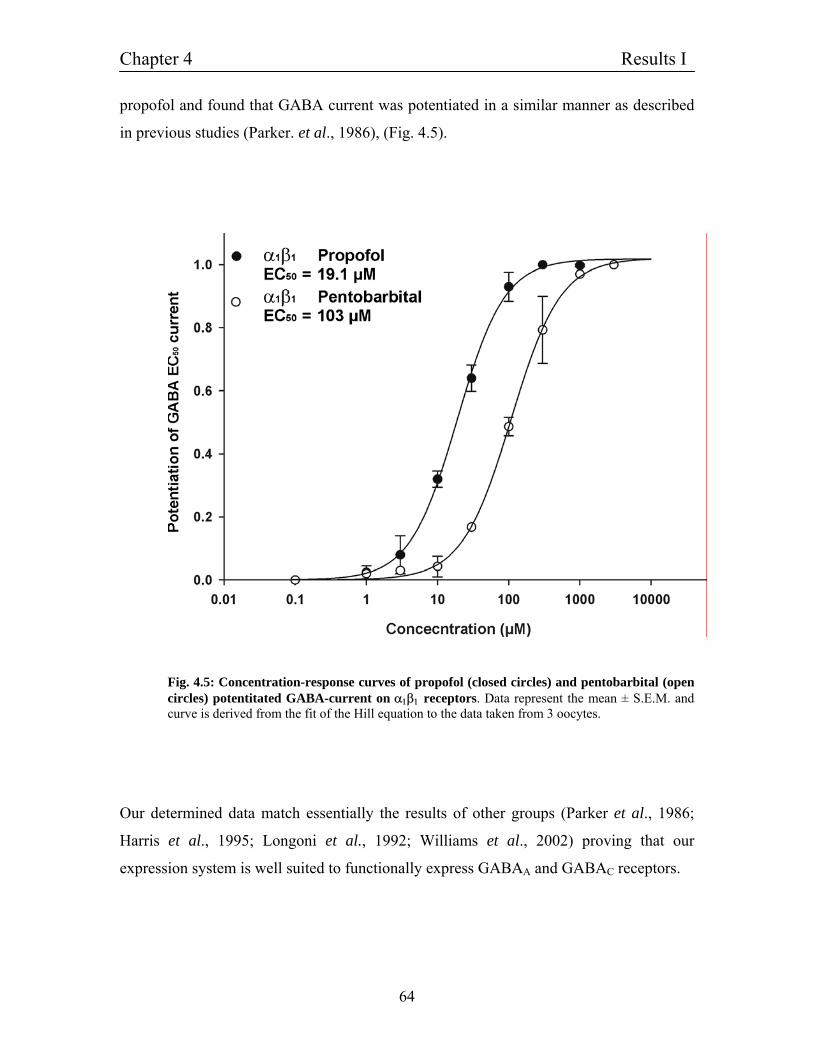
Chapter 4 Results I
64
propofol and found that GABA current was potentiated in a similar manner as described
in previous studies (Parker. et al., 1986), (Fig. 4.5).
Fig. 4.5: Concentration-response curves of propofol (closed circles) and pentobarbital (open circles) potentitated GABA-current on α1β1 receptors. Data represent the mean ± S.E.M. and curve is derived from the fit of the Hill equation to the data taken from 3 oocytes.
Our determined data match essentially the results of other groups (Parker et al., 1986;
Harris et al., 1995; Longoni et al., 1992; Williams et al., 2002) proving that our
expression system is well suited to functionally express GABAA and GABAC receptors.

Chapter 4 Results I
65
4.4: Direct effects of histamine on heteromultimeric GABAA receptors
Next we examined the effect of histamine on GABAA receptors. We checked α1β1, α1β2,
α1β3 and α5β1 GABAA receptor subunit combinations. In most tested oocytes, application
of histamine up to 10 mM concentration did not evoke any response or responses below 1
% of the maximal current evoked by saturating concentrations of GABAA (100 µM - 1
mM). These experiments show that under normal conditions, GABAA receptors are not
activated by histamine. In few tested GABAA expressing oocytes, histamine evoked
small currents especially in the subunit combination α1β2γ2. These few special unusual
cases are described later (Chapter 7) in the results section.
4.5: Modulation of heteromultimeric GABAA receptors by histamine GABAA receptors are modulated by various chemical agents like barbiturates (Sigel,
2002; Boileau and Czajkowski, 1999), neurosteroids (Rick et al., 1998) and anesthetic.
(Krasowski et al., 1998) At lower concentration they show no activity on GABAA
receptor, though co-application of them with GABA modulates GABA-evoked current
(Yamakura et al., 2001; Belelli et al., 1999). Therefore, we decided to check the effect of
co-application of histamine with GABA on GABAA receptors.
4.5.1: Potentiation of α1β1 GABAA receptors by histamine Application of GABA to oocytes injected with rat α1β1 subunits cRNA elicited inward
membrane currents (at -60 mV), which are due to the opening of membrane channels
permeable mainly to chloride ions. The amplitude of the responses evoked by 2 µM
GABA in injected oocytes was greatly potentiated when GABA was applied together
with histamine. Control oocytes showed no response to histamine. Fig. 4.6A shows
potentiation of GABA responses by different concentration of histamine. Measurements
were made by exposing oocyte to increasing concentration of histamine (1 µM - 30 mM)
at EC10 GABA (2 µM). The onset of this potentiating effect was rapid. For example, the
oocyte illustrated in Fig. 4.6A showed a maximal potentiation with in 10 sec of adding

Chapter 4 Results I
66
histamine and recovered to the control value within a similar time after washing out the
histamine. Histamine potentiates α1β1 with an EC50 of 0.49 mM, (n = 7).
(Huang and Dillon, 1999) indicated that GABAergic signaling in the CNS may be
significantly altered during conditions that increase or decrease pH. Histamine is a
polyamine so there is a possibility that prolong standing at room temperature may alter its
pH, too. To avoid such situation we measured the pH (7.4) of histamine solution before
and after the experiment and did not find any alteration in the pH of histamine solution
that ruled out the possibility that potentiation by histamine might be just because of
alteration in pH.
To further characterize the nature of potentiation with GABA we checked potentiation
mediated by histamine at various concentration of GABA. Fig. 4.6B and C shows the
histamine potentiated GABA-evoked current at EC50 (10 µM GABA) and EC90 GABA (1
mM), respectively. The variation in the GABA concentration affects the degree of
potentiation. At EC50 of GABA, the histamine potentiated GABA-evoked current with an
EC50 of 710 ± 60 µM, (n = 7). In case of EC90 of GABA, we found that potentiation
mediated by histamine decreased remarkably. α1β1 receptors were potentiated with much
higher EC50 of histamine than the EC50 calculated for it at EC10 and EC50 of GABA. The
EC50 of histamine at EC90 of GABA is 1.9 ± 0.2 mM, (n = 4).
We next inquired whether evoked potentiation was histamine specific or any other
neurotransmitter or substances can modulate GABAA receptors too. We checked large
variety of known neurotransmitters as well as other substances at concentration up to 1
mM and never found any potentiation of the current evoked by 2 µM GABA (Table 3).
Finally, to check that potentiation was not due to the polyamine nature of histamine, we
checked spermidine, a biogenic polyamine and found no change in GABA evoked
currents. Therefore, we confirmed that GABAA receptors are specifically modulated by
histamine.
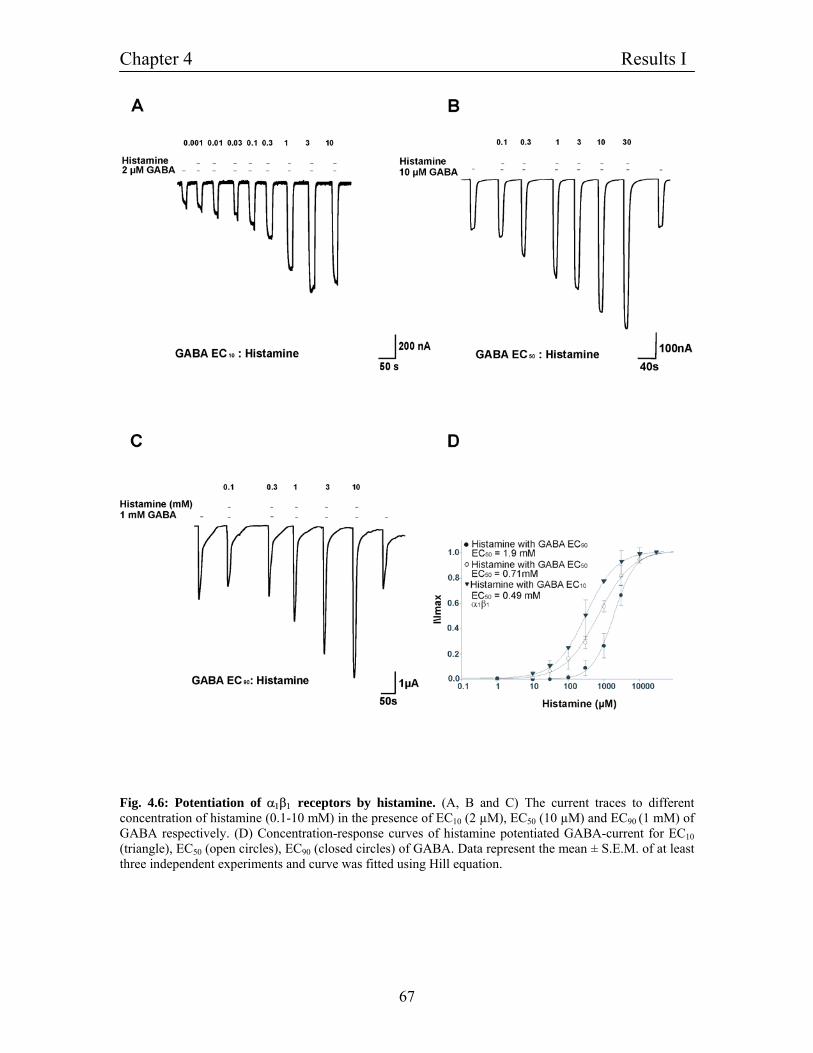
Chapter 4 Results I
67
Fig. 4.6: Potentiation of α1β1 receptors by histamine. (A, B and C) The current traces to different concentration of histamine (0.1-10 mM) in the presence of EC10 (2 µM), EC50 (10 µM) and EC90 (1 mM) of GABA respectively. (D) Concentration-response curves of histamine potentiated GABA-current for EC10 (triangle), EC50 (open circles), EC90 (closed circles) of GABA. Data represent the mean ± S.E.M. of at least three independent experiments and curve was fitted using Hill equation.

Chapter 4 Results I
68
Table 3: Various chemicals checked on GABAA receptors.
We investigated if the co-application of histamine with GABA alters the permeability of
the receptor to ions. Therefore, we injected α1β1 subunits cRNAs into Xenopus oocytes.
The membrane current activated by GABA decreased in amplitude as the oocyte was
depolarized and inverted direction at about –25 mV (Fig. 4.7), which corresponds to the
chloride equilibrium potential in Xenopus oocytes under experimental conditions (Barish
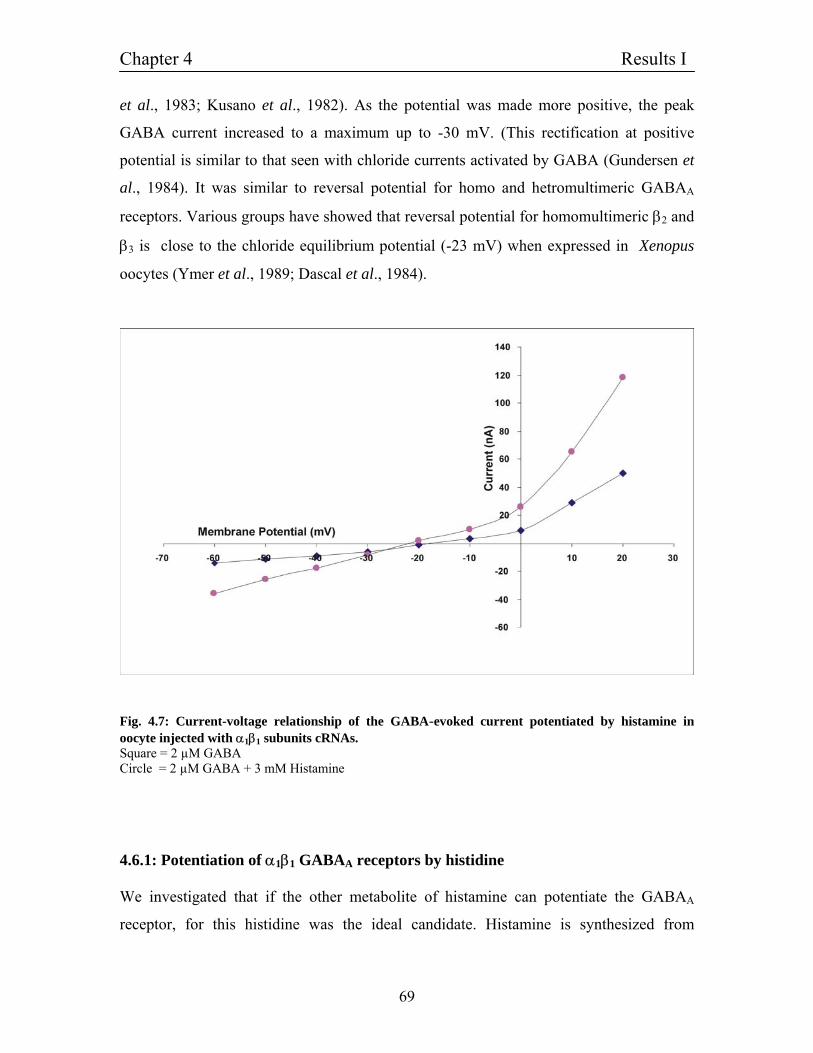
Chapter 4 Results I
69
et al., 1983; Kusano et al., 1982). As the potential was made more positive, the peak
GABA current increased to a maximum up to -30 mV. (This rectification at positive
potential is similar to that seen with chloride currents activated by GABA (Gundersen et
al., 1984). It was similar to reversal potential for homo and hetromultimeric GABAA
receptors. Various groups have showed that reversal potential for homomultimeric β2 and
β3 is close to the chloride equilibrium potential (-23 mV) when expressed in Xenopus
oocytes (Ymer et al., 1989; Dascal et al., 1984).
Fig. 4.7: Current-voltage relationship of the GABA-evoked current potentiated by histamine in oocyte injected with α1β1 subunits cRNAs. Square = 2 µM GABA Circle = 2 µM GABA + 3 mM Histamine 4.6.1: Potentiation of α1β1 GABAA receptors by histidine We investigated that if the other metabolite of histamine can potentiate the GABAA
receptor, for this histidine was the ideal candidate. Histamine is synthesized from

Chapter 4 Results I
70
histidine, which is transported in to neurons by L-amino acid transporter. Histidine
decarboxylase converts histidine to histamine (Haas and Panula, 2003). (Fig. 4.8) shows
potentiation of GABA-evoked membrane currents by histidine. Records are from oocyte
injected with α1β1 subunit cRNA. Oocytes were voltage clamped at -60 mV and the
traces show clamp currents. Inward membrane currents correspond to downward
deflection in this figure. Fig. 4.8A shows response evoked by GABA EC10 (2 µM)
combined with the increasing concentration of histidine (1 µM - 1 mM). Fig. 4.8D shows
dose-response curve for histidine potentiated GABA-evoked currents. Histidine
potentiated GABA-evoked current with an EC50 of 168 ± 14 µM. Data were obtained
from 7 oocytes and are normalized as a fraction of the response in each oocyte to 2 µM
GABA alone. The curve was fitted by Hill equation. In the 3 oocytes, where the
maximum response to both GABA (2 µM) and histidine (1 mM) were determined, the
maximal current evoked by histidine plus GABA was 2-2.5 folds of that evoked by
GABA.
4.6.2: Characterization of histidine potentiation
To characterize the nature of potentiation with GABA we checked potentiation mediated
by histidine at various concentration of GABA. Fig. 4.8B and C shows the histidine
potentiated GABA-evoked current at EC50 and EC90 GABA concentrations, respectively.
We found that the variation in the GABA concentration affects the degree of potentiation.
At 10 µM GABA (EC50), histidine potentiated GABA-evoked current with an EC50 of
78.7 µM, (n = 5). In case of higher concentrations of GABA (1 mM, EC90) we found
quite it interesting that histidine potentited α1β1 receptors with much lower EC50 for
histidine than calculated for it at lower EC10 and EC50 GABA concentrations. The EC50
for histidine at 1 mM of GABA (EC90) is 32.4 ± 5 µM, (n = 5) in all cases, in contrast to
histamine, histidine potentiation induced higher robust inward currents.
Fig. 4.8D shows the dose-response relationship of histidine potentiated GABA-evoked
currents. Here, at EC90 GABA, histidine shows much lower EC50 for potentiation than
EC10. Moreover, at EC90 α1β1 receptors were nearly 10 folds more sensitive to the
histidine.
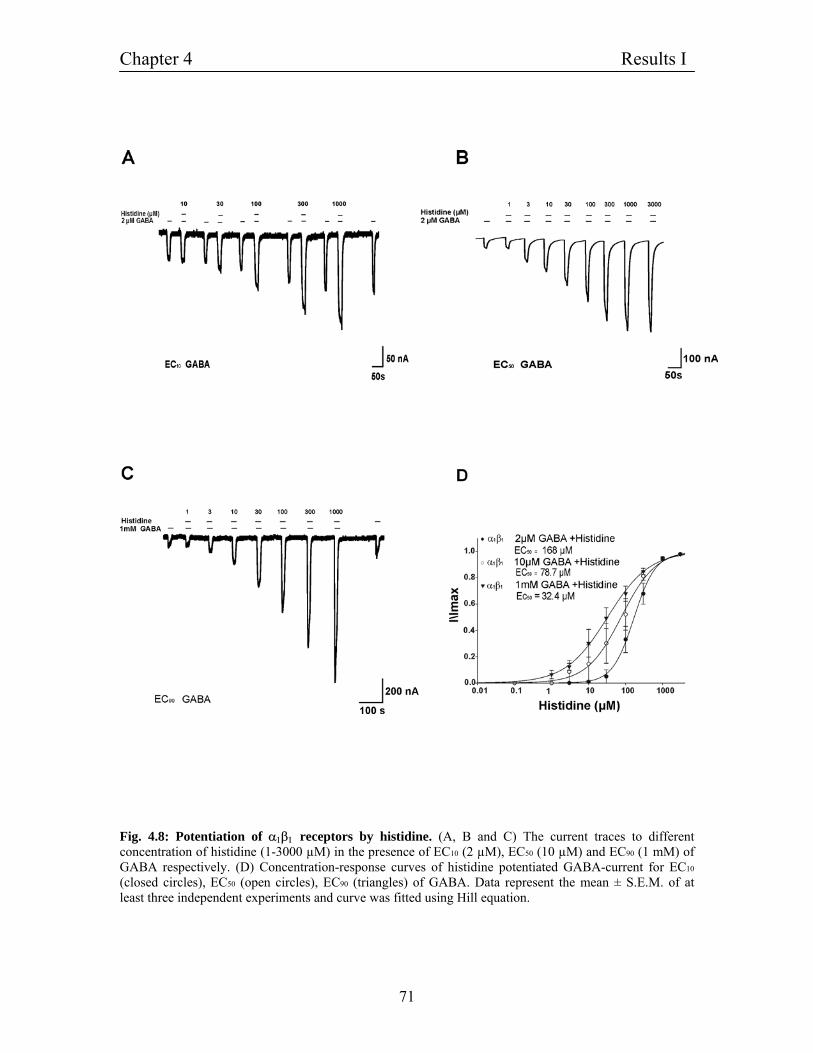
Chapter 4 Results I
71
Fig. 4.8: Potentiation of α1β1 receptors by histidine. (A, B and C) The current traces to different concentration of histidine (1-3000 µM) in the presence of EC10 (2 µM), EC50 (10 µM) and EC90 (1 mM) of GABA respectively. (D) Concentration-response curves of histidine potentiated GABA-current for EC10 (closed circles), EC50 (open circles), EC90 (triangles) of GABA. Data represent the mean ± S.E.M. of at least three independent experiments and curve was fitted using Hill equation.
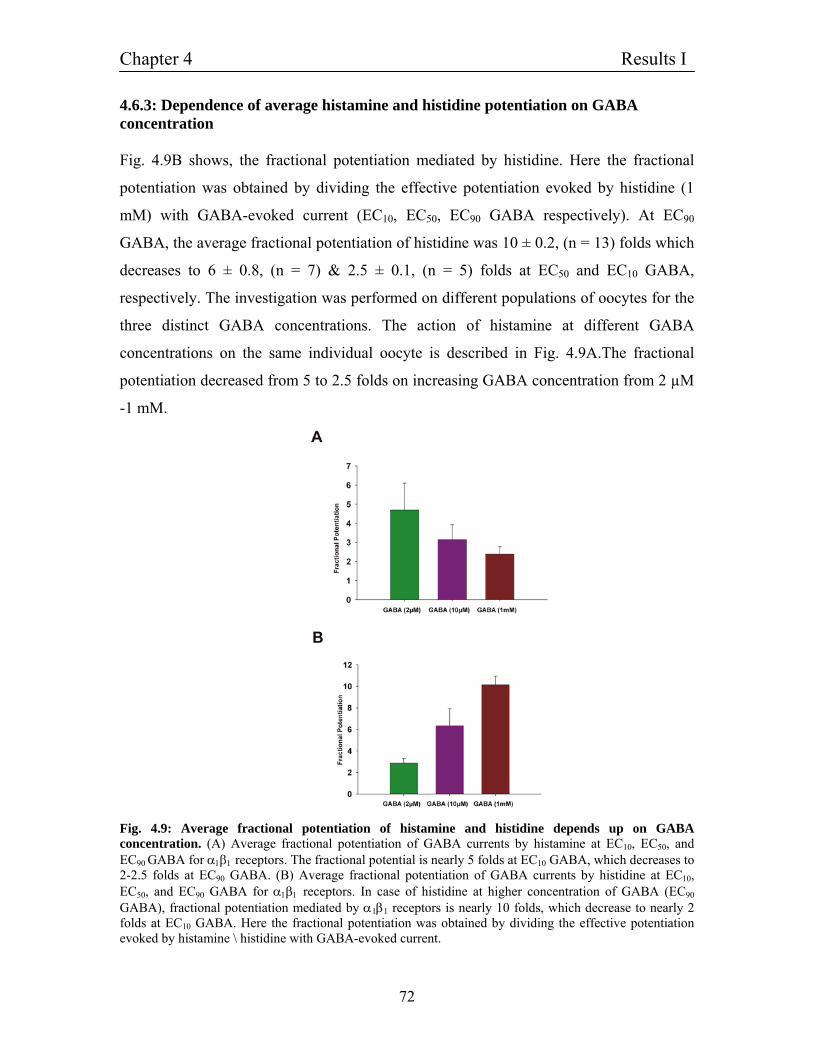
Chapter 4 Results I
72
4.6.3: Dependence of average histamine and histidine potentiation on GABA concentration Fig. 4.9B shows, the fractional potentiation mediated by histidine. Here the fractional
potentiation was obtained by dividing the effective potentiation evoked by histidine (1
mM) with GABA-evoked current (EC10, EC50, EC90 GABA respectively). At EC90
GABA, the average fractional potentiation of histidine was 10 ± 0.2, (n = 13) folds which
decreases to 6 ± 0.8, (n = 7) & 2.5 ± 0.1, (n = 5) folds at EC50 and EC10 GABA,
respectively. The investigation was performed on different populations of oocytes for the
three distinct GABA concentrations. The action of histamine at different GABA
concentrations on the same individual oocyte is described in Fig. 4.9A.The fractional
potentiation decreased from 5 to 2.5 folds on increasing GABA concentration from 2 µM
-1 mM.
Fig. 4.9: Average fractional potentiation of histamine and histidine depends up on GABA concentration. (A) Average fractional potentiation of GABA currents by histamine at EC10, EC50, and EC90 GABA for α1β1 receptors. The fractional potential is nearly 5 folds at EC10 GABA, which decreases to 2-2.5 folds at EC90 GABA. (B) Average fractional potentiation of GABA currents by histidine at EC10, EC50, and EC90 GABA for α1β1 receptors. In case of histidine at higher concentration of GABA (EC90
GABA), fractional potentiation mediated by α1β1 receptors is nearly 10 folds, which decrease to nearly 2 folds at EC10 GABA. Here the fractional potentiation was obtained by dividing the effective potentiation evoked by histamine \ histidine with GABA-evoked current.
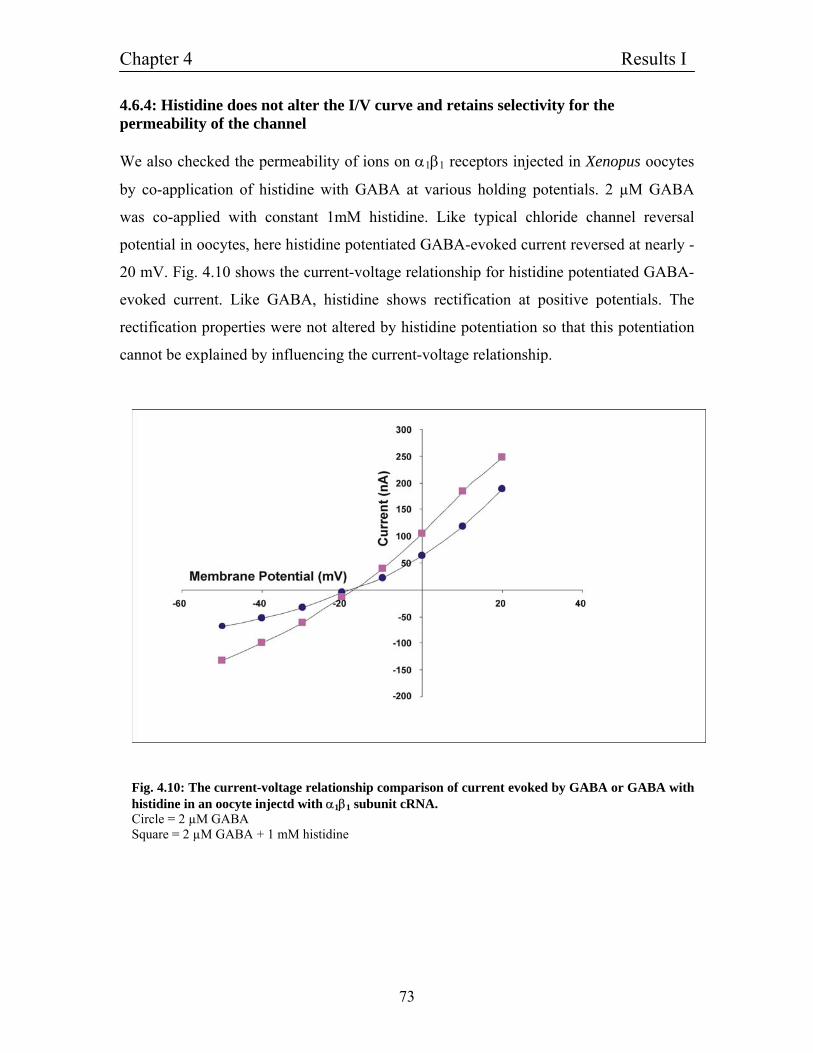
Chapter 4 Results I
73
4.6.4: Histidine does not alter the I/V curve and retains selectivity for the permeability of the channel We also checked the permeability of ions on α1β1 receptors injected in Xenopus oocytes
by co-application of histidine with GABA at various holding potentials. 2 µM GABA
was co-applied with constant 1mM histidine. Like typical chloride channel reversal
potential in oocytes, here histidine potentiated GABA-evoked current reversed at nearly -
20 mV. Fig. 4.10 shows the current-voltage relationship for histidine potentiated GABA-
evoked current. Like GABA, histidine shows rectification at positive potentials. The
rectification properties were not altered by histidine potentiation so that this potentiation
cannot be explained by influencing the current-voltage relationship.
Fig. 4.10: The current-voltage relationship comparison of current evoked by GABA or GABA with histidine in an oocyte injectd with α1β1 subunit cRNA. Circle = 2 µM GABA Square = 2 µM GABA + 1 mM histidine

Chapter 4 Results I
74
4.7: Potentiation of GABAA receptors in HEK 293 cells It is a common fact that the choice of the recombinant expression system can severely
influence the properties of the expressed channels. Therefore, to check whether the
potentiation observed in Xenopus oocytes can be repeated in other cell culture system,
various combinations of GABAA receptors were expressed in HEK 293 cells (3.6.1) and
investigated by patch clamp measurements (3.6.2). To achieve a functional expression of
the α5β1 receptor, the plasmids pCDNA3-rat-G-α5 and pCDNA3-rat-G-β1 were co-
transfected in a 1:1 ratio in HEK 293 cells (3.6.1). The potentiation of histamine–
potentiated currents was determined using brief application of 2 µM GABA alone and 1
mM histamine in combination with 2 µM GABA in whole cell patch-clamp mode.
Potentiation of α5β1 GABAA receptors with histamine produced a substantial increase of
2.5 ± 0.2, (n = 8) folds in the amplitude of currents activated with 2 µM GABA.
Experiments performed with α1β1 receptors in cooperation with Angela Vogt-Eisele and
Katja Erlkamp of our department confirmed these findings. They found that also in α1β1
subunit combinations 1 mM histamine potentiated the current evoked by 2 µM GABA up
to 5 folds (Fig. 4.11). In later experiments with GABAA receptors in a primary culture of
native neurons from the olfactory bulb, they also confirmed that histamine and histidine
potentiated the GABA evoked currents. A proof that the potentiation also occurs in native
neurons and is not limited to artificial recombinant expression systems.
Therefore, my own patch-clamp measurements and those performed in cooperation with
other groups of our department confirm that the potentiation of GABAA receptors
observed in Xenopus oocytes can be repeated in other cell systems, too.
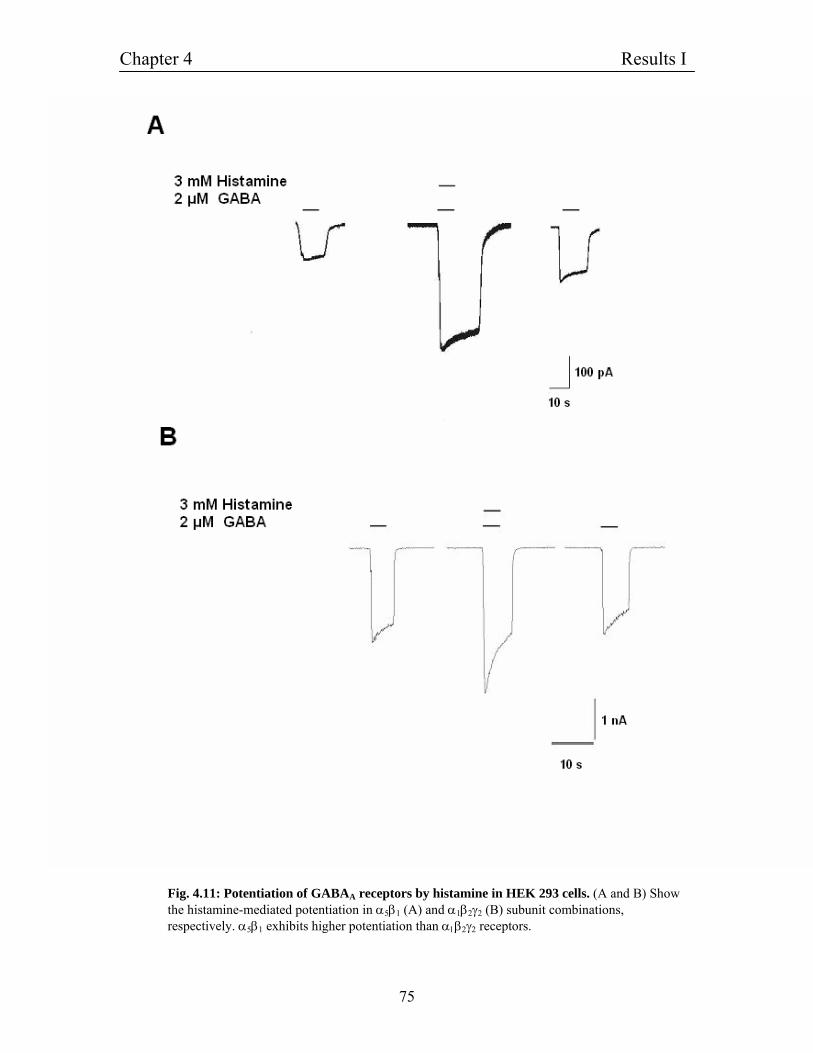
Chapter 4 Results I
75
Fig. 4.11: Potentiation of GABAA receptors by histamine in HEK 293 cells. (A and B) Show the histamine-mediated potentiation in α5β1 (A) and α1β2γ2 (B) subunit combinations, respectively. α5β1 exhibits higher potentiation than α1β2γ2 receptors.

Chapter 4 Results I
76
4.8: Dependence of histamine potentiation on GABAA receptor subunit combinations
We started to investigate two major questions, (1) if substitution of various subunits
affects the affinity for the potentiation mediated by histamine and (2) which subunit has
the binding site for histamine. For this we first of all replaced β subunit keeping the α1
subunits constant. Fig. 4.12A and B shows the current traces for α1β2 and α1β3 subunit
combinations expressed in oocytes respectively. At a concentration of 2 µM GABA,
histamine potentiates α1β2 and α1β3 subunit combinations with an EC50 of 2.15 ± 0.16
mM and 335 ± 0.02 µM, (n = 3) respectively. α1β3 was more sensitive for histamine and
in this subunit combination; the potentiation starts at 100µM histamine whereas α1β2 was
significantly less sensitive though it is potentiated by histamine. The average amplitude
of histamine-mediated potentiation in α1β2 is 2 folds, whereas α1β3 shows more
pronounced potentiation with amplitude of 5 folds. Based on the EC50 values of various
beta subunit combinations, the order of sensitivity for histamine is α1β3 > α1β1 > α1β2.
Therefore, from these experiments we were able to show that affinity for potentiation by
histamine depends up on the subunit combination. Moreover, we started to assume that at
least β subunit should have the binding site for the histamine, as substitution of β
subunits did not abolish potentiation mediated by histamine.
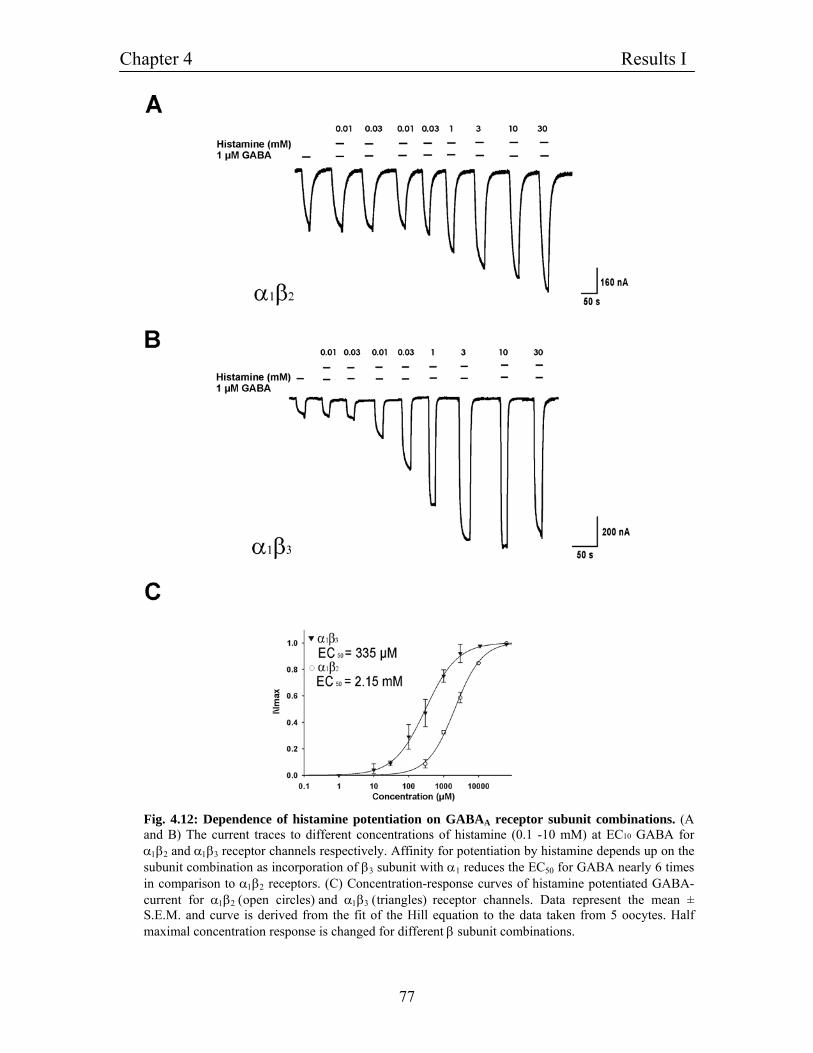
Chapter 4 Results I
77
Fig. 4.12: Dependence of histamine potentiation on GABAA receptor subunit combinations. (A and B) The current traces to different concentrations of histamine (0.1 -10 mM) at EC10 GABA for α1β2 and α1β3 receptor channels respectively. Affinity for potentiation by histamine depends up on the subunit combination as incorporation of β3 subunit with α1 reduces the EC50 for GABA nearly 6 times in comparison to α1β2 receptors. (C) Concentration-response curves of histamine potentiated GABA-current for α1β2 (open circles) and α1β3 (triangles) receptor channels. Data represent the mean ± S.E.M. and curve is derived from the fit of the Hill equation to the data taken from 5 oocytes. Half maximal concentration response is changed for different β subunit combinations.

Chapter 4 Results I
78
In the second series of subunit shuffling experiment, we replaced α1 with α5 keeping the
β1 subunit constant. α1 and α5 have very less sequence homology, so we presumed that if
α subunit has the binding site for histamine than shuffling of α1 with α5 should vanish or
decrease the potentiation meditated by histamine. Fig. 4.13A shows potentiation of
GABA (2 µM) evoked current by histamine on oocyte injected with α5β1 subunit cRNA.
The marked difference between α1β1 and α5β1 is that the onset of potentiation starts at
10-30 µM in α1β1 but in α5β1 it starts at 100 µM histamine concentrations. Also,
comparing with α1β1, the average fractional potentiation mediated by α5β1 is 2.5 lesser
than that of α1β1.
α1β2γ2 is the most prominent GABA subunit combination in the CNS; it accounts nearly
70 % of the total GABAA receptors. We checked α1β2γ2 subunit combinations and indeed
it is potentiatited by histamine Fig. 4.13B. Also like α1β1, the onset of potentiation starts
at 30 µM histamine concentration. Fig. 4.13C shows the dose response curves for all
these different combinations. The EC50 for α5β1 and α1β2γ2 are 1.8 and 1.2 mM, (n = 4)
respectively.
The table 4 shows the EC50 for histamine potentiated GABA-current. The most sensitive
combination for histamine was α1β3 whereas α1β2 was found to be the least sensitive for
histamine comparing to all the subunit combinations checked in our experiments.
Fig. 4.14A and B depicts the bar-diagram of EC50 and fractional potentiation for
histamine on various subunit combinations of GABAA receptors respectively. It is
evident from the data that subunit combinations, which showed less EC50 for histamine,
enhanced maximum histamine-mediated potentiation also.
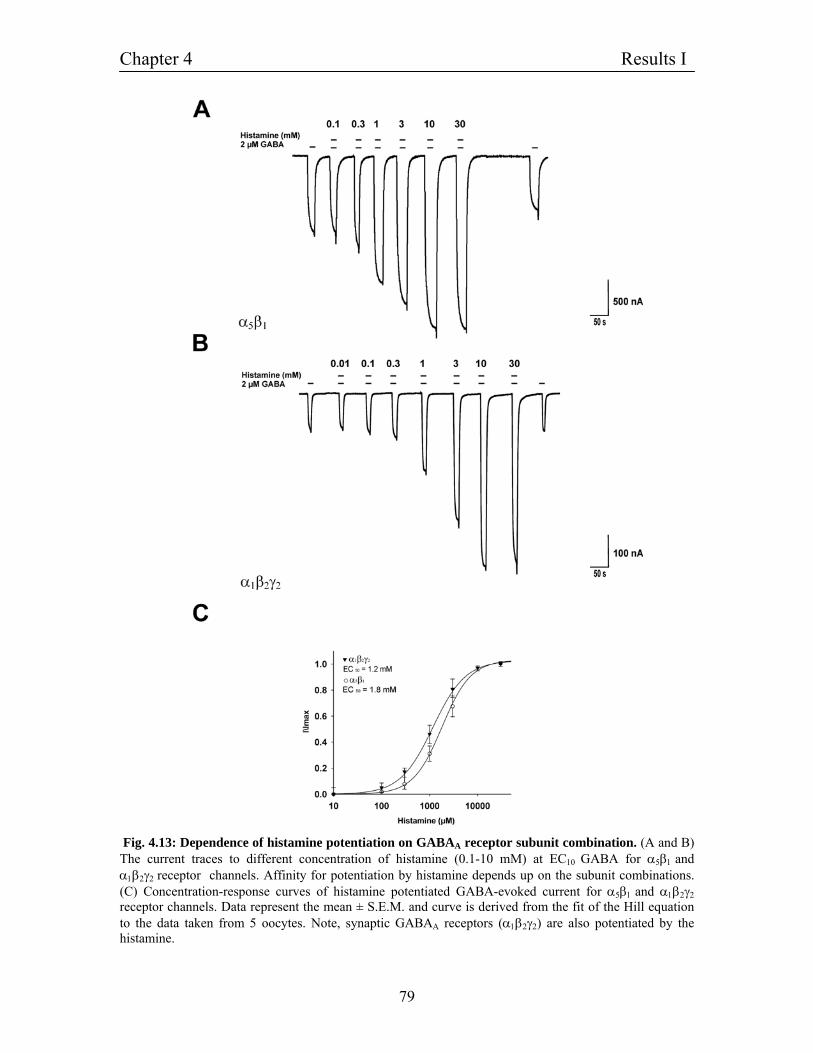
Chapter 4 Results I
79
Fig. 4.13: Dependence of histamine potentiation on GABAA receptor subunit combination. (A and B) The current traces to different concentration of histamine (0.1-10 mM) at EC10 GABA for α5β1 and α1β2γ2 receptor channels. Affinity for potentiation by histamine depends up on the subunit combinations. (C) Concentration-response curves of histamine potentiated GABA-evoked current for α5β1 and α1β2γ2 receptor channels. Data represent the mean ± S.E.M. and curve is derived from the fit of the Hill equation to the data taken from 5 oocytes. Note, synaptic GABAA receptors (α1β2γ2) are also potentiated by the histamine.
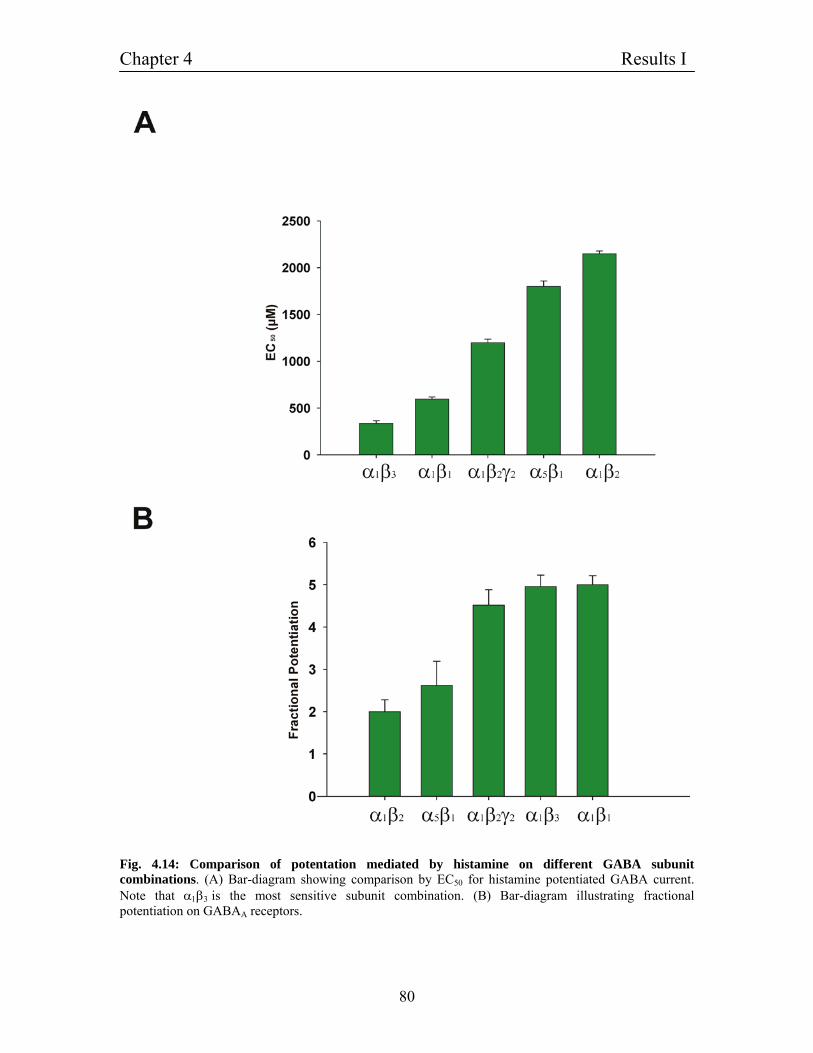
Chapter 4 Results I
80
Fig. 4.14: Comparison of potentation mediated by histamine on different GABA subunit combinations. (A) Bar-diagram showing comparison by EC50 for histamine potentiated GABA current. Note that α1β3 is the most sensitive subunit combination. (B) Bar-diagram illustrating fractional potentiation on GABAA receptors.
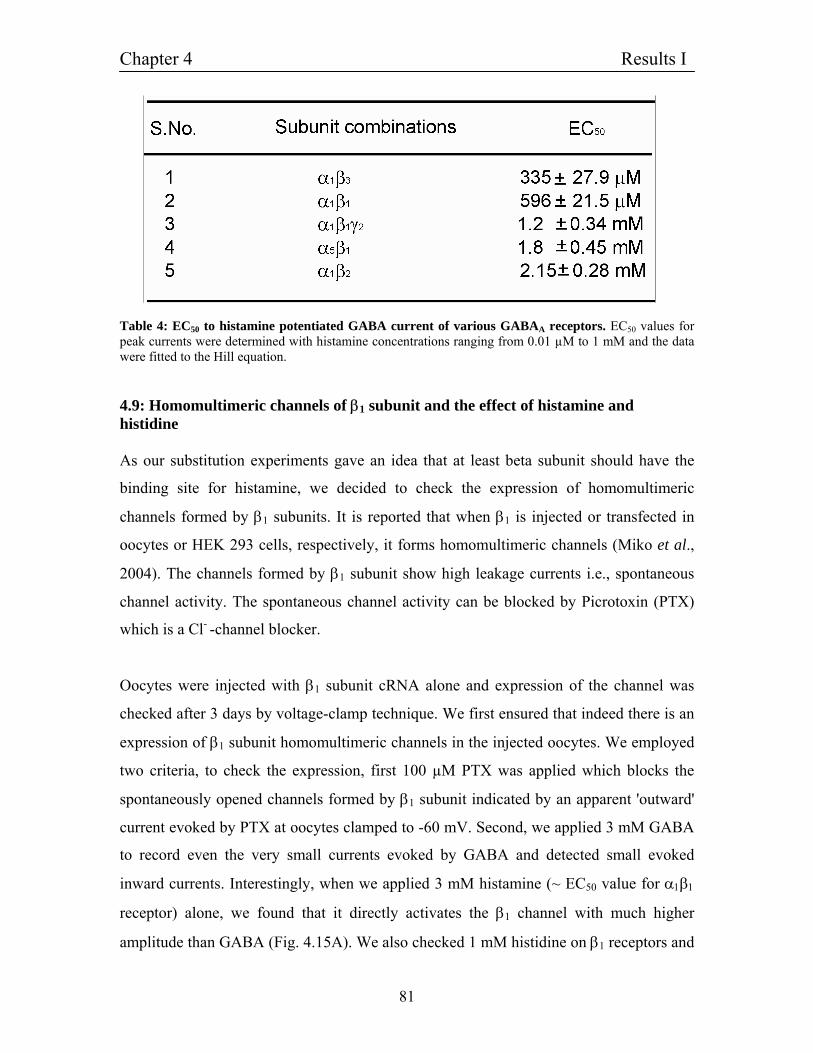
Chapter 4 Results I
81
Table 4: EC50 to histamine potentiated GABA current of various GABAA receptors. EC50 values for peak currents were determined with histamine concentrations ranging from 0.01 µM to 1 mM and the data were fitted to the Hill equation.
4.9: Homomultimeric channels of β1 subunit and the effect of histamine and histidine As our substitution experiments gave an idea that at least beta subunit should have the
binding site for histamine, we decided to check the expression of homomultimeric
channels formed by β1 subunits. It is reported that when β1 is injected or transfected in
oocytes or HEK 293 cells, respectively, it forms homomultimeric channels (Miko et al.,
2004). The channels formed by β1 subunit show high leakage currents i.e., spontaneous
channel activity. The spontaneous channel activity can be blocked by Picrotoxin (PTX)
which is a Cl- -channel blocker.
Oocytes were injected with β1 subunit cRNA alone and expression of the channel was
checked after 3 days by voltage-clamp technique. We first ensured that indeed there is an
expression of β1 subunit homomultimeric channels in the injected oocytes. We employed
two criteria, to check the expression, first 100 µM PTX was applied which blocks the
spontaneously opened channels formed by β1 subunit indicated by an apparent 'outward'
current evoked by PTX at oocytes clamped to -60 mV. Second, we applied 3 mM GABA
to record even the very small currents evoked by GABA and detected small evoked
inward currents. Interestingly, when we applied 3 mM histamine (~ EC50 value for α1β1
receptor) alone, we found that it directly activates the β1 channel with much higher
amplitude than GABA (Fig. 4.15A). We also checked 1 mM histidine on β1 receptors and

Chapter 4 Results I
82
like histamine, histidine directly gated β1 homomultimeric channels (Fig. 4.15B). Co-
application of 3 mM histamine with 3 mM GABA evoked almost same amplitude of
currents compared to the current induced by 3 mM histamine alone. Moreover, co-
application of 1 mM histidine with 3 mM GABA showed the same amplitude of current
like current mediated by 1 mM histidine alone. In homomultimeric β1 channels, the
amplitude of histidine-gated current was higher than the histamine gated currents up to
3/4. We tried 1 mM and 10 mM histamine concentration with 3 mM GABA to check if
the lower or higher concentration of histamine induces any potentiation. For both
concentrations, we found the same amplitude of current compared to histamine alone.
Therefore, at least at the concentrations of histamine and GABA tested, there was no
potentiation effect of histamine but rather a direct opening of the channel. The high
spontaneous activity of β1 channels caused a general instability of oocytes during
measurement of injected oocytes. Therefore, we could not check the lower concentration
of histamine on β1 receptors and extended the characterization of the receptor. A more
detailed characterization was done with the other homomultimeric β2 and β3 receptors as
described later (Chapter 5 and 6).
We investigated if is there any possibility of contribution of homomultimeric channels
formed by β1 in α1β1 subunit combination for histamine direct activation. Does
expression of β1 alone in α1β1 can cause artefacts or misinterpretation of our results? We
ruled out this possibility for two reasons: firstly, in α1β1 2 µM GABA induces a huge
GABA-evoked current whereas in case of β1 there is no response in our experimental
conditions and second, there is no spontaneous channel activity in case of α1β1 receptors
which can be confirmed by application of PTX, which does not show any blocking
activity when applied alone on α1β1 receptors.
These experiments gave us the clear idea that indeed the β1 subunit has a binding site for
histamine. The explanation for not direct activation by histamine on α1β1 receptors is
discussed in the discussion section.
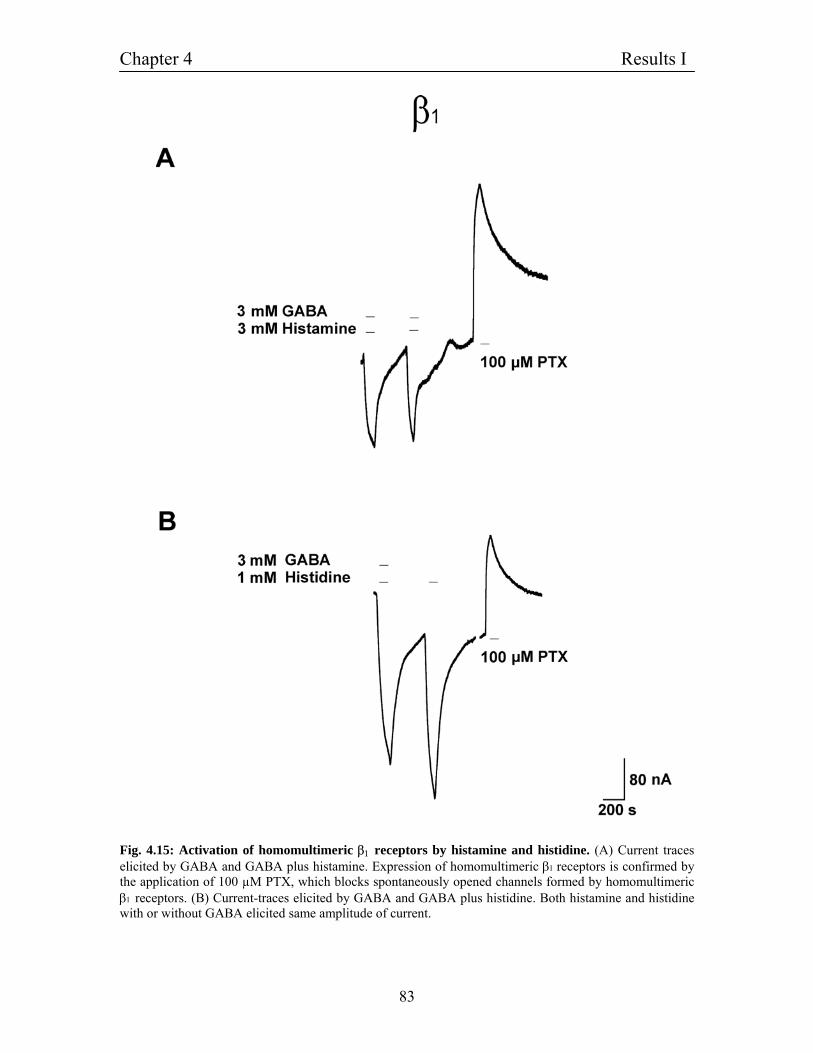
Chapter 4 Results I
83
Fig. 4.15: Activation of homomultimeric β1 receptors by histamine and histidine. (A) Current traces elicited by GABA and GABA plus histamine. Expression of homomultimeric β1 receptors is confirmed by the application of 100 µM PTX, which blocks spontaneously opened channels formed by homomultimeric β1 receptors. (B) Current-traces elicited by GABA and GABA plus histidine. Both histamine and histidine with or without GABA elicited same amplitude of current.

Chapter 4 Results I
84
4.9.1: Homomultimeric channels of β3 subunit and effect of histamine There are subtle differences in the expression of β1 and β3 homomultimeric channels.
GABAA receptors that contain β3 subunits are not activated in the absence of GABA.
They do not have any spontaneously opened channel activity and on the other hand, 100
µM bicuculline nearly completely inhibits current activated by an EC50 concentration of
GABA in cells expressing homomultimeric β3 subunits but had a very small inhibitory
effect in cells expressing homomultimeric β1 subunits (Miko et al., 2004). As in our
previous experiments we found that α1β3 is the most sensitive subunit combination for
histamine potentiation, we looked for the expression of homomultimeric β3 subunit and
effect of histamine on it. Like β1 we found that β3 is also activated by 3 mM histamine
alone. The co-application of 3 mM histamine with 3 mM GABA had the same amplitude
of current mediated by 3 mM histamine alone. The characterization of β3 is presented in
more detail in Chapter 5.
4.10: Molecular cloning of ρ1 subunit of GABAC receptors We asked could GABAC receptor like GABAA receptors be potentiated by histamine. For
construction of an expression plasmid, rat GABAC receptor ρ1 subunit cDNA was cloned
in to pSGEM vector. The plasmid containing ρ1 cDNA as a template was amplified by
PCR (2.10) using oligonucleotides rho1-ATG and rho1-stop (3.2.2). A PCR fragment of
1500 bp of ρ1 subunit was generated, having BamH1 site at 5'-terminal and Xho1
restriction site at 3'-terminal. The pSGEM plasmid was digested by restriction Enzyme
BamHI and Xho1 and PCR fragment by BamHI and XhoI (3.3.1). Fragments were
separated using gel electrophoresis (3.1.2) and purified using a gel extraction kit (3.1.5).
After ligation (3.3.5) and transformation into E. coli XL1-blue (3.3.7), plasmids isolated
from several colonies (3.1.8) were analyzed by restriction mapping (3.3.1) and from
clones with the correct insert, large amounts of plasmid was prepared by the maxi prep
method (3.1.9). The identity and integrity of the cDNA insert was confirmed by
sequencing (3.3.8) using primers flanking the polylinker site (T7 and pSGEM-down) (Fig.
4.16).
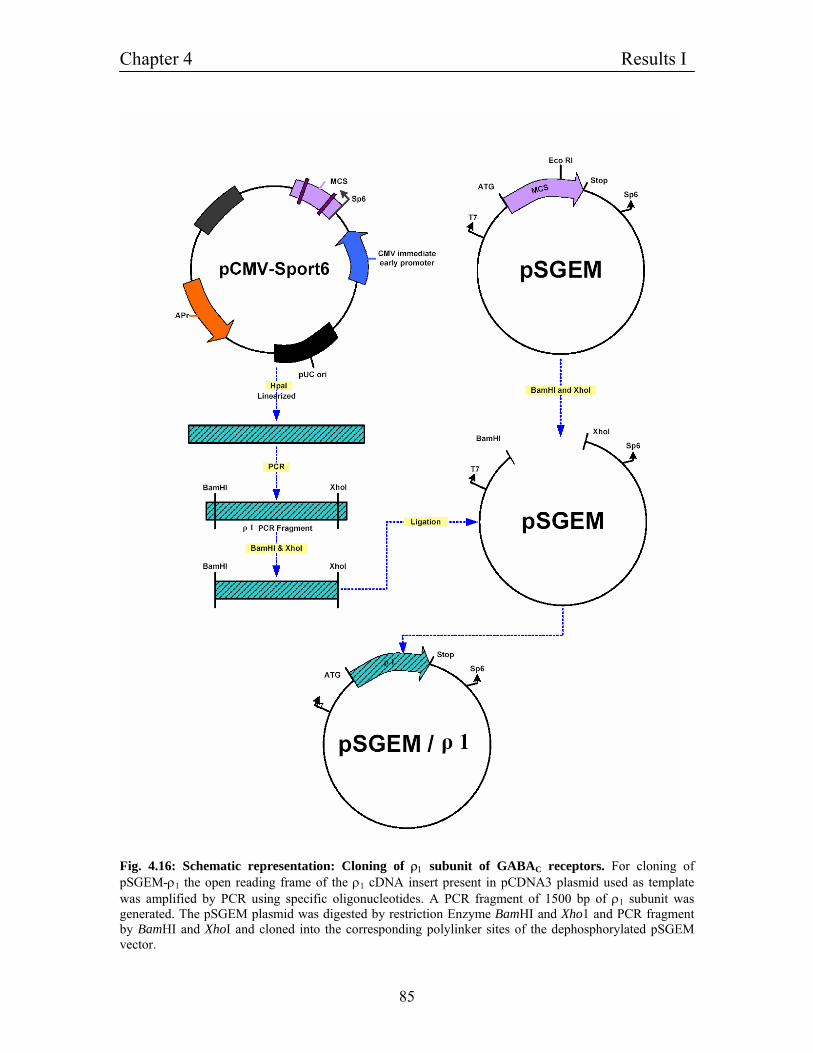
Chapter 4 Results I
85
Fig. 4.16: Schematic representation: Cloning of ρ1 subunit of GABAC receptors. For cloning of pSGEM-ρ1 the open reading frame of the ρ1 cDNA insert present in pCDNA3 plasmid used as template was amplified by PCR using specific oligonucleotides. A PCR fragment of 1500 bp of ρ1 subunit was generated. The pSGEM plasmid was digested by restriction Enzyme BamHI and Xho1 and PCR fragment by BamHI and XhoI and cloned into the corresponding polylinker sites of the dephosphorylated pSGEM vector.

Chapter 4 Results I
86
4.10.1: GABAC receptors: No potentiation by histamine and histidine We analysed GABAC receptors to test if GABAC receptors can also be potentiated by
histamine. Interestingly, histamine up to 10 mM did not modulate the GABA-elicited
currents at 200 nM GABA (EC10 ρ1 receptor channels). Fig. 4.17 shows insensitivity of
ρ1 receptor channels for modulation by histamine. In contrast to GABAA receptors, ρ1
was not potentiated by histamine at any GABA concentration tested. Therefore, we
conclude here that potentiation by histamine is specific only for GABAA receptors.
Fig. 4.17: No potentiation by histamine and histidine on GABAC receptors. The current traces for the EC50 GABA alone and in the presence of histamine and histidine. Both histamine and histidine do not show any potentiation effect on GABA-evoked current.

Chapter 4 Results I
87
4.11: Possible mechanisms of the histamine action In the next set of experiments, the nature of the histamine action on GABAA receptors
was examined in greater detail. In the first set of experiments, possible binding sites were
narrowed down by a comparison of the histamine action with other potentiating agents
(4.11.3), then the action of histamine on the GABA affinity of α1β1 receptors was
characterized (4.11.4) and finally, the binding site for histamine on the β1 subunit was
identified by the investigation of mutated GABA receptors (4.11.5, 4.11.8).
4.11.1: Histamine binding site is different from pentobarbital binding site on β3 subunit of GABAA receptors β subunits of GABAA receptors have binding site for various modulators like
pentobarbital and propofol (Bali and Akabas 2004; Karsowksi et al., 1998; Sanna et al.,
1995). The following experiments were performed with the β3 subunit because of better
expression success compared to β1 homomultimeric channels. We first checked any
similarity of histamine binding site with pentobarbital binding site. Pentobarbital
activated homomultimeric β3 subunit when applied alone (30-100 µM). Fig. 4.18A shows
the traces for current evoked by 100 µM pentobarbital. Bemegride is the potent
antagonist for pentobaribital. When 3 µM bemegride was applied with 100 µM
pentobarbital it inhibited pentobarbital-evoked current more than 50 % (Fig. 4.18A).
Bemegride acts as a reversible antagonist because the GABA-evoked current returned to
its normal level after washing out the bemegride within 1 minute. Fig. 4.18B shows the
traces of histamine-evoked currents on β3 subunit. 500 µM histamine-evoked current
(~EC50 of histamine on α1β1 receptors) is not blocked by 3 µM bemegride. To exclude
the possibility that 3 µM bemegride was a very small concentration for higher histamine
(500 µM) concentration, we either decreased histamine concentration and increased
bemegride concentration and vice-versa. Even in these conditions we did not find any
inhibition for histamine-evoked current by bemegride. The next question was if
bemegride was a specific blocker for pentobarbital. Therefore, bemegride action on
propofol activation was investigated. Application of 50 µM propofol, on homomultimeric
β3 subunit activated the channel. Fig. 4.18C shows the current traces of 50 µM propofol

Chapter 4 Results I
88
and effect of 3 µM bemegride on propofol evoked current. 3 µM bemegride did not block
the propofol-evoked current, we also applied higher concentration of bemegride (10 µM)
but found no inhibition by bemegride. All these experiments suggested that pentobarbital
should act on a different binding site compared to histamine and propofol.
4.11.2: Histamine binding site is similar to propofol binding site on β3 subunit of GABAA receptors - Experiments on homomultimeric β3 subunit We decided to check the possibility of any similarity between propofol and histamine
binding site. There is no specific propofol antagonist existing in our knowledge so, we
could not proceed in previous experimental way. But for the histamine action on GABA
receptors there are several candidates existing, namely the histamine analogs acting on
metabotropic histamine receptors. We first checked those antagonists for histamine-
evoked current. Screening of various metabotropic histamine receptor antagonists pointed
thioperamide (H3 antagonist) as the potent antagonist for histamine-evoked current on
homomultimeric β3 subunits. Fig. 4.18E shows the inhibition of histamine-evoked current
by thioperamide. 30 µM thioperamide inhibits more than 50 % of the current evoked by
500 µM histamine. Remarkably, when 30 µM thioperamide was co-applied with 300 µM
propofol it blocked the propofol-evoked current also more than 50 % (Fig. 14.8F).
Furthermore, we checked the activity of thioperamide on pentobarbital-evoked current.
Application of 30 µM thioperamide did not show any inhibition on pentobarbital-evoked
current Fig. 4.18D. All these experimental data revealed three important things. First time
we showed that thioperamide can act as an antagonist for propofol and hence we identify
the new antagonist for propofol action. Second, these experiments give further arguments
that pentobarbital has a different binding site than histamine. Third, as both propofol and
histamine are blocked by thioperamide with same efficacy, which lead to
pharmacological confirmation that propofol and histamine may have the same binding
site. In the later described pharmacological characterization of β3 homomultimeric
channels (5.4.1) it was found that even the IC50 for thioperamide was comparable for the
block of propofol as well as histamine-evoked currents.
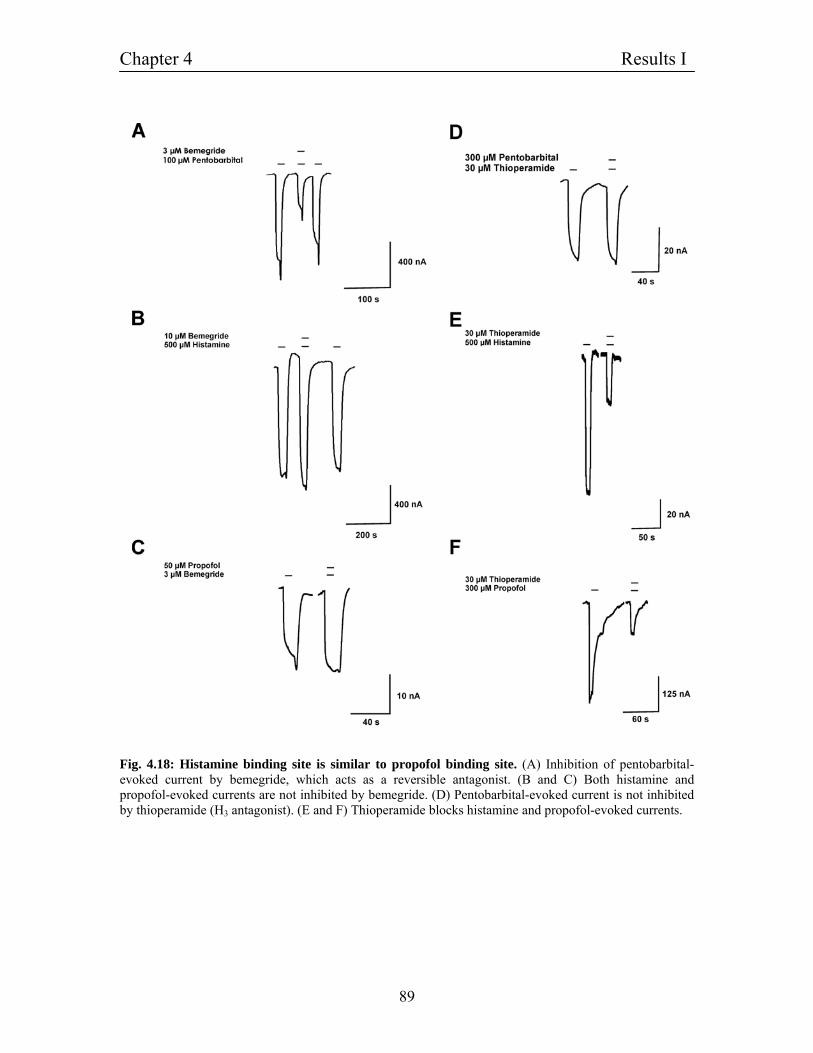
Chapter 4 Results I
89
Fig. 4.18: Histamine binding site is similar to propofol binding site. (A) Inhibition of pentobarbital- evoked current by bemegride, which acts as a reversible antagonist. (B and C) Both histamine and propofol-evoked currents are not inhibited by bemegride. (D) Pentobarbital-evoked current is not inhibited by thioperamide (H3 antagonist). (E and F) Thioperamide blocks histamine and propofol-evoked currents.

Chapter 4 Results I
90
4.11.3: Histamine binding site is similar to propofol binding site on β3 subunit of GABAA receptors - Experiments on heteromultimeric α1β1 receptors The preliminary electrophysiological experiments gave us an idea that histamine may
have the similar binding site like propofol. We asked the question if this is the case then,
if propofol and histamine are mixed together at their EC50 they should potentiate the
GABA-evoked current like if one of them is applied at nearly to its EC80-90. As having the
same binding site means, at EC50 this binding site will be half occupied by one of the
modulators, so addition of another agonist should lead in the enhancement of GABA-
evoked current. Second, if both of them are mixed together at their EC90 and co-applied
with 2 µM GABA, then there should not be any additional potentiation. The reason
behind it was that at saturation concentration, the modulator should almost occupy the
modulator’s binding site so addition of a modulator having the similar binding site should
not enhance any more potentiation. If it is not the case then in EC90 mixing experiment,
addition of another modulator should lead more enhancement of GABA-evoked current
as presence of two modulators with different binding sites should potentiates GABA-
evoked current more efficiently.
Fig. 4.19A shows the current-response relationship for propofol and histamine
potentiation on α1β1 receptor at EC10 GABA. When 3 µM propofol (~ EC50 of propofol
for α1β1 receptor) was co-applied with 2µM GABA, it enhanced the GABA current up to
170 %. With 2 µM GABA, 1 mM histamine (~ EC50 value of histamine potentiation on
α1β1 receptors) evoked almost same amplitude (155 %) of potentiation, similar to
propofol. When 3µM propofol and 1 mM histamine were co-applied with 2 µM GABA,
they increased the GABA-evoked current amplitude up to 250 %; that means simply both
of them showed an additive effect, which was an indication that they somehow may act
on the similar binding site.
Fig. 4.19B depicts the similar experimental strategy but here the parameters were
modified. We checked the effect of EC90 of both modulators alone and in combination of
each other with 2 µM GABA. 10 µM propofol (~ EC90 of propofol for α1β1 receptor)
increased the amplitude of GABA-evoked current at EC10 GABA nearly 300 %. 10 mM

Chapter 4 Results I
91
histamine (~ EC90 of histamine for α1β1 receptor) induced nearly 200 % increase in
amplitude for GABA-evoked current. As expected, the combination of both 10 µM
propofol and 10 mM histamine at EC10 GABA did not enhance the GABA-evoked
current more than 300 %. So, co-application of both modulators at their EC90 did not
enhance more amplitude of GABA-evoked current w.r.t when they applied alone. From
these electrophysiological experiments we were able to get the notion that both
modulators are acting on the same binding site.
In the next series of experiments we investigated the different GABAA receptor subunit
combination. Oocytes injected with α5β1 subunit cRNA were measured after 2-3 days.
We employed the same strategy like previous α1β1 experiments. First we checked the
effect of propofol, histamine alone and in combination with the co-application of 2 µM
GABA at their EC50 concentration. The combination of histamine and propofol showed
the additive effect and increased the GABA-current nearly two fold comparing to current
evoked by histamine or propofol with 2 µM GABA (Fig. 4.20A). Next, we checked the
co-application of histamine and propofol alone and histamine plus propofol with 2 µM
GABA at their EC90 concentration. Interestingly, there was no additive effect. The
amplitude of the current evoked by histamine or propofol with GABA alone or histamine
plus propofol with GABA remained almost same (Fig. 4.20B). This experiment also gave
us the further proof that histamine and propofol have same binding sites.
4.11.4: Effect of histamine on EC50 of GABA on GABAA receptors Fig. 4.21B shows dose-response curves, plotted using hill equation, for GABA alone
(filled symbols) and GABA plus 3 mM histamine (open symbols) for α1β1 receptors. The
data for GABA alone at concentration below about 0.1 µM are on a straight line. At
higher concentration, the increments in response with increase in concentration declined,
and 1 mM GABA gave a nearly maximal response. A further increase in the
concentration from 1 mM to 3 mM caused a slight decrease in the response. However,
this may have arisen because of desensitization during relatively slow drug application.
When histamine (3 mM) was applied together with GABA, the dose-response curve for
GABA was remained same.
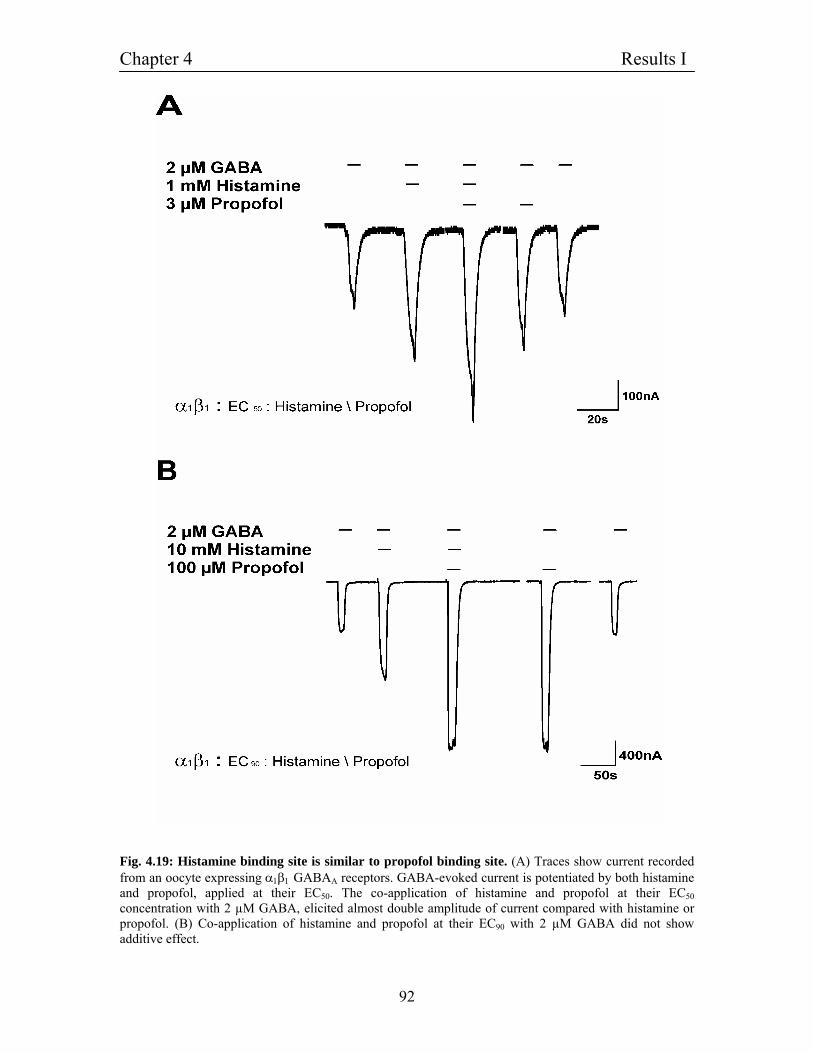
Chapter 4 Results I
92
Fig. 4.19: Histamine binding site is similar to propofol binding site. (A) Traces show current recorded from an oocyte expressing α1β1 GABAA receptors. GABA-evoked current is potentiated by both histamine and propofol, applied at their EC50. The co-application of histamine and propofol at their EC50 concentration with 2 µM GABA, elicited almost double amplitude of current compared with histamine or propofol. (B) Co-application of histamine and propofol at their EC90 with 2 µM GABA did not show additive effect.
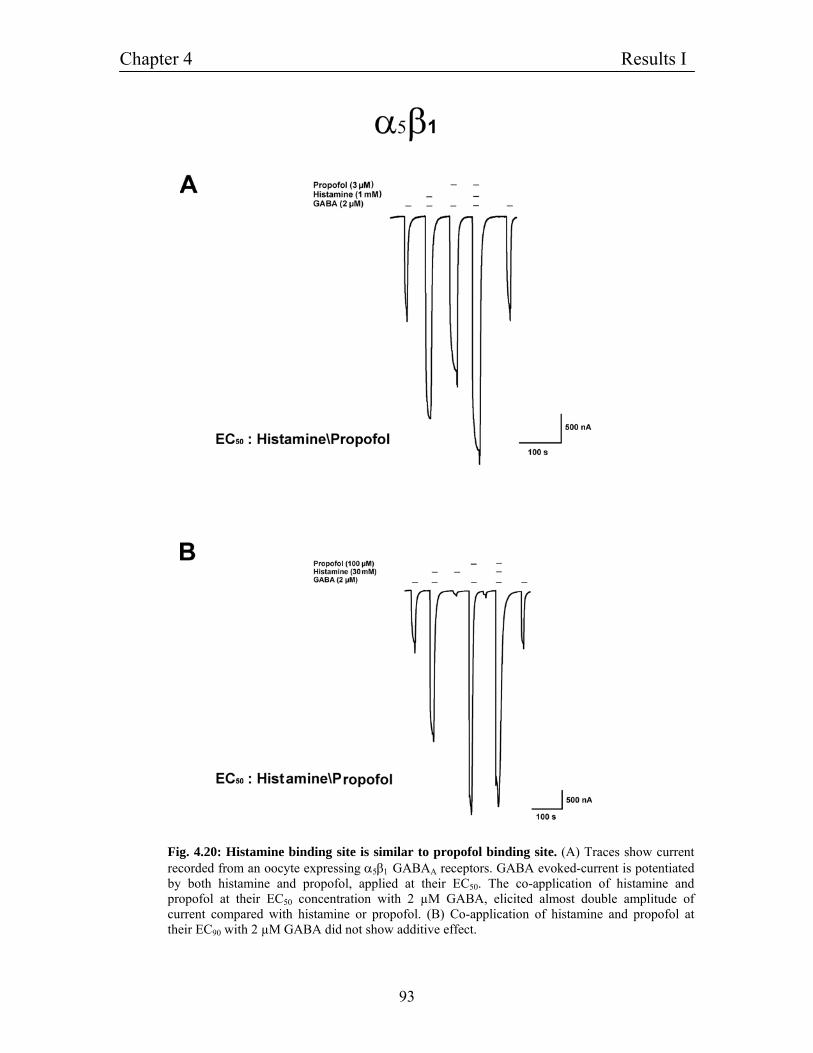
Chapter 4 Results I
93
Fig. 4.20: Histamine binding site is similar to propofol binding site. (A) Traces show current recorded from an oocyte expressing α5β1 GABAA receptors. GABA evoked-current is potentiated by both histamine and propofol, applied at their EC50. The co-application of histamine and propofol at their EC50 concentration with 2 µM GABA, elicited almost double amplitude of current compared with histamine or propofol. (B) Co-application of histamine and propofol at their EC90 with 2 µM GABA did not show additive effect.
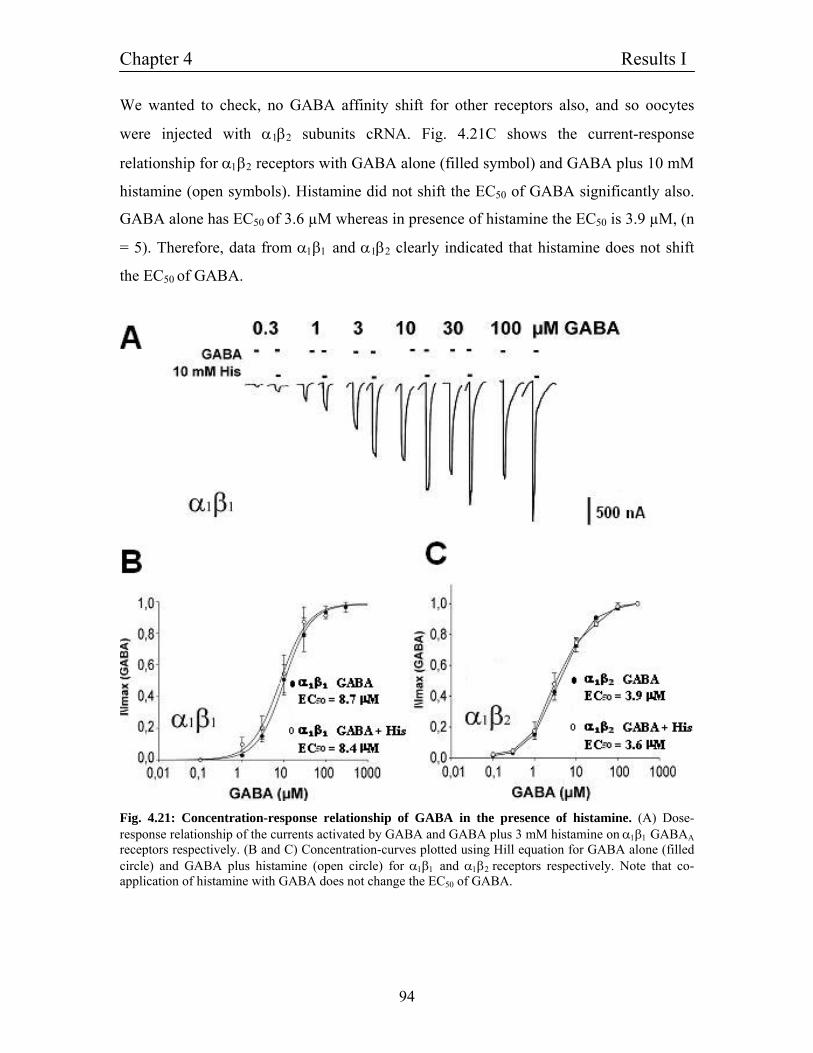
Chapter 4 Results I
94
We wanted to check, no GABA affinity shift for other receptors also, and so oocytes
were injected with α1β2 subunits cRNA. Fig. 4.21C shows the current-response
relationship for α1β2 receptors with GABA alone (filled symbol) and GABA plus 10 mM
histamine (open symbols). Histamine did not shift the EC50 of GABA significantly also.
GABA alone has EC50 of 3.6 µM whereas in presence of histamine the EC50 is 3.9 µM, (n
= 5). Therefore, data from α1β1 and α1β2 clearly indicated that histamine does not shift
the EC50 of GABA.
Fig. 4.21: Concentration-response relationship of GABA in the presence of histamine. (A) Dose-response relationship of the currents activated by GABA and GABA plus 3 mM histamine on α1β1 GABAA receptors respectively. (B and C) Concentration-curves plotted using Hill equation for GABA alone (filled circle) and GABA plus histamine (open circle) for α1β1 and α1β2 receptors respectively. Note that co-application of histamine with GABA does not change the EC50 of GABA.
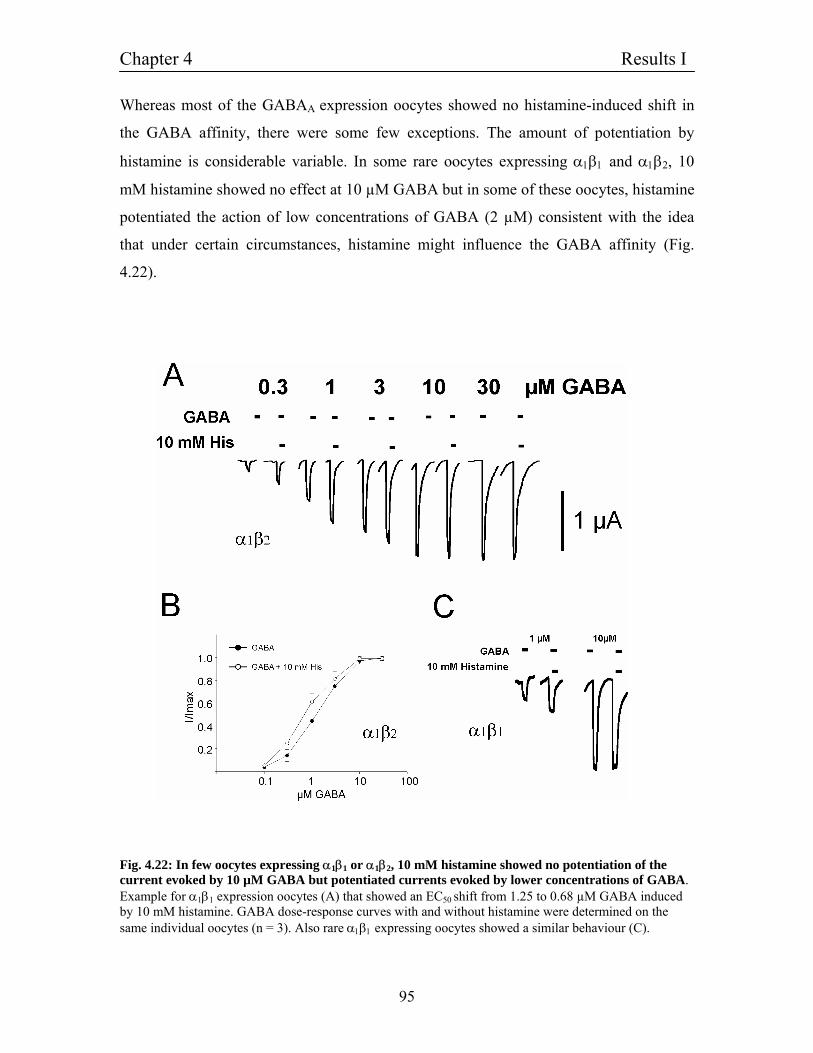
Chapter 4 Results I
95
Whereas most of the GABAA expression oocytes showed no histamine-induced shift in
the GABA affinity, there were some few exceptions. The amount of potentiation by
histamine is considerable variable. In some rare oocytes expressing α1β1 and α1β2, 10
mM histamine showed no effect at 10 µM GABA but in some of these oocytes, histamine
potentiated the action of low concentrations of GABA (2 µM) consistent with the idea
that under certain circumstances, histamine might influence the GABA affinity (Fig.
4.22).
Fig. 4.22: In few oocytes expressing α1β1 or α1β2, 10 mM histamine showed no potentiation of the current evoked by 10 µM GABA but potentiated currents evoked by lower concentrations of GABA. Example for α1β1 expression oocytes (A) that showed an EC50 shift from 1.25 to 0.68 µM GABA induced by 10 mM histamine. GABA dose-response curves with and without histamine were determined on the same individual oocytes (n = 3). Also rare α1β1 expressing oocytes showed a similar behaviour (C).

Chapter 4 Results I
96
4.11.5: Molecular cloning of point mutation in β1 subunits Previous reports have suggested that Methionine at 286 position (M286) of the GABAA
receptors β1 subunit is involved in propofol potentiation (Krasowski et al., 1998). To
determine whether this domain in the GABAA β1 subunit plays a similar role in histamine
mediated potentiation, we decided to create a point mutation (M286W) at position
corresponding to the mutation in the GABAA receptor β1 subunit that is important for the
propofol binding. Rat GABAA receptor β1 subunit cDNA was cloned into pSGEM vector
(4.2). To mutate the methionine to tryptophan in the GABAA receptor β1 subunit at 286
position, site-directed mutagenesis (3.2.3) was performed by using mutated
oligonucleotides in conjunction with a primer-directed polymerase chain reaction (3.2.2).
A mutated DNA fragment of the β1 subunit coding TM3 to C-terminal end including the
mutation M286W was PCR amplified using primers having a BamHI site at the 3´end. In
the amplified PCR fragment at 5' end the EcoRV site was kept preserved. The plasmid
containing cDNA of β1 subunit was digested by restriction enzymes BamHI and EcoRV
fragments were separated using gel electrophoresis (3.1.2) and purified using a gel
extraction kit (3.1.5). The mutated, BamHI-EcoRV restricted PCR fragment was ligated
into the pSGEM vector. After ligation (3.3.5) and transformation into E. coli XL1-blue
(3.3.7), plasmids isolated from several colonies (3.1.8) were analyzed by restriction
mapping (3.3.1) and from clones with the correct insert, large amounts of plasmid was
prepared by the maxi prep method (3.1.9). The identity and integrity of the cDNA insert
was confirmed by sequencing (3.3.8) using primers flanking the polylinker site (T7 and
pSGEM-down) (Fig. 4.23).
4.11.6: β1 (M286W) mutation completely abolishes potentiation mediated by histamine Previous reports have suggested that Methionine at 286 position (M286) of the GABAA
receptors β1 subunit is involved in propofol potentiation (Krasowski et al., 1998) To
determine whether this domain in the GABAA β1 subunit plays a similar role in
histamine-medited potentiation, we created point mutation (M286W) at position 286
(4.11.6) corresponding to the mutation in the GABAA receptor β1 subunit that is
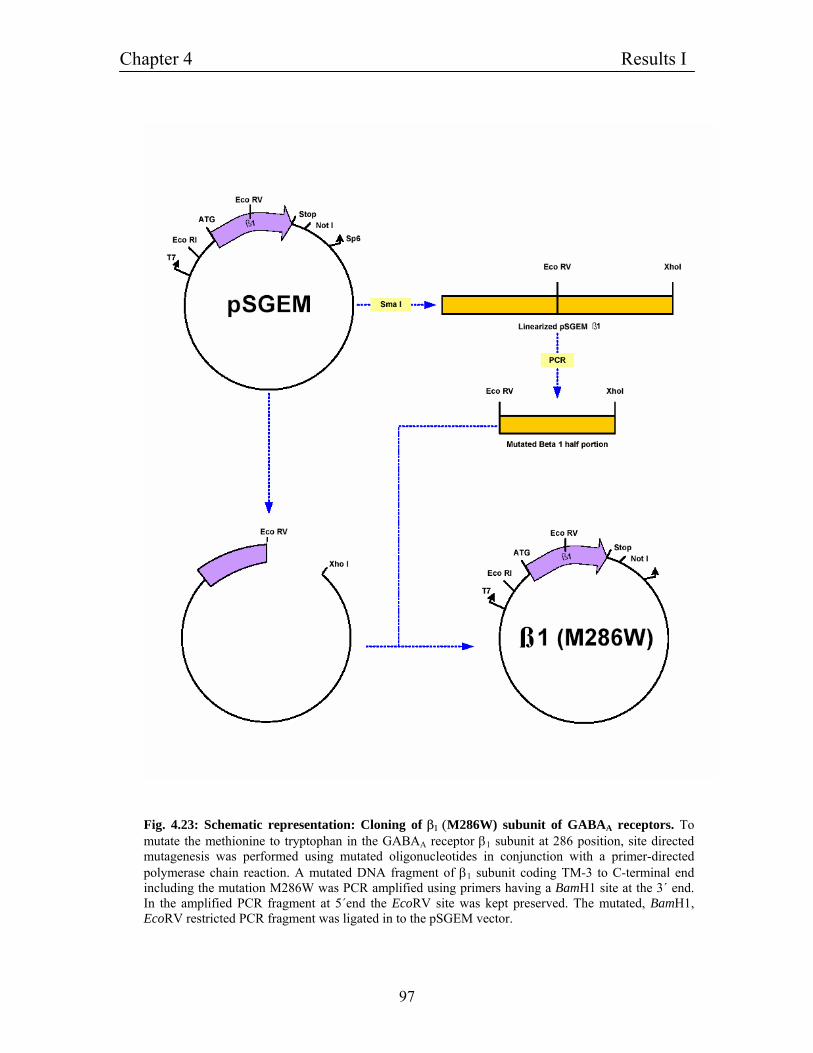
Chapter 4 Results I
97
Fig. 4.23: Schematic representation: Cloning of β1 (M286W) subunit of GABAA receptors. To mutate the methionine to tryptophan in the GABAA receptor β1 subunit at 286 position, site directed mutagenesis was performed using mutated oligonucleotides in conjunction with a primer-directed polymerase chain reaction. A mutated DNA fragment of β1 subunit coding TM-3 to C-terminal end including the mutation M286W was PCR amplified using primers having a BamH1 site at the 3´ end. In the amplified PCR fragment at 5´end the EcoRV site was kept preserved. The mutated, BamH1, EcoRV restricted PCR fragment was ligated in to the pSGEM vector.

Chapter 4 Results I
98
important for the propofol binding. The mutant receptors were then characterized by
determining the potentiation by histamine on GABA-evoked current. Fig. 4.24A
represents a schematic representation for the mutation done on β1 subunit. This mutation
lies in between the TM2 and TM3 domains and specifically to the exterior or outer most
portion of TM2 domain.
Fig. 4.24B shows the electrophysiological characterization of the mutation. Oocytes were
injected with α1β1 (M286W) subunit combinations. Firstly, we checked the potentiation
mediated by propofol on mutant receptor. At EC10 GABA (2 µM), propofol was checked
with increasing concentration (1-10 µM). No potentiation of GABA-evoked current by
propofol was in accordance with previous mutational study done by other groups
(Karsowski et al., 1998). Interestingly when histamine was checked at EC10 GABA, it did
also not induce any potentiation. Fig. 4.24B describes the current traces for histamine. In
α1β1 (M286W), three different concentration dependent effects were checked. We tested
various concentration of histamine (0.1 - 30 mM) at EC50 (~10 µM) and EC90 (~1 mM) of
GABA to completely rule out any potentiation that could be attributed by these
concentrations. Therefore, from these experiments we confirmed that histamine binds to
the similar binding site of propofol.
4.11.7 Histamine and propofol have similar binding sites Fig. 4.25 depicts the effect of β1 subunit mutation on propofol and histamine mediated
potentiation in α1β1 (M286W) expression oocytes. At EC10 GABA, 30 mM histamine did
not show any potentiation that indicates even at that high concentration mutation is so
pronounced that it abolishes all the activity of histamine. It is of special interest also, as in
some studies it has been shown that at very higher concentration of agonist, there is some
degree of activation or potentiation like in pentobarbital mutational study. Induction of no
potentiation by histamine on α1β1 (M286W) shows clearly that M286 is the vital amino
acid for the histamine binding on β1 subunit. Moreover, we also found that 10 µM
propofol with 2 µM GABA does not induce any potentiation. We also did some mixing
experiments where 30 mM histamine was co-applied in the presence of 2 µM GABA and
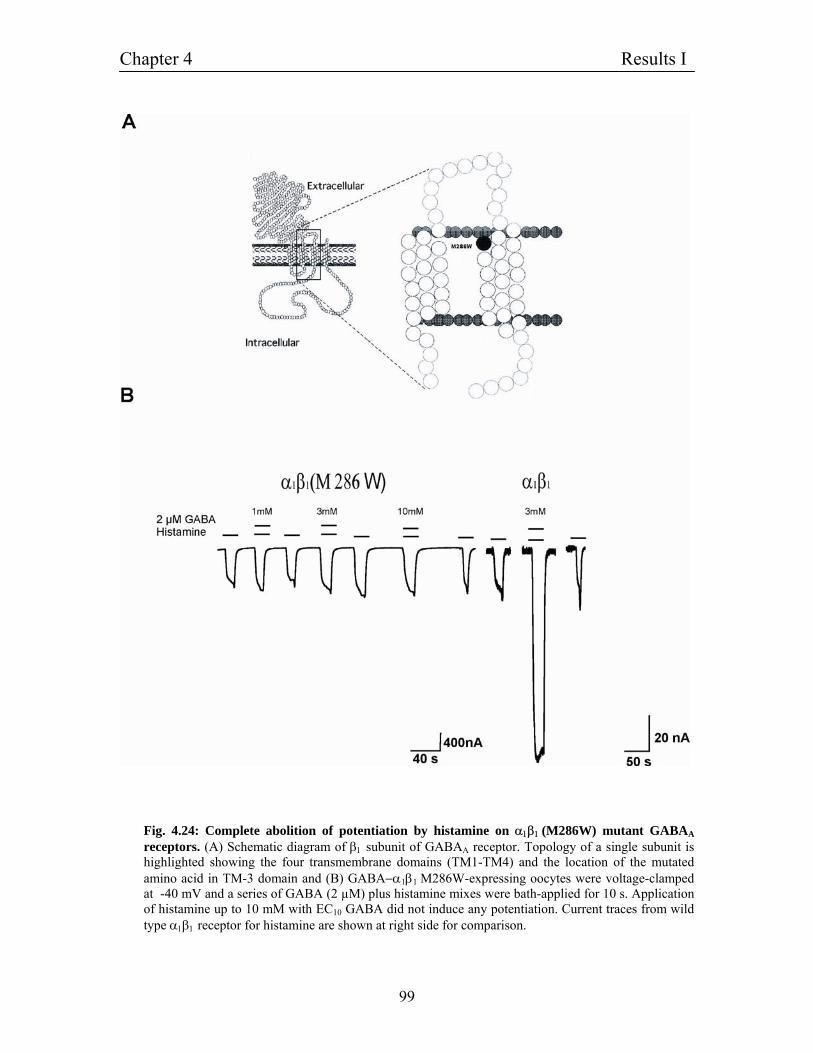
Chapter 4 Results I
99
Fig. 4.24: Complete abolition of potentiation by histamine on α1β1 (M286W) mutant GABAA receptors. (A) Schematic diagram of β1 subunit of GABAA receptor. Topology of a single subunit is highlighted showing the four transmembrane domains (TM1-TM4) and the location of the mutated amino acid in TM-3 domain and (B) GABA−α1β1 M286W-expressing oocytes were voltage-clamped at -40 mV and a series of GABA (2 µM) plus histamine mixes were bath-applied for 10 s. Application of histamine up to 10 mM with EC10 GABA did not induce any potentiation. Current traces from wild type α1β1 receptor for histamine are shown at right side for comparison.
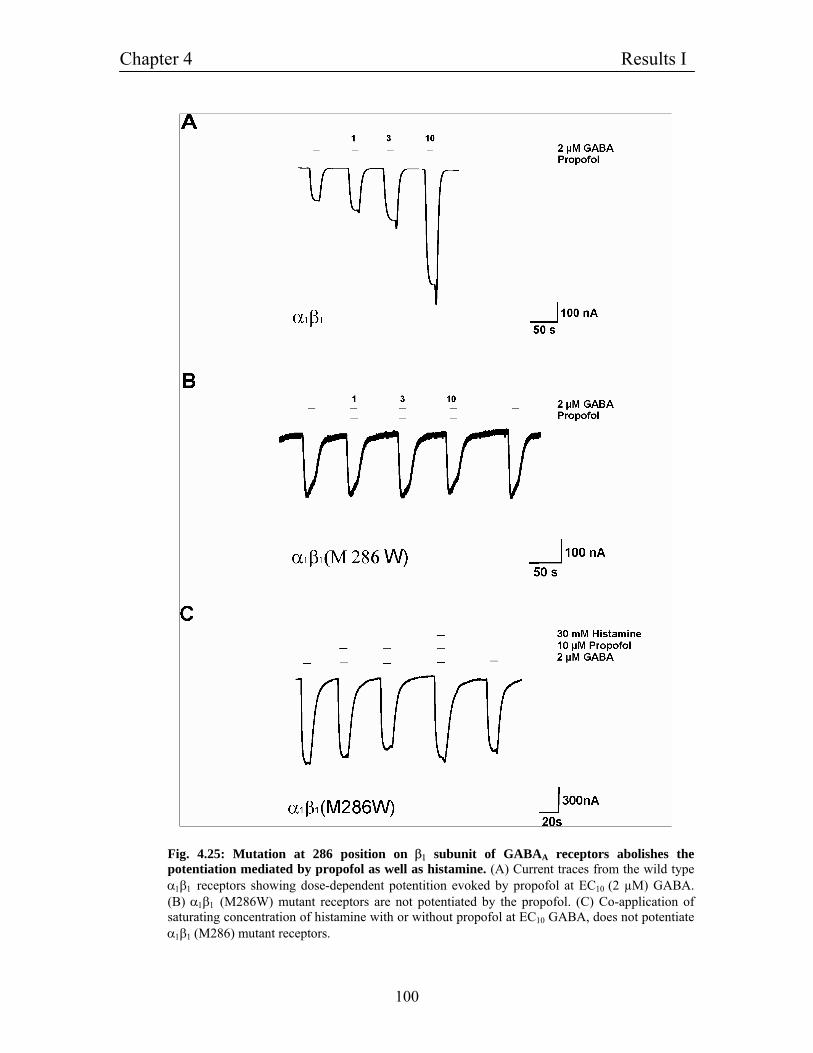
Chapter 4 Results I
100
Fig. 4.25: Mutation at 286 position on β1 subunit of GABAA receptors abolishes the potentiation mediated by propofol as well as histamine. (A) Current traces from the wild type α1β1 receptors showing dose-dependent potentition evoked by propofol at EC10 (2 µM) GABA. (B) α1β1 (M286W) mutant receptors are not potentiated by the propofol. (C) Co-application of saturating concentration of histamine with or without propofol at EC10 GABA, does not potentiate α1β1 (M286) mutant receptors.

Chapter 4 Results I
101
10 µM propofol. Even at that high concentrations, we did not observe any potentiation,
whereas in our previous results we have shown that mixing experiment in α1β1
potentiated GABA-evoked current in the presence of 30 mM histamine and 10 µM
propofol with 2 µM GABA. These experimental data also give further proof that
histamine and propofol has same binding site.
4.11.8: β1 (M286W) mutation completely abolishes potentiation mediated by histidine We also investigated the effect of β1 subunit mutation on the potentiation of histidine.
Fig. 4.26A shows the current-traces where oocytes were injected with α1β1 (M286W)
subunit combination. Application of 1 mM histidine (~ EC99 in α1β1 receptors) with 2 µM
GABA, did not show any potentiation. We checked various concentration of histidine (1-
1000 µM) to clearly characterise the abolition of histidine-mediated potentiation by
mutation. Also we did experiments with different GABA concentration like EC50 and
EC90 GABA, there also keeping all the parameters similar but we could not detect any
potentiation (Fig. 4.26B and C). From these data we demonstrate here that like histamine,
histidine has the same binding site on β1 subunit at the same location similar to propofol
binding site.
4.11.9: Sequence alignment with GABAC receptors depicts that histamine has similar binding site as propofol Moreover, sequence alignment of ρ1 subunit with various beta subunits of GABAA
receptors showed that at position 286 in ρ1 subunit there is tryptophan (W) instead of
methionine (M), which makes ρ1 subunit insensitive to propofol. From this analysis two
important conclusions were derived. First, GABAC receptors are not potentiated by
histamine and histidine; second, having tryptophan instead of methionine at position 286
makes ρ1-receptor insensitive for propofol as well as histamine that give also another
proof for same binding site for propofol and histamine.

Chapter 4 Results I
102
4.11.10: β1 (M286W) mutation does not interfere with the potentiaion mediated by other modulators We also examined mutant receptor sensitivity to other modulators like neurosteroids and
pentobarbital, which have the binding site on β1 subunit. Fig.4.27A shows the current
traces from the oocytes injected with α1β1 subunits cRNA. Application of 50 µM
pentobarbital and 50 µM of the 5α-Pregnane-3α-ol-11, enhanced GABA evoked currents
up to 2–2.5 and 3–3.5 respectively. Fig. 4.27B, shows the current-traces for the response
of the Neurosteroide and pentobarbital on α1β1 (M286W) receptor. Both modulators
potentiated the mutant receptor at same concentration, checked with wild type α1β1 with
almost same efficacy. Therefore, mutation is specific only for the propofol and histamine.
It does not interfere in the activity of other modulators at least for neurosteroids and
pentobarbital.
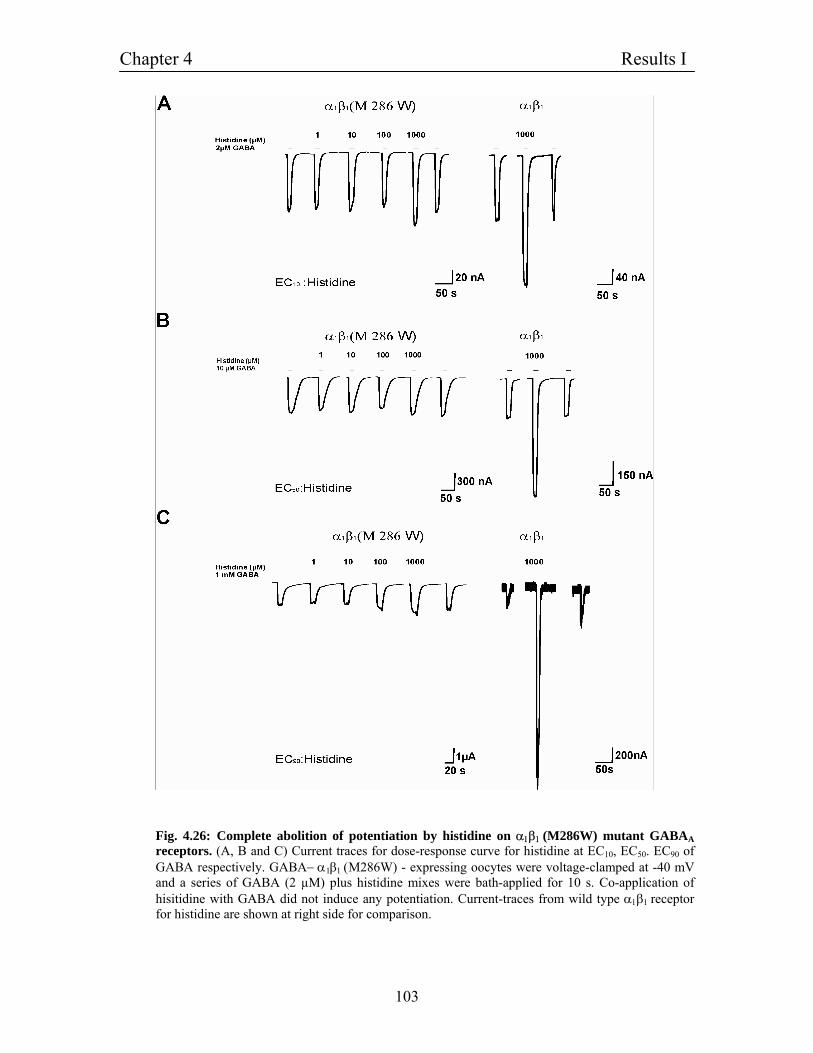
Chapter 4 Results I
103
Fig. 4.26: Complete abolition of potentiation by histidine on α1β1 (M286W) mutant GABAA receptors. (A, B and C) Current traces for dose-response curve for histidine at EC10, EC50. EC90 of GABA respectively. GABA− α1β1 (M286W) - expressing oocytes were voltage-clamped at -40 mV and a series of GABA (2 µM) plus histidine mixes were bath-applied for 10 s. Co-application of hisitidine with GABA did not induce any potentiation. Current-traces from wild type α1β1 receptor for histidine are shown at right side for comparison.
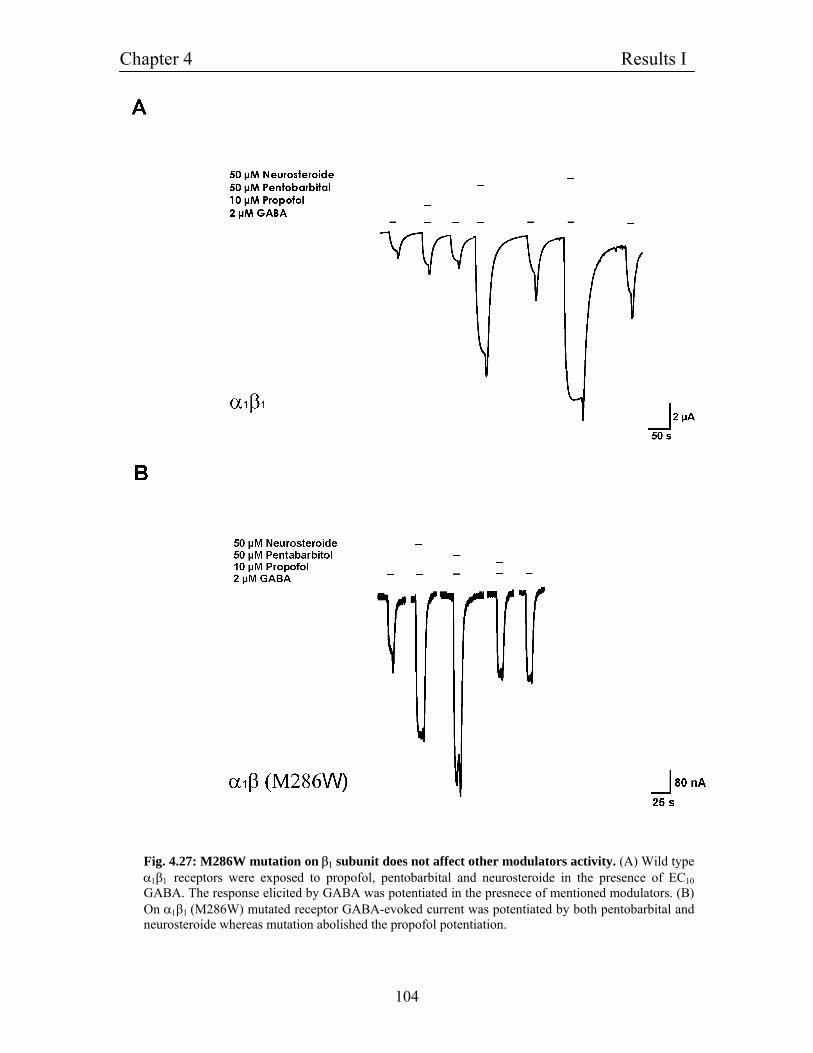
Chapter 4 Results I
104
Fig. 4.27: M286W mutation on β1 subunit does not affect other modulators activity. (A) Wild type α1β1 receptors were exposed to propofol, pentobarbital and neurosteroide in the presence of EC10 GABA. The response elicited by GABA was potentiated in the presnece of mentioned modulators. (B) On α1β1 (M286W) mutated receptor GABA-evoked current was potentiated by both pentobarbital and neurosteroide whereas mutation abolished the propofol potentiation.

Chapter 4 Results I
105
Summary of major findings of the first part of my thesis:
1. Identification of two new modulators for GABAA receptors. 2. GABAA receptors are potentiated by both histamine and histidine. 3. Potentiation of GABAA receptors by histamine does not arise from any shift in
equilibrium potential or change in form of the current-voltage relationship. 4. Histamine-mediated potentiation is not restricted to the oocyte recombinant
system but also shown in HEK 293 cells.
5. Affinity for potentiation by histamine depends up on the subunit combinations. 6. Our experiments show that histamine-binding site is located on β subunits of
GABAA receptors. 7. Our pharmacological and mutational studies show that indeed histamine binding
site is located at M286 on β1 subunit of GABAA receptors, which is the binding site for propofol also.
8. GABAC receptors are not modulated by histamine.

Chapter 5 Results II
106
Chapter 5 5: Characterization of homomultimeric β3 channels
In our previous experiments, we found that both β1 and β3 are activated by histamine
alone. We asked the question, do β3 subunits of GABAA receptors behave like possible
histamine-gated chloride channels? For this reason, we decided to characterize β3
subunits in detail. Homomultimeric β1 subunits form spontaneously opened channel,
which have very high leakage current, because of which we could not fully characterize
β1 subunit. Homomultimeric β3 receptors were more stable and showed very less leakage
current, so we extended our experiments on β3 subunit of GABAA receptors.
5.1: Homomultimeric β3 receptors behave like histamine-gated ion channels Injection of β3 cRNA in to Xenopus oocytes resulted in expression of substantial
histamine-activated currents not seen in uninjected oocytes. At a holding potential of -50
mV, micro molar concentrations of histamine rapidly activated inward currents that
deactivated quickly after washout of histamine, (Fig. 5.1A). As the histamine
concentration was raised from 10 µM to 10 mM, there was a progressive increase in
histamine-evoked current that seemed to reach at plateau at about 10 mM histamine.
Representative histamine dose response curves are shown in Fig. 5.1C. Fitting the data to
the Hill equation yielded an EC50 of 597 ± 5 µM, (n = 7) for the β3 subunit injected
oocytes.
The effect of histidine, the histamine metabolite, was also checked on β3 homomultimeric
channels. Results from five oocytes, exposed to a range of histidine concentrations are
shown in Fig. 5.1B. As the histidine concentration was raised from 10 µM to 10 mM,
histidine-evoked currents increased in amplitude in a dose-dependent manner. The
histidine-evoked current has an EC50 of 1.135 ± 0.15 mM, (n = 5). There are subtle
differences in the activation by histamine and histidine. First, histamine activates β3

Chapter 5 Results II
107
homomultimeric channels with less than the half maximal concentration than histidine.
Second, histidine induces higher amplitude current (2-4 folds larger) than histamine at
saturating concentrations determined in the same individual oocytes.
We also checked another metabolite in histamine pathway. Telemethyhistamine (t-MHA)
is produced in synapses by the methylation of histamine by histaminemethyltransferase
(Haas and Panula, 2003). t-MHA-currents were first detectable at about 30 µM
concentration, increased steeply in size as the t-MHA concentration was raised and
reached a maximum with about 30 mM t-MHA (Fig. 5.2). The EC50 derived from fitting
data to Hill equation was 1.1 ± 0.13 mM, (n = 3).
5.2: Histamine-gated homomultimeric β3 receptors behave like typical ligand-gated chloride channels To identify the ion permeating through the receptor channel, the current reversal potential
was determined while activating the β3 receptor with histamine (Fig. 5.3). The membrane
current activated by GABA decreased in size as the oocyte was depolarized and inverted
direction at about -20 mV, which corresponds to the chloride equilibrium potential in
Xenoups oocytes (Barish et al., 1983; Kusano et al., 1982). Moreover our current-voltage
relationship fits with β2 and β3 reversal potential reported by various groups where they
showed that reversal potential for β2 and β3 was close to the chloride equilibrium
potential (-23 mV) when expressed in Xenopus oocytes (Ymer et al., 1989; Dascal et al.,
1984).
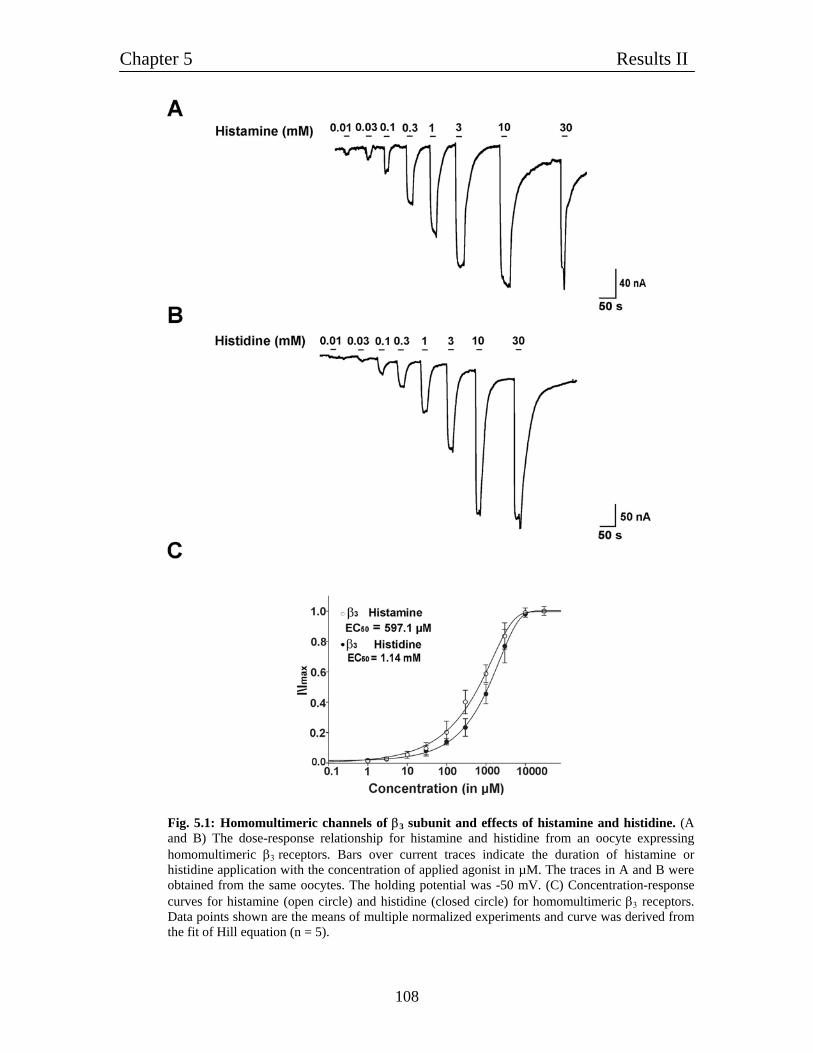
Chapter 5 Results II
108
Fig. 5.1: Homomultimeric channels of β3 subunit and effects of histamine and histidine. (A and B) The dose-response relationship for histamine and histidine from an oocyte expressing homomultimeric β3 receptors. Bars over current traces indicate the duration of histamine or histidine application with the concentration of applied agonist in µM. The traces in A and B were obtained from the same oocytes. The holding potential was -50 mV. (C) Concentration-response curves for histamine (open circle) and histidine (closed circle) for homomultimeric β3 receptors. Data points shown are the means of multiple normalized experiments and curve was derived from the fit of Hill equation (n = 5).
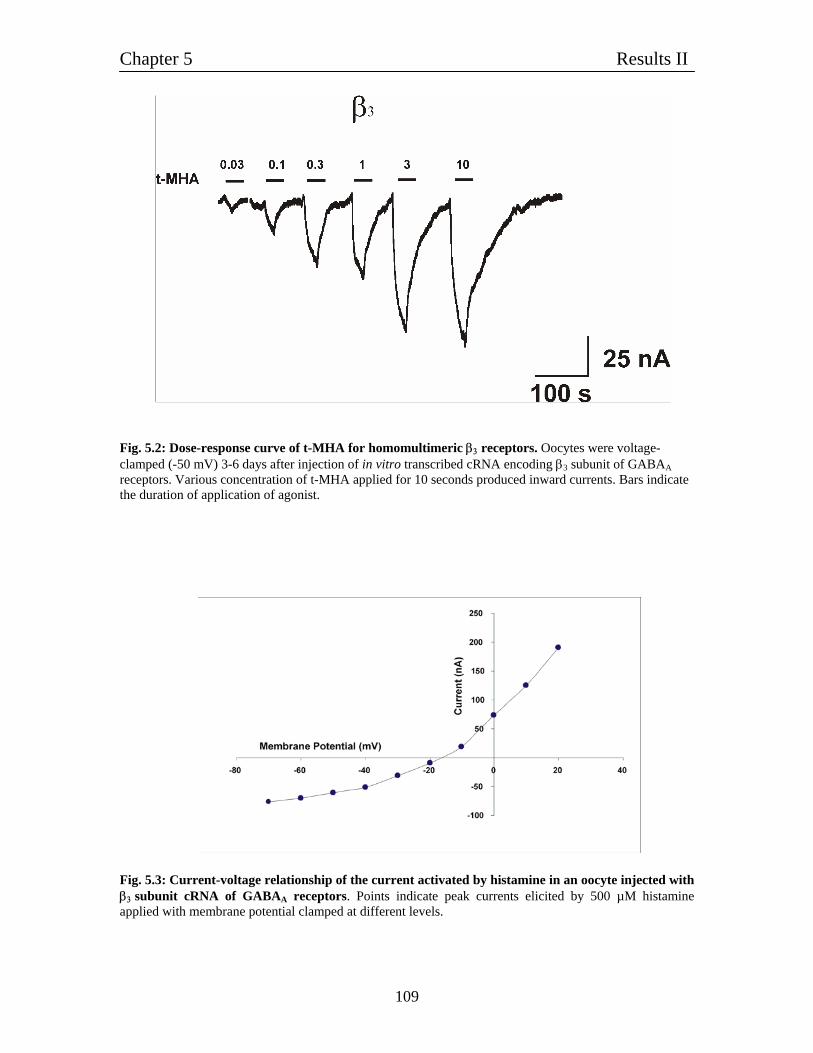
Chapter 5 Results II
109
Fig. 5.2: Dose-response curve of t-MHA for homomultimeric β3 receptors. Oocytes were voltage-clamped (-50 mV) 3-6 days after injection of in vitro transcribed cRNA encoding β3 subunit of GABAA receptors. Various concentration of t-MHA applied for 10 seconds produced inward currents. Bars indicate the duration of application of agonist.
Fig. 5.3: Current-voltage relationship of the current activated by histamine in an oocyte injected with β3 subunit cRNA of GABAA receptors. Points indicate peak currents elicited by 500 µM histamine applied with membrane potential clamped at different levels.

Chapter 5 Results II
110
5.3: Relative comparison of various agonist of histamine with GABA - Relative agonists efficacy compared to GABA To find the relative comparison between maximal current induced by GABA or
histamine, we expressed β3 subunit cRNA in Xenopus oocytes. We could not detect any
current of GABA up to 1 mM GABA concentration but from 3 mM GABA we could
detect a very small GABA-evoked current. Oocytes exhibiting inward current in response
to 3 mM GABA greater than 25 nA (at -50 mV) in amplitude were selected for this
experiment. So β3 homomultimeric channels are also activated by GABA but with very
small amplitude. Next, we determined the sensitivity of these homomultimeric receptors
to histamine, histidine and t-MHA. Fig. 5.4A shows the current traces for GABA,
histamine, histidine and t-MHA applied to the same individual oocyte. The bar graphs in
Fig. 5.4B shows the average percentage activation of β3 homomultimeric channel by
histamine and its precursors. We compared 3 mM GABA with saturating concentration
(10 mM) of histamine, histidine and t-MHA. 10 mM t-MHA induces 3 folds higher
amplitude of current than 3 mM GABA. 10 mM histidine showed the highest amplitude
(9 folds) of current w.r.t. 3 mM GABA whereas, 10 mM histamine showed moderate
current enhancement up to 6 folds.
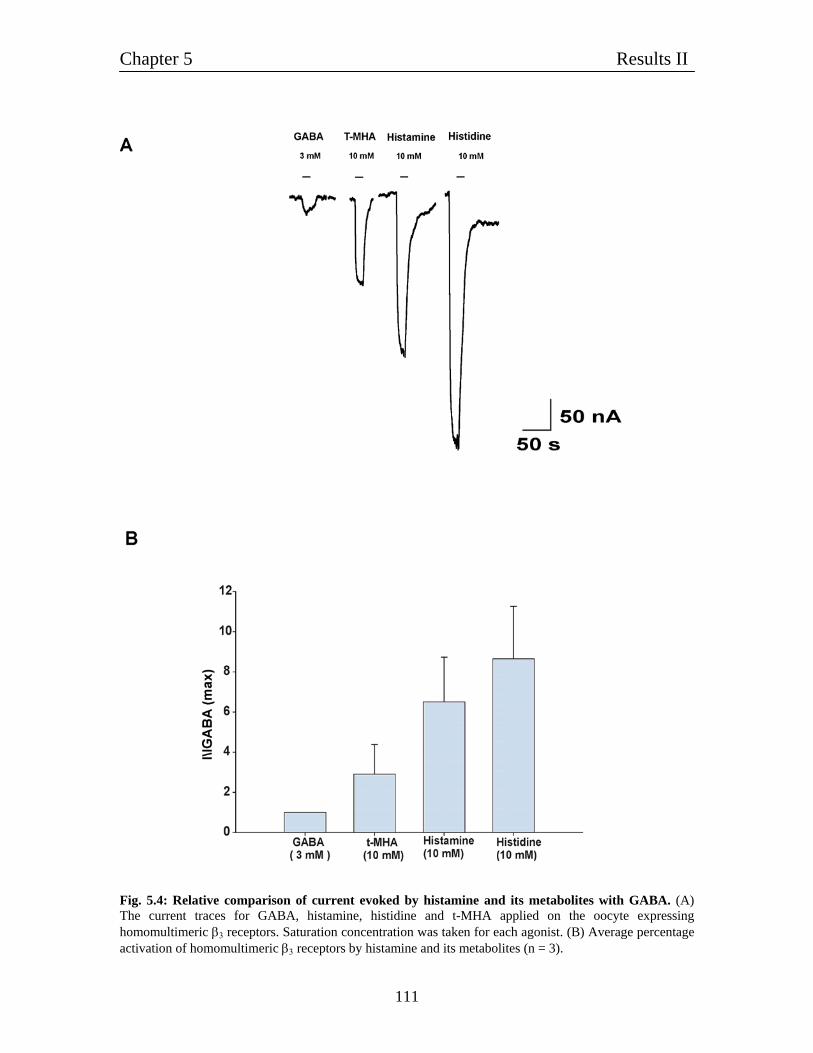
Chapter 5 Results II
111
Fig. 5.4: Relative comparison of current evoked by histamine and its metabolites with GABA. (A) The current traces for GABA, histamine, histidine and t-MHA applied on the oocyte expressing homomultimeric β3 receptors. Saturation concentration was taken for each agonist. (B) Average percentage activation of homomultimeric β3 receptors by histamine and its metabolites (n = 3).

Chapter 5 Results II
112
5.4: Pharmacological characterization of histamine-gated homomultimeric β3 receptors After establishing that β3 is indeed behaves like histamine-gated ion channels, we asked
whether the histamine-evoked current on homomultimeric β3 receptors, can be blocked
by known antagonists of metabotropic histamine receptors. Fig. 5.5A is a plot of sub
maximal histamine-activated currents for β3 homomultimeric channels and effect of
thioperamide (H3 receptor antagonist) on it. Thioperamide significantly reduced the half
maximum histamine-current amplitude evoked by 500 µM histamine in a dose-dependent
manner. The IC50 value for thioperamide is 9.7 ± 0.8 µM, (n = 5). Also, at high
concentration 300 µM thioperamide blocks the spontaneously open channels formed by
β3 subunit. Homomultimeric β3 currents evoked by 500 µM histamine were inhibited in
concentration-dependent manner by famotidine (Fig. 5.5B). Famotidine (H2 receptor
antagonist) maximally inhibited the histamine-activated current, with an IC50 of 77.1 ±
8.9 µM, (n = 5). The affinity of thioperamide was higher than the famotidine. We also
checked cimetidine (another H2 receptor antagonist) up to 500 µM concentration but did
not find any inhibition of the current evoked by 500 µM histamine. Also pyrilamine (H1
receptor antagonist) did not block any histamine-evoked current on homomultimeric
β3 receptors up to 500 µM concentration. Fig. 5.5C shows current-dose inhibition by
thoperamide and famotidine dose-response curve was fitted by Hill equation.
We observed that β3 homomultimeric channels were strongly picrotoxin sensitive.
Picrotoxin inhibition was fully recovered. Picrotoxin inhibited histamine-activated
current with an IC50 of 300 nM, (n = 3). HTMT, an agonist for homomultimeric receptors
also reduced the histamine activated current with an IC50 48.7 ± 2.5 µM, (n = 3) (Fig.
5.6A). Harmane and related alkaloids, such as harmine and harmaline, are agents with a
β-carboline structure. These compounds are also found endogenously in mammalian
tissues, including central nervous system. These harmala alkaloids have some
pharmacologic effects involved in convulsive or anticonvulsive actions. Harmane and
other related β-carbolines are putative endogenous ligands of the benzodiazepine
receptor. Harmane is a competitive inhibitor of benzodiazepine receptor binding in vitro.
We checked the activity of harmane on histamine-evoked currents on β3 receptors. Fig.

Chapter 5 Results II
113
5.6B shows the dose-response current inhibiton by harmane. The amplitude of histamine
evoked current decreased with increasing concentration of harmane. Harmane blocks
histamine current with an IC50 of 82.7 ± 6.3 µM, (n = 4). Like also thioperamide and
famotidine at higher concentration it shows outward current which indicates the
inhibition of spontaneously activated open channels.
5.4.1: Thioperamide acts as a competitive blocker for histamine-evoked current The tested antagonists like thioperamide, famotidine and pyrilamine are competitive
antagonists for the metabotropic histamine receptors. In the next set of experiments it was
tested if thioperamide acted as a competitive antagonist for the histamine action on
homomultimeric GABA β3 receptors also. The current evoked by different concentration
of histamine was effectively blocked when thioperamide was given in 30 µM
concentration. At rising concentrations of histamine the thiopermide block was less
pronounced and at 10 mM histamine, 30 µM thioperamide didn't cause any block of the
histamine induced current at all (Fig. 5.7A). A dose-response curve constructed from
three of such measurements showed that thioperamide shifted the dose-response curve to
the rightward direction and the EC50 of histamine shifted from 597 µM to 2.72 mM (Fig.
5.7B), a clear indication that the blocking mechanism is competitive. Competitive
blockers can act in two different ways. First, competitive blockers acting also in the
absence of the agonist by blocking for example population of spontaneously active
receptors or channels are also called 'inverse agonists'. Second, blockers showing no
action in the absence of the agonist are called 'neutral antagonist'. It was found that
thioperamide also blocked the population of spontaneously open channels in β2 and β3
homomultimeric channels (data not shown). Such action of competitive antagonist
classifies thioperamide as an 'inverse agonist'.
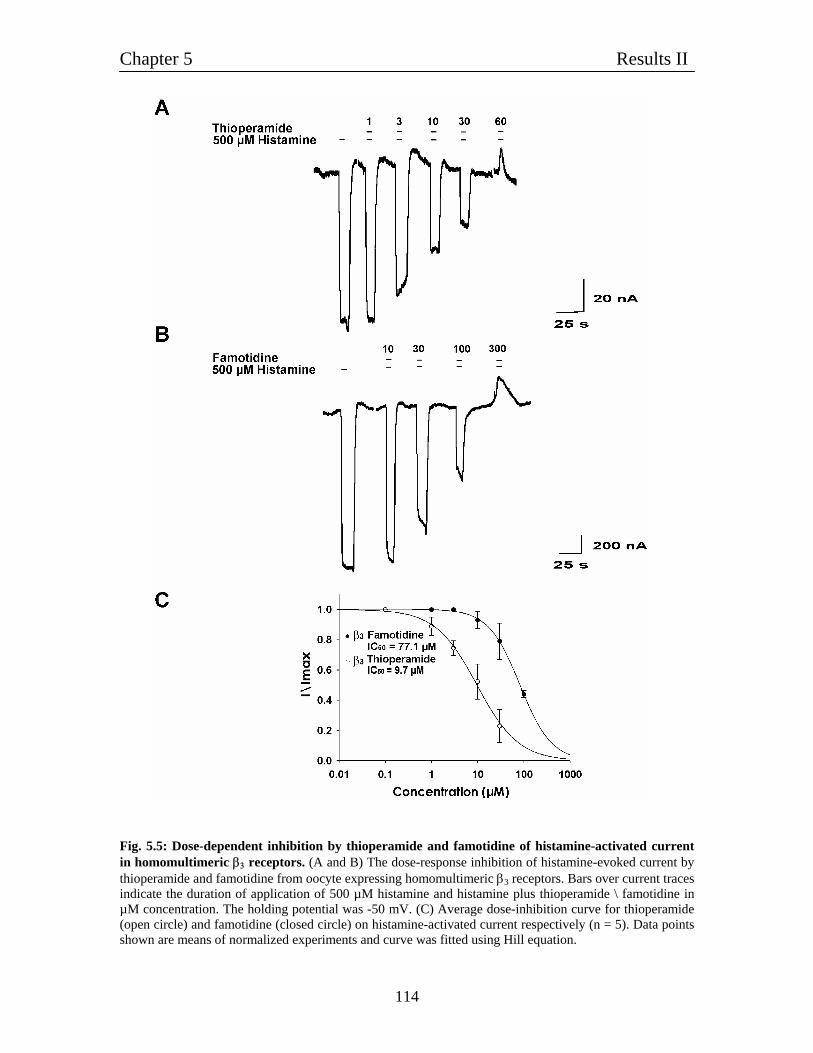
Chapter 5 Results II
114
Fig. 5.5: Dose-dependent inhibition by thioperamide and famotidine of histamine-activated current in homomultimeric β3 receptors. (A and B) The dose-response inhibition of histamine-evoked current by thioperamide and famotidine from oocyte expressing homomultimeric β3 receptors. Bars over current traces indicate the duration of application of 500 µM histamine and histamine plus thioperamide \ famotidine in µM concentration. The holding potential was -50 mV. (C) Average dose-inhibition curve for thioperamide (open circle) and famotidine (closed circle) on histamine-activated current respectively (n = 5). Data points shown are means of normalized experiments and curve was fitted using Hill equation.
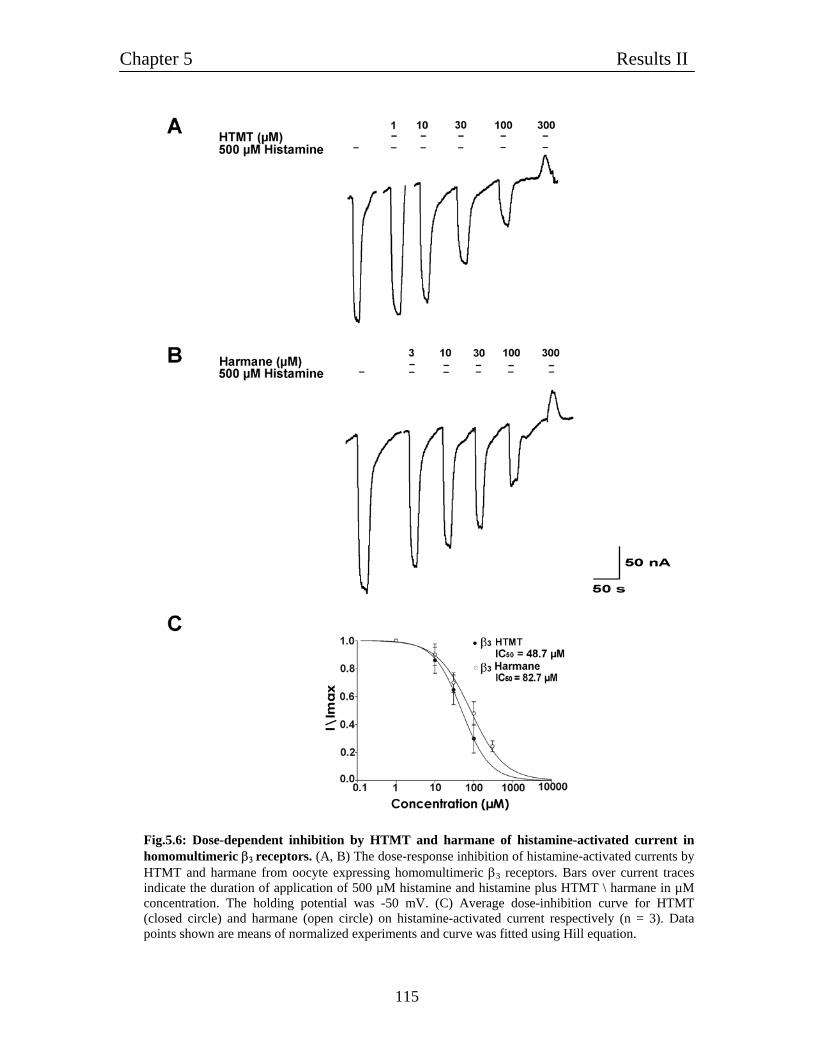
Chapter 5 Results II
115
Fig.5.6: Dose-dependent inhibition by HTMT and harmane of histamine-activated current in homomultimeric β3 receptors. (A, B) The dose-response inhibition of histamine-activated currents by HTMT and harmane from oocyte expressing homomultimeric β3 receptors. Bars over current traces indicate the duration of application of 500 µM histamine and histamine plus HTMT \ harmane in µM concentration. The holding potential was -50 mV. (C) Average dose-inhibition curve for HTMT (closed circle) and harmane (open circle) on histamine-activated current respectively (n = 3). Data points shown are means of normalized experiments and curve was fitted using Hill equation.
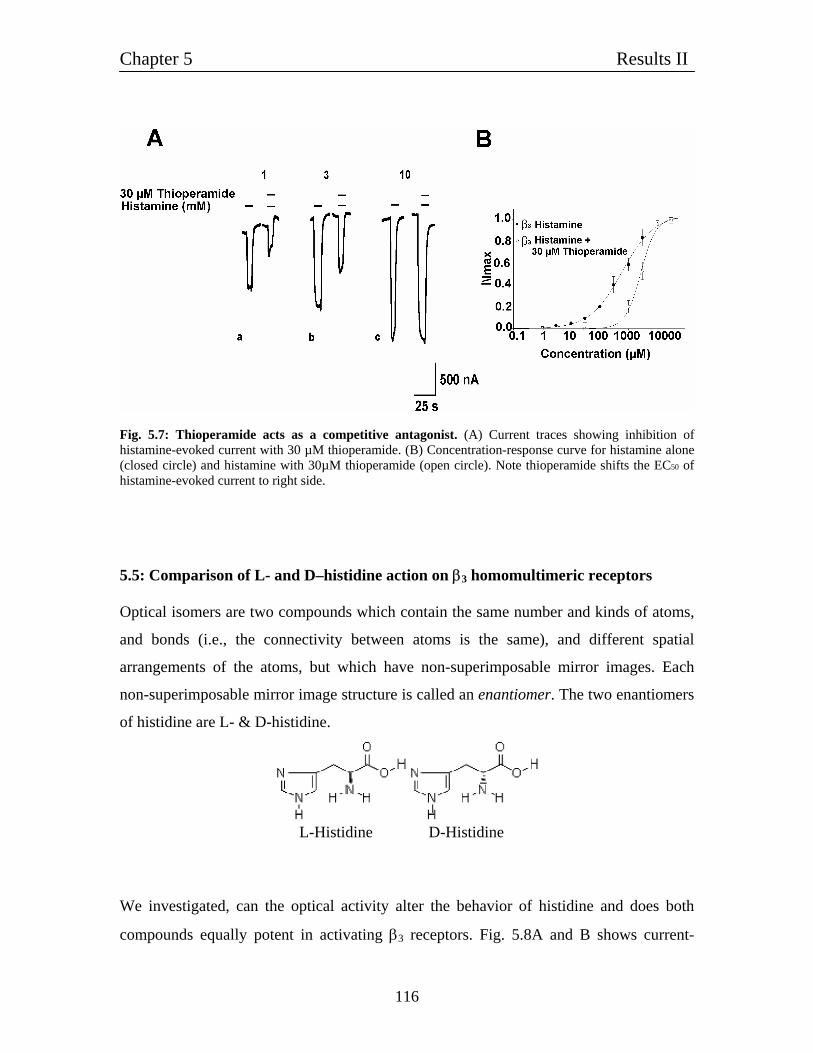
Chapter 5 Results II
116
Fig. 5.7: Thioperamide acts as a competitive antagonist. (A) Current traces showing inhibition of histamine-evoked current with 30 µM thioperamide. (B) Concentration-response curve for histamine alone (closed circle) and histamine with 30µM thioperamide (open circle). Note thioperamide shifts the EC50 of histamine-evoked current to right side.
5.5: Comparison of L- and D–histidine action on β3 homomultimeric receptors Optical isomers are two compounds which contain the same number and kinds of atoms,
and bonds (i.e., the connectivity between atoms is the same), and different spatial
arrangements of the atoms, but which have non-superimposable mirror images. Each
non-superimposable mirror image structure is called an enantiomer. The two enantiomers
of histidine are L- & D-histidine.
L-Histidine D-Histidine
We investigated, can the optical activity alter the behavior of histidine and does both
compounds equally potent in activating β3 receptors. Fig. 5.8A and B shows current-
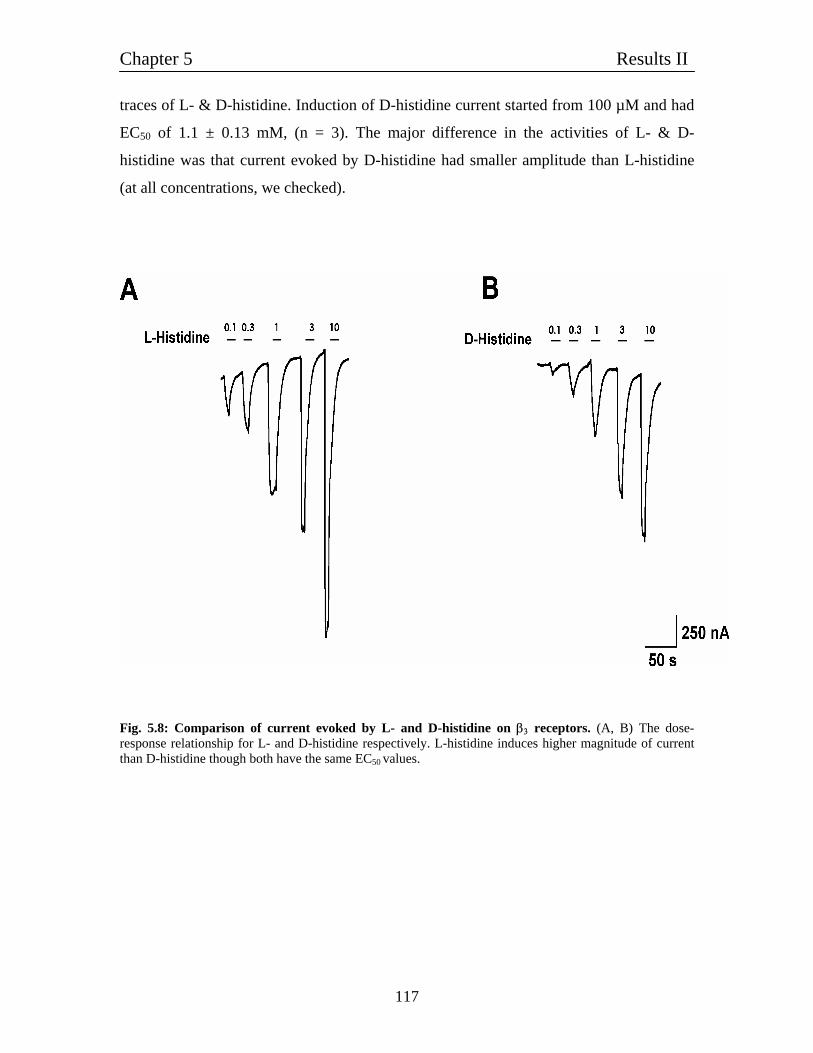
Chapter 5 Results II
117
traces of L- & D-histidine. Induction of D-histidine current started from 100 µM and had
EC50 of 1.1 ± 0.13 mM, (n = 3). The major difference in the activities of L- & D-
histidine was that current evoked by D-histidine had smaller amplitude than L-histidine
(at all concentrations, we checked).
Fig. 5.8: Comparison of current evoked by L- and D-histidine on β3 receptors. (A, B) The dose-response relationship for L- and D-histidine respectively. L-histidine induces higher magnitude of current than D-histidine though both have the same EC50 values.

Chapter 5 Results II
118
5.6: Inhibition of propofol-induced current by thioperamide on homomultimeric β3 receptors In our previous experiments on α1β1 receptors, we found that histamine and propofol
have similar binding site. After characterization of β3 receptors pharmacology for
histamine, we looked for the activity of propofol on β3 receptors. We hypothesized that
like histamine, propofol should behave like histamine and propofol alone should induce
propofol-evoked current. Indeed, when propofol alone was applied on β3 receptors, it
started to induce a propofol-evoked current at 30 µM propofol concentration. We could
not make the dose-response curve for propofol. However, gradual increase of propofol
from 30 µM - 10 mM mediated propofol-evoked current in a dose dependent manner but
did not lead to the maximal saturation of the receptor. This can be described as that in
hetromultimeric recombinant receptors; higher concentration of propofol starts the direct
activation of the channel. In our experiments for dose-response curve, this can be the
case. Therefore we decided to use equivalent concentration of propofol w.r.t. histamine.
300 µM propofol (~ EC50 of histamine = 597 µM on β3 receptors) was taken for further
experiment, which produced current > 1 µA. In β3 receptors, thioperamide acts as a
competitive antagonist and inhibits 500 µM histamine-evoked current at IC50 of 9.7 µM.
Therefore, we checked the pharmacology of propofol by thioperamide. Fig. 5.9 shows,
dose-dependent inhibition of propofol-evoked current by thioperamide. Thioperamide
inhibits the propofol-evoked current with an IC50 of 9.5 ± 0.05 µM, (n = 5). Remarkably,
this IC50 value for propofol-evoked current is almost similar to histamine-evoked current
on β3 receptor for thioperamide. This pharmacological experiment gives another strong
support for similar binding site for histamine and propofol.
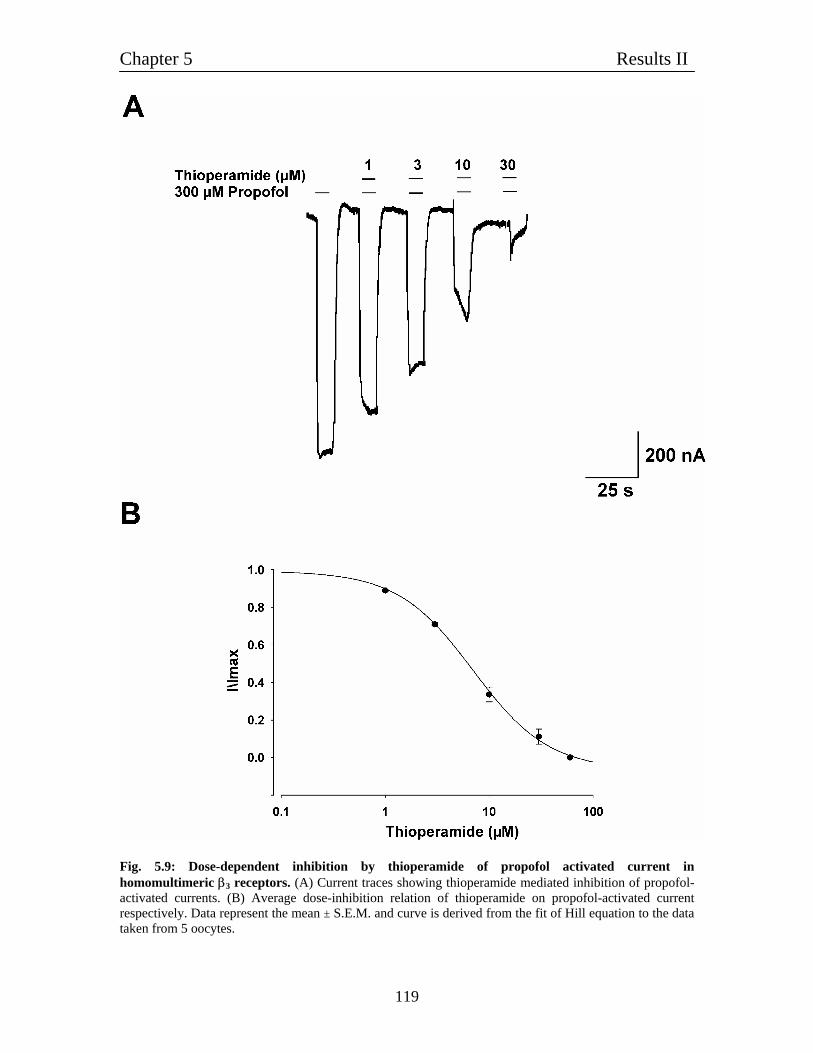
Chapter 5 Results II
119
Fig. 5.9: Dose-dependent inhibition by thioperamide of propofol activated current in homomultimeric β3 receptors. (A) Current traces showing thioperamide mediated inhibition of propofol-activated currents. (B) Average dose-inhibition relation of thioperamide on propofol-activated current respectively. Data represent the mean ± S.E.M. and curve is derived from the fit of Hill equation to the data taken from 5 oocytes.

Chapter 5 Results II
120
Summary of major findings:
1. Homomultimeric β3 subunit forms histamine-gated ion channel with an EC50 of 597 µM.
2. Homomultimeric β3 receptors are also activated by histidine and t-MHA with an
EC50 of 1.1 and 1.1 mM respectively.
3. Metabotropic histamine H3 receptor antagonist thioperamide acts as a competitive inhibitor for histamine-evoked current with an IC50 of 9.7 µM.
4. Histamine evoked current can be inhibited by famotidine (H2 receptor antagonist),
harmane and HTMT with a relative order of HTMT > famotidine > harmane.
5. Homomultimeric β3 receptors are also activated by propofol alone. Propofol-evoked current can be blocked by thioperamide.
6. Inhibition of both histamine and propofol-evoked current by thioperamide gives
also evidence that both histamine and propofol have same binding site.

Chapter 6 Results III
121
Chapter 6
6: Characterization of homomultimeric β2 channels
Our previous results have shown that β1 and β3 behaved like histamine-gated chloride
channels. In the literature, until now to our knowledge there is no report about the
successful expression of β2 homomeric channels. We decided to re-examine first the
result of injecting β2 alone into Xenopus oocytes. Unexpectedly, the human β2 cRNA
expressed histamine-gated chloride channels, suggesting the formation of
oligohomomeric β2 receptors.
6.1: Homomeric β2 receptors behave like histamine-gated ion channels Fig. 6.1A shows the histamine generated membrane currents in oocytes injected with β2
cRNA. Histamine generated substantial membrane currents that resembled closely the
currents generated by oocytes injected with β3 subunit. The currents generated by low
concentration of histamine applied to β2 injected oocytes were fairly well maintained; but
the current elicited by high concentrations desensitized rapidly and recovered well after
washing. The histamine currents of β2 injected oocytes were fast desenzitising and
occasionally had amplitude of > 1.5 µA with 3 mM histamine. The histamine dose-
current amplitude relationships were fitted well by Hill equation with a resulting EC50 of
280 ± 2.1 µM, (n = 7), (Fig. 6.1C). The effect of histidine, the histamine metabolite, was
also checked on β2 homomultimeric channels. β2 homomultimeric channels were
activated by histidine in a dose dependent manner. Also histidine-evoked currents had
amplitude > 2.5 µA with 3 mM histidine. Fig. 6.1B shows the dose-response curve where
the induction of histidine currents start from 10 µM and reached to saturation at 10 mM
histidine. The histidine current had EC50 of 480 ± 5.8 µM, (n = 7). Thus, our results show
that β2 can form homomultimeric channels when injected into oocytes, which are

Chapter 6 Results III
122
activated, by histamine and histidine. t-MHA directly activated inward currents in a
concentrationdependent manner over a concentration range of 0.1 - 30 mM (Fig. 6.2).
The efficacy of t-MHA at these homomultimeric receptors was quite less compared to
histamine, as t-MHA induced currents of very less amplitude. 6.2: Histamine-gated homomultimeric β2 receptors behave like typical ligand-gated chloride channels
To identify the ion permeability through the receptor channel, the current reversal
potential was determined while activating the receptors with histamine. The voltage-
current relation indicates a rectifying channel with a reversal potential around -20 mV
(Fig. 6.3), as expected for chloride ions in Xenopus oocytes using Ringer as external
solution.
6.3: Relative comparison of various agonist of histamine with GABA
It has been controversial whether or not β2 homomultimers can form functional GABA-
gated ion channels. In one report it has been shown that both β1 and β3 can form
homomultimeric channels (Miko et al., 2004; Sigel et al., 1989; Krishek et al., 1997). To
address the same question for the β2 subunit, we further examined the β2 homomers. Fig.
6.4A and B shows the relative comparison of histamine and its metabolites with GABA.
At 10 mM saturating concentration histidine-evoked current was 8 folds higher than 3
mM GABA.
6.4: Pharmacological characterization of histamine-gated homomultimeric β2 receptors Thioperamide, H3 receptor antagonist, inhibited histamine-gated ion channels formed by
β2 receptors in a concentration-dependent manner over a range of 1-60 µM (Fig. 6.5A)
The IC50 value of thioperamide concentration-response curve for homomultimeric β2
subunit were 28.4 ± 1.2 µM, (n = 5). At 100 µM thioperamide showed outward current,
indicating the blockage of spontaneously activated channel-current. Next, H2 receptor
antagonist, famotidine was investigated for its activity on β2 subunit receptors.
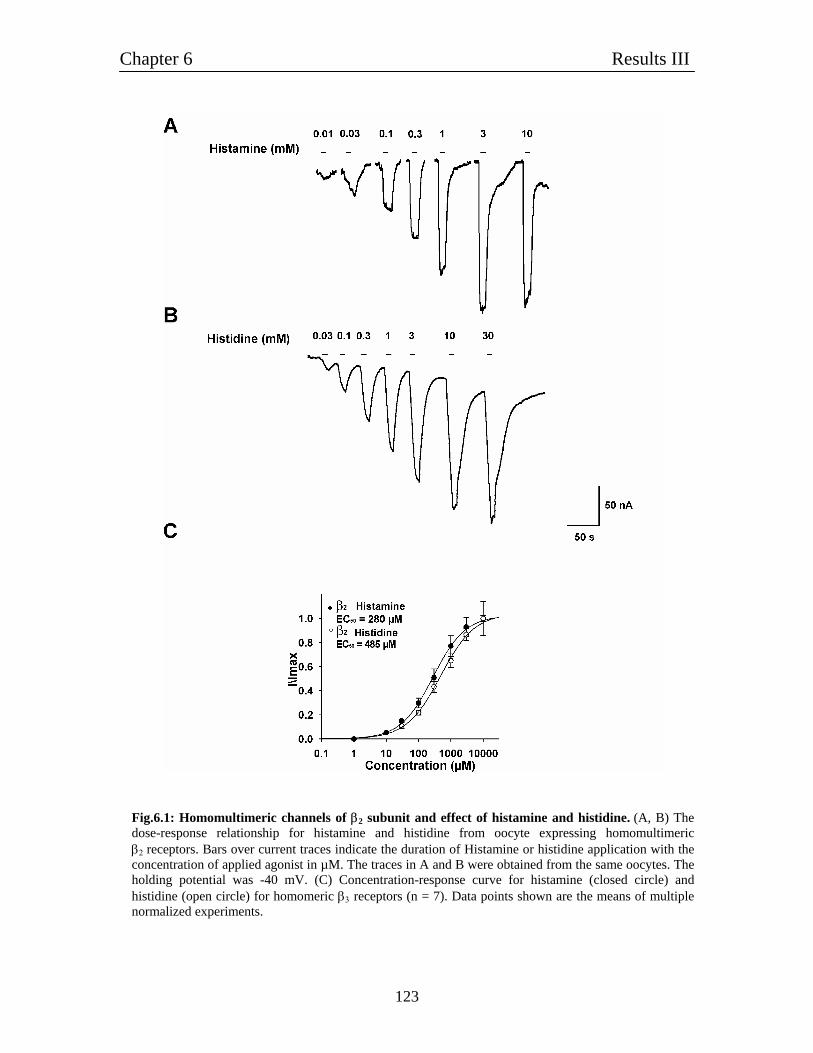
Chapter 6 Results III
123
Fig.6.1: Homomultimeric channels of β2 subunit and effect of histamine and histidine. (A, B) The dose-response relationship for histamine and histidine from oocyte expressing homomultimeric β2 receptors. Bars over current traces indicate the duration of Histamine or histidine application with the concentration of applied agonist in µM. The traces in A and B were obtained from the same oocytes. The holding potential was -40 mV. (C) Concentration-response curve for histamine (closed circle) and histidine (open circle) for homomeric β3 receptors (n = 7). Data points shown are the means of multiple normalized experiments.
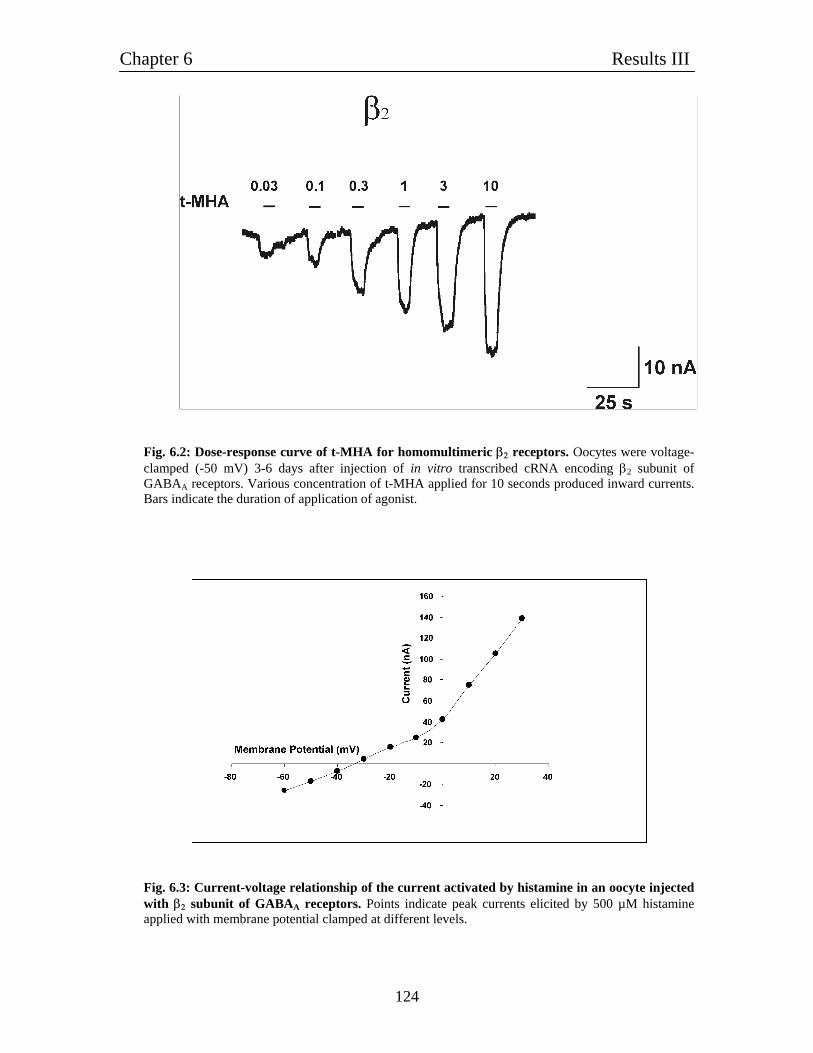
Chapter 6 Results III
124
Fig. 6.2: Dose-response curve of t-MHA for homomultimeric β2 receptors. Oocytes were voltage-clamped (-50 mV) 3-6 days after injection of in vitro transcribed cRNA encoding β2 subunit of GABAA receptors. Various concentration of t-MHA applied for 10 seconds produced inward currents. Bars indicate the duration of application of agonist.
Fig. 6.3: Current-voltage relationship of the current activated by histamine in an oocyte injected with β2 subunit of GABAA receptors. Points indicate peak currents elicited by 500 µM histamine applied with membrane potential clamped at different levels.
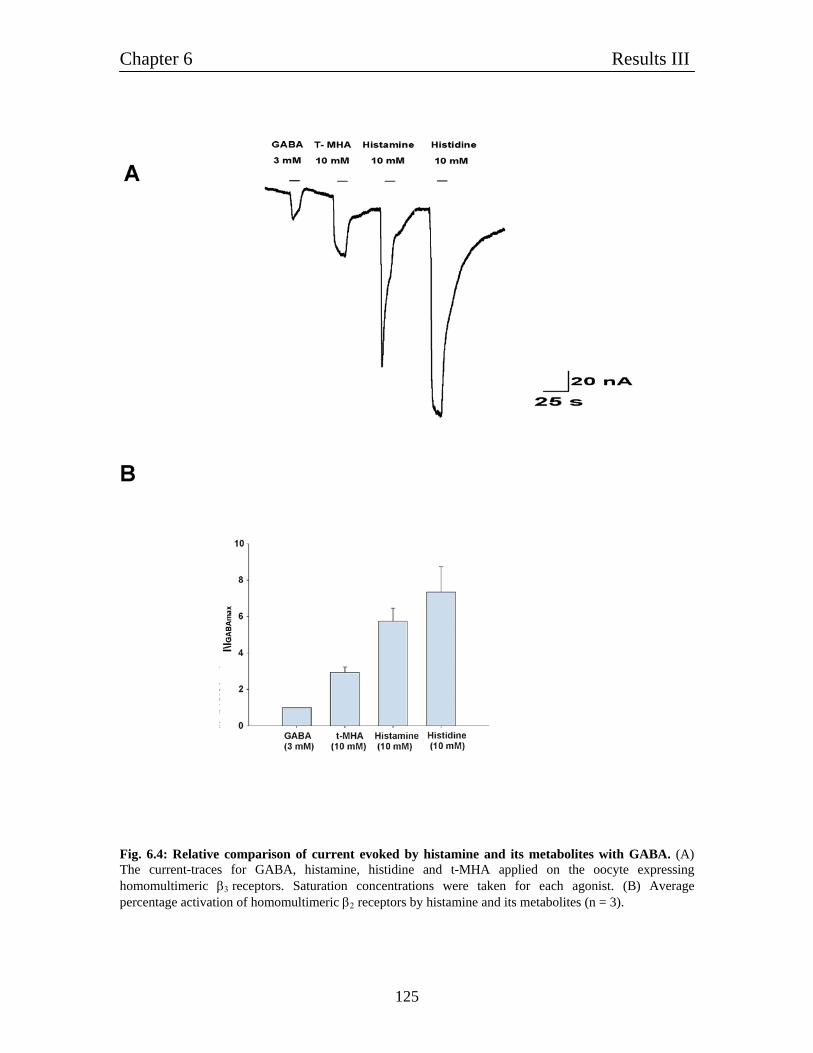
Chapter 6 Results III
125
Fig. 6.4: Relative comparison of current evoked by histamine and its metabolites with GABA. (A) The current-traces for GABA, histamine, histidine and t-MHA applied on the oocyte expressing homomultimeric β3 receptors. Saturation concentrations were taken for each agonist. (B) Average percentage activation of homomultimeric β2 receptors by histamine and its metabolites (n = 3).

Chapter 6 Results III
126
Famotidine inhibited histamine current with an IC50 of 70.5 ± 1.8 µM, (n = 3). At 300µM
concentration it also blocked the spontaneously active currents (Fig. 6.5B). The dose-
response curve was fitted with hill equation (Fig. 6.5C). Thioperamide is a more potent
antagonist than famotidine on β2 receptors. Pyrilamine, H1 receptor antagonist, at a
concentration up to 300 µM did not affect current evoked by 500 µM Histamine. The
affinity and efficacy of thioperamide block were much greater than the famotidine.
Control β2 currents evoked by 500 µM histamine were inhibited in concentration-
dependent manner by HTMT (Fig. 6.6A). HTMT maximally inhibited the histamine-
activated current, with an IC50 of 100 ± 1.2 µM, (n = 3).
Fig. 6.6B is a plot of sub maximal histamine-activated currents for β2 homomultimeric
channels and effect of harmane on it. Harmane significantly reduced the half maximum
histamine-current amplitude in a dose-dependent manner. The IC50 value for harmane is
36 ± 1.6 µM, (n = 3). Also, at high concentration 300 µM harmane blocks the
spontaneously open channels formed by β2 subunit.
6.5: Relative comparison of homomultimeric β2 and β3 receptors
Fig. 6.7 shows the relative comparison of β2 and β3 homomultimeric channels by their
pharmacology. β3 is 3 times more sensitive to thioperamide for histamine-evoked current
than β2. Also for HTMT, β2 was 2 folds more sensitive than β3. Famotidine, H2
antagonist, however, blocked histamine-evoked current with same efficacy on β2 and β3
homomultimeric receptors. Only harmane, was found to be more sensitive on β2 than β3
ranging up to 2-3 folds higher sensitivity.
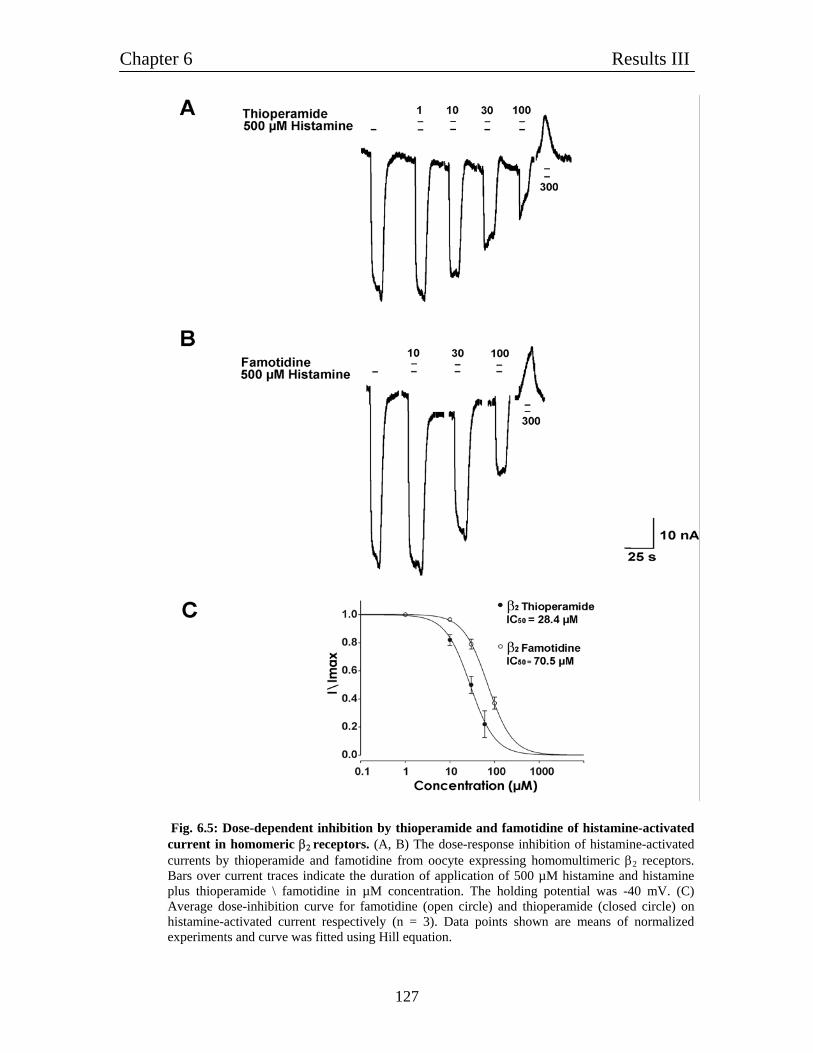
Chapter 6 Results III
127
Fig. 6.5: Dose-dependent inhibition by thioperamide and famotidine of histamine-activated current in homomeric β2 receptors. (A, B) The dose-response inhibition of histamine-activated currents by thioperamide and famotidine from oocyte expressing homomultimeric β2 receptors. Bars over current traces indicate the duration of application of 500 µM histamine and histamine plus thioperamide \ famotidine in µM concentration. The holding potential was -40 mV. (C) Average dose-inhibition curve for famotidine (open circle) and thioperamide (closed circle) on histamine-activated current respectively (n = 3). Data points shown are means of normalized experiments and curve was fitted using Hill equation.
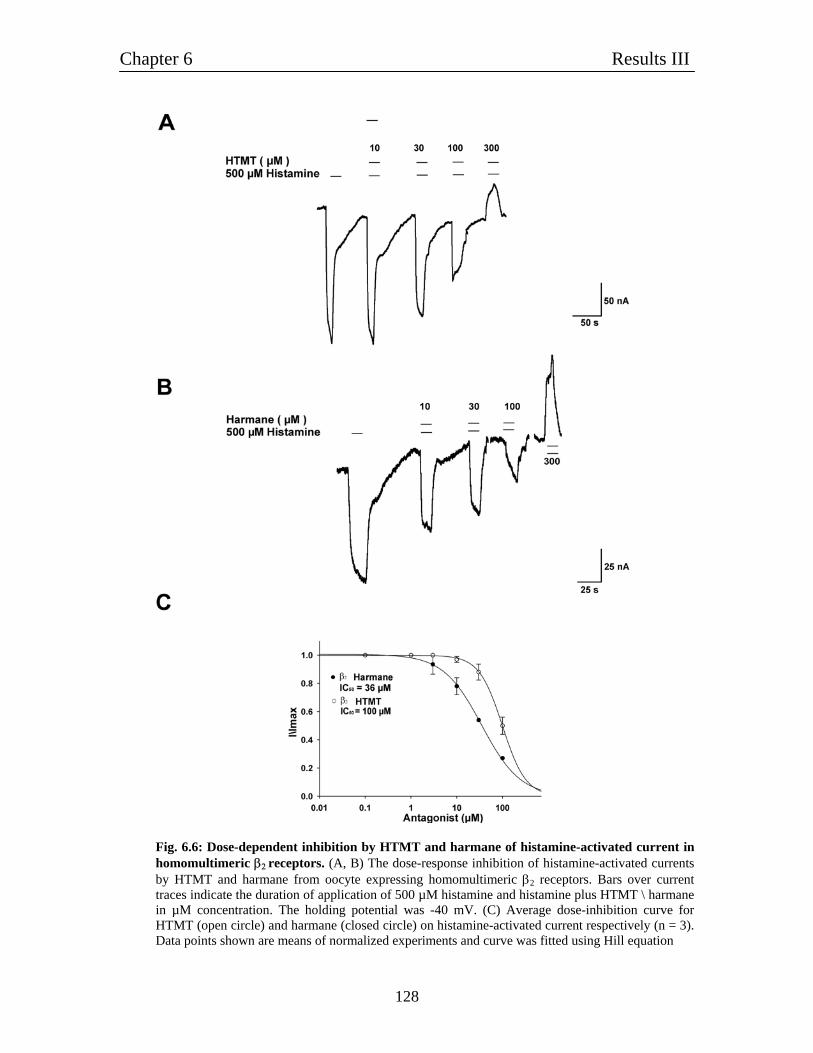
Chapter 6 Results III
128
Fig. 6.6: Dose-dependent inhibition by HTMT and harmane of histamine-activated current in homomultimeric β2 receptors. (A, B) The dose-response inhibition of histamine-activated currents by HTMT and harmane from oocyte expressing homomultimeric β2 receptors. Bars over current traces indicate the duration of application of 500 µM histamine and histamine plus HTMT \ harmane in µM concentration. The holding potential was -40 mV. (C) Average dose-inhibition curve for HTMT (open circle) and harmane (closed circle) on histamine-activated current respectively (n = 3). Data points shown are means of normalized experiments and curve was fitted using Hill equation
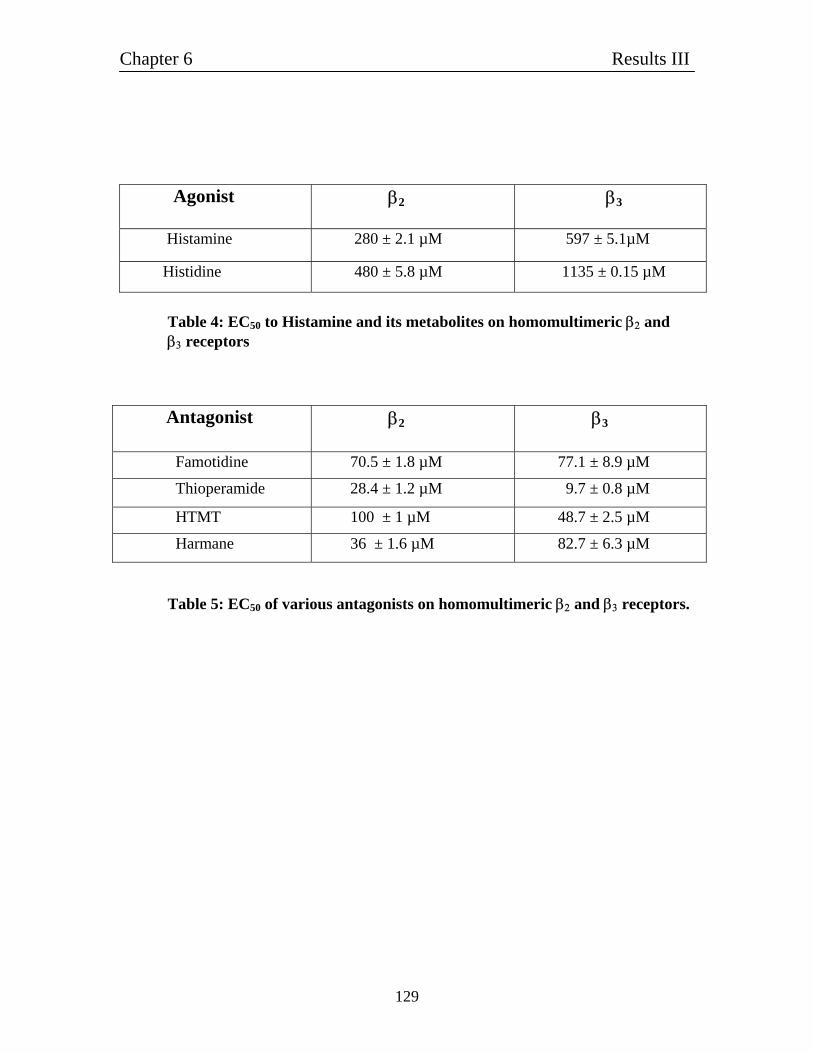
Chapter 6 Results III
129
Agonist β2 β3
Histamine 280 ± 2.1 µM 597 ± 5.1µM
Histidine 480 ± 5.8 µM 1135 ± 0.15 µM
Table 4: EC50 to Histamine and its metabolites on homomultimeric β2 and β3 receptors
Antagonist β2 β3
Famotidine 70.5 ± 1.8 µM 77.1 ± 8.9 µM
Thioperamide 28.4 ± 1.2 µM 9.7 ± 0.8 µM
HTMT 100 ± 1 µM 48.7 ± 2.5 µM
Harmane 36 ± 1.6 µM 82.7 ± 6.3 µM
Table 5: EC50 of various antagonists on homomultimeric β2 and β3 receptors.
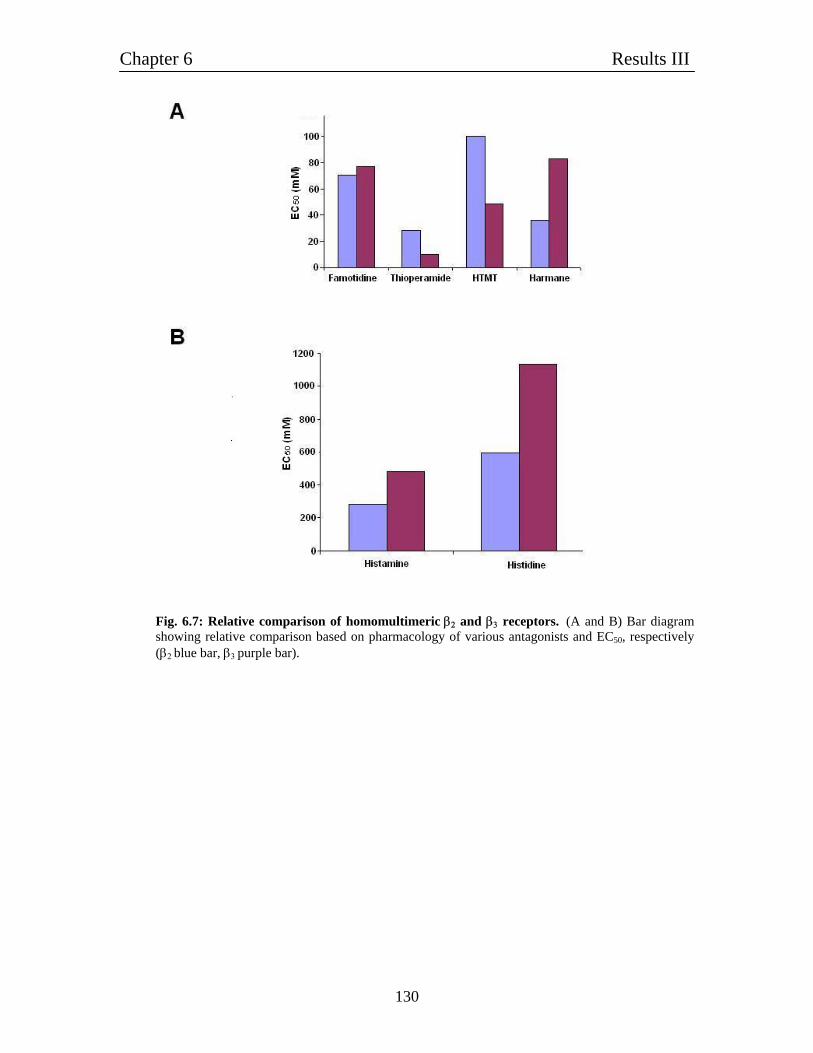
Chapter 6 Results III
130
Fig. 6.7: Relative comparison of homomultimeric β2 and β3 receptors. (A and B) Bar diagram showing relative comparison based on pharmacology of various antagonists and EC50, respectively (β2 blue bar, β3 purple bar).

Chapter 6 Results III
131
Summary of major findings:
1. Homomultimeric β2 subunit forms histamine-gated ion channel with an EC50 of 280 µM.
2. Homomultimeric β2 receptors are also activated by histidine (EC50 0.48 mM) and t-
MHA. 3. Metabotropic histamine H3 receptor antagonist thioperamide inhibits histamine-
evoked current with an IC50 of 28.4 µM.
4. Histamine evoked current can be inhibited by famotidine (H2 receptor antagonist), harmane and HTMT with a relative order of harmane > famotidine > HTMT.
5. Homomultimeric β2 receptors are more sensitive to histamine than β3 receptors.
6. Pharmacologically homomultimeric β2 and β3 receptors are quite distinct to each
other.

Chapter 7 Results IV
132
Chapter 7 7.1: Characterization of ε-subunit containing receptors
The ε-subunit of the GABAA receptor was independently cloned and functionally
characterized in recombinant expression systems by two groups (Davies et al., 1997;
Whiting et al., 1997). Both groups showed that co-expression of alpha beta epsilon
subunits produced functional receptors; however the sensitivity of these receptors to the
potentiating effects of general anesthetic agents differed. In human and rat tissues,
complex patterns of transcripts are derived from the genes that encode the gamma-
aminobutyric acid GABAA receptor epsilon subunit. A ε subunit transcript
(approximately 3.6 kb) is expressed at relatively high levels in regions of the human brain
and heart, but is not detected in most other major tissues. The ε subunit has greater
amino-acid sequence similarity to the γ subunits than to any other classes of GABAA
receptor polypeptides. Furthermore, the ε and γ subunits both require the presence of α
and β subunits to incorporate within functional receptors (Davies et al., 1997). Therefore
it was hypothesized that ε and γ subunits compete for the common site in the GABA
receptor complex. It has demonstrated that expression of ε subunit does indeed modify
the functional properties of α1β3γ2 receptors stably expressed in WSS-1 cells (Davies et
al., 1997). Presence of ε subunit in GABAA receptor complex causes the spontaneous
current activity. Moreover, our initial experiments in bioinformatics revealed some
distinctive sequence homology characteristic for histamine-gated chloride channels. We
decided to examine the properties of ε subunits in Xenopus oocytes.

Chapter 7 Results IV
133
7.2: ε-containing receptors behave like histamine-gated ion channels It was noted that oocytes injected with ε subunit cRNAs required an unusually large
holding current to maintain a potential of -50 mV and the resulting baseline value was
relatively unstable. We injected different subunit combinations to check the effect of ε
subunit on receptor complex.
Oocytes co-expressing α1β1ε subtypes were activated by histamine (30 - 10,000 µM) in a
concentration-dependent manner (Fig. 7.1A). The α1β1ε had histamine EC50 of 3 mM,
(n = 9). We also observed that α1β1ε subunit combination was highly sensitive to GABA.
The comparison of GABA-evoked current is also shown in Fig. 7.1A.
Oocytes injected with β1ε subunits cRNA did not give any response even at the 1 mM
GABA concentration. It is very different from the α1β1ε receptor where even 1 µM
GABA concentration gives large current response. As α subunit is responsible for the
binding of GABA to GABAA receptor complex we concluded it as logical for no
response to GABA. But what was quite interesting also here that β1ε subunit combination
formed highly spontaneously active channel which were activated by histamine in a dose
dependent manner Fig. 7.1B.
In case of oocyte injected with ε subunit cRNA showed spontaneous current in the
absence of any agonist. Oocyte clamped at -50 mV usually had leakage current up to 1
µA. Interestingly; also application of histamine activated the ε subunit in a dose-response
manner (Fig. 7.1C). When GABA was applied on homomeric ε receptors it blocked the
spontaneously opened channel. From all our experiments, we concluded that ε was also a
similar histamine-gated ion channel.
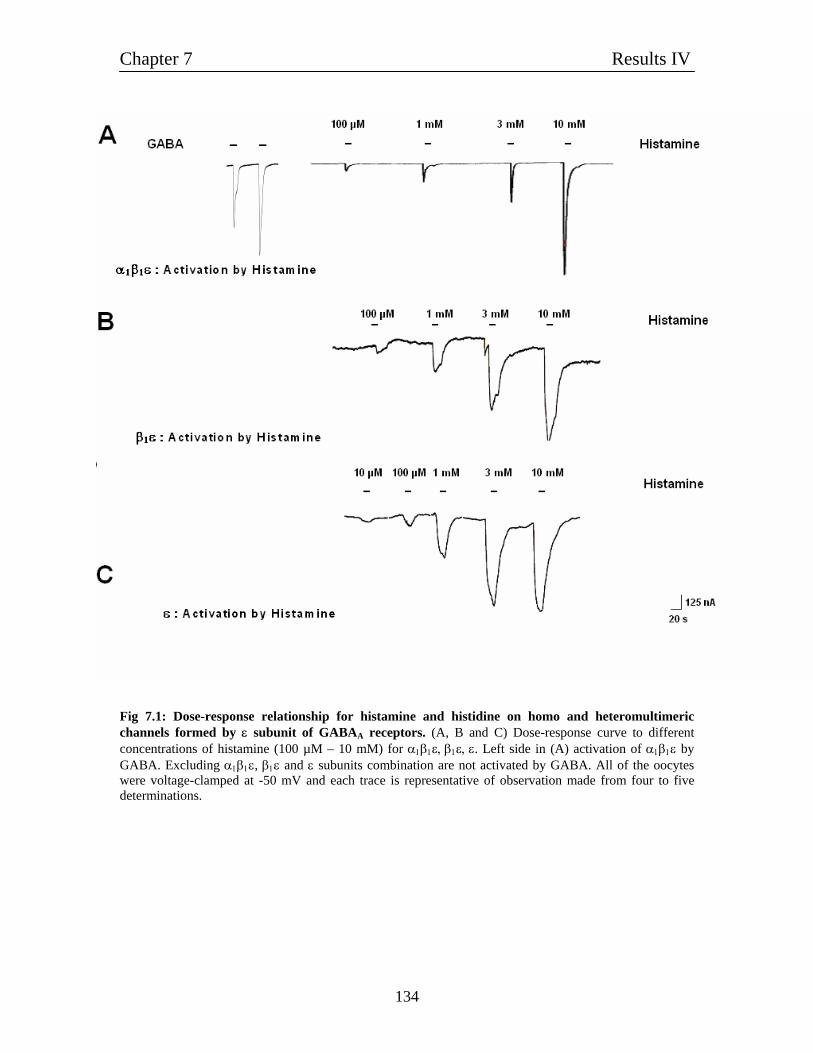
Chapter 7 Results IV
134
Fig 7.1: Dose-response relationship for histamine and histidine on homo and heteromultimeric channels formed by ε subunit of GABAA receptors. (A, B and C) Dose-response curve to different concentrations of histamine (100 µM – 10 mM) for α1β1ε, β1ε, ε. Left side in (A) activation of α1β1ε by GABA. Excluding α1β1ε, β1ε and ε subunits combination are not activated by GABA. All of the oocytes were voltage-clamped at -50 mV and each trace is representative of observation made from four to five determinations.

Chapter 7 Results IV
135
7.3: Pharmacology of ε subunit To characterize the channel pharmacologically various antagonists for metabotropic
histamine receptors were checked. As the EC50 of ε is nearly 3 mM, so in our further
experiments we always took 3 mM histamine. We found that cimetidine - H2 receptor
antagonist - inhibited histamine-evoked current potently. It inhibits the histamine-evoked
current in a dose dependent manner with an IC50 of 3-5 µM (Fig 7.2A). We checked the
H1 receptor antagonist pyrilamine and observed that it was less sensitive antagonist.
Pyrilamine inhibited histamine-evoked current at IC50 value of 300-400 µM.
Thioperamide blocked the histamine-evoked current with an IC50 of 3 µM. The
histamine-evoked current was blocked in highly selective manner by PTX, a chloride
channel blocker, at nM concentration.
Fig. 7.2: Pharmacological characterization of homomeric ε subunit receptors. (A and B) Dose-response inhibition by cimetidine and pyrilamine. Cimetidine (H2-receptor antagonist) inhibited histamine-evoked current more effectively than pyrilamine (H1-receptor antagonist). All of the oocytes were voltage clamped at -50 mV and each trace is representative of observation made from four to five determinations.

Chapter 7 Results IV
136
7.4: Expression of α1β1ε, β1ε and ε in HEK 293 cells
To check that the high EC50 value of histamine for homomultimeric ε subunit can be
reduced in different cell culture system, we decided to functionally express and
characterize ε subunit in HEK 293 cells. As we found other two subunit combinations
α1β1ε, β1ε were also sensitive to histamine, we transfected HEK 293 cells with α1β1ε, β1ε
as well as ε subunit cDNAs. First of all, we checked α1β1ε subunit combinations, which
like in oocytes were highly sensitive for GABA. Interestingly, we found histamine did
not show any response even up to 3 mM concentration. What was evident that we did not
observe any spontaneous channel activity and channels were highly stable with almost no
leakage current, which was in contradiction with other ε subunit combination (for e.g.
α1β3ε) by other research group. We also checked β1ε and ε receptors in HEK 293 cells
but never found any response for histamine or GABA. Interestingly, there was also no
leakage current in any of the combination tested. We were curious for this anomalous
behaviour of the receptor and asked if ε subunit is really transported to the cell membrane
of the HEK 293 cells as it is documented in several reports that the single subunits are
retained in the endoplasmic reticulum of the cell and are not transported to the cell
membrane (Bollan et al., 2003; Gorrie et al., 1997). The reason and explanation is
already being discussed in the introduction section.
7.5: Molecular cloning of GFP-tagged subunits of GABAA receptors
The negative results obtained in the HEK 293 cells transfected with ε subunit alone, lead
us to check first the expression of ε subunit at the cell membrane. We addressed the
question, does ε able to be transported to the cell membrane or not. For this we tagged
ε subunit with GFP as well as to further get more information for the transportation of the
subunits we decided to tag α1 and β1 subunits of GABAA receptor with GFP.

Chapter 7 Results IV
137
7.5.1: Molecular cloning of GFP- tagged α1 and β1 subunits of GABAA receptors
In the next set of cloning steps pCDNA3 vectors were cloned that allowed the expression
of GABA receptors fused with a GFP on the C-terminal end. In the first step, the open
reading frame of the rat GABA α1 and β1 cDNA was PCR amplified using the plasmid
pSGEM-rat-α1 / β1 as templates (3.2.2). Primer pairs used were rGA-α1-4-stop codon,
GABA α1-Not1-Stop-Ev and RR-GABA-β2-up, RR-GABA-β2-do for α1 or β2,
respectively (2.3). In the corresponding 'down' primer, an EcoRV site was inserted
upstream of the stop codon that served for later insertion of the GFP fragment. After gel-
purification (3.1.2 and 3.1.5) the PCR fragments were cut by EcoRI and NotI (flanking
sites present in the used PCR primers for cloning) and ligated into the EcoRI and NotI cut
pCDNA3 vector (3.3.5). From clones with the correct insert, plasmid was prepared
(3.1.8) and the insert verified by sequencing (3.3.8). The resulting plasmids were named
pCDNA3-α1-EcoRV-stop and pCDNA3-β1-EcoRV-stop. pCDNA3-GFP was digested by
enzymes EcoRV and HpaI to take out the GFP open reading frame. The insert isolated
from this plasmid consists of the entire GFP open reading frame without start and stop
codon. The plasmids pCDNA3-α1-EcoRV-stop and pCDNA3-β1-EcoRV-stop were cut
by restriction enzyme EcoRV (3.3.1) and dephosphorylated (3.3.2). Restrction by EcoRV
caused the linearization of plasmids with blunt ends. These blunt-end plasmids could be
ligated with the EcoRV- HpaI-GFP fragment as HpaI generates blunt ends, too. From
clones with the correct insert, plasmid was prepared (3.1.8) and the sequence and
orientation of the fused GFP verified by sequencing (3.3.8). The resulting plasmids were
named pCDNA3-α1-N-GFP and pCDNA3-β1-N-GFP. Fig. 7.3 shows the schematic
representation of GFP tagged α1 subunit of GABAA receptor. For the GFP tagged β1
subunit the same procedure was employed.
7.5.2: Molecular cloning of GFP-tagged ε subunit of GABAA receptor
To tag the ε subunit C-terminal with GFP we employed different cloning strategy which
required 3 sub cloning steps. As ε subunit contains EcoRV, restriction site cloning could
not be done like in α1 and β1-GFP tagged experiment.
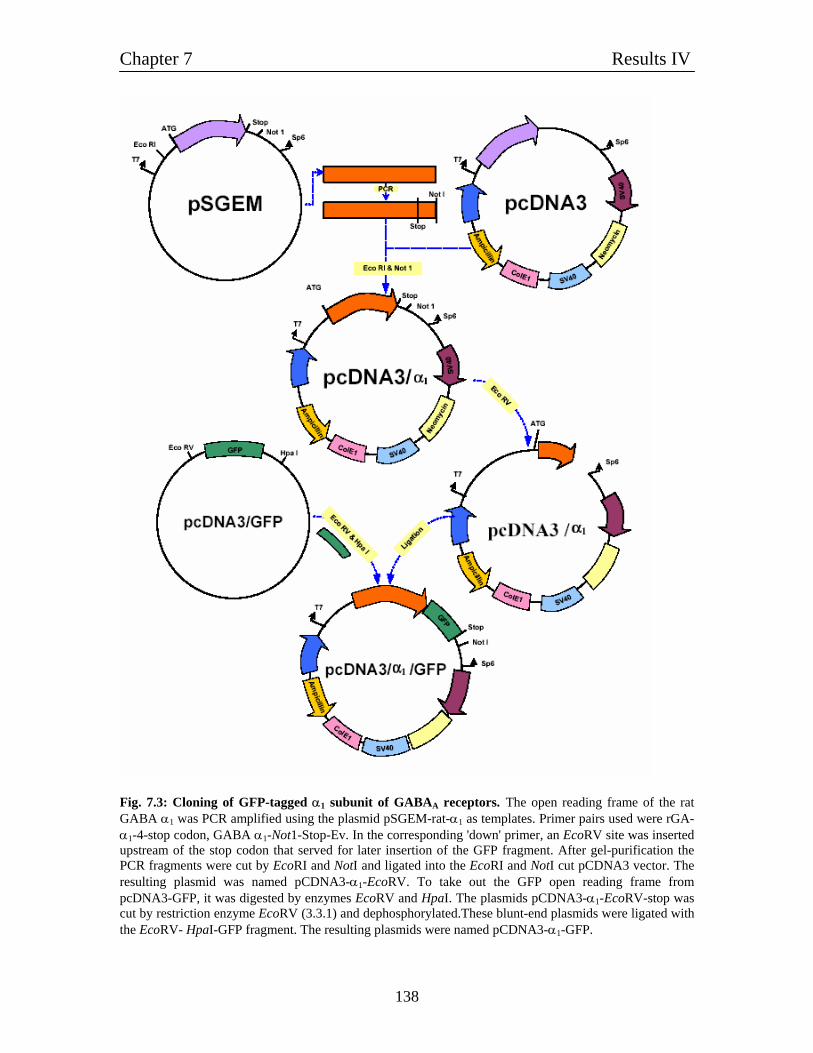
Chapter 7 Results IV
138
Fig. 7.3: Cloning of GFP-tagged α1 subunit of GABAA receptors. The open reading frame of the rat GABA α1 was PCR amplified using the plasmid pSGEM-rat-α1 as templates. Primer pairs used were rGA-α1-4-stop codon, GABA α1-Not1-Stop-Ev. In the corresponding 'down' primer, an EcoRV site was inserted upstream of the stop codon that served for later insertion of the GFP fragment. After gel-purification the PCR fragments were cut by EcoRI and NotI and ligated into the EcoRI and NotI cut pCDNA3 vector. The resulting plasmid was named pCDNA3-α1-EcoRV. To take out the GFP open reading frame from pcDNA3-GFP, it was digested by enzymes EcoRV and HpaI. The plasmids pCDNA3-α1-EcoRV-stop was cut by restriction enzyme EcoRV (3.3.1) and dephosphorylated.These blunt-end plasmids were ligated with the EcoRV- HpaI-GFP fragment. The resulting plasmids were named pCDNA3-α1-GFP.

Chapter 7 Results IV
139
In first step, by rGABA ε-Not1-Stop–EV in conjuction with a primer-directed polymerase
chain reaction, restriction sites for EcoRV and NotI were introduced, before and after the
stop codon of ε subunit. The elongation conditions were as follows: 94o C for 2 min,
followed by 20 cycles at 94° C for 1min, 59º C for 1 min, and 72º C for 1 min, and
ending with an incubation at 72º C for 10 minutes.
The pCDNA3 vector was digested by restriction enzymes EcoRV whereas SalI restricted
PCR fragment. Fragments were separated using gel electrophoresis and purified using a
gel extraction kit (3.1.5). Chimeric fragments were ligated using the rapid ligation kit.
Purified plasmid DNA for transfection was made using the plasmid-maxi kit (3.1.8) and
the entire coding region was sequenced.
In the second step of cloning, to take out the GFP open reading frame from pCDNA3 /
GFP, the vector was digested by enzymes EcoRV and HpaI. The plasmids containing ε-
EcoRV-stop-NotI was cut by restriction enzyme EcoRV. This restriction of plasmid
produced two fragments large and small. This large fragment was ligated with GFP
fragment, cloned and sequenced.
In the third step of cloning, plasmid containing the large fragment of ε subunit with GFP
was digested by EcoRV which linearised the plasmid with blunt ends. In this linearised
plasmid, the small ε fragment was ligated as a result after the ligation we obtained the
complete reading frame of ε subunit tagged with GFP. Purified plasmid DNA for
transfection were made using the plasmid-maxi kit (3.1.8) and the entire coding region of
all mutant was sequenced using Big dye ready reaction mix and an ABI automated DNA
sequencer. Fig. 7.3A and B shows the schematic representation of cloning strategy for
GFP tagged ε subunit. Fig. 7.4A and B shows the schematic representation of the GFP-
tagged ε subunit of GABAA receptors.
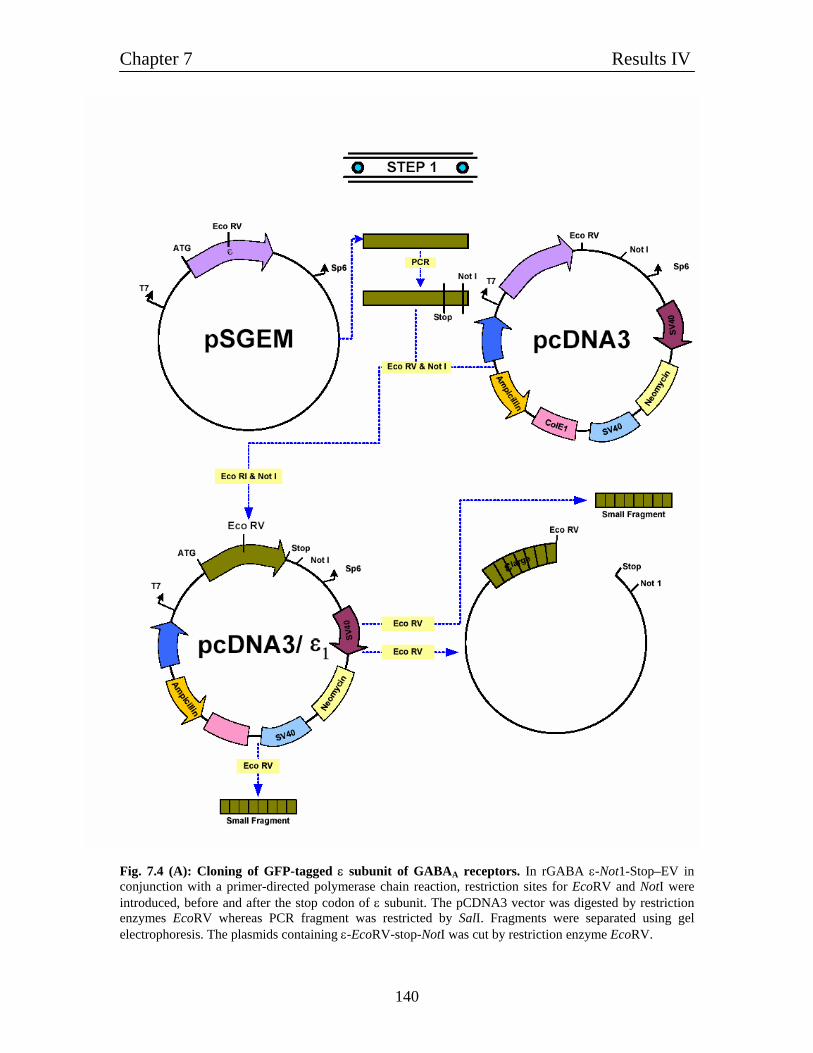
Chapter 7 Results IV
140
Fig. 7.4 (A): Cloning of GFP-tagged ε subunit of GABAA receptors. In rGABA ε-Not1-Stop–EV in conjunction with a primer-directed polymerase chain reaction, restriction sites for EcoRV and NotI were introduced, before and after the stop codon of ε subunit. The pCDNA3 vector was digested by restriction enzymes EcoRV whereas PCR fragment was restricted by SalI. Fragments were separated using gel electrophoresis. The plasmids containing ε-EcoRV-stop-NotI was cut by restriction enzyme EcoRV.
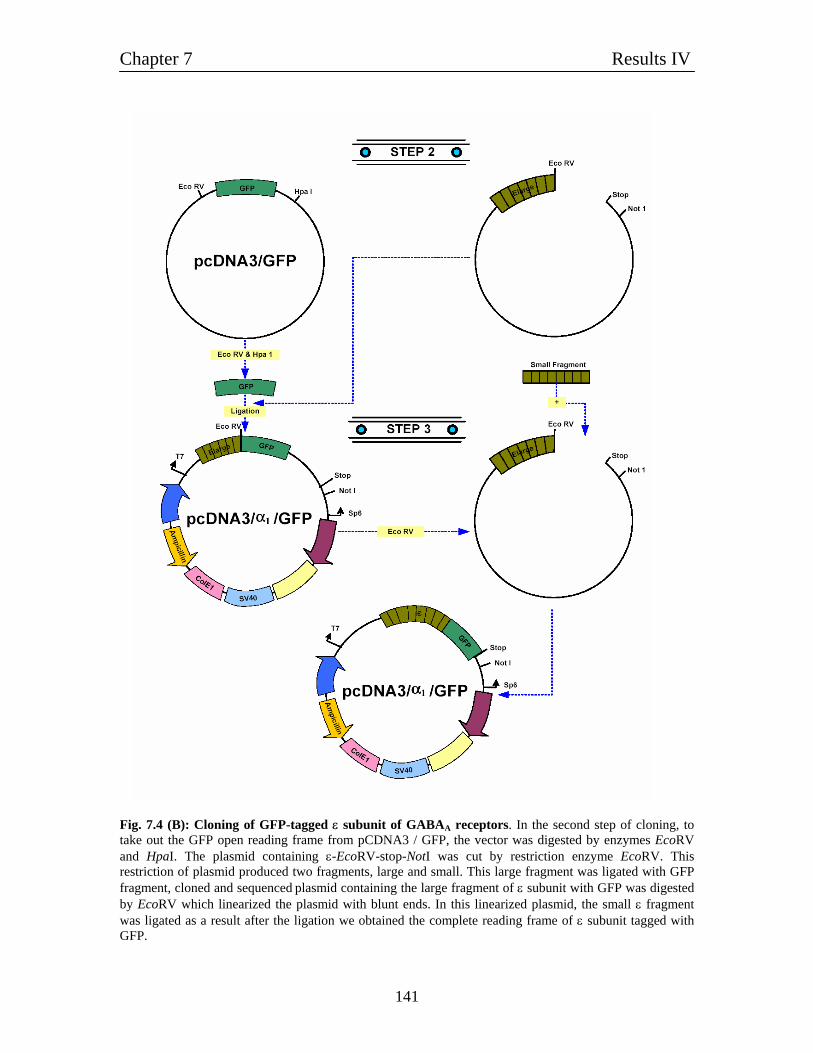
Chapter 7 Results IV
141
Fig. 7.4 (B): Cloning of GFP-tagged ε subunit of GABAA receptors. In the second step of cloning, to take out the GFP open reading frame from pCDNA3 / GFP, the vector was digested by enzymes EcoRV and HpaI. The plasmid containing ε-EcoRV-stop-NotI was cut by restriction enzyme EcoRV. This restriction of plasmid produced two fragments, large and small. This large fragment was ligated with GFP fragment, cloned and sequenced plasmid containing the large fragment of ε subunit with GFP was digested by EcoRV which linearized the plasmid with blunt ends. In this linearized plasmid, the small ε fragment was ligated as a result after the ligation we obtained the complete reading frame of ε subunit tagged with GFP.
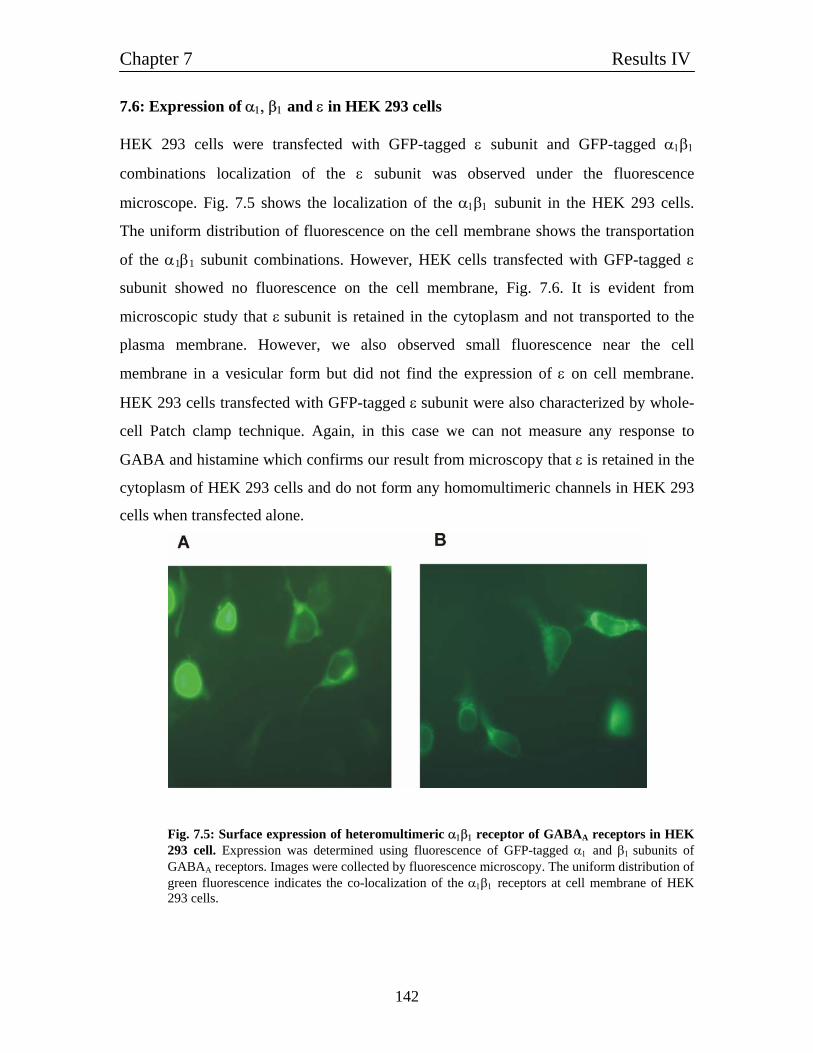
Chapter 7 Results IV
142
7.6: Expression of α1, β1 and ε in HEK 293 cells HEK 293 cells were transfected with GFP-tagged ε subunit and GFP-tagged α1β1
combinations localization of the ε subunit was observed under the fluorescence
microscope. Fig. 7.5 shows the localization of the α1β1 subunit in the HEK 293 cells.
The uniform distribution of fluorescence on the cell membrane shows the transportation
of the α1β1 subunit combinations. However, HEK cells transfected with GFP-tagged ε
subunit showed no fluorescence on the cell membrane, Fig. 7.6. It is evident from
microscopic study that ε subunit is retained in the cytoplasm and not transported to the
plasma membrane. However, we also observed small fluorescence near the cell
membrane in a vesicular form but did not find the expression of ε on cell membrane.
HEK 293 cells transfected with GFP-tagged ε subunit were also characterized by whole-
cell Patch clamp technique. Again, in this case we can not measure any response to
GABA and histamine which confirms our result from microscopy that ε is retained in the
cytoplasm of HEK 293 cells and do not form any homomultimeric channels in HEK 293
cells when transfected alone.
Fig. 7.5: Surface expression of heteromultimeric α1β1 receptor of GABAA receptors in HEK 293 cell. Expression was determined using fluorescence of GFP-tagged α1 and β1 subunits of GABAA receptors. Images were collected by fluorescence microscopy. The uniform distribution of green fluorescence indicates the co-localization of the α1β1 receptors at cell membrane of HEK 293 cells.
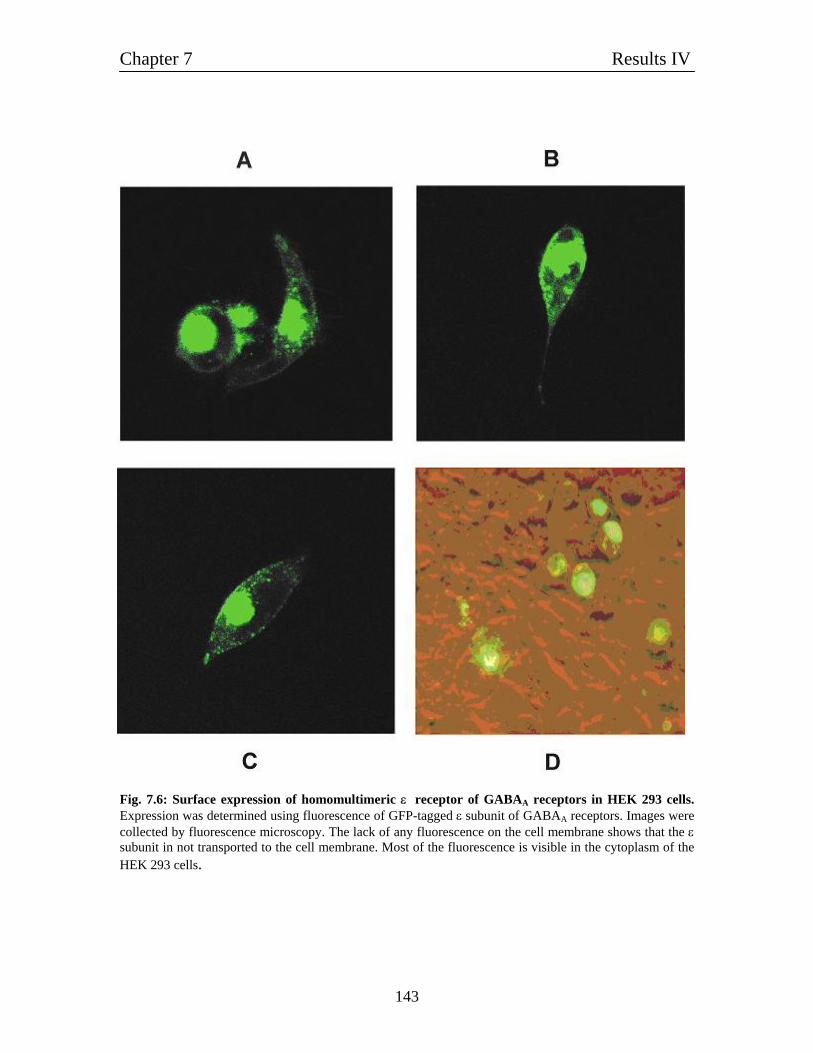
Chapter 7 Results IV
143
Fig. 7.6: Surface expression of homomultimeric ε receptor of GABAA receptors in HEK 293 cells. Expression was determined using fluorescence of GFP-tagged ε subunit of GABAA receptors. Images were collected by fluorescence microscopy. The lack of any fluorescence on the cell membrane shows that the ε subunit in not transported to the cell membrane. Most of the fluorescence is visible in the cytoplasm of the HEK 293 cells.

Chapter 7 Results IV
144
Summary of Major Findings
1. Homomultimeric ε subunit forms histamine-gated ion channel with an EC50 of 3 mM histamine when injected in Xenopus oocytes.
2. Incorporation of different subunits with ε reduces the spontaneously opened channel frequency of the receptors. The order of decreasing spontaneous channel activity in our subunit combinations checked was ε > β1ε > α1β1ε.
3. Histamine evoked current can be inhibited by pyrilamine, cimetidine,
thioperamide. The increasing order of sensitivity of antagonist was pyrilamine < thioperamide < cimetidine.
4. In HEK 293 cells the ε subunit is not transported to the cell membrane.

Chapter 8 Results V
145
Chapter 8 8.1: Properties of α1β2γ2 receptors and direct activation by histamine As described in (4.8) α1β2γ2 composed receptors the GABA response was potentiated by
histamine. In contrast to the findings at α1β1 receptors, histamine alone (without GABA)
was activating small currents in α1β2γ2 receptors when expressed in Xenopus oocytes. In
most tested oocytes, these current evoked by high concentrations of histamine (10 - 30
mM) were rather small reaching typically 5-10 % of the maximally current evoked by
saturating concentrations of GABA (Fig. 8.1A). In some extreme instances, they were
reaching up to 35 % of the maximal GABA-induced currents (Fig. 8.1B). In few cases,
histamine-induced currents were detected in other subunit combinations like α1β1 also,
but in these few cases such currents were detectable at all, they had typical currents as
low as 1-3 % of the GABA induced currents only.
It was tested, whether such histamine-activated currents were detectable in other
expression systems also. Therefore, α1β2γ2 receptors were expressed in HEK 293 cells
and transfected cells were exposed to GABA and histamine (Fig. 8.1C). At a holding
potential of -50 mV, GABA induced a desensitizing inward current that was not blocked
by 20 µM Zn2+, a proof, that the cell expressed α1β2γ2 receptors because α1β2 receptors
would have been blocked under these conditions. Application of 3 mM histamine to the
same individual cell didn't induce any detectable current (Fig. 8.1C). My findings that
histamine didn't directly activate heteromultimeric GABA receptors expressed in HEK
293 cells was supported by the experiments performed by Angela Vogt-Eisele and Katja
Erlkamp, who found that histamine also evoked no detectable current in HEK 293 cells
expressing α1β1 (personal communication).

Chapter 8 Results V
146
What could be the reason for the pronounced histamine activated current found in the
oocyte expression system? As it is described later, homomultimeric channels composed
of β subunits behave like histamine-activated channels (5.1.7). Though the formation of
heteromultimeric channels composed of α-β-γ subunits is strongly favoured, there is a
high chance of getting also homomultimeric β or γ channels if the pool of available
channel subunits is extremely unbalanced. Recently it was shown that GABA-gated γ2
homumultimeric channel could also be functionally expressed in Xenopus oocytes
(Ataulfo et al., 2004). Therefore, it was tested whether histamine might activate those γ2
channels also and indeed it was found, that 10 mM histamine activated currents in
Xenopus oocytes expressing γ2 subunits. So, in oocytes injected with a combination of α,
β, and γ subunits we have at least two possible channel populations, namely β and γ
homomultimeric channels that are known to be activated by histamine. The variation in
the amount of histamine-induced current might reflect different amounts of
'contaminating' homomultimeric β or γ channels. The reason that there are no histamine-
evoked currents detectable in α1β2γ2 expressing HEK 293 cells lies in the fact that
homomultimeric β2 channels are not targeted to the cell membrane, also the expression of
homomultimeric γ2 receptors was never reported in HEK 293 cells but only in Xenopus
oocytes. Therefore, possibly, that the level of potential homomultimeric channels is much
lower or not even existing in HEK 293 cells and, as consequence, there is also no current
evocable by histamine in contrast to Xenopus oocytes.
For the next experiment it was aimed to check whether the nature of action of histamine
varies with its concentration. At low histamine concentrations of 0.3 mM histamine
activated nearly no currents but had a clear potentiating effect on the current evoked by 2
µM GABA (Fig. 8.1E). At higher concentrations of 10 mM histamine, the effect of the
direct activation predominates and the current evoked by the mix of (10 mM histamine +
2 µM GABA) seems to be the simple summation of the current evoked by 10 mM
histamine and 2 µM GABA alone (Fig. 8.1E). In this oocyte it seems that the potentiating
effect of histamine at low concentration is replaced by a direct activation of the receptor
at high concentrations of histamine. This fits to the finding that potentiating agents like

Chapter 8 Results V
147
propofol also show potentiating effects at low and direct activating effects at high
concentrations.
The last experiment shows that we can not rule out that at least a part of the histamine-
evoked current in Xenopus oocytes is mediated by α1β2γ2 receptors oocytes directly and
not by possible heteromultimeric β and γ channels.
In the next set of experiments, the influence of the GABA concentration was investigated
in greater detail. For the potentiation experiments, we used rather low concentrations of 1
mM histamine to avoid the direct activation of currents, further; oocytes were chosen that
had a small fraction histamine activated currents (> 5 % of currents evoked by 2 µM
GABA) only. In these oocytes (n = 3), histamine potentiated the current evoked by 2 µM
GABA up to 2.5 folds but the potentiation decreased with higher GABA concentrations
and no potentiation was detectable at 100 µM GABA. This behaviour fits to the idea that
1 mM histamine lowers the EC50 for GABA in these oocytes. All investigated oocytes
described in this chapter were verified to express α1β2γ2 receptors due to the absence of
the block with 10 - 30 µM Zn2+ (Fig. 8.1G).
8.2: Homomultimeric γ2 receptors: Activation by histamine and its metabolites We also checked the expression of homomeric γ2 subunit receptors. Oocytes injected with
γ2 showed activation by histamine. Comparing to saturating concentration of 3 mM
GABA, current evoked by 10 mM histamine was 1/2 fold smaller. We also found that
like homomeric β subunit receptor histidine and t-MHA activated the homomeric
γ2 subunit receptors. The relative order of current amplitude is histidine > histamine > t-
MHA.
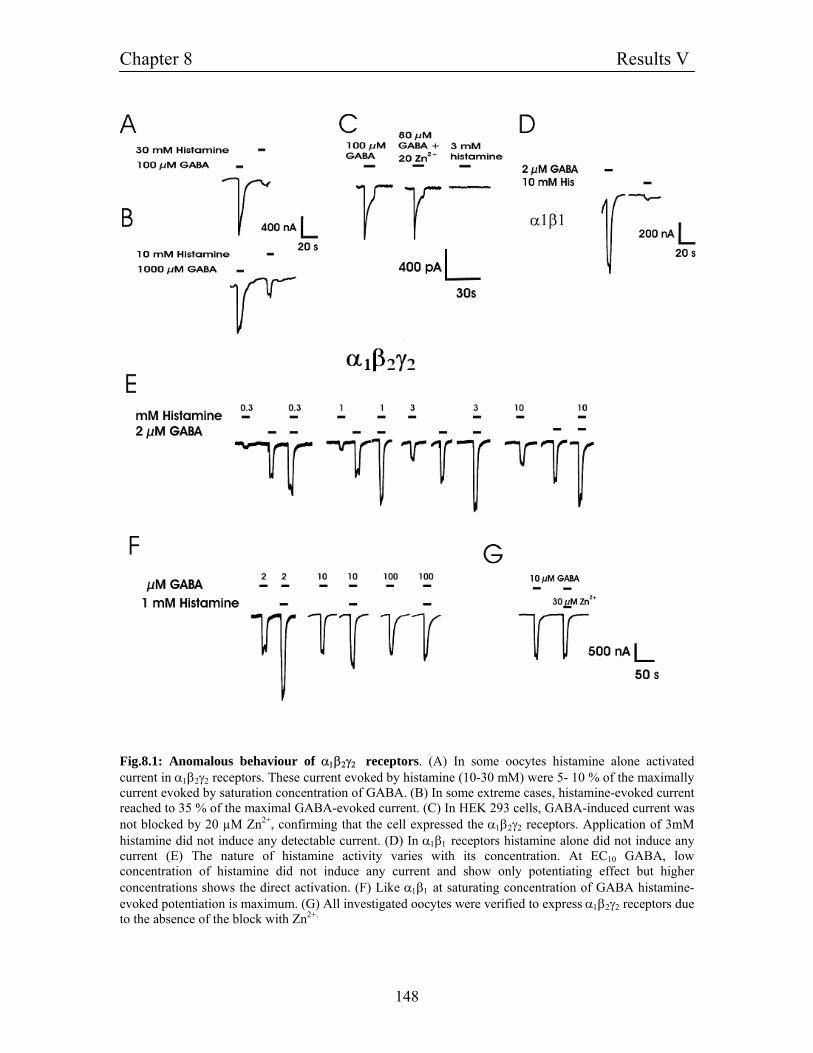
Chapter 8 Results V
148
Fig.8.1: Anomalous behaviour of α1β2γ2 receptors. (A) In some oocytes histamine alone activated current in α1β2γ2 receptors. These current evoked by histamine (10-30 mM) were 5- 10 % of the maximally current evoked by saturation concentration of GABA. (B) In some extreme cases, histamine-evoked current reached to 35 % of the maximal GABA-evoked current. (C) In HEK 293 cells, GABA-induced current was not blocked by 20 µM Zn2+, confirming that the cell expressed the α1β2γ2 receptors. Application of 3mM histamine did not induce any detectable current. (D) In α1β1 receptors histamine alone did not induce any current (E) The nature of histamine activity varies with its concentration. At EC10 GABA, low concentration of histamine did not induce any current and show only potentiating effect but higher concentrations shows the direct activation. (F) Like α1β1 at saturating concentration of GABA histamine-evoked potentiation is maximum. (G) All investigated oocytes were verified to express α1β2γ2 receptors due to the absence of the block with Zn2+.

Chapter 8 Results V
149
Fig. 8.2: Activation of homomultimeric γ2 receptors by GABA, histamine and its metabolites. Oocytes injected with γ2 subunit alone showed activation by histamine and its metabolites. Compared to saturating concentration of 3 mM GABA, current evoked by 10 mM histamine was nearly 1 \ 2 fold smaller. The relative order of amplitude of histamine and its metabolites was histidine > histamine > t-MHA. Bars over current traces indicate the duration of applied agonist in mM concentration.

Chapter 9 Discussion
150
Chapter 9 Discussion We were interested to find the histamine-gated ion channel in vertebrates. Two major
documentations, first the identification of histamine-gated ion channel in Drosophila
melanogaster (Gisselmann et al., 2002) and second electrophysiological evidence for a
possible histamine-gated ion channel in the SON nucleus (Hatton and Yang, 2001), lead
us to search for the gene of histamine-gated ion channels in mammals. A systematic
scanning of various vertebrate genome and EST databases from zebra fish to human
using the sequence information of newly identified Drosophila HisClα1 and HisClα2
genes and looking for various characteristic features of ligand-gated ion channels like
signal peptides, binding and transmembrane domains did not identify any novel gene for
ligand-gated ion channels having similarity with known histamine-gated ion channels.
However, histamine-gated channels from insects showed some homology with known
GABAA receptors subunits as well as some homology to glycine receptors also.
We found the homology to GABAA receptors interesting because of the various reasons.
The histamine containing neurons are found exclusively in the TM nucleus of the
hypothalamus in the brain (Bekkers, 1993). GABA containing neurons in the
ventrolateral preoptic nucleus innervates the TM nucleus and form symmetrical synapses
with histaminergic neurons. The GABA synthesizing enzyme, glutamate acid
decarboxylase (GAD) and GABA are also found in most TM neurons (Vincet et al.,
1983; Takeda et al., 1984; Senba et al., 1987). Interestingly, many neurons in TM
nucleus contain both GABA and histamine. In invertebrates, GABA and histamine can
act at the same ionotropic histamine receptor (Gisselmann et al., 2004). Therefore, we
supposed that histamine was candidate for acting on vertebrate GABAA receptor also. To
prove our hypothesis, we investigated the action of histamine on recombinant and native
GABAA receptors. Various chemical agents like benzodiazepines, barbiturates, steroids,
convulsants and anesthetics modulate GABAA receptors (Sigel et al., 2002; Boileau et
al., 1999; Rick et al., 1998; Olsen et al., 1997; Kraswoski et al., 1998). Modulation of

Chapter 9 Discussion
151
GABAA receptors influences several functions such as epilepsy; sleep disorders, anxiety
and pain (Kash et al., 1997; Delory et al., 1988; McKernan et al., 2000; Nelson et al.,
2002).
Histamine potentiates GABA-evoked currents in GABAA receptors We found that GABAA receptors are potentiated by histamine. The currents elicited by 2
µM GABA (a concentration near EC10 for α1β1 receptors) was potentiated by histamine
up to 3-4 folds. Potentiation by histamine does not affect the permeability of the ion
across the channel pore. This suggests that general property of the channel is not altered
in the presence of the histamine. The M2 pore-forming domain determines ion selectivity
of ligand-gated ion channels. The M2 domain possesses three rings of charge called the
cytoplasmic, intermediate and extracellular charged domains (Imoto et al., 1988). The
cytoplasmic ring is negative in all ligand-gated ion channels, whereas the intermediate
and extracellular rings are negative in cation-selective and positive in anion-selective
channels (Karlin and Akabas, 1995). The intermediate ring of charge is located near the
intracellular mouth of the channel, where the pore is most constricted, and is thought to
provide the electrostatic interaction for cation or anion selectivity (Imoto et al., 1988;
Lester, 1992; Wilson and Karlin, 1998; Xu and Akabas, 1996; Keramidas et al., 2002).
As the channel formed by the α1β1 subunit combinations has the intermediate and
extracellular ring which is positive charged so no alteration in the permeability is logical
during the potentiation mediated by the histamine.
On the level of a single GABAA channel, the increase in the amplitude of GABA-evoked
current by histamine may arise by several possibilities, like increase in the frequency of
open probability of the channel, increase in open time period and alteration of the single
channel conductance. In oocytes expressing α1β1 GABAA receptors the potentiation of
high concentrations of histamine persisted even at high, saturating GABA concentrations
and histamine potentiated the GABA-evoked current beyond the maximal current evoked
by GABA. In those oocytes showing a robust potentiation by histamine at 10 µM GABA,
no affinity change for GABA in the presence of histamine occurs, pointing out that the
overall the tendency of the receptor to open at various concentrations of GABA is the

Chapter 9 Discussion
152
same which suggests that the potentiation by histamine does not change the open
probability of the receptor. It may alter the open frequency, the open time duration or the
conductance of the channel, but future work has to be done on single channels to find out
the nature of histamine action. In virtually all oocytes expressing αβ subunit
combinations where the effect was systematically tested, GABA-potentiation was
independent on the GABA concentration (except for some few rare oocytes where
histamine didn't potentiate well. In these oocytes, histamine had a detectable effect at low
GABA concentrations only). This was different in oocytes expressing α1β2γ2 receptors.
Here, the potentiating effect was higher at lower GABA-concentrations and the nearly
vanished at high, saturating GABA concentrations. This is consistent with the idea that
histamine affects the GABA affinity and shifts the dose response curve to the left site.
The direct histamine-activated current component prevented a reliable quantitative
analysis of the of the EC50 shift so far that therefore remains to be determine in future but
the fact itself could be clearly demonstrated. Our findings suggest that γ2 subunits
influence the potentiation by histamine in heteromultimeric channels. This is not
unexpected as γ2 subunits also alter the effect of other potentiating agents like
benzodiazepines, too. In addition, we demonstrated that γ2 subunits have a histamine-
binding site itself as indicated by the fact that the corresponding γ2 homomultimeric
channels are gated by histamine.
Different subunit combinations of GABAA receptors are potentiated by histamine;
however they exhibit different sensitivity to histamine potentiation. For example
α1β3 subunit combinations were found to be very sensitive to histamine. α1β2, though
potentiated by histamine, showed higher concentration dependability for histamine. This
is also consistent with other reports, (Walters et al., 2000) showed the biphasic
potentiation of α1β2γ2 receptors by diazepam. At nM concentrations, diazepam
potentiated α1β2γ2 receptors, which leads to saturation at 1 µM. On the other hand, µM
concentration of diazepam (20 µM and above) evoked a second component of
potentiation, further increasing GABA-elicited current from 3 fold (the nM component)
to approximately eight folds. In contrast, α1β2 receptors show monophasic potentiation.

Chapter 9 Discussion
153
Diazepam elicited only a single component of potentiation with in the µM concentration
range for α1β2-receptor channel. Also the lack of α1β2 receptor channel sensitivity to
diazepam with in the nM concentration range has been documented previously, as the
presence of γ subunit is essential in conferring diazepam nM action (Pritchett et al.,
1989).
Experiments done in HEK 293 cells clearly show that the observed effect was not an
artifact of the Xenopus oocyte system and can also be observed in HEK 293 cells.
Potentiation mediated by histamine shows the same degree of potentiation in different
cell culture system. Our experiments in HEK 293 cells reveal that the degree of
potentiation mediated by histamine remains unchanged. Histamine showed similar
potentiating effects as in oocytes for e.g. at 2 µM GABA, 1 mM histamine evoked an up
to 5-folds potentiation of the GABA current (2.9 folds potentiation on an average) which
is in accordance with the potentiation in oocyte where we observed an up to 5 folds
potentiation. Also histamine-mediated potentiation of GABAA receptors is applicable to
bulbous neurons, which indicate that not in artificial recombinant system but in also
native neurons this effect can be reproduced.
We observed on α1β2γ2 receptors a considerable direct activation by high concentrations
of histamine in some batches of oocytes. The relative amplitude of the histamine-evoked
current to the maximum-evoked current of GABA or histamine potentiated GABA-
evoked current was typically up to 10 %. We found in HEK 293 cells that application of
histamine up to 3 mM does not show any activity. The direct activation by histamine can
be described like that in the α1β2γ2 receptors there is strong chance that a strong
expression of β or γ subunits can cause a considerable population of histamine-activated
homomultimeric receptors in addition to the expression of heteromeric receptors. As we
have clearly shown that both homomultimeric β and γ channels can be activated by
histamine alone, so the occurrence of histamine-activated currents in oocytes injected
with α1β2γ2 subunits should not be a surprise. But we cannot rule out the possibility that
at least a fraction of the observed histamine-activated current is carried by directly
activated heteromeric α1β2γ2 receptors.

Chapter 9 Discussion
154
We found that the metabolites in histamine pathways also potentiate GABA-evoked
current. L-histidine is a water-soluble amino acid. Histidine potentiated 2 µM GABA
current up to two folds. The in vivo actions of supplemental L-histidine are entirely
unclear. Our experiments give an idea that one of the possible mechanisms of histidine in
the body could be the interaction of histidine with GABAA receptors. The potentiation
mediated by histamine and histidine has intrinsic differences. First, histidine potentiates
GABAA receptors with lower EC50 than histamine in α1β1 receptors. The difference of
EC50 in between them is 1/10. Second, the amplitude of potentiation evoked by histamine
at 2 µM GABA concentration is higher (4-5 folds) than histidine (2 folds). Third, the
average fractional potential decreases for histamine from lower to higher GABA
concentration whereas in case of histidine it increases.
Histamine acts on GABAA β-subunits on the propofol binding site The subunit responsible for the histamine-binding site was determined by our subunit
substitution experiments and functional expression of homomultimeric channels of
GABAA receptors. Substitution experiments where we shuffled subtypes of one subunit
keeping another subunit constant, gave us an idea that β subunit is the possible candidate
for the histamine binding site. To get the clear indication for the subunit responsible for
histamine binding, we checked the homomultimeric channel expression of β subunit. It
has been well documented that homomultimeric β subunits can form functional channels
that either can open spontaneously or can be directly activated by some general anesthetic
(Sigel et al., 1990; Cestari et al., 1996; Krishek et al., 1996; Davies et al., 1997; Sanna et
al., 1995). However, whether or not homomeric β subunits can form functional GABA-
gated ion channels is still controversial and appears to depend on different species. For
instance, while human and bovine β receptors were found to form channels that can be
gated by GABA (Sanna et al., 1995; Blair et al., 1988; Pritchett et al., 1988; Krishek et
al., 1994) rat and mouse β1 and β3 subunits were insensitive to GABA (Sigel et al., 1990;
Krishek et al., 1996; Davies et al., 1997). We confirm by our experiments that rat β1
forms homomultimeric receptors, which are spontaneously active and can be blocked by
PTX. Our results are further extension for the view that β1 subunit can form
homomultimeric channel, which has already shown by (Miko et al., 2004). Although the

Chapter 9 Discussion
155
precise reason behind discrepancy in the results from different group is not clearly
understood but there are a number of possibilities that could attribute, in this discrepancy.
First, it can be because of different expression level of β1 subunit. Second, it may depend
on the level of posttranslational modulation of the subunits, which can vary pronouncedly
among different batches of Xenopus oocytes. Another possibility to reconcile this
discrepancy can be due to different levels of spontaneous activity of the β1 subunits
expressed in oocytes in different experimental conditions.
β subunit has the binding site for various modulators like pentobarbital and propofol. By
our pharmacological and electrophysiological results first we discriminated any
possibility of similarity between the pentobarbital and the histamine-binding site. That
was achieved by using antagonists for pentobarbital and histamine, respectively. First,
bemegride an antagonist of pentobarbital, inhibited pentobarbital-evoked current strongly
but did not have any influence on histamine-evoked currents. Second, thioperamide the
H3 antagonist competitively blocked the histamine-evoked current but was insensitive for
pentobarbital. Interestingly, in the same approach we found similarity in between
propofol and histamine binding site. We hypothesized that histamine should have the
binding site similar to propofol because of various reasons. Thioperamide inhibited
propofol-evoked current with same efficiency like its activity on histamine-evoked
current. Bemegride didn’t block propofol-evoked current, which was consistent with
earlier findings that pentobarbital and propofol have different binding sites. The
bemegride experiment was a clear indication to hypothesize that the different binding site
of different modulators need different antagonists. Having the same efficiency for the
same antagonist indicates that two different modulators can have the same binding site.
Moreover, in experiments where we mixed propofol and histamine to their EC50 and EC90
concentrations and co-applied them with 2 µM GABA on α1β1 and α5β1 receptors gave
another support for our hypothesis.
Propofol binding alters the GABAA receptor structure in the M3 membrane-spanning
segment region (Williams and Akabas, 2002). In several pioneering studies using
mutated recombinant receptors, several groups identified amino acid residue M286 the

Chapter 9 Discussion
156
binding site for propofol on β subunit. (Krasowski et al., 1998) has shown that on α2β1
(M286W) mutant receptor, propofol concentration greater than 10 µM failed to enhance
sub maximal GABA-currents. In contrast to lack of the potentiation, propofol still
directly activates this mutant receptor, with a concentration-response relationship that
overlaps with that for the wild-type α2β receptors. Moreover, mutation of a GABA
binding site residue, β2W157, reduced direct activation but did not affect propofol´s
modulatory actions (Fukumi et al., 1999). These results suggest for propofol, potentiation
and direct activation may involve binding to distinct sites. Sub maximal GABA currents
at wild type α1β1 receptors were potentiated by propofol. Thus, in agreement with the
other published studies, the presence of γ subunit is not required for the potentiation by
propofol. Our mutagenesis result also strongly proves hypothesis that (M286W) residue
on β subunit is indeed the binding site for propofol. In α1β1 (M286) mutant receptors,
propofol did not enhance the propofol-evoked currents at the same anesthetic
concentration tested in wild type α1β receptors. However, this mutation leads to the
spontaneous activity in the α1β1 (M286W) mutant receptor, which can be illustrated by
large leakage current at -60 mV holding potentials.
Remarkably the mutation in α1β (M286W) mutant receptor abolished the potentiation
mediated by histamine and histidine. No potentiation of mutant receptor by histamine up
to 10 mM and histidine up to 1 mM concentration was detected which strongly suggest
that both have the same binding site like propofol. We checked the effect of mutation at
different GABA concentration from 2 µM to 1 mM GABA with various concentrations
of histamine and histidine and found that even varying the concentration so much does
not lead still any potentiation. It is interesting to note also that this mutation does not
affect the activity of other modulators on β subunits. Modulation by pentobarbital and
neurosteroid remained same in α1β (M286W) mutant receptors. These data, first, provide
functional correlates for the receptor binding data, demonstrating vanished efficacies in
anesthetic–induced enhancement of ligand binding with mutant α1β1 (M286W) receptors;
and second strongly support the hypothesis that this methionine residue at the TM3
domain is important for the allosteric actions of propofol, histamine as well as histidine.

Chapter 9 Discussion
157
It is somewhat perplexing that methionine substitution at the aligned β3 M2 position,
β3N265, virtually eliminates propofol´s anesthetic efficacy in a knock-in mouse (Jurd et
al., 2003). Methionine occupies a volume 43 Ao greater than aspargine and is more
hydrophobic. Thus, steric bulk at position 265 can alter propofol binding, perhaps by
inducing a conformational change at the propofol binding site ~ 10 Ao away. Moreover,
we show that GABAC receptors are not potentiated by histamine. This is in accordance
with the previous findings for the action of other modulators on GABAC receptors
(Bormann et al., 1995; Feigenspan et al., 1998). Ionotropic GABAC receptors composed
of ρ subunits are insensitive to benzodiazepines, anesthetic and barbiturates (Polenzani
et al., 1991; Shimada et al., 1992). So, histamine behaves somehow like other modulators
already characterized. Sequence alignment show that the amino acid present at 286
location on ρ1 subunit of GABAC receptor is tryptophan instead of methionine which
gives the reason for no potentiation by histamine on GABAC receptors.
Homomultimeric receptors composed of β subunits are histamine-gated channels Our experiments on the homomultimeric β1 and β3 receptors support further the idea that
homomultimeric β and β3 receptors can be functionally expressed at least in Xenopus
oocytes forming histamine-gated channels. We contradict that β3 subunits do not form
spontaneously active channels shown by (Miko et al., 2004). We used human and they
used rat β3 subunits, this can be a plausible reason for having spontaneous channel
activity we observed in our experiments. Human homomultimeric β3 receptor is highly
sensitive for PTX, which blocks the open channel with in nM concentration.
Homomultimeric β3 receptors exhibit profound differences in the activity of histamine
and histidine. First, histamine activates homomultimeric β3 receptors with less half
maximal concentration than histidine. Second, histidine induces higher amplitude of
current (2-4 folds larger) than histamine at same concentration. Another metabolite of
histamine, t-MHA activates homomultimeric β3 receptors but with higher EC50 and lower
amplitude of currents. Histamine retained the ion permeability of homomultimeric β3
receptors. Homomultimeric β3 receptors show the pharmacology like H2 and H3
metabotropic histamine receptors. The histamine antagonists thioperamide and

Chapter 9 Discussion
158
famotidine inhibit histamine-evoked current. In case of homomultimeric β3 receptors
thioperamide acts as a competitive antagonist and is more active than famotidine. This is
the first report, which shows that homomultimeric β3 receptors behave like histamine-
gated ion channels.
We also investigated if β2 receptors are functionally expressed. Until now, nobody has
reported that β2 subunit of GABAA receptors can form homomultimeric functional
channels even though there are some reports which point out the spontaneous activity
without any ligand activation by homomultimeric β2 receptors. We stated that possible
homomultimeric β2 receptors may act like histamine-gated ion channels rather than
GABA. In this study we have demonstrated the functional expression of homomultimeric
β2 receptors in Xenopus oocytes. GABA as well as histamine and its metabolites activate
homomultimeric β2 receptors. The effect of activation by GABA is nearly 1\8 fold less
than histamine and its metabolites. In addition, we found that the magnitude of
spontaneous opening of homomultimeric β2 receptor channel was predominant and the
spontaneous channel activity accounts for open probability.
The inability of other groups to access the functional expression of homomultimeric
β2 receptors could be due to the different level of spontaneous activity of
homomultimeric β2 receptors in oocytes under different experimental conditions. We
have found that the magnitude of the spontaneous activity appeared to be so predominant
that it nearly overshadowed the magnitude of GABA-activated current in cells expressing
homomultimeric β2 subunits. Overall the magnitude of the PTX-sensitive current was 5-9
folds larger than that of the GABA-activated current. This indicates that the tendency of
homomultimeric β2 subunits to open spontaneously increase with a decrease in the
sensitivity of these receptors to GABA. It is therefore plausible to predict that when
homomultimeric β2 receptors channel open spontaneously at the maximal probability,
these receptors might no longer respond to activation by GABA.

Chapter 9 Discussion
159
The most important finding for the homomultimeric β2 receptors is that they are activated
by histamine and its metabolites. The amplitude of current activated by them is much
higher than the GABA-evoked current. Comparing it with PTX-evoked 'outward' current,
histamine-evoked current showed higher amplitude of the current also. Our different
approach to look for functional expression of homomultimeric β2 receptors lead us to be
able to measure the activity of these homomultimeric receptors. Also pharmacologically,
we were able to block the histamine-evoked current by various antagonists like
thioperamide, famotidine, HTMT and harmane.
We confirmed our finding that β subunits of GABAA receptors can form functional
homomultimeric channels by our electrophysiological, pharmacological studies. We ruled
out the possibility of any artifact or contamination of any other subunit as all 3 different β
subunits exhibited the histamine-evoked current and could be studied by further by using
histamine antagonists.
β2 and β3 are pharmacologically different. We found that β2 is more active than β3
subunit of GABAA receptors w.r.t. histamine. Also we have already shown that though
all these β subunits were activated by histamine they exhibited different pharmacology.
This may have various important aspects. Although there is evidence suggesting that
other type of GABAA receptors subunits apart from β subunits also could be involved in
spontaneous channel activity of the wild type of GABAA receptors, it may be at partly
rely on the presence of distinct β subunits (Sigel et al., 1990; Krishek et al., 1996). There
is evidence showing that spontaneous opening of GABAA receptor channels can be
detected in-vivo in spinal cord neurons (Mathers, 1985) and in pituitary cells (Taleb et al.,
1987; Hamann et al., 1990).
Different actions of histamine on homo- and heteromultimeric GABAA receptors We were also interested to further characterize the homomultimeric β receptors. Why
does histamine activate homomultimeric β receptors directly but does not directly act on
heteromultimeric α1β1 receptors? Here it is only potentiating the GABA response but not

Chapter 9 Discussion
160
opening the channel directly. This can be explained by looking into the structural
arrangement of subunits in heteromultimeric receptor complexes of GABAA receptors.
The minimal structural requirement for GABAA receptors gated by GABA is a
heteropentamer built from two different subunits with one peptide derived from the
α class and the other from the β class of variants (Schofield et al., 1987). Thus it was
expected that both subunit classes contribute to the formation of the binding pocket.
Indeed, a number of amino acids are members of both classes and most of them,
conserved within a subunit class, have been identified as being involved in high-affinity
agonist binding. The first one in this series was recognized by the F to L mutation at
position 64 in the rat α1 subunit in an electrophysiological assay (Sigel et al., 1990), later
confirmed by direct photolabelling of the site with (3H muscimol) (Smith and Olsen,
1994) whereas the homologous residue in the α5 variant has shown to be involved in the
formation of the GABA binding pocket. The equivalent residues in the β2 and γ2 subunits
do not have GABA binding properties (Sigel et al., 1992). The two neighboring amino
acids R66, corresponding to R70 in α5, and S68 (T in α6) in the α variant have been
reported to contribute to the GABA binding domain (Boileau et al., 1999) as well as
R120 in α (Hartvig et al., 2000). We suggest that the in the α1β1 receptor the histamine /
propofol binding domain defined by M286, may not be accessible to the histamine in the
same manner as in homomultimeric β receptors, as in the α1β1 heteropentamer there are
lot of chances for different subunit combinations to occur like 3 α and 2 β, 2 α and 3 β or
4 α and 1 β so it seems that the presence of α subunits in the α1β receptors hinders the
accessibility of histamine to the domain of β responsible for histamine gating in
homomultimeric β1.
The finding that histamine does not activate the α1β1 receptors directly can be also
explained by the earlier studies, which indicate that the pentameric GABAA receptors
undergo dynamic structural arrangement during their assembly in the endoplasmic
reticulum, transportation to the cell membrane and activation in the presence of a ligand.
Addition of the ligand triggers a small rotation of the extracellular domains of the
receptor subunits (Unwin, 1995), which then opens the channel pore formed by the

Chapter 9 Discussion
161
adjoining TM2 regions of the five subunits (predicted from the data obtained with
nicotinic acetylcholine receptors (Unwin, 1993)). Using disulphide bond mapping in
recombinant GABAA α1β1 mutant receptors, (Horenstein et al., 2001) could demonstrate
that the extracellular portion of the TM2–lined pore is more flexible than the intracellular
portion and that these domains of the α1 and β1 subunits may rotate asymmetrically, since
homologous residues (α1T261C and β1T261C form disulphide bonds only when the
receptors are activated by GABA. The resulting covalent modification keeps the channel
open. In the pentameric structural arrangement of the α1β1 receptors there may be the
possibility that presence of histamine doesn't change the alteration in the dynamic of the
receptor as a result the TM2 domain is not exposed to the ligand and hence the receptor
in the presence of histamine is not activated. In the case of homomultimeric β receptors
the pentameric arrangement of the β1 subunits is not hindered and only the TM2 domain
of the β1 subunit forms the pore, so there is more accessibility of this region for the
histamine, which leads to its structural rearrangement and eventually leads to the
activation by the histamine.
Also in the case of direct propofol activation it is reported that the direct activation is
absent in α4β1γ2 receptors (Wafford et al., 1996), and greater in α6β3γ2 than in α1β3γ2
receptors though propofol shows a higher efficiency in the potentiation of the GABA
effect (Krasowski et al., 1997). It is clearly evident that subunit structural arrangement
changes the accessibility of propofol to the GABAA receptor. As histamine and propofol
share the same binding site, the different actions of histamine on homo-and
heteromultimeric receptors can be explained in a parallel manner. Histamine action on further GABAA subunits
Our idea that spontaneous activity can be mediated by other subunits of GABAA
receptors lead us to further check some of the newly identified GABAA receptors
subunits. We extended our search for the histamine-activated homomultimeric channels
for GABAA receptors. The ε subunit indeed was the ideal candidate to go further using
this approach as it is well established that ε subunit causes the spontaneous activity when

Chapter 9 Discussion
162
it is incorporated with different subunits of GABAA receptors to form heteromultimeric
receptors (Davies et al., 1997; Whiting et al., 1997). The ε subunit was also important in
our study as we found that this subunit behaves somewhat differently from other GABAA
receptors subunits. For instance, it is not potentiated by barbiturates and benzodiazepines
and its current-voltage relationship does not show outward rectification at the positive
holding potential that are two important characteristics features of other GABAA subunits
(Davies et al., 1997; Whiting et al., 1997).
There are several reports about the heteromultimeric receptor containing ε subunit but till
now nobody has shown the homomultimeric expression of ε subunit. Here we show also
that ε subunit can form functional homomultimeric ε receptors when injected in oocytes.
These homomultimeric receptors show high spontaneous activity and from our
experience, are difficult to measure as the histamine-evoked current has very small
amplitude and because of high spontaneous activity the histamine-evoked currents are
difficult to measure.
We also report here some of the new combinations active at least in recombinant system
which can also be activated by histamine. α1β1ε containing receptors have become the
ideal choice for the characterization of ε subunit further. It is documented that ε competes
for the γ subunit and there are several reports indicating that βγ can form functional
heteromultimeric channels. Our result also indicate that replacing the γ subunit with ε
facilitate the functional expression of heteromultimeric β1ε receptors. Comparing them
with α1β1ε receptor, we found that they are highly spontaneously active and GABA does
not activate them up to 1 mM concentration. Heteromultimeric α1β1ε receptors have very
high affinity for GABA application of GABA on β1ε receptors blocks the spontaneously
active channels. The homomultimeric ε receptors are highly sensitive for histamine
receptor antagonist as most of them have roughly the IC50 of 3-5 µM excluding
pyrilamine which requires very high concentration (~ 300-500 µM) to block the
histamine-evoked currents. Quite interestingly comparing the pharmacology of ε subunit
with various homomultimeric β subunits receptors show marked differences. Cimetidine

Chapter 9 Discussion
163
was not able to inhibit histamine-evoked current in various homomultimeric β subunits
receptors whereas it is highly active antagonist for homomultimeric ε receptors.
Possible functional consequences of histamine potentiation Various subunit combinations of GABAA receptors exhibit GABA-evoked current
potentiation by histamine. Potentiation by histamine on various subunit combinations
gives indication that histamine potentiation is not restricted to a specific location in brain
area. As both GABAergic and histaminergic systems are widely distributed, so our
current finding indicates the global interaction between histamine and GABA in brain.
This can have several pivotal effects. As synaptic GABAA receptors are also potentiated
with histamine to same extent (for example α1β2γ2 were potentiated by histamine up to 4
folds), it proves that synaptically GABA receptor can be targeted by histamine. This
gives an idea of the importance of the localization of the GABAA receptors in the TM
nucleus of the hypothalamus. There can be several possible sources of the histamine to
the GABAA receptors. As it is documented that one possible source of histamine
targeting GABAA receptors are TM neurons. As some of these neurons are containing
both GABA and histamine, one cannot exclude that both neurotransmitters are even co-
released at the same synapse. In addition, histamine can diffuse out of a histaminergic
synapse by a `spill-out` effect as described for GABA-synapses (Rossi and Hamann,
1998) and thus may act on neighboring synaptic or extra synaptic GABAA receptors.
Further, mast cells in the brain are an additional source for histamine. Mast cells occur in
the CNS of many species and up to 50 % of the brain histamine is attributable to the
presence of these cells. By selectively enhancing the GABAA receptors, histamine should
enhance process in which GABA currents participate, especially those related to
histaminergic neurons in the TM nucleus like sleep and wakefulness (Lin et al., 1986 and
1988; Paramentier et al., 2002).
Modulation of GABAA receptors at synapses can have profound effects. For example in
epilepsy, compounds that increase or reduce GABAergic inhibition are used, respectively
as anti-epileptic or pro-convulsive drugs for many forms of human or experimental
epilepsy (Prince, 1978). Impaired inhibition is thought to be important in temporal lobe

Chapter 9 Discussion
164
epilepsy, the most common feature of epilepsy in human. Enhancement in GABA current
by histamine can possibly be used for epilepsy treatment.
Dependency of potentiation by histamine on various subunits combination may lead to
alteration in the function differently in different location of the brain, which means the
histamine-evoked potentiation should enhance more the brain’s area activity where α1β3
receptors are predominantly expressed. This is also an important aspect as GABAA
receptors exhibit different activity for GABA in the various brain area depending on the
subunit combinations. This is also consistent with other modulators like propofol, which
show at lower concentrations a sedative effect in the frontal cortex while at higher
concentration it causes hypnosis in thalamus and at even more higher concentration leads
to immobility due to the action of propofol on spinal cord (Rudolph and Antkowiak,
2004).
Our current finding indicate a possible resolution for a quite interesting but vague
hypothesis which depicts that hypothalamic networks that are involved in sleep
regulation might have a key role in mediating anesthetic induced hypnosis. Although not
identical, sleep and general anesthesia share common EEG patterns. General anesthesia
produces a variety of useful effects, including sedation (defined as a decrease in activity)
and hypnosis (defined as promoting the onset of sleep). (Nelson et al., 2002) showed that
the same brain region may be important for both sleep and anesthetic. The authors report
that TM nucleus is also a key target for at least two anesthetics that are known to act at
GABAA receptors. During deep anesthesia (so called stage III), EEG recordings resemble
those associated with non-REM sleep, suggesting that they are generated by similar
patterns of cortical and thalamocortical activity. In their study the authors discovered the
anatomical sites responsible for the hypnotic action of propofol and found that it is
located in TM nucleus. Our findings lead into the same approach though we did not
perform experiments in TM nucleus but it is quite evident that physiological localization
of propofol and histamine at least in one brain region show their common mode of
activity.

Chapter 9 Discussion
165
Our results report for the first time that antagonist defined for histamine can have the
possible side effects in the body via interaction with GABAA receptors. So, it should be
taken in care when these antagonists are administrated in the body. Our result may give a
clue about some of the side effects observed during the therapy in which histamine
antagonist are used. It is also of importance that thioperamide acts on propofol and
inhibits propofol-evoked current in the range of histamine IC50. As far as from our
knowledge, this is the first time report for the thioperamide acting as a potent antagonist
for propofol. So anesthesia mediated by propofol can be overcome by using antagonists
on the basis of thioperamide in the future.
Histamine and neurological diseases We assume that the current findings may lead to a new approach about interaction
between GABA and histamine in certain brain disorders like schizophrenia.
Abnormalities in GABA activity in schizophrenia have been consistently shown in the
last ten years. Schizophrenia is associated with both decreased numbers and
abnormalities in the distribution of GABAergic neurons in the cortex, particularly in the
cortical laminae (Kaplen & Saock, 1995). GAD1, the GABA precursor, has also been
shown to decrease activity in schizophrenia. This decreased activity has been found in
nucleus accumbens, putaman, amaygdala and hippocampus. This suggests that loss of
neuroinhibitory control of GABA, in specific regions of the brain, may be responsible for
some of schizophrenia’s symptoms. There is growing evidence to suggest the
involvement of histaminergic pathways in the pathophysiology of schizophrenia.
Overactive histamine activity is thought to contribute significantly to deficit symptoms
such as apathy and social withdrawal associated with the disorder, while the efficacy of
certain atypical antipsychotic may include their action at histamine receptors (Rauscher et
al., 1980; Nakai et al., 1991). Many patients diagnosed as schizophrenic have either a
chronic excess or deficiency of blood histamine and its end product t-MHA (Schwartz et
al., 1971). Nutritional treatment correcting these imbalances has led to great
improvement or recovery for most such patients. One of histamine’s many roles in the
body is to act as on inhibitory neurotransmitter. It is used to promote alpha wave activity
in the brain which enables an individual to handle anxiety and stress easier (McLeod et

Chapter 9 Discussion
166
al., 1998). If the person is deficient in histidine, it leads to a lack of histamine and creates
unbalances in calming alpha-rhythms in the brain allowing the excitatory beta waves
(responsible for the brain activity that leads to anger and tension) to promote (McLeod et
al., 1998). We speculate, during schizophrenia, when the GABA level in brain is altered,
brain tries to compensate the loss of inhibitory neurotransmission. Increase in histamine
level in schizophrenia supports our idea. In schizophrenia, GABAA receptors at the soma
and axon initial segments of pyramidal neurons might be locally unregulated in response
to a reduction in perisomatic inhibitory input from chandlier and wide arbor neurons.
Brain tries to overcome the reduced level of GABA, by promoting higher expression of
GABAA receptors. There should be the enhancement for the affinity of GABA for
GABAA receptors. As our results show that histidine increases affinity of GABAA
receptors for GABA and hence leads to hyperpolarisation in neurons to minimize the
reduction of GABA in brain. Our result shows that even in the presence of low GABA,
histamine potentiates GABAA receptor and may show anxiolytic effects.

Chapter 10 Summary
167
Chapter 10 Summary
In this thesis we demonstrate for the first time that histamine effectively potentiates
GABA-evoked current in heteromultimeric GABAA receptors. Further it directly opens
homomultimeric GABAA receptors that thus function as histamine-gated channels. A
typical 2-4 folds potentiation was found for various combinations of GABAA receptors
expressed in Xenopus oocytes and HEK 293 cells, an effect not restricted to recombinant
receptors but present in native neurons of the olfactory bulb, too. In homomultimeric β
channels, histamine opened the channel with a far better efficacy than GABA. A
pharmacological characterization in combination with the investigation of mutated
β1 subunits (M286W) indicates that histamine acts on the propofol-binding site. We
found that the histamine metabolite histidine had similar potentiating effects. Histamine
and histidine are good candidates for the endogenous ligand acting at the allosteric
modulatory propofol binding site and thus may modulate neuronal events by acting
directly on GABAA receptors in addition to the known histamine action on metabotropic
H1-H3 receptors. We identified further GABAA β subunits as potential components of
mammalian histamine-gated channels, whose existence has been proposed for many
years.
Direct activation by histamine on homomultimeric GABAA receptor could be a missing
link in the various physiological processes like sleep and wake. It is tempting to check the
β subunit expression in the SON nucleus where reports indicate the presence of the
possible ionotropic histamine-gated channel.
The direct activation of histamine on GABAA receptors points the possible answer for
one of the evolutionary questions yet to be answered. Why during the evolution, the
nature just produced metabotropic histamine receptors and not a distinct class of
ionotropic histamine channels in vertebrates? Other neurotransmitters like GABA and

Chapter 10 Summary
168
ACh have both metabotropic as well as ionotropic receptors, then what was the need not
go in the same trend for the nature to produce ionotropic histamine receptors? Maybe our
bioinformatics approaches are still far from identifying the 'real' histamine-gated ion
channel of vertebrates as it might belong to a yet unidentified class of ion channels
distinct from the superfamily of ligand-gated ion channels. Nevertheless, various GABAA
receptors behave like histamine-gated ion channels in recombinant expression systems
and are thus good candidates for being components of the long sought-after histamine
gated-channels in vertebrates.

Chapter 11 References
169
Chapter 11 References
Alkire,M.T. (1998) Quantitative EEG correlations with brain glucose metabolic rate during anesthesia in volunteers. Anesthesiology 89, 323-333.
Alkire,M.T. and Haier,R.J. (2001) Correlating in vivo anaesthetic effects with ex vivo receptor density data supports a GABAergic mechanism of action for propofol, but not for isoflurane. Br.J.Anaesth. 86, 618-626.
Almeida,A.P. and Beaven,M.A. (1981) Phylogeny of histamine in vertebrate brain. Brain Res. 208, 244-250.
Altschul,S.F., Gish,W., Miller,W., Myers,E.W., and Lipman,D.J. (1990) Basic local alignment search tool. J.Mol.Biol. 215, 403-410.
Amin,J. and Weiss,D.S. (1993) GABAA receptor needs two homologous domains of the beta-subunit for activation by GABA but not by pentobarbital. Nature 366, 565-569.
Armstrong,W.E. and Sladek,C.D. (1985) Evidence for excitatory actions of histamine on supraoptic neurons in vitro: mediation by an H1-type receptor. Neuroscience 16, 307-322.
Arrang,J.M., Devaux,B., Chodkiewicz,J.P., and Schwartz,J.C. (1988) H3-receptors control histamine release in human brain. J.Neurochem. 51, 105-108.
Arrang,J.M., Garbarg,M., and Schwartz,J.C. (1983) Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 302, 832-837.
Asada,H., Kawamura,Y., Maruyama,K., Kume,H., Ding,R.G., Kanbara,N., Kuzume,H., Sanbo,M., Yagi,T., and Obata,K. (1997) Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc.Natl.Acad.Sci.U.S.A 94, 6496-6499.
Ash, A.S. and Schild,H.O. (1966) Receptors mediating some actions of histamine. Br.J.Pharmacol. 27, 427-439.
Attwell, D., Barbour,B., and Szatkowski,M. (1993) Nonvesicular release of neurotransmitter. Neuron 11, 401-407.
Awapara, J.Landua, A.J. Fuerst, and Seale,B. (1950) Free gamma-aminobutyric acid in brain. J.Biol.Chem. 187, 35-39.
Bali,M. and Akabas,M.H. (2004) Defining the propofol binding site location on the GABAA receptor. Mol.Pharmacol. 65, 68-76.

Chapter 11 References
170
Ballivet, M., Alliod,C., Bertrand,S., and Bertrand,D. (1996) Nicotinic acetylcholine receptors in the nematode Caenorhabditis elegans. J.Mol.Biol. 258, 261-269.
Bekkers, J.M. (1993) Enhancement by histamine of NMDA-mediated synaptic transmission in the hippocampus. Science 261, 104-106. Belelli, D. and J. J. Lambert. "Neurosteroids: endogenous regulators of the GABAA
Belelli, D. and J.J. Lambert. Neurosteroids: endogenous regulators of the GABAA receptors. Nat. Rev. NEurosci. 6.7 (2005) : 565-75.
Baumann, S.W., Baur, R., and Sigel, E. (2002) Forced subunit assembly in a1b2g2 GABAA receptors.Insight into the absolute arangement. J. Biol.Chem. 277, 46020-46025.
Baylis, H.A., Matsuda,K., Squire,M.D., Fleming,J.T., Harvey,R.J., Darlison,M.G., Barnard,E.A., and Sattelle,D.B. (1997) ACR-3, a Caenorhabditis elegans nicotinic acetylcholine receptor subunit. Molecular cloning and functional expression. Receptors.Channels 5, 149-158.
Bedford, F.K., Kittler,J.T., Muller,E., Thomas,P., Uren,J.M., Merlo,D., Wisden,W., Triller,A., Smart,T.G., and Moss,S.J. (2001) GABA(A) receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nat.Neurosci. 4, 908-916.
Beg, A.A. and Jorgensen,E.M. (2003) EXP-1 is an excitatory GABA-gated cation channel. Nat.Neurosci 6, 1145-1152.
Bekkers, J.M. (1993) Enhancement by histamine of NMDA-mediated synaptic transmission in the hippocampus. Science 261, 104-106.
Belelli, I., Pistis,I., Peters,J.A., and Lambert,J.J. (1999) General anaesthetic action at transmitter-gated inhibitory amino acid receptors. Trends Pharmacol.Sci. 20, 496-502.
Benke, D. (1994) Distribution, prevalence, and drug binding profile of gamma-aminobutyric acid type A receptor subtypes differing in the beta-subunit variant. J.Biol.Chem. 269, 27100-07.
Berghammer,H. and Auer,B. (1993) Easypreps: fast and easy plasmid minipreparation for analysis of recombinant clones in E. coli. Biotechniques 14, 524, 528.
Bhargava, K.P., Gupta,M.B., and Tangri,K.K. (1973) Mechanism of ulcerogenic activity of indomethacin and oxyphenbutazone. Eur.J.Pharmacol. 22, 191-195.
Blair,L.A., Levitan,E.S., Marshall,J., Dionne,V.E., and Barnard,E.A. (1988) Single subunits of the GABAA receptor form ion channels with properties of the native receptor. Science 242, 577-579.

Chapter 11 References
171
Bloom, F.E. and Iversen,L.L. (1971) Localizing 3H-GABA in nerve terminals of rat cerebral cortex by electron microscopic autoradiography. Nature 229, 628-630.
Bohme, I., Rabe,H., and Luddens,H. (2004) Four amino acids in the alpha subunits determine the gamma-aminobutyric acid sensitivities of GABAA receptor subtypes. J.Biol.Chem. 279, 35193-35200.
Boileau, A.J. and Czajkowski,C. (1999) Identification of transduction elements for benzodiazepine modulation of the GABA(A) receptor: three residues are required for allosteric coupling. J.Neurosci. 19, 10213-10220.
Boileau, A.J., Evers,A.R., Davis,A.F., and Czajkowski,C. (1999) Mapping the agonist binding site of the GABAA receptor: evidence for a beta-strand. J.Neurosci. 19, 4847-4854.
Bollan,K., King,D., Robertson,L.A., Brown,K., Taylor,P.M., Moss,S.J., and Connolly,C.N. (2003) GABA(A) receptor composition is determined by distinct assembly signals within alpha and beta subunits. J.Biol.Chem. 278, 4747-4755.
Bonhomme,V., Fiset,P., Meuret,P., Backman,S., Plourde,G., Paus,T., Bushnell,M.C., and Evans,A.C. (2001) Propofol anesthesia and cerebral blood flow changes elicited by vibrotactile stimulation: a positron emission tomography study. J.Neurophysiol. 85, 1299-1308.
Bonnert,T.P., McKernan,R.M., Farrar,S., le Bourdelles,B., Heavens,R.P., Smith,D.W., Hewson,L., Rigby,M.R., Sirinathsinghji,D.J., Brown,N., Wafford,K.A., and Whiting,P.J. (1999) theta, a novel gamma-aminobutyric acid type A receptor subunit. Proc.Natl.Acad.Sci.U.S.A 96, 9891-9896.
Bormann,J. (2000) The 'ABC' of GABA receptors. Trends Pharmacol.Sci. 21, 16-19.
Bormann, J. and A. Feigenspan. (1995) GABAC receptors. Trends Neurosci. 18, 515-19.
Bormann, J., Hamill,O.P., and Sakmann,B. (1987) Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J.Physiol 385, 243-286.
Bowery, N.G., Hill,D.R., Hudson,A.L., Doble,A., Middlemiss,D.N., Shaw,J., and Turnbull,M. (1980) (-)Baclofen decreases neurotransmitter release in the mammalian CNS by an action at a novel GABA receptor. Nature 283, 92-94.
Brandon, N.J., Uren,J.M., Kittler,J.T., Wang,H., Olsen,R., Parker,P.J., and Moss,S.J. (1999) Subunit-specific association of protein kinase C and the receptor for activated C kinase with GABA type A receptors. J.Neurosci. 19, 9228-9234.
Brown, N., Kerby,J., Bonnert,T.P., Whiting,P.J., and Wafford,K.A. (2002) Pharmacological characterization of a novel cell line expressing human alpha(4)beta(3)delta GABA(A) receptors. Br.J.Pharmacol. 136, 965-974.

Chapter 11 References
172
Buckingham, S., Lapied,B., Corronc,H., and Sattelle,F. (1997) Imidacloprid actions on insect neuronal acetylcholine receptors. J.Exp.Biol. 200, 2685-2692.
Bugajski,J. and Janusz,Z. (1981) Lipolytic responses induced by intracerebroventricular administration of histamine in the rat. Agents Actions 11, 147-150.
Burg, M.G., Sarthy,P.V., Koliantz,G., and Pak,W.L. (1993) Genetic and molecular identification of a Drosophila histidine decarboxylase gene required in photoreceptor transmitter synthesis. EMBO J. 12, 911-919.
Casale,T.B., Rodbard,D., and Kaliner,M. (1985) Characterization of histamine H-1 receptors on human peripheral lung. Biochem.Pharmacol. 34, 3285-3292.
Cestari,I.N., Uchida,I., Li,L., Burt,D., and Yang,J. (1996) The agonistic action of pentobarbital on GABAA beta-subunit homomeric receptors. Neuroreport 7, 943-947.
Chang,B.Y., Conroy,K.B., Machleder,E.M., and Cartwright,C.A. (1998) RACK1, a receptor for activated C kinase and a homolog of the beta subunit of G proteins, inhibits activity of src tyrosine kinases and growth of NIH 3T3 cells. Mol.Cell Biol. 18, 3245-3256.
Chang,Y., Wang,R., Barot,S., and Weiss,D.S. (1996) Stoichiometry of a recombinant GABAA receptor. J.Neurosci. 16, 5415-5424.
Cherubini,E. and Conti,F. (2001) Generating diversity at GABAergic synapses. Trends Neurosci. 24, 155-162.
Collinson, N. et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J.Neurosci. 22.13 (2002): 5572-80.
Connolly, C.N., Wooltorton, J.R., Smart,T.G., and Moss,S.J. (1996) Subcellular localization of gamma-aminobutyric acid type A receptors is determined by receptor beta subunits. Proc.Natl.Acad.Sci.U.S.A 93, 9899-9904.
Connors, B.W., Malenka,R.C., and Silva,L.R. (1988) Two inhibitory postsynaptic potentials, and GABAA and GABAB receptor-mediated responses in neocortex of rat and cat. J.Physiol 406, 443-468.
Cooper,E., Couturier,S., and Ballivet,M. (1991) Pentameric structure and subunit stoichiometry of a neuronal nicotinic acetylcholine receptor. Nature 350, 235-238.
Corringer,P.J., Le Novere,N., and Changeux,J.P. (2000) Nicotinic receptors at the amino acid level. Annu.Rev.Pharmacol.Toxicol. 40, 431-458.
Costa, E. (1982) Perspectives in the molecular mechanisms of antidepressant action. Adv.Biochem.Psychopharmacol. 31, 21-26.

Chapter 11 References
173
Costa, E., Guidotti,A., and Mao,C.C. (1975) Evidence for involvement of GABA in the action of benzodiazepines: studies on rat cerebellum. Adv.Biochem.Psychopharmacol. 113-130.
Crestani, F.; Keist R.; Fritschy,J.M.; Benke,D.; Vogt,K.; Prut,L.; Bluthmann,H.; Mohler,H.; Rudolph,U. (2002) Contribution of the alpha1-GABA(A) receptor subtype to the pharmacological actions of benzodiazepine site inverse agonists. Neuropharmacology 43, 679-84.
Cully, D.F., Vassilatis,D.K., Liu,K.K., Paress,P.S., Van der Ploeg,L.H., Schaeffer,J.M., and Arena,J.P. (1994) Cloning of an avermectin-sensitive glutamate-gated chloride channel from Caenorhabditis elegans. Nature 1994 371, 707-711.
Davies, P.A., Kirkness,E.F., and Hales,T.G. (1997) Modulation by general anaesthetics of rat GABAA receptors comprised of alpha 1 beta 3 and beta 3 subunits expressed in human embryonic kidney 293 cells. Br.J.Pharmacol. 120, 899-909.
Dazzi, L. (1995) Modulation of basal and stress-induced release of acetylcholine and dopamine in rat brain by abecarnil and imidazenil, two anxioselective gamma-aminobutyric acidA receptor modulators. J.Pharmacol.Exp.Ther. 273, 241-47.
DeLorey, T.M., Handforth,A., Anagnostaras,S.G., Homanics,G.E., Minassian,B.A., Asatourian,A., Fanselow,M.S., Delgado-Escueta,A., Ellison,G.D., and Olsen,R.W. (1998) Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J.Neurosci. 18, 8505-8514.
Drutel, G., Peitsaro,N., Karlstedt,K., Wieland,K., Smit,M.J., Timmerman,H., Panula,P., and Leurs,R. (2001) Identification of rat H3 receptor isoforms with different brain expression and signaling properties. Mol.Pharmacol. 59, 1-8.
Duncan, P.G., Brink,C., Adolphson,R.L., and Douglas,J.S. (1980) Cyclic nucleotides and contraction/relaxation in airway muscle: H1 and H2 agonists and antagonists. J.Pharmacol.Exp.Ther. 215, 434-442.
Enz, R. and Cutting,G.R. (1998) Molecular composition of GABAC receptors. Vision Res. 38, 1431-1441.
Ericson, H., Blomqvist,A., and Kohler,C. (1991) Origin of neuronal inputs to the region of the tuberomammillary nucleus of the rat brain. J.Comp Neurol. 311, 45-64.
Essrich, C., Lorez,M., Benson,J.A., Fritschy,J.M., and Luscher,B. (1998) Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat.Neurosci. 1, 563-571.
Farrar,S.J., Whiting,P.J., Bonnert,T.P., and McKernan,R.M. (1999) Stoichiometry of a ligand-gated ion channel determined by fluorescence energy transfer. J.Biol.Chem. 274, 10100-10104.

Chapter 11 References
174
Feng, G., Tintrup,H., Kirsch,J., Nichol,M.C., Kuhse,J., Betz,H., and Sanes,J.R. (1998) Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science 282, 1321-1324.
Feng, H.J. and Macdonald,R.L. (2004) Multiple actions of propofol on alphabetagamma and alphabetadelta GABAA receptors. Mol.Pharmacol. 66, 1517-1524.
Fiset, P., Paus,T., Daloze,T., Plourde,G., Meuret,P., Bonhomme,V., Hajj-Ali,N., Backman,S.B., and Evans,A.C. (1999) Brain mechanisms of propofol-induced loss of consciousness in humans: a positron emission tomographic study. J.Neurosci. 19, 5506-5513.
Fisher, J.L. and Macdonald,R.L. (1997) Single channel properties of recombinant GABAA receptors containing gamma 2 or delta subtypes expressed with alpha 1 and beta 3 subtypes in mouse L929 cells. J.Physiol 505 ( Pt 2), 283-297.
Fleming, J.T., Squire,M.D., Barnes,T.M., Tornoe,C., Matsuda,K., Ahnn,J., Fire,A., Sulston,J.E., Barnard,E.A., Sattelle,D.B., and Lewis,J.A. (1997) Caenorhabditis elegans levamisole resistance genes lev-1, unc-29, and unc-38 encode functional nicotinic acetylcholine receptor subunits. J Neurosci 17, 5843-5857.
Fon, E.A. and Edwards,R.H. (2001) Molecular mechanisms of neurotransmitter release. Muscle Nerve 24, 581-601.
Friedl, W., Lentes,K.U., Schmitz,E., Propping,P., and Hebebrand,J. (1988) Limited tryptic proteolysis of the benzodiazepine binding proteins in different species reveals structural homologies. J.Neurochem. 51, 1877-1881.
Fritschy, J.M. and Mohler,H. (1995) GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J.Comp Neurol. 359, 154-194.
Gengs, C., Leung,H.T., Skingsley,D.R., Iovchev,M.I., Yin,Z., Semenov,E.P., Burg,M.G., Hardie,R.C., and Pak,W.L. (2002) The target of Drosophila photoreceptor synaptic transmission is a histamine-gated chloride channel encoded by ort (hclA). J.Biol.Chem. 277, 42113-42120.
Gisselmann, G., Plonka,J., Pusch,H., and Hatt,H. (2004) Drosophila melanogaster GRD and LCCH3 subunits form heteromultimeric GABA-gated cation channels. Br.J Pharmacol. 142, 409-413.
Gisselmann, G., Plonka,J., Pusch,H., and Hatt,H. (2004) Unusual functional properties of homo- and heteromultimeric histamine-gated chloride channels of Drosophila melanogaster: spontaneous currents and dual gating by GABA and histamine. Neurosci.Lett. 372, 151-156.
Gisselmann, G., Pusch,H., Hovemann,B.T., and Hatt,H. (2002) Two cDNAs coding for histamine-gated ion channels in D. melanogaster. Nat.Neurosci. 5, 11-12.

Chapter 11 References
175
Gorrie, G.H., Vallis,Y., Stephenson,A., Whitfield,J., Browning,B., Smart,T.G., and Moss,S.J. (1997) Assembly of GABAA receptors composed of alpha1 and beta2 subunits in both cultured neurons and fibroblasts. J.Neurosci. 17, 6587-6596.
Gundersen, C. B., R. Miledi, and I. Parker. (1984) Properties of human brain glycine receptors expressed in Xenopus oocytes. Proc.R.Soc.Lond B Biol.Sci. 221, 235-44.
Gunther, U., Benson,J., Benke,D., Fritschy,J.M., Reyes,G., Knoflach,F., Crestani,F., Aguzzi,A., Arigoni,M., Lang,Y., and . (1995) Benzodiazepine-insensitive mice generated by targeted disruption of the gamma 2 subunit gene of gamma-aminobutyric acid type A receptors. Proc.Natl.Acad.Sci.U.S.A 92, 7749-7753.
Haas, H. And Panula. (2003) The role of histamine and the tuberomamillary nucleus in the nervous system. Nat. Rev.Neurosci. 4, 121-130.
Haas, H.L., Wolf,P., and Nussbaumer,J.C. (1975) Histamine: action on supraoptic and other hypothalamic neurones of the cat. Brain Res. 88, 166-170.
Hamann, M., Desarmenien,M., Vanderheyden,P., Piguet,P., and Feltz,P. (1990) Electrophysiological study of tert-butylbicyclophosphorothionate-induced block of spontaneous chloride channels. Mol.Pharmacol. 37, 578-582.
Hamill, O.P., Marty,A., Neher,E., Sakmann,B., and Sigworth,F.J. (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pfluegers Arch. 391, 85-100.
Hardie, R.C. (1989) A histamine-activated chloride channel involved in neurotransmission at a photoreceptor synapse. Nature 339, 704-706.
Hartvig, L., Lukensmejer,B., Liljefors,T., and Dekermendjian,K. (2000) Two conserved arginines in the extracellular N-terminal domain of the GABA(A) receptor alpha(5) subunit are crucial for receptor function. J.Neurochem. 75, 1746-1753.
Hashemzadeh-Gargari, H. and Freschi,J.E. (1992) Histamine activates chloride conductance in motor neurons of the lobster cardiac ganglion. J.Neurophysiol. 68, 9-15.
Hatton, G.I. and Yang,Q.Z. (2001) Ionotropic histamine receptors and H2 receptors modulate supraoptic oxytocin neuronal excitability and dye coupling. J.Neurosci. 21, 2974-2982.
Heinen, K. et al. (2003) Impaired dendritic spine maturation in GABAA receptor alpha1 subunit knock out mice. Neuroscience 122, 699-705.
Hevers, W. and Luddens,H. (1998) The diversity of GABAA receptors. Pharmacological and electrophysiological properties of GABAA channel subtypes. Mol.Neurobiol. 18, 35-86.

Chapter 11 References
176
Hill, D.R. and Bowery,N.G. (1981) 3H-baclofen and 3H-GABA bind to bicuculline-insensitive GABA B sites in rat brain. Nature 290, 149-152.
Hill, D. R. and N.G. Bowery. (1981) 3H-GABA bind to bicuculline-intsensitive GABA(B) sites in rat brain. Nature 290, 149-52.
Hill-Venning, C., Belelli,D., Peters,J.A., and Lambert,J.J. (1997) Subunit-dependent interaction of the general anaesthetic etomidate with the gamma-aminobutyric acid type A receptor. Br.J.Pharmacol. 120, 749-756.
Hofbauer, R.K., Fiset,P., Plourde,G., Backman,S.B., and Bushnell,M.C. (2004) Dose-dependent effects of propofol on the central processing of thermal pain. Anesthesiology 100, 386-394.
Hofstra, C.L., Desai,P.J., Thurmond,R.L., and Fung-Leung,W.P. (2003) Histamine H4 receptor mediates chemotaxis and calcium mobilization of mast cells. J.Pharmacol.Exp.Ther. 305, 1212-1221.
Horenstein, J., Wagner,D.A., Czajkowski,C., and Akabas,M.H. (2001) Protein mobility and GABA-induced conformational changes in GABA(A) receptor pore-lining M2 segment. Nat.Neurosci. 4, 477-485.
Huang, R.Q. and Dillon,G.H. (1999) Effect of extracellular pH on GABA-activated current in rat recombinant receptors and thin hypothalamic slices. J.Neurophysiol. 82, 1233-1243.
Imoto,K., Busch,C., Sakmann,B., Mishina,M., Konno,T., Nakai,J., Bujo,H., Mori,Y., Fukuda,K., and Numa,S. (1988b) Rings of negatively charged amino acids determine the acetylcholine receptor channel conductance. Nature 335, 645-648.
Imoto, K., Methfessel,C., Sakmann,B., Mishina,M., Mori,Y., Konno,T., Fukuda,K., Kurasaki,M., Bujo,H., Fujita,Y., and . (1986) Location of a delta-subunit region determining ion transport through the acetylcholine receptor channel. Nature 324, 670-674.
Inagaki, N., Yamatodani,A., Shinoda,K., Panula,P., Watanabe,T., Shiotani,Y., and Wada,H. (1988) Histaminergic nerve fibers in the median eminence and hypophysis of rats demonstrated immunocytochemically with antibodies against histidine decarboxylase and histamine. Brain Res. 439, 402-405.
Johnston, G.A. (1996) GABAc receptors: relatively simple transmitter -gated ion channels? Trends Pharmacol.Sci. 17, 319-323.
Jones, A.K. and Sattelle,D.B. (2004) Functional genomics of the nicotinic acetylcholine receptor gene family of the nematode, Caenorhabditis elegans. Bioessays 26, 39-49.

Chapter 11 References
177
Jovanovic, J. N. et al. (2004) Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. J.Neurosci. 24, 522-30.
Jurd,R., Arras,M., Lambert,S., Drexler,B., Siegwart,R., Crestani,F., Zaugg,M., Vogt,K.E., Ledermann,B., Antkowiak,B., and Rudolph,U. (2003) General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 17, 250-252.
Kaila, K. (1994) Ionic basis of GABAA receptor channel function in the nervous system. Prog.Neurobiol. 42, 489-537.
Kaisti, K.K., Metsahonkala,L., Teras,M., Oikonen,V., Aalto,S., Jaaskelainen,S., Hinkka,S., and Scheinin,H. (2002) Effects of surgical levels of propofol and sevoflurane anesthesia on cerebral blood flow in healthy subjects studied with positron emission tomography. Anesthesiology 96, 1358-1370.
Kang, M., Yoshimatsu, H., Ogawa,R., Kurokawa,M., Oohara,A., Tamari,Y., and Sakata,T. (1994) Thermoregulation and hypothalamic histamine turnover modulated by interleukin-1 beta in rats. Brain Res.Bull. 35, 299-301.
Karlin, A. (1993) Structure of nicotinic acetylcholine receptors. Curr.Opin.Neurobiol. 3, 299-309.
Karlin, A. and Akabas, M.H. (1995) Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron 15, 1231-1244.
Krasowski, M.D., O'Shea, S.M., Rick, C.E., Whiting, P.J., Hadingham,K.L., Czajkowski,C., and Harrison,N.L. (1997) Alpha subunit isoform influences GABA(A) receptor modulation by propofol. Neuropharmacology 36, 941-949.
Kash, S.F., Tecott, L.H., Hodge,C., and Baekkeskov,S. (1999) Increased anxiety and altered responses to anxiolytics in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc.Natl.Acad.Sci.U.S.A 96, 1698-1703.
Kehoe, J. and McIntosh, J.M. (1998) Two distinct nicotinic receptors, one pharmacologically similar to the vertebrate alpha7-containing receptor, mediate Cl currents in aplysia neurons. J Neurosci 18, 8198-8213.
Keramidas, A., Moorhouse, A.J., Pierce,K.D., Schofield,P.R., and Barry,P.H. (2002) Cation-selective mutations in the M2 domain of the inhibitory glycine receptor channel reveal determinants of ion-charge selectivity. J.Gen.Physiol 119, 393-410.
Kittler, J.T. and Moss, S.J. (2003) Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr.Opin.Neurobiol. 13, 341-347.

Chapter 11 References
178
Kneussel, M., Brandstatter, J.H., Gasnier,B., Feng,G., Sanes,J.R., and Betz,H. (2001) Gephyrin-independent clustering of postsynaptic GABA(A) receptor subtypes. Mol.Cell Neurosci. 17, 973-982.
Knoflach, F., Benke, D., Wang,Y., Scheurer,L., Luddens,H., Hamilton,B.J., Carter,D.B., Mohler,H., and Benson,J.A. (1996) Pharmacological modulation of the diazepam-insensitive recombinant gamma-aminobutyric acidA receptors alpha 4 beta 2 gamma 2 and alpha 6 beta 2 gamma 2. Mol.Pharmacol. 50, 1253-1261.
Korpi, E.R. (1994) Role of GABAA receptors in the actions of alcohol and in alcoholism: recent advances. Alcohol Alcohol 29, 115-129.
Krasowski, M.D., Koltchine, V.V., Rick,C.E., Ye,Q., Finn,S.E., and Harrison,N.L. (1998) Propofol and other intravenous anesthetics have sites of action on the gamma-aminobutyric acid type A receptor distinct from that for isoflurane. Mol.Pharmacol. 53, 530-538.
Krishek, B.J., Moss, S.J., and Smart,T.G. (1996) Homomeric beta 1 gamma-aminobutyric acid A receptor-ion channels: evaluation of pharmacological and physiological properties. Mol.Pharmacol. 49, 494-504.
Krishek, B.J., Xie, X., Blackstone,C., Huganir,R.L., Moss,S.J., and Smart,T.G. (1994) Regulation of GABAA receptor function by protein kinase C phosphorylation. Neuron 12, 1081-1095.
Lambert, J.J., Belelli, D., Hill-Venning,C., and Peters,J.A. (1995) Neurosteroids and GABAA receptor function. Trends Pharmacol.Sci. 16, 295-303.
Le Novere, N., Corringer, P.J., and Changeux,J.P. (2002) The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J.Neurobiol. 53, 447-456.
Lester, H.A. (1992) The permeation pathway of neurotransmitter-gated ion channels. Annu.Rev.Biophys.Biomol.Struct. 21, 267-292.
Leurs, R., Brozius, M.M., Jansen,W., Bast,A., and Timmerman,H. (1991) Histamine H1-receptor-mediated cyclic GMP production in guinea-pig lung tissue is an L-arginine-dependent process. Biochem.Pharmacol. 42, 271-277.
Lin, J. S. (2000) Effects of amphetamine and modafinil on the sleep/wake cycle during experimental hypersomnia induced by sleep deprivation in the cat. J.Sleep Res. 9, 89-96
Lin, J. S., K. Sakai, and M. Jouvet. (1986) Role of hypothalamic histaminergic systems in the regulation of vigilance states in cats. C.R.Acad.Sci.III 303, 469-74.
Lin, J. S., K. Sakai, and M. Jouvet. (1988) Evidence for histaminergic arousal mechanisms in the hypothalamus of cat. Neuropharmacology 27, 111-22.

Chapter 11 References
179
Liman, E.R., Tytgat, J., and Hess, P. (1992) Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9, 861-871.
Low, K. Crestani, F.; Keist,R.; Benke,D.; Brunig,I.; Benson,J.A.; Fritschy,J.M.; Rulicke,T.; Bluethmann,H.; Mohler,H.; Rudolph,U (2000) Molecular and neuronal substrate for the selective attenuation of anxiety. Science 290, 131-34.
Luscher, B. and Keller,C.A. (2004) Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol.Ther. 102, 195-221.
Macdonald, R.L. and Olsen,R.W. (1994) GABAA receptor channels. Annu.Rev.Neurosci. 17, 569-602.
Masaki,T., Yoshimatsu,H., Chiba,S., Watanabe,T., and Sakata,T. (2001) Targeted disruption of histamine H1-receptor attenuates regulatory effects of leptin on feeding, adiposity, and UCP family in mice. Diabetes 50, 385-391.
Mathers, D.A. (1985) Spontaneous and GABA-induced single channel currents in cultured murine spinal cord neurons. Can.J.Physiol Pharmacol. 63, 1228-1233.
Matsuda, K., Buckingham, S.D., Kleier,D., Rauh,J.J., Grauso,M., and Sattelle,D.B. (2001) Neonicotinoids: insecticides acting on insect nicotinic acetylcholine receptors. Trends Pharmacol.Sci. 22, 573-580.
McClintock, T.S. and Ache, B.W. (1989) Histamine directly gates a chloride channel in lobster olfactory receptor neurons. Proc.Natl.Acad.Sci.U.S.A 86, 8137-8141.
McKernan, R.M., Rosahl, T.W., Reynolds,D.S., Sur,C., Wafford,K.A., Atack,J.R., Farrar,S., Myers,J., Cook,G., Ferris,P., Garrett,L., Bristow,L., Marshall,G., Macaulay,A., Brown,N., Howell,O., Moore,K.W., Carling,R.W., Street,L.J., Castro,J.L., Ragan,C.I., Dawson,G.R., and Whiting,P.J. (2000) Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat.Neurosci. 3, 587-592.
Martinez-Torres, A. and R. Miledi. (2004) Expression of functional receptors by the human gamma-aminobutyric acid A gamma 2 subunit. Proc.Natl.Acad.Sci.U.S.A 101, 3220-23.
McKernan, R.M. and Whiting, P.J. (1996) Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 19, 139-143.
McLeod, R.L., Aslanian, R., del Prado,M., Duffy,R., Egan,R.W., Kreutner,W., McQuade,R., and Hey,J.A. (1998) Sch 50971, an orally active histamine H3 receptor agonist, inhibits central neurogenic vascular inflammation and produces sedation in the guinea pig. J.Pharmacol.Exp.Ther. 287, 43-50.
Mehta, A.K. and Ticku, M.K. (1999) An update on GABAA receptors. Brain Res.Brain Res.Rev. 29, 196-217.

Chapter 11 References
180
Mihalek, R.M., Banerjee, P.K., Korpi,E.R., Quinlan,J.J., Firestone,L.L., Mi,Z.P., Lagenaur,C., Tretter,V., Sieghart,W., Anagnostaras,S.G., Sage,J.R., Fanselow,M.S., Guidotti,A., Spigelman,I., Li,Z., DeLorey,T.M., Olsen,R.W., and Homanics,G.E. (1999) Attenuated sensitivity to neuroactive steroids in gamma-aminobutyrate type A receptor delta subunit knockout mice. Proc.Natl.Acad.Sci.U.S.A 96, 12905-12910.
Mihic, S.J., Ye, Q., Wick,M.J., Koltchine,V.V., Krasowski,M.D., Finn,S.E., Mascia,M.P., Valenzuela,C.F., Hanson,K.K., Greenblatt,E.P., Harris,R.A., and Harrison,N.L. (1997) Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature 389, 385-389.
Miko, A., Werby, E., Sun,H., Healey,J., and Zhang,L. (2004) A TM2 residue in the beta1 subunit determines spontaneous opening of homomeric and heteromeric gamma-aminobutyric acid-gated ion channels. J.Biol.Chem. 279, 22833-22840.
Miledi, R., I .Parker and K. Sumikawa. (1982) Synthesis of chick brain GABA receptors by frog oocytes. Proc.R.Soc.Lond B Biol.Sci. 216, 509-15.
Minier, F. and Sigel,E. (2004) Positioning of the alpha-subunit isoforms confers a functional signature to gamma-aminobutyric acid type A receptors. Proc.Natl.Acad.Sci.U.S.A 101, 7769-7774.
Mochly-Rosen, D. and Gordon, A.S. (1998) Anchoring proteins for protein kinase C: a means for isozyme selectivity. FASEB J. 12, 35-42.
Mongan, N.P., Baylis,H.A., Adcock,C., Smith,G.R., Sansom,M.S., and Sattelle,D.B. (1998) An extensive and diverse gene family of nicotinic acetylcholine receptor alpha subunits in Caenorhabditis elegans. Receptors.Channels 6, 213-228.
Mongan, N.P., Jones, A.K., Smith,G.R., Sansom,M.S., and Sattelle,D.B. (2002) Novel alpha7-like nicotinic acetylcholine receptor subunits in the nematode Caenorhabditis elegans. Protein Sci. 11, 1162-1171.
Monti, J.M., Jantos, H., Boussard,M., Altier,H., Orellana,C., and Olivera,S. (1991) Effects of selective activation or blockade of the histamine H3 receptor on sleep and wakefulness. Eur.J.Pharmacol. 205, 283-287.
Moss, S. J. and T. G. Smart. (2001) Constructing inhibitory synapses. Nat.Rev.Neurosci. 2, 240-50.
Mullis, K.B. and Faloona, F.A. (1987) Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 155, 335-350.
Nakai, T., Kitamura, N., Hashimoto,T., Kajimoto,Y., Nishino,N., Mita,T., and Tanaka,C. (1991) Decreased histamine H1 receptors in the frontal cortex of brains from patients with chronic schizophrenia. Biol.Psychiatry 30, 349-356.

Chapter 11 References
181
Nakamura, S. (1993) Loss of large neurons and occurrence of neurofibrillary tangles in the tuberomammillary nucleus of patients with Alzheimer's disease. Neurosci.Lett. 151, 196-99.
Neelands, T.R., Fisher, J.L., Bianchi,M., and Macdonald,R.L. (1999) Spontaneous and gamma-aminobutyric acid (GABA)-activated GABA(A) receptor channels formed by epsilon subunit-containing isoforms. Mol.Pharmacol. 55, 168-178.
Neelands, T.R. and Macdonald, R.L. (1999) Incorporation of the pi subunit into functional gamma-aminobutyric Acid(A) receptors. Mol.Pharmacol. 56, 598-610.
Nelson, L.E., Guo, T.Z., Lu,J., Saper,C.B., Franks,N.P., and Maze,M. (2002) The sedative component of anesthesia is mediated by GABA(A) receptors in an endogenous sleep pathway. Nat.Neurosci. 5, 979-984.
Nicoll, R.A. (1988) The coupling of neurotransmitter receptors to ion channels in the brain. Science 241, 545-551.
Nusser, Z., Roberts, J.D., Baude,A., Richards,J.G., and Somogyi,P. (1995) Relative densities of synaptic and extrasynaptic GABAA receptors on cerebellar granule cells as determined by a quantitative immunogold method. J.Neurosci. 15, 2948-2960.
O'Reilly, M., Alpert, R., Jenkinson,S., Gladue,R.P., Foo,S., Trim,S., Peter,B., Trevethick,M., and Fidock,M. (2002) Identification of a histamine H4 receptor on human eosinophils--role in eosinophil chemotaxis. J.Recept.Signal.Transduct.Res. 22, 431-448.
Olsen, R.W., Yang, J., King,R.G., Dilber,A., Stauber,G.B., and Ransom,R.W. (1986) Barbiturate and benzodiazepine modulation of GABA receptor binding and function. Life Sci. 39, 1969-1976.
Ookuma, K., Yoshimatsu, H., Sakata,T., Fujimoto,K., and Fukagawa,F. (1989) Hypothalamic sites of neuronal histamine action on food intake by rats. Brain Res. 490, 268-275.
Orange, P.R., Heath, P.R., Wright,S.R., Ramchand,C.N., Kolkeiwicz, L., and Pearson,R.C. (1996) Individuals with schizophrenia have an increased incidence of the H2R649G allele for the histamine H2 receptor gene. Mol.Psychiatry 1, 466-469.
Panula, P., Flugge,G., Fuchs,E., Pirvola,U., Auvinen,S., and Airaksinen,M.S. (1989) Histamine-immunoreactive nerve fibers in the mammalian spinal cord. Brain Res. 484, 234-239.
Panula, P., Pirvola, U., Auvinen, S., and Airaksinen,M.S. (1989) Histamine-immunoreactive nerve fibers in the rat brain. Neuroscience 28, 585-610.
Parker, I., C. B. Gundersen, and R. Miledi. (1986) Actions of pentobarbital on rat brain receptors expressed in Xenopus oocytes. J.Neurosci. 6, 2290-97.

Chapter 11 References
182
Parmentier, R., Ohtsu, H., Djebbara-Hannas,Z., Valatx,J.L., Watanabe,T., and Lin,J.S. (2002) Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. J.Neurosci. 22, 7695-7711.
Peran, M., Hooper, H., Rayner,S.L., Stephenson,F.A., and Salas,R. (2004) GABAA receptor alpha1 and alpha6 subunits mediate cell surface anchoring in cultured cells. Neurosci.Lett. 364, 67-70.
Pirker, S., Schwarzer, C., Wieselthaler,A., Sieghart,W., and Sperk,G. (2000) GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101, 815-850.
Prell, G. D. (1995) Histamine metabolites in cerebrospinal fluid of patients with chronic schizophrenia: their relationships to levels of other aminergic transmitters and ratings of symptoms. Schizophr.Res. 14, 93-104.
Prince, D.A. (1978) Neurophysiology of epilepsy. Annu.Rev.Neurosci. 1, 395-415.
Prior, P., Schmitt, B., Grenningloh,G., Pribilla,I., Multhaup,G., Beyreuther,K., Maulet,Y., Werner,P., Langosch,D., Kirsch,J., and . (1992) Primary structure and alternative splice variants of gephyrin, a putative glycine receptor-tubulin linker protein. Neuron 8, 1161-1170.
Pritchett, D.B., Sontheimer, H., Gorman, C.M., Kettenmann,H., Seeburg,P.H., and Schofield,P.R. (1988) Transient expression shows ligand gating and allosteric potentiation of GABAA receptor subunits. Science 242, 1306-1308.
Pritchett, D.B., Sontheimer, H., Shivers,B.D., Ymer,S., Kettenmann,H., Schofield,P.R., and Seeburg,P.H. (1989) Importance of a novel GABAA receptor subunit for benzodiazepine pharmacology. Nature 338, 582-585.
Quinlan, J. J., L. L. Firestone, and G. E. Homanics. (2000) Mice lacking the long splice variant of the gamma 2 subunit of the GABA(A) receptor are more sensitive to benzodiazepines. Pharmacol.Biochem.Behav. 66, 371-74.
Ranganathan, R., Cannon,S.C., and Horvitz,H.R. (2000) MOD-1 is a serotonin-gated chloride channel that modulates locomotory behaviour in C. elegans. Nature 408, 470-475.
Rauscher,F.P., Nasrallah,H.A., and Wyatt,R.J. (1980) Cutaneous histamine response in schizophrenia. J.Clin.Psychiatry 41, 44-51.
Raymond,V., Mongan,N.P., and Sattelle,D.B. (2000) Anthelmintic actions on homomer-forming nicotinic acetylcholine receptor subunits: chicken alpha7 and ACR-16 from the nematode Caenorhabditis elegans. Neuroscience 101, 785-791.
Reite, O.B. (1972) Comparative physiology of histamine. Physiol Rev. 52, 778-819.

Chapter 11 References
183
Rick, C.E., Ye, Q., Finn,S.E., and Harrison,N.L. (1998) Neurosteroids act on the GABA(A) receptor at sites on the N-terminal side of the middle of TM2. Neuroreport 9, 379-383.
Roberts, E. and Frankel, S. (1950) gamma-Aminobutyric acid in brain: its formation from glutamic acid. J.Biol.Chem. 187, 55-63.
Rossi, D.J. and Hamann, M. (1998) Spillover-mediated transmission at inhibitory synapses promoted by high affinity alpha6 subunit GABA(A) receptors and glomerular geometry. Neuron 20, 783-795.
Rudolph, U. and Antkowiak,B. (2004) Molecular and neuronal substrates for general anaesthetics. Nat.Rev.Neurosci. 5, 709-720.
Rudolph,U.; Crestani,F.; Benke,D.; Brunig,I.; Benson,J.A.; Fritschy,J.M.; Martin,J.R.; Bluethmann,H.; Mohler,H. (1999) Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature 401, 796-800.
Ryu, J.H., Yanai, K., and Watanabe,T. (1994) Marked increase in histamine H3 receptors in the striatum and substantia nigra after 6-hydroxydopamine-induced denervation of dopaminergic neurons: an autoradiographic study. Neurosci.Lett. 178, 19-22.
Sakata, T., Ookuma, K., Fukagawa,K., Fujimoto,K., Yoshimatsu,H., Shiraishi,T., and Wada,H. (1988) Blockade of the histamine H1-receptor in the rat ventromedial hypothalamus and feeding elicitation. Brain Res. 441, 403-407.
Sambrook, J. and M. J. Gething. (1989) Protein structure. Chaperones, paperones. Nature 342, 224-25.
Sanna, E., Garau,F., and Harris,R.A. (1995) Novel properties of homomeric beta 1 gamma-aminobutyric acid type A receptors: actions of the anesthetics propofol and pentobarbital. Mol.Pharmacol. 47, 213-217.
Schneider, C., Risser, D., Kirchner,L., Kitzmuller,E., Cairns,N., Prast,H., Singewald,N., and Lubec,G. (1997) Similar deficits of central histaminergic system in patients with Down syndrome and Alzheimer disease. Neurosci.Lett. 222, 183-186.
Schofield, P.R., Darlison, M.G., Fujita,N., Burt,D.R., Stephenson,F.A., Rodriguez,H., Rhee,L.M., Ramachandran,J., Reale,V., Glencorse,T.A., (1987) Sequence and functional expression of the GABA A receptor shows a ligand-gated receptor super-family. Nature 328, 221-227.
Schwartz, J.C., Arrang, J.M., Garbarg,M., Pollard,H., and Ruat,M. (1991) Histaminergic transmission in the mammalian brain. Physiol Rev. 71, 1-51.

Chapter 11 References
184
Schwartz, J.C., Pollard, H., Bischoff,S., Rehault,M.C., and Verdiere-Sahuque,M. (1971) Catabolism of 3 H-histamine in the rat brain after intracisternal administration. Eur.J.Pharmacol. 16, 326-335.
Senba, E., Daddona, P.E., and Nagy,J.I. (1987) Adenosine deaminase-containing neurons in the olfactory system of the rat during development. Brain Res.Bull. 18, 635-648.
Serra, M. et al. (1995) Antagonism of isoniazid-induced convulsions by abecarnil in mice tolerant to diazepam. Pharmacol.Biochem.Behav. 52, 249-54.
Sieghart, W. and Sperk, G. (2002) Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr.Top.Med.Chem. 2, 795-816.
Siegwart, R., Jurd, R., and Rudolph, U. (2002) Molecular determinants for the action of general anesthetics at recombinant alpha(2)beta(3)gamma(2)gamma-aminobutyric acid(A) receptors. J.Neurochem. 80, 140-148.
Sigel, E. (1995) Functional modulation of ligand-gated GABAA and NMDA receptor channels by phosphorylation. J.Recept.Signal.Transduct.Res. 15, 325-332.
Sigel, E. (2002) Mapping of the benzodiazepine recognition site on GABA(A) receptors. Curr.Top.Med.Chem. 2, 833-839.
Sigel, E., Baur, R., Kellenberger,S., and Malherbe,P. (1992) Point mutations affecting antagonist affinity and agonist dependent gating of GABAA receptor channels. EMBO J. 11, 2017-2023.
Sigel, E., Baur, R., Malherbe,P., and Mohler,H. (1989) The rat beta 1-subunit of the GABAA receptor forms a picrotoxin-sensitive anion channel open in the absence of GABA. FEBS Lett. 257, 377-379.
Sigel,E., Baur,R., Trube,G., Mohler,H., and Malherbe,P. (1990) The effect of subunit composition of rat brain GABAA receptors on channel function. Neuron 5, 703-711.
Smith, C., McEwan, A.I., Jhaveri,R., Wilkinson,M., Goodman,D., Smith,L.R., Canada,A.T., and Glass,P.S. (1994) The interaction of fentanyl on the Cp50 of propofol for loss of consciousness and skin incision. Anesthesiology 81, 820-828.
Smith, G.B. and Olsen, R.W. (1994) Identification of a [3H]muscimol photoaffinity substrate in the bovine gamma-aminobutyric acidA receptor alpha subunit. J.Biol.Chem. 269, 20380-20387.
Soll, A.H. and Walsh,J.H. (1979) Regulation of gastric acid secretion. Annu.Rev.Physiol 41, 35-53.
Squire, M.D., Tornoe, C., Baylis,H.A., Fleming,J.T., Barnard,E.A., and Sattelle,D.B. (2000) Molecular cloning and functional co-expression of a Caenorhabditis elegans nicotinic acetylcholine receptor subunit (acr-2). Receptors.Channels 1995. 3, 107-115.

Chapter 11 References
185
Study, R.E. and Barker, J.L. (1981) Diazepam and pentobarbital: fluctuation analysis reveals different mechanisms for potentiation of gamma-aminobutyric acid responses in cultured central neurons. Proc.Natl.Acad.Sci.U.S.A 78, 7180-7184.
Suzdak, P.D., Schwartz, R.D., Skolnick,P., and Paul,S.M. (1986) Ethanol stimulates gamma-aminobutyric acid receptor-mediated chloride transport in rat brain synaptoneurosomes. Proc.Natl.Acad.Sci.U.S.A 83, 4071-4075.
Takeda, N., Inagaki, S., Shiosaka,S., Taguchi,Y., Oertel,W.H., Tohyama,M., Watanabe,T., and Wada,H. (1984) Immunohistochemical evidence for the coexistence of histidine decarboxylase-like and glutamate decarboxylase-like immunoreactivities in nerve cells of the magnocellular nucleus of the posterior hypothalamus of rats. Proc.Natl.Acad.Sci.U.S.A 81, 7647-7650.
Taleb, O., Trouslard,J., Demeneix,B.A., Feltz,P., Bossu,J.L., Dupont,J.L., and Feltz,A. (1987) Spontaneous and GABA-evoked chloride channels on pituitary intermediate lobe cells and their internal Ca requirements. Pflugers Arch. 409, 620-631.
Taylor, J. and Gordon-Weeks, P.R. (1991) Calcium-independent gamma-aminobutyric acid release from growth cones: role of gamma-aminobutyric acid transport. J.Neurochem. 56, 273-280.
Thompson, J.D., Higgins, D.G., and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673-4680.
Toyota,H., Dugovic,C., Koehl,M., Laposky,A.D., Weber,C., Ngo,K., Wu,Y., Lee,D.H., Yanai,K., Sakurai,E., Watanabe,T., Liu,C., Chen,J., Barbier,A.J., Turek,F.W., Fung-Leung,W.P., and Lovenberg,T.W. (2002) Behavioral characterization of mice lacking histamine H(3) receptors. Mol.Pharmacol. 62, 389-397.
Treinin, M., Gillo, B., Liebman,L., and Chalfie,M. (1998) Two functionally dependent acetylcholine subunits are encoded in a single Caenorhabditis elegans operon. Proc.Natl.Acad.Sci.U.S.A 95, 15492-15495.
Tretter, V., Ehya, N., Fuchs,K., and Sieghart,W. (1997) Stoichiometry and assembly of a recombinant GABAA receptor subtype. J.Neurosci. 17, 2728-2737.
Tung, A., Bergmann, B.M., Herrera,S., Cao,D., and Mendelson,W.B. (2004) Recovery from sleep deprivation occurs during propofol anesthesia. Anesthesiology 100, 1419-1426.
Tuomisto, L., Eriksson, L., and Fyhrquist,F. (1980) Vasopressin release by histamine in the conscious goat. Eur.J.Pharmacol. 63, 15-24.
Unwin, N. (1993) Nicotinic acetylcholine receptor at 9 A resolution. J.Mol.Biol. 229, 1101-1124.

Chapter 11 References
186
Unwin, N. (1995) Acetylcholine receptor channel imaged in the open state. Nature 373, 37-43.
Velazquez, J.L., Thompson, C.L., Barnes,E.M., Jr., and Angelides,K.J. (1989) Distribution and lateral mobility of GABA/benzodiazepine receptors on nerve cells. J.Neurosci. 9, 2163-2169.
Veselis, R.A., Reinsel, R.A., Feshchenko,V.A., and Dnistrian,A.M. (2002) A neuroanatomical construct for the amnesic effects of propofol. Anesthesiology 97, 329-337.
Vincent, S.R., Hokfelt, T., Skirboll, L.R., and Wu, J.Y. (1983) Hypothalamic gamma-aminobutyric acid neurons project to the neocortex. Science 220, 1309-1311.
Wachowiak, M., Cohen, L.B., and Ache, B.W. (2002) Presynaptic inhibition of olfactory receptor neurons in crustaceans. Microsc.Res.Tech. 58, 365-375.
Wafford, K.A., Thompson, S.A., Thomas,D., Sikela,J., Wilcox,A.S., and Whiting,P.J. (1996) Functional characterization of human gamma-aminobutyric acidA receptors containing the alpha 4 subunit. Mol.Pharmacol. 50, 670-678.
Walters, R.J., Hadley,S.H., Morris,K.D., and Amin,J. (2000) Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat.Neurosci. 3, 1274-1281.
Wan,Q., Xiong,Z.G., Man,H.Y., Ackerley,C.A., Braunton,J., Lu,W.Y., Becker,L.E., MacDonald,J.F., and Wang,Y.T. (1997) Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature 388, 686-690.
Wang, H., Bedford, F.K., Brandon, N.J., Moss,S.J., and Olsen,R.W. (1999) GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature 397, 69-72.
Wang, H. and Olsen, R.W. (2000) Binding of the GABA(A) receptor-associated protein (GABARAP) to microtubules and microfilaments suggests involvement of the cytoskeleton in GABARAPGABA(A) receptor interaction. J.Neurochem. 75, 644-655.
Wei, W., Zhang, N., Peng,Z., Houser,C.R., and Mody,I. (2003) Perisynaptic localization of delta subunit-containing GABA(A) receptors and their activation by GABA spillover in the mouse dentate gyrus. J.Neurosci. 23, 10650-10661.
Whiting, P.J. (2003) GABA-A receptor subtypes in the brain: a paradigm for CNS drug discovery? Drug Discov.Today 8, 445-450.
Whiting, P. J. (1997) Neuronally restricted RNA splicing regulates the expression of a novel GABAA receptor subunit conferring atypical functional properties. J.Neurosci. 17, 5027-37.

Chapter 11 References
187
Wilson, G.G. and Karlin, A. (1998) The location of the gate in the acetylcholine receptor channel. Neuron 20, 1269-1281.
Wisden, W., Laurie, D.J., Monyer,H., and Seeburg,P.H. (1992) The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J.Neurosci. 12, 1040-1062.
Witte, I., Kreienkamp,H.J., Gewecke,M., and Roeder,T. (2002) Putative histamine-gated chloride channel subunits of the insect visual system and thoracic ganglion. J.Neurochem. 83, 504-514.
Xu, M. and Akabas,M.H. (1996) Identification of channel-lining residues in the M2 membrane-spanning segment of the GABA(A) receptor alpha1 subunit. J.Gen.Physiol 107, 195-205.
Xue, H. (1998) Identification of major phylogenetic branches of inhibitory ligand-gated channel receptors. J.Mol.Evol. 47, 323-333.
Yamakura, T., Bertaccini,E., Trudell,J.R., and Harris,R.A. (2001) Anesthetics and ion channels: molecular models and sites of action. Annu.Rev.Pharmacol.Toxicol. 41, 23-51.
Yamashita, M., Fukui, H., Sugama,K., Horio,Y., Ito,S., Mizuguchi,H., and Wada,H. (1991) Expression cloning of a cDNA encoding the bovine histamine H1 receptor. Proc.Natl.Acad.Sci.U.S.A 88, 11515-11519.
Yarwood, S.J., Steele, M.R., Scotland,G., Houslay,M.D., and Bolger,G.B. (1999) The RACK1 signaling scaffold protein selectively interacts with the cAMP-specific phosphodiesterase PDE4D5 isoform. J.Biol.Chem. 274, 14909-14917.
Yassin, L., Gillo, B., Kahan, T., Halevi,S., Eshel,M., and Treinin,M. (2001) Characterization of the deg-3/des-2 receptor: a nicotinic acetylcholine receptor that mutates to cause neuronal degeneration. Mol.Cell Neurosci. 17, 589-599.
Yee, B.K. (2004) GABA receptors containing the alpha5 subunit mediate the trace effect in aversive and appetitive conditioning and extinction of conditioned fear. Eur.J.Neurosci. 20, 1928-36.
Yoshimatsu, H., Itateyama, E., Kondou,S., Tajima,D., Himeno,K., Hidaka,S., Kurokawa,M., and Sakata,T. (1999) Hypothalamic neuronal histamine as a target of leptin in feeding behavior. Diabetes 48, 2286-2291.
Zhang, Y., Proenca, R., Maffei,M., Barone,M., Leopold,L., and Friedman,J.M. (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425-432.
Zheng, Y., Hirschberg, B., Yuan,J., Wang,A.P., Hunt,D.C., Ludmerer,S.W., Schmatz,D.M., and Cully,D.F. (2002) Identification of two novel Drosophila

Chapter 11 References
188
melanogaster histamine-gated chloride channel subunits expressed in the eye. J.Biol.Chem. 277, 2000-2005.
Zorumski, C.F. and Isenberg, K.E. (1991) Insights into the structure and function of GABA-benzodiazepine receptors: ion channels and psychiatry. Am.J.Psychiatry 148, 162-173.

Appendix
189
Appendix I J. Biol. Chem., Vol. 280, Issue 16, 16254-16262, April 22, 2005
A Novel Chloride Channel in Drosophila melanogaster Is Inhibited by Protons*
Katrin Schnizler , Beate Saeger ¶, Carsten Pfeffer , Alexander Gerbaulet , Ulrich Ebbinghaus-Kintscher¶, Christoph Methfessel , Eva-Maria Franken¶, Klaus Raming¶, Christian H. Wetzel||, Arunesh Saras||, Hermann Pusch||, Hanns Hatt||, and Günter Gisselmann||** Summary
A systematic analysis of the Drosophila genome data reveals the existence of pHCl, a
novel member of ligand-gated ion channel subunits. pHCl shows nearly identical
similarity to glutamate-, glycine- and histamine-gated ion channels, does however not
belong to any of these ion channel types. We identified three different sites, where
splicing generates multiple transcripts of the pHCl mRNA. The pHCl is expressed in
Drosophila embryo, larvae, pupae and the adult fly. In embryos, in situ hybridization
detected pHCl in the neural cord and the hindgut. Functional expression of the three
different splice variants of pHCl in oocytes of Xenopus laevis and Sf9 cells induces a
chloride current with a linear current-voltage relationship that is inhibited by extracellular
protons and activated by ivermectin in a pH-dependent manner. Further, currents through
pHCl channels were induced by a raise in temperature. Our data give genetic and
electrophysiological evidence that pHCl is a member of a new branch of ligand-gated ion
channels in invertebrates with however a hitherto unique combination of pharmacological
and biophysical properties.

Appendix
190
Appendix II Identification of a novel branch of invertebrate ionotropic
acetylcholine receptors in C. elegans Günter Gisselmann, Christian H. Wetzel, Michael Kuczkowiak, Arunesh Saras, Hermann Pusch & Hanns Hatt Lehrstuhl für Zellphysiologie, Ruhr-Universität Bochum, Universitätsstraße 150, 44780 Bochum, Germany
Abstract The wealth of the complete C. elegans genome sequence has given us the possibility to
describe all members of the superfamily of ligand gated ion channels present in an
individual species. Homology analysis of candidate genes for ligand gated cation
channels revealed that the previously described gene ACR-22 defines a new subfamily
consisting of 14 putative gene coding for ligand-gated ion channel subunits with weak
sequence homology to nicotinic acetylcholine receptors. By an functional screening in
Xenopus oocytes we found that ACR-22 and R13A5.4, a close homologe to ACR-22,
were able to build functional homomultimeric ion channels activated by acetylcholine
(ACR-22A: EC50 = 15 µM, R13A5.4: EC50 = 95 µM). proteins. Whereas all general
characteristic sequence features of ligand gated ion channels were found in ACR-22 and
R13A5.4 there were some pronounced sequence differences to typical acetylcholine
gated cation channels. Especially the two adjacent cysteins were absent that are present in
nearly all ligand binding acetylcholine receptor α-subunits. Further, the pore forming
M2-region critical for the ion selectivity of the ion channel showed an untypical charge
distribution. Investigation of ACR-22 channels expressed in Xenopus oocytes reveals that
they were permeable for monovalent cations and calcium. At ACR-22, carbachole and
choline were agonists but nicotine failed to activate ACR-22. d-Tubocurarine, levamisole
and strychnine blocked acetycholine induced currents. The electrophysiological
properties and the sequence analysis suggest that ACR-22 and R13A5.4 define a novel
branch of acetylcholine receptors in C. elegans.

Acknowledgments
191
Acknowledgements This work would have not been conceivable without enthusiastic help and support of many people. Prof. Dr. Hans Hatt, Dr. Günter Gisselmann, Dr. Hermann Pusch, and Mary Grace Lucero, participated directly in the experiments. I would like to give my sincere thanks to Prof. Dr. Hanns Hatt for a very kind, motivating and guidance during my whole study, for support, valuable advice and comments. Words are less to express my gratitude to Dr. Gisselmann for helping me in every aspect of my doctoral work. Especially during our project meetings, his criticisms, the appreciation, and the noble approach given by him are highly appreciated. I show my gratitude to Dr. Pusch for teaching me the basic electrophysiology and giving me always friendly and warmth encouragement. Thomas Lichleitner for help and advice on computer programming and for providing software and H. Bartel and W. Garbowski for technical assistance during the experiments. I ought to give my special thanks to the post docs in our lab specially Christian Wetzel for giving me in depth knowledge for patch clamp techniques; Angela for our collaborative work; Ulrike Thomas for an exceptionally kind parental care and always giving me support and help. On this moment my heartiest thanks to my coach Slavko for help, support and a congenial atmosphere. I would like to mention specially the name of Mary Grace Lucero for her dedication and endless patience during the proof reading of my thesis and for making this thesis to finish on time. I show all my respect, gratitude to my parents, brother and sisters for their benediction, inspiration, for letting me to finish my doctoral study. I want to thank my friends namely, Nils, Jennet, Christian, who made my stay in Germany as one of the memorable days of my life.

1
Curriculum Vitae Name Arunesh Saras Title PhD Date of Birth 20.10.1978 Place of Birth New Delhi, India Nationality Indian Education 1998-2000 BSc (H), Zoology, Kirori Mal College, (New Delhi, India)
2000- 2002 MSc (H), Biotechnology Madurai Kamraj University
(Madurai, India)
2002-2003 Junior Research fellow (JRF) All India Institute of Medical Science (AIIMS) (New Delhi, India).
2003 onwards Graduate Student at the International Graduate School of Neuroscience Ruhr University Bochum, (Bochum, Germany). Scholarships
1991 5th standard : Merit Scholarship given by Govt of India 1992-1997 6th-12th standard : Merit Scholarship given by Govt of India 1998-2000 BSc (1st-3rd year) Merit Scholarship given by Govt of India 2001-2002 MSc (1st-2nd year) Merit Scholarship given by Department of
Biotechnology, India.
2002-2003 Council of Scientific & Industrial Reasearch (CSIR), Joint CSIR-UGC Junior Research Fellowship (JRF) , New Delhi, India.

2
2003 onwards International Graduate School of Neuroscience (IGSN) International Graduate School of Neuroscience Scholarship.
Academic Awards
1983 Gold Medal given by the former President of India, Mr. Gyani Zail Singh 1989 Delhi (Zone Level) Science Fair
First Prize for the Model : Different Surfaces have different friction The model was selected for the State Level Science Fair
Model was telecasted on Indian National Television channel, Delhi Doordarshan,
Programme name – New Scientist
1994 Science Open Merit Test
First prize (among particitpating candiates from school), Organised by Delhi State Science Teacher´s Forum, New Delhi, India.
1994 Lion Pratibha ( General Knowledge Contest) Merit Certificate
1995 Lion Pratibha ( General Knowledge Contest) Merit Certificate
1998 India Quiz Competition First Prize in All India Radio (AIR) Quiz Competition, the program was telecasted on international channel of AIR
1999 All India Biomedical Entrace Examination All India Second Rank Conducted by the Delhi University
2000 Summer Training Award The Second the best Summer Trainee from the MKU since 1990 under the supervision of Vice - Chancellor of Delhi Prof. Deepak Pental
Research Training May-June 2000 Transformation and Regenaration in Arabibopsis by Agrobacterium and Cloning work Mentor : Prof. Deepak Pental Vice Chancellor Delhi University, Delhi

3
January-July 2001 Identification of Superior Biotypes of Casuarina equesatiafolia using RAPD analysis Mentor: Prof. A.K. Gutta Center of Plant Molecular Biology, School of Biotechnoloy, MKU July 2001–June 2002 Effect of Prenatal sound over stimulation on synapses of chick brain stem auditory nuclei: A Transmission Electron Microscopic study Mentor: Prof Shashi Wabhwa Neurobiology Lab Department of Anatomy All India Institute of medical Sciences New Delhi, India October 2002 onwards Functional Expresion and Characterization of histamine - gated chloride channels Mentor: Prof. Dr Hans Hatt Ruhr Univertiy Bochum, International Graduate School of Neuroscience,Bochum, Germany Conferences\ Workshops attended 1. National Symposium on Relevance of Plant Biochemistry and Biotechnology –
Modern trends. Paper presentation.
The American College, Madurai, India. March 1 – 3, 2001
2. Summer cource in Bioinformatics Nijemegen, Netherland, August 18 – 29, 2003 3. SFB – symposium Neuro-vision, Ruhr University Bochum, Germany. March 21 – 23, 2004 4. GRK workshop Borkum `Disease of the Brain` Borkum, Germany October 2 – 5, 2004 5. American Society for Cell Biology Meeting, Poster presentation, Washington DC, USA December 4 – 8, 2004

4
6. Berlin Brain Day Berlin, Germany November 18 – 23, 2004 7. 6th NRW Neuroscience Conference
Gottingen, Germany Febuary 17-20, 2005
Extracurricular Activities 1. Hindi Academy – Poem Competition
Consulation Prize for the poem. 1992-93, New Delhi, India
2. Inter – College Health Awareness Competition
Cartoon Making Competition, Ist prize, New Delhi, India. October 2 – 6, 1996
3. Inter – College Health Awareness Competition Extrempore speech Competition, III rd prize, New Delhi, India. October 2 – 6, 1996
4. Perfect Health Mela Special certificate from Ministry of Health and Welfare, Govt. of India and Ministries of Transport, Environment and and Prohibition, Govt. of Delhi October 2 – 6 , 1996 New Delhi, India
5. Lions Club International VI Dr. N.S. Pradhan Memeorial Debate, 3rd Prize in Inter – University Hindi Debate, January 28th , 1998
6. Inter College Chess Competition
Certificate of Merit 3rd Place University of Delhi, 1998-99

5
Publication
1. A Novel Chloride Channel in Drosophila melanogaster Is Inhibited by Protons*
Katrin Schnizler, Beate Saeger, Carsten Pfeffer, Alexander Gerbaulet, Ulrich Ebbinghaus-Kintscher, Christoph Methfessel, Eva-Maria Franken, Klaus Raming,
Christian H. Wetzel, Arunesh Saras, Hermann Pusch, Hanns Hatt, and Günter Gisselmann.
J. Biol. Chem., Vol. 280, Issue 16, 16254-16262, April 22, 2005
Top Related