
WNT/β-Catenin Signaling Regulates Multiple Steps of Myogenesis ...
14
WNT/-Catenin Signaling Regulates Multiple Steps of Myogenesis by Regulating Step-Specific Targets Akiko Suzuki, a,b Richard C. Pelikan, b Junichi Iwata a,b,c Department of Diagnostic and Biomedical Sciences a and Center for Craniofacial Research, b The University of Texas Health Science Center at Houston School of Dentistry, Houston, Texas, USA; The University of Texas Graduate School of Biomedical Sciences at Houston, Houston, Texas, USA c Molecules involved in WNT/-catenin signaling show specific spatiotemporal expression and play vital roles in myogenesis; however, it is still largely unknown how WNT/-catenin signaling regulates each step of myogenesis. Here, we show that WNT/ -catenin signaling can control diverse biological processes of myogenesis by regulating step-specific molecules. In order to identify the temporally specific roles of WNT/-catenin signaling molecules in muscle development and homeostasis, we used in vitro culture systems for both primary mouse myoblasts and C2C12 cells, which can differentiate into myofibers. We found that a blockade of WNT/-catenin signaling in the proliferating cells decreases proliferation activity, but does not induce cell death, through the regulation of genes cyclin A2 (Ccna2) and cell division cycle 25C (Cdc25c). During muscle differentiation, the inhibi- tion of WNT/-catenin signaling blocks myoblast fusion through the inhibition of the Fermitin family homolog 2 (Fermt2) gene. Blocking WNT/-catenin signaling in the well-differentiated myofibers results in the failure of maintenance of their structure by disruption of cadherin/-catenin/actin complex formation, which plays a crucial role in connecting a myofiber’s cytoskeleton to the surrounding extracellular matrix. Thus, our results indicate that WNT/-catenin signaling can regulate multiple steps of myogenesis, including cell proliferation, myoblast fusion, and homeostasis, by targeting step-specific molecules. S keletal muscle plays highly specialized roles in the generation of force and is capable of extensive metabolic and functional plasticity. Skeletal muscle also exhibits robust regenerative capac- ity, and its formation is composed of complex and highly regu- lated processes among embryonic development and regeneration in mature skeletal musculature (1). Muscle precursor cells (also known as satellite cells in the adult) lie under, or are embedded in, the basal lamina of the myofiber in skeletal muscles and are the major source of regeneration and growth of skeletal muscles (2). During development and regeneration in response to injury or recovery from atrophy, muscle precursor cells start to proliferate, at which stage they are referred to as myoblasts, and subsequently differentiate to form new myofibers or fuse with existing myofi- bers (2). These processes require fine-tuned regulations of prolif- eration and differentiation that are likely regulated by signal trans- duction pathways. The WNT family (wingless-type mouse mammary tumor virus [MMTV] integration site family) of signaling molecules consists of 21 secreted glycoprotein ligands. WNT ligands are essential to activate downstream pathways (canonical -catenin-dependent and/or noncanonical -catenin-independent pathways) in phys- iological and pathological conditions. WNT/-catenin signaling can regulate a variety of genes in a specific spatiotemporal manner (3). In the absence of WNT ligands, -catenin is incorporated into a protein complex containing axin, adenomatous polyposis coli (APC), and the serine-threonine kinase glycogen synthase kinase 3 (GSK3), which is here termed the “destruction complex.” The destruction complex phosphorylates -catenin and leads it to a degradation process by the ubiquitin-proteasome (4). In the pres- ence of WNT ligands, WNT ligands bind to a Frizzled receptor (FZD) and the low-density lipoprotein receptor-related protein 5/6 (LRP5/6), followed by the recruitment of axin to LRP5/6 (5). Subsequently, the destruction complex is inactivated, and -catenin can escape from its incorporation into the destruction complex, resulting in its stabilization and translocation into the nucleus (4). Increased nuclear concentrations of -catenin result in induction of target genes by interacting with transcriptional coactivators such as members of the T-cell factor/lymphocyte- enhancement factor 1 (TCF/LEF-1) family (6). In addition, cyto- plasmic accumulated -catenin is involved in cell-cell interactions in combination with cadherin and actin (7). WNT signaling is essential for a variety of developmental and regenerative processes, including embryonic muscle development and maintenance of skeletal muscle homeostasis in the adult (8– 10). For example, WNT ligands regulate the specification of skel- etal myoblasts in the paraxial mesoderm and induce location- specific expression of muscle regulatory factors (11). Treatment with secreted Frizzled-related protein 3 (sFRP3), a soluble WNT antagonist, reduces skeletal myogenesis in a dose-dependent fash- ion (12). Indeed, mice with a deficiency of WNT/-catenin sig- naling (Ctnnb1 / mice) are embryonic lethal by E8.5 with in- creased cell death (13). Mice with conditional depletion of -catenin in the muscle precursor Pax7 cell lineage (Ctnnb1 F/F ; Pax7-Cre mice) show reduced muscle mass and slow myofibers (14). Mice with expression of a constitutively active stabilized form of -catenin in the Pax7 lineage (Ctnnb ex3 ; Pax7-Cre mice) exhibit reduced myogenesis but increased slow myofibers (14, 15). Thus, dysregulation of WNT/-catenin signaling can Received 20 September 2014 Returned for modification 19 October 2014 Accepted 27 February 2015 Accepted manuscript posted online 9 March 2015 Citation Suzuki A, Pelikan RC, Iwata J. 2015. WNT/-catenin signaling regulates multiple steps of myogenesis by regulating step-specific targets. Mol Cell Biol 35:1763–1776. doi:10.1128/MCB.01180-14. Address correspondence to Junichi Iwata, [email protected]. Copyright © 2015, American Society for Microbiology. All Rights Reserved. doi:10.1128/MCB.01180-14 May 2015 Volume 35 Number 10 mcb.asm.org 1763 Molecular and Cellular Biology on February 19, 2018 by guest http://mcb.asm.org/ Downloaded from
Transcript of WNT/β-Catenin Signaling Regulates Multiple Steps of Myogenesis ...

WNT/�-Catenin Signaling Regulates Multiple Steps of Myogenesis byRegulating Step-Specific Targets
Akiko Suzuki,a,b Richard C. Pelikan,b Junichi Iwataa,b,c
Department of Diagnostic and Biomedical Sciencesa and Center for Craniofacial Research,b The University of Texas Health Science Center at Houston School of Dentistry,Houston, Texas, USA; The University of Texas Graduate School of Biomedical Sciences at Houston, Houston, Texas, USAc
Molecules involved in WNT/�-catenin signaling show specific spatiotemporal expression and play vital roles in myogenesis;however, it is still largely unknown how WNT/�-catenin signaling regulates each step of myogenesis. Here, we show that WNT/�-catenin signaling can control diverse biological processes of myogenesis by regulating step-specific molecules. In order toidentify the temporally specific roles of WNT/�-catenin signaling molecules in muscle development and homeostasis, we used invitro culture systems for both primary mouse myoblasts and C2C12 cells, which can differentiate into myofibers. We found thata blockade of WNT/�-catenin signaling in the proliferating cells decreases proliferation activity, but does not induce cell death,through the regulation of genes cyclin A2 (Ccna2) and cell division cycle 25C (Cdc25c). During muscle differentiation, the inhibi-tion of WNT/�-catenin signaling blocks myoblast fusion through the inhibition of the Fermitin family homolog 2 (Fermt2) gene.Blocking WNT/�-catenin signaling in the well-differentiated myofibers results in the failure of maintenance of their structure bydisruption of cadherin/�-catenin/actin complex formation, which plays a crucial role in connecting a myofiber’s cytoskeleton tothe surrounding extracellular matrix. Thus, our results indicate that WNT/�-catenin signaling can regulate multiple steps ofmyogenesis, including cell proliferation, myoblast fusion, and homeostasis, by targeting step-specific molecules.
Skeletal muscle plays highly specialized roles in the generationof force and is capable of extensive metabolic and functional
plasticity. Skeletal muscle also exhibits robust regenerative capac-ity, and its formation is composed of complex and highly regu-lated processes among embryonic development and regenerationin mature skeletal musculature (1). Muscle precursor cells (alsoknown as satellite cells in the adult) lie under, or are embedded in,the basal lamina of the myofiber in skeletal muscles and are themajor source of regeneration and growth of skeletal muscles (2).During development and regeneration in response to injury orrecovery from atrophy, muscle precursor cells start to proliferate,at which stage they are referred to as myoblasts, and subsequentlydifferentiate to form new myofibers or fuse with existing myofi-bers (2). These processes require fine-tuned regulations of prolif-eration and differentiation that are likely regulated by signal trans-duction pathways.
The WNT family (wingless-type mouse mammary tumor virus[MMTV] integration site family) of signaling molecules consistsof 21 secreted glycoprotein ligands. WNT ligands are essential toactivate downstream pathways (canonical �-catenin-dependentand/or noncanonical �-catenin-independent pathways) in phys-iological and pathological conditions. WNT/�-catenin signalingcan regulate a variety of genes in a specific spatiotemporal manner(3). In the absence of WNT ligands, �-catenin is incorporated intoa protein complex containing axin, adenomatous polyposis coli(APC), and the serine-threonine kinase glycogen synthase kinase3 (GSK3�), which is here termed the “destruction complex.” Thedestruction complex phosphorylates �-catenin and leads it to adegradation process by the ubiquitin-proteasome (4). In the pres-ence of WNT ligands, WNT ligands bind to a Frizzled receptor(FZD) and the low-density lipoprotein receptor-related protein5/6 (LRP5/6), followed by the recruitment of axin to LRP5/6 (5).Subsequently, the destruction complex is inactivated, and�-catenin can escape from its incorporation into the destructioncomplex, resulting in its stabilization and translocation into the
nucleus (4). Increased nuclear concentrations of �-catenin resultin induction of target genes by interacting with transcriptionalcoactivators such as members of the T-cell factor/lymphocyte-enhancement factor 1 (TCF/LEF-1) family (6). In addition, cyto-plasmic accumulated �-catenin is involved in cell-cell interactionsin combination with cadherin and actin (7).
WNT signaling is essential for a variety of developmental andregenerative processes, including embryonic muscle developmentand maintenance of skeletal muscle homeostasis in the adult (8–10). For example, WNT ligands regulate the specification of skel-etal myoblasts in the paraxial mesoderm and induce location-specific expression of muscle regulatory factors (11). Treatmentwith secreted Frizzled-related protein 3 (sFRP3), a soluble WNTantagonist, reduces skeletal myogenesis in a dose-dependent fash-ion (12). Indeed, mice with a deficiency of WNT/�-catenin sig-naling (Ctnnb1�/� mice) are embryonic lethal by E8.5 with in-creased cell death (13). Mice with conditional depletion of�-catenin in the muscle precursor Pax7� cell lineage (Ctnnb1F/F;Pax7-Cre mice) show reduced muscle mass and slow myofibers(14). Mice with expression of a constitutively active stabilizedform of �-catenin in the Pax7� lineage (Ctnnb�ex3; Pax7-Cremice) exhibit reduced myogenesis but increased slow myofibers(14, 15). Thus, dysregulation of WNT/�-catenin signaling can
Received 20 September 2014 Returned for modification 19 October 2014Accepted 27 February 2015
Accepted manuscript posted online 9 March 2015
Citation Suzuki A, Pelikan RC, Iwata J. 2015. WNT/�-catenin signaling regulatesmultiple steps of myogenesis by regulating step-specific targets. Mol Cell Biol35:1763–1776. doi:10.1128/MCB.01180-14.
Address correspondence to Junichi Iwata, [email protected].
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
doi:10.1128/MCB.01180-14
May 2015 Volume 35 Number 10 mcb.asm.org 1763Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

lead to severe developmental defects and perturbation of musclehomeostasis. Nevertheless, the temporally specific roles of WNT/�-catenin signaling during myogenesis remain unknown.
The multiple steps of muscle development and regeneration,beginning with muscle progenitor cell activation and ending withmyofiber formation, are all subject to separate levels of regulationand are affected by a variety of muscle disorders and atrophy (2,14). In the present study, we investigated the role of WNT/�-catenin signaling in muscle biology, including cell proliferation,differentiation, and homeostasis of muscle cells. We used bothprimary myoblasts and C2C12 cells (a myoblast cell line) that havethe ability to differentiate into myofibers in the culture with dif-ferentiation inducers. This process provides an opportunity toinvestigate the temporally specific role of WNT/�-catenin signal-ing during myogenesis. We found that WNT/�-catenin signalingcan regulate multiple steps of muscle development, ranging fromcell proliferation to homeostasis, through the regulation of step-specific targets. Knowledge of the temporally specific regulatorymechanism may be applied to therapeutic approaches to stimulateeffective skeletal muscle regeneration following muscle trauma oratrophy.
MATERIALS AND METHODSCell culture. C2C12 cells, a murine skeletal muscle cell line, were obtainedfrom the American Type Culture Collection (CRL-1772). Primary myo-blasts were isolated from the limb and tongue of C57B6/J mice, as de-scribed previously (16). Briefly, for preparation of primary myoblasts,hind limb muscle and tongue were dissected from embryonic day 15.5(E15.5) C57B6/J mouse embryos and digested by a 1.8-U/ml dispase so-lution (Gibco) for 1 h at 37°C and 5% CO2. Digested tissues were thensuspended with growth medium (Dulbecco modified Eagle medium[DMEM] supplemented with 10% fetal bovine serum, penicillin, strepto-mycin, 2 mM L-glutamate, 1 mM sodium pyruvate, and nonessentialamino acids), and cells were collected by centrifugation. Resuspendedcells in growth medium were placed into a cell culture dish coated withhuman fibronectin (BD Biocoat; BD Falcon) and cultured at 37°C and 5%CO2 in a humidified incubator. Cell proliferation assays were performedusing a cell counting kit (Dojindo Molecular Technologies). Cells weretreated with IWR1-endo (Tocris Bioscience, Bristol, United Kingdom) atthe indicated concentration (0 to 80 �M) for 24 to 48 h. Bromodeoxyu-ridine (BrdU) incorporation assays (BrdU incorporation for the last 1 h)were performed using cells treated with vehicle or 80 �M IWR1-endo for36 h. Incorporated BrdU was stained with a rat polyclonal antibodyagainst BrdU (Abcam), as described previously (17, 18). Myogenic differ-entiation was induced in muscle differentiation medium (DMEM supple-mented with 2% horse serum, 2 mM L-glutamate, penicillin, streptomy-cin, and insulin [100 ng/ml]) for the indicated number of days. Toexamine the effect of IWR1-endo on myogenic differentiation, cells weretreated with IWR1-endo for 3 to 5 days at the indicated concentrations (0to 10 �M). To investigate the effect of IWR1-endo on maintenance ofmyofibers, well-differentiated C2C12 cells were cultured with vehicle or 1�M IWR1-endo in differentiation medium for another 3 to 5 days. Thesmall interfering RNA (siRNA) knockdown for Ctnnb1 (Invitrogen),Ccna2, Cdc25c, and Fermt2 (Sigma-Aldrich) was performed as describedpreviously (17, 19). The overexpression of Fermt2 (OriGene Technolo-gies, Inc., Rockville, MD) was also performed as described previously(17).
Tongue organ culture. Timed-pregnant C57B6/J mice were sacrificedat E14.5. Each tongue was microdissected and cultured in serum-freechemically defined BGJb medium supplemented with penicillin, strepto-mycin, and 100 �g of ascorbic acid/ml (16, 17). Tongue explants weretreated with vehicle or 80 �M IWR1-endo for 24 h for the BrdU incorpo-ration assay or for 72 h for the differentiation assay and then harvested,
fixed in 4% paraformaldehyde– 0.1 M phosphate buffer (pH 7.4), andprocessed to analyze histology. BrdU staining was performed as describedpreviously (17). Immunohistological staining for myosin heavy chain(MYH) was also performed as described previously (16, 20).
Evaluation of myoblast fusion. After a 5-day culture in differentiationmedium with vehicle or IWR1-endo (1 �M), C2C12 cells were stained forMYH and counterstained with DAPI (4=,6=-diamidino-2-phenylindole)for nuclei. The extent of fusion was calculated by using the fusion index(21, 22) as follows: % fusion � (the number of nuclei in multiple-nucleus myotubes/total number of nuclei in MYH-positive cells andmyotubes) � 100.
Immunoprecipitation. Extracts in cell lysis buffer containing 10 mMTris (pH 7.5), 100 mM NaCl, 1% Triton X-100, and Complete proteaseinhibitor (Roche Diagnostics) were lysed. Immunoprecipitation assayswere performed as described previously (17). A mouse monoclonal anti-body against pan-cadherin (Abcam) and a rabbit polyclonal antibodyagainst actin (Abcam) were used for immunoprecipitation assays.
Immunoblotting. Immunoblots were performed, as described previ-ously (18), using rabbit polyclonal antibodies against cleaved caspase 3,active �-catenin, and phosphorylated �-catenin (Cell Signaling Technol-ogy), actin (Abcam), mouse monoclonal antibodies against pan-cadherin(Abcam), and GAPDH (glyceraldehyde-3-phosphate dehydrogenase;Millipore).
Immunofluorescence analysis. Immunofluorescence analysis wasperformed, as described previously (20), using the following antibodies:rabbit polyclonal antibody against active �-catenin (Cell Signaling Tech-nology) and mouse monoclonal antibodies against MYH (Sigma-Aldrich)and pan-cadherin (Abcam). DyLight 554-conjugated phalloidin was usedfor staining F-actin (Cell Signaling Technology). Fluorescent images weretaken by an inverted fluorescence microscope (IX71; Olympus). Confocalimages were taken by a laser confocal scanning microscope (IX81 Flu-oveiw FV1000; Olympus).
Quantitative reverse transcription-PCR (RT-PCR). For cell prolifer-ation assays, total RNAs isolated from C2C12 cells treated with 80 �MIWR1-endo or control vehicle for 36 h (n � 6 per group) were dissectedwith a QIAshredder and RNeasy mini-extraction kit (Qiagen), as de-scribed previously (19, 23). Gene expression profiling for cell cycle regu-lators was performed by PCR arrays (PrimePCR assay; Bio-Rad Labora-tories). The PCR primers were purchased from Bio-Rad. For myoblastfusion assays, total RNAs isolated from C2C12 cells cultured in differen-tiation medium with 1 �M IWR1-endo or control vehicle for 12 h to 5days (n � 6 per group) were dissected with the QIAshredder and RNeasymini-extraction kit (Qiagen), as described previously (16). For timecourse experiments, C2C12 cells were treated with 0.1 �g of WNT3A/mlfor the indicated times with 80 �M IWR1-endo or control vehicle for cellproliferation and with 1 �M IWR1-endo or control vehicle for differen-tiation. The following PCR primers were used for further specific analysis:Ccna2, 5=-TTGCTCCTCTTAAGGACCTTCC-3= and 5=-CATTTAACCTCCATTTCCCTAAGGT-3=; Cdc25c, 5=-TGACGTCTATAGCCCCACC-3= and 5=-TAAGCGGAGAGGCAGACATC-3=; Fermt2, 5=-GATCACTTTGGAAGGCGGGA-3= and 5=-GCGCGTACTGCTTCTCGTTA-3=;and Gapdh, 5=-AACTTTGGCATTTGGAAGG-3= and 5=-ACACATTGGGGGTAGGAACA-3=. The PCR primers for Ctnnb1 were purchased fromSanta Cruz Biotechnology, Inc.
Reporter assay. A Cignal TCF/LEF reporter construct (Qiagen) wastransfected in C2C12 cells using Lipofectamine 3000 (Invitrogen).C12C12 cells were cultured in growth medium or differentiation mediumwith or without 0.1 �g of WNT3A (R&D Systems)/ml and IWR1-endo(80 �M for cell proliferation and 1 �M for differentiation) for 24 h ac-cording to the manufacturer’s protocol. Luciferase assays were performedby the Dual-Glo luciferase assay system (Promega).
Comparative analysis of transcription factor binding site. TheUCSC genome browser was used to obtain the genomic sequences of thefollowing murine genes, including 5 kbp upstream of the respective tran-scription start site: Ccna2 (NM_009828.2), Ccnb1 (NM_172301.3),
Suzuki et al.
1764 mcb.asm.org May 2015 Volume 35 Number 10Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

Cdc25c (NM_009860.2), Ccnd2 (NM_009829.3), Cdk1 (NM_007659.3),Cdk4 (NM_009870.3), Cdkn1a (NM_007669.4), Dmd (NM_007868.5),E2f5 (NM_007892.2), and Fermt2 (NM_146054.2). These sequences werethen mapped to seven additional mammalian genomes (human [build38], chimpanzee [build 2.1.4], orangutan [build 2.0.2], rhesus macaque[build 1.0], rat [build 5], dog [build 3.1], and horse [build equCab2])through the BLAST tool, as described previously (16, 19). Multiple align-ments for these sequences were obtained using the Clustal Omega toolwith default parameters and settings (24). Binding motifs of LEF1 (min-imal core sites: 5=-CTTTG-3=or 5=-CAAAG-3=; optimal sites: 5=-CTTT-GWW-3= or 5=-WWCAAAG-3=, W � A/T) (25–27) were searched in thealigned DNA sequences.
Chromatin immunoprecipitation (ChIP) assay. Cell extracts wereincubated with �-catenin antibody (Abcam) overnight at 4°C, followed byprecipitation with magnetic beads. Washing and elution of the immunecomplexes, as well as precipitation of DNA, were performed according tostandard procedures, as described previously (16, 19). The putative LEF1target sites of the genes in the immune complexes were detected by PCRusing the following primers: Ccna2 gene, 5=-TGGTGTTGCAGATCTACCGT-3= and 5=-TCTGCTAACAAAATGGCAATGC-3= (�2364 to�2154); Cdc25c gene, 5=-TTCATCGGTCTCAGCTTCCC-3= and 5=-AGTCACCACTGAGCCTTGTC-3= (�3164 to �3029); and Fermt2 gene, 5=-TAGAGGTTCTAGCGGGGGTT-3= and 5=-TTCAGGCCTTGGCTTTGAGT-3= (�3138 to �2949). The positions of PCR fragments correspondto NCBI mouse genome build 38 (mm10).
Statistical analysis. Two-tailed Student t tests were applied for sta-tistical analysis. A P value of �0.05 was considered statistically signif-icant. For all graphs, data are represented as means the standarddeviations (SD).
RESULTSWNT/�-catenin signaling regulates cell proliferation activitythrough Ccna2 and Cdc25c expression. We have recently re-ported that upregulation of Dkk1/4, WNT/�-catenin antagonists,inhibits muscle development (20), suggesting that WNT/�-catenin signaling plays a role in muscle development. To explorehow WNT/�-catenin signaling regulates myogenesis, we used acell culture system of C2C12 cells, which are an immortalizedmouse myoblast cell line originally established from normal adultC3H mouse hind limb muscle. C2C12 cells express WNT ligandsduring cell proliferation and differentiation (28). We performedcell proliferation assays using C2C12 cells treated with or withoutWNT/�-catenin inhibitor IWR1-endo (0 to 80 �M) that can pro-mote axin stabilization and thereby inhibit WNT signaling (29).Cell proliferation activity was decreased by IWR1-endo treatmentin the dose-dependent manner (Fig. 1A and B). To validate cellproliferation defects after treatment with IWR1-endo (80 �M) for36 h, we performed a BrdU incorporation assay. Cells incorpo-rated BrdU were reduced by the treatment with IWR1-endo (Fig.1C and D). We further confirmed that IWR1-endo had no toxicityon C2C12 cells at the concentrations we tested. We detected noapoptosis with treatment of 80 �M IWR1-endo (Fig. 1E). IWR1-endo specifically inhibits WNT/�-catenin signaling by inductionof the �-catenin destruction complex formation (29). As ex-pected, nonphosphorylated (active) �-catenin was decreased, andphosphorylated (inactive) �-catenin was increased at 12 and 24 hafter the treatment with 80 �M IWR1-endo (Fig. 1F). To testwhether IWR1-endo specifically blocks WNT/�-catenin signalingcascade through transcriptional response elements of LEF/TCF,which is a well-known �-catenin cotranscription factor, we per-formed LEF/TCF luciferase reporter assays after treatment withWNT3A (0.1 �g/ml) and IWR1-endo (80 �M) for 24 h using
proliferating cells. WNT3A induced LEF/TCF-mediated pro-moter activity approximately six times, and IWR1-endo (80 �M)completely inhibited the induction of LEF/TCF-mediated pro-moter activity (Fig. 1G). These results indicate that IWR1-endoinhibits canonical WNT/�-catenin signaling.
Next, to identify downstream targets of WNT/�-catenin sig-naling, we performed PCR array analyses of C2C12 cells treatedwith or without 80 �M IWR1-endo for 36 h (n � 5 per group). Inthis analysis, we uncovered seven genes that were differentiallyexpressed (�1.5-fold, P 0.05), two genes that were more abun-dant in the treated group, and five genes that were more abundantin the control group (Table 1). The genes altered in the treatedgroup were consistent with cell proliferation defects, with signifi-cant reductions in the expression levels of transcripts related tocell cycle transition. We analyzed the promoter sequence (includ-ing 5 kbp upstream of the transcription start site) of the candidatetarget genes. We found that both Ccna2 and Cdc25c genomic re-gions have a phylogenetically conserved LEF1 recognition se-quence (Tables 2 and 3). To test whether �-catenin could bind tothe promoter regions of Ccna2 and Cdc25c, we performed chro-matin immunoprecipitation (ChIP) assays. The binding site forLEF1 in each gene was immunoprecipitated with �-catenin anti-body but not with control IgG. Furthermore, the immunoprecipi-tated amounts were decreased after the treatment with 80 �MIWR1-endo for 24 h in both Ccna2 and Cdc25c, indicating that thebinding site for LEF1 in each Ccna2 and Cdc25c gene is functionaland responsive to WNT/�-catenin signaling activity (Fig. 1H andI). Importantly, gene expression of Ccna2 and Cdc25c was inducedby WNT3A ligands (0.1 �g/ml) in a time-dependent manner (Fig.1J and K). In addition, we analyzed cell proliferation activity aftertreatment with siRNA knockdown for Ccna2, Cdc25c, and Ccna2/Cdc25c combination. Gene expression of Ccna2 and Cdc25c wassignificantly reduced after the siRNA knockdown by 80 and 60%reduction, respectively (Fig. 1L). Cell proliferation activity wassignificantly reduced after the knockdown of Ccna2, Cdc25c, orCcna2/Cdc25c combination (Fig. 1M). Furthermore, we analyzedgene expression of Ccna2 and Cdc25c after siRNA knockdown forCtnnb1 (Fig. 1N and O). Cell proliferation activity was signifi-cantly reduced after the knockdown for Ctnnb1 (Fig. 1P and Q).Taken together, our results indicate that gene expression of Ccna2and Cdc25c is regulated by the WNT/�-catenin pathway, andCCNA2 and CDC25C play a crucial role during myoblast prolif-eration.
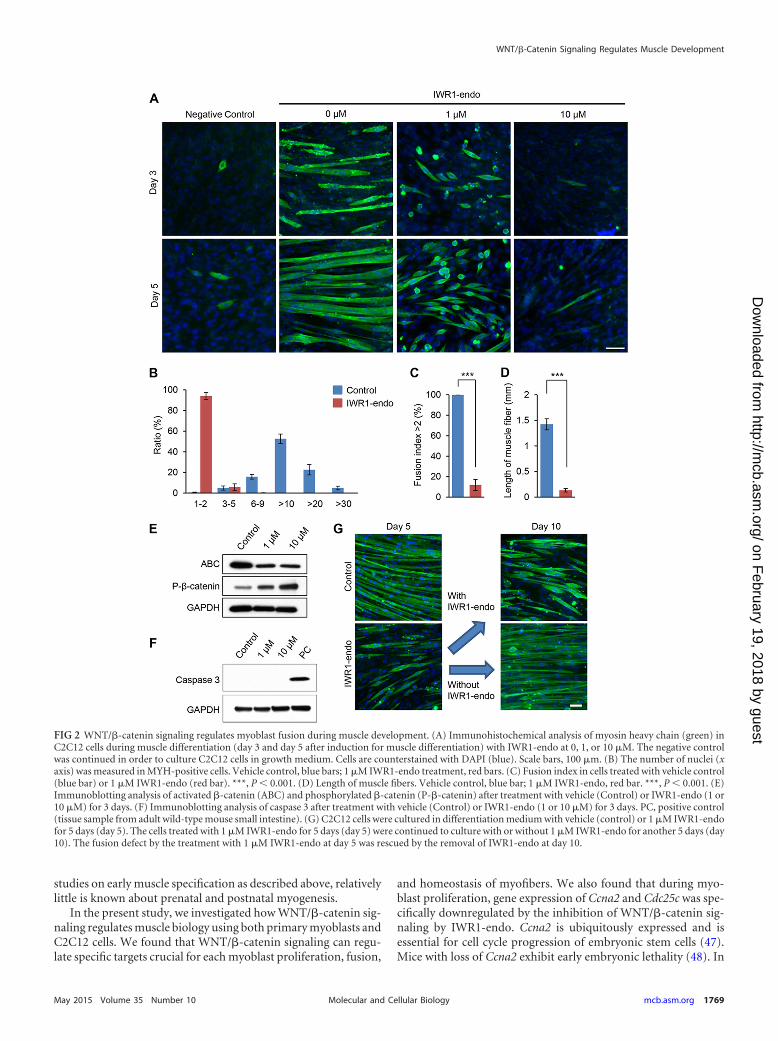
WNT/�-catenin signaling regulates muscle differentiationthrough Fermt2 expression. Next, we investigated myogenic dif-ferentiation of C2C12 cells in differentiation medium with (atboth 1 and 10 �M) and without IWR1-endo. Without the IWR1-endo treatment, we could detect myofibers after a 3-day culture indifferentiation medium. After a 5-day culture in differentiationmedium without IWR1-endo, C2C12 cells were well developedinto multinucleated myofibers. In contrast, C2C12 cells culturedin differentiation medium with 1 �M IWR1-endo were mostlymononucleated and failed to differentiate into myotubes. Thissuggests that WNT/�-catenin signaling is crucial for myoblast fu-sion, a critical process for the terminal differentiation of skeletalmuscle. We further tested higher concentrations of IWR1-endo.We found that treatment with IWR1-endo at 10 �M and higherblocked myoblast differentiation of C2C12 cells (Fig. 2A). Toquantify the fusion defect with treatment of 1 �M IWR1-endo, wecounted the number of nuclei in the cells stained with a myosin
WNT/�-Catenin Signaling Regulates Muscle Development
May 2015 Volume 35 Number 10 mcb.asm.org 1765Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
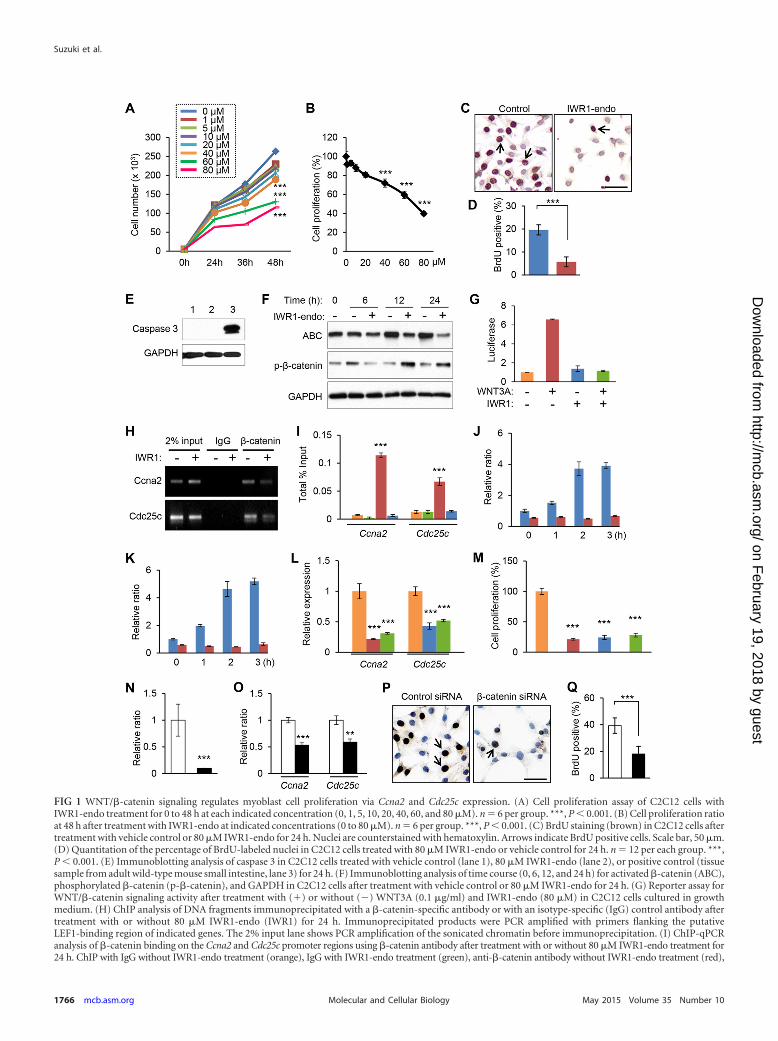
FIG 1 WNT/�-catenin signaling regulates myoblast cell proliferation via Ccna2 and Cdc25c expression. (A) Cell proliferation assay of C2C12 cells withIWR1-endo treatment for 0 to 48 h at each indicated concentration (0, 1, 5, 10, 20, 40, 60, and 80 �M). n � 6 per group. ***, P 0.001. (B) Cell proliferation ratioat 48 h after treatment with IWR1-endo at indicated concentrations (0 to 80 �M). n � 6 per group. ***, P 0.001. (C) BrdU staining (brown) in C2C12 cells aftertreatment with vehicle control or 80 �M IWR1-endo for 24 h. Nuclei are counterstained with hematoxylin. Arrows indicate BrdU positive cells. Scale bar, 50 �m.(D) Quantitation of the percentage of BrdU-labeled nuclei in C2C12 cells treated with 80 �M IWR1-endo or vehicle control for 24 h. n � 12 per each group. ***,P 0.001. (E) Immunoblotting analysis of caspase 3 in C2C12 cells treated with vehicle control (lane 1), 80 �M IWR1-endo (lane 2), or positive control (tissuesample from adult wild-type mouse small intestine, lane 3) for 24 h. (F) Immunoblotting analysis of time course (0, 6, 12, and 24 h) for activated �-catenin (ABC),phosphorylated �-catenin (p-�-catenin), and GAPDH in C2C12 cells after treatment with vehicle control or 80 �M IWR1-endo for 24 h. (G) Reporter assay forWNT/�-catenin signaling activity after treatment with (�) or without (�) WNT3A (0.1 �g/ml) and IWR1-endo (80 �M) in C2C12 cells cultured in growthmedium. (H) ChIP analysis of DNA fragments immunoprecipitated with a �-catenin-specific antibody or with an isotype-specific (IgG) control antibody aftertreatment with or without 80 �M IWR1-endo (IWR1) for 24 h. Immunoprecipitated products were PCR amplified with primers flanking the putativeLEF1-binding region of indicated genes. The 2% input lane shows PCR amplification of the sonicated chromatin before immunoprecipitation. (I) ChIP-qPCRanalysis of �-catenin binding on the Ccna2 and Cdc25c promoter regions using �-catenin antibody after treatment with or without 80 �M IWR1-endo treatment for24 h. ChIP with IgG without IWR1-endo treatment (orange), IgG with IWR1-endo treatment (green), anti-�-catenin antibody without IWR1-endo treatment (red),
Suzuki et al.
1766 mcb.asm.org May 2015 Volume 35 Number 10Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

heavy chain (MYH), a differentiation marker for myofibers. In thecontrol, almost 100% of muscle cells had more than three nuclei(as shown by the fusion index [i.e., the ratio of muscle cells withmultiple nuclei to all muscle cells]). In contrast, ca. 90% of thecells treated with 1 �M IWR1-endo had single nuclei in each cell(Fig. 2B and C). Consistent with the IWR1-endo induced fusiondefect, the length of muscle fibers in the treated group was signif-icantly shorter than those in the control group (�1.4 mm in con-trol versus �0.1 mm in the treated group) (Fig. 2D). Furthermore,we confirmed that IWR1-endo treatment decreased nonphospho-rylated (active) �-catenin and increased phosphorylated (inac-tive) �-catenin at 1 and 10 �M (Fig. 2E). Importantly, the treat-ment with either 1 or 10 �M IWR1-endo induced no apoptosis inthe process of muscle differentiation (Fig. 2F). To validate furtherthat IWR1-endo treatment had no toxic effects on myoblasts, wecultured C2C12 cells with 1 �M IWR1-endo in differentiationmedium for 5 days (fusion defects at day 5 treated with IWR1-endo) and then switched the medium with or without 1 �MIWR1-endo for another 5 days (Fig. 2G). The inhibited myo-blast fusion at day 5 was completely restored at day 10 withoutIWR1-endo (IWR1-endo was removed from the culture me-dium after 5 days of the culture), indicating that WNT/�-catenin signaling specifically inhibits myoblast fusion but doesnot induce apoptosis.
After this, we explored target molecules which were involved inmyoblast fusion and altered by the 1 �M IWR1-endo treatment atday 5. Among 11 candidate molecules (Nphs1, Ptk2, Fermt2,Adam12, Itga3, Itgb1, Cdh2, Cdh15, Myof, Dysf, and Tmem8c),gene expression of Fermitin family homolog 2 (Fermt2; alsoknown as Kindlin2) was significantly decreased by the treatmentof 1 �M IWR1-endo (1.8-fold reduction, P 0.01) (Fig. 3A).Decreased Fermt2 expression at day 2 was restored at day 7 after
the depletion of 1 �M IWR1-endo, indicating that Fermt2 expres-sion is regulated by WNT/�-catenin signaling (Fig. 3B). To testwhether IWR1-endo specifically blocks WNT/�-catenin signalingcascade through transcriptional response elements of LEF/TCF,we performed LEF/TCF luciferase reporter assays after treatmentwith WNT3A (0.1 �g/ml) and IWR1-endo (1 �M) at the muscledifferentiation stage. WNT3A induced LEF/TCF-mediated pro-moter activity approximately six times, and IWR1-endo (1 �M)completely inhibited the induction of LEF/TCF-mediated pro-moter activity (Fig. 3C). These results indicate that IWR1-endoinhibits canonical WNT/�-catenin signaling. FERMT2, a focal ad-hesion protein, can regulate integrin activation and mediate cell-cell or cell-matrix adhesion (30, 31). We analyzed the promotersequence of the Fermt2 gene (including 5 kbp upstream of thetranscription start site). We found that the murine Fermt2genomic region has a conserved LEF1 recognition sequence (Ta-bles 1 and 2). To test whether �-catenin could bind to the pro-moter region of Fermt2, we performed ChIP assays. The bindingsite for LEF1 in the promoter region of Fermt2 was immunopre-cipitated with �-catenin antibody but not with control IgG. Inaddition, the immunoprecipitated amounts were decreased bytreatment with 1 �M IWR1-endo for 48 h, indicating that thebinding site for LEF1 in the Fermt2 promoter region is functionaland responsible for WNT/�-catenin signaling activity (Fig. 3Dand E). Gene expression of Fermt2 was induced by WNT3A li-gands (0.1 �g/ml) in the time dependent manner (Fig. 3F). Fur-thermore, gene expression of Fermt2 was significantly reducedafter siRNA knockdown for Ctnnb1 (Fig. 3G and H). Myoblastfusion was disrupted by the knockdown of Ctnnb1 (Fig. 3I). Takentogether, our results indicate that gene expression of Fermt2 isregulated by the WNT/�-catenin pathway.
To validate the function of Fermt2 during muscle differentia-tion, we cultured C2C12 cells in differentiation medium withtreatment of siRNA knockdown for Fermt2. Gene expression of
TABLE 1 Mean ratios of differentially expressed genes related to cellproliferation based on PCR array analysis in C2C12 cells treated withvehicle control or IWR1-endo for 24 ha
Gene
Mean ratio SDb
Control Treatment
Ccnd2 1.00 0.37 �4.10 1.40*Cdkn1a 1.00 0.12 �1.73 0.23*Ccnb1 1.00 0.23 –1.56 0.18*Cdk4 1.00 0.35 –1.73 0.09*Ccna2 1.00 0.36 –1.88 0.14***Cdc25c 1.00 0.34 –2.06 0.13**Cdk1 1.00 0.57 –3.33 0.12*a Cell cycle regulators altered with IWR1-endo treatment compared to controlsexpressed as a ratio are listed.b *, P 0.05; **, P 0.01; ***, P 0.001. Ratios are based on the geometric meanexpression scores derived from control and treatment groups. All relevant transcriptswith a �1.5-fold change with P 0.05 are shown.
TABLE 2 Distribution of potential LEF1 recognition sequences inpromoters of selected genes of interest
Gene
No. of sites
Minimal core Optimal Conserved
Ccna2 16 6 1Ccnb1 16 5 0Ccnd2 5 1 0Cdc25c 18 7 1Cdk1 14 4 0Cdk4 13 5 0Cdkn1a 10 3 0Dmd 14 8 0E2f5 22 8 0Fermt2 16 7 1
and anti-�-catenin antibody with IWR1-endo treatment (blue). (J and K) Quantitative RT-PCR analysis of gene expression of Ccna2 (J) and Cdc25c (K) aftertreatment with WNT3A (0.1 �g/ml) for 0, 1, 2, and 3 h (h) with or without 80 �M IWR1-endo. **, P 0.01; ***, P 0.001. (L) Quantitative RT-PCR analysisafter treatment with siRNA for control (orange), Ccna2 (red), Ccna2/Cdc25c combination (green), and Cdc25c (blue). ***, P 0.001. (M) Cell proliferation assayof C2C12 cells treated with siRNA for control (orange), Ccna2 (red), Cdc25c (blue), and Ccna2/Cdc25c combination (green). ***, P 0.001. (N) Ctnnb1expression after treatment with siRNA for control (open bar) and Ctnnb1 (closed bar). ***, P 0.001. (O) Quantitative RT-PCR analysis of Ccna2 and Cdc25cexpression after treatment with siRNA for control (open bars) and Ctnnb1 (closed bars). ***, P 0.001. **, P 0.01. (P) BrdU staining (brown) in C2C12 cellsafter treatment with control or Ctnnb1 siRNA knockdown for 24 h. Nuclei are counterstained with hematoxylin. Arrows indicate BrdU-positive cells. Scale bar,50 �m. (Q) Quantitation of the percentage of BrdU-labeled nuclei in C2C12 cells after treatment with control or Ctnnb1 siRNA knockdown for 24 h. n � 12 pergroup. ***, P 0.001.
WNT/�-Catenin Signaling Regulates Muscle Development
May 2015 Volume 35 Number 10 mcb.asm.org 1767Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

Fermt2 was significantly reduced after the siRNA knockdown at80% reduction (Fig. 3J). Myoblast fusion was disrupted by theknockdown of Fermt2 (Fig. 3K). In addition, overexpression ofFermt2 under the condition of 1 �M IWR1-endo treatment re-stored the myofiber fusion defect (Fig. 3L and M). Altogether, ourresults indicate that gene expression of Fermt2 is regulated byWNT/�-catenin pathway, and Fermt2 plays a crucial role in myo-blast fusion.
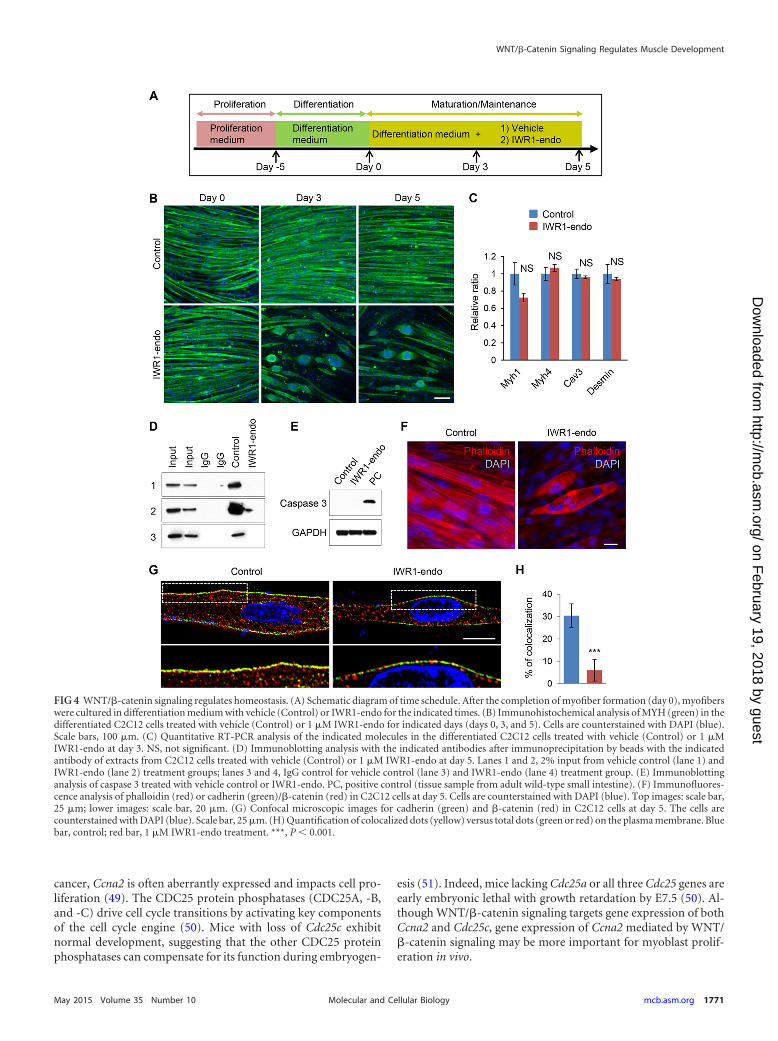
The cadherin/�-catenin/actin complex formation mediatedby WNT/�-catenin signaling is crucial for muscle homeostasis.To test if WNT/�-catenin signaling is involved in muscle homeo-stasis, we treated well-differentiated C2C12 cells with or without 1�M IWR1-endo (Fig. 4A and B). The long and straightened myo-fibers were disrupted and changed their form to a round, multi-nucleated shape after the IWR1-endo treatment (Fig. 4B). Next,we analyzed gene expression of the molecules related to cytoskel-etal structure formation (MYH1/4, caveolin 3, and desmin); how-ever, we failed to identify genes altered by the treatment with 1 �MIWR1-endo (Fig. 4C), which suggests that the WNT/�-cateninpathway may regulate cytoskeletal structure formation at the post-transcriptional level. Previous studies have demonstrated that�-catenin can bind to both cadherin and actin (32, 33). An extra-cellular matrix niche is crucial for regulating the movement andmorphology of skeletal muscle cells (34). We hypothesized thatthe cadherin/�-catenin/actin complex plays a crucial role in myo-fiber morphogenesis by anchoring skeletal structure molecules tothe connective tissue (niche) composed of an extracellular matrix.To test the molecular interaction among them, we performed im-munoprecipitation for actin, cadherin, and �-catenin (Fig. 4D).Our results indicate that cadherin/�-catenin/actin complex ex-isted in myofibers and that it was disrupted by the treatment with1 �M IWR1-endo. Thus, �-catenin is crucial for forming the cad-herin/�-catenin/actin complex. In addition, we confirmed that 1�M IWR1-endo induced no apoptosis at the maturation/mainte-nance stage of muscle cells (Fig. 4E). To further validate our find-ings of the complex formation regulated by WNT/�-catenin sig-naling, we performed immunofluorescence analyses of thesecomplex molecules with or without treatment of 1 �M IWR1-endo. Actin stained with phalloidin was straightened and paral-leled each other in control. In contrast, actin formation wasdisrupted and accumulated after the treatment with 1 �M IWR1-endo (Fig. 4F). Without IWR1-endo treatment, cadherin and�-catenin were colocalized in the myofibers. In contrast, there waspoor colocalization of cadherin and �-catenin after the treatmentwith 1 �M IWR1-endo (Fig. 4G and H). Thus, our data indicatethat �-catenin is an essential binding partner for both the cyto-plasmic tail of cadherin and actin and is crucial for the mainte-nance of myofiber morphology.
Molecular mechanisms of myogenesis mediated by WNT/�-catenin signaling are conserved in primary muscle cells. Wetested whether the mechanisms we found in C2C12 cells were
conserved in primary muscle cells from C57B6/J mice. We isolatedprimary muscle cells from the limb and tongue of mice. We per-formed BrdU incorporation assays to test cell proliferation activ-ity with or without treatment of 80 �M IWR1-endo. In primarymyoblasts from both the limb and tongue, IWR1-endo reducedcell proliferation activity (Fig. 5A to C). Gene expression of Ccna2and Cdc25c was significantly decreased by treatment of IWR1-endo in both primary limb and tongue myoblasts (Fig. 5D). Then,we investigated the muscle differentiation of primary myoblastsfrom both the limb and the tongue. The treatment with 1 �MIWR1-endo strikingly inhibited myoblast fusion in both primarylimb and tongue myoblasts. Gene expression of Fermt2 was sig-nificantly reduced by the treatment with IWR1-endo (Fig. 5F).Myoblasts differentiated into myofibers within 7 days in a culturein differentiation medium in the control. In contrast, treatmentwith 1 �M IWR1-endo blocked myotube formation by inhibitingmyoblast fusion (Fig. 5G). Next, we investigated the maintenanceability of muscle cells. The well-differentiated and straightenedmyofibers changed their shape after treatment with 1 �M IWR1-endo, as seen in C2C12 cells (Fig. 5H). These results indicate thatWNT/�-catenin-mediated signaling mechanisms are well con-served in primary muscle cells. Furthermore, to test whether themechanisms were conserved in a muscular tissue, we culturedtongue explants from C57B6/J mice with or without IWR1-endo.In the tongue explants, IWR1-endo treatment (80 �M, 24 h) re-sulted in decreased cell proliferation, as seen in our cell culturesystem (Fig. 6A and B). In addition, IWR1-endo treatment (80�M, 72 h) resulted in defects in the elongation of muscle fibers(Fig. 6C). Collectively, these results suggest that WNT/�-cateninsignaling can regulate multiple steps of muscle development, in-cluding proliferation, differentiation, and structural homeostasis,by regulating step-specific targets (Fig. 6D).
DISCUSSION
Mice with deficiency in WNT ligands and their receptors FZDsshow early embryonic lethality, often affecting multiple tissues (3,10). WNT/�-catenin signaling is active within myogenic cells, asshown by BAT-gal mice, in which seven TCF/LEF-binding sitesdrive nuclear LacZ expression when bound by transcriptionallyactive TCF/LEFs (35). During axial myogenesis, WNT/�-cateninsignaling is crucial for dermomyotome and myotome formation(14, 36–38). WNT ligands can positively regulate the number ofdermomyotomal Pax3� and Pax7� progenitors (39–41). Duringfetal myogenesis, �-catenin affects myofiber number and posi-tively regulates the number of slow fibers in mice with conditionaldeletion of Ctnnb1 in muscle progenitor cells (Pax7iCre/�;Ctnnb1�/fl2-6 mice) (14). Subsequently, WNT ligands are neces-sary and sufficient to promote normal Myf5 and MyoD expression(42–45). WNT/�-catenin activation also induces satellite cell pro-liferation during skeletal muscle regeneration (46). Compared to
TABLE 3 Evolutionarily conserved LEF1 recognition sites within Ccna2, Cdc25c, and Fermt2 promoters
GeneBinding site positionrelative to TSS Binding site sequence context (5=–3=)a Extent of conservation
Ccna2 –2311 GGAAGCAGCTTTGAAAACCAA In all eight speciesCdc25c –3095 GTAGCCATCAAAGCATAAGAA In all eight speciesFermt2 –2973 CTTTAATCTTCAAAGCAGCAA In all species except doga Binding site motifs are underlined.
Suzuki et al.
1768 mcb.asm.org May 2015 Volume 35 Number 10Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
studies on early muscle specification as described above, relativelylittle is known about prenatal and postnatal myogenesis.
In the present study, we investigated how WNT/�-catenin sig-naling regulates muscle biology using both primary myoblasts andC2C12 cells. We found that WNT/�-catenin signaling can regu-late specific targets crucial for each myoblast proliferation, fusion,
and homeostasis of myofibers. We also found that during myo-blast proliferation, gene expression of Ccna2 and Cdc25c was spe-cifically downregulated by the inhibition of WNT/�-catenin sig-naling by IWR1-endo. Ccna2 is ubiquitously expressed and isessential for cell cycle progression of embryonic stem cells (47).Mice with loss of Ccna2 exhibit early embryonic lethality (48). In
FIG 2 WNT/�-catenin signaling regulates myoblast fusion during muscle development. (A) Immunohistochemical analysis of myosin heavy chain (green) inC2C12 cells during muscle differentiation (day 3 and day 5 after induction for muscle differentiation) with IWR1-endo at 0, 1, or 10 �M. The negative controlwas continued in order to culture C2C12 cells in growth medium. Cells are counterstained with DAPI (blue). Scale bars, 100 �m. (B) The number of nuclei (xaxis) was measured in MYH-positive cells. Vehicle control, blue bars; 1 �M IWR1-endo treatment, red bars. (C) Fusion index in cells treated with vehicle control(blue bar) or 1 �M IWR1-endo (red bar). ***, P 0.001. (D) Length of muscle fibers. Vehicle control, blue bar; 1 �M IWR1-endo, red bar. ***, P 0.001. (E)Immunoblotting analysis of activated �-catenin (ABC) and phosphorylated �-catenin (P-�-catenin) after treatment with vehicle (Control) or IWR1-endo (1 or10 �M) for 3 days. (F) Immunoblotting analysis of caspase 3 after treatment with vehicle (Control) or IWR1-endo (1 or 10 �M) for 3 days. PC, positive control(tissue sample from adult wild-type mouse small intestine). (G) C2C12 cells were cultured in differentiation medium with vehicle (control) or 1 �M IWR1-endofor 5 days (day 5). The cells treated with 1 �M IWR1-endo for 5 days (day 5) were continued to culture with or without 1 �M IWR1-endo for another 5 days (day10). The fusion defect by the treatment with 1 �M IWR1-endo at day 5 was rescued by the removal of IWR1-endo at day 10.
WNT/�-Catenin Signaling Regulates Muscle Development
May 2015 Volume 35 Number 10 mcb.asm.org 1769Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
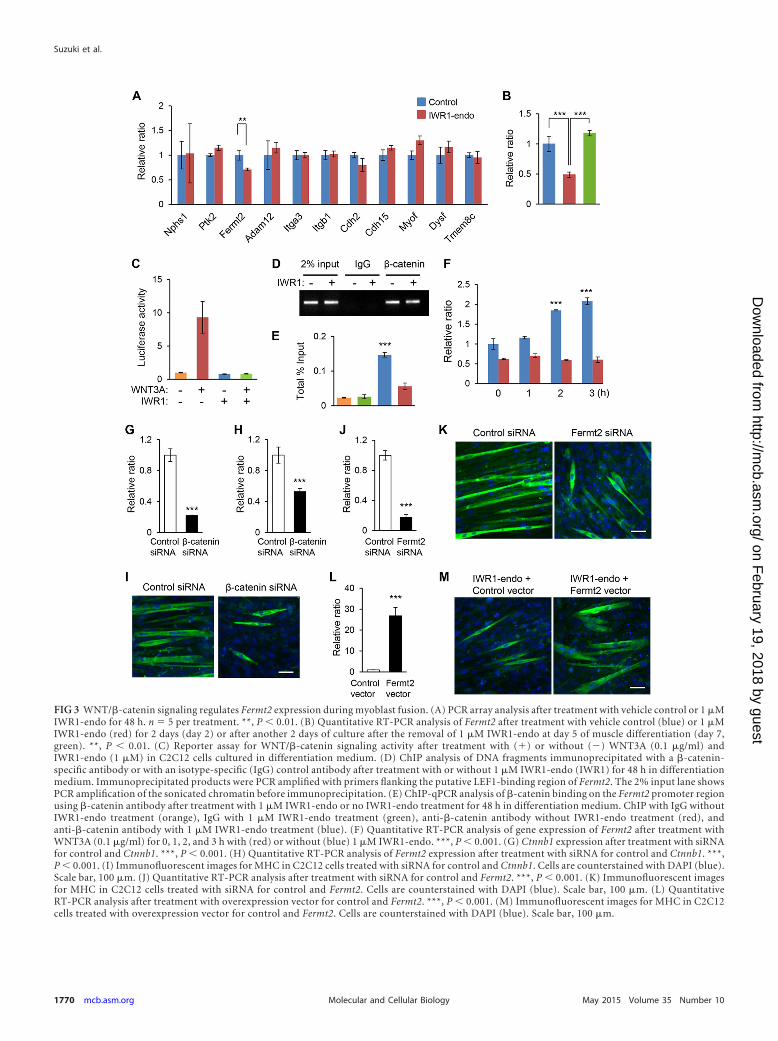
FIG 3 WNT/�-catenin signaling regulates Fermt2 expression during myoblast fusion. (A) PCR array analysis after treatment with vehicle control or 1 �MIWR1-endo for 48 h. n � 5 per treatment. **, P 0.01. (B) Quantitative RT-PCR analysis of Fermt2 after treatment with vehicle control (blue) or 1 �MIWR1-endo (red) for 2 days (day 2) or after another 2 days of culture after the removal of 1 �M IWR1-endo at day 5 of muscle differentiation (day 7,green). **, P 0.01. (C) Reporter assay for WNT/�-catenin signaling activity after treatment with (�) or without (�) WNT3A (0.1 �g/ml) andIWR1-endo (1 �M) in C2C12 cells cultured in differentiation medium. (D) ChIP analysis of DNA fragments immunoprecipitated with a �-catenin-specific antibody or with an isotype-specific (IgG) control antibody after treatment with or without 1 �M IWR1-endo (IWR1) for 48 h in differentiationmedium. Immunoprecipitated products were PCR amplified with primers flanking the putative LEF1-binding region of Fermt2. The 2% input lane showsPCR amplification of the sonicated chromatin before immunoprecipitation. (E) ChIP-qPCR analysis of �-catenin binding on the Fermt2 promoter regionusing �-catenin antibody after treatment with 1 �M IWR1-endo or no IWR1-endo treatment for 48 h in differentiation medium. ChIP with IgG withoutIWR1-endo treatment (orange), IgG with 1 �M IWR1-endo treatment (green), anti-�-catenin antibody without IWR1-endo treatment (red), andanti-�-catenin antibody with 1 �M IWR1-endo treatment (blue). (F) Quantitative RT-PCR analysis of gene expression of Fermt2 after treatment withWNT3A (0.1 �g/ml) for 0, 1, 2, and 3 h with (red) or without (blue) 1 �M IWR1-endo. ***, P 0.001. (G) Ctnnb1 expression after treatment with siRNAfor control and Ctnnb1. ***, P 0.001. (H) Quantitative RT-PCR analysis of Fermt2 expression after treatment with siRNA for control and Ctnnb1. ***,P 0.001. (I) Immunofluorescent images for MHC in C2C12 cells treated with siRNA for control and Ctnnb1. Cells are counterstained with DAPI (blue).Scale bar, 100 �m. (J) Quantitative RT-PCR analysis after treatment with siRNA for control and Fermt2. ***, P 0.001. (K) Immunofluorescent imagesfor MHC in C2C12 cells treated with siRNA for control and Fermt2. Cells are counterstained with DAPI (blue). Scale bar, 100 �m. (L) QuantitativeRT-PCR analysis after treatment with overexpression vector for control and Fermt2. ***, P 0.001. (M) Immunofluorescent images for MHC in C2C12cells treated with overexpression vector for control and Fermt2. Cells are counterstained with DAPI (blue). Scale bar, 100 �m.
Suzuki et al.
1770 mcb.asm.org May 2015 Volume 35 Number 10Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
cancer, Ccna2 is often aberrantly expressed and impacts cell pro-liferation (49). The CDC25 protein phosphatases (CDC25A, -B,and -C) drive cell cycle transitions by activating key componentsof the cell cycle engine (50). Mice with loss of Cdc25c exhibitnormal development, suggesting that the other CDC25 proteinphosphatases can compensate for its function during embryogen-
esis (51). Indeed, mice lacking Cdc25a or all three Cdc25 genes areearly embryonic lethal with growth retardation by E7.5 (50). Al-though WNT/�-catenin signaling targets gene expression of bothCcna2 and Cdc25c, gene expression of Ccna2 mediated by WNT/�-catenin signaling may be more important for myoblast prolif-eration in vivo.
FIG 4 WNT/�-catenin signaling regulates homeostasis. (A) Schematic diagram of time schedule. After the completion of myofiber formation (day 0), myofiberswere cultured in differentiation medium with vehicle (Control) or IWR1-endo for the indicated times. (B) Immunohistochemical analysis of MYH (green) in thedifferentiated C2C12 cells treated with vehicle (Control) or 1 �M IWR1-endo for indicated days (days 0, 3, and 5). Cells are counterstained with DAPI (blue).Scale bars, 100 �m. (C) Quantitative RT-PCR analysis of the indicated molecules in the differentiated C2C12 cells treated with vehicle (Control) or 1 �MIWR1-endo at day 3. NS, not significant. (D) Immunoblotting analysis with the indicated antibodies after immunoprecipitation by beads with the indicatedantibody of extracts from C2C12 cells treated with vehicle (Control) or 1 �M IWR1-endo at day 5. Lanes 1 and 2, 2% input from vehicle control (lane 1) andIWR1-endo (lane 2) treatment groups; lanes 3 and 4, IgG control for vehicle control (lane 3) and IWR1-endo (lane 4) treatment group. (E) Immunoblottinganalysis of caspase 3 treated with vehicle control or IWR1-endo. PC, positive control (tissue sample from adult wild-type small intestine). (F) Immunofluores-cence analysis of phalloidin (red) or cadherin (green)/�-catenin (red) in C2C12 cells at day 5. Cells are counterstained with DAPI (blue). Top images: scale bar,25 �m; lower images: scale bar, 20 �m. (G) Confocal microscopic images for cadherin (green) and �-catenin (red) in C2C12 cells at day 5. The cells arecounterstained with DAPI (blue). Scale bar, 25 �m. (H) Quantification of colocalized dots (yellow) versus total dots (green or red) on the plasma membrane. Bluebar, control; red bar, 1 �M IWR1-endo treatment. ***, P 0.001.
WNT/�-Catenin Signaling Regulates Muscle Development
May 2015 Volume 35 Number 10 mcb.asm.org 1771Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
A recent study suggests a potential role of WNT/�-catenin sig-naling in myoblast fusion (52). Members of the R-spondin family(RSPO) of secreted cysteine-rich proteins (RSPO1, -2, -3, and -4)activate the WNT/�-catenin signaling pathway at the receptorlevel (53, 54). RSPO2 promotes myogenic differentiation andmyocyte fusion via activation of WNT/�-catenin signaling inC2C12 cells (55). RSPOs can regulate WNT receptor expressionand control myogenesis through gene expression of the trans-forming growth factor � (TGF�) antagonist Fst (56). Thus, WNT
signaling is activated through WNT ligands, RSPO, and nonca-nonical (�-catenin-independent) WNT signaling activators dur-ing muscle development. During myoblast fusion, the cell signal-ing pathway is tightly regulated and controls downstream targetmolecules involved in membrane fusion and cytoskeletal remod-eling (57). For example, a recent study indicates that Tmem8c(also known as Myomaker) specifically expresses in skeletal mus-cles and plays a crucial role in myoblast fusion during muscledevelopment (58). In the present study, the bioinformatics anal-
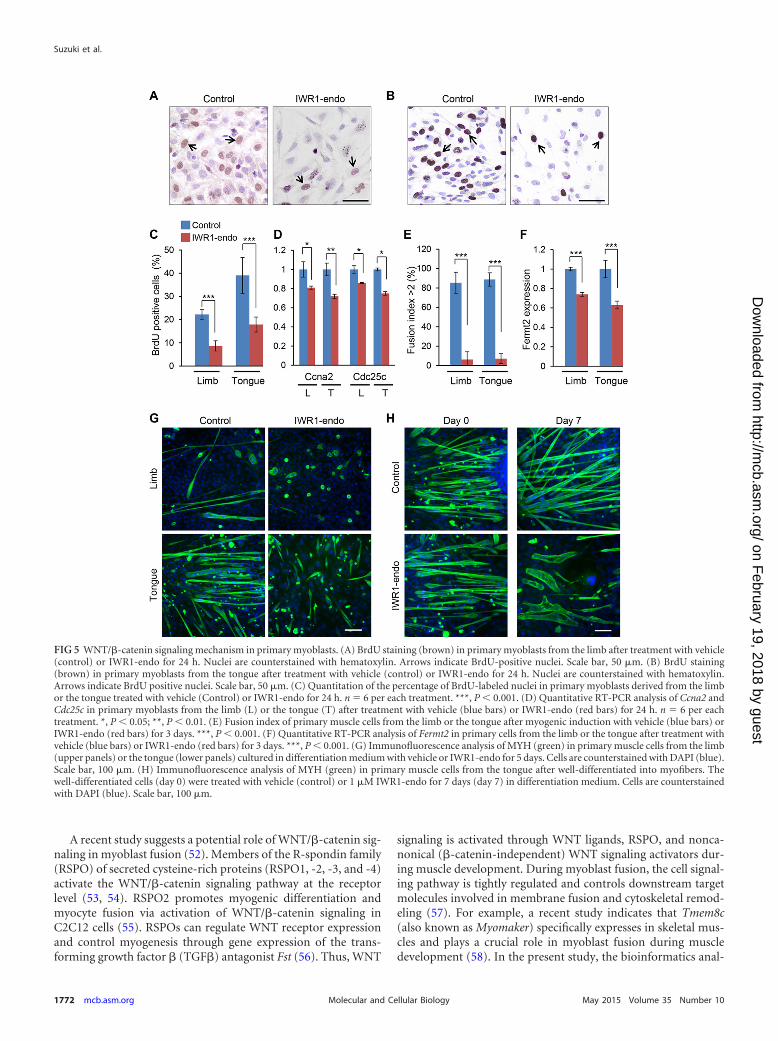
FIG 5 WNT/�-catenin signaling mechanism in primary myoblasts. (A) BrdU staining (brown) in primary myoblasts from the limb after treatment with vehicle(control) or IWR1-endo for 24 h. Nuclei are counterstained with hematoxylin. Arrows indicate BrdU-positive nuclei. Scale bar, 50 �m. (B) BrdU staining(brown) in primary myoblasts from the tongue after treatment with vehicle (control) or IWR1-endo for 24 h. Nuclei are counterstained with hematoxylin.Arrows indicate BrdU positive nuclei. Scale bar, 50 �m. (C) Quantitation of the percentage of BrdU-labeled nuclei in primary myoblasts derived from the limbor the tongue treated with vehicle (Control) or IWR1-endo for 24 h. n � 6 per each treatment. ***, P 0.001. (D) Quantitative RT-PCR analysis of Ccna2 andCdc25c in primary myoblasts from the limb (L) or the tongue (T) after treatment with vehicle (blue bars) or IWR1-endo (red bars) for 24 h. n � 6 per eachtreatment. *, P 0.05; **, P 0.01. (E) Fusion index of primary muscle cells from the limb or the tongue after myogenic induction with vehicle (blue bars) orIWR1-endo (red bars) for 3 days. ***, P 0.001. (F) Quantitative RT-PCR analysis of Fermt2 in primary cells from the limb or the tongue after treatment withvehicle (blue bars) or IWR1-endo (red bars) for 3 days. ***, P 0.001. (G) Immunofluorescence analysis of MYH (green) in primary muscle cells from the limb(upper panels) or the tongue (lower panels) cultured in differentiation medium with vehicle or IWR1-endo for 5 days. Cells are counterstained with DAPI (blue).Scale bar, 100 �m. (H) Immunofluorescence analysis of MYH (green) in primary muscle cells from the tongue after well-differentiated into myofibers. Thewell-differentiated cells (day 0) were treated with vehicle (control) or 1 �M IWR1-endo for 7 days (day 7) in differentiation medium. Cells are counterstainedwith DAPI (blue). Scale bar, 100 �m.
Suzuki et al.
1772 mcb.asm.org May 2015 Volume 35 Number 10Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
ysis of Tmem8c promoter region (up to 5 kb upstream of ttheranscription start site of Tmem8c gene) detected no TCF/LEF op-timal binding site (WNT/�-catenin response element). In addi-tion, gene expression of Tmem8c was not altered by IWR1-endotreatment. These data suggest that gene expression of Tmem8c isregulated by the other signaling pathways such as TGF�, fibroblastgrowth factor, and hedgehog. Among 11 candidate molecules,only Fermt2 expression was significantly (�1.5-fold change, P 0.05) decreased by IWR1-endo treatment. The other molecules
involved in myoblast fusion tested in this study (Nphs1, Ptk2,Adam12, Itga3, Itgb1, Cdh2, Cdh15, Myof, and Dysf) may be alsocontrolled by the other signaling pathways.
A previous study shows that RNA interference knockdown forFermt2 in C2C12 cells results in significant abnormalities duringthe early stages of myogenesis (31). In addition, targeted disrup-tion of the murine Fermt2 gene results in early embryonic lethalitywith musculature developmental defects (59). These findings,coupled with our results, indicate that WNT/�-catenin-mediatedFermt2 expression is crucial for myogenic fusion. AlthoughFermt2-null mice are lethal by the stage of muscle fusion, the fu-ture study using mice with conditional deletion of Fermt2 willallow us to investigate the role of Fermt2 during muscle fusion.Moreover, Fermt2 may play additional roles aside from myoblastfusion during muscle development. Interestingly, different stepsof muscle differentiation are also regulated by WNT/�-cateninsignaling. For example, inhibition of GSK3� results in activatedWNT/�-catenin signaling and leads to enhanced differentiationof C2 myogenic cells (60). The RSPO family of proteins, WNT/�-catenin signaling agonists, can promote myogenic differentiationin cell culture (55). Pharmacological activations of WNT/�-catenin signaling can also facilitate the differentiation of satellitecells and myoblasts (60–62). Thus, WNT/�-catenin signaling mayplay a pivotal role for the volume and size of skeletal muscles byregulating various steps of myoblast differentiation and fusion.
In this study, we found that during muscle maturation/main-tenance, a blockade of WNT/�-catenin signaling resulted in dis-rupted cell adhesion via loss of cadherin/�-catenin/actin complexformation. Previous studies indicate that �-catenin plays dualroles in WNT/�-catenin signaling and in cadherin-mediated celladhesion (32, 63). �-Catenin colocalized with cadherin in mem-branes may be essential for proper cell structural formation. Cad-herin family molecules exhibit distinctly regulated tissue distribu-tion and control morphogenesis. Skeletal muscle cells expressboth N-cadherin (encoded by Cdh2) and M-cadherin (encoded byCdh15) throughout myogenesis, from progenitors to myofibers,in both development and regeneration (64, 65). Mice with loss ofCdh2 are embryonic lethal by E10 with disorganized somites (66).The in vitro studies of Cdh2�/� cells indicate that N-cadherin isnot essential for myogenesis, but other cadherins may compensatefor the lack of N-cadherin (67). M-cadherin is a classical calcium-dependent cell adhesion molecule that is highly expressed in de-veloping skeletal muscle, satellite cells, and cerebellum (65). Micewith loss of M-cadherin exhibit no gross developmental defects(68). These results strongly suggest that loss of the combination ofN- and M-cadherins or other cadherins, or both, is essential formyogenic defects. The actin family is categorized into four mus-cle-specific isoforms: -skeletal, -cardiac, -smooth, and�-smooth. It is further categorized into two ubiquitously ex-pressed isoforms: �-cytoplasmic and �-cytoplasmic (69). Skeletalmyoblasts almost exclusively express �- and �-cytoplasmic actins,but on differentiation, these cytoplasmic isoforms are drasti-cally downregulated and sequentially replaced by -cardiac and -smooth actins, and ultimately -skeletal actin (69). Mice withdeficiency of -skeletal actin are indistinguishable from their lit-termates at birth but die in the early neonatal period (postnataldays 1 to 9) with skeletal muscle defects (70). Recent studies indi-cate that �-cytoplasmic actin is required for cytoskeletal mainte-nance but not development (71, 72). Thus, actin isoforms mayalso largely compensate for the lack of an individual actin. Com-
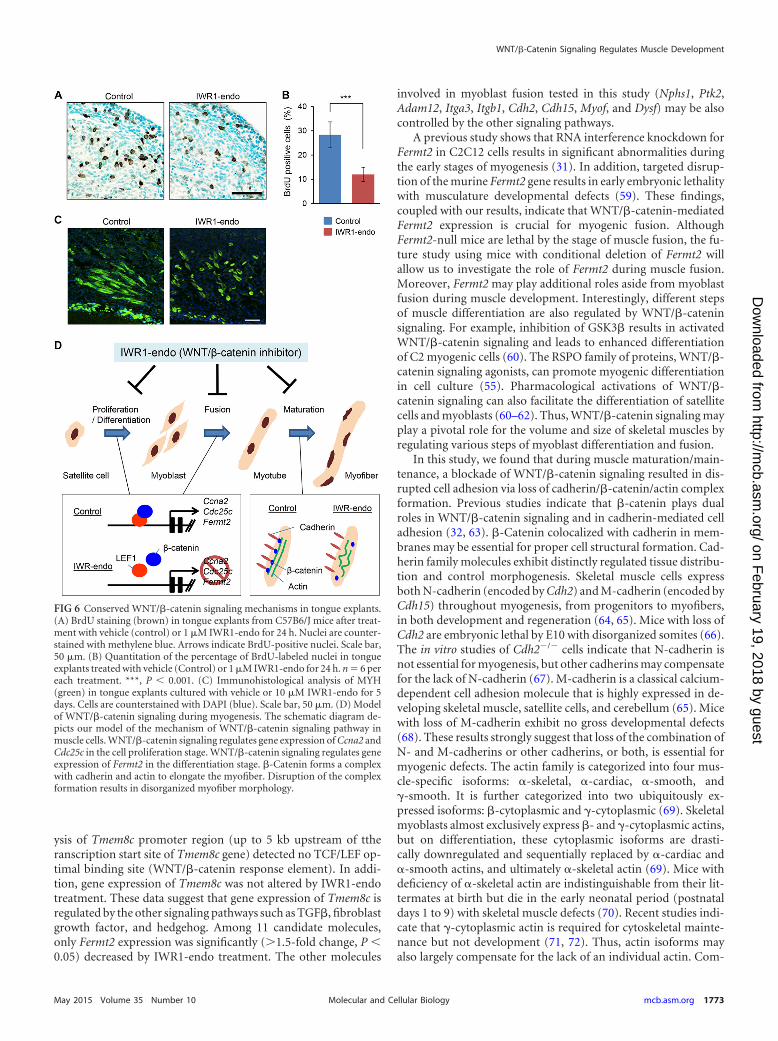
FIG 6 Conserved WNT/�-catenin signaling mechanisms in tongue explants.(A) BrdU staining (brown) in tongue explants from C57B6/J mice after treat-ment with vehicle (control) or 1 �M IWR1-endo for 24 h. Nuclei are counter-stained with methylene blue. Arrows indicate BrdU-positive nuclei. Scale bar,50 �m. (B) Quantitation of the percentage of BrdU-labeled nuclei in tongueexplants treated with vehicle (Control) or 1 �M IWR1-endo for 24 h. n � 6 pereach treatment. ***, P 0.001. (C) Immunohistological analysis of MYH(green) in tongue explants cultured with vehicle or 10 �M IWR1-endo for 5days. Cells are counterstained with DAPI (blue). Scale bar, 50 �m. (D) Modelof WNT/�-catenin signaling during myogenesis. The schematic diagram de-picts our model of the mechanism of WNT/�-catenin signaling pathway inmuscle cells. WNT/�-catenin signaling regulates gene expression of Ccna2 andCdc25c in the cell proliferation stage. WNT/�-catenin signaling regulates geneexpression of Fermt2 in the differentiation stage. �-Catenin forms a complexwith cadherin and actin to elongate the myofiber. Disruption of the complexformation results in disorganized myofiber morphology.
WNT/�-Catenin Signaling Regulates Muscle Development
May 2015 Volume 35 Number 10 mcb.asm.org 1773Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

pared to cadherin and actin, �-catenin may play the most criticalrole in the cadherin/�-catenin/actin complex formation.
Among skeletal muscles, tongue muscles are the most devel-oped muscle at birth for suckling, compared to the other muscles(73, 74). Interestingly, mice lacking Myf5 and Pax3 do not developskeletal muscle in the trunk or limb, and yet head muscles formnormally (75). In chicken embryo, WNT signals, which promotetrunk myogenesis, have been shown to block myogenesis in bran-chial arches (76). These findings suggest that craniofacial muscledevelopment may be differently regulated compared to that oftrunk and limb muscles. We investigated primary myoblasts fromthe tongue compared to that of the limb. We found that require-ment of WNT/�-catenin signaling was similar among tongue andlimb myogenesis.
Our findings may be relevant in human disease, given thataltered WNT/�-catenin signaling is implicated in multiple mal-formations and syndromes, including muscle disorders in hu-mans (77–79). For example, in patients with muscular defects,such as myopathies and atrophy, WNT/�-catenin signaling ismost likely altered due to a genetic and/or epigenetic cause(s)(80). Our results highlight that WNT/�-catenin signaling is awidely used mechanism in skeletal muscles which regulate step-specific targets during myogenesis. Our findings on the mecha-nisms of action of WNT/�-catenin signaling offer several intrigu-ing possibilities into the potential for therapeutic interventions.
ACKNOWLEDGMENTS
We thank Yoshihiro Komatsu for discussion.This study was supported by the UT Houston School of Dentistry
faculty startup fund to J.I.
REFERENCES1. Tajbakhsh S. 2009. Skeletal muscle stem cells in developmental versus
regenerative myogenesis. J Intern Med 266:372–389. http://dx.doi.org/10.1111/j.1365-2796.2009.02158.x.
2. Bentzinger CF, Wang YX, Rudnicki MA. 2012. Building muscle: molec-ular regulation of myogenesis. Cold Spring Harbor Perspect Biol4:a008342. http://dx.doi.org/10.1101/cshperspect.a008342.
3. Grigoryan T, Wend P, Klaus A, Birchmeier W. 2008. Deciphering thefunction of canonical Wnt signals in development and disease: condi-tional loss- and gain-of-function mutations of beta-catenin in mice.Genes Dev 22:2308 –2341. http://dx.doi.org/10.1101/gad.1686208.
4. MacDonald BT, Tamai K, He X. 2009. Wnt/beta-catenin signaling:components, mechanisms, and diseases. Dev Cell 17:9 –26. http://dx.doi.org/10.1016/j.devcel.2009.06.016.
5. Song X, Wang S, Li L. 2014. New insights into the regulation of Axinfunction in canonical Wnt signaling pathway. Protein Cell 5:186 –193.http://dx.doi.org/10.1007/s13238-014-0019-2.
6. Cisternas P, Henriquez JP, Brandan E, Inestrosa NC. 2014. Wnt signal-ing in skeletal muscle dynamics: myogenesis, neuromuscular synapse andfibrosis. Mol Neurobiol 49:574 –589. http://dx.doi.org/10.1007/s12035-013-8540-5.
7. Kaufmann U, Kirsch J, Irintchev A, Wernig A, Starzinski-Powitz A.1999. The M-cadherin catenin complex interacts with microtubules inskeletal muscle cells: implications for the fusion of myoblasts. J Cell Sci112(Pt 1):55– 68.
8. Cisternas P, Vio CP, Inestrosa NC. 2014. Role of Wnt signaling in tissuefibrosis, lessons from skeletal muscle and kidney. Curr Mol Med 14:510 –522. http://dx.doi.org/10.2174/1566524014666140414210346.
9. Tran T, Andersen R, Sherman SP, Pyle AD. 2013. Insights into skeletalmuscle development and applications in regenerative medicine. Int RevCell Mol Biol 300:51– 83. http://dx.doi.org/10.1016/B978-0-12-405210-9.00002-3.
10. von Maltzahn J, Chang NC, Bentzinger CF, Rudnicki MA. 2012. Wntsignaling in myogenesis. Trends Cell Biol 22:602– 609. http://dx.doi.org/10.1016/j.tcb.2012.07.008.
11. Cossu G, Borello U. 1999. Wnt signaling and the activation of myogenesisin mammals. EMBO J 18:6867– 6872. http://dx.doi.org/10.1093/emboj/18.24.6867.
12. Borello U, Coletta M, Tajbakhsh S, Leyns L, De Robertis EM, Buck-ingham M, Cossu G. 1999. Transplacental delivery of the Wnt antagonistFrzb1 inhibits development of caudal paraxial mesoderm and skeletalmyogenesis in mouse embryos. Development 126:4247– 4255.
13. Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R.1995. Lack of beta-catenin affects mouse development at gastrulation.Development 121:3529 –3537.
14. Hutcheson DA, Zhao J, Merrell A, Haldar M, Kardon G. 2009. Embry-onic and fetal limb myogenic cells are derived from developmentally dis-tinct progenitors and have different requirements for beta-catenin. GenesDev 23:997–1013. http://dx.doi.org/10.1101/gad.1769009.
15. Liu Y, Sugiura Y, Wu F, Mi W, Taketo MM, Cannon S, Carroll T, LinW. 2012. �-Catenin stabilization in skeletal muscles, but not in motorneurons, leads to aberrant motor innervation of the muscle during neu-romuscular development in mice. Dev Biol 366:255–267. http://dx.doi.org/10.1016/j.ydbio.2012.04.003.
16. Iwata J, Suzuki A, Pelikan RC, Ho TV, Chai Y. 2013. Noncanonicaltransforming growth factor beta (TGF�) signaling in cranial neural crestcells causes tongue muscle developmental defects. J Biol Chem 288:29760 –29770. http://dx.doi.org/10.1074/jbc.M113.493551.
17. Iwata J, Hacia JG, Suzuki A, Sanchez-Lara PA, Urata M, Chai Y. 2012.Modulation of noncanonical TGF-beta signaling prevents cleft palate inTgfbr2 mutant mice. J Clin Invest 122:873– 885. http://dx.doi.org/10.1172/JCI61498.
18. Iwata J, Hosokawa R, Sanchez-Lara PA, Urata M, Slavkin H, Chai Y.2010. Transforming growth factor-beta regulates basal transcriptionalregulatory machinery to control cell proliferation and differentiation incranial neural crest-derived osteoprogenitor cells. J Biol Chem 285:4975–4982. http://dx.doi.org/10.1074/jbc.M109.035105.
19. Iwata J, Suzuki A, Pelikan RC, Ho TV, Sanchez-Lara PA, Chai Y. 2014.Modulation of lipid metabolic defects rescues cleft palate in Tgfbr2 mutantmice. Hum Mol Genet 23:182–193. http://dx.doi.org/10.1093/hmg/ddt410.
20. Iwata J, Suzuki A, Yokota T, Ho TV, Pelikan R, Urata M, Sanchez-Lara PA, Chai Y. 2014. TGF� regulates epithelial-mesenchymal inter-actions through WNT signaling activity to control muscle develop-ment in the soft palate. Development 141:909 –917. http://dx.doi.org/10.1242/dev.103093.
21. Brustis JJ, Elamrani N, Balcerzak D, Safwate A, Soriano M, Poussard S,Cottin P, Ducastaing A. 1994. Rat myoblast fusion requires exteriorizedm-calpain activity. Eur J Cell Biol 64:320 –327.
22. Honda H, Rostami A. 1989. Expression of major histocompatibility com-plex class I antigens in rat muscle cultures: the possible developmental rolein myogenesis. Proc Natl Acad Sci U S A 86:7007–7011. http://dx.doi.org/10.1073/pnas.86.18.7007.
23. Iwata J, Suzuki A, Pelikan RC, Ho TV, Sanchez-Lara PA, Urata M,Dixon MJ, Chai Y. 2013. Smad4-Irf6 genetic interaction and TGF�-mediated IRF6 signaling cascade are crucial for palatal fusion in mice.Development 140:1220 –1230. http://dx.doi.org/10.1242/dev.089615.
24. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R,McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011.Fast, scalable generation of high-quality protein multiple sequence align-ments using Clustal Omega. Mol Syst Biol 7:539. http://dx.doi.org/10.1038/msb.2011.75.
25. van Beest M, Dooijes D, van De Wetering M, Kjaerulff S, Bonvin A,Nielsen O, Clevers H. 2000. Sequence-specific high mobility group boxfactors recognize 10-12-base pair minor groove motifs. J Biol Chem 275:27266 –27273.
26. Tetsu O, McCormick F. 1999. Beta-catenin regulates expression of cyclinD1 in colon carcinoma cells. Nature 398:422– 426. http://dx.doi.org/10.1038/18884.
27. Yochum GS, Cleland R, Goodman RH. 2008. A genome-wide screen forbeta-catenin binding sites identifies a downstream enhancer element thatcontrols c-Myc gene expression. Mol Cell Biol 28:7368 –7379. http://dx.doi.org/10.1128/MCB.00744-08.
28. Tanaka S, Terada K, Nohno T. 2011. Canonical Wnt signaling is involvedin switching from cell proliferation to myogenic differentiation of mousemyoblast cells. J Mol Signaling 6:12. http://dx.doi.org/10.1186/1750-2187-6-12.
29. Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, Wei S, Hao W,
Suzuki et al.
1774 mcb.asm.org May 2015 Volume 35 Number 10Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

Kilgore J, Williams NS, Roth MG, Amatruda JF, Chen C, Lum L. 2009.Small molecule-mediated disruption of Wnt-dependent signaling in tis-sue regeneration and cancer. Nat Chem Biol 5:100 –107. http://dx.doi.org/10.1038/nchembio.137.
30. Shi X, Ma YQ, Tu Y, Chen K, Wu S, Fukuda K, Qin J, Plow EF, Wu C.2007. The MIG-2/integrin interaction strengthens cell-matrix adhesionand modulates cell motility. J Biol Chem 282:20455–20466. http://dx.doi.org/10.1074/jbc.M611680200.
31. Dowling JJ, Vreede AP, Kim S, Golden J, Feldman EL. 2008. Kindlin-2is required for myocyte elongation and is essential for myogenesis. BMCCell Biol 9:36. http://dx.doi.org/10.1186/1471-2121-9-36.
32. Kemler R. 1993. From cadherins to catenins: cytoplasmic protein inter-actions and regulation of cell adhesion. Trends Genet 9:317–321. http://dx.doi.org/10.1016/0168-9525(93)90250-L.
33. Nelson WJ. 2008. Regulation of cell-cell adhesion by the cadherin-catenincomplex. Biochem Soc Trans 36:149 –155. http://dx.doi.org/10.1042/BST0360149.
34. Ten Broek RW, Grefte S, Von den Hoff JW. 2010. Regulatory factors andcell populations involved in skeletal muscle regeneration. J Cell Physiol224:7–16. http://dx.doi.org/10.1002/jcp.22127.
35. Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, HassanAB, Volpin D, Bressan GM, Piccolo S. 2003. Mapping Wnt/beta-cateninsignaling during mouse development and in colorectal tumors. Proc NatlAcad Sci U S A 100:3299 –3304. http://dx.doi.org/10.1073/pnas.0434590100.
36. Ikeya M, Takada S. 1998. Wnt signaling from the dorsal neural tube isrequired for the formation of the medial dermomyotome. Development125:4969 – 4976.
37. Linker C, Lesbros C, Stark MR, Marcelle C. 2003. Intrinsic signalsregulate the initial steps of myogenesis in vertebrates. Development 130:4797– 4807. http://dx.doi.org/10.1242/dev.00688.
38. Schmidt M, Patterson M, Farrell E, Munsterberg A. 2004. Dynamicexpression of Lef/Tcf family members and beta-catenin during chick gas-trulation, neurulation, and early limb development. Dev Dyn 229:703–707. http://dx.doi.org/10.1002/dvdy.20010.
39. Galli LM, Willert K, Nusse R, Yablonka-Reuveni Z, Nohno T, Denet-claw W, Burrus LW. 2004. A proliferative role for Wnt-3a in chicksomites. Dev Biol 269:489 –504. http://dx.doi.org/10.1016/j.ydbio.2004.01.041.
40. Schmidt C, Stoeckelhuber M, McKinnell I, Putz R, Christ B, Patel K.2004. Wnt 6 regulates the epithelialisation process of the segmental platemesoderm leading to somite formation. Dev Biol 271:198 –209. http://dx.doi.org/10.1016/j.ydbio.2004.03.016.
41. Otto A, Schmidt C, Patel K. 2006. Pax3 and Pax7 expression and regu-lation in the avian embryo. Anat Embryol 211:293–310. http://dx.doi.org/10.1007/s00429-006-0083-3.
42. Munsterberg AE, Kitajewski J, Bumcrot DA, McMahon AP, Lassar AB.1995. Combinatorial signaling by Sonic hedgehog and Wnt family mem-bers induces myogenic bHLH gene expression in the somite. Genes Dev9:2911–2922. http://dx.doi.org/10.1101/gad.9.23.2911.
43. Maroto M, Reshef R, Munsterberg AE, Koester S, Goulding M, LassarAB. 1997. Ectopic Pax-3 activates MyoD and Myf-5 expression in embry-onic mesoderm and neural tissue. Cell 89:139 –148. http://dx.doi.org/10.1016/S0092-8674(00)80190-7.
44. Borello U, Berarducci B, Murphy P, Bajard L, Buffa V, Piccolo S,Buckingham M, Cossu G. 2006. The Wnt/beta-catenin pathway regulatesGli-mediated Myf5 expression during somitogenesis. Development 133:3723–3732. http://dx.doi.org/10.1242/dev.02517.
45. Brunelli S, Relaix F, Baesso S, Buckingham M, Cossu G. 2007. Betacatenin-independent activation of MyoD in presomitic mesoderm re-quires PKC and depends on Pax3 transcriptional activity. Dev Biol 304:604 – 614. http://dx.doi.org/10.1016/j.ydbio.2007.01.006.
46. Otto A, Schmidt C, Luke G, Allen S, Valasek P, Muntoni F, Lawrence-Watt D, Patel K. 2008. Canonical Wnt signaling induces satellite-cellproliferation during adult skeletal muscle regeneration. J Cell Sci 121:2939 –2950. http://dx.doi.org/10.1242/jcs.026534.
47. Kalaszczynska I, Geng Y, Iino T, Mizuno S, Choi Y, Kondratiuk I, SilverDP, Wolgemuth DJ, Akashi K, Sicinski P. 2009. Cyclin A is redundant infibroblasts but essential in hematopoietic and embryonic stem cells. Cell138:352–365. http://dx.doi.org/10.1016/j.cell.2009.04.062.
48. Murphy M, Stinnakre MG, Senamaud-Beaufort C, Winston NJ,Sweeney C, Kubelka M, Carrington M, Brechot C, Sobczak-Thepot J.1997. Delayed early embryonic lethality following disruption of the mu-
rine cyclin A2 gene. Nat Genet 15:83– 86. http://dx.doi.org/10.1038/ng0197-83.
49. Gopinathan L, Tan SL, Padmakumar VC, Coppola V, Tessarollo L,Kaldis P. 2014. Loss of cdk2 and cyclin A2 impairs cell proliferation andtumorigenesis. Cancer Res 74:3870 –3879. http://dx.doi.org/10.1158/0008-5472.CAN-13-3440.
50. Lee G, White LS, Hurov KE, Stappenbeck TS, Piwnica-Worms H. 2009.Response of small intestinal epithelial cells to acute disruption of cell di-vision through CDC25 deletion. Proc Natl Acad Sci U S A 106:4701– 4706.http://dx.doi.org/10.1073/pnas.0900751106.
51. Chen MS, Hurov J, White LS, Woodford-Thomas T, Piwnica-WormsH. 2001. Absence of apparent phenotype in mice lacking Cdc25C proteinphosphatase. Mol Cell Biol 21:3853–3861. http://dx.doi.org/10.1128/MCB.21.12.3853-3861.2001.
52. Pansters NA, van der Velden JL, Kelders MC, Laeremans H, Schols AM,Langen RC. 2011. Segregation of myoblast fusion and muscle-specificgene expression by distinct ligand-dependent inactivation of GSK-3�. CellMol Life Sci 68:523–535. http://dx.doi.org/10.1007/s00018-010-0467-7.
53. de Lau WB, Snel B, Clevers HC. 2012. The R-spondin protein family.Genome Biol 13:242. http://dx.doi.org/10.1186/gb-2012-13-3-242.
54. Jin YR, Yoon JK. 2012. The R-spondin family of proteins: emergingregulators of WNT signaling. Int J Biochem Cell Biol 44:2278 –2287. http://dx.doi.org/10.1016/j.biocel.2012.09.006.
55. Han XH, Jin YR, Seto M, Yoon JK. 2011. A WNT/beta-catenin signalingactivator, R-spondin, plays positive regulatory roles during skeletal myo-genesis. J Biol Chem 286:10649 –10659. http://dx.doi.org/10.1074/jbc.M110.169391.
56. Han XH, Jin YR, Tan L, Kosciuk T, Lee JS, Yoon JK. 2014. Regulationof the follistatin gene by RSPO-LGR4 signaling via activation of the WNT/beta-catenin pathway in skeletal myogenesis. Mol Cell Biol 34:752–764.http://dx.doi.org/10.1128/MCB.01285-13.
57. Abmayr SM, Pavlath GK. 2012. Myoblast fusion: lessons from flies andmice. Development 139:641– 656. http://dx.doi.org/10.1242/dev.068353.
58. Millay DP, O’Rourke JR, Sutherland LB, Bezprozvannaya S, SheltonJM, Bassel-Duby R, Olson EN. 2013. Myomaker is a membrane activatorof myoblast fusion and muscle formation. Nature 499:301–305. http://dx.doi.org/10.1038/nature12343.
59. Dowling JJ, Gibbs E, Russell M, Goldman D, Minarcik J, Golden JA,Feldman EL. 2008. Kindlin-2 is an essential component of intercalateddiscs and is required for vertebrate cardiac structure and function. CircRes 102:423– 431. http://dx.doi.org/10.1161/CIRCRESAHA.107.161489.
60. Rochat A, Fernandez A, Vandromme M, Moles JP, Bouschet T, CarnacG, Lamb NJ. 2004. Insulin and wnt1 pathways cooperate to induce reservecell activation in differentiation and myotube hypertrophy. Mol Biol Cell15:4544 – 4555. http://dx.doi.org/10.1091/mbc.E03-11-0816.
61. Polesskaya A, Seale P, Rudnicki MA. 2003. Wnt signaling induces themyogenic specification of resident CD45� adult stem cells during mus-cle regeneration. Cell 113:841– 852. http://dx.doi.org/10.1016/S0092-8674(03)00437-9.
62. Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA. 2008. A temporalswitch from notch to Wnt signaling in muscle stem cells is necessary fornormal adult myogenesis. Cell Stem Cell 2:50 –59. http://dx.doi.org/10.1016/j.stem.2007.10.006.
63. Gates J, Peifer M. 2005. Can 1000 reviews be wrong? Actin, alpha-catenin,and adherens junctions. Cell 123:769 –772.
64. Krauss RS, Cole F, Gaio U, Takaesu G, Zhang W, Kang JS. 2005. Closeencounters: regulation of vertebrate skeletal myogenesis by cell-cell con-tact. J Cell Sci 118:2355–2362. http://dx.doi.org/10.1242/jcs.02397.
65. Krauss RS. 2010. Regulation of promyogenic signal transduction by cell-cell contact and adhesion. Exp Cell Res 316:3042–3049. http://dx.doi.org/10.1016/j.yexcr.2010.05.008.
66. Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M,Hynes RO. 1997. Developmental defects in mouse embryos lacking N-cadherin. Dev Biol 181:64 –78. http://dx.doi.org/10.1006/dbio.1996.8443.
67. Charlton CA, Mohler WA, Radice GL, Hynes RO, Blau HM. 1997.Fusion competence of myoblasts rendered genetically null for N-cadherinin culture. J Cell Biol 138:331–336. http://dx.doi.org/10.1083/jcb.138.2.331.
68. Hollnagel A, Grund C, Franke WW, Arnold HH. 2002. The cell adhesionmolecule M-cadherin is not essential for muscle development and regen-eration. Mol Cell Biol 22:4760 – 4770. http://dx.doi.org/10.1128/MCB.22.13.4760-4770.2002.
69. Jaeger MA, Sonnemann KJ, Fitzsimons DP, Prins KW, Ervasti JM.
WNT/�-Catenin Signaling Regulates Muscle Development
May 2015 Volume 35 Number 10 mcb.asm.org 1775Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

2009. Context-dependent functional substitution of alpha-skeletal actinby gamma-cytoplasmic actin. FASEB J 23:2205–2214. http://dx.doi.org/10.1096/fj.09-129783.
70. Crawford K, Flick R, Close L, Shelly D, Paul R, Bove K, Kumar A, LessardJ. 2002. Mice lacking skeletal muscle actin show reduced muscle strength andgrowth deficits and die during the neonatal period. Mol Cell Biol 22:5887–5896. http://dx.doi.org/10.1128/MCB.22.16.5887-5896.2002.
71. Belyantseva IA, Perrin BJ, Sonnemann KJ, Zhu M, Stepanyan R, McGeeJ, Frolenkov GI, Walsh EJ, Friderici KH, Friedman TB, Ervasti JM.2009. Gamma-actin is required for cytoskeletal maintenance but not de-velopment. Proc Natl Acad Sci U S A 106:9703–9708. http://dx.doi.org/10.1073/pnas.0900221106.
72. Sonnemann KJ, Fitzsimons DP, Patel JR, Liu Y, Schneider MF, MossRL, Ervasti JM. 2006. Cytoplasmic gamma-actin is not required for skel-etal muscle development but its absence leads to a progressive myopathy.Dev Cell 11:387–397. http://dx.doi.org/10.1016/j.devcel.2006.07.001.
73. Noden DM, Francis-West P. 2006. The differentiation and morphogen-esis of craniofacial muscles. Dev Dyn 235:1194 –1218. http://dx.doi.org/10.1002/dvdy.20697.
74. Yamane A. 2005. Embryonic and postnatal development of masticatoryand tongue muscles. Cell Tissue Res 322:183–189. http://dx.doi.org/10.1007/s00441-005-0019-x.
75. Tajbakhsh S, Rocancourt D, Cossu G, Buckingham M. 1997. Redefiningthe genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5act upstream of MyoD. Cell 89:127–138. http://dx.doi.org/10.1016/S0092-8674(00)80189-0.
76. Tzahor E, Kempf H, Mootoosamy RC, Poon AC, Abzhanov A, TabinCJ, Dietrich S, Lassar AB. 2003. Antagonists of Wnt and BMP signalingpromote the formation of vertebrate head muscle. Genes Dev 17:3087–3099. http://dx.doi.org/10.1101/gad.1154103.
77. Al-Qattan MM. 2011. WNT pathways and upper limb anomalies. J HandSurg 36:9 –22. http://dx.doi.org/10.1177/1753193410380502.
78. Kim N, Vu TH. 2006. Parabronchial smooth muscle cells and alveolarmyofibroblasts in lung development: birth defects research, part C. Em-bryo Today Rev 78:80 – 89. http://dx.doi.org/10.1002/bdrc.20062.
79. He F, Chen Y. 2012. Wnt signaling in lip and palate development. FrontOral Biol 16:81–90. http://dx.doi.org/10.1159/000337619.
80. Alexander MS, Kawahara G, Motohashi N, Casar JC, Eisenberg I,Myers JA, Gasperini MJ, Estrella EA, Kho AT, Mitsuhashi S, ShapiroF, Kang PB, Kunkel LM. 2013. MicroRNA-199a is induced in dystro-phic muscle and affects WNT signaling, cell proliferation, and myo-genic differentiation. Cell Death Differ 20:1194 –1208. http://dx.doi.org/10.1038/cdd.2013.62.
Suzuki et al.
1776 mcb.asm.org May 2015 Volume 35 Number 10Molecular and Cellular Biology
on February 19, 2018 by guest
http://mcb.asm
.org/D
ownloaded from


















