Walleye stereo view of the DELFWT-PHDELWT (Refmac) omit ... · S3 Supplementary Figure S3:...
48
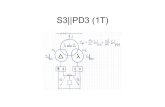
Supplementary Figure S1: Stereo view of EPZ004777 in 4ER3 Walleye stereo view of the DELFWT-PHDELWT (Refmac) omit map for EPZ004777 to 2.40 Å resolution, contoured at 3σ. The figure was prepared using PyMOL.
Transcript of Walleye stereo view of the DELFWT-PHDELWT (Refmac) omit ... · S3 Supplementary Figure S3:...

Supplementary Figure S1: Stereo view of EPZ004777 in 4ER3
Walleye stereo view of the DELFWT-PHDELWT (Refmac) omit map for EPZ004777 to 2.40 Å
resolution, contoured at 3σ. The figure was prepared using PyMOL.

S2
Supplementary Figure S2: Electron density map of inhibitors and activation loop
Electron density of the inhibitors and activation loop for the 7 structures presented in this work

S3
Supplementary Figure S3: Inhibition and selectivity profiles of EPZ004777 and analogues
(a) A radioactivity-based assay is used to follow the dose response effect of chemical inhibitors
on DOT1L activity. (b) EPZ004777 and SGC0946 are inactive against a panel of 12 protein
methyltransferases and DNMT1. Assay details are presented in Supplementary Methods
and Supplementary Table S3.

S4
Supplementary Figure S4: Cellular activity of EPZ004777 and SGC0946
(a) Comparative immunoblot of EPZ004777 and SGC0946 in Molm13 MLL cells, demonstrating
reduced H3K79me2. (b) Inhibition of H3K79me2 by a series of DOT1L inhibitors in A431 cells
(4 day treatment). (c,d) SGC0946 (1µM, 7 days) more effectively inhibited MLL target genes
HOXA9 and Meis1 than EPZ004777. Note that Meis1 expression in THP1 cells was too low for
reliable detection.

S5
Supplementary Figure S5: Binding affinity measured by SPR
(a) Biacore SPR sensorgrams of the tested compounds from single cycle kinetics runs with
5 concentrations. (b) Summary of DOT1L SPR data.

S6
Supplementary Figure S6: ITC profile of EPZ004777 binding to DOT1L
Isothermal titration calorimetry indicates a 1:1 binding stoichiometry between EPZ004777 and
DOT1L in solution.

S7
Supplementary Figure S7: Molecular dynamics simulations
Molecular dynamics RMS values for (a) all residues and (b) loop regions of apo DOT1L and
SAM-bound DOT1L.

S8
Supplementary Figure S8: Conformational flexibility of DOT1L determined by HX MS
(a) Coverage map of all the peptic peptides in DOT1L. 98% total protein coverage was obtained
under the experimental conditions described. Not all these peptides were followed with HX MS.
Amino acids that were not covered by digestion are represented with an X. (b) Representative

S9
mass spectra of a peptide (188-199) showing that upon drug binding the isotopic distribution
changes showing less deuterium incorporation due to protection by the drug. (c) HX MS
Relative Difference Plots for two peptides (as labeled) from DOT1L in the presence of
EPZ004777, FED2 and SAH. The differences in deuterium incorporation between the bound
protein and the native protein are plotted for deuterium exchange time points of 10 s (blue bars),
1 min (red bars), 10 min (green bars), and 1 hr (purple bars). The data shown are for peptides
common to all three experiments, ordered from N to C terminus (top to bottom).

S10
Supplementary Figure S9: Hydrophobicity of the DOT1L cofactor binding site
The enclosed portion of the cofactor site is more hydrophobic in complex with FED2 than with
SAM and close analogs: SAM (PDB codes 1NW3 and 3QOW), SAH (PDB code 3QOX), a
methylated SAH analog (PDB codes 3SR4), Bromo-deaza-SAH (PDB code 3SX0).

S11
Supplementary Table S1a: Data Collection, Phasing, and Refinement Statistics
EPZ004777 SGC0946 FED1 SGC0947 FED2 two EPZ004777 molecules 5-iodotubercidin
Data Collection
Space Group P65 P65 P65 P65 P65 P65 P65
Cell Dimensions
a = b (A) 155.71 149.91 150.59 150.62 150.01 150.61 149.78
c (A) 48.49 52.84 52.88 52.80 53.24 53.18 53.52
Data Indexing and Scaling
Wavelength 0.97931 0.97931 0.97907 0.97932 0.97944 0.97931 0.91963
Resolution 45.0 - 2.40 (2.53) 50.0 - 2.3 (2.42) 40 - 2.5
(2.54) 43.0 - 2.2 (2.32) 45.0 - 2.15 (2.27)
45.0 - 2.57 (2.71)
50.0 - 2.05 (2.12)
Measured reflections 29179 337447 845351 397116 422310 597179 256567
Unique reflections 26567 30402 24065 34959 37551 22277 43269
Rmerge 0.106 (0.715) 0.083 (0.986) 0.070
(0.825) 0.116 (0.835) 0.112
(0.815) 0.094 (0.939) 0.098 (0.973)
I/sigI 12.7 (3.0) 18.3 (2.8) 21.2 (2.58) 13.2 (3.2) 15.2 (3.1) 21.3 (4.2) 27.1 (2.0)
Completeness (%) 99.7 (99.6) 99.9 (99.9) 99.9 (100.0) 99.7 (100) 100 (100) 100 (100) 99.6 (96.7)
Redundancy 11.0 (10.6) 11.1 (11.3) 11.1 (11.0) 11.4 (11.4) 11.2 (11.0) 11.0 (11.3) 5.9 (4.5)
PDB code 4ER3 4ER6 4ER0 4ER7 4EQZ 4ER5 3UWP
Refinement
Resolution 35.0 - 2.40 35.0 - 2.30 35.0 - 2.50 35.0 - 2.20 25.0 - 2.15 49.0 - 2.57 40.0 - 2.05
Number of Reflections 26518 28891 22790 33201 35653 21147 41035
Test Set 1310 1479 1189 1722 1850 1100 2092
Rwork/Rfree 0.2503 / 0.2819 0.217 / 0.230 0.237 /
0.272 0.218 / 0.242 0.199 /
0.221 0.195 / 0.218 0.195 / 0.223

S12
Number of Atoms 2557 2688 2498 2757 2874 2647 2801
Mean Bfactor 44.47 56.33 43.25 37.99 27.56 43.16 35.82
Ramachandran Favored 96.7 98.19 97.09 98.75 98.17 98.14 97.94
Ramachandran Disallowed 0 0 0 0 0 0 0
RMS deviations
Bond lengths (A) 0.010 0.012 0.017 0.014 0.015 0.011 0.013
Bond Angles () 1.154 1.044 1.02 1.38 1.387 1.307 1.327
Data
collection
Beamline 31ID 31ID 19ID 31ID 23ID 31ID 23ID
Collection/Index Mosflm XDS HKL Mosflm XDS XDS HKL
Geometric
Restraints Elbow Elbow PRODRG PRODRG PRODRG Elbow PRODRG
Supplementary Table S1b: Number of atoms/average B-factors (Å2)/average occupancies for some atom selections with MOLEMAN
Complex EPZ004777 SGC0946 FED1 SGC0947 FED2 two EPZ004777
molecules 5-iodotubercidin
Activation loop (residues 122-
140) 54/71.342/1.000 114/75.823/1.000 23/71.211/1.000 69/86.735/1.000 144/52.656/0.939 142/79.199/0.986 143/46.601/1.000
Binding loop (residues 301-
311) 20/57.954/1.000 79/76.894/0.924 61/80.202/1.000 75/61.265/1.000 75/39.540/1.000 45/77.019/1.000 75/45.681/1.000
Protein others 2393/53.547/0.979 2422/57.540/0.996 2369/51.769/0.984 2475/45.016/0.968 2510/32.566/0.959 2364/50.828/0.988 2491/42.579/0.984
Inhibitor 39/44.927/1.000 40/64.464/0.988 39/56.235/1.000 74/45.576/0.496 36/26.539/1.000
2409/51.318/0.988;
39/59.566/0.700 20/40.187/0.965
Regarding inclusion of weak diffraction data for crystallographic model refinement, we refer to a recent paper29. Similar to a method described therein, we investigated the effect of weak higher resolution diffraction data on model quality using the 5-iodotubercidin complex as an example. We performed refinement both at “full resolution” (2.05Å) and “reduced resolution” (2.21Å, coinciding with the boundary of a lower resolution shell for which separate statistics are available) and compared the fit of either model version to the “reduced resolution” data, and model geometry. Refmac-5.7.0027 and phenix.model_vs_data_1.8-1069 were used for this analysis.

S13
Supplementary Table S1c: The effect of higher resolution data on fit and geometry of the 5-iodotubercidin complex model
High resolution limit for refinement (Å) 2.05 2.21
High resolution shell range (Å) 2.12-2.05 2.31-2.21
High resolution shell Rmerge (%) 97.3 53.4
Rwork/Rfree for data to high resolution limit for refinement (%) 19.2/21.9 18.5/21.1
Rwork/Rfree for data to 2.21Å (%) 18.7/21.3 18.5/21.1

S14
Supplementary Table S2: Caco-2 permeability and efflux of EPZ004777 and SGC0946
Compound (10 µM)
Papp
A to B
(permeability)
Papp
B to A
(efflux)
EPZ04777 0 5.4 ± 0.6
SGC0946 5.7 ± 0.7 10.6 ± 1.9
Digoxin (Pgp control) 0.4 ± 0.0 13.2 ± 1.7
Atenolol (neg.control) 0.1 ± 0.1 0.2 ± 0.1
Metoprolol (pos. control) 68.2 ± 1.3 31.1 ± 1.1
Permeability of EPZ004777 and SGC0946 was measured in Caco-2 cells as described in the
Supplementary Methods section. This is an in vitro assay to test for intestinal absorption and
efflux of compounds that is also indicative of cell permeability. Both compounds were tested in
triplicate along with metoprolol (a positive control with high permeability and low efflux), atenolol
(low permeability) and digoxin (low permeability and high efflux). SGC0946 had a higher
apparent permeability (5.7x10-6 cm/sec) than EPZ004777 (not detectable). Papp: Apparent
Permeability/Efflux (rate) coefficient (x10-6 cm/sec), n=3. A: Apical. B: Basolateral

S15
Supplementary Table S3: Activity profile of SGC0946 against a panel of 29 diverse
receptors from the Ricerca selectivity panel
Cat # Assay Name Batch* Spec. Rep. Conc. % Inh.
200510 Adenosine A1 312075 hum 2 1 µM -4
200610 Adenosine A2A 312076 hum 2 1 µM 0
203100 Adrenergic α1A 312078 rat 2 1 µM 22
203200 Adrenergic α1B 312079 rat 2 1 µM 17
203620 Adrenergic α2A 3 12080 hum 2 1 µM 7
204010 Adrenergic β1 312081 hum 2 1 µM -1
204110 Adrenergic β2 312061 hum 2 1 µM -1
214600 Calcium Channel L-Type, Dihydropyridine 312056 rat 2 1 µM 2
217030 Cannabinoid CB1 312084 hum 2 1 µM 5
219500 Dopamine D1 312085 hum 2 1 µM 7
219700 Dopamine D2S 312086 hum 2 1 µM -3
226600 GABAA, Flunitrazepam, Central 312088 rat 2 1 µM 8
226500 GABAA, Muscimol, Central 312087 rat 2 1 µM 19
233000 Glutamate, NMDA, Phencyclidine 312091 rat 2 1 µM 0
239610 Histamine H1 312092 hum 2 1 µM -1
241000 Imidazoline I2, Central 312093 rat 2 1 µM 10
252710 Muscarinic M2 312095 hum 2 1 µM 7
252810 Muscarinic M3 312096 hum 2 1 µM 19
258590 Nicotinic Acetylcholine 312032 hum 2 1 µM 13
258700 Nicotinic Acetylcholine α, Bungarotoxin 312097 hum 2 1 µM -1
260410 Opiate µ(OP3, MOP) 312067 hum 2 1 µM -3
264500 Phorbol Ester 312098 mouse 2 1 µM -1
265600 Potassium Channel [KATP] 312099 ham 2 1 µM -15
265900 Potassium Channel hERG 312102 hum 2 1 µM -15
268420 Prostanoid EP4 312103 hum 2 1 µM -5
270000 Rolipram 312105 rat 2 1 µM 24
271700 Serotonin (5-Hydroxytryptamine) 5-HT2B 312106 hum 2 1 µM 8
278110 Sigma σ1 312057 hum 2 1 µM -10
204410 Transporter, Norepinephrine (NET) 312038 hum 2 1 µM 13

S16
Supplementary Table S4: Experimental conditions used for selectivity profiling
Protein Protein conc. (µM)
Peptide Peptide conc. (µM)
SAM (µM)
SUV39H2 0.02 H3 1-25a 3 3.5
G9a 0.02 H3 1-25 5 2
EHMT1 0.02 H3 1-25 5 2
SETDB1 0.02 H3 1-25 10 15
PRMT3 0.1 H4 1-24 7 9
SETD7 0.02 H3 1-25 90 1
MLL 1 H3 1-25 10 2
SETD8 0.05 H4 1-24b 50 1.5
SUV420H1 0.5 H4 K20Me1c 3 12
SUV420H2 1 H4 K20Me1 5 9
PRC2 0.1 H3 21-44d 5 2
DNMT1 0.1 ds-DNAe 0.1 2
PRMT5 0.2 H4 1-24 10 2
(a) A peptide corresponding to the first 25 residues of histone H3; (b) first 24 residues of histone H4; (c) a peptide corresponding to the first 24 residues of histone H4 with lysine 20 monomethylated; (d) residues 21 to 44 of histone H3; (e) double stranded DNA.

S17
Supplementary Methods
Protein expression and purification
To produce recombinant DOT1L fragments containing N-terminal 351 and 420 amino acids in E. coli, the
corresponding cDNA fragments were amplified by PCR and cloned into a modified pET28-MHL vector with an N-
terminal 6-His tag. The proteins were overexpressed in E.coli BL21 (DE3) V2R-pRARE in Terrific Broth medium
in the presence of 50 µg/mL kanamycin and chloramphenicol. Cells were grown at 37°C to an OD600 of 1.5 and
induced by isopropyl-1-thio-D-galactopyranoside (IPTG, final concentration 1 mM) and incubated overnight at
15°C. The cell pellets were frozen in liquid nitrogen and stored at -80°C. For purification, the cell paste was thawed
and resuspended in lysis buffer with 1mM phenylmethyl sulfonyl fluoride (PMSF). Slightly different purification
protocols were used for DOT1L (1-420) and DOT1L (1-351). DOT1L (1-420) was purified by Ni-NTA column
(Qiagen) and processed by in-house produced TEV protease to remove the His tag. The protein was then incubated
in 50 mM Tris-HCl pH 8.0, 1 mM MgCl2 with benzonase nuclease for 2 hours at room temperature to remove DNA
which binds to the C-terminal region of DOT1L (1-420), but not DOT1L (1-351). The filtered protein sample was
diluted with 50 mM K2HPO4/ KH2PO4 pH 7.0, and further purified by HiTrap-SP (GE Healthcare). The protein
was finally purified by gel filtration (Superdex 200, GE Healthcare). For purification of DOT1L (1-351), only Ni-
NTA affinity chromatography and size exclusion chromatography were used.
DOT1L purified by above-mentioned methods always contains co-purified SAM as reported6. This partially
occupied form of DOT1L is applicable for use in the enzyme assay, crystallography and SPR experiment. However,
for ITC and hydrogen-deuterium exchange experiments, DOT1L(1-420) in apo form is required. During the ITC
experiment, replacement of SAM by EPZ004777 was endothermic, and produced a distinct profile, very different
from the exothermic profile obtained with apo-DOT1L. To obtain SAM-free DOT1L, 2 mL of 10mg/mL partially
occupied form of DOT1L (1-420) was incubated with 1.0 mM 5-iodotubercidin for one hour at room temperature.
The sample was then concentrated to 0.5 mL and then diluted with 50 mM K2HPO4/ KH2PO4 pH 7. The diluted
sample was loaded onto a HiTrap SP FF column (GE Healthcare), and then washed with 4 liters of buffer (50 mM
K2HPO4/ KH2PO4, pH 7.0) with a flow rate of 5 mL/min to remove SAM before elution with NaCl. The collected
protein fraction was further purified by gel filtration (Superdex 200, GE Healthcare). SAM-free DOT1L(1-420)
shows an A260/A280 around 0.57, while the partially occupied form of DOT1L shows an A260/A280 around 0.66.
Data Collection and Indexing
Crystallographic data collection and indexing information for the structures presented herein are presented in Table
S1. The following beam lines at the Advanced Photon Source were used for data collection: 31ID (LRL-CAT,
http://lrlcat.lilly.com/), 23ID (GMCA-CAT, www.gmca.anl.gov), and 19ID (SBC-CAT, 222.sbc.anl.gov).
Data were indexed using the XDS program30 or within the HKL suite of programs31 or using Mosflm32. Original
index and test set reflections were selected from the 2.1 Å DOT1L structure, PDB accession number 3QOW and
implemented using the pointless program followed by scala from the CCP4i suite of programs33. A paired down

S18
model of DOT1L was then used for rigid body refinement directly into each dataset. Missing loops and ligands
were then manually built and real space refined within COOT34, followed by iterative rounds of refinement using
refmac 535. Geometric restraints for compounds were created using the program Elbow from the phenix suite of
programs36 or using the online program PRODRG37. All models were refined with good geometric restraints and
excellent clash and Molprobity scores and deposited in the PDB.
Isothermal titration calorimetry
SAM-free DOT1L(1-420), purified by above-mentioned method, was used in the ITC experiment with a VP-ITC
calorimeter (MicroCal, LLC). The enthalpy of binding of DOT1L (15 µM in cell) and EPZ004777 (0.2 mM in
syringe) was measured at 25°C in 20 mM Tris-HCl pH 8.0, 200 mM NaCl, 5% DMSO. A clear 1:1 stoichiometry
was observed. We did not attempt to calculate the KD by fitting the binding isotherms since the binding was too
tight to be determined directly by ITC, instead, we used SPR to measure the KD.
Cellular H3K79 methylation assay in A431 cells
A431 cells were plated at 1,000 cells/well in 50µL in 384-well clear bottom plates (Corning 3712) and incubated
overnight. Cells were treated with compound with an automated pin transfer instrument (Janus, Perkin Elmer) and
incubated for 3-4 days. Following incubation with compound, media was aspirated (EL406, BioTek), cells were
fixed in 50 µL formaldehyde solution (3.7% formaldehyde in PBS) and incubated 10 minutes at room temperature.
Fixation solution was aspirated and cells were rinsed in 50µL of blocking solution (1% BSA in PBS) twice. Then
50µL permeabilization solution was added (1% SDS in blocking solution) for 2 minutes, then aspirated. After rinse
with blocking solution, cells were incubated in 50µL of blocking solution for 30 minutes at room temperature.
Blocking solution was aspirated, and cells were incubated for 1+ hour at room temperature in 10 µL of primary
antibody for dimethylated histone 3 K79 (ab1791) at a 1:5000 dilution in blocking solution. Primary antibody
solution was aspirated, and cells were washed twice in 50µL of blocking solution. Following the second wash, cells
were incubated in 10 µL for 60 minutes at room temperature in secondary antibody (Invitrogen A-21244) and
nuclear staining (Invitrogen H3570) solution at 1:1000 dilution in blocking solution. Secondary antibody and
nuclear staining solution was aspirated and cells were washed two times in 50µL of blocking solution. Then 50µL of
PBS was added to each well. Image acquisition was performed on a high content screening microscope
(ImageXpress Micro, Molecular Devices), and image analysis (MetaXpress3.0, Molecular Devices) was performed
to obtain average dimethylated lysine 79 and acetylated-tubulin signal per cell based on treatment. Replicate
experimental data from incubations with inhibitor were normalized to DMSO controls. Dose response data was
generated (Graphpad Prism) by normalization of maximum and minimum dimethylated lysine 79 compared to
EPZ004777.
Caco-2 permeability
Human, epithelial Caco-2 cells were grown as monolayers, fully polarized and differentiated. All compounds were

S19
tested under non-gradient pH conditions (pH 7.4/7.4) for 90 minutes as previously described38, 39. LC/MS analysis
was performed for all samples using a Waters Xevo QTof and an ACQUITY UPLC system, to determine relative
peak areas of parent compound. The percent of transported drug was calculated based on these peak areas, relative
to the initial, dosing concentration.
Hydrogen-deuterium exchange
Hydrogen exchange experiments were performed essentially as described in Iacob et al.36 70 pmol of DOT1L
protein were incubated with different concentrations of inhibitor for a final protein:inhibitor concentration ratio of
1:6 during deuterium labeling. Under these conditions, >98% of the DOT1L protein molecules were bound based
on the Kds described in Figure 3b. All mixtures were incubated for 30 min at room temperature before deuterium
labeling. As a control, DOT1L was incubated in 20 mM Tris, 150 mM NaCl, 3 mM DTT (pH 8.0) buffer and treated
exactly as the inhibitor bound protein. Deuterium exchange was initiated by dilution of each protein with 15-fold 20
mM Tris, 150 mM NaCl, 3 mM DTT (pD 8.0), D2O buffer at room temperature. At each deuterium exchange time
point (from 10 s to 4 hours) an aliquot from the exchange reaction was removed and labeling was quenched by
adjusting the pH to 2.5 with an equal volume of quench buffer (0.8M GnHCl, 0.8% Formic Acid, H2O). Quenched
samples were immediately frozen on dry ice and stored at -80°C until analysis.
Each frozen sample was thawed rapidly and injected into a custom Waters nanoACQUITY UPLC HDX
ManagerTM and analyzed as previously described40. The protein samples were digested using a Poroszyme
immobilized pepsin cartridge (Applied Biosystems) which was accommodated within the UPLC system. The
cooling chamber of the UPLC system, which housed all the chromatographic elements, was held at 0.1°C for the
entire time of the measurements. The injected peptides were trapped and desalted for 3 min at 100 µL/min and then
separated in 6 min by an 8–40% acetonitrile:water gradient at 40 µL/min. The separation column was a 1.0×100.0
mm ACQUITY UPLC C18 BEH (Waters) containing 1.7 µm particles and the back pressure averaged 8800 psi at
0.1°C. The average amount of back-exchange using this experimental setup was 18% to 25%, based on analysis of
highly deuterated peptide standards. Deuterium levels were not corrected for back-exchange and are therefore
reported as relative24; however, all comparison experiments were done under identical experimental conditions thus
negating the need for back exchange correction24. The UPLC step was performed with protonated solvents, thereby
allowing deuterium to be replaced with hydrogen from side chains and the amino/carboxyl terminus that exchange
much faster than amide linkages41. All experiments were performed in duplicate. The error of determining the
deuterium levels was ± 0.20 Da in this experimental setup consistent with previously obtained values28. Mass spectra
were obtained with a Waters XEVO G2 TOF equipped with standard ESI source (Waters Corp., Milford, MA,
USA). The instrument configuration was the following: capillary was 3.2 kV, trap collision energy at 6 V, sampling
cone at 35 V, source temperature of 80°C and desolvation temperature of 175°C. Mass spectra were acquired over
an m/z range of 100 to 2000. Mass accuracy was ensured by calibration with 500 fmol/µL GFP, and was less than
10 ppm throughout all experiments. The mass spectra were processed with the software DynamXTM42(Waters
Corp., Milford, MA, USA) by centroiding an isotopic distribution corresponding to the +2, +3, or +4 charge state of
each peptide. Deuteration levels were calculated by subtracting the centroid of the isotopic distribution for peptide

S20
ions of undeuterated protein from the centroid of the isotopic distribution for peptide ions from the deuterium
labeled sample. The resulting relative deuterium levels were automatically plotted versus the exchange time.
Identification of the peptic fragments was accomplished through a combination of exact mass analysis and MSE43
using Identity Software (Waters Corp., Milford, MA, USA). MSE was performed by a series of low-high collision
energies ramping from 5–30 V, therefore ensuring proper fragmentation of all the peptic peptides eluting from the
LC system. DOT1L peptic peptide map was prepared using MSTools44.
Molecular dynamics simulations
The DOT1L protein structures were isolated from the relevant crystal structures (PDBs: 1NW3 and 3QOW) and
prepared using the Protein Preparation Module from Schrödinger/2011 (Schrodinger, NY). All crystallographic
waters and counterions were removed. The Ligprep Module was then used to obtain minimized 3D structures of
SAM.
The resulting structures for the proteins and small molecules served as the starting point for the molecular dynamics
(MD) simulations, which were carried out using the PMEMD version included in the AMBER11 Molecular
Dynamics Package. These MD simulations were performed after careful relaxation of the systems using
minimization and equilibration protocols. The ionizable residues were set to their normal ionization states at neutral
pH. The protein atoms were surrounded by a periodic box of TIP3P45 water molecules that extended 10 Å from the
protein. Na+ counterions were placed by LEaP REF to neutralize the system.
The ff03.r1 version of the all-atom AMBER force field was used to model the protein, and the GAFF force field was
used for the SAM structure46. After geometry optimization at the B3LYP/6-31G* level, atom-centered partial
charges were derived using the AMBER antechamber program (RESP methodology)47. In the MD simulation
protocol, the time step was chosen to be 2 fs and the SHAKE algorithm48 was used to constrain all bonds involving
hydrogen atoms. A non-bonded cutoff of 10.0 Å was used and the non-bonded pair list was updated every 25 time
steps. Langevin dynamics was used to control the system temperature (300K) using a collision frequency of 1.0 ps-
1. Isotropic position scaling was used to maintain the system pressure at 1 atm. Periodic boundary conditions were
applied to simulate a continuous system. To include the contributions of long-range interactions, the Particle-Mesh-
Ewald (PME) REF method was used with a grid spacing of ~1 Å combined with a fourth-order B-spline
interpolation. PME also enabled computation of the potential and forces in between grid points. The trajectories
were analyzed using the PTRAJ module of AMBER. The same module was used to calculate the theoretical B-
factors. They were normalized by taking the difference between the raw B-factor and the average B-factor and
dividing this by the standard deviation.
Pocket hydrophobicity
Structures of DOT1L in complex with SAM (PDB codes 1NW3 and 3QOW), SAH (PDB code 3QOX), Bromo-
deaza-SAH (PDB code 3SX0), a methylated SAH analog (PDB code 3SR4), and compound 4 were loaded in ICM
version 3.7-2b (Molsoft, San Diego, CA), and the adenosine end common to all compounds was deleted from the

S21
structures. The molecular surface of the receptor surrounding the non-adenosine portion of the compound was
generated with Molsoft’s ICM (San Diego, CA), and all DOT1L atoms within 1.5 Å of this surface that were lining
the binding pocket were selected. Oxygen, nitrogen atoms, and hydrogen atoms attached to them were considered
polar. Other atoms were considered hydrophobic. The hydrophobicity was quantified as the ratio of hydrophobic
over polar atoms.

S22
Synthesis of EPZ004777
Reactions were run as described in the individual procedures using standard double manifold and syringe techniques; glassware was dried by baking in an oven at 130 °C for 12h prior to use. Solvents for reactions were purchased anhydrous from Sigma-Aldrich and used as received; the only exception being EtOH, which was stored over 4 Å molecular sieves. HPLC grade solvents were used for aqueous work ups and chromatography. Reagents were used as received. Reactions were monitored by thin-layer chromatography using EMD silica gel 60 F254 (250-micron) glass-backed plates (visualized by UV fluorescence quenching and staining with KMnO4) and by LC-MS using a Waters Aquity BEH C18 2 x 50 mm 1.7 µm particle column (50 °C) eluting at 1 mL/min with H2O/acetonitrile [0.2% v/v added formic acid or concentrated NH4OH(aq) solution; 95:5(0min)→1:99(3.60min)→1:99(4.00min)] using alternating positive/negative electrospray ionization (125-1000 amu) and UV detection (210-350 nm). Flash column chromatography was carried out using Merck grade 9385 silica gel 60 Å pore size (230-400 mesh). Melting points were obtained using a capillary melting point apparatus and are uncorrected. 1H NMR spectra were recorded at 400 MHz on a Bruker spectrometer and are reported in ppm using the residual solvent signal (dimethylsulfoxide-d6 = 2.50 ppm; chloroform-d = 7.27 ppm; methanol-d4 = 3.31 ppm; dichloromethane-d2 = 5.32 ppm) as an internal standard. Data are reported as: {(δ shift), [(s = singlet, d = doublet, dd, doublet of doublets, ddd = doublet of a dd, t = triplet, quin = quintet, sept = septet, br = broad, ap = apparent), (J = coupling constant in Hz) and (integration)]}. Proton-decoupled 13C NMR specta were recorded at 100 MHz on a Bruker spectrometer and are reported in ppm using the residual solvent signal (chloroform-d = 77.0 ppm; dimethylsulfoxide-d6 = 39.51 ppm; methanol-d4 = 49.15 ppm) as an internal standard. Infrared spectra were recorded using an ATR-FTIR instrument. High resolution mass spectra were acquired by flow injection on a qTOF Premiere Mass Spectrometer operating in ES+ ionization with resolution ~15,000.
Retrosynthesis.

S23
Synthetic Routes.

S24

S25
NH
N
NH
O
N
N
NH
Cl
5
4-chloro-7H-pyrrolo[2,3-d]pyrimidine (5). A 100 mL flask equipped with a reflux condenser was charged with 1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (7.11 g, 52.6 mmol) and POCl3 (100 mL, 1.07 mol). The mixture was heated at reflux for 1 h. On cooling to room temperature the majority of POCl3 was distilled through a short-path apparatus under high-vacuum (<1torr) and trapped in a receiver flask cooled with an acetone/dry-ice bath; slight warming of the flask with a heat gun was necessary towards the end of the distillation. The remaining mobile brown oil was cooled to 0 °C and small pieces of ice were slowly added until the remaining POCl3 had quenched; extracted the cold mixture with Et2O (4 x 100 mL). The organic extracts were pooled, dried (MgSO4) and filtered. Concentration under vacuum left ~7.5 g of a white solid. Recrystallization from EtOAc (~100 mL; filtered while hot using house-vacuum through a medium porosity sintered glass funnel to remove an insoluble yellow precipitate) yielded 6.14 g (76%) of the title compound
as white plates: mp 184–186 °C dec (lit.1 mp 189–190 °C dec); 1H NMR (400 MHz, DMSO-d6) δ 12.56 (br s, 1H), 8.59 (s, 1H), 7.69 (d, J = 3.4 Hz, 1H), 6.60 (d, J = 3.4 Hz, 1H).
N
N
NH
Cl
N
N
NH
ClBr
5 6
5-bromo-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (6). A 200 mL flask equipped with a 25 mL pressure-equalizing addition funnel was charged with 5 (2.51 g, 16.3 mmol) and DMF (50 mL). To the colorless solution was added N-bromosuccinimide (2.91 g, 16.4 mmol) in DMF (20 mL) dropwise over 30 min via the addition funnel. After stirring for an additional 30 min, the reaction was poured into H2O (350 mL; rapidly stirred) and the precipitate collected by vacuum filtration. The solid was air dried for 16 h to
afford 3.63 g (96%) of the title compound as a white powder: mp 205–210 °C dec (lit.2 mp 215–217 °C dec); 1H
NMR (400 MHz, DMSO-d6) δ 12.98 (br s, 1H), 8.62 (s, 1H), 7.95 (s, 1H).
O OH
HO OH
HOO OH
TBSO
O O
O OHHO
O O
7D-Ribose
(3aR,4R,6R,6aR)-6-(((tert-butyldimethylsilyl)oxy)methyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-ol
(7). An oven-dried 200 mL flask under N2 was charged with D-(−)-ribose (5.00 g, 33.3 mmol), acetone (70 mL) and conc. H2SO4 (0.15 mL, 2.8 mmol). The mixture was stirred for 3 h (all solids dissolved in ~30 min), neutralized with solid NaHCO3, filtered through Celite and concentrated under vacuum to leave 6.39 g of a colorless syrup. To
1 Davoll, J. J. Chem. Soc. 1960, 131. 2 Pudlo, J. S.; Saxena, N. K.; Nassiri, M. R.; Turk, S. R.; Drach, J. C.; Townsend. L. B. J. Med. Chem. 1988, 31, 2086.

S26
a solution of the syrup in CH2Cl2 (80 mL; under N2) was added imidazole (5.44 g, 79.9 mmol) and TBSCl (6.02 g, 39.9 mmol). The resulting white mixture was stirred for 14 h then concentrated to a white semi-solid. The semi-solid was suspended in H2O (50 mL) and extracted with EtOAc (3 x 50 mL). The organic extracts were pooled, washed with brine (50 mL), dried (Na2SO4) and filtered. Concentration gave 11.6 g of a colorless oil which was purified using a Biotage flash system with a 65i silica (350 g) cartridge (470 mL = 1 CV), eluting at 65 mL/min, detecting at 220 nm and collecting 51 mL fractions; solvent A = 99:1 hexanes/Et3N, solvent B = 94:5:1 hexanes/EtOAc/Et3N. Method: 10% B (3 CV; equilibration), 10→100% B (4 CV; linear gradient), then 100% B (11 CV; isocratic). 6.06 g (60% over 2-steps) of the title compound was obtained as a white solid (β:α = ~7:1 by 1H
NMR integration): mp 47–48 °C (lit.3 mp 50 °C); 1H NMR (400 MHz, methanol-d4) δ 5.25 (s, 1H), 4.72 (d, J = 6.0
Hz, 1H), 4.51 (d, J = 6.0 Hz, 1H), 4.13-4.10 (m, 1H), 3.73-3.65 (m, 2H), 1.44 (s, 3H), 1.31 (s, 3H), 0.93 (s, 9H), 0.11 (s, 6H).
O ClTBSO
O O
O OHTBSO
O O
87
tert-butyl(((3aR,4R,6R,6aR)-6-chloro-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methoxy)dimethyl-
silane (8).
An oven-dried flask under N2 was charged with 7 (609 mg, 2.00 mmol), THF (8 mL) and CCl4 (290 µL, 3.01 mmol). The colorless solution was cooled to -70 °C (2-propanol/dry-ice) and P(NMe2)3 (400 µL, 2.20 mmol) was added dropwise over 5 min via syringe. The mixture was stirred with warming to -40 °C over 45 min and then to 0
°C (5 min) prior to use in a reaction; treated as a 0.25M theoretical solution of 8. An aliquot (~0.2 mL) can be transferred via syringe to an oven-dried NMR tube (sealed with rubber septum and under N2) and concentrated under high vacuum to leave a colorless oil: 1
H NMR (400 MHz, dichloromethane-d2) δ 6.16-6.15 (m, 1H), 4.76 (ap d, J = 1.6 Hz, 2H), 4.35 (d, J = 1.6 Hz, 1H), 3.80 (ap d, J = 3.2 Hz, 2H), 1.61 (s, 3H), 1.37 (s, 3H), 0.89 (s, 9H), 0.071 (s, 3H), 0.056 (s, 3H); HMPA is present in the NMR and appears at 2.60 (d, J = 9.2 Hz).
N
N
NH
Cl
N
N
N
Cl
Na
OTBSO
O O
N N
N
Cl
8
5 95a
7-((3aR,4R,6R,6aR)-6-(((tert-butyldimethylsilyl)oxy)methyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-
yl)-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (9).
To an oven-dried flask equipped with a septum and N2 inlet adapter was added NaH (78.0 mg, 60% dispersion in mineral oil, 3.25 mmol), which was then washed with pentane (3 x 1 mL). After drying the NaH to a fine powder under a stream of N2, DMF (6 mL) was introduced and the suspension cooled to 0 °C. Pyrimidine 5 (491 mg, 3.20 mmol) was added over 2-3 min as a solution in DMF (2 mL) via syringe; CAUTION: H2 gas released. The resulting clear solution (5 min) was allowed to warm to room temperature and the previously prepared solution of 8 (8.00 mL, 2.00 mmol theoretical) was added over 5 min via cannula; followed by washing of the flask and cannula with THF (2 mL). The reaction mixture was stirred at room temperature for 22 h (turned cloudy and pale-green after 2 h) then poured into H2O (50
3 Rosemeyer, H.; Seela, F. Helv. Chim. Acta 1988, 71, 1573.

S27
mL) and extracted with EtOAc (3 x 50 mL).The organic extracts were pooled, washed with H2O (3 x 30 mL) and brine (50 mL), dried (MgSO4) and filtered. Concentration under vacuum gave 1.03 g of a yellow semi-solid, which was triturated with 9:1 hexanes/EtOAc (~15 mL), the solid was removed by filtration and the filtrate concentrated under vacuum to leave 911 mg of a yellow oil. Purification by silica gel (30 g) flash column (3 x 12 cm) chromatography, eluting with 87.5:12.5 hexanes/EtOAc (500 mL) yielded 616 mg (70%) of the title compound as a colorless, viscous oil: [α]D
25 –33.4° (c 0.94, MeOH); 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 7.58 (d, J =
3.6 Hz, 1H), 6.63 (d, J = 3.6 Hz, 1H), 6.42 (br s, 1H), 5.06 (br s, 1H), 4.96 (br d, J = 4.8 Hz, 1H), 4.36 (d, J = 2.8 Hz, 1H), 3.89 (dd, J = 3.0, 11.0 Hz, 1H), 3.80 (dd, J = 3.4, 11.0 Hz, 1H), 1.66 (s, 3H), 1.40 (s, 3H), 0.90 (s, 9H), 0.070 (s, 6H); 13
C NMR (100 MHz, CDCl3) δ 152.2, 151.0, 150.9, 127.1, 118.3, 114.2, 100.4, 90.6, 86.2, 85.1, 80.9,
63.4, 27.4, 25.9, 25.5, 18.4, -5.4, -5.5; IR (film) ν 2954, 2932, 2858, 1587, 1547, 1258, 1203, 1137, 1086 cm-1; HRMS (ESI) calcd for C20H31N3O4ClSi [M+H]+: 440.1772, found: 440.1772.
OTBSO
O O
N N
N
Cl
9
OHO
O O
N N
N
Cl
11
((3aR,4R,6R,6aR)-6-(4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-
4-yl)methanol (11). To a solution of silyl-ether 9 (616 mg, 1.40 mmol) and THF (8 mL) was added TBAF (1.7 mL, 1.0 M solution in THF, 1.7 mmol) in one portion via syringe. The resulting pale-yellow solution was stirred 30 min then poured into H2O (50 mL) and extracted with EtOAc (3 x 50 mL). The organic extracts were pooled, washed with H2O (1 x 25 mL) and brine (1 x 50 mL), dried (MgSO4) and filtered. Concentration under vacuum gave 557 mg of an oil. Purification by silica gel (20 g) flash column (3 x 10 cm) chromatography, eluting with 80:20 (300 mL) and then 75:25 (200 mL) CH2Cl2/EtOAc yielded 447 mg (98%) of the title compound as a colorless glass: [α]D
25 –54.2° (c
0.52, MeOH); 1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 7.34 (d, J = 3.6 Hz, 1H), 6.64 (d, J = 3.8 Hz, 1H), 5.88
(d, J = 4.9 Hz, 1H), 5.30 (dd, J = 2.0, 11.0 Hz, 1H), 5.24 (dd, J = 5.5, 5.5 Hz, 1H), 5.12 (dd, J = 1.7, 6.1 Hz, 1H), 4.50 (ap d, J = 1.8 Hz, 1H), 3.98 (ddd, J = 1.9, 1.9, 12.6, 1H), 3.82 (ddd, J = 1.9, 11.0, 12.8 Hz, 1H), 1.65 (s, 3H), 1.38 (s, 3H); 13
C NMR (100 MHz, CDCl3) δ 153.3, 150.3, 149.6, 129.6, 119.9, 114.2, 100.2, 95.7, 85.6, 83.0, 81.3, 63.3, 27.6, 25.3; IR (film) ν 3352, 2989, 2937, 1588, 1549, 1200, 1081 cm-1; HRMS (ESI) calcd for C14H17N3O4Cl [M+H]+: 326.0908, found: 326.0908.
OHO
O O
N N
N
Cl
11
OI
O O
N N
N
Cl
13
4-chloro-7-((3aR,4R,6S,6aS)-6-(iodomethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-7H-
pyrrolo[2,3-d]pyrimidine (13). An oven-dried flask under N2 was charged with I2 (218 mg, 0.859 mmol) and THF (3 mL). To the dark solution was added PPh3 (226 mg, 0.862 mmol) in portions over 5 min. The resulting yellow-orange mixture was stirred for 10 min then alcohol 11 (139 mg, 0.427 mmol) and imidazole (61.2 mg, 0.899 mmol) were added as a solution in THF

S28
(2 mL) over 2-3 min via syringe. The resulting red-brown mixture was stirred for 1 h, diluted with hexanes (15 mL; stirred mixture 20 min) and filtered through a pad (4 x 2 cm) of Celite under vacuum. The filter cake was washed with 4:1 hexanes/Et2O (2 x 10 mL) and the combined filtrates concentrated under vacuum to leave 361 mg of a white semi-solid. Purification by silica gel (20 g) flash column (3 x 8 cm) chromatography, eluting with 85:15 (400 mL) hexanes/EtOAc yielded 185 mg (99%) of the title compound as a colorless wax: [α]D
25 –45.5° (c 0.62, MeOH); 1
H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 7.46 (d, J = 3.6 Hz, 1H), 6.68 (d, J = 3.6 Hz, 1H), 6.29 (d, J = 2.8 Hz, 1H), 5.35 (dd, J = 2.8, 6.5 Hz, 1H), 5.01 (dd, J = 3.4, 6.5 Hz, 1H), 4.31 (ddd, J = 3.2, 4.4, 7.6 Hz, 1H), 3.44 (dd, J = 7.1, 10.4, 1H), 3.33 (dd, J = 4.8, 10.4 Hz, 1H), 1.64 (s, 3H), 1.40 (s, 3H); 13
C NMR (100 MHz, CDCl3) δ 152.6, 151.0, 150.6, 128.1, 118.6, 115.0, 100.9, 91.2, 85.1, 84.4, 84.3, 27.1, 25.4, 5.96; IR (film) ν 2988, 2938, 1587, 1548, 1202, 1083 cm-1; HRMS (ESI) calcd for C14H16N3O3ClI [M+H]+: 435.9925, found: 435.9926.
OI
O O
N N
N
Cl
13
ON3
O O
N N
N
N3
15
4-azido-7-((3aR,4R,6R,6aR)-6-(azidomethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-7H-
pyrrolo[2,3-d]pyrimidine (15). A flask was charged with iodide 13 (122 mg, 0.280 mmol), NaN3 (91.0 mg, 1.40 mmol) and DMF (1.5 mL). The stirred reaction mixture was heated at 85 °C for 5 h. The resulting pale-yellow solution was cooled to room temperature, diluted with EtOAc (20 mL) and filtered through a plug (1 x 2 cm) of Celite under vacuum, followed by a wash of the plug with EtOAc (10 mL). The combined filtrates were diluted with EtOAc (15 mL), washed with H2O (1 x 15 mL) and brine (1 x 15 mL), dried (MgSO4) and filtered. Concentration under vacuum gave 101 mg of a cloudy oil. Purification by silica gel (6 g) flash column (1.5 x 10 cm) chromatography, eluting with 95:5 (200 mL) CH2Cl2/EtOAc yielded 97.7 mg (>99%) of the title compound as a white foam (could not be separated from 5-10% of the α-epimer): 1
H NMR (400 MHz, CDCl3) δ 9.39 (s, 1H), 7.55 (br s, 1H), 7.19 (br s, 1H), 6.44 (br s, 1H), 5.19-5.18 (m, 1H), 4.99 (dd, J = 4.7, 4.7 Hz, 1H), 4.39 (ap d, J = 3.5 Hz, 1H), 3.71 (dd, J = 2.6, 13.1 Hz, 1H), 3.59 (dd, J = 4.8, 13.1 Hz, 1H), 1.67 (s, 3H), 1.40 (s, 3H); 13
C NMR (100 MHz, CDCl3) δ 151.4, 146.6, 141.0, 131.7, 125.8, 115.4, 103.2, 90.7, 84.8, 83.9, 81.0, 52.3, 27.3, 25.4; HRMS (ESI) calcd for C14H16N9O3 [M+H]+: 358.1376, found: 358.1376.
ON3
O O
N N
N
N3
1 5
OH2N
O O
N N
N
NH2
17
7-((3aR,4R,6R,6aR)-6-(aminomethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-7H-pyrrolo[2,3-
d]pyrimidin-4-amine (17). To a 50 mL round-bottomed flask was added diazide 15 (95.4 mg, 0.267 mmol), palladium (8.5 mg, 10% w/w on activated carbon, 0.80 mmol, 30 mol%) and MeOH (5 mL). The flask was sealed with a septum and while stirring H2 gas was bubbled through the reaction mixture from a balloon attached to a 20-gauge needle (6 inch), while venting with another 20-gauge needle (1.5 inch). After 5 min the venting needle was removed and the reaction stirred under a positive pressure (1 atm) of H2 gas for 1.5 h. The mixture was filtered through a pad (2 x 2 cm) of

S29
Celite under vacuum. The filter cake was washed with MeOH (2 x 5 mL) and the filtrate concentrated under vacuum to give 98.4 mg of a slightly cloudy oil. Purification by silica (15 g) flash column (3 x 6 cm) chromatography, eluting with 91:8:1 (300 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 79.8 mg (98%) of the title compound as a colorless wax: [α]D
25 –35.8° (c 0.52, MeOH); 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.07 (d, J = 3.8 Hz, 1H), 6.37 (d, J = 3.6 Hz, 1H), 6.16 (d, J = 3.3 Hz, 1H) 5.35-5.31 (m, 3H), 4.97 (dd, J = 4.0, 6.7 Hz, 1H), 4.17 (ddd, J = 4.3, 4.3, 5.6 Hz, 1H), 3.04 (dd, J = 4.4, 13.3 Hz, 1H), 2.94 (dd, J = 5.8, 13.3 Hz, 1H), 1.62 (s, 3H), 1.42 (br s, 2H), 1.38 (s, 3H); 13
C NMR (100 MHz, CDCl3) δ 156.8, 152.1, 150.5, 123.4, 114.5, 104.0, 98.8, 90.5,
86.5, 84.1, 81.6, 43.9, 27.3, 25.4; IR (film) ν 3344, 3196, 2988, 2937, 1641, 1592, 1563, 1078 cm-1; HRMS (ESI) calcd for C14H20N5O3 [M+H]+: 306.1566, found: 306.1566.
NH2
H2N OHOHN
HNH
O
19
NNH
O
N
1-(4-(tert-butyl)phenyl)-3-(3-hydroxypropyl)urea (19). An oven-dried 50 mL round-bottomed flask under N2 was charged with carbonyldiimidazole (897 mg, 5.53 mmol) and THF (15 mL). The solution was cooled to 0 °C and 4-tert-butylaniline (800 µL, 5.02 mmol) was added as a solution in THF (2 mL) dropwise over 5 min via syringe. After 5 min the reaction was allowed to warm to room temperature and stir 45 min before adding 3-aminopropanol (460 µL, 6.05 mmol) over 1 min via syringe. The solution was stirred for 1h and the THF removed by evaporation. The remaining red oil was dissolved in EtOAc (100 mL), washed with 1M HCl (3 x 50 mL), saturated NaHCO3 solution (1 x 50 mL) and brine (1 x 50 mL), dried (MgSO4) and filtered. Concentration under vacuum gave 1.27 g of a red-orange foam. Purification by silica (30 g) flash column (3 x 12 cm), eluting with 50:50 (300 mL) EtOAc/CH2Cl2 and then EtOAc (300 mL) yielded 1.11 g (88%) of the title compound as a viscous, colorless oil: 1
H NMR (400 MHz, DMSO-d6) δ 8.32 (s, 1H), 7.28 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 6.05 (br t, J = 5.0 Hz, 1H), 4.47 (t, J = 5.0 Hz, 1H), 3.45 (dt, J = 5.7, 5.7 Hz, 2H), 3.13 (dt, J = 6.2, 6.2 Hz, 2H), 1.57 (quin, J = 6.3 Hz, 2H), 1.24 (s, 9H); 13
C NMR (100 MHz, DMSO-d6) δ 155.4, 143.1, 137.9, 125.1, 117.5, 58.5, 36.4, 33.8, 32.9, 31.3; IR (film) ν 3328, 2961, 1649, 1598, 1553, 1517 cm-1; HRMS (ESI) calcd for C14H23N2O2 [M+H]+: 251.1760, found: 251.1761.
NH
O
OH
N
20
OHNH
NH
O
19
(Z)-2-((4-(tert-butyl)phenyl)imino)-1,3-oxazinan-6-ol (20). An oven-dried 100 mL round-bottomed flask under N2 was charged with CH2Cl2 (10 mL) and oxalyl chloride (205 µL, 2.39 mmol). The solution was cooled to -78 °C (acetone/dry ice) and dimethylsulfoxide (230 µL, 3.24 mmol) was added over 5 min via syringe. The solution was stirred 20 min then alcohol 19 (400mg, 1.60 mmol) was added as a solution in CH2Cl2 (6 mL) over 10 min via syringe. Stirred for 1 h with warming to -50 °C, added diisopropylethylamine (1.40 mL, 8.04 mmol) in a steady stream over 1 min, stirred 5 min and then removed the cooling bath. After 30 min (20 min to reach room temperature) the resulting solution was poured into ice cold H2O (50 mL) and extracted with CH2Cl2 (2 x 50 mL). The organic extracts were pooled, washed with ice cold 0.1 M HCl (50 mL), saturated NaHCO3 solution (50 mL), H2O (50 mL) and brine (2 x 50 mL), dried (Na2SO4) and filtered. Concentration under vacuum gave 403 mg of an off-white solid. The crude material was refluxed with EtOAc (25

S30
mL) for 20 min, allowed to cool to room temperature and then cooled to 0 °C. The white solid was isolated by vacuum filtration through a medium porosity funnel (60 mL) and then dried under high vacuum to yield 294 mg (74%) of the title compound as a white powder: mp 174–175 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.32 (d, J = 7.9 Hz, 2H), 7.18 (d, J = 7.9 Hz, 2H), 6.63 (br s, 1H; slowly exchanges on adding 1 drop D2O), 5.91 (d, J = 5.6 Hz, 1H; rapidly exchanges on adding 1 drop D2O), 5.09 (d, J = 3.1 Hz, 1H), 3.47 (ddd, J = 3.2, 12.2, 12.2 Hz, 1H), 3.12-3.09 (m, 1H), 1.95 (br dd, J = 12.5, 12.5 Hz, 1H), 1.81 (d, J = 12.9 Hz, 1H), 1.28 (s, 9H); 1
H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 8.2 Hz, 2H), 7.24 (d, J = 8.2 Hz, 2H), 5.30 (d, J = 2.6 Hz, 1H), 5.02 (br s, 1H), 3.73 (ddd, J = 4.0, 12.2, 12.2 Hz, 1H), 3.34-3.31 (m, 1H), 2.84 (d, J = 4.1 Hz, 1H), 2.19-2.06 (m, 2H), 1.32 (s, 9H); 13
C NMR (100 MHz, DMSO-d6) δ 153.9, 147.4, 140.5, 126.9, 124.8, 79.2, 34.7, 34.1, 31.2, 29.5; IR (film) ν 3402, 3238, 3071, 2959, 2905, 2868, 1641, 1506 cm-1; HRMS (ESI) calcd for C14H21N2O2 [M+H]+: 249.1603, found: 249.1603; COSY (see pg. S31–S32); NOESY (see pg. S33–S34).
OH2N
O O
N N
N
NH2
17
ONH
O O
N N
N
NH2
21
7-((3aR,4R,6R,6aR)-6-((isopropylamino)methyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-7H-
pyrrolo[2,3-d]pyrimidin-4-amine (21). A 2-dram vial was charged sequentially with amine 17 (35.1 mg, 0.115 mmol), EtOH (0.75 mL), acetone (110 µL, 10% v/v solution in EtOH, 0.15 mmol), NaBH3CN (0.35 mL, 0.50 M solution in EtOH, 0.18 mmol) and HOAc (70 µL, 10% v/v solution in EtOH, 0.12 mmol). The solution was stirred at room temperature 1.5h then added via pipet to half saturated NaHCO3 (10 mL) solution and extracted with CHCl3 (3 x 10 mL). The organic extracts were pooled, washed with brine (2 x 10 mL), dried (Na2SO4) and filtered. Concentration under vacuum gave 40.5 mg of a cloudy residue. Purification by silica pipet (4 mL; ¾ full) column chromatography, eluting with 94:5:1 (30 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 37.5 mg (94%) of the title compound as a colorless wax: [α]D
25 –18.5° (c 0.78, MeOH); 1
H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 7.08 (d, J = 3.5 Hz, 1H), 6.38 (d, J = 3.4 Hz, 1H), 6.16 (br s, 1H), 5.34 (d, J = 4.9 Hz, 1H), 5.19 (br s, 2H), 4.99 (dd, J = 5.0, 5.0 Hz, 1H), 4.29 (ap d, J = 4.4 Hz, 1H), 2.93 (dd, J = 3.9, 12.2 Hz, 1H), 2.84 (dd, J = 6.1, 12.2 Hz, 1H), 2.75 (ap quin, J = 6.2 Hz, 1H), 1.62 (s, 3H), 1.38 (s, 3H), 1.03 (ap t, J = 7.3 Hz, 6H); 13
C NMR (100 MHz, CDCl3) δ 156.7, 152.1, 150.5, 123.5, 114.5, 104.0, 98.7, 90.7,
84.9, 84.0, 82.1, 49.1, 48.7, 27.3, 25.6, 22.9, 22.7; IR (film) ν 3330, 3186, 2966, 1642, 1591, 1563, 1474, 1078 cm-1; HRMS (ESI) calcd for C17H26N5O3 [M+H]+: 348.2036, found: 348.2036.
OHN
O O
N N
N
NH2
NH
O
NH
OH2N
O O
N N
N
NH2
17 22
20
1-(3-((((3aR,4R,6R,6aR)-6-(4-amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrofuro[3,4-
d][1,3]dioxol-4-yl)methyl)amino)propyl)-3-(4-(tert-butyl)phenyl)urea (22).

S31
A 2-dram vial was charged with amine 17 (23.7 mg, 77.4 µmol), oxazinanol 20 (18.3 mg, 73.7 µmol), 4 Å molecular sieves (ca. 150 mg; finely powdered with a mortar and pestle) and EtOH (0.8 mL). The mixture was stirred 5 min then NaBH3CN (295 µL, 0.50 M solution in EtOH, 0.15 mmol) and HOAc (155 µL, 1.0 M solution in EtOH, 0.16 mmol) were added. The reaction mixture was heated at 40°C for 20 h, cooled to room temperature and filtered through a pad (2 x 2 cm) of Celite under vacuum; washed pad with CHCl3 (2 x 5 mL). The filtrate was diluted with saturated NaHCO3 solution (10 mL) and extracted with CHCl3 (3 x 10 mL). The organic extracts were pooled, dried (Na2SO4) and filtered. Concentration of the filtrate gave 38.8 mg of a cloudy residue. Purification by silica (7 g) flash column (1.5 x 13 cm) chromatography, eluting with 91:8:1 (150 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 26.5 mg (67%) of the title compound as a white solid: mp 90–94 °C; [α]D
25 –22.9° (c 0.34, MeOH); 1
H NMR (400 MHz, methanol-d4) δ 8.11 (s, 1H), 7.28-7.21 (m, 5H), 6.64 (d, J = 3.6 Hz, 1H), 6.21 (d, J = 3.2 Hz, 1H), 5.29 (dd, J = 3.2, 6.5 Hz, 1H), 4.92 (dd, J = 3.8, 6.5 Hz, 1H), 4.26 (ddd, J = 3.5, 4.5, 8.0 Hz, 1H), 3.18 (t, J = 6.7 Hz, 2H), 2.89-2.79 (m, 2H), 2.66-2.55 (m, 2H), 1.63 (quin, J = 7.0 Hz, 2H), 1.59 (s, 3H), 1.36 (s, 3H), 1.28 (s, 9H); 13
C NMR (100 MHz, methanol-d4) δ 159.2, 158.7, 152.7, 151.3, 146.6, 138.3, 126.7, 124.2, 120.5, 115.9, 105.1, 101.6, 91.2, 85.7, 85.4, 83.9, 52.8, 48.1, 38.8, 35.2, 32.0, 31.0, 27.7, 25.8; IR (film) ν 3333, 3200, 2960, 2868, 1645, 1593, 1561, 1081 cm-1; HRMS (ESI) calcd for C28H40N7O4 [M+H]+: 538.3142, found: 538.3141.
1-(3-((((2R,3S,4R,5R)-5-(4-amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-3,4-dihydroxytetrahydrofuran-2-
yl)methyl)amino)propyl)-3-(4-(tert-butyl)phenyl)urea (SGC0928). Acetonide 22 (14.2 mg, 26.4 µmol) and 9:1 TFA/H2O (1 mL) were stirred together for 1.5 h and then concentrated to dryness from toluene (3 x 2 mL) to leave 23.2 mg of a colorless residue. Purification by silica column chromatography, eluting with 89:10:1 (10 mL) and 83.5:15:1.5 (20 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 12.1 mg (92%) of the title compound as a white solid: mp 132–141 °C; [α]D
25 –19.0° (c 0.58, MeOH); 1H NMR
(400 MHz, methanol-d4) δ 8.09 (s, 1H), 7.28-7.21 (m, 5H), 6.63 (d, J = 3.8 Hz, 1H), 6.10 (d, J = 5.0 Hz, 1H), 4.55 (dd, J = 5.3, 5.3 Hz, 1H), 4.20-4.12 (m, 2H), 3.23 (t, J = 6.7 Hz, 2H), 2.99-2.90 (m, 2H), 2.72 (t, J = 7.2 Hz, 2H), 1.72 (quin, J = 6.9 Hz, 2H), 1.28 (s, 9H); 13
C NMR (100 MHz, methanol-d4) δ 159.2, 158.8, 152.5, 151.4, 146.7, 138.3, 126.7, 123.9, 120.5, 105.2, 101.3, 90.4, 83.9, 75.4, 73.6, 53.0, 48.2, 38.9, 35.2, 32.0, 31.0; IR (film) ν 3338, 3214, 2959, 2867, 1645, 1596, 1558 cm-1; HRMS (ESI) calcd for C25H36N7O4 [M+H]+: 498.2829, found: 498.2829.
ONH
O O
N N
N
NH2
21
ON
O O
N N
N
NH2
NH
O
NH
23
20
1-(3-((((3aR,4R,6R,6aR)-6-(4-amino-5-bromo-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydro-
furo[3,4-d][1,3]dioxol-4-yl)methyl)(isopropyl)amino)propyl)-3-(4-(tert-butyl)phenyl)urea (23).

S32
A 2-dram vial was charged with amine 21 (24.8 mg, 71.4 µmol), oxazinanol 20 (19.8 mg, 79.7 µmol), 4 Å molecular sieves (ca. 150 mg; finely powdered with a mortar and pestle) and EtOH (1.0 mL). After stirring the mixture at room temperature for 5 min, NaBH3CN (0.30 mL, 0.50 M solution in EtOH, 0.15 mmol) and HOAc (0.16 mL, 1.0 M solution in EtOH, 0.16 mmol) were added via sequentially via syringe. The reaction mixture was heated at 45 °C for 4 h. On cooling to room temperature the mixture was filtered through a pad (2 x 2 cm) of Celite and the filter cake washed with CHCl3 (2 x 5 mL). The filtrate was diluted with half saturated NaHCO3 (10 mL) and extracted with CHCl3 (3 x 10 mL). The organic extracts were pooled, dried (Na2SO4) and filtered. Concentration under vacuum gave 67 mg of a cloudy residue. Purification by silica (8 g) flash column (1.5 x 12 cm), eluting with 91.5:7.5:1 (150 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 36.0 mg (87%) of the title compound as a white solid: mp 96–104 °C; [α]D
25 +1.92° (c 0.52, MeOH); 1H NMR (400 MHz, methanol-d4) δ 8.09 (s, 1H), 7.29-7.22 (m,
5H), 6.63 (d, J = 3.6 Hz, 1H), 6.22 (d, J = 2.6 Hz, 1H), 5.34 (dd, J = 2.7, 6.5 Hz, 1H), 4.95 (dd, 3.6, 6.5 Hz, 1H), 4.23 (ddd, J = 3.2, 5.8, 7.7 Hz, 1H), 3.17 (t, J = 6.5 Hz, 2H), 2.94 (ap quin, J = 6.4 Hz, 1H), 2.70 (dd, J = 5.4, 13.7 Hz, 1H), 2.59 (dd, J = 7.8, 13.4 Hz, 1H), 2.50 (t, J = 6.7 Hz, 2H), 1.64-1.51 (m, 5H), 1.37 (s, 3H), 1.29 (s, 9H), 0.98 (d, J = 6.6 Hz, 3H), 0.85 (d, J = 6.5 Hz, 3H); 1
H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 6.96 (d, J = 3.8 Hz, 1H), 6.84 (br s, 1H), 6.63 (br s, 1H), 6.37 (d, J = 3.6 Hz, 1H), 6.14 (d, J = 1.6 Hz, 1H), 5.54 (dd, 1.6, 6.4 Hz, 1H), 5.32 (br s, 2H), 5.09 (dd, 3.2, 6.4 Hz, 1H), 4.37-4.33 (m, 1H), 3.19 (br s, 2H), 2.92 (sept, 6.6 Hz, 1H), 2.67-2.57 (m, 2H), 2.49 (ddd, J = 3.9, 7.8, 13.3 Hz, 1H), 2.39 (ddd, J = 3.8, 6.8, 13.2 Hz, 1H), 1.62 (s, 3H), 1.40 (s, 3H), 1.29 (s, 9H), 0.92 (d, J = 6.5 Hz, 3H), 0.77 (d, J = 6.5 Hz, 3H); 13
C NMR (100 MHz, methanol-d4) δ 159.2, 158.7, 152.7, 151.2, 146.6, 138.3, 126.7, 124.3, 120.6, 115.6, 105.1, 101.6, 91.2, 86.6, 85.3, 84.6, 53.8, 52.1, 39.7, 35.2, 32.0, 29.4, 27.6, 25.8, 18.6, 17.7 (one signal missing due to overlap with solvent); 13
C NMR (100 MHz, CDCl3) δ 156.8, 156.7, 155.8, 151.9, 149.9, 145.1, 137.0, 125.7, 124.5, 119.0, 118.9, 114.1, 104.2, 98.9, 91.7, 85.8, 84.3, 83.6, 52.2, 49.1, 40.1, 34.2, 31.4, 27.1, 25.4, 17.3, 17.0; IR (film) ν 3330, 3206, 2963, 2869, 1644, 1593, 1561, 1517, 1084 cm-1; HRMS (ESI) calcd for C31H46N7O4 [M+H]+: 580.3611, found: 580.3610.
OH2N
O O
N N
N
NH2
17
ON
O O
N N
N
NH2
NH
O
NH
23
20ONH
O O
N N
N
NH2
21
Single-pot conversion of 17 to 23. A flask was charged with amine 17 (58.8 mg, 0.192 mmol), EtOH (0.5 mL) and acetone (0.20 mL, 10% v/v solution in EtOH, 0.27 mmol). After the solution had stirred 5 min, NaBH3CN (0.58 mL, 0.50 M solution in EtOH, 0.29 mmol) and HOAc (0.30 mL, 1.0 M solution in EtOH, 0.30 mmol) were added sequentially. The reaction was stirred 30 min, then oxazinanol 20 (50.4 mg, 0.203 mmol) was added in one portion followed by 4 Å molecular sieves (ca. 200 mg; finely powdered with a mortar and pestle). After 5 min, NaBH3CN (0.58 mL, 0.50 M solution in EtOH, 0.29 mmol) and HOAc (0.30 mL, 1.0 M solution in EtOH, 0.30 mmol) were added sequentially and the reaction mixture stirred rapidly at 45 °C for 24 h. The mixture was allowed to cool to room temperature and filtered through a pad (2 x 4 cm) of Celite and the filter cake washed with CHCl3 (2 x 10 mL). The filtrate was diluted with half saturated NaHCO3 solution (25 mL) and extracted with CHCl3 (3 x 25 mL). The organic extracts were pooled, dried (Na2SO4) and filtered. Concentration under vacuum gave 144 mg of a viscous oil. Purification with a Biotage flash system using a 25+M silica (20 g) cartridge (24 mL = 1 CV), eluting at 20 mL/min, detecting at 230nm and collecting 12 mL fractions (40 total); solvent A = 98.5:1.0:0.5 CHCl3/MeOH/conc. NH4OH(aq), solvent B = 90:9:1 CHCl3/MeOH/conc. NH4OH(aq). Method: 100% A (3 CV; equilibration), 0→75% B (15 CV; linear gradient), 75% B (2 CV; isocratic), 75→100% B (1 CV; linear gradient) and 100% B (1 CV); obtained 88.9 mg (80%) of 23 as a white solid.

S33
1-(3-((((2R,3S,4R,5R)-5-(4-amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-3,4-dihydroxytetrahydrofuran-2-
yl)methyl)(isopropyl)amino)propyl)-3-(4-(tert-butyl)phenyl)urea (EPZ004777). The acetonide (23; 46.1 mg, 79.5 µmol) was stirred with 9:1 TFA/H2O (2 mL) for 2.5 h. The solution was concentrated to dryness from toluene (3 x 3 mL) and dried further under high vacuum to leave 70 mg of a glass. Purification by silica (10 g) flash column (1.5 x 17 cm), eluting with 90:9:1 (200 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 39.3 mg (92%) of the title compound as a white solid: mp 105–122 °C; [α]D
25 –4.62° (c 0.52,
MeOH); 1H NMR (400 MHz, methanol-d4) δ 8.06 (s, 1H), 7.26 (d, J = 3.6 Hz, 1H), 7.24 (d, J = 8.8 Hz, 2H), 7.17
(d, J = 8.8 Hz, 2H), 6.63 (d, J = 3.8 Hz, 1H), 6.15 (d, J = 4.6 Hz, 1H), 4.47 (dd, J = 5.1, 5.1 Hz, 1H), 4.18 (dd, 5.4, 5.4 Hz, 1H), 4.13-4.09 (m, 1H), 3.27-3.16 (m, 2H), 3.05 (sept, J = 6.5 Hz, 1H), 2.84 (dd, J = 4.4, 13.9 Hz, 1H), 2.71 (dd, J = 7.2, 14.0 Hz, 1H), 2.59 (t, J = 6.7 Hz, 2H), 1.67 (quin, J = 6.6 Hz, 2H), 1.27 (s, 9H), 1.03 (d, J = 6.5 Hz, 3H), 1.00 (d, J = 6.5 Hz, 3H); 13
C NMR (100 MHz, methanol-d4) δ 159.1, 158.7, 152.5, 151.4, 146.6, 138.2, 126.6, 123.3, 120.5, 105.0, 101.4, 89.8, 84.1, 75.4, 73.8, 54.0, 52.1, 39.7, 35.1, 32.0, 29.3, 18.4, 18.0 (one signal missing due to overlap with solvent); IR (film) ν 3334, 3210, 2963, 2869, 1645, 1596, 1558, 1517, 1476 cm-1; HRMS (ESI) calcd for C28H42N7O4 [M+H]+: 540.3298, found: 540.3297.
N
N
NH
Cl
N
N
N
Cl
K
OTBSO
O O
N N
N
Cl
8
6 10
Br Br
Br
6a
5-bromo-7-((3aR,4R,6R,6aR)-6-(((tert-butyldimethylsilyl)oxy)methyl)-2,2-dimethyltetrahydrofuro[3,4-
d][1,3]dioxol-4-yl)-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (10). Potassium hydride (14 mg, 30% dispersion in mineral oil, 0.35 mmol) was weighed into a 2-dram vial (oven dried and cooled under a stream of N2), which was then sealed using a screw-cap with Teflon coated rubber septum insert and purged with N2. The KH was washed with pentane (3 x 1 mL) and dried to a fine powder under a stream of N2. The vial was cooled to -50 °C (2-PrOH/dry ice chunks) and the KH powder suspended in a small amount of THF (0.2 mL). Pyrimidine 6 (81.4 mg, 0.350 mmol) was added by syringe as a solution in 4:1 THF/DMF (1 mL) dropwise over 3 min; CAUTION: H2 gas released. Allowed the mixture to warm to -40 °C (over 20 min) and stir
10 min, producing a clear solution. The solution was warmed to 0 °C then chloride 8 (1.00mL, 0.25 M solution in THF, 0.25 mmol; theoretical) was added via syringe over 1 min. The cooling bath was removed and the pale-yellow reaction mixture stirred at room temperature 24 h. The mixture was poured into half saturated NaCl solution (50 mL) and extracted with EtOAc (3 x 30 mL). The organic extracts were pooled, washed with H2O (2 x 30 mL) and brine (30 mL), dried (MgSO4) and filtered. Concentration under vacuum gave 158 mg of a yellow semi-solid. Purification by silica gel (12 g) flash column (1.5 x 19 cm) chromatography, eluting with 91:8:1 (200 mL) hexanes/EtOAc/Et3N yielded 57.7 mg (44%) of the title compound as a colorless, viscous oil: [α]D
25 –50.9° (c 0.64, MeOH); 1
H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 7.72 (s, 1H), 6.46 (d, J = 2.8 Hz, 1H), 4.94 (dd, J = 2.8,

S34
6.2 Hz, 1H), 4.91 (dd, J = 2.4, 6.1 Hz, 1H), 4.41 (ddd, J = 2.6, 2.6, 2.6 Hz, 1H), 3.94 (dd, J = 2.6, 11.4 Hz, 1H), 3.82 (dd, J = 3.1, 11.4 Hz, 1H), 1.66 (s, 3H), 1.39 (s, 3H), 0.92 (s, 9H), 0.125 (s, 3H), 0.119 (s, 3H); 13
C NMR (100 MHz, CDCl3) δ 152.2, 151.4, 150.1, 126.7, 115.5, 114.2, 90.8, 88.8, 86.4, 85.6, 80.9, 63.6, 27.3, 25.9, 25.4, 18.4, -
5.3, -5.5; IR (film) ν 3131, 2954, 2931, 2858, 1579, 1538, 1450, 1210, 1125, 1086 cm-1; HRMS (ESI) calcd for C20H30N3O4SiClBr [M+H]+: 518.0877, found: 518.0878.
OTBSO
O O
N N
N
Cl
9
OTBSO
O O
N N
N
Cl
10
Br
Direct synthesis of 10 from 9.
A 25 mL round-bottomed flask was charged sequentially with 9 (504 mg, 1.15 mmol), DMF (4 mL) and NBS (215 mg, 1.21 mmol) as a solution in DMF (1.5 mL) over 1 min. The solution was heated at 35 °C for 30 min, allowed to cool to room temperature, poured into H2O and extracted with EtOAc (3 x 40 mL). The organic extracts were pooled, washed with H2O (3 x 40 mL) and brine (2 x 40 mL), dried (MgSO4) and filtered. Concentration under vacuum gave 593mg (>99%) of 10 as a viscous oil; homogeneous by 1H and 13C NMR.
OTBSO
O O
N N
N
Cl
10
OHO
O O
N N
N
Cl
12
Br Br
((3aR,4R,6R,6aR)-6-(5-bromo-4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrofuro[3,4-
d][1,3]dioxol-4-yl)methanol (12). To a 0 °C solution of silyl-ether 10 (300 mg, 0.578 mmol) and THF (5 mL), in a 25 mL round-bottomed flask, was added TBAF (0.64 mL, 1.0 M solution in THF, 0.64 mmol) over 1 min via syringe. The resulting pale-yellow solution was stirred 30 min then poured (still cold) into half saturated NaHCO3 solution (50 mL) and extracted with CH2Cl2 (1 x 50 mL, 2 x 25 mL). The organic extracts were pooled, washed with H2O (1 x 30 mL) and brine (1 x 30 mL), dried (Na2SO4) and filtered. Concentration gave 367 mg of an orange oil. Purification by silica gel (20 g) flash column (3 x 8 cm) chromatography, eluting with 95:5 (250 mL) and then 80:20 (250 mL) CH2Cl2/EtOAc yielded 215 mg (98%) of the title compound as a white solid: mp 80–83 °C; [α]D
25 –64.3° (c 0.94, MeOH); 1H
NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.44 (s, 1H), 5.90 (d, J = 4.6 Hz, 1H), 5.18 (dd, J = 5.4, 5.4 Hz, 1H), 5.08 (dd, J = 1.9, 6.2 Hz, 1H), 4.69 (dd, J = 2.4, 10.2 Hz, 1H), 4.47 (ap d, J = 2.0 Hz, 1H), 3.95 (ddd, J = 2.2, 2.2, 12.5 Hz, 1H), 3.81 (ddd, J = 2.2, 10.3, 12.5, 1H), 1.63 (s, 3H), 1.37 (s, 3H); 13
C NMR (100 MHz, CDCl3) δ 153.4, 150.9, 149.1, 129.1, 116.9, 114.4, 95.0, 88.5, 85.7, 83.1, 81.1, 63.1, 27.5, 25.2; IR (film) ν 3362, 3124, 2989, 2936, 1580, 1541, 1452, 1210, 1109, 1080 cm-1; HRMS (ESI) calcd for C14H16N3O4ClBr [M+H]+: 404.0013, found: 404.0011.
OHO
O O
N N
N
Cl
12
OI
O O
N N
N
Cl
14
Br Br

S35
5-bromo-4-chloro-7-((3aR,4R,6S,6aS)-6-(iodomethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-7H-
pyrrolo[2,3-d]pyrimidine (14). An oven-dried 25 mL round-bottomed flask under N2 was charged with I2 (241 mg, 0.950 mmol) and THF (4 mL). To the dark solution was added PPh3 (250 mg, 0.953 mmol) in portions over 5 min. The resulting yellow-orange mixture was stirred 15 min and then alcohol 12 (192 mg, 0.475 mmol) and imidazole (68.0 mg, 0.999 mmol) were added in THF (2 mL) over 2-3 min via syringe. The resulting red-brown mixture was stirred 45 min, diluted with hexanes (15 mL; stirred mixture 15 min) and filtered through a pad (4 x 3 cm) of Celite under vacuum. The pad was washed with 4:1 hexanes/Et2O (2 x 15 mL) and the combined filtrates concentrated under vacuum to leave 524 mg of a white semi-solid. Purification by silica gel (30 g) flash column (3 x 12 cm) chromatography, eluting with 80:20 (500 mL) hexanes/Et2O yielded 239 mg (98%) of the title compound as a white solid: mp 114–115 °C dec; [α]D
25 –49.0° (c 0.60, MeOH); 1
H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 7.54 (s, 1H), 6.29 (d, J = 2.8 Hz, 1H), 5.25 (dd, J =2.8, 6.5 Hz, 1H), 4.94 (dd, J = 3.4, 6.4 Hz, 1H), 4.28 (ddd, J = 3.1, 4.2, 7.2 Hz, 1H), 3.43 (dd, J = 6.9, 10.4 Hz, 1H), 3.35 (dd, 4.6, 10.5, 1H), 1.64 (s, 3H), 1.39 (s, 3H); 13
C NMR (100 MHz, CDCl3) δ 152.8, 151.5, 149.9, 127.7,
115.9, 115.2, 90.8, 89.4, 84.8, 84.4, 84.2, 27.1, 25.3, 5.9; IR (film) ν 3126, 2987, 2937, 1579, 1539, 1452, 1211, 1085 cm-1; HRMS (ESI) calcd for C14H15N3O3ClBrI [M+H]+: 513.9030, found: 513.9030.
OI
O O
N N
N
Cl
14
ON3
O O
N N
N
N3
16
Br Br
4-azido-7-((3aR,4R,6R,6aR)-6-(azidomethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-5-bromo-7H-
pyrrolo[2,3-d]pyrimidine (16). A 20 mL vial was charged with iodide 14 (220 mg, 0.428 mmol), NaN3 (115 mg, 1.77 mmol) and DMF (5 mL). Sealed the vial with a screw-cap and heated the mixture at 60 °C for 1.5 h; all solids dissolved within 30 min. The resulting pale-yellow solution was allowed to cool to room temperature, diluted with EtOAc (15 mL) and filtered through a pad (2 x 3 cm) of Celite under vacuum; washed filter cake with EtOAc (2 x 10 mL). The combined filtrates were washed with H2O (3 x 10 mL) and brine (10 mL), dried (MgSO4) and filtered. Concentration under vacuum gave 184 mg of an oil. Purification by silica gel (20 g) flash column (3 x 8 cm) chromatography, eluting with 98:2 (150 mL), then 95:5 (100mL) and finally 90:10 (100 mL) CH2Cl2/EtOAc yielded 177 mg (95%) of the title compound as a colorless wax (could not be separated from 5-10% of the α-epimer): 1
H NMR (400 MHz, CDCl3) δ 9.39 (s, 1H), 7.60 (s, 1H), 6.43 (br s, 1H), 5.11 (m, 1H), 4.96 (m, 1H), 4.40 (ap d, J = 3.6 Hz, 1H), 3.73
(dd, J = 3.1, 13.2 Hz, 1H), 3.61 (dd, J = 4.2, 13.0 Hz, 1H), 1.66 (s, 3H), 1.39 (s, 3H); 13C NMR (100 MHz, CDCl3) δ
152.0, 145.9, 140.2, 132.7, 125.0, 115.5, 91.7, 90.7, 84.9, 83.9, 80.9, 52.3, 27.2, 25.4; HRMS (ESI) calcd for C14H15N9O3Br [M+H]+: 436.0481, found: 436.0480.
ON3
O O
N N
N
N3
16
OH2N
O O
N N
N
NH2
18
Br Br
7-((3aR,4R,6R,6aR)-6-(aminomethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-5-bromo-7H-
pyrrolo[2,3-d]pyrimidin-4-amine (18).

S36
To an oven-dried 10 mL round-bottomed flask under N2, containing a solution of diazide 16 (39.5 mg, 90.6 µmol) and THF (1 mL), was added PMe3 (0.19 mL, 1.0 M solution in THF, 0.19 mmol) over 1 min via syringe. The resulting pale-yellow mixture was stirred 1 h at room temperature and then a 2:1:1 mixture of H2O (1 mL) and THF (0.5mL) and MeOH (0.5 mL) was added. A water-cooled condenser was attached to the flask and the solution heated at reflux for 12 h. After cooling to room temperature the solution was diluted with brine (10 mL) and extracted with 95:5 CHCl3/MeOH (3 x 10 mL). The organic extracts were pooled, dried (Na2SO4) and filtered. Concentration under vacuum gave 38 mg of a cloudy residue. Purification by silica pipet (4 mL; ¾ full) column chromatography, eluting with 94:5:1 (40 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 30.0 mg (86%) of the title compound as a colorless wax: [α]D
25 –42.0° (c 0.30, MeOH); 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 7.11 (s,
1H), 6.13 (d, J = 3.1 Hz, 1H), 5.75 (br s, 2H), 5.23 (dd, J = 3.2, 6.6 Hz, 1H), 4.93 (dd, J = 4.0, 6.5 Hz, 1H), 4.17 (ddd, J = 4.3, 4.3, 5.4, 1H), 3.04 (dd, J = 4.3, 13.3 Hz, 1H), 2.94 (dd, J = 5.7, 13.4 Hz, 1H), 1.61 (s, 3H), 1.54 (br s, 2H), 1.37 (s, 3H); 13
C NMR (100 MHz, CDCl3) δ 156.9, 152.9, 149.6, 122.1, 114.7, 102.7, 90.3, 88.0, 86.7, 84.2, 81.5, 43.8, 27.3, 25.4; IR (film) ν 3471, 3312, 3132, 2987, 2936, 1629, 1585, 1559, 1078 cm-1; HRMS (ESI) calcd for C14H19N5O3Br [M+H]+: 384.0671, found: 384.0670.
OH2N
O O
N N
N
NH2
18
ONH
O O
N N
N
NH2
24
Br Br
5-bromo-7-((3aR,4R,6R,6aR)-6-((isopropylamino)methyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-
7H-pyrrolo[2,3-d]pyrimidin-4-amine (24). A 2-dram vial was charged with amine 18 (14.7 mg, 38.3 µmol), EtOH (0.5 mL) and acetone (50 µL, 10% v/v solution in EtOH, 68 µmol). The solution was stirred 5 min then NaBH3CN (115 µL, 0.50 M solution in EtOH, 58 µmol) and HOAc (60 µL, 1.0 M solution in EtOH, 60 µmol) were added. The reaction was stirred 1.5 h at room temperature, diluted with half saturated NaHCO3 solution (3 mL) and extracted with CHCl3 (4 x 3 mL). The organic extracts were pooled, dried (Na2SO4) and filtered. Concentration under vacuum gave 21.4 mg of a colorless residue. Purification by silica pipet (4 mL; 3/4 full) column chromatography, eluting with 96:3:1 (30 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 14.5 mg (89%) of the title compound as a colorless wax: 1
H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 7.14 (s, 1H), 6.14 (d, J = 3.0 Hz, 1H), 5.69 (br s, 2H), 5.23 (dd, J = 3.1, 6.6 Hz, 1H), 4.94 (dd, J = 3.9, 6.5 Hz, 1H), 4.28 (ddd, J = 4.0, 4.0, 6.1 Hz, 1H), 2.93 (dd, J = 4.0, 12.4 Hz, 1H), 2.84-2.72 (m, 2H), 1.61, (s, 3H), 1.37 (s, 3H), 1.05 (d, J = 5.9 Hz, 3H), 1.04 (d, J = 5.9 Hz, 3H); 13
C NMR (100 MHz, CDCl3) δ 156.8, 153.0, 149.6, 122.2, 114.6, 102.7, 90.4, 87.9, 85.2, 84.2, 82.0, 49.0, 48.8, 27.3, 25.5, 22.9, 22.7; HRMS (ESI) calcd for C17H25N5O3Br [M+H]+: 426.1141, found: 426.1139.
OHN
O O
N N
N
NH2
NH
O
NH
OH2N
O O
N N
N
NH2
18 25
20
Br Br
1-(3-((((3aR,4R,6R,6aR)-6-(4-amino-5-bromo-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydro-
furo[3,4-d][1,3]dioxol-4-yl)methyl)amino)propyl)-3-(4-(tert-butyl)phenyl)urea (25).

S37
A 2-dram vial was charged with amine 18 (14.1 mg, 36.7 µmol), oxazinanol 20 (10.0 mg, 40.3 µmol), 4 Å molecular sieves (ca. 75 mg; finely powdered with a mortar and pestle) and EtOH (0.5 mL). The mixture was stirred 10 min then NaBH3CN (150 µL, 0.50 M solution in EtOH, 75 µmol) and HOAc (80 µL, 1.0 M solution in EtOH, 80 µmol) were added. The mixture was heated at 45 °C for 14 h, allowed to cool to room temperature, filtered through a plug of Celite (1 x 2 cm) and the filter cake washed with CHCl3 (2 x 5 mL). The combined filtrates were diluted with saturated NaHCO3 solution (10 mL) and extracted with CHCl3 (3 x 5 mL). The organic extracts were pooled, dried (Na2SO4) and filtered. Concentration under vacuum gave 24.0 mg of a colorless residue. Purification by silica gel (5 g) flash column (1.5 x 8) chromatography, eluting with 94:5:1 (100 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 14.1 mg (62%) of the title compound as a colorless residue: 1
H NMR (400 MHz, methanol-d4) δ 8.12 (s, 1H), 7.45 (s, 1H), 7.27 (d, J = 8.8 Hz, 2H), 7.23 (d, J = 8.8 Hz, 2H), 6.19 (d, J = 3.0 Hz, 1H), 5.25 (dd, J = 3.0, 6.5 Hz, 1H), 4.90 (dd, J = 3.8, 6.5 Hz, 1H), 4.26 (ddd, J = 3.3, 4.7, 8.0 Hz, 1H), 3.20 (t, J = 6.7 Hz, 2H), 2.88-2.79 (m, 2H), 2.68-2.57 (m, 2H), 1.65 (quin, J = 6.9 Hz, 2H), 1.58 (s, 3H), 1.36 (s, 3H), 1.28 (s, 9H); 13
C NMR (100 MHz, methanol-d4) δ 158.8, 158.7, 153.7, 150.8, 146.6, 138.3, 126.7, 124.1, 120.5, 115.9, 103.4, 91.2, 89.5, 86.1, 85.5, 83.9, 52.7, 48.1, 38.8, 35.2, 32.0, 31.1, 27.6, 25.8; HRMS (ESI) calcd for C28H39N7O4Br [M+H]+: 616.2247, found: 616.2248.
1-(3-((((2R,3S,4R,5R)-5-(4-amino-5-bromo-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-3,4-dihydroxytetrahydrofuran-
2-yl)methyl)amino)propyl)-3-(4-(tert-butyl)phenyl)urea (Compound 3). Acetonide 25 (6.9 mg, 11.2 µmol) and 9:1 TFA/H2O (0.5 mL) were stirred for 45 min then concentrated to dryness from toluene (4 x 1 mL) and CHCl3 (1 mL) to leave 9.8 mg of a white solid. Purification by silica column chromatography, eluting with 90:9:1 (10 mL), 87:12:1 (10 mL) and 84:15:1 (10 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 5.7 mg (92%) of the title compound as a white solid: mp 121–130 °C dec; [α]D
25 –15.3° (c 0.34, MeOH);
1H NMR (400 MHz, methanol-d4) δ 8.11 (s, 1H), 7.43 (s, 1H), 7.27 (d, J = 8.9 Hz, 2H), 7.22 (d, J = 8.9 Hz, 2H),
6.09 (d, J = 5.0 Hz, 1H), 4.51 (dd, J = 5.2, 5.2 Hz, 1H), 4.18-4.11 (m, 2H), 3.24 (t, J = 6.7 Hz, 2H), 2.98-2.90 (m, 2H), 2.73 (t, J = 7.1 Hz, 2H), 1.73 (quin, J = 6.9 Hz, 2H), 1.28 (s, 9H); 13
C NMR (100 MHz, methanol-d4) δ 158.8, 153.5, 151.0, 146.7, 138.3, 126.7, 123.7, 120.5, 103.5, 90.2, 89.2, 84.2, 75.4, 73.5, 52.9, 48.2, 38.8, 35.2, 32.0, 31.0 (one signal missing); IR (film) ν 3474, 3320, 2961, 2869, 1633, 1594, 1558, 1520 cm-1; HRMS (ESI) calcd for C25H35N7O4Br [M+H]+: 576.1934, found: 576.1934.
ONH
O O
N N
N
NH2
24
ON
O O
N N
N
NH2
NH
O
NH
26
20
Br Br
1-(3-((((3aR,4R,6R,6aR)-6-(4-amino-5-bromo-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydro-
furo[3,4-d][1,3]dioxol-4-yl)methyl)(isopropyl)amino)propyl)-3-(4-(tert-butyl)phenyl)urea (26).

S38
A 2-dram vial was charged with amine 24 (10.9 mg, 25.6 µmol), oxazinanol 20 (7.0 mg, 28.2 µmol), 4 Å molecular sieves (ca. 100 mg; finely powdered with a mortar and pestle) and EtOH (0.6 mL). After stirring the mixture at room temperature for 5 min, NaBH3CN (0.10mL, 0.50 M solution in EtOH, 50 µmol) and HOAc (55 µL, 1.0 M solution in EtOH, 55 µmol) were added sequentially via syringe. The vial was sealed with a screw-cap and the mixture heated at 55 °C. After 22 h let the reaction cool to room temperature, filtered through a pad (2 x 2 cm) of Celite under vacuum and washed the filter cake with CHCl3 (2 x 5 mL). The filtrate was diluted with half saturated NaHCO3 (10 mL) and extracted with CHCl3 (3 x 10 mL). The organic extracts were pooled, dried (Na2SO4) and filtered. Concentration under vacuum gave 20.5 mg of a slightly cloudy residue. Purification by silica (5 g) flash column (1.5 x 8 cm) chromatography, eluting with 96:3:1 (150 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 14.4 mg (86%) of the title compound as a colorless glass: 1
H NMR (400 MHz, methanol-d4) δ 8.10 (s, 1H), 7.41 (s, 1H), 7.27 (d, J = 8.8 Hz, 2H), 7.22 (d, J = 8.8 Hz, 2H), 6.20 (d, J = 2.6 Hz, 1H), 5.30 (dd, J = 2.5, 6.5 Hz, 1H), 4.92 (dd, J = 3.5, 6.4 Hz, 1H), 4.22 (ddd, J = 3.6, 6.0, 7.5 Hz, 1H), 3.24-3.13 (m, 2H), 2.93 (sept, J = 6.5 Hz, 1H), 2.67 (dd, J = 5.8, 13.8 Hz, 1H), 2.57 (dd, 7.5, 13.8 Hz, 1H), 2.49 (t, J = 6.8 Hz, 2H), 1.64-1.52 (m, 2H), 1.55 (s, 3H), 1.36 (s, 3H), 1.28 (s, 9H), 0.98 (d, J = 6.5 Hz, 3H), 0.86 (d, J = 6.5 Hz, 3H); 13
C NMR (100 MHz, methanol-d4) δ 158.8, 158.6, 153.6, 150.7, 146.6, 138.3, 128.6, 126.7, 124.2, 120.6, 115.6, 103.4, 91.3, 89.5, 86.9, 85.3, 84.5, 53.8, 52.0, 39.7, 35.2, 32.0, 29.5, 27.6, 25.7, 18.7, 17.7; HRMS (ESI) calcd for C31H45N7O4Br [M+H]+: 658.2716, found: 658.2718.
OH2N
O O
N N
N
NH2
18
ON
O O
N N
N
NH2
NH
O
NH
26
20ONH
O O
N N
N
NH2
24
Br BrBr
Single-pot conversion of 18 to 26. A 10 mL round-bottomed flask was charged with amine 18 (33.4 mg, 86.9 µmol), EtOH (1.0 mL) and acetone (0.13 mL, 10% v/v solution in EtOH, 0.18 mmol). Stirred solution 5 min then NaBH3CN (0.26 mL, 0.50 M solution in EtOH, 0.13 mmol) and HOAc (0.13 mL, 1.0 M solution in EtOH, 0.13 mmol) were added sequentially via syringe. The reaction was stirred at room temperature 45 min, then oxazinanol 20 (50.4 mg, 0.203 mmol) was added in one portion followed by 4 Å molecular sieves (ca. 200 mg; finely powdered with a mortar and pestle). After 5 min additional NaBH3CN (0.26 mL, 0.50 M solution in EtOH, 0.13 mmol) and HOAc (0.13 mL, 1.0 M solution in EtOH, 0.13 mmol) were added, the vial was sealed (screw-cap) and the mixture heated at 55 °C for 24 h. Let cool to room temperature, diluted with 1:1 CHCl3/MeOH (10 mL) and filtered through a pad (2 x 4 cm) of Celite under vacuum; washed filter cake with CHCl3 (2 x 5 mL). The combined filtrates were diluted with saturated NaHCO3 (15 mL) and extracted with CHCl3 (3 x 5 mL). The organic extracts were pooled, dried (Na2SO4) and filtered. Concentration under vacuum gave 79.3 mg of a cloudy wax. Purification by silica gel (12 g) flash column chromatography, eluting with 96:3:1 (200 mL) and 94:5:1 (100 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 45.3 mg (79%) of the title compound as a colorless glass.
1-(3-((((2R,3S,4R,5R)-5-(4-amino-5-bromo-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-3,4-dihydroxytetrahydrofuran-
2-yl)methyl)(isopropyl)amino)propyl)-3-(4-(tert-butyl)phenyl)urea (SGC0946).

S39
Acetonide 26 (11.4 mg, 17.3 µmol) was stirred with 9:1 TFA/H2O (0.5 mL) for 45 min. The solution was then concentrated under vacuum from toluene (3 x 1 mL) and CHCl3 (1 mL) to leave 14.6 mg of a colorless residue. Purification by silica chromatography, eluting with 91:8:1 (25 mL) CHCl3/MeOH/conc. NH4OH(aq) yielded 8.9 mg (83%) of the title compound as a white solid: mp 120–125 °C dec; [α]D
25 –1.15° (c 0.52, MeOH); 1H NMR (400
MHz, methanol-d4) δ 8.07 (s, 1H), 7.44 (s, 1H), 7.24 (d, J = 8.8 Hz, 2H), 7.18 (d, J = 8.8 Hz, 2H), 6.14 (d, J = 4.8 Hz, 1H), 4.44 (dd, J = 5.1, 5.1 Hz, 1H), 4.17 (dd, J = 5.3, 5.3 Hz, 1H), 4.09 (ddd, 5.0, 5.0, 6.5 Hz, 1H), 3.28-3.16 (m, 2H), 3.06 (ap quin, J = 6.5 Hz, 1H), 2.84 (dd, J = 3.9, 13.8Hz, 1H), 2.71 (dd, J = 6.8, 13.8 Hz, 1H), 2.60 (t, J = 6.6 Hz, 2H), 1.68 (quin, J = 6.8 Hz, 2H), 1.28 (s, 9H), 1.04 (d, J = 6.5 Hz, 3H), 1.02 (d, J = 6.5 Hz, 3H); 13
C NMR
(100 MHz, methanol-d4) δ 158.74, 158.66, 153.5, 151.0, 146.6, 138.2, 126.6, 123.3, 120.5, 103.4, 89.8, 89.3, 84.3,
75.4, 73.6, 53.9, 52.1, 39.7, 35.1, 32.0, 29.3, 18.4, 18.0 (one signal missing); IR (film) ν 3476, 3322, 2962, 2869, 1631, 1592, 1556, 1518 cm-1; HRMS (ESI) calcd for C28H41N7O4Br [M+H]+: 618.2403, found: 618.2403.
The synthesis of all the analogues started from the commercially available adenosine or 7-deazaadenosine (SI-1), which was converted to acetal followed by chlorination of primary alcohol to produce chloro derivative SI-2. Amination of compound SI-2 with 1, 3-diaminopropane afforded the diamine SI-3 in 78% yield. The primary amino group of the resulting product was selectively protected with di-tert-butyl carbonate, followed by alkylation of the secondary amino group with 2-iodopropane to generate tertiary amine SI-4. Global deprotection of compound SI-4 was proceeded under acidic conditions to generate free primary amine SI-5. Coupling of SI-5 with various isocynates or acids afforded desired final analogues SI-6.
Reagents. Commercial reagents and solvents were used as received. Ammonia saturated dichloromethane was obtained by agitation of dichloromethane in the presence of ammonium hydroxide followed by drying over anhydrous sodium sulfate. Instrumentation. Proton and carbon-13 nuclear magnetic resonance (1H NMR and 13C NMR) spectra were recorded with a Varian inverse probe 600 INOVA spectrometer at the Harvard Medical School East Quad NMR Facility. Chemical shifts are recorded in parts per million on the δ scale and are referenced to the residual protium in the NMR solvent (Methanol-d4: δ 3.31) for 1H NMR, the carbon resonances of the solvent (Methanol-d4: δ 49.75) for 13C NMR respectively. Data is reported as follows: chemical shift [multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant(s) in Hertz, integration]. High resolution mass spectra (HRMS) were recorded on a Bruker APEX 4.7 Tesla FTMS spectrometer using eletronspray ion source (ESI) at the Instrumentation Facility of the Department of Chemistry, Massachusetts Institute of Technology. The intermediates and final product were purified with a CombiFlash RF system (Teledyne Isco) and a Varian ProStar HPLC using a Dynamax 21.4X250mm 100Å C18 column with water-acetonitrile

S40
containing 0.1% TFA as the mobile phase eluting at 20 mL/min and UV monitoring at 220 nm. The fractions were collected and the solvent removed on a lyophilizer. Organic solutions were concentrated on Büchi R-205 rotary evaporators.
7-((3aR,4R,6S,6aS)-6-(chloromethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-7H-pyrrolo[2,3-
d]pyrimidin-4-amine (SI-2a)
9-((3aR,4R,6S,6aS)-6-(chloromethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-amine
(SI-2b)
General Procedure: 2,2-Dimethoxypropane (8.3 mL, 68 mmol, 18 equiv) was added as a suspension of adenosine or 7-deazaadenosine in acetone (1.0 g, 3.7 mmol, 1 equiv), followed by the addition of p-Toluenesulfonic acid (713 mg, 3.70 mmol, 1.00 equiv). The mixture was then stirred at 23 °C for 2 h. Solid sodium bicarbonate (945 mg, 11.2 mmol, 3.00 equiv) was then added, and the reaction was stirred for another 30 min. The solvent was then removed under reduce pressure. The residue was dissolved in dichloromethane followed by addition of saturated aqueous NaHCO3 solution. The layers were separated and the aqueous layer was extracted with dichloromethane (3 × 150 mL). The combined organic layers were washed with brine (50 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The residue was purified by flash column chromatography (Combiflash RF system, 40 gram silica gel, gradient 0 to 15% methanol-dichloromethane) to afford acetal SI-7 (1.034 g, 90%) as white solid.
4-Dimethylaminopyridine (825 mg, 6.72 mmol, 2.0 equiv) was added to a solution of acetal SI-7 in dry acetonitrile (16.8 mL, 0.2 M), followed by slow addition of thionyl chloride (1.22 mL, 16.8 mmol, 5.0 equiv) at 0 °C. The resulted mixture was allowed to warm to 23 °C in 4 h, and stirred at 23 °C for 8 h. Methanol (20 mL), water (20 mL) and ammonium hydroxide solution (2 mL) were added sequentially at 0 °C. The mixture was concentrated to dryness under reduce pressure. The residue was dissolved in dichloromethane followed by the addition of saturated aqueous NaHCO3 solution. The layers were separated and the aqueous layer was extracted with dichloromethane (3 × 150 mL). The combined organic layers were washed with brine (50 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The residue was purified by flash column chromatography (Combiflash RF system, 40 gram silica gel, gradient 0 to 15% dichloromethane-methanol) to afford SI-2 (0.86 g, 79%) as yellow solid.
SI-2a
1H NMR (400 MHz, Methanol-d4, 25 °C) δ 8.01 (s, 1H), 7.16 (d, J = 3.7 Hz, 1H), 6.53 (d, J = 3.7 Hz, 1H), 6.15 (d, J = 3.2 Hz, 1H), 5.17 (dd, J = 6.5, 3.2 Hz, 1H), 4.94 (dd, J = 6.5, 3.1 Hz, 1H), 4.24 (ddd, J = 6.4, 5.0, 3.1 Hz, 1H), 4.09 – 3.99 (m, 1H), 3.75 – 3.48 (m, 2H), 1.49 (s, 3H), 1.26 (s, 3H).
13C NMR (100 MHz Methanol-d4, 25 °C) ä 157.9, 150.8, 149.6, 122.4, 114.2, 103.4, 100.1, 89.8, 84.8, 84.0, 82.2, 43.7, 26.0, 24.1.

S41
IR (film) ν 3335, 3197, 2989, 1639, 1591, 1564, 1475, 1376, 1271, 1212, 1084, 976, 725 cm-1.
HRMS(ESI) calc’d for C14H18ClN4O3 [M+H]+: 325.1062, found 325.1077 m/z.
SI-2b
1H NMR (400 MHz, Methanol-d4, 25 °C) δ 8.13 (d, J = 10.6 Hz, 1H), 6.11 (d, J = 2.5 Hz, 1H), 5.38 (d, J = 2.5 Hz, 1H), 5.02 (dd, J = 6.3, 2.9 Hz, 1H), 3.70 (dd, J = 11.3, 7.0 Hz, 1H), 3.57 (dd, J = 11.3, 5.5 Hz, 1H), 1.49 (s, 2H), 1.28 (s, 1H).
13C NMR (100 MHz, Methanol-d4, 25 °C) δ 156.0, 152.6, 148.7, 140.3, 119.1, 114.1, 90.5, 86.4, 83.8, 82.6, 43.3, 25.9, 24.0.
IR (film) ν 3328, 3179, 2989, 1651, 1599, 1477, 1375, 1298, 1210, 1089, 871, 798 cm-1.
HRMS(ESI) calc’d for C13H17ClN5O3 [M+H]+: 326.1014, found 326.1011 m/z.
N1-(((3aR,4R,6R,6aR)-6-(4-amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrofuro[3,4-
d][1,3]dioxol-4-yl)methyl)propane-1,3-diamine (SI-3a)
N1-(((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-
yl)methyl)propane-1,3-diamine (SI-3b)
General Procedure: Compound SI-2 (400 mg, 1.1 mmol) was dissolved in dry 1, 3-diamino propane (11 mL, 0.1 M). The resulting solution was heated at 65 °C for 16 h. The solvent was removed in vacuo by short path distillation. The residue was purified by flash column chromatography (5 gram silica gel, gradient 0 to 15% dichloromethane-methanol with saturated ammonium) to afford SI-3 (0.86 g, 78%) as yellow oil.
SI-3a
1H NMR (400 MHz, Methanol-d4, 25 °C) δ 8.02 (s, 1H), 7.22 (d, J = 3.7 Hz, 1H), 6.58 (d, J = 3.7 Hz, 1H), 6.12 (d, J = 3.1 Hz, 1H), 5.20 (dd, J = 6.6, 3.1 Hz, 1H), 4.90 (dd, J = 6.6, 4.0 Hz, 1H), 3.44 – 3.29 (m, 2H), 3.06 – 2.99 (m, 2H), 2.89 (t, J = 7.2 Hz, 2H), 2.76 (td, J = 7.1, 2.6 Hz, 2H), 2.03 – 1.84 (m, 1H), 1.84 – 1.66 (m, 3H), 1.50 (s, 3H), 1.27 (s, 3H).
13C NMR (100 MHz, Methanol-d4, 25 °C) δ 159.5, 153.3, 151.7, 125.1, 116.7, 105.8, 102.3, 86.0, 85.0, 84.2, 52.6, 48.3, 39.8, 28.2, 27.4, 26.4, 19.9.

S42
IR (film) ν 3372, 3197, 2989, 1643, 1593, 1563, 1479, 1377, 1273, 1212, 1079, 865, 726 cm-1.
HRMS(ESI) calc’d for C17H27N6O3 [M+H]+: 363.2139, found 363.2148 m/z.
SI-3b
1H NMR (400 MHz, Methanol-d4, 25 °C) δ 8.28 (s, 1H), 8.22 (s, 1H), 6.16 (d, J = 2.8 Hz, 1H), 5.50 (dd, J = 6.4, 2.8 Hz, 1H), 5.01 (dd, J = 6.4, 3.4 Hz, 1H), 4.34 (td, J = 6.2, 3.4 Hz, 1H), 2.85 (d, J = 6.2 Hz, 2H), 2.65 (t, J = 7.1 Hz, 2H), 2.58 (q, J = 6.9 Hz, 2H), 1.60 (s, 3H), 1.38 (s, 3H).
13C NMR (100 MHz, Methanol-d4, 25 °C) δ 158.2, 154.8, 151.1, 142.7, 121.4, 116.4, 92.4, 87.5, 85.6, 84.7, 53.2, 41.2, 33.4, 28.2, 26.3.
IR (film) ν 3333, 3179, 3000, 2935, 1650, 1601, 1478, 1377, 1300, 1211, 1083, 869 cm-1.
HRMS(ESI) calc’d for C16H26N7O3 [M+H]+: 364.2092, found 364.2098 m/z.
N
NX
NO
NH2
OO
HN
H2N
(Boc)2O, DCM
23 C
82%
N
NX
NO
NH2
OO
N
BocHN
SI-3 SI-4
CH3CHICH3
DIPEA, CH3CN
65 C
50%
with 40% r.c.s.m.
N
NX
NO
NH2
OO
HN
BocHN
SI-8
N
N
NO
NH2
OO
N
BocHN
SI-4a
N
NN
NO
NH2
OO
N
BocHN
SI-4b
a: X = C-Hb: X = N
tert-Butyl (3-((((3aR,4R,6R,6aR)-6-(4-amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-
dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(isopropyl)amino)propyl)carbamate (SI-4a)
tert-Butyl (3-((((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-
yl)methyl)(isopropyl)amino)propyl)carbamate (SI-4b)
General Procedure: Compound SI-3 (278 mg, 0.7 mmol, 1 equiv) was dissolved into acetonitrile and THF (1: 1, 7 mL, 0.1 M) at 23 °C. Di-tert-butyl dicarbonate (153 mg, 0.7 mmol, 1.0 equiv) was added to the reaction mixture at 0 °C, and the reaction was further stirred another 30 min. The saturated aqueous sodium bicarbonate solution (15 mL) was added, and was extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with brine (25 mL), were dried over anhydrous sodium sulphate, were filtered, and were concentrated under reduced pressure. The residue was purified by flash column chromatography (Combiflash RF system, 12 gram silica gel, gradient 0 to 15% methanol-dichloromethane) to afford free amine SI-8 (266 mg, 82%) as yellow oil.
Diisopropylethylamine (2.16 mL, 12.4 mmol, 20.0 equiv) and 2-iodopropane (1.2 mL, 12.4 mmol, 20.0 equiv) were added sequentially to a solution of compound SI-8 in dry acetonitrile (288 mg, 0.62 mmol, 1 equiv in 4 mL, 0.16 M). The mixture was then stirred and heated at 65 °C for 16 h. The solvent was then removed in vacuo. The residue was purified by flash column chromatography (Combiflash RF system, 4 gram silica gel, gradient 0 to 15% methanol-dichloromethane) to afford desired acetal SI-4 (154 mg, 50 %) as white solid with recover starting material SI-3 (57.6 mg, 40%).
SI-8a

S43
1H NMR (400 MHz, Methanol-d4, 25 °C) δ 8.11 (s, 1H), 7.27 (d, J = 3.7 Hz, 1H), 6.64 (d, J = 3.7 Hz, 1H), 6.29 – 6.10 (m, 1H), 5.37 – 5.25 (m, 1H), 4.98 – 4.89 (m, 1H), 4.26 (dt, J = 9.0, 4.4 Hz, 1H), 3.02 (t, J = 7.0 Hz, 2H), 2.87 (d, J = 7.4 Hz, 2H), 2.60 (q, J = 6.6 Hz, 2H), 1.60 (s, 6H), 1.40 (s, 9H), 1.37 (s, 3H).
13C NMR (100 MHz, Methanol-d4, 25 °C) δ 159.8, 153.3, 151.8, 124.9, 116.5, 102.2, 86.0, 84.5, 53.2, 48.6, 39.8, 31.1, 29.5, 28.3, 26.4.
IR (film) ν 3338, 3187, 2971, 2928, 1690, 1684, 1637, 1586, 1565, 1475, 1277, 1165, 1080 cm-1.
HRMS(ESI) calc’d for C22H35N6O5 [M+H]+: 463.2663, found 463.2684 m/z.
SI-8b
1H NMR (400 MHz, Methanol-d4, 25 °C) δ 8.28 (s, 1H), 8.22 (s, 1H), 6.16 (d, J = 2.8 Hz, 1H), 5.49 (dd, J = 6.4, 2.8 Hz, 1H), 5.01 (dd, J = 6.4, 3.4 Hz, 1H), 4.34 (td, J = 6.1, 3.4 Hz, 1H), 3.02 (td, J = 6.6, 2.4 Hz, 2H), 2.86 (d, J = 6.1 Hz, 2H), 2.69 – 2.46 (m, 2H), 1.66 – 1.49 (m, 6H), 1.39 (d, J = 5.3 Hz, 12H).
13C NMR (100 MHz, Methanol-d4, 25 °C) δ 158.2, 154.7, 151.1, 142.7, 121.4, 116.4, 92.4, 87.5, 85.7, 84.7, 53.2, 48.7, 39.8, 31.3, 29.5, 28.3, 26.3.
IR (film) ν 3333, 3193, 2989, 1692, 1643, 1590, 1249, 1216, 1166, 1078 cm-1.
HRMS(ESI) calc’d for C21H34N7O5 [M+H]+: 464.2616, found 464.2613 m/z.
SI-4a
1H NMR (400 MHz, Methanol-d4, 25 °C) ä 8.11 (s, 1H), 7.25 (d, J = 3.7 Hz, 1H), 6.64 (d, J = 3.7 Hz, 1H), 6.20 (d, J = 2.8 Hz, 1H), 5.32 (dd, J = 6.7, 2.8 Hz, 1H), 4.94 (dd, J = 6.7, 3.7 Hz, 1H), 4.17 (td, J = 6.7, 3.6 Hz, 1H), 3.04 (td, J = 6.6, 3.5 Hz, 1H), 2.95 (p, J = 6.7 Hz, 1H), 2.68 (dd, J = 13.7, 6.4 Hz, 1H), 2.55 (dd, J = 13.8, 7.0 Hz, 1H), 2.46 (t, J = 6.9 Hz, 1H), 1.60 (s, 3H), 1.58 – 1.48 (m, 1H), 1.41 (s, 9H), 1.38 (s, 3H), 1.01 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.5 Hz, 3H).
13C NMR (100 MHz, Methanol-d4, 25 °C) δ 159.8, 153.2, 151.8, 125.0, 116.2, 105.7, 102.1, 91.8, 87.1, 86.0, 85.2, 53.9, 52.2, 40.5, 29.6, 28.3, 26.4, 19.7, 17.8.
IR (film) ν 3290, 2965, 2960, 1692, 1632, 1587, 1511, 1364, 1268, 1167, 1078 cm-1.
HRMS(ESI) calc’d for C25H41N6O5 [M+H]+: 505.3133, found 505.3133 m/z.
SI-4b
1H NMR (400 MHz, Methanol-d4, 25 °C) ä 8.27 (s, 1H), 8.24 (s, 1H), 6.22 (s, 1H), 5.53 (d, J = 6.1 Hz, 1H), 5.09 (s, 1H), 4.58 (s, 0H), 4.33 (s, 1H), 3.73 (p, J = 6.9 Hz, 1H), 3.23 (q, J = 7.8 Hz, 1H), 3.04 (s, 2H), 1.61 (s, 3H), 1.54 – 1.31 (m, 16H), 1.06 (s, 3H), 0.85 (s, 3H).
13C NMR (100 MHz, Methanol-d4, 25 °C) δ 158.2, 154.8, 150.9, 143.0, 121.4, 116.3, 92.5, 85.8, 85.3, 56.6, 53.7, 44.6, 29.6, 28.2, 26.3, 17.7, 14.0.
IR (film) ν 3295, 2927, 2989, 1672, 1210, 1153, 1078, 668 cm-1.

S44
HRMS(ESI) calc’d for C24H40N7O5 [M+H]+: 506.3085, found 506.3087 m/z.
(2R,3R,4S,5R)-2-(4-amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-(((3-
aminopropyl)(isopropyl)amino)methyl)tetrahydrofuran-3,4-diol (SI-5a)
(2R,3R,4S,5R)-2-(6-amino-9H-purin-9-yl)-5-(((3-aminopropyl)(isopropyl)amino)methyl)tetrahydrofuran-3,4-
diol (SI-5b)
General Procedure: Hydrogen chloride solution in dioxane (5 mL, 4.0 M) was added to the solution of Boc-protected diamine SI-4 in acetonitrile (80 mg, 0.15 mmol in 5 mL, 0.03 M). After stirring at 23 °C for 6 h, solvents was removed under reduce pressure. The residue was used directly in the next step without further purification.
1-(3-((((2R,3S,4R,5R)-5-(4-amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-3,4-dihydroxytetrahydrofuran-2-
yl)methyl)(isopropyl)amino)propyl)-3-(4-(tert-butyl)phenyl)urea (EPZ004777)
1-(3-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-
yl)methyl)(isopropyl)amino)propyl)-3-(4-(tert-butyl)phenyl)urea (FED1)
1-(3-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-
yl)methyl)(isopropyl)amino)propyl)-3-(p-tolyl)urea (FED2)
General Procedure: The isocynate (0.074 mmol, 1.0 equiv) was added to a solution of SI-5 (27 mg, 0.74 mmol, 1 equiv) in acetonitrile and methanol (1:1, 3.7 mL, 0.02 M). The reaction mixture was stirred at 23 °C for 30 min, and the solvent was removed under reduced pressure. The residue was purified by preparative-HPLC eluting with a

S45
gradient from 5% to 95% acetonitrile in water with 0.1% TFA, and product containing fractions were collected. All solvents were removed with lyophilizer to afford final urea compounds as a TFA salt. The TFA residue was removed with flash column chromatography (0.5 gram silica gel, 10% methanol-Chloroform with 1% aq. NH4OH) to afford free amine.
EPZ004777
1H NMR (400 MHz, Methanol-d4, 25 °C) δ 8.07 (s, 1H), 7.90 (s, 1H), 7.33 – 7.22 (m, 2H), 7.22 – 7.12 (m, 2H), 6.63 (d, J = 3.7 Hz, 1H), 6.12 (d, J = 4.4 Hz, 1H), 4.50 (dd, J = 5.5, 4.4 Hz, 1H), 4.31 – 4.10 (m, 2H), 3.22 (td, J = 6.4, 2.1 Hz, 2H), 3.13 – 2.97 (m, 2H), 2.97 – 2.58 (m, 3H), 1.81 – 1.65 (m, 2H), 1.29 (s, 9H), 1.24 – 0.97 (m, 6H).
IR (film) ν 3295, 2927, 2989, 1672, 1210, 1153, 1078, 668 cm-1.
HRMS(ESI) calc’d for C28H42N7O4 [M+H]+: 540.3293, found 540.3294 m/z.
FED1
1H NMR (400 MHz, Methanol-d4, 25 °C) δ 8.16 (s, 1H), 7.92 (s, 1H) 7.05 (dd, J = 14.1, 8.2 Hz, 2H), 6.98 – 6.80 (m, 2H), 5.90 (dd, J = 22.2, 3.6 Hz, 2H), 4.56 (dt, J = 21.7, 4.4 Hz, 1H), 4.43 – 4.17 (m, 2H), 3.88 – 3.33 (m, 6H), 3.19 – 2.97 (m, 5H), 2.05 – 1.66 (m, 2H), 1.41 – 1.04 (m, 6H), 1.33 (s, 9H).
13C NMR (100 MHz, Methanol-d4, 25 °C) δ 180.9, 167.6, 141.7, 133.0, 128.9, 119.1, 118.8, 106.7, 84.3, 73.3, 72.1, 36.3, 34.4, 25.6, 19.3, 15.5, 14.5.
IR (film) ν 3299, 2991, 2989, 2965, 1663, 1652, 1210, 1153, 1073, 668 cm-1.
HRMS(ESI) calc’d for C27H40N8O4 [M+H]+: 541.3245, found 541.3247 m/z.
FED2
1H NMR (400 MHz, Methanol-d4, 25 °C) δ 8.19 (d, J = 17.4 Hz, 2H), 7.05 (dd, J = 14.1, 8.2 Hz, 2H), 6.98 – 6.80 (m, 2H), 5.92 (dd, J = 22.2, 3.6 Hz, 2H), 4.56 (dt, J = 21.7, 4.4 Hz, 1H), 4.43 – 4.17 (m, 2H), 3.88 – 3.33 (m, 6H), 3.19 – 2.97 (m, 5H), 2.16 (s, 3H), 2.05 – 1.66 (m, 2H), 1.41 – 1.04 (m, 6H)
13C NMR (100 MHz, Methanol-d4, 25 °C) δ 181.8, 167.6, 141.6, 132.0, 128.9, 119.1, 118.7, 107. 9, 84.3, 73.1, 72.0, 35.9, 34.4, 25.6, 19.3, 15.5, 14.5, 14.2.
IR (film) ν 3298, 2993, 2989, 1682, 1652, 1210, 1153, 1073, 665 cm-1.
HRMS(ESI) calc’d for C24H34N8O4 [M+H]+: 499.2776, found 499.2784 m/z.

S46
N
NN
NO
NH2
OHHO
N
HN
O
N
NN
NO
NH2
OHHO
N
H2N
t-Bu-Ph-COOH,
iPr2NEt, HCTU
23 C
85%
SI-5EJC1
N-(3-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-
yl)methyl)(isopropyl)amino)propyl)-4-(tert-butyl)benzamide (EJC1)
Diisopropylethylamine (0.018 mL, 0.10 mmol, 10 equiv) and 2 - (6 - Chloro - 1H - benzotriazole - 1 - yl) - 1,1,3,3 - tetramethylaminium hexafluorophosphate (HCTU) (8.9 mg, 0.022 mmol, 1.05 equiv) were added sequencially to a solution of SI-5 (7.5 mg, 0.0205 mmol, 1 equiv) and p-tert-butylbenzoic acid (3.83 mg, 0.0215 mmol, 1.05 equiv) in N,N-dimethylformamide (DMF, 0.5 mL, 0.04 M). The reaction mixture was further stirred at 23 °C for 2 h. The reaction was directly purified by preparative-HPLC eluting with a gradient from 5% to 95% acetonitrile in water with 0.1% TFA, and product containing fractions were collected. All solvents were removed with lyophilizer to afford final amide compound EJC1 (9.2 mg, 85%) as white solid.
EJC1
1H NMR (400 MHz, Methanol-d4, 25 °C) ä 8.10 (s, 1H), 7.79 (d, J = 0.7 Hz, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.43 – 7.36 (d, J = 8.2 Hz, 2H), 5.91 (d, J = 48.9 Hz, 2H), 4.56 (s, 1H), 4.35 (dd, J = 8.4, 2.3 Hz, 2H), 3.93 – 3.29 (m, 10H), 1.95 (d, J = 7.7 Hz, 6H), 1.25 (s, 12H).
13C NMR (100 MHz, Methanol-d4, 25 °C) δ 169.4, 141.9, 141.3, 133.4, 128.6, 126.7, 126.0, 125.2, 119.6, 109.0, 90.8, 73.2, 72.1, 51.9, 34.4, 30.1.
IR (film) ν 3291, 2927, 2989, 1652, 1632, 1210, 1153, 1073, 666 cm-1.
HRMS(ESI) calc’d for C27H40N7O4 [M+H]+: 526.3136, found 526.3125 m/z.

S47
Supplementary References:
29. Karplus, P.A. & Diederichs, K. Linking crystallographic model and data quality. Science 336,
1030-1033.
30. Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta
Crystallogr D Biol Crystallogr 66, 133-144 (2010).
31. Otwinowski, z. & Minor, w. Processing of X-ray diffraction data collected in oscillation mode.
Methods in Enzymology 276, 307-326 (1997).
32. Battye, T.G., Kontogiannis, L., Johnson, O., Powell, H.R. & Leslie, A.G. iMOSFLM: a new
graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol
Crystallogr 67, 271-281 (2011).
33. Collaborative Computational Project, N. The CCP4 suite: programs for protein
crystallography. Acta Crystallogr D Biol Crystallogr 50, 760-763 (1994).
34. Emsley, P., Lohkamp, B., Scott, W.G. & Cowtan, K. Features and development of Coot. Acta
Crystallogr D Biol Crystallogr 66, 486-501 (2010).
35. Murshudov, G.N., Vagin, A.A. & Dodson, E.J. Refinement of macromolecular structures by the
maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240-255 (1997).
36. Moriarty, N.W., Grosse-Kunstleve, R.W. & Adams, P.D. electronic Ligand Builder and
Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation.
Acta Crystallogr D Biol Crystallogr 65, 1074-1080 (2009).
37. Schuttelkopf, A.W. & van Aalten, D.M. PRODRG: a tool for high-throughput crystallography
of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr 60, 1355-1363 (2004).
38. Artursson, P. Epithelial transport of drugs in cell culture. I: A model for studying the passive
diffusion of drugs over intestinal absorptive (Caco-2) cells. Journal of pharmaceutical
sciences 79, 476-482 (1990).
39. Yee, S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small
intestinal) absorption in man--fact or myth. Pharmaceutical research 14, 763-766 (1997).
40. Wales, T.E., Fadgen, K.E., Gerhardt, G.C. & Engen, J.R. High-speed and high-resolution UPLC
separation at zero degrees Celsius. Anal Chem 80, 6815-6820 (2008).
41. Englander, S.W. & Kallenbach, N.R. Hydrogen exchange and structural dynamics of proteins
and nucleic acids. Q Rev Biophys 16, 521-655 (1983).
42. Wei, H. et al. Using hydrogen/deuterium exchange mass spectrometry to study
conformational changes in granulocyte colony stimulating factor upon PEGylation. J Am Soc
Mass Spectrom 23, 498-504 (2012).
43. Plumb, R.S. et al. UPLC/MS(E); a new approach for generating molecular fragment
information for biomarker structure elucidation. Rapid Commun Mass Spectrom 20, 1989-
1994 (2006).
44. Kavan, D. & Man, P. MSTools-Web based application for visualization and presentation of
HXMS data. Internat J Mass Spectrom 302, 52-58 (2011).
45. Jorgensen, W.L.C., Madura, J., Impey, R.W. & Klein, M.L. Comparison of simple potential
functions for the simulation of liquid water. J Chem Phys. 79, 926-935 (1983).
46. Wang, J., Wang, W., Kollman, P.A. & Case, D.A. Automatic atom type and bond type
perception in molecular mechanical calculations. J Mol Graph Model 25, 247-260 (2006).

S48
47. Bayly, C.A., Cieplak, P., Cornell, W.D. & Kollman, P.A. A well behaved elestrostatic potential
based method using charge restraints for deriving atomic charges: The RESP model. J. Phys.
Chem 97, 10269-10280 (1993).
48. Ryckaert, J.P., Ciccotti, G. & Berendsen, H.J.C. Numerical integration of the cartesian
equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J
Comput Phys 23, 327-341 (1977).