Theoretical Study of the Dynamics of Collisions Between HCl and ω-Hydroxylated Alkanethiol...
11
Published: January 18, 2011 r2011 American Chemical Society 2273 dx.doi.org/10.1021/jp1106422 | J. Phys. Chem. C 2011, 115, 2273–2283 ARTICLE pubs.acs.org/JPCC Theoretical Study of the Dynamics of Collisions Between HCl and ω-Hydroxylated Alkanethiol Self-Assembled Monolayers William A. Alexander and Diego Troya* Department of Chemistry, Virginia Tech, Blacksburg, Virginia 24061-0212, United States b S Supporting Information ABSTRACT: We present a classical-trajectory study of collisions of HCl with hydroxylated alkanethiol self-assembled monolayers. The potential-energy surface used in the trajectory propagation is a combi- nation of the standard OPLS force field to describe the surface and an analytical potential for the gas/surface interaction developed in this work. The gas/surface potential has been derived based on high-quality electronic-structure calculations of model HCl-alcohol systems in the gas phase and includes a flexible Buckingham term and a Coulombic term. The results of the trajectories calculations are in good agreement with recent molecular-beam experiments on the same system, thereby lending support to the accuracy of the calculations. The collision dynamics differ vastly from prior scattering studies involving rare gases and CO, primarily because the gas/surface attraction governed by hydrogen bonding dramatically increases the ability of the gas mole- cules to trap on the surface for extended times. The properties of the desorbing HCl molecules are largely insensitive to the initial collision energy and are only mildly affected by the incident angle. An analysis of the reaction mechanism reveals the distinct dynamics of trajectories that either recoil from the surface directly or undergo multiple collisions with the surface and result in thermalization. ’ INTRODUCTION Studying energy transfer in collisions of gases with surfaces yields information important to a fundamental understanding of gas/surface chemical dynamics, which is relevant in a variety of fields. For instance, the accommodation and transport of gases on hydrocarbon surfaces plays an important role in atmospheric chemistry. Surfactant-coated aerosols are abundant in the tropo- sphere, and these coatings represent a barrier for gaseous species to enter and react with species interior to the droplet. 1 The oxidation of these organic surfaces by various reactive pathways typically confers hydrophilic character to the surface, which may aid in dissolution of polar atmospheric molecules into the aerosol. Once in the aerosol, the gas species may undergo subsequent reaction. While HCl is a sink species for catalytic, ozone-degrading Cl radicals, HCl dissolved into aerosol particles, or other particles such as water droplets and ice crystals, may react to recover Cl 2 , which upon photolysis restarts the ozone-destroying catalytic process. Even before dissolution into the particle, hydroxyl sites on the surfaces can bind the gas-phase HCl through hydrogen-bonding interactions, initiate reaction by dissociating HCl, or serve as a surface-bound reagent for other reactions. 2 Each of these scenarios is mediated by the initial gas/ surface collision, where the interactions of HCl with the organic surface determine whether the molecule will scatter away directly or accommodate on the surface. The dynamics of this initial collision between HCl and organic sur- faces has recently been studied using time-of-flight molecular-beam scattering techniques by Lohr et al. 3,4 Hydrophobic and hydrophilic surfaces were modeled with straight-chain CH 3 -(CH 2 ) 15 -SH and terminally hydroxylated OH-(CH 2 ) 16 -SH alkanethiol self-as- sembled monolayer surfaces on gold, respectively. In those experi- mental studies, it was found that Ar and HCl had similar accom- modation fractions and energy-transfer dynamics when colliding with the nonpolar hydrocarbon surface. However, these gases showed remarkably di fferent dynamics when scattering from the hydroxylated surface. Using the traditional separation of the products ’ energy distribution into an impulsive scattering (IS) and thermal desorption (TD) channel, 5 the experiments indicate that while 43% of the Ar atoms recoil from the OH-SAM with a thermal energy distribution at the surface temperature when colliding at 85 kJ 3 mol -1 and 30°, 73% of the HCl thermalize at the same conditions, showing the effect of gas/surface hydrogen bonding on the dynamics of interfacial scattering. The results that Ar and HCl exhibit similar scattering dynamics in collisions with CH 3 -SAMs imply that kinematics, rather than the weak gas/surface chemical forces in that system, determine the scattering outcome, such that gases with similar size, mass, and incident energy follow similar scattering pathways regardless of their electronic structure. This result is consistent with recent studies of Ar and DCl scattering from squalane (a liquid alkane at room Received: November 7, 2010 Revised: December 11, 2010
Transcript of Theoretical Study of the Dynamics of Collisions Between HCl and ω-Hydroxylated Alkanethiol...

Published: January 18, 2011
r 2011 American Chemical Society 2273 dx.doi.org/10.1021/jp1106422 | J. Phys. Chem. C 2011, 115, 2273–2283
ARTICLE
pubs.acs.org/JPCC
Theoretical Study of the Dynamics of Collisions Between HCland ω-Hydroxylated Alkanethiol Self-Assembled MonolayersWilliam A. Alexander and Diego Troya*
Department of Chemistry, Virginia Tech, Blacksburg, Virginia 24061-0212, United States
bS Supporting Information
ABSTRACT: We present a classical-trajectory study of collisions ofHCl with hydroxylated alkanethiol self-assembled monolayers. Thepotential-energy surface used in the trajectory propagation is a combi-nation of the standard OPLS force field to describe the surface and ananalytical potential for the gas/surface interaction developed in this work.The gas/surface potential has been derived based on high-qualityelectronic-structure calculations of model HCl-alcohol systems in thegas phase and includes a flexible Buckingham term and a Coulombicterm. The results of the trajectories calculations are in good agreementwith recent molecular-beam experiments on the same system, therebylending support to the accuracy of the calculations. The collisiondynamics differ vastly from prior scattering studies involving rare gasesand CO, primarily because the gas/surface attraction governed byhydrogen bonding dramatically increases the ability of the gas mole-cules to trap on the surface for extended times. The properties of the desorbing HCl molecules are largely insensitive to the initialcollision energy and are onlymildly affected by the incident angle. An analysis of the reactionmechanism reveals the distinct dynamics oftrajectories that either recoil from the surface directly or undergo multiple collisions with the surface and result in thermalization.
’ INTRODUCTION
Studying energy transfer in collisions of gases with surfacesyields information important to a fundamental understanding ofgas/surface chemical dynamics, which is relevant in a variety offields. For instance, the accommodation and transport of gaseson hydrocarbon surfaces plays an important role in atmosphericchemistry. Surfactant-coated aerosols are abundant in the tropo-sphere, and these coatings represent a barrier for gaseous species toenter and react with species interior to the droplet.1 The oxidation ofthese organic surfaces by various reactive pathways typically confershydrophilic character to the surface, which may aid in dissolution ofpolar atmospheric molecules into the aerosol. Once in the aerosol,the gas species may undergo subsequent reaction. While HCl is asink species for catalytic, ozone-degradingCl radicals, HCl dissolvedinto aerosol particles, or other particles such as water droplets andice crystals, may react to recover Cl2, which upon photolysis restartsthe ozone-destroying catalytic process. Even before dissolution intothe particle, hydroxyl sites on the surfaces can bind the gas-phaseHCl through hydrogen-bonding interactions, initiate reaction bydissociating HCl, or serve as a surface-bound reagent for otherreactions.2 Each of these scenarios is mediated by the initial gas/surface collision, where the interactions of HCl with the organicsurface determine whether the molecule will scatter away directly oraccommodate on the surface.
The dynamics of this initial collision betweenHCl and organic sur-faces has recently been studied using time-of-flight molecular-beam
scattering techniques by Lohr et al.3,4 Hydrophobic and hydrophilicsurfaces were modeled with straight-chain CH3-(CH2)15-SH andterminally hydroxylated OH-(CH2)16-SH alkanethiol self-as-sembled monolayer surfaces on gold, respectively. In those experi-mental studies, it was found that Ar and HCl had similar accom-modation fractions and energy-transfer dynamics when collidingwiththe nonpolar hydrocarbon surface. However, these gases showedremarkably different dynamicswhen scattering from the hydroxylatedsurface. Using the traditional separation of the products’ energydistribution into an impulsive scattering (IS) and thermal desorption(TD) channel,5 the experiments indicate that while 43% of the Aratoms recoil from the OH-SAM with a thermal energy distributionat the surface temperature when colliding at 85 kJ 3mol
-1 and 30�,73% of theHCl thermalize at the same conditions, showing the effectof gas/surface hydrogen bonding on the dynamics of interfacialscattering. The results that Ar and HCl exhibit similar scatteringdynamics in collisionswithCH3-SAMs imply that kinematics, ratherthan the weak gas/surface chemical forces in that system, determinethe scattering outcome, such that gases with similar size, mass, andincident energy follow similar scattering pathways regardless of theirelectronic structure. This result is consistent with recent studies ofAr and DCl scattering from squalane (a liquid alkane at room
Received: November 7, 2010Revised: December 11, 2010

2274 dx.doi.org/10.1021/jp1106422 |J. Phys. Chem. C 2011, 115, 2273–2283
The Journal of Physical Chemistry C ARTICLE
temperature)6 andwith earlier experimental work on the scattering ofNe, H2O, NH3, and CH4, also from squalane.7 The trend thatmolecules with similar mass display similar behavior when scatteringfrom the nonpolar surfaces has also been corroborated by studiesusing alkanethiol self-assembled monolayers.8
Another revealing result of Lohr and co-workers was that theOH-SAM is a more rigid collision partner than a regularCH3-SAM surface due to the extensive hydrogen-bondingnetwork formed by the terminal OH groups. The larger rigidityof the OH-SAM can be appreciated by the percentage ofcollisions in which Ar atoms recoil directly from the surface(57%), which is notably larger than for a less rigid regularCH3-SAM (39%). The extensive hydrogen-bonding networkin the OH-SAM has been recently thoroughly studied viamolecular-dynamics simulations by the Hase group.9 The simu-lations reveal that, in comparison to a normal alkanethiol SAM,the OH-SAM is disordered, with extensive, dynamic hydrogenbonding between the OH groups, giving rise to local areas on thesurface where clusters and chains of hydrogen-bonded hydroxylgroups form a rigid “glassy” surface. To form these networks, thehydroxyl groups can approximate to an O-O distance of ∼3 Å,in comparison to the 5 Å nearest-neighbor spacing of the sulfuranchor groups. This clustering results in small, unordereddomains in which the underlying hydrocarbon regions of thealkyl chain are exposed. Trajectory calculations of Ar scatteringfrom the CH3- and OH-SAM surfaces agree with the experi-mental findings that Ar recoils from the OH-SAM with higheraverage final energies than from the CH3-SAM and thatpenetration into the monolayer is decreased on the hydroxylsurface.9 Although ab initio calculations of the potential-energysurface show the Ar/OH-SAM potential to be more attractivethan the Ar/CH3-SAM potential, the rigidity of the hydrogen-bonding network results in Ar scattering with slightly higher finalenergies in direct scattering events.
A number of other dynamics investigations of HCl-surfacecollisions involving hydroxylated systems have been performedrecently using both experiment and theory. The Nathansongroup provided extensive insight into the scattering, trapping,and dissociation channels when HCl collides with sulfuricacid10,11 and glycerol.6,12-14 The liquid scattering experimentsreveal high trapping probabilities, in qualitative agreement withthe work of Lohr et al. on SAM surfaces. Pettersson and co-workers performed molecular-dynamics and molecular-beamstudies of HCl and Ar in collision with water ice surfaces15-18
and demonstrated that these species transfer energy efficiently tothe surface. Additionally, they observed distinct scattering chan-nels for the recoiling HCl molecule: direct inelastic scattering,trapping followed by prompt desorption, and long-time up-take.18 The first channel is analogous to the aforementionedimpulsive scattering (IS) channel observed in rare-gas scattering,while the second and third are due tomolecules which thermalizeon the surface, with the third being characterized by the forma-tion of realtively strong hydrogen bonds between the gas and thesurface. The studies of Lohr and co-workers of HCl scatteringfrom OH-SAM surfaces also showed evidence for this third,long-lived trapping channel.3
With this study, we aim to investigate the mechanistic detailsof HCl’s initial collision with a model hydroxylated organicsurface via use of classical trajectories. We propagate the trajec-tories using our own potential-energy surface (PES) derivedfrom high-accuracy ab initio calculations, the implementation ofwhich is extensively detailed within. Our general approach is
analogous to work we and others have done previously in studiesof rare-gas9,19-25 and small-molecule26-30 collisions with modelorganic surfaces. In all of those studies, the organic surface wasdesigned to mimic the self-assembled monolayers (SAMs) usedextensively in many experimental scattering studies.9,21,31-38 How-ever, the increased complexity of describing the hydrogen-bondingattractions between the gas and the surface makes the HCl/OH-SAM system substantially more challenging to model thanthe previous work involving straight-chain alkanethiol surfaces andrare gases. An accurate description of a hydrogen-bonding systemsuch as HCl/OH-SAM requires that the long-range character ofthe gas/surface interactions must be captured within the PES. Thisproblem is not as apparent in simpler systems, such as rare gasesscattering from alkanethiol SAMs, where short-range repulsions andkinematic effects dominate the scattering outcome19,21 and theattractive dispersion interactions decrease rapidly with the separa-tion between the gas and the surface.
The remainder of the paper is divided into two parts. In thefirst, we describe our methods for deriving an analytical two-bodypairwise potential for the HCl/OH-SAM system derived byfitting high-accuracy electronic-structure calculations to modelgas-phase systems. In the second section, we enlist our gas/surface model to perform classical trajectory simulations of HClscattering from hydroxyl-terminated self-assembled monolayers.We validate our model with comparison to existing molecular-beam scattering experiments, subsequently investigate the influ-ence of collision energy and incidence angle on the scatteringdynamics, and provide details of the microscopic collisionmechanism.
’COMPUTATIONAL DETAILS
Potential-Energy Surfaces. The potential-energy surfacesemployed to evolve the HCl/SAM trajectories are analgous tothose described in detail in our prior work on gas/SAMcollisions.19-21,26,27 Briefly, we divide the global potential intothree terms: the potential describing the organic monolayer(surface potential), the potential describing the HCl molecule(gas potential), and the potential for the HCl/SAM interactions(gas/surface potential).We employ an explicit-atom model using the OPLSAA force
field39 for the surface potential, as this standard force field bearsout the experimental structure of regular SAMs,40 with somemodifications, as listed in Table S1 of the Supporting Informa-tion. With this parameter set, the OH-SAM behaves similarly tothe behavior described by Hase and co-workers,9 with localizedhydrogen-bonding clusters and chains which break and reformover time. While the nature of the hydrogen-bonding network isdynamic, the overall extent of hydrogen bonding is consistentthroughout the simulation. To form hydrogen bonds, the OHgroups move closer together (∼3 Å O-O distance) than thenormal SAM nearest neighbor spacing of 5 Å for the sulfur headgroups. As such, the normal ordered and periodic nature of theSAM surface is disrupted, creating domains of glassy hydrogen-bonded islands alternating with crevices in which the interiorhydrocarbon backbone is exposed. These two different domaintypes represent quite different collision partners and shouldgive rise to substantially different scattering dynamics whenHCl impinges on each. In the experiment these effects will beaveraged, but in the current study, we can differentiate betweenthese two impact domains to gain further insight into thecollision dynamics.

2275 dx.doi.org/10.1021/jp1106422 |J. Phys. Chem. C 2011, 115, 2273–2283
The Journal of Physical Chemistry C ARTICLE
A standard Morse function is used to describe the gaspotential
VHCl ¼ D 1- e- β r- reð Þh i2
ð1Þ
where D, β, and re are the bond dissociation energy, curvature ofthe potential, and equilibrium bond length, respectively. Toappropriately describe the HCl molecule, D and re are taken tobe 103.15 kJ 3mol
-1 41 and 1.2750 Å.42 The value of β = 1.9005Å-1 was determined by fitting to established experimental valuesof HCl’s spectroscopic constants.42 The resulting fundamentalfrequency and rotational constant obtained with our Morsepotential are ωe = 2990.9 cm-1 and Be = 10.582 cm-1, whichare in excellent agreement with the experimental values of ωe =2990.9 cm-1 and Be = 10.596 cm-1.42
Regarding the gas/surface potential, earlier work demon-strated that popular force fields commonly fail to model accu-rately intermolecular interactions involving gases with SAMs.19
Instead, analytic potentials must be derived from high-quality abinitio calculations26,43 for adequate accuracy, and we followedsuch strategy in this work for the HCl/OH-SAM system. In thefollowing, we first describe the ab initio calculations used tocapture the intermolecular interactions of HCl with OH-SAMsand then detail how we use those electronic-structure calcula-tions to develop pairwise analytical potentials that can beconveniently used to propagate classical trajectories.Ab Initio Calculations. To model the interactions between
HCl and theOH-SAMs, we calculated intermolecular potential-energy curves for the HCl-CH3OH system by scanning theHCl-CH3OH center-of-mass coordinate from the asymptote torepulsive energies of about 400 kJ 3mol
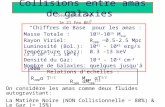
-1 using electronic-structuremethods. The separation between the points of each scan is 0.1 Å.The equilibrium geometry of the CH3OH molecule as well as theequilibrium HCl bond length have been held fixed throughoutthe scans. For adequate coverage of the potential-energy surface, weinvestigated a total of eight different approaches of HCl to theCH3OH molecule, which are depicted in Figure 1. In the “facial”approaches, f(H-C) and f(Cl-C), the HCl approaches the centerof the triangle formed by the three methyl hydrogens, with the scanaxis collinear to the C-O bond. In “bisect” approaches, b(H-O)and b(Cl-O), HCl approaches the hydroxyl oxygen collinearly tothe scan axis that bisects the methanol C-O-H angle (—(C-O-H) =108.8�, so the angle formed by theH-Cl andC-Obondaxes is 125.6�). In the configurations denoted h(H-H) andh(Cl-H), HCl approaches the hydroxyl hydrogen collinear tothe O-H bond. Finally, we scanned the HCl molecule collinearto the methyl hydrogen located anti to the hydroxyl, denoteda(H-H) and a(Cl-H) approaches.
The electronic Schr€odinger equation has been solved at eachstep of the scans using second-order M€oller-Plesset perturbationtheory in combination with the double-, triple-, and quadruple-ζfamily of correlation-consistent basis sets of Dunning,44-46 aug-mented with diffuse functions (aug-cc-pVDZ, aug-cc-pVTZ, andaug-cc-pVQZ, respectively). Coupled cluster calculations withexplicit single and double excitations and perturbative treatmentof triple excitations (CCSD(T)) have also been carried out with theaug-cc-pVDZ basis set. The focal-point approach of Allen and co-workers47,48 has been used to estimate CCSD(T) energies with theaug-cc-pVTZ and aug-cc-pVQZ basis sets from MP2 calculationswith those basis sets. (Hereafter, focal-point CCSD(T) energieswill be referred to as fp-CCSD(T).) The focal-point approach isbased on the observation that the difference between MP2 andCCSD(T) energies is essentially independent of basis set for high-quality basis sets. By calculating the differences between MP2and CCSD(T) energies with an affordable basis set (e.g., aug-cc-pVDZ), for a variety of intermolecular geometries, CCSD(T)energies with larger basis sets (e.g., aug-cc-pVTZ, aug-cc-pVQZ)can be estimated from MP2 calculations at those geometries (fp-CCSD(T) data). The legitimacy of this approach has been demon-strated by our group and others in the description of various pairs ofinteracting species.43,49,50 Complete basis set (CBS) estimates areobtained for both MP2 and fp-CCSD(T) calculations using thetwo-point extrapolation procedure of Halkier et al.51 We removedthe basis-set superposition error using the standard counterpoisemethod52 in all of the points of the calculated potential-energysurfaces. The electronic-structure calculations have been carriedout with the Gaussian03 suite of programs.53
Figure 2 shows the intermolecular potential-energy curve ofthe HCl-CH3OH approach, b(H-O), that yields the deepest wellas a function of the separation between the closest interatomicdistance of the interactingmolecules as predicted by various ab initiomethods. The intermolecular potential-energy curve shows theexpected features of a steep repulsive wall at short distances and arelatively deep attractive well at longer separations due to stabilizingintermolecular interactions dominated by the formation of a hydro-gen bond. An examination of the dependence of the intermolecularpotential on the basis set reveals that, as expected, for MP2calculations, an increase in the size of the basis set results in lowerintermolecular energies. In addition, the location of the potential
Figure 1. Schematic of the HCl-CH3OH orientations investigatedwith ab initio calculations in this work.
Figure 2. Calculated intermolecular potential energy for the HCl-O-(H)CH3 system as a function of theH(Cl)-Odistance for the b(H-O)approach orientation (see Figure 1) with various basis sets.

2276 dx.doi.org/10.1021/jp1106422 |J. Phys. Chem. C 2011, 115, 2273–2283
The Journal of Physical Chemistry C ARTICLE
wells occurs at shorter approach distances with larger basis sets. TheMP2method converges quickly with increasing basis set size, so thatthe difference between aug-cc-pVDZ and aug-cc-pVTZ energies issubstantially larger than that between the aug-cc-pVTZ and aug-cc-pVQZ data. Quantitatively, the root-mean-square deviation(rmsd) between MP2/aug-cc-pVDZ and aug-cc-pVTZ energies is6.80 kJ 3mol
-1 for the overall 315 points calculated for all eightapproaches, while the rmsd between aug-cc-pVTZ and aug-cc-pVQZ data decreases to 1.93 kJ 3mol
-1.Comparison of the CCSD(T) and MP2 data with the aug-
cc-pVDZ basis set reveals how treating electron correlation influ-ences the characteristics of the potential-energy surface. We find thedifferences between MP2 and CCSD(T) data to be sizable, with armsd of 3.23 kJ 3mol
-1 over all approaches, which is nearly twice theinfluence of increasing the basis from the triple to the quadruple basisset. This indicates that for this system, inclusion of CCSD(T)-leveldata will greatly increase the accuracy of our potential.Analytic HCl/OH-SAM Potentials. Using the highest level
ab initio information for the HCl-CH3OH system (fp-CCSD(T)/CBS), we constructed two-body analytic potentials that can be usedto describe the HCl/OH-SAM gas/surface potential-energysurface. We considered two different potential forms constructedas sums of two-body functions. The first functional form is thefamiliar Buckingham potential, where each two-body term isexpressed in the form
Vij ¼ Aije- Bijrij þCij
rnijij
ð2Þ
where rij is the internuclear distance between the atoms of each pairand Aij, Bij, Cij, and nij are adjustable parameters specific to each pairof atoms.Wewill refer to this function hereafter as “Fit A.”Weused anonlinear least-squares procedure to obtain the values of the Aij, Bij,Cij, and nij parameters that minimize the relative differences betweenanalytic energies obtained with the Buckingham potentials and thefp-CCSD(T)/CBS data for all of the points contained in thepotential-energy-surface scans described before. While the Bucking-ham function normally includes an inverse r6 term, designed todescribe attraction due to dispersion, by allowing this exponent tovary, the generalized exponential function is in principle flexibleenough to incorporate longer-ranged interactions which arrise frompartial localization of charge within the molecules. We tested thisidea with a second fit which includes addition of an explicitCoulombic term, making the function of the form
Vij ¼ Aije-Bijrij þ Cij
rnijij
þ qiqj4πε0rij
ð3Þ
where the additional terms qi and qj are the partial charges of theinteracting atomic pair and ε0 is the permittivity of free space.We will denote this function “Fit B.” Atomic partial charges forthe methanol molecule were taken from the OPLSAA forcefield and are-0.683, þ0.145, þ0.418, and þ0.060 e for the O,C, H(O), and H(C) atoms, respectively.39 Charges on the HClmolecules were taken to beþ0.176 and-0.176 e for the H andCl atoms, respectively.41 We found that with the inclusion ofthe Coulombic term, allowing the nij exponent to vary did notresult in significant improvement of the overall fit and addi-tionally resulted in no significant differences in the moleculardynamics results of test runs. We did find, however, slightimprovement if we allowed nij to vary only for the Cl-H(C)and Cl-H(O) pairs. For both functional forms, we found theH-H(O) and H-H(C) terms to be almost entirely repulsive,
so for those terms, either the Aij (for Fit A) or the Cij (for Fit B)parameter has been set to zero to simplify the function.The two-body Buckingham potentials provide an adequate
representation of all eight approaches of HCl to the CH3OHmolecules. Each of the fits includes a total of about 315 pointsdistributed roughly evenly between the eight approaches and coversregions of the potential-energy surface from the asymptote up to∼400 kJ 3mol
-1. Because we would like to implement thesepotentials in molecular dynamics simulations at superthermal (∼1eV) scattering energies, particular care was taken to maintain anaccurate description of the repulsive walls while ensuring accuracy inthe region of the attractive well. To achieve this, different weightingschemes were implemented for each fit. In Fit A, we weighed pointswhose energies were >350 kJ 3mol
-1 and <-0.5 kJ 3mol-1 by a
factor of 2.0. In Fit B, those points with energies <-0.5 kJ 3mol-1
were also given increased weight by a factor of 2.0 during the fit.Additionally, in Fit B, points in the h(Cl-H) approach wereweighed by a factor of 4.0, as this approach was difficult to fitotherwise. In both fits, for points between 0.40 and-0.40 kJ 3mol
-1,absolute, as opposed to relative, differences were taken for our fittingprocedure. This procedure ensured a minimization of root-mean-square deviation values in the repulsive region of the potential whilemaintaining a good fit globally for the eight approaches. The averagerelative errors between fp-CCSD(T)/CBS calculations and ourfitted values in the repulsive region are 5.0% and 16.8% for Fits Aand B, respectively. The majority of the deviation in the fit arises inthe facial approaches, especially f(H-C). As this approach is notsampled in our application of these potentials to the HCl/OH-SAM system, this discrepancy is not a concern for this study.Omission of the facial approach data lowers the average relative errorfor the repulsive region of Fit B to 7.9%. Average absolute deviationsin the region of the attractivewell are 0.18 and 0.10 kJ 3mol
-1 for FitsA and B, respectively.The optimum parameters of the analytic potentials we
obtained for the systems studied in this work are shown inTables 1 and 2. A direct comparison between the analytic and abinitio data for the eight approaches is displayed in Figure 3, and thelocation and depth of the potential minima for all approaches areshown in Table 3. The table shows that the separation betweenHCland CH3OH at the minima furnished by the analytic potential-energy surface is within 0.1 Å of the fp-CCSD(T)/CBS estimates.The agreement between the analytic and ab initio energy minimais overall quite good, with average deviations of only 1.27 and0.28 kJ 3mol
-1 over all approaches for Fits A andB, respectively, with
Table 1. Parameters of the Analytic Potentials Describing theHCl/OH-SAM Interactions for Fit Aa
pair Aij Bij Cij nij
Cl-C 322 819.9 3.747 -30 770.076 6.379
Cl-O 218 063.1 4.294 -20 791.229 10.070
Cl-H(C) 4205.6 3.113 -29.980 7.22
Cl-H(O) 4863.8 3.253 -52.781 6.857
H-C 4912.4 3.326 -118.257 9.569
H-O 4743.5 3.516 -111.921 1.549
H-H(C)b 0.0 n/a 24.112 16.046
H-H(O)b 0.0 n/a 30.932 6.012aUnits are such that if internuclear distances are given in Angstroms,then the potential energy is in kcal 3mol-1. bThe H-H(C) and H-H-(O) pairs were found to be almost wholly repulsive; so, we simplified thisterm by setting Aij to zero; when Cij is positive the inverse rij
n term inthe gas/surface potential becomes repulsive.

2277 dx.doi.org/10.1021/jp1106422 |J. Phys. Chem. C 2011, 115, 2273–2283
The Journal of Physical Chemistry C ARTICLE
Fit A having a maximum deviation of 3.98 kJ 3mol-1 for the
b(H-O) approach. The deviations in Fit B are substantially smallerthan the collision energy of the scattering calculations (60-110 kJ 3mol
-1), and therefore, the small size of the fit error shouldhave a minor impact. However, the inability of Fit A to describe wellthe hydrogen-bonding b(H-O) approach is expected to have amarked effect on the scattering dynamics.Classical-TrajectoryCalculations. Using the analytical potential-
energy surfaces described in the prior section, we performed classical-trajectory calculations of collisions of the HCl molecule withterminally hydroxylated alkanethiol self-assembled monolayers.Our trajectory integration scheme is analogous to that reported
previously,19,20,26 so only a brief description of details relevant tothis work is provided here.Unless otherwise noted, we integrated batches of 3000 trajec-
tories for each set of collision energy (Ecoll) and incident angle(θi) explored in this work. Sample work with more than twicethat number of trajectories was used to establish the statisticalsignificance of the results obtained with 3000 trajectories. Initialconditions for the HCl molecule were determined via quasi-classicalsampling of selected vibrational and rotational states as implementedin the VENUS96 computer program.54 We chose to propagate alltrajectories with the HCl molecule in the v = 0, j = 0 state, whichshould closely match the low rotational states populated in theexperiment after the cooling associatedwith the supersonic expansionin the molecular beam. At the beginning of each trajectory, HCl wasplaced above the surface at a separation of at least 10 Å from theclosest surface atom. The initial conditions (coordinates andmomenta) of the surface are taken as intermediate steps of a 0.5 nsisothermal simulation of the SAM at 300 K. In the scattering calcula-tions, we use SAMs composed of 36 thiolate chains (S-(CH2)8-OH) that are replicated in two dimensions using the periodic-boundary-conditions algorithm of the TINKER package of pro-grams.55While the experimentswe comparewith employ SAMswithsomewhat longer chain lengths, it has been shown that the scatteringbehavior is independent of methylene chain length for straight-chainalkyl SAMs of more than 6 methylene units.31 The initial azimuthalangle formedby the incidentHCl velocity vector and the tilt directionof the SAM chains is randomly selected to establish fair comparisonwith experiment. The initial spatial orientation of HCl was assignedvia rotation through random Euler’s angles.The trajectories were stopped postcollision when either the
gas recoiled to a distance of 12Å from the closest atomof the surfaceor the HCl molecule had not desorbed from the surface after 24 ps.In those trajectories stopped as a result of long trapping times, HClis assumed to be fully thermalized with the surface and is assignedfinal translational energies, E0T, and rovibrational states, v0, j0, basedon Boltzmann distributions at the surface temperature, and a finalpolar angle, θf, based on a random distribution.
’RESULTS AND DISCUSSION
Comparison with Experimental Translational-Energy Distri-butions. In Figure 4, we plot the calculated product translational-energy distributions (PTDs) of HCl after collision with the
Table 2. Parameters of the Analytic Potentials Describing theHCl/OH-SAM Interactions for Fit Ba
pair Aij Bij Cij nij
Cl-C -1385.3 1.792 5001.252 6.000
Cl-O 60 766.5 3.088 -6675.732 6.000
Cl-H(C) 6036.7 3.327 -67.191 9.480
Cl-H(O) 16.4 0.923 223.492 5.123
H-C 68 206.9 4.162 -1266.895 6.000
H-O 4296.7 4.158 -7.884 6.000
H-H(C)b 359.9 2.853 0.0 n/a
H-H(O)b 704.9 3.915 0.0 n/aaUnits are such that if internuclear distances are given in Angstroms,then the potential energy is in kcal 3mol-1. bThe H-H(C) and H-H-(O) pairs were found to be almost wholly repulsive; so, we simplified thisterm by setting Cij to zero.
Figure 3. Comparison of fp-CCSD(T)/CBS and analytic intermolec-ular potential-energy surfaces for the HCl-CH3OH system. Ap-proaches are denoted in each panel (see Figure 1). Distances r(X-Y)are defined as the distance between the two closest atoms in eachapproach (i.e., H-O distance in the b(H-O) approach).
Table 3. Comparison of ab Initio and Analytic Energy andGeometry of the van derWaalsMimima for theHCl-CH3OHSystema
approachb ab initio fit A fit B
f(H-C) 1.20 (2.7) 1.76 (2.7) 1.61 (2.6)
f(Cl-C) 3.11 (3.5) 2.64 (3.4) 2.96 (3.5)
b(H-O) 25.31 (1.9) 29.20 (1.8) 25.45 (1.8)
b(Cl-O) 4.39 (3.1) 2.97 (3.1) 5.27 (3.1)
h(H-H) wholly repulsive
h(Cl-H) 1.14 (3.0) 0.36 (3.0) 1.09 (3.1)
a(H-H) wholly repulsive
a(Cl-H) 1.08 (3.1) 0.58 (3.1) 1.11 (3.1)a Energies below the asymptote in kJ 3mol-1. Values in parenthesescorrespond to the distance between the two closest atoms (i.e., H-Odistance in the b(H-O) approach) in Angstroms. The ab initio datacorrespond to fp-CCSD(T)/CBS values. bThe approach geometries aredepicted in Figure 1

2278 dx.doi.org/10.1021/jp1106422 |J. Phys. Chem. C 2011, 115, 2273–2283
The Journal of Physical Chemistry C ARTICLE
OH-SAM surface at Ecoll = 85 kJ 3mol-1 with a 30� incident angle
for both of the fitted analytical potentials derived above. Extensiveloss of initial translational energy to the surface is seen with both fits.However, use of Fit A in our molecular dynamics trajectories resultsin HCl scattering with much higher final translational energies thanwith Fit B.While the average final translational energy, ÆE0Tæ, is 14.2kJ 3mol
-1 for Fit A, for Fit B ÆE0Tæ = 7.7 kJ 3mol-1.
The figure also shows the experimentally determined PTD ofLohr et al.3 We note that the experimental distributions corre-spond to a specific final recoil angle with respect to the surfacenormal (θf = 30�), and the calculations consider all of thescattered flux. However, as we will show later, the amount offinal translational energy of HCl is essentially independent of itsrecoil angle, thereby validating the comparison in Figure 4.Excellent agreement between theory and experiment is foundfor the results of dynamics trajectories employing the Fit Bparameter set. Results of the Fit A potential, however, are clearlyin disagreement with experiment.In searching for the origins of the different results obtained
with Fits A and B, we find that trajectories in which HClundergoes a “direct” collision with the surface actually result insimilar average translational energies for either fit, ÆE0Tæ is 17.5and 16.3 kJ 3mol-1 for Fits A and B, respectively. We take here“direct” trajectories to mean those trajectories in which the HClcenter-of-mass has only one turning point in the surface-normaldirection during the course of the simulation. The scatteringbehavior of directly scattered HCl is mostly governed by therepulsive regions of the potential-energy surface. As such, thesimilar energy transfer characteristics of directly scattered HClfor the two fits is not surprising as both employ the same generalform in the description of the repulsive region of the HCl/OH-SAM potential. On the other hand, the number of mole-cules undergoing a direct vs nondirect mechanism is vastlydifferent on both surfaces. Fit A is a generally more repulsivesurface. In effect, while 69% of the HCl molecules scatter directlywhen employing this potential, only 22% of trajectories displaydirect scattering with Fit B. Additionally, if HCl does not scatterdirectly, it may become trapped on the surface for times longerthan feasible to integrate. For these trajectories, we set a cutoff
time of 24 ps. We deem these timed-out trajectories “longtrapping” and observe that the fraction of long trapping issubstantial, 69%, with Fit B, while Fit A has less than 5% oftrajectories that time out. The main reason for long trapping inthe HCl/OH-SAM system is due to strong hydrogen-bondinginteractions between the gas and the surface. As such, the absenceof long trapping trajectories for the Fit A parameter set is anindicator that this potential may not describe these interactionswell. Fits A and B differ in the attractive part of the potential inthat Fit B incorporates a long-ranged Coulombic term withexplicit partial charges on the HCl atoms. The level of agreementbetween theory and experiment seen in Figure 4 therefore seemsto indicate that the inclusion of a Coulombic term in the potentialis necessary for an accurate description of the experimentalscattering behavior. Since it has been shown to be superior atprediction of experimental scattering trends, we used Fit B forfurther investigation into the dynamics of the initial HCl collisionwith the OH-SAM surface. Hereafter, reference to simulationsand data shall be exclusively for the Fit B parameter set.Effect of Incident Translational Energy. We focus here on
the correlation between initial translational energy and finaltranslation (T f T0 energy retention), final HCl rotation (T fR0 energy transfer), and final HCl vibration (T f V0 energytransfer). In Figure 5, we plot the calculated final translational-energy probability distributions for HCl scattering fromOH-SAM with Ecoll = 56, 85, and 101 kJ 3mol-1 and θi = 30�.Scattering properties from these initial conditions are summa-rized in Table 4. Over this range of collision energies, transfer ofthe initial translational energy is quite efficient, and in a largefraction of trajectories HCl loses enough energy to thermalizewith the surface. On average, the translational energy-transferratios, defined as (Ecoll - ÆE0Tæ)/Ecoll are 0.89, 0.91, and 0.91 atEcoll = 56, 85, and 101 kJ 3mol-1, respectively. As the collisionenergy increases, the PTDs shift to slightly higher values, with aporportional decrease in the low-energy scattering component,pointing to a rather weak T f T0 energy correlation.Table 4 also lists the final average rotational and vibrational
energies of HCl at the three collision energies. With the relativelyclose energetic spacing of the HCl rotational states, one mayexpect the opportunity for high rotational states to be populatedat these collision energies. Instead, on average we see relativelylittle rotational excitation of HCl after collision. The average final
Figure 4. Calculated HCl final translational energy distributions for FitsA and B compared to the experimental translational energy distributionobserved by Lohr et al.3 for 85 kJ 3mol-1 HCl scattering from a 300 KOH-SAM with a θi = 30� angle of incidence. Note that the experi-mental distribution is recorded at a detection angle of θf = 30�, while thecalculated distributions are integrated over all final scattering angles.
Figure 5. Calculated HCl final translational-energy distributions scat-tering from an OH-SAM with a θi = 30� angle of incidence for variouscollision energies.

2279 dx.doi.org/10.1021/jp1106422 |J. Phys. Chem. C 2011, 115, 2273–2283
The Journal of Physical Chemistry C ARTICLE
rotational energy for all three collision energies is about 2.5 kJ 3mol-1, corresponding to only between 4.2% and 2.5% of theinitial translational energy available. In contrast, previous work26
on CO scattering from the rigid CF3-SAM surface showed thatCO channeled nearly 8% of its 86 kJ 3mol-1 collision energy intorotations, three times the amount of Tf R0 energy transfer seenin 85 kJ 3mol
-1 HCl scattering from the OH-SAM. The gas/surface collisions are seen to be not at all efficient in promotingHCl vibrational excitation; HCl always emerges vibrationallycold, even though the amount of energy available during thecollisions is sufficient for vibrational excitation.While collision energy seems to have little influence on the
final rovibrational state of the scattered HCl molecule, it does seemto influence themechanismof the collision. Separationof trajectoriesinto direct and indirect pathways is an important measure, as thedifferent average final scattering characteristics are determinedmainly by the branching ratio between thosemoleculeswhich scatterdirectly after only a few encounters with the surface and thosemolecules which trap on or within the SAM surface. This separationis qualitatively analogous to the common practice of fitting IS andTD components to experimentally observed final energy distribu-tions of scattered molecular beams.We find the percentage of directtrajectories to increase substantially from 15% to 26% for HClscattering from the OH-SAM at Ecoll = 56 and 101 kJ 3mol
-1,respectively (see Table 4). The enhancement of a direct mechanismwith increasing collision energy is also apparent when examining thepercentage of trajectories that remain trapped on the SAM surfaceafter 24 ps, which clearly decreases with collision energy. We notethat while not rigorously comparable, our simulations show that 22%of trajectories participate in a direct mechanism for 85 kJ 3mol
-1
HCl scattering, which is consistent with the experimentally deter-mined IS fraction of 26%.4 Additionally, the translational energytransfer fraction for direct trajectories of 0.81 agrees well with theexperimentally determined IS energy transfer fraction of 0.78.4
We also investigated whether the recoil direction of HCl isinfluenced by the initial collision energy. Examination of the HClpolar-angle distributions displayed in Figure 6 (lines, left axis)reveals that collision energy has virtually no effect on the recoildirection of the scattered HCl. As seen in Table 4, the averagefinal polar scattering angles (angle formed by HCl’s final velocityvector and the surface normal) do not change appreciably in therange of collision energies explored. Also plotted in Figure 6 are
average final translational energy values as a function of polarscattering angle (symbols, right axis). Interestingly, the averagefinal translational energy is not heavily influenced by scatteringangle, in contrast to previous work on rare gases and diatomicsscattering from straight-chain hydrocarbon SAMs, which show apronounced increase in final translational energy as the polarangle increases.21,26 In those previous studies, quantitative agree-ment with experimental results required selecting for analysisonly a small subset of the simulated trajectories grouped aroundthe experimental detection angle of 30�. However, with theinsensitivity to polar scattering angle for the HCl/OH-SAMsystem, we found good agreement with experimental resultswithout this angle resolution being necessary.Effect of Incident Angle. Thus far, our discussion has been
concerned with HCl/OH-SAM collisions in which the anglebetween the initial HCl velocity vector and the surface normalwas 30�, which corresponds to the experimental geometricalconstraints of the molecular-beam scattering experiments byLohr and co-workers.3 In this section, we examine the depen-dence of the scattering properties on the angle of incidence. Finaltranslational-energy distributions for HCl collisions with Ecoll =85 kJ 3mol-1 at three different incidence angles of 30�, 45�, and60� are plotted in Figure 7. The results are summarized inTable 4.Energy transfer out of initial translation is overall less efficient
as the angle of incidence increases from θi = 30� to 60�. Averagetranslational energy-transfer fractions decrease from 0.91 to 0.90to 0.86 for θi = 30�, 45�, and 60�, respectively. This result hasbeen observed before in other scattering work21,26 and iscommonly understood as being due to the fact that it is moredifficult to transfer an impinging molecule’s momentum directedparallel to the surface than perpendicular momentum. Over therange of energies explored, the influence of the angle of incidencehas amuchmoremarked effect in determining the final scatteringproperties than collision energy. For instance, taking the data atEcoll = 85 kJ 3mol-1 and θi = 30� as a reference, we see that anincrease of 15� in the incident angle bears essentially the sameeffect as an increase of 16 kJ 3mol-1 in the collision energy. Withincreasing incident angle, a larger component of the HCl’stranslational momentum is sequestered in the surface-paralleldirection, effectively leaving less energy available to participate inefficient energy-transfer pathways.
Table 4. Calculated Scattering Properties As a Function ofCollision Energy and Angle of Incidence in Collisions of HClwith OH-SAMsa
Ecollb θi
c ÆE0Tæd ÆE0ROTæe ÆE0VIBæf Æθfæg % directh % longi
56 30 6.4(13.2) 2.4 15.9 44 14.6 77
85 30 7.7(16.3) 2.4 15.8 45 22.4 69
85 45 8.9(19.7) 2.5 16.4 46 24.1 68
85 60 12.0(25.6) 2.6 16.9 48 31.1 60
101 30 8.9(18.8) 2.5 16.2 45 26.2 66a Initial rovibrational states: v = 0, j = 0; all energies are in kJ 3mol-1.Values within parentheses correspond to averages considering onlythose molecules undergoing a direct mechanism, as defined in the text.b Initial gas translational energy. c Incident collision angle defined indegrees from the surface normal. dAverage final HCl translationalenergy. eAverage final HCl rotational energy. fAverage final HClvibrational energy. gAveragepolar scattering angle in degrees. hPercent-age of trajectories experiencing only one turning point. iPercentage oftrajectories that do not desorb the surface after 24 ps.
Figure 6. Calculated HCl final angular distributions (lines, left axis) andaverage final translational energies as a function of polar scattering angle(symbols, right axis) for a range of collision energies andθi = 30� angle ofincidence.

2280 dx.doi.org/10.1021/jp1106422 |J. Phys. Chem. C 2011, 115, 2273–2283
The Journal of Physical Chemistry C ARTICLE
The data in Table 4 show some indication that rovibrationalexcitation is enhanced atmore grazing angles, but this effect is weakat best. However, the effect of the angle of incidence on thecollision mechanism is more pronounced. This is seen mostclearly in the fraction of trajectories undergoing a direct mechanism(‘%direct’ inTable 4). Increasing the incident angle from30� to 60�increases the probability of direct scattering from 22% to 31%. Thisincrease in directly scatteredmolecules is near fully compensated forby the decrease in the fraction of trajectories which trapon/in the surface for longer than 24 ps, which diminishes from69% to 60% with the same increase in the incident angle.Finally, we examined the effect of the incident angle on the
polar recoil angle. Final polar angle distributions are shown inFigure 8 (lines, left axis) along with angle-resolved average finaltranslational energy values (symbols, right axis). While final angledistributions were insensitive to changes in translational energy(see Figure 6), the distributions clearly shift to higher scatteringangles as the angle of incidence is increased. Average θf values are45�, 46�, and 48� for HCl scattering with Ecoll = 85 kJ 3mol-1 andθi = 30�, 45�, and 60�, respectively. Additionally, at θi = 45� and60�, a pronounced increase in final HCl translational energy isseen as a function of scattering angle. While at 30� the differences
in final energies over the range of scattering angles shows only anincrease of 2 kJ 3mol-1 or less, when HCl scatters from theOH-SAM with θi = 60�, ÆE0Tæ increases from∼7 kJ 3mol
-1 fornear-normal (∼0�) scattering angles to ∼18 kJ 3mol-1 for near-parallel (∼90�) scattering angles.Mechanistic Details of the Collision. We will now focus our
discussion on the microscopic mechanistic details of the HCl/OH-SAM collision. Important quantities commonly reportedin experimental studies of gas/surface scattering dynamics are thefractions of molecules included in the impulsive scattering andtrapping desorption components of the scattered molecule’stranslational energy distribution as well as the average fractionalenergy transfer in the impulsive scattering channel. A complica-tion in comparing these experimental values to results of com-putational simulations is the determination of what constituteswhether a trajectory should be defined as participating in the“impulsive scattering” or “trapping desorption” channels. In thissection, we employ various rubrics of impulsive scattering in thehopes of adding additional insight into the mechanism of HCl/OH-SAM collisions. During the course of a trajectory, if theHCl molecule does not scatter directly, it may experience multi-ple encounters with the SAM surface. As a way to track theseencounters, we monitor both the linear and the angular momen-tumof theHClmolecule during the trajectory and identify changesin the x,y,z components of these vectors. We denote any change insign in a component of the linear momentum as a “hop” and anysign change in a component of the angular momentum as a “kick”.For instance, the number of inner turning points experienced bythe HCl center of mass along the surface normal (z axis) will givethe value of hopsz for a particular trajectory. A hopsz value of 1corresponds to the “direct” trajectories described in previoussections. If the sign of any component (x,y,z) of the linearmomentum vector changes, it is included in the hopsxyz valuefor that trajectory. During the trajectory, the sign of the angularmomentum vector components can also change sign, and wecount these changes in the “kicks” value for the trajectory.In Figures 9 and 10 we plotted hopsz probability distributions
for various collision energies and angles of incidence, respec-tively, along with the average final translational energies as afunction of hopsz. Average energy transfer fractions, (Ecoll -ÆE0Tæ)/Ecoll, for the overall distributions and for the hopsz = 1
Figure 7. CalculatedHCl final translational-energy distributions for 85 kJ 3mol-1 HCl scattering from OH-SAM with various angles of incidence.
Figure 8. Calculated HCl final angular distributions (lines, left axis) andaverage final translational energies as a function of polar scattering angle(symbols, right axis) over a range of incident collision angles for85 kJ 3mol
-1 HCl scattering from an OH-SAM.
Figure 9. Calculated HCl hopsz distributions (lines, left axis) andaverage final translational energies as a function of hopsz (symbols,right axis) for a range of collision energies scattering with a θi = 30� angleof incidence.

2281 dx.doi.org/10.1021/jp1106422 |J. Phys. Chem. C 2011, 115, 2273–2283
The Journal of Physical Chemistry C ARTICLE
fractions are included in Table 5. It is seen that the fraction ofone-hop trajectories increases with more glancing angles andwith collision energy. Additionally, the energy-transfer fractionincreases with Ecoll and decreases with incident angle. All theseresults are expected from earlier work on simpler rare-gas/SAMsystems. The general shape of the hop distributions is similarfor all initial conditions, with the probability sharply peaked athopsz = 1 and quickly decreasing with increasing number of hops.Within about five hops, the probability is almost negligible.However, all distributions have a broad peak around hopsz =35 that corresponds to molecules that remain trapped on thesurface when the time cutoff (24 ps) terminates the trajectory.Note that due to this simulation time cutoff, the maximum valueof hopsz that can be obtained by HCl seems to be around 50hops. An important observation is the near absence of populationat hop values intermediate to these two peaks. The implication isthat if the HCl molecule does not scatter immediately, it willbecome trapped for long times. Examination of the average finaltranslational energies as a function of time shows that, onaverage, the HCl molecule needs only a few encounters withthe surface to dissipate enough of its energy and become fullyaccommodated on the surface. In effect, after about five hops, theaverage translational energies are thermal.
Figure 11 shows hopsxyz (bottom panel) and kicksxyz (top panel)probability distributions forHCl scattering with Ecoll = 85 kJ 3mol
-1
and θi = 30�. Comparison of the two distributions reveals that theoverall features are similar to that of the hopsz distributions, with asharp peak at low hops or kicks, and a second broad feature at highervalues.Within the 24 ps trajectory time limit, themaximum numberof sign changes in the linearmomentum is around 120hopsxyz, whilethe maximum number of sign changes in the angular momentum isabout 800 kicks. As analysis of the hopsxyz and kicksxyz distributionsyields nearly equivalent results, we have chosen to discuss resultswith the kicks distributions exclusively. Since the peaks in the kicksdistributions are well separated and resolved, we have been able tointegrate the area under each curve as a way to assign fractionsanalgous to the common experimental determination of impulsivescattering and trapping desorption components. Integration of the“low-kicks” peak yields fractions analogous to the impulsive scatter-ing component in experiment. We included these fractions alongwith the average energy transfer fractions in the low-kick peak inTable 5. The trends with incident energy and angle are the same aswhen examining hopz = 1 trajectories. However, as would beexpected with inclusion of molecules which undergo a few en-counters with the surface, average energy transfer fractions areoverall higher thanwhen examining one-hopz trajectories.While useof the hopsz metric gives values a direct fraction of 22%, 28% oftrajectories are included in the low-kick region of the distribution, aresult that seems in slightly better agreement with the experimen-tally determined IS fraction of 26%.4 The hopsz rubric does howeverreturn a slightly better energy transfer fraction value of 0.81 than thelow-kicks value of 0.83 (experimentally determined value is 0.78). Itis obvious however that regardless of our choice in the analysis of thesimulations, agreement with experiment is very good. Also includedin the top panel of Figure 11 are average final translational energyvalues as a function of kicks. From the figure, it is seen that by about100 angular momentum kicks the impinging molecule will have lostenough energy to thermalize with the SAM surface.With such well-resolved and distinct features in the hops and
kicks distributions, it remains a question as to what determinesthe partitioning between these two channels for this system. Ithas been suggested that the hydrogen-bonding OH networkshould present a more rigid scattering partner, therefore yieldinga higher propensity for impulsive scattering when the impingingmolecule impacts the hydroxyl groups than when colliding withexposed sections of the hydrocarbon backbone.3,4,9 We investi-gated whether the point of impact on the surface determines the
Figure 10. Calculated HCl hopsz distributions (lines, left axis) andaverage final translational energies as a function of hopsz (symbols, rightaxis) over a range of incident collision angles for 85 kJ 3mol
-1 HClscattering from OH-SAM.
Table 5. Comparison of Scattering Properties When Various Definitions of “Impulsive Scattering” Are Used in Collisions of HClwith OH-SAMsa
hopsz kicks
Ecollb θi
c (Ecoll - ÆE0Tæ)/Ecolld (Ecoll - ÆE0T,directæ)/Ecolle % direct f (Ecoll - ÆE0T,low-kickæ)/Ecollg % low-kickh
56 30 0.89 0.76 15 0.80 22
85 30 0.91 0.81 22 0.83 28
85 45 0.90 0.77 24 0.79 30
85 60 0.86 0.70 31 0.73 38
101 30 0.91 0.81 26 0.84 33a Initial rovibrational states: v=0, j = 0. Energies are in kJ 3mol-1. b Initial gas translational energy in kJ 3mol-1. c Incident collision angle defined indegrees from the surface normal. dOverall average energy transfer fraction. eAverage energy transfer fraction for those trajectories in which theHCl center-of-mass coordinate along the surface normal experiencess only one inner turning point. fPercentage of trajectories experiencing onlyone turning point. gAverage energy transfer fraction for those trajectories included in the “low-kick” peak of the kick distribution, as defined inthe text. h Percentage of trajectories included in the “low-kick” peak of the kick distribution, as defined in the text.

2282 dx.doi.org/10.1021/jp1106422 |J. Phys. Chem. C 2011, 115, 2273–2283
The Journal of Physical Chemistry C ARTICLE
scattering outcome by analysis and inspection of trajectoryanimations. Our observations are summarized in Table 6 andsuggest that whether HCl collides first with the hydroxyl groupsor the hydrocarbon backbone has very little effect on itssubsequent dynamics before desorption. In fact, our simulationspoint slightly to the opposite effect for the HCl/OH-SAMsystem in trajectories undergoing a direct mechanism. WhenHCl impacts the surface such that its center-of-mass is closest to ahydroxyl hydrogen, average energy transfer out of translation isenhanced relative to other impact sites. Consistently, overallaverage trajectory time is also slightly increased when HClimpacts the hydroxyl hydrogen. A rationalization for this slighttrend would be that even if the surface hydrogen-bondingnetwork is more rigid than the hydrocarbon backbone, HClcolliding in the OH network has a larger probability of beingattracted to the surface via partial hydrogen bonds prior todesorption. This attraction, which would not be as probable ifHCl scatters from the hydrocarbon backbone, would act to slowdown the recoiling HCl. Regardless of this slight trend, we
believe that HCl’s actual impact site on the SAM is less importantthan the initial energy transfer in the first encounter with thesurface and the local geometry and environment HCl finds itselfin immediately after this encounter. HCl can lose a substantialfraction of its energy in its first encounter with the surface, andthis seems to be independent of where it impacts. Whether themolecule then scatters directly or becomes trapped for some timewill be dependent on the gas/surface potential experienced afterthis encounter.
’CONCLUDING REMARKS
The energy transfer dynamics in collisions of HCl withhydroxylated self-assembled monolayers have been investigatedwith the use of classical trajectories with the goal of under-standing the factors that control the scattering properties ofhydrogen-bonding gases in collisions with hydrogen-bondingsurfaces. Analytic potential-energy surfaces required to de-scribe the HCl/OH-SAM scattering have been derived fromextensive electronic-structure calculations of the interactionpotentials between the HCl and the CH3OH molecules.Highly accurate focal-point-CCSD(T) calculations extrapo-lated to the complete basis set limit have been performedalong multiple approaches of HCl to the methanol molecule toobtain good coverage of the global potential-energy surface.Using these ab initio data, we constructed analytic potential-energy surfaces based on two-body Buckingham functions.Inclusion of a Coulombic term was found to be necessary foraccurate description of the HCl/OH-SAM collision dy-namics. The analytic potentials accurately reproduce the abinitio data and result in a model applicable to describe HClscattering from hydroxylated alkanes.
These analytic potentials have been used in molecular-dy-namics simulations of HCl collisions with terminally hydroxy-lated self-assembled monolayer surfaces over a range of collisionenergies and angles of incidence. Our calculated scattering datawith Ecoll = 85 kJ 3mol-1 and θi = 30� are in excellent agreementwith previous molecular-beam scattering data at similar condi-tions. At all angles and collision energies examined, energytransfer to the surface is extensive. While previous studies of raregases and CO colliding with OH-SAM surfaces showed largeimpulsive-scattering components due to interactionwith the glassyhydrogen-bonding network, the increased attractive hydrogen-bonding interactions present due to HCl’s large dipole seem tocounteract the influence of these rigid regions on the surface andresult in extensive accommodation of the gas on the surface.
Collision with the OH-SAM surface is not very efficient atpromoting rotational excitation, and increasing the initial colli-sion energy of the HCl has little effect on rotational excitation;vibrational excitation is wholly inefficient, even though theavailable energy in the collision is sufficient for vibrationalpopulation. However, increased collision energy does shift thescattering mechanism to more direct-type scattering behavior.Increasing the angle of incidence from 30� (near surface-normal)to 60� (near surface-parallel) results in small increases in theproportion of directly scattered molecules and slightly higheraverage final translational energies. On the whole, the propertiesof the scattered HCl molecule are insensitive to changes over theinvestigated range of energies (56-101 kJ 3mol-1) and are onlymildly affected by changes in the incident angle, and this is in partdue to the fact that in most trajectories HCl undergoes extensivetrapping under the conditions studied.
Table 6. Scattering Properties As a Function of HCl ImpactSite on the OH-SAM Surfacea
impact point ÆE0Tæb Ætimeæc ÆE0ROTæd
All Trajectories
backbone H or C 7.9 18.6 2.2
hydroxyl H or O 7.1 18.5 2.3
Direct Trajectoriese
backbone H or C 17.2 2.7 2.5
hydroxyl H or O 14.6 2.8 3.1a Ecoll = 85 kJ 3mol-1, θi = 30�. Energies are in kJ 3mol-1. bAveragefinal translational energy. cAverage simulation time in picoseconds.dAverage final rotational energy. eTrajectories with hopsz = 1.
Figure 11. (a) Calculated HCl kicks distribution (lines, left axis) andaverage final translational energies (kJ 3mol
-1) as a function of kicks (sym-bols, right axis) for Ecoll = 85 kJ 3mol
-1 HCl scattering from OH-SAMwith a θi = 30� angle of incidence. Low-kicks region detailed in inset.(b) Calculated HCl hopsxyz distribution for the same system; note thequalitatively similar shape to the kicks distribution, above.

2283 dx.doi.org/10.1021/jp1106422 |J. Phys. Chem. C 2011, 115, 2273–2283
The Journal of Physical Chemistry C ARTICLE
’ASSOCIATED CONTENT
bS Supporting Information. Table S1 containing the param-eters of the force field used to describe the SAM in this work.This material is available free of charge via the Internet at http://pubs.acs.org.
’AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
’ACKNOWLEDGMENT
This work has been supported by NSF Grant Nos. CHE-0547543 and CHE-0741927 and by AFOSR Grant No. FA9550-06-1-0165. D.T. is a Cottrell Scholar of Research Corporation.
’REFERENCES
(1) Ellison, G. B.; Tuck, A. F.; Vaida, V. J. Geophys. Res. Atmos. 1999,104, 11633.(2) Robinson, G. N.; Worsnop, D. R.; Jayne, J. T.; Kolb, C. E.;
Swartz, E.; Davidovits, P. J. Geophys. Res. Atmos. 1998, 103, 25371.(3) Lohr, J. R.; Day, B. S.; Morris, J. R. J. Phys. Chem. B 2005, 109,
15469.(4) Lohr, J. R.; Day, B. S.;Morris, J. R. J. Phys. Chem. A 2006, 110, 1645.(5) Saecker, M. E.; Govoni, S. T.; Kowalski, D. V.; King, M. E.;
Nathanson, G. M. Science 1991, 252, 1421.(6) Brastad, S. M.; Albert, D. R.; Huang, M.; Nathanson, G. M. J.
Phys. Chem. A 2009, 113, 7422.(7) Saecker, M. E.; Nathanson, G. M. J. Chem. Phys. 1993, 99, 7056.(8) Bennett, M. E.; Alexander, W. A.; Lu, J. W.; Troya, D.; Morris,
J. R. J. Phys. Chem. C 2008, 112, 17272.(9) Tasic, U.; Day, B. S.; Yan, T.-Y.; Morris, J. R.; Hase, W. L. J. Phys.
Chem. C 2008, 112, 476.(10) Morris, J. R.; Antman, M. D.; Nathanson, G. M. J. Phys. Chem. A
2000, 104, 6738.(11) Behr, P.; Morris, J. R.; Antman, M. D.; Ringeisen, B. R.; Splan,
J. R.; Nathanson, G. M. Geophys. Res. Lett. 2002, 28, 1961.(12) Ringeisen, B. R.; Muenter, A. H.; Nathanson, G. M. J. Phys.
Chem. B 2002, 106, 4999.(13) Ringeisen, B. R.; Muenter, A. H.; Nathanson, G. M. J. Phys.
Chem. B 2002, 106, 4988.(14) Chorny, I.; Benjamin, I.; Nathanson, G. M. J. Phys. Chem. B
2004, 108, 995.(15) Bolton, K.; Svanberg, M.; Pettersson, J. B. C. J. Chem. Phys.
1999, 110, 5380.(16) Andersson, P. U.; Nagard, M. B.; Bolton, K.; Svanberg, M.;
Pettersson, J. B. C. J. Phys. Chem. A 2000, 104, 2681.(17) Svanberg, M.; Pettersson, J. B. C.; Bolton, K. J. Phys. Chem. A
2000, 104, 5787.(18) Andersson, P. U.; Nagard, M. B.; Pettersson, J. B. C. J. Phys.
Chem. B 2000, 104, 1596.(19) Day, B. S.; Morris, J. R.; Troya, D. J. Chem. Phys. 2005, 122,
214712.(20) Day, B. S.; Morris, J. R.; Alexander, W. A.; Troya, D. J. Phys.
Chem. A 2006, 110, 1319.(21) Alexander, W. A.; Day, B. S.; Moore, H. J.; Lee, T. R.; Morris,
J. R.; Troya, D. J. Chem. Phys. 2008, 128, 014713.(22) Vazquez, S. A.; Morris, J. R.; Rahaman, A.; Mazyar, O. A.;
Vayner, G.; Addepalli, S. V.; Hase, W. L.; Martinez-Nunez, E. J. Phys.Chem. A 2007, 111, 12785.(23) Bosio, S. B. M.; Hase, W. L. J. Chem. Phys. 1997, 107, 9677.(24) Yan, T.-Y.; Hase, W. L. Phys. Chem. Chem. Phys. 2000, 4, 901.(25) Yan, T.-Y.; Isa, N.; Gibson, K. D.; Sibener, S. J.; Hase, W. L. J.
Phys. Chem. A 2003, 107, 10600.
(26) Alexander, W. A.; Morris, J. R.; Troya, D. J. Chem. Phys. 2009,130, 084702.
(27) Alexander, W. A.; Morris, J. R.; Troya, D. J. Phys. Chem. A 2009,113, 4155.
(28) Perkins, B. G., Jr.; Nesbitt, D. J. Proc. Natl. Acad. Sci. 2008, 105,12684.
(29) Martinez-Nunez, E.; Rahaman, A.; Hase, W. L. J. Phys. Chem. C2007, 111, 354.
(30) Noguerira, J. J.; Vazquez, S. A.; Mazyar, O. A.; Hase, W. L.;Perkins, B. G., Jr.; Nesbitt, D. J.; Martinez-Nunez, E. J. Phys. Chem. A2009, 113, 3850.
(31) Day, B. S.; Morris, J. R. J. Phys. Chem. B 2003, 107, 7120.(32) Day, B. S.; Shuler, S. F.; Ducre, A.; Morris, J. R. J. Chem. Phys.
2003, 119, 8084.(33) Day, B. S.; Morris, J. R. J. Chem. Phys. 2005, 122, 234714.(34) Isa, N.; Gibson, K. D.; Sibener, S. J. J. Chem. Phys. 2004, 120,
2417.(35) Gibson, K. D.; Isa, N.; Sibener, S. J. J. Chem. Phys. 2003, 119,
13083.(36) Gibson, K. D.; Isa, N.; Sibener, S. J. J. Phys. Chem. A 2006, 110,
1469.(37) Fogarty, D. P.; Kautz, N. A.; Kandel, S. A. Surf. Sci. 2007, 601,
2117.(38) Fogarty, D. P.; Kandel, S. A. J. Chem. Phys. 2006, 125, 174710.(39) Jorgensen, W. L.; Tirado-Rives, J. J. Am. Chem. Soc. 1988, 118,
11225.(40) Hautman, J.; Klein, M. L. J. Chem. Phys. 1989, 91, 4994.(41) http://webbook.nist.gov/chemistry.(42) Huber, K. P.; Herzberg, G. Molecular Spectra and Molecular
Structure: IV. Constants of Diatomic Molecules; Van Nostrand Reinhold:New York, 1979.
(43) Alexander, W. A.; Troya, D. J. Phys. Chem. A 2006, 110, 10834.(44) Dunning, T. H., Jr. J. Chem. Phys. 1989, 90, 1007.(45) Woon, D. E.; Dunning, T. H., Jr. J. Chem. Phys. 1993, 98, 1358.(46) Wilson, A. K.; Woon, D. E.; Peterson, K. A.; Dunning, T. H., Jr.
J. Chem. Phys. 1999, 110, 7667.(47) East, A. L. L.; Allen, W. D. J. Chem. Phys. 1993, 99, 4638.(48) Csaszar, A. G.; Allen, W. D.; Schaefer, H. F., III J. Chem. Phys.
1998, 108, 9751.(49) Tasic, U.; Hein, P.; Troya, D. J. Phys. Chem. A 2007, 111, 3618.(50) Cooper, J. R.; Moazzen-Ahmadi, N. J. Chem. Phys. 2007, 444,
28.(51) Halkier, A.; Helgaker, T.; Jorgensen, P.; Klopper, W.; Koch, H.;
Olsen, J.; Wilson, A. K. Chem. Phys. Lett. 1998, 286, 243.(52) Boys, S. F.; Bernardi, F. Mol. Phys. 1970, 19, 553.(53) Frisch, M. J. et al. Gaussian 03, Revision C.02, Gaussian, Inc.:
Wallingford, CT, 2004.(54) Hase,W. L.; Duchovic, R. J.; Hu, X.; Komornicki, A.; Lim, K. F.;
Lu, D.; Peslherbe, G. H.; Swamya, K. N.; Linde, S. R. V.; Varandas, A.;Wang, H.; Wolf, R. J.Quantum Chem. Program Exch. Bull. 1996, 16, 671.
(55) Ponder, J. W.; Richards, F. M. J. Comput. Chem. 1987, 8, 1016.