The Androgen Receptor in Prostate Cancer - ASBMB...The positions of the two trinucleotide repeats...
4
The Androgen Receptor in Prostate Cancer Margaret Centenera θ , Eleanor Need θ , Lisa Butler and Wayne Tilley* Dame Roma Mitchell Cancer Research Laboratories, Department of Medicine, University of Adelaide and Hanson Institute, Adelaide, SA 5000 θ Contributed equally to this work *Corresponding author: [email protected] Page 12 AUSTRALIAN BIOCHEMIST Vol 38 No 1 April 2007 SHOWCASE ON RESEARCH Prostate cancer is the most commonly diagnosed cancer in Australian men, accounting for approximately 23% of all new male cancer cases in 2001 (1). Despite its prevalence, the aetiology of prostate cancer remains to be fully elucidated, with increasing age, race and a family history of prostate cancer being the only clear risk factors (2). The emergence of biochemical testing (utilising serum prostate specific antigen - PSA - measurement) has resulted in the majority of prostate cancers detected being confined to the prostate; the treatment for these localised cancers is effective and involves either surgical removal of the gland or radiotherapy. Once the cancer metastasises beyond the prostatic capsule, treatment involves the utilisation of agents that specifically block various aspects of androgen signalling (i.e. androgen ablation therapy - AAT) (Fig. 1). While AAT is initially effective, tumours invariably regrow due to the acquisition of a castrate- resistant state, beyond which there are limited treatment options. Recent studies have revealed that androgen signalling, and in particular the intracellular mediator of androgen action, the androgen receptor (AR), plays a critical role in prostate cancer development and progression. This represents a paradigm shift in our understanding of resistance to AAT and provides opportunities for developing new treatment strategies for metastatic prostate cancer that target the AR. Genetic Factors and Serum Hormone Levels in Prostate Cancer Risk Genetic factors clearly play a role in the development and/or progression of prostate cancer; men who have a first degree relative with prostate cancer have a 2-3 fold higher risk of prostate cancer development (3). Moreover, twin studies suggest that as much as 42% of the risk of prostate cancer may be due to genetic factors (4). The precise genetic changes that contribute to risk, however, remain to be determined and are most likely to be weakly penetrant genes or the effects of genes acting together to contribute to risk. Just as the precise genetic changes in prostate cancer remain elusive, the precise role of serum levels of androgens and the role they play in the risk of developing prostate cancer remain unsolved. Several studies have demonstrated that there is no correlation between serum levels of androgens and the risk of developing prostate cancer. More recently, we have reported that higher serum androgen levels are more closely related to less aggressive prostate cancer rather than more aggressive prostate cancer (5). Androgen Receptor Signalling in Prostate Cancer Androgens and the AR play key roles in the proliferation and inhibition of apoptosis in normal prostate cells and in prostate cancer (6); castrated Fig. 1. Targeting the AR at multiple points in the androgen signalling axis. In the cytoplasm, the AR is held in an immature complex that includes heat-shock proteins, p23 and a tetratricopeptide (TPR) containing protein. Upon binding the metabolite of testosterone, DHT, the AR dissociates from the complex and is transported into the nucleus. In the nucleus, the AR is phosphorylated (p) and binds as a dimer to DNA where it recruits cofactor molecules, such as the p160 and p300/CBP coactivators, and the transcriptional machinery (TM) required to activate target genes. Currently, hormonal therapies are aimed at (i) reducing circulating levels of androgens (eg. through the use of luteinising hormone releasing hormone (LHRH) agonists/antagonists or estrogens) or (ii) blocking androgens binding the AR (AR antagonists). Novel AR targeting agents that are now emerging include: (iii) histone deacetylase inhibitors (HDACIs), (iv) antisense oligonucleotides, (v) ansamycin antibiotics and (vi) dominant negative androgen receptors.
Transcript of The Androgen Receptor in Prostate Cancer - ASBMB...The positions of the two trinucleotide repeats...

The Androgen Receptor in Prostate CancerMargaret Centeneraθ, Eleanor Needθ, Lisa Butler and Wayne Tilley*
Dame Roma Mitchell Cancer Research Laboratories, Department of Medicine,University of Adelaide and Hanson Institute, Adelaide, SA 5000
θContributed equally to this work*Corresponding author: [email protected]
Page 12 AUSTRALIAN BIOCHEMIST Vol 38 No 1 April 2007
SHOWCASE ON RESEARCH
Prostate cancer is the most commonly diagnosed cancer in Australian men, accounting for approximately 23% of all new male cancer cases in 2001 (1). Despite its prevalence, the aetiology of prostate cancer remains to be fully elucidated, with increasing age, race and a family history of prostate cancer being the only clear risk factors (2). The emergence of biochemical testing (utilising serum prostate specific antigen − PSA − measurement) has resulted in the majority of prostate cancers detected being confined to the prostate; the treatment for these localised cancers is effective and involves either surgical removal of the gland or radiotherapy. Once the cancer metastasises beyond the prostatic capsule, treatment involves the utilisation of agents that specifically block various aspects of androgen signalling (i.e. androgen ablation therapy − AAT) (Fig. 1). While AAT is initially effective, tumours invariably regrow due to the acquisition of a castrate-resistant state, beyond which there are limited treatment options. Recent studies have revealed that androgen signalling, and in particular the intracellular mediator of androgen action, the androgen receptor (AR), plays a critical role in prostate cancer development and progression. This represents a paradigm shift in our understanding of resistance to AAT and provides opportunities for developing new treatment strategies for metastatic prostate cancer that target the AR.
Genetic Factors and Serum Hormone Levels in Prostate Cancer RiskGenetic factors clearly play a role in the development
and/or progression of prostate cancer; men who have a first degree relative with prostate cancer have a 2-3 fold higher risk of prostate cancer development (3). Moreover, twin studies suggest that as much as 42% of the risk of prostate cancer may be due to genetic factors (4). The precise genetic changes that contribute to risk, however, remain to be determined and are most likely to be weakly penetrant genes or the effects of genes acting together to contribute to risk. Just as the precise genetic changes in prostate cancer remain elusive, the precise role of serum levels of androgens and the role they play in the risk of developing prostate cancer remain unsolved. Several studies have demonstrated that there is no correlation between serum levels of androgens and the risk of developing prostate cancer. More recently, we have reported that higher serum androgen levels are more closely related to less aggressive prostate cancer rather than more aggressive prostate cancer (5).
Androgen Receptor Signalling inProstate CancerA n d r o g e n s a n d t h e A R p l a y k e y r o l e s i n t h e
proliferation and inhibition of apoptosis in normal prostate cells and in prostate cancer (6); castrated
F i g . 1 . Targeting the AR at multiple points in the androgen signalling axis.In the cytoplasm, the AR is held in an immature complex that includes heat-shock proteins, p23 and a tetratr icopeptide (TPR) containing protein. Upon b i n d i n g t h e m e t a b o l i t e o f t e s t o s t e r o n e , D H T , t h e A R dissociates from the complex and i s t ransported in to the nucleus. In the nucleus, the AR is phosphorylated (p) and binds as a dimer to DNA where i t recruits cofactor molecules, such a s t h e p 1 6 0 a n d p 3 0 0 / C B P c o a c t i v a t o r s , a n d t h e transcriptional machinery (TM) required to activate target genes. Currently, hormonal therapies are aimed at (i) reducing circulating levels of androgens (eg. through the use of luteinising hormone releasing hormone (LHRH) agonists/antagonists or estrogens) or (ii) blocking androgens binding the AR (AR antagonists). Novel AR targeting agents that are now emerging include: (iii) histone deacetylase inhibitors (HDACIs), (iv) antisense oligonucleotides, (v) ansamycin antibiotics and (vi) dominant negative androgen receptors.

SHOWCASE ON RESEARCH
Vol 38 No 1 April 2007 AUSTRALIAN BIOCHEMIST Page 13
The Androgen Receptorin Prostate Cancer
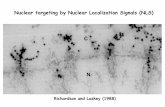
Fig. 2. Schematic structure of wild type androgen receptor and dominant negative androgen receptor proteins.A. Structure of the wild type androgen receptor with broad functional domains indicated, including the amino-terminal transactivation domain (NTD), the DNA-binding domain (DBD), the hinge region (H), and the ligand-binding domain (LBD). The positions of the two trinucleotide repeats (polyQ and polyG) in the NTD are shown. Activation functions AF-1 and AF-5 in the NTD and activation function AF-2 in the LBD are shown.B. Structure of the dominant negative androgen receptor with deletion of amino acids 39-410 indicated.
individuals or individuals with loss of function germline mutations in the AR do not develop prostate cancer (7). Ligand-dependent transcriptional regulation of genes involved in growth of prostate tumours by the AR involves interaction with a complement of cofactors that mediate nuclear-cytoplasmic movement, recognition of response elements, chromatin remodelling, and the recruitment of basal transcription factors (Fig. 1). For many nuclear receptors, cofactor recruitment is mediated by the highly conserved ligand-induced AF2 structure in the ligand-binding domain (LBD). However, the structural constraints of high-affinity hormone binding have restricted AF2 to interaction with a limited number of proteins. Consequently, the divergence of the oxy-steroid receptors (i.e. AR, progesterone receptor and glucocorticoid receptor) for specificity of gene regulation resulted in a weakening of this capacity in AF2 and a strengthening of amino terminal domain (NTD)-cofactor interactions. Interestingly, as the oxy-steroid receptors share the same consensus DNA response element, a major determinant of promoter and cell-specific transcriptional activity of each of these receptors is determined by the high variability of their NTDs and the cofactors that they recruit (8). Disruption of the AR-NTD and/or of aberrant NTD-cofactor interactions therefore has the potential to perturb AR signalling in prostate tumours by altering AR-regulated gene transcription.The AR gene conta ins two long polymorphic
trinucleotide repeats in its NTD, a poly-glutamine coding region close to the N-terminus of the NTD, and a poly-glycine coding region close to the DNA binding domain of the AR (Fig. 2A). Shorter poly-glutamine r e p e a t s h a v e b e e n s h o w n t o h a v e i n c r e a s e d transcriptional activity (9) and to be weakly positively associated with prostate cancer risk in some studies, while not associated in others (2). More recently, it has been indicated that polymorphisms within androgen response elements may contribute additively along with decreased poly-glutamine length to increase prostate cancer risk by as much as five-fold (10), demonstrating an interplay of genetic effects which together contribute to increased prostate cancer risk.
Aberrant AR Signalling is Oncogenic inthe ProstatePerhaps more compelling evidence for the involvement
of AR in prostate cancer development is provided by characterisation of a mouse model we recently developed in collaboration with Professor Norman Greenberg (11). The role of normal and aberrant AR-NTD function in the development of prostate cancer was demonstrated utilising the prostate and epithelial cell-specific probasin promoter to create transgenic mouse lines overexpressing wild-type AR (wtAR) or one of two AR variants with promiscuous ligand activation (AR-T877A (mouse T857A)) or altered AR coregulator interactions (AR-E231G) specifically in the prostate. Whereas overexpression of wtAR and AR-T877A had no discernable effect on prostate morphology, 100% of mice expressing AR-E231G developed lesions similar to prostatic intraepithelial neoplasia at 12 weeks of age, and advanced prostate cancer with lung metastases by 50 weeks (11). This finding highlights the oncogenic potential of the AR.
AR and the Involvement of the Microenvironment in the Progression of Localised Prostate CancerMeasurement of AR levels by immunohistochemistry
on a large prostate cancer cohort demonstrated that high levels of AR are associated with increased cell proliferation, clinical stage of prostate cancer, lymph node involvement, extension beyond the prostate basement membrane and invasion into the surrounding seminal vesicles (12). In addition, high AR expression predicted a higher probability of recurrence and in multivariate analysis, high AR expression was an independent prognostic indicator of survival. While this demonstrates the importance of epithelial AR in the progression of prostate cancer to metastasis, there is also evidence that the tumour microenvironment is important in prostate cancer progression. High AR expression in the malignant epithelial cells and low AR expression in the adjacent periepithelial stroma are

associated with higher clinical stage, and earlier relapse after radical prostatectomy (13). Furthermore, while androgen-responsive growth of normal prostate epithelial cells is initially paracrine, in malignant prostate cancer cells, androgen-responsive growth is governed by autocrine mechanisms (14). These findings indicate that the androgen receptor and androgen signalling play a fundamental role in both normal and malignant growth of the prostate.
Continued AR Signalling During Prostate Cancer ProgressionAR expression has been demonstrated in virtually all
clinical states of human prostate cancer, including those that have failed AAT (15), which is indicative of the critical role the AR plays throughout the progression of this disease. Recently it has been recognised that failure of AAT is not due to a decreased requirement for androgen signalling by emerging castrate-resistant tumours, but that the AR remains the major determinant of disease progression and is able to maintain signalling in a castrate environment due to mechanisms that include increased levels of AR, acquisition of AR gene mutations, and/or altered levels of crit ical AR coregulators (6,15).
Increased Levels of ARAmplification of the AR gene is a process that leads to
increased expression of the receptor thereby allowing continued cell growth in castrate levels of androgens. AR gene amplification has been reported to be present in approximately 30% of recurrent tumours, compared with no amplification found in untreated prostate cancers (16). Recent evidence suggests that an increase in AR level is actually sufficient to mediate disease progression in a castrate environment (15). Studies from our laboratory have documented increased AR and AR signalling in human metastatic tumours using array profiling and microarray analysis (15), while the Sawyers laboratory demonstrated that increased AR expression was the only consistent change in the transition of hormone-sensitive to castrate-resistant disease (17).
AR Gene MutationsStudies of AR gene mutations have provided important
insights into AR function and its role in prostate cancer. AR mutations occur more frequently in advanced disease and following AAT (15), and typically exhibit one of two main phenotypes: (a) activation by alternative ligands, as demonstrated by the androgen-responsive LNCaP cell line containing a single point mutation (Thr-877-Ala, located in the LBD) that allows the receptor to be activated not only by androgens but also by progestins, estrogens, glucocorticoids and anti-androgens (18); and (b) greater transactivation capacity via altered interaction with coregulator molecules, as demonstrated by the AR-E231G variant discussed above that exhibits increased basal activity in the absence of ligand compared to wtAR, and a greater capacity to respond to the coregulator ARA70 (11).
Altered Expression of AR CoregulatorsAR s ignal l ing is h ighly mediated by speci f ic
interactions with coregulators that enhance (coactivators) or repress (corepressors) AR transactivation activity. Amplification and/or overexpression of key coregulators (including the p160 coactivators) has been demonstrated following AAT, resulting in increased transactivation capacity in a castrate environment or in the presence of non-classical ligands (19).
Targeting the ARThe finding that the AR is the major mediator of
prostate cancer cell growth and survival following failure of androgen deprivation therapy suggests that the AR remains a viable target for the treatment of this disease. Part of our research group's focus, therefore, is on developing a targeted strategy that more effectively inhibits activity of the AR, irrespective of its level of expression or mechanism of activation.Several different aspects of the AR signalling pathway
have been targeted and tested for their ability to suppress proliferation of prostate cancer cells. A reduced AR level has been achieved through the use of mRNA hammerhead ribozymes and antisense oligonucleotides, with pre-clinical studies demonstrating that lowering AR resulted in reduced proliferation of prostate cancer cells (15). More recently, AR short hairpin RNAs have been used to knock down AR expression in LNCaP xenograft tumours resulting in significantly retarded tumour growth and an extensive delay in the progression to a castrate-resistant state (20). Maturation of the AR complex has been targeted by use of the ansamycin antibiotic geldanamycin analogue, 17-AAG. This heat shock protein (Hsp90) inhibitor has been shown to reduce AR levels as well as inhibit prostate tumour growth in vivo, and is currently in use in phase II clinical trials (6). Also in clinical trials is the use of the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA), which acts on multiple protein and DNA targets to affect AR transcriptional competence (21). SAHA has been successful in inhibiting prostate tumour growth both in vitro and in vivo and is currently being trialled for a number of tumours, including prostate cancer (6).One approach that is currently being investigated in
our laboratory is the use of AR variants (ARis) that act in a dominant negative manner to inhibit AR activity in prostate cancer cells. A number of potential ARis were created by deleting or mutating specific activation function domains in the AR. The most efficient of the AR variants was ARi-410 that contains a deletion (amino acids 39-410) of the entire AF-1 and part of AF-5 domain of the NTD (Fig. 2B). This construct has no intrinsic activity in the presence of DHT and is able to inhibit both wtAR and mutant ARs in vitro by up to 95% (22). In addition, ectopic expression of the ARi in the LNCaP prostate cancer cell line reduced both the level of cell proliferation and the level of PSA secreted by the cells. This AR inhibitor has the potential to block AR signalling in prostate cancer cells, irrespective of the level of expression or structure of the AR.
SHOWCASE ON RESEARCH
The Androgen Receptorin Prostate Cancer
Page 14 AUSTRALIAN BIOCHEMIST Vol 38 No 1 April 2007

SHOWCASE ON RESEARCH
Vol 38 No 1 April 2007 AUSTRALIAN BIOCHEMIST Page 15
The Androgen Receptorin Prostate Cancer
Combination TherapiesAs described above, various aspects of the androgen
signalling axis can be targeted to achieve significant inhibition of AR activity and suppression of cell growth. Importantly, there is potential to achieve a more complete blockade of AR signalling compared to AAR by using a combination of agents that target multiple points in the AR pathway. Such a strategy may prevent the outgrowth of therapy-resistant cells and therefore prevent the regrowth of tumours following treatment. Indeed, our laboratory has shown that treatment of prostate cancer cells in vitro with low levels of the AR antagonist, bicalutamide, combined with the histone deacetylase inhibitor, SAHA, resulted in LNCaP cells that were sensitised to growth inhibition and death by low doses of SAHA that are ineffective if given alone (21). That the low doses of bicalutmide and SAHA used do not affect LNCaP growth alone, and that the combination did not affect growth of the AR-negative PC-3 prostate cancer cells, indicates that the combination therapy has the potential to target only those cells that are dependent on AR signalling. Clinically, this approach offers the potential to more specifically target prostate cancer cells, the majority of which are AR dependent at all stages of progression (15), while reducing toxicity in other androgen-dependent tissues including bone, muscle and brain. This would provide a significant advantage over high-dose monotherapy in both treatment efficacy and quality of life.
ConclusionResearch into the role of the AR in prostate cancer
highlights the importance of nuclear receptor signalling in biology and in disease. There is now compelling evidence that the AR is involved in all stages of prostate tumorigenesis including initiation, progression and treatment resistance, which represents a paradigm shift away from the idea that prostate tumours develop 'androgen-independence'. Whereas current treatments for prostate cancer typically involve androgen withdrawal (i.e. AAT), what has now emerged as the challenge in this field of research is the need to develop effective clinical treatment strategies that directly target the AR.
References1. Australian Institute of Health and Welfare (AIHW)
Australasian Association of Cancer Registries (AACR) (2003), AIHW (Cancer Series no. 23), Canberra
2. Hsing, A.W., and Chokkalingam, A.P. (2006) Front. Biosci. 11, 1388-1413
3. Stanford, J.L., and Ostrander, E.A. (2001) Epidemiol. Rev. 23, 19-23
4. Lichtenstein, P., Holm, N.V., Verkasalo, P.K., Iliadou, A., Kaprio, J., Koskenvuo, M., Pukkala, E., Skytthe, A., and Hemminki, K. (2000) N. Engl. J. Med. 343, 78-85
5. Severi, G., Morris, H.A., MacInnis, R.J., English, D.R., Tilley, W., Hopper, J.L., Boyle, P., and Giles, G.G. (2006) Cancer Epidemiol. Biomarkers Prev. 15, 86-91
6. Scher, H.I., and Sawyers, C.L. (2005) J. Clin. Oncol. 23, 8253-8261
7. Huggins, C., Stephens, R.C., and Hodges, C.V. (1941) Arch. Surg. 43, 209
8. Scheller, A., Hughes, E., Golden, K.L., and Robins, D.M. (1998) J. Biol. Chem. 273, 24216-24222
9. Buchanan, G., Yang, M., Cheong, A., Harris, J.M., Irvine, R.A., Lambert, P.F., Moore, N.L., Raynor, M., Neufing, P.J., Coetzee, G.A., and Tilley, W.D. (2004) Hum. Mol. Genet. 13, 1677-1692
10.Xue, W., Irvine, R.A., Yu, M.C., Ross, R.K., Coetzee, G.A., and Ingles, S.A. (2000) Cancer Res. 60, 839-841
11.Han, G., Buchanan, G., Ittmann, M., Harris, J.M., Yu, X., Demayo, F.J., Tilley, W., and Greenberg, N.M. (2005) Proc. Natl. Acad. Sci. USA 102, 1151-1156
12.Li, R., Wheeler, T., Dai, H., Frolov, A., Thompson, T., and Ayala, G. (2004) Am. J. Surg. Pathol. 28, 928-934
13.Henshall, S.M., Quinn, D.I., Lee, C.S., Head, D.R., Golovsky, D., Brenner, P.C., Delprado, W., Stricker, P.D., Grygiel, J.J., and Sutherland, R.L. (2001) Cancer Res. 61, 423-427
14.Gao, J., Arnold, J.T., and Isaacs, J.T. (2001) Cancer Res. 61, 5038-5044
15.Scher, H.I., Buchanan, G., Gerald, W., Butler, L.M., and Tilley, W.D. (2004) Endocr. Relat. Cancer 11, 459-476
16.Visakorpi, T., Hyytinen, E., Koivisto, P., Tanner, M., Keinanen, R., Palmberg, C., Palotie, A., Tammela, T., Isola, J., and Kallioniemi, O.P. (1995) Nat. Genet. 9, 401-406
17.Chen, C.D., Welsbie, D.S., Tran, C., Baek, S.H., Chen, R., Vessella, R., Rosenfeld, M.G., and Sawyers, C.L. (2004) Nat. Med. 10, 33-39
18.Veldscholte, J., Ris-Stalpers, C., Kuiper, G.G., Jenster, G., Berrevoets, C., Claassen, E., van Rooij, H.C., Trapman, J., Brinkmann, A.O., and Mulder, E. (1990) Biochem. Biophys. Res. Commun. 173, 534-540
19.Gregory, C.W., He, B., Johnson, R.T., Ford, O.H., Mohler, J.L., French, F.S., and Wilson, E.M. (2001) Cancer Res. 61, 4315-4319
20.Cheng, H., Snoek, R., Ghaidi, F., Cox, M.E., and Rennie, P.S. (2006) Cancer Res. 66, 10613-10620
21.Marrocco, D.L., Tilley, W.D., Bianco-Miotto, T., Evdokiou, A., Scher, H.I., Rifkind, R.A., Marcs, P.A., Richon, V.M., and Butler, L.M. (2007) Mol. Cancer Ther. in press
22.Butler, L.M., Centenera, M.M., Neufing, P.J., Buchanan, G., Choong, C.S., Ricciardelli, C., Saint, K., Lee, M., Ochnik, A., Yang, M., Brown, M.P., and Tilley, W.D. (2006) Mol. Endocrinol. 20, 1009-1024