
TEORIE DELLE REAZIONI CHIMICHE - scienze.uniroma2.it · Qualunque teoria delle reazioni chimiche...
94
1 TEORIE DELLE REAZIONI CHIMICHE Qualunque teoria delle reazioni chimiche deve spiegare la dipendenza dalla temperatura delle costanti di equilibrio e delle costanti di velocità. - Dipendenza degli equilibri dalla temperatura K = e -ΔG o /RT ΔG o RT e poiché ΔG o = -TΔS o ΔH o ΔH o R 1 T + ΔS o R ln K = - ln K = - Se ΔH o e ΔS o sono indipendenti dalla temperatura (vero per un intervallo di temperatura limitato), riportando in grafico ln K in funzione di 1/T, si ha una retta, di pendenza - ΔH o /R Dalla misura delle costanti di equilibrio a due temperature si può determinare ΔH o se T 2 > T 1 1 T 2 1 T 1 - > 0 ln K 2 K 1 = ΔH o R 1 T 2 1 T 1 - K 2 K 1 dipende dal segno di ΔH o K 2 K 1 > cioè se la reazione è endotermica se ΔH o > 0 K 2 K 1 < cioè se la reazione è esotermica se ΔH o < 0 - Dipendenza della velocità dalla temperatura La velocità di una reazione semplice aumenta con l'aumentare della temperatura. un aumento di 10 K provoca un aumento di due o tre volte della costante di velocità La relazione quantitativa tra la costante di velocità (k) della reazione e la tempe- ratura assoluta (K) è formulata nell'equazione di Arrhenius (empirica) ln k = ln A - R E a 1 T R = 1.987 cal K -1 moli -1 (8.314 J K -1 moli -1 ) T in gradi Kelvin k = A e -E a /RT A fattore pre-esponenziale E a energia di attivazione - E a = RT ln (k/A) Riportando in grafico log k (o ln k) in funzione di 1/T, si ottiene E a dalla pendenza della retta l'intercetta è log A (o ln A) Si sottintende che A sia indipendente dalla temperatura
Transcript of TEORIE DELLE REAZIONI CHIMICHE - scienze.uniroma2.it · Qualunque teoria delle reazioni chimiche...

1
TEORIE DELLE REAZIONI CHIMICHEQualunque teoria delle reazioni chimiche deve spiegare la dipendenza dalla temperatura delle costanti di equilibrio e delle costanti di velocità.
- Dipendenza degli equilibri dalla temperatura
K = e-ΔGo/RT ΔGo
RT
e poiché ΔGo = -TΔSoΔHo ΔHo
R1T + ΔSo
R
ln K = -
ln K = -
Se ΔHo e ΔSo sono indipendenti dalla temperatura (vero per un intervallo ditemperatura limitato), riportando in grafico ln K in funzione di 1/T, si ha una retta,di pendenza -ΔHo/R
Dalla misura delle costanti di equilibrio a due temperature si può determinareΔHo
se T2 > T11T2
1T1
- > 0ln
K2
K1= ΔHo
R1T2
1T1
-
K2
K1dipende dal segno di ΔHo
K2 K1> cioè se la reazione è endotermicase ΔHo > 0K2 K1< cioè se la reazione è esotermicase ΔHo < 0
- Dipendenza della velocità dalla temperatura
La velocità di una reazione semplice aumenta con l'aumentare della temperatura.un aumento di 10 K provoca un aumento di due o tre volte della costantedi velocità
La relazione quantitativa tra la costante di velocità (k) della reazione e la tempe-ratura assoluta (K) è formulata nell'equazione di Arrhenius (empirica)
ln k = ln A -R
Ea 1T R = 1.987 cal K-1 moli-1 (8.314 J K-1 moli-1)
T in gradi Kelvin
k = A e-Ea/RT A fattore pre-esponenzialeEa energia di attivazione - Ea = RT ln (k/A)
Riportando in grafico log k (o ln k) in funzione di 1/T, si ottiene Ea dallapendenza della rettal'intercetta è log A (o ln A)
Si sottintende che A sia indipendente dalla temperatura
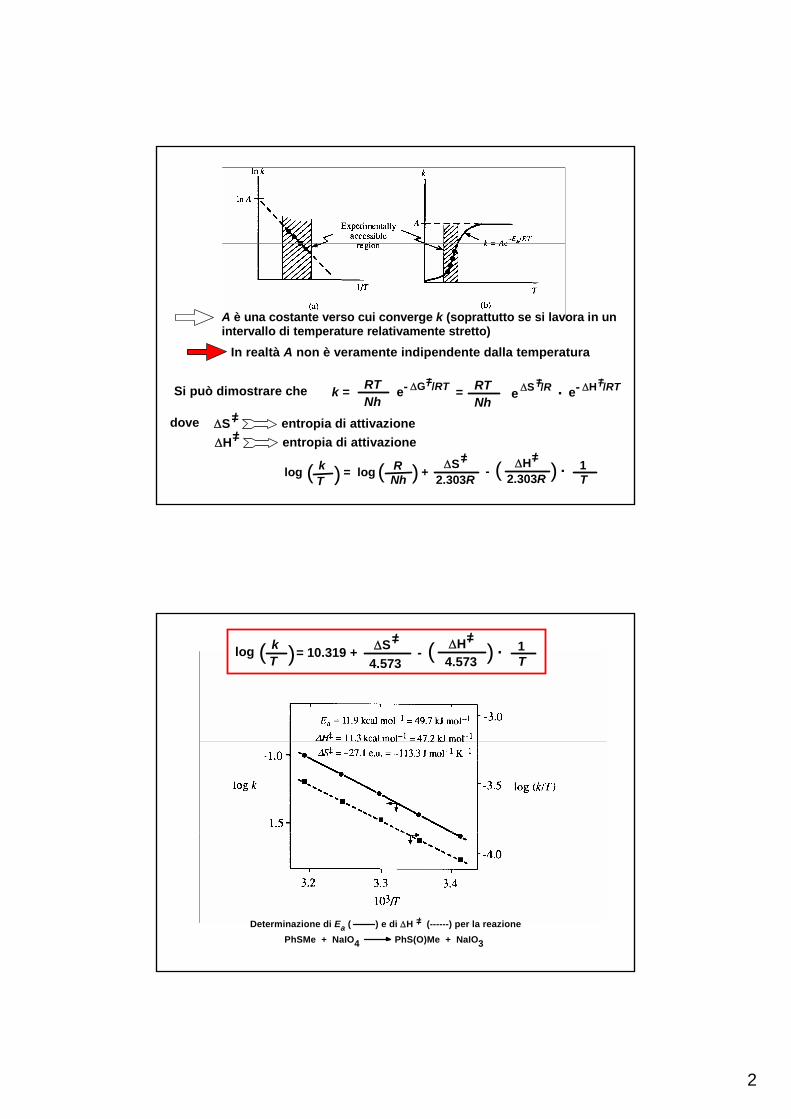
2
In realtà A non è veramente indipendente dalla temperatura
Si può dimostrare che
=/ΔSdove entropia di attivazione=/ΔH entropia di attivazione
k = RTNh
e- ΔG /RT=/= RT
Nhe ΔS /R=/ . e- ΔH /RT=/
RNhlog k
T( ) = log ( ) +=/ΔS
2.303R- ( 2.303R
=/ΔH ) . 1T
A è una costante verso cui converge k (soprattutto se si lavora in unintervallo di temperature relativamente stretto)
=log k T( ) 10.319
=/ΔS+4.573
- (=/ΔH ) . 1
4.573 T
PhSMe + NaIO4 PhS(O)Me + NaIO3
Determinazione di Ea ( ) e di ΔH (------) per la reazione=/

3
Se le concentrazioni sono espresse in M (moli/litro), il tempo in secondi e la temperatura in gradi Kelvin
=/ΔH in cal moli-1
in cal moli-1K-1 = u.e. (unità entropiche) =/ΔS
ΔH =/Ea = + RT
di solito RT è piccolo (~ 0.6 kcal/mole a temp. ambiente)
in condizioni di pseudo-ordine, usando kψ, ΔH è lo stesso (pendenza), ma ΔS no (intercetta). Va perciò determinata usando k e non kψ.
=/
=/
Normalmente i grafici di Arrhenius sono lineari (o almeno non ci sono motivi convincenti per non ritenerli tali)
Grafico di Arrhenius per la conversione sedia-sediadell'anello del cicloesano
In alcune reazioni si osservano grafici di Arrhenius curvi. i possibili motivi sono tre
La curvatura è un artefatto, introdotto da un errore sistematico della misura
Non improbabile: le costanti di velocità nell'intervallo di temperatura possono variare di ordini di grandezza, richedendo metodi analitici diversi in regioni di temperature diverse.
lavoro sperimentale accuratola curvatura è dovuta a parametri di attivazione che dipendono dalla temperatura
Non tutti accettano questa possibilità
La curvatura è la conseguenza di una reazione complessaPer reazioni reversibili e parallele: kexp = k1 + k2 non si può usare kexp
= RNh
e-ΔS1 /R=/
e ΔS2 /R=/ .ΔH1 /RT=/kexpT ( + e e- ΔH1 /RT=/ )log (k/T) in funzione di (1/T) non è una retta curvatura diagnosticadi situazione complessa
RTNhPer reazioni parallele
k1 k2 =[X]
[Y] =[X][Y] RT
Nh
e-ΔS1 /R=/
e ΔH1 /RT=/
ΔS2 /=/ .e e- ΔH1 /RT=/

4
=[X][Y]
log=/ΔS2ΔS1 -=/( )
2.303R- ( =/ΔH2ΔH1 -=/
2.303R ) .T1
=/ΔS2ΔS1 -=/( )=/ΔH2ΔH1 -=/
Riportando in grafico log [X] / [Y] in funzione di - (1/T) si ha una rettapendenza
( ) intercetta
Nel caso di pre-equilibrio kexp = K1 k2
= RNh
(ΔS°1 /Rekexp
Te-ΔS2 /R)=/ (ΔH°1 /RT ΔH2 /RT)=/+ +
Riportando in grafico log ( ) in funzione di (1/T) kexp /T
pendenza variazione complessiva di entalpia=/ΔS2ΔS°1
+( )=/ΔH2ΔH°1
+
( )variazione complessiva di entropiaintercetta
Determinando K1 in funzione della temperatura si determinano ΔH°1 e ΔS°2 dalla variazione complessiva si calcolano ΔH2 e ΔS2
=/ =/
Nel caso di condizioni di stato stazionario kexp = k1k2
k-1 + k2
Sostituendo in k = RTNh
e- ΔG /RT=/= RT
Nhe ΔS /R=/ . e- ΔH /RT=/
e facendo il logaritmo curvatura diagnostica
Se uno dei termini al denominatore può essere trascurato, si hanno semplifica-zioni
k-1 << k2 kexp = k1Se
Se k-1 >> k2 kexp = k1 k2 k-1
= K1k2

5
Grafico di Arrhenius curvo per l'idrolisi del trifluoroacetatodi metile in DMSO
TEORIA DELLA COLLISIONEAnalizza la dipendenza della velocità di reazione dalla temperatura sulla base delnumero di collisioni che avvengono nell'unità di tempo.Le collisioni si dividono in due classi
COLLISIONI ELASTICHE: cambiano solo la direzione e la velocità delmovimento delle particelle
COLLISIONI ANELASTICHE (o REATTIVE): cambia in qualche modo lacomposizione chimica delle molecole che collidono
Collisioni fisiche (a) e chimiche (b) delle molecole A e B

6
Dalla teoria cinetica dei gas si ottiene un'espressione per il numero di collisioni (Z') che avvengono tra due molecole A e B in una unità di volume nel periodo diuna unità di tempo.
nA e nB numero di molecole di massa mA e mB in 1 cm3
μ massa ridotta 1μ
= 1 1+mA mB
molecola cm-3 s-1Z' = 8πRTNμ( )1/2
(rA + rB)2 nAnB
quando nA = nB = 1 cm3 molecola s-1Z = 8πRTNμ
(rA + rB)2
La collisione è possibile solo all'interno di (rA + rB)
dove rA è il raggio di A e rBquello di B
per ottenere Z in unità di k, bisogna moltiplicare per il numero di Avogadro edividere per 1000
M-1 s-1Z = 8πRTNμ( 1/2
(rA + rB)2 )N1000
a temperatura ambiente, per molecole con peso molecolare < 100
Z ~ 1011 M-1 s-1
Valore molto maggiore dei k solitamente osservati, dovuto al fatto chenon tutte le collisioni portano a reazione
Perché si abbia reazione, le molecole che collidono devono avere un surplus di energia (energia di attivazione)
Z deve essere moltiplicato per un fattore pre-esponenziale,in accordo con la legge di distribuzione di Boltzmann
k = p Z e-Ea/RT
confrontando con l'equazione di Arrhenius A = pZp <_ 1 per tener conto dell'orientamento
Un test della teoria della collisione consiste nel confrontare i valori osservato ecalcolato del fattore pre-esponenziale A
P =A (osservato)A (calcolato)
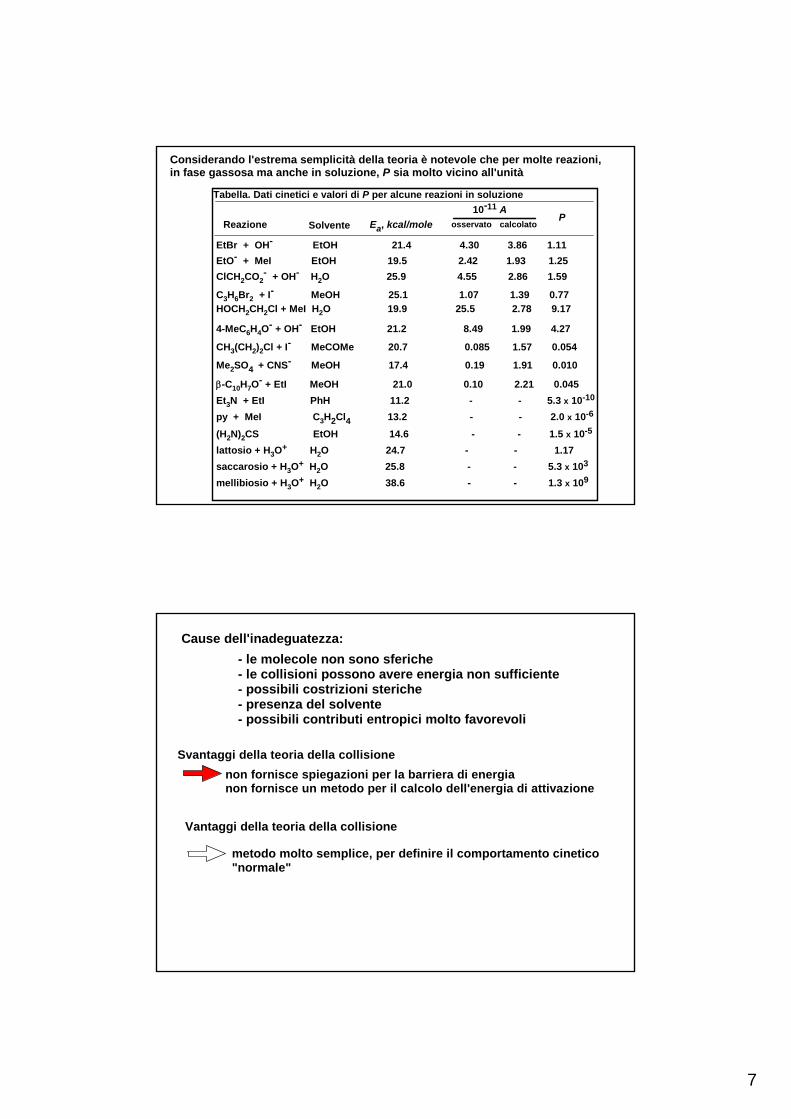
7
Considerando l'estrema semplicità della teoria è notevole che per molte reazioni,in fase gassosa ma anche in soluzione, P sia molto vicino all'unità
EtBr + OH- EtOH 21.4 4.30 3.86 1.11EtO- + MeI EtOH 19.5 2.42 1.93 1.25ClCH2CO2
- + OH- H2O 25.9 4.55 2.86 1.59
C3H6Br2 + I- MeOH 25.1 1.07 1.39 0.77HOCH2CH2Cl + MeI H2O 19.9 25.5 2.78 9.17
4-MeC6H4O- + OH- EtOH 21.2 8.49 1.99 4.27
CH3(CH2)2Cl + I- MeCOMe 20.7 0.085 1.57 0.054
Me2SO4 + CNS- MeOH 17.4 0.19 1.91 0.010
β-C10H7O- + EtI MeOH 21.0 0.10 2.21 0.045
Et3N + EtI PhH 11.2 - - 5.3 x 10-10
py + MeI C3H2Cl4 13.2 - - 2.0 x 10-6
(H2N)2CS EtOH 14.6 - - 1.5 x 10-5
lattosio + H3O+ H2O 24.7 - - 1.17saccarosio + H3O+ H2O 25.8 - - 5.3 x 103
mellibiosio + H3O+ H2O 38.6 - - 1.3 x 109
Tabella. Dati cinetici e valori di P per alcune reazioni in soluzione
Ea, kcal/mole10-11 A
osservato calcolatoP
Reazione Solvente
Cause dell'inadeguatezza:- le molecole non sono sferiche- le collisioni possono avere energia non sufficiente- possibili costrizioni steriche- presenza del solvente- possibili contributi entropici molto favorevoli
Svantaggi della teoria della collisionenon fornisce spiegazioni per la barriera di energianon fornisce un metodo per il calcolo dell'energia di attivazione
Vantaggi della teoria della collisione
metodo molto semplice, per definire il comportamento cinetico"normale"

8
TEORIA DELLO STATO DI TRANSIZIONE
Quando due molecole collidono in una collisione che porta ai prodotti (o una singola molecola esegue i movimenti che provocano la variazione chimica), passano attraverso una configurazione di massima energia potenziale, chiamata STATO DI TRANSIZIONE.
La premessa fondamentale della teoria dello stato di transizione è che i reagentiformino una specie di complesso con una struttura tra lo stato dei reagenti (RS) e lo stato dei prodotti (PS) di una reazione
Lo stato di transizione (o struttura di transizione o complesso attivato)si trova lungo la coordinata di reazione (q) e la sua energia rappresentail massimo del profilo di energia
L'assunzione fondamentale della teoria dello stato di transizione e che A e B (RS)sono in equilibrio con il complesso attivato-
L'equilibrio è caratterizzato da una costante di pseudo-equilibrio, K=/
Si assume anche che la velocità complessiva (cioè la la formazione del prodotto)è influenzata anche dalla velocità di decomposizione del complesso attivato, chepuò essere caratterizzata dalla costante di velocità k =/
A + B AB X + YK k =/=/ =/
PEA [AB] = K k [A][B]=/ =/ =/velocità = k [AB ]=/=/
velocità = k2 [A][B]
per la reazione bimolecolare: k2 = K k=/ =/
k =/

9
AB ha una frequenza vibrazionale (ν) associata al modo di formazione del prodotto lungo la coordinata di reazione
=/
k = κν=/
Se tutte le elongazioni vibrazionali portassero alla decomposizione del complessoattivato verso la formazione del prodotto, k sarebbe numericamente uguale a ν=/
invece
κ = coefficiente di trasmissione 0 < κ < 1_
=/K
La costante di pseudo-equilibrio è legata alle funzioni di partizione (Q) ed alla differenza di energia tra complesso attivato e reagenti allo zero assoluto
=/K =QAB =/QAQB
e - ΔE0 /RT=/
Separando la porzione vibrazionale della funzione di partizione di AB associataalla decomposizione del complesso attivato lungo la coordinata di reazione
=/
si arriva all'espressione
ν non è presentek2 κ = RTNh
=/K'
=/K' è legata all'energia libera di attivazione =/=/ΔG = -RT ln K'
la costante di velocità k2 può essere espressa mediante una delle seguentiformule (equivalenti)
k2 κ = RTNh e
=/- ΔG /RT k2 κ = RTNh e e
=/ΔS /R =/- ΔH /RT
Equazioni di Eyring
Il valore numerico del coefficiente di trasmissione ( ) è incognito e si sceglie 1κ
Le funzioni termodinamiche di attivazione sono la conseguenza della struttura del complesso attivato.
=/=/ =/k2 è grande se ΔG e ΔH sono piccole e ΔS è grande
Dai valori numerici sperimentali di ΔH e ΔS si possono avere indicazioni sugli stati di transizione e, di conseguenza, sul meccanismo.
=/ =/
STRUTTURA DEL COMPLESSO ATTIVATO
In casi semplici si possono avere indicazioni sulla simmetria dello stato di transizione
La struttura di un complesso attivato non si può determinare sperimentalmente, perché non ha una vita misurabile. La sua geometria molecolare può essere determinata teoricamente (metodi di ottimizzazione di gradiente quantomeccanico)

10
Esempio:
In casi più complessi si possono fare ipotesi sulla base del postulato di Hammond
I- + -------------- II1/2- 1/2-
=/
+ I-C IH
HH
CIH
HH
CH
HH
con energia di attivazione piccola, l'energia e la geometria di TS sarà vicina a quella del reagente (TS reagent like) (Fig.a)
con energia di attivazione elevata ed energie simili per reagenti e prodottiTS sarà circa a metàstrada (Fig. b)
con energia di attivazione elevata e reazione endotermica, TS saràsimileal prodotto (TS productlike) (Fig. c)
SUPERFICI DI ENERGIA POTENZIALE
Restano 3N-6 GRADI DI LIBERTA', che sono i movimenti vibrazionali interni degli atomi uno rispetto all'altro (3N-5 se la molecola è lineare).
Gli N atomi di una molecola si possono muovere in tre direzioni mutua-mente perpendicolari e perciò indipendenti e quindi una molecola ha untotale di 3N gradi di libertà.
Se gli atomi fossero fissi, gli uni rispetto agli altri, la posizione dellamolecola rigida nello spazio sarebbe definita dalle tre coordinatecartesiane del suo centro di massa e dai tre angoli rotazionali per indicareil suo orientamento nello spazio
la molecola costituisce un'unità
La vibrazione molecolare totale è complessa, ma con buona approssima-zione si può dividere in 3N-6 modi normali indipendenti
Ciascun modo si può considerare equivalente alla vibrazione di stretching di unamolecola biatomica
modello oscillatore armonico funzione parabolicaPer una molecola poliatomica si ha una curva di energia potenziale per ciascunodei 3N-6 modi vibrazionali
L'energia potenziale è caratterizzata da una superficie in uno spazio a 3N-6+1 dimensioni.
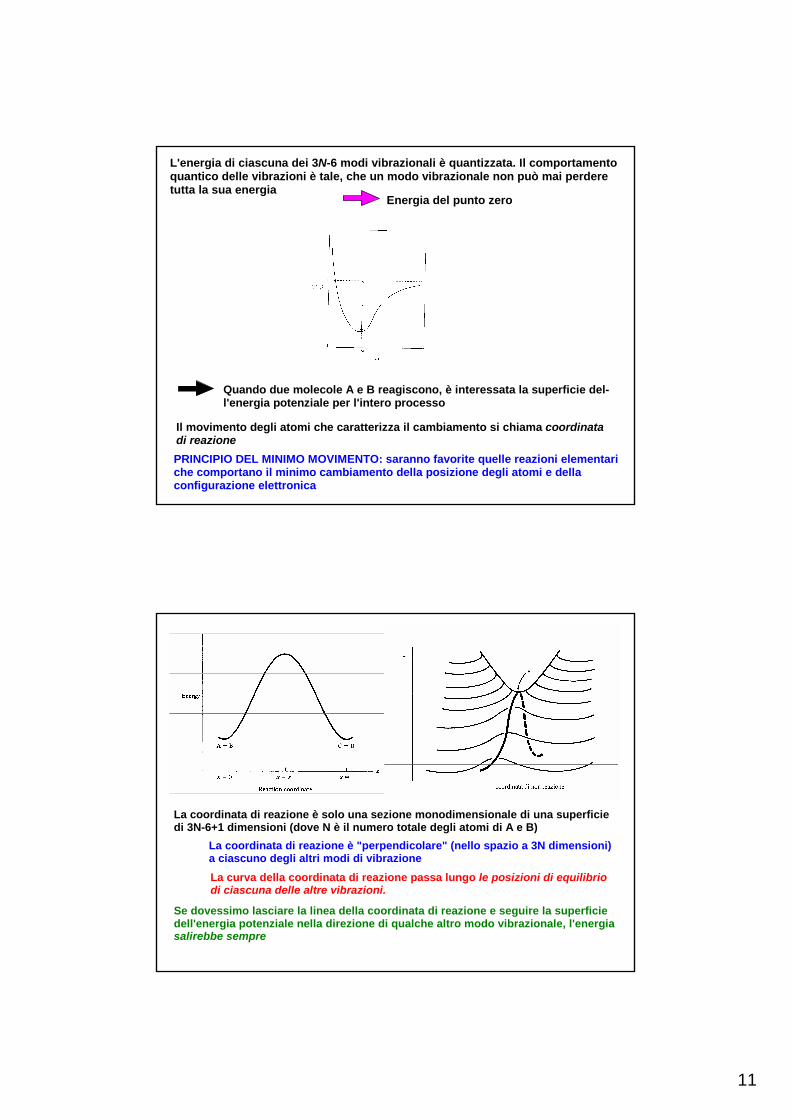
11
L'energia di ciascuna dei 3N-6 modi vibrazionali è quantizzata. Il comportamentoquantico delle vibrazioni è tale, che un modo vibrazionale non può mai perdere tutta la sua energia
Energia del punto zero
Quando due molecole A e B reagiscono, è interessata la superficie del-l'energia potenziale per l'intero processo
Il movimento degli atomi che caratterizza il cambiamento si chiama coordinatadi reazionePRINCIPIO DEL MINIMO MOVIMENTO: saranno favorite quelle reazioni elementariche comportano il minimo cambiamento della posizione degli atomi e dellaconfigurazione elettronica
La coordinata di reazione è solo una sezione monodimensionale di una superficiedi 3N-6+1 dimensioni (dove N è il numero totale degli atomi di A e B)
La coordinata di reazione è "perpendicolare" (nello spazio a 3N dimensioni)a ciascuno degli altri modi di vibrazioneLa curva della coordinata di reazione passa lungo le posizioni di equilibriodi ciascuna delle altre vibrazioni.
Se dovessimo lasciare la linea della coordinata di reazione e seguire la superficie dell'energia potenziale nella direzione di qualche altro modo vibrazionale, l'energiasalirebbe sempre

12
La superficie mostra solo l'energia potenziale.
L'energia totale del sistema molecolare è la somma delle sue energie:vibrazionale, rotazionale, cinetica, potenziale e interna.
Per calcolare la superficie di energia potenziale ci sono tre metodimetodo puramente teorico: usa solo grandezze fisiche fondamentali,come la carica elettricametodo semiempirico: introduce nei calcoli un numero limitato di datisperimentali
metodo empirico: fa largo uso di dati sperimentali
Per una reazione in cui stato iniziale e finale abbiano energia diversa, la superficiedell'energia potenziale non sarà simmetrica
Stato dei reagenti più stabile dello stato dei prodotti
Stato di transizione "repulsivo"o "tardo" (late) o "simile ai prodotti"Il legame AB è molto formato nello stato di transizione
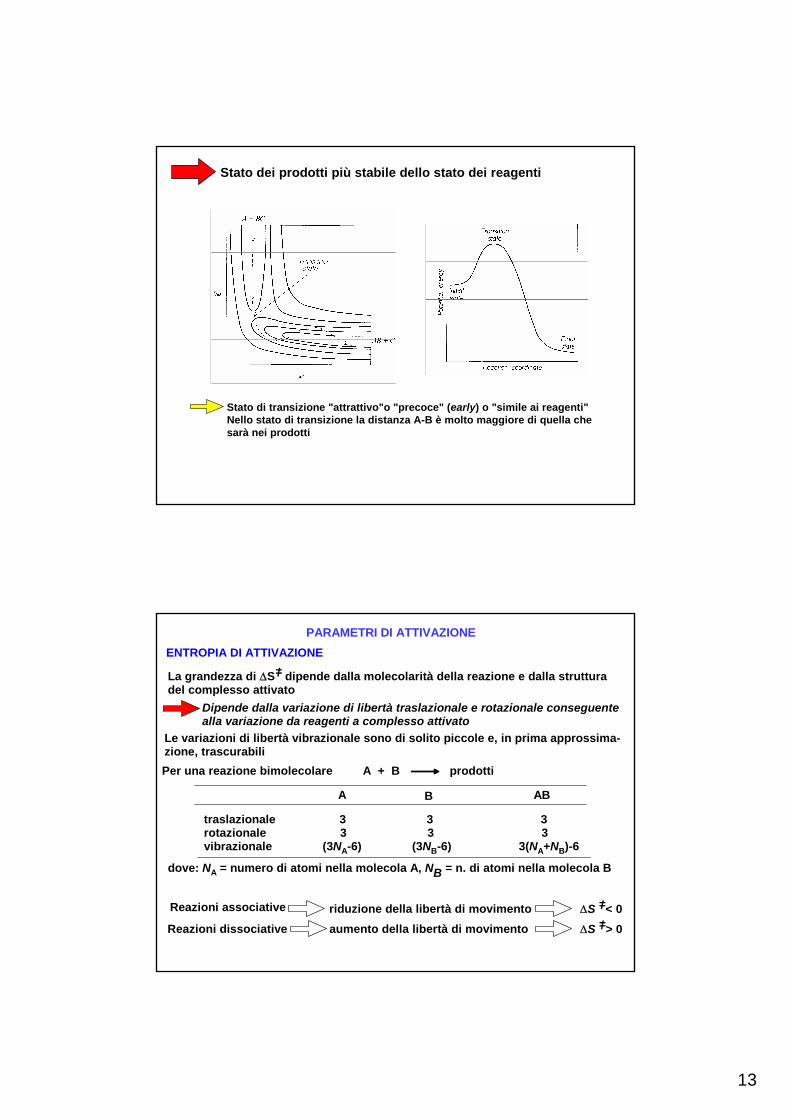
13
Stato dei prodotti più stabile dello stato dei reagenti
Stato di transizione "attrattivo"o "precoce" (early) o "simile ai reagenti"Nello stato di transizione la distanza A-B è molto maggiore di quella che sarà nei prodotti
PARAMETRI DI ATTIVAZIONEENTROPIA DI ATTIVAZIONE
La grandezza di ΔS dipende dalla molecolarità della reazione e dalla strutturadel complesso attivato
=/
Dipende dalla variazione di libertà traslazionale e rotazionale conseguentealla variazione da reagenti a complesso attivato
Le variazioni di libertà vibrazionale sono di solito piccole e, in prima approssima-zione, trascurabili
dove: NA = numero di atomi nella molecola A, NB = n. di atomi nella molecola B
Per una reazione bimolecolare A + B prodotti
traslazionale 3 3 3rotazionale 3 3 3vibrazionale (3NA-6) (3NB-6) 3(NA+NB)-6
A B AB
Reazioni associative riduzione della libertà di movimento =/ΔS < 0Reazioni dissociative aumento della libertà di movimento =/ΔS > 0
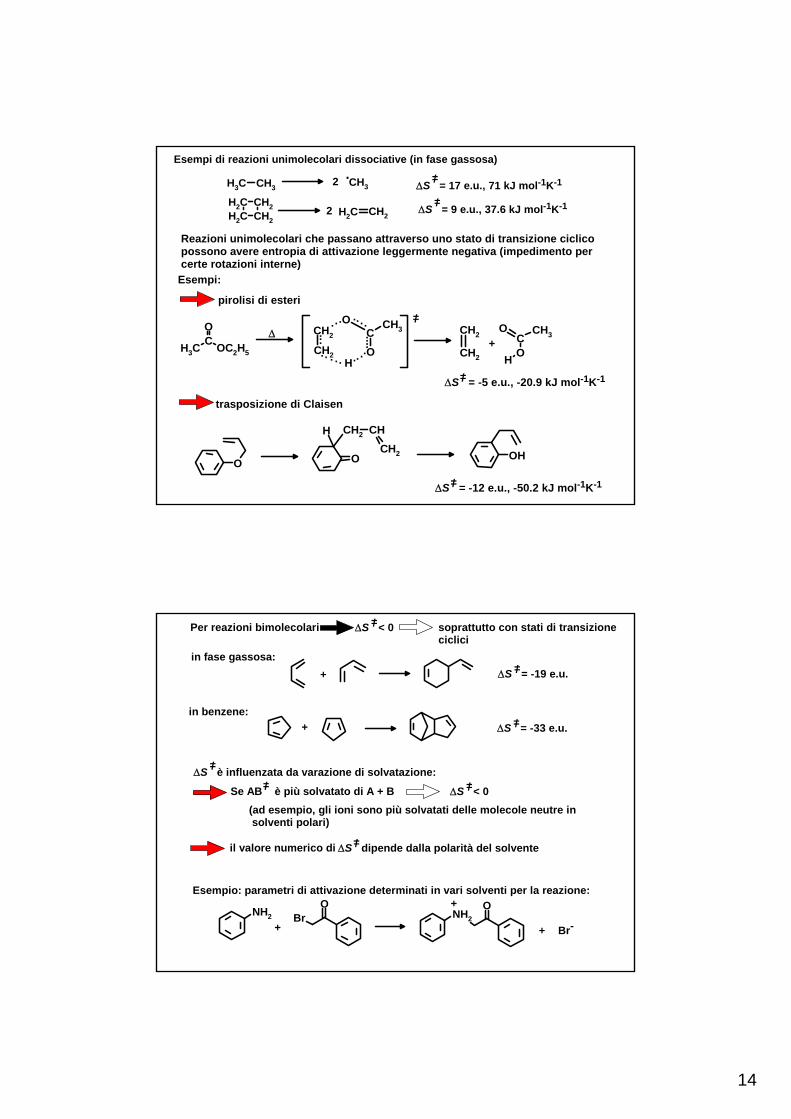
14
Esempi di reazioni unimolecolari dissociative (in fase gassosa)
2 .ΔS = 17 e.u., 71 kJ mol-1K-1=/
2 ΔS = 9 e.u., 37.6 kJ mol-1K-1=/CH2CH2 CH2
CH2
CH3 CH3 CH3
CH2 CH2
Reazioni unimolecolari che passano attraverso uno stato di transizione ciclicopossono avere entropia di attivazione leggermente negativa (impedimento percerte rotazioni interne)
Esempi:
pirolisi di esteri
Δ
CH2
CH2
OC
CH3O
H.. ..
.
.
. . ..
...... .. ...
.. =/
+
ΔS = -5 e.u., -20.9 kJ mol-1K-1=/
trasposizione di Claisen
CH2
CH2
OH
CH3C OC2H5
OC
CH3O
OH
O
CH2H CHCH2
O
ΔS = -12 e.u., -50.2 kJ mol-1K-1=/
Per reazioni bimolecolari soprattutto con stati di transizioneciclici
ΔS < 0=/
in fase gassosa:+ ΔS = -19 e.u.=/
in benzene:+ ΔS = -33 e.u.=/
ΔS è influenzata da varazione di solvatazione:=/
Se AB è più solvatato di A + B ΔS < 0=/=/
(ad esempio, gli ioni sono più solvatati delle molecole neutre in solventi polari)
il valore numerico di ΔS dipende dalla polarità del solvente=/
Esempio: parametri di attivazione determinati in vari solventi per la reazione:
+
+
+ Br-NH2
OBr NH2
O
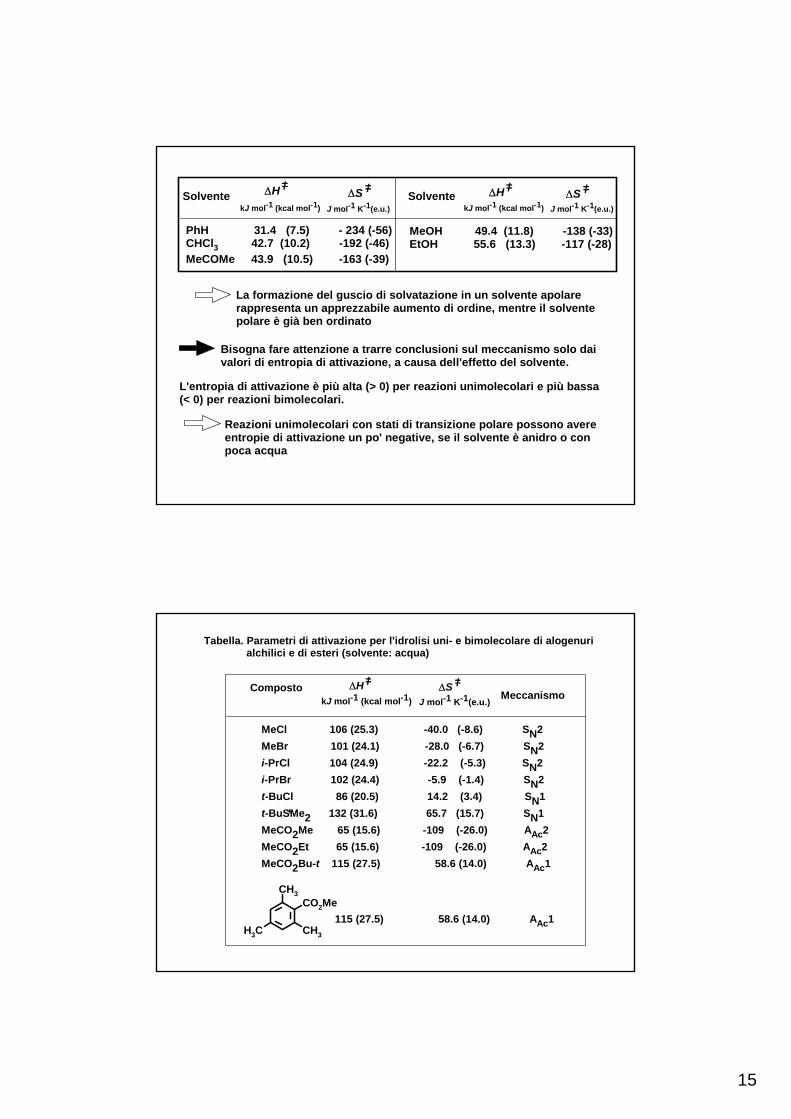
15
Solvente
PhH 31.4 (7.5) - 234 (-56)CHCl3 42.7 (10.2) -192 (-46)MeCOMe 43.9 (10.5) -163 (-39)
SolventeΔH =/
kJ mol-1 (kcal mol-1)
ΔS =/
J mol-1 K-1(e.u.)
ΔH =/
kJ mol-1 (kcal mol-1)ΔS =/
J mol-1 K-1(e.u.)
MeOH 49.4 (11.8) -138 (-33)EtOH 55.6 (13.3) -117 (-28)
La formazione del guscio di solvatazione in un solvente apolarerappresenta un apprezzabile aumento di ordine, mentre il solventepolare è già ben ordinato
Bisogna fare attenzione a trarre conclusioni sul meccanismo solo dai valori di entropia di attivazione, a causa dell'effetto del solvente.
L'entropia di attivazione è più alta (> 0) per reazioni unimolecolari e più bassa(< 0) per reazioni bimolecolari.
Reazioni unimolecolari con stati di transizione polare possono avere entropie di attivazione un po' negative, se il solvente è anidro o conpoca acqua
Tabella. Parametri di attivazione per l'idrolisi uni- e bimolecolare di alogenuri alchilici e di esteri (solvente: acqua)
ΔH =/
kJ mol-1 (kcal mol-1)ΔS =/
J mol-1 K-1(e.u.)Composto
Meccanismo
MeCl 106 (25.3) -40.0 (-8.6) SN2MeBr 101 (24.1) -28.0 (-6.7) SN2i-PrCl 104 (24.9) -22.2 (-5.3) SN2i-PrBr 102 (24.4) -5.9 (-1.4) SN2t-BuCl 86 (20.5) 14.2 (3.4) SN1t-BuSMe2 132 (31.6) 65.7 (15.7) SN1MeCO2Me 65 (15.6) -109 (-26.0) AAc2MeCO2Et 65 (15.6) -109 (-26.0) AAc2MeCO2Bu-t 115 (27.5) 58.6 (14.0) AAc1
115 (27.5) 58.6 (14.0) AAc1CO2Me
CH3
CH3 CH3
+

16
ENTALPIA DI ATTIVAZIONE
L'entalpia di attivazione (o energia di attivazione) è la differenza di entalpia (o di energia) tra complesso attivato e reagenti
ΔH =/Il valore numerico di o di Ea è più difficile da prevedere menodiagnostico
Ci si può aspettare un valore più alto per reazioni unimolecolari che per reazionibimolecolari
Esempio:
2
Ea = 258 kJ mol-1
ΔS =/ = + 41.8 J mol-1 K-1
Ea = 96.6 kJ mol-1
ΔS =/ = - 79.5 J mol-1 K-1
=/
in soluzione ΔH =/ è molto influenzato dalla solvatazione
Se la solvatazione stabilizza lo stato di transizione più dei reagenti, ci si aspetta una diminuzione di ΔH
+ + Br-Tabella. Parametri di attivazione per la reazione SN2
RBr I- RI
R voluminosi possono impedire che il reagente raggiunga il centro di reazionepiù difficile raggiungere lo stato di transizione aumento di ΔH =/
R
Me 68.2 (16.3) - 33.5 (-8)Et 78.6 (18.8) -41.8 (-10)
ΔH =/
kJ mol-1 (kcal mol-1)
ΔS =/
J mol-1 K-1(e.u.)
ΔH =/kJ mol-1 (kcal mol-1)
ΔS =/J mol-1 K-1(e.u.)
i-Pr 85.8 (20.5) -58.6 (-14)t-BuCH2 100.4 (24.0) -58.6 (-14)
R
Il gruppo voluminoso nello stato di transizione avrà movimento vibrazionale limitato, a causa dell'affollamento diminuzione di ΔS =/
RELAZIONE ISOCINETICAIn alcuni casi i parametri di attivazione per una serie di reazioni possono esserein relazione
ΔS =/ΔH =/ = β + c relazione isocinetica
per una serie di composti che variano solo per il sostituente, fatti reagirenelle stesse condizioniper una reazione di un solo sistema molecolare, misurata in più solventi

17
Quando la relazione isocinetica è valida, si assume che il meccanismo sia lo stesso in tutta la serie
β dimensioni di temperatura assoluta
Quando T = β K, la velocità di ogni termine della serie è la stessa
sostituendo a ΔH il secondo termine della relazione isocinetica:=/
ln k = ln RTNh( ) + -
=/ΔS (T - β)RT
cRT
ln k = ln RTNh( ) +
=/ ΔH TΔS - =/
RT
= 0 a T = β
ln kik = ln RTNh
- cRTkik costante di velocità isocinetica
(isokinetic)
La situazione è illustrata in figura
All'interno della serie, la reazione che è più veloce a T < β, è la più lentaa T > β
ΔH =/Al di sotto della temperatura isocinetica, è più veloce la reazione con piùpiccola; al di sopra della temperatura isocinetica, è più veloce la reazione dellaserie con ΔH più grande.=/
Prendendo due reazione, A e B, di una data serie (due rette della figura) per cuisia valida la relazione isocinetica, con valori diversi di entropia di attivazione, illogaritmo del rapporto delle costanti di velocità è dato da:
kA
kBlog( ) =
(ΔSA - ΔSB )(T-β)=/ =/
RT se ΔSA > ΔSB =/ =/ kA < kB a T < β
kA > kB a T > β

18
Poiché ΔH e ΔS non si possono determinare molto accuratamente, per stimareβ senza conoscerli esplicitamente
=/ =/
metodo di EXNER
Si prende una serie di composti e si misurano, a due diverse temperature(T1 e T2) due costanti di velocità (k1 e k2) per ciascun termine della serie
log k2 = b log k1 + a
esprimendo ln k1 e ln k2 in funzione dei parametri di attivazione si ha:
log k1 = log RTNh( ) T1ΔS - ΔH =/ =/
RT1
T1T2 (1 - b)
β
=/
+
log k2 = logRTNh( ) T2ΔS - ΔH =/ =/
RT2+
ΔH =(T1- bT2)
=/ΔS -T1T2
(T1- bT2){aR + (b-1)R ln RNh
+ R(b ln T1- lnT2)}=
T1T2 (1 - b)(T1- bT2)
occorre determinare i valori di ln k1 e ln k2 per la serie di composti (o di solventi) e determinare b con i minimi quadrati, da b si calcola β usandodue temperature sperimentali
Solvolisi di cloruri di benzoile sostituiti in paracorrelazione di log k a 25°C in funzione di log k a 0°C
Non tutte le reazioni seguono la relazione entalpia-entropiasono pochi gli studi cinetici a temperatura superiore a β
β può assumere valori estremi (0 o ∞ ) quando per una seriecambia solo una delle funzioni termodinamiche, mentre l'altrarimane praticamente costante

19
poiché la reattività relativa (effetto del sostituente o del solvente) allatemperatura isocinetica si inverte, i risultati ottenuti ad una solatemperatura vanno trattati con molta cautela
INADEGUATEZZA DELLO STATO DI TRANSIZIONE
La teoria dello stato di transizione è stata molto usata per interpretare le costantidi velocità, ma rimangono dubbi sulla sua validità
nella forma solita non è corretta dal punto di vista quantomeccanico
non spiega l'effetto tunnel
la teoria assume che la coordinata di reazione possa essere separatadagli altri movimenti
calcoli dettagliati per reazioni semplici H. + H2 H2 + H.
hanno dimostrato che l'errore introdotto nella costante di velocità è significativoa temperature fino a 1000K ed è più importante per il movimento di atomi leggeri
sembra che la teoria possa essere inadeguata per processi in cui la coordinata di reazione comporti movimento in più di una dimensione
GRANDEZZA DELLE QUANTITA' CINETICHE
κTh
~ 1012.8 s-1 a 298 K (κ è la costante di Boltzmann e h quella di Planck)
questo valore dovrebbe rappresentare la velocità attesa per una reazione mono-stadio unimolecolare in fase gassosa, con entalpia ed entropia di attivazione zero
in soluzione, le velocità delle reazioni bimolecolari sono limitate dallavelocità con cui le specie si diffondono tra le molecole di solvente
velocità controllata dalla diffusione
varia con la viscosità del solvente e con la temperaturaper i solventi comuni a temperatura ambiente ~ 1010 M-1s-1
ΔH =/ kJ mol-1 (kcal mol-1)
ΔS =/
J mol-1 K-1(e.u.)κT /h e-ΔH /RT=/
s-1e ΔS /R=/
4.2 (1) 1.15 x 1012 -120 (-30) 2.75 x 10-7
8.4 (2) 2.12 x 1011 -105 (-25) 3.41 x 10-6
21 (5) 1.33 x 109 -84 (-20) 4.23 x 10-5
42 (10) 2.85 x 105 -63 (-15) 5.25 x 10-4
63 (15) 6.10 x 101 -42 (-10) 6.50 x 10-3
84 (20) 1.31 x 10-2 -21 ( -5) 8.07 x 10-2
105 (25) 2.80 x 10-6 0 1126 (30) 6.00 x 10-10 21 (5) 2.14 x 101
146 (35) 1.29 x 10-13 42 (10) 1.54 x 102
167 (40) 2.76 x 10-17 63 (15) 1.91 x 103
La tabella dà un'idea della relazione tra parametri di attivazione e costanti di velocità

20
STRUTTURA DELLO STATO DI TRANSIZIONE E DIAGRAMMA TRIDIMENSIONALE DELLA COORDINATA DI REAZIONEANALISI DI THORNTON
Approssimiamo la curva dell'energia potenziale nella regione dello stato di transizione ad una parabola. Supponiamo di fare un piccolo cambiamento della struttura, che renda più difficile procedere verso destra (cambiamentoequivalente ad aumentare l'energia della parte destra della curva più della sinistra)
Si ottiene aggiungendo all'energia in ciascun punto lungo la curva un incremento dΔE°, che aumenta verso destra
δΔE° = mxdove m (in questo caso) è positivo e x è il parametro "coordinata di reazione"
la retta rappresenta la perturbazione δΔE°Se si mette l'origine al vertice della parabola, è facile vedere che il risultato dell'aggiunta della perturbazione alla curva dell'energia potenziale è spostare il suo massimo (e quindi lo stato di transizione) verso destra (curva tratteggiata)
Una perturbazione che renda più difficile il movi-mento da sinistra verso destra, sposta lo TS verso destra lungo la coordinata direazione.Una perturbazione con pendenza negativa, cioèche renda più facile il movimento da sinistra a destra, sposterà la curva (e TS) verso sinistra.
Consideriamo come le variazioni strutturali influenzano la posizione dellostato di transizione sulla superficie di energia potenziale rispetto ai gradidi libertà diversi dalla coordinata di reazione.
La curva è una parabola con l'aperura verso l'alto
Se si fa una variazione strutturale che renda più difficile l'allungamento di un legame diverso da quello che si rompe
δΔE° = mz
dove m è positivo e z è una parametro "coordinata non di reazione"
coordinata di vibrazione, z
La curva dell'energia potenzialeperturbata si sposta verso sinistra
Rendere più difficile l'allungamentodel legame ha cambiato la strutturadello stato di transizione in modotale che la distanza di equilibrio dellegame è più corta

21
Le variazioni di energia dello stato di transizione hanno i seguenti effetti
1. LUNGO LA COORDINATA DI REAZIONE la struttura dello stato di transizione si sposta verso l'estremità che viene innalzata, allon- tanandosi dall'estremità che si abbassa2. PERPENDICOLARMENTE ALLA COORDINATA DI REAZIONE la struttura dello stato di transizione si sposta verso il lato che si abbassa, allontanandosi dal lato che si alza.
Una base strappa un protone da un C ed un gruppo uscente si allontanadal C adiacente
la reazione può seguire meccanismi diversi:
a) Processo sincrono (E2)
B:- + BH + + :X-
stato di transizione C C...........
.....X
B......H
δ-
δ- =/
C CC CX
H
b) il protone viene strappato prima dell'uscita del gruppo uscente (E1cb)
B:- + C CX
HBH + C C
X
..-
C CX
..-C C + :X-
c) il gruppo uscente si allontana per primo (E1)
+ :X-
B: + C CH
BH + C C+-
C CX
HC C
H +
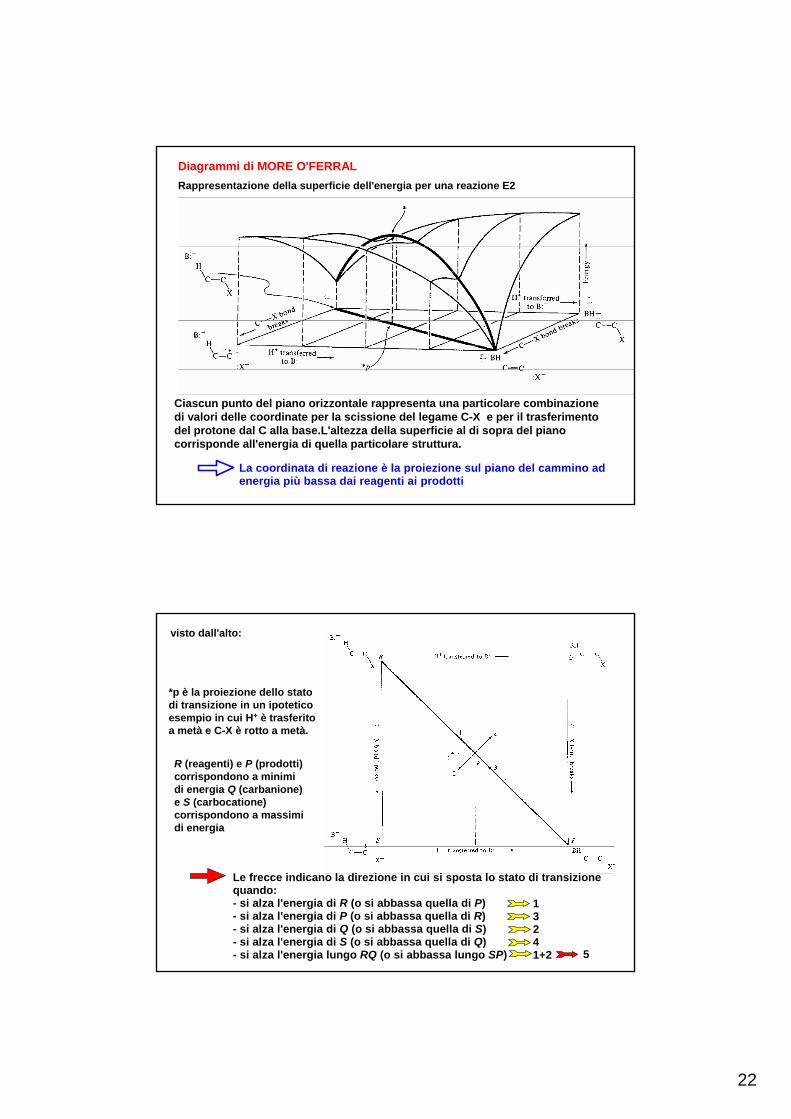
22
Diagrammi di MORE O'FERRALRappresentazione della superficie dell'energia per una reazione E2
Ciascun punto del piano orizzontale rappresenta una particolare combinazione di valori delle coordinate per la scissione del legame C-X e per il trasferimento del protone dal C alla base.L'altezza della superficie al di sopra del piano corrisponde all'energia di quella particolare struttura.
La coordinata di reazione è la proiezione sul piano del cammino ad energia più bassa dai reagenti ai prodotti
visto dall'alto:
*p è la proiezione dello stato di transizione in un ipotetico esempio in cui H+ è trasferito a metà e C-X è rotto a metà.
R (reagenti) e P (prodotti) corrispondono a minimi di energia Q (carbanione) e S (carbocatione) corrispondono a massimi di energia
Le frecce indicano la direzione in cui si sposta lo stato di transizionequando:- si alza l'energia di R (o si abbassa quella di P)- si alza l'energia di P (o si abbassa quella di R)- si alza l'energia di Q (o si abbassa quella di S)- si alza l'energia di S (o si abbassa quella di Q)- si alza l'energia lungo RQ (o si abbassa lungo SP)
13241+2 5

23
Superficie dell'energia e coordinata di reazione per un'ipotetica E2, in cui l'energia dei reagenti è più elevata rispetto all'esempio precedente, mentre l'energia degli altri angoli è invariata
La forma della superficie è variata così da portare la struttura dello stato di transizione più vicino ai reagenti (freccia 1 nella proiezione)
Superficie dell'energia e coordinata di reazione per un'ipotetica E2, in cui, rispetto al primo esempio, è stata innalzata l'energia lungo lo spigolo posteriore (X miglior gruppo uscente)
La forma della superficie cambia, così da portare lo stato di transizionepiù vicino allo spigolo di sinistra (freccia 5)
H+ viene meno trasferito
24
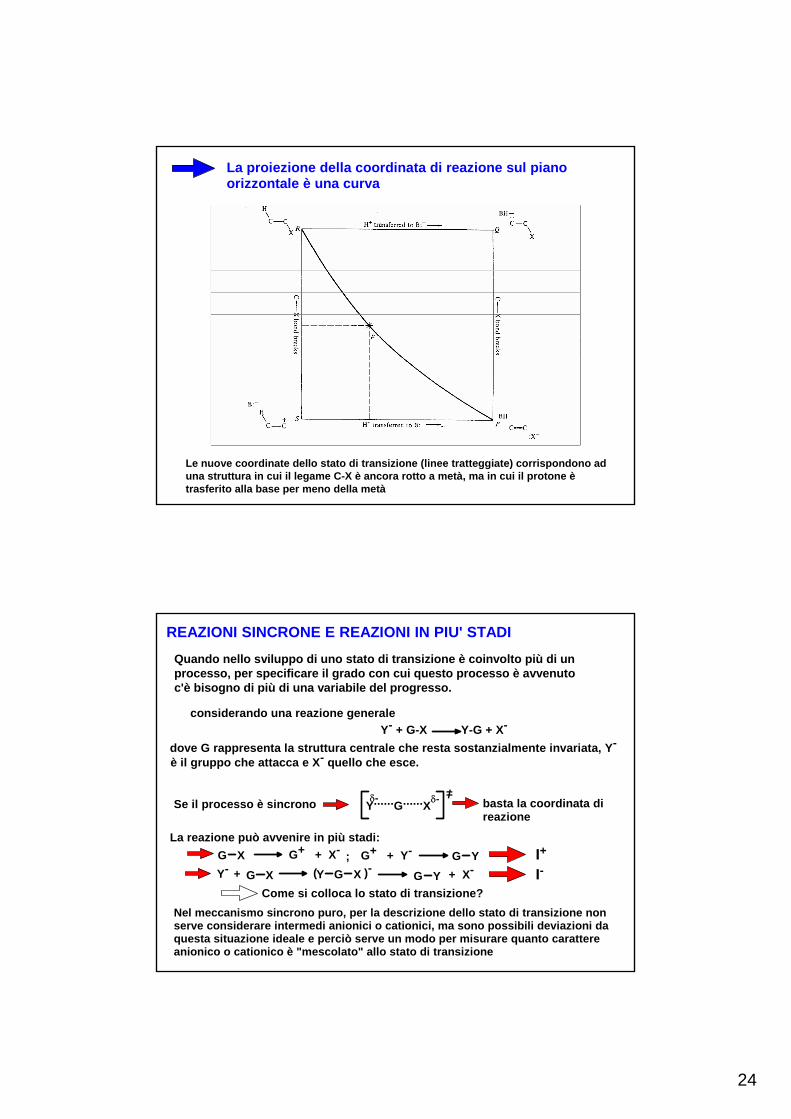
La proiezione della coordinata di reazione sul pianoorizzontale è una curva
Le nuove coordinate dello stato di transizione (linee tratteggiate) corrispondono ad una struttura in cui il legame C-X è ancora rotto a metà, ma in cui il protone ètrasferito alla base per meno della metà
REAZIONI SINCRONE E REAZIONI IN PIU' STADI
Quando nello sviluppo di uno stato di transizione è coinvolto più di un processo, per specificare il grado con cui questo processo è avvenuto c'è bisogno di più di una variabile del progresso.
considerando una reazione generaleY- + G-X Y-G + X-
dove G rappresenta la struttura centrale che resta sostanzialmente invariata, Y- è il gruppo che attacca e X- quello che esce.
Se il processo è sincrono Y......G......Xδ- δ- =/
basta la coordinata di reazione
La reazione può avvenire in più stadi:
Come si colloca lo stato di transizione?
G X G+ + X- ; G+ + Y- G Y I+ Y- + G X G XY( )- G Y + X- I-
Nel meccanismo sincrono puro, per la descrizione dello stato di transizione nonserve considerare intermedi anionici o cationici, ma sono possibili deviazioni daquesta situazione ideale e perciò serve un modo per misurare quanto carattereanionico o cationico è "mescolato" allo stato di transizione

25
tipo di reazioneDescrizione dello stato
R I+ I- P
Y- R-X Y-R+X- Y-R-X+ Y-R X-
B- H-R-R-X B- H-R-R+X- BH -R-R-X BH R=R X-
Y- RCOX Y-RCO+X- RC(O-)XY RCOY X-
Y+ Ar-X (Y-Ar-X)+ Y+Ar-X+ Y-Ar X+
Y- Ar-X Y-Ar+X- (Y-Ar-X)- Y-Ar X-
Sostituzione nucleofila alifatica
Sostituzione nucleofila acilica
β-Eliminazione
Sostituzione elettrofila aromatica
Sostituzione nucleofila aromatica
Diagrammi RPI (reagent-intermediate-product)
METODO DI Pross e Shaik (o DEL LEGAME DI VALENZA)
Le teorie MO e VB sono teoricamente equivalenti, dato che entrambeportano alla stessa funzione d'onda.
il metodo di Pross e Shaik consiste nell'esaminare gli stati elettronici dei legami di valenza di reagenti e prodotti e di connetterli su un diagramma dell'energia infunzione della coordinata di reazione, chiamato diagramma di correlazione distato.
Consideriamo la rottura del legame polare R-X (con X più elettronegativodi R)
In prima approssimazione lo stato fondamentale ed il primo stato eccitato di RXsi possono rappresentare:
R-Xstato fondamentale = R. .X
R-Xprimo stato eccitato = R+ :X-

26
Variazione delle due configurazioni con la distanza R-X in FASE GASSOSA(Figura a sinistra):
Una descrizione molto migliore di questi due stati si ottiene mescolando partedella configurazione ad energia più elevata a quella più bassa (in modo legante)e parte di quella ad energia più bassa con quella più alta (in modo antilegante)
R-Xstato fondamentale = R. .X + (R+ :X-)λ
R-Xprimo stato eccitato = R+ :X- + (R. .X)λ
L'energia di questi stati corretti varia dando la linea tratteggiata (figura a sinistra)
All'aumentare della distanza tra R e X, la regione dello spazio in cui c'èun'elevata probabilità di trovare il secondo elettrone su X ha unasovrapposizione MOLTO PICCOLA con la regione dello spazio in cui c'è un'elevata probabilità di trovare l'elettrone dispari su R
In soluzione (figura di destra) la configurazione R+:X- è fortementestabilizzata per solvatazione
primo stato eccitato = R. .X configurazione delle particelle dissociate: stato fondamentale = R+ :X-
Se non ci fosse nessuna interazione di configurazione, le curve delle dueconfigurazioni si incrocerebbero
nella realtà, questo incrocio "nelle intenzioni" non avviene
Man mano che il legame R-X è stirato, l'energia della configurazione fondamentaleaumenta, fino a che diventa quasi uguale all'energia della configurazione del primostato eccitato
Man mano che le energie delle due configurazioni diventano quasi uguali,aumenta l'interazione tra loro.
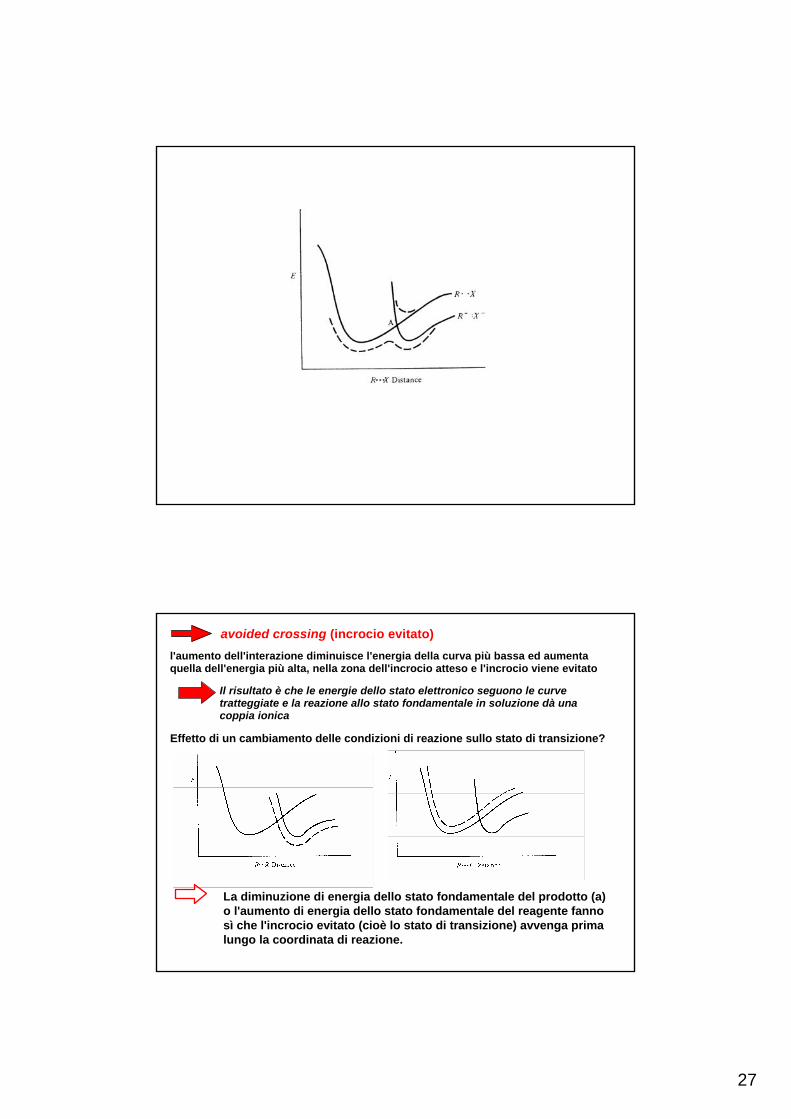
27
avoided crossing (incrocio evitato)l'aumento dell'interazione diminuisce l'energia della curva più bassa ed aumentaquella dell'energia più alta, nella zona dell'incrocio atteso e l'incrocio viene evitato
Il risultato è che le energie dello stato elettronico seguono le curvetratteggiate e la reazione allo stato fondamentale in soluzione dà unacoppia ionica
Effetto di un cambiamento delle condizioni di reazione sullo stato di transizione?
La diminuzione di energia dello stato fondamentale del prodotto (a) o l'aumento di energia dello stato fondamentale del reagente fanno sì che l'incrocio evitato (cioè lo stato di transizione) avvenga prima lungo la coordinata di reazione.

28
Nel modello di Pross e Shaik anche le energie del primo stato eccitato direagenti e prodotti influisce sulla posizione dello stato di transizione, in-fluenzando la pendenza della curva
Effetto, sulla posizione dello stato di transizione per la rottura di R-X, dell'aumento di energia del primo stato eccitato dei reagenti (curva tratteggiata)
il modello di Pross e Shaik rende molto semplice che cosa è che provocala barriera di energia
Si forma una barriera, perché i reagenti devono aprire i loro gusci di valenza(stirare o rompere legami) e perciò aumentare di energia, fino a che incontranouno stato elettronico decrescente, nel quale possono passare.
Applicando il modello ad una reazione SN
Si sviluppa una descrizione VB scrivendo una combinazione linearedi un insieme ragionevole di configurazioni
Y:- + R-X R-Y + X:-
lo stato iniziale è dominato dalla configurazione R e lo stato finale da P,anche se ciascuno stato comprende contributi da tutte le configurazioni
L'energia della configurazione di R aumenta, perché è energeticamente sfavorevole mantenere la configurazione dello stato iniziale man mano che si procede lungo la coordinata di reazioneL'energia dello stato P diminuisce, msan mano che ci si avvicina allostato finaleNel punto in cui queste configurazioni si incontrano, si ha un "incrocioevitato" ed il sistema in reazione segue il cammino ad energia più bassa.
Y:- R. .X R, simile ai reagentiY. . R :X- P, simile ai prodotti
Y:- R+ :X- I+, simile al carbocationeY:- R:- X+ I-, simile al carbanione

29
Una variazione strutturale che stabilizzi P rispetto a R abbassa il profilodi energia di P, dando uno stato di transizione anticipato (comportamentosecondo Hammond)
Supponendo che la configurazione I+ non contribuisca in modo significativo aglistati iniziale e finale, ma sia significativa nella zona di mezzo della coordinata di reazione:

30
TEORIA DELLA VELOCITA' DI MarcusLa teoria di Marcus considera che la barriera di energia della reazione siacostituita da due contributi
una parte termodinamica, da ΔG°una parte cinetica intrinseca, che è la barriera che esisterebbese reagenti e prodotti avessero la stessa energia libera
Considerando la reazione:HO- + H3CBr HOCH3 + Br-
Marcus descrive la coordinata di reazione per questo processo come due parabole che si intersecano
una che rappresenta la superficie dell'energia potenzialeper lo stiramento del legame C-Br nei reagentiuna che rappresenta la superficie dell'energia potenzialeper lo stiramento del legame C-OH nel prodotto
La forma e la collocazione di queste parabole sono determinate dalla variazionecomplessiva di energia libera e dalla barriera intrinseca.
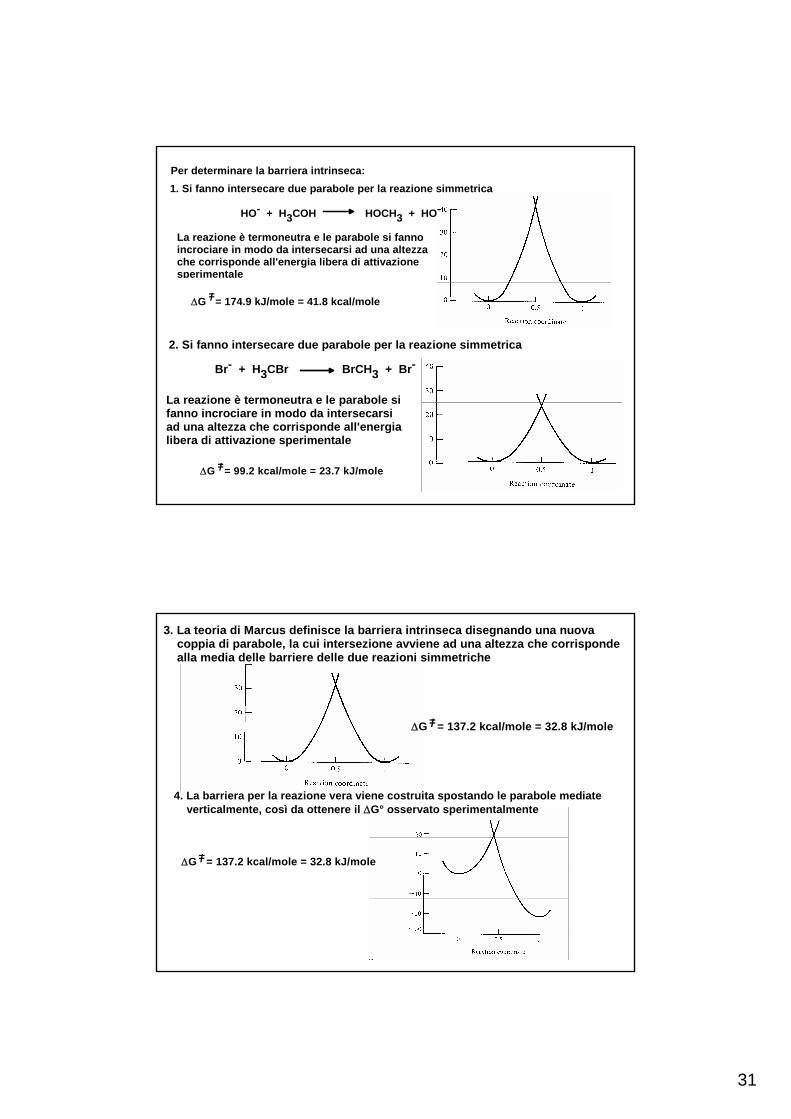
31
2. Si fanno intersecare due parabole per la reazione simmetrica
Br- + H3CBr BrCH3 + Br-
La reazione è termoneutra e le parabole si fanno incrociare in modo da intersecarsi ad una altezza che corrisponde all'energia libera di attivazione sperimentale
Per determinare la barriera intrinseca:1. Si fanno intersecare due parabole per la reazione simmetrica
HO- + H3COH HOCH3 + HO-
La reazione è termoneutra e le parabole si fannoincrociare in modo da intersecarsi ad una altezzache corrisponde all'energia libera di attivazione sperimentale
ΔG = 174.9 kJ/mole = 41.8 kcal/mole=/
ΔG = 99.2 kcal/mole = 23.7 kJ/mole =/
3. La teoria di Marcus definisce la barriera intrinseca disegnando una nuova coppia di parabole, la cui intersezione avviene ad una altezza che corrisponde alla media delle barriere delle due reazioni simmetriche
ΔG = 137.2 kcal/mole = 32.8 kJ/mole =/
4. La barriera per la reazione vera viene costruita spostando le parabole mediate verticalmente, così da ottenere il ΔG° osservato sperimentalmente
ΔG = 137.2 kcal/mole = 32.8 kJ/mole =/
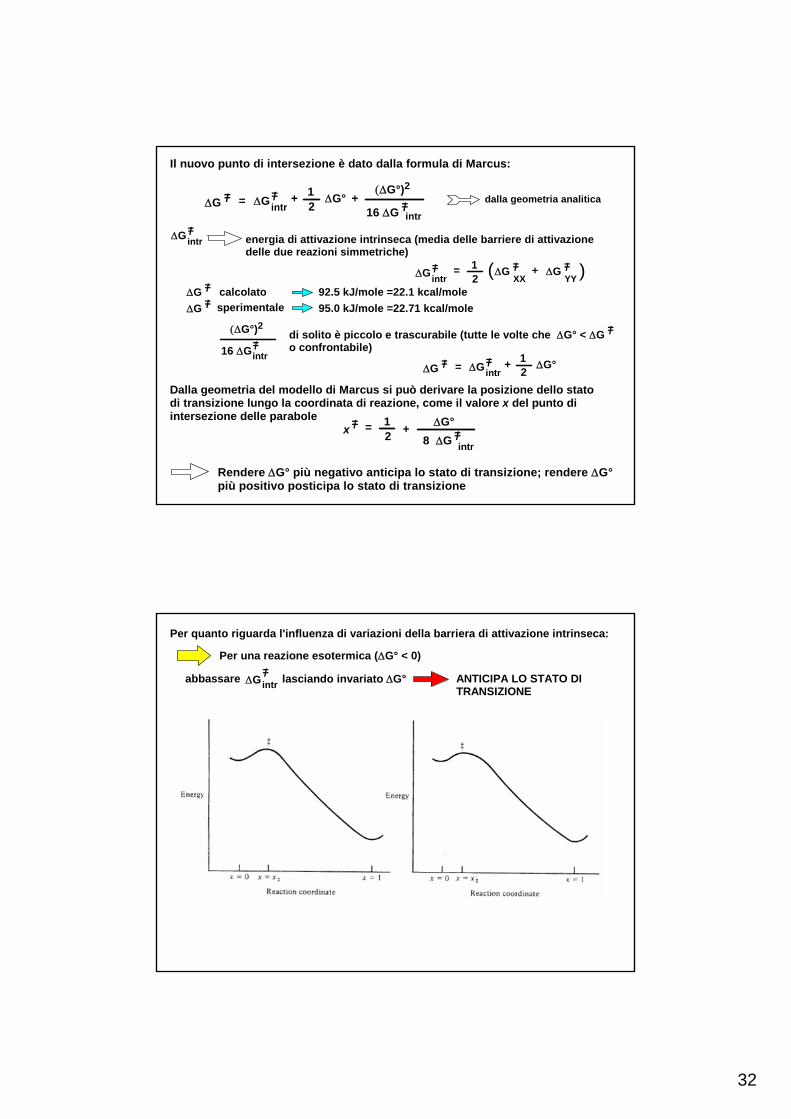
32
Il nuovo punto di intersezione è dato dalla formula di Marcus:
dalla geometria analiticaΔG= =/intr
+ 12
ΔG° +(ΔG°)2
16 ΔG =/intr
=/ΔG
ΔG=/intr energia di attivazione intrinseca (media delle barriere di attivazione
delle due reazioni simmetriche)
ΔG=/intr
= 12
=/ΔG( XX+ =/ΔG
YY )=/ΔG calcolato=/ΔG sperimentale 95.0 kJ/mole =22.71 kcal/mole
92.5 kJ/mole =22.1 kcal/mole
(ΔG°)2
16 ΔG=/intr
di solito è piccolo e trascurabile (tutte le volte che ΔG° < ΔGo confrontabile)
=/
ΔG= =/intr
+ 12
ΔG°=/ΔG
Dalla geometria del modello di Marcus si può derivare la posizione dello statodi transizione lungo la coordinata di reazione, come il valore x del punto diintersezione delle parabole =/
8 ΔG =/intr
= +12
ΔG°x
Rendere ΔG° più negativo anticipa lo stato di transizione; rendere ΔG°più positivo posticipa lo stato di transizione
Per quanto riguarda l'influenza di variazioni della barriera di attivazione intrinseca:
Per una reazione esotermica (ΔG° < 0)
abbassare ΔG=/intr lasciando invariato ΔG° ANTICIPA LO STATO DI
TRANSIZIONE
33
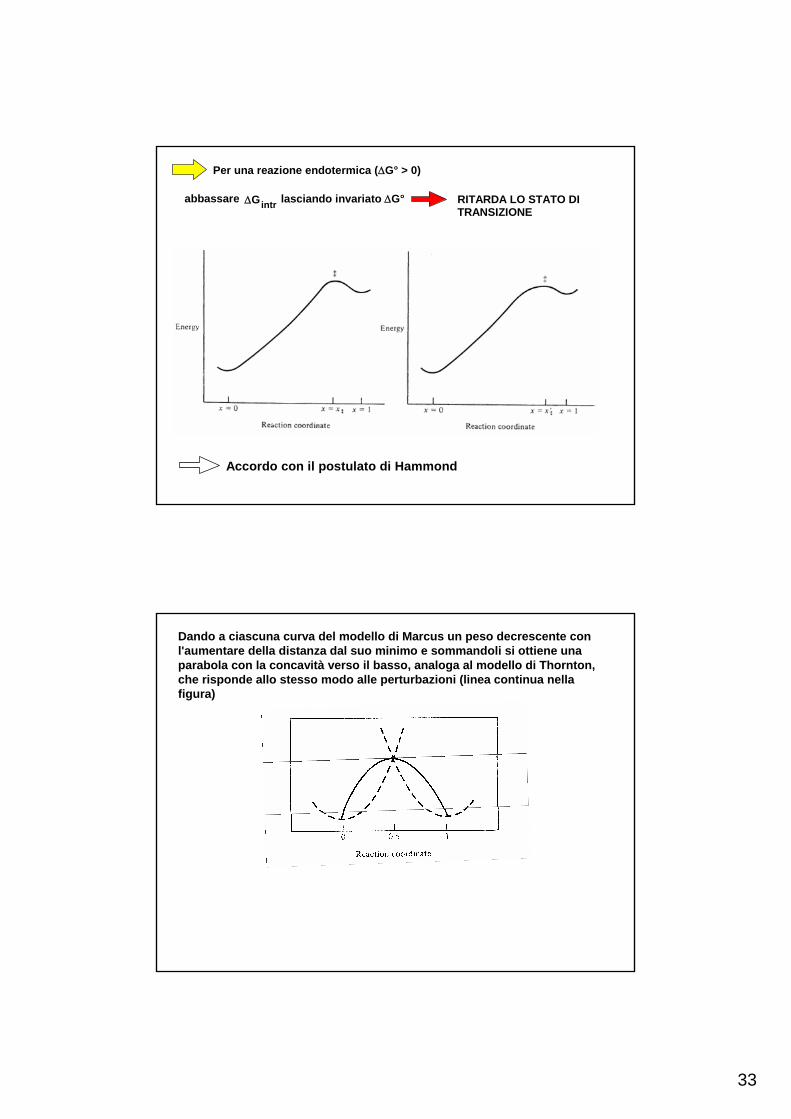
Per una reazione endotermica (ΔG° > 0)
abbassare ΔGintr lasciando invariato ΔG° RITARDA LO STATO DITRANSIZIONE
Accordo con il postulato di Hammond
Dando a ciascuna curva del modello di Marcus un peso decrescente con l'aumentare della distanza dal suo minimo e sommandoli si ottiene una parabola con la concavità verso il basso, analoga al modello di Thornton, che risponde allo stesso modo alle perturbazioni (linea continua nella figura)

34
CORRELAZIONI LINEARI DI ENERGIA LIBERA
Una serie di variazioni nelle condizioni di reazione (per esempio l'elettrone-gatività dei sostituenti del substrato o il potere ionizzante del solvente) quasisempre provocherà una serie di cambiamenti nella velocità di una reazione chimica o nella posizione di un equilibrio chimico.Se la stessa serie di variazioni di condizione influenza in modo analogo la velocità o l'equilibrio di una seconda reazione, esiste una correlazione lineare di energia libera tra i due insiemi di effetti.
Quando una tale relazione esiste, può essere molto utile per chiarire il mec-canismo, permettendo di prevedere le velocità di reazione, e di scoprire inquali condizioni avvenga un cambiamento di meccanimo.
Problema: se il sostituente è vicino al centro di reazione influenza la reazionemediante processi puramente sterici, che mascherano gli effetti elettronicise il sostituente è lontano dal centro di reazione, gli effetti sonomolto attenuati
EQUAZIONE DI HAMMETTLa prima formulazione di una relazione quantitativa tra struttura e reattività si devead Hammett (1937).
L'effetto elettronico di un sostituente può essere definito confrontando i Ka di acidi benzoici sostituiti in meta ed in para con il Ka dell'acido benzoico.
KX-meta
+ H2O + H3O+
25°C
-CO2H
X
CO2
X
KX-para + H3O++ H2O
25°C
-CO2H
X
CO2
X
Ko
-
+ H2O
CO2H CO2
X è separato fisicamente da -CO2H, ma è collegato attraverso un sistema dielettroni π relativamente polarizzabile.

35
Dopo aver determinato i Ka degli acidi benzoici in acqua a 25°C, Hammett ha asse-gnato a ciascun sostituente due parametri, σmeta e σpara, definiti come:
σmeta = logKX-meta
Koσpara = log
Ko
KX-para
La costante del sostituente, σ, misura la grandezza dell'effetto elettronico del sosti-tuente. Per come sono definiti, sostituenti ad attrazione elettronica hanno σ > 0,sostituenti a rilascio elettronico hanno σ < 0.
Dalla definizione log (KX/Ko) vs. σ è una retta con pendenza 1
Per definizione: σH = 0.00 la retta deve passare per il punto di coordinate (0,0)
Per determinare se i σ hanno validità generale, Hammett ha studiato le ionizzazioni di altre famiglie di acidi.
acidi fenilacetici
+ H2O
-+ H3O+
K'o
K'XX = H
Riportando in grafico log (K'X/K'o) in funzione di σ si ottiene ancora una retta, ma con pendenza minore di 1. Questa diminuzione dell'effetto del sostituente rispetto agli acidi benzoici è dovuta alla maggiore separazione tra sostituente e centro acido.
CH2 CO2CH2 CO2H
X X
σ
logK'XK'o
= ρσ
Grafico di log (K'X/K'o) in funzione di σ per la dissociazione di acidi fenilacetici (+) e 3-fenilpropanoici (o)
CO
OH
X
CO
OH
X
CO
OH
NO2
X
OHX
Acido SolventeTemp.(°C) ρ
25
25
25
25
1.000
1.957
0.905
2.113
H2O
CH3CH2OH
H2O
H2O
Valori di ρ per la dissociazione di alcune famiglie di acidi
Le correlazioni lineari in seguito all'effetto di variazioni strutturali sull'equilibrio o sulla velocità non sono limitati alle reazioni di trasferimento del protone.

36
Esempi: il log della costante di velocita della reazione tra esteri e trietilammina:
+ N(CH3)3MeOH
100°C
..... .- + +N(CH3)4
è stato correlato con il log della costante di dissociazione degli acidi corrispondenti:
+ H2O..... .- + H3O+
R COH
O
R CO
O
CH3
R CO
O
R CO
O
Le correlazioni di GettlerEsiste una correlazione tra il log delle costanti di velocità di reazioni dei chetonicon vari nucleofili all'N ed il log della costante di velocità della reazione con idrossilammina degli stessi chetoni:
asse X:+C O
R'
R"NH2 OH
kC N
R'
R"OH ossima
asse Y: C OR'
R"+ NH2 NH C NH2
OC N
R'
R"NH C NH2
O
semicarbazone(S)
+tiosemicarbazone
(T)
+guanidilidrazone
(G)
C NR'
R"NH C NH2
S
NH2 NH C NH2
S
C NR'
R"NH C NH2
NH
NH2 NH C NH2
NH
C OR'
R"
C OR'
R"
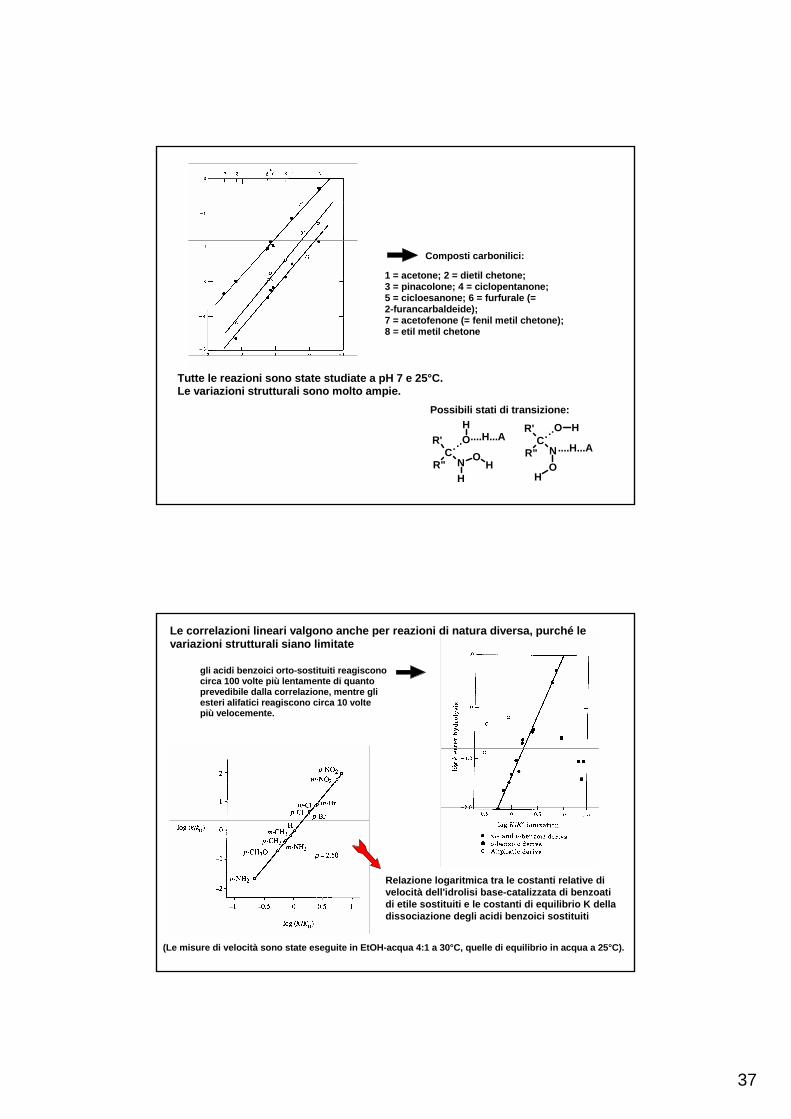
37
Composti carbonilici:
1 = acetone; 2 = dietil chetone;3 = pinacolone; 4 = ciclopentanone;5 = cicloesanone; 6 = furfurale (=2-furancarbaldeide);7 = acetofenone (= fenil metil chetone);8 = etil metil chetone
Possibili stati di transizione:
COR'
R" N
H
O
HH
.......H...A C
OR'
R" N
H
OH
.......H...A
Tutte le reazioni sono state studiate a pH 7 e 25°C.Le variazioni strutturali sono molto ampie.
gli acidi benzoici orto-sostituiti reagisconocirca 100 volte più lentamente di quantoprevedibile dalla correlazione, mentre gliesteri alifatici reagiscono circa 10 voltepiù velocemente.
Relazione logaritmica tra le costanti relative di velocità dell'idrolisi base-catalizzata di benzoatidi etile sostituiti e le costanti di equilibrio K della dissociazione degli acidi benzoici sostituiti
(Le misure di velocità sono state eseguite in EtOH-acqua 4:1 a 30°C, quelle di equilibrio in acqua a 25°C).
Le correlazioni lineari valgono anche per reazioni di natura diversa, purché levariazioni strutturali siano limitate

38
CO
O CH2CH3
XC
O
O
X
CO
OH
XH2O
CH3CH2OH+ OHk
+
+ CO
O
X+
KOH3
+
log k = ρ log K + Clog ko= ρ log Ko+ C log k
ko= ρσ
log K
Ko= ρσ
costante del SOSTITUENTE
costante della REAZIONE
σ
ρ
σ misura la grandezza dell'effetto elettronico del sostituente
Gruppi ad attrazione elettronica KX/Ko > 1 σ > 0Gruppi a rilascio elettronico KX/Ko < 1 σ < 0
σ viene dalla combinazione di effetto induttivo e coniugativo. L'effetto induttivo èpraticamente lo stessoo in meta ed in para. L'effetto coniugativo è molto maggiorein para. Per i sostituenti con effetto coniugativo a rilascio elettronico (-NH2, -OH, alogeno) σp < σmCon gli alogeni (forte effetto induttivo ad attrazione elettronica): σm > 0, σp > 0
Sostituenti elettronattrattori sia per effetto induttivo che coniugativo (-CO2R, SO2R, NO2) hanno σp > σm > 0
ρ è una misura della sensibilità di una reazione agli effetti del sostituente
Se la velocità di una reazione è aumentata dai sostituenti ad attrazione elettronicae rallentata da quelli a rilascio elettronico:
k > ko con σ > 0, k < ko con σ < 0,
ρ > 0
Nel caso di reazioni favorite da sostituenti a rilascio elettronico e sfavorite da quelli ad attrazione: k < ko con σ > 0,
k > ko con σ < 0, ρ < 0
Il segno di ρ è indicativo di una variazione di carica sul centro di reazione

39
ArCO2H ArCO2- + H+
ArCH2CO2H ArCH2CO2- + H+
ArCH2CH2CO2H ArCH2CH2CO2- + H+
ArOH ArO- + H+
ArNH3+ ArNH2 + H+
ArCH2NH3+ ArCH2NH2 + H+
ArCO2Et + OH- ArCO2- + EtOH
ArCH2CO2Et + OH- ArCH2CO2- + EtOH
ArCH2CH2CO2Et + OH- ArCH2CH2CO2- + EtOH
ArCO2Me + PhNH2 ArCONHPh + MeOH
ArCO2Et + H2O ArCO2H + EtOH
ArCO2H + MeOH ArCO2Me + H2O
ArCO2H + Ph2CN2 ArCO2CHPh2 + N2ArCONH2 + OH- ArCO2
- + NH3
ArCONH2 + H2O ArCO2H + NH3
H2OEtOH
H2O
ArCOCl + MeOH ArCO2Me + HCl
1.
H2O
H2O
H2O
H2O
EtOH 87.8%EtOH 87.8%EtOH 87.8%
PhNO2EtOH 60%
MeOH
EtOH
H2O
H2O
MeOH
25
No. Reazione Solvente °C ρ
25
25
25
2525
25
30
3030
80
100
25
30
100
100
0
1.0001.96
0.490.21
0.472.11
2.77
0.72
ArCH=CHCO2H ArCH=CHCO2- + H+
H2O
25
2.430.82
0.59
0.52
0.14
-0.23
0.94
1.07
0.12
1.47
2.
3.
4.
5.
6.7.
8.9.
10.
11.
12.
13.
14.
15.
16.
17.
H+
H+
H+
|ρ| è tanto più grande quanta più carica si sviluppa nello stato di transizione
ArCHO + NH2NHCONH2 ArCH=NNCONH2 + H2O
ArCH2Cl + H2O ArCH2OH + HCl
Ar(Ph)CHCl + EtOH Ar(Ph)CHOEt + HCl
ArO- + EtI ArOEt + I-
ArNMe2 + MeI ArNMe3+ I-
ArH + NO2+ ArNO2 + H+
ArBr + (CH2)5NH (CH2)5NAr + HBr
ArCO2Et + H2O ArCO2H + EtOH
ArCH3 + Br2 ArCH2Br + HBr
pH 1.75; EtOH 25%18.
Me2CO 90%
PhH
EtOH
25
No. Reazione Solvente °C ρ
25
25
35
30
18
80
120
25
99
0.910.07
-3.30
-2.78
-5.93
-0.73
-0.62
pH 7.0; EtOH 25%
19. EtOH 47.7% -2.18
20. -5.09
21. EtOH 42.4 -0.99
22. ArOH + MeCOBr MeCO2Ar + HBr MeCO2Et 0 -1.45
23.
24. ArNH2 + PhCOCl PhCONHAr + HCl PhH
25.
26.
27.
28.
29.
30.
31. + diossano
PhBr
Ac2O
4.87
CCl480 -1.39CCl4
40 -0.76
ArCO2Et + H2O ArCO2H + CH2=CH2 gas 515 0.20
+ Ar-CH=CH2
Ar O
O
O
O
Ar O
O
C OPh
Ph
OPhPh
Ar

40
ρ dipende anche dal solvente:
ArCO2H ArCO2- ρEtOH = 1.96, ρH2O = 1,00
La solvatazione dell'anione è meno buona in EtOH rispetto all'acqua eperciò l'effetto del sostituente si sente di più.
Gli effetti elettronici si attenuano se ci sono atomi di C tra l'anello aromatico sosti-tuito ed il centro di reazione.
Considerando il valore medio delle costanti ρ per alcune reazioni dei compostiX-C6H4-Y-CO2R, sono stati calcolati i seguenti valori di attenuazione per vari Y:
Y = CH2 0.43Y = CH2CH2 0.22Y = CH=CH 0.48Y = C=C 0.39Y = p-C6H4 0.24
In pratica: dai valori di k (o K) e le costanti note dei sostituenti, σ, si calcola ρ per la reazione. Poi, conoscendo ρ della reazione, si possono ottenere i valori σ per nuovi sostituenti.
? Perché correlazioni lineari di energia libera?
KK0
( )B
B
B BΔG e ΔG0 sono le variazioni di energia libera degli equilibri degli acidi benzoici
log KK0
( ) = ρσ = ρ log KK0
( )B
B
dove: K costante di equilibrio per l'equilibrio in esameK B costante di equilibrio per la ionizzazione degli acidi benzoici
moltiplicando entrambi i termini per -2.303 RT
-2.303 RT log KK0
( ) = -2.303 RT ρ log
siccome -2.303 RT log K = ΔG
ΔG e ΔG0 sono le variazioni di energia libera degli equilibri studiati
ΔG di un composto sostituito, che dà una certa reazione, è linearmenteproporzionale al valore ΔG dell'equilibrio di ionizzazione dell'acidobenzoico sostituito con lo stesso sostituente
B
ΔG - ΔG0 = ρ (ΔG - ΔG0 )B B
ΔG = ρ ΔG + (ΔG0 - ρ ΔG0 )B B

41
Analogamente, per le costanti di velocità:
log k = log ( RTNh ) ΔG /=
2.303 RT -
/= /=ΔG - ΔG0 = ρ (ΔG - ΔG0 )B B
ΔG = ρ ΔG + (ΔG0 - ρ ΔG0 )B B/= /=
L'energia libera di attivazione, ΔG , di una data reazione è correlata linearmente con la variazione di energia libera della ionizzazione degliacidi benzoici sostituiti
/=
ΔG0/=ΔG - /= = - 2.303 ρ RT log K
K0( )B
B
= -log kk0
( ) = ρ log KK0
( )B
BΔG0/=
2.303 RTΔG - /=
Un dato sostituente esercita lo stesso effetto (stabilizzante o destabi-lizzante), sia sugli stati reagenti dei sistemi in equilibrio, sia sugli statidi transizione
L' equazione di Hammett e quelle analoghe si basano perciò sulla interdipendenzalineare delle variazioni di energia libera
LFER (linear free energy relationship)
Le correlazioni lineari di energia libera si basano su osservazioni empiri-che e non si possono derivare né dai principi della termodinamica, né dalla teoria della cinetica di reazione
Le LFER sono valide per serie di reazioni isocinetiche, oppure isoentropiche, oisoentalpiche.
se una serie di reazioni di composti analoghi segue una LFER,è una prova che le reazioni hanno lo stesso meccanismo
APPLICAZIONE DELL'EQUAZIONE DI HAMMETT A REAZIONI MULTISTADIOLa costante di velocità determinata sperimentalmente per reazioni multistadio (kexp) è di solito una combinazione di varie costanti
di conseguenza ρexp è una combinazione di varie ρ
non sempre utile- Approssimazione PEA
Per il composto sostituito: kexp = K1k2
per il composto non sostituito: kexp0 = K1
0k20
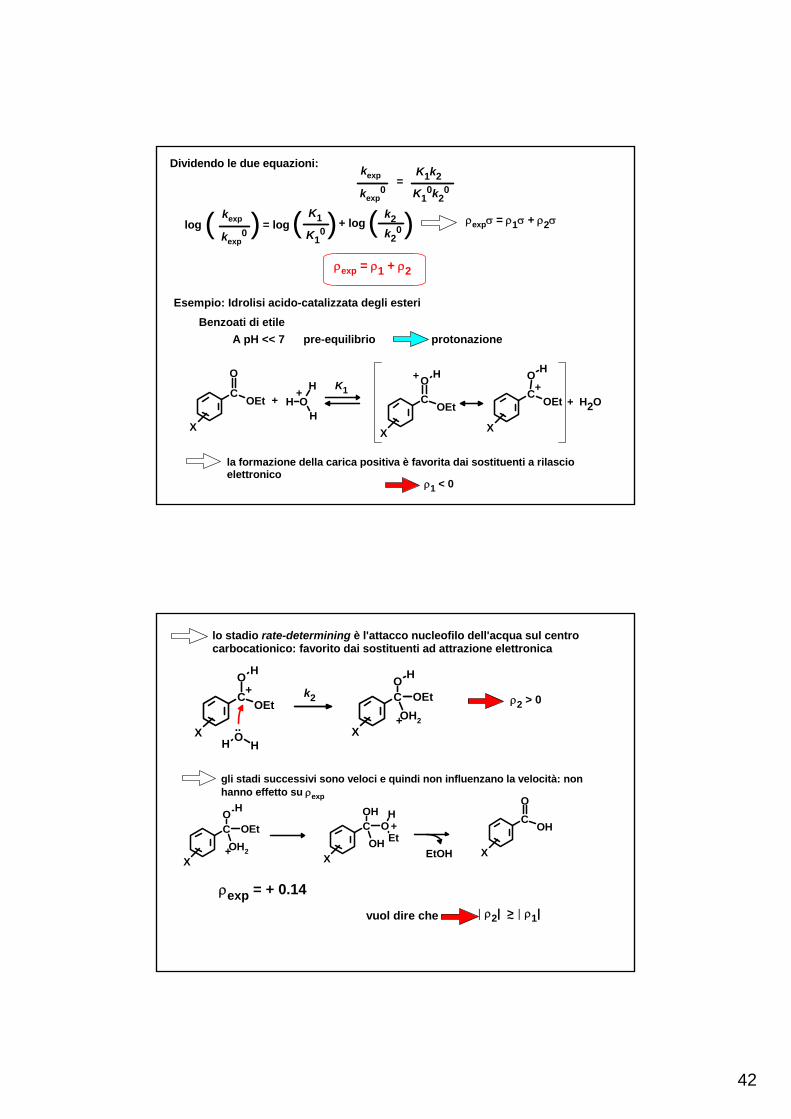
42
Dividendo le due equazioni: kexp
kexp0
= K1k2
K10k2
0
log ( )kexp
kexp0
= log( K1
K10)+ log( k2
k20) ρexpσ = ρ1σ + ρ2σ
ρexp = ρ1 + ρ2
Esempio: Idrolisi acido-catalizzata degli esteri
A pH << 7 pre-equilibrio protonazioneBenzoati di etile
+ + K1 ++
+ H2O
la formazione della carica positiva è favorita dai sostituenti a rilascioelettronico
ρ1 < 0
COEt
O
X
OH
HH C
OEt
O H
X
COEt
O H
X
lo stadio rate-determining è l'attacco nucleofilo dell'acqua sul centrocarbocationico: favorito dai sostituenti ad attrazione elettronica
+
OHH
..
k2
+
ρ2 > 0COEt
O H
X
C OEtO H
OH2X
gli stadi successivi sono veloci e quindi non influenzano la velocità: nonhanno effetto su ρexp
+
+
EtOH
C OEtO H
OH2X
C OOH
OH Et
H
X
COH
O
X
ρexp = + 0.14vuol dire che | ρ2| >~ | ρ1|

43
- Approssimazione SSA
La forma più semplice di kexp è:kexp =
k1k2k-1 + k2
Se k1, k-1 e k2 rispondono in modo diverso al sostituente
log (kexp/kexp0) non è lineare al variare di σ non vale Hammett
kexp ~k1k2~
k2
kexp
kexp0log ( )= k1 = log
k1( )k10
Assumiamo che all'estremità di destra, cioè con σ >>0, sia k-1 << k2
ρexp = ρ1
all'estremità sinistra, cioè con σ <<0, sarà k-1 >> k2
kexp k1k2k-1
~~ = K1k2 ρexp = ρequ + ρ2
Se ρ1, ρequ e ρ2 hanno valori diversi, il grafico di Hammett avrà una curvatura nei pressi di σ = 0
Esempio: formazione dei semicarbazioni
+ H2NNH-CO-NH2k1
k-1
+ H+ + H3O+
In soluzione acida lo stadio più lento è la formazione dell'addotto
k2
k1
CHNHNH-CO-NH2
OH
X
CH N NH C NH2
O
C H
O
CHNHNH-CO-NH2
OH
X
X X
grafici con la concavità verso il basso sono diagnostici di un cambiamentodello stato rate-determining entro lo stesso meccanismo

44
L'attacco nucleofilo della semicarbazide sull'aldeide è favorito dai gruppiad attrazione elettronica e quindi si aspetta ρexp positivo
ρexp ~ ρ1
a pH 1.75 ρexp = 0.91 = ρ1
A pH 7 la formazione dell'addotto è un equilibrio rapido e lo stadio rate-determining diventa la disidratazione
ρexp = ρequ + ρ2
k2
ρequ ha un valore positivo (1.81) e ρ2 un valore negativo (-1.74): in parte sicompensano e quindi ρexp ha un valore piccolo
La reazione è favorita dai gruppi a rilascio elettronico e quindi si aspetta ρ2 negativo
ρexp = 1.81 - 1.74 = 0.07
In soluzione neutra la reazione è pochissimo influenzata dai sostituenti sull'anello aromatico
Ad un valore di pH intermedio tra 1.75 e 7 non si possono usare le semplificazionidi SSA
a pH 3.9 che lo stadio rate-determining sia la formazione dell'addotto (k1)o la disidratazione (k2) DIPENDE DAL SOSTITUENTE
Con i sostituenti a rilascio elettronico la disidratazione è veloce ρexp > 0
ρexp = ρequ + ρ2
Con i sostituenti ad attrazione elettronica la disidratazione è lenta
piccolo

45
- Reazioni parallelekexp = k1 + k2
Se k1 e k2 rispondono diversamente ai sostituenti, ci si deve aspettare una curvatura nel grafico di Hammett
Per esempio, assumiamo che sia k1 >> k2 a σ << 0k1 << k2 a σ >> 0
k1 >> k2
k1 << k2
si ottengono due casi limite per kexp/kexp0
k1
k10 + k2
0=
k1
k10 ( )1+
k20
k10
-1
k2
k10 + k2
0=
k2
k20 ( )1+
k10
k20
-1kexp
0
kexp k1 + k2
k10 + k2
0= =
passando ai logaritmi:
k2
k20 1+
k10
k20
log ( ) - log ( )
k1 >> k2
k1 << k2
=logkexp
0
kexp )(1+
k20
k10
logk1
k10( ) - log ( )
logkexp
0
kexp )( =
k1 >> k2
k1 << k2
ρ1σ - costante
ρ2σ - costante'
Se ρ1 e ρ2 sono diversi, il grafico di Hammett ha una curvatura, con la concavitàRIVOLTA VERSO L'ALTO
Esempio: idrolisi di benzoati di etile in H2SO4 99.9%L'acqua è in concentrazione molto bassa (0.1%) ed è completamenteprotonata. in queste condizioni non sono operativi i meccanismi chesi hanno in soluzione acquosa
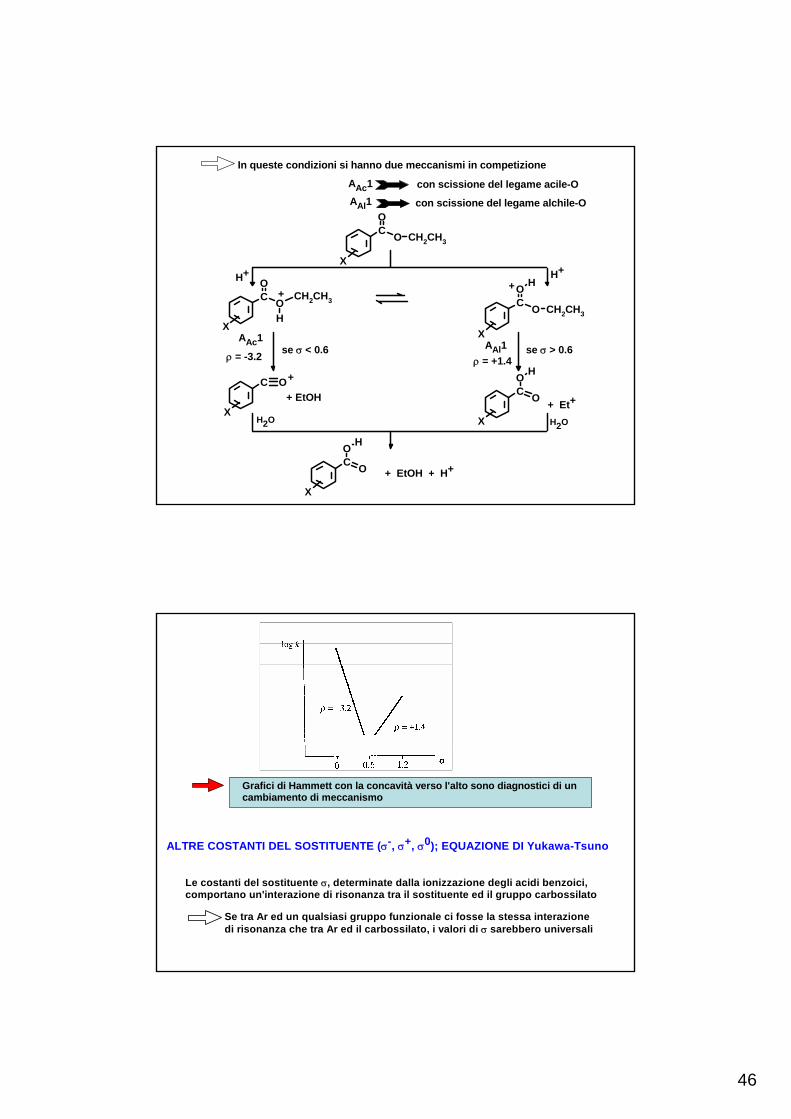
46
In queste condizioni si hanno due meccanismi in competizione
AAc1 con scissione del legame acile-OAAl1 con scissione del legame alchile-O
H+ H+
AAl1AAc1
ρ = -3.2 ρ = +1.4se σ < 0.6 se σ > 0.6
+ Et+
C O
O
CH2CH3
X
C O
OCH2CH3
HX
+C O
O
CH2CH3
H
X
+
C O
X
+
+ EtOH C O
O H
X
C O
O H
X
H2OH2O
+ EtOH + H+
Grafici di Hammett con la concavità verso l'alto sono diagnostici di uncambiamento di meccanismo
ALTRE COSTANTI DEL SOSTITUENTE (σ-, σ+, σ0); EQUAZIONE DI Yukawa-Tsuno
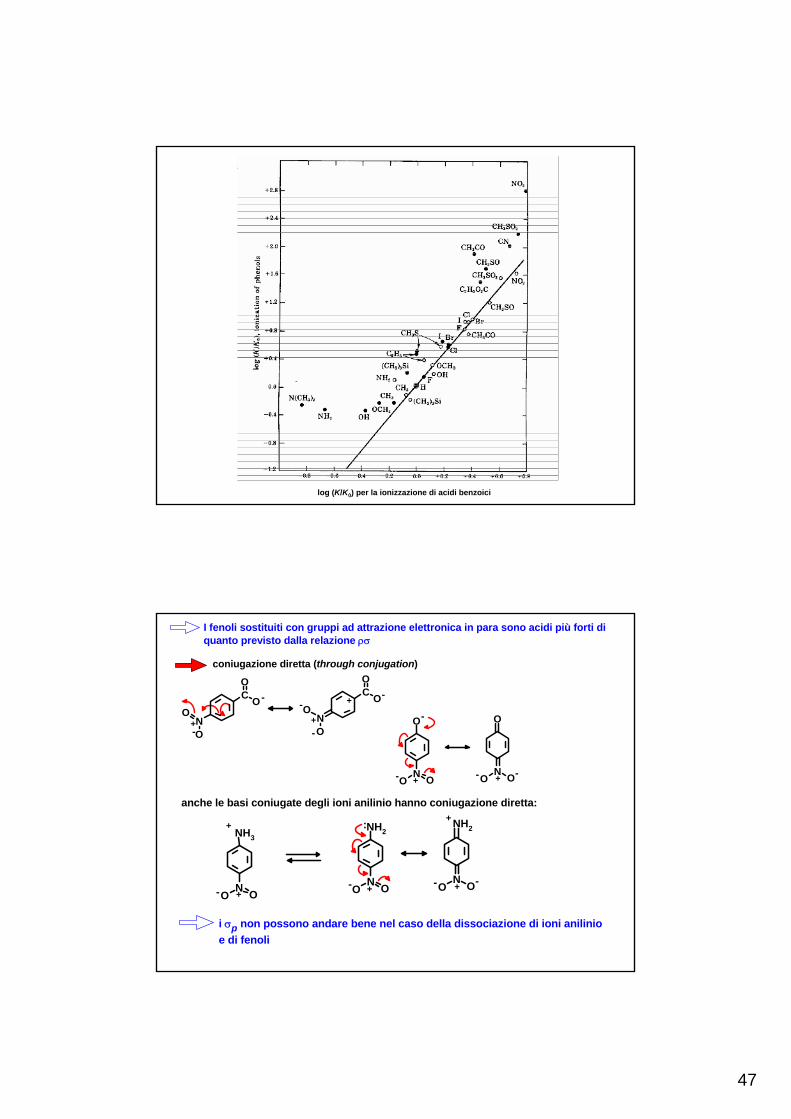
Le costanti del sostituente σ, determinate dalla ionizzazione degli acidi benzoici, comportano un'interazione di risonanza tra il sostituente ed il gruppo carbossilato
Se tra Ar ed un qualsiasi gruppo funzionale ci fosse la stessa interazione di risonanza che tra Ar ed il carbossilato, i valori di σ sarebbero universali
47
log (K/K0) per la ionizzazione di acidi benzoici
I fenoli sostituiti con gruppi ad attrazione elettronica in para sono acidi più forti di quanto previsto dalla relazione ρσ
coniugazione diretta (through conjugation)
-+
--
-
+ -
+ -
- + + --
C
NO
O
O
OC
NO
O
O
O
NO O
O
NO O
O
anche le basi coniugate degli ioni anilinio hanno coniugazione diretta:
NO O
NH2
- + +N
O O
NH2
--
: +
NO O
NH3+
+-
i σp non possono andare bene nel caso della dissociazione di ioni anilinio e di fenoli
48
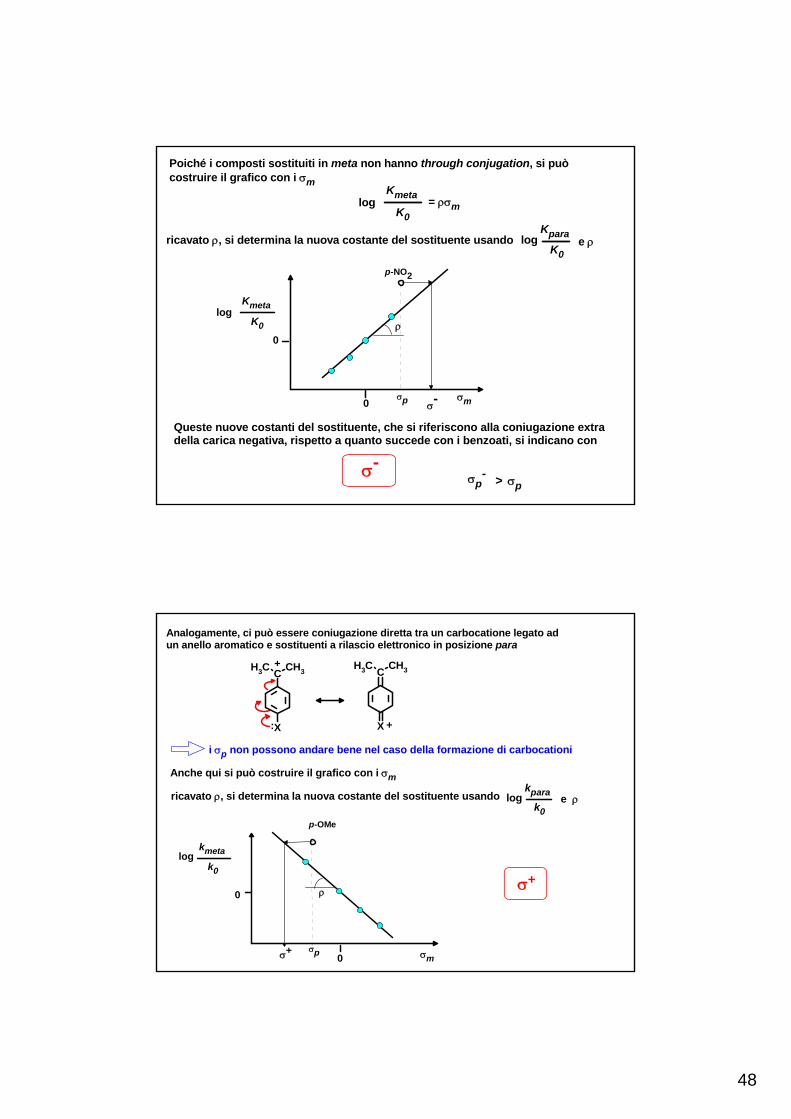
Poiché i composti sostituiti in meta non hanno through conjugation, si puòcostruire il grafico con i σm
logKmeta
K0= ρσm
ricavato ρ, si determina la nuova costante del sostituente usando log e ρKpara
K0
logKmeta
K0
σm0
0ρ
p-NO2
σp σ-
Queste nuove costanti del sostituente, che si riferiscono alla coniugazione extradella carica negativa, rispetto a quanto succede con i benzoati, si indicano con
σ-σp
- > σp
Analogamente, ci può essere coniugazione diretta tra un carbocatione legato adun anello aromatico e sostituenti a rilascio elettronico in posizione para
+
: +
C CH3CH3
X
C CH3CH3
X
i σp non possono andare bene nel caso della formazione di carbocationi
Anche qui si può costruire il grafico con i σm
ricavato ρ, si determina la nuova costante del sostituente usando log e ρkpara
k0
σ+ρ
σm0
0
p-OMe
σpσ+
logkmeta
k0
49
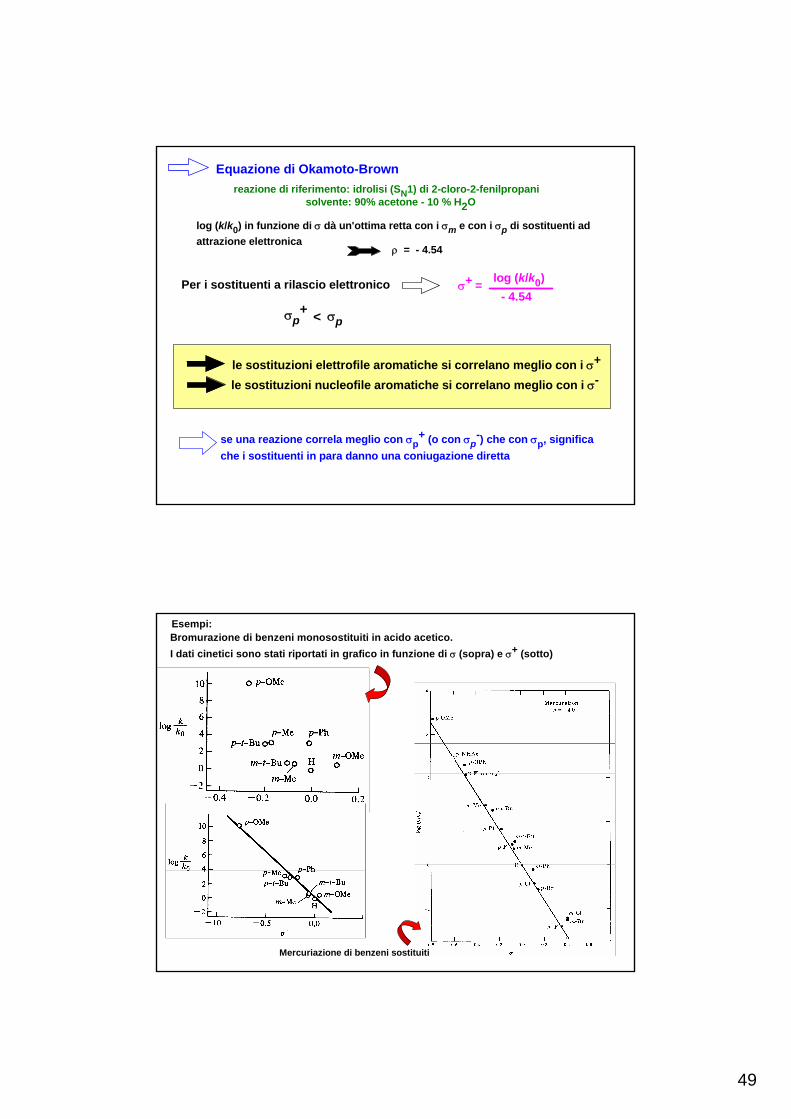
Equazione di Okamoto-Brownreazione di riferimento: idrolisi (SN1) di 2-cloro-2-fenilpropani
solvente: 90% acetone - 10 % H2O
log (k/k0) in funzione di σ dà un'ottima retta con i σm e con i σp di sostituenti adattrazione elettronica
ρ = - 4.54
Per i sostituenti a rilascio elettronico σ+ = log (k/k0)- 4.54
σp+ < σp
le sostituzioni elettrofile aromatiche si correlano meglio con i σ+
le sostituzioni nucleofile aromatiche si correlano meglio con i σ-
se una reazione correla meglio con σp+ (o con σp
-) che con σp, significache i sostituenti in para danno una coniugazione diretta
Esempi:Bromurazione di benzeni monosostituiti in acido acetico. I dati cinetici sono stati riportati in grafico in funzione di σ (sopra) e σ+ (sotto)
Mercuriazione di benzeni sostituiti

50
La coniugazione diretta è un fenomeno piuttosto diffuso (anche negli acidibenzoici), ma con estensione diversa in reazioni diverse
:
-
+
E' sorprendente che bastino i σ ed i σ- o i σ ed i σ+ per correlare moltidati cinetici
C OH
O
X
C OH
O
X
Equazione di Yukawa-TsunoYukawa e Tsuno hanno proposto una relazione più generale.
Dalle costanti di equilibrio della reazione in acido solforico:
+ 2 H2SO4+ + H3O+ + HSO4
-
sono state ricavate delle costanti del sostituente, che sono state riportate ingrafico in funzione delle costanti ricavate per l'alogenazione di benzeni sosti-tuiti, in AcOH
X X
OHC CX X
X X
La relazione è stata espressa con l'equazione:= ρ {σ + r (σ+ - σ)}( )k0
klog
r (σ+ - σ)} fattore di correzione, che misura l'estensione della coniugazione diretta
se r = 0 log (k/k0) = ρσ
se r = 1equazione di Hammett
log (k/k0) = ρσ+ equazione di Okamoto-Brown
Non c'è nessuna relazione tra ρ e r: ρ grande non comporta necessaria-mente r grande.
Yukawa e Tsuno hanno verificato l'applicabilità della loro relazione con le reazionidi sostituzione elettrofila aromatica, ottenendo in molti casi una correlazionemigliore che con ρσ+
ArH + Br2AcOH, 25°C ArBr + HBr ρ = -8.8; r = 1.7
+ AcCl ρ = -2.5; r = 1.6C CAr
Ar
OH OH
ArAr
C CAr
Ar
Ar O
Ar

51
Una reazione analoga è stata applicata con successo a reazioni con reagenti nucleofili:
= ρ {σ + r (σ- - σ)}( )k0
klog
Esempio: Reazione del cloruro di picrile con aniline meta- e para-sostituite
σ ; σ- ;0.76 σ- + 0.24σ
Poiché i valori originali di σ devono avere almeno un residuo di coniugazionediretta, sono stati fatti tentativi di ottenere costanti del sostituente privi ditale contributo
σo
Taft ha determinato questi valori dalle costanti di ionizzazione di acidi fenilaceticie fenilpropanoici e dalle costanti di velocità dell'idrolisi dei rispettivi esteri etilici
Problemi data la separazione tra sostituente e centro di reazione,ρ piccoli ed accuratezza inferiore
Per i sostituenti in para che donano elettroni per risonanza σo > σ
conferma della presenza di coniugazione diretta residua negli acidi benzoici
Oggi si cerca il modo di descrivere gli effetti dei sostituenti con metodi computazionali. Non è facile trovare un descrittore che funzioni bene.
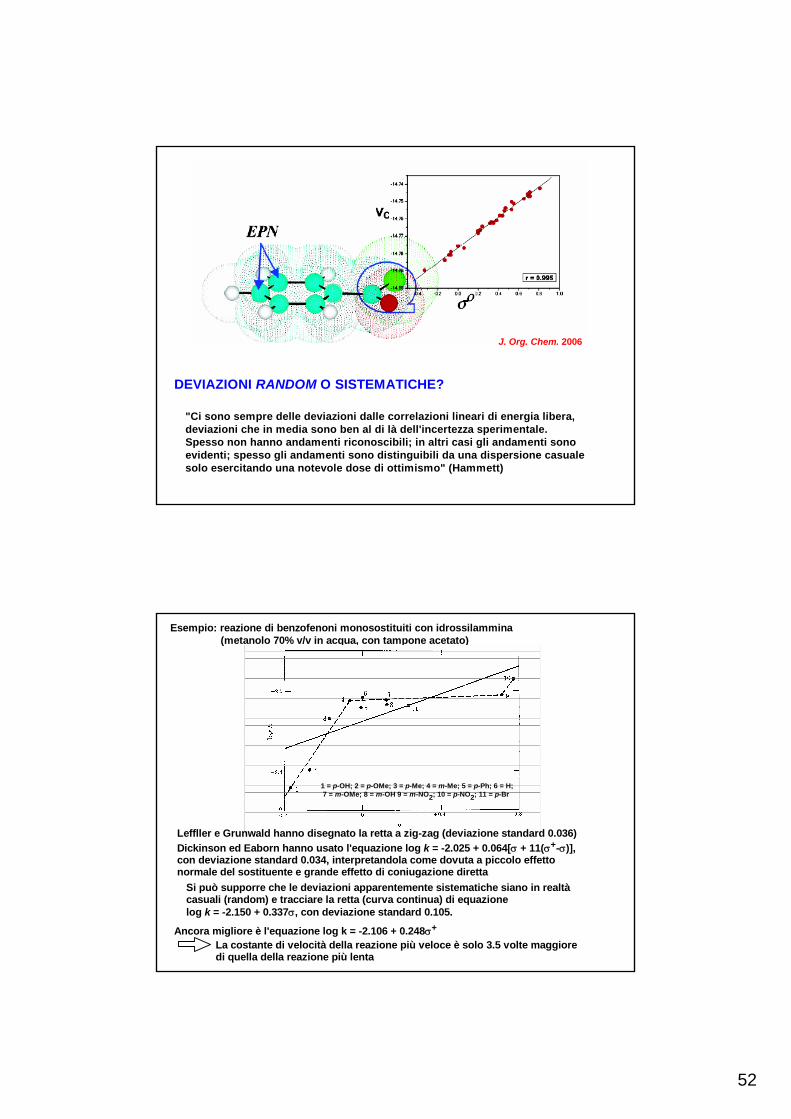
Un esempio recente utilizza i valori dei potenziali elettrostatici (EPN) agli atomi di carbonio nelle posizioni orto e para di un benzene sostituito, calcolati con metodo DFT (density functional theory)
52
DEVIAZIONI RANDOM O SISTEMATICHE?
"Ci sono sempre delle deviazioni dalle correlazioni lineari di energia libera,deviazioni che in media sono ben al di là dell'incertezza sperimentale.Spesso non hanno andamenti riconoscibili; in altri casi gli andamenti sonoevidenti; spesso gli andamenti sono distinguibili da una dispersione casualesolo esercitando una notevole dose di ottimismo" (Hammett)
J. Org. Chem. 2006
Esempio: reazione di benzofenoni monosostituiti con idrossilammina (metanolo 70% v/v in acqua, con tampone acetato)
1 = p-OH; 2 = p-OMe; 3 = p-Me; 4 = m-Me; 5 = p-Ph; 6 = H; 7 = m-OMe; 8 = m-OH 9 = m-NO2; 10 = p-NO2; 11 = p-Br
Leffller e Grunwald hanno disegnato la retta a zig-zag (deviazione standard 0.036)Dickinson ed Eaborn hanno usato l'equazione log k = -2.025 + 0.064[σ + 11(σ+-σ)],con deviazione standard 0.034, interpretandola come dovuta a piccolo effettonormale del sostituente e grande effetto di coniugazione diretta
Si può supporre che le deviazioni apparentemente sistematiche siano in realtàcasuali (random) e tracciare la retta (curva continua) di equazionelog k = -2.150 + 0.337σ, con deviazione standard 0.105.
Ancora migliore è l'equazione log k = -2.106 + 0.248σ+
La costante di velocità della reazione più veloce è solo 3.5 volte maggioredi quella della reazione più lenta

53
SEPARAZIONE DEGLI EFFETTI INDUTTIVO E DI RISONANZA
- Equazione di Taft a due parametriPer descrivere separatamente gli effetti induttivo (polare) e di risonanza (mesomerico), Taft ha introdotto un'equazione con due costanti del sostituente
log k0
k = ρIσI + ρRσR
σI
ρI
è la costante del sostituente che ne descrive l'effetto induttivoσR
ρR
è la costante del sostituente che ne descrive l'effetto coniugativoè la costante della reazione che misura la sensibilità della reazioneall'effetto induttivoè la costante della reazione che misura la sensibilità della reazioneall'effetto coniugativo
σI e σR sono numericamente gli stessi per i sostituenti meta e para
l'equazione di Taft si deve applicare separatamente ai compostimeta e para sostituiti: cambia la sensibilità
ρIm ρR
pρIp ρR
m, , ,
Per determinare sono stati usati sistemi senza coniugazione σI
Da misure di pKa di acidi biciclo[2.2.2]-ottancarbossilici, sostituiti in 4,in acqua ed in etanolo-acqua 1:1 v/v
-+ H+
X COH
OX C
O
O
geometria simile a quella degli acidi benzoici, con solo effetto induttivo

54
Per calcolare i valori di σI si possono usare sia i valori di pKa determinatiin acqua, sia quelli determinati in etanolo
ρ = 1 in H2O a 25°Cρ = 1.56 in EtOH/H2O 1:1 v/v a 25°C
σI =ΔpKa
ρ
I valori di σI per altri gruppi sono stati determinati usando i pKa di acidi aceticisostituiti in α
+ H+-
σI = 0.246 (pKa0- pKa)
X CH2CO
OX CH2C
O
OH
I valori di σI per altri gruppi sono stati determinati dai chemical shifts di 19F NMRdi fluorobenzeni meta sostituiti
55
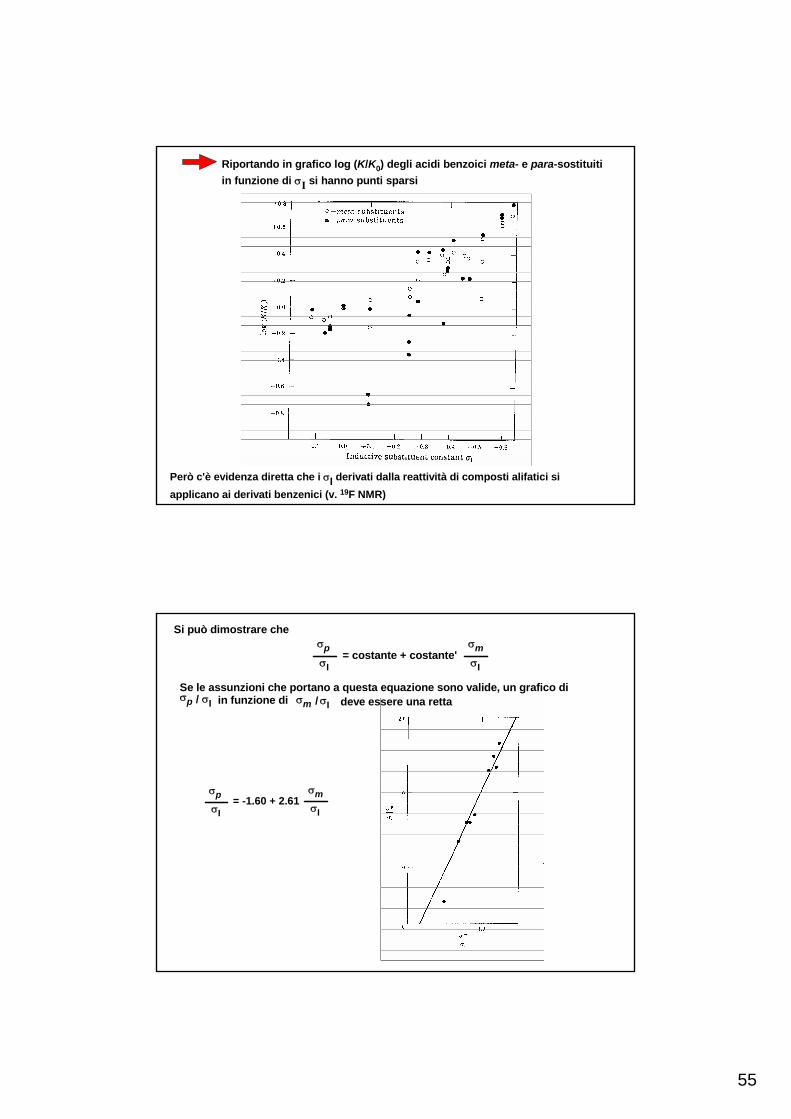
Riportando in grafico log (K/K0) degli acidi benzoici meta- e para-sostituitiin funzione di σI si hanno punti sparsi
Però c'è evidenza diretta che i σI derivati dalla reattività di composti alifatici siapplicano ai derivati benzenici (v. 19F NMR)
σpσI
= -1.60 + 2.61σmσI
Si può dimostrare cheσpσI
= costante + costante'σmσI
Se le assunzioni che portano a questa equazione sono valide, un grafico di σp σI σm σI/ in funzione di / deve essere una retta
56
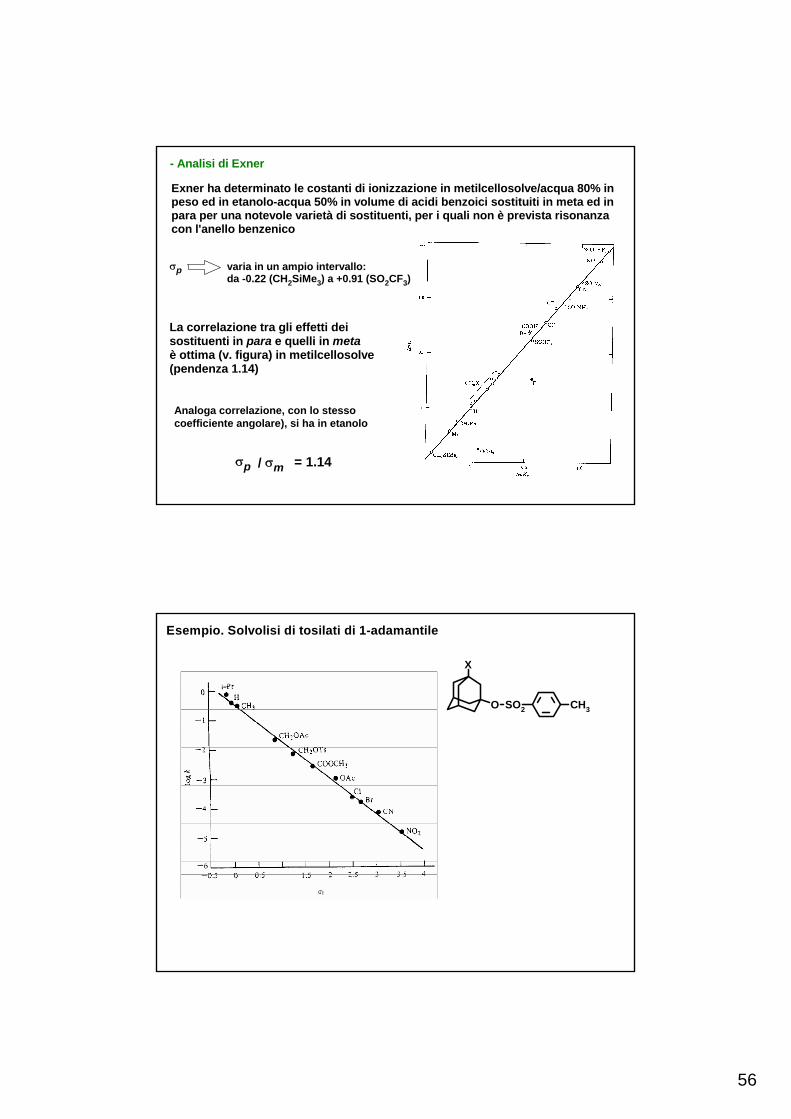
- Analisi di Exner
Exner ha determinato le costanti di ionizzazione in metilcellosolve/acqua 80% inpeso ed in etanolo-acqua 50% in volume di acidi benzoici sostituiti in meta ed inpara per una notevole varietà di sostituenti, per i quali non è prevista risonanzacon l'anello benzenico
σp / σm = 1.14
σp varia in un ampio intervallo: da -0.22 (CH2SiMe3) a +0.91 (SO2CF3)
La correlazione tra gli effetti dei sostituenti in para e quelli in metaè ottima (v. figura) in metilcellosolve(pendenza 1.14)
Analoga correlazione, con lo stesso coefficiente angolare), si ha in etanolo
Esempio. Solvolisi di tosilati di 1-adamantile
O
X
SO2 CH3

57
Esempio. Correlazione della basicità di 3,7-diazabiciclo[3.3.1]nonani con σI
NHNH
una correlazione con tre punti non è significativa !!
J. Org Chem., 2006

58
Assunzioni di Taft: 1. gli effetti induttivo e coniugativo sono additivi2. l'effetto induttivo di un sostituente è praticamente lo stesso in meta ed in para3. l'effetto coniugativo in meta è considerevolmente più piccolo che in para
Le tre condizioni sono incorporate nelle equazioni:σp = σI + σRσm = σI + α σR
α e σR si possono calcolare da σp, σm e σI
poiché ci sono quattro serie di σp
σpDa σp-, σp
+, , σp0 σR σR
-, σR+, , σR
0
Quale serie di σR va meglio, si ricava per tentativi, dai graficilog (k/k0) = ρIσI + ρRσR
Per determinare σR0 si possono usare anche metodi spettroscopici
Per la determinazione della costante di risonanza, σR, non è disponibilenessuna reazione (effetto coniugativo sempre insieme all'effetto induttivo)
Dai chemical shifts 13C NMR del C in para (δpC) in un benzene mono-
sostituito rispetto al benzene non sostituito (δC)
ΔδpC = δp
C (C6H5X) - δC (C6H6)
che si possono correlare con i valori σI e σR0 Δδp
C = 4.0 σI + 19.8 σR
0
Dall'intensità dell'assorbimento integrata (A) della banda a circa 1600 cm-1nello spettro IR di benzeni monosostituiti
|σR0| = 0.0075 (A - 100)1/2
il sostituente aumenta il momento dipolare e questo aumenta l'intensità dell'as-sorbimento, sia per sostituenti elettron-attrattori che per sostituenti elettron-donatori (di qui, il valore assoluto)
con questo metodo determinati i σR0 per circa 200 sostituenti
Le costanti del sostituente σI e σR forniscono informazioni sugli effetti induttivo econiugativo che il sostituente esercita sullo scheletro

59
Gruppi in cui il primo atomo è più elettronegativo del C sono ad attrazione elettro-nica per effetto induttivo, ma elettrondonatori per effetto coniugativo.
F, OMe, OAc, NMe2, NHAc σI > 0, σR0 < 0
invece CO2R, CN, NO2, SO2Q σI > 0, σR0 > 0
ALCHILITaft ha concluso (da idrolisi di esteri e da misure di basicità di alcooli ed ammine)che l'effetto induttivo degli alchili cresce nell'ordine:
Me < Et < i-Pr < t-BuσI -0.04 -0.05 -0.06 -0.07
Secondo altri autori l' effetto induttivo è praticamente lo stesso e, poichéil numero è piccolo, si può porre uguale a zero
L'equazione a due parametri di Taft è meno usata dell'equazione di Hammett
Secondo Taft, l'uso della sua equazione ha il vantaggio di distinguere il ruolodegli effetti induttivo e coniugativo
fornisce una "regolazione fine" alla comprensione degli effettielettronici nei dettagli della reazione
Esempio: Esterificazione degli acidi benzoici para-sostituiti in MeOH a 25°C
C OH
OC OH
OH+
MeOH C OHOH
OH
CH3+
H+
ecc.
ρI = -0.53; ρR = +0.19X X X
ρR > 0
ρI < 0 negli intermedi si sviluppa una carica positiva
la coniugazione è in grado di stabilizzare il secondo intermediopositivo meno di quanto stabilizzi l'acido benzoico
Esempio: decomposizione termica dei sali di diazonio
non si può descrivere con l'equazione di Hammetti sali di diazonio sostituiti in para deviano notevolmente dalla linearità
l'equazione di Taft dà una buona correlazione se si usano σR+
per i composti meta sostituiti ρI = -4.5; ρR = -1.8
per i composti para sostituiti ρI = -3.7; ρR = +2.4
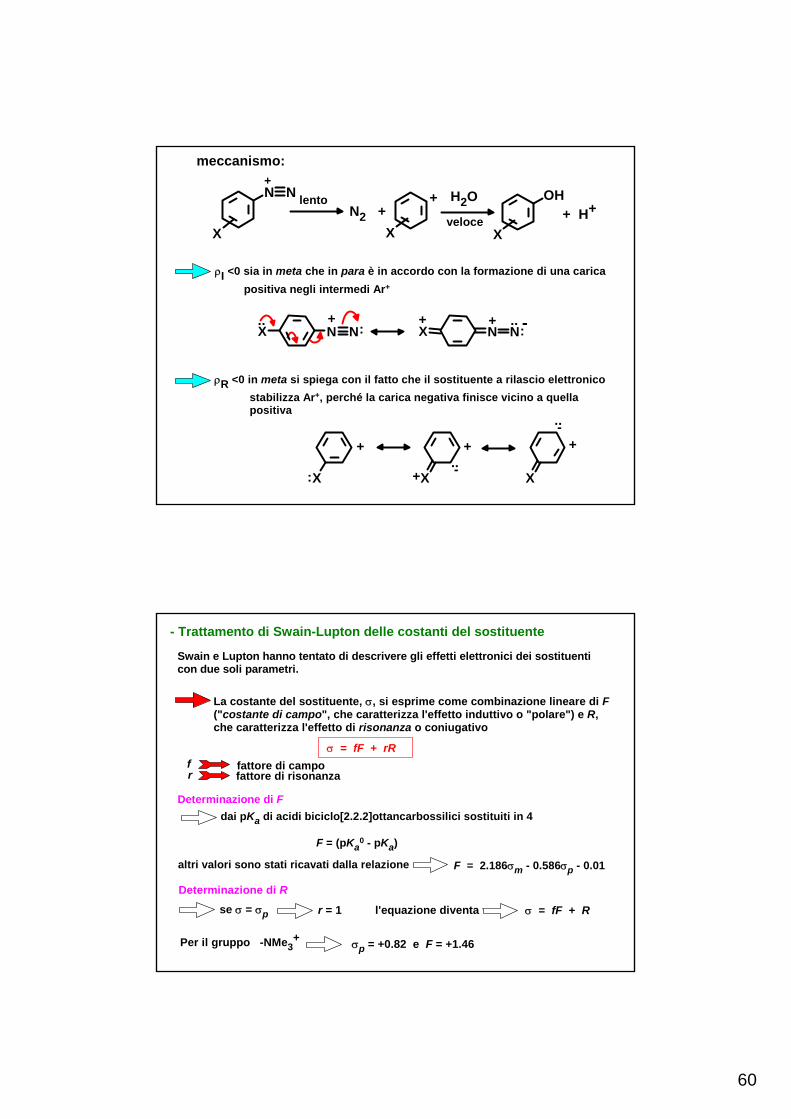
60
meccanismo:+
lentoN2 +
+ H2O
veloce + H+N N
X X
OH
X
ρI <0 sia in meta che in para è in accordo con la formazione di una carica positiva negli intermedi Ar+
+:..
X N N++
:.. -X N N
ρR <0 in meta si spiega con il fatto che il sostituente a rilascio elettronico stabilizza Ar+, perché la carica negativa finisce vicino a quella positiva
X:
+
X+
+
X..-
..-+
- Trattamento di Swain-Lupton delle costanti del sostituente
Swain e Lupton hanno tentato di descrivere gli effetti elettronici dei sostituenti con due soli parametri.
La costante del sostituente, σ, si esprime come combinazione lineare di F ("costante di campo", che caratterizza l'effetto induttivo o "polare") e R,che caratterizza l'effetto di risonanza o coniugativo
σ = fF + rRfr
fattore di campofattore di risonanza
Determinazione di Fdai pKa di acidi biciclo[2.2.2]ottancarbossilici sostituiti in 4
F = (pKa0 - pKa)
altri valori sono stati ricavati dalla relazione F = 2.186σm - 0.586σp - 0.01
Determinazione di Rse σ = σp r = 1 l'equazione diventa σ = fF + R
Per il gruppo -NMe3+ σp = +0.82 e F = +1.46

61
Questo gruppo viene preso come riferimento, assumendo che non abbia contri-buto coniugativo (R = 0.00) e permette di determinare f
f = 0.56
conosciuto f, il valore di R per un qualsiasi sostituente si ricava dal suo valore diσp R = σp - 0.56F
Swain e Lupton hanno messo in relazione F ed R con altre costanti del sostituenteσm = 0.60F + 0.27R + 0.00 (r = 1.00)σp = 0.56F + 1.00R + 0.00 (r = 1.00)σp
0 = 0.60F + 0.70R + 0.01 (r = 0.993)σp
+ = 0.51F + 1.58R - 0.07 (r = 0.939)σp
- = 0.75F + 1.52R + 0.09 (r = 0.970)
Le correlazioni con σm e σp con F e R sono perfette per via della definizione; conσ0 la correlazione è ancora buona, con σ+ e σ- la correlazione è scarsa)
I valori originali di F sono stati corretti, dividendo i ΔpKa ottenuti dagliacidi biciclo[2.2.2]ottancarbossilici per la costante della reazione (ρ =1.65)
I valori di F e di R sono stati espressi in funzione delle costanti di Hammett
Le costanti di Swain e Lupton (F e R) sono state usate per correlare misure NMR e IR, equilibri acido-base, velocità di reazioni elettrofile e nucleofile ed ancheattività biologiche
F = 1.369 σm - 0.373 σp - 0.009
R = σp - 0.921 F
successo variabile
Critiche: 1. la maggior parte dei valori di F non è stata ottenuta da misure dirette di pKa2. R è stato basato sull'assunzione che NMe3
+ non abbia contributo di risonanza.Però σm > σp NMe3
+ ha un piccolo effetto elettronico a rilascioperciò i valori di R non sono validi
3. E' discutibile che un solo insieme di R sia soddisfacente per tutti i tipi di riso- nanza

62
- Equazione di Drago-Dadmun (1993)
Per correlare le misure chimico-fisiche Drago e Dadmun hanno usato l'equazione:
ΔχX - ΔχH = dEΔEX + dCΔCX
dC
ΔEX
ΔCX
dE
costante elettrostatica del sostituentecostante covalente del sostituente
costante covalente della reazionecostante elettrostatica della reazione
ΔχX simbolo dei dati chimico-fisici
Si può applicare a reazioni di un elettrofilo con vari nucleofili o di un nucleofilo con vari elettrofili
Il nuovo insieme di valori ΔEX e ΔCX descrive in modo soddisfacente fenomenicon diversi tipi di contributi di risonanza
SEPARAZIONE DEGLI EFFETTI ELETTRONICI E STERICI
- Equazione di TaftVisto il successo dell'equazione di Hammett per i composti aromatici, sembròragionevole cercare una correlazione analoga sostituente-reattività per i com-posti alifatici
problemi problemi sterici (i sostituenti sono più vicini ai siti di reazione)
Per determinare le costanti del sostituente in sistemi alifatici, Taft ha usato l'idro-lisi di esteri alifatici, sulla base delle seguenti assunzioni.
1. Nei composti alifatici l'effetto coniugativo è trascurabile2. Nel caso dell'idrolisi acido-catalizzata, l'effetto elettronico è trascurabile e la costante di velocità dipende dall'effetto sterico di R in RCO2Et3. La reazione base-catalizzata è influenzata dall'effetto elettronico e dall'effetto sterico. La grandezza dell'effetto sterico è la stessa dell'idrolisi acido-cataliz- zata
Assunzione 1 non richiede giustificazioni, se il sostituente non ha doppi legami che possano coniugare con il sito di reazione

63
Assunzione 2 per giustificarla bisogna conoscere il meccanismo
R C OEt
O+ H3O+ K1
R C OEt
OH
+ H2O+
R C OEt
OH
+ H2O+
R C OEtOH
OH2+
k2
veloce
lentoprodotti
utilizzando l'approssimazione del pre-equilibrio kexpA = K1k2
ρexp = ρ1 + ρ2
ρ1 < 0, ρ2 > 0 ρexp ~ 0 se | ρ1 | ~ | ρ2 |
Questo ragionamento trova sostegno nel valore prossimo a zero di ρ per l'idrolisi acido-catalizzata dei benzoati di etile meta e para sostituiti
Nel caso dell'idrolisi acida degli esteri, il logaritmo del rapporto dellecostanti di velocità si pone uguale al requisito sterico di R
kA k0
A log( ) = ES
ES costante sterica del sostituente
Assunzione 3 si basa sull'esame delle differenze strutturali degli intermedidell'idrolisi in ambiente acido e basico
+C OEtROH2
OHC OEtROH
O-
intermedio dellaidrolisi acida
intermedio dellaidrolisi basica
le due strutture differiscono per due soli protoni
Lo stadio rate-determining dell'idrolisi basica è il primo stadio

64
+ HO-lento
prodotti-k1
kexpB = k1R C OEt
OR C OEt
O
OH
Questa costante di velocità ( ) è influenzata sia dagli effetti elettronici che da quelli sterici
kexpB
kB k0
B log ( ) = ρ*σ* + ES
elettronico sterico
= ρ*σ* -kB k0
B log ( ) kA
k0A
log ( )σ* misura l'effetto elettronico induttivo del sostituenteρ* costante della reazione, che misura la sensibilità della velocità all'effetto
elettronico induttivo del sostituente
k0B k0
A e sono misurati con l'acetato di etile
σ*Me = 0per definizioneCH3CO2Et
kB kA e sono misurati con acetati di etile sostituitireazione di riferimento
CH2CO2EtX
per questa reazione Taft ha scelto arbitrariamente ρ* = 2.46 (in modo da avere σ* ~ σ di Hammett)
σ* = 12.48
-kB k0
B log ( ) kA
k0A
log ( )
Tabella. Costanti induttive del sostituente di Taft
Gruppo σ* Gruppo σ*
Me 0.00 CH2OH +0.55Et -0.10 CH2OMe +0.52Pr -0.10 CH2F +1.10Bu -0.13 CH2Cl +1.05i-Pr -0.19 CH2Br +1.00t-Bu -0.30 CH2I +0.85Ph +0.60 CH2NO2 +1.40PhCH2 +0.215 CH2CN +1.30PhCH2CH2 +0.08 CH2CF3 +0.92OMe -0.22 CH2COCH3 +0.60
In seguito è stata stabilita una relazione tra i valori σI di X ed i valori σ* dei gruppi CH2X
σI(X) = 0.45 σ*(XCH2)

65
La costante sterica, ES, si può determinare dall'equazione
kA log( ) = ESk0A
I valori ES misurano il requisito sterico del sostituente rispetto al metile
ES(Me) = 0Se R è più piccolo di Me ES > 0Se R è più grande di Me ES< 0
Taft ha suggerito, per descrivere gli effetti elettronici induttivi dei sostituenti sureazioni di composti alifatici, l'equazione:
klog( )k0
= ρ*σ*
In molti casi l'equazione dà risultati soddisfacenti
RCO2Et + OH- RCO2- + EtOH ρ* = +2.48
RCO2H + H2O RCO2- + H3O+ ρ* = +1.72
RCH2Br + PhS- RCH2SPh + Br- ρ* = -0.61RR'R"CCl + EtOH RR'R"COEt + HCl ρ* = -3.29
In altri casi, per ottenere una buona correlazione si è dovuta includere la costantesterica del sostituente, ES, insieme alla costante sterica della reazione, δ.
klog( )k0
= ρ*σ* + δES
La validità delle costanti σ* ed ES è stata criticata da molti ricercatori
CRITICHE:Il metodo non separa in modo appropriato gli effetti induttivo e sterico: EScontiene ancora un effetto induttivo residuo e, soprattutto, σ* contiene uneffetto sterico residuo
vengono preferiti i σIA FAVORE: è stato dimostrato che i valori ES sono proporzionali ai raggi di van der Waals dei sostituenti
Più recentemente i valori ES originali (relativi a Me) sono stati sostituiti con una serie di ES modificati, relativi all'H
ES(H) = 0

66
equazione di Taft modificata
ρIρS
+ ρSESk
log( )k0= ρIσI
costante induttiva della reazionecostante sterica della reazione
EFFETTO ortoUn sostituente in orto può esercitare sulla reazione più di un effetto
- effetto di campo (polarizzazione diretta del centro di reazione attraverso lo spazio)- effetto di impedimento sterico di risonanza (può non essere coplanare con il benzene e quindi non dare coniugazione completa)- effetto sterico (può ostacolare l'avvicinamento del reagente al centro di rea- zione o la formazione del guscio di solvatazione)- possibilità di legame idrogeno- possibilità di effetto del gruppo adiacente
difficile da trattare quantitativamente
Taft ha determinato valori σo* e ES dall'idrolisi acida e basica di esteri benzoici sostituiti in orto
Le costanti steriche sono state chiamate ES, anche se numericamente sono diver-se dalle ES alifaticheIl riferimento per σo* è stato Me, ma con l'aiuto di σp(Me) è stato riscalato ad H come sostituente di riferimento
+ δESk
log( )k0= ρoσo
H - 0.00 -OMe -0.22 -0.39 +0.99Me 0.00 -0.17 0.00F +0.41 -0.24 -0.49Cl +0.37 +0.20 +0.18Br +0.38 +0.21 0.00I +0.38 +0.21 -0.20NO2 +0.97 +0.80 -0.75
gruppo σ0* σ0 ES
Costanti del sostituente di Taft, per sostituenti orto
successo molto limitato

67
CRITICHE:- un solo insieme di valori σo non è soddisfacente per rappresentare una varietà di effetti elettronici- ES contiene ancora un piccolo contributo di effetti coniugativi
- Equazione di Charton
+ βσR + ψυ + h= ασI
Per l'interpretazione dell'effetto orto, Charton ha introdotto l'equazione:
klog( )k0
σI costante induttiva del sostituenteσR costante coniugativa del sostituenteυ costante sterica del sostituente
α sensibilità della reazione all'effetto induttivoβ
ψ
sensibilità della reazione all'effetto coniugativosensibilità della reazione all'effetto sterico
Come e sono stati presi i valori calcolati in precedenzaσI σR
Per sostituenti simmetrici, υ è stata determinata dai raggi di van der Waals
υX = rX - 1.20
Per sostituenti non simmetrici, Charton ha calcolato υ da misure cineticheυ e ES sono in buona correlazione
Charton ha applicato la sua equazione con successo per interpretare molte reazioni. in molti casi ha ottenuto valori di ψ molto piccoli(impedimento sterico molto piccolo)
- Equazione di Fujita-Nishioka
Fujita e Nishioka nella descrizione dell'effetto orto hanno considerato tre effetti
- effetto polare (induttivo) e coniugativo ordinario- effetto di campo di prossimità- effetto sterico dei sostituenti orto
per l'effetto elettronico normale hanno usato le costanti di Hammett, conl'assunzione che
σorto = σpara
per la descrizione dell'effetto polare attraverso lo spazio dei sostituentiorto hanno usato i parametri F di Swain-Lupton

68
per la caratterizzazione degli effetti sterici hanno usato i valori ES di Taft
In questo modo la reattività di composti orto, meta e para sostituiti si può rac-chiudere in una sola equazione
log ko,m,p = ρ σo,m,p + δES + fF + c
ρ sensibilità della reazione agli effetti elettronici
δ
f sensibilità della reazione all'effetto campo
sensibilità della reazione all'effetto sterico
Con sostituenti in meta e para δES e fF vanno a zero e l'equazione diventaquella di Hammett
Per σ bisogna scegliere sempre la scala più adatta (σ, σ0, σ+, σ-)
CRITICHE:- Le costanti del sostituente non sono in grado di descrivere l'effetto del legame idrogeno o l'effetto di partecipazione del gruppo adiacente- non necessariamente
σorto = σpara
EFFETTO ISOTOPICOCon buona approssimazione, la sostituzione in un composto di un isotopo con un altro non altera la superficie dell'energia potenziale.
Gli isotopi sono isoelettronici: la struttura elettronica (e quindi tutte le forze di legame) rimane la stessa.
--------
_ _ _ _ _ _ _ _ _ _ _ _ _ _ _Energia
coordinata di vibrazione
C H
C D
Le differenze di reattività (K o k) conseguenti alla sostituzione isotopica sono attribuibili solo alla variazione della massa, che si manifesta nellafrequenza dei modi vibrazionali

69
Per un atomo piccolo, di massa m, legato ad un atomo di massa molto maggiore:
ν = 12π
fm
f costante di forza
se le masse sono confrontabili, si deve usare la massa ridotta μ =m1m2m1+ m2il trattamento quantomeccanico porta ai livelli di energia
εn = n + 12( )hν n = 0, 1, 2, .....
ed alle separazioni dei livelli energetici Δε = hν
Le energie sono misurate dal punto più basso sulla curva dell'energia potenziale
con n = 0 ε = 12
hν ε energia del punto zeroν vibrazione del punto zero (ZPV)
L'energia del punto zero è inversamente proporzionale alla massa
La molecola che contiene l'isotopo più pesante (di solito 2H al posto di 1H)nel livello fondamentale si trova ad un'energia più bassa della molecolacon l'analogo isotopico più leggero
E' da questa differenza di energia che nasce l'effetto isotopico
EFFETTO ISOTOPICO SUGLI EQUILIBRIConsideriamo l'equilibrio:
AH + BD AD + BHK
Poiché la sostituzione isotopica non altera la superficie dell'energia potenziale,AH e AD differiscono solo nella loro energia di vibrazione del punto zero (ZPV)e così fanno BH e BD.
La differenza tra le energie del punto zero tra stato finale e stato iniziale ΔE0 = { E0(AD) + E0(BH) - E0(AH) - E0(BD)}
si può esprimere come differenze di ZPV delle frequenze fondamentali di vibra-zione
ΔE0 = 12 Nhc Σ
3n-6
i=1
{ νi(AD) + νi(BH) - νi(AH) - νi(BD)}
E' possibile dimostrare che la costante di equilibrio dipende principalmentedalla differenza di energia di ZPV
Σ3nB-6
{ νi(BH) - νi(BD)}i=1
Σ3nA-6
{ νi(AH) - νi(AD)}i=1
0.7195T
0.7195T
exp
exp
K =

70
E' possibile (per molecole piccole) calcolare la costante di equilibrio dalle frequen-ze vibrazionali
La percentuale di deuterio in A sarà più pronunciata se K > 1, cioè se:
Σ3nA-6
{ νi(AH) - νi(AD)}i=1
Σ3nB-6
{ νi(BH) - νi(BD)}i=1
>
La somma delle differenze delle frequenze può essere influenzata da due fattorin il numero di atomi nella molecola aumenterà il numero di termini nella
sommatoriaf un legame più forte ha una costante di forza maggiore ed il valore
della frequenza è proporzionale alla radice quadrata della costante diforza
ν = 12πc
fμ
ciascun Δνi è proporzionale alla radice quadrata della sua costante di forza
In un equilibrio l'isotopo più pesante sarà più abbondante nella molecola con il maggiornumero di atomi e preferisce occupare la posizione che governa il legame più forte.
νi(AH) - νi(AD) Δνi = = 12πc fi-1 1
μAHμAD( )
Esempio
+ + + +K
CCD3CD3
CD3
C ClCH3CH3
CH3CCH3
CH3
CH3CCD3CD3
CD3Cl
Kcalc = 2.35 (25°C)Kexp = 2.39
Le relazioni valgono per qualunque coppia di isotopi. Però l'effetto isotopico perH e D è il più grande, perché l'incremento di massa è circa 100%.Da 12C a 13C la massa aumena solo dell'8.33%: il rapporto delle costanticonverge ad un valore non lontano dall'unità
K +CC Cl
K12/K13 = 0.983
Esempio: equilibrio di protonazione di una base (B) in acqua ed in acqua deuterata
H3O+ + BKb
H
KbD
B-H+ + H2O
D3O+ + B B-D+ + D2O

71
Si dimostra che la differenza osservata tra le costanti di equilibrio misurate in acqua ed in acqua deuterata è il risultato della differenza delle energie di vibrazione del punto zero.
/ KbHKb
D 3.26 (calcolato)3.0 (sperimentale)~~
EFFETTO ISOTOPICO CINETICO
Il livello dell'energia del punto zero per la vibrazione di stiramento (stretching) del legame C-H è più alto di quello del legame C-D
Se il legame C-H (e C-D) si rompe nello stadio rate-determining, la vibrazione di stretching dei reagenti si trasforma in moto traslazionale al di sopra della barriera e, per quel particolare grado di libertà, l'energia del punto zero sparisce
--------
_ _ _ _ _ _ _ _ _ _ _ _ _ _
stato dei reagenti
stato ditransizione
E
coordinata di vibrazione
C HC D
Poiché la molecola con C-H parte da un'energia più alta, la sua energia di attivazione è minore e kH/kD sarà maggiore di 1

72
Dalle frequenze di H2 e D2 si possono calcolare le loro energie vibrazionali del
E0 =12 Nhcν
punto zero (ZPV)
r rCurve potenziali per le molecole H2 e D2, che specificano l'energia della vibrazione al punto zero (E0) e l'energia di dissociazione (D0)
E D2 2 D
E0D2
457.6 kJ moli-1D0
D2
EH2 2 H
457.6 kJ moli-1
E0H2
D0H2
H2 D2D2D2
H2H2
ν
E0
D0
Per H2 = 4370 cm-1
= 25.9 kJ moli-1
= 431.7 kJ moli-1
ν
E0
D0
= 3091 cm-1
= 18.4 kJ moli-1
= 439.2 kJ moli-1
Per D2
più grande perché l'energia ZPV è più bassa
Si può calcolare l'effetto isotopico atteso, se il solo contributo fosse la perditadella differenza di energisa del punto zero (rottura completa del legame)
La frequenza di C-D dovrebbe essere più piccola di quella di C-H di un fattorecirca
12
= 11.41
sperimentale 11.35
Esprimendo la frequenza in cm-1, la differenza di energia di attivazione è:
Dν- E =/ = 1
2 hc ( H - )E =/H νD
Considerando la differenza di energia di attivazione approssimativamente uguale alla differenza di entalpia di attivazione, facendo il rapporto delle costanti di velocità ed assumendo che ΔS sia uguale per due molecole che differisconosolo per la sostituzione di un H con un D:
kHkD
= exp ( )- D- E =/E =/H
κT(dove κ è la costante di Boltzmann)
kHkD
~~ exphc
2κT (1 - 11.35 ) = exp ( 0.1865
T )νH νH

73
Nello stato di transizione ci sarà di solito uno o più modi vibrazionali (oltre al movimento lungo la coor-dinata di reazione) che comportano il movimento dell'idrogeno
La vibrazione di stretching di C-H nello spettro IR è attorno a 3000 cm-1; a T = 298 K, l'effetto isotopico sarà:
kHkD
~~ 0.1865T
(3000) = 6.5
kH / kD (e quindi l'effetto isotopico) diminuisce con l'aumento della T
Questo modello semplice è troppo grossolano per spiegare l'intervallo osservatodi effetti isotopici.
in molti casi l'idrogeno, piuttosto che dissociarsi come atomo libero, sitrasferisce da una molecola ad un'altra e rimane parzialmente legato pertutto il processo
Stato di transizione in cui l'idrogeno rimane legato e contribuisce all'energia del punto zeroattraverso un modo vibrazionale indipendente dalla coordinata di reazione. La differenza di energia tra H e D può cancellare in parte o tutta la differenza che viene dallo stato di transizione
Coordinata di reazione
Energia
L'effetto isotopico cinetico è determinato dalla differenza di energiavibrazionale del punto zero tra reagenti e stato di transizione
AH + B (AHB) =/ prodotti
AD + B (ADB) =/ prodotti
Si può dimostrare che:
exp
exp
= =/
kHkD
νi (AH) e νi (AD) misurabili sperimentalmente
νi (H) e νi (D) =/=/ si possono fare solo "ipotesi ragionevoli"
0.7195T Σ
3nA-6
{νi (AH) - νi (AD)}i =1
0.7195T Σ
3n -7 {νi (H) - νi (D) }
i =1
=/=/

74
b) Se nello stato di transizione il legame di H(D) è allentato:
kHkD
> 1
a) Se nello stato di transizione il legame di H(D) è completamente rotto:
νi (H) ~ νi (D) =/=/
kHkD
>> 1
c) se nello stato di transizione il legame di H(D) è invariato rispetto ai reagenti, non si osserva effetto isotopico cinetico
kHkD
= 1
kHkD
< 1
d) se, per qualche motivo, nello stato di transizione la differenza tra le energie del punto zero è maggiore che non nei reagenti
in questo caso si parla di
effetto isotopico cinetico inverso

75
Esempio
+ + .Mn(CO)5
νC-H = 2900 cm-1
{ νi (Mn-H) - νi (Mn-D) }
C CH2R2 L
< 1
C CH2R2LMn(CO)5
.
νMn-H = 1800 cm-1
{ νi (C-H) - νi (C-D) } > kHkD
=/=/
Qualche volta si può osservare effetto isotopico intramolecolarequando con due isotopi diversi si formano due siti, altrimentiequivalenti, ed il reagente può attaccarli entrambi
Esempio: clorurazione del metano-d1CH3D + .Cl CH2D. + HClCH3D + .Cl CH3
. + DCl
L'energia del punto zero per i reagentidelle due reazioni è la stessa
l'effetto isotopico viene dalle energie delpunto zero degli stati di transizione che portano ai due radicali CH2D. e CH3
.
L'energia del punto zero dello stato di transizione che porta al radicale .CH2D è <
la reazione che comporta rottura del legame C-H è più veloce
- EFFETTO ISOTOPICO CINETICO PRIMARIO
L'effetto isotopico cinetico primario si ha quando il legame che si rompe (o si forma) è marcato isotopicamente (per es., con H e D).
l'effetto maggiore si ha se C-H (C-D) è completamente rottonello stato di transizione
frequenza di stretching νC-H = 2900 cm-1 νC-D = 2150 cm-1
frequenza di bending δC-H = 1400 cm-1(2) δC-D = 1050 cm-1(2)
Se nelle strutture dello stato di transizione i legami C-H e C-D sono completamenterotti, spariscono le frequenze ad essi associate
νi (H) = νi (D) = 0=/=/
A 298 K, il massimo effetto isotopico cinetico sarà:
= exp { 2900 + 1400 + 1400 - 2150 - 1050 - 1050} = 33kHkD
0.7195298
Questo valore superiore è troppo alto, l'effetto isotopico cinetico osservato sperimentalmente è
1 < < 9kHkD

76
Spiegazione il legame C-H (C-D) non è completamente rotto
Un modo più realistico di stimare la grandezza dell'effetto isotopico cineticocomporta il processo di trasferimento di H o D da un atomo A ad un atomo B
AH + B A ----- H ------- B A + BH
In questo trasferimento, illustrato come disposizione lineare, nellostato di transizione ci sono due modi di stretching e due modi dege-nerati di bending (identici, ma in due piani perpendicolari)
(L = H, D)
νas(L) νs(L)
A L B A L B
+ +-
A L B A L B=/ =/
δ(L) δ(L)=/ =/
l'effetto isotopico cinetico è dato dall'equazione:
=
La coordinata di reazione per il trasferimento di H (D) dall'atomo A all'atomoB comprende νas nello stato di transizione
nessun contributo al rapporto
kHkD
exp { ν(AH) - ν(AD) + 2 δ(AH) - 2 δ(AD)}
exp { νas(H) - νas(D) + νs(H) - νs(D) + 2 δ(H) - 2 δ(D) }
νas(H) = νas(D) kHkD
0.7195298
0.7195298
=/ =/ =/ =/ =/ =/
=/ =/
L H o D
La frequenza della vibrazione di stretching simmetrico è data da:
νs =
fBm
fA e costanti di forza dei legami A-L e L-B nello stato di transizione masse
12πc
fA fB - 2+ fA fBmL
fBmB
+fAmA
+

77
Se L (H o D) occupa una posizione simmetricamente centrale (fA = fB), noncambia la posizione nella vibrazione simmetrica
νs è indipendente dalla differenza di massa tra H e D e perciònello stato di transizione:
Se nello stato di transizione le vibrazioni di bending sono sostanzialmente ridotteνs(H) = νs(D)
limite superiore realistico per l'effetto isotopico cinetico primario, se illegame C-D (C-D) si rompe nello stato di transizione
=/ =/
(per esempio, δ(H) = 1050 cm-1, δ(D) = 780 cm-1)
exp { 2900 - 2150 + 1400 + 1400 - 1050 - 1050}=
exp {1050 + 1050 - 780 - 780}
= 9
=/ =/
kHkD
0.7195298
0.7195298
Applicando questo metodo, si trovano i seguenti limiti superiori:
= 11 per N-H; 13 per O-H; 8 per S-HkHkD
In molte reazioni, il valore misurato di kH/kD è considerevolmente più piccolo
oppure
νs non è più indipendente dalla differenza di massa tra H e D
Se H(D) è più vicino a B fBfA<< stato di transizione simileai prodotti
Se H(D) è più vicino ad A fBfA>> stato di transizione simileai reagenti
νs(H) > νs(D)=/ =/
Perciò kH/kD è più piccolo del limite superiore: più lo stato di transizione è vicinoai reagenti (o ai prodotti), più piccolo è l'effetto isotopico cinetico
Se H(D) è simmetricamente centrale, ma lo stato di transizione non è lineare
L
A B
kH/kD > 5 per stati di transizione lineari
kH/kD tra 1 e 3 per stati di transizione non lineariνs(L)
kH/kD si abbassa notevolmente
=/
νs(H) > νs(D)=/ =/
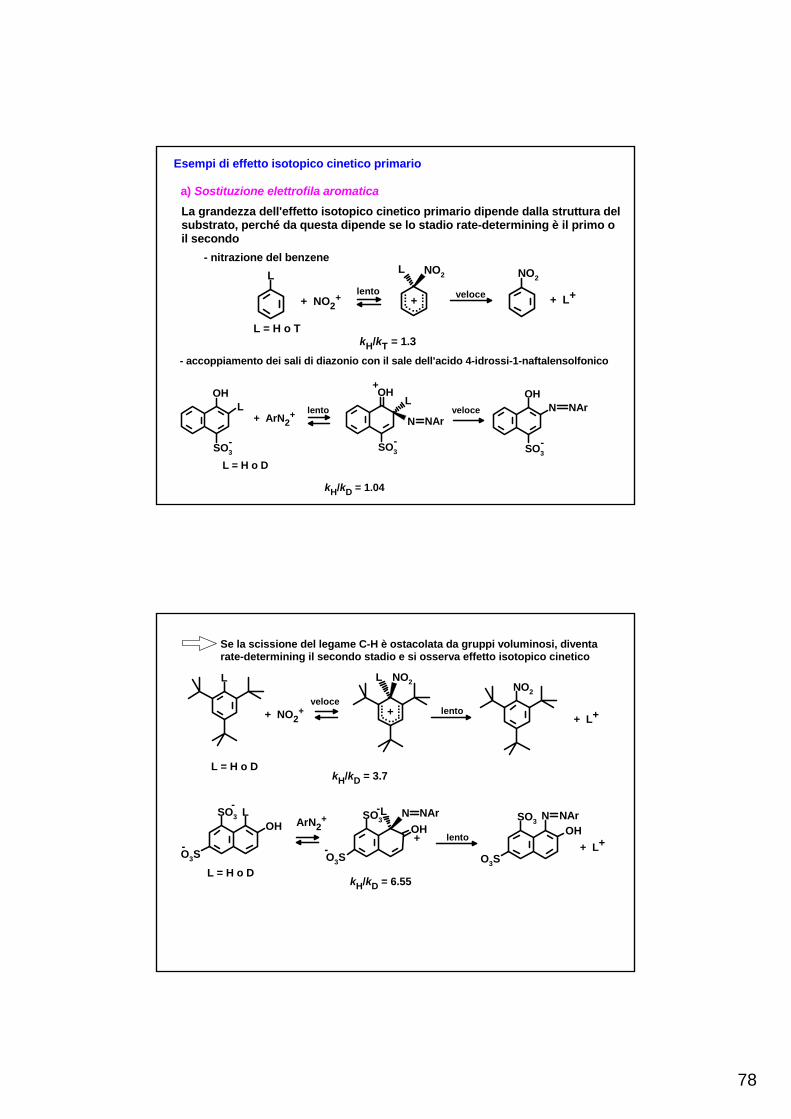
78
Esempi di effetto isotopico cinetico primario
a) Sostituzione elettrofila aromaticaLa grandezza dell'effetto isotopico cinetico primario dipende dalla struttura delsubstrato, perché da questa dipende se lo stadio rate-determining è il primo oil secondo
- nitrazione del benzene
+ NO2+ lento veloce + L+
L = H o TkH/kT = 1.3
+
L NO2
............ ..
L NO2
- accoppiamento dei sali di diazonio con il sale dell'acido 4-idrossi-1-naftalensolfonico
-
lento+ ArN2
+
-
+
veloce
-
L = H o D
kH/kD = 1.04
OH
SO3
LOH
SO3
N
L
NAr
OH
SO3
N NAr
Se la scissione del legame C-H è ostacolata da gruppi voluminosi, diventarate-determining il secondo stadio e si osserva effetto isotopico cinetico
+ NO2+ + lento
veloce
+ L+
L = H o DkH/kD = 3.7
............ ..
-
-
L = H o D
ArN2+
-
-
lento+
L NO2 NO2
SO3
SO3
OHN NAr
L
SO3
SO3
LOH
N NAr
+ L+
kH/kD = 6.55
SO3
SO3
LOH
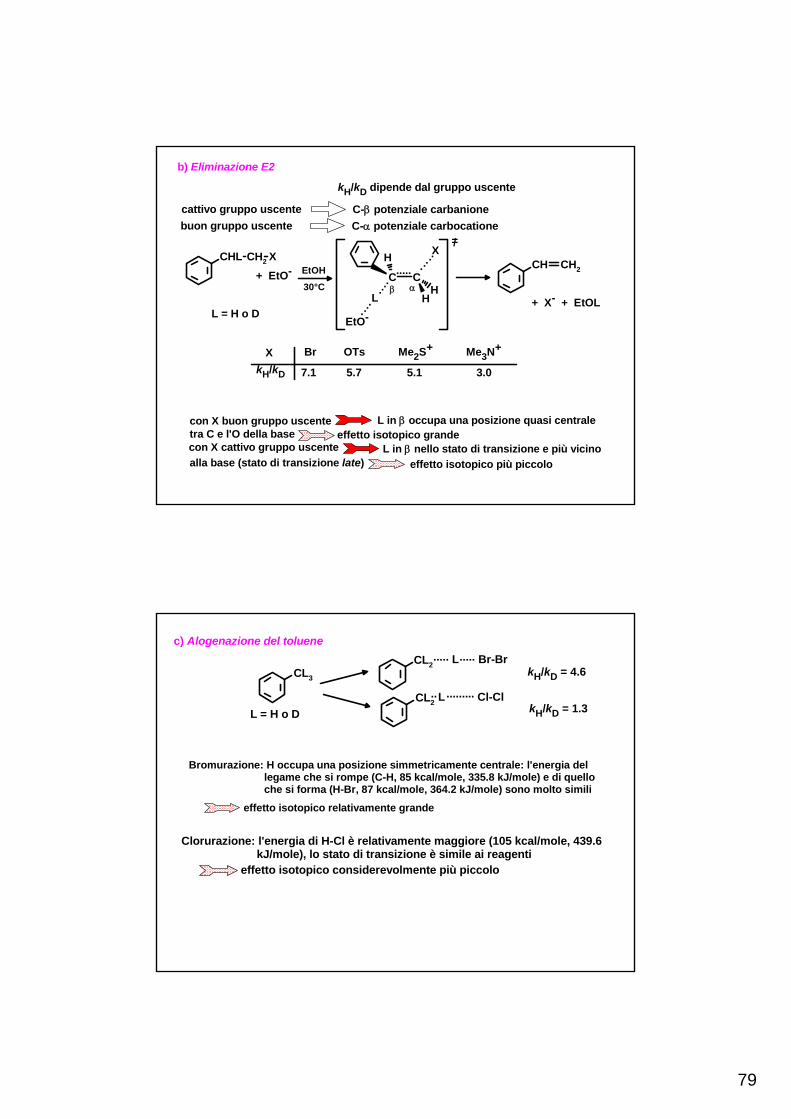
79
b) Eliminazione E2
kH/kD dipende dal gruppo uscente
cattivo gruppo uscente C-β potenziale carbanione
XkH/kD
Br OTs Me2S+ Me3N+
7.1 5.7 5.1 3.0
buon gruppo uscente C-α potenziale carbocatione
CHL CH2 X+ EtO- EtOH C C
H
HH
...
..L...
...
EtO-
β α
.......X
=/
30°C
L = H o D
CH CH2
+ X- + EtOL
con X buon gruppo uscente L in β occupa una posizione quasi centraletra C e l'O della base effetto isotopico grandecon X cattivo gruppo uscente L in β nello stato di transizione e più vicinoalla base (stato di transizione late) effetto isotopico più piccolo
c) Alogenazione del toluene
L = H o D
..... L..... Br-BrkH/kD = 4.6
..L.........kH/kD = 1.3
Cl-Cl
CL3
CL2
CL2
Bromurazione: H occupa una posizione simmetricamente centrale: l'energia del legame che si rompe (C-H, 85 kcal/mole, 335.8 kJ/mole) e di quello che si forma (H-Br, 87 kcal/mole, 364.2 kJ/mole) sono molto simili
effetto isotopico relativamente grande
Clorurazione: l'energia di H-Cl è relativamente maggiore (105 kcal/mole, 439.6 kJ/mole), lo stato di transizione è simile ai reagenti
effetto isotopico considerevolmente più piccolo
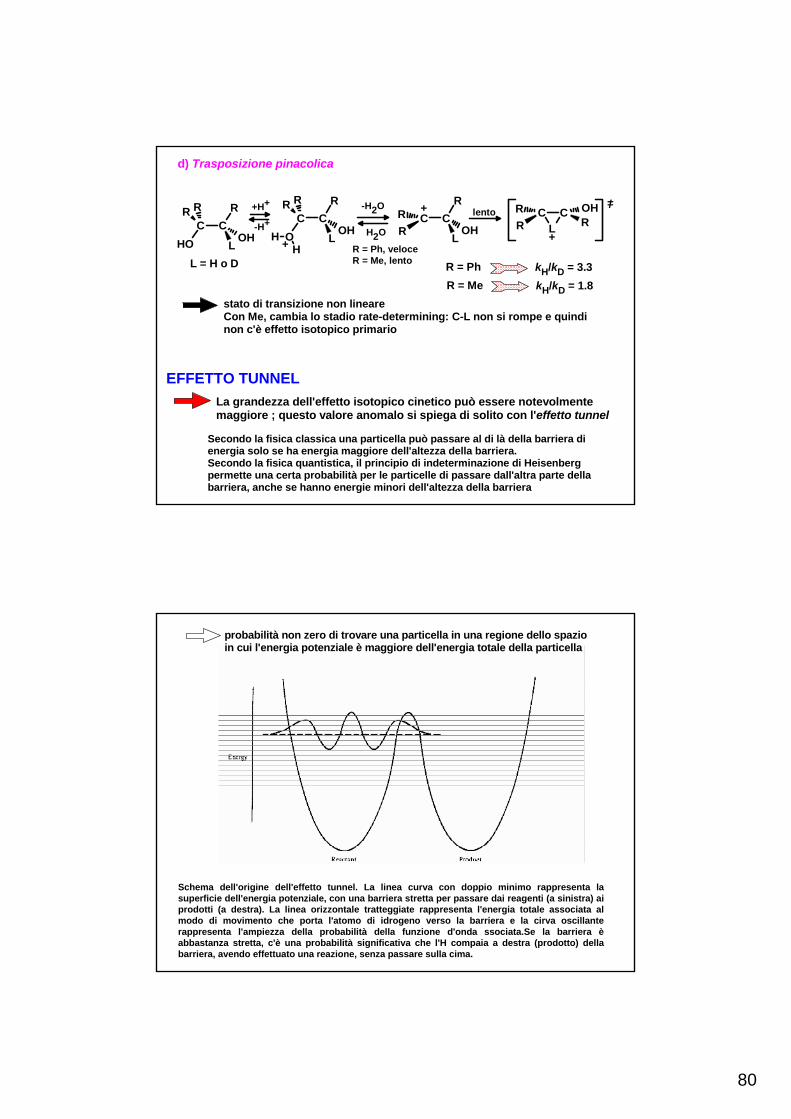
80
stato di transizione non lineareCon Me, cambia lo stadio rate-determining: C-L non si rompe e quindi non c'è effetto isotopico primario
-H++H+
+
-H2O
H2O
+
R = Ph, veloceR = Me, lento
lento C CR OHRR L
+
=/
L = H o D R = Ph kH/kD = 3.3R = Me kH/kD = 1.8
C CR
OH OH
RR
L
C CR
O OH
RR
LHH
C CROH
R
R L
d) Trasposizione pinacolica
EFFETTO TUNNELLa grandezza dell'effetto isotopico cinetico può essere notevolmentemaggiore ; questo valore anomalo si spiega di solito con l'effetto tunnel
Secondo la fisica classica una particella può passare al di là della barriera dienergia solo se ha energia maggiore dell'altezza della barriera.Secondo la fisica quantistica, il principio di indeterminazione di Heisenbergpermette una certa probabilità per le particelle di passare dall'altra parte della barriera, anche se hanno energie minori dell'altezza della barriera
Schema dell'origine dell'effetto tunnel. La linea curva con doppio minimo rappresenta la superficie dell'energia potenziale, con una barriera stretta per passare dai reagenti (a sinistra) ai prodotti (a destra). La linea orizzontale tratteggiate rappresenta l'energia totale associata al modo di movimento che porta l'atomo di idrogeno verso la barriera e la cirva oscillante rappresenta l'ampiezza della probabilità della funzione d'onda ssociata.Se la barriera èabbastanza stretta, c'è una probabilità significativa che l'H compaia a destra (prodotto) della barriera, avendo effettuato una reazione, senza passare sulla cima.
probabilità non zero di trovare una particella in una regione dello spazio in cui l'energia potenziale è maggiore dell'energia totale della particella

81
Me2CLNO2 + 2,4,6-Me3C5H5N Me2CNO2 + 2,4,6-Me3C5H5NH- +Me2CLNO2 + C5H5N Me2CNO2 + C5H5NH- +
PhCL(OL)CF3 + 2MnO4- + 2 OL- PhCOCF3 + 2 MnO4
2- + 2 L2O
+ EtO--
+ EtOL
+ (Me2N)2C=NL
-
+ (Me2N)2C=N+L2
kH/kD °C solvente
10 25 t-BuOL/L2O
24 25 t-BuOL/L2O
16 25 L2O
10 -60 EtOL28 -90 EtOL
45 25 C6H5Me12 25 CH2Cl2
Effetto tunnel in reazioni di trasferimento di H (L = H o D)
N
CL2CN
O2 N
CLCN
O2
N
CL2CN
O2N
CLCN
O2
Il ruolo dell'effetto tunnel è meno pronunciato per i composti con il Dperché l'energia del punto zero è più bassa e quinsi la barriera daattraversare è più largaperché l'incertezza per la posizione del D è minore (massa maggiore)
Un effetto cinetico isotopico insolitamente alto (>9) insieme ad un grafico diArrhenius curvo (a basse temperature reazioni più veloci di quanto atteso) si può considerare una prova di effetto tunnel
L'effetto tunnel è più pronunciato a temperatura minore (relativamentepoche molecole possiedno l'energia di attivazione
L'effetto tunnel dipende dal solventesi osserva solo se H non è solvatato nello stato di transizione
EFFETTO ISOTOPICO DELL'ATOMO PESANTEGli elementi più pesanti di H hanno isotopi che differiscono molto meno nella massa e di conseguenza l'effetto isotopico è più basso
Per un legame C-O:
ν14 = 12πc m14m16/(m14+m16)
f
ν12 = 12πc m12m16/(m12+m16)
f
c è la velocità della luce, f è la costante di forza e m è la massa atomica dei vari isotopi
ν12
ν14 m12(m14+m16)
m14(m12+m16)= = 0.95831
Considerando solo le frequenze di stretching dei reagenti e prendendo un valoremedio per ν(12C-O) = 1050 cm-1
k12
k14= exp
0.7195298
ν12( )1 -ν14ν12
= 1.1114

82
l'effetto dello scambio isotopico 12C/13C è ancora più piccolo
k12 / k13 = 1.0579Per altri atomi pesanti:
ν(C-X)mlight/mheavyklight/kheavy
1100 1050 700 600
14/15 16/18 32/34 79/81
1.042 1.063 1.0137 1.00236
C-X C-N C-O C-S C-Br
Di tutti gli effetti isotopici de atomo pesante, il più grande è 12C/14C,che però difficilmente supra il 10%
k12/k14 < 1.10_
E' molto difficile misurare accuratamente la diminuzione della velocità della reazione che deriva dalla sostituzione con l'atomo pesante
Si preferisce usare un metodo cinetico competitivo, per esempio misurando il rapporto degli isotopi nei reagenti e nei prodotti conla spettrometria di massa
con isotopi radioattivi (per esempio, 14C) si può usare la radioattività (metodomeno accurato)
metodo importante per chiarire il meccanismo
EsempiIdrolisi basica del benzoato di metile e reazione con l'idrazina
* * + OH- lento-* * *
+ -*OCH3
COMe
O
C OMeO
OH
COH
O
k12/k13 = 1.043k16/k18 = 1.006 effetto isotopico cinetico solo per 12C/13C
+ NH2NH2* ** * lento *
+ CH3OH*
COMe
O
C OMeOH
NHNH2
CNHNH2
O
k12/k13 = 1.041k16/k18 = 1.041 effetto isotopico sia per C che per O

83
Eliminazione base-catalizzata del bromuro di dimetil(2-feniletil)solfonio
*+
OH-
H2O
+ Me2S*CH CH2
C C
H
H
SMe2
HH
*+
..
.
...H
OH- =/
C CH SMe2
HH.....
k32/k34 = 1.0064Il valore è quasi a metà strada tra il valore massimo (1.0137) e 1.000 (cioè nessuneffetto)
Si è concluso che si tratta di un meccanismo E2 con un certo carattere di E1cb
EFFETTO ISOTOPICO CINETICO SECONDARIOCambiamenti di reattività si possono osservare anche quando il legame con unatomo sostituito con un suo isotopo non si rompe nello stato di transizione
effetto isotopico secondario
a causa del suo valore piccolo, si può misurare solo per H/D e solo se sonovicini al centro di reazione
In realtà il minimo di potenziale è anarmonico: è più facile allungareun legame che comprimerlo
la distanza media R-H è maggiore della distanza media R-D, a causa della lorodifferenza di energia del punto zero
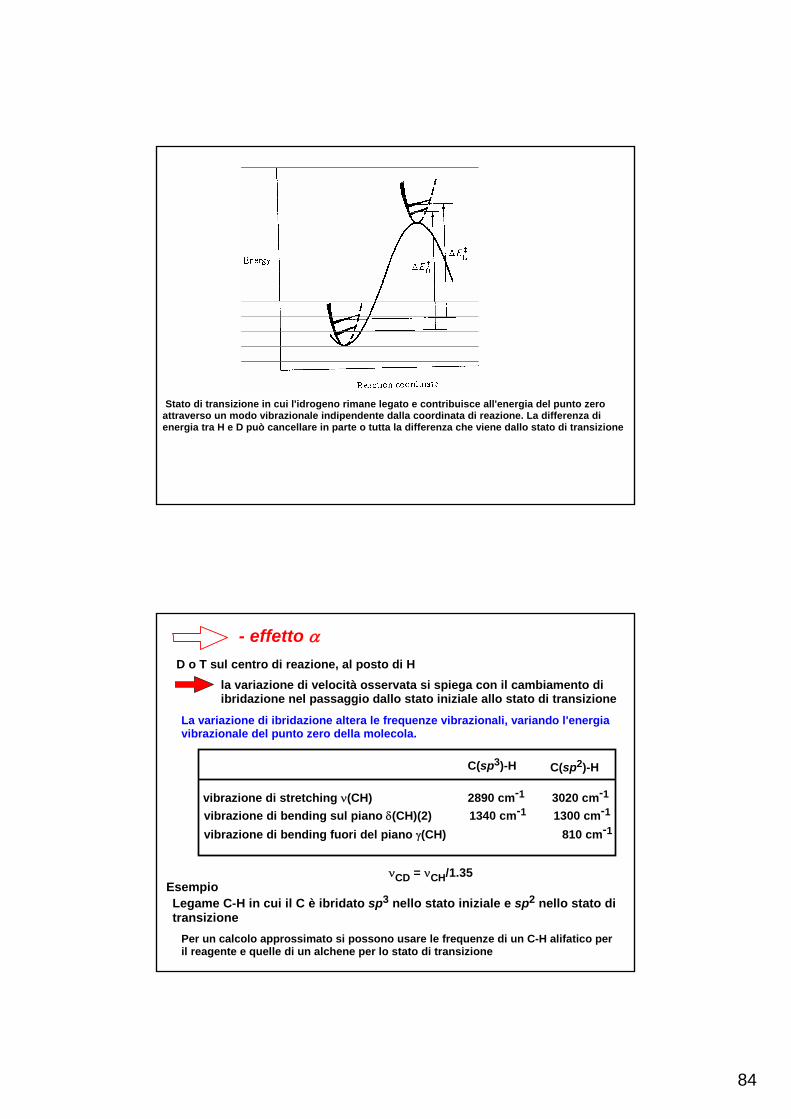
84
Stato di transizione in cui l'idrogeno rimane legato e contribuisce all'energia del punto zeroattraverso un modo vibrazionale indipendente dalla coordinata di reazione. La differenza di energia tra H e D può cancellare in parte o tutta la differenza che viene dallo stato di transizione
- effetto αD o T sul centro di reazione, al posto di H
la variazione di velocità osservata si spiega con il cambiamento di ibridazione nel passaggio dallo stato iniziale allo stato di transizione
La variazione di ibridazione altera le frequenze vibrazionali, variando l'energiavibrazionale del punto zero della molecola.
vibrazione di stretching ν(CH) 2890 cm-1 3020 cm-1
vibrazione di bending sul piano δ(CH)(2) 1340 cm-1 1300 cm-1
vibrazione di bending fuori del piano γ(CH) 810 cm-1
C(sp2)-HC(sp3)-H
νCD = νCH/1.35EsempioLegame C-H in cui il C è ibridato sp3 nello stato iniziale e sp2 nello stato ditransizione
Per un calcolo approssimato si possono usare le frequenze di un C-H alifatico peril reagente e quelle di un alchene per lo stato di transizione

85
La variazione più significativa è la deformazione fuori del piano γ(CH)dell'H alchenico rispetto al bending δ(CH) del gruppo CH alifatico
Usando queste frequenze, il valore-limite superiore calcolato per l'effetto isotopicosecondario secondario di D in α è:
kHkD
=exp
exp {2890 + 1340 + 1340 - 2140 - 993 - 993}
{3020 + 1300 + 810 - 2237 - 963 - 600}= 1.32
0.7195298
0.7195298
sperimentalekHkD
= 1.15 ÷ 1.20
Se la variazione di geometria durante la reazione è da C sp2 a sp3, si ha un effettoisotopico cinetico inverso
limite inferiore calcolatokHkD
= 11.32
= 0.76
sperimentalekHkD
= 0.85
Un effetto isotopico cinetico inverso si osserva nell'addizione a doppi legami C=C
Esempio: reazione di Diels-Alder
+kH/kD= 1.18
kH/kD= 0.88
L = H o D
L
LC
CCC N
C N
N
CN L
CNCNCN CN
L
kH/kD normale e inverso di queste reazioni valori reciproci, entro gli errorisperimentali
Esempio: SN1 e SN2
SN1 variazione della geometria da tetraedrica a trigonale planareeffetto isotopico secondario α relativamente grande
SN2 variazione della geometria da tetraedrica a bipiramide trigonale

86
Nello stato di transizione (bipiramide trigonale) le vibrazioni C-H (C-D) nel pianoequatoriale sono accoppiate con le frequenze del nucleofilo che sta entrando inposizione apicale e del gruppo uscente in posizione apicale, con conseguenteaumento delle frequenze di deformazione fuori dal piano dei leganti equatoriali.
effetto isotopico secondario α piccolo e qualche volta diventa inverso
Cl3OTs 0.96 70CH3CL2OTs 1.038 54(CH3)2CLOTs 1.134 30CL3CH2OTs 1.018 60(CL3)2CHOTs 1.551 30(CL3)3CCl 2.54 20
Composto kH/kD °C
L = H o D
Esempio: solvolisi del p-toluensolfonato di isopropile
TFA
EtOH-H2O 80%
TsOH
TFACF3CO2-CLMe2
TsOH
EtO-CLMe2L = H o D
+ -OTs
OTs........ ........δ-δ+
C OL
CH3
CH3
SO
OCH3
CL
CH3CH3
CL
CH3CH3
OEt
H
in TFA kH/kD = 1.22 solvente molto ionizzante SN1
in EtOH kH/kD = 1.09 solvente più nucleofilo SN2
La grandezza dell'effetto isotopico secondario α dipende dal gruppo uscente e dal numero di sostituenti sul C centro di reazione
Nella solvolisi di R-Q:
kH/kD Q: I < Br < Cl < OTsR: Me < Et < i-Pr < t-Bu
Per un meccanismo SN2 puro su un C primario kH/kD = 0.95 ÷ 1.04

87
- effetto β
L'effetto della sostituzione di H con D sul C β rispetto al centro di reazione èapprezzabile, soprattutto quando si forma un carbocatione (SN1 o E1)
La grandezza dell'effetto β può essere uguale o anche maggiore di quella del-l'effetto α.
Passando da H a D, la riduzione della velocità può essere del 10-30%
kH/kD = 1.1 ÷ 1.3L'effetto isotopico secondario di D in β è legato all'interazione iperconiu-gativa tra l'orbitale p vuoto del carbocatione ed il legame C-H(D) in β.
+CH3 C CH2 C
H+
H2C CH.
... ....
δ+
δ+
IR le frequenze di CH3 in cationi del tipo H3C-C+ sono 150-250 cm-1 piùpiccole di quelle dei corrispondenti alogenuri.C'è una differenza tra la variazione di frequenza di CH3 e CD3
Se il meccanismo è SN2, non c'è carica positiva sul C e kH/kD ~ 1
L'importanza dell'iperconiugazione è sottolineata dal fatto che il valore kH/kD varia con la conformazione molecolare.
Effetto isotopico massimo quando l'orbitale p che si sta liberando sulcentro carbocationico è parallelo al legame C-H (C-D) in β.
12,12d2
kH/kD = 1.14
αβ
entrambi i legami C-H (D) sonoquasi paralleli all'orbitale p sulC adiacente
L = H o D
ClCH3L L
9,10d2
il legame C-H (D) è a circa 90°rispetto all'orbitale p sul C adiacente
L = H o D
kH/kD = 0.986
α
β
ClCH3
L
L
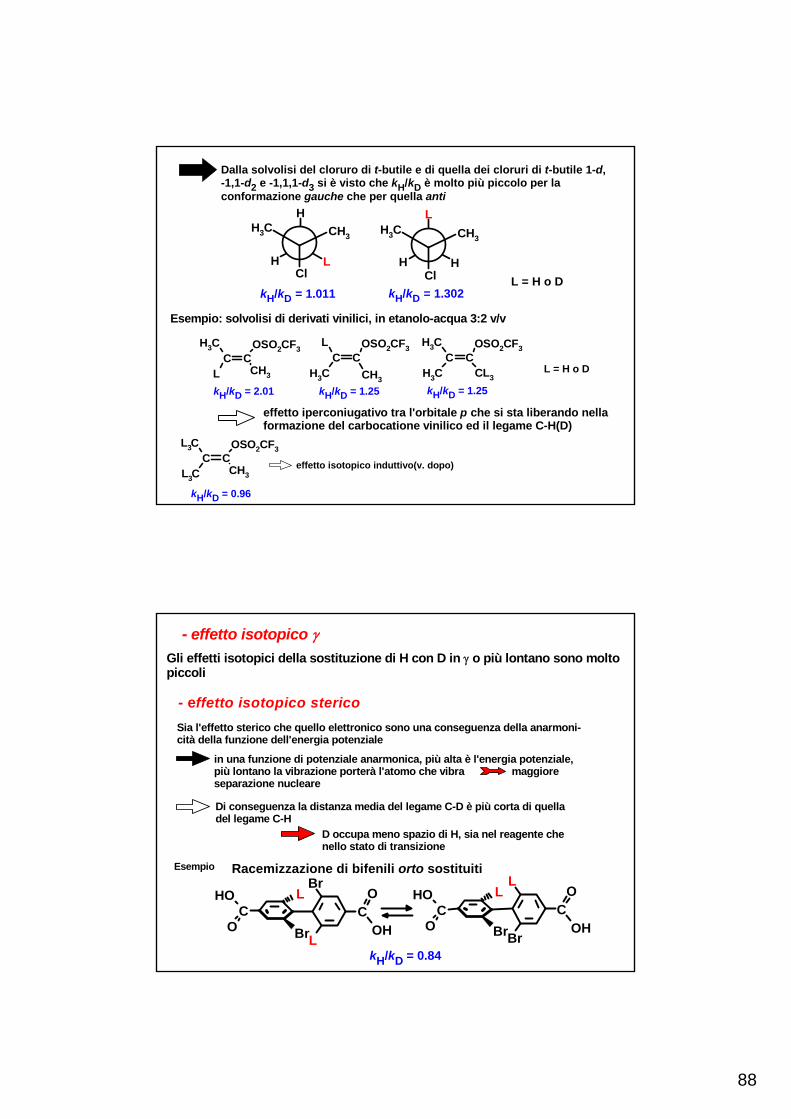
88
Dalla solvolisi del cloruro di t-butile e di quella dei cloruri di t-butile 1-d,-1,1-d2 e -1,1,1-d3 si è visto che kH/kD è molto più piccolo per laconformazione gauche che per quella anti
L = H o DkH/kD = 1.011 kH/kD = 1.302
LH
HCH3
CH3
ClHH
LCH3
CH3
Cl
Esempio: solvolisi di derivati vinilici, in etanolo-acqua 3:2 v/v
kH/kD = 2.01 kH/kD = 1.25 kH/kD = 1.25
L = H o DC C
CH3
L CH3
OSO2CF3C C
L
CH3 CH3
OSO2CF3C C
CH3
CH3 CL3
OSO2CF3
effetto iperconiugativo tra l'orbitale p che si sta liberando nella formazione del carbocatione vinilico ed il legame C-H(D)
kH/kD = 0.96
effetto isotopico induttivo(v. dopo)C CC
CH3
OSO2CF3L3
CL3
- effetto isotopico γGli effetti isotopici della sostituzione di H con D in γ o più lontano sono moltopiccoli
- effetto isotopico sterico Sia l'effetto sterico che quello elettronico sono una conseguenza della anarmoni-cità della funzione dell'energia potenziale
in una funzione di potenziale anarmonica, più alta è l'energia potenziale,più lontano la vibrazione porterà l'atomo che vibra maggioreseparazione nucleare
Di conseguenza la distanza media del legame C-D è più corta di quella del legame C-H
D occupa meno spazio di H, sia nel reagente che nello stato di transizione
Esempio Racemizzazione di bifenili orto sostituiti
kH/kD = 0.84
BrC
LOH
OBr
L
COH
O
BrC
LOH
OL
Br
COH
O
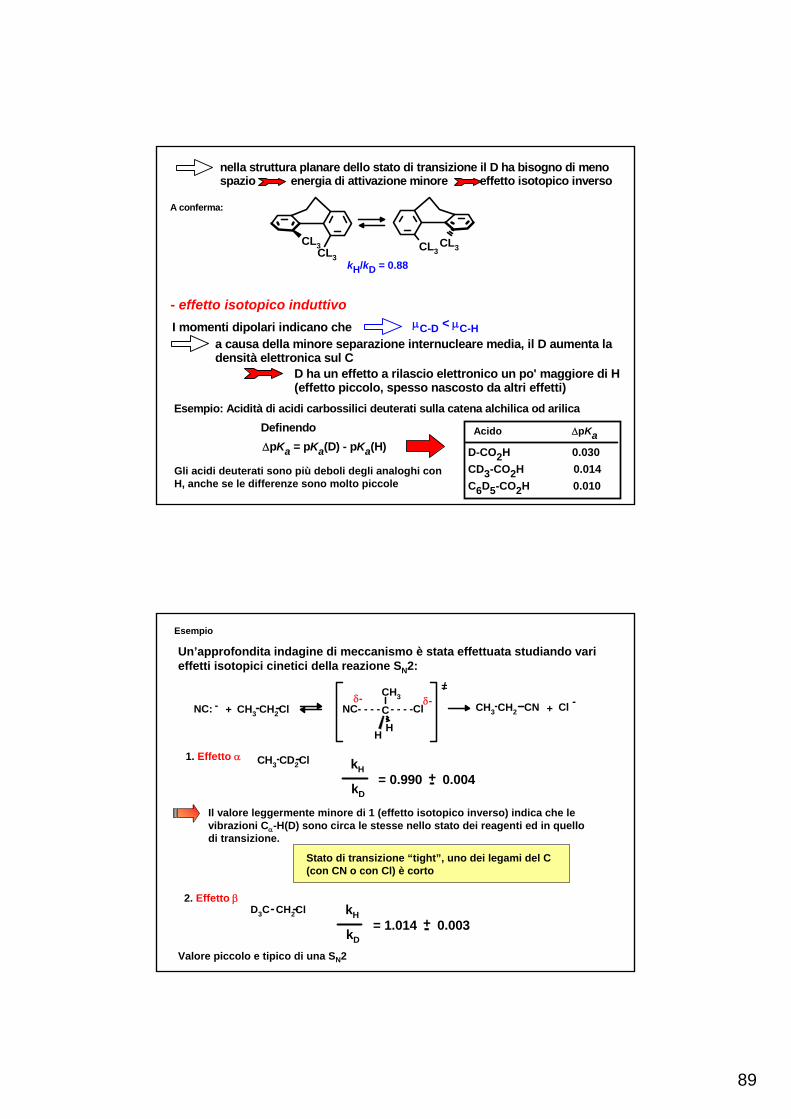
89
nella struttura planare dello stato di transizione il D ha bisogno di menospazio energia di attivazione minore effetto isotopico inverso
A conferma:
kH/kD = 0.88
CL3CL3
CL3CL3
- effetto isotopico induttivoI momenti dipolari indicano che μC-D < μC-H
a causa della minore separazione internucleare media, il D aumenta ladensità elettronica sul C
D ha un effetto a rilascio elettronico un po' maggiore di H(effetto piccolo, spesso nascosto da altri effetti)
Esempio: Acidità di acidi carbossilici deuterati sulla catena alchilica od arilica
Acido ΔpKa
D-CO2H 0.030CD3-CO2H 0.014C6D5-CO2H 0.010
DefinendoΔpKa = pKa(D) - pKa(H)
Gli acidi deuterati sono più deboli degli analoghi con H, anche se le differenze sono molto piccole
Esempio
Un’approfondita indagine di meccanismo è stata effettuata studiando vari effetti isotopici cinetici della reazione SN2:
CH3 CH2ClNC: -CH3
C
HH
=/
CH3 CH2 CN+δ-δ- -
NC- - - - - - - -Cl + Cl
1. Effetto α CH3 CD2Cl+-
kH
kD= 0.990 0.004
Il valore leggermente minore di 1 (effetto isotopico inverso) indica che le vibrazioni Cα-H(D) sono circa le stesse nello stato dei reagenti ed in quello di transizione.
Stato di transizione “tight”, uno dei legami del C (con CN o con Cl) è corto
2. Effetto βD3C CH2Cl
+-kH
kD= 1.014 0.003
Valore piccolo e tipico di una SN2

90
Il valore piccolo indica che nello stato di transizione sul Cα c’è pochissima carica positiva
Contributo iperconiugativo nullo
Effetto induttivo e/o sterico
3. Effetto isotopico del Cl
CH3 CH2 Cl35/37
+-kCl
35
kCl37 = 1.0070 0.0003
è circa il 50% del valore teorico massimo per la rottura di C-Cl; è analogo a quello di altre reazioni in cui il legame C- - - - -Cl è piuttosto lungo
Nello stato di transizione si ha significativa rottura del legame Cα-Cl
4. Effetto isotopico del Cα
CH3-C11/14H2-Cl
+-kC
11
kC14 = 1.218 0.019
E’ in accordo sia con uno stato di transizione simmetrico, che conTS early e con TS late
Effetto grande e vicino al valore massimo
5. Effetto isotopico dell’N del nucleofilo
6. Effetto isotopico del C del nucleofiloNC12/13
+-kC
12
kC13 = 1.0009 0.0007 (a 30°C)
CN14/15
+-kN
14
kN15 = 1.0002 0.0006
NESSUN effetto isotopico
Nello stato di transizione non c’è variazione nel legame dell’N in CN, rispetto allo stato dei reagenti
Valore piccolo e tipico di una SN2 (atteso inverso per NC- - - - C lungo)
Per confronto con dati di letteratura, nello stato di transizione N- - - - -Cα è corto
CH3
H
CH
=/δ-δ-
NC- - - - - - - - - - - - - - -ClChem. Eur. J, 2003, in DMSOJ. Org. Chem. 2006, in THF

91
EFFETTO ISOTOPICO DEL SOLVENTELa marcatura isotopica del solvente può influenzare la velocità della reazione espesso danno informazioni utili per la comprensione del meccanismo.La maggior parte dei dati sperimentali riportati in letteratura riguardano l'uso diH2O e D2O, ma sono disponibili anche dati con miscele di solventi, contenentiH2O e D2O.
Lo scambio H/D in solventi protici può portare a quattro diversi effetti:1. I valori di Ka in mezzi deuterati sono più piccoli e quindi l'equilibrio è spostato
verso la forma non ionizzata.
4. Con la sostituzione isotopica, cambiano anche le caratteristiche fisiche del solvente.
3. Effetto isotopico secondario se protoni dei reagenti, facilmente scam- biabili, sono sostituiti con D, ma il legame C-D non si rompe nello stadio lento
2. Effetto isotopico cinetico primario In reazioni acido- o base-catalizzate in cui si ha trasferimento del protone nello stadio rate-determining.
se il reagente entra nello stadio rate-determining in forma protonata, lavelocità aumenta nel solvente deuterato kH/kD < 1
Di conseguenza saranno un po' diverse le interazioni solvente-soluto, alterando per esempio la forza del legame idrogeno o la nucleofilità deireagenti. Lo stato di transizione verrà influenzato e la velocità di reazionecambierà.La grandezza dell'effetto isotopico in questo caso è simile a quella del-l'effetto isotopico secondario.
I calcoli approssimati permettono di stimare l'effetto isotopico del solvente daglieffetti su equilibrio, primario e secondario:
k H2O
k D2O KD
KH= ( )eq kD
kH( )seckD
kH( )pri
Non esiste un modo semplice per stimare il contributo del fattore 4.
- Fattore di frazionamento isotopico (isotope fractionating factor)
Un substrato (XH) che contenga un solo idrogeno scambiabile ed un solvente checontenga un isotopo (ROD) stabiliranno un equilibrio.
XH + ROD XD + ROH
La costante di equilibrio, φ, si indica come "fattore di frazionamento isotopico"

92
φ =[XH][ROD][XD][ROH]
= [XD]/[XH][ROD]/[ROH]
φ si può considerare come il rapporto relativo di D e H nel substrato enel solvente
La grandezza di φ dipende soprattutto dai tipi di legame coinvolti ed è relativa-mente indipendente dalla struttura dettagliata del substrato e del solvente, comepure dal numero di idrogeni sostituibili.
I valori di φ per alcuni tipi di legame sono:
X-L φ
O L
O L-
O L+
C(sp3)-L
C(sp2)-L
C(sp)-L
N L
+N L
S L
X-L φ
1.0
0.69
0.50
0.92
0.90
0.69
0.92
0.97
0.43
Per stimare l'effetto isotopico su un equilibrio chimico, si deve dividere il prodotto dei valori di φ dei reagenti per quello dei prodotti
KD
KH =( )eq
reagenti
Πφii
Π φjj
prodotti
Esempio: calcoliamo l'effetto isotopico sulla dissociazione degli acidi carbossilici
Q CO
O L+ L-O-L Q C
O
O -+ O
L
LL+
L = H o D
KaD
KaH
=φOL+φOL+ φOL+
φOL φOL φOL = 13
(0.69)3= 3.04
valore molto simile ai rapporti KaH/Ka
D sperimentali

93
Gli acidi carbossilici sono tre volte più acidi in H2O che in D2OD3O+ in D2O è un acido più forte di H3O+ in H2O
Esempio: calcolo della protonazione di una base
Q-O-L + L-O- Q-O- + L-O-L
KD
KH =φOL φOL
= 0.5φOL φOL
-
OD- è una base due volte più forte di OH-
Per calcolare l'effetto isotopico secondario si devono usare i valori di φ deireagenti e dello stato di transizione
seckD
kH( ) =
reagenti
Πφii
Π φjj
stato di transizione=
φrφ =/
Nell'equazione vanno introdotti i valori di φ di atomi di H che scambiano nelsolvente, ma non si rompono nello stadio rate-determining del meccanismo
φr φ dei reagenti valori noti
media pesata dei valori φ di reagenti (φr) e prodotti (φp)
φ /= = φr(1-x) φp
x
dove x è il valore numerico della coordinata di reazione in corrispondenza dellostato di transizione (tra 0 e 1)
seckD
kH( ) = ( φrφp)
x
φ /=
Esempi di effetto isotopico del solventea) Sostituzione nucleofilaNel meccanismo SN1 la molecola di acqua entra nella reazione solo dopo lo stadiorate-determining
RY R+x ....... Y-x /= R+ + Y-
effetto isotopico atteso nessunoNel meccanismo SN2, se l'acqua funziona da nucleofilo, ci si aspetta un effettoisotopico cinetico secondario
L2O + RY +x ............ R Y-x/= + + Y-
L = H o DO
L
LO
L
LR

94
k H2O
k D2O= ( φOL
2 )x
φOL+2= ( )x12
0.692= (2.1)x
per x = 0.5 kH/kD = 1.4
RX
MeCl 1.28EtCl 1.24i-PrCl 1.32t-BuCl 1.30
SN2 quasi puro
SN1 quasi puro
k H2O/kD2O
kH/kD quasi identico
(probabilmente, a causa della solvatazione)
Nel meccanismo SN2, se OH- (OD-) è il nucleofilo, ci si aspetta un effettoisotopico cinetico inverso
LO- + PhCH2SMe2+ -(1-x) ..............O
LCPh
HHSMe2
+(1-x)/=
PhCH2OL + Me2S
k H2O
k D2O( ) = ( φOL- )x
φOL= (0.5)x
a metà della coordinata di reazione (cioè a x = 0.5)
k H2O/kD2O = 0.7
sperimentale k H2O/kD2O = 0.77





![Gli esperimenti. Gli esperimenti radiochimici [A,Z] (ν,e - )[A,Z+1] Apparentemente, molte reazioni possibili; condizioni di diversa natura limitano.](https://static.fdocument.org/doc/165x107/5542eb5a497959361e8c7a0b/gli-esperimenti-gli-esperimenti-radiochimici-az-e-az1-apparentemente-molte-reazioni-possibili-condizioni-di-diversa-natura-limitano.jpg)












