TC Vortrag 04 Waschen-Bleichen-Faerben 03 Anlagen 2 Chemie
96
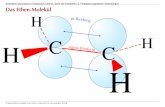
TC-Vortrag 04 66/161 Waschen – Bleichen – Färben 5.2 weitere chemische Hintergrundinformationen 5.2.1 Carotin und Carotinoide Strukturformel β-Carotin Strukturformel α-Carotin Lycopin (Tomate: Solanum lycopersicum) Hagebutte Canthaxanthin
-
Upload
awrcats-writer -
Category
Documents
-
view
156 -
download
2
description
Anlage 2 Organische Chemie zum TC-Vortrag 04 Waschen - Bleichen - FärbenDiese Anlage ist für diejenigen gedacht, welche aufgrund des Vortrags oder auch allgemein einen Einstieg bzw. Hintergrundinformation zur Organischen Chemie im vortrag selbst vermissen. Aus zeitlichen wie aus thematischen Gründen stelle ich daher hiermit eine separate zweite Anlage zum Vortrag zur Verfügung. Beachten Sie auch die Anlage 1 sowie das Scriptum zum zum öffentlichen Vortrag vom 10.11.2013 in der Wäscherei von Tucson auf AZ Arizona, Teil der größten deutschsprachigen WildWest-Simulation auf SecondLife
Transcript of TC Vortrag 04 Waschen-Bleichen-Faerben 03 Anlagen 2 Chemie
TC-Vortrag 04 66/161 Waschen – Bleichen – Färben
5.2 weitere chemische Hintergrundinformationen
5.2.1 Carotin und Carotinoide
Strukturformel β-Carotin
Strukturformel α-Carotin
Lycopin (Tomate: Solanum lycopersicum) Hagebutte
Canthaxanthin

TC-Vortrag 04 67/161 Waschen – Bleichen – Färben
5.2.2 Benzol und Benzolderivate
Benzol, Benzen Toluol (Methylbenzen, Methylbenzol, Phenylmethan)
Benzol (auch Benzen) ist eine flüssige organische Verbindung mit einem charakteristischen aromatischen Geruch. Die Verbindung mit der Summenformel C6H6 ist ein aromatischer Kohlenwasserstoff und das einfachste und zugleich klassische Beispiel für die Aromatizität bestimmter Verbindungen. Benzol ist mischbar mit fast allen organischen Solventien, jedoch kaum mit Wasser. Als Lösungsmittel hat Benzol seine Bedeutung verloren, da es krebserregend ist.
Benzol wird zur Herstellung wichtiger Industriechemikalien wie Ethylbenzol, Cumol, Cyclohexan, sowie Nitrobenzol verwendet.
Der Name Benzol wurde im Jahr 1843 erstmals von Justus von Liebig gebraucht. Liebig änderte die Namensgebung von Eilhard Mitscherlich von 1833, der das Benzol als Benzin bezeichnet hatte, um. Im angelsächsischen und französischen Sprachbereich wurde die adaptierte Bezeichnung (franz: benzène, engl: benzene) von Mitscherlich jedoch weiterhin benutzt.
In der 2. Hälfte des 17. Jahrhunderts wurde Benzol von Johann Rudolph Glauber, der auch das Glaubersalz entdeckte, bei der Destillation von Steinkohleteer entdeckt. Die Zusammensetzung war für ihn jedoch unbekannt und so nannte er es ein „subtiles und liebliches Oleum“. Im Jahre 1825 wurde Benzol von dem englischen Physiker Michael Faraday im Leuchtgas entdeckt, nämlich dadurch, dass er dieses Öl aus flüssigen Rückständen isolierte, die sich beim Verbrennen von Walölen in den Londoner Straßenlaternen aus der Gasphase abschieden. Er schlug deshalb den Namen „Pheno“ (gr. phainein = leuchten) vor.
Ein Jahr später erkannte man dieses Öl als Kohlenwasserstoff. Im Jahre 1834 erhielt der deutsche Chemiker Eilhard Mitscherlich Benzol aus Benzoesäure und Calciumoxid, des Weiteren setzte er Benzol zu Nitrobenzol, Azobenzol und Benzolsulfonsäure um. Er benannte den Stoff wegen seiner Verwandtschaft zu Benzoesäure als „Benzin“. Außerdem erstellte er die richtige Summenformel C6H6. Im gleichen Jahr wurde „Benzin“ von Justus von Liebig in Benzol umbenannt. 1845 isolierte der englische Chemiker Charles Mansfield während seiner Arbeit unter Leitung von August Wilhelm von Hofmann Benzol aus Steinkohleteer.
Im Jahr 1849 begann die industrielle Herstellung des Benzols auf der Basis von Steinkohle. Es wurde sorglos mit ihm umgegangen, bis Kampagnen schließlich über 100 Jahre später über die Gefahren des Benzols aufklärten, als die Giftigkeit des Benzols bekannt wurde.
Um die korrekte Strukturformel des Benzols schwelte ein langer Gelehrtenstreit. Erste Vorschläge wie die Prisman-Struktur, die des Benzvalen, des Dicyclopropenyl sowie das Dewar-Benzol (von James Dewar) stellten sich als falsch heraus.
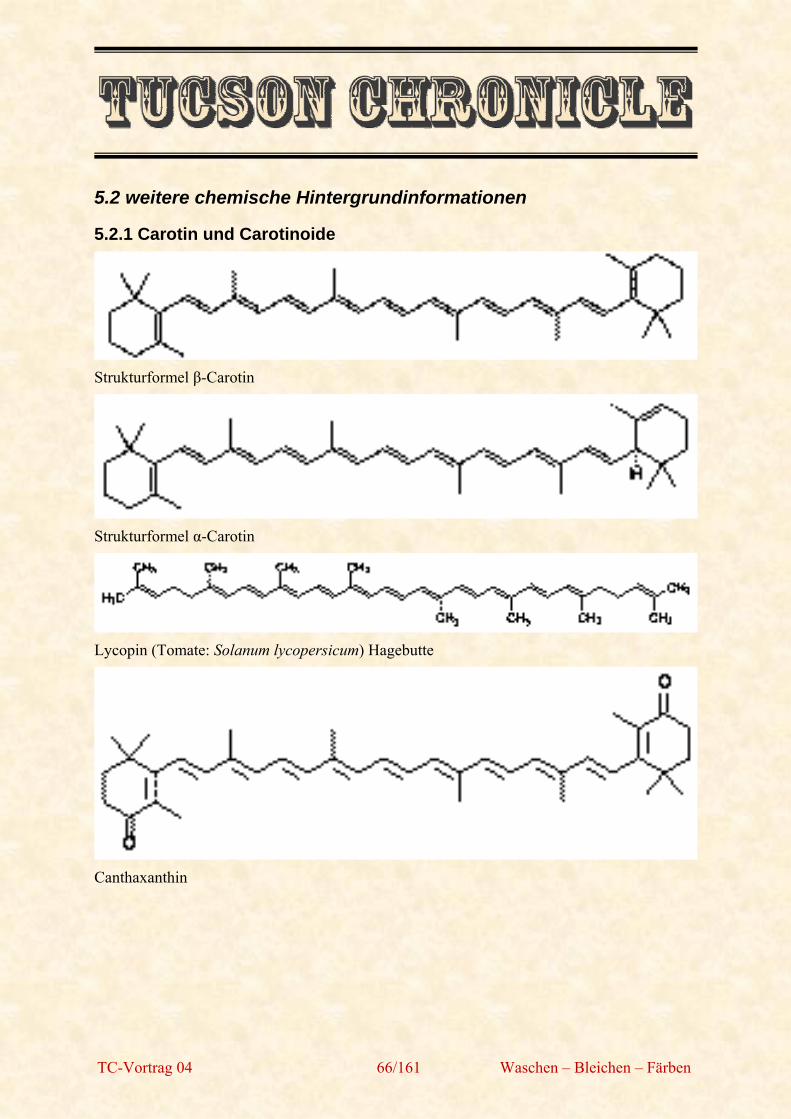
TC-Vortrag 04 68/161 Waschen – Bleichen – Färben
Erst im Jahre 1861 formulierte der österreichische Chemiker Johann Josef Loschmidt, damals noch Schullehrer, einige mögliche Strukturformeln des Benzols, die der deutsche Chemiker und Professor für Chemie August Kekulé dann 1865 – möglicherweise als Anregung für seine Kekulé-Strukturformel (siehe rechte untere Abbildung) – übernahm. Einer Legende nach kam Kekulé dieser Einfall im Traum. Er träumte von einer Schlange, die sich selbst in den Schwanz biss. Kekulé beschreibt dies in seiner Rede zum 25-jährigen Jubiläum des Benzolrings 1890. Die sechs Affen, die sich abwechselnd entweder mit beiden oder mit einer Hand an den Füßen fassten und so die Ringstruktur bildeten, beruhen auf einem 1886 bei einem Bierabend der Deutschen Chemischen Gesellschaft gemachten Scherz.
Sie trägt jedoch dem experimentellen Befund Rechnung, dass im Benzol alle Kohlenstoffatome gleichwertig sind. Mit dieser Formel konnten allerdings noch nicht alle Besonderheiten des Benzols erklärt werden, wie beispielsweise seine ungewöhnlich niedrige Reaktivität. Rätselhaft war insbesondere das Ausbleiben einer Additionsreaktion mit Bromwasser, wie sie nach der Kekulé-Strukturformel eigentlich zu erwarten wäre. Der Beweis der Gleichwertigkeit der Wasserstoffe im Benzolmolekül konnte von 1869 bis 1874 erbracht werden. Im Jahre 1872 formulierte Kekulé seine Oszillationshypothese eines dauernden Platzwechsels von Einfach- und Doppelbindungen.
Erst im 20. Jahrhundert konnte das Phänomen der delokalisierten Elektronenwolken, die dem Benzolmolekül eine besondere Stabilität verleihen, über Röntgenstrukturanalyse nachgewiesen werden. 1925 führten Armit und Robinson die vereinfachte Schreibweise mit dem konzentrischen Kreis in der Formel ein, welcher zum Ausdruck bringen soll, dass alle Bindungen absolut gleichwertig sind und keine lokalisierbaren Doppelbindungen existieren.

TC-Vortrag 04 69/161 Waschen – Bleichen – Färben
Benzol ist der einfachste benzoiden aromatischen Kohlenwasserstoffe, die auch Arene genannt werden. Die besonderen Bindungsverhältnisse dieser Stoffgruppe wird Aromatizität genannt und hier anhand des Benzols als Prototyp der Arene beschrieben:
Jedes Kohlenstoffatom verfügt über vier Valenzelektronen, von denen zwei das Atom mit den benachbarten C-Atomen verbinden. Ein Elektron bindet das zugehörige Wasserstoffatom. Die verbleibenden sechs π-Elektronen ergeben formal drei π-Bindungen, wie sie in der Strukturformel mit drei Doppelbindungen ausgedrückt wurden. In dem heute gültigen Orbitalmodell bilden die sechs π-Elektronen eine delokalisierte Ladungswolke (delokalisiertes 6-π-Elektronensystem) über und unter der Ebene des Kohlenstoffrings.
Da in der systematischen chemischen Nomenklatur die Endung -ol für Alkohole verwendet wird, ist die im Deutschen meist verwendete, historisch bedingte Bezeichnung Benzol irreführend; der Name Benzen wurde von der IUPAC als offizielle Nomenklatur für diesen Kohlenwasserstoff bestimmt.
Viele wichtige Chemikalien sind Derivate des Benzols, haben also einen Benzolring als Grundgerüst. Dazu gehören beispielsweise Verbindungen mit Alkylgruppen, wie Toluol und die Xylole. Technisch bedeutend sind Ethylbenzol und Cumol, sowie Styrol mit einer Alkenylgruppe.
Wichtige Verbindungen mit funktionelle Gruppen sind Phenol mit einer Hydroxygruppe (–OH), Anilin mit einer Aminogruppe (–NH2), Benzoesäure mit einer Carboxygruppe (–COOH), sowie Chlorsubstituierte Benzole.
Benzoldämpfe sind beim Einatmen giftig; die Symptome akuter Vergiftungen treten erst bei relativ hohen Konzentrationen ein. Leichte Vergiftungen äußern sich in Schwindelgefühl, Brechreiz, Benommenheit und Apathie. Bei einer schweren Vergiftung kommt es zu Fieber und Sehstörungen bis hin zu vorübergehender Erblindung und Bewusstlosigkeit. Bei der so genannten Benzolsucht, die beim Einatmen von Benzol eintreten kann, kommt es zu Trunkenheits- und Euphoriegefühlen. Benzol kann bei längerer Einwirkung auf den Organismus zum Tod führen.
Die Giftwirkung ebenso wie die karzinogene Wirkung ist auf die Bildung eines karzinogenen Metaboliten zurückzuführen. Im Körper wird Benzol am Ring oxidiert. Das entstehende hochreaktive Epoxid reagiert mit zahlreichen biologischen Verbindungen und kann auch das Erbgut schädigen. Eine langzeitige Aufnahme kleinerer Benzolmengen führt vor allem zu Schädigungen der inneren Organe und des Knochenmarks. Letzteres resultiert in einer Abnahme der Zahl der roten Blutkörperchen (Anämie), was sich in Herzklopfen, Augenflimmern, Müdigkeit, Schwindel, Blässe und Kopfschmerzen äußert. Benzol wird im Gehirn, Knochenmark und Fettgewebe gespeichert. Es wird nur langsam über die Niere ausgeschieden. Der Abbau erfolgt über verschiedene Umbauprodukte wie das Brenzcatechin, Phenol, Hydrochinon und Benzochinon. Das Hauptausscheidungsprodukt ist schließlich die Phenylmercaptursäure (N-Acetyl-S-phenyl-cystein).
Bei zwei Prozent Luftvolumenanteil Benzol in der Atemluft kommt es nach fünf bis zehn Minuten zum Tod. Die akute letale Dosis (oral) beträgt beim Menschen 50 Milligramm pro Kilogramm Körpergewicht. Zwischen einem Luftvolumenanteil von 1,4 bis 8 Prozent bildet Benzol explosive Gemische.

TC-Vortrag 04 70/161 Waschen – Bleichen – Färben
Benzol ist aufgrund dieser Gefahren mit besonderer Vorsicht zu handhaben. Benzol muss bei 15 °C bis 25 °C gelagert werden. Der TRK-Wert lag bei 1 Milliliter pro Kubikmeter Luft (bzw. 3,25 mg/m³ Luft). Jede Exposition gegenüber Benzol sollte möglichst vermieden oder verringert werden; vor dem Gebrauch von Benzol sind besondere Anweisungen einzuholen. Bei Unfall oder Unwohlsein sollte sofort ein Arzt hinzugezogen werden. Orte, an denen Benzol austritt oder austreten könnte, sollten sofort verlassen und nur in Vollschutzanzügen wieder betreten werden. Benzol ist stark wassergefährdend.
Xylol (ortho, meta, para)
Dimethylbenzol 1,2 1,3 1,4
Die Xylole (von griechisch ξύλον (xýlon) = „Holz“) (auch Xylene oder nach der IUPAC-Nomenklatur Dimethylbenzene) sind flüssige organisch-chemische Verbindungen mit einem charakteristischen aromatischem Geruch und der allgemeinen Summenformel C8H10. Sie zählen zu den aromatischen Kohlenwasserstoffen und bestehen jeweils aus einem Benzolring mit zwei Methylsubstituenten (–CH3). Durch unterschiedliche Anordnung der Methylgruppen ergeben sich drei Konstitutionsisomere des Xylols: 1,2-Xylol (ortho-Xylol), 1,3-Xylol (meta-Xylol) und 1,4-Xylol (para-Xylol). In der Technik (z. B. als Lösungsmittel) werden sie meist als (ungetrenntes) Isomerengemisch verwendet und setzen sich in der Regel aus 60 % m-Xylol, 10–25 % o-Xylol und 10–25 % p-Xylol zusammen. Als Lösungsmittel verwendete Xylolmischungen enthalten häufig auch Ethylbenzol, das im gleichen Temperaturbereich siedet und ähnliche Lösungseigenschaften besitzt.
p-Xylol ist Ausgangsstoff für die Darstellung von Terephthalsäure (> 1 Million Jahrestonnen) und o-Xylol in ähnlichen Mengen zur Gewinnung von Phthalsäure für die Kunststoffindustrie. Durch Nitrierung erhält man die Nitroxylole, die durch anschließende Reduktion zur Darstellung der Xylidine dienen.
Xylole sind entzündlich und wirken gesundheitsschädigend bei Aufnahme über die Haut und die Atemwege. Sie können zum Beispiel Kopfschmerzen, Gedächtnis- und Orientierungsstörungen, Schwindel und Atemnot hervorrufen. Xylole sind wassergefährdend (WGK 2). Zwischen einem Luftvolumenanteil von 1 bis 8 % bilden sie explosive Gemische. Emissionen von Xylolen sind hauptsächlich auf den Kfz-Verkehr zurückzuführen. In den letzten Jahren ist ein Rückgang der Xylolemissionen zu verzeichnen.
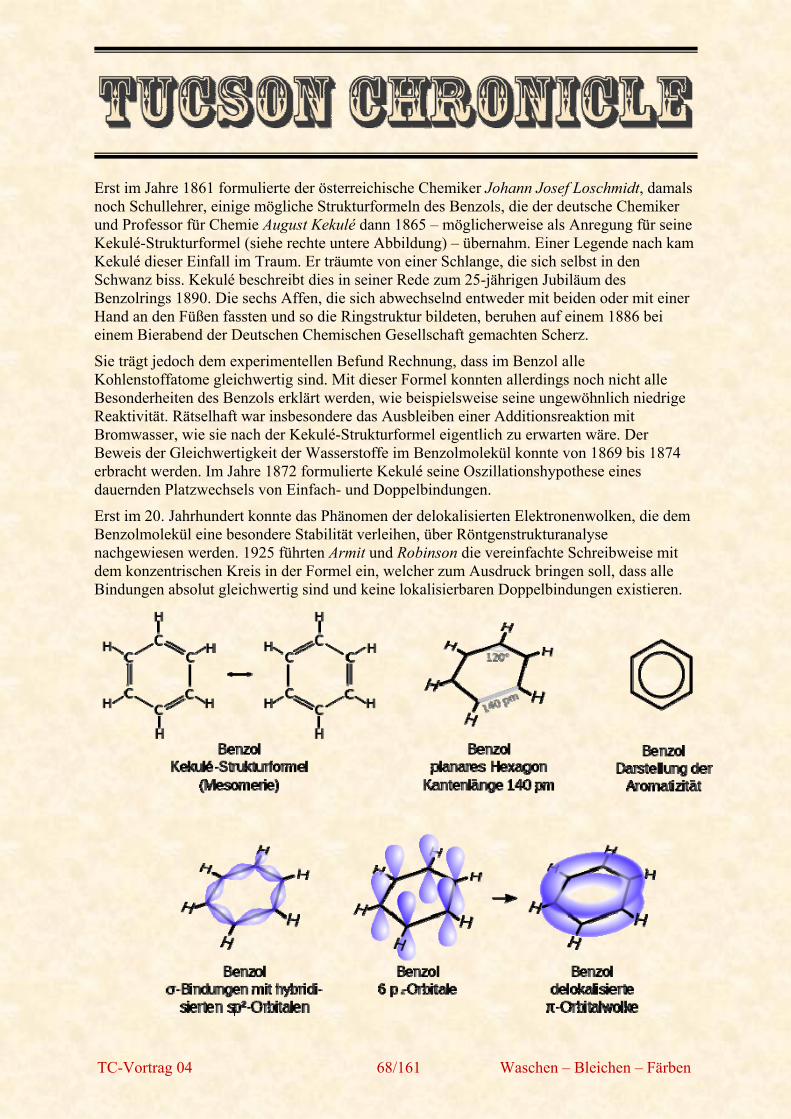
TC-Vortrag 04 71/161 Waschen – Bleichen – Färben
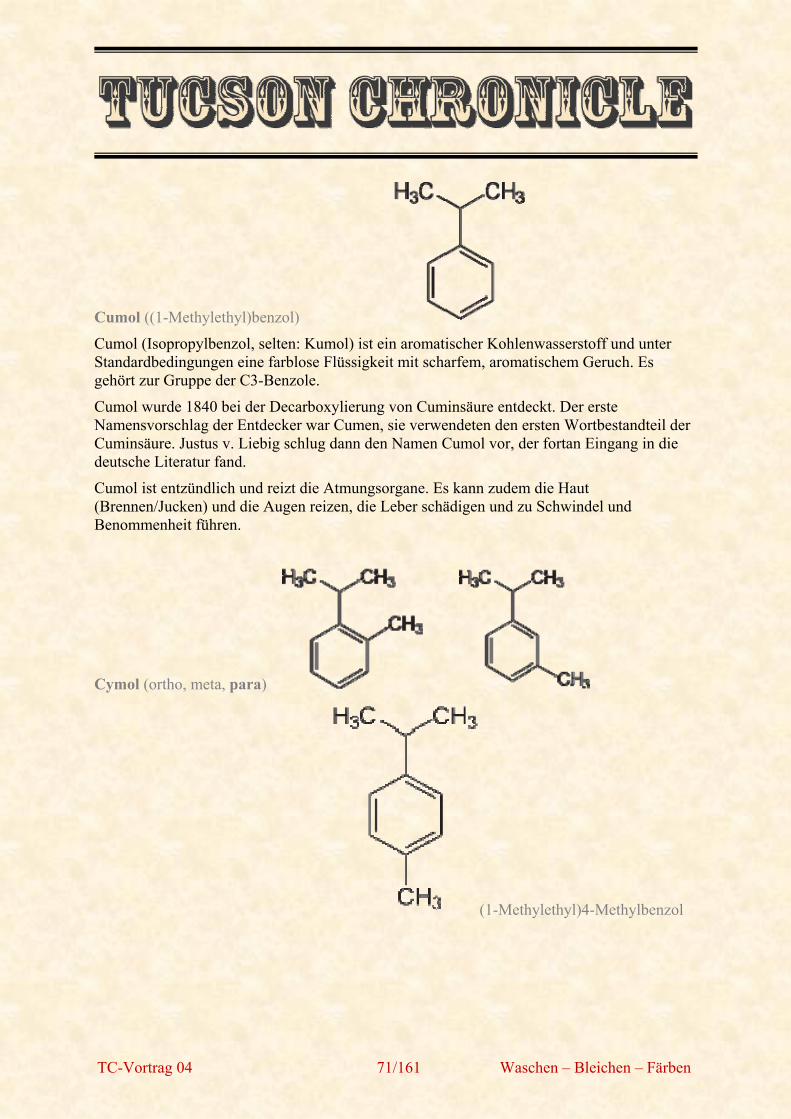
Cumol ((1-Methylethyl)benzol)
Cumol (Isopropylbenzol, selten: Kumol) ist ein aromatischer Kohlenwasserstoff und unter Standardbedingungen eine farblose Flüssigkeit mit scharfem, aromatischem Geruch. Es gehört zur Gruppe der C3-Benzole.
Cumol wurde 1840 bei der Decarboxylierung von Cuminsäure entdeckt. Der erste Namensvorschlag der Entdecker war Cumen, sie verwendeten den ersten Wortbestandteil der Cuminsäure. Justus v. Liebig schlug dann den Namen Cumol vor, der fortan Eingang in die deutsche Literatur fand.
Cumol ist entzündlich und reizt die Atmungsorgane. Es kann zudem die Haut (Brennen/Jucken) und die Augen reizen, die Leber schädigen und zu Schwindel und Benommenheit führen.
Cymol (ortho, meta, para)
(1-Methylethyl)4-Methylbenzol

TC-Vortrag 04 72/161 Waschen – Bleichen – Färben
5.2.3 Anilin und weitere künstliche Farbstoffe
Nitrobenzol Anilin
Nitrobenzol ist die einfachste aromatische organische Nitroverbindung mit der Summenformel C6H5NO2. Es wurde erstmals im Jahr 1834 durch Eilhard Mitscherlich dargestellt. Nitrobenzol ist bei Raumtemperatur flüssig und in saurem und überwiegend auch in alkalischem Milieu beständig. Es ist giftig und steht im Verdacht, Krebs zu erzeugen.
Nitrobenzol wird technisch durch Nitrierung von Benzol unter Einsatz von Nitriersäure hergestellt. Sie bildet durch Dehydratisierung von Salpetersäure mit konzentrierter Schwefelsäure zunächst reaktive Nitroniumionen.
Anilin [aniˈliːn] (nach spanisch oder auch arabisch: an-nil = blau = Indigo-Farbe) ist eine hellbraune Flüssigkeit mit aminartigem Geruch, die an der Luft leicht bräunlich wird. Es handelt sich um einen Benzolring mit einer Aminogruppe (–NH2) und damit um eine aromatische Verbindung. Mit Säuren versetzt bildet es Anilinsalze. Die basische Wirkung von Anilin wird durch den mesomeren Effekt verringert, da dieser die Elektronendichte der Aminogruppe verringert.
Anilin kann krebserregend wirken und ist in größeren Mengen auftretend ein Blutgift. Es oxidiert den roten Blutfarbstoff Hämoglobin zu Methämoglobin und verhindert damit den Sauerstofftransport im Blut. Das Gift kann durch Schlucken, Einatmen und durch die Haut aufgenommen werden. Bei leichten Vergiftungen kommt es zur Blaufärbung der Haut und der Fingernägel, zu Schwindelanfällen und Erregungszuständen. Bei höherer Konzentration treten Kopfschmerzen, Schwindel, Bewusstseinsstörungen und Atemnot auf. Letzteres kann den Tod verursachen. Langfristige Vergiftungserscheinungen zeigen sich in Schwächegefühl, Appetitlosigkeit und Blasenkrebs.
Anilin wurde 1826 von Otto Unverdorben erstmals durch Kalkdestillation aus Indigo hergestellt. Er nannte das erhaltene Öl Crystallin (eine charakteristische Eigenschaft ist die Bildung kristallisierbarer Salze mit Säuren). 1834 isolierte Friedlieb Ferdinand Runge erstmals Anilin aus der lange Zeit wichtigsten Quelle, dem Steinkohlenteer, und nannte es Kyanol (Blauöl, nach dem Verhalten der Substanz gegenüber Chlorkalklösung). Fritzsche hatte 1840 das Anilin aus der Destillation von Anthranilsäure erhalten, er konnte auch die Identität des von Zinin dargestellten Produktes nachweisen. Zinin erhielt Anilin (von ihm Benzidam genannt) aus Nitrobenzol durch Reduktion mit Schwefelwasserstoff. A. W. Hofmann zeigte, dass diese Verbindungen identisch sind, er konnte ferner das Nitrobenzol mit einem deutlich verbesserten Verfahren (Zink + Säure) zu Anilin reduzieren.
Seit 1897 wird Anilin von der Badischen Anilin- und Soda-Fabrik (BASF) zur Synthese des vorher nur aus pflanzlichen Rohstoffen gewonnenen Farbstoffs Indigo eingesetzt (Heumann-Synthese). Schon vorher wurde Anilin in großem Maßstab hergestellt, etwa von der Agfa (Aktien-Gesellschaft für Anilin-Fabrikation) ab 1873. Eine bekannte Anwendung des

TC-Vortrag 04 73/161 Waschen – Bleichen – Färben
Farbstoffes war Anilinleder. Auch in der Drucktechnik wurde Anilin verwendet, u. a. bekam der Flexodruck den auch heute noch verwendeten Beinamen Anilindruck, da erst durch das Anilin eine gute Druckqualität erzeugt werden konnte.
In der Technik gewinnt man Anilin durch eine Reduktion von Nitrobenzol mit Eisen in Gegenwart von Salzsäure (Bechamp-Reduktion).
Anschließend wird mit Branntkalk (CaO) neutralisiert, und das Anilin zusammen mit dem Wasser abdestilliert. Das als Nebenprodukt entstehende Eisen(II,III)-oxid kann als Pigment eingesetzt werden.
Erkennungsmittel für Anilin, nach Fr. Field.
Löst man Anilin in Wasser und leitet dann die röthlichen Gase hindurch, welche man durch Erwärmen von Stärke oder Zucker mit starker Salpetersäure erhält, so entsteht bald ein gelber Farbstoff (Vogel's Zinalin), welcher sich als feines Pulver niederschlägt.
Das beste Mittel, um bei Vorlesungsversuchen die Bildung von Anilinroth zeigen zu können, besteht darin, daß man 1 Theil Jod und 2 Theile Anilin mit einander in einem Reagensglase gelinde erhitzt. Das Product in Alkohol gelöst, erscheint sehr intensiv roth und kann, in Wasser gegossen, gleich zum Färben verwendet werden. Daß die Anilinfarben, bei den früheren zahlreichen Untersuchungen über Anilin und seine Zersetzungsproducte, nicht früher gefunden, hat wahrscheinlich seinen Grund darin, daß man früher das Anilin meistentheils aus Indigo darstellte, welcher es frei von Toluidin liefert. Die Gegenwart von Toluidin ist aber unentbehrlich zur Entstehung der Farben. Merkwürdig ist ferner, daß die Basen selbst meist farblos oder schwach gefärbt sind, während die Salze die wundervollsten Farben zeigen. Löst man das reine Rosanilin einmal in kaltem, dann in heißem Wasser, und setzt zu beiden Lösungen gleich viel verdünnte Schwefelsäure, so tritt die Färbung nur in der heißen Lösung ein. Umgekehrt wird eine heiße Lösung von essigsaurem Rosanilin durch Zusatz von Aetznatron sofort entfärbt, eine kalte nicht. Diese Farblosigkeit der Basen kann man zu einem hübschen Experiment benutzen. Man schreibt die Buchstaben ANILINE, jeden mit einer anderen Basis auf ein weißes Blatt Papier. A z.B. mit Aethyl-Rosanilin (Violett), N mit Phenyl-Violett (Indigo), I mit Phenylblau (Blau), L mit Anilingrün, I mit Anilingelb, N mit Chrysanilin (Anilinorange), E endlich mit Rosanilin (Roth.) Spritzt man nun das Papier mit einer Mischung von Essigsäure und Alkohol an, so erhält man das Wort in Regenbogenfarben. (Breslauer Gewerbeblatt, 1865, Nr. 17.)
Quelle: Polytech. Journal 1865, Band 177/Miszelle 9 (S. 410) URL: http://dingler.culture.hu-berlin.de/article/pj177/mi177mi05_9
Anilinfarben zum Aquarelliren und Coloriren von Photographien; von Dr. E. Jacobsen in Berlin.
Einen wesentlichen Vorzug besitzen die Anilinfarben vor anderen, z.B. den meisten Honigfarben, darin, daß alle Lasurfarben (d.h. durchsichtig) und nicht Körperfarben sind, daß mithin auch die unter der Farbe liegende photographische Zeichnung in ihren feinsten Tönen und Conturen zur Geltung kommt.

TC-Vortrag 04 74/161 Waschen – Bleichen – Färben
Die Anilinfarben erreichen bei einer schönen Brillanz und Feinheit fast die Töne der Oelfarben; sie mischen sich leicht unter einander und auch mit anderen Farben, und wenn man schließlich die fertig colorirten Photographien mit Wachsfirniß überreibt, so erhalten sie auch eine genügende Haltbarkeit und Schutz gegen Wasserflecken und dergl. Ein weiterer Vortheil ist, daß die Farben nicht zu schnell beim Arbeiten trocknen und, wenn sie gehörig verdünnt sind, sehr hübsche Effecte liefern. Die nöthigen praktischen Vortheile findet man bald beim Arbeiten selbst. Die Haltbarkeit läßt nichts zu wünschen übrig. Die Farben wurden sämmtlich fast 8 Tage lang hinter Glasscheiben dem Sonnenlichte direct ausgesetzt, ohne in dieser Zeit, Grün ausgenommen, eine Veränderung zu zeigen; an einer Zimmerwand ohne Sonne zeigten sie auch später noch dieselben Töne wie frisch aufgetragene Anilinfarben. Das Grün kann leicht ersetzt werden durch Mischen von Indigcarmin und Anilingelb.
Beim Auftragen selbst vermeide man die Conturen zu überschreiten, da sich einige der Anilinfarben nicht abwaschen lassen, namentlich ist dieß bei Roth I., Violett, Gelb und Braun (Lichtbraun I.) der Fall. Beim Coloriren des Gesichtes thut man wohl, zuerst das Roth der Wangen anzulegen und darnach den Localton; überhaupt verdünne man das Roth und Violett sehr stark, lege erst einmal den betreffenden Gegenstand blaß-roth an und übergehe mit ebenso schwacher Farbe bis zur gewünschten Dunkelheit. Alle übrigen Farben verhalten sich im Allgemeinen ganz so wie gewöhnliche Aquarellfarben und haften direct auch auf Albuminbildern; sind letztere indeß zu tief copirt oder mit den Fingern befaßt worden, so muß man das Albuminbild vor dem Auftragen der Farben mit einem Tropfen Glycerin abreiben. Um den Hintergrund ein wenig abzutönen, verdünne man die betreffende Farbe sehr stark. Lichter auf Goldsachen und dergl. lassen sich ebenso leicht anfertigen, wenn man ein wenig Permanent- (Baryt-) Weiß (kein Bleiweiß) mit den passenden Anilinfarben abtönt; der Wachsfirniß verreibt diese Theile nicht; man kann dieses Aufsetzen von Lichtern aber auch nach dem Wichsen vornehmen, denn auf dem überzogenen Bilde kann man ebenso leicht malen wie vorher. Für Fleischtöne eignen sich namentlich Roth I., II., III., Violett I. und Lichtbraun I.; diese erfordern aber auch die größte Vorsicht beim Auftragen, d.h. man muß sie sehr stark mit Wasser verdünnen. Das dunkle Braun (Neutraltinte) ist sehr geeignet zum Retouchiren und Ausflecken der Bilder; man gibt ihm, je nach der Farbe der Photographie, entweder mit ein wenig von Roth II. und Lichtbraun I. einen bräunlichen, oder mit Violett II. einen violetten Ton. Dabei ist zu bemerken, daß für diesen Zweck die Farben in Schaukastenbildern viel länger dem Lichte widerstehen, als z.B. Carmin und Pariserblau mit chinesischer Tusche gemischt. Beim Aquarelliren und Coloriren von Photographien auf gewöhnlichem Papier muß man das Papier, um die Farben zu fixiren und haltbarer gegen Licht zu machen, zuerst mit einer schwachen Leimung (von Gelatine, Hausenblase oder Eiweiß) versehen. Da die Farben glänzend auftrocknen, so überzieht man am besten solche Aquarellbilder mit einer geeigneten durchsichtigen Wachsmasse. (Deutsche Industriezeitung, 1864, Nr. 46.)
Quelle: Polytech. Journal 1864, Band 174/Miszelle 9 (S. 405–406) URL: http://dingler.culture.hu-berlin.de/article/pj174/mi174mi05_9

TC-Vortrag 04 75/161 Waschen – Bleichen – Färben
Ueber eine neue Methode zur Werthbestimmung der Anilinfarbstoffe; von Armand Müller.
Es dürfte in der Färberwelt schon längst das Bedürfniß gefühlt worden seyn, ein einfaches, von Jedem ausführbares Verfahren zu kennen, mit dessen Hülfe man die Anilinpigmente, welche in enormen Quantitäten und in den verschiedensten Nüancen sowohl, als im wechselndsten Gehalt alljährlich dargestellt und consumirt werden, so auf ihren Gehalt an färbendem Princip sowohl als auf ihre Nüance prüfen könnte, daß die Resultate der Untersuchung es dem Farbentechniker ermöglichten, in kurzer Zeit, mit Bestimmtheit und direct procentisch über den Handelswerth einer Waare ein Urtheil zu gewinnen. Diese Bedingungen aber sind bis jetzt noch durch keine vorgeschlagene Methode erfüllt worden. Das Colorimeter, nur bei Intensitätsbestimmungen einigermaßen befriedigende Resultate ergebend, und für den praktischen Färber etwas zu complicirt, sowie das Probefärben lassen Demjenigen, der sich häufig mit den Theerpigmenten beschäftigt, und dem oft die kleinsten Differenzen in Färbekraft oder Nüance höchst unliebsam, ja geradezu nachtheilig sind, sehr viel zu wünschen übrig.
Der Verfasser hat sich daher schon längere Zeit mit der Auffindung eines Verfahrens beschäftigt, welches den gestellten Forderungen möglichst Rechnung tragen sollte und gibt nun in Nachstehendem seine Resultate in einer neuen Methode, welche, sollte sie vielleicht auch noch wesentlicher Verbesserungen bedürfen, immerhin interessant genug ist, um vor das Forum eines größeren Publicums gebracht zu werden.
Die Basis desselben ist: Fixirung des zu untersuchenden Farbstoffes auf einer durchsichtigen Glasplatte mittelst Collodium als dünnes lasirendes Häutchen, behufs Vergleichung desselben mit einem ebenso behandelten Normalpigment derselben oder einer ähnlichen Nüance.
Eine solche Schicht gibt, wenn sie nach untenstehenden Verhältnissen, die sich nach langen Versuchsreihen als die günstigsten erwiesen haben, dargestellt wird, die Möglichkeit an die Hand, auf's Schärfste die kleinsten Differenzen zwischen zwei oder mehreren Farbstoffen zu erkennen.
Um nun überhaupt eine genaue Vergleichung möglich zu machen, ist es nothwendig, ein für alle Versuche gleichbleibendes "„Normalcollodium“" anzuwenden, um Schichten zu erzeugen, die an und für sich egal sind, und genau dieselben Dimensionen bezüglich Dicke wie alle anderen haben; denn es ist ganz klar, daß ein dickflüssiges Collodium mit derselben Menge Farbstoff weit dunklere Schichten erzeugt, als ein mehr äther- oder alkoholhaltiges, also dünneres. – Die Möglichkeit einer klaren Haut und folglich einer Vergleichung hängt ferner ab von dem Verhältniß des Alkohols zur Quantität der Collodiumwolle einerseits und andererseits von demjenigen zwischen Alkohol und Farbstoff. Bei zu wenig Weingeist scheidet sich nämlich das Pigment krystallisirt aus, was auch bei zu dünnem Collodium überhaupt eintritt, und die Fläche wird undurchsichtig. Ist dagegen zu viel vorhanden, so kann, wenn das Pyroxylin nicht sehr löslich ist, leicht eine flockenartige Ausscheidung des Collodiums mit dem allmählichen Verdampfen des Aethers stattfinden, was die Schicht Zur Vergleichung ebenfalls untauglich macht. Es dürfte sich vielleicht mit größerem Vortheil an Stelle der gewöhnlichen Collodiumwolle Sutton's Alkolen, das in absolutem Weingeist löslich ist, anwenden lassen. Da dem Verfasser solches Material mangelte, könnten darüber keine Versuche angestellt werden.

TC-Vortrag 04 76/161 Waschen – Bleichen – Färben
Das Normal-Collodium wird bereitet durch Lösen von 12 Grammen bester Schießbaumwolle (Pyroxylin) in 600 Kubikcentimeter Aether, und Zufügen von 350 Kubikcentimeter Weingeist vom spec. Gewicht 0,8156 (96 Proc. Tr.).
Man bewahrt die Solution in einem nach Art der Gay-Lussac'schen Bürette construirten Glasgefäß im Dunkeln sorgsam auf, um jede Zersetzung oder Verdampfung von Aether und Alkohol, wodurch der Titer der Lösung gestört würde, während und nach dem Gebrauch zu verhindern.
Zur Intensitätsbestimmung aller (spritlöslichen) Farbstoffe, z.B. eines gelblichen, krystallisirten Fuchsins, eines Teiges oder einer Lösung derselben Nüance, verfährt man wie folgt:
Man nimmt aus einer früheren Sendung krystallisirten Pigmentes, von der die Erfahrung gelehrt hat, daß sie in jeder Hinsicht den Zwecken der Färberei diente, eine kleine Probe heraus, wiegt genau 0,2 Gramm von derselben ab und bringt sie in ein circa 120 Kubikcentimeter haltendes, und mit einem gut eingeschliffenen Glasstöpsel verschließbares Gefäß. Hierauf läßt man etwas rasch aus einer Gay-Lussac'schen Bürette genau 100 Kubikcentimeter Normalcollodium in dasselbe einfließen, und schüttelt, indem man das Fläschchen mit dem Stöpsel deckt, einigemale tüchtig um. Die Lösung darf nicht durch Wärme unterstützt werden. Sie wird nun, sobald sie beendet ist, was man mit einem Glasspatel, den man nach Verlauf von einer halben Stunde (um Verlust zu vermeiden) am inneren Boden des Gefäßes reibt und dann herauszieht, leicht erkennt, rasch auf den oberen Rand einer überall gut gereinigten und klaren Glasplatte gegossen. Damit die Schichten sich bei allen Versuchen genau gleich dick anlegen, läßt man die Tafel zweckmäßig auf der Seite eines hölzernen Prisma's ruhen, dessen Basis, um einen constanten Neigungswinkel (60°) gegen die Tischplatte zu erzielen, ein gleichseitiges Dreieck bildet, und, auf den Arbeitstisch aufgeschraubt, dachartig anzusehen ist. Zum Abfluß der im Ueberfluß aufgegossenen Mischung gräbt man längs den beiden Seiten des Prisma's Rinnen in den Tisch ein, welche, an einzelnen Stellen durchbohrt, die Flüssigkeit in zweckmäßig darunter befestigte Gefäße leiten.
Nachdem die Haut vollkommen angetrocknet ist, wird ein regelmäßiger Theil derselben auf der Glasplatte reservirt, indem man das Uebrige mit einem feuchten Tuch leicht wegwischt. – Die Normalfläche für gelbliches Fuchsin ist mit diesen Operationen zur Vergleichung mit anderen Schichten fertig.
Auf ganz dieselbe Weise werden eventuell Normaltafeln aus krystallisirtem Violett (Jod- und Spritviolett), Blau, Grün (nur in höchst concentrirtem Zustand), Phosphin, Vesuvin, Nigros in den verschiedenen Anilinbraun's, aus "„La Phénicienne“" (Rothëin), Corallin, Saffranin, Rouge coquelicot, African Red, und aus allen anderen Anilin- und Phenylfarben, die in Weingeist löslich sind, dargestellt. – Pikrinsäure und Dinitronaphtylsäure (Martiusgelb) können mit der Collodiummethode nicht bestimmt werden; denn sie krystallisiren leider während des Trocknens der Schicht aus. Eine solche Platte steht unter dem Mikroskop durch Bildung der zierlichsten Krystallgruppen, deren Fond dann in den Newton'schen Farben glänzt, wunderschön aus, und läßt dem staunenden Auge im Entstehungsmoment dieser Gebilde am besten einen wenn auch nur kurzen und schwachen Einblick thun in die geheimnißvollen Werkstätten der Naturkräfte.
Die Normalplatten halten sich im Dunkeln und von schädlichen Ausdünstungen unberührt, lange Zeit ohne sich zu verändern; sie müssen indessen dennoch sehr sorgfältig behandelt

TC-Vortrag 04 77/161 Waschen – Bleichen – Färben
werden, und ziehe ich daher stets vor, den krystallisirten Normalfarbstoff vorräthig zu halten und die Platte jedesmal vor Untersuchung eines Farbstoffes frisch zu bereiten, d.h. wenn monatlich nur 1 bis 2 Proben zu machen sind.
Die Schichten unbrauchbar gewordener Tafeln entfernt man leicht mit einem in concentrirte Schwefelsäure getauchten Bürstchen.
Es kommt den Consumenten von Anilinfarben häufig vor, daß eine nach Muster beorderte Farbstoffsendung von weit geringerer Qualität ist, als man nach dem Muster hätte erwarten sollen; ebenso differiren nicht selten größere Bezüge unter sich. Um das fragliche Pigment zu untersuchen, wägt man von demselben genau so viel ab, als oben bei Fuchsin angegeben, nämlich 0,2 Gramm, bringt diese Menge in ein ebenfalls 120 Kubikcentim. haltendes, gut verschließbares Glasgefäß und läßt 50 Kubikcentimeter Normalcollodium einfließen. Bis die völlige Lösung erreicht ist, wird einigemale tüchtig umgeschüttelt.
Es ist nun klar, daß wenn der Farbstoff denselben Gehalt hat wie der normale, es noch 50 Kubikcentimeter Normalcollodium bedarf, um eine ebenso helle Platte wie die des letzteren zu geben; im anderen Falle wird die Zahl der Kubikcentimeter, welche bis zur Erreichung der Normalintensität noch auslaufen müssen, plus 50 Kubikcentimeter (d.h. der Lösungsmenge) direct proportional seyn der Intensität resp. dem Werth des Farbstoffes gegenüber demjenigen der Normalplatte, und ihn procentisch angeben, ähnlich wie dieß bei den verschiedensten alkalimetrischen und acidimetrischen Proben ebenfalls geschieht.
Man nimmt nach vollständiger Lösung des Farbstoffes (von oben) aus den Fläschchen, das man ganz nahe an die Normalplatte, die auf das Prisma gelegt wurde, bringt, möglichst rasch einen Tropfen Flüssigkeit heraus, und läßt ihn auf die Tafel, nahe der Normalfläche, jedoch auf unbedeckten Grund fallen. Das Gefäß mit der Lösung schließt man inzwischen zu, und vergleicht dann die Schichten mit einander nach dem vollständigen Trocknen, was in 1 bis 2 Minuten erfolgt ist. Ist die Normalfläche noch heller, so werden vorsichtig, aber schnell einige weitere Kubikcentimeter hinzugebracht und auf's Neue getupft.
Sobald die Nüance der beiden Schichten genau dieselbe ist, notirt man die Zahl der verbrauchten Kubikcentimeter Normalcollodium und hat dann, nach Zuzählung der Lösungsmenge, die man übrigens aus derselben Bürette fließen lassen und so direct ablesen kann, die Bestimmung vollendet.
Diese Collodiumlösung läßt bei einiger Uebung noch Differenzen von 1/2 und 1/4 Proc. erkennen. Soll ein Teig auf seinen Gehalt geprüft werden, so wägt man ebenfalls die bekannte Normalmenge (0,2 Gramm für je 100 Kubikcentimeter Normal-Collodium) ab und vergleicht ihn mit der Platte aus krystallisirtem Farbstoff; nur titrirt man statt von 50 Kubikcentimetern von 10 K. C. aus.
Wässerige oder weingeistige Lösungen werden eingedampft und wie Teige bestimmt.
Es gibt noch ein zweites Princip, nach welchem die Collodiummethode für Gehaltsprüfungen ausgeführt werden kann, darauf basirend, Normalplatten herzustellen, auf denen der Normalfarbstoff so aufgetragen ist, daß Decimalreihen von Intensitäts-Nüancen zur Vergleichung und directen procentischen Bestimmung entstehen, ohne daß es nöthig wäre, das zu untersuchende Pigment auf die Normal-Nüance zu titriren.
Derartige Platten, deren Rückseiten zur Erlangung einer bestimmten Menge von Decimalreihen nach einem eigenthümlichen Schema ebenfalls collodionirt werden, verlangen

TC-Vortrag 04 78/161 Waschen – Bleichen – Färben
indessen eine etwas zu sorgfältige Behandlung und Aufbewahrung, um der letzten Methode eine allgemeine Anwendung zu verschaffen.
Ich beschreibe sie aus diesem Grunde nicht eingehender, trotzdem die Tafeln, einmal hergestellt, was allerdings viel Mühe und Zeit in Anspruch nimmt, den positiven Gehalt eines zu untersuchenden Pigmentes mit großer Sicherheit angeben.
Noch sey kurz erwähnt die Anwendung der Collodiummethode zur Nüancebestimmung eines Farbstoffes ohne Rücksicht auf dessen Gehalt.
Es leuchtet ein, daß die auf einer Glasplatte fixirte Schicht, verglichen mit einer anderen, nicht nur eventuell eine Differenz in der Farbentiefe, sondern mit derselben Genauigkeit auch Nüancenunterschiede anzeigt, und es ist oft für den Fabrikanten von größter Wichtigkeit nach dieser Seite hin ebenfalls genauen Aufschluß zu erhalten.
Schon bei der Intensitätsbestimmung wird man also über diesen Punkt wenigstens annähernd klar werden.
Die Collodiummethode ermöglicht es jedoch, die meisten Farbstoffe nach ihren Nüancen ebenfalls in ein (relatives) Zahlensystem zu bringen, so daß der Werth derselben auch nach dieser Hinsicht gewissermaßen quantitativ, wenn der Ausdruck erlaubt ist, bestimmt werden kann.
Man bereitet sich Normallösungen aus krystallisirtem Fuchsin extragelblich, Reinviolett (sprit- oder wasserlöslich), Reinblau und Grün (ebenso eventuell Braun als Cerise oder Havanna, Phosphine u.s.w.) durch Abwägen von je 0,2 Grm. und Zusammenbringen mit 100 K. C. Normalcollodium.
Setzt man nun die Nüance obigen Fuchsins = 1, die des Violetts = 101, des Blaus = 202, des Grüns = 303 u.s.w. so liegen jedesmal in der Mitte zweier sich hier folgender Lösungen je 100 Uebergangsstufen, wovon jedem nicht normalen Farbstoff, als Mischung zweier anderer betrachtet, irgend eine zugehört.
Angenommen, es sey ein bläuliches Fuchsin, krystallisirt oder in Teigform, zu untersuchen.
Man wägt von demselben nach Bestimmung seiner Intensität ein Aequivalent der Normalmenge (0,2 Grm.) ab, löst in 100 K. C. Collodium auf, und macht auf einer Glasplatte einen Abguß (Nr. 1).
Hierauf werden zu 100 K. C. der Normal-Fuchsinlösung so lange Normal-Violettsolution aus einer Gay-Lussac'schen Bürette unter Beobachtung der bezüglichen Vorschriften hinzugefügt, bis ein letzter Tropfen der so veränderten Flüssigkeit eine, der Fläche Nr. 1 genau gleichgefärbte Schicht (Nr. 2) gibt.
Die verbrauchten K. C. Violettlösung sind nun, nach Zuzählung der Nüancezahl des Normalfarbstoffes, der einfachste Ausdruck der Stellung des zu untersuchenden Farbstoffes in der Zahlenreihe. Wenn bei obigem Beispiel die Normalfuchsinsolution 14 K. C. erfordert hätte, um eine Schicht gleich der des zu untersuchenden Farbstoffes zu erzeugen, so würde die Nüance des letzteren = 15 seyn. Ein zweites Fuchsin erfordere bloß 5 K. C. Violettlösung; seine Stellung in der Zahlenreihe wird also = 6 seyn und von dem vorhergehend untersuchten um eine bestimmte Nüance, deren relativer Ausdruck = 10 ist, nach Gelb zu abweichen.

TC-Vortrag 04 79/161 Waschen – Bleichen – Färben
Ist daher in der angedeuteten Weise die Nüance eines jeden Pigmentes, welches in größerer Menge verwendet werden soll, bestimmt worden, so hat man in den bezüglichen Resultaten einen ganz genauen Maaßstab für seine praktische Anwendbarkeit.
Aus Reimann's Färberzeitung, 1871, Nr. 38–46. Müller, Armand, Methode zur Werthbestimmung der Anilinfarbstoffe. Quelle: Polytech. Journal 1871, Band 202, Nr. CIX. (S. 458–463) URL: http://dingler.culture.hu-berlin.de/article/pj202/ar202109
Anilinrot
Fuchsin, Azaleïn, Mauve, Magenta, Roseïn, Tyralin, Rosanilin
Fuchsin ist ein roter Triphenylmethanfarbstoff, der in Alkohol (Ethanol) gelöst in der Mikroskopie und Histologie zum Färben verwendet wird. Fuchsin wurde 1858 von dem deutschen Chemiker August Wilhelm von Hofmann und fast zur gleichen Zeit von dem Lyoner Chemiker François-Emmanuel Verguin entdeckt und nach der amerikanischen Zierpflanze Fuchsie benannt, deren blaurote Blüten einen ähnlichen Farbton aufweisen. Es war der zweite großtechnisch hergestellte Teerfarbstoff.
Fuchsin wird durch Reaktion von 4-Aminobenzaldehyd, 4-Aminobenzylalkohol oder 4,4′-Diaminodiphenylmethan mit Anilin in Gegenwart von Oxidationsmitteln und Eisen(II)-chlorid hergestellt. Dabei entsteht außer dem Fuchsin als Verunreinigung auch Parafuchsin, welches ähnliche Eigenschaften besitzt.
Fuchsin bildet grüngelb metallisch glänzende Kristalle, die sich in Wasser und Alkohol langsam mit intensiv roter Farbe auflösen.
Das Fuchsin-Molekül stellt ein mesomeriestabilisiertes Kation dar, mit den Eigenschaften eines Cyaninfarbstoffs. Dies begründet die intensive Farbigkeit des Fuchsins.
Fuchsin wurde lange als Färbemittel für Wolle und Leder verwendet.
1895 wurde Fuchsin erstmals als Auslöser für Blasenkrebs beschrieben.
Fuchsin wirkt antimykotisch und antiseptisch bei grampositiven Bakterien, wird aber wegen des Verdachts auf Kanzerogenität weder am Menschen noch in der Veterinärmedizin eingesetzt.

TC-Vortrag 04 80/161 Waschen – Bleichen – Färben
Es wird in der Feulgenschen Nuclealreaktion eingesetzt, um DNA in Zellkernen oder im Kernäquivalent von Bakterien nachzuweisen. Es kann auch zur Chromosomenfärbung verwendet werden.
Fuchsin wurde auch in der Farbfotografie eingesetzt.
Basisches Fuchsin wird seit den 1960er Jahren verwendet, um Mikrorisse in Knochen in vitro zu färben und mit der Fluoreszenz des Farbstoffs zu identifizieren.
Fuchsin wird in der Technik zur Feststellung von Rissen in Bauteilen, z. B. Faserverbundbauteilen, verwendet.
Aufgrund der vorhandenen offenen Restporosität in Steinzeug, die durch den "Fuchsintest" nachgewiesen werden kann, lässt sich eine klare Unterscheidung zum Porzellan hin machen.
Als Fuchsinschweflige Säure wird es als Nachweismittel für Aldehyde verwendet. Dabei wird zu der magentafarbenen Lösung von Fuchsin etwas verdünnte Schweflige Säure zugegeben, bis sich die Flüssigkeit entfärbt hat. Beim Zugeben zu einem Aldehyd färbt sich die Lösung über blau und rot zu violett.
Ueber die Anwendung des Fuchsins in der Scharlachfärberei; von Carl Bulkowsky Assistent für chemische Technologie am k. k. Polytechnicum zu Wien.
Die Auffindung rother Pigmente oder einer einfachen Färbemethode, mittelst welcher man schnell und wohlfeil den Fasern einige der schönsten rothen Farbentöne von ziemlicher Beständigkeit ertheilen könnte, ist ein Problem, dessen Lösung eine tief empfundene Lücke der Colorie ausfüllen würde. Die Herstellung jener Farbentöne ist gegenwärtig entweder mit einem großen Aufwande an Zeit und Arbeit verknüpft, oder es sind hierzu sehr kostspielige Farbmaterialien erforderlich. Dieß gilt insbesondere für Scharlach, Amaranth und Nelkenroth; Farbentöne, welche durchgehends gelber als das Roth des Fuchsins sind. Zur Erzielung genannter Farben bedarf es heut zu Tage noch immer eines sehr geschickten und erfahrenen Färbers, also selbst zu einer Zeit wo die Chemie demselben rathend und helfend zur Seite steht; es darf uns daher nicht Wunder nehmen, daß dieser Theil der Färberei in früheren Zeiten geradezu als Kunst betrachtet wurde.
Die hellrothen Farbentöne sind für die farbigen Dessins der Gewebe ganz unentbehrlich; sie sind es ja vorzugsweise, welche ihnen Leben und Frische ertheilen, und daraus erklärt es sich, daß der auf die Herstellung dieser Farben bezugnehmende Theil der Färberei und des Zeugdruckes seit jeher von den Coloristen mit Vorliebe gepflegt wurde.
Die Natur der zu färbenden Faser bedingt die Anwendung eines ganz speciellen Farbstoffes und einer besonderen Färbe- oder Druckmethode. Zum Färben der Seide in den Nuancen Ponceau bis Nelkenroth steht die Carthaminsäure ausschließlich in Verwendung und kann trotz ihres hohen Preises nicht entbehrt werden, weil nur sie allein der Seide diese Farbentöne in erforderlicher Schönheit zu ertheilen vermag. Die Nachtheile, welche durch ihre Unechtheit bedingt sind, kommen viel weniger in Betracht, wie denn überhaupt die Mode an den Färber Anforderungen stellt, welche nicht immer die Echtheit, sondern großentheils die Schönheit einer Farbe betreffen.
Für Baumwolle werden der Krapp und die verschiedenen Krapppräparate benutzt, sobald es sich um die Herstellung der hellrothen Farbentöne handelt. In der Wollfärberei werden Cochenille, Lac-dye als die einzig tauglichen Farbstoffe für Scharlach, Amaranth und

TC-Vortrag 04 81/161 Waschen – Bleichen – Färben
Nelkenroth verwendet. Obgleich dieselben seit einiger Zeit bedeutend im Preise gesunken sind, so lassen sie sich doch noch immer nicht für wohlfeile Waaren gebrauchen.
Seltsamer Weise findet sich unter den so zahlreichen Theerfarben bis jetzt kein einziges Präparat, vor, welches einen der früher genannten Farbstoffe in dieser Richtung zu ersetzen oder ihre Anwendung in merkbarer Weise zu beschränken vermochte. In Bezug auf die vorhin erwähnten rothen Farbentöne unterscheidet sich die moderne Colorie von der älteren so gut wie gar nicht.
Die rothen Farbentöne, welche man mit Fuchsin und Peonin (rothem Corallin) erhält, nähern sich mehr dem Purpur und sind außerdem gegen die Einwirkung des Lichtes sehr empfindlich, während die mit Krapp und Cochenille erzeugten Farben frei von diesen Nachtheilen sind.
Bekanntlich war es der berühmte englische Farbenchemiker Bancroft, welcher dargethan hatte, daß die echte Scharlachfarbe mit Cochenille nur unter dem Einflusse gewisser Salze als Beizmittel hergestellt werden kann, und daß das unveränderte Pigment der Cochenille die Wollfaser nur carmoisin- und nicht scharlachroth färbe. Er stellte die Ansicht auf, daß diese Salze einen Theil desselben in einen gelben Farbstoff überführen, welcher mit dem ungeänderten Pigment vereint, das Scharlachroth auf der Faser erzeuge.
Diese Ansicht schienen auch seine Versuche zu bestätigen, und darauf gestützt, gelang es ihm ein neues Princip in der Scharlachfärberei einzuführen, nach welchem die Cochenille nicht für sich allein, sondern unter Mitbenutzung gelber Farbstoffe verwendet wird, und wobei alle jene Substanzen weggelassen werden, welche dem carmoisinrothen Cochenillepigment den gelblichen Ton verleihen.
Die Billigkeit des Fuchsins, die Leichtigkeit mit welcher dasselbe von der Thierfaser fixirt wird, gaben den Anstoß zu ähnlichen Experimenten. Man versuchte mit Fuchsin und Pikrinsäure (oder anderen gelben Farbstoffen) ein Scharlachroth von gleicher Schönheit zu erzielen, wodurch es möglich geworden wäre, auch billige Stoffe mit dieser Farbe zu versehen.
Es läßt sich a priori behaupten, daß eine aus Fuchsin und irgend einem gelben Pigment erhaltene Mischfarbe, bezüglich ihrer Echtheit der Cochenillefarbe nachstehen muß; dagegen erscheint es ebenso gewiß, daß sich auf diesem Wege beständigere Farben erhalten ließen, wenn wir einen dem Fuchsin in der Farbe gleichkommenden, jedoch echteren Theerfarbstoff besäßen, da es uns an echten gelben Pigmenten nicht mangelt.
Die Lücke in der Farbenreihe der künstlichen Pigmente, unter denen sich kein zweckentsprechendes Scharlachpräparat vorfindet, wäre sodann gewissermaßen ausgefüllt, weil man auf indirectem Wege mit einem purpurrothen Farbstoffe denselben Effect erzielen könnte. Leider besitzen wir unter den rothen Theerfarbstoffen keinen, welcher die Echtheit des Cochenillecarmoisins besäße, und mit Fuchsin lassen sich somit nur unechte Mischfarben erhalten.
Um an Cochenille zu sparen, haben die Wollfärber die hellrothen Farbenabstufungen häufig in folgender Weise hervorgebracht: Der Strangwolle wurde ein aurorafarbiger Grund mit Cochenille ertheilt und der gewünschte Farbenton durch Nachfärben mit Fuchsin gegeben. Diese Methode erfüllt ihren Zweck nur theilweise, weil diese Farben den mit Cochenille erhaltenen an Feuer und Reinheit bedeutend nachstehen.

TC-Vortrag 04 82/161 Waschen – Bleichen – Färben
seit einiger Zeit wird jedoch scharlach- und amaranthrothes Tuch aus England importirt, dessen Farbenton nichts zu wünschen übrig läßt, und welches dennoch nicht mit Cochenille, sondern mit Fuchsin gefärbt seyn soll. Die Billigkeit dieser Fabricate zwang die Schönfärber zu Versuchen, das Fuchsin in die Scharlachfärberei einzuführen, welche aber wie es scheint nicht zum Ziele geführt haben, weil meines Wissens weder in den technischen Journalen noch in den Kreisen competenter Fachmänner etwas über die Verwendbarkeit des Fuchsins in genannter Richtung verlautete. Im Gegentheile, ich wurde von Färbern und Fabrikanten öfter zu Rathe gezogen, welche sich mit derartigen Versuchen befaßten und zu ihrer größten Ueberraschung die unangenehme Entdeckung machten, daß das Fuchsin mit gelben Pigmenten combinirt, dem Tuche in den meisten Fällen eine Mißfarbe ertheilt, ohne daß sie die Ursache dieses eigenthümlichen Verhaltens ergründen konnten. In manchen Fällen gelang es allerdings, dem Stoffe die gewünschte Farbe zu ertheilen; es glückte jedoch nicht, den Bedingungen auf die Spur zu kommen, unter welchen stets dasselbe Resultat zu erlangen ist.
Ich hatte mich mit diesem Gegenstande schon früher beschäftigt und stieß genau auf dieselben Schwierigkeiten, so daß ich wahrscheinlich von weiteren Versuchen abgestanden wäre, wenn ich nicht in Erfahrung gebracht hätte, daß das Fuchsin in einigen englischen Etablissements für die Scharlachfärberei Verwendung finde. Durch fortgesetzte Versuche bin ich endlich dahin gelangt, Schafwollentuch mit Fuchsin in den Nuancen: Scharlach, Amaranth und Nelkenroth ebenso schön als mit Cochenille zu färben.39)
Ohne in die Einzelheiten meiner Untersuchungen einzugehen, will ich hier nur deren Resultate kurz erwähnen:
Eine wässerige Fuchsinlösung, von etwas beträchtlicher Concentration, ertheilt dem mit Pikrinsäure grundirten Tuche immer eine Mißfarbe, namentlich dann, wenn die Temperatur der Färbeflotte gering ist. Wendet man hingegen eine stark verdünnte Fuchsinlösung an, so erhält Man eine ziemlich hübsche Scharlachfarbe. Tiefere Nuancen, wie z. B. Amaranth, lassen sich auf diese Weise nicht erhalten. Genau dieselben Erscheinungen finden statt, wenn man zum Grundiren anstatt der Pikrinsäure irgend ein Salz des Dinitronaphtols (Naphtalingelb, Martiusgelb) verwendet.
Schon in mäßig concentrirter Fuchsinlösung schlägt sich auf der Faser ein schwer lösliches Rosanilinsalz der Pikrinsäure, beziehungsweise des Dinitronaphtols nieder; jedes Fäserchen erscheint stellenweise mit einem bronzeartigen Ueberzug versehen. Die Mißfarbe, welche hierdurch zum Vorschein kommt, ist offenbar durch die Farbe und den Metallglanz des niedergeschlagenen Rosanilinsalzes bedingt.
Obwohl man diesen Uebelstand durch Anwendung sehr verdünnter Fuchsinlösungen vermeiden kann, so ist diese Färbemethode denn doch nicht für die Praxis geeignet. Das Färben erfordert zu lange Zeit und ist zu umständlich. Man muß mit der Zugabe von Fuchsin in die erschöpfte Färbeflotte äußerst behutsam zu Werke gehen, wenn man nicht Gefahr laufen will, die Waare gänzlich zu verderben.
Es ist eine bekannte Thatsache, daß das Fuchsin nur in alkalischer Lösung der Woll- und Seidenfaser eine schöne, feurige und satte Farbe ertheilt. Von diesem Kunstgriffe machen die Seidenfärber auch wirklich einen ausgedehnten Gebrauch, denn sie setzen ihren Färbebädern immer eine gewisse Menge Marseiller Seife zu.

TC-Vortrag 04 83/161 Waschen – Bleichen – Färben
Wird Tuch mit Pikrinsäure oder Naphtalingelb grundirt, so ist die Anwendung alkalischer Fuchsinlösungen ausgeschlossen, denn in einem solchen Falle werden beide von der Faser fast ganz abgezogen.
Will man einen seifenechten Grund, so muß man zu anderen gelben Pigmenten seine Zuflucht nehmen, und insbesondere auf jene Rücksicht nehmen mit welchen man das glänzendste Goldgelb erzielen kann. Das Waugelb ist somit ganz unbrauchbar, weil es bekanntlich einen grünlichen Stich besitzt. Das reine Goldgelb, also ein Gelb mit einer ganz kleinen Beimischung von Orange, läßt die rothen Farben am reinsten erscheinen.
Diesen Farbenton erhält man, wie zahlreiche Versuche gezeigt haben, am besten aus dem Farbstoff der Kreuzbeeren; auch mit Flavin lassen sich ziemlich schöne Effecte erzielen, jedoch muß dem ersteren Farbmateriale der Vorzug gegeben werden.
Der gelbe Grund wird dem Tuche in folgender Weise ertheilt:
Zuerst wird dasselbe einer sorgfältigen Reinigung durch Waschen u. s. w. unterzogen, dann ungefähr eine Stunde lang mit raffinirtem Weinstein, Zinnchlorid und Alaun angesotten. Der angebeizte Stoff wird nachher gereinigt und bei einer Temperatur von etwa 80° C. in einer mit Kreuzbeerextract, beziehungsweise mit Flavin versetzten Färbeflotte bis zu der erforderlichen Farbentiefe ausgefärbt, sodann in Wasser gut gespült, bis das Waschwasser vollkommen klar ablauft.
Alle vorher geschilderten Schwierigkeiten beim sogenannten Rötyen des gelb grundirten Tuches fallen hinweg, wenn man das Färbebad in nachstehender Weise bereitet:
Auf 1000 Gewichtstheile Wasser, welches in der Färbekufe durch einen Dampfstrom auf 50 bis 60° C. erwärmt werden muß, gibt man 1,7 Gewichtstheile krystallisirte Soda und 0,145 Gewichtstheile Diamantfuchsin. Letzteres wird in Form einer weingeistigen oder wässerigen Lösung zugefügt. Die Soda ist für die Erzielung schöner Farben unerläßlich; sie bewirkt eine Zersetzung des Fuchsins, die Base wird in Freiheit gesetzt, verbleibt jedoch bei diesem Grade der Verdünnung gelöst. Die Fuchsinlösung verliert hierdurch ihre tiefrothe Farbe und erhält eine dem lichten Biere ähnliche Färbung. Diese Flüssigkeit besitzt nicht die unangenehme Eigenschaft rein wässeriger Fuchsinlösungen, den Farbstoff beim Erkalten in Form eines metallisch-glänzenden Häutchens an der Oberfläche auszuscheiden, welches sich beim Herausziehen der Gewebe an der Faser anlegt, durch kein Mittel zu entfernen ist und die Waare verdirbt.
In dem auf die angegebene Weise bereiteten Färbebade wird das Tuch bei einer Temperatur von 55° C. mittelst des Haspels hin- und herbewegt. Im Anfange erhält dasselbe eine Mißfarbe, erst später, nachdem die Flüssigkeit bis in das. Innerste der Faser gedrungen, kommt ein äußerst lebhaftes und glänzendes Scharlach zum Vorschein, welches, wenn die Operation nicht unterbrochen wird, in Amaranth und endlich in Nelkenroth übergeht.
Es ist sehr interessant zu sehen, wie eine Flüssigkeit von so geringer Färbung, so intensive Farben zu geben im Stande ist. Nachdem aber bekanntlich das Rosanilinhydrat einen farblosen Körper darstellt, so ist dieser Umstand einigermaßen befremdend und es hat den Anschein, als ob sich im Inneren der Faser der ursprüngliche Farbstoff regeneriren würde; wenigstens läßt diese Erscheinung vorderhand keine andere Deutung zu.
Es ist selbstverständlich, daß in dem Maaße als das Färbebad erschöpft wird, eine Nachspeisung desselben mit Fuchsinlösung, nöthigenfalls auch mit Soda vorgenommen

TC-Vortrag 04 84/161 Waschen – Bleichen – Färben
werden muß. Die Einhaltung der Temperatur zwischen 50 bis 55° C. ist für das Gelingen dieser Operation sehr wesentlich. Kochhitze ist ganz zu vermeiden, weil durch sie nur magere Farben zum Vorschein kommen. Bei niederer Temperatur geht das Färben zu langsam von Statten, in Folge dessen der gelbe Farbstoff von der alkalischen Flüssigkeit in bemerkbarer Weise abgezogen wird. Die Erzielung gelbrother Farbentöne erscheint dann nicht mehr möglich.
Nach erfolgter Färbung muß das Tuch mit Wasser gewaschen werden und hierbei zeigt sich die interessante Erscheinung, daß der in dieser Weise fixirte Farbstoff der lösenden Einwirkung des Wassers großen Widerstand entgegensetzt, während ein mit Fuchsin substantiv gefärbtes Wollgewebe das Waschwasser sehr stark röthet. Im ersteren Falle laufen die Waschwässer fast wasserklar ab.
Die nächste Operation, das warme Pressen, welchem das Tuch unterworfen werden muß, bildet eine gefährliche Klippe, woran viele Bemühungen gescheitert sind, welche die Einführung des Fuchsins in die Scharlachfärberei zum Zwecke hatten. Eine der unangenehmsten Eigenschaften des Fuchsins ist nämlich die, daß es auf den Stoffen bedeutend nachdunkelt und an Feuer einbüßt, sobald die Temperatur beim Pressen eine gewisse Höhe überschreitet; ein Nachtheil, welcher sich schwer vermeiden läßt, wenn die Preßplatten mit Ofen und nicht mittelst Dampf erhitzt werden. Im vorliegenden Falle dürfen nur hydraulische Pressen zur Anwendung kommen, bei welchen die Preßplatten mit Dampf von geringer Spannung erwärmt werden können; der Ausfall an Wärme muß durch einen höheren Druck ersetzt werden.
Unter Einhaltung aller der früher genannten Bedingungen kann das Fuchsin zur Erzielung der hellrothen Farbentöne verwendet werden, welche man bisher in dieser Reinheit nur mit Cochenille erhalten konnte. Dieß gilt jedoch nur für Tuch; denn ungefilzte Gewebe, wie z. B. Wollmousseline, in der nämlichen Weise behandelt, erhalten äußerst magere Farben.
Emil Saloschin in Brighouse (Yorkshire) gibt eine Erklärung, warum das Fuchsin nahezu alles Gelb, wenn es auch noch so voll gefärbt war, gewissermaßen verzehrt, so daß die resultirende Farbe dünn und fadenscheinig aussieht. Die Ursache dessen liegt seiner Ansicht zufolge in dem violetten Stiche des Fuchsinrothes, welcher einen großen Theil des Gelb zu Weiß ergänzt; der Rest ist ein mageres Roth, welches nur bei gefilzten Geweben in Folge größerer Massenwirtung voll und satt erscheinen kann.
Die Farbe welche das Fuchsinroth zu Scharlach ergänzt, ist eigentlich ein hohes Orange und nicht Goldgelb. Für Seide, wo jene Farbe leicht mit Orlean gegeben werden kann, läßt sich eine Scharlachfarbe durch Decken mit Fuchsin leicht hervorbringen. Für Wolle haben wir keinen Farbstoff, der ein schönes, glänzendes Orange liefern würde (Krapp und Garancin geben viel zu matte Farbentöne); wir sind daher noch nicht im Stande, für ungefilzte Gewebe das Fuchsin in genannter Richtung zu verwenden.
Wird ein mit Kreuzbeerextract grundirtes Tuch mit rothem Corallin (Peonin) nachgefärbt, so erhält man ein äußerst lebhaftes Mennigroth (Tunis). Der Unterschied zwischen diesem und dem früher angeführten Verfahren ist durch die Natur dieses Farbstoffes bedingt, und besteht nur darin, daß man die Färbeflotte anstatt mit Soda, mit etwas Marseiller Seife alkalisch machen, und den Farbstoff in Form einer weingeistigen Lösung zufügen muß.

TC-Vortrag 04 85/161 Waschen – Bleichen – Färben
Bulkowsky, Carl, über die Anwendung des Fuchsins in der Scharlachfärberei. Quelle: Polytech. Journal 1869, Band 192, Nr. XXXIV. (S. 142–148) URL: http://dingler.culture.hu-berlin.de/article/pj192/ar192034
Anilinviolett
Anlilinpurpur, Anileïn, Indisin, Phenameïn, Harmalin, Violin, Rosolan, Mauveïn
Mauveïn ist ein basischer Azinfarbstoff in der namensgebenden Farbe mauve. Chemisch handelt es sich um ein Phenazin-Derivat, das dem Safranin eng verwandt ist. Die Verbindung dient(e) in erster Linie als Textilfarbstoff.
William Henry Perkin entdeckte es im Alter von nur 18 Jahren bei dem Versuch, Chinin zu synthetisieren, im Jahr 1856. Er stellte diese Substanz aus Anilin her, das mit Kaliumdichromat oxidiert wurde. Das von ihm verwendete Anilin enthielt allerdings erhebliche Mengen an o- und p-Toluidin, so dass das erhaltene Produkt ein Gemisch aus Mauvein und Pseudomauvein war.
Perkins erstes gefärbtes Stück Stoff (so will es die Legende), war eine vorher weiße Bluse seiner Schwester, die dann in schönster leuchtender Malvenfarbe erstrahlte. Bis Ende des 19., Anfang des 20. Jahrhunderts wurden die englischen Penny-Briefmarken mit Mauvein gefärbt. Heute hat Mauveïn keine Bedeutung mehr als Farbstoff.
Anilinblau
Azulin, Azurin
XVII. Ueber die Darstellung von Anilinblau für die Färberei und den Zeugdruck, durch Behandlung von Anilinroth mit Aldehyd; von C. Lauth.
Aus dem Répertoire de Chimie appliquée, Juli 1861, t. III p. 273.
Die Vergleichung der Formel des Anilinroths C³⁶ H²⁰ N⁴ O⁵ derjenigen des Anilinvioletts C³⁶ H¹⁷ N³ O² brachte mich auf den Gedanken, daß man letzteres durch Behandlung des Anilinroths mit reducirenden Substanzen erhalten könne. Nachdem eine Reihe von Reactionen kein günstiges Resultat gegeben hatte, stellte ich Versuche mit dem durch Erhitzen des salpetersauren Anilins bereiteten Anilinroth, dem sogenannten Anilein, an: alle gewöhnlichen Reductionsmittel zerstören diesen Farbstoff, keines veranlaßt jedoch die Bildung von Anilinviolett.

TC-Vortrag 04 86/161 Waschen – Bleichen – Färben
Versetzt man aber das Anilein (oder das Azalein, das Fuchsin, das durch Behandlung des Anilins mit Arsensäure oder mit gewissen Reductionsmitteln dargestellte Anilinroth) als Auflösung in käuflichem Alkohol oder Holzgeist, oder auch in käuflicher Essigsäure, mit Zinnchlorür (Zinnsalz in Stücken) und erhitzt zum Kochen, so verschwindet die rothe Farbe nach und nach, und wird durch eine violette ersetzt, welche endlich vollständig blau wird.
Wie das Zinnfalz wirken auf das in Alkohol aufgelöste Anilein eine Menge von Substanzen, die meisten mineralischen und organischen Säuren, und die sauren Salze, welche durch Wasser in basische Salze und freie Säure zerlegt werden.
Es war nun klar, daß die Eigenschaft das Roth in Blau umzuwandeln, nicht dem Alkohol oder den ihn stets begleitenden Substanzen, angehört; von mir angestellte Versuche ergaben, daß die Wirkung durch eine fremde Substanz hervorgebracht wird.
Diese Substanz ist das Aldehyd C⁴ H⁴ O². Man macht eine Auflösung von Anilein (Fuchsin oder irgend einem Anilinroth) in Schwefelsäure und aetzt ihr eine kleine Menge reines Aldehyd zu; nach einstündiger Berührung neutralisirt man die Flüssigkeit mit reinem Natron, und erhält dann ein außerordentlich blaues Violett. Bei länger fortdauernder Berührung dieser Substanzen verschwindet die rothe Nüance vollständig und wird durch ein sehr reines Blau ersetzt.
Die Mutterlauge enthält eine beträchtliche Menge Ammoniak. Wie das Aldehyd wirken, ebenfalls in der Kälte, mehr oder weniger rasch, die meisten natürlichen wesentlichen Oele, das Rautenöl, Anisöl etc.
Das so erhaltene Anilinblau hat folgende Eigenschaften: es ist in Wasser, Alkohol, Essigsäure, sowie in Glycerin vollständig löslich, welchen es eine blaue, in Violett stechende Farbe ertheilt; aus diesen Auflösungen setzt es sich in Form sehr glänzender bronzefarbiger Blättchen ab. Es löst sich mit gelber Farbe in concentrirter Schwefelsäure, concentrirter Salzsäure, in verdünnter Salpetersäure, überhaupt in allen Säuren auf; aus diesen Auflösungen wird es durch die Alkalien gefällt. In den ätzenden und kohlensauren Alkalien löst es sich ebenfalls mit gelber Farbe auf, ohne durch dieselben verändert zu werden; diese Auflösungen werden durch die Säuren gefällt. In wässerigen Kochsalzlösungen ist es vollkommen unauflöslich; aus seinen Auflösungen in Wasser und sogar in Alkohol wird es durch Zusatz einer geringen Menge Gerbstoff gefällt, mit welchem es eine Verbindung eingeht.
Das Anilinblau wird durch eine Temperatur von 200° C. zerstört.
Es ist für die Färberei und den Zeugdruck ausgezeichnet geeignet, und gibt auf Seide, Wolle und Baumwolle sehr lebhafte Nuancen.
Lauth und Depouilly ließen sich bekanntlich das durch Behandlung des Anilins mit Salpetersäure erhaltene Roth unter dem Namen Anilein patentiren. Nach den Analysen von E. Kopp und Jacquemain ist bei der Erzeugung dieses Farbstoffs in 3 Aeq. Anilin 1 Aeq. NO⁴ Nitryl eingetreten, dagegen 1 Aeq. Wasserstoff ausgetreten; Lauth benennt daher dieses Anilinroth trianiline mononitrée. Man sehe über die Darstellung des Anileins die Angaben im polytechn. Journal Bd. CLVIII S. 147 und Bd. CLIX S. 451.
A. d. Red.
Lauth hat die beschriebenen Verfahrungsarten zur Darstellung von Anilinviolett und Anilinblau, um sich die Priorität der Entdeckung zu sichern, in drei versiegelten Packeten am

TC-Vortrag 04 87/161 Waschen – Bleichen – Färben
24. December 1860, 10. Januar und 4. März 1861 bei der Mülhauser Industriegesellschaft hinterlegt. Dieselben wurden am 26. Juni d. J. auf sein Verlangen geöffnet und deren Inhalt im Bulletin de la Société industrielle de Mulhouse t. XXXI p. 374–376 mitgetheilt.
Quelle: Lauth, über Darstellung von Anilinblau für die Färberei und den Zeugdruck. Polytechnisches Journal 1861, Band 162, Nr. XVII. (S. 55–57) URL: http://dingler.culture.hu-berlin.de/article/pj162/ar162017
Der neue Farbstoff hat wegen seiner bequemen Handhabung sehr bald Eingang in den Wollfärbereien gefunden; er eignet sich zu vielen anderen Zwecken, z.B. zum Färben des Papiers in der Masse, zum Farben von Horn u.s.w. – Das ebenfalls zur Vorlage kommende "„wasserlösliche Jodviolett“" oder "„Primula“" von derselben Firma scheint in ähnlicher Weise dargestellt zu seyn.
Um lösliches Indigblau von dem von mir untersuchten wasserlöslichen Anilinblau zu unterscheiden, zwei Körper, welche gegen sehr viele Reagentien sich ganz ähnlich verhalten, so z.B. von Zinkfeile, besser noch von Zinkstaub in saurer, neutraler und alkalischer Lösung, ferner von Schwefelammonium leicht reducirt werden, obgleich in der Schnelligkeit, womit dieß geschieht, schon eine Verschiedenheit sich zeigt, wende ich mit bestem Erfolg 8–10 procentige Natronlauge an, welche bei anhaltendem Kochen die beiden löslichen Indigblau so verändert, daß beim Ansäuren mit Essigsäure die blaue Farbe nicht wieder erscheint, sondern zuweilen eine grünliche, meist aber kirschrothe Färbung an deren Stelle tritt, während das lösliche Anilinblau nur schwierig seine Farbe verliert, und dieselbe sofort oder nach mehrtägigem Stehen auf Zusatz von Essigsäure oder Salzsäure wieder annimmt. (Vom Verf. aus den "„Sitzungsberichten der Isis zu Dresden“" mitgetheilt.)
aus: Ueber die Darstellung und die Eigenschaften von in Wasser löslichem Anilinblau; von Assistent Naschold. 1868, Band 187/Miszelle 5 (S. 356–358)
URL: http://dingler.culture.hu-berlin.de/article/pj187/mi187mi04_5
Anilinblau auf Seide und Baumwolle.
Das Anilinblau kommt jetzt im Handel in metallisch glänzenden feinen Krystallen vor und liefert leicht eine prachtvolle Farbe auf Seide und Wolle, auch, wenn auch schwieriger, auf Baumwolle. Die Farben sind echt und mehr blau als die, welche das bekannte Chinolinblau liefert. Der Farbstoff ist in Wasser nicht löslich und muß daher vorher in 90 bis 95grädigem Spiritus aufgelöst werden.
Die Seide wird mit Alaun und Weinstein gebeizt und dann in dem Bade von Anilinblau bei 40–50° R. ausgefärbt. Einige Färber begnügen sich damit, die Seide in einem schwachen Soda- oder Seifenbade zu waschen und dann direct zu färben; dieß ist jedoch nicht zu empfehlen, da die Verbindung des Farbstoffes mit der Faser nicht fest genug wird.
Um Baumwolle anilinblau zu färben, muß man dieselbe recht stark beizen. Sie wird zuerst durch ein Sodabad genommen, sodann mit Thonerdenatron ungefähr 3 Stunden lang gebeizt und zuletzt durch eine Salmiaklösung genommen, um die Thonerde frei zu machen. Nach zweistündigem Liegen kann man in gewöhnlicher Weise zum Ausfärben schreiten. (Deutsche Musterzeitung, 1863, Nr. 1.)
Methode zum Färben mit wasserlöslichem Anilinblau auf Wolle; von Lachmann und Breuninger in Glauchau (Sachsen).

TC-Vortrag 04 88/161 Waschen – Bleichen – Färben
Ein Haupterforderniß beim Färben von Stoffen, sey es nun von Wolle, Seide, Baumwolle oder Leinen, ist, daß die Farbe nicht bloß rein und glänzend, sondern auch vollkommen gleichartig auf die Faser aufgebracht werde. Die Stoffe sollen, wie man sich auszudrücken pflegt, egal gefärbt seyn.
Beim Färben mit wasserlöslichen Anilinfarben, namentlich mit wasserlöslichem Anilinblau auf Wolle, bietet obiger Umstand nun eine Schwierigkeit, welche bis jetzt die Veranlassung war, daß das letztere das in Spiritus auflösliche Anilinblau noch nicht verdrängen konnte, weil in vielen Färbereien die Mehrausgabe für den Spiritus der Möglichkeit eines Mißlingens beim Färben mit wasserlöslichem Blau vorgezogen wird.
Forscht man der Ursache obiger Schwierigkeit nach, so findet man, daß sie in der zu großen Verwandtschaft der Wollfaser zu dem wasserlöslichen Blau liegt.
Ein Zusatz von in Spiritus gelöstem Blau zu einem Färbebade zeigt zunächst die Erscheinung, daß der Farbstoff als in Wasser unlöslich, oder vielmehr sehr schwer löslich, in den kleinsten Atomen sich ausscheidet und in der Flotte suspendirt schwimmt, so daß diese Atome nur ganz langsam und bloß bei längerem Kochen sich auflösen und nach und nach sich mit der Wolle vereinigen, wodurch die Egalität erzielt wird. Das wasserlösliche Blau dagegen fällt, da es vollständig in der sauren Flotte aufgelöst ist, sofort auf die Wolle; ein gleichförmiges Durchdringen der Wollpartikelchen mit der Farbstofflösung ist nicht möglich, da die Farbe auf ihrem Wege an den zunächst liegenden Theilen der Faser abgesetzt wird, ehe sie zu den weniger zugänglichen Partien gelangt. Die Folge ist Unegalität.
Wenn es nun eine Methode gäbe, welche den Farbstoff des wasserlöslichen Blau's, wie den des spirituslöslichen langsam aufgehen ließe, so sollte man denken, daß ebenfalls eine Egalität erzielt werden müßte, und diese Schlußfolgerung hat sich auch bei untenstehender Behandlungsweise bestätigt.
Das wasserlösliche Blau zeigt in seiner chemischen Constitution das gleiche Verhältniß zum spirituslöslichen Blau, wie der Indigocarmin zum Indigo. Es ist ein anilinblauschwefelsaures Salz, wie der Indigocarmin ein indigoschwefelsaures Salz ist. Das neutrale anilinblauschwefelsaure Salz ist aber nicht rein blau gefärbt, sondern erhält diesen Farbeton erst, wenn durch Zusatz einer stärkeren Säure die Basis dieses Salzes weggenommen wurde und reine Anilinblauschwefelsäure sich abgeschieden hat, wie es stets beim Zusatz desselben zu den sauren Färbeflotten der Fall ist.
Die neutrale, nicht mit Säure versetzte Auflösung des wasserlöslichen Anilinblau's hat nun die Eigenschaft, langsam und deßhalb ganz egal auf die Wolle aufzugehen. Wolle, in solche Lösung getaucht, braucht längere Zeit, um sich mit demselben zu verbinden.
Da das neutrale Salz aber nicht blau ist, sondern einen lichtgrauen Ton besitzt, so ist natürlich dann auch die Wolle bloß licht-graublau gefärbt. Taucht man jedoch die so gefärbte Wolle nun in ein saures Bad, so zeigt sich dieselbe wie mit einem Zauberschlage auf einmal blaugefärbt, und zwar, weil das Auffärben des neutralen Salzes langsam geschah, ganz egal, und was die Dunkelheit des Tons betrifft, so ist derselbe entsprechend der Zeitdauer der Einwirkung der neutralen Flotte.
Will man nun diese Methode in der Praxis anwenden, so sind zwei Gefäße erforderlich. In dem einen ist eine ziemlich concentrirte neutrale Auflösung von wasserlöslichem Anilinblau befindlich.

TC-Vortrag 04 89/161 Waschen – Bleichen – Färben
Es ist gut, dieselbe immer stark zu halten, damit die Dauer der Einwirkung, um den gewünschten Ton zu erreichen, verkürzt wird.
Man löst zu diesem Behufe wenigstens 1 Pfund auf circa 500 Pfd. reinen warmen Wassers auf, rührt in das ganz säurefreie Wasser, das nicht kochend, sondern bloß warm zu seyn braucht, ein, und läßt nun die Wolle eintauchen. Bei einiger Uebung ist man bald im Klaren über den Zeitpunkt, bis zu welchem die Einwirkung stattzufinden hat; eine kleine Probe, in heißes saures Wasser getaucht, belehrt alsbald darüber. Ist derselbe erreicht, so läßt man die Wolle über der neutralen Flotte etwas abtropfen, um davon, da sie noch bedeutend Farbstoff enthält, nichts zu verlieren, und bringt sie dann in das Gefäß, welches kochende saure Flotte enthält. Ein kurzes Kochen, und die Arbeit des Färbens ist ohne irgend welche Mängel fertig.
Entspricht die Nüance des wasserlöslichen Blau's nicht dem gewünschten Muster, will man z.B. röthlich nüanciren, so läßt man die Einwirkung der ersten Flotte nicht bis zur verlangten Dunkelheit des Tons währen, sondern hält etwas lichter, und nüancirt in der zweiten sauren Flotte mit dem entsprechenden spirituösen Rothstichblau aus.
Quelle: Methode zum Färben mit wasserlöslichem Anilinblau auf Wolle. Lachmann, Breuninger, Polytechnisches Journal 1866, Band 182, Nr. LXV. (S. 235–237) URL: http://dingler.culture.hu-berlin.de/article/pj182/ar182065
Anilingrün
Aldehydgrün, Emeraldin, Jodgrün
Färben von Anilingrün auf Wolle.
Zum Färben von Anilingrün auf Wolle empfiehlt Ch. Lauth, diese in einem Bade vorzubereiten, dem unterschwefligsaures Natron und eine Säure oder ein saures Salz zugesetzt wird; dabei schlägt sich auf der Wolle Schwefel nieder, welcher dieselbe zur Aufnahme des Anilingrüns geeignet macht. Die Wolle verliert dabei, jedenfalls in Folge des Eindringens des weichen, zähen Schwefels in die Fasern, ihre Elasticität, wird weich und zieht sich stark zusammen; dieß läßt sich aber dadurch vermeiden, daß man dem Bade eine kleine Menge Alaun oder eines Zinksalzes zusetzt. Nicht alle Formen des Schwefels besitzen die Eigenschaft, als Beize für Anilingrün dienen zu können; so ist z.B. eine Lösung von Schwefel in Schwefelkohlenstoff in dieser Beziehung, ganz wirkungslos. Vor der Behandlung mit unterschwefligsaurem Natron muß die Wolle entfettet und durch schwache Salzsäure von allen Metallverbindungen gereinigt werden, die sie beim Spinnen und Weben aufgenommen haben könnte; wird dieß übersehen, so entstehen im Schwefelbade leicht braune Flecken in Folge der Bildung von Schwefelmetallen. Das Färben erfolgt einfach in der Weise, daß man die nach dem Beizen gut ausgewaschene Wolle in eine Lösung von Anilingrün in warmem Wasser bringt, die allmählich auf ca. 100°C. erwärmt wird. (Deutsche Industriezeitung, 1873, Nr. 41.)
Anilingelb
Anilionorange – Aurin Chrysanilin,
Anilingelb (p-Aminoazobenzol) ist ein gelber Azofarbstoff. Es ist ein Derivat des Azobenzols und gleichzeitig ein aromatisches Amin.

TC-Vortrag 04 90/161 Waschen – Bleichen – Färben
Darstellung des Anilingelb; von Dr. Hugo Schiff.
Es ist mir gelungen, das Anilingelb durch Einwirkung der Hydrate von Antimonsäure und Zinnsäure auf Anilin in größerer Menge darzustellen.
Man reibt ein gepulvertes Alkalisalz einer dieser Säuren mit dem halben Gewicht Anilin zu einem dünnen Brei an, und versetzt denselben allmählich unter Umrühren so lange mit Salzsäure, bis die Flüssigkeit stark sauer reagirt. Das Anilin wird sogleich in den scharlachrothen Farbstoff umgewandelt, und dieser läßt sich nach dem Eintrocknen der Masse mit Aether-Alkohol ausziehen. Ich umgehe hier das weitere Reinigungsverfahren und gebe nur an, daß die rothe ätherische Lösung der Salzsäureverbindung beim Verdunsten kantharidenglänzende Blättchen des Salzes liefert. Andere Salze werden auf ähnliche Weise erhalten. Letztere lösen sich in Alkohol, in Aether und in angesäuertem Wasser, während eine größere Menge reinen Wassers zersetzend wirkt. Alkalien zersetzen die Salze unter Abscheidung eines intensiv gelben flockigen Körpers, welcher mit Säuren wieder die rothen Verbindungen entstehen läßt.
Tränkt man Seide oder Wolle mit der rothen schwach sauren Lösung und bringt den Stoff dann in eine verdünnte heiße Sodalösung, so erhält man eine intensiv gelbe Färbung, welche sehr haltbar ist und etwa die Nüance des Pikringelb zeigt. Da das als Zinnbeize käufliche zinnsaure Natron zur Darstellung der rothen Verbindung dienen kann und diese schon bei gewöhnlicher Temperatur entsteht, so zweifle ich nicht, daß dieses Verfahren zur technischen Gewinnung eines Anilingelb Anwendung finden könnte. Das Verfahren würde besonders geeignet seyn, um die gelbe Färbung sogleich auf dem Stoffe selbst zu erzeugen. – Analysen der rothen und gelben Verbindung habe ich bis jetzt noch nicht ausführen können. (Annalen der Chemie und Pharmacie, September 1863, Bd. CXXVII S. 345.)
Quelle: Polytech. Journal 1863, Band 170/Miszelle 11 (S. 157–158) URL: http://dingler.culture.hu-berlin.de/article/pj170/mi170mi02_11
Pseudo-Anilinfarben (sogenanntes "„Anilingelb“" und "„Anilingrün“"); von Dr. Emil Jacobsen.
Es werden gegenwärtig in Berlin gelbe und grüne Farbstofflösungen fälschlich als Anilinfarben angeboten; die eine, das "„Anilingelb“", angeblich ein englisches Fabrikat, stellt eine stark nach Safran riechende, orangegelbe Flüssigkeit dar, die beim Stehen einen flockigen, schmutzig gelben Bodensatz abgibt. Sie besteht aus einer spirituösen Lösung von Pikrinsäure, durch Zusatz von Safrantinktur orange gefärbt. Das sogenannte "„Anilingrün“" ist ein Gemisch von Pikrinsäure und Anilinblau, in Alkohol gelöst. Es ist eine dunkel saftgrüne Flüssigkeit. Der bittere Geschmack läßt in beiden vermeintlichen Anilinfarben die Pikrinsäure erkennen. Setzt man zu einer Probe dieses Anilingrüns einige Tropfen Salzsäure, so schwindet die gelbe Farbe (Pikrinsäure erscheint in salzsäurehaltigem Wasser fast farblos) und der blaue Farbstoff wird sichtbar. Setzt man zum sogenannten Anilingelb in gleicher Weise Salzsäure, so verliert er sichtlich an Intensität und erscheint rein safrangelb. (Aus des Verf. chemisch-technischem Repertorium, II. Jahrg. 1. Halbj. S. 16.)
Quelle: Polytech. Journal 1864, Band 171/Miszelle 9 (S. 155) URL: http://dingler.culture.hu-berlin.de/article/pj171/mi171mi02_9

TC-Vortrag 04 91/161 Waschen – Bleichen – Färben
Anilingelb, nach C. A. Martius und P. Grieß.
Ein Anilinproduct, das durch Einwirkung von salpetriger Säure auf Anilin dargestellt wird und zuerst vor etwa zwei Jahren von Simson, Maule und Nicholson in London unter dem Namen Anilingelb in den Handel gebracht wurde, hielten diese Fabrikanten für identisch mit dem von Gries ausführlich beschriebenen Diazoamidobenzol (C²⁴H¹¹N³). Um sich zu überzeugen, ob dem wirklich so sey, untersuchten Martius und Grieß (Bericht über die Sitzung der Berliner Akademie vom 7. December 1865) das Verhalten dieses Körpers, den sie käuflich als ein braungelbes, lockeres, krystallinisches Pulver erhielten, gegen kochende Chlorwasserstoffsäure, durch welche das Diazoamidobenzol eine sehr charakteristische Zersetzung nach der Formel
erleidet. Es fand aber dabei nicht die allergeringste Gasentwickelung statt und ebenso wenig konnten in der tief roth gefärbten chlorwasserstoffsauren Auflösung Phenol oder Anilin aufgefunden werden. Wurde dagegen die salzsaure Lösung mit Ammoniak übersättigt, nachdem sie durch Filtration von einer Spur eines löslichen Harzes befreit worden war, so entstand eine reichliche Menge eines gelben krystallinischen Niederschlages, während sich in der Mutterlauge beträchtliche Mengen Oxalsäure nachweisen ließen. Das Anilingelb ist, abgesehen von der Spur harziger Substanz, das Oxalat einer organischen Base, des Amidodiphenylimid (C²⁴H¹¹N³), die mit dem ihr isomeren Diazoamidobenzol nichts gemein hat. Daß dieselbe in ähnlicher Weise wie das Diazoamidobenzol durch Einwirkung von salpetriger Säure auf alkoholische Lösungen von Anilin entsteht, haben M. und G. im Laufe ihrer Untersuchungen bestätigt gefunden; es hängt nur von der Temperatur ab, ob der eine oder der andere dieser beiden Körper gebildet wird; zur Bildung des Amidodiphenylimid ist eine höhere Temperatur erforderlich.
Bei der Untersuchung eines anderen gelben Farbstoffes, der durch Einwirkung von zinnsaurem Natron auf salzsaures Anilin entsteht und dessen Bildung zuerst in der Fabrik von J. J. Müller und Comp. in Basel, später auch von H. Schiff beobachtet wurde, fanden M. und G., daß derselbe mit dem Amidodiphenylimid identisch ist.
Fast alle schwachsauren Auflösungen des Amidodiphenylimid färben Wolle und Seide intensiv citronengelb. Aus einer Lösung der Pikrinsäureverbindung kann Wolle mit einer Farbe gefärbt werden, die dem Cochenilleroth, was Schönheit und Tiefe des Tones anlangt, wenig nachsteht. Dessenungeachtet haben diese Farben eine sehr untergeordnete praktische Bedeutung, weil sie flüchtig sind und in Folge dessen von den damit gefärbten Stoffen, namentlich in höherer Temperatur, nach und nach wegsublimiren. (Deutsche Industriezeitung, 1866, Nr. 17.)
Quelle: Polytech. Journal 1866, Band 180/Miszelle 9 (S. 326) URL: http://dingler.culture.hu-berlin.de/article/pj180/mi180mi04_9

TC-Vortrag 04 92/161 Waschen – Bleichen – Färben
Anilinbraun
Havannabraun, Bismarkbraun
Bismarckbraun Y oder Vesuvin ist ein Farbstoff aus der Reihe der basischen (kationischen) Azofarbstoffe. Benannt wurde der Farbstoff nach Otto von Bismarck, dem Reichsgründer und ersten Kanzler des Deutschen Reichs. Entdeckt wurde es 1863 von Carl Alexander von Martius als der erste Diazofarbstoff.
Verfahren zur Darstellung von Anilinbraun; von Georges de Laire in Paris.
Um nach dieser Erfindung (patentirt in England am 17. März 1863) Anilinbraun zu bereiten, behandelt man Anilinviolett oder Anilinblau mit einem Anilinsalz, am besten chlorwasserstoffsaurem Anilin.
Man bringt 1 Theil trockenes Anilinviolett oder Anilinblau zum Schmelzen und setzt es dann sogleich 4 Theilen wasserfreiem chlorwasserstoffsaurem Anilin zu. Nachdem der Anilinfarbstoff sich ganz aufgelöst hat, erhöht man die Temperatur des Gemisches rasch auf den Siedepunkt des chlorwasserstoffsauren Anilins, beiläufig 240° Cels. Die Masse wird auf dieser Temperatur erhalten, bis ihre Farbe, welche anfangs keine Veränderung zu erleiden scheint, plötzlich in Braun übergeht. Die Operation dauert 1–2 Stunden und ist als beendigt zu betrachten, wenn sich gelbe Dämpfe an den Seiten des Apparats verdichten; gleichzeitig ist ein starker und charakteristischer Knoblauchgeruch zu bemerken.
Die so erhaltene braune Farbe ist in Wasser, Alkohol und Säuren löslich, und kann unmittelbar zum Färben benutzt werden. – Man kann sie auch reinigen, indem man sie aus ihrer wässerigen Lösung durch Kochsalz fällt.
Anstatt schon gebildetes Anilinviolett oder Anilinblau anzuwenden, kann man dieselben durch das zur Erzeugung des Farbstoffs dienende Material ersetzen; wenn man z.B. arsensaures Anilin (welches für sich erhitzt, Anilinroth liefert) mit chlorwasserstoffsaurem Anilin behandelt, so entsteht Anilinbraun.
Aus dem London Journal of arts, December 1863, S. 348. Quelle: Polytech. Journal 1864, Band 171, Nr. XVII. (S. 72–73) URL: http://dingler.culture.hu-berlin.de/article/pj171/ar171017
Anilinbraun zum Coloriren von Photographien etc.
Ein sehr schönes, sattes Anilinbraun wird erhalten durch Erhitzen von einem Theil salzsaurem Anilin und drei Theilen irgend eines Anilin-Violetts. Fuchsin gibt ein Braun, welches mehr in das Gelbliche zieht, während Violett ein tiefes Braun erzielen läßt. Das Erhitzen geschieht in einer Porzellanschale auf dem Sandbade und ist nur darauf zu sehen, daß das Gemisch fortwährend flüssig erhalten werde. Tüchtiges Umrühren befördert die Bildung der braunen Farbe Von Zeit zu Zeit wird eine kleine Probe in Spiritus gelöst und wenn die so erhaltene verdünnte Lösung weder einen Stich in's Rothe oder Blaue besitzt, sondern rein braun erscheint, wird die Operation unterbrochen. Die Temperatur darf 250° Cels. nicht überschreiten. Nach dem Erkalten läßt sich das Braun leicht aus der Schale entfernen und löst sich fast ohne Rückstand in Weingeist von 90 Proc. Tr. Die weingeistige Lösung verträgt eine Verdünnung mit der Hälfte Wasser und dient nach dem Filtriren zum Färben. Mit Glycerin

TC-Vortrag 04 93/161 Waschen – Bleichen – Färben
versetzt, kann sie mit Erfolg zum Coloriren von Photographien benutzt werden. Wegen der sich entwickelnden Dämpfe geschieht das Erhitzen des Gemisches unter einem gut ziehenden Rauchfange. Die Ausbeute beträgt 3 3/4 bis 3 4/5 Theile.
Quelle: Polytech. Journal 1867, Band 184/Miszelle 8 (S. 376) URL: http://dingler.culture.hu-berlin.de/article/pj184/mi184mi04_8
Anilinschwarz
Indigschwarz, schwarzer Indig, Lukasschwarz
Anilinschwarz ist eines der ältesten synthetischen Farbmittel. Da bei der Synthese von Anilinschwarz ein Reaktionsgemisch unterschiedlicher Farbstoffe entsteht, ist die eindeutige Zuordnung einer Strukturformel nicht möglich. Ein Beispiel eines Farbstoffes sei im Folgenden dargestellt:
Anilinschwarz bezeichnet einerseits einen Farbstoff, der durch Oxidation von Anilinsalzen mit Kaliumchlorat, Bichromat und dergleichen erzeugt wird, und zwar fast immer direkt auf der Faser (Baumwolle, seltener auf Seide oder Halbseide). Anilinschwarz gehört zu den echtesten und schönsten schwarzen Farbstoffen und hat daher, besonders in der Baumwollfärberei, große Bedeutung.
Andererseits wurde es als Pigment erfolgreich im Lackbereich eingesetzt, dann aber vom heute dominanten Pigmentruß abgelöst und hat inzwischen eine geringe Bedeutung. Anilinschwarz gilt als ältestes synthetisch hergestelltes organisches Pigment.
Der Farbton von Anilinschwarz wird als tiefes, neutrales Schwarz beschrieben. Anilinschwarz verfügt über ein hohes Deckvermögen und eine gute Dispergierbarkeit, weist aber im Vergleich zu Ruß eine weitaus geringere Farbstärke auf. Das Pigment ist leicht leitfähig. Die Licht- und Wetterechtheit ist im Vollton gut, nimmt aber in Weißabmischungen stark ab. C.I. Pigment Black 1 kann die Oberfläche von Lacken beeinflussen und erzeugt ein Erscheinungsbild, das als matt und samtartig beschrieben wird.
Anilinschwarz wird besonders in der Baumwollfärberei eingesetzt. In anderen Bereichen wird Anilinschwarz heutzutage nur noch eingesetzt, wenn Pigmentruß zu Problemen führt oder die Mattierung der Oberfläche gezielt erzeugt werden soll. Im Lack- und Druckfarbengebiet wird es eingesetzt, wenn Verarbeitungsprobleme durch Rußpigmente verursacht werden. Im Kunststoffbereich ist dies der Fall, wenn Probleme beim Verschweißen durch Pigmentruß entstehen.
5.2.4 Phenol
Den Anilinfarben nahestehend sind die Farben welche man aus der Carbolsäure (Phenylsäure, Phenol) herstellt.

TC-Vortrag 04 94/161 Waschen – Bleichen – Färben
Phenol
Hieraus entstehen u. a. die Farbstoffe: Pikrinsäure, Phenylbraun, Granatbraun, Corallin und Azulin.
Das Phenol (IUPAC: Benzenol, Veraltet: Karbolsäure oder kurz Karbol) ist der einfachste Vertreter der Phenole. Phenole sind organische Verbindungen, in denen mindestens eine Hydroxygruppe direkt an einem aromatischen Ring gebunden ist. Phenol ist ein Derivat des Benzols. Phenol wurde im Jahr 1834 vom Chemiker Friedlieb Ferdinand Runge bei der Destillation von Steinkohlenteer entdeckt; er bezeichnete die Substanz jedoch als „Carbolsäure“. Auguste Laurent entdeckte sie 1841 erneut und ermittelte die Summenformel als C6H6O. Charles Gerhardt nannte sie Phenol. Der Name weist auf das Leuchtgas hin, welches neben Steinkohlenteer bei der Produktion von Koks entstand. Leuchtgas (Stadtgas) diente damals zur Beleuchtung der Städte (gr. phainomei: leuchten).
Phenol ist hydroxysubstituiertes Benzol. Sein Schmelzpunkt liegt bei 41 °C und der Siedepunkt bei 182 °C. Reines Phenol bildet bei Zimmertemperatur farblose Kristallnadeln, jedoch ist das kommerziell erhältliche Produkt i. d. R. durch geringe, aber intensiv gefärbte Verunreinigungen rosa bis rötlich-braun gefärbt. Es besitzt einen charakteristischen, aromatischen Geruch. Die Hydroxy-Gruppe des Phenols reagiert im Gegensatz zu Alkoholen sauer, womit Phenol eine schwache Säure ist. Die Ursache ist die Mesomeriestabilisierung der korrespondierenden Base des Phenolations. Die negative Ladung kann in den Ring delokalisiert werden.
Eine technisch bedeutende nukleophile Substitutionsreaktion ist die Darstellung von Anilin aus Phenol bei 250 °C und Anwesenheit eines Oxid-Katalysators (Aluminium-, Silicium- oder Magnesiumoxid, auch Borsäure) als „Halcon-Prozess“.
Phenol verursacht Verätzungen und ist ein Nerven-/Zellgift.
Phenol setzt sich durch katalytische Hydrierung zu Cyclohexanol um.
Sir Joseph Lister setzte es 1865 – in fünfprozentiger Lösung – als Antiseptikum bei der Wunddesinfektion ein; damals war die Carbolsäure nahezu das einzig verfügbare Mittel gegen Wundinfektionen. Wegen seiner hautirritierenden Nebenwirkung wurde es aber bald durch andere Antiseptika ersetzt. Wegen seiner bakteriziden Wirkung wird es noch heute – wenngleich seltener – als Desinfektionsmittel eingesetzt.
In Merck’s Warenlexikon findet 1884 sich zur Karbolsäure folgender Hinweis:
„Es handelt sich um eine Substanz, die für die Farbenindustrie und Medizin (als Desinfektionsmittel) eine außerordentliche Bedeutung erlangt hat. Der Handelsname ist immer noch Karbolsäure, während der wissenschaftliche Name jetzt Phenol ist; diese

TC-Vortrag 04 95/161 Waschen – Bleichen – Färben
Substanz besitzt zwar die Eigenschaften einer schwachen Säure und ist imstande, sich mit Basen zu verbinden, wird deshalb auch Phenylsäure oder Phensäure genannt, zugleich aber und in noch höherem Grade spielt sie die Rolle eines Alkohols, daher auch der Name Phenylalkohol, den man in Phenol gekürzt hat. Man gewinnt die Karbolsäure hauptsächlich aus dem Steinkohlenteer und dem Braunkohlenteer, im Holzteer sind nur äußerst geringe Mengen davon enthalten, denn das Kreosot des Holzteers besteht nicht, wie man eine Zeit lang glaubte, aus Phenol, sondern aus dem strukturell ähnlichen Kresol, sowie noch einigen anderen Stoffen. Um die Karbolsäure zu gewinnen, behandelt man den zwischen 150–200 °C übergehenden Teil des Teers mit Natronlauge, welche sich mit der Karbolsäure und dem Kresol, das auch im Steinkohlenteer enthalten ist, verbindet, trennt diese Lösung von den übrigen Teerbestandteilen und zersetzt sie mit einer Säure. Man destilliert dann das abgeschiedene ölige Produkt und fängt das, was über 190 °C übergeht, besonders auf; letzteres wird als rohes Kresol, das, was unter 190 °C übergeht, als rohe Karbolsäure verkauft. Beide Substanzen sind in diesem Zustande noch braune, sehr übel riechende, ölige Flüssigkeiten. Diese rohe Karbolsäure (acidum carbolicum crudum) wird teils weiter gereinigt, teils wird sie zur Konservierung von Holz und zum Desinfizieren von Abtrittgruben verwendet.“
Phenol wirkt sowohl lokal als auch systemisch stark toxisch; bei dermaler Exposition besitzt es eine reizende bis ätzende Wirkung auf Schleimhäute, Haut und Augen. Die Augen können Schäden in Form einer Trübung der Hornhaut, Schwellungen und Verwachsung der Lider bis zur Erblindung erleiden. Hautkontakt führt zuerst zu Hautrötung, später zu einer Weißverfärbung; längere Einwirkungszeit verursacht eine Dunkelfärbung bis zur Bildung von Nekrosen.
Phenol wird vorwiegend über die Haut resorbiert, aber auch inhalative oder orale Aufnahme ist möglich. Im menschlichen Organismus schädigt die Substanz akut Nieren, Blut, Zentralnerven- und Herz-Kreislauf-System. Bei chronischer Exposition sind auch gastrointestinale und nervale Störungen, weiterhin Schädigung von Leber, Nieren und Hautveränderungen bekannt. Bei Inhalation wurden als Vergiftungssymptome Schwindel, Kopfschmerz und Störungen der Ohren, Erbrechen, Schlaflosigkeit und Nierenreizung beschrieben. Die Aufnahme hoher Mengen führte innerhalb weniger Stunden zu massiven Nierenfunktionsstörungen bis zu akutem Nierenversagen. Orale Aufnahme bewirkt Verätzungen im Mund, Rachen, Speiseröhre und Magen; weiterhin sind Schluckstörungen und Störungen im Magen-Darm-Trakt bekannt.
Die Toxizität wird auf reaktive Metaboliten des Phenol zurückgeführt, die an die DNA und andere Makromoleküle binden und dabei Brüche in den Chromosomen und mutagene Effekte auslösen können. Eine orale Dosis ab 1 g kann vereinzelt für einen Menschen tödlich sein; individuell wurden aber auch wesentlich höhere Dosen überlebt. Der orale LDLo-Wert für den Menschen liegt zwischen 140 und 1400 mg/kg Körpergewicht; bei Kindern beträgt die orale minimale letale Dosis 10 mg/kg Körpergewicht.
5.2.5 Pikrinsäure
Auf die gelbe Pikrinsäure will ich besonders hinweisen. Ihre chemische Struktur eine besondere – es handelt sich um Trinitrophenylsäure, sie ist also dem Trinitrotoluol nahestehend. Durch die hohe Anzahhl der Nitrogruppen weist Pikrinsäure eine nicht zu unterschätzende Reaktivität auf, weshalb sie wie andere brisante Sprengstoffe mit

TC-Vortrag 04 96/161 Waschen – Bleichen – Färben
entsprechender Vorsicht zu behandeln ist. Aber sie wird auch gern bei der Herstellung gelber Farbstoffe eingesetzt.
Pikrinsäure (gr. πικρος, pikros = bitter) ist der Trivialname für 2,4,6-Trinitrophenol (TNP). Die Struktur besteht aus einem Benzolring mit einer Hydroxygruppe (–OH) und drei Nitrogruppen (–NO2) als Substituenten. Es gehört zur Stoffgruppe der Trinitrophenole, einer Gruppe von sechs Konstitutionsisomeren. Ihre Salze heißen Pikrate.
Pikrinsäure
Durch Behandlung von Indigo mit Salpetersäure konnte Peter Woulfe als erster 1771 Pikrinsäure darstellen. Neben der Gelbfärbung von Seide hatte sie jedoch zunächst noch keine größere Bedeutung.
Pikrinsäure ist giftig. Auf der Haut kann sie starke allergische Reaktionen hervorrufen. Die Kontamination mit Stäuben oder Dämpfen ist daher zu vermeiden. 1864 verfasste der deutsche Arzt Wilhelm Erb eine Arbeit über Physiologische und therapeutische Wirkungen der Pikrin-Säure. 1865 habilitierte er sich auch mit einer Arbeit zu dieser Thematik.
An der Luft verbrennt Pikrinsäure mit starker Rauchentwicklung; bei sehr raschem Erhitzen oder durch eine Initialzündung erfolgt eine Detonation. Pikrinsäure ist empfindlich gegen thermische (Hitze, Feuer) und mechanische (Schlag, Reibung) Belastung und gilt im Sinne des Sprengstoffgesetzes als explosionsgefährlicher Stoff. Für den Versand zur Verwendung als Laborchemikalie (siehe unten) wird die kristallisierte Säure durch Zugabe von etwas Wasser stabilisiert („phlegmatisiert“).
Die Substanz war das erste detonierende, brisante Geschoss-Füllmittel und wurde als Lyddit, Ekrasit, Schimose oder Melinit ab 1886 so verwendet, nachdem der Franzose Eugène Turpin die Sprengstoffeigenschaften der lange zuvor bekannten Säure entdeckt hatte.
Für die katastrophale Halifax-Explosion im Jahr 1917 waren 2.300 Tonnen Pikrinsäure verantwortlich.
Die Verwendung von Pikrinsäure zur Anfärbung von Backwaren im ausgehenden 19. Jahrhundert war weit verbreitet und war als Weltersches Bitter bekannt, was nach einer Häufung von Vergiftungsfällen jedoch unterbunden wurde.
Die Pikrinsäure wird über die Sulfonierung von Phenol und nachfolgende Behandlung mit Salpetersäure hergestellt. Alternativ bietet sich die Darstellung aus Chlorbenzol über 2,4-Dinitrochlorbenzol, 2,4-Dinitrophenol und dessen erneute Nitrierung an. Eine direkte Herstellung der Substanz gelingt durch die Oxynitrierung von Benzol durch konzentrierte Salpetersäure in Gegenwart von Quecksilber(II)-nitrat. Früher wurde Pikrinsäure auch aus Akaroidharz hergestellt.
Pikrinsäure bildet farblose bis leicht gelbe, stark bitter schmeckende Kristalle, die nur schwer in kaltem Wasser, hingegen besser löslich in siedendem Wasser und leicht löslich in Ethanol

TC-Vortrag 04 97/161 Waschen – Bleichen – Färben
und Benzol sind. Bedingt durch die Häufung elektronenziehender Nitrogruppen (–NO2) ist die Pikrinsäure durch ihre phenolische Hydroxygruppe eine starke Säure (pKs = 0,29). Daher bildet sie zahlreiche Salze mit anorganischen und organischen Basen. Die Salze werden Pikrate genannt. So entsteht mit Ammoniak das Ammoniumpikrat.
Als starke Säure greift wässrige Pikrinsäure unedle Metalle unter Pikratbildung an.
Einige der Salze z. B. Bleipikrat sind extrem empfindlich gegenüber Schlag, Reibung und Funken. Sie verhalten sich somit wie Initialsprengstoffe. Ammoniumpikrat wurde als Sprengstoff verwendet.
Ebenfalls als Pikrate bezeichnet man die Charge-Transfer-Komplexe, die Pikrinsäure mit Aromaten bildet. Diese Feststoffe sind oft schwerlöslich und farbig. Wegen der charakteristischen und scharfen Schmelzpunkte (z. B. Benzol-Pikrat 84 °C, Toluol-Pikrat 88 °C, Anthracen-Pikrat 138 °C) wurde Pikrinsäure vor allem früher als Nachweisreagenz für Aromaten verwendet.
Primär dient die Pikrinsäure der Farbstoffindustrie zur Herstellung von 2-Amino-4,6-dinitrophenol (Pikraminsäure). Sie wurde früher zusammen mit arabischem Gummi und destilliertem Wasser zur Herstellung gelber Tinte verwendet. Ein weiteres Einsatzgebiet ist die organische Analytik zum Nachweis von Aminen, Alkaloiden und Kreatinin. Diese basischen Stoffe bilden gelbe Salze, welche durch ihren Schmelzpunkt charakterisiert wurden (Derivat-Bildung).
Die Verwendung von Pikrinsäure als Füllmaterial für Granaten (wie im Ersten Weltkrieg) wurde wegen der unkontrollierten Bildung von sehr stoßempfindlichen Schwermetallpikraten eingestellt. Die Pikrinsäure wurde hier durch TNT ersetzt. In der Mikroskopie verwendet man Pikrinsäure als Bestandteil von Fixierflüssigkeiten (zur Konservierung zellulärer Strukturen) und zum Anfärben von Präparaten. Ein weiteres Einsatzgebiet von Pikrinsäure ist die Metallografie. Hier wird die Substanz zum Ätzen metallischer Oberflächen verwendet, z. B. bei der Präparation von Magnesiumlegierungen oder bei Seigerungsuntersuchungen an Stählen. Die Ätzung der Stähle wird mit Igeweskys-Reagenz, einer 5-%igen Lösung von Pikrinsäure in wasserfreiem Alkohol, durchgeführt. Pikrinsäure dient auch der Kreatinin-Konzentrationsmessung: Kreatinin bildet in alkalischer Lösung mit Pikrinsäure einen Meisenheimer-Komplex (Jaffé-Reaktion), dessen rote Farbe photometrisch gemessen wird.
Pikrinsäure ist im Sinne des Sprengstoffgesetzes als explosionsgefährlicher Stoff der Stoffgruppe A (trocken) bzw. C (mit 25 % Wasser angefeuchtet) gemäß § 1 Abs. 3 Sprengstoffgesetz eingestuft. Für Privatpersonen ist trockene Pikrinsäure somit nach § 27 SprengG erlaubnispflichtig. Trocken ist Pikrinsäure in Lagergruppe 1.1 oder I bzw. als Gefahrgut in Klasse 1.1 (Stoffe, die massenexplosionsfähig sind) eingestuft, angefeuchtet mit 30 % Wasser in Lagergruppe 1.4.
Als handelsübliches Produkt ist Pikrinsäure mit > 30 % Wasser angefeuchtet und damit phlegmatisiert. Angefeuchtet (> 30 % Wasser) verhält sich Pikrinsäure wie ein entzündlicher Feststoff und wird zum Transport als Entzündbarer fester Stoff der Gefahrgutklasse 4.1 nach ADR gekennzeichnet.
5.2.6 Naphthalin
Aus Benzol und Azetylen stellt man Naphthalin her (Naphtalin).

TC-Vortrag 04 98/161 Waschen – Bleichen – Färben
Naphthalin
Naphthalin (von griechisch naphtha = Erdöl, Name nach IUPAC: Naphthalen) ist ein farbloser Feststoff mit der Summenformel C10H8, der schon bei Raumtemperatur sublimiert. Es ist ein bicyclischer aromatischer Kohlenwasserstoff mit charakteristischem Geruch nach Mottenpulver/Teer. Naphthalin ist gesundheitsschädlich und umweltgefährlich.
1819 wurde Naphthalin vom britischen Chemiker Alexander Garden aus Steinkohleteer isoliert. 1866 wurde von Emil Erlenmeyer zum ersten Mal die Naphthalinformel aufgestellt. Traditionell wird Naphthalin auch zu den polycyclischen aromatischen Kohlenwasserstoffen (PAK) gezählt. Das Naphthalinmolekül besteht aus zwei anellierten Benzolringen, sein chemisches Verhalten ähnelt dem der anderen PAKs
Spuren von Naphthalin werden von Magnolien und einigen Hirscharten produziert. Außerdem wurde der Stoff bei einer Termitenart nachgewiesen, die es offenbar als Abwehrstoff gegen natürliche Feinde wie Ameisen und giftige Pilze verwendet.
Naphthalin wird aus der Mittelölfraktion des Steinkohlenteers (bis zu 11 %), sowie Braunkohlen- und Holzteer, Crackgasöl oder auch aus Kohle, wenn diese verkokt wird, gewonnen. Im Steinkohleteer ist es mengenmäßig die größte Komponente. Es kommt auch in Petroleum und anderen fossilen Energieträgern vor und entsteht auch bei der Verbrennung von Holz oder Tabak. Naphthalin wird von Gaswerksstandorten und Holzimprägnierwerken emittiert, des Weiteren entsteht es auch auf Mülldeponien.
Früher war Naphthalin der Hauptbestandteil von Mottenkugeln, wird aber heute wegen seines unangenehmen Geruchs oft durch andere Substanzen ersetzt. Des Weiteren ist Naphthalin kaum insektizid wirksam. Auch zur Desinfektion von Insektensammlungen ist es kaum wirksam, obwohl man es lange Zeit dazu verwendete.
Naphthalin kam auch in dem Anfang des 20. Jahrhunderts verwendeten Leuchtgas vor und verstopfte oft die Gasleitungen, da es sich als Feststoff abschied. Trotz seines gesundheitlichen Gefährdungspotenzials wurde es zum Beispiel medizinisch zur Darmdesinfektion verwendet.
Hauptsächlich wird Naphthalin zur Synthese von Phthalsäureanhydrid verwendet, das zu Lösungsmitteln, Kunststoffen und Kraftstoffen weiterverarbeitet wird. Auch zur Herstellung der Lösungsmittel und Kraftstoffzusätze Decalin und Tetralin wird es benötigt, für die Herstellung von Azofarbstoffen, zur Synthese des Holzschutzmittels Chlornaphthalin, von Insektiziden (Carbamaten) sowie von PVC-Weichmacher-Zwischenprodukten, außerdem zur Herstellung von Alkylnapththalinsulfaten, die als Seifen benutzt werden. Weitere industriell wichtige Abkömmlinge sind die Naphthole, Bromnaphthaline, Naphthylamine und Nitronaphthaline.
Phthalsäure
Unter Salpetersäure entsteht Phtalsäure. Phthalsäure ist eine Chemikalie, die in der Chemie zu den Carbonsäuren, genauer den Dicarbonsäuren, zählt. Üblicherweise wird mit Phthalsäure

TC-Vortrag 04 99/161 Waschen – Bleichen – Färben
die ortho-Phthalsäure bezeichnet, die neben der Terephthalsäure die größte technische Bedeutung hat.
o-Phthalsäure
Phthalsäure wurde 1836 von Auguste Laurent bei der Oxidation von 1,2,3,4-Tetrachlor-1,2,3,4-tetrahydronaphthalin mit Salpetersäure entdeckt, trägt also ihren Namen nach diesem Kohlenwasserstoff.
Der mengenmäßig größte Teil der Phthalsäuren wird zur Herstellung von Kunstharzen oder Kunstfasern verwendet. Die Salze und Ester der Phthalsäuren werden Phthalate genannt. Die Ester werden unter Phthalsäureester näher beschrieben. Aus dem Phthalsäureanhydrid gelangt man durch Friedel-Crafts-Acylierung von Benzol zu Anthrachinon, aus dem Küpenfarbstoffe hergestellt werden können.
Phthalsäureanhydrid (kurz PSA) ist das Anhydrid der Phthalsäure:
Diese organische Verbindung ist ein wichtiger Ausgangsstoff für die Herstellung von Kunstharzen, daneben auch von Farbstoffen oder Farbpigmenten. Bis in die 1960er Jahre wurde Phthalsäureanhydrid fast ausschließlich durch Luft-Oxidation von Naphthalin aus Steinkohlenteer gewonnen (Gibbs-Wohl-Naphthalin-Oxidation).
Naphthylamin
Aus dem Naphthalin lässt sich analog zum Anilin die Base Naphtylamin herstellen.
1-Naphthylamin
Daneben gibt es auch eine isomere Form,
das 2-Naphthylamin
Nikolai Nikolajewitsch Sinin erhielt 1-Naphthylamin (von ihm Naphtalidam genannt) aus 1-Nitronaphthalin durch Reduktion mit Schwefelwasserstoff oder salpetriger Säure.

TC-Vortrag 04 100/161 Waschen – Bleichen – Färben
Großtechnisch wird es gewonnen, indem Naphthalin mit Nitriersäure (Salpetersäure mit Schwefelsäure) zu 1-Nitronaphthalin im diskontinuierlichen Rührkessel nitriert wird. Nach dem Abtrennen wird das Nitronaphthalin mit Eisen zum 1-Naphthylamin reduziert.
1-Naphthylamin wird für die Synthese von Azofarbstoffen und Herbiziden, Aminonaphthalinsulfonsäuren, -Naphthol sowie Phenylnaphthylamin und als Antioxidationsmittel für Kautschuk verwendet.
Hieraus entstehen die Farben
Martiusgelb – Manchestergelb, Naphtalingelb
Magdalarot – Naphtalinrot
Naphtalinviolett
Naphtalinblau.
Ueber Naphtalin und Anthracen (Paranaphtalin); von Dr. Herrn. Vohl in Cöln.
Die hohe Wichtigkeit, welche in jüngster Zeit das Naphtalin und Anthracen in der Farbstofftechnik erlangt haben, lassen es einem jeden Farbstofffabrikanten erwünscht erscheinen, diese beiden Substanzen möglichst rein im Handel beziehen zu können. Ganz so wie bei den Anilinfarben der Farbstofffabrikant das Anilin, resp. das Nitrobenzol oder Benzol nicht selbst darstellt, sondern diese Arbeit anderen Fabriken überläßt, welche diese Substanzen in der geeigneten Reinheit zu verhältnißmäßigen nicht zu hohen Preisen beschaffen, muß die Darstellung des reinen Naphtalins und Anthracens von der eigentlichen Farbendarstellung getrennt und besonderen Fabriken zugewiesen bleiben.
Wie ich schon in meiner Abhandlung "„über das Naphtalin und seine Verwendung in der Technik“" (polytechn. Journal Bd. CLXXXVI S. 138) bemerkte, ist die Reinigung des Naphtalins mit besonderen Schwierigkeiten verbunden. Die Sublimation, welche bei den wenigen Fabriken, die Naphtalin liefern, fast allgemein noch in Gebrauch ist, liefert kein reines kreosot- und ölfreies Product, wie es erheischt wird (Siedepunkt zwischen + 216 und 218° C., Schmelzpunkt = + 79° C. und spec. Gew. = 1,1517 bei + 15° C.), und erfordert einen großen Zeitaufwand; ferner verlangt das Fabricat in dieser voluminösen Form große Verpackungsgesäße, wodurch das Brutto-Gewicht und die Fracht unnütz vermehrt wird. Allen diesen Uebelständen wird durch meine Reinigungsmethode und Darstellung in Stangen- oder Ziegelform entgegengetreten. Im Interesse der Farbstofffabrikanten mache ich auf die Firma Friedrich Gerhartz in Cöln aufmerksam, welche das Naphtalin in fester Form (Stangen und Ziegel) in großer Schönheit und chemisch rein liefert. Die Proben, welche mit zugeschickt wurden, habe ich geprüft und sie vollständig kreosot- und ölfrei gefunden.
Auch liefert Gerhartz Anthracen in derselben Form und voraussichtlich nitrirte Derivate desselben. Das Anthracen hat durch die Entdeckung von C. Gräbe und C. Liebermann, aus demselben den Farbstoff der Krappwurzel darzustellen, eine große Wichtigkeit erlangt. (Journal für praktische Chemie, 1869, Bd. CVII S. 188.)
Quelle: Polytech. Journal 1869, Band 193/Miszelle 12 (S. 436–437) URL: http://dingler.culture.hu-berlin.de/article/pj193/mi193mi05_12

TC-Vortrag 04 101/161 Waschen – Bleichen – Färben
5.2.7 Anthracenfarben
Anthracen (Paranaphtalin, Photen) kann aus Kohlenteer gewonnen werden. 1869 wurde hieraus das Anthtracenrot, künstliches Alizarin hergestellt, welches wohl als Konkurrenz zur Krappfärbung aufkommt.
Verfahren zur Darstellung von Anthracen aus dem Pech von Steinkohlentheer, und zur Darstellung von Farbstoffen aus Anthracen; von J. Brönner und H. Gutzkow in Frankfurt a. M.
Bayerisches Patent vom 29. September 1869 und 26. Januar 1870.
I. Um das Anthracen und ähnliche Stoffe aus dem Asphalt, resp. Pech von Steinkohlentheer zu erhalten, destilliren wir dasselbe mit Hülfe von überhitztem Wasserdampf in einer Gasretorte und leiten die Dämpfe durch ein weites nur wenig aufsteigendes Rohr auf dem kürzesten Weg zuerst in ein Zwischengefäß, aus dem die darin condensirten rohen Anthracenmassen abgelassen werden können. Aus diesem Zwischengefäß leiten wir die noch nicht condensirten Gase durch eine geräumige Kühlvorrichtung entweder in einen Gasbehälter oder in's Freie, oder benutzen sie direct zur Heizung.
Aus dem auf diese Weise, und aus dem aus dem Schweröl des Steinkohlentheeres dargestellten, hinlänglich gereinigten Anthracen stellen wir die beiden Farbstoffe in folgender Weise dar:
Wir verwandeln das Anthracen durch Oxydationsmittel in ein neues Product und bedienen uns hierzu aller bekannten Oxydationsmittel, welche fähig sind diese Umwandlung herbeizuführen, z. B. des zweifachchromsauren Kalis und Schwefelsäure, oder der krystallisirten Essigsäure, vorzugsweise aber der Salpetersäure, und zwar entweder der verdünnten oder der concentrirten Salpetersäure. Das so erhaltene neue Product reinigen wir entweder durch Sublimation, Krystallisation oder auf andere Weise, und stellen durch Oxydation desselben die beiden Farbstoffe her, oder wir thun dieß, indem wir sofort das ungereinigte Product dazu verwenden.
In folgender Weise führen wir diese Sache vorzugsweise aus: Wir behandeln Anthracen in der Kälte oder auch bei erhöhter Temperatur mit seinem doppelten Gewicht einer Salpetersäure von 1,3 bis 1,5 spec. Gewicht, waschen das neue Product mit Wasser, und lösen es gereinigt oder ungereinigt in der erforderlichen Menge Schwefelsäure. Wir erwärmen zur besseren Lösung, und setzen dann die erforderliche Menge eines Quecksilbersalzes, z. B. des salpetersauren Oxyduls oder Oxyds hinzu. Nachdem sich die Farbstoffe gebildet haben, bringen wir sie auf beliebige Weise, sey es durch kaltes oder kochendes Wasser, oder Alkohol, Aether, Schwefelkohlenstoff, Alaunlösungen, wässerige Alkalien oder durch sonstige Mittel in Lösung, doch behalten wir uns vor, die beiden Farbstoffe ehe wir sie in Lösung bringen zur vollständigen Entwickelung der Farbe, wenn es uns nöthig erscheint, zuvor noch mit Alkalien zu behandeln. Wir können sie sodann eindampfen, oder durch Säuren niederschlagen, und endlich durch Krystallisation aus ihren Lösungen, oder durch Sublimation oder auf andere Weise reinigen.
Je nach der Wahl der oxydirenden Stoffe die wir anwenden, oder nach ihren Mengen, oder nach der Höhe der Temperatur bei der Darstellung, erhalten wir einen Farbstoff der sich in Alkohol, Aether etc. mit gelber, und einen solchen welcher sich darin mit rother Farbe auflöst.

TC-Vortrag 04 102/161 Waschen – Bleichen – Färben
Unser Verfahren unterscheidet sich von dem der HHn. Graebe und Liebermann115 erstens dadurch, daß wir das aus dem Anthracen erhaltene erste Product oxydiren, während Graebe und Liebermann Wasserstoff durch Brom ersetzen und sodann zur Vertretung des Broms zweimal HO durch Alkalien einführen, und zweitens dadurch, daß wir nicht nur Alizarin, sondern auch einen Farbstoff erhalten, der sich ähnlich wie das Purpurin in Alkohol etc. mit rother Farbe, nicht wie das Alizarin mit gelber auflöst.
II. Nach dem neueren bayerischen Patent wenden I. Brönner und H. Gutzkow zur Darstellung des Alizarins aus dem Anthracen nachfolgendes verbessertes Verfahren an. Das Anthracen wird in oben angegebener Weise durch oxydirend wirkende Substanzen in Oxanthracen verwandelt, letzteres durch Sublimation gereinigt und daraus Alizarin dargestellt, indem man es mit concentrirter Natron- oder Kalilauge auf 200–250° C. erhitzt. Zu dem Ende wird circa das doppelte Gewicht an Natron- oder Kalilauge angewendet, nach Beendigung der Reaction mit Wasser verdünnt, das Alizarin mittelst einer Säure niedergeschlagen, dann filtrirt und so lange ausgewaschen, bis alle Säure entfernt ist. (Bayerisches Industrie- und Gewerbeblatt, 1870 S. 214.) 115Mitgetheilt im polytechn. Journal, 1870, Bd. CXCVI S. 359.
Quelle: Polytech. Journal 1871, Band 201, Nr. CXXV. (S. 545–546) URL: http://dingler.culture.hu-berlin.de/article/pj201/ar201125
Mittheilungen aus dem technischen Laboratorium des k. ungarischen Polytechnicums in Ofen; von V. Martha.
Aus den Berichten der deutschen chemischen Gesellschaft zu Berlin, 1870, Nr. 10.
I. Beiträge zur Kenntniß der Anthracen-Farbstoffe.
Die HHrn. Grabe und Liebermann erwähnen in ihrer Abhandlung "„über Anthracen und Alizarin,“" daß das Anthrachinon der Einwirkung von Oxydationsmitteln mit ungeheurer Energie widersteht; daß selbst alkoholische Kalilauge in zugeschmolzenem Glasrohr bei 200° C. nicht auf dasselbe einwirkt, und auch schmelzendes Kali keine Veränderung hervorbringt. Ich kann nun mittheilen, daß es mir gelungen ist, das Antrachinon trotz seiner auffallenden Beständigkeit direct zu oxydiren, und will die hierauf bezüglichen Versuche kurz beschreiben.
Erhitzt man eine absolut alkoholische Lösung von reinem, mittelst Chromsäure erhaltenem, wiederholt sublimirtem, fast farblosem Anthrachinon mit festem Aetzkali in einem Probirrohr zum Kochen, so bemerkt man bald, daß die Lösung gelb wird und zwei Schichten bildet: eine untere, bestehend aus geschmolzenem Netzkali, und eine obere, alkoholische Anthrachinonlösung. Bei fortgesetztem Erhitzen färbt sich die obere Lösung immer dunkler, schließlich wird dieselbe, wenn nur noch wenig Alkohol vorhanden ist, ganz braunschwarz, und nun mischen sich die zwei Flüssigkeiten unter starker Gasentwickelung, die Masse wird schön grün, dann dunkelblau, und bei weiterem Erhitzen tritt endlich die charakteristische violette Farbe des Alizarin-Kalis auf. Nun läßt man erkalten, löst in Wasser, fällt die purpurviolette Lösung mit Schwefelsäure, extrahirt mit Aether, und kann nun aus der ätherischen Lösung mit wässerigem Kali das Alizarin mit allen seinen charakteristischen Eigenschaften abscheiden. Doch wird bei dieser Operation bei weitem der größere Theil des Anthrachinons nicht angegriffen; man kann dasselbe nur durch wiederholtes Schmelzen mit neuen Kali- und Alkohol-Mengen in Farbstoff umwandeln. Der Vorgang ist hier derselbe, wie bei der Darstellung der sogenannten Chinonsäure von Schoonbroodt, welcher beobachtet hat,

TC-Vortrag 04 103/161 Waschen – Bleichen – Färben
daß diese Verbindung durch Erhitzen des Chinons mit Aetzkali unter Wasserstoffentwickelung entsteht. Dieser Körper hat die Zusammensetzung C⁶H²(OH)²O² also Bioxychinon. Setzt man der alkoholischen Anthrachinon-Lösung etwas Zinnchlorür hinzu und erhitzt auf gleiche Weise mit festem Kali bis zum Schmelzen, so beobachtet man eine eigenthümliche Erscheinung. Ist Zinnchlorür im Ueberschuß vorhanden, so wird die untere geschmolzene Kalischicht viel früher grün, als beim Schmelzen ohne Zinnchlorür; die obere, alkoholische Flüssigkeit aber wird nach kurzer Zeit feurig blutroth, und bedeckt sich beim Erkalten und Offenstehenlassen oder beim Durchleiten eines Luftstromes mit einer braunschwarzen Haut, die durch Schütteln der Flüssigkeit entfernt, sich immer so lange von Neuem bildet, als die Lösung noch jene intensiv rothe Farbe zeigt. Der abgeschiedene braunschwarze Niederschlag, abfiltrirt und gewaschen, wird durch festes Kali allein theilweise zu Alizarin oxydirt. Mit der näheren Untersuchung der dabei gebildeten Körper bin ich gegenwärtig beschäftigt. Läßt man die erwähnte blutrothe Kalilösung nicht erkalten, sondern erhitzt bis zum Schmelzen, so wird die ganze Masse grün, dann blau, schließlich violett. Hat man Ueberschuß von Zinnchlorür zugesetzt, so bildet sich weniger, und manchmal gar kein Alizarin; in geringer Menge jedoch beigemengt, steigert Zinnchlorür die Ausbeute an Farbstoff. Die erwähnten Operationen indessen lassen sich mit nur kleinen Portionen sicher ausführen; arbeitet man mit größeren Mengen, so erhält man häufig nur braune oder rothbraune huminartige Substanzen, aber keinen, oder sehr wenig Farbstoff. Weit ergiebiger läßt sich die directe Oxydation des Anthrachinons so ausführen, daß man dasselbe wohl gemengt mit ungefähr dem doppelten Gewichte Natrium-Aethylat in schmelzendes Kali einträgt. Nach dem Zusatz der einzelnen Portionen dieses Gemenges findet starkes Aufschäumen statt, die Masse wird braunschwarz, später fast ganz schwarz. Man trägt nun unter fortwährendem Umrühren so lange ein, bis das Product ganz dick wird und am Rande der Porzellanschale die Schmelze in dünnen Partien schwarzviolett erscheint. Nach ungefähr viertelstündigem Schmelzen läßt man erkalten, fällt die braunviolette Lösung mit Schwefelsäure, und erhält so den unreinen Farbstoff in Form brauner Flocken, welchem noch unzersetztes Anthrachinon, sowie bei der Oxydation gebildete humusartige Körper beigemengt sind. Man schüttelt nun mit Aether, welcher nur Alizarin und etwas Anthrachinon aufnimmt, entzieht der ätherischen Lösung den reinen Farbstoff mit Natronlauge, und wiederholt mit derselben Portion Aether die angegebenen Operationen, so lange derselbe noch Farbstoff aufnimmt. Nun vereinigt man die Farbstofflösungen, fällt mit Schwefelsäure, filtrirt, wäscht und sublimirt nach dem Trocknen.
Die geringe Menge des dem Alizarin noch anhängenden Anthrachinons läßt sich leicht durch vorsichtiges Erhitzen im Sandbade bis auf 180 bis 200° C. sicher und vollständig entfernen; erst über diese Temperatur hinaus und bis gegen 300° sublimirt das Alizarin in den charakteristischen orangerothen federartigen Nadeln. Ich verglich nun dieses Product der directen Oxydation des Anthrachinons mit reinem Pflanzen-Alizarin und dem sublimirten Präparate der HHrn. Meister, Lucius und Comp. in Höchst und konnte die vollständige Identität jener drei Präparate constatiren. Da ich mir größere Quantitäten reines Pflanzen-Alizarin darstellen wollte, schlug ich ein Verfahren ein, das ich seiner ungemeinen Einfachheit wegen kurz anführen will.
Türkischroth gefärbte Baumwollstoffe werden am besten mit einem Gemisch von Alkohol und starker Salzsäure im Wasserbad ausgezogen, die Lösung mit Kali gefällt, der prächtig purpurviolette Niederschlag abfiltrirt, gewaschen und auf dem Filter mit verdünnter Salzsäure zersetzt; die so erhaltene orangegelbe Masse gewaschen und nach dem Trocknen sublimirt.

TC-Vortrag 04 104/161 Waschen – Bleichen – Färben
Man erhält so in einer halben Stunde größere Mengen vom reinsten Alizarin. Auch kann man die gefärbten Stoffe nur kurze Zeit (2 bis 3 Minuten) mit concentrirter Schwefelsäure behandeln und dann die blutrothe Lösung mit Wasser fällen, um den Farbstoff zu erhalten, dem aber hartnäckig eine fettige Substanz, von der Beize herrührend, anhängt, die sich nur durch Behandeln mit Kali, abermaliges Zersetzen und nachheriges Sublimiren vollständig entfernen läßt.
Behandelt man mit Krapp gefärbte Stoffe mit concentrirter Schwefelsäure längere Zeit und verdünnt dann mit Wasser, so scheidet sich ein wolliger, holzgelber Niederschlag ab, der sich, gut gewaschen, ganz ausgezeichnet zum Färben gebeizter Stoffe verwenden läßt. Vielleicht könnte man dieses höchst einfache Verfahren im Großen anwenden, um auf diese Weise aus alten, unbrauchbaren Stoffen ein ziemlich concentrirtes Farbmaterial in außergewöhnlich reinem Zustand zu erhalten.
In ganz frisch bereiteter, absolut kohlensäurefreier Natronlauge löst sich das Pflanzen-Alizarin und mein directes Oxydationsproduct mit Prächtig rein indigoblauer Farbe; die Lösung in einem Uhrglas auf weißer Unterlage betrachtet zeigt eine eigenthümlich rasche Veränderung; dieselbe wird an den Rändern sehr schnell violett, nach einigen Augenblicken schon zeigen sich carminviolette Flecken, welche dann rasch durch die ganze Flüssigkeit zunehmen und schon nach wenigen Minuten ist die Lösung rein carminroth, welche Farbe dann längere Zeit ansteht. Das Höchster Product zeigte gleich im ersten Moment der Lösung schon einen violetten Ton und wurde dann gleichfalls rasch carminroth; im nicht sublimirten Zustande löst sich dieses Präparat mit stark rothvioletter Farbe, welche beim Stehen über Nacht im offenen Uhrglase in einen gelbbraunen Ton übergeht.
Schließlich noch einige Worte über Anthracen selbst. Nach Fritzsche's Angabe soll das aus einer im Sonnenlicht gebleichten Benzollösung abgeschiedene Anthracen prachtvolle violettblaue Fluorescenz zeigen; ich konnte das Entstehen dieser Krystalle ganz ausgezeichnet beobachten, als ich Anthracen mit geschmolzenem Schwefel behandelte, wobei Ströme von Schwefelwasserstoff entwichen. Bei dieser Operation sublimirte nur ein Theil des überschüssigen Anthracens in schönen Krystallen, welche nun jene violettblaue Fluorescenz zeigten, während dasselbe Anthracen ohne Behandlung mit Schwefel wiederholt umkrystallisirt, dann im luftleeren Raume sublimirt, schneeweise oder ganz farblose Krystalle lieferte die absolut nichts von jener schönen Fluorescenz zeigten, und nur an einigen Krystallblättchen konnte man an den Kanten bei günstiger Beleuchtung den bekannten grünen Reflex beobachten. Die Sublimation im luftleeren Raume kann ich überhaupt nicht genug anempfehlen, besonders für größere Mengen von Alizarin; ein weites Rohr, dessen Hinterer Theil mit der zu sublimirenden Substanz gefüllt ist, liegt von heißem Sand umgeben und ist mit einer Sprengel'schen Quecksilber-Pumpe oder Bunsen'schen Wasserpumpe in Verbindung gesetzt. Es geht nun so die Sublimation ganz ausgezeichnet vor sich; ich erhielt reines Alizarin in 3–4 Linien dicken Rinden mit schön facettirter Oberfläche und dunkelfeuerrother Farbe; bei langsamem, vorsichtigem Erhitzen bilden sich manchmal halbzolllange, ziemlich dicke, stängelige Nadeln von derselben dunklen Farbe, die zerrieben ein orangegelbes Pulver liefern.
II. Ueber Reindarstellung des Anthracens (von A. Schuller).
Hat man größere Quantitäten von rohem, noch mit öligen Substanzen verunreinigtem Anthracen zu reinigen, so läßt sich dieß durch Umkrystallisiren aus Benzol oder Alkohol

TC-Vortrag 04 105/161 Waschen – Bleichen – Färben
(seiner relativ geringen Löslichkeit wegen) nur mit großem Zeit- und Mühe-Verlust ausführen. Auch Sublimation jener unreinen Masse ist bei größerer Quantität nur schwer auszuführen; am besten gelingt die Reinigung durch folgende Operation: In einer geräumigen Retorte wird Anthracen vorsichtig bis zum beginnenden Sieden erhitzt, die Retorte mit einer großen tubulirten Glasglocke oder einem ähnlichen irdenen Gefäße, dessen Bodenöffnung mit einem feinen Drahtgitter verschlossen ist, in Verbindung gesetzt. Nun bläst man mittelst eines starken Blasebalges einen kräftigen Luftstrom in die Retorte und treibt auf diese Weise das Anthracen in ganz erstaunlich kurzer Zeit fast vollständig rein und trocken ab. Es verdichtet sich in der Glocke als schwachgelbliche, schneeartige Masse; dieselbe Menge Anthracen, zu deren Reinigung durch Umkrystallisiren und Sublimiren auf gewöhnliche Weise einige Tage erforderlich sind, erhält man nach dem eben mitgetheilten Verfahren in eben so viel Stunden; dabei bekommt man das Anthracen in einer ganz pulverigen Form, in der es besonders leicht Oxydationsmitteln zugänglich ist. Auch Anthrachinon, aus rohem Anthracen dargestellt, kann man auf diese Weise als hellgelbes, den Schwefelblumen ähnliches Pulver erhalten; nur verstopft Anthrachinon den Hals der Retorte leichter als Anthracen, was durch entsprechende Vorsichtsmaßregeln zu beseitigen ist.
Annalen der Chemie und Pharmacie, VII. Supplementband S. 286. Quelle: Polytech. Journalö 1870, Band 197, Nr. XVII. (S. 58–62) URL: http://dingler.culture.hu-berlin.de/article/pj197/ar197017
5.2.8 Chinonin-Farbstoffe
Auch der Chininfabrikation bekannte Stoffe können auch aus dem Teer gewonnen werden. Das Chinonin als Abfallprodukt der Chininfabrikation kann zu einem lebhaft blauen Farbstoff, dem Cyanin oder Lepidinblau umgesetzt werden. Auch: Chinolin
Ueber die Farbstoffe der Blumen; von den HHrn. E. Frémy und Cloëz.
Aus dem Journal de Pharmacie, April 1854, S. 249.
Unsere chemischen Kenntnisse über die Farbstoffe der Blumen sind noch sehr unvollständig. Auch hat das Studium derselben große Schwierigkeiten, weil sie nicht krystallisirbar sind und sich durch die zu ihrer Abscheidung angewandten Agentien sehr oft verändern, ferner weil selbst solche Blumen, welche eine sehr lebhafte Farbe besitzen, ihre Färbung oft sehr geringen Mengen von Farbstoff verdanken.
Ueber die Natur der Farbstoffe der Blumen wurden verschiedene Meinungen aufgestellt. Mehrere Chemiker nahmen an, daß die Blumen ihre Farbe nur zwei Farbstoffen verdanken, einem blauen, Anthokyan (Blumenblau, bleu cyanique), und einem gelben, Anthoxanthin (Blumengelb, jaune xanthique) genannt. Andere wollten eine Beziehung zwischen dem grünen Farbstoff der Blätter, Chlorophyll, und den Farbstoffen der Blumen erkennen, und gründeten ihre Ansicht im Allgemeinen auf Betrachtungen, welche sie aus der Elementar-Analyse dieser näheren Bestandtheile schöpften; nun ist aber bekanntlich das Chlorophyll in reinem Zustande noch nicht dargestellt worden, es enthält wahrscheinlich wandelbare Mengen von fetten und eiweißartigen Substanzen, überdieß kannte man die Farbstoffe der Blumen selbst nur ungenügend.
Eine Zeit lang wollte man die blaue Farbe der Blumen der Gegenwart von Indigo zuschreiben; allein Chevreul wies mit Bestimmtheit nach, daß die blaue Substanz der Blumen

TC-Vortrag 04 106/161 Waschen – Bleichen – Färben
von den Säuren stets geröthet wird und ganz verschieden vom Indigo ist, welcher bekanntlich bei Behandlung mit den stärksten Säuren seine blaue Farbe behält.
Das Studium der Farbstoffe der Blumen war sonach bisher nur ein oberflächliches und mußte ganz von vorn begonnen werden. Diese Stoffe sind für den Chemiker von Interesse, weil sie in den Laboratorien als Reagens auf Alkalien dienen, und eine nähere Kenntniß derselben würde es vielleicht dem Gärtner ermöglichen bei den Blumen, die er zieht, die gewünschten Farben hervorzubringen.
Wir wollten vorerst die Verfahrungsweisen zur Darstellung der Farbstoffe aus den Blumen sorgfältig erforschen, und untersuchen ob diese Substanzen als besondere nähere Bestandtheile zu betrachten sind, oder ob sie von einem und demselben Körper herrühren, welcher von den Säften der Gewächse auf verschiedene Weise modificirt würde.
Blauer Farbstoff der Blumen (Cyanin).
Die blaue Substanz der Blumen nennen wir Cyanin. Um sie zu erhalten, behandeln wir die Blumenblätter der Veilchen, der blauen Kornblumen oder der Schwertlilien zuerst mit kochendem Alkohol; die Blume entfärbt sich und die Flüssigkeit nimmt sogleich eine schöne blaue Farbe an.
Läßt man den Farbstoff einige Zeit mit dem Alkohol in Berührung, so verschwindet allmählich die blaue Farbe der Flüssigkeit und wird bald durch eine braungelbe ersetzt; der Farbstoff erlitt in diesem Fall durch die längere Einwirkung des Alkohols eine wirkliche Reduction, er kann aber, wenn man den Alkohol in Berührung mit der Luft abdampft, seine anfängliche Farbe wieder annehmen; man darf aber den Alkohol nicht zu lang mit dem Farbstoff in Berührung lassen, weil sonst der alkoholische Auszug seine blaue Färbung durch die Einwirkung des Sauerstoffs nicht mehr bekäme.
Der nach Abdampfung des Alkohols bleibende Rückstand wird mit Wasser behandelt, welches eine fette und harzige Substanz absondert; die wässerige Lösung, welche nun den Farbstoff enthält, wird mit neutralem essigsaurem Blei gefällt; dieser Niederschlag, welcher eine schöne grüne Farbe besitzt, kann mit viel Wasser ausgewaschen und dann mit Schwefelwasserstoff zersetzt werden; der Farbstoff bleibt dann in Wasser aufgelöst. Diese Flüssigkeit wird im Wasserbad vorsichtig abgedampft, der Rückstand mit absolutem Alkohol behandelt und endlich die alkoholische Lösung mit Aether gefällt, welcher das Cyanin in bläulichen Flocken abscheidet.
Das Cyanin ist unkrystallisirbar, in Wasser und Alkohol löslich, in Aether unlöslich; von Säuren und sauren Salzen wird es augenblicklich roth gefärbt; Alkalien färben es bekanntlich grün. Es scheint die Rolle einer Säure zu spielen, wenigstens bildet es mit Kalk, Baryt, Strontian, Bleioxyd u.s.w. in Wasser unlösliche, grüne Verbindungen.
Die den Sauerstoff begierig anziehenden Körper, wie schweflige Säure, phosphorige Säure, Alkohol, wirken entfärbend auf dasselbe; in Berührung mit Sauerstoff nimmt es seine Farbe wieder an.
Rosenrother Farbstoff.
Zum Ausziehen der Substanz welche mehrere Dahlienarten, die Rose, die Pfingstrose etc. rosenroth färbt, wandten wir den Alkohol an, indem wir genau dasselbe Verfahren befolgten

TC-Vortrag 04 107/161 Waschen – Bleichen – Färben
wie zur Darstellung des Cyanins; die rosenrothe Substanz wurde mit neutralem essigsaurem Blei gefällt, dann mittelst absoluten Alkohols und Aethers gereinigt.
Bei Vergleichung der Eigenschaften dieses Farbstoffs mit jenen des Cyanins, erkannten wir daß der rosenrothe Farbstoff derselbe ist, wie der blaue, oder doch nur eine Modification desselben; er entsteht, wenn die Pflanzensäfte, womit der blaue Farbstoff in Berührung kommt, sauer reagiren. Wir haben diese saure Reaction bei den Säften mit rother oder rosenrother Färbung stets beobachtet, während der Saft von blauen Blumen immer neutral reagirte.
Wir behandelten die meisten von den rosenroth und roth gefärbten Blumen, welche im Museum zu Paris gezogen werden, mit Alkalien, wobei sie sich anfangs blau und hernach schön grün färbten.
Nicht selten sieht man rosenrothe Blumen, wie Malven und namentlich den Hibiscus syriacus, beim Abwelken eine blaue und hernach eine grüne Farbe annehmen; diese Veränderung rührt, wie wir fanden, von der Zersetzung einer stickstoffhaltigen organischen Substanz her, welche in den Blumenblättern in reichlicher Menge enthalten ist. Dieser Körper erzeugt bei seiner Zerstörung Ammoniak, welches den Blumen beim Welken die blaue oder grüne Farbe gibt; eine schwache Säure ertheilt übrigens den Blumenblättern ihre rosenrothe Farbe wieder.
Bei mehreren rosenrothen Blumen beobachtet man auch eine Farbenveränderung, wenn die Blumenblätter (z.B. im luftleeren Raum) schnell austrocknen; es läßt sich hier nicht wohl annehmen, daß eine stickstoffhaltige organische Substanz sich soweit zersetzte, daß sie Ammoniak lieferte; in diesem Fall bemerkt man aber, daß die Farbenveränderungen ins Violette stechen und nie bis in Grün übergehen, ferner daß sie stets mit Kohlensäure-Entwickelung verbunden sind, wovon wir uns durch einen directen Versuch überzeugten. Die anfangs rosenrothen Blumenblätter, welche durch Trocknen violett werden, entwickeln also Kohlensäure; man kann daher annehmen, daß diese Kohlensäure die rosenrothe Farbe in der Blume erhielt und daß sie bei deren Entweichen die blaue Farbe annehmen, welche diejenigen Blumen charakterisirt, deren Saft neutral ist.
Wir glauben sonach mit Gewißheit behaupten zu können, daß die rosenrothen, violetten und blauen Blumen ihre Farbe derselben Substanz verdanken, welche nur von dem Saft dieser Blumen verschieden modificirt wurde.
Die scharlachrothen Blumen enthalten ebenfalls das durch eine Säure geröthete Cyanin, welches aber mit den sogleich zu beschreibenden gelben Farbstoffen gemengt ist.
Gelbe Farbstoffe.
Die einfachsten Versuche beweisen, daß nicht die geringste Analogie zwischen der die Blumen gelbfärbenden Substanz und der oben besprochenen besteht; die Reagentien können niemals den aus den Blumen gezogenen gelben Stoffen die blauen, rosenrothen oder grünen Farben ertheilen, welche sich mit dem Cyanin so leicht hervorbringen lassen.
Bei unserer Untersuchung der verschiedenen gelb gefärbten Blumen fanden wir, daß sie ihre Farbe Stoffen verdanken, deren Eigenschaften sehr von einander abweichen und welche nicht von demselben näheren Bestandtheil abgeleitet werden können; der eine dieser Stoffe ist in Wasser ganz unauflöslich und wir nennen ihn Xanthin. Den andern, sehr auflöslichen gelben Farbstoff nennen wir Xantheïn.

TC-Vortrag 04 108/161 Waschen – Bleichen – Färben
In Wasser unlöslicher gelber Farbstoff (Xanthin).
Wir haben diesen Farbstoff aus mehreren gelben Blumen, vorzüglich aber aus der Sonnenblume (Helianthus annuus) gezogen.
Um ihn zu erhalten, behandeln wir die Blumen mit kochendem absolutem Alkohol, welcher den Farbstoff auflöst, ihn aber beim Erkalten fast vollständig wieder fallen läßt. Der so erhaltene gelbe Niederschlag ist nicht das reine Xanthin, sondern enthält eine beträchtliche Menge Oel; um diesen Fettkörper abzusondern, kochen wir den gelben Niederschlag mit einer kleinen Menge Alkali, damit das dem Xanthin beigemengte Oel, welches diesen Farbstoff sogar aufgelöst erhält, verseift werde; da aber das Xanthin in seifehaltigem Wasser auflöslich ist, so verdünnen wir die Masse nicht mit Wasser, sondern zersetzen sie durch eine Säure, welche die bei der Verseifung entstandenen Fettsäuren und das Xanthin abscheidet; diesen Niederschlag behandeln wir mit kaltem Alkohol, welcher die Fettsäuren auflöst und das Xanthin zurückläßt. Letzteres ist eine schöngelbe Substanz, welche in Wasser unlöslich, in Alkohol und Aether aber auflöslich ist und dieselben goldgelb färbt.
Sie scheint unkrystallisirbar zu seyn und besitzt die allgemeinen Eigenschaften der Harze.
Das Xanthin, in veränderlichen Mengen dem, durch die Pflanzensäfte verschiedentlich modificirten Cyanin beigemengt, ertheilt den Blumen orangegelbe, rothe und scharlachrothe Farben.
In Wasser löslicher gelber Farbstoff (Xantheïn).
Wenn man die Substanz auszieht, welche gewisse Dahlienarten gelb färbt, so erkennt man leicht, daß sie mit dem Xanthin keine Aehnlichkeit hat. Das Xanthin ist bekanntlich in Wasser unauflöslich, hingegen das Xantheïn (der neue Farbstoff) in Wasser sehr leicht löslich.
Um letzteres zu erhalten, behandeln wir die gelben Dahlienblätter mit Alkohol, welcher den gelben Farbstoff rasch auflöst, nebst den fetten und harzigen Stoffen; die Flüssigkeit wird zur Trockne abgedampft und der Rückstand in Wasser aufgenommen, welches die Harze und Fettsubstanzen fällt; diese Flüssigkeit wird neuerdings zur Trockne abgedampft und der Rückstand mit absolutem Alkohol behandelt; diese Auflösung wird mit Wasser verdünnt und mit neutralem essigsaurem Blei versetzt, welches den Farbstoff niederschlägt; das Bleisalz wird hernach mit Schwefelsäure zersetzt; das Xantheïn bleibt im Wasser aufgelöst; man reinigt es endlich mit Alkohol.
Das Xantheïn ist in Wasser, Alkohol und Aether löslich, krystallisirt aber aus keinem dieser Lösungsmittel. Die Alkalien ertheilen ihm eine sehr satte braune Färbung; sein Färbevermögen ist beträchtlich; es liefert auf den verschiedenen Geweben gelbe Farben, denen es nicht an Lebhaftigkeit fehlt.
Die Säuren machen die durch Alkalien hervorgebrachte braune Färbung verschwinden. Das Xantheïn verbindet sich mit den meisten Metalloxyden und bildet mit denselben unlösliche gelbe oder braune Lacke.
Dieses sind die Eigenschaften der Farbstoffe, welche wir aus den Blumen gezogen haben. Unsere bisherigen Versuche beweisen, daß die gelben Farbstoffe von denjenigen Pigmenten welche die Blumen blau und rosa färben, ganz verschieden sind, was auch mit allen bisher hierüber angestellten Beobachtungen übereinstimmt; denn bekanntlich können die blauen Blumen roth werden und sogar weiß, wenn sich die Farbe ganz zersetzt; niemals aber werden

TC-Vortrag 04 109/161 Waschen – Bleichen – Färben
sie gelb, sowie umgekehrt die gelbe Blume niemals blau wird. Nicht selten wird eine orangegelbe Blume roth; alsdann hat sich das Xanthin zersetzt und das Cyanin, durch die Pflanzensäfte geröthet, ist vorherrschend geworden.
Wir haben somit das Vorkommen dreier Farbstoffe in den Blumen nachgewiesen; sie sind: das Cyanin seine blaue oder rosenrothe Substanz), das Xanthin (eine in Wasser unlösliche gelbe Substanz) und das Xantheïn (eine in Wasser lösliche gelbe Substanz).
Diese drei Stoffe können im reinen Zustand und durch ihre Vermischung die Farben der meisten Blumen hervorbringen; doch getrauen wir uns zur Zeit noch nicht zu behaupten, daß diese von uns isolirt dargestellten Stoffe die einzigen sind, welche alle Blumen färben.
Wir werden nun die Elementar-Zusammensetzung dieser drei Stoffe bestimmen.
Quelle: Polytech. Journal 1854, Band 132, Nr. CIV. (S. 377–382) URL: http://dingler.culture.hu-berlin.de/article/pj132/ar132104
5.3 Quellen
Handbuch der chemischen Technologie; Wagner, Rudolf; 8.A; Leipzig 1871, S. 653 ff.
http://kremer-pigmente.de
Wikipedia (diverse Fundstellen)
http://www.planet-schule.de/sf/multimedia-simulationen-detail.php?projekt=farben_ausbleichen
http://www.planet-schule.de/warum_chemie/farbe/themenseiten/t_index/s1.html
Polytechnisches Journal (diverse Fundstellen) siehe Dingler Online: http://dingler.culture.hu-berlin.de
W. Stein, Polyt. Zentralblatt von 1868, S.190
u. a.

TC-Vortrag 04 110/161 Waschen – Bleichen – Färben
5.4 Glossar chemischer Fachbegriffe insb. Organische Chemie
Aldehyde
Aldehyde (aus neulateinisch alcoholus dehydrogenatus, „dehydrierter Alkohol“ oder „Alkohol, dem Wasserstoff entzogen wurde“) sind chemische Verbindungen mit der funktionellen Gruppe −CHO, die Aldehydgruppe oder auch Formylgruppe genannt wird.
Die Carbonylgruppe (>C=O) der Aldehyde trägt im Unterschied zu den Ketonen einen Wasserstoff- und einen Kohlenstoffsubstituenten. Eine Ausnahme bildet der einfachste Aldehyd Methanal (Formaldehyd), der zwei Wasserstoffsubstituenten trägt. Aldehyde mit einem Alkylrest (also Alkan-Derivate) werden als Alkanale bezeichnet; deren homologe Reihe leitet sich nomenklatorisch entsprechend von der homologen Reihe der Alkane ab. Weiter existieren Mehrfachaldehyde – wie beispielsweise das Glyoxal, der einfachste Dialdehyd.
z. B.
Allgemeine Struktur eines Aldehyds: Der Rest R kann ein Wasserstoffatom oder ein Organyl-Rest (Alkyl-, Aryl-, Alkenyl-Rest etc.) sein. Die Aldehydgruppe (Formylgruppe) ist blau gekennzeichnet. Beispiele: Formaldehyd (Methanal, links), Acetaldehyd (Ethanal, Mitte) und Propionaldehyd (Propanal, rechts) mit blau gekennzeichneter Aldehydgruppe (Formylgruppe)
Alkanale erhalten nach der IUPAC-Nomenklatur den Namen des Alkans mit derselben Anzahl an Kohlenstoff-Atomen mit dem Suffix -al oder -carbaldehyd. Dementsprechend heißt der vom Methan abgeleitete Aldehyd Methanal, der vom Ethan abgeleitete Ethanal. Falls eine andere funktionelle Gruppe eine höhere Priorität aufweist, wird das Präfix „Formyl-“ verwendet. Ist die Verbindung hingegen ein Naturstoff oder eine Carbonsäure, so wird das Präfix „Oxo-“ gewählt.
Der Trivialname leitet sich von der lateinischen Bezeichnung für die bei Hinzufügen eines Sauerstoffatoms jeweils entstehende Carbonsäure her. Für Methanal (H–CHO) ist das die Methansäure (lat. acidum formicum, H–COOH), daher Formaldehyd, für Ethanal die Ethansäure (lat. acidum aceticum, CH3–COOH), daher Acetaldehyd. Entsprechend leiten sich die anderen Trivialnamen ab. Dicarbonsäuren, bei denen eine Carbonsäuregruppe zu einer Aldehydgruppe reduziert wurde, werden gelegentlich Semialdehyde genannt.
Zwischen den Aldehydgruppen von Alkanalen kommt es zu Dipol-Dipol-Kräften, da die C=O-Doppelbindung sehr polar ist. Wasserstoffbrückenbindungen bilden sich nicht, weil kein sauerstoffgebundenes Wasserstoffatom vorhanden ist. Deswegen liegen die Siedepunkte der Aldehyde zwischen denen der Alkohole und Alkane. Mit Wasser können Aldehyde Wasserstoffbrückenbindungen eingehen, weil das Sauerstoffatom zwei freie Elektronenpaare hat und negativ polarisiert ist. Deswegen sind kurzkettige Aldehyde gut wasserlöslich. Bei längerkettigen Aldehyden überwiegt die Wirkung der unpolaren Alkylreste, was die Verbindungen unlöslich in Wasser macht. Viele Aldehyde haben einen charakteristischen Geruch.

TC-Vortrag 04 111/161 Waschen – Bleichen – Färben
Aldehyde und Ketone werden außerdem zur Herstellung von Kunststoffen, Lösungsmitteln, Farbstoffen, Parfums und Medikamenten verwendet. Ausgehend von Acrolein wird DL-Methionin, ein Futtermittelzusatzstoff, in Mengen von mehr als 100.000 Tonnen pro Jahr hergestellt. In der Medizin werden Formaldehyd und Glutaraldehyd als Flächen- und Instrumentendesinfektionsmittel eingesetzt. Beide Aldehyde haben eine gute Wirksamkeit gegen viele verschiedene Mikroorganismen. Insbesondere unbehüllte Viren und sporenbildende Bakterien (z. B. Milzbrand), die nur wenigen Desinfektionsmitteln zugänglich sind, können so erreicht werden. Da Aldehyde irritierend auf Haut und Schleimhäute wirken und gelegentlich Allergien auslösen, muss mit diesen Mitteln sorgfältig umgegangen werden.
Alkane
Mit Wasserstoff gesättigte Kohlenwasserstoff-Kette, aliphatischer Kohlenwasserstoff . Der vierbindige Kohlenstoff ist hier entweder an einen weiteren Kohlenstoff oder an Wasserstoff gebunden und bildet so unverzweigte oder verzweigte Ketten und Ringe. Die niedermolekularen Alkane sind bei Zimmertemperatur gasförmig: Methan, Ethan, Propan, Butan. Mit zunehmender Molekülschwere werden die Alkane flüssig und dann fest: Pentan, Hexan, Heptan, Octan, Nonan, Decan, Undecan, Dodecan, ….
Die Namen leiten sich bis auf ein paar besondere Namen jeweils von der Zahl der Kohlenstoffatome der längsten Kette ab und tragen die Namensendung -an. z. B. Hexan C6H14 (CH3-CH2-CH2-CH2-CH2-CH3)
Ethan C2H6
Als Alkane (Grenzkohlenwasserstoffe, früher Paraffine) bezeichnet man in der organischen Chemie die Stoffgruppe der gesättigten, acyclischen Kohlenwasserstoffe, deren Vertreter nur aus den beiden Elementen Kohlenstoff (C) und Wasserstoff (H) bestehen. Damit sind sie eine Untergruppe der aliphatischen Kohlenwasserstoffe. Für sie gilt die allgemeine Summenformel CnH2n+2 mit n = 1, 2, 3, …
Das Grundgerüst der Alkane kann aus unverzweigten (linearen) wie aus verzweigten Kohlenstoffketten bestehen. Die unverzweigten Verbindungen werden als n-Alkane bezeichnet und bilden eine homologe Reihe der Alkane.
n-Butan
Die verzweigten Alkane werden Isoalkane (i-Alkane) genannt.
iso-Butan (2-Methylpropan)

TC-Vortrag 04 112/161 Waschen – Bleichen – Färben
Mit steigender Anzahl an Kohlenstoffatomen steigt auch die Anzahl der Möglichkeiten für deren kovalente Verknüpfung. Deswegen kommen alle Alkane mit einer höheren Zahl an Kohlenstoffatomen als Propan in einer Vielzahl von Konstitutionsisomeren – Molekülen mit der gleichen Summenformel, aber unterschiedlichem Aufbau (Konstitution) – vor. Diese werden als Isomere bezeichnet.
Beim Butan tritt der Fall ein, dass bei gleicher Summenformel C4H10 zwei unterschiedliche Anordnungsmöglichkeiten für die Kohlenstoffatome im Alkanmolekül möglich sind. Butan existiert also in zwei verschiedenen Konstitutionen: n-Butan und iso-Butan (isomeres Butan). Davon leitet sich der Begriff iso-Alkane – abgekürzt i-Alkane – ab.
Pentan tritt bereits in drei verschiedenen Konstitutionen auf, dem n-Alkan mit einer unverzweigten Kette, dem iso-Pentan mit einer Verzweigung am zweiten Kohlenstoffatom und dem neo-Pentan mit zwei Verzweigungen am zweiten Kohlenstoffatom.
Mit wachsender Anzahl der Kohlenstoffatome steigt rasch auch die Zahl der möglichen Isomere, von denen die meisten allerdings nur theoretisch bestehen – in Natur und Technik sind nur wenige von Bedeutung. Icosan (ehemals Eicosan) mit einer Kette aus zwanzig Kohlenstoffatomen besitzt bereits 366.319 verschiedene Konstitutionsisomere. Bei Alkanen mit 167 Kohlenstoffatomen übersteigt die Anzahl der theoretisch möglichen Isomere die geschätzte Zahl der Teilchen im sichtbaren Universum.
Gesättigte cyclische Kohlenwasserstoffe haben eine abweichende allgemeine Summenformel und bilden die Gruppe der Cycloalkane und werden dort beschrieben.
Nomenklatur - Namensbildung
Die Nomenklatur der Alkane ist durch die International Union of Pure and Applied Chemistry (IUPAC) genau festgelegt. Nachfolgend erläutert mit einem Beispiel zur Bezeichnung eines iso-Alkans:
Alle Stammnamen weisen die Endung -an auf. Dieser Endung wird ein griechisches Zahlenwort vorangestellt, das auf die Anzahl der Kohlenstoffatome hinweist. Für die ersten vier Alkane, hierbei handelt es sich um Trivialnamen, werden stattdessen historisch bedingt die Namen Methan, Ethan (vormals Äthan), Propan und Butan vergeben. Wie die Namen von Alkanen mit mehr als zehn Kohlenstoffatomen gebildet werden, findet sich im Artikel Nomenklatur http://de.wikipedia.org/wiki/Nomenklatur_(Chemie)#Lineare_Ketten.
Für verzweigte Alkane gelten die folgenden Benennungsregeln:
1. Die Kohlenstoffatome der längsten durchgehenden Kohlenstoffkette werden so durchnummeriert, dass die tertiären bzw. quartären Kohlenstoffatome jeweils eine möglichst niedrige Zahl erhalten. Dies ist der Fall, wenn die Summe aller dieser Zahlen am niedrigsten ist (Beispielmolekül oben: 2+3+4=9). Entsprechend dieser längsten Kette erhält das Molekül seinen Stammnamen (Beispielmolekül oben: 6 Kohlenstoffatome → Hexan).
2. Die Namen der abzweigenden Alkylgruppen (Seitenketten) werden ebenfalls durch ihre Länge bestimmt und alphabetisch aufsteigend sortiert dem Stammnamen des Alkans vorangestellt (s. u. 4. Zusatzregel a).

TC-Vortrag 04 113/161 Waschen – Bleichen – Färben
3. Diesen Alkylgruppennamen werden die Nummern, durch Bindestriche von diesen getrennt, der Kohlenstoffatome, an denen sie abzweigen, vorangestellt (s. u. 5. Zusatzregel b).
4. Zusatzregel a) Zweigt mehr als eine Alkylgruppe mit gleichem Namen von der Hauptkette ab, werden diesen Alkylgruppennamen deren Anzahl in der griechischen Schreibweise (di =zwei, tri =drei, etc.) als Zahlwort vorangestellt. Beachte: Diese Zahlenwörter werden bei der alphabetischen Sortierung nicht berücksichtigt.
5. Zusatzregel b) Gibt es mehrere abzweigende Alkylgruppen mit gleichem Namen, werden die Zahlen mit aufsteigendem Wert durch Kommata getrennt notiert. Zweigen zwei gleiche Alkylgruppen an einem quartären Kohlenstoffatom ab, dann wird die Nummer des Kohlenstoffatoms doppelt notiert.
Ein Beispiel für die Zusatzregeln a) und b) ist das 3-Ethyl-2,2,4-trimethylhexan: Am oben abgebildeten 3-Ethyl-2,4-dimethylhexan wäre am zweiten Kohlenstoffatom das Wasserstoffatom durch eine Methyl-Gruppe ersetzt.
Früher wurden Alkane als „Grenzkohlenwasserstoffe“ oder Paraffine bezeichnet. Letzteres leitet sich von lateinisch parum affinis ab, was sich mit „wenig verwandt“ übersetzen lässt – man glaubte früher, dass Stoffe, die miteinander reagieren, irgendeine Art von „Verwandtschaft“ aufweisen müssten – und brachte damit die relative Reaktionsträgheit dieser Verbindungen zum Ausdruck. Heute bezeichnet der Name meist nur noch ein Stoffgemisch aus bestimmten festen Alkanen.
Die Molekülstruktur, speziell die Größe der Oberfläche der Moleküle, bestimmt den Siedepunkt des zugehörigen Stoffes: je kleiner die Fläche, umso niedriger ist der Siedepunkt, da so die zwischen den Molekülen wirkenden Van-der-Waalsschen Kräfte kleiner sind; eine Verkleinerung der Oberfläche kann dabei durch Verzweigungen oder durch eine ringförmige Struktur erreicht werden. Das bedeutet in der Praxis, dass Alkane mit höherem Kohlenstoffanteil in der Regel einen höheren Siedepunkt als Alkane mit geringerem Kohlenstoffanteil haben; unverzweigte Alkane haben höhere Siedepunkte als verzweigte und ringförmige wiederum höhere Siedepunkte als die unverzweigten. Ab fünf Kohlenstoffatomen sind unverzweigte Alkane unter Normalbedingungen flüssig, ab siebzehn fest. Der Siedepunkt nimmt pro CH2-Gruppe um zwischen 20 und 30 Grad zu.
Auch der Schmelzpunkt der Alkane steigt mit zwei Ausnahmen bei Ethan und Propan bei Zunahme der Anzahl der Kohlenstoffatome; allerdings steigen die Schmelzpunkte insbesondere bei den höheren Alkanen langsamer als die Siedepunkte. Außerdem steigt der Schmelzpunkt von Alkanen mit ungerader Kohlenstoffzahl zu Alkanen mit gerader Kohlenstoffzahl stärker als umgekehrt. Die Ursache dieses Phänomens ist die größere Packungsdichte der Alkane mit geradzahliger Kohlenstoffzahl.
Der Schmelzpunkt der verzweigten Alkane kann sowohl ober- als auch unterhalb des entsprechenden Wertes für die unverzweigten Alkane liegen. Je sperriger das Molekül ist, desto schwieriger lässt sich die entsprechende Substanz eng packen und desto niedriger liegt folglich auch der Schmelzpunkt. Umgekehrt existiert eine Reihe von Isoalkanen, die eine wesentlich kompaktere Struktur einnehmen als die korrespondierenden n-Alkane; in diesem Fall liegen ihre Schmelzpunkte daher über denjenigen ihrer geradlinigen Isomere.

TC-Vortrag 04 114/161 Waschen – Bleichen – Färben
Alkane leiten weder den elektrischen Strom noch sind sie dauerhaft elektrisch polarisiert. Aus diesem Grund bilden sie keine Wasserstoffbrückenbindungen aus und lassen sich in polaren Lösungsmitteln wie Wasser sehr schlecht lösen. Da die Wasserstoffbrückenbindungen zwischen den einzelnen Wassermolekülen in der unmittelbaren Nähe eines Alkans von diesem wegweisen und daher nicht isotrop ausgerichtet sind, also nicht gleichmäßig in alle Richtungen zeigen, wäre eine Mischung beider Substanzen mit einer Zunahme der molekularen Ordnung verbunden. Da dies nach dem Zweiten Hauptsatz der Thermodynamik verboten ist, bilden sich bei dem Versuch einer Mischung immer zwei separate Schichten (Phasen). Man bezeichnet Alkane daher als wasserabweisend oder hydrophob. Ihre Löslichkeit in unpolaren Lösungsmitteln ist dagegen gut, ein Umstand, der als lipophil bezeichnet wird. Untereinander sind sie beispielsweise bei gleichem Aggregatzustand in jedem Verhältnis mischbar.
Bis auf einige Ausnahmen nimmt die Dichte der Alkane mit zunehmender Zahl der Kohlenstoffatome zu. Da sie geringer ist als diejenige des Wassers, schwimmen Alkane bei versuchter Mischung immer oben, weshalb brennende, flüssige Alkane nicht mit Wasser gelöscht werden können.
Alkene
Mit Wasserstoff größtenteils gesättigte Kohlenwasserstoff-Kette, welche eine oder mehrere Doppelbindungen zwischen Kohlenstoffatomen aufweist. Mehrere Doppelbindungen treten in den Alkenen selten direkt nebeneinander auf. Durch Wechsel der Position der Doppelbindungen entstehen ggf. unterschiedliche Grenzformen (s. Mesomerie).
Alkene (früher auch Olefine) sind chemische Verbindungen aus der Gruppe der aliphatischen Kohlenwasserstoffe, die an beliebiger Position mindestens eine Kohlenstoff-Kohlenstoff-Doppelbindung im Molekül besitzen. Ein Beispiel mit mehreren C=C-Doppelbindungen ist Butadien, das zwei Doppelbindungen im Molekül besitzt.
Propen, oft Propylen genannt. Allgemeine Strukturformel für Alkene mit der charakteristischen C=C-Doppelbindung zwischen zwei sp2-hybridisierten Kohlenstoffatomen (blau markiert). Dabei gilt: R1 bis R4 sind Wasserstoffatome oder Alkyl-Reste. In Dienen ist einer der Reste R1 bis R4 eine Alkenylgruppe.
Alkene sind ungesättigte Verbindungen im Gegensatz zu den Alkanen, bei denen alle Valenzen des Kohlenstoffatoms abgedeckt (gesättigt) sind. Alkene kommen im geringen Maßstab im Erdöl vor, in der Natur werden sie als Pheromone und Phytohormone verwendet. Sie sind die wichtigsten Basisprodukte der industriellen organischen Chemie.
Alkene mit einer Doppelbindung bilden eine homologe Reihe mit der allgemeinen Summenformel CnH2n beginnend mit dem Ethen. Der veraltete Name Olefine ergibt sich aus dem alten Namen Olefin von Ethen, da es mit Halogenen ölige, wasserunlösliche Flüssigkeiten bildet, die aus Halogenalkanen bestehen. Es gibt auch cyclische Alkene, die Cycloalkene, deren wichtigster Vertreter das Cyclohexen ist.Die Namen leiten sich bis auf ein paar besondere Namen wie bei den Alkanen jeweils von der Zahl der Kohlenstoffatome der längsten Kette ab, wobei das Suffix -an durch -en ersetzt wird.

TC-Vortrag 04 115/161 Waschen – Bleichen – Färben
Die Position der Doppelbindung in der Kohlenstoffkette wird im Namen durch eine Zahl angegeben, die das Kohlenstoffatom bezeichnet, an dem die Doppelbindung beginnt. Sie gilt dabei als funktionelle Gruppe und muss für die Reihenfolge der Nummerierung berücksichtigt werden, also eine möglichst kleine Ziffer erhalten. Bei Molekülen mit mehreren funktionellen Gruppen wird die Zahl direkt vor das -en , sonst auch vor den Namen gestellt. Mehrfache Doppelbindungen erhalten vor das Suffix das entsprechende griechische Zahlwort gestellt.
2-Hexen: CH3-CH=CH-CH2-CH2-CH3 3-Hexen: CH3-CH2-CH=CH-CH2-CH3
1,4-Hexadien: CH2=CH-CH=CH-CH2-CH3 Ethen: C2H4 bzw. CH2=CH2
Neben der Strukturisomerie, bei der die Kohlenstoffatome unterschiedlich angeordnet sind, kann bei Alkenen an der C=C-Doppelbindung auch noch die cis-trans-Isomerie auftreten.
Da die Doppelbindung im Gegensatz zur Einfachbindung nicht frei drehbar ist, kann es bei anhängenden Atomen oder Atomgruppen an der Doppelbindung zu zwei möglichen Anordnungen kommen. Cis-trans-Isomere unterscheiden sich in ihren physikalischen und chemischen Eigenschaften. Sie lassen sich über das Dipolmoment und über IR-Spektroskopie unterscheiden. Während das cis im Verbindungsnamen erwähnt wird, kann man das trans auch weglassen.
Am Beispiel der isomeren But-2-ene lässt sich die cis-trans-Isomerie nachvollziehen. Beim cis-But-2-en liegen beide Methylgruppen als Kettenreste diesseits (lat. cis), das heißt auf der gleichen Seite. Beim trans-But-2-en liegen die Methylgruppen auf der jeweils anderen (lat. trans) Seite der Doppelbindung.
cis-2-Buten oder (Z)-2-Buten trans-2-Buten oder 2-Buten oder (E)-2-Buten
Von der IUPAC wurde die cis/trans-Bezeichnung ersetzt (da sie bei mehr als zwei Substituenten leicht in die Irre führt; man betrachte nur (E)-2-Brom-1-chlor-1-fluor-ethen!) durch E/Z, wobei (E) (entgegengesetzt) meist – aber nicht immer – für trans steht und (Z) (zusammen) für cis. Dabei wird die gegenseitige Lage der Substituenten höchster CIP-Priorität angegeben. Genauere Ausführungen siehe (E,Z)-Isomerie.
(E,Z)-Nomenklatur bei Alkenen: Die CIP-Priorität der vier Substituenten ist a > b und c > d.
E: a und d nebeneinander Z: a und c nebeneinander
Traditionell haben einige einfache Substanzen mit (E,Z)-Isomerie unterschiedliche Namen: Fumarsäure [(E)-Butendisäure] und Maleinsäure [(Z)-Butendisäure] sowie deren Derivate sind dafür Beispiele.
CIP-Priorität
Die Cahn-Ingold-Prelog-Konvention (kurz: CIP-Konvention oder (RS)-System) dient zur eindeutigen Beschreibung der räumlichen Anordnung der unterschiedlichen Substituenten an Atomen oder an Doppelbindungen.

TC-Vortrag 04 116/161 Waschen – Bleichen – Färben
Die CIP-Konvention wurde 1966 von Robert Sidney Cahn, Christopher Kelk Ingold und dem Schweizer Nobelpreisträger Vladimir Prelog vorgeschlagen und 1982 von Vladimir Prelog und Günter Helmchen überarbeitet. Zweck der CIP-Nomenklatur ist:
die Bestimmung der absoluten Konfiguration [(R)- oder (S)-Deskriptor] der Substituenten am Stereozentrum eines Moleküls mit Chiralitätszentren
die Bestimmung der geometrischen Anordnung [(E)- oder (Z)-Notation] – auch (E)- oder (Z)-Deskriptor – der Substituenten an der Doppelbindung eines cis-trans-Isomers
die Bestimmung der Anordnung [(RA)- oder (SA)-Deskriptor] an kumulierten Doppelbindungen. (Allene)
Komplexe Moleküle mit mehreren Stereozentren und/oder mehreren Doppelbindungen mit cis-trans-Isomerie können in ihrem geometrischen Aufbau durch den systematischen IUPAC-Namen mit den vorangestellten CIP-Deskriptoren eindeutig benannt werden.
Identifizierung der Chiralitätszentren
Zuerst werden die Chiralitätszentren des Moleküls identifiziert. Ein Chiralitätszentrum ist ein Atom, das vier verschiedene Substituenten trägt. An den meisten Molekülen finden sich Stereozentren an Kohlenstoffatomen. Sie können aber auch an Stickstoff-, Schwefel-, Silicium- oder Phosphoratomen auftreten. Als Substituenten zählen Atome, Atomgruppen oder freie Elektronenpaare. Man markiert die Stereozentren in der Strukturformel durch Sternchen. Jedes Chiralitätszentrum wird einzeln betrachtet.
Priorisierung der Substituenten
Es werden die Substituenten am Chiralitätszentrum betrachtet. Ziel ist es, den vier verschiedenen Substituenten die Prioritäten 1 bis 4 zuzuordnen.
1. Die Substituenten, die direkt am Chiralitätszentrum gebunden sind (man bezeichnet diese als Substituenten der ersten Sphäre) werden nach ihrer Ordnungszahl geordnet. Freie Elektronenpaare erhalten die fiktive Ordnungszahl 0. Die Prioritäten werden von hoher Ordnungszahl nach niedriger Ordnungszahl vergeben (Priorität 1: höchste Ordnungszahl, Priorität 2: zweithöchste Ordnungszahl etc.).
2. Sind zwei oder mehr Substituenten identisch, werden deren Substituenten durch eine Liste aller in der zweiten Sphäre an sie gebundenen Substituenten ersetzt, wieder in Reihenfolge der Ordnungszahl. Die Listen werden miteinander verglichen, wobei der erste unterschiedliche Substituent den Ausschlag gibt. Wieder werden die Prioritäten an die Substituenten der Sphäre 1 entsprechend der Ordnungszahl (diesmal des ersten unterschiedlichen Substituenten) vergeben.
3. Sind die Listen der Substituenten in der zweiten Sphäre identisch, werden die einzelnen Substituenten in dieser Sphäre durch eine Liste derer in der dritten Sphäre ersetzt, in Reihenfolge ihrer Ordnungszahlen. Es wird wieder entsprechend Punkt 2. vorgegangen.
4. Punkt 3. wird solange in der jeweils nächsten Sphäre wiederholt, bis eine Unterscheidung getroffen ist.

TC-Vortrag 04 117/161 Waschen – Bleichen – Färben
5. Ist selbst bei Betrachtung der letzten Sphäre (dem Molekülende, oder bei Cyclen dem Ausgangsatom (s. u.)) keine Unterscheidung möglich, müssen weitere Unterscheidungskriterien in folgender Reihenfolge untersucht werden:
1. Sind im Molekül Isotope vorhanden, so hat an erster unterschiedlicher Stelle das schwerere Isotop höhere Priorität.
2. Sind im Molekül unterschiedlich konfigurierte Doppelbindungen vorhanden, so hat an erster unterschiedlicher Stelle das (Z)-Isomer höhere Priorität als das (E)-Isomer.
3. Gleiche Deskriptorenpaare in den substituierenden Atomgruppen haben Priorität vor unterschiedlichen [beispielsweise (SS) vor (RS)].
4. Handelt es sich um ein Pseudochiralitätszentrum, so haben (R)-konfigurierte Atomgruppen Priorität vor (S)-konfigurierten.
Bestimmung des Deskriptors
„rechtsdrehend“ oder „im Uhrzeigersinn“
„linksdrehend“ oder „gegen den Uhrzeigersinn“
Der Substituent mit der niedrigsten Priorität 4 wird unter die Bildebene gestellt. Anschließend zählt man kreisförmig um das aktive Zentrum vom Substituenten mit der Priorität 1 bis zur Priorität 3. Läuft diese Kreisbewegung rechtsherum, also im Uhrzeigersinn, so liegt eine (R)-Konfiguration vor, läuft sie linksherum (gegen den Uhrzeigersinn), so liegt eine (S)-Konfiguration vor. (R) ist die Abkürzung von lateinisch rectus (rechts) und (S) von lateinisch sinister (links).
Doppelbindungen und konjugierte Systeme
Doppel- und Dreifachbindungen werden so behandelt, als ob das jeweilige Atom bzw. die jeweilige Gruppe doppelt bzw. dreifach vorhanden wäre (Duplikatatome). Duplikatatome besitzen konventionsgemäß keine Substituenten in der nächsten Sphäre. Dabei ist zu beachten, dass Doppelbindungen zwischen Heteroatomen mit wenigstens einem Element ab der dritten Pheriode konventionsgemäß als Einfachbindungen betrachtet werden (beispielsweise wird P=O als P–O interpretiert). In konjugierten Systemen (wie Aromaten) wird anstelle des Duplikatatoms ein fiktives Duplikatatom, dessen Ordnungszahl dem Mittelwert der Ordnungszahlen der Atome entspricht, zu denen in mesomeren Grenzstrukturen Doppelbindungen gezeichnet werden können, verwendet.
(Carbo-)Cyclen
An Chiralitätszentren an Carbocyclen wird jeder Zweig des Rings in allen Sphären betrachtet, bis der Ausgangspunkt erreicht wird, dieser wird nur noch als Duplikatatom berücksichtigt.

TC-Vortrag 04 118/161 Waschen – Bleichen – Färben
Beispiele für die Priorität der Substituenten in abfallender Priorität:
–I > –Cl > –S–CH3 > –SH > –F > –O–CH3 > –OH > –N3 > –N (CH3)2 > –NH–C6H5 > –NH2 > –COOH > –CONH2 > –CONH2 > –CHO > –CH2OH > –CD3 > –CD2H > –CDH2 > –CH3 > –D > –H > freies Elektronenpaar (D = Deuterium, schweres H-Isotop, vgl. CIP-Regel 5.1.)
Die CIP-Regeln können auch zur eindeutigen Bestimmung der Konfiguration von Molekülen mit Chiralitätsachsen, Chiralitätsebenen oder helikalen Strukturen verwendet werden. Wenn ein Molekül mehrere Chiralitätszentren aufweist, so wird jedes einzelne gemäß den oben genannten Regeln charakterisiert und im systematischen Namen aufgeführt.
Alkine
Mit Wasserstoff größtenteils gesättigte KW-Kette, welche eine oder mehrere Dreifachbindungen zwischen Kohlenstoffatomen aufweist. Diese Bindung ist sehr reaktiv.
Die Namen leiten sich bis auf ein paar besondere Namen wie bei den Alkanen jeweils von der Zahl der Kohlenstoffatome der längsten Kette ab und tragen die Namensendung -in.
Ethin, Azetylen, C2H2
Alkine (früher Acetylene und Acetylenkohlenwasserstoffe) sind chemische Verbindungen aus der Gruppe der aliphatischen Kohlenwasserstoffe, die an beliebiger Position mindestens eine Kohlenstoff-Kohlenstoff-Dreifachbindung im Molekül besitzen. Die Alkine mit nur einer solchen Dreifachbindungen bilden eine homologe Reihe mit der allgemeinen Summenformel CnH2n-2 (mit n = 2, 3, 4, ...), die mit Ethin beginnt.
Verbindungen mit zwei oder mehreren Kohlenstoff-Kohlenstoff-Dreifachbindungen werden Polyine genannt; im erweiterten Sinne können acyclische Kohlenwasserstoffe mit mehreren Kohlenstoff-Kohlenstoff-Dreifachbindungen zu den Alkinen gezählt werden.
Cyclische Verbindungen mit einer Kohlenstoff-Kohlenstoff-Dreifachbindung werden hingegen zu den Cycloalkinen gezählt und aromatische Kohlenwasserstoffe mit formaler Dreifachbindung im Ring Arine genannt.
Alkine (von oben nach unten):
(a) Ethin
(b) 1-Butin
(c) das dazu isomere 2-Butin
(d) Allgemeine Strukturformel eines Alkins mit einer endständigen Dreifachbindung.
Die typische Kohlenstoff-Kohlenstoff-Dreifachbindung ist zusammen mit den beiden beteiligten sp-hybridisierten Kohlenstoffatomen jeweils blau markiert.

TC-Vortrag 04 119/161 Waschen – Bleichen – Färben
Zum ersten Mal wurde Acetylen (Ethin), das einfachste Alkin, in Jahr 1836 von Edmund Davy erhalten, einem Chemieprofessor an der Royal Dublin Society und Vetter des berühmten englischen Chemikers Humphry Davy. Beim Versuch, Kalium in metallischer Form darzustellen, erhielt er Ethin durch Erhitzen von Kaliumsalzen wie Kaliumacetat oder Kaliumcarbonat mit Kohle, gefolgt von der Reaktion des gebildeten Kaliumcarbids mit Wasser. Die Entdeckung geriet jedoch in Vergessenheit. Im Jahr 1862 gelang dem deutscher Chemiker und Arzt Friedrich Wöhler die Darstellung von Acetylen durch die Reaktion von Wasser mit Calciumcarbid. Im Jahr 1863 gelang dem französischen Chemiker Marcelin Berthelot die Darstellung aus den Elementen über den Lichtbogen zwischen Graphitelektroden in einer Wasserstoffatmosphäre. Er gab dem Gas den Namen Acetylen. 1895 entdeckte Henry Le Chatelier, dass Acetylen mit Sauerstoff mit einer sehr heißen Flamme (bis 3100 °C) verbrennt. Damit war die Grundlage des Acetylen-Schweißens und -Schneidens gelegt.
Alkyl‐Rest/Alkyl‐Gruppe
Wird einem Alkanmolekül ein Wasserstoffatom entzogen, entsteht ein Radikal, ein Molekül mit einem ungebundenen Elektron, das man als Alkylradikal bezeichnet. Den Namen dieses Alkylrestes erhält man, wenn man bei der Endung des Alkans, dem das Wasserstoffatom entzogen wurde, das -an durch ein -yl ersetzt, z. B. Propan => Propyl. Symbolisch werden Alkyle häufig mit R notiert; sind die Alkylreste unterschiedlich, wird dieses durch R1, R2, R3, usw. kenntlich gemacht. Alkane lassen sich auch als dimerisierte Alkyle auffassen.
Alkohole
Alkane mit der funktionellen OH-Gruppe bezeichnet man als Alkohol. Die Namensbildung erfolgt durch anhängen des Suffixes –ol an den Namen des zugrundeliegenden Alkans, z. B. Ethan => Ethanol.
Polyalkohole verfügen über mehrere OH-Gruppen. Zucker können zu den Polyakoholen gerechnet werden und weisen einen Ringschluss über ein Sauerstoffatom auf.
Alkohole (arabisch الكحول , DMG al-kuḥūl, oder الغول / al-ġawl: das Allerfeinste, reine Substanz, eigentlich: feines Antimonpulver) sind chemische Verbindungen, die eine oder mehrere an aliphatische Kohlenstoffatome gebundene Hydroxygruppen (–O–H) haben. Alkohole, die sich von den Alkanen ableiten, werden Alkanole genannt. Um eine klare Abgrenzung der Alkohole von Halbacetalen oder Carbonsäuren sicherzustellen, kann man ergänzen, dass das Kohlenstoffatom (sp3-hybridisiert, siehe auch Enole) mit der Hydroxygruppe nur noch mit Kohlenstoff- oder Wasserstoffatomen gebunden sein darf.
Ist die Hydroxygruppe an ein Kohlenstoffatom gebunden, das Teil eines aromatischen Ringes ist, so werden die Verbindungen als Phenole bezeichnet. Sie zählen nicht zu den Alkoholen, da diese Hydroxygruppen analog einer Carboxygruppe sauer regieren. Funktionelle Gruppe der Alkohole ist die blau markierte Hydroxygruppe. R ist ein Alkyl-Rest, jedoch kein Aryl-Rest,
Acyl-Rest oder ein Heteroatom. Der HOC-Bindungswinkel in einem Alkohol beträgt 109°.

TC-Vortrag 04 120/161 Waschen – Bleichen – Färben
Der Name einfacher Alkohole ergibt sich als Zusammensetzung aus dem Namen des ursprünglichen Alkans und der Endung -ol. Zusätzlich wird die Position der OH-Gruppe durch eine vorangestellte Zahl verdeutlicht, zum Beispiel Propan-2-ol. Eine veraltete, bis 1957 gültige Bezeichnung für Alkohole ist – nach einem Vorschlag von Hermann Kolbe – Carbinole.
Die Alkohole werden nach verschiedenen Kriterien (Zahl der Nichtwasserstoffnachbarn, Wertigkeit, Vorhandensein von Doppel-/Dreifachbindungen und Kettenlänge) eingeteilt.
Zahl der Nichtwasserstoffnachbarn: Man unterscheidet Alkohole nach der Zahl der Nichtwasserstoffnachbarn des Kohlenstoffatoms, an welchem sich die Hydroxygruppe befindet. Bei primären Alkoholen trägt es zwei, bei sekundären ein und bei tertiären kein Wasserstoffatom. Ein Sonderfall ist das Methanol, das neben der Hydroxygruppe drei Wasserstoffatome am Kohlenstoffatom trägt.
Von links nach rechts: Methanol ein primärer Alkohol; die allgemeine Formel eines primären Alkohols, sowie die allgemeinen Formeln eines sekundären Alkohols und eines tertiären Alkohols. R1 bis R3 ist ein Organyl-Rest (Alkyl-Rest, Alkenyl-Rest, Aryl-Rest, Benzyl-Rest etc.). Blau markiert ist die kennzeichnende Gruppierung für primäre, sekundäre und tertiäre Alkohole.
Wertigkeit der Alkohole: Ist mehr als eine Hydroxygruppe in einem Alkoholmolekül vorhanden, wird deren Anzahl durch Einfügen einer der Anzahl der Hydroxygruppen entsprechenden griechischen Silbe (-di-, -tri-, usw.) vor der Endung -ol angegeben und man spricht von mehrwertigen Alkoholen. Ein Alkandiol ist das Ethan-1,2-diol (Trivialname Ethylenglycol), ein Alkantriol das Propan-1,2,3-triol (Trivialname Glycerin). Die Zahl vor der Endung -ol gibt die Position der funktionellen Gruppe(n) an. Dies gilt auch für einwertige Alkohole, zum Beispiel Propan-2-ol (Trivialname Isopropanol).
Doppel- bzw. Dreifachbindungen: In Bezug auf das Vorhandensein von Doppel- bzw. Dreifachbindungen unterscheidet man Alkanole, Alkenole und Alkinole sowie den Spezialfall der meist instabilen Enole.
Über die Kettenlänge werden Alkohole ebenfalls unterschieden. Die Bezeichnung Fettalkohole verwendet man für Alkohole mit endständiger primärer –OH-Gruppe mit gerader Kette und einer Länge von sechs (Hexanol) bis hin zu 22 (Behenylalkohol) Kohlenstoffatomen. Sie werden meist durch Reduktion der –COOH-Gruppe aus Fettsäuren gewonnen. Die höheren primären Alkohole mit 24 bis 36 Kohlenstoffatome bezeichnet man als Wachsalkohole.

TC-Vortrag 04 121/161 Waschen – Bleichen – Färben
Niedrigmolekulare Alkohole sind Flüssigkeiten, die einen charakteristischen Geruch und einen brennenden Geschmack besitzen. Höhere Alkohole sind meist feste Verbindungen mit nur schwach ausgeprägtem Geruch. Aufgrund von intermolekularen Wasserstoffbrücken-bindungen besitzen die Alkohole im Vergleich zu Kohlenwasserstoffen gleicher Molekül-masse relativ hohe Schmelz- und Siedepunkte. Wichtigstes gemeinsames Merkmal der Alkohole ist die Hydrophilie. Diese Eigenschaft nimmt mit zunehmender Länge des Alkylrestes ab und mit der Anzahl der Hydroxygruppen zu. Besonders die kurzkettigen Alkohole werden aufgrund ihres amphiphilen Charakters oft als Lösungsmittel verwendet.
Hohe Siedepunkte: Sauerstoff ist elektronegativer als Wasserstoff und Kohlenstoff, d. h. er zieht Elektronen stärker an als diese. Das führt zu einer unsymmetrischen Verteilung der Elektronen entlang der C–O–H-Bindung, man spricht von einer polaren Bindung, es bildet sich ein molekularer Dipol aus. Diese Dipole können untereinander Wasserstoffbrücken-bindungen ausbilden, die die Anziehung der einzelnen Moleküle untereinander drastisch verstärken. Dies führt für Alkohole zu relativ hohen Siedepunkten gegenüber den um eine Methyleneinheit verlängerten Homologen ihrer Stammverbindung, die eine annähernd gleiche molarer Masse besitzen. So hat beispielsweise das unpolare Ethan (C2H6) (M = 30) einen Siedepunkt von −89 °C, während Methanol (CH3OH) (M = 32) diesen erst bei 65 °C erreicht.
Wasserstoffbrückenbindung zwischen zwei Alkohol-Molekülen
1. Im Vergleich zu Alkanen mit einer vergleichbaren molaren Masse haben Alkohole einen höheren Schmelz- und Siedepunkt, da die Hydroxygruppe (OH-Gruppe) Wasserstoffbrückenbindungen ausbildet.
2. Je mehr Hydroxygruppen ein Molekül aufweist, desto mehr Wasserstoffbrücken-bindungen können ausgebildet werden und desto höher ist der Siedepunkt.
3. Zwischen den Alkylresten bilden sich zusätzlich Van-der-Waals-Kräfte aus. Deswegen steigt der Siedepunkt mit der Länge des Alkylrestes
4. Da die Stärke der Van-der-Waals-Wechselwirkungen nicht nur von der Größe des Alkylrestes, sondern auch von dessen Oberfläche abhängig ist, weisen stark verzweigte, eher kugelförmige Moleküle mit einer mittelständigen Hydroxygruppe einen niedrigeren Siedepunkt als unverzweigte, langgestreckte, primäre Alkohole auf.
Hydrophilie: Die OH-Gruppe ist ebenfalls in der Lage, Wasserstoffbrückenbindungen mit Wasser einzugehen. Sie erhöht damit die Hydrophilie, die Wasserlöslichkeit, der Verbindung. Organische Alkylreste selbst sind nicht wasserlöslich, also hydrophob. Die Wasserlöslichkeit sinkt daher mit der Größe des organischen Anteils und steigt mit der Zahl der Hydroxygruppen. Die Propanole und tert.-Butanol sind bei Raumtemperatur noch in jedem Verhältnis mit Wasser mischbar, alle langkettigeren Alkohole lösen sich nur noch in zunehmend kleinen Mengen. Größere Mengen gelöster anorganischer Salze können auch bei den kurzkettigen Alkoholen eine Phasentrennung bewirken („Salzfracht“).
1. Die Hydroxygruppe eines Alkohols ist aufgrund der ungleichen Ladungsverteilung polar. Somit ist die Fähigkeit derselben, auch zu ebenfalls polaren Wassermolekülen Wasserstoffbrückenbindungen ausbilden zu können, für die gute Löslichkeit vor allem kurzkettiger Alkohole verantwortlich.

TC-Vortrag 04 122/161 Waschen – Bleichen – Färben
2. Je mehr Hydroxygruppen ein Alkohol aufweist, desto mehr Wasserstoffbrücken können diese mit dem Wasser ausbilden. Daher steigt mit wachsender Anzahl der hydrophilen Hydroxygruppen die Wasserlöslichkeit.
3. Diesem Effekt wirkt allerdings der hydrophobe, also wasserabweisende, unpolare Alkylrest entgegen: Je länger er ist, desto geringer ist die Wasserlöslichkeit des Alkohols.
Amine
Als Amine bezeichnet man organische Abkömmlinge (Derivate) des Ammoniaks (NH3), bei dem ein oder mehrere Wasserstoffatome durch Alkyl- oder Arylgruppen ersetzt sind.
Man teilt die Amine, je nachdem, wie viele Wasserstoff-Atome des Ammoniaks gegen organische Molekülgruppen ausgetauscht wurden, in primäre R-NH2, sekundäre R-NH-R oder tertiäre Amine NR3ein. Eine vierbindige Variante am positiv geladenen Stickstoffion (Ammoniumion) stellen die quartären Ammoniumverbindungen NR4
+(X-) dar.
Dimethylamin Trimethylamin Betain
Die Alkylgruppen sekundärer oder tertiärer Amine können zu einem Ring geschlossen sein. Es liegen dann cyclische sekundäre oder tertiäre Amine vor, wie z. B. Piperidin.
Wie auch Ammoniak reagieren Amine basisch, indem ein Proton an das freie Elektronenpaar des Stickstoffs angelagert wird (Die einfachste Methode zur Trennung von Aminen ist die Extraktion mit wässriger Salzsäure.):
Protonierung eines primären Amins durch eine Säure HX
Der Substitutionsgrad von Aminen beeinflusst deren Basizität. Alkylgruppen konzentrieren die Elektronendichte auf dem Stickstoffatom, so dass das freie Elektronenpaar leichter mit einer Säure geteilt werden kann. Bei reinen (wasserfreien) Aminen nimmt daher die Basizität mit zunehmendem Substitutionsgrad zu. In Gegenwart von Wasser wird das Amin hydratisiert, wobei Wärme freigesetzt wird. Diese Hydratationswärme ist bei primären Aminen am größten und bei tertiären am geringsten. Mit zunehmendem Substitutionsgrad findet jedoch auch eine zunehmende sterische Hinderung bei der Hydratation statt. Beides zusammen führt zu einer veränderten Reihenfolge der Basizität in wässrigen Lösungen:

TC-Vortrag 04 123/161 Waschen – Bleichen – Färben
Die Basizität aliphatischer Amine ist wegen des +I-Effekts größer als die von Ammoniak, die aromatischer Amine ist deutlich geringer, da hier der –I-Effekt der aromatischen Ringe zum Tragen kommt. In wässriger Lösung zeigen Alkylamine folgende Reihenfolge der Basizität: tertiär < primär < sekundär. Der Abfall der Basizität tertiärer Amine liegt an der schlechteren Solvatation der Ammoniumionen durch das Wasser; in der Gasphase sind sie stärker basisch als sekundäre Amine.
Elektronenanziehende Gruppen am organischen Rest oder am Stickstoffatom verringern die Basizität weiter.Bei biologischen Vorgängen entstehen Amine unter Anderem durch Decarboxylierung von Aminosäuren oder durch Transaminierung von Aldehyden. Präparativ sind Amine durch Alkylierung von Ammoniak, z. B. mit Alkylhalogeniden, zugänglich, wobei man hierbei Gemische aller Alkylierungsstufen erhält, da die zunächst gebildeten primären Amine ebenfalls alkyliert werden, ebenso die sekundären usw..
Die in Pflanzen, Tieren und dem Menschen natürlich vorkommenden, biogenen Amine besitzen große Bedeutung als Gewebshormone, Transmethylierungspartner oder Transmitter-Substanzen. Außerdem findet man Amin-Derivate als Basen in der DNA. Auch Aminosäuren, die Bausteine von Peptiden, sind Amine. Die im Eiweiß von Fischen peptidisch gebundenen Aminosäuren werden nach dem Tod der Tiere biochemisch zu Aminen und Kohlendioxid abgebaut. Die entstandenen Amine sind verantwortlich für den charakteristischen Geruch des Fisches, der bisweilen als unangenehm empfunden wird. Deshalb wird Fisch oft mit einem Stückchen Zitrone serviert. Zitronensäure protoniert (wie alle anderen Säuren auch) Amine und vermindert so den intensiven Amingeruch.
Amine reagieren mit salpetriger Säure in Abhängigkeit vom eingesetzten Amin zu unterschiedlichen Produkten. Primäre Amine reagieren zu Diazoniumverbindungen. Alkyldiazonium-Ionen sind auch bei Temperaturen von 0 °C relativ instabil. Sie spalten Stickstoff ab und reagieren zu einem Carbokation. Anschließend reagieren sie zu einem Alken weiter. Sekundäre Amine reagieren zu Nitrosaminen und tertiäre Amine können langsam unter Abspaltung einer Alkylgruppe ebenfalls zu Nitrosaminen reagieren; bei aromatischen tertiären Aminen findet eine Reaktion am aromatischen Ring statt und es entsteht eine aromatische Nitrosoverbindung. Besonders wichtig ist die Reaktion primärer aromatischer Amine. Amine werden zur Herstellung von Azoverbindungen verwendet, z. B. bei der Herstellung von Farbstoffen.

TC-Vortrag 04 124/161 Waschen – Bleichen – Färben
Aromate
Aromatische Verbindungen, kurz auch Aromaten, sind eine Stoffklasse in der organischen Chemie. Ihr Name stammt vom aromatischen Geruch der zuerst entdeckten Verbindungen dieser Stoffklasse.
Aromatische Moleküle besitzen mindestens ein Ringsystem, das nach der Hückel-Regel in konjugierten Doppelbindungen, freien Elektronenpaaren oder unbesetzten p-Orbitalen eine Anzahl von 4n+2 (n = 0, 1, 2, …) delokalisierten Elektronen enthält. Diese Delokalisierung führt zu einem besonderen Bindungssystem, in dem im Ring nicht zwischen Einzel- und Doppelbindungen unterschieden werden kann. In einfachen, symmetrischen Ringsystemen wie beim Benzol sind damit alle Bindungen identisch. In Strukturformeln werden hilfsweise entweder die mehreren mesomeren Grenzstrukturen dargestellt oder die Einfachbindungen werden mit einem (manchmal gestrichelten) Ring versehen, der die delokalisierten Elektronen symbolisiert. Aromaten sind im Vergleich zu nichtaromatischen Doppelbindungssystemen energieärmer und deshalb weniger reaktiv. Insbesondere neigen sie nicht zu Additionsreaktionen.
Benzol ist die einfachste aromatische Verbindung, mit dem alle anderen Aromaten durch die Struktur verwandt sind. Sie besitzen oft angenehmen, aromatischen Geruch. Von diesem typischen Geruch leitet sich die Bezeichnung Aromat, aus gr. ‚aroma‘=‚Duft‘, ab.
Benzol: (a) einfache mesomere Strukturformel, (b) Historische Kekulé-Benzol-Formel aus der Originalpublikation, (c) mesomere Strukturformel, (d) Benzolmolekül mit Darstellung der delokalisierten Elektronen und der Gleichheit der Bindungen durch einen Ring. (Die rechts wiedergegebene Präsentation findet man bisweilen in Schulbüchern.)
Benzol mit sechs Elektronen (genauer sechs π-Elektronen) in delokalisierten Doppelbindungen, einer der einfachsten aromatischen Verbindungen, hier durch mesomere Grenzstrukturen dargestellt. (Hinweis: Die Präsentationen oben und unten sind gleichwertig.)
Allerdings lassen sich die Aromaten nicht über den Geruch definieren, da bei hoher molarer Masse oder stark polaren Substituenten oft kein Geruch wahrnehmbar ist.

TC-Vortrag 04 125/161 Waschen – Bleichen – Färben
Aromaten sind mehrfach ungesättigte Verbindungen, die gegenüber der Addition an der Doppelbindung relativ reaktionsträge sind und die stattdessen relativ leicht direkt an einer Doppelbindung eine Substitution eingehen.
Diese Namensbestimmung, die eine experimentelle Unterscheidung erlaubt, war beispielsweise im 20. Jahrhundert gültig, schon bevor die Struktur- und Bindungsverhältnisse geklärt waren. Heute wird in der Regel eine allgemeinere Definition über die elektronische Struktur bevorzugt. Die angegebenen Eigenschaften – kurz: Substitution statt Addition – sind natürlich dennoch charakteristische und sehr wichtige Merkmale.
Das Bindungssystem der Aromaten zeigt eine besondere Stabilität, die zum Beispiel durch den Vergleich der Hydrierungsenthalpie des Aromaten mit einer entsprechenden nichtaromatischen und hypothetischen Bezugsverbindung (im Fall von Benzol Cyclohexatrien) als Resonanzenergie bestimmt werden kann.
Die Resonanzfrequenz der Wasserstoffatome im Kernresonanzexperiment ist charakteristisch. Diese äußert sich in einer starken Tieffeldverschiebung für Protonen außerhalb des aromatischen Systems und einer Hochfeldverschiebung für Protonen innerhalb des aromatischen Systems.
Notwendige, aber nicht hinreichende Voraussetzungen für einen Aromaten:
Ein cyclisches Molekül, das heißt, es hat mindestens einen Ring, der in vielen Fällen ein Benzolring ist.
Ein vollständig über den Ring konjugiertes Doppelbindungs-System. Das sind entweder
mehrere Doppelbindungen, die bei Kohlenwasserstoffen jeweils durch genau eine Einfachbindung getrennt sind (Im Sonderfall der Arine kann auch eine Dreifachbindung auftreten.) oder
eine oder mehrere Doppelbindungen, die durch positiv oder negativ geladene Kohlenstoffatome oder durch Heteroatome getrennt sind.
Gleichbedeutend und kürzer lautet diese Bedingung:
alle Atome des Rings sind sp2-hybridisiert (Siehe auch: Orbital).
Ein Aromat liegt dann vor, wenn auch die folgenden Bedingungen erfüllt sind:
o Das Doppelbindungssystem ist planar; in Ausnahmefällen sind leichte Abweichungen von der Ebene gestattet. Zum Beispiel ist in einigen Cyclophanen die Benzoleinheit in einem Winkel von bis zu 30° bootförmig deformiert.
o Die Zahl der delokalisierten Elektronen muss der Hückel-Regel genügen, das heißt im konjugierten Elektronensystem müssen 2 oder 6 oder 10 oder 14… Elektronen vorliegen:
Die von Erich Hückel aufgestellte Hückel-Regel wird meist durch die Formel (4n + 2) π-Elektronen (n = 0, 1, 2, 3, …), delokalisiert über alle Ringatome des Systems, wiedergegeben. Cyclisch konjugierte π-Systeme mit 4n π-Elektronen (n = 1, 2, 3, …) heißen Antiaromaten.

TC-Vortrag 04 126/161 Waschen – Bleichen – Färben
Die Grundstruktur vieler aromatischer Verbindungen ist das Benzol C6H6. (Die Hückel-Regel ist hier mit n=1 erfüllt: Benzol besitzt 6 π-Elektronen.) Das Benzol wird daher als einer der einfachsten aromatischen Kohlenwasserstoffe angesehen – insbesondere da die besonderen Eigenschaften aromatischer Verbindungen am Benzol und dessen Derivaten entdeckt wurden. Benzol ist gegenüber einem hypothetischen (das heißt nicht herstellbaren) Cyclohexatrien mit lokalisierten Doppelbindungen stabiler und damit weniger reaktiv.
Aryl‐Rest/Aryl‐Gruppe
Eine Arylgruppe (abgekürzt: Ar) ist ein organisch-chemischer Rest mit einem aromatischen Grundgerüst. Aryl ist somit die allgemeine Bezeichnung für eine einwertige Atomgruppe, die sich von aromatischen Kohlenwasserstoffen durch Entzug eines an den Ring gebundenen Wasserstoffatoms ableiten. Die meisten Aryl-Reste leiten sich vom Benzol (C6H6) ab, die einfachste Arylgruppe ist die Phenylgruppe (Ph), (–C6H5). Aryl-Reste können entweder als Fragment eines Moleküls (siehe Tabelle) oder als instabiles freies Radikal auftreten.
Aryl-Kationen entstehen als reaktionsfreudige Zwischenprodukte bei der Stickstoffabspaltung aus Aryl-Diazoniumsalzen, beim sogenannten „Verkochen“ von Aryl-Diazoniumsalzen bilden sich aromatische Alkohole (z. B. Phenole). Aryl-Anionen treten ebenso als reaktive Zwischenprodukte in organischen Synthesen auf und sind stabiler als Aryl-Kationen.
Die Bezeichnung Arylgruppe wird vor allem dann verwendet, wenn man allgemein formuliert und nicht genauer spezifizieren will, um welche aromatische Gruppe es sich handelt.
Aryl-Rest Name Strukturformel Bemerkungen
Phenyl-Rest Toluol Phenyl-Rest: blau
Phenyl-Rest Phenol Phenyl-Rest: blau
Phenyl-Rest Benzoesäure Phenyl-Rest: blau
1-Naphthyl-Rest 1-Naphthol 1-Naphthyl-Rest: blau
2-Naphthyl-Rest 2-Naphthylamin 2-Naphthyl-Rest: blau

TC-Vortrag 04 127/161 Waschen – Bleichen – Färben
9-Anthryl-Rest 9-Anthracencarbaldehyd 9-Anthryl-Rest: blau
9-Phenanthryl-Rest
Phenanthren-9-boronsäure
9-Phenanthryl-Rest: blau
Azo‐Gruppe
In der Chemie bezeichnet die Azogruppe eine funktionelle Gruppe, die aus zwei durch eine Doppelbindung verbundenen Stickstoff-Atomen (–N=N–) besteht. Organische Verbindungen mit Azogruppe nennt man Azoverbindungen, bekannte Vertreter dieser Stoffgruppe sind die Azofarbstoffe. Der Name Azo leitet sich vom französischen Wort Azote für Stickstoff ab.
Die einfachste Azoverbindung ist das leuchtend gelbe Diimin (H–N=N–H, Diazen, Diimid, Azowasserstoff). Es ist nicht stabil, kann aber bei sehr tiefen Temperaturen synthetisiert werden. Die einfachsten aliphatischen bzw. aromatischen Azoverbindungen sind das farblose Azomethan (H3C–N=N–CH3) bzw. das rote Azobenzol (H5C6–N=N–C6H5). Generell sind aromatische Azoverbindungen stabiler als aliphatische. Während aromatische Azoverbindungen in der Regel farbig sind, sind aliphatische farblos. Je stärker die π-Elektronen delokalisiert sind, desto größer ist die Wellenlänge der maximalen Absorption.
Bei geeigneter Substitution erliegen Azo-Verbindungen der Azo-Hydrazo-Tautomerie.
Carbonsäuren
Carbonsäuren (veraltet auch Karbonsäuren) sind organische Verbindungen, die eine oder mehrere Carboxygruppen (–COOH) tragen. Die Carbonsäuresalze werden Carboxylate und ihre Ester Carbonsäureester genannt.
Die systematische Benennung der Carbonsäuren erfolgt im Deutschen durch die Endung „-säure“ und dem vorangestellten Grundgerüst, bei den Säuren der Alkane zum Beispiel Ethansäure (Essigsäure), Methansäure (Ameisensäure).
Viele Carbonsäuren tragen unsystematische Namen (Trivialnamen), die ebenfalls mit „-säure“ enden. Der Trivialname weist zumeist auf die Quelle hin, woraus die Carbonsäure gewonnen werden kann; einige Beispiele sind Ameisensäure, Apfelsäure (auch: Äpfelsäure), Buttersäure, Weinsäure, Zitronensäure oder Essigsäure.
Nach der chemischen Struktur des Rests R, an welchen die Gruppe –COOH gebunden ist, unterscheidet man zwischen aliphatischen, aromatischen und heterocyclischen Carbonsäuren.

TC-Vortrag 04 128/161 Waschen – Bleichen – Färben
Die aliphatischen Carbonsäuren lassen sich in Alkansäuren, Alkensäuren und Alkinsäuren unterteilen. Alkansäuren nennt man auch gesättigte Carbonsäuren. Alkensäuren, also Carbonsäuren mit mindestens einer Doppelbindung im Rest und Alkinsäuren mit mindestens einer Dreifachbindung im Rest, nennt man hingegen ungesättigte Carbonsäuren. Neben der Struktur des Restes lassen sich die Carbonsäuren nach Anzahl der Carboxygruppen unterscheiden. Monocarbonsäuren verfügen über eine Carboxygruppe, während Dicarbonsäuren zwei und Tricarbonsäuren drei Carboxygruppen tragen.
Allgemeine Struktur der Monocarbonsäuren mit der blau markierten Carboxy-Funktion.
Außerdem gibt es auch Gruppen von Carbonsäuren, die weitere funktionelle Gruppen tragen, wie Ketocarbonsäuren, Hydroxycarbonsäuren und Aminosäuren (eigentlich: Aminocarbonsäuren).
Fettsäuren sind unverzweigte, aliphatische Monocarbonsäuren mit mindestens vier Kohlenstoffatomen, z. B. Ölsäure.
Als Harzsäuren bezeichnet man Carbonsäuren, die in Naturharzen vorkommen. Metallacarbonsäuren sind Komplexe mit einem Carboxyligand.
Cellulose
Die Cellulose (häufig auch Zellulose) ist der Hauptbestandteil von pflanzlichen Zellwänden (Massenanteil etwa 50 %) und damit die häufigste organische Verbindung und auch das häufigste Polysaccharid (Vielfachzucker). Sie ist unverzweigt und besteht aus mehreren hundert bis zehntausend β-D-Glucose-Molekülen (β-1,4-glykosidische Bindung) bzw. Cellobiose-Einheiten. Die Cellulosemoleküle lagern sich zu höheren Strukturen zusammen, die als reißfeste Fasern in Pflanzen häufig statische Funktionen haben. Cellulose ist bedeutend als Rohstoff zur Papierherstellung, aber auch in der chemischen Industrie und anderen Bereichen.
Cellulose wurde im Jahr 1838 von dem französischen Chemiker Anselme Payen entdeckt, der diese aus Pflanzen isolierte und deren chemische Formel bestimmte. Cellulose wurde im Jahr 1870 von Hyatt Manufacturing Company dazu genutzt, um das erste Plastomer, Zelluloid, herzustellen. Hermann Staudinger ermittelte im Jahr 1920 die Struktur von Cellulose. 1992 wurde Cellulose zum ersten Mal von Kobayashi und Shoda chemisch synthetisiert (ohne die Hilfe biologisch basierender Enzyme)
Ein Glucosedimer, dargestellt in Sesselkonformation (Cellobiose-Einheit)
Cellulose ist ein Polymer (Polysaccharid ‚Vielfachzucker‘) aus dem Monomer Cellobiose, einem Disaccharid (‚Zweifachzucker‘). Die Monomere sind durch β-1,4-glycosidische Bindungen miteinander verknüpft.

TC-Vortrag 04 129/161 Waschen – Bleichen – Färben
Die Cellobiose selbst besteht aus zwei Molekülen des Monosaccharids (‚Einfachzuckers‘) Glucose. Hier liegt ebenfalls eine β-1,4-glycosidische Bindung vor, so dass häufig auch die Glucose als Monomer der Cellulose definiert wird.
Die Verknüpfung der Monomere erfolgt durch eine Kondensationsreaktion, bei der zwei Hydroxygruppen (-OH) ein Wassermolekül (H2O) bilden und das verbleibende Sauerstoffatom die ringförmige Grundstruktur (Pyranring) der beiden Monomere verbindet. Neben dieser starken, kovalenten Bindung werden intramolekular zusätzlich die weniger starken Wasserstoffbrücken ausgebildet. Häufig besteht ein Cellulosemolekül aus mehreren tausend Glucoseeinheiten.
Eigenschaften: Cellulose ist in Wasser und in den meisten organischen Lösungsmitteln unlöslich. Lösungsmittel wie Dimethylacetamid/Lithiumchlorid oder Dimethylsulfoxid/Tetrabutylammoniumfluorid sowie Ammoniak/Cu2+ (Schweizers Reagens) vermögen jedoch Cellulose zu lösen. Sie kann durch starke Säuren gespalten werden. Mit konzentrierten Säuren bei erhöhter Temperatur kann die Cellulose zu Glucose abgebaut werden, indem die glycosidischen Bindungen gespalten werden.
Der Chemiekonzern BASF hat ein Einblasverfahren entwickelt, bei dem Cellulose in einer ionischen Flüssigkeit rein physikalisch gelöst wird. Diese Lösung kann für chemische Synthesen verwendet werden, die bisher nicht möglich waren.
Hauptsächlich aus Cellulose bestehendes Pflanzenmaterial wird vom Menschen mindestens seit der Altsteinzeit als Brennstoff zum Kochen und Heizen genutzt. Cellulose ist daneben ein wichtiger Rohstoff für stoffliche Nutzungen, aber auch als natürlicher oder zugesetzter Bestandteil von Nahrungs- und Futtermitteln von Bedeutung. Da Cellulose zudem in fast allen Arten pflanzlicher Biomasse vorkommt, ist sie auch in vielen anderen Bereichen wichtig, wie z. B. in Holz (Lignocellulose) als Baustoff etc.
Cellulose als Rohstoff: Cellulose ist ein wichtiger Rohstoff zur Papierherstellung. Als Ausgangsrohstoff dient das lignin- und cellulosereiche Holz. Aus diesem wird Holzschliff hergestellt, das für Papier weniger hoher Qualität verwendet wird. Durch Entfernen des Ligninanteils kann Zellstoff verwendet werden, der hauptsächlich aus Cellulose besteht und für Papiere höherer Qualität verwendet werden kann.
In der Bekleidungsindustrie werden die hauptsächlich aus Cellulose bestehenden Pflanzenfasern für verschiedene Stoffe verwendet. Beispiele sind Baumwolle sowie Bastfasern des Lein (Flachs), die zu Leinen verarbeitet werden.
Die Samenhaare des Baumwollstrauches (Gossypium herbaceum)
bestehen aus fast reiner Cellulose.
Ein weiteres Cellulose-Regenerat ist Cellophan (Cellulosehydrat), das in Form von Folien ein verbreitetes Verpackungsmaterial ist. Auch synthetische Cellulosefasern („Zellwolle“) können hergestellt werden. Dazu wird eine alkalische Lösung von xanthogenierter Cellulose („Viscose-Lösung“) zu Fäden verarbeitet, der sogenannten Regeneratfaser (z. B. Viskose).

TC-Vortrag 04 130/161 Waschen – Bleichen – Färben
Unterschiedlichste Cellulosederivate finden vielfältige Anwendung, wie z. B. Methylcellulose, Celluloseacetat und Cellulosenitrat in der Bau-, Textil- und chemischen Industrie. Vom Cellulosenitrat abgeleitet ist Zelluloid, der erste Thermoplast.
Cellulose als Nahrung: Fast alle Tiere – mit Ausnahme weniger Mollusken, wie einiger Schnecken, etwa der Weinbergschnecke und weniger Termitenarten – einschließlich der meisten Pflanzenfresser können Cellulose im Gegensatz zu Stärke nicht durch eigene Stoffwechselleistungen abbauen, obwohl beide Moleküle aus Traubenzuckermolekülen aufgebaut sind. Diese Tiere besitzen nur die Enzyme, die α-1,4- oder α-1,6-glycosidische Bindungen (z. B. in Stärke) spalten können (Amylasen), nicht aber β-1,4-glycosidische Bindungen der Cellulose. Deshalb können diese Tiere (z. B. Kühe) den hohen Energiegehalt dieses Kohlenhydrates nur mit Hilfe von endosymbiontischen Prokaryoten erschließen, die in ihrem Darm leben. Cellulose essende Tiere nähren sich dann von der stetig nachwachsenden Symbiontenmasse in ihrem Verdauungssystem.
Wiederkäuer verdauen einen großen Teil der Cellulose und anderer Polysaccharide im Pansen mithilfe anaerober Prokaryoten, die die Cellulose zu Fettsäuren umsetzen. Ähnliches gilt für Pferde und Wassergeflügel, bei denen die Verarbeitung jedoch im Dickdarm stattfindet.
Auch der Mensch besitzt keine Verdauungsenzyme für den Abbau von Cellulose. Mit Hilfe anaerober Bakterien im ersten Teil des Dickdarms, dem Blinddarm und dem aufsteigenden Colon wird ein Teil der Cellulose aus der Nahrung zu kurzkettigen Fettsäuren abgebaut. Über die Colonschleimhaut werden sie resorbiert und vom Stoffwechsel verwertet. Cellulose ist somit, neben Hemicellulosen, Pektin und Lignin, ein wichtiger pflanzlicher Ballaststoff in der menschlichen Nahrung.
Einige terrestrische Krebse wie die Isopoda können Cellulose mit der Unterstützung endosymbiotischer Bakterien abbauen. Dasselbe gilt für Insekten wie Silberfischchen, fast alle Termiten, Schaben oder Blattschneiderameisen. In 200 untersuchten Termitenspezies wurden mehr als 450 unterschiedliche Endosymbionten identifiziert. Endosymbionten fossilierter Termiten wurden bereits aus der Kreidezeit direkt (in burmesischem Bernstein) nachgewiesen.
Viele Bakterien, Pilze und Flagellaten können über ihre Cellulasen die Cellulose nur bis zum Glucosedimer Cellobiose zersetzen. Einige wenige Protozoen und Pilze wie Aspergillus-, Penicillium- und Fusarium-Arten besitzen zusätzlich die notwendigen β-1,4-Glucosidasen oder Cellobiasen, welche die Cellobiose in Glucose aufspalten. Manche holzzersetzenden Pilze wie Ceriporiopsis subvermispora können Cellobiose auch über die Cellobiosedehydrogenase (CDH), ein extrazelluläres Hämoflavoenzym, oxidativ abbauen. Dabei entsteht statt der Glucose Gluconsäure.
Cycloalkane
Die Cycloalkane (Cyclane, ältere Bezeichnung: Naphthene, Cycloparaffine) sind eine Stoffgruppe von ringförmigen, gesättigten Kohlenwasserstoffen. Die Ringe können Seitenketten tragen.In der Systematik der organischen Chemie zählt man sie zu den alicyclischen Verbindungen. Die Cycloalkane ohne Seitenketten bilden eine homologe Reihe mit der allgemeinen Summenformel CnH2n, wobei n ≥ 3 ist. Somit ist das kleinste vorkommende Cycloalkan das Cyclopropan.

TC-Vortrag 04 131/161 Waschen – Bleichen – Färben
Natürlich vorkommende Cycloalkane (Cyclopentan, Cyclohexan, Cycloheptan) wurden als erstes vom Chemiker Wladimir Wassiljewitsch Markownikow in der Rohbenzin-Fraktion, auch Naphtha genannt, des kaukasischen Erdöls gefunden. Die Bezeichnung Naphthen, die gelegentlich für alle Cycloalkane verwendet wird, stammt daher. Meist wird diese unpräzise Bezeichnung aber nur auf die Derivate des Cyclopentans und -hexans angewandt. In der Erdölindustrie ist sie noch heute gebräuchlich.
In Strukturformelzeichnungen der Cycloalkane werden die Ringe verkürzt durch Vielecke dargestellt. Cycloalkane kann man sich als eine Form der Alkane vorstellen, bei der die beiden Enden der Kohlenstoffkette miteinander verknüpft sind. Es handelt sich nicht um Isomere der Alkane. Sie sind wie die Alkane gesättigte Verbindungen.
Cis-trans-Isomerie bei den Cycloalkanen
Da die Rotation eines Substituenten um ein Ringkohlenstoffatom unmöglich ist, tritt eine spezielle Form der Isomerie auf, die cis-trans-Isomerie. Bei cis-trans-Isomeren ist die räumliche Anordnung von Substituenten unterschiedlich. Die Substituenten können auf der gleichen Seite (cis) oder auf unterschiedlichen Seiten (trans) der Ringbindung liegen.
Cis-trans-Isomerie beim 1,2-Dimethylcyclopropan
Konformationen
Um Molekülspannungen zu umgehen, sind die Cycloalkane nicht, wie häufig dargestellt, planar. Sie liegen vielmehr in Konformationen vor, in denen der Innenwinkel der Tetraederform (109,45 °) möglichst erhalten bleibt. Bei Cyclopropan, Cyclobutan und Cyclopentan wird dieser Winkel nicht ganz erreicht, es kommt zur sogenannten Baeyer-Spannung; wegen dieser Spannung sind diese Moleküle reaktiver. Im Cyclohexanmolekül hat diese Spannung aber bereits nahezu keinen Einfluss mehr.
Sesselkonformation beim Cyclohexan
Ester
Ester bilden in der Chemie eine Stoffgruppe chemischer Verbindungen, die formal oder tatsächlich durch die Reaktion einer Säure und eines Alkohols oder Phenols unter Abspaltung von Wasser (eine Kondensationsreaktion) entstehen. Es gibt Ester von organischen Säuren (z. B. Carbonsäuren wie Essigsäure, Sulfonsäuren) und solche von anorganischen Säuren (z. B. Phosphorsäure, Schwefelsäure, Borsäure, Kohlensäure).
Die Bezeichnung Ester wurde von dem Chemiker Leopold Gmelin im Jahre 1850 aus dem Begriff „Essigäther“, einem historischen Namen für Ethylacetat, gebildet. Die Dämpfe von Ethylacetat wirken betäubend, ähnlich denen von „Äther“ (Diethylether), daher der Begriff „Essigäther“, da „Äther“ (auch heute noch) etwas leicht Flüchtiges bedeutet, aber nicht ausschließlich mit Ether gleichzusetzen ist (anderes Beispiel: „Salpetersäureethyläther“).
Die Herstellung von Estern wird als Veresterung oder Esterbildung bezeichnet.

TC-Vortrag 04 132/161 Waschen – Bleichen – Färben
Carbonsäreester
Carbonsäureester sind Ester der Carbonsäuren mit der funktionellen Gruppe –COOR. Sie setzen sich aus einem Säureteil und einem Alkoholteil zusammen. Sie bilden eine in der Organischen Chemie häufig anzutreffende Stoffgruppe.
Biologisch bedeutende Ester sind die Triglyceride (auch „Glycerol-Triester“, seltener veraltet „Neutralfette“). Diese natürlichen Öle (flüssiger Aggregatzustand) oder Fette (fest) sind fast alle schlecht wasserlöslich, obwohl sie polar sind, denn sie besitzen (bis auf wenige Ausnahmen) drei hydrophobe Alkylgruppen. Je langkettiger die Alkylgruppen sind, desto schlechter ist die Löslichkeit des Triesters in Wasser.
Fette und fette Öle (Ester von Glycerin und Fettsäuren, z. B. Triglyceride) sind wichtiger Nahrungsbestandteil und Energiespeicherstoff für die meisten tierischen Organismen.
Die Herstellung von Estern wird als Veresterung oder Esterbildung bezeichnet. Die Veresterung (auch Esterbildung oder Fischer-Veresterung genannt) ist eine chemische Reaktion und wurde von Emil Fischer entdeckt. Sie ist eine Gleichgewichts- und Kondensationsreaktion, bei der ein Alkohol oder Phenol mit einer organischen oder anorganischen Säure zu einem Ester reagiert. Zudem wird ein Katalysator verwendet (z. B. Schwefelsäure oder Salzsäure).
Alkohol und Nitryl-Kation reagiert unter Deprotonierung zu einem Salpetersäureester Schwefelsäure wirkt hier als Katalysator
Ether
Als Ether(veraltet Äther) werden in der Chemie organische Verbindungen bezeichnet, die als funktionelle Gruppe eine Ethergruppe – ein Sauerstoffatom, das mit zwei Organylresten substituiert ist – besitzen (R1–O–R2). Die Alkyl- oder Aryl-Reste sind im Fall eines cyclischen Ethers miteinander verbunden. In der Umgangssprache bezeichnet Ether oft auch den Diethylether (H5C2–O–C2H5), einen der wichtigsten und einfachsten Ether. Sind beide Reste an der Sauerstoffbrücke aliphatisch, so werden diese Ether nach der IUPAC auch als Alkoxyalkane bezeichnet.
Allgemeine Strukturformel eines Ethers: R1 und R2 sind Organylgruppen. Das Sauerstoffatom des Ethers ist blau markiert.

TC-Vortrag 04 133/161 Waschen – Bleichen – Färben
Ether sind in der Natur weit verbreitete Verbindungen. Die glycosidische Bindung der Polysaccharide ist eine Sauerstoffbrücke zwischen zwei Kohlenstoffatomen; diese Acetale sind faktisch intramolekulare, geminal angeordnete Di-Ether. Auch viele andere Naturstoffe, wie z. B. die Aromastoffe Anethol, 1,8-Cineol, Eugenol, Anisol und Vanillin, sowie die Gruppen der Ubichinone und Strobilurine und viele Arzneistoffe sind Ether.
Die Bindungsverhältnisse in Ethern ähneln denen in Alkoholen und im Wasser, das als Grundkörper dieser beiden Verbindungsklassen aufgefasst werden kann. Kohlenstoff- und Sauerstoff-Atome sind jeweils sp3 hybridisiert. Dies führt zu einer tetraedrischen Anordnung der Atomorbitale um alle beteiligten Atome. Der Bindungswinkel des Sauerstoffs ist aufgrund der gegenüber Wasserstoff (104,5° im H2O) voluminöseren Alkylsubstituenten mit 112° erweitert. Die C–O-Bindungen sind mit ca. 143 pm so lang wie in Alkoholen.
Bei dem Umgang mit niederen Ethern sollte deren niedriger Siedepunkt und leichte Entflammbarkeit nie unterschätzt werden. (Diethyl)Ether-Luft-Gemische sind zwischen 2 und 36 Vol% explosiv. Wichtig ist, dass Ether-Dämpfe nicht nur farblos, sondern auch schwerer als Luft sind. Sie sammeln sich also an tiefgelegenen Stellen. Aufgrund dieser Tatsache und der narkotisierenden Wirkung von Ethern sind sie nur in gut funktionierenden Abzügen zu verwenden.
Aufgrund ihres ambivalenten Charakters sind die meisten Ether hervorragende Lösungsmittel und lösen viele wasserunlösliche Verbindungen. So wird der Großteil des produzierten Diethylethers als Lösungsmittel im Umfeld der chemischen und medizinischen Industrie sowie im Laborbedarf verbraucht.
Das Wort „Ether“ stammt aus dem Griechischen (aither, ᾿αιθήρ) und bedeutet in etwa „obere Luft“ oder „Feuerluft“.
Diethylether diente zur Zeit der Prohibition teilweise als Ethanol-Ersatz (orale Einnahme) und vereinzelt sind auch noch heute Fälle des Etherkonsums bekannt. Die physiologische Wirkung ist ähnlich der des Alkohols. Beim Abbau des Diethylethers im Körper entsteht unter anderem auch Ethanol.
Diethylether diente als einer der ersten Stoffe als Anästhetikum. Aufgrund der starken Nebenwirkungen (ausgeprägte Erregungsphase, Übelkeit, Brennbarkeit), ist er jedoch in der Anästhesie nur noch von historischem Interesse.
Gefahrenhinweise und Lagerung von Ethern - Autoxidation
Bei Aufbewahrung von Ethern an Licht bilden diese mit Luftsauerstoff Peroxide. Diese können sich bei der (Vakuum-)Destillation eines Ethers im Rückstand ansammeln und zu Explosionen führen.

TC-Vortrag 04 134/161 Waschen – Bleichen – Färben
Autoxidation von Ethern, gezeigt am Beispiel von Diethylether. Das Diethyletherradikal (1-Ethoxyethylradikal) A reagiert mit Luftsauerstoff zum Peroxid-Radikal B und anschließend zum Peroxid und dem Radikal A, das die Kettenreaktion weiterführt.
Dabei wird in α-Stellung zum Sauerstoff-Atom ein Wasserstoff-Atom unter Bildung eines Radikals abstrahiert und es bildet sich mit Sauerstoff ein Peroxid. Eine Ausnahme stellt Methyl-tert-butylether (MTBE) dar, da dieser auf der tert-Butyl-Seite kein α-ständiges Wasserstoff besitzt und auf der Methylseite die Entstehung eines Radikals zu ungünstig ist.
Die Peroxide können meist durch die Braunverfärbung von essigsauren Iodid-Lösungen nachgewiesen werden. Dabei wird Iodid zu Iod oxidiert, welches mit Iodid zu I3− reagiert und für die braune Farbe verantwortlich ist. Im Handel sind zudem spezielle Teststäbchen erhältlich. Die Vernichtung von Peroxiden kann beispielsweise mit Eisen(II)-Salzen erfolgen. Die Lagerung von Ethern für den Labor-Gebrauch sollte daher nur in kleinen Gebinden von maximal 1 Liter über Kaliumhydroxid-Plätzchen in Braunglasflaschen erfolgen.
Glycosidische Bindung
Als glycosidische Bindung oder glykosidische Bindung bezeichnet man die chemische Bindung zwischen dem anomeren Kohlenstoffatom eines Kohlenhydrats (Glycon) und dem Hetero- oder selten Kohlenstoffatom eines Aglycons oder einem zweiten Zuckermolekül. Verbindungen, die eine glycosidische Bindung enthalten, sind in der Natur weit verbreitet und werden Glycoside genannt.
Grundstruktur eines Glycosids: R im Aglycon XR kann ein beliebiger organischer Rest sein
(mit XR ≠ OH, Acylrest O-(CO)-R')
Wenn das Aglycon ein Alkohol oder Phenol ist, dann stammt das Brücken-Sauerstoffatom vom Aglycon. Glycoside können auch Bindungen zu anderen Heteroatomen wie Schwefel, Selen, Stickstoff und Phosphor oder selten zu Kohlenstoff („C-Glycoside“) aufweisen. Die glycosidische Bindung ist hydrolytisch spaltbar, wobei das Reaktionsgleichgewicht auf Seiten

TC-Vortrag 04 135/161 Waschen – Bleichen – Färben
der Spaltungsprodukte liegt. Die Bindung ist kinetisch aber sehr stabil. Sie wird mit geringem Energieaufwand unter Wasserabspaltung durch eine Kondensationsreaktion gebildet. In der Natur erfolgt die als Glycosylierung bezeichnete Bildung enzymatisch über ein aktiviertes Saccharid, im Labor durch spezielle Aktivierungsmethoden oder durch Reaktion eines Zuckers mit einem großen Überschuss des Alkohols unter Säurekatalyse.
Das α- und β-Anomer der D-Glucose in der Fischer-Schreibweise. Die Position der anomeren Kohlenstoffatome (rot oder grün) relativ zur CH2OH-Gruppe
am C-5 bestimmen, ob es sich um die α-Verbindung (verschiedene Ringseiten) oder die β-Verbindung (dieselbe Seite des Rings) handelt.
Die Position der Hydroxygruppe des an der Bindung beteiligten anomeren Kohlenstoffatoms bestimmt den Namen der Verbindung und auch den Typ der Bindung: Glucose kommt in der cyclischen Pyranoseform als α-D-Glucopyranose oder β-D-Glucopyranose vor. Dies wird bei der Nomenklatur einer Bindung als α- oder β-glycosidische Bindung berücksichtigt. Bei einem einfachen Glycosid, etwa aus dem Monosaccharid Glucose und einem Alkohol wie Ethanol, wird dieses entweder als Ethyl-α-D-glucopyranosid oder Ethyl-β-D-glucopyranosid bezeichnet. Während eine glycosidische Bindung im Normalfall eine C-O-Bindung ist, wird bei Glycosiden mit Heteroatom (S, Se, N, P) dies durch kursive Nennung im Namen berücksichtigt, z.B. Ethyl-S-α-D-glucopyranosid.
Definition α und β: Durch die Präfixe α und β wird die anomere Konfiguration definiert. Entscheidend ist die sterochemische Relation des exocyclischen Sauerstoffs zum konfigurationsbestimmenden C-Atom des Zuckers (α = cis, β = trans). Dabei ist α nicht immer axial und β nicht immer äquatorial zum Ring. Beispiele: Bei α-D-Glucopyranose steht der exocyclische Sauerstoff cis zum konfigurations-bestimmenden C-5: das Aglycon steht axial (β ist hier äquatorial). Bei dem Paradebeispiel β-D- oder L-Arabinopyranose steht der exocyclische Sauerstoff cis zum konfigurationsbestim-menden C-4. Hier ist β also axial. Die Unterscheidung ist am besten in der Fischer-Schreib-weise zu sehen.
Di-, Oligo- und Polysaccharide
Bei der Kondensation zweier Kohlenhydrate über eine glycosidische Bindung zu einem Disaccharid berücksichtigt die Nomenklatur der Bindung zwei verschiedene Faktoren:
Einerseits je nach Ausgangverbindung die α- oder β-Stellung des an der Bindung beteiligten ersten anomeren Kohlenstoffatoms.
Der zweite Teil der Benennung stammt aus der Position der Kohlenstoffatome, die an die Sauerstoffbrücke direkt gebunden sind. Bei der Saccharose (Rohrzucker) kommt die Bindung über das 1. Kohlenstoffatom der Glucose mit einer Hydroxygruppe in α-Stellung zum 2. Kohlenstoffatom der β-D-Fructose zustande. Die Bindung ist also eine α-1→2-glycosidische Bindung und Saccharose heißt

TC-Vortrag 04 136/161 Waschen – Bleichen – Färben
demnach korrekt 1-α-D-Glucopyranosyl-2-β-D-fructofuranosid oder auch 2-β-D-Fructofuranosyl-1-α-D-glucopyranosid.
Es ist ebenfalls möglich, dass zwei gleiche Monosaccharide zu einem Disaccharid wie der Cellobiose verbunden werden. Dabei verbrückt das Sauerstoffatom das C1-Atom des ersten β-D-Glucosemoleküls mit dem 4. Kohlenstoffatom eines zweiten, identischen Monomers. Die Verbindung ist eine β-1→4-glycosidische Bindung und die Cellobiose demnach das 1-β-D-Glucopyranosyl-4-β-D-glucopyranosid. Da das anomere erste Kohlenstoffatom des zweiten Glucosemoleküls frei bleibt, kann dieses mit weiteren Monomeren zu einem Polysaccharid wie Cellulose weiterreagieren. Analog können zwei Glucosemoleküle α-1→4-glycosidisch zu Maltose (1-α-D-Glucopyranosyl-4-α-D-Glucopyranose) verknüpft werden.
Eine weitere Verbindungsmöglichkeit kommt in der Gentiobiose vor: zwei Glucoseeinheiten sind über das 1. C-Atom des ersten Monomers und das 6. C-Atom des zweiten β-1→6-verbrückt, die Verbindung heißt auch 1-β-D-Glucopyranosyl-6-β-D-Glucose. In Laminaribiose sind ebenfalls zwei Glucosemoleküle β-1→3-verknüpft.
Bei Polysacchariden kommen verschiedene glycosidische Bindungen vor:
Amylopektin, der Hauptbestandteil der Stärke, ist aus α-1→4-verbundenen Glucosemonomeren aufgebaut.
Amylose, die ebenfalls in Stärke vorkommt, enthält nur α-1→4-verbrückte Glucose, die eine Helixstruktur bildet.
Cellulose besteht aus Glucose, die β-1,4-verbunden ist.
Glycogen besteht aus Glucose, wobei die Hauptkette vorwiegend α-1→4-Bindungen aufweist, aber auch Verzweigungen mit α-1→6-Struktur.
Komplexe Glycoside und Heteroglycoside
Neben einfachen Alkoholen und anderen Sacchariden kann der Zuckeranteil auch mit einem komplexeren Alkohol (wie einem Steroidalkohol), einem Phenol oder einem anderen Hetero-atom wie Stickstoff, Schwefel oder Phosphor verbunden sein. Dieser Fall tritt beispielsweise in den Nukleosiden und Nukleotiden auf, bei denen eine N-glycosidische Bindung über eine Stickstoffbrücke vorliegt. Der Zuckeranteil ist hier meist die Ribose oder die Desoxyribose. In der Natur kommen daneben sehr viele O- und N-Glycoside, seltener S-Glycoside vor, die verschiedenste Funktionen wie Aromastoffe (Amygdalin, Glycyrrhizin), Abwehrstoffe (Senfölglycoside, Barbaloin, Cycasin), Energiespeicher und -übertrager (Adenosindi- bzw. –triphosphat ADP, ATP), Farbstoffe (Azalein, Betanin, Karmin, Malvin) und Speicherstoffe (Coniferin) erfüllen. Auch einige natürliche und künstliche Antibiotika wie Azithromycin, Bleomycin, Cethromycin, Doramectin und Streptomycin sind Glycoside.
Induktiver Effekt, I‐Effekt, +I‐Effekt, ‐I‐Effekt
Der Induktive Effekt oder I-Effekt ist in der organischen Chemie ein ladungsverändernder Effekt, der sowohl als +I-Effekt („elektronenschiebend“) als auch als −I-Effekt ("elektronenziehend") auftritt. Er wird durch elektrostatische Induktion durch funktionelle Gruppen entlang einer oder mehrerer chemischer Bindungen ausgelöst.

TC-Vortrag 04 137/161 Waschen – Bleichen – Färben
Die Ursache dieser Effekte ist eine Asymmetrie in der Verteilung der Elektronen in einer Elektronenpaarbindung zwischen zwei gleichen, aber unterschiedlich substituierten Atomen oder zwischen zwei unterschiedlichen Atomen.
Zwei Atome, die durch eine Elektronenpaarbindung gebunden sind, teilen sich zwei Elektronen. Diesen Elektronen ist kein fester Platz zugewiesen, sondern sie sind innerhalb dieser Bindung frei beweglich. Die Elektronen werden im Fall einer Asymmetrie der Elektronenverteilung zu dem Atom hingezogen, dessen Elektronegativität größer ist.
Man unterscheidet zwei Arten von I-Effekten: den +I-Effekt (sprich: positiver induktiver Effekt) und den −I-Effekt (sprich: negativer induktiver Effekt). Ein elektronegativeres Atom übt einen −I-Effekt aus, sodass sich die Elektronendichte bei dem anderen Atom verringert. Bei einem +I-Effekt werden die Elektronen von dem einen Atom weggeschoben und somit die Elektronendichte an dem anderen Atom erhöht. Die Bindung weist nun einen Dipolcharakter auf, der durch δ+ am Atom mit der geringeren Ladungsdichte und δ− am Atom mit der hohen Ladungsdichte gekennzeichnet wird. Um die Stärke des induktiven Effektes von Atomen oder Atomgruppen zu vergleichen, wird die Elektronegativität des Substituenten mit der Elektronegativität des Wasserstoffs verglichen. Je größer die Differenz der Elektronegativitäten, desto stärker ist der induktive Effekt.
Atombindungen können, je nach Elektronegativität der Bindungspartner, polarisiert sein, also als polare Atombindungen vorliegen. Ist eines der Elemente elektronegativer als sein Bindungspartner, so halten sich die Elektronen häufiger in seiner Nähe auf. Dadurch verschiebt sich die Ladungsverteilung, sodass das elektronegativere Element mehr oder weniger stark negativ polarisiert ist. Entlang der Bindung liegt ein Bindungsdipolmoment vor.
Als Beispiel ist hier Wasser (H2O) anzuführen. Durch die höhere Elektronegativität halten sich die Elektronen häufiger beim Sauerstoff-Atom auf. Im Wassermolekül wird dies durch δ− in der Nähe des O-Atoms, sowie durch jeweils ein δ+ neben jedem der beiden H-Atome ausgedrückt. Oft wird das δ− beim Sauerstoff etwas größer geschrieben. Dies ist üblich, da die δ−-Ladung des Sauerstoff-Atoms doppelt so hoch ist wie die jedes einzelnen Wasserstoff-Atoms. Durch Vektoraddition der einzelnen Bindungsdipolmomente ergibt sich das elektrische Dipolmoment des Moleküls.
Bindungsdipolmomente und elektrisches Dipolmoment sollten nicht miteinander verwechselt werden: So haben symmetrisch gebaute Moleküle wie z. B. Kohlenstoffdioxid (O=C=O) polare Bindungen, aber kein elektrisches Dipolmoment.
Bild rechts: Bindungsdipolmomente und elektrisches Dipolmoment eines H2O-Moleküls. In
grün: elektrisches Dipolmoment .

TC-Vortrag 04 138/161 Waschen – Bleichen – Färben
Der induktive Effekt kann sich über mehrere Bindungen hinweg auf andere Atome oder Atomgruppen auswirken. Die Stärke nimmt jedoch mit dem Quadrat der Entfernung ab. Man geht davon aus, dass sich induktive Effekte nicht weiter als drei benachbarte Bindungen auswirken. Tritt ein induktiver Effekt in einem Molekül wie beispielsweise 1-Fluorpropan auf, so wirkt die induktive Kraft auch auf die in einer Kette folgenden Atome:
Das Fluor-Atom löst einen Induktionseffekt aus, der sich auf die drei folgenden Kohlenstoff-atome auswirkt. Am stärksten ist der Induktionseffekt auf das erste Kohlenstoffatom, das direkt an das Fluoratom gebunden ist, dies wird durch das Symbol δ+ indiziert. Die Stärke nimmt allerdings ab, je weiter das betroffene Kohlenstoffatom vom Fluoratom entfernt ist. Auf das zweite Kohlenstoffatom in der Alkyl-Kette ist der Induktionseffekt geringer, was durch die Markierung δδ+ ausgedrückt wird. Wiederum sehr viel geringer wirkt sich der induktive Effekt des Fluoratoms auf das noch weiter vom Fluoratom entfernte dritte Kohlenstoffatom aus, was durch die Markierung δδδ+ ausgedrückt wird.
In der Regel betrachtet man I-Effekte bei komplexeren Verbindungen. Dadurch ist es möglich, das Verhalten der Verbindungen zu analysieren. Beispielsweise hat der −I-Effekt bei Trichloressigsäure weitergehende Auswirkungen. In dieser Verbindung üben drei Cl-Atome am C-Atom einen −I-Effekt aus. Dadurch zieht das C-Atom die Elektronen des ihm benachbarten C-Atoms zu sich, wodurch dieses C-Atom Elektronen vom benachbarten und einfach gebundenen Sauerstoff zu sich zieht. Die Bindung zwischen dem O-Atom und dem mit ihm verbundenen H-Atoms ist dadurch geschwächt und das H+-Ion (Proton) sehr leicht abspaltbar.
Einen +I-Effekt haben Teilchen, die elektronenschiebend wirken. Dies geschieht z. B., wenn das Teilchen negativ geladen ist oder eine niedrige Elektronegativität besitzt. Ebenso ist der +I-Effekt bei der Ausbildung von Hybridorbitalen zu beobachten, so wirkt z. B. die Methylgruppe CH3 elektronenschiebend, auch wenn das aufgrund der C–C-Einfachbindung nicht einzusehen ist.
Den −I-Effekt haben Atome, die elektronenziehend wirken. Dies kommt in der Regel durch hohe Elektronegativität oder eine positive Ladung zustande. Stark elektronegative Teilchen ziehen besonders stark Elektronen an.
Die Auswirkungen des Induktionseffektes sind, dass andere polare Moleküle sich nun am besagten Molekül ausrichten und es angreifen können. Zudem hat der Induktionseffekt Einfluss auf die Lage der Zweit-Substituenten am Benzol. Radikale oder Carbeniumionen (Carbokationen), also Teilchen mit Elektronenmangel, werden durch Substituenten mit +I-Effekt stabilisiert und durch solche mit -I-Effekt destabilisiert. Abgesehen davon hat der induktive Effekt entscheidenden Einfluss auf die Säurestärke eines Moleküls. Verfügt ein Molekül beispielsweise über einen stark elektronegativen (elektronenanziehenden) Substituenten, wird die Abspaltung eines Protons erleichtert (−I-Effekt), die Säurestärke ist entsprechend groß. Umgekehrt führt ein elektronenschiebender Substituent zu einer geringen Säurestärke (+I-Effekt).
Beispiele für induktiv wirkende Gruppen:

TC-Vortrag 04 139/161 Waschen – Bleichen – Färben
+I (positiver induktiver Effekt)
t-Butylgruppe –C(CH3)3 i-Propylgruppe –CH(CH3)2 Alkylrest –R
I=0 (kein Induktiver Effekt)
Wasserstoffatom -H
−I (negativer induktiver Effekt)
Sauerstoff in der Carbonylgruppe –C=O
Hydroxygruppe –OH Iodatom –I Bromatom –Br Chloratom –Cl Nitrogruppe –NO2 Aminogruppe –NH2 Carboxygruppe –COOH Fluoratom –F Cyanogruppe –CN
Isomerie – gleiche Anzahl der Atome in einem Molekül
Isomerie ist das Auftreten von zwei oder mehreren chemischen Verbindungen mit gleicher Summenformel und Molekülmasse, die sich jedoch in der Verknüpfung oder der räumlichen Anordnung der Atome unterscheiden. Die entsprechenden Verbindungen werden Isomere genannt und lassen sich durch unterschiedliche Strukturformeln darstellen. Sie unterscheiden sich in ihren chemischen oder/und physikalischen, und oft auch in ihren biochemischen Eigenschaften. Isomerie tritt vor allem bei organischen Verbindungen, aber auch bei (anorganischen) Koordinationsverbindungen auf. Die Isomerie wird in verschiedene Bereiche unterteilt.
Konstitutionsisomere besitzen die gleiche allgemeine Summenformel, unterscheiden sich aber in der Reihenfolge der Atome und Bindungen. Die Isomere sind daher im Allgemeinen verschiedene Substanzen mit unterschiedlichen chemischen (u. a. Reaktivität) und physikalischen Eigenschaften (u. a. Schmelz- und Siedepunkt, Löslichkeit). Man kann mehrere Fälle unterscheiden:
Funktionsisomere besitzen unterschiedliche funktionelle Gruppen, z. B. Ethanol (CH3–CH2–OH) und Dimethylether CH3–O–CH3.
Skelettisomere haben verschieden verzweigte Kohlenstoffgrundgerüste. Sie werden oft in Stoffgruppen zusammengefasst. Bei Kohlenwasserstoffen sind dies z. B. die Pentane oder die Hexane. Ähnliches gilt für Verbindungen mit einer funktionellen Gruppe. So zählen Butanol und 2-Methyl-1-propanol zu den Butanolen.
Bei Stellungsisomeren (auch Ortsisomere oder Regioisomere genannt) liegt die gleiche funktionelle Gruppe an verschiedenen Positionen, ein Beispiel ist 1,2-Propandiol und 1,3-Propandiol. Von Tautomerie spricht man, wenn die beiden Isomere in einer reversiblen chemischen Reaktion ineinander übergehen, indem Teile des Moleküls (meist Wasserstoffatome) ihren Platz wechseln und sich gleichzeitig Bindungen verschieben. Aufgrund des schnellen Erreichens des chemischen Gleichgewichtes lassen sich die einzelnen Tautomere zumeist nicht isolieren.
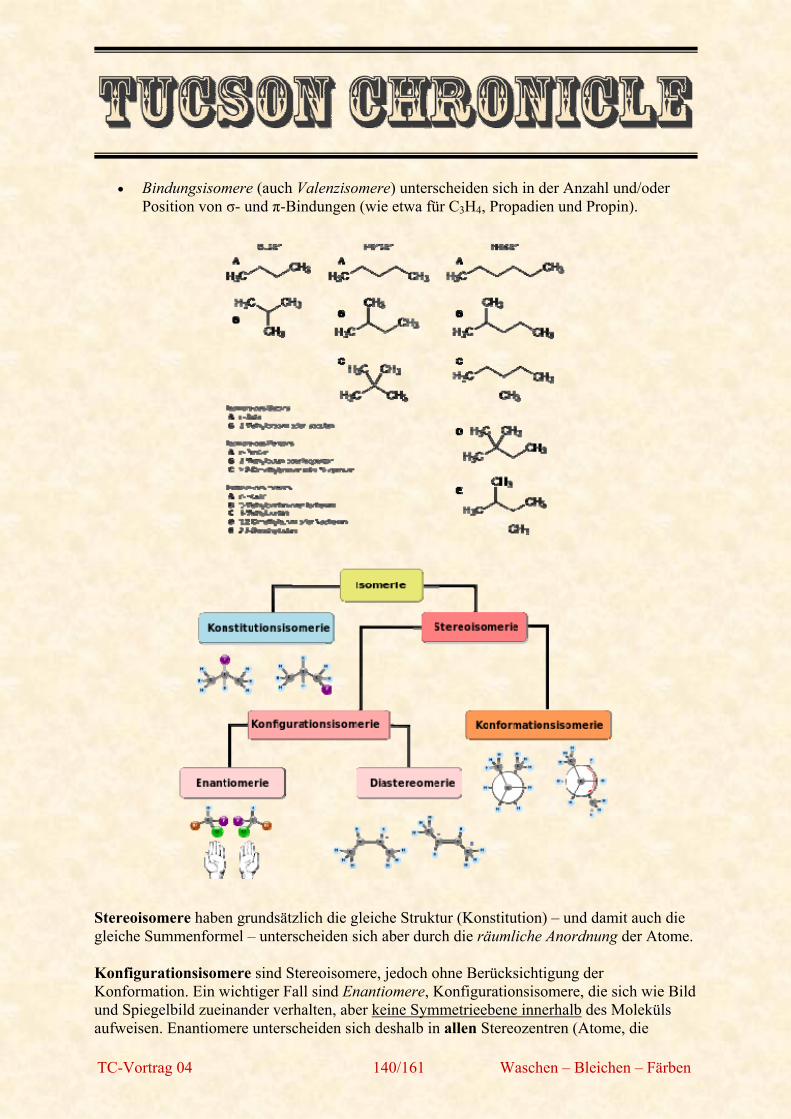
TC-Vortrag 04 140/161 Waschen – Bleichen – Färben
Bindungsisomere (auch Valenzisomere) unterscheiden sich in der Anzahl und/oder Position von σ- und π-Bindungen (wie etwa für C3H4, Propadien und Propin).
Stereoisomere haben grundsätzlich die gleiche Struktur (Konstitution) – und damit auch die gleiche Summenformel – unterscheiden sich aber durch die räumliche Anordnung der Atome.
Konfigurationsisomere sind Stereoisomere, jedoch ohne Berücksichtigung der Konformation. Ein wichtiger Fall sind Enantiomere, Konfigurationsisomere, die sich wie Bild und Spiegelbild zueinander verhalten, aber keine Symmetrieebene innerhalb des Moleküls aufweisen. Enantiomere unterscheiden sich deshalb in allen Stereozentren (Atome, die

TC-Vortrag 04 141/161 Waschen – Bleichen – Färben
aufgrund von vier unterschiedlichen Substituenten unter diesen zwei verschiedene Reihenfolgen erlauben). Wichtige Beispiele sind Zucker, Aminosäuren und viele chirale Arzneistoffe.
Alle Konfigurationsisomere, die keine Enantiomere sind, bezeichnet man als Diastereomere. Diastereoisomere unterteilen sich wie folgt:
Eine Form ist die cis-trans-Isomerie an unsymmetrisch substituierten Doppelbindungen. Diese Isomerie liegt z. B. zwischen Maleinsäure (cis-Form) und Fumarsäure (trans-Form) vor. Lt. IUPAC-Regeln gebildete Namen werden mit E und Z gebildet: (E/Z)-Isomere, s. Alkene.
Als Epimere bezeichnet man Paare von Diastereoisomeren eines Moleküls mit mehreren Stereozentren, die sich in einem dieser Zentren unterscheiden (z. B. Isoleucin und Alloisoleucin oder auch Glucose und Galactose), in den anderen jedoch gleich sind. Epimere sind somit immer auch Diastereomere, aber nicht umgekehrt.
In der Zuckerchemie benutzt man den Begriff Anomer als Spezialfall eines Epimers, dessen Unterschied am ersten Kohlenstoff liegt (relevant bei Bildung der α- oder β-Form eines Zuckers wie der Glucose).
Eine meso-Form besitzt zwar mehrere Chiralitätszentren, aber auch eine Symmetrie-ebene und lässt sich daher mit dem Spiegelbild zur Deckung bringen. Allgemein gültig ist die Formulierung, dass eine meso-Form äquivalente (mit den gleichen Resten substituierte) Chiralitätszentren mit entgegengesetzter Konfiguration (z. B. ein Stereozentrum mit (R)- und ein gleichsubstituiertes mit (S)-Konfiguration) besitzt. Zwei meso-Formen sind also entgegen dem ersten Anschein keine Enantiomere und optisch inaktiv (optischer Drehwert α = 0 Grad). Die Anzahl der Stereozentren in meso-Verbindungen ist geradzahlig (2, 4, 6, 8 usw.).
eine Sonderform der Diastereomerie ist Endo-exo-Isomerie, die nur bei substituierten verbrückten bicyclischen Kohlenwasserstoffen auftritt.
Berechnung der maximalen Anzahl an Stereoisomeren eines Moleküls: 2n bei n Chiralitäts-zentren. Wenn meso-Formen vorhanden sind, verringert sich die Anzahl der Isomere jeweils pro meso-Form um eins. Bsp: Cyclohexan mit sechs Substituenten hat sechs Stereozentren, also maximal 26 = 64 Stereoisomere. Im Zusammenhang mit Stereoisomeren tritt oft optische Aktivität auf, d. h., Enantiomere drehen die Ebene von linear polarisiertem Licht um den gleichen Betrag in entgegengesetzte Richtungen.
Allen diesen Gruppen ist gemeinsam, dass ein Isomer nur durch Bindungsbruch in eine andere Form überführt werden kann. Das trifft auf die letzte Gruppe von Isomeren, die auch unter den Oberbegriff Stereoisomere fallen, nicht zu: Konformationsisomere (Konformere) sind Stereoisomere, die sich schon durch die Drehung von Einfachbindungen ineinander überführen lassen. Daher wird häufig auch von Rotameren gesprochen. Die thermische Energie bei Raumtemperatur reicht für die Überführung meist aus. Ein Beispiel ist das ekliptische und das gestaffelte Ethan (gut sichtbar in der Newman-Projektion). Die beiden Gruppen des Ethans können im Prinzip in jedem beliebigen Winkel zueinander stehen, wobei die Energiedifferenz geringer als die thermische Energie ist, so dass in einer Lösung die isomeren Formen kontinuierlich ineinander übergehen und sich normalerweise nicht isolieren lassen. Ein Sonderfall ist die Atropisomerie, bei der eine axiale Chiralität auftritt.

TC-Vortrag 04 142/161 Waschen – Bleichen – Färben
Weitere Begriffe und tabellarische Übersicht
Der Begriff der Mesomerie gehört trotz der sprachlichen Ähnlichkeit nicht in diesen Themenbereich.
Isomere Gemeinsamkeiten Unterschiede
Konstitutionsisomere Summenformel Struktur
Stereo-isomere
Konfiguration-sisomere
Diastereomere Summenformel
+ Struktur
räumliche Anordnung
Enantiomere räumliche Anordnung, aber wie
Bild und Spiegelbild
Konformationsisomere räumliche Stellung
Isomere Unterschiedliche chemische
und physikalische Eigenschaften
Überführung ohne Bindungsbruch
möglich
Konstitutionsisomere Ja nein
Stereo-isomere
Konfigurations-isomere
Diastereomere Ja nein
Enantiomere
optisch aktiv, chemische Unterschiede nur bei chiralen
Reaktionspartnern (z. B. Enzymen)
nein
Konformationsisomere Nein ja
Ketone
Ketone sind chemische Verbindungen, die als funktionelle Gruppe eine nicht endständige Carbonylgruppe (>C=O) enthalten. Eine Ketongruppe [C–C(O)–C] enthält drei Kohlenstoffatome. Alle Ketone enthalten mindestens drei Kohlenstoffatome.
Allgemeine Struktur der Ketone
[R1 und R2 sind organische Reste (Alkyl, Aryl o. ä.)]. Die Carbonylgruppe ist blau markiert.
Im Gegensatz zu den Aldehyden ist hier der Carbonylkohlenstoff in beide Bindungs-richtungen mit Kohlenstoffatomen verbunden. Ketone kann man als Oxidationsprodukte sekundärer Alkohole auffassen. Die von den Alkanen ableitbaren Ketone nennt man auch Alkanone. Entsprechend bezeichnet man die auf Alkene und Alkine zurückzuführenden Ketone als Alkenone (siehe Enone) bzw. Alkinone. Das einfachste Keton ist Aceton. Ein einfaches aromatisches Keton ist Benzophenon (Diphenylketon). Ein gemischtes Keton ist Acetophenon (Methylphenylketon).
Nach der IUPAC-Nomenklatur erhalten Ketone das Suffix -on, sofern die Carbonylgruppe die im Molekül enthaltene Gruppe mit höchster Priorität ist. Dementsprechend heißt das vom Propan abgeleitete Keton Propanon (CH3–CO–CH3, Trivialname: Aceton).

TC-Vortrag 04 143/161 Waschen – Bleichen – Färben
Bei Ketonen mit mehr als drei C-Atomen befindet sich die Carbonylgruppe in der Stammkette und erhält die kleinstmögliche Nummer, diese kann entweder vor dem Namen oder, bei komplizierteren Molekülen unumgänglich, direkt vor der Endung -on stehen.
Beispielsweise heißt: CH3–CH2–CH2–CO–CH3 dementsprechend 2-Pentanon oder Pentan-2-on. Falls die Carbonylgruppe nicht die höchste Priorität besitzt, verwendet man das Präfix Oxo-. Zum Beispiel heißt CH3–CO–CH2–CHO 3-Oxobutanal.
Sehr häufig tragen die Ketone jedoch Namen, welche aus der Bezeichnung der Alkylreste gefolgt von der Endung -keton bestehen (Radikofunktionelle Nomenklatur). So wird Butanon auch Methylethylketon (MEK) genannt. Falls der eine Substituent eine Phenylgruppe ist, ist die Endung -phenon gebräuchlich, wie zum Beispiel bei Acetophenon.
Ketone gehen, ähnlich wie Aldehyde, Additions- und Kondensationsreaktionen ein.
Ketone (R1, R2, R3 = Organyl-Rest) stehen mit ihrer tautomeren Form, den Enolen im Gleichgewicht (Keto-Enol-Tautomerie), wenn an das α-Kohlenstoffatom ein Wasserstoffatom gebunden ist.
Ketone lassen sich im Gegensatz zu den Aldehyden nur unter drastischen Reaktions-bedingungen oxidativ angreifen. Dabei wird eine Bindung zwischen Carbonylkohlenstoff und einem Nachbarkohlenstoff gespalten. Die entstehenden Molekülbruchstücke sind Carbonsäuren.
Wichtige Ketone: Aceton, Cyclohexanon, Fructose, Himbeerketon
Aceton [at͡ səˈtoːn] (auch: Azeton) ist der Trivialname für die organisch-chemische Verbindung Propanon bzw. Dimethylketon. Aceton ist eine farblose Flüssigkeit und findet Verwendung als polares, aprotisches Lösungsmittel und als Ausgangsstoff für viele Synthesen der organischen Chemie. Es ist mit seinem Strukturmerkmal der Carbonylgruppe (>C=O), die zwei Methylgruppen trägt, das einfachste Keton.
Aceton wurde 1606 zum ersten Mal von Andreas Libavius durch Erhitzen von Blei(II)-acetat hergestellt. 1661 konnte Robert Boyle es durch die trockene Destillation von Holz gewinnen. Bis in die Mitte des 20. Jahrhunderts war die Aceton-Butanol-Fermentation ein wichtiges Verfahren zur Gewinnung auch von Aceton. Für die industrielle Produktion kam hierbei das anaerobe Bakterium Clostridium acetobutylicum zum Einsatz. Dieses ist in der Lage, Zucker zu den Lösemitteln Aceton, 1-Butanol, Ethanol und zu den organischen Säuren Essigsäure und Buttersäure zu vergären. Aufgrund dieser Eigenschaften besteht ein großes Interesse daran, das Bakterium in der industriellen Biotechnologie zur Produktion dieser Produkte zu nutzen. Das wichtigste Herstellungsverfahren von Aceton ist heutzutage das Cumolhydroperoxid-Verfahren, das auch als Phenolsynthese nach Hock bekannt ist:

TC-Vortrag 04 144/161 Waschen – Bleichen – Färben
Hier werden Benzol und Propen zunächst durch eine Friedel-Crafts-Alkylierung im Sauren in Isopropylbenzol (Cumol) überführt. Dieses reagiert dann mit Sauerstoff in einer Radikalreaktion zum Hydroperoxid, welches sich während der sauren Aufarbeitung zu Phenol und Aceton zersetzt.
Als weiteres Herstellungsverfahren wird die Dehydrierung bzw. Oxidehydrierung von Isopropanol durchgeführt. Eine weitere Möglichkeit der Acetonherstellung besteht darin, Calciumacetat (1) zu erhitzen, wobei es in Aceton (2) und Calciumoxid zerfällt. (Bekannt unter der Bezeichnung "Kalksalzdestillation")
Aceton ist eine farblose, niedrigviskose Flüssigkeit mit einem charakteristischen, leicht süßlichen Geruch, leicht entzündlich und bildet mit Luft ein explosives Gemisch. Der Siedepunkt bei Normaldruck beträgt 56 °C. Es ist in jedem Verhältnis mit Wasser und den meisten organischen Lösungsmitteln mischbar. Aceton kann aufgrund seiner polaren Carbonylgruppe mit Kationen auch Komplexverbindungen bilden. Das Acetonmolekül zeigt Keto-Enol-Tautomerie; sein pKs-Wert beträgt 20.
Die Verbindung bildet mit einer Reihe anderer Lösungsmittel azeotrop siedende Gemische. Die azeotropen Zusammensetzungen und Siedepunkte finden sich in der folgenden Tabelle. Keine Azeotrope werden mit Wasser, Ethanol, 1-Propanol, 2-Propanol, n-Butanol, Benzol, Toluol, Ethylbenzol, Diethylether, Ethylacetat und Acetonitril gebildet.
Lösungs-mittel
n-Pentan
n- Hexan
n- Heptan
Cyclo-hexan
MethanolChloro-
form
Tetra-chlor-
methan
Diiso-propyl-ether
Methyl-acetat
Gehalt Aceton
in Ma%
21 59 90 67 88 22 89 61 50
Siede-punkt
in °C 32 50 56 53 55 64 56 54 55

TC-Vortrag 04 145/161 Waschen – Bleichen – Färben
Ein Azeotrop oder azeotropes Gemisch (griechisch a- „nicht“, zeo- „siedend“, tropos „Wendung“, hier im Sinne von „Änderung“) ist ein Stoffgemisch, das man nicht durch Destillation trennen kann. Die Zusammensetzung des Gemischs bleibt beim Phasenübergang von flüssig zu gasförmig gleich, es verhält sich also wie ein Reinstoff. Azeotropie ist das Gegenteil von Zeotropie.
Niedermolekulare Ketone sind farblose, leichtbewegliche Flüssigkeiten und aufgrund der Polarität der Carbonylgruppe in Wasser löslich. Niedermolekulare Ketone zeichnen sich durch einen meist angenehmen, fruchtigen Geruch aus. Höhermolekulare Ketone sind feste Stoffe.
Ketone besitzen auf Grund des +I-Effekts der Alkylsubstituenten am Carbonyl-Kohlenstoff-atom eine niedrigere Reaktionsfreudigkeit als Aldehyde und neigen nicht zur Polymerisation. Im Unterschied zu den Aldehyden erfolgt mit Ketonen keine Reaktion bei der Fehlingschen Probe, da die Ketongruppe unter Erhalt des Kohlenstoffgerüsts nicht weiter oxidiert werden kann.
Aceton reagiert in Gegenwart von Benzaldehyd in alkalischer Lösung zum Dibenzalaceton. Die Reaktion findet nach dem allgemeinen Mechanismus der Aldol-Kondensation statt.
Aceton wird u. a. als gängiges Lösungs- und Extraktionsmittel für Harze, Fette und Öle, Kolophonium, Celluloseacetat sowie als Nagellackentferner und Plastikkleber eingesetzt.
Cyclohexanon ist im reinen Zustand eine farblose, wasserklare Flüssigkeit, deren Geruch ein wenig an Aceton erinnert. Es gehört zur Gruppe der zyklischen Ketone. Zur technischen Synthese von Cyclohexanon stehen hauptsächlich zwei Verfahren zur Verfügung:
1) Die katalytische Oxidation von Cyclohexan mit Luftsauerstoff, die über das instabile Cyclohexylhydroperoxid verläuft und einem radikalischen Mechanismus folgt. Dabei wird ein Gemisch aus Cyclohexanon und Cyclohexanol gebildet, welche man auch abgekürzt als Anon und Anol bezeichnet. Das Gemisch kann destillativ aufgetrennt werden.

TC-Vortrag 04 146/161 Waschen – Bleichen – Färben
2) Die Oxidation von Cyclohexanol mit verdünnter Salpetersäure oder in der Gasphase an Zinkoxid-Katalysatoren. Das Cyclohexanol wird zuvor durch Hydrierung von Phenol an einem Nickel-Katalysator hergestellt:
Aufgrund seiner vielfältigen Reaktionsmöglichkeiten als Keton wird es oft in der organischen Synthesechemie eingesetzt, wenn Sechsring-Strukturelemente in Molekülgerüste eingebaut werden sollen. Weiterhin ist Cyclohexanon ein sehr gutes Lösungsmittel für Lackrohstoffe, Polyvinylchlorid, basische Farbstoffe sowie Natur- und Kunstharze. Auch in Deckfarben für Leder, Druckfarben und Abbeizmitteln ist der Stoff enthalten.
Fructose
Fructose (umgangssprachlich Fruchtzucker, oft auch Fruktose, von lateinisch fructus ‚Frucht‘, veraltet Lävulose) ist eine natürlich vorkommende chemische Verbindung mit der Summenformel C6H12O6. Fructosen gehören als Monosaccharide (Einfachzucker) zu den Kohlenhydraten. Sie kommt in mehreren isomeren (anomeren) Formen vor. In diesem Artikel betreffen die Angaben zur Physiologie allein die D-Fructose. L-Fructose besitzt physiologisch keine und auch sonst nur geringe Bedeutung.
D-(−)-Fructose
Lävulos
Laevulose
L,D,D-Ketohexose
α-Acrose
Fructose kommt in der Natur vor allem in Früchten wie Kernobst (Äpfel und Birnen zu je etwa 6 g/100 g), Beeren (beispielsweise Weintrauben 7,5 g/100 g) sowie in manchen exotischen Früchten (Granatapfel und Kaki) und im Honig (35,9–42,1 g/100 g) vor.
Im Haushaltszucker (hergestellt aus Zuckerrüben oder Zuckerrohr) ist Fructose in gebundener Form enthalten: Rohr- oder Rübenzucker (Saccharose) ist ein Zweifachzucker, der aus je einem Molekül Glucose (Traubenzucker) und Fructose zusammengesetzt ist. Ein bedeutsamer Anteil bei der Zuckeraufnahme kommt aus industriell gefertigten Nahrungsmitteln, die mit Fructose angereicherten Sirup aus Maisstärke (high-fructose corn syrup, HFCS) enthalten.
Fructose ist eine farb- und geruchlose, leicht wasserlösliche, sehr süß schmeckende Verbindung, die prismen- oder nadelförmige, stark hygroskopische Kristalle bildet. Bei 60 %

TC-Vortrag 04 147/161 Waschen – Bleichen – Färben
Luftfeuchtigkeit nimmt sie innerhalb einer Stunde 0,28 % Wasser auf, innerhalb von 9 Tagen 0.6 %. Das Monosaccharid ist optisch aktiv und kommt daher in zwei spiegelbildichen Isomeren, den sogenannten Enantiomeren vor. Fructose gehört zur Gruppe der Hexosen und – wegen der Ketogruppe – zu den Ketosen. In kristalliner Form liegt sie als Sechsring (Fructopyranose) vor, gebunden als Fünfring (Fructofuranose).
Die α- und β-Anomere der jeweiligen Ringformen können in wässriger Lösung ineinander umgewandelt werden und stehen untereinander in einem Gleichgewicht. Bei 20 °C liegt in Wasser gelöste D-Fructose zu 76 % in der β-Pyranoseform, zu 4 % in der α-Furanoseform und zu 20 % in der β-Furanoseform vor.
Keilstrichformel der D-Fructose
Haworth-Schreibweise von
α-D-Fructofuranose β-D-Fructofuranose α-D-Fructopyranose β-D-Fructopyranose
Im Darm wird Fructose von Menschen unterschiedlich gut, vor allem langsamer als Glucose, resorbiert. Dies liegt am passiven Transport durch spezielle Proteine, zum einen durch das so genannte GLUT5 (apikal, d. h. an der dem Darmlumen zugewandten Zelloberfläche), das der Fructose Zutritt zu den Darmzellen (Enterocyten) gewährt und zum anderen durch GLUT2 (basolateral, d. h. dem Blutkreislauf zugewandt), das der Fructose erlaubt, von den Darm-zellen ins Blut zu gelangen.
Glucose wird hingegen sekundär-aktiv (SGLT1, apikal), also unter Energieverbrauch, in die Zelle gepumpt. Dies geschieht reguliert über eine rückgekoppelte Hemmung. Im Gegensatz dazu fließt Fructose unreguliert ohne Energieaufwand entlang ihres Konzentrationsgradienten. Dies führt dazu, dass Fructose niemals vollständig aus der Nahrung aufgenommen wird. Vor allem bei Kleinkindern besteht daher die Gefahr, dass es bei zu hohen Fructosemengen in der Nahrung zu osmotischer Diarrhoe kommt.

TC-Vortrag 04 148/161 Waschen – Bleichen – Färben
D-Fructose wird in Zellen der Leber durch das Enzym Ketohexokinase in D-Fructose-1-phosphat umgewandelt, so kann sie die Zelle nicht mehr verlassen. Der Vorrat an energie-reichen Phosphaten wird durch die Ketohexokinase „geplündert“: ATP → ADP → AMP und die AMP-Desaminase hochreguliert. Es fällt IMP an, das über den Purinabbau die Konzentration der Harnsäure ansteigen lässt. Fructose-1-phosphat zerfällt durch die Fructose-1-phosphat-Aldolase vermittelt in Glycerinaldehyd und Dihydroxyacetonphosphat. Nach Phosphorylierung kann Glycerinaldehyd (dann als Glycerinaldehyd-3-phosphat) in die Glyko-lyse eintreten. Bedeutsamer ist der Abfluss der Zerfallsprodukte in die Triglyceridsynthese. Triglyceride lagern sich als Depotfett an, aber auch als Fetttröpfchen zwischen den Myo-fibrillen der Muskulatur. Im Fettgewebe kann Fructose auch als Fructose-6-phosphat in die Glycolyse eintreten, wenn die Glykogenreserven erschöpft sind.
Bezogen auf Saccharose (also Haushaltszucker) hat eine 10-prozentige D-Fructoselösung eine Süßkraft von 114 Prozent. Der Blutzucker steigt bei Zufuhr von Fruchtzucker deutlich langsamer an als bei Zufuhr der in der Küche üblicherweise verwendeten Saccharose; der glykämische Index liegt mit 20 auf einem recht niedrigen Niveau. Fructose anstelle von handelsüblicher Saccharose ist aus ernährungsmedizinischer Sicht aber nicht sinnvoll, da sich eine erhöhte Fruktoseaufnahme ungünstig auf den Stoffwechsel auswirkt und zudem ein Übermaß an Fruktose in der Ernährung die Entwicklung von Adipositas sowie des metabolischen Syndroms begünstigt. Das metabolische Syndrom (manchmal auch als tödliches Quartett, Reavan-Syndrom oder Syndrom X bezeichnet) wird heute als der entscheidende Risikofaktor für koronare Herzkrankheiten angesehen. Es ist charakterisiert durch diese vier Faktoren: abdominelle Fettleibigkeit, Bluthochdruck (Hypertonie), veränderte Blutfettwerte (Dyslipidämie) und Insulinresistenz. Die Erkrankung entwickelt sich aus einem Lebensstil, der durch permanente Überernährung und Bewegungsmangel gekennzeichnet ist, und betrifft einen hohen Anteil der in Industriestaaten lebenden Bevölkerung.
Himbeerketon
Himbeerketon (Rheosmin, nach IUPAC 4-(4-Hydroxyphenyl)-butan-2-on) ist ein natürlich in der Himbeere vorkommendes Phenol, das den wesentlichen Geruchseindruck der Beere ausmacht (eine sogenannte „character-impact-Verbindung“).
Himbeerketon ist die Hauptgeruchskomponente in Himbeeren (Rubus idaeus). Das natürliche Himbeeraroma wird jedoch durch etwa 250 weitere Substanzen (meist Alkohole wie Linalool und Geraniol, Ketone wie Jonon, Aldehyde oder Lactone) hervorgerufen. Rheosmin liegt in Himbeeren, Großfrüchtigen Moosbeeren (Vaccinium macrocarpon), Brombeeren (Rubus) und Loganbeeren (Rubus loganobaccus) daneben auch als Glucosid Lindleyin bzw. Isolindleyin vor. Das Keton kommt auch im Tabakrauch vor, wobei es im Tabak selbst nicht gefunden wurde. Himbeerketon wird zur Aromatisierung von Lebensmitteln, etwa Süßwaren, eingesetzt.

TC-Vortrag 04 149/161 Waschen – Bleichen – Färben
Kohlenstoff
Atom mit vier freien Bindungsachsen, welche einen Teraeder bilden. Der Kohlenstoff kann sich mit sich selbst verbinden und anorganische Gitterstrukturen bilden (Graphit, Diamant). Die meisten Verbindungen des Kohlenstoffs werden zu den organischen Verbindungen gezählt (Bsp. reine Kohlenwasserstoffe: Alkane, Alkene, Alkine und deren weitere Reaktions-produkte mit Sauerstoff (Alkohole, organische Säuren, Ether, Ester, Anhydride…) oder weiteren Atomen oder Molekülgruppen (vor allem mit Stickstoff (Nitro-, Amino-, Azo-Verbindungen).
Kohlenwasserstoffe
Kohlenstoff-Wasserstoff-Verbindungen mit mehr oder weniger starker Sättigung mit Wasserstoff (s. Alkan, Alken, Alkin).
Kohlenhydrate
Kohlenhydrate oder Saccharide, zu denen vor allem die Zucker und Stärken gehören, bilden eine biologisch und chemisch bedeutsame Stoffklasse. Als Produkt der Photosynthese machen Kohlenhydrate den größten Teil der Biomasse aus. Mono-, Di- und Polysaccharide (u. a. Stärke) stellen zusammen mit den Fetten und Proteinen den quantitativ größten verwertbaren und nicht-verwertbaren (Ballaststoffe) Anteil an der Nahrung. Neben ihrer zentralen Rolle als physiologischer Energieträger spielen sie als Stützsubstanz vor allem im Pflanzenreich und in biologischen Signal- und Erkennungsprozessen (z. B. Zell-Zell-Erkennung, Blutgruppen) eine wichtige Rolle. Die Wissenschaft, die sich mit der Biologie der Kohlenhydrate beschäftigt, heißt Glykobiologie.
Chemisch handelt es sich um mehrwertige Alkohole mit einer reaktiven Carbonylfunktion (>C=O), also Hydroxyaldehyde (Aldosen) oder Hydroxyketone (Ketosen). Die Carbonyl-gruppe liegt meist als stabileres Acetal oder Halbacetal vor. Die Reaktion mit den enthaltenen OH-Gruppen ermöglicht einen Ringschluss bzw. die Bildung von Oligo- und Poly-kondensaten. Am weitesten verbreitet sind Monosaccharide mit entweder fünf oder sechs C-Atomen, da hier ein solcher Ringschluss leicht möglich ist. Einfachzucker können über glykosidische Bindungen durch eine Kondensationsreaktion zu Zwei- und Mehrfachzuckern verketten.
Ausschnitt aus einem Amylopektinpolymer, dem Hauptbestandteil der Stärke. Die Glucopyranose-Einheiten der Hauptkette sind α-1,4-glykosidisch miteinander verbunden. Etwa alle 25 Monomere erfolgt eine α-1,6-glykosidische Verzweigung.
Die Monosaccharide (Einfachzucker, z. B. Trauben-zucker, Fruchtzucker), Disaccharide (Zweifach-zucker, z. B. Kristallzucker, Milchzucker, Malz-zucker) und Oligosaccharide (Mehrfachzucker, z. B. Raffinose) sind wasserlöslich, haben einen süßen Geschmack und werden im engeren Sinne als Zucker bezeichnet.

TC-Vortrag 04 150/161 Waschen – Bleichen – Färben
Die Polysaccharide (Vielfachzucker, z. B. Stärke, Cellulose, Chitin) sind hingegen oftmals schlecht oder gar nicht in Wasser löslich und geschmacksneutral.
Mesomerie
Als Mesomerie (auch Resonanz oder Resonanzstruktur) wird in der Chemie das Phänomen bezeichnet, dass die Bindungsverhältnisse in einem Molekül, oder mehratomigen Ion, nicht durch eine einzige Strukturformel, sondern nur durch mehrere Grenzformeln dargestellt werden können. Keine dieser Grenzformeln beschreibt die Bindungsverhältnisse und damit die Verteilung der Elektronen in ausreichender Weise. Die tatsächliche Elektronenverteilung im Molekül bzw. Ion liegt zwischen den von den Grenzformeln angegebenen Elektronenverteilungen. Dies wird durch den Mesomeriepfeil (Resonanzpfeil) ↔ symbolisiert. Der Mesomeriepfeil darf nicht mit dem Doppelpfeil Gleichgewichtspfeil ⇌ verwechselt werden, der ein chemisches Gleichgewicht kennzeichnet. Der Begriff der Mesomerie wurde 1933 von Christopher Kelk Ingold eingeführt.
Der wirkliche Zustand eines Moleküls, also der Zwischenzustand zwischen den Grenzstrukturen wird als mesomerer Zustand bezeichnet.
Ein Beispiel für eine mesomere Verbindung ist Benzol (siehe Abbildung). Auch andere Aromaten sind mesomere Verbindungen.
mesomere Grenzstrukturen des Benzols
delokalisierte Doppelbindungen
Gemäß der Oktettregel sind diejenigen Moleküle besonders stabil, bei denen jedes Atom von acht Valenzelektronen umgeben ist. Für das Benzol lassen sich zwei Strukturformeln aufstellen, bei denen dies der Fall ist (mesomere Grenzstrukturen).
Dass keine der beiden Grenzformeln des Benzols korrekt ist, lässt sich aus den Bindungslängen der Bindungen zwischen den Kohlenstoffatomen ableiten. Die durch Doppelbindungen miteinander verbundenen Kohlenstoffatome müssten geringere Abstände haben als jene, die nur durch eine einfache Atombindung miteinander verbunden sind. Das ist jedoch nicht der Fall. Die Bindungslängen zwischen den Kohlenstoffatomen betragen einheitlich 139 pm.
Im Benzolring verfügt jedes Kohlenstoffatom über vier Valenzelektronen, von denen zwei das Atom mit den benachbarten C-Atomen verbinden. Ein Elektron bindet das zugehörige Wasserstoffatom. Die verbleibenden sechs π-Elektronen ergeben formal drei π-Bindungen, wie sie in der Strukturformel mit drei Doppelbindungen ausgedrückt werden. In dem heute gültigen Orbitalmodell bilden die sechs π-Elektronen eine

TC-Vortrag 04 151/161 Waschen – Bleichen – Färben
delokalisierte Ladungswolke (delokalisiertes 6-π-Elektronensystem) über und unter der Ebene des Kohlenstoffrings (Mehrzentrenbindung).
Mesomerie-Energie:
Daraus ergibt sich ein um 151 kJ/mol abgesenkter Energiezustand, welcher die Bindungsenergie um denselben Betrag erhöht, woraus eine größere Stabilität gegenüber den hypothetischen Grenzformeln (Cyclohexatrien) mit drei isolierten Doppelbindungen resultiert. Diese Energie-differenz wird als Mesomerie- bzw. Resonanzenergie bezeichnet und ergibt sich aus der Differenz der Hydrierungsenergien des hypothetischen Cyclohexatriens und des Benzols. Der gleiche Wert ergibt sich aus der Differenz der Verbrennungsenergien beider Verbindungen.
Es lässt sich allerdings auch eine andere Bezugssubstanz für die Mesomerieenergie benutzen. Im Falle des Vergleichs der Hydrierungsenergien bzw. Verbrennungsenergien von Benzol mit dem entsprechenden linearen Molekül (Hexatrien) ergibt sich ein etwas niedrigerer Wert. Diese Resonanzenergie wird als „Dewar Resonance Energy (DRE)“ bezeichnet.
Allgemein gilt: Je mehr mesomere Grenzstrukturen ein Molekül oder Ion besitzt, desto stabiler ist es. Die Energiedifferenz zwischen den Grenzstrukturen und dem tatsächlichen mesomeren Zustand, die in vielen Fällen abgeschätzt werden kann, wird als Mesomerieenergie oder Resonanzenergie bezeichnet.
Nitrate
Nitrate sind die Salze und Ester der Salpetersäure (HNO3). Die Salze haben die allgemeine Zusammensetzung MINO3 (MI: einwertiges Kation). Einige der Salze werden mit dem historischen Trivialnamen Salpeter bezeichnet. Das planare Anion NO3− trägt eine negative Ladung. Die Ester der Salpetersäure werden auch Salpetersäureester genannt und haben die allgemeine Struktur R–O–NO2 (R: organischer Rest). Einige Salpetersäureester werden fälschlicherweise als Nitroverbindung bezeichnet, so z. B. Glycerintrinitrat als Nitroglyzerin. Nitroverbindungen (R–NO2) haben jedoch im Gegensatz zu Nitraten eine C-N-Bindung.
Das mesomeriestabilisierte Nitrat-Anion. Die Gesamtladung ist –1.

TC-Vortrag 04 152/161 Waschen – Bleichen – Färben
Salpetersäureester mit vereinfachter Formel (links) und der Strukturformel (rechts). Der Rest R ist ein Organyl-Rest (Aryl-Rest, Alkyl-Rest,Arylalkyl-Rest etc.). Die Salpetersäureestergruppe (Nitratgruppe) ist blau markiert.
Das Nitration ist planar gebaut. Alle Bindungswinkel O–N–O betragen 120°. Ebenso sind die Bindungslängen der N–O-Bindungen gleich lang und liegen zwischen den Längen für Einfach- bzw. Doppelbindungen. Die reale Struktur des Nitrations muss deshalb zwischen drei mesomeren Grenzstrukturen existieren:
Stickstoff verfügt als Element der zweiten Periode über keine Oktettaufweitung durch d-Orbitale, so dass die auf den ersten Blick ungünstig erscheinenden mesomeren Grenz-strukturen mit positiven und negativen Ladungen vorliegen.
Nitrat wirkt als effizienter Sauerstoffspender. Deshalb ist Kaliumnitrat Bestandteil des Schwarzpulvers (Sprengsalpeter). Es werden gegebenenfalls auch Nitrate anderer Kationen verwendet, wenn farbige Lichteffekte in der Pyrotechnik gewünscht werden.
Als Lebensmittelzusatzstoff wird Natriumnitrat (E 251) und Kaliumnitrat (E 252) als Konservierungsmittel z. B. zum Pökeln von Fleisch- und Wurstwaren verwendet, da es die Bildung anaerober Keime hemmt.
Nitro‐Gruppe
Eine Nitrogruppe ist in der organischen Chemie die funktionelle Gruppe –NO2. Der Bindungspartner kann Kohlenstoff oder Sauerstoff sein. Ist der Bindungspartner Kohlenstoff, zählt die Verbindung zu den Nitro-Verbindungen. Ist der Bindungspartner Sauerstoff (R–O–NO2), zählt die Verbindung zu den Salpetersäureestern (Nitraten).
Nitroverbindung mit vereinfachter Formel (links) und der Strukturformel (rechts). Der Rest R ist ein Organyl-Rest (Aryl-Rest, Alkyl-Rest, Arylalkyl-Rest etc.). Die Nitrogruppe ist blau markiert.
In der mesomeriestabilisierten –NO2-Gruppe liegt ein negatives Sauerstoffatom und ein positives Stickstoffatom vor. Zwei Doppelbindungen am Stickstoff sind ungünstig, so dass

TC-Vortrag 04 153/161 Waschen – Bleichen – Färben
man von einem neutralen und einem geladenem Sauerstoffatom ausgeht. Die –NO2-Gruppe hat bei aromatischen Verbindungen einen −M-Effekt und desaktiviert daher einen Benzolring bei chemischen Reaktionen.
Nitrogruppen haben durch den 3-wertigen Stickstoff eine oxidierende Wirkung. Dies kommt zum Beispiel bei vielen Sprengstoffverbindungen zum Tragen. TNT (Trinitrotoluol) ist deshalb so brisant, weil es auf sieben Kohlenstoffatome drei Nitrogruppen hat. Das Molekül ist in sich selbst ein Oxidations- (Nitrogruppen) und ein Reduktionsmittel (Kohlenstoff-atome). Auf dem -M-Effekt der Nitrogruppe beruht die antiauxochrome Wirkung in farbigen Verbindungen, z.B. bei der Pikrinsäure. Die bei der Behandlung von Proteinen mit konzentrierter Salpetersäure auftretende Gelbfärbung (Xanthoproteinreaktion) kommt durch die Nitrierung der aromatischen Aminosäuren zustande, deren Absorption dabei in den sichtbaren Bereich des Lichtes verschoben wird.
Organische Chemie
Die organische Chemie (kurz: OC), häufig auch kurz Organik, ist ein Teilgebiet der Chemie, in dem die chemischen Verbindungen behandelt werden, die auf Kohlenstoff basieren, mit Ausnahme einiger anorganischer Kohlenstoffverbindungen und elementarem (reinem) Kohlenstoff.
Die große Bindungsfähigkeit des Kohlenstoffatoms ermöglicht eine Vielzahl von unter-schiedlichen Bindungen zu anderen Atomen. Während viele anorganischen Stoffe durch Temperatureinfluss, katalytische Reagenzien nicht verändert werden, finden viele organische Reaktionen bei Raumtemperatur oder leicht erhöhter Temperatur mit katalytischen Mengen an Reagenzien statt.
Die Entstehung der Vielzahl der Naturstoffe (pflanzliche, tierische Farbstoffe, Zucker, Fette, Proteine, Nukleinsäuren) und letztlich der bekannten Lebewesen basiert auf der Bindungs-fähigkeit des Kohlenstoffatoms.
Organische Moleküle enthalten als Elemente neben Kohlenstoff häufig Wasserstoff, Sauerstoff, Stickstoff, Halogene; die chemische Struktur und die funktionellen Gruppen sind die Grundlage für die Verschiedenartigkeit der Einzelmoleküle.
Die Chemie ist nach Georg Ernst Stahl die Wissenschaft der Analyse und Synthese von Stoffen. In der organischen Analytik erfolgt zunächst aus einem Gemisch von Stoffen eine physikalische Trennung und Charakterisierung (Schmelzpunkt, Siedepunkt, Brechungsindex) von Einzelstoffen, dann wird die elementare Zusammensetzung (Elementaranalyse), Molekülmasse, funktionellen Gruppen (chemische Reagenzien, NMR-, IR-, UV-Spektroskopie) bestimmt, so dass sich bei wenig komplexen Verbindungen die Struktur der organischen Verbindung sicher angeben lässt.
Im Bereich der Synthese untersuchen organische Chemiker die Einwirkung von Reagenzien (Säuren, Basen, anorganischen und organischen Stoffen) auf organische Stoffe, um Gesetzmäßigkeiten von chemischen Reagenzien auf bestimmte funktionelle Gruppen und Stoffgruppen herauszufinden.
Aus der Kenntnis der Vielzahl von Gesetzmäßigkeiten kann ein organischer Chemiker eigenständig Synthesen von organischen Naturstoffen (z. B. Zucker, Peptide, Naturfarbstoffe, Vitamine) planen oder in der Natur unbekannte organische Stoffe (Kunststoffe, Ionen-

TC-Vortrag 04 154/161 Waschen – Bleichen – Färben
austauscher, Arzneistoffe, Pflanzenschutzmittel, Kunstfasern für Kleidungsstücke) synthetisieren, die den Wohlstand einer Gesellschaft erheblich beeinflussen.
Abgrenzung zur anorganischen Chemie
Mit wenigen Ausnahmen umfasst die Organik die Chemie aller Verbindungen, die der Kohlenstoff mit sich selbst und anderen Elementen eingeht. Dazu gehören auch alle Bausteine des derzeit bekannten Lebens. Es sind etwa 40 Millionen organische Verbindungen bekannt (2012).
Ausnahmen sind formal zunächst die elementaren Formen des Kohlenstoffs (Graphit, Diamant) und systematisch alle zur anorganischen Chemie zählenden wasserstofffreien Chalkogenide des Kohlenstoffs (Kohlenstoffmonoxid, Kohlenstoffdioxid, Schwefelkohlenstoff), die Kohlensäure und Carbonate, die Carbide sowie die ionischen Cyanide, Cyanate und Thiocyanate (siehe Kohlenstoff-Verbindungen).
Die Blausäure gehört zum Grenzgebiet der anorganischen und organischen Chemie. Obwohl man sie traditionell zur anorganischen Chemie zählen würde, wird sie als Nitril (organische Stoffgruppe) der Ameisensäure aufgefasst. Die Cyanide werden in der Anorganik behandelt, wobei hier nur die Salze der Blausäure gemeint sind, wohingegen die unter selbigem Namen bekannten Ester als Nitrile zur Organik gehören. Auch die Cyansauerstoffsäuren, Thiocyansäuren und deren Ester gelten als Grenzfälle. Weiter ist die metallorganische Chemie (Metallorganyle) nicht konkret der organischen oder anorganischen Chemie zuzuordnen.
Auch völlig unnatürlich wirkende Stoffe, wie Kunststoffe und Erdöl, zählen zu den organischen Verbindungen, da sie wie die Substanzen von Lebensformen aus Kohlenstoffverbindungen bestehen. Erdöl, Erdgas und Kohle, die Ausgangsstoffe für viele synthetische Produkte, sind letztlich organischen Ursprungs.
Alle Lebewesen enthalten organische Stoffe: Aminosäuren, Proteine, Kohlenhydrate und die DNA (desoxyribose nucletid acid, DNS Desoxribonucleinsäure). Das Teilgebiet der organischen Chemie, das sich mit den Stoffen und Stoffwechselprozessen in Lebewesen befasst, ist die Biochemie (oder auch Molekularbiologie).
Die Sonderstellung des Kohlenstoffs beruht darauf, dass das Kohlenstoffatom vier Bindungselektronen hat, wodurch es unpolare Bindungen mit ein bis vier weiteren Kohlenstoffatomen eingehen kann. Dadurch können lineare oder verzweigte Kohlenstoffketten sowie Kohlenstoffringe entstehen, die an den nicht mit Kohlenstoff besetzten Bindungselektronen mit Wasserstoff und anderen Elementen (vorwiegend Sauerstoff, Stickstoff, Schwefel, Phosphor) verbunden sind, was zu großen und sehr großen Molekülen (z. B. Homo- und Heteropolymere) führen kann und die riesige Vielfalt an organischen Molekülen erklärt. Von dem ebenfalls vierbindigen Silicium gibt es auch eine große Anzahl Verbindungen, aber bei Weitem keine solche Vielfalt.
Die Eigenschaften organischer Substanzen werden sehr stark von ihrer jeweiligen Molekülstruktur bestimmt. Selbst die Eigenschaften von einfachen organischen Salzen wie den Acetaten werden deutlich von der Molekülform des organischen Teils geprägt. Es gibt auch viele Isomere, das sind Verbindungen mit der gleichen Gesamtzusammensetzung (Summenformel), aber unterschiedlicher Struktur (Strukturformel).

TC-Vortrag 04 155/161 Waschen – Bleichen – Färben
Dagegen bestehen die Moleküle in der anorganischen Chemie meist nur aus einigen wenigen Atomen, bei denen die allgemeinen Eigenschaften von Festkörpern, Kristallen und/oder Ionen zum Tragen kommen. Es gibt aber auch Polymere, die keinen Kohlenstoff enthalten (oder nur in Nebengruppen), z. B. die Silane.
Organische Synthesestrategien unterscheiden sich von Synthesen in der anorganischen Chemie, da organische Moleküle meist Stück für Stück aufgebaut werden können. Etwa 60 % der Chemiker in Deutschland und den USA haben als Schwerpunktfach die organische Chemie gewählt.
Viele organische Naturstoffe wurden schon in der Frühzeit der menschlichen Entwicklung genutzt (die Farbstoffe Indigo, Alizarin, die ätherischen Öle, Weingeist). Eine künstliche Darstellung von organischen Stoffen durch Menschenhand ist jedoch in sehr früher Zeit nicht beschrieben worden.
Johann Rudolph Glauber beschrieb in seinen Werken eine Vielzahl von selbst dargestellten organischen Verbindungen, da jedoch die Elementaranalyse noch nicht entwickelt war, kann nur vermutet werden, welche Stoffe er damals erhalten hatte. Weingeist und Essig reinigte Glauber über eine fraktionierte Destillation, Ethylchlorid erhielt er aus Weingeist, Essigsäure aus der Holzdestillation, Aceton aus der Erhitzung von Zinkazetat, Acrolein entstand bei der Destillation von Rüb-, Nuss- und Hanföl Benzol aus Steinkohle. Alkaloide fand er durch eine Salpetersäure-Trennung.
Lemery schrieb 1675 das Buch Cours de Chymie. In diesem Werk wurden die Stoffe in drei Gebiete eingeteilt: Mineralreich (Metalle, Wasser, Luft, Kochsalz, Gips), Pflanzenreich (Zucker, Stärke, Harze, Wachs, Pflanzenfarbstoffe), Tierreich (Fette, Eiweiße, Hornsubstanzen). Lemery unterschied auch die Stoffe des Pflanzen- und Tierreiches als organische Stoffe im Gegensatz zu den Stoffen der unbelebten Natur des Mineralreiches.
Bereits im 18. Jahrhundert war eine beträchtliche Zahl von organischen Substanzen als Reinstoff isoliert worden.
Beispiele sind der Harnstoff (1773 von Hilaire Rouelle) und viele Säuren, wie die von Ameisen erhaltene Ameisensäure (1749 von Andreas Sigismund Marggraf), die Äpfelsäure aus Äpfeln, und die aus dem Weinstein gewonnene Weinsäure (1769), die Citronensäure (1784), das Glycerin (1783), die Oxalsäure, die Harnsäure (von Carl Wilhelm Scheele).
Antoine Laurent de Lavoisier bestimmte erstmals qualitativ die in organischen Stoffen enthaltenen chemischen Elemente: Kohlenstoff, Wasserstoff, Sauerstoff, Stickstoff. Joseph Louis Gay-Lussac und Louis Jacques Thenard führten erste Elementaranalysen zur Ermittlung der quantitativen Zusammensetzung von Elementen in organischen Stoffen aus. Die Elementaranalyse wurde 1831 von Justus von Liebig verbessert. Nun konnte die elementare Zusammensetzung von organischen Stoffen schnell bestimmt werden.
Jöns Jakob Berzelius stellte die These auf, dass organische Stoffe nur durch eine besondere Lebenskraft im pflanzlichen, tierischen oder menschlichen Organismus geschaffen werden kann. Berzelius wendete auch das Gesetz der multiplen Proportionen – mit dem er im Bereich der anorganischen Verbindungen Atomgewichte und Zusammensetzung, d. h. deren chemische Formeln, bestimmen konnte auch auf organische Verbindungen an.

TC-Vortrag 04 156/161 Waschen – Bleichen – Färben
Die Struktur und Zusammensetzung von organischen Verbindungen war um 1820 noch sehr ungeklärt. Gay-Lussac glaubte, dass das Ethanol eine Verbindung aus einem Teil Ethen und einem Teil Wasser sei.
Weiterhin glaubten die Chemiker damals, dass bei gleicher qualitativer und quantitativer Zusammensetzung (Summenformel) der Elemente einer Verbindung (Elementaranalyse) die Stoffe auch identisch sein müssen. Erste Zweifel traten im Jahr 1823 auf, als Justus von Liebig und Friedrich Wöhler das knallsaure Silber sowie das cyansaure Silber untersuchten. Sie fanden bei gleicher chemischer Zusammensetzung sehr unterschiedliche Stoffe.
Im Jahr 1828 erhitzte Friedrich Wöhler Ammoniumcyanat und erhielt einen ganz andersartigen Stoff, den Harnstoff. Ausgangsprodukt und Endprodukt haben die gleiche chemische Summenformel (Isomerie), sie besitzen jedoch sehr unterschiedliche Eigenschaften: das Ammoniumcyanat ist eine anorganische Verbindung, der Harnstoff ist eine organische Verbindung. Damit war die Hypothese von Berzelius, dass organische Verbindungen nur durch eine besondere Lebenskraft entstehen können, widerlegt.
Hermann Kolbe formulierte 1859 die These, dass alle organischen Stoffe Abkömmlinge der anorganischen Stoffe – insbesondere des Kohlenstoffdioxids – sind. So ergibt der Ersatz einer Hydroxygruppe durch Alkylreste oder Wasserstoff Carbonsäuren, der Ersatz zweier Hydroxygruppen durch Alkylgruppen oder Wasserstoff die Aldehyde, Ketone.
Kolbe gebrauchte auch das Wort Synthese im Zusammenhang mit der künstlichen Darstellung von organischen Naturstoffen. Chemiker konnten bald durch eigene Forschungen neue organische Moleküle synthetisieren.
In Analogie zu positiv und negativ geladenen Ionen in der anorganischen Chemie vermutete Berzelius sogenannte Radikale in der organischen Chemie; darauf basierte seine Radikaltheorie. Ein Radikalteil des organischen Moleküls sollte eine positive, der andere Teil eine negative Ladung besitzen. Einige Jahre später untersuchten Jean Baptiste Dumas, Auguste Laurent, Charles Gerhardt und Justus von Liebig die Substitution bei organischen Verbindungen. Die Wasserstoffatome in organischen Verbindungen wurden durch Halogenatome ersetzt. Die alte Radikaltheorie von Berzelius, nach der sich positiv und negativ geladene Radikalteile in organischen Molekülen zusammenlagern, musste verworfen werden. In der Folge wurde von August Wilhelm von Hofmann, Hermann Kolbe, Edward Frankland, Stanislao Cannizzaro weitere Grundlagen über die Zusammensetzung von organischen Stoffen gefunden. 1857 veröffentlichte Friedrich August Kekulé seine Arbeit „Über die s. g. gepaarten Verbindungen und die Theorie der mehratomigen Radikale“ in Liebigs Annalen der Chemie (Bd. 104, Nr. 2, S. 129 ff.), die als Ausgangspunkt der organischen Strukturchemie gesehen wird. In dieser Arbeit wird der Kohlenstoff erstmals als vierwertig beschrieben.
Adolf von Baeyer, Emil Fischer, August Wilhelm von Hofmann erforschten Synthesen von Farbstoffen, Zuckern, Peptiden und Alkaloiden.
Ein Großteil der Arbeitszeit der früheren Chemiker lag in der Isolierung eines Reinstoffes.
Der Prüfung der Stoffidentität von organischen Stoffen erfolgte über Siedepunkt, Schmelzpunkt, Löslichkeit, Dichte, Geruch, Farbe, Brechungsindex.
Besonders wichtig wurde der Rohstoff Kohle für die organische Chemie. Ihren Aufschwung nahm die organische Chemie mit der Untersuchung der bei der Leuchtgaserzeugung

TC-Vortrag 04 157/161 Waschen – Bleichen – Färben
entstehenden Abfallprodukte, als der deutsche Chemiker Friedlieb Ferdinand Runge (1795–1867) im Steinkohlenteer die Stoffe Phenol und Anilin entdeckt hatte. William Henry Perkin – ein Schüler August Wilhelm von Hofmann – entdeckte im Jahr 1856 den ersten synthetischen Farbstoff – das Mauvein. Von Hofmann und Emanuel Verguin führten das Fuchsin in die Färberei ein. Johann Peter Grieß entdeckte die Diazofarbstoffe. Die organische Chemie gewann nun zunehmende wirtschaftliche Bedeutung.
Chemische Strukturformel und Reaktionsmechanismus
Grundlage der Stoffkenntnis ist die chemische Strukturformel. Dies ist der Bauplan eines organischen Moleküls. Die Strukturformel eines Stoffes muss immer gedanklich aus Ergebnissen der Stoffanalyse abgeleitet werden. Die Stoffanalyse umfasst mindestens den korrekten Kohlenstoff-, Wasserstoff-, Sauerstoff- und Stickstoffgehalt eines Moleküls (Elementaranalyse), die Art der funktionellen Gruppen und die Bestimmung der molaren Masse.
Eine sehr wichtige Reaktionsklasse bezieht sich auf den Ersatz eines Wasserstoffatoms im Molekül durch ein Halogen, eine Nitrogruppe, eine Sulfongruppe, man bezeichnet diese Reaktion als Substitution. Zu Beginn dieses Abschnittes wurden einige Beispiele aus dieser Reaktionsklasse genannt. Eine weitere wichtige Reaktionsklasse ist die Eliminierung. Die Abspaltung von Hydroxygruppen und Halogenen und der Ausbildung von Doppelbindungen im Molekül bezeichnet man als Eliminierung. Die Wasserabspaltung bei Pinakol zu 2,3-Dimethyl-1,3-butadien ist eine Eliminierung. Andere sehr wichtige Umsetzungen sind die Oxidation und die Reduktion von organischen Molekülen. Die Reduktion von Nitrobenzol zu Anilin durch Zink oder Eisenspäne in Anwesenheit einer Säure oder die Oxidation von Ethanol zu Acetaldehyd oder Essigsäure mittels Kaliumpermanganat sind Beispiele für diese Reaktionsklassen.
Grundchemikalien der organisch-chemischen Industrie
Basis für alle wichtigen synthetischen Stoffe sind die Grundchemikalien. Sie werden in großen Chemieanlagen aus Erdöl, Erdgas oder Kohle hergestellt.
Bis zum Zweiten Weltkrieg war die Kohle die Basis für die Grundchemikalien der organischen Chemie. Aus der Kohle konnte Benzol, Toluol, Xylol – Bausteine für organische Farbstoffe – gewonnen werden. Mit einem elektrischen Lichtbogen kann aus Kohle und Kalk das Kalziumcarbid (großtechnisch seit 1915) gewonnen werden. Kalziumcarbid lässt sich in Acetylen umwandeln und bildete damals nach Verfahren von Walter Reppe (Reppe Chemie) das Ausgangsprodukt für Acetaldehyd, Essigsäure, Aceton, Butylenglykol, Butadien, Acrylsäure, Acrylnitril. Aus Kohle ließ sich auch Methanol (Synthese nach Pier) und Dieselöl (nach Bergius) gewinnen. Auch nach dem Zweiten Weltkrieg wurden viele Grundchemikalien noch aus Kohle hergestellt. Zwischen 1960 bis 1970 wurden die Verfahren in den westlichen Industriestaaten durch modernere Verfahren auf Basis von Erdöl ersetzt.

TC-Vortrag 04 158/161 Waschen – Bleichen – Färben
Stoffgruppen der organischen Chemie
Es ergeben sich zwei Möglichkeiten für eine systematische Einteilung der einzelnen Substanzen der organischen Chemie in Stoffgruppen:
Kohlenwasserstoffe ohne funktionelle Gruppe bilden die Ausgangsstoffe der organischen Chemie und die Grundlage ihrer Nomenklatur.
Sauerstoff- und Hydroxyverbindungen
o Alkohole
o Aldehyde
o Ester
o Ether
o Ketone
o Carbonsäuren
Stickstoffverbindungen
o Amine
o Amide
o Nitroverbindungen, beispielsweise TNT
o Nitrile
Schwefelverbindungen
o Alkanthiole
o Sulfide
o Disulfide
o Ester der Schwefelsäure
o Sulfone
o Sulfoxide
o Thionamide
o Thiolester
o Thiosäure
Phosphorverbindungen
o Phosphate
o Phosphine
Metallorganische Verbindungen, beispielsweise Ferrocen

TC-Vortrag 04 159/161 Waschen – Bleichen – Färben
Einteilung nach Kohlenstoffgerüst:
aliphatische Kohlenwasserstoffe (Aliphaten)
acyclische Kohlenwasserstoffe
o gesättigt (Alkane)
o ungesättigt (Alkene und Alkine)
cyclische Kohlenwasserstoffe
aromatische Kohlenwasserstoffe (Aromaten)
einfache Aromaten
kondensierte Aromaten
Heterocyclen
biochemische Verbindungen (Alkaloide, Aminosäuren, Kohlenhydrate, Proteine, Steroide, Terpene, Vitamine )
Reaktionen
Die Reaktionen in der organischen Chemie lassen sich größtenteils in die folgenden Grundtypen einordnen:
Radikalische Substitution (SR)
Nukleophile Substitution (SN):
o Nukleophile aliphatische Substitution
o Nukleophile aromatische Substitution
Elektrophile Substitution (SE):
o Elektrophile aliphatische Substitution
o Elektrophile aromatische Substitution
Eliminierung
Nukleophile Addition (AN)
Elektrophile Addition (AE)
Radikalische Addition (AR)
Perizyklische Reaktionen
Umlagerung (sofern sie nicht zu oben genannten Reaktionstypen gehören)
Oxidation sowie Reduktion
Darüber hinaus sind viele Reaktionen unter dem Namen ihres Entdeckers bekannt (Namensreaktionen). Eine Einteilung nach dem entstehenden Bindungstyp bzw. Baustein findet sich in der Liste von Reaktionen in der organischen Chemie.

TC-Vortrag 04 160/161 Waschen – Bleichen – Färben
Organyl‐Rest
Organischer Rest einer Verbindung s. auch: Alkyl-Rest
Strukturformel und Textdarstellung von Formeln
Der Begriff Strukturformel stellt in der Chemie einen Sammelbegriff für chemische Darstellungsweisen dar, die Information darüber liefern, wie Atome in einem Molekül verbunden und im Raum angeordnet sind. Strukturformeln zeigen die Atombindungen und – teilweise – die chemische Struktur. Sie werden besonders in der organischen Chemie verwendet.
Als Strukturformeln zunehmender Komplexität können Elektronenformeln, Valenzstrichformeln, Keilstrichformeln und Skelettformeln angesehen werden. Eine Sonderstellung nehmen zudem Projektionsformeln (z. B. die Fischerprojektion und die Newmanprojektion) ein.
Nach Vorarbeiten von Charles Frédéric Gerhardt, Hermann Kolbe, Edward Frankland konnte August von Kekulé im Jahr 1857 für viele chemische Elemente die Zahl der möglichen Bindungen zu anderen Atomen angeben. Die Zahl der Bindungen eines Atomes zu anderen Atomen wird seit 1860 (nach einem Vorschlag von Emil Erlenmeyer) Wertigkeit genannt. Kekulé formulierte für das Kohlenstoffatom die Vierwertigkeit. Im Jahr 1865 erkannte Kekulé die chemische Struktur von aromatischen Verbindungen wie Benzol und Naphthalin.
Alexander Butlerow benutzte auf der Naturforscherversammlung in Speyer im Jahr 1861 den Namen Chemische Struktur. Alexander Crum Brown verwendete im Jahr 1864 erstmals eine Strukturformel (für Essigsäure) in der die Bindungen zwischen den Atomen durch Striche gekennzeichnet wurde.Seit 1864 wurden neben den Summenformeln für organische Stoffe auch Strukturformeln verwendet.
In der organischen Chemie wird häufig die sogenannte Skelettformel verwendet, bei der C- und H-Atome nicht ausgeschrieben sondern impliziert (vorausgesetzt) werden. Die Darstellung des Kohlenstoffgerüstes erfolgt über das Zeichnen der Bindungen zwischen den Kohlenstoffatomen. Für jedes Kohlenstoffatom wird eine Ecke gezeichnet. Da Kohlenstoffatome normalerweise 4 Atombindungen ausbilden, wird die Anzahl der angelagerten Wasserstoffatome berechnet, indem die Anzahl der Bindungen des Kohlenstoffatoms von 4 subtrahiert wird. In aromatischen Ringen ist zudem die Zahl der delokalisierten Elektronen zu berücksichtigen. Im Benzolring befinden sich beispielsweise 6 delokalisierte Elektronen. Folglich ist an jedem der sechs Kohlenstoffatome des Rings nur ein Wasserstoffatom gebunden und nicht zwei wie beim Cyclohexan.

TC-Vortrag 04 161/161 Waschen – Bleichen – Färben
Vergleich verschiedener Strukturformeln unterschiedlicher Abstraktionsgrade
Molekül- name
Summen-Formel
Elektronen- formel
Valenzstrich- formel
Keilstrich- formel
Skelett- formel
Methan
CH4
existiert nicht
Propan
C3H8
Essigsäure
C2H4O2
Wasser
H2O
existiert nicht
Neben den Strukturformeln werden in der Chemie oft auch andere Darstellungsweisen genutzt, insbesondere wenn die Darstellung nur in reiner Textform erfolgen kann.
Molekül- name
Konstitutions- formel
Summen- formel
Verhältnis- formel
Methan CH4 CH4 CH4
Propan CH3–CH2–CH3 C3H8 C3H8
Essigsäure CH3–COOH C2H4O2 CH2O
Wasser existiert nicht H2O H2O
Während die Konstitutionsformel die Anordnung der Gruppen im Molekül angeben kann, beschränkt sich die Summenformel auf die Aufzählung der im Molekül beteiligten Atome inkl. deren Anzahl. Die Verhältnisformel reduziert die Information auf die Darstellung der beteiligten Atome in ihrem Verhältnis.