
Supporting Information - Proceedings of the National … were detected using BAS-2500 phosphorimager...
9
Supporting Information Huh et al. 10.1073/pnas.1214287110 SI Materials and Methods ELISA. ELISA was performed as previously described (1). Briefly, total protein was extracted from Cucumber mosaic virus (CMV)- inoculated leaves using a disposable grinder in 200 μL of extraction buffer (0.1 M Tris·HCl with 1% (wt/vol) sodium sulfite, pH 7.4) at the indicated time points. Forty-five micrograms of each protein extract was incubated overnight at 4 °C with 50 μL of coating buffer (Sigma-Aldrich) in an ELISA plate (Nunc). The plates were washed with TBST buffer (0.01 M Tris·HCl, 0.85% NaCl, and 0.05% Tween- 20, pH 7.4) and then blocked for 1 h with 0.1 M TBS buffer (0.1 M Tris·HCl and 0.85% NaCl, pH 7.4) containing 1% (wt/vol) BSA. After washing with TBST buffer, a 1:500 dilution in 0.1 M TBS buffer containing 0.1% BSA of polyclonal anti-CMV was used as the detection antibody during incubation for 2 h at room temperature. A 1:2,000 dilution in 0.1 M TBS buffer containing 0.1% BSA of alka- line phosphatase goat anti-rabbit IgG (Sigma-Aldrich) was used as a secondary antibody. For color development, p-nitrophenyl phos- phate (Sigma-Aldrich) was used as the substrate, and the reaction was visualized and quantified by measuring absorbance at wave- length of 405 nm (A 405 ) in an ELISA reader (Bio-Rad). RNA Blot Analysis. Total RNA was prepared from CMV-infected plants as described previously (2). Five micrograms of total RNA were electrophoresed in 1.5% (wt/vol) agarose gel containing 6% (vol/vol) formaldehyde in 1× MOPS buffer (pH 7.0) and transferred to Hybond N membranes (Amersham). Hybridizations with a 32 P-labeled CMV-specific3′ UTR probe were performed with PerfectHyb (Sigma-Aldrich) at 65 °C. Membranes were washed twice at room temperature with 2× SSC containing 0.1% SDS for 5 min, followed by a 20-min wash in 0.5× SSC containing 0.1% SDS at 65 °C. Finally, a high-stringency wash was performed in 0.1× SSC containing 0.1% SDS at room temperature for 20 min. Signals were detected using BAS-2500 phosphorimager (Fujifilm). qRT-PCR Analysis for the Gene Expression upon Salicylic Acid Application and CMV Inoculation. Seeds were sown on 1/2 Murashige and Skoog medium and 10-d-old seedlings were sprayed with 1 mM salicylic acid (SA). At ∼3.5 wk, Col-0 leaves were dusted with carborundum and gently rubbed with CMV-Kor using brushes as described (1). The samples were collected at the indicated time points and RNA was purified using a modified version of the hot-phenol method (2). qRT-PCR experiments were carried out using the LightCycler Real-Time PCR Systems (Roche) with KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems) following the manufacturer’s pro- tocol. The thermal cycling program was 55 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Actin7 (At5g09810) was used as the internal reference. The copy number of target genes was determined by the comparative CT method (Applied Bio- systems). Primers used in these experiments are listed in Table S1. Histological Assays. For histochemical analysis of β-glucuronidase (GUS) activity, seedlings or plants inoculated with CMV together with control plants, were vacuum-infiltrated with X-Gluc staining solution [0.5 M sodium phosphate buffer (pH 7.0), 10% (vol/vol) Triton X-100, 0.1 M K 3 Fe(CN) 6 , 0.1 MK 4 Fe(CN) 6 , and 2 mM X- Glc A], incubated at 37 °C overnight, and then transferred into 70% (vol/vol) ethanol to eliminate chlorophyll (3). GUS activity was observed using an Axioplan 2 imaging microscope (Carl Zeiss). GST–APUM5–PHD and GST–APUM6–PHD Protein Purification and EMSA. To obtain the PHD region of APUM5 and APUM6, the PHD-con- taining sequence from APUM5 and APUM6 cDNA was PCR- amplified using Pfu DNA polymerase and TA-cloned into the T- easy vector following the manufacturer’s protocol (Promega). The clone was then digested with BamHI and subcloned into a modified pET vector containing the gene coding for the N-terminal GST (GST) tag. Escherichia coli BL21-CodonPlus (DE3)-RIL cells were used to express the recombinant protein. Cells were grown in LB with kanamycin (50 μg/mL) to an optical density at 600 nm (OD 600 ) of 0.5. The overexpression of GST-fused APUM5–PHD and APUM6–PHD was achieved by adding 1 mM isopropyl-β-D-thio- galactoside to the medium and growing the cells at 28 °C for 6 h. Bacteria were pelleted after induction, suspended in lysis buffer [20 mM Tris (pH 8.0), 120 mM NaCl, 10% (vol/vol) glycerol, 0.1% Triton X-100, 5 mM DTT, and 50 μg/mL PMSF], and subjected to sonication on ice for 1 min with a Vibra cell sonifier (Sonics & Materials). Bacterial lysates were then centrifuged and the su- pernatants were incubated with glutathione-linked resin. The resin was washed with PBS and proteins were eluted from the resin with elution buffer [100 mM Tris (pH 8.0), 120 mM NaCl, 10% glyc- erol, 0.1% Triton X-100, 5 mM DTT, 50 μg/mL PMSF, and 20 mM glutathione]. For EMSA, synthetic RNAs were radiolabeled with [ 32 P]ATP (GE Healthcare) using T4 polynucleotide kinase (TaKaRa) and purified from free nucleotides using Illustra ProbeQuant G-50 Micro Columns (GE Healthcare). The indicated concentrations of purified GST–APUM5–PHD and GST–APUM6–PHD pro- teins were incubated in binding buffer [10 mM Hepes (pH 7.4), 1 mM EDTA, 50 mM KCl, 1 mM DTT, 0.01% BSA, and 0.01% Tween-20] with 10 fmol of labeled synthetic RNAs at 28 °C for 1 h. Electrophoresis was performed on a 5% (vol/vol) native acrylamide gel at 100 V for 50 min at 4 °C. Gels were dried. Signals were detected using a BAS-2500 phosphorimager. In Vitro Replication Assay. The putative APUM5 binding motif “UGUACUUCUA” in 3′ UTR regions of CMV RNA 1, RNA 2, and RNA 3 in pUC-T7 vector was mutated to “ AAAACUUCUA” using a QuikChange XL Site-Directed Mutagenesis Kit (Stratagene). Wild-type and mutant CMV RNAs were linearized by digestion with MluI and then in vitro transcripts were obtained using T7 mMES- SAGE mMACHINE kit (Ambion). These in vitro transcripts were cotransformed by polyethylene glycol-mediated protoplast trans- formation in Arabidopsis protoplasts prepared from wild-type and 35S-APUM5 #1 transgenic plants. After 24 h, RNA samples were extracted from protoplasts and RNA blot analysis was performed. Cap-Binding Assay. N-APUM5 (1–525 amino acids), eIF4E, and CBP80 proteins were synthesized via an in vitro translation sys- tem (Promega) using [ 35 S]methionine. The synthesized proteins were added to 20 μL of a 50% (vol/vol) slurry of 7-methyl-GTP- Sepharose (Amersham) and incubated for 1 h at 4 °C. The resin was washed six times with washing buffer [50 mM Hepes (pH 7.0), 40 mM NaCl, 2 mM EDTA, and 0.1% Triton X-100]. The bound proteins were eluted with 2× SDS sample buffer and then electrophoresis was performed using 12% SDS-polyacrylamide gel electrophoresis. Gels were dried and the signals were de- tected using a BAS-2500 phosphorimager. 1. Kim MJ, Huh SU, Ham BK, Paek KH (2008) A novel methyltransferase methylates Cucumber mosaic virus 1a protein and promotes systemic spread. J Virol 82(10):4823–4833. 2. Verwoerd TC, Dekker BM, Hoekema A (1989) A small-scale procedure for the rapid isolation of plant RNAs. Nucleic Acids Res 17(6):2362. 3. Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: Beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6(13):3901–3907. Huh et al. www.pnas.org/cgi/content/short/1214287110 1 of 9
Transcript of Supporting Information - Proceedings of the National … were detected using BAS-2500 phosphorimager...

Supporting InformationHuh et al. 10.1073/pnas.1214287110SI Materials and MethodsELISA. ELISA was performed as previously described (1). Briefly,total protein was extracted from Cucumber mosaic virus (CMV)-inoculated leaves using a disposable grinder in 200 μL of extractionbuffer (0.1 M Tris·HCl with 1% (wt/vol) sodium sulfite, pH 7.4) atthe indicated time points. Forty-five micrograms of each proteinextract was incubated overnight at 4 °C with 50 μL of coating buffer(Sigma-Aldrich) in an ELISA plate (Nunc). The plates were washedwithTBST buffer (0.01MTris·HCl, 0.85%NaCl, and 0.05%Tween-20, pH 7.4) and then blocked for 1 h with 0.1 M TBS buffer (0.1 MTris·HCl and 0.85% NaCl, pH 7.4) containing 1% (wt/vol) BSA.After washing with TBST buffer, a 1:500 dilution in 0.1 M TBSbuffer containing 0.1%BSAof polyclonal anti-CMVwas used as thedetection antibody during incubation for 2 h at room temperature. A1:2,000 dilution in 0.1 M TBS buffer containing 0.1% BSA of alka-line phosphatase goat anti-rabbit IgG (Sigma-Aldrich) was used asa secondary antibody. For color development, p-nitrophenyl phos-phate (Sigma-Aldrich) was used as the substrate, and the reactionwas visualized and quantified by measuring absorbance at wave-length of 405 nm (A405) in an ELISA reader (Bio-Rad).
RNA Blot Analysis. Total RNA was prepared from CMV-infectedplants as described previously (2). Five micrograms of total RNAwere electrophoresed in 1.5% (wt/vol) agarose gel containing6% (vol/vol) formaldehyde in 1× MOPS buffer (pH 7.0) andtransferred to Hybond N membranes (Amersham). Hybridizationswith a 32P-labeled CMV-specific 3′ UTR probe were performedwith PerfectHyb (Sigma-Aldrich) at 65 °C. Membranes werewashed twice at room temperature with 2× SSC containing 0.1%SDS for 5 min, followed by a 20-min wash in 0.5× SSC containing0.1% SDS at 65 °C. Finally, a high-stringency wash was performedin 0.1× SSC containing 0.1% SDS at room temperature for 20 min.Signals were detected using BAS-2500 phosphorimager (Fujifilm).
qRT-PCR Analysis for the Gene Expression upon Salicylic Acid Applicationand CMV Inoculation. Seeds were sown on 1/2 Murashige and Skoogmediumand10-d-oldseedlingsweresprayedwith1mMsalicylicacid(SA). At ∼3.5 wk, Col-0 leaves were dusted with carborundum andgently rubbed with CMV-Kor using brushes as described (1). Thesamples were collected at the indicated time points and RNA waspurified using a modified version of the hot-phenol method (2).qRT-PCR experiments were carried out using the LightCyclerReal-Time PCR Systems (Roche) with KAPA SYBR FAST qPCRMaster Mix (Kapa Biosystems) following the manufacturer’s pro-tocol. The thermal cycling program was 55 °C for 10 min, followedby 40 cycles of 95 °C for 15 s and 60 °C for 1min.Actin7 (At5g09810)was used as the internal reference. The copy number of target geneswas determined by the comparative CT method (Applied Bio-systems). Primers used in these experiments are listed in Table S1.
Histological Assays. For histochemical analysis of β-glucuronidase(GUS) activity, seedlings or plants inoculated with CMV togetherwith control plants, were vacuum-infiltrated with X-Gluc stainingsolution [0.5 M sodium phosphate buffer (pH 7.0), 10% (vol/vol)Triton X-100, 0.1 M K3Fe(CN)6, 0.1 MK4Fe(CN)6, and 2 mM X-Glc A], incubated at 37 °C overnight, and then transferred into70% (vol/vol) ethanol to eliminate chlorophyll (3). GUS activitywas observed using an Axioplan 2 imaging microscope (Carl Zeiss).
GST–APUM5–PHDandGST–APUM6–PHDProteinPurificationandEMSA.Toobtain the PHD region of APUM5 and APUM6, the PHD-con-taining sequence from APUM5 and APUM6 cDNA was PCR-amplified using Pfu DNA polymerase and TA-cloned into the T-easy vector following the manufacturer’s protocol (Promega). Theclone was then digested with BamHI and subcloned into a modifiedpET vector containing the gene coding for the N-terminal GST(GST) tag. Escherichia coli BL21-CodonPlus (DE3)-RIL cells wereused to express the recombinant protein. Cells were grown in LBwith kanamycin (50 μg/mL) to an optical density at 600 nm (OD600)of 0.5. The overexpression of GST-fused APUM5–PHD andAPUM6–PHD was achieved by adding 1 mM isopropyl-β-D-thio-galactoside to the medium and growing the cells at 28 °C for 6 h.Bacteria were pelleted after induction, suspended in lysis buffer[20 mM Tris (pH 8.0), 120 mMNaCl, 10% (vol/vol) glycerol, 0.1%Triton X-100, 5 mMDTT, and 50 μg/mL PMSF], and subjected tosonication on ice for 1 min with a Vibra cell sonifier (Sonics &Materials). Bacterial lysates were then centrifuged and the su-pernatants were incubated with glutathione-linked resin. The resinwas washed with PBS and proteins were eluted from the resin withelution buffer [100 mM Tris (pH 8.0), 120 mM NaCl, 10% glyc-erol, 0.1% Triton X-100, 5 mM DTT, 50 μg/mL PMSF, and20 mM glutathione].For EMSA, synthetic RNAs were radiolabeled with [32P]ATP
(GE Healthcare) using T4 polynucleotide kinase (TaKaRa) andpurified from free nucleotides using Illustra ProbeQuant G-50Micro Columns (GE Healthcare). The indicated concentrationsof purified GST–APUM5–PHD and GST–APUM6–PHD pro-teins were incubated in binding buffer [10 mM Hepes (pH 7.4),1 mM EDTA, 50 mM KCl, 1 mM DTT, 0.01% BSA, and 0.01%Tween-20] with 10 fmol of labeled synthetic RNAs at 28 °C for1 h. Electrophoresis was performed on a 5% (vol/vol) nativeacrylamide gel at 100 V for 50 min at 4 °C. Gels were dried.Signals were detected using a BAS-2500 phosphorimager.
In Vitro Replication Assay. The putative APUM5 binding motif“UGUACUUCUA” in 3′ UTR regions of CMV RNA 1, RNA 2,and RNA 3 in pUC-T7 vector was mutated to “AAAACUUCUA”usingaQuikChangeXLSite-DirectedMutagenesisKit (Stratagene).Wild-type andmutant CMVRNAswere linearized by digestion withMluI and then in vitro transcripts were obtained using T7 mMES-SAGE mMACHINE kit (Ambion). These in vitro transcripts werecotransformed by polyethylene glycol-mediated protoplast trans-formation in Arabidopsis protoplasts prepared from wild-type and35S-APUM5 #1 transgenic plants. After 24 h, RNA samples wereextracted from protoplasts and RNA blot analysis was performed.
Cap-Binding Assay. N-APUM5 (1–525 amino acids), eIF4E, andCBP80 proteins were synthesized via an in vitro translation sys-tem (Promega) using [35S]methionine. The synthesized proteinswere added to 20 μL of a 50% (vol/vol) slurry of 7-methyl-GTP-Sepharose (Amersham) and incubated for 1 h at 4 °C. The resinwas washed six times with washing buffer [50 mM Hepes (pH7.0), 40 mM NaCl, 2 mM EDTA, and 0.1% Triton X-100]. Thebound proteins were eluted with 2× SDS sample buffer and thenelectrophoresis was performed using 12% SDS-polyacrylamidegel electrophoresis. Gels were dried and the signals were de-tected using a BAS-2500 phosphorimager.
1. KimMJ, Huh SU, Ham BK, Paek KH (2008) A novel methyltransferasemethylates Cucumbermosaic virus 1a protein and promotes systemic spread. J Virol 82(10):4823–4833.
2. Verwoerd TC, Dekker BM, Hoekema A (1989) A small-scale procedure for the rapidisolation of plant RNAs. Nucleic Acids Res 17(6):2362.
3. Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: Beta-glucuronidase asa sensitive and versatile gene fusion marker in higher plants. EMBO J 6(13):3901–3907.
Huh et al. www.pnas.org/cgi/content/short/1214287110 1 of 9
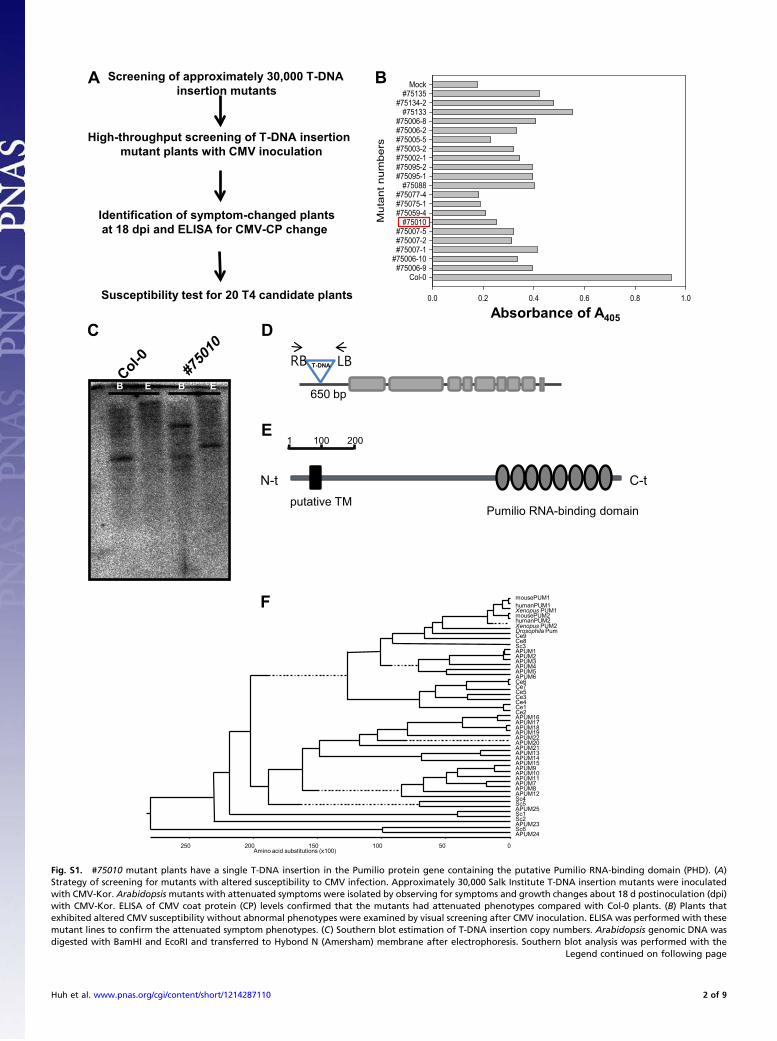
0.0 0.2 0.4 0.6 0.8 1.0
Mut
ant n
umbe
rs
Col-0#75006-9
#75006-10#75007-1#75007-2#75007-5
#75010#75059-4#75075-1#75077-4
#75088#75095-1#75095-2#75002-1#75003-2#75005-5#75006-2#75006-8
#75133#75134-2
#75135Mock
Absorbance of A405
A Screening of approximately 30,000 T-DNA
insertion mutants
High-throughput screening of T-DNA insertion
mutant plants with CMV inoculation
Identification of symptom-changed plants
at 18 dpi and ELISA for CMV-CP change
Susceptibility test for 20 T4 candidate plants
B
B E B E
650 bp
C D
E1 100 200
putative TMPumilio RNA-binding domain
N-t C-t
Amino acid substitutions (x100)050100150200250
mousePUM1humanPUM1Xenopus PUM1mousePUM2humanPUM2Xenopus PUM2Drosophila PumCe9Ce8Sc3APUM1APUM2APUM3APUM4APUM5APUM6Ce6Ce7Ce5Ce3Ce4Ce1Ce2APUM16APUM17APUM18APUM19APUM22APUM20APUM21APUM13APUM14APUM15APUM9APUM10APUM11APUM7APUM8APUM12Sc4Sc5APUM25Sc1Sc2APUM23Sc6APUM24
F
Fig. S1. #75010 mutant plants have a single T-DNA insertion in the Pumilio protein gene containing the putative Pumilio RNA-binding domain (PHD). (A)Strategy of screening for mutants with altered susceptibility to CMV infection. Approximately 30,000 Salk Institute T-DNA insertion mutants were inoculatedwith CMV-Kor. Arabidopsis mutants with attenuated symptoms were isolated by observing for symptoms and growth changes about 18 d postinoculation (dpi)with CMV-Kor. ELISA of CMV coat protein (CP) levels confirmed that the mutants had attenuated phenotypes compared with Col-0 plants. (B) Plants thatexhibited altered CMV susceptibility without abnormal phenotypes were examined by visual screening after CMV inoculation. ELISA was performed with thesemutant lines to confirm the attenuated symptom phenotypes. (C) Southern blot estimation of T-DNA insertion copy numbers. Arabidopsis genomic DNA wasdigested with BamHI and EcoRI and transferred to Hybond N (Amersham) membrane after electrophoresis. Southern blot analysis was performed with the
Legend continued on following page
Huh et al. www.pnas.org/cgi/content/short/1214287110 2 of 9
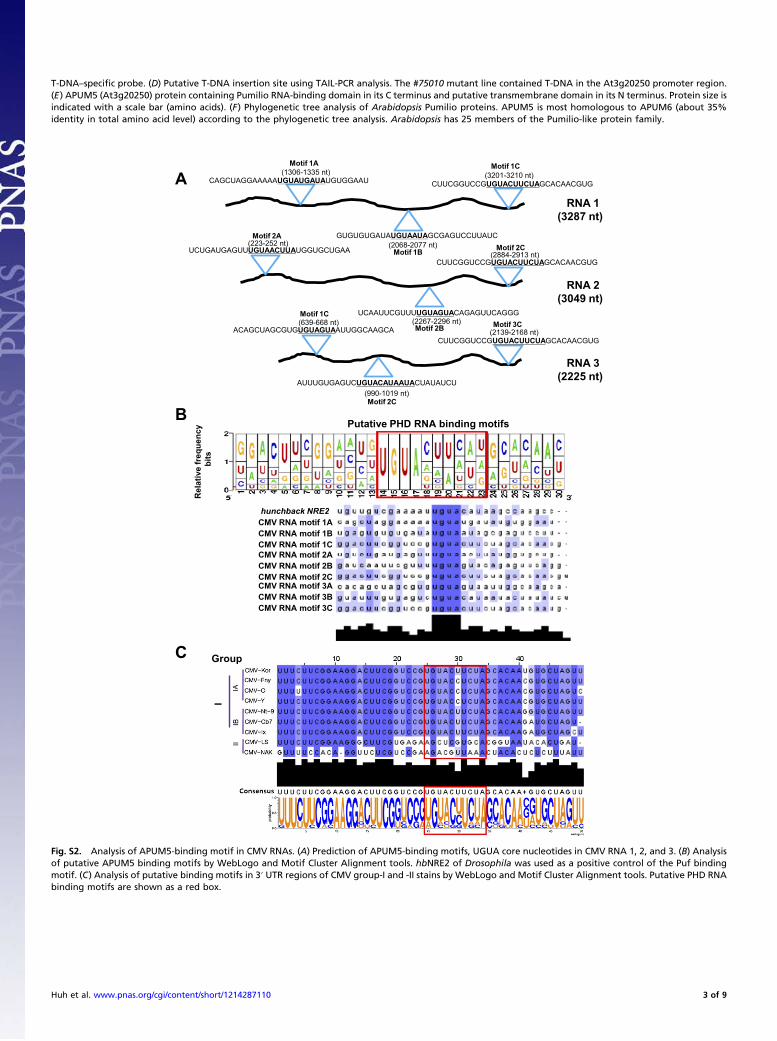
T-DNA–specific probe. (D) Putative T-DNA insertion site using TAIL-PCR analysis. The #75010 mutant line contained T-DNA in the At3g20250 promoter region.(E) APUM5 (At3g20250) protein containing Pumilio RNA-binding domain in its C terminus and putative transmembrane domain in its N terminus. Protein size isindicated with a scale bar (amino acids). (F) Phylogenetic tree analysis of Arabidopsis Pumilio proteins. APUM5 is most homologous to APUM6 (about 35%identity in total amino acid level) according to the phylogenetic tree analysis. Arabidopsis has 25 members of the Pumilio-like protein family.
B
Re
la
tive
fre
qu
en
cy
bits
Putative PHD RNA binding motifs
A CAGCUAGGAAAAAUGUAUGAUAUGUGGAAU
GUGUGUGAUAUGUAAUAGCGAGUCCUUAUC
CUUCGGUCCGUGUACUUCUAGCACAACGUG
UCUGAUGAGUUUGUAACUUAUGGUGCUGAA
UCAAUUCGUUUUGUAGUACAGAGUUCAGGG
ACAGCUAGCGUGUGUAGUAAUUGGCAAGCA
AUUUGUGAGUCUGUACAUAAUACUAUAUCU
RNA 1
(3287 nt)
RNA 2
(3049 nt)
RNA 3
(2225 nt)
CUUCGGUCCGUGUACUUCUAGCACAACGUG
CUUCGGUCCGUGUACUUCUAGCACAACGUG
(1306-1335 nt)
(2068-2077 nt)
(3201-3210 nt)
(223-252 nt)
(2267-2296 nt)
(2884-2913 nt)
(639-668 nt)
(990-1019 nt)
(2139-2168 nt)
Motif 1A
Motif 1B
Motif 1C
Motif 2A
Motif 2B
Motif 2C
Motif 1C
Motif 2C
Motif 3C
C
hunchback NRE2
CMV RNA motif 1A
CMV RNA motif 1B
CMV RNA motif 1C
CMV RNA motif 2A
CMV RNA motif 2B
CMV RNA motif 2C
CMV RNA motif 3A
CMV RNA motif 3B
CMV RNA motif 3C
I
Group
Fig. S2. Analysis of APUM5-binding motif in CMV RNAs. (A) Prediction of APUM5-binding motifs, UGUA core nucleotides in CMV RNA 1, 2, and 3. (B) Analysisof putative APUM5 binding motifs by WebLogo and Motif Cluster Alignment tools. hbNRE2 of Drosophila was used as a positive control of the Puf bindingmotif. (C) Analysis of putative binding motifs in 3′ UTR regions of CMV group-I and -II stains by WebLogo and Motif Cluster Alignment tools. Putative PHD RNAbinding motifs are shown as a red box.
Huh et al. www.pnas.org/cgi/content/short/1214287110 3 of 9
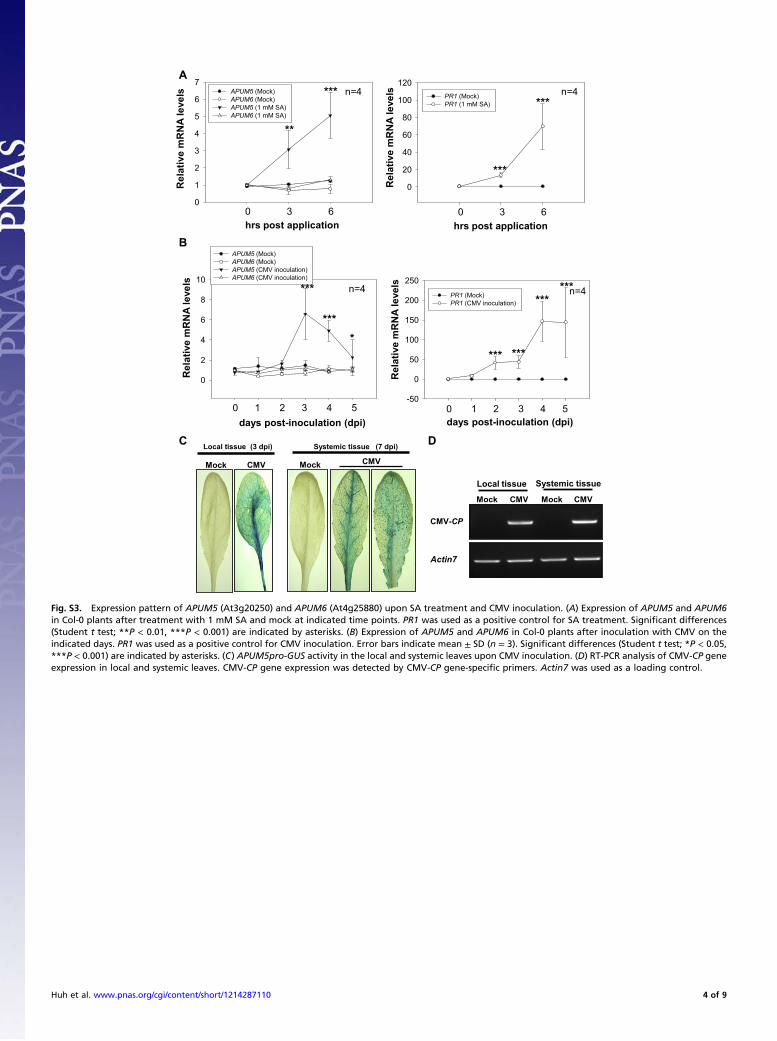
0
2
4
6
8
10
APUM5 (Mock) APUM6 (Mock) APUM5 (CMV inoculation) APUM6 (CMV inoculation)
0 1 2 3 4 5
Re
la
tive
m
RN
A le
ve
ls
-50
0
50
100
150
200
250
PR1 (Mock) PR1 (CMV inoculation)
Re
la
tive
m
RN
A le
ve
ls
0 1 2 3 4 5days post-inoculation (dpi) days post-inoculation (dpi)
n=4 n=4
B
0
1
2
3
4
5
6
7APUM5 (Mock) APUM6 (Mock) APUM5 (1 mM SA) APUM6 (1 mM SA)
Re
la
tive
m
RN
A le
ve
ls
0 3 6hrs post application
0
20
40
60
80
100
120PR1 (Mock) PR1 (1 mM SA)
Re
la
tive
m
RN
A le
ve
ls
0 3 6hrs post application
n=4n=4A
**
***
***
***
***
***
*
*** ***
***
***
Local tissue (3 dpi)
Mock CMV
Systemic tissue (7 dpi)
MockCMV
CMV-CP
Actin7
Local tissue Systemic tissue
Mock CMV Mock CMV
DC
Fig. S3. Expression pattern of APUM5 (At3g20250) and APUM6 (At4g25880) upon SA treatment and CMV inoculation. (A) Expression of APUM5 and APUM6in Col-0 plants after treatment with 1 mM SA and mock at indicated time points. PR1 was used as a positive control for SA treatment. Significant differences(Student t test; **P < 0.01, ***P < 0.001) are indicated by asterisks. (B) Expression of APUM5 and APUM6 in Col-0 plants after inoculation with CMV on theindicated days. PR1 was used as a positive control for CMV inoculation. Error bars indicate mean ± SD (n = 3). Significant differences (Student t test; *P < 0.05,***P < 0.001) are indicated by asterisks. (C) APUM5pro-GUS activity in the local and systemic leaves upon CMV inoculation. (D) RT-PCR analysis of CMV-CP geneexpression in local and systemic leaves. CMV-CP gene expression was detected by CMV-CP gene-specific primers. Actin7 was used as a loading control.
Huh et al. www.pnas.org/cgi/content/short/1214287110 4 of 9
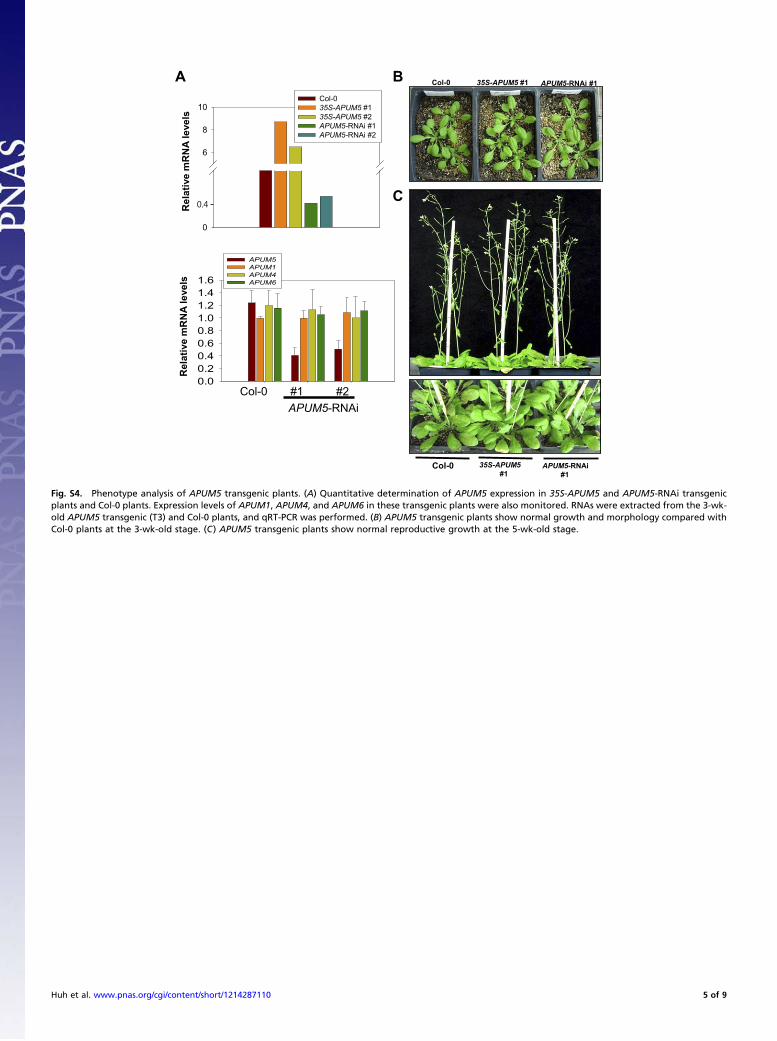
0.00.20.40.60.81.01.21.41.6
APUM5
APUM1
APUM4
APUM6
A
Col-0APUM5-RNAi#1 #2
Re
la
tive
mR
NA
le
ve
ls
Rel
ativ
e m
RN
A le
vels
0
6
8
10Col-0 35S-APUM5 #135S-APUM5 #2APUM5-RNAi #1 APUM5-RNAi #2
Re
la
tive
mR
NA
le
ve
ls
BCol-0 35S-APUM5 #1 APUM5-RNAi #1
C
Col-0 35S-APUM5
#1
APUM5-RNAi
#1
Fig. S4. Phenotype analysis of APUM5 transgenic plants. (A) Quantitative determination of APUM5 expression in 35S-APUM5 and APUM5-RNAi transgenicplants and Col-0 plants. Expression levels of APUM1, APUM4, and APUM6 in these transgenic plants were also monitored. RNAs were extracted from the 3-wk-old APUM5 transgenic (T3) and Col-0 plants, and qRT-PCR was performed. (B) APUM5 transgenic plants show normal growth and morphology compared withCol-0 plants at the 3-wk-old stage. (C) APUM5 transgenic plants show normal reproductive growth at the 5-wk-old stage.
Huh et al. www.pnas.org/cgi/content/short/1214287110 5 of 9
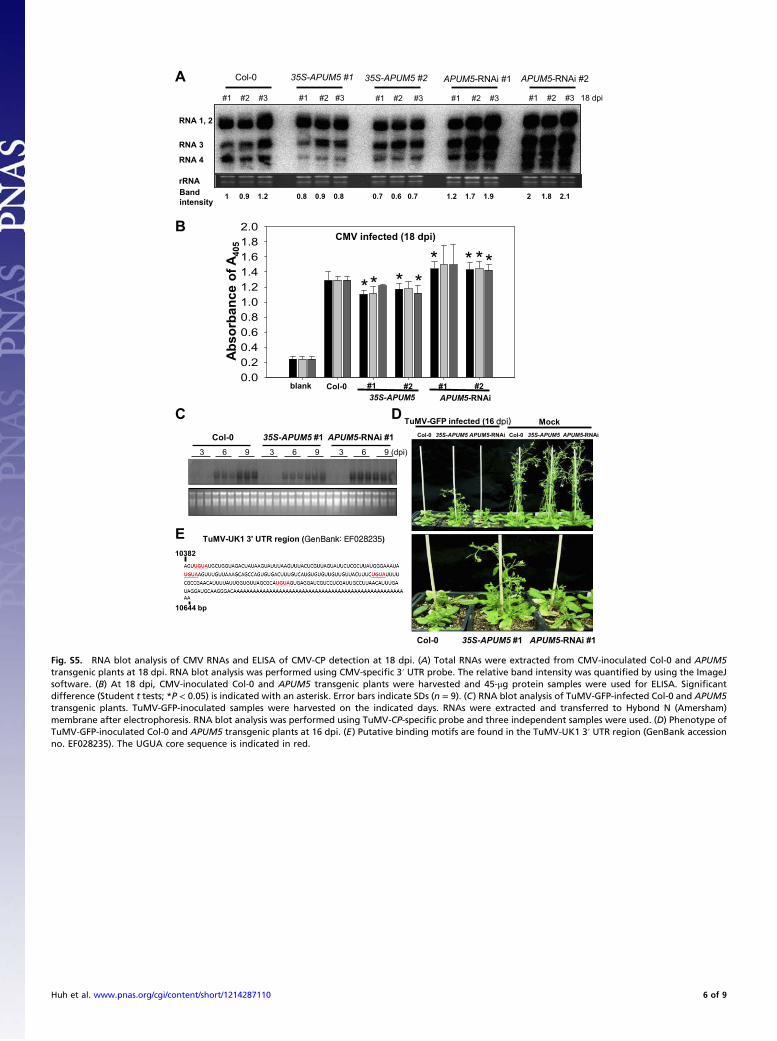
0.00.20.40.60.81.01.21.41.61.82.0
Ab
so
rb
an
ce o
f A
405
Col-0
35S-APUM5 APUM5-RNAi
blank
CMV infected (18 dpi)
** *** **
#1 #2 #1 #2
*
A Col-0 35S-APUM5 #1 APUM5-RNAi #1
#1 #2 #3 #1 #2 #3 #1 #2 #3 #1 #2 #3 #1 #2 #3 18 dpi
rRNA
RNA 1, 2
RNA 3
RNA 4
35S-APUM5 #2 APUM5-RNAi #2
Band
intensity
1 0.9 1.2 0.8 0.9 0.8 0.7 0.6 0.7 1.2 1.7 1.9 2 1.8 2.1
B
CTuMV-GFP infected (16 Mock
Col-0 35S-APUM5 APUM5-RNAi Col-0 35S-APUM5 APUM5-RNAi
Col-0 35S-APUM5 #1 APUM5-RNAi #1
Col-0 35S-APUM5 #1 APUM5-RNAi #1
3 6 9 3 6 9 3 6 9 (dpi)
D
TuMV-UK1 3' UTR regionE
10644 bp
10382
Fig. S5. RNA blot analysis of CMV RNAs and ELISA of CMV-CP detection at 18 dpi. (A) Total RNAs were extracted from CMV-inoculated Col-0 and APUM5transgenic plants at 18 dpi. RNA blot analysis was performed using CMV-specific 3′ UTR probe. The relative band intensity was quantified by using the ImageJsoftware. (B) At 18 dpi, CMV-inoculated Col-0 and APUM5 transgenic plants were harvested and 45-μg protein samples were used for ELISA. Significantdifference (Student t tests; *P < 0.05) is indicated with an asterisk. Error bars indicate SDs (n = 9). (C) RNA blot analysis of TuMV-GFP-infected Col-0 and APUM5transgenic plants. TuMV-GFP-inoculated samples were harvested on the indicated days. RNAs were extracted and transferred to Hybond N (Amersham)membrane after electrophoresis. RNA blot analysis was performed using TuMV-CP-specific probe and three independent samples were used. (D) Phenotype ofTuMV-GFP-inoculated Col-0 and APUM5 transgenic plants at 16 dpi. (E) Putative binding motifs are found in the TuMV-UK1 3′ UTR region (GenBank accessionno. EF028235). The UGUA core sequence is indicated in red.
Huh et al. www.pnas.org/cgi/content/short/1214287110 6 of 9
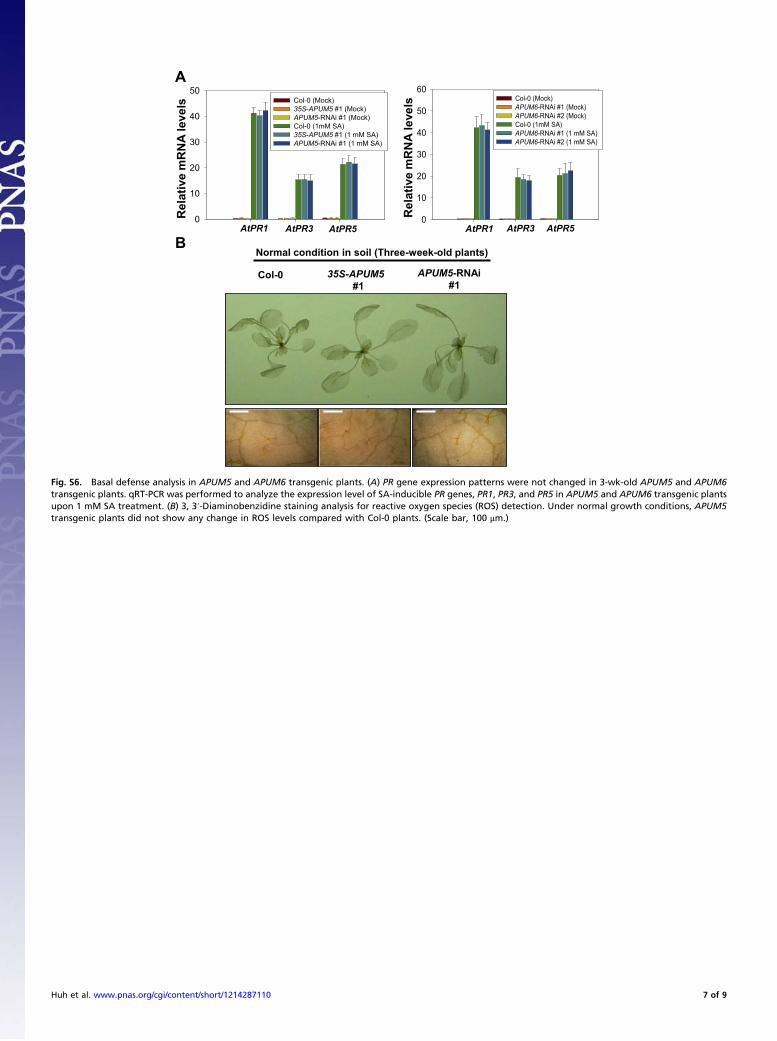
Col-0 35S-APUM5
#1
APUM5-RNAi
#1
Normal condition in soil (Three-week-old plants)
B
0
10
20
30
40
50
60Col-0 (Mock) APUM6-RNAi #1 (Mock) APUM6-RNAi #2 (Mock) Col-0 (1mM SA) APUM6-RNAi #1 (1 mM SA) APUM6-RNAi #2 (1 mM SA)
AtPR1 AtPR3 AtPR5
Re
la
tive
mR
NA
le
vels
0
10
20
30
40
50Col-0 (Mock) 35S-APUM5 #1 (Mock) APUM5-RNAi #1 (Mock) Col-0 (1mM SA) 35S-APUM5 #1 (1 mM SA) APUM5-RNAi #1 (1 mM SA)
AtPR1 AtPR3 AtPR5
Relative
mR
NA
le
vels
A
Fig. S6. Basal defense analysis in APUM5 and APUM6 transgenic plants. (A) PR gene expression patterns were not changed in 3-wk-old APUM5 and APUM6transgenic plants. qRT-PCR was performed to analyze the expression level of SA-inducible PR genes, PR1, PR3, and PR5 in APUM5 and APUM6 transgenic plantsupon 1 mM SA treatment. (B) 3, 3′-Diaminobenzidine staining analysis for reactive oxygen species (ROS) detection. Under normal growth conditions, APUM5transgenic plants did not show any change in ROS levels compared with Col-0 plants. (Scale bar, 100 μm.)
Huh et al. www.pnas.org/cgi/content/short/1214287110 7 of 9
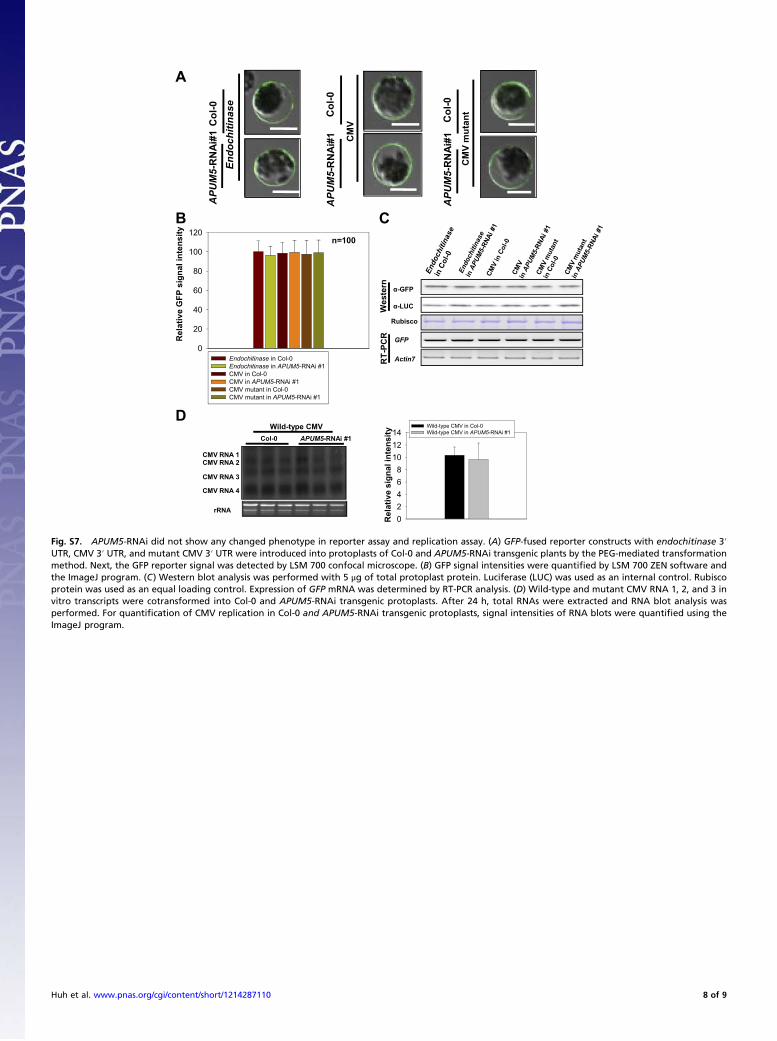
A
0
20
40
60
80
100
120
Endochitinase in Col-0Endochitinase in APUM5-RNAi #1CMV in Col-0CMV in APUM5-RNAi #1CMV mutant in Col-0CMV mutant in APUM5-RNAi #1
Re
la
tiv
e G
FP
sig
na
l in
te
ns
ity
n=100
B
Rubisco
α-GFP
α-LUC
GFP
Actin7
RT
-P
CR
Western
C
Wild-type CMV
Col-0 APUM5-RNAi #1
CMV RNA 1
rRNA
CMV RNA 2
CMV RNA 3
CMV RNA 4
02468
101214
Wild-type CMV in Col-0Wild-type CMV in APUM5-RNAi #1
Relativ
e s
ig
nal in
ten
sity
D
En
do
ch
itin
as
e
Co
l-0
AP
UM
5-R
NA
i#1
CM
V m
utan
t
Co
l-0
AP
UM
5-R
NA
i#1
CM
V
Co
l-0
AP
UM
5-R
NA
i#1
Fig. S7. APUM5-RNAi did not show any changed phenotype in reporter assay and replication assay. (A) GFP-fused reporter constructs with endochitinase 3′UTR, CMV 3′ UTR, and mutant CMV 3′ UTR were introduced into protoplasts of Col-0 and APUM5-RNAi transgenic plants by the PEG-mediated transformationmethod. Next, the GFP reporter signal was detected by LSM 700 confocal microscope. (B) GFP signal intensities were quantified by LSM 700 ZEN software andthe ImageJ program. (C) Western blot analysis was performed with 5 μg of total protoplast protein. Luciferase (LUC) was used as an internal control. Rubiscoprotein was used as an equal loading control. Expression of GFP mRNA was determined by RT-PCR analysis. (D) Wild-type and mutant CMV RNA 1, 2, and 3 invitro transcripts were cotransformed into Col-0 and APUM5-RNAi transgenic protoplasts. After 24 h, total RNAs were extracted and RNA blot analysis wasperformed. For quantification of CMV replication in Col-0 and APUM5-RNAi transgenic protoplasts, signal intensities of RNA blots were quantified using theImageJ program.
Huh et al. www.pnas.org/cgi/content/short/1214287110 8 of 9

Human Pum
Xenopus Pum
Mouse Pum
APUM5
APUM6
Conservation
A
B
C
N-t C-tPumilio-RNA binding domainPutative TM
(kDa)
Input
7-methyl-GTP
Sepharose beads
Elution
Fig. S8. N-terminal region of APUM5 did not bind to m7G Cap structure in vitro. (A) Schematic diagram of N-terminal fragment of APUM5 used for in vitrotranslation. (B) Cap-binding activity motif is conserved in the N-terminal region of mammalian and Xenopus Pufs. Tryptophan is a key amino acid for cap-binding activity (red box). Arabidopsis APUM5 and APUM6 were aligned with mammalian Pufs using the W Cluster program. (C) N-APUM5, eIF4E, and CBP80proteins were synthesized via an in vitro translation system (Promega) using [35S]methionine. The synthesized proteins were incubated with 7-methly-GTP-Sepharose beads (Amersham) for 1 h at 4 °C. The resin was washed with washing buffer and 12% SDS/PAGE was performed. Gels were dried and signals weredetected using a BAS-2500 phosphorimager. N-APUM5 protein size is ∼55 kDa. The eIF4E (∼25 kDa) was used as a positive control and CBP80 (∼80 kDa) wasused as a negative control for the cap-binding assay.
Table S1. Primer pairs used in real-time qRT-PCR and RT-PCR
Gene product Forward primer (5′ to 3′) Reverse primer (5′ to 3′)
APUM5 GGTCAAGTCGCCACTCTTTC CTCCAGGACATGCTGAGTGA
APUM6 CATTGTTCAGCCGAGTGAGA TGAGTCAGCCCACTGTCTTG
AtPR1 CGGAGCTACGCAGAACAACT CGTTCACATAATTCCCACGA
AtPR2 CAGCTACATGGGAGACACGG TTGCGTCGAATAGGTTTTGG
AtPR3 CACTCCCGGTGGTACTCCTC GACAAGCGGCATCATTCCTA
AtPR4 CTCGTGGTCAAGCTTCTTGC ACATCCAAATCCAAGCCTCC
AtPR5 AGTTCCTCCCGTCACTCTGG TCCTCCGGATGGTCTTATCC
AtPDF1.2 AGTTGTGCGAGAAGCCAAGT CACACGATTTAGCACCAAAGA
AtActin7 AATGGTGAAGGCTGGTTTTG TGCCTCTGTGAGTAGAACTG
GFP AAGTCGTGCTGCTTCATGTG ACGTAAACGGCCACAAGTTC
CMV-CP TGTGGGTGACAGTCCGTAAA TCGTCTTTTGCGTACACGAG
NtActin CGGAATCCACGAGACTACATAC GGGAAGCCAAGATAGAGC
CMV RNA1 (+) CCATTTTTGAGCCATCCACT CCTACTCACGGTCGGACATT
CMV RNA2 (+) GGATGTCAGCGAGAGTGTCA CATCGTCTCAAAAGCGACAA
CMV RNA3 (+) TGTGGGTGACAGTCCGTAAA TCGTCTTTTGCGTACACGAG
CMV RNA1 (−) CTGACCAAAATGAAGCAGCA TATGATCGTTACCGCGAACA
CMV RNA2 (−) CATCGTCTCAAAAGCGACAA GGATGTCAGCGAGAGTGTCA
CMV RNA3 (−) TCGTCTTTTGCGTACACGAG TGTGGGTGACAGTCCGTAAA
Huh et al. www.pnas.org/cgi/content/short/1214287110 9 of 9














