
Supporting Information - PNAS on poly-L-lysine–coated glass ... PAGE and transferred to...
9
Supporting Information Bakhti et al. 10.1073/pnas.1220104110 SI Materials and Methods Antibodies, Plasmids, and Other Reagents. Dextran, dextran sulfate, and protamine sulfate were purchased from Sigma-Aldrich. The following primary antibodies were used: mouse anti–α-actin (AC-40), mouse anti-CNPase (2′,3′-Cyclic-nucleotide 3′-phospho- diesterase), and rabbit anti-myc (Sigma-Aldrich); mouse anti-A2B5, mouse anti-MAG (myelin-associated glycoprotein), and mouse anti-MOG (myelin oligodendrocyte glycoprotein) (Millipore); rabbit anti-MBP (myelin basic protein) (DakoCytomation); mouse anti-MBP (Sternberger); mouse anti-GFAP (Santa Cruz Bio- technology); rabbit anti-Iba1 (Wako Chemicals); mouse anti-gal- actocerebroside (O1) and mouse anti-sulfatide (O4) (1); rabbit anti- PLP (proteolipid protein) (P6) (2); and mouse anti-PLP (AA3) (K.-A.N. laboratory); secondary antibodies were purchased from Dianova and Invitrogen. Alexa Fluor 647, Con A, and CellMask Orange were purchased from Invitrogen. The following expression plasmid was used: PLP-myc (3). Cell Culture and Transfection. Primary cultures of mouse oligoden- drocytes from postnatal day 1 were prepared as described previously (4, 5). Neonatal brain hemispheres were stripped free of meninges and then digested and cultured in 10% (vol/vol) horse serum in Eagle’s basal medium in a poly-L-lysine–coated flask at 37 °C. After 8–10 d, oligodendrocyte cells were harvested from mixed glia culture using mechanical dissociation. Isolated cells were then cultured in DMEM containing B27 supplement, 1% horse serum, triiodothy- ronine, L-thyroxine, pyruvate, glutamine, and penicillin/strepto- mycin on poly-L-lysine–coated glass coverslips or dishes. Microglia and astrocytes were isolated from the same mixed glia culture and used for oligodendrocyte purification. After 10 d of culture and removal of oligodendrocytes, the remaining cells were trypsinized and plated on poly-L-lysine–coated glass coverslips in DMEM containing 10% FCS, glutamine, and penicillin/strepto- mycin. After 3 d, the cells were used for further analysis. The potoroo kidney (PtK2) cell line was cultured as previously described (6). The cells were seeded on glass coverslips and cul- tured at 37 °C with 5% CO 2 in DMEM with 10% FCS. Transient transfection of PtK2 cells was performed using TransIT-LT1 (Mirus) transfection reagent according to the manufacturer’s protocol. Myelin Purification. The preparation of myelin membrane was performed as described previously (7). The brains from 2- to 6-mo- old mice were homogenized in 0.32 M sucrose solution (containing 5 mM EDTA, 10 mM Hepes, pH 7.4, and protease inhibitors) by tip sonication. The homogenates were applied to a two-step su- crose gradient (0.32 and 0.85 M sucrose in Hepes-EDTA buffer). The gradients were centrifuged at 74,000 × g for 30 min, and the interfaces (crude myelin) were collected, diluted with H 2 O, and centrifuged at 74,000 × g for 15 min. The pellet fraction was washed twice with ice-cold H 2 O and recovered by a 10-min centrifugation at 12,500 × g, and the procedure was repeated further to obtain purified myelin. The pure myelin fraction was resuspended in Hepes-EDTA buffer and stored at -20 °C for further analysis. Myelin Particle Preparation. Myelin particles were purified using a ConA Glycoprotein Isolation Kit (Thermo Scientific) according to the manufacturer’s instructions. Briefly, a fraction of myelin mem- brane was washed and resuspended with H 2 O. After being passed through a 27-gauge needle and sonication for 3 min, the myelin membrane was then applied to Con A beads for 30 min at room temperature (RT). The flow-through was collected using centrifu- gation at 1,000 × g for 1 min. To remove MBP, the particles were resuspended in a buffer solution that was composed of two volumes of 10 mM Tris buffer (pH 7.4) and one volume of 450 mM Tris buffer (pH 7.4), 6 mM CaCl 2 , and 6 mM MgCl 2 for 3 h at 37 °C with gentle shaking (8, 9). The final pellet was resuspended in PBS and kept at 4 °C for further analysis. Myelin Particle Labeling and Binding Assay. Purified myelin particles were fluorescently labeled using two lipophilic dyes (PKH26, red; PKH67, green). Briefly, 100 μL of myelin particles was mixed with 900 μL of diluent C (Sigma-Aldrich) and 1 μL of PKH dyes. The mixture was vigorously vortexed and incubated for 5 min at RT. Two hundred microliters of 100% FCS was added to the samples to stop the reaction, and the mixture was centrifuged at 12,000 × g with a 5417R microcentrifuge (Eppendorf) for 10 min followed by two washes with PBS. The final pellet was resuspended in the cell medium and kept at 4 °C. For the binding assay, 100 μL of labeled particles was added on top of the cells and cultured on coverslips. The binding procedure was carried out for 30 min at 37 °C and the cells were directly fixed and processed for immunolabeling. Using confocal imaging, the number of myelin particles per cell area was calculated (20 confocal images were quantified per condition in each independent experiment). Lectin Staining. All lectins were obtained from Vector Laboratories. Fixed cells were incubated with 5–10 μg/mL lectins for 10 min at RT and then mounted on glass slides. For MAL II (Maackia amurensis lectin) staining, cells were incubated with 10 μg/mL biotinylated MAL II for 30 min at RT, followed by washing with PBS and in- cubating with fluorescent streptavidin for 10 min before mounting. Free-floating brain sections were incubated with 15 μg/mL MAL II at 4 °C overnight. The samples were then stained by fluorescent streptavidin for 5 h at RT and mounted. For wheat germ agglutinin (WGA) staining, sections were incubated with 10 μg/mL fluores- cent lectin for 5 h at RT before mounting. The brain slices were also costained with antibody against MOG to label myelinated areas. Neuraminidase Treatment. Neuraminidase (sialidase) from Clos- tridium perfringens (Sigma-Aldrich) was dissolved in PBS (pH 6.5) containing 0.05% BSA (10). Enzyme (5 units/mL) was added to cells for 1 h at 37 °C before performing the particle binding assay. The efficiency of sialic acid removal by sialidase was assessed using WGA lectin staining. Metabolic Labeling and Click Chemistry Detection. Tetraacetylated azido-mannosamine (Ac4ManNAz) (Invitrogen) was used to mea- sure the synthesis of sialic acid (11). Cells were incubated with 50 μM Ac4ManNAz for 24 h at 37 °C. The azido-mannosamine was met- abolically incorporated into glycoproteins and glycolipids through the permissive feature of oligosaccharide synthesis. The metabolic incorporation of modified sugar into sialic acid biosynthesis was detected using a fluorophore-conjugated alkyne that specifically interacts with the azido-sugar. The amount of sialic acid in- corporation into cells was analyzed using ImageJ software (National Institutes of Health). Exogenous Ganglioside Incorporation. A stock solution of total brain gangliosides was prepared in PBS containing 7 mM defatted BSA. Twelve microliters of solution was added to 1 mL of culture medium to obtain 80 μM gangliosides and BSA. After incubation for 1 h at 37 °C, cells were washed extensively with PBS containing 2 mg/mL fatty acid-free BSA to remove excess lipids (12, 13). The cells were immediately used for the myelin particle binding assay. Bakhti et al. www.pnas.org/cgi/content/short/1220104110 1 of 9
Transcript of Supporting Information - PNAS on poly-L-lysine–coated glass ... PAGE and transferred to...

Supporting InformationBakhti et al. 10.1073/pnas.1220104110SI Materials and MethodsAntibodies, Plasmids, and Other Reagents.Dextran, dextran sulfate,and protamine sulfate were purchased from Sigma-Aldrich. Thefollowing primary antibodies were used: mouse anti–α-actin(AC-40), mouse anti-CNPase (2′,3′-Cyclic-nucleotide 3′-phospho-diesterase), and rabbit anti-myc (Sigma-Aldrich);mouse anti-A2B5,mouse anti-MAG (myelin-associated glycoprotein), and mouseanti-MOG (myelin oligodendrocyte glycoprotein) (Millipore);rabbit anti-MBP (myelin basic protein) (DakoCytomation); mouseanti-MBP (Sternberger); mouse anti-GFAP (Santa Cruz Bio-technology); rabbit anti-Iba1 (Wako Chemicals); mouse anti-gal-actocerebroside (O1) andmouse anti-sulfatide (O4) (1); rabbit anti-PLP (proteolipid protein) (P6) (2); and mouse anti-PLP (AA3)(K.-A.N. laboratory); secondary antibodies were purchased fromDianova and Invitrogen. Alexa Fluor 647, Con A, and CellMaskOrange were purchased from Invitrogen. The following expressionplasmid was used: PLP-myc (3).
Cell Culture and Transfection. Primary cultures of mouse oligoden-drocytes from postnatal day 1 were prepared as described previously(4, 5). Neonatal brain hemispheres were stripped free of meningesand then digested and cultured in 10% (vol/vol) horse serum inEagle’s basal medium in a poly-L-lysine–coated flask at 37 °C. After8–10 d, oligodendrocyte cells were harvested frommixed glia cultureusing mechanical dissociation. Isolated cells were then cultured inDMEM containing B27 supplement, 1% horse serum, triiodothy-ronine, L-thyroxine, pyruvate, glutamine, and penicillin/strepto-mycin on poly-L-lysine–coated glass coverslips or dishes.Microglia and astrocytes were isolated from the samemixed glia
culture and used for oligodendrocyte purification. After 10 d ofculture and removal of oligodendrocytes, the remaining cells weretrypsinized and plated on poly-L-lysine–coated glass coverslips inDMEM containing 10% FCS, glutamine, and penicillin/strepto-mycin. After 3 d, the cells were used for further analysis.The potoroo kidney (PtK2) cell line was cultured as previously
described (6). The cells were seeded on glass coverslips and cul-tured at 37 °C with 5% CO2 in DMEM with 10% FCS. Transienttransfection of PtK2 cells was performed using TransIT-LT1(Mirus) transfection reagent according to the manufacturer’sprotocol.
Myelin Purification. The preparation of myelin membrane wasperformed as described previously (7). The brains from 2- to 6-mo-oldmice were homogenized in 0.32M sucrose solution (containing5 mM EDTA, 10 mM Hepes, pH 7.4, and protease inhibitors) bytip sonication. The homogenates were applied to a two-step su-crose gradient (0.32 and 0.85 M sucrose in Hepes-EDTA buffer).The gradients were centrifuged at 74,000 × g for 30 min, and theinterfaces (crude myelin) were collected, diluted with H2O, andcentrifuged at 74,000× g for 15min. The pellet fractionwaswashedtwice with ice-cold H2O and recovered by a 10-min centrifugationat 12,500 × g, and the procedure was repeated further to obtainpurified myelin. The pure myelin fraction was resuspended inHepes-EDTA buffer and stored at −20 °C for further analysis.
Myelin Particle Preparation. Myelin particles were purified usingaConAGlycoprotein IsolationKit (ThermoScientific) according tothe manufacturer’s instructions. Briefly, a fraction of myelin mem-brane was washed and resuspended with H2O. After being passedthrough a 27-gauge needle and sonication for 3 min, the myelinmembrane was then applied to Con A beads for 30 min at roomtemperature (RT). The flow-through was collected using centrifu-
gation at 1,000 × g for 1 min. To remove MBP, the particles wereresuspended in a buffer solution that was composed of two volumesof 10 mM Tris buffer (pH 7.4) and one volume of 450 mM Trisbuffer (pH 7.4), 6 mMCaCl2, and 6mMMgCl2 for 3 h at 37 °C withgentle shaking (8, 9). The final pellet was resuspended in PBS andkept at 4 °C for further analysis.
Myelin Particle Labeling and Binding Assay. Purified myelin particleswere fluorescently labeled using two lipophilic dyes (PKH26, red;PKH67, green). Briefly, 100 μL of myelin particles was mixed with900 μL of diluent C (Sigma-Aldrich) and 1 μL of PKH dyes. Themixture was vigorously vortexed and incubated for 5 min at RT.Two hundredmicroliters of 100%FCSwas added to the samples tostop the reaction, and the mixture was centrifuged at 12,000 × gwith a 5417R microcentrifuge (Eppendorf) for 10 min followed bytwo washes with PBS. The final pellet was resuspended in the cellmedium and kept at 4 °C. For the binding assay, 100 μL of labeledparticles was added on top of the cells and cultured on coverslips.The binding procedure was carried out for 30 min at 37 °C and thecells were directly fixed and processed for immunolabeling. Usingconfocal imaging, the number of myelin particles per cell area wascalculated (20 confocal images were quantified per condition ineach independent experiment).
Lectin Staining.All lectins were obtained fromVector Laboratories.Fixed cellswere incubatedwith 5–10μg/mL lectins for 10min atRTand thenmounted on glass slides. ForMAL II (Maackia amurensislectin) staining, cells were incubated with 10 μg/mL biotinylatedMAL II for 30 min at RT, followed by washing with PBS and in-cubating with fluorescent streptavidin for 10 min before mounting.Free-floating brain sections were incubated with 15 μg/mLMAL
II at 4 °C overnight. The samples were then stained by fluorescentstreptavidin for 5 h at RT andmounted. For wheat germ agglutinin(WGA) staining, sections were incubated with 10 μg/mL fluores-cent lectin for 5 h atRTbeforemounting. The brain sliceswere alsocostained with antibody against MOG to label myelinated areas.
Neuraminidase Treatment. Neuraminidase (sialidase) from Clos-tridium perfringens (Sigma-Aldrich) was dissolved in PBS (pH 6.5)containing 0.05%BSA (10). Enzyme (5 units/mL)was added to cellsfor 1 h at 37 °C before performing the particle binding assay. Theefficiency of sialic acid removal by sialidasewas assessed usingWGAlectin staining.
Metabolic Labeling and Click Chemistry Detection. Tetraacetylatedazido-mannosamine (Ac4ManNAz) (Invitrogen) was used to mea-sure the synthesis of sialic acid (11).Cellswere incubatedwith 50 μMAc4ManNAz for 24 h at 37 °C. The azido-mannosamine was met-abolically incorporated into glycoproteins and glycolipids throughthe permissive feature of oligosaccharide synthesis. The metabolicincorporation of modified sugar into sialic acid biosynthesis wasdetected using a fluorophore-conjugated alkyne that specificallyinteracts with the azido-sugar. The amount of sialic acid in-corporation into cells was analyzed using ImageJ software (NationalInstitutes of Health).
Exogenous Ganglioside Incorporation.A stock solution of total braingangliosides was prepared in PBS containing 7 mM defatted BSA.Twelvemicrolitersof solutionwasadded to1mLof culturemediumto obtain 80 μM gangliosides and BSA. After incubation for 1 h at37 °C, cells were washed extensively with PBS containing 2 mg/mLfatty acid-free BSA to remove excess lipids (12, 13). The cells wereimmediately used for the myelin particle binding assay.
Bakhti et al. www.pnas.org/cgi/content/short/1220104110 1 of 9

Lipid Extraction and Liposome Preparation and Aggregation. Lipidsfrom myelin membrane were extracted using the chloroform-methanol approach (14). Briefly, 350 μL of myelin was added to1,050 μL of chloroform:methanol (ratio 2:1). The mixture wasthen vortexed vigorously and centrifuged at 3,800 × g witha 5417R microcentrifuge (Eppendorf) for 5 min at RT. Theorganic phase (lower fraction) was recovered and added to 70 μLof methanol and 475 μL of 50 mM NaCl followed by mixing andcentrifuging at 3,800 × g for 5 min. The organic phase containinglipids was collected and subjected to speed vacuum for 45 min at37 °C to obtain a dry lipid fraction.Liposomes from myelin lipids with or without exogenous lipids
were prepared as described previously (15, 16). Lipids (includingproteolipid proteins) extracted from myelin membrane were dis-solved in 2-chloroethanol to obtain the total concentration of 1 mg/mL lipid/protein. The lipid/protein suspensions were briefly centri-fuged and dialyzed at RT for 6 h against 2 L of reconstitution buffer(5 mMHepes, 1 mMEDTA, and 10 mMNaCl, pH 5.0) using a 10-kDa molecular-weight cutoff Slide-A-Lyzer Mini dialysis device(Pierce/Thermo Scientific). After changing the dialysis buffer threetimes, the samples were collected and analyzed. To measure lipo-some aggregation, the optical density of samples was monitored at405 nm using a Biophotometer Plus (Eppendorf) (17).
Immunoblot Analysis. Samplesweremixedwith lysis buffer (100mMTris, pH 7.5, 300 mM NaCl, 2 mM EDTA, and 2% Triton X-100supplemented with Complete protease inhibitor mixtures) andincubated for 10 min on ice. After centrifugation at 10,000 × g for10 min, the protein concentration was measured using a Bradfordassay. A fraction of the supernatant was then mixed with samplebuffer (20% glycerol, 4 mM EDTA, 4% SDS, 4% 2-mercaptoe-thanol, and 100 mMTris·HCl, pH 6.8) and subjected to 12% SDS/PAGE and transferred to nitrocellulose membranes using stan-dard protocols. Western blots were revealed by enhanced chem-iluminescence (Pierce/Thermo Scientific).To immunolabel lipids in myelin liposomes, a dot-blot assay
was performed. Eight microliters of liposomes was applied anddried onto a nitrocellulose membrane. The membrane was thenincubated in blocking solution (5% skim milk powder in PBS) for30 min at RT and incubated with primary antibody in PBS at 4 °Covernight. After being washed with PBS, the membrane wasincubated with the appropriate HRP-secondary antibodies for40 min at RT and the corresponded lipids were detected byenhanced chemiluminescence.
Immunostaining and Confocal Microscopy. For immunofluorescence,cells were fixed with 4% paraformaldehyde (PFA) for 15 min andpermeabilized with 0.1% Triton X-100 in PBS. Fixed cells werethen incubated with blocking solution (2%BSA, 0.2% fish gelatin,and 2% FCS in PBS) for 30 min at room temperature. Next, cellswere incubated with primary antibodies diluted in blocking solu-tion, washed with PBS, and then incubated with secondary anti-bodies and mounted. The morphological analysis of fixed cellsamples was performed using laser scanning confocal microscopy.Fluorescent images were acquired with a Leica DMIRE2 micro-scope and a Leica TCS SP2 AOBS confocal laser scanning setup.
Atomic Force Microscopy. Gold-coated cantilevers (Arrow TL2Au;NanoWorld) were used for single-particle force spectroscopymeasurements and calibrated before each measurement using thethermal noise method (18). The cantilevers were washed twice inisopropanol and ultrapure water for 1 min each. Thereafter, thecantilevers were cleaned using a plasma cleaner for 1 min. Forbinding of the myelin particles, the cantilevers were incubated in a10 μg/mL poly-D-lysine solution for 10 min and washed in ultrapure
water afterward. Measurements were performed using a CellHesion200 setup (JPK Instruments) combined with an Olympus IX81microscope and a Petri dish heater, assuring a constant tempera-ture of 37 °C throughout the measurement. A contact time of 30 sand a force of 500 pN were used to bind the particles to the poly-D-lysine–coated cantilever. For investigation of myelin–oligoden-drocyte interaction, the time of particle–cell contact (5 s), contactforce (500 pN), and velocity of cantilever retraction (3 μm/s) werekept constant throughout the measurements. To quantify the in-teraction between myelin particles and oligodendrocytes, the maxi-mumadhesion force (minimum in force during cantilever retraction)from the recorded force retraction curves was analyzed. To evaluatethe statistics of the obtained data, a nonparametric statisticalhypothesis test, namely the Wilcoxon rank-sum test, was used.
Electron Microscopy. Sample preparation for electron microscopyanalysis was performed as described previously (19). Briefly, thedissected nerves were chemically fixed by immersion fixation in 4%PFAand2.5%glutaraldehydefor4h.Thesampleswerethenwashedwith 0.1 M phosphate buffer and postfixed with 2% OsO4 for 4 hat 4 °C. They were then dehydrated in a graded series of ethanolfollowed by propylene oxide and were infiltrated with Epon-propylene oxide mixtures and pure Epon. The nerves were fi-nally transferred into embedding molds and polymerized for24 h at 60 °C.The high-pressure freezing of freshly extracted optic nerves was
performed with an HPM100 system (Leica). The samples wereplacedina2-mminner-diameterspecimencarrierwith0.2-mmdepth(Microscopy Services) surrounded by 20% polyvinylpyrrolidone inPBStoexcludeairaroundthesamplesandthenfrozenat2,000barfora few milliseconds.Freeze substitution was carried out for high-pressure frozen
samples using a Leica AFS2 unit. First, the samples were placedin tannic acid (0.1% in acetone) for 100 h at −90 °C, and thenwashed with acetone at −90 °C and subsequently incubated witha new substitution solution containing OsO4 (2% OsO4 in ace-tone, −90 °C) and 0.1% uranyl acetate. Afterward, the temper-ature was increased from −90 °C to −20 °C in increments of 5 °C/h,and the samples were kept for 16 h at −20 °C. The temperature wasraised further from −20 °C to 4 °C in increments of 10 °C/h. Thesamples were next washed with acetone at 4 °C, allowed to adjustto room temperature for 1 h, and then transferred into Epon (50%Epon in acetone for 3 h, 90%Epon in acetone overnight, and 100%Epon for 6 h). Finally, the samples were placed in embeddingmoldsand polymerized for 24 h at 60 °C.Ultrathin sections for imaging were prepared using a Leica
Ultracut S ultramicrotome set and sections 50-nm-thick were cut.The sections were incubated in 2% aqueous uranyl acetate for 30minforcontrastingandthenwashedinwater followedbyleadcitratefor 5 min and again washed in water and dried. The imaging wasperformed using a LEOEM912ABelectronmicroscope (Zeiss) at80 kV, and images were recorded with a 2,048 × 2,048-pixel CCDcamera (Proscan) (19).To assess myelin lamella splitting, areas of 76 μm2 in five ran-
domly selected areas throughout the optic nerves of littermatemice were analyzed. All axons within these areas were quantified(100 axons per animal). A theoretical cross was set along the longaxis of the axons and was centered at the middle of the axons toobtain four intersections (opposing two by two) of myelin sheaths.The distance between lamellae was measured by dividing the totalmyelin thickness by the number of myelin lamellae. An average offour measurements was calculated for each axon.Negative-staining electron microscopy was performed as de-
scribed (3).
1. Sommer I, Schachner M (1981) Monoclonal antibodies (O1 to O4) to oligodendrocyte cellsurfaces: An immunocytological study in the central nervous system.DevBiol 83(2):311–327.
2. Linington C, Waehneldt TV (1990) Conservation of the carboxyl terminal epitope of myelinproteolipid protein in the tetrapods and lobe-finned fish. J Neurochem 54(4):1354–1359.
Bakhti et al. www.pnas.org/cgi/content/short/1220104110 2 of 9

3. Trajkovic K, et al. (2008) Ceramide triggers budding of exosome vesicles intomultivesicular endosomes. Science 319(5867):1244–1247.
4. Trajkovic K, et al. (2006) Neuron to glia signaling triggers myelin membraneexocytosis from endosomal storage sites. J Cell Biol 172(6):937–948.
5. Bakhti M, Winter C, Simons M (2011) Inhibition of myelin membrane sheath formationby oligodendrocyte-derived exosome-like vesicles. J Biol Chem 286(1):787–796.
6. Eggeling C, et al. (2009) Direct observation of the nanoscale dynamics of membranelipids in a living cell. Nature 457(7233):1159–1162.
7. Larocca JN, Norton WT (2007) Isolation of myelin. Curr Protoc Cell Biol 33:3.25.1–3.25.19.
8. Glynn P, Chantry A, Groome N, Cuzner ML (1987) Basic protein dissociating frommyelin membranes at physiological ionic strength and pH is cleaved into three majorfragments. J Neurochem 48(3):752–759.
9. Earl C, Chantry A, Mohammad N, Glynn P (1988) Zinc ions stabilise the association ofbasic protein with brain myelin membranes. J Neurochem 51(3):718–724.
10. Finkelstein EI, Chao PH, Hung CT, Bulinski JC (2007) Electric field-induced polarizationof charged cell surface proteins does not determine the direction of galvanotaxis. CellMotil Cytoskeleton 64(11):833–846.
11. Schwetz TA, Norring SA, Ednie AR, Bennett ES (2011) Sialic acids attached to O-glycansmodulate voltage-gated potassium channel gating. J Biol Chem 286(6):4123–4132.
12. Schwarzmann G, Hofmann P, Pütz U, Albrecht B (1995) Demonstration of directglycosylation of nondegradable glucosylceramide analogs in cultured cells. J BiolChem 270(36):21271–21276.
13. Simons M, et al. (1999) Exogenous administration of gangliosides displaces GPI-anchored proteins from lipid microdomains in living cells. Mol Biol Cell 10(10):3187–3196.
14. Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification.Can J Biochem Physiol 37(8):911–917.
15. Boggs JM, Vail WJ, Moscarello MA (1976) Preparation and properties of vesicles ofa purified myelin hydrophobic protein and phospholipid. A spin label study. BiochimBiophys Acta 448(4):517–530.
16. ter Beest MB, Hoekstra K, Sein A, Hoekstra D (1994) Reconstitution of proteolipidprotein: Some properties and its role in interlamellar attachment. Biochem J 300(Pt 2):545–552.
17. Johnson CP, Chapman ER (2010) Otoferlin is a calcium sensor that directly regulatesSNARE-mediated membrane fusion. J Cell Biol 191(1):187–197.
18. Hutter JL, Bechhoefer J (1993) Calibration of atomic-force microscope tips. Rev SciInstrum 64(7):1868–1873.
19. Möbius W, et al. (2010) Electron microscopy of the mouse central nervous system.Methods Cell Biol 96:475–512.
Bakhti et al. www.pnas.org/cgi/content/short/1220104110 3 of 9

Fig. S1. Preparation and characterization of myelin particles and binding assay. (A and B) Schemes of CNS myelin (A) and particle structure (B) and the in-teracting oligodendrocyte are shown. (C) The procedure of preparation and labeling of myelin particles is presented. First, we remove the glycoprotein-richfraction, which represents membrane enriched in the paranodal and ad- and abaxonal domains of myelin. The flow-through was then incubated with a buffercontaining 2 mM calcium to wash MBP from the surface of the particles. (D) Each fraction was then analyzed for different proteins using Western blotting. (E)Control myelin and purified particles were immunolabeled with antibodies against PLP and CNPase and stained with the lipophilic dye PKH26 as the reference.(F) Myelin particles were added to the culture of different glia including astrocytes, microglia, and undifferentiated (A2B5-positive) and differentiated (MBP-positive) oligodendrocytes. Whereas particles attached to differentiated cells, little binding was observed when using undifferentiated cells. Few particlesattached to astrocytes (GFAP-positive). Particles were internalized by microglia (Iba1-positive) (arrowheads). (G) Myelin particles were added to fully differ-entiated oligodendrocytes and confocal microscopy was used to determine whether the particles attached to the surface or were internalized. Image analysisrevealed the localization of myelin particles to the surface of oligodendrocytes; green, MBP; red, myelin particle. (Scale bars, 20 μm.)
Bakhti et al. www.pnas.org/cgi/content/short/1220104110 4 of 9
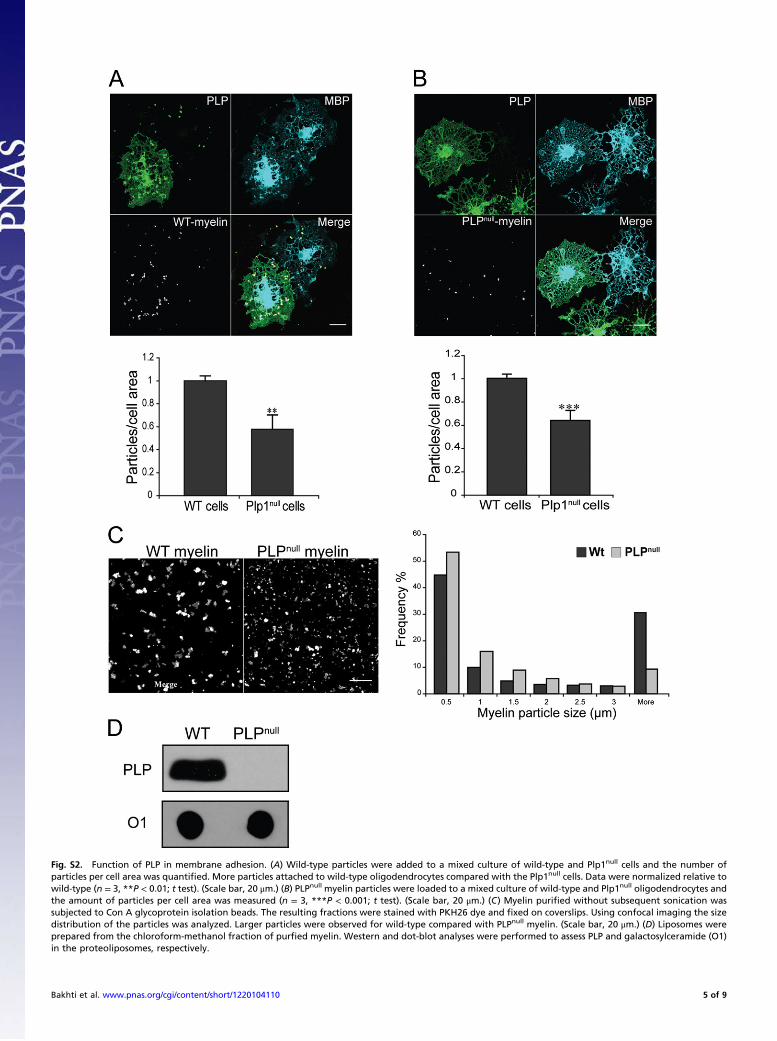
Fig. S2. Function of PLP in membrane adhesion. (A) Wild-type particles were added to a mixed culture of wild-type and Plp1null cells and the number ofparticles per cell area was quantified. More particles attached to wild-type oligodendrocytes compared with the Plp1null cells. Data were normalized relative towild-type (n = 3, **P < 0.01; t test). (Scale bar, 20 μm.) (B) PLPnull myelin particles were loaded to a mixed culture of wild-type and Plp1null oligodendrocytes andthe amount of particles per cell area was measured (n = 3, ***P < 0.001; t test). (Scale bar, 20 μm.) (C) Myelin purified without subsequent sonication wassubjected to Con A glycoprotein isolation beads. The resulting fractions were stained with PKH26 dye and fixed on coverslips. Using confocal imaging the sizedistribution of the particles was analyzed. Larger particles were observed for wild-type compared with PLPnull myelin. (Scale bar, 20 μm.) (D) Liposomes wereprepared from the chloroform-methanol fraction of purfied myelin. Western and dot-blot analyses were performed to assess PLP and galactosylceramide (O1)in the proteoliposomes, respectively.
Bakhti et al. www.pnas.org/cgi/content/short/1220104110 5 of 9
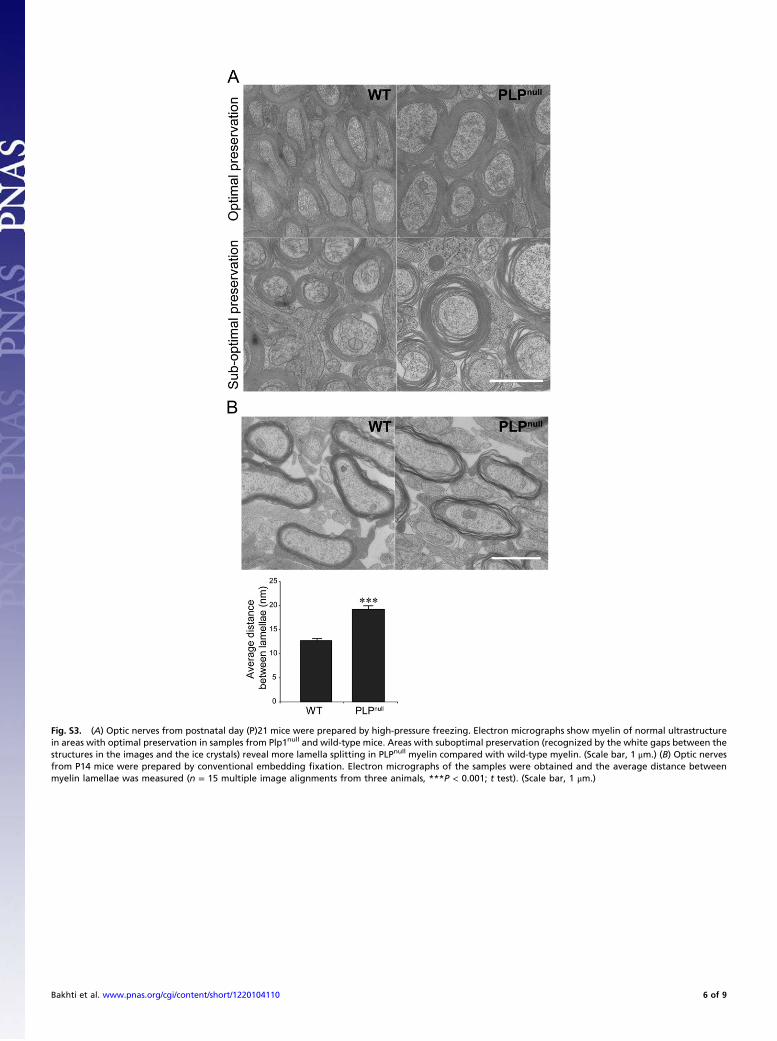
Fig. S3. (A) Optic nerves from postnatal day (P)21 mice were prepared by high-pressure freezing. Electron micrographs show myelin of normal ultrastructurein areas with optimal preservation in samples from Plp1null and wild-type mice. Areas with suboptimal preservation (recognized by the white gaps between thestructures in the images and the ice crystals) reveal more lamella splitting in PLPnull myelin compared with wild-type myelin. (Scale bar, 1 μm.) (B) Optic nervesfrom P14 mice were prepared by conventional embedding fixation. Electron micrographs of the samples were obtained and the average distance betweenmyelin lamellae was measured (n = 15 multiple image alignments from three animals, ***P < 0.001; t test). (Scale bar, 1 μm.)
Bakhti et al. www.pnas.org/cgi/content/short/1220104110 6 of 9

Fig. S4. Down-regulation of glycocalyx components during oligodendrocyte maturation. (A) Oligodendroglial cells at day in culture (DIV) 2 and 5 were stainedusing different lectins. Con A, peanut agglutinin (PNA), and WGA staining revealed a reduction in cell-surface carbohydrates upon maturation of the cells.(Scale bar, 20 μm.) (B) Microglia, astrocytes, and oligodendrocytes at DIV 2 and 5 after plating were labeled using the lectin MAL II, which specifically binds tosialic acid residues. A significant reduction in cell-surface sialic acids was observed in oligodendrocytes at DIV 5 compared with cells at DIV 2 and with astrocytesand microglia. (Scale bar, 20 μm.) (C) WGA and MAL II staining of brain sections from 2-mo-old wild-type mice revealed a significant reduction in lectin bindingin myelinated areas, which are shown by an antibody directed against MOG. (Scale bar, 200 μm.)
Bakhti et al. www.pnas.org/cgi/content/short/1220104110 7 of 9
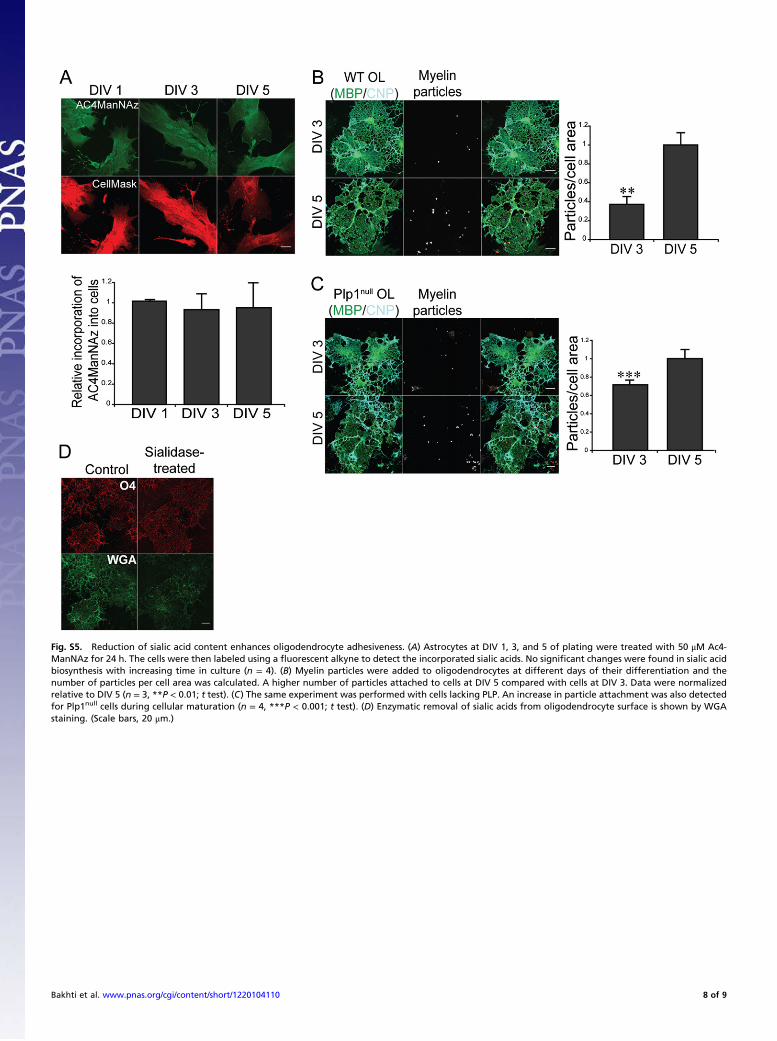
Fig. S5. Reduction of sialic acid content enhances oligodendrocyte adhesiveness. (A) Astrocytes at DIV 1, 3, and 5 of plating were treated with 50 μM Ac4-ManNAz for 24 h. The cells were then labeled using a fluorescent alkyne to detect the incorporated sialic acids. No significant changes were found in sialic acidbiosynthesis with increasing time in culture (n = 4). (B) Myelin particles were added to oligodendrocytes at different days of their differentiation and thenumber of particles per cell area was calculated. A higher number of particles attached to cells at DIV 5 compared with cells at DIV 3. Data were normalizedrelative to DIV 5 (n = 3, **P < 0.01; t test). (C) The same experiment was performed with cells lacking PLP. An increase in particle attachment was also detectedfor Plp1null cells during cellular maturation (n = 4, ***P < 0.001; t test). (D) Enzymatic removal of sialic acids from oligodendrocyte surface is shown by WGAstaining. (Scale bars, 20 μm.)
Bakhti et al. www.pnas.org/cgi/content/short/1220104110 8 of 9
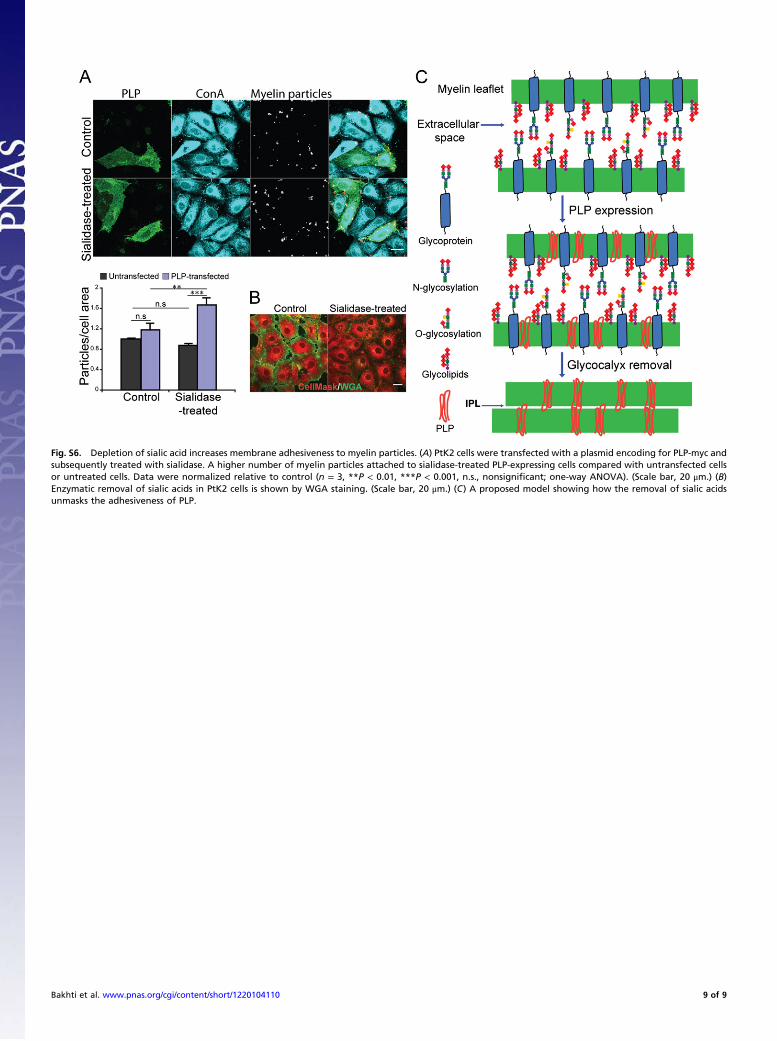
Fig. S6. Depletion of sialic acid increases membrane adhesiveness to myelin particles. (A) PtK2 cells were transfected with a plasmid encoding for PLP-myc andsubsequently treated with sialidase. A higher number of myelin particles attached to sialidase-treated PLP-expressing cells compared with untransfected cellsor untreated cells. Data were normalized relative to control (n = 3, **P < 0.01, ***P < 0.001, n.s., nonsignificant; one-way ANOVA). (Scale bar, 20 μm.) (B)Enzymatic removal of sialic acids in PtK2 cells is shown by WGA staining. (Scale bar, 20 μm.) (C) A proposed model showing how the removal of sialic acidsunmasks the adhesiveness of PLP.
Bakhti et al. www.pnas.org/cgi/content/short/1220104110 9 of 9


















