
Supplementary information - Nature · 2,5 0,4 0,002268 0,002215 0,002250 0,002245 1,816 e-05...
52
1 Supplementary information Supplementary Figure S1 Supplementary Figure S1- DLS results. (a) Z-average size, count rate (in kilocounts/s) and polydispersity index (PDI = σ 2 /Z D 2 , with σ = standard deviation and Z D = Z-average size) as function of reaction time, insert: Number average size distribution at t = 8 and 12 minutes. (b) Correlation data of mineralizing solution at 4, 8 and 12 min, insert shows the relative increase of the contribution of the spheres during time as indicated by the arrows. The raw correlation data shows the correlation time - a measure of particle size – plotted against the correlation coefficient which is a measure of intensity but standardized at 0.7-0.9. The drops in the graphs correspond to the presence of a particle with a certain size and can be used to describe the presence of large particles inside the solution, as in contrast to the data on number-, volume- and intensity-averaged size, it does not have a cut-off at 4 µm. The correlation data of the mineralization reaction after 4 min shows us 3 distinct drops at around 200 nm, 40 µm and even larger species. The smallest size corresponds to the collapsed polymer aggregates, the larger sizes correspond to the low-density polymeric superstructures observed in cryo-TEM.
Transcript of Supplementary information - Nature · 2,5 0,4 0,002268 0,002215 0,002250 0,002245 1,816 e-05...

1
Supplementary information
Supplementary Figure S1
Supplementary Figure S1- DLS results. (a) Z-average size, count rate (in kilocounts/s) and
polydispersity index (PDI = σ2/ZD
2, with σ = standard deviation and ZD = Z-average size) as
function of reaction time, insert: Number average size distribution at t = 8 and 12 minutes. (b)
Correlation data of mineralizing solution at 4, 8 and 12 min, insert shows the relative increase of
the contribution of the spheres during time as indicated by the arrows. The raw correlation data
shows the correlation time - a measure of particle size – plotted against the correlation
coefficient which is a measure of intensity but standardized at 0.7-0.9. The drops in the graphs
correspond to the presence of a particle with a certain size and can be used to describe the
presence of large particles inside the solution, as in contrast to the data on number-, volume-
and intensity-averaged size, it does not have a cut-off at 4 µm. The correlation data of the
mineralization reaction after 4 min shows us 3 distinct drops at around 200 nm, 40 µm and even
larger species. The smallest size corresponds to the collapsed polymer aggregates, the larger
sizes correspond to the low-density polymeric superstructures observed in cryo-TEM.

2
Supplementary Figure S2
Supplementary Figure S2 - Close-up of pH/Ca2+
-concentration upon stepwise adding the
phosphate stock to the calcium-stock. During this particular reaction the electrode was
immersed into the calcium stock before the addition of the phosphate stock. The addition of the
phosphate stock leads to a drop in the calcium concentration in the reaction solution, while the
pH stays constant. The calcium electrode responds immediately and is also immediately stable.
After the addition no changes in the calcium concentration or pH are observed with in the first
10 minutes, during the development of the branched polymeric assemblies.

3
Supplementary Figure S3
Supplementary Figure S3 - Representation of the mineralization experiment, showing the
different stages with corresponding time points, chemical formulas or names and the amounts
of Ca bound (∆Ca1-4), phosphate bound (∆P1-4) and H+-released (∆H1-4) in between the different
stages.
4
Supplementary Figure S4
y = 9,993E-05x + 2,520E-07
R2 = 1,000E+00
y = 6,829E-05x + 9,885E-06
R2 = 9,998E-01
y = 3,164E-05x - 9,633E-06
R2 = 9,991E-01
0
0,0005
0,001
0,0015
0,002
0,0025
0,003
0 2 4 6 8 10 12 14 16 18 20
time (min)
calc
ium
/ a
dd
ed
NaO
H(m
mo
l)
total calcium
bound calcium
free calcium
added NaOH
Linear (total
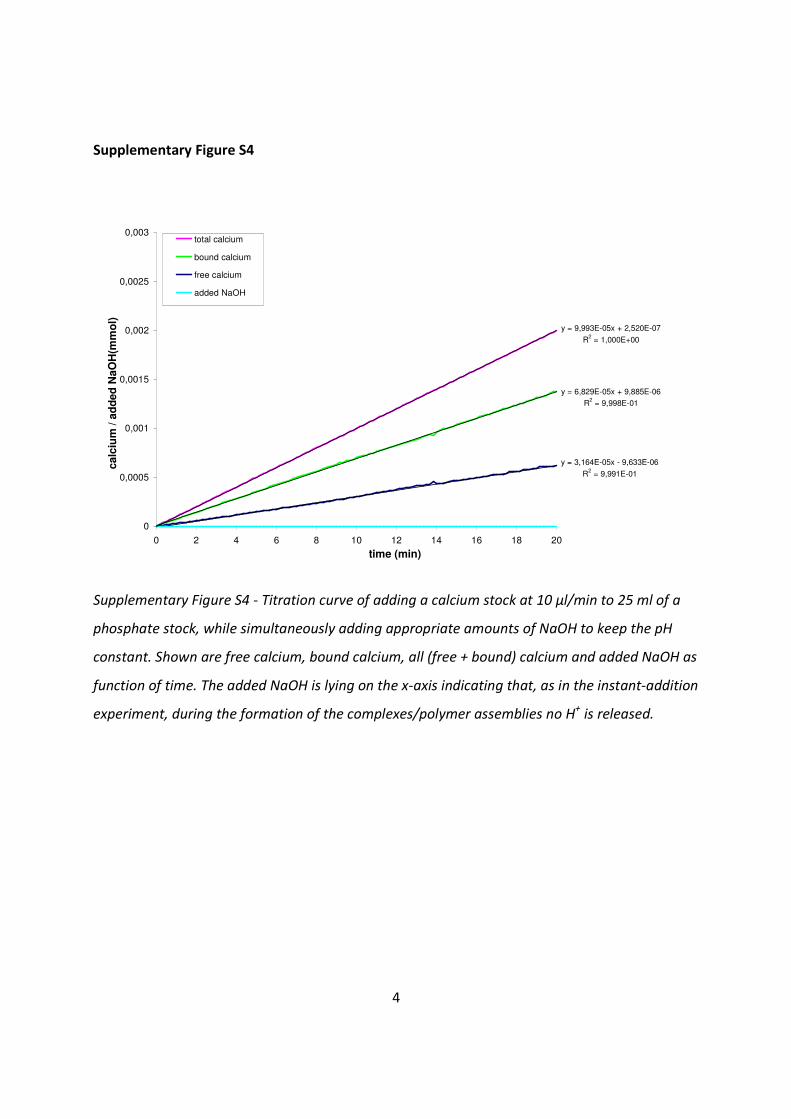
Supplementary Figure S4 - Titration curve of adding a calcium stock at 10 µl/min to 25 ml of a
phosphate stock, while simultaneously adding appropriate amounts of NaOH to keep the pH
constant. Shown are free calcium, bound calcium, all (free + bound) calcium and added NaOH as
function of time. The added NaOH is lying on the x-axis indicating that, as in the instant-addition
experiment, during the formation of the complexes/polymer assemblies no H+ is released.
5
Supplementary Figure S5
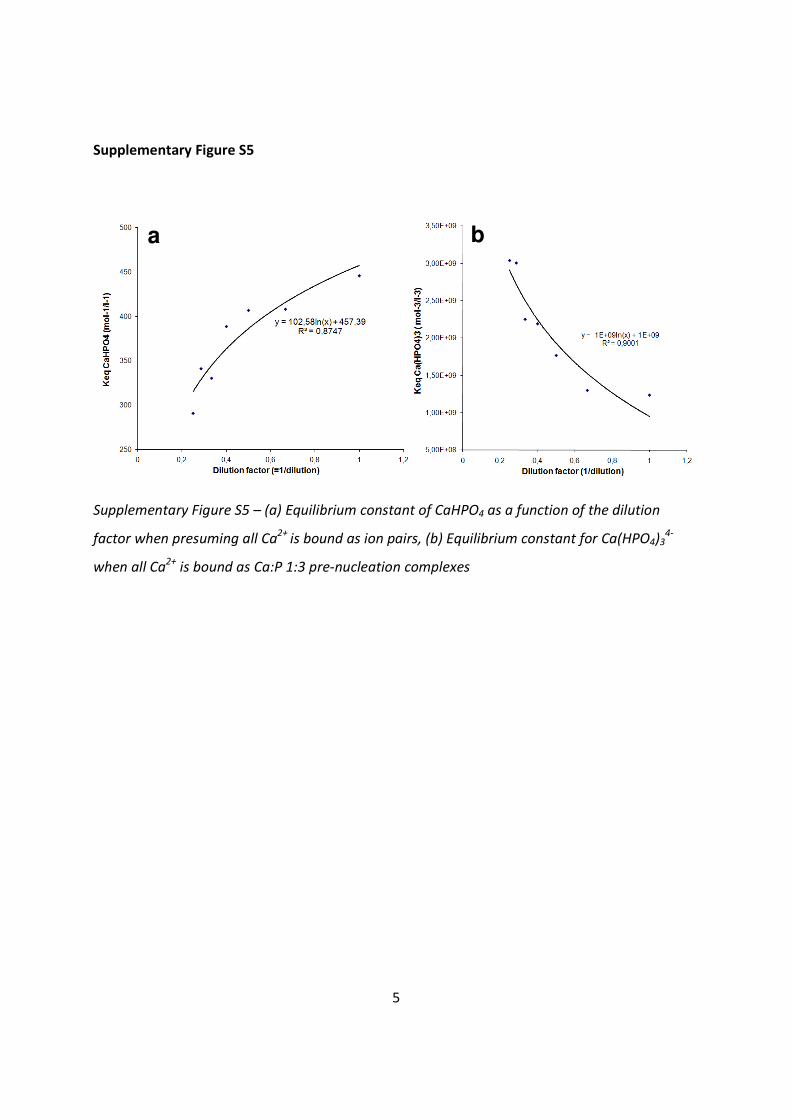
Supplementary Figure S5 – (a) Equilibrium constant of CaHPO4 as a function of the dilution
factor when presuming all Ca2+
is bound as ion pairs, (b) Equilibrium constant for Ca(HPO4)34-
when all Ca2+
is bound as Ca:P 1:3 pre-nucleation complexes
a b
6
Supplementary Figure S6
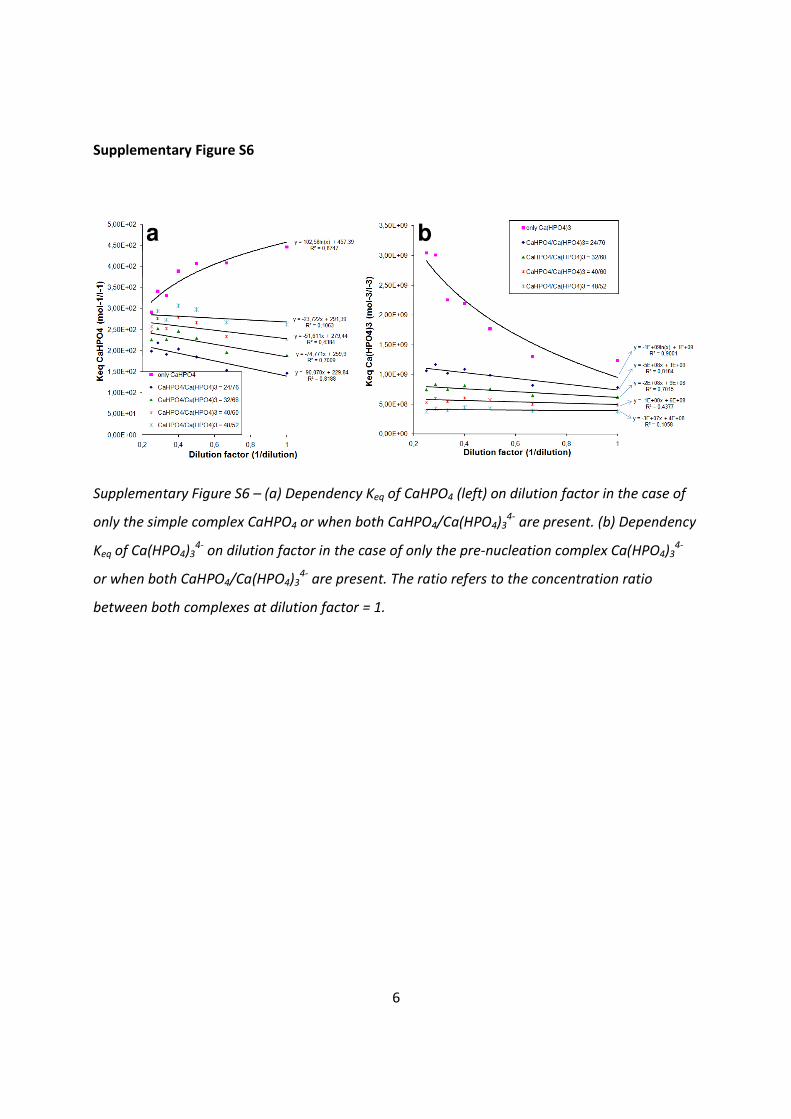
Supplementary Figure S6 – (a) Dependency Keq of CaHPO4 (left) on dilution factor in the case of
only the simple complex CaHPO4 or when both CaHPO4/Ca(HPO4)34-
are present. (b) Dependency
Keq of Ca(HPO4)34-
on dilution factor in the case of only the pre-nucleation complex Ca(HPO4)34-
or when both CaHPO4/Ca(HPO4)34-
are present. The ratio refers to the concentration ratio
between both complexes at dilution factor = 1.
b a

7
Supplementary Figure S7
Supplementary Figure S7 - Structure pre-nucleation complex as derived from ab-initio
calculations, projected from top view (a) or side view (b). a* and b* show the conformation of
the hydration waters surrounding the complex. The planar arrangement of the phosphates can
be explained by the significant repulsion between individual phosphates. As represented in
figure a* and b*, water binds preferably by H-bonds to the oxygens of the phosphate, whereas
the sides of the calcium are rather bare only binding to 1 H2O molecule. The size of the complex
and the corresponding atomic distances derived from the ab-initio calculations correspond well
to values calculated for the complex structure according to various effective ionic radii from
literature74
(See also supplementary Table S3). The coordination number of Ca2+
was found to be
7, which is within the range of reported coordination numbers74
for Ca2+
(6-12).
= Ca2+
= P5+
= O2-
= H+
a b
a* b*
double bond
single bond
H-bond
8
Supplementary Figure S8A
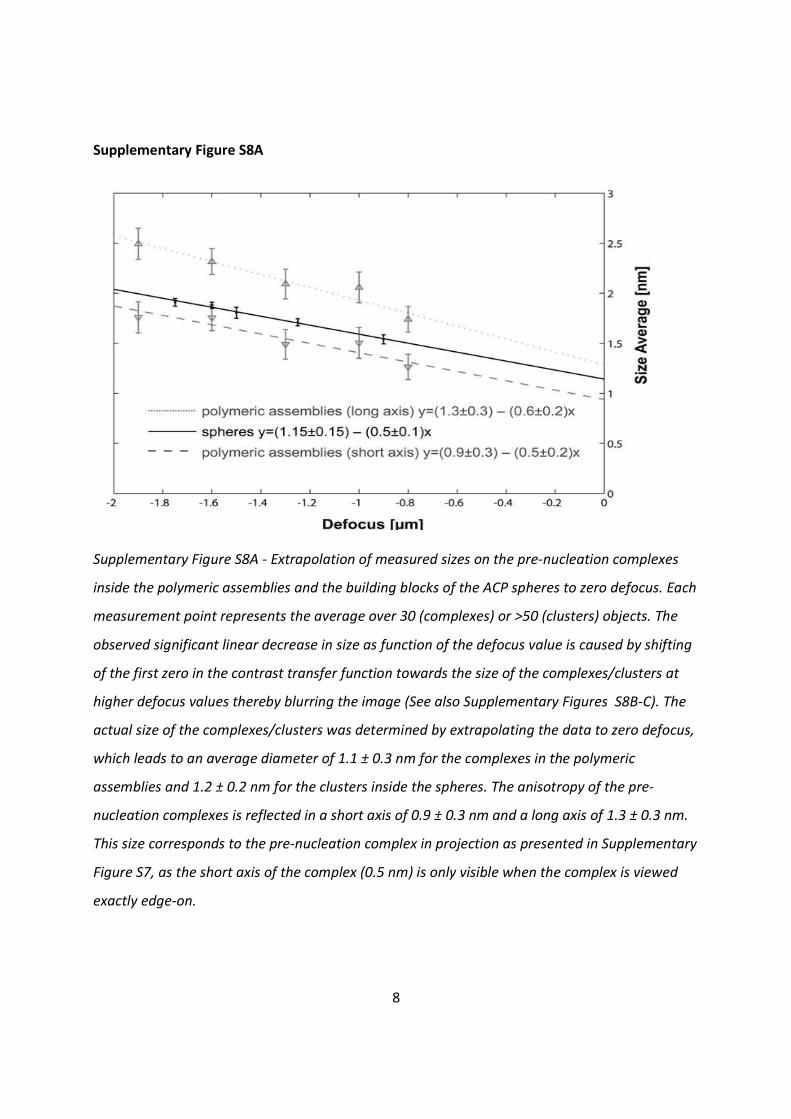
Supplementary Figure S8A - Extrapolation of measured sizes on the pre-nucleation complexes
inside the polymeric assemblies and the building blocks of the ACP spheres to zero defocus. Each
measurement point represents the average over 30 (complexes) or >50 (clusters) objects. The
observed significant linear decrease in size as function of the defocus value is caused by shifting
of the first zero in the contrast transfer function towards the size of the complexes/clusters at
higher defocus values thereby blurring the image (See also Supplementary Figures S8B-C). The
actual size of the complexes/clusters was determined by extrapolating the data to zero defocus,
which leads to an average diameter of 1.1 ± 0.3 nm for the complexes in the polymeric
assemblies and 1.2 ± 0.2 nm for the clusters inside the spheres. The anisotropy of the pre-
nucleation complexes is reflected in a short axis of 0.9 ± 0.3 nm and a long axis of 1.3 ± 0.3 nm.
This size corresponds to the pre-nucleation complex in projection as presented in Supplementary
Figure S7, as the short axis of the complex (0.5 nm) is only visible when the complex is viewed
exactly edge-on.

9
Supplementary Figure S8B
Supplementary Figure S8B - Complex size measurement from cryo-TEM defocus series. (a)
Average over all images of the series with measured complexes numbered and circled. (b) Cryo-
TEM image at -800 nm defocus, (c) Cryo-TEM image at -1000 nm defocus, (d) Cryo-TEM image
at -1300 nm defocus, (e) Cryo-TEM image at -1600 nm defocus, (f) Cryo-TEM image at -1900 nm
defocus. Scale bars are 20 nm. The yellow lines in panels (b-f) represent the long axis and the red
lines the short axis of the measured complexes.

10
Supplementary Figure S8C
Supplementary Figure S8C - Close-ups of particle 2 from defocus series with horizontal and
vertical line scans through the center of the complex. Performing these size measurements on
the same objects at different defocus values rules out that the anisotropy is due to the
visualization of different populations while changing focus.

11
Supplementary Figure S8D
Supplementary Figure S8D - Cryo-TEM images of mineralization product and initial buffers
acquired at -1000 nm defocus. A) CaP polymer, b) Tris buffer and 10 mM CaCl2, c) Tris buffer and
10 mM K2HPO4. Both Tris buffered-saline controls do not show any signs of polymeric
assemblies. Scale bars are 50 nm.

12
Supplementary Figure S9
Supplementary Figure S9 - Fractal dimension analysis on Cryo-Tomograms. (a-d) cross-sections
of tomograms of branched polymeric assemblies and spheres, before (a+c) and after (b+d)
segmentation. (e+f) 3D surface rendering of segmented tomograms of polymer assemblies and
spheres, respectively. (g) box size vs. box number plot with corresponding fractal dimensions.
The fractal dimension was determined from a linear fit in the log-log plot using box sizes
between 4 and 128 pixel edge length to avoid artifacts due to noise and defocus effects,
respectively.

13
Supplementary Figure S10
Supplementary Figure S10 - FTIR spectra on samples from AFM experiments quenched at times
shown in figure. Solution supersaturations were σHAP = 3.36, σOCP = 1.76 and σACP = 0.04. Peaks
at 560, 599, 877 and 1036 are characteristic of OCP. FTIR was performed on samples preserved
as described in the Methods section.
14
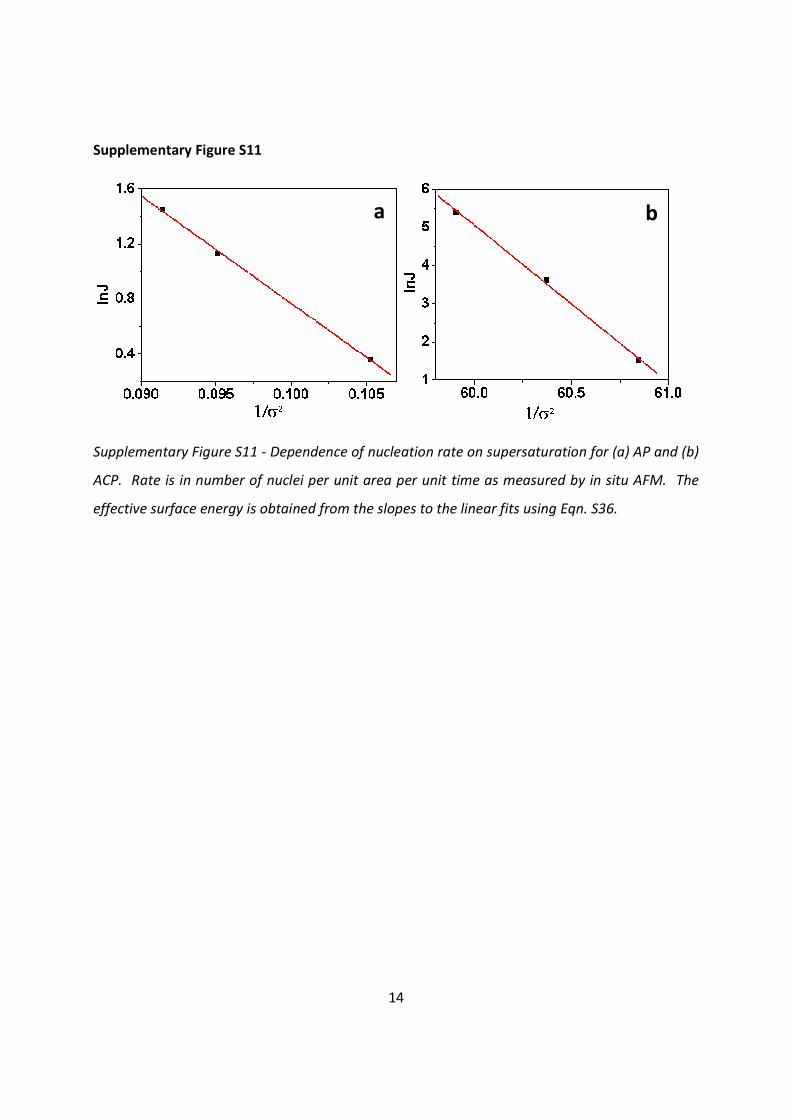
Supplementary Figure S11
Supplementary Figure S11 - Dependence of nucleation rate on supersaturation for (a) AP and (b)
ACP. Rate is in number of nuclei per unit area per unit time as measured by in situ AFM. The
effective surface energy is obtained from the slopes to the linear fits using Eqn. S36.
a b

15
Supplementary Figure S12
Supplementary Figure S12 - SEM-images of filtrated + dried calcium phosphate samples at t = 18
min, 51 min, 106 min and 183 min showing typical morphologies expected for ACP and
OCP/HAP.

16
Supplementary Figure S13
Supplementary Figure S13: Cryo-TEM images of structures formed during calcium phosphate
mineralization without the use of Tris-buffer. Left: polymeric assemblies (15 min), middle:
spheres (35 min) and right: OCP-like ribbons (45 min). Scale bar = 50 nm. Note that other than a
slightly faster kinetics, expected when the ionic strength decreases in the absence of Tris-buffer,
there is hardly a difference in morphology between these structures and the ones observed in
the presence of Tris-buffer. Furthermore, any significant binding of Tris-buffer with Ca2+
in the
form of Ca2+
-Tris complexes (log K = 0.25 ± 0.02 at 25°C see reference 76) was calculated to
correspond to less than 0.25 % of the total amount of calcium at all time points during the
mineralization reaction.
17
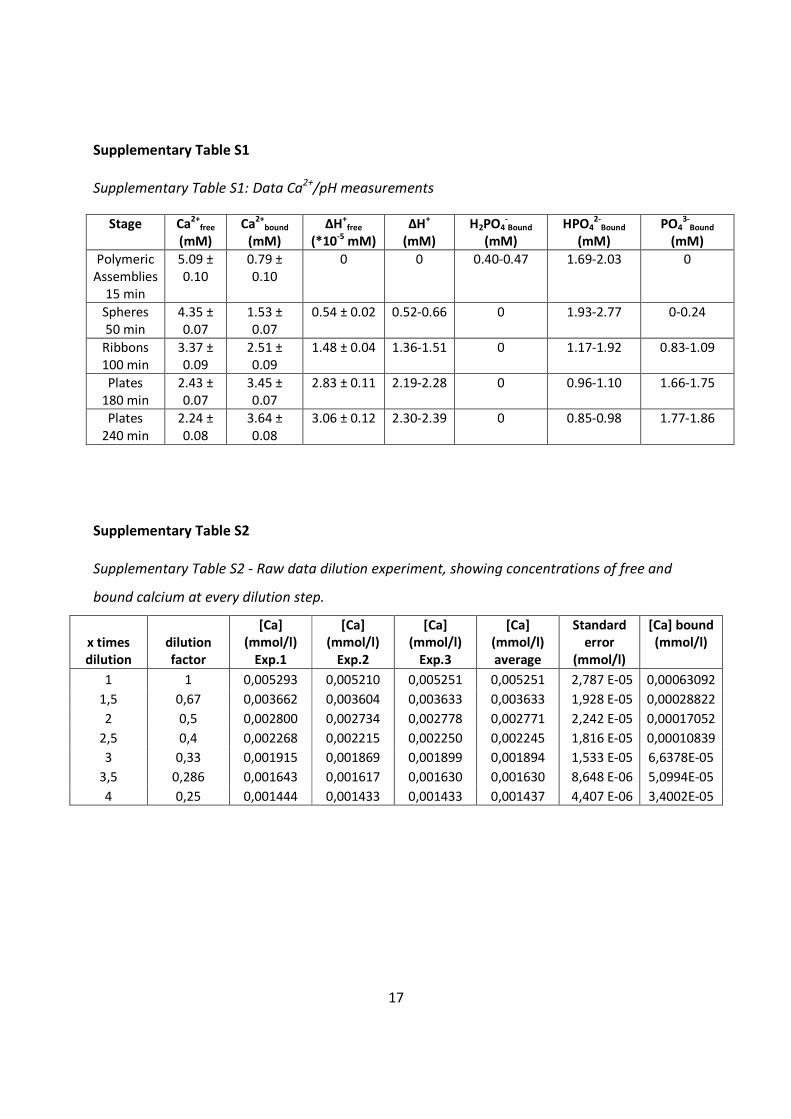
Supplementary Table S1
Supplementary Table S1: Data Ca2+
/pH measurements
Stage Ca2+
free
(mM)
Ca2+
bound
(mM)
ΔH+
free
(*10-5
mM)
ΔH+
(mM)
H2PO4-Bound
(mM)
HPO42-
Bound
(mM)
PO43-
Bound
(mM)
Polymeric Assemblies
15 min
5.09 ± 0.10
0.79 ± 0.10
0 0 0.40-0.47 1.69-2.03 0
Spheres 50 min
4.35 ± 0.07
1.53 ± 0.07
0.54 ± 0.02 0.52-0.66 0 1.93-2.77 0-0.24
Ribbons 100 min
3.37 ± 0.09
2.51 ± 0.09
1.48 ± 0.04 1.36-1.51 0 1.17-1.92 0.83-1.09
Plates 180 min
2.43 ± 0.07
3.45 ± 0.07
2.83 ± 0.11 2.19-2.28 0 0.96-1.10 1.66-1.75
Plates 240 min
2.24 ± 0.08
3.64 ± 0.08
3.06 ± 0.12 2.30-2.39 0 0.85-0.98 1.77-1.86
Supplementary Table S2
Supplementary Table S2 - Raw data dilution experiment, showing concentrations of free and
bound calcium at every dilution step.
x times
dilution
dilution
factor
[Ca]
(mmol/l)
Exp.1
[Ca]
(mmol/l)
Exp.2
[Ca]
(mmol/l)
Exp.3
[Ca]
(mmol/l)
average
Standard
error
(mmol/l)
[Ca] bound
(mmol/l)
1 1 0,005293 0,005210 0,005251 0,005251 2,787 E-05 0,00063092
1,5 0,67 0,003662 0,003604 0,003633 0,003633 1,928 E-05 0,00028822
2 0,5 0,002800 0,002734 0,002778 0,002771 2,242 E-05 0,00017052
2,5 0,4 0,002268 0,002215 0,002250 0,002245 1,816 E-05 0,00010839
3 0,33 0,001915 0,001869 0,001899 0,001894 1,533 E-05 6,6378E-05
3,5 0,286 0,001643 0,001617 0,001630 0,001630 8,648 E-06 5,0994E-05
4 0,25 0,001444 0,001433 0,001433 0,001437 4,407 E-06 3,4002E-05

18
Supplementary Table S3
Supplementary Table S3 - Size of pre- nucleation complexes and corresponding atomic distances
according to ab-initio calculations (bold) and from calculations on the structure using reported
ionic radii from literature74
. Radial distribution function data on ACC by Posner41
are given in
brackets.
Atomic distances (Å) Structure Size (Å)
Ca-O Ca-P P-O O-O P-P
11.0 (l)
12.0-13.4 (l) Pre-nucleation
complex 5.0 (h)
5.6 (h)
2.47 ± 0.06
2.4 (2.3)
3.03 ± 0.03
3.2-3.9 (3-6)
1.55 ± 0.02
1.5 (1.5)
2.45 ± 0.01
2.5 (2.3)
5.1 ± 0.3
5.6-6.8 (3-6)
11.0 (l)
11.9-12.6 (l) Pre-nucleation
complex-H+
5.0 (h)
5.6 (h)
2.47 ± 0.06
2.4 (2.3)
3.03 ± 0.03
3.2-3.9 (3-6)
1.59 ± 0.01
1.5 (1.5)
2.45 ± 0.01
2.5 (2.3)
5.1 ± 0.3
5.5-6.8 (3-6)
Supplementary Table S4
Supplementary Table S4 - Ca/P ratio measured by EDX of filtrated + dried sample at different
stages (n = 10)
Sample Ca/P ratio (± error)
18 min 1.48 ± 0.06 51 min 1.45 ± 0.02 106 min 1.45 ± 0.03 183 min 1.51 ± 0.09 239 min 1.46 ± 0.02

19
SUPPLEMENTARY METHODS
DLS investigation + zeta potential measurements
Dynamic light scattering (DLS) and zeta potential measurements were performed on a Malvern
Zetasizer Nano ZS Series (Malvern Instruments Limited, UK) with a 633 nm, 4mW HeNe laser,
measurement angle 173°; 20 °C). Measurements were averaged over 11 runs of 10 s. The
refractive index of apatite (n = 1.63) and viscosity and refractive index of phosphate buffer
saline (PBS, η = 1.0041 (20 °C), n = 1.33) were used for the calculations. Zeta potentials were
measured in transmission at a 50V effective voltage (average over 100 runs). The Von
Smoluchowski equation55 was applied, considering the high ionic strength used (I = 0.2 M) and
size of the polymeric aggregates (> 100 nm).
Calculation chemistry of calcium phosphate
Control over physiochemical conditions/ process parameters
Ca2+-ISE
To enable a quantitative interpretation of the Ca2+-ISE measurements, the physicochemical
conditions during the experiment regarding pH, temperature and ionic strength (pH = 7.4-7.2, t
= 20 °C, I = 0.2-0.22) were kept as close as possible to the Ca2+ - calibration (pH = 7.4, t = 20 °C, I
= 0.2, R2 calibration curve = 1). Furthermore, as the Ca2+-ISE is pH-dependent and formed
calcium phosphate could interfere with the Ca2+-ISE increasingly during time, the Ca2+-ISE
measurement was stopped after 4h, where both pH and Ca2+ measurements reach a stable
plateau (pH = 7.167, Ca2+ concentration = 2.24 ± 0.22 mmol/l). An additional experiment was

20
performed where it was observed that in the used pH-range, the Ca2+-electrode is independent
of the pH.
As a further control, the Ca2+-concentration at t = 4h was validated using ICP-OES
measurements of the supernatant (Ca2+-concentration = 2.38 ± 0.05 mmol/l). As at this time
point concentrations of ions/complexes are dictated by the precipitated OCP-phase, the Ksp of
the OCP-phase does not allow high concentrations of these complexes as was also calculated
using equilibrium constants derived from the dilution experiment described later in the
Supplementary Methods section (max. 4% of complexes). The ICP measurement of the solution
therefore should correspond to the Ca2+-ISE measurements. Although differences in ionic
strength between the calibration experiment and the actual precipitation reaction are small, to
correct the Ca2+-activity for this small difference, the ionic strength (I) of the solution at any
time point was calculated according to the following equation where ci = concentration of
species i with charge zi.
2
12
1i
n
i
i zcI ⋅⋅= ∑=
[S1]
Accordingly, while the total ionic strength (I = 0.2-0.22) surpasses the limit for extended Debye-
Hückel61, activity coefficients (γ) were calculated according to a modified version of the Davies
Extended Debye-Hückel Equation at 20°C48,62 that has reported to work well for aqueous
solutions in the range of 10-3 to 1 M62 .

21
( )
⋅−
+⋅⋅=− Ib
IBa
Izii
15046.0log
2γ [S2]
with Ba = 1.0 and b = 0.2
As especially the value for the linear coefficient (b) is reported to be dependent on, for
example, the type of electrolyte used62, activity coefficients were calculated upon varying b
from 0.1 to 0.3, showing no significant differences in species concentration. Similarly, the value
for Ba was varied from 1.0 to 1.5 resulting only in minor changes in species concentration.
Furthermore, as this equation is only valid for free ions in solution, the ionic strength of the
solution (I) was corrected for the formation of ion complexes/precipitates by subtracting the
complexed/precipitated ions from the original concentration. Due to the high concentration of
especially NaCl and Trizma-buffer in solution, contribution of ion complexes to the total ionic
strength of the solution was calculated to be negligibly small (max. 0.0064 M on a total of 0.22
M).
pH-electrode
As the pH-electrode used is accurate to +/- 0.001 pH unit, this ensures us of enough sensitivity
to visualize even small changes in phosphate chemistry. For example, the pH drop from
polymeric assemblies to spheres (7.425 � 7.367) corresponds to the release of 0.55 – 0.65 mM
H+, which is 58x the accuracy limit.

22
Control over thermodynamic pathway
As the pH does not drop below 7.0, the thermodynamically stable end product of the
mineralization reaction is apatite17. This structure was also confirmed by Cryo-TEM, electron
diffraction and in-situ FTIR on samples > 3days of incubation. The calcium phosphate phase
after 4h of reaction is OCP, as confirmed by in-situ SAXS and electron diffraction, in situ FTIR
and ICP-OES (Ca/P = 1.33 ± 0.02). OCP is described as an intermediate for apatite formation in
solution23-24.
Control over phosphate species in solution
During the whole mineralization reaction, from the observed pH-range (pH = 7.2-7.4) we can
calculate that > 99.9% of the phosphate in solution (H2PO4-, HPO4
2-, PO43-) is described by the
following equilibrium:
H2PO4- HPO4
2- + H+ (pKa = 7.21). [S3]
Chemical reactions
During the mineralization experiment a pH decrease was observed. Based on the phosphate
equilibrium, this pH-decrease can be caused by different types of reactions in which calcium
phosphate is involved. As the model can only calculate the average calcium phosphate
chemistry, the end-products of the reactions as described in this section do not have to be one
specific calcium phosphate but can represent a mixture of multiple calcium phosphate species.

23
Formation of a calcium phosphate species at time t=0
During the formation of a calcium phosphate species, the pH will decrease when Ca2+
effectively binds with PO43-,HPO4
2- or with H2PO4- and HPO4
2- in a ratio lower than the ratio
between both species present in solution (see Equation [S3]). Conversely, a pH increase will be
observed when calcium binds phosphate in a higher H2PO4-/HPO4
2- ratio or solely with H2PO4-.
As upon the formation of complexes or polymeric assemblies at t = 0 the Ca2+ is bound without
any observable pH-decrease (Supplementary Figure S2), this phenomenon can only be
described by a reaction of Ca2+ with H2PO4-/HPO4
2- where the ratio between H2PO4- and HPO4
2-
corresponds to the exact ratio of both species in solution (Equation [S4]). At the average pH =
7.425, this ratio is H2PO4-/HPO4
2- = 0.190/0.810, leading to the overall chemistry of
Ca((H2PO4)0.19(HPO4)0.81)x2-1.81*x
, according to the following reaction scheme:
Overall reaction
Ca2+ + x*0.19*H2PO4- + x*0.81*HPO4
2- � Ca((H2PO4)0.19(HPO4)0.81)x 2-1.81*x
[S4]
We want to note that:
1. Theoretically, Ca2+ can also bind to a certain mixture of H2PO4-/HPO4
2-/PO43- in such a
way that the pH stays constant as well. However, from in-situ IR the first signal that
exclusively can be attributed to PO43- (1021 cm-1) arises when the ribbon-like aggregates
appear. The pattern of the calcium phosphate species at t=0 can be attributed to the
overlap / broadening of the signals of free HPO42-/H2PO4
- in solution. Furthermore, to
keep the pH stable at the used pH, the formation of a significant amount of Cax(PO4)y

24
species implies the formation of even more species containing H2PO4-. As we have a
solution with mostly HPO42- ions, the presence of significant amounts of both Cax(PO4)y
and Cax(H2PO4)y species is very unlikely due to the large difference in the pKa of the
bound phosphates. This behavior was observed for the known CaP-ion pairs (CaPO4-,
CaHPO4 and CaH2PO4+), where the predicted pH-dependency of these ion pairs63 shows
only limited overlap between CaPO4- and CaH2PO4
+.
2. The overall reaction as described above can be divided into the concomitant formation
of multiple calcium phosphate species like the earlier described calcium phosphate ion
pairs 63,64.
3. In the case of the formation of an equilibrium species, the one-way arrow as described
above can also be read as a two-way arrow.
Reaction of calcium phosphate species with Ca2+ from the solution
After the formation of the pre-nucleation species, a simultaneous decrease of pH and Ca2+ was
observed upon the formation of the spheres, ribbons and finally OCP plates. Such a behavior
can be explained by a reaction in which bound phosphate releases protons upon the attraction
of Ca2+ from the solution (Equations [S5-S6]).
Cax(H2PO4)y(HPO4)z2x-y-2z + a Ca2+ � Cax+a (H2PO4)y-b (HPO4)z+b
2x-(y-b)-2(z+b) + b H+ [S5]
Cax(HPO4)y(PO4)z2x-2y-3z + a Ca2+
� Cax+a(HPO4)y-b(PO4)z+b2x -2(y-b)-3(z+b) + b H+ [S6]

25
Reaction of calcium phosphate species with calcium and phosphate from solution
In addition to reactions [S5] and [S6], calcium phosphate species can also attract new
phosphate from the solution at every phase transition. Taking into account the overall pH
decrease, such an event effectively will remove HPO42- from the equilibrium (Equation [S3]) and
release of H+. The amount of H+ released here is equal to the fraction of H2PO4- in solution and
therefore depends on the pH. Furthermore, as binding of PO43- is equivalent to binding HPO4
2-
and concomitant proton release from the bound HPO42- (Equation [S6]), for the average
chemistry only reactions [S7-S8] are of importance.
Cax(H2PO4)y(HPO4)z2x-y-2z + a HPO4
2- + b Ca2+ � Cax+b(H2PO4)y(HPO4)z+a2(x+b)-y-2(z+a) [S7]
Cax(HPO4)y(PO4)z2x-2y-3z + a HPO4
2- + b Ca2+ � Cax+b(HPO4)y+a(PO4)z2(x+b)-2(y+a)-3z [S8]
Side reactions
Upon designing the experiment, the possible incorporation of CO2 into the system is taken into
account. The used reaction solutions as well as both stocks are equilibrated with the CO2 from
the air, which enables us to calculate the concentrations of carbonate inside the solution. At
the starting pH of 7.4, the total concentration of HCO3- will amount to ~ 0.15 mM HCO3. This
value is very small compared to the concentrations of Tris-buffer (50 mM) and phosphate
buffer (1.6-4.12 mM), and will decrease even further upon the pH decrease that was observed
in the experiment.

26
Due to the abundance of Tris-buffer and phosphate-buffer, and taking into account the values
of the dissociation constants of Tris-buffer (pKa = 8.2) and phosphate (pKa2 = 7.21), at the used
pH range (pH 7.2-7.4) the small amount of HCO3- (pKapp,1 = 6.35, pKa2 = 10.33) does not have a
significant influence on the buffer capacity or pH of the solution.
A pH of 7.2-7.4 is too low to form any calcium carbonate bulk phase. Binding of Ca2+ with
carbonate in the pre-nucleation stage is restricted to the formation of the CaHCO3+-complex.
Using formation constants from literature ranging between (2.4-18.2),65 there is at maximum
4.6*10-6M of CaHCO3+, which is 170 x lower than the total amount of bound Ca2+ in the pre-
nucleation stage (7.9*10-4M). From quantitative FTIR analysis at comparable conditions52 we
know that the total amount of incorporated carbonate into the eventual apatite phase typically
represents less than 1.0% of the precipitate, and therefore does not imply significant
differences to our calculations.
Calculation of released H+
Effect of the buffer
Due to the high concentration of buffer used, the H+ released during the experiment is taken
up either by the Tris-buffer (H+ + Tris ↔ TrisH+, pKa = 8.2 (20 °C)) or by the remaining
phosphate buffer, which changes the equilibrium of the Tris/phosphate-buffer in the direction
of TrisH+/H2PO4-. The amount of H+ that is released at any time interval during the experiment
can be obtained by adding up these pH-induced changes in HTris+, H2PO4- and free H+, of which
the last term is negligibly small when compared to the first two.

27
ΔH+ = ΔHTris+ + ΔH2PO4- + ΔH+(free) [S9]
Correction for ionic strength
Next to the pH, the buffer equilibria are also dependent on changes in ionic strength. To correct
for this, activity coefficients for TrisH+, H2PO4- and HPO4
2- were calculated according to the
earlier described Davies Extended Debye-Hückel Equation48,62.
Correction due to depletion of phosphate buffer
Due to the formation of calcium phosphate, the buffer capacity of the phosphate buffer
decreases in time. To compensate for this, the amount of phosphate buffer in solution at every
time point was calculated by deducing the amount of bound phosphate from the total amount
of phosphate added initially. In terms of Equation [S9], this means that the value for ΔH2PO4-
decreases when more phosphate is bound from the solution, leading to a lower value of ΔH+.
Definition of the model
Based on the in-situ Ca2+/pH measurements we can give the following representation of the
mineralization experiment (Supplementary Figure S3).
For calculation purposes, we consider the chemistry at the plateaus at t = 15 min (pre-
nucleation species), t = 50 min (spheres), t = 100 min (ribbons), t = 180 min (plates) and t = 240
min (OCP) as the average chemistry of the four different stages (1-4). The amount of calcium

28
and phosphate bound (ΔCa2+1-4, ΔP1-4) and protons released (ΔH+
1-4) in between the different
time points represents the change in chemistry.
From this scheme, the ΔCa2+ (free calcium) is measured by the Ca2+-ISE and bound calcium is
the result of subtracting the initial concentration of calcium (= 5.88 mmol/l) from the
concentration measured at t = 0-4 h. Combined with the precise determination of the calcium
phosphate at t = 4h as OCP, we know the concentration of bound OCP in solution at this time
point, which is 3.638 mM/8 = 0.455 mM.
Finally, the values for ΔH+1-4 can be found by filling in Equation [S9], so the unknown
parameters in this model are the ΔP1-4 and the amount of phosphate (x) reacting with Ca2+ in
the pre-nucleation stage.
In formula, we have to solve the following equation:
Ca((H2PO4)0.19(HPO4)0.81)x2-1.81x + (ΔCa2+) + (ΔP) + (ΔH+) = 0.455 mM Ca8(HPO4)2(PO4)4 [S10]
Using this equation, the number of phosphates bound to calcium in the pre-nucleation stage (x)
can now be determined by choosing values for ΔP. The ranges for ΔP can be found by looking at
the possible reactions between the complex and OCP stage (Eqns. [S4-S8]) and filling in mass
balances for bound P and H+.

29
Mass and charge balances
Mass balance for bound P
All the phosphate (P) that is bound in OCP (6*0.455 mM) at the end of the reaction is equal to
the P bound in the beginning (x) + the P that is bound from the solution during the different
phase transitions (ΔP).
Pbound end = Pcomplexes + ΔP
2.729 mM = x + ΔP = x + ΔP1 + ΔP2 + ΔP3 + ΔP4 = x + (r1 + r2 + r2 + r4)* ΔP [S11]
Here r1 to r4 represent the fraction of the total phosphate bound from the solution between
each time point where r1 + r2 + r3 + r4 = 1.
Mass balance ΔH+
The ΔH+ can be divided into the ΔH+ released upon proton release from a bound phosphate
(ΔH+bound, Eqn. [S5] and [S6]) and the ΔH+ released upon binding a HPO4
2- group from the
solution (ΔH+sol, Eqn. [S7] and [S8]).
ΔH+ = ΔH+bound + ΔH+
sol
The ΔH+bound is only dependent on the amount and type of phosphate in the beginning
(x(H2PO4)0.19(HPO4)0.81-1.81) and the end stage ((HPO4)2(PO4)4) and can be defined in the
following way:

30
ΔH+bound = 0.190*x (H2PO4
- � HPO4
2-) + 4*0.455 mM (HPO42-
� PO43-) = 0.190*x + 1.819 mM
The ΔH+sol can be written as an average exchange rate α (amount of H+ released/HPO4
2- bound)
times the total amount of phosphate bound in between the complex and OCP stage (ΔP).
ΔH+sol = α* ΔP = α1*ΔP1 + α2*ΔP2 + α3*ΔP3 + α4*ΔP4 = (α1*r1 + α2*r2 + α3*r3 + α4*r4)*ΔP
Here α1-4 are the average exchange ratios (amount of H+ released/HPO42- bound) in between
the subsequent stages, which can be obtained experimentally.
The total mass balance for ΔH+ therefore reduces to:
ΔH+ = ΔH+bound + ΔH+
sol = 0.190*x + 1.819 mM + (α1*r1 + α2*r2 + α3*r3 + α4*r4)*ΔP [S12]
Charge balance
As the charge balance is already integrated in the P and H+ balance (the only two species that
are exchanging charges), this doesn’t lead to a unique correlation and therefore is not
necessary for the solution of the model.
Solution of the model
Combining both mass balances we end up with the following 2 equations:
2.729 mM = x + (r1 + r2 + r2 + r4)* ΔP

31
ΔH+ = 0.190*x + 1.819 mM + (α1*r1 + α2*r2 + α3*r3 + α4*r4)*ΔP
Here ΔH+ can be retrieved from Equation S9. By choosing when and how much of the
phosphate is bound at each phase transition (by choosing values for r1 to r4) one can now
calculate the upper and lower estimates for ΔP and x. For example, when all ΔP is bound during
the first phase transition then r1 = 1 and r2 = r3 = r4 =0 etc..
Accordingly, the chemistry in the pre-nucleation stage can be determined by filling in Equation
[S10]:
Ca2+t = 15 min + x*(H2PO4)0.19(HPO4)0.81
-1.81
By choosing the pathway (by filling in r1 to r4) we also know the ΔP at the different intermediate
stages as ΔP1 = r1* ΔP. As ΔH+ can be obtained by Equation S9 we can also calculate the
chemistry of all the intermediate stages. For example, for the spheres (50 min) the ΔH+ sol 1=
α1*r1*ΔP and the ΔH+bound1 = ΔH+
1- ΔH+ sol 1. Starting with the chemistry of the pre-nucleation
complexes, the ΔP1 tells how much HPO42- has to be added to the phosphate chemistry,
whereas the ΔH+bound1 tells how much of the H2PO4
- is transformed into HPO42- (Equation S5)
and when the H2PO4- is depleted how much of the bound HPO4
2- is transformed into PO43-
(Equation S6).
So for ΔH+bound1 < 0.19x the chemistry of the spheres is:
Ca2+t = 50 min + H2PO4
-*(0.19x-ΔH+bound1) + HPO4
2-*(0.81x + ΔH+bound1 + ΔP1)

32
And for ΔH+bound1 > 0.19x the chemistry of the spheres is:
Ca2+t = 50 min + HPO4
2-*(x - ΔH+bound1
E + ΔP1) + PO4
3-*(ΔH+bound1
E)
where ΔH+bound1
E = ΔH+bound1 - 0.19x , representing the excess of H+ released over the H+
released upon transforming all bound H2PO4- into HPO4
2- (= 0.19x). As there is no H2PO4- in the
ribbons and plates, we can use the same equation here:
Ribbons
Ca2+t = 100 min + HPO4
2-*(x - ΔH+bound1 + 2
E + ΔP1 + ΔP2) + PO4
3-*(ΔH+bound1 + 2
E)
where ΔH+bound1 + 2
E = ΔH+bound1 + ΔH+
bound2 - 0.19x
Plates
Ca2+t = 180 min + HPO4
2-*(x - ΔH+bound1 + 2 + 3
E + ΔP1 + ΔP2 + ΔP3) + PO4
3-*(ΔH+bound1 + 2 + 3
E)
where ΔH+bound1 + 2 + 3
E = ΔH+bound1 + ΔH+
bound2 + ΔH+bound3
- 0.19x
Next to the exact solutions of this model, the ranges in chemistry (as given in Figure 2 of the
manuscript) also reflect variations in the measured pH within the standard error and a variation
in PO43-/HPO4
2- ratio of the obtained OCP at t = 4h between 1.8 and 2.2. Measured Ca2+free,
calculated Ca2+bound and ΔH+
free at the different time points with corresponding standard errors
as well as calculated range in ΔH+ according to Equation S9 and bound phosphate species at
every time point taking into account the variation in pH and PO43-/HPO4
2- ratio of the OCP are
tabulated in Supplementary Table S1.

33
Specification of the calcium phosphate phases
Titration experiment
As upon the formation of the sphere stage a clear phase separation was observed, from here on
the different calcium phosphate stages represent bulk phases, where the concentration of ions
(or complex species) in solution is dictated by the solubility of these phases. For the pre-
nucleation stage this is not so clear and therefore a titration experiment was performed to
determine the character of these calcium phosphate species. The titration experiment was
performed by titration of a 10 mM calcium stock at a rate of 10 µl/min to the 10 mM phosphate
stock, monitoring the Ca2+-concentration and pH at a constant pH = 7.40, using the same
procedures, ionic strength, pH, temperature and stock concentrations as described for the
mineralization reaction in the methods section.
Supplementary Figure S4 shows plots of [Ca2+free] and [Ca2+
bound] vs. time during the first 20 min
of titrating a 10 mM calcium stock solution to the 10 mM phosphate stock. As this period
represents the pre-nucleation stage, it shows that a constant percentage of approximately 68%
of all the added calcium is bound (ratio slopes bound Ca/total calcium = 0.68), further implying
a constant ratio between [Ca2+free] and [Ca2+
bound]. Using this titration curve and the chemistry
calculations, we can assume an equilibrium behavior for the calcium phosphate species in
solution, with an average chemistry of Cax(HPO4)y(H2PO4)z2x-2y-z in the following way:
Keq = [Cax(HPO4)y(H2PO4)z2x-2y-z]/[Ca2+
free]x⋅[HPO42-
free]y⋅[H2PO4-free]z [S13]

34
Considering the high excess of phosphate in solution with respect to the added Ca2+-ions –
particularly in the early stages, the concentration of phosphate species [HPO42-
free], [H2PO4-free]
may be considered constant, reducing the formula to:
[Cax(HPO4)y(H2PO4)z2x-2y-z]/[Ca2+
free]x = constant [S14]
As the [Cax(HPO4)y(H2PO4)z2x-2y-z] represents all Ca2+ that is bound in solution, from the constant
ratio between [Ca2+free] and [Ca2+
bound], it automatically follows that x = 1 and that the behavior
in the titration curve corresponds to an equilibrium calcium phosphate complex with only 1
Ca2+ incorporated (i.e. Ca (HPO4)y(H2PO4)z2x-2y-z).
Now the average chemistry does not refer only to the pre-nucleation complexes but also to the
earlier described ion pairs, i.e. CaHPO4 , CaH2PO4+ and CaPO4
- 63-64, 66. The average chemistry can
therefore be divided into the chemistry of the ion pairs (fraction x1) and of the pre-nucleation
complexes (fraction x2).
[Ca(HPO4)y(H2PO4)z2-2y-z ] = x1*[ION PAIR] + x2*[PRENUCLEATION-COMPLEX] [S15]
As the difference between the ion pairs and prenucleation complexes is only the amount/type
of phosphates the calcium binds to, the following relationship between the activities of both
complexes can be derived:

35
[PRENUCLEATION COMPLEX]/[ION PAIR] = KPRE-COMPLEX/KION PAIR* [HPO42-]a*[H2PO4
-]b [S16]
Here the factors a and b represent the stoichiometric excesses of [HPO42-] or [H2PO4
-] groups in
the pre-nucleation complex with respect to the ion pair.
Because [HPO42-] and [H2PO4
-] are constant during the titration experiment (i.e. are present in
excess), the ratio between the ion pairs and pre-nucleation complexes during the titration
experiment is also constant. Hence, the calcium phosphate indeed behaves as a complex with a
chemistry that is an average between the ion pair and pre-nucleation complex. So taking into
account the presence of CaP-ion pairs, we can conclude from these data that the pre-nucleation
complexes are equilibrium species containing only 1 Ca2+-ion.
Dilution experiment
A dilution experiment was performed to investigate the equilibrium behavior of the proposed
ion pair / calcium phosphate complex mixture. Starting in the pre-nucleation stage, dilution
should change the concentration of the ionic species directed by the equilibrium constants at a
constant pH/temperature. As during dilution the concentration of free Ca2+ is measured, and
the expected concentrations of free [HPO42-] and [H2PO4
-] can be calculated for a given calcium
phosphate chemistry of the complexes, an equilibrium constant can be calculated for every
dilution step. Finally, in an iterative process the composition was determined for which the
calculated equilibrium constants did not change during the different dilution steps, which then
corresponds to the average composition of the equilibrium species in solution.

36
For the dilution experiment, the sample at t = 5 min reaction (100 ml) was diluted 1.5, 2, 2.5, 3,
3.5 and 4 times through the addition 50 ml of Tris-buffered saline (I=0.2, pH = 7.45). The Ca2+-
concentration and pH were monitored (section 1.8) for a total time span of 2h. After each
dilution step approximately 150-200 s equilibration time was allowed. The Ca2+-concentrations
used for calculating the equilibrium constants are averaged values from a triplo. Standard
deviations at each concentration were between 0.5 - 1.5%.
Results (Supplementary Table S2) shows a high reproducibility between the three experiments
performed, resulting in a low standard error on the free Ca-concentration and, at the same
time an acceptable standard error on the Ca2+ bound to complexes or ion pairs (= calculated
concentration of free Ca2+ without complexes or ion pairs – measured free Ca2+). Furthermore,
as the pH during the experiment remained constant (pH = 7.456-7.459), this indicates that
ratio’s between H2PO4-(aq) and HPO4
2-(aq) do not change upon dilution and, corresponding to
the mineralization experiment described in the manuscript, that the ratio of H2PO4-/HPO4
2-
inside the formed calcium phosphate complexes or ion pairs should correspond to this ratio.
The value for bound Ca2+ before dilution as obtained in this experiment (0.63 ± 0.03) is lower
than the original experiment (0,79 ± 0,1) , which is likely to be due to the difference in pH
(7.458 vs 7.425).
For the fitting of the complex chemistry, we first investigate two extreme situations: a situation
where all bound calcium in the pre-nucleation stage is described by the ion pairs (100% ion
pairs) and a situation in which the ion pairs are disregarded, and the average chemistry

37
corresponds to the pre-nucleation complex chemistry (100% pre-nucleation complexes).
Secondly, we investigate the situation in which there are both ion pairs + pre-nucleation
complexes in solution (ion pairs + pre-nucleation complexes).
100% ion pairs or 100% pre-nucleation complexes
In Supplementary Figure S5a the Keq for CaHPO4 is shown as a function of 1/dilution, presuming
all complexes in solution are represented by the ion pairs CaHPO4 (82%) + CaH2PO4+ (18%). As is
visible from the curve, the Keq decreases substantially upon dilution and therefore is not a good
estimate for the chemistry of the bound species in the pre-nucleation stage. The values
obtained by dilution, however, are in correspondence to the wide range of equilibrium values
that are reported in literature63-64, 66, indicating that also in many of these experiments the
complex species in solution cannot be described by ion pairs only.
In Supplementary Figure S5b the Equilibrium constant for the calcium phosphate complex is
shown as a function of 1/dilution, presuming all complexes in solution are either Ca(HPO4)34- or
Ca(HPO4)2(H2PO4)3-. Results show that also here the Keq is not constant upon dilution and shows
an opposite behavior as the CaHPO4 ion pair where the Keq increases. Because the activity
coefficient of the complex is unknown (γcomplex) the Keq should be read as Keq/γcomplex. The value
of the activity coefficient, however, does not affect the behavior of the curve while the dilution
with tris-buffered saline assures us of an approximate constant ionic strength.

38
Ion pairs + prenucleation complexes
When presuming a mixture of both ion pairs and pre-nucleation complexes, we can use
equation S16 to calculate the contribution of both complexes at the different dilution steps.
This results in the following curves for the Keq of CaHPO4 and Keq/γ of Ca(HPO4)34- using initial
concentration ratios (dilution =1) between CaHPO4/Ca(HPO4)34- of 24/76, 32/68, 40/60 and
48/52 (Supplementary Figures S6a+b). As can be observed from both graphs, the dependency
of the Keq on the dilution (as represented in the slope of the curves) as well as the absolute
value of Keq reduces significantly when both ion pairs and pre-nucleation complexes are
present. As a consequence of equation S16, the CaHPO4/Ca(HPO4)34- ratio increases upon
dilution, favoring the formation of the ion pairs. Starting from an initial CaHPO4/Ca(HPO4)34-
ratio of 40/60 and using a 95% confidence interval, the dependency of Keq of CaHPO4 and Keq/γ
of Ca(HPO4)34- on the dilution is not significant anymore (p = 0.106 > 0.05) and we can conclude
that at these conditions and at the ratio of 48/52 the chosen composition fits the equilibrium
behavior well.
Using Equation S15, we can calculate that in the mineralization experiment (Figure 2
manuscript), the maximum CaHPO4/Ca(HPO4)34- ratio is about 14/86. Though both data suggest
a significant amount of ion pairs to be present, the difference between the absolute numbers of
both experiments is likely due to the pH difference.
ICP-OES
Inductively coupled plasma optical emission spectroscopy (ICP-OES) was performed to measure
the concentration of the calcium and phosphorus inside the supernatant at the end of the

39
reaction (4h), using a Spectro CirosCCD spectrometer (Spectro Analytical Instruments GmbH,
Germany). Samples were centrifuged and the supernatant was decanted and stored at -20 °C
until measurement. Calibration curves were prepared for Ca (2.5 to 10 mg/l) and P (1.25 to 5
mg/ml), and samples were diluted 25x.
Ab-initio calculations
Possible geometries and reciprocal arrangements of the ionic moieties in the pre-nucleation
complexes were obtained by ab initio geometry optimizations at the Hartree-Fock level with a
6-31G** (dp) basis set using the GAMESS software. The solvent (water) was described both
explicitly, by surrounding the complex with 18 water molecules to account for the first
hydration shell, and implicitly, using the conductor-like polarizable continuous model (CPCM) to
describe the bulk solvent around the explicitly solvated complex. The geometry was optimized
starting from different initial configurations and the equilibrium geometry with the lower
energy was taken for visualization. However, all the optimized geometries gave similar
distances (maximum 2% of variation).
Pre-nucleation complex and post-nucleation cluster size measurements
To determine the sizes of pre-nucleation complexes/post-nucleation clusters a line was drawn
from one side of the object to the opposite side and in the perpendicular direction, for multiple
objects in the same image (Magnification 61,000 ×, pixel size 0.14 nm). To prevent overlap in
the projections, only complexes/clusters at the edge of the polymeric aggregates/spheres were
used. Images were taken at a decreasing defocus value after which the size of the clusters was

40
extrapolated to a defocus value of 0. The position of the first 0 of the CTF in the Fourier
transform of the electron micrographs was used to determine the defocus value of the image
by comparison to simulated CTFs (CTF explorer©, Max V. Sidorov, 2000-2002; estimated error ±
100 nm). Fitting was done by linear regression following the method of York67.
Thickness of ribbons by cryo-TEM
In correspondence to SAXS measurements an estimate of the thickness of the ribbon-like
structures was also done by cryo-TEM image analysis, where the ribbon was positioned in such
a way that the edge was visible, which was measured at 20 points. This resulted in an average
thickness of 2.1 nm at -1.5 µm underfocus, which after extrapolation to 0 defocus, using the
same slope as found for the polymeric assemblies and spheres (y = 0.5-0.6 x), results in a
thickness of 1.3-1.6 nm, corresponding well to the SAXS measurements.
Fractal dimension analysis
Fractal dimension analysis of the cryo-tomograms was done using a 3D boxcounting technique
in Matlab. The analysis included: 1) segmentation of the tomograms by thresholding using the
Isodata algorithm68; 2) extraction of the aggregates using morphological filters (closing, erosion,
dilation) in combination with size criteria; 3) determination of the fractal dimension by the
boxcounting method. The final scripts are implemented using in part functions (or parts
thereof) from the TOM toolbox69, the Delft Image processing library [www.diplib.org] and the
Matlab file exchange [File ID: #13063]. To account for defocus and in particular noise that is

41
present in cryo tomograms the fractal dimension was determined from a linear fit in the log-log
plot using box sizes between 4 and 128 pixel edge length.
Zeta potential calculations
To correlate the measured zeta potential as given in Figure 6 of the manuscript with the
chemistry calculations and morphological transitions, a theoretical value of the zeta potential
for the polymeric assemblies and spheres was derived using data from the size measurements
(DLS) and the obtained level of fractality, i.e. the fractal dimension. For estimating the
theoretical zeta potential, three different representations were taken into account: a bare
complex model, an opaque spherical aggregate model and a partially penetrable spherical
aggregate model. As the ion pairs present inside the solution are mostly represented by
CaHPO4, they do not have a high overall charge and therefore we can ignore them for the
calculations.
Bare complexes in solution
If the complexes are not part of aggregates and are able to move freely with respect to each
other, the zeta potential measurements should correspond to that of a solution containing
bare, charged particles. Under these conditions the zeta potential, presumed in value close to
the actual surface potential, is given by the following equation70:
( )aa
z B
D
a
λ
λ
⋅
+
⋅=Ψ
1
1 [S22]

42
Where ( )aΨ = Dimensionless Potential, Ψ(a) = Potential (V), a = radius sphere (nm), z = number
of elementary charges on each sphere, λD = Debye length (0.68 nm) and λB = Bjerrum length
(0.71 nm).
Hydrodynamically opaque structures
If the pre-nucleation complexes form sufficiently dense aggregates, they behave as more or less
spherical entities in which only the complexes situated at the outside of the structure
contribute to the effective surface potential (Structure 2, Fig. 6). Obviously, this is only true if
the hydrodynamic flow field of the fluid cannot substantially enter the aggregate when it moves
through the fluid. Only charges that are hydrodynamically coupled to the bulk fluid contribute
to the electrophoretic mobility. The charge density at the surface of our effective,
representative sphere is much lower than that of the bare complexes it is made up of, because
in a fractal the volume is not completely filled with complexes. This means that the surface
charge density observed in an electrophoretic experiment is a lot lower than that of the
complexes. A simple calculation confirms this.
The radius of the sphere surrounding the structure (R) then can be described by the following
expression:
aNR d ⋅≅1
[S23]

43
where N = amount of pre-nucleation complexes in the spherical volume, d = fractal dimension
of the complex aggregate, a = radius of an individual complex (nm). Using this formula, the pre-
nucleation complex density ρ (complexes/m3) of the sphere is represented by:
dddd
aR
R
aR
R
N −−−
⋅⋅⋅
=
⋅⋅
⋅=
⋅⋅
= 3
33 4
3
3
4
3
4 πππρ [S24]
As only the outer layer of pre-nucleation complexes is presumed to be hydrodynamically
coupled to the moving fluid, the number of complexes that contribute to the zeta potential (Na)
is the following:
dd
a aRN−− ⋅⋅= 113 [S25]
For the measurement of the dimensionless potential, we can use the formula for the bare pre-
nucleation complexes, but we have to multiply z by the amount of contributing complexes (Na)
and replace a by R. Furthermore, we can presume that the radius is much higher than the
Debye length, so R>> λD, resulting in the following equation:
( ) BD
ddBD
a
B
D
aR zaRR
zNRR
zN λλλλλ
λ
⋅⋅⋅⋅⋅≅⋅
⋅⋅≅⋅
+
⋅⋅=Ψ −− 13
23
1
1 [S26]

44
Partially penetrable structures
In reality, the flow field resulting from the polymeric assemblies moving in the electric field is
likely to penetrate this aggregate, but only to an amount that is a function of the density of pre-
nucleation complexes in it. Deep in the core the fluid co-moves with the moving aggregate
resulting from hydrodynamic interaction between the complexes that make up the aggregate.
Only charges on the pre-nucleation complexes in the transition zone, the so-called skin,
contribute to the electrophoretic mobility of the aggregate (Structure 1, Fig. 6). The extent to
which the medium can penetrate depends on the complex density, which is directly related to
the fractality of the polymeric assembly.
To define to which depth pre-nucleation complexes can contribute to the zeta potential, the
hydrodynamic skin depth (ξ) is introduced71:
21
⋅=
ρζ
ηξ s [S27]
where ηs = viscosity solvent, afactorfriction s ⋅⋅⋅== ηπζ 6_ and ρ = “density of Stokeslets”,
i.e., the density of the complexes that make up the aggregate. Replacing the density by
Equation [S24] and inserting the equation for the friction factor results in the following
equation,
21
13
2
9
⋅⋅= −− ddaRξ [S28]

45
The number of complexes inside the skin of the aggregates (Nξ) is defined in a similar way as
was done for the opaque structure, but then replacing a for ξ,
ρξπξ ⋅⋅⋅⋅= 24 RN [S29]
Inserting in the equations for ξ (Equation S28) and ρ (Equation S24) results in the following
expression:
+⋅−
+⋅
⋅⋅= 2
1
2
1
2
1
2
1
2dd
aRNξ [S30]
For the dimensionless potential, also the same formula can be applied as derived for the
opaque structure, replacing Na by Nξ, i.e.:
( )( ) ( )( ) 2
113
22 +−− ⋅⋅⋅⋅⋅=
⋅⋅⋅≅Ψ dd
BD
BD
R aRzR
zN λλλλ
ξ [S31]
Note that for d = 3, i.e., for a completely dense packing, eqns. S26 and S31 become equal apart
from a numerical prefactor. This is to be expected as in that case the penetration length should
be of the order of the size of the pre-nucleation complexes. Complete penetration of the
aggregates by the flow field, implying R ≈ξ, occurs only if small enough, if R ≈ a. Obviously the
assumption of a quasi-spherical object breaks down for rod-like assemblies with d = 1, in which

46
all particles do couple to the flow field. Our fractal dimensions are all in excess of 2 where this
analysis should be reasonably accurate.
Taking into account these three different regimes, we can conclude the following:
• The polymeric assemblies seem to correspond well to a partially penetrable structure
(Structure 1, fig. 6). For a polymeric assembly (z = −3.5, Df = 2.2, a = 0.55 nm) with a
radius of 20 µm, inserting the variables into Equation S31 leads to ( )RΨ = 0.118 and ψ(R) =
3.0 mV, a good match of the measured zetapotential (−3.5 mV).
• The appearance of the spherical nodules leads to a decrease in the zeta potential. As
this is also accompanied by a decrease in overall size (which should increase the zeta
potential), the spherical nodules must represent opaque structures, shielding a higher
amount of negative charge.
• The aggregated spheres after the phase transition, are composed of these opaque
spherical nodules (Structure 3, fig. 6). Of the aggregated spheres we know the fractality
(Df = 2.7), cluster charge (z = 2) and cluster size (a = 0.6 nm) and overall size (R > 10 µm,
in DLS). However, as the aggregated spheres are in fact also a fractal of the 200 nm sized
spheres, this fractality is also requisite to calculate the zeta potential. Though this
fractality is above the size limits for cryo-tomography, we can assume the following:
a. The aggregation process is likely to occur in the diffusion-limited regime, while
due to the size of the invidual units (200 nm) diffusion speeds will be rather low.
Cryo-TEM images show a chain-like aggregation of spheres, which is typical for a
diffusion-limited structure.

47
b. A structure with a diffusion limited fractality (Df = 1.7) is so diffuse that a partially
penetrable model is much more likely than an opaque structure model.
So for the calculation of the theoretical zeta potential, first the amount of charge on
each 200 nm sphere was calculated by Equation S26. Accordingly, an estimate of the
potential of the spherical aggregate (Df = 1.7, a = 200 nm) is acquired by applying
Equation S31 in which case we obtain values of 0.7-2.9 mV for 100-10 µm structures.
Analysis of nucleation data: Fitting of classical nucleation rate equation
To analyze the AFM data using classical nucleation theory72, we took the nucleation rate Jn to
be given by:
[S32]
For creation of a hemispherical nucleus of radius R on a foreign substrate, the change in free
energy is given by:
[S33]
where the subscripts ML, MS, and LS refer to the mineral-liquid, mineral-substrate and liquid-
substrate interfaces, respectively. The maximum in ∆G occurs at the critical radius Rc given by:
Rc =2ωαeff
kTσ, αeff = αML +
1
2(αMS − αLS ) Rc =
2ωαeff
kTσ, αeff = αML +
1
2(αMS − αLS ) [S34]

48
and has a value of:
[S35]
Substituting Eqn. S32 into Eqn. S35 and taking the logarithm of both sides gives:
ln(Jn ) = A −B
σ 2, B =
8πω 2αeff
3
3 kT( )3 ln(Jn ) = A −B
σ 2, B =
8πω 2αeff
3
3 kT( )3 [S36]
Supplementary Figs. S11A and S11B show the dependence of nucleation rate on σ-2 for AP and
of ACP, respectively. To extract the effective surface energies from this data, values of ω were
obtained as follows: In the case of AP (Ca10(PO4)6(OH)2) the lattice constant is a = b = 9.423 Å, c
=6.883 Å, α=β=90°, and γ=120° 73. The volume of the growth unit is 5.288×10-28/18 =2.937×10-29
m3.
ACP was previously reported to be composed of a basic unit (Posner cluster) with chemical
composition of Ca9(PO4)6 and diameter of 9.5 Å 41. From this, the volume of the growth unit of
ACP would be estimated as 4.489×10-28/15 =2.993×10-29 m3. However, the ACP (spheres)
measured in this study consists of Ca2(HPO4)32- units, and thus are not at the Posner-cluster
stage. When the disk-like structure is the building block for the ACP, the volume of growth
should be represented as a 1.1*0.5 nm disk in the most optimal packing of the complex. This is
actually when it packs as OCP, though we can take this as a lower estimate for ω. The volume of
a growth unit then would be 4.75×10-28/5 = 9.5×10-29 m3, which is about 3 times the value of ω
of apatite. Another approach to estimating the volume is to use values of ionic radii74. A fully
hydrated Ca2(HPO4)32- unit then has a volume of approx. 4.9×10-28 m3 and ω = (1/5)•4.9×10-28

49
= 9.8×10-29 though would also contain 26 waters, which is unrealistic. A non-hydrated
Ca2(HPO4)32- has a volume of 1.8×10-28 m3 giving ω = 3.6×10-29 m3. Although this comes close to
the value for a Posner-cluster, based on the size and stoichiometry of the complexes, a
reasonable estimate is somewhat larger. The precise choice of the value for ω has no effect on
the estimated magnitude of the free energy barrier and a minor effect on the estimated critical
radius for classical ion-by-ion nucleation, because, as can be seen from Eqn. S35, the value of
αeff determined from analysis of the nucleation rate data scales inversely with the value of ω2/3,
while the magnitude of the barrier scales with α3ω2 and the critical radius depends on the
product of the two terms. However, as is shown below, the magnitudes of both ∆GC and RC do
depend on the choice of ω. So to be conservative, we have done our calculations using an
intermediate value of 5x10-29 m3 for ω. Linear fits to the data give values for αeff of 90 mJ•m-2
and 40 mJ•m-2, respectively.
Analysis of calcium phosphate precipitate
The mineralization reaction was performed according to the methods section of the manuscript
and. Samples at the different stages were collected by filtration and drying after which SEM
images were taken (Supplementary Figure S12) and Ca/P ratio was measured by Energy-
Dispersive X-ray spectroscopy (EDX) (Supplementary Table S4). Calibration was performed on
standards with known Ca/P ratio (HAP (Ca/P = 1.67), DCPD (Ca/P = 1.00), and OCP (Ca/P = 1.33).

50
SUPPLEMENTARY NOTE 1
Extended classical nucleation theory for homogenous nucleation
The extended classical nucleation theory as described in the manuscript for heterogenous
nucleation on a collagen substrate can easily be adapted to the case of homogenous nucleation
observed in cryo-TEM. Here nucleation of the spherical nodules proceeds via densification of
the polymeric assembly of complexes (Fig. 1c-d). This process is therefore better described by a
colloidal aggregation model, where upon aggregation of complexes into the reaction-limited
fractal aggregate (i.e. polymeric assembly), a structure with a surface energy αpol is created with
a much higher surface/volume ratio than a spherical nucleus75.
pol
nucl
polpol SR
RkTR
Gρ
ρα
παπσ
ω
π
3
44
3
4 32
3
−+−=∆ (S37)
Here Spol is the total amount of polymer surface/volume of solution, including the inner surface
of the polymer due to a less optimal packing of complexes. The ratio ρnucl/ρpol reflects the
difference in density (clusters/volume of solution) between the nucleus and a spherical
selection (with radius Rpol) of the polymer assembly containing the same amount of complexes
according to Rpol = R·( ρnucl/ρpol)1/3. In analogy to the case of heterogeneous nucleation on
collagen analyzed above, the surface energy of the nuclei 4πR2α is now reduced in value due to
the loss of polymer surface energy 4/3πR3αpolSpol (ρnucl/ρpol). However, in contrast to
heterogeneous nucleation from complexes, the remaining unknown variables do not allow for
quantification of the barriers for this case.

51
SUPPLEMENTARY REFERENCES
61) Debye, P. & Hückel , E. Zur Theorie der Electrolyte. Physik. Zeitschrift 24, 185-206
(1923).
62) Marcus, Y. Introduction to Liquid State Chemistry, p. 231-245 (John Wiley &
Sons, Chichester, 1977).
63) Chughtai, A., Marchall, R. & Nancollas, G. H. Complexes in calcium phosphate
solutions. J. Phys. Chem. 72, 208-211 (1968).
64) Zhang, J., Ebrahimpour, A. & Nancollas, G. H. Ion association in calcium
phosphate solutions at 37 °C. J. Solution Chem., 20, 455-465 (1991).
65) Moore, E.W., Verne H.J. Pancreatic calcification constants of CaHCO3+ and
CaCO30 complexes determined wit Ca2+-electrode. Am. J. Physiol., 241, G182-
G190 (1998).
66) Martell, A. E., Smith, R. & Motekaitis, R. NIST Critically Selected Stability
Constants of Metal Complexes, Database Version 5.0 NIST Standard Reference
Database 46 (Texas A&M University, College Station, TX, 1998).
67) D. York, N. Evensen, M. Martinez, Unified equations for the slope, intercept, and
standard errors of the best straight line. Am. J. Phys. 72, 367-375 (2004).
68) Sezgin, M. & Sankur, B. Survey over image thresholding techniques and
quantitative performance evaluation. J. Electron. Imaging. 13, 146 (2004).
69) Nickell, S. et al. TOM software toolbox: acquisition and analysis for electron
tomography. J. Struct. Biol. 149, 227 (2005).

52
70) Young, H. D. & Freedman, R. A. University Physics 9th
Edition, p. 738 (Addison-
Wesley Publishing Company Inc., USA, 1996).
71) Grosberg, A. Y. & Khokhlov, A. R. Statistical Physics of Macromolecules, p. 238
(AIP Press, New York, 1994).
72) Kashchiev, D. Nucleation: Basic theory with applications (Butterworths,
Heinemann, Oxford, 1999).
73) Wilson, R., Elliott, J. & Dowker, S. Rietveld Refinement of the Crystallographic
Structure of Human Dental Enamel Apatites. Am. Mineral. 84, 1406-1414 (1999).
74) Barsoum, M. Fundamentals of Ceramics, p. 86-89 (The McGraw-Hill Companies
Inc., USA, 2000).
75) Kalinin, S. V., Vertegel, A. A., Oleynikov, N. N. & Tetryakov, Y. D. Kinetics of Solid
State Reactions with Fractal Reagent. J. Mater. Synth. Process. 6, 305-309 (1998).
76) Sigel, H., Scheller, K.H., Prijs, B. "Metal Ion/Buffer Interactions- Stability of Alkali
and Alkaline Earth Ion Complexes with Triethanolamine (Tea), 2-Amino-
2(hidroxymetyl)-1,3-propanediol (Tris) and 2-[Bis(2-hidroxyethyl)-amino] 2
(hidroxymethyl)- 1,3 propanediol (Bistris) in Aqueous and Mixed Solvents"
Inorganica Chimica Acta 66, 147-155 (1982).







![05-Penurunan Pondasi Dangkalwidodosuyadi.lecture.ub.ac.id/files/2012/05/05-Penurunan...Title 05-Penurunan Pondasi Dangkal [Compatibility Mode] Author WIDODO SUYADI Created Date 5/14/2012](https://static.fdocument.org/doc/165x107/5c85e47409d3f2e9068b9fe5/05-penurunan-pondasi-05-penurunan-pondasi-dangkal-compatibility-mode-author-widodo.jpg)
![Ν-3325/05 (ΦΕΚ-68/Α/11-3-05)€¦ · Web viewΝ-3325/05 (ΦΕΚ-68/Α/11-3-05) [ ΙΣΧΥΕΙ από 11-3-05] (ΦΕΚ-68/Α/05) Ίδρυση και λειτουργία βιομηχανικών,](https://static.fdocument.org/doc/165x107/601ade5699d7095f7870ece3/-332505-6811-3-05-web-view-332505-6811-3-05-.jpg)









