
STRUCTURE ÉLECTRONIQUE DES...
28
JL 05-06 HdB-2PC F:\Documents and Settings\Julien\Mes documents\Chimie\chimie cours\cours orga 2006\molécules-2006-v1.doc 27/11/05 CHAPITRE O2 STRUCTURE ÉLECTRONIQUE DES MOLÉCULES En première année, nous nous sommes intéressés aux molécules diatomiques par la méthode des orbitales moléculaires. Dans ce chapitre, nous étendons cette étude aux molécules polyatomiques. Dans le modèle CLOA, les orbitales moléculaires sont des combinaisons linéaires de toutes les orbitales atomiques de tous les atomes engagés dans la molécule. Ainsi les électrons sont délocalisés sur l’ensemble de la molécule et nous perdons la notion-même de liaison entre deux atomes. Nous nous limiterons à l’étude des polyènes conjugués et des composés analogues dans le formalisme simplifié de Hückel.
Transcript of STRUCTURE ÉLECTRONIQUE DES...

JL 05-06 HdB-2PC
F:\Documents and Settings\Julien\Mes documents\Chimie\chimie cours\cours orga 2006\molécules-2006-v1.doc 27/11/05
CHAPITRE O2
STRUCTURE ÉLECTRONIQUE DES MOLÉCULES
En première année, nous nous sommes intéressés aux molécules diatomiques
par la méthode des orbitales moléculaires. Dans ce chapitre, nous étendons
cette étude aux molécules polyatomiques. Dans le modèle CLOA, les orbitales
moléculaires sont des combinaisons linéaires de toutes les orbitales atomiques
de tous les atomes engagés dans la molécule. Ainsi les électrons sont
délocalisés sur l’ensemble de la molécule et nous perdons la notion-même de
liaison entre deux atomes. Nous nous limiterons à l’étude des polyènes
conjugués et des composés analogues dans le formalisme simplifié de Hückel.

2 LES OUTILS DE LA CHIMIE ORGANIQUE
1. MISE EN PLACE DU MODÈLE
La structure électronique des molécules polyatomiques est extrêmement complexe. Rappelons que toute étude repose sur la connaissance de la structure géométrique, accessible par l’expérience.
Nous ferons usage des mêmes approximations que celles qui ont été utilisées pour les molécules diatomiques :
l’approximation de Born-Oppenheimer, qui consiste à figer les noyaux et à ne considérer que le mouvement des électrons,
l’approximation orbitale où nous négligeons les interactions entre électrons. Ainsi la fonction d’onde polyélectronique s’écrit sous la forme d’un produit de fonctions d’onde monoélectroniques. De même, l’énergie électronique est égale à la somme des énergies des électrons individualisés.
Nous nous plaçons dans le cadre de la méthode CLOA : chaque fonction d’onde moléculaire (ou orbitale moléculaire) est une combinaison linéaire des orbitales atomiques des atomes mis en jeu. Nous savons alors que seules se combinent entre elles des orbitales atomiques ayant des niveaux relativement proches en énergie et qui ont le même comportement vis-à-vis des opérations de symétrie qui laissent la molécule inchangée.
La suite de l’étude sera limitée aux molécules planes ou localement planes pour lesquelles il est possible, dans un formalisme relativement aisé à mettre en œuvre, d’obtenir les orbitales moléculaires essentielles à l’interprétation de la réactivité chimique. Ainsi nous nous intéresserons aux alcènes – éventuellement substitués par des hétéroatomes –, aux polyènes conjugués et, dans le chapitre 8, aux composés aromatiques comme le benzène et ses dérivés.
Les niveaux d’énergie électronique sont expérimentalement accessibles à l’aide de la spectroscopie de photo-électrons et les résultats obtenus par le calcul sont toujours à comparer avec les données expérimentales.
1.1. Utilisation des symétries
Le plan de la molécule (ou de la zone plane) est plan de symétrie pour la densité électronique. Nous choisissons l’axe Oz du repère orthogonal à ce plan. Ainsi les fonctions d’onde doivent être soit symétriques, soit antisymétriques dans la réflexion sur ce plan. Seules des orbitales ayant un comportement identique vis-à-vis de cette opération de symétrie peuvent alors se combiner.
Les orbitales symétriques par rapport au plan donnent lieu à la formation d’un premier jeu d’orbitales moléculaires, admettant le plan de la molécule comme plan de symétrie, constitué d’orbitales de type σ (obtenues par recouvrement axial) et π (obtenues par recouvrement latéral dans le plan).
Les orbitales antisymétriques, orthogonales à ce plan, donnent lieu à la formation d’un autre jeu d’orbitales moléculaires, par recouvrement latéral, donc de type π. Ces orbitales admettent le plan de la molécule comme plan d’anti-symétrie.
Le calcul montre que les électrons π sont, en général, moins liés aux noyaux que les autres électrons. Plus polarisables, ils sont donc souvent responsables de la réactivité chimique. C’est pourquoi le système antisymétrique d’orbitales π sera le seul à faire l’objet d’une étude attentive dans le cas des alcènes et des polyènes.

[2] structure électronique des molécules 3
NOTE : les orbitales symétriques ne peuvent être obtenues « manuellement » que par des méthodes brutales d’approximation, revenant à relocaliser les électrons entre deux atomes (méthode de l’hybridation) ou à l’aide d’ordinateurs équipés des logiciels appropriés.
Il faut néanmoins garder présente l’idée que la structure géométrique d’une molécule résulte d’un ensemble de facteurs et que les électrons du système symétrique peuvent jouer un rôle essentiel. Ainsi des théoriciens ont montré que la structure du benzène était régulière non pas à cause des interactions entre électrons π mais par suite des interactions entre électrons du système symétrique.
1.2. Système antisymétrique ππππ de l’éthène
Comme nous l’avons indiqué en première année dans l’étude des alcènes, l’éthène
42HC est une molécule plane [ figure 2.1].
C C
H
HH
H
116,6 °
121,7 °
d(C C) = 133,0 pm
d(C H) = 107,6 pm
Figure 2.1 – Paramètres géométriques de l’éthène
Le système π est constitué à partir des deux orbitales zp2 des deux atomes de carbone, antisymétriques dans la réflexion sur le plan. Ici, la détermination des orbitales moléculaires est élémentaire, puisque le système est formellement analogue à celui de l’ion moléculaire +
2H : compte tenu de la présence d’un plan de symétrie orthogonal à l’axe CC− , les orbitales π sont soit symétriques, soit antisymétriques dans la réflexion sur ce plan. Nous trouvons donc les expressions suivantes :
][ 21 ppaSS +=Ψ ][ 21 ppaAA −=Ψ
L’allure de la distribution spatiale des électrons est représentée par des courbes ou des surfaces d’iso-densité électronique, obtenues à l’aide d’un logiciel [ figure 2.2]. Ces courbes mettent en évidence le caractère liant de l’orbitale SΨ (augmentation de la densité électronique entre les atomes) et le caractère antiliant de l’orbitale ΨA (existence d’un plan nodal entre les atomes).
Nous utiliserons souvent les représentations des parties angulaires des orbitales atomiques [ figure 2.3] mettant simplement en évidence la symétrie des fonctions et la présence des plans nodaux (la taille des lobes est proportionnelle à la valeur du coefficient de l’orbitale atomique dans l’orbitale moléculaire).

4 LES OUTILS DE LA CHIMIE ORGANIQUE
Figure 2.2 – Surfaces d’isodensité électronique des orbitales π et π* de l’éthène (vue dans le plan orthogonal au plan de la molécule)
orbitale ΨS liante (π) orbitale ΨA antiliante (π*)
Figure 2.3 – Représentations schématiques des orbitales π et π* de l’éthène
Il reste à déterminer les valeurs des niveaux d’énergie associés, c’est l’objet de la méthode de Hückel (ou de toute autre méthode de calcul).
1.3. Méthodes de détermination des OM
Plusieurs méthodes existent, plus ou moins complexes à mettre en œuvre. Les méthodes dites ab initio permettent d’obtenir des résultats quantitativement très corrects, c’est-à-dire proches des valeurs expérimentales mais sont très coûteuses en temps de calcul. � Erich Hückel,
physicochimiste allemand, (1896-1980), co-auteur d’une théorie électrostatique des écarts à l’idéalité en solution aqueuse, avec Petrus Debye, physico-chimiste néerlandais, prix Nobel de chimie en 1936.
Les chercheurs ont donc développé des méthodes beaucoup plus rustiques mais aisées à mettre en œuvre. Certes, les résultats numériques peuvent assez éloignés des valeurs expérimentales, mais l’accord qualitatif est bon et nous nous en contenterons, à ce premier niveau d’étude.
La méthode dite « Hückel simple » � permet d’accéder sans trop de difficultés aux énergies et aux coefficients des OM de type π dans les molécules planes conjuguées. C’est elle qui est étudiée dans la section suivante. L’avantage de cette méthode réside dans la simplicité de sa mise en œuvre, liée à sa rusticité. Elle est en particulier utilisable avec un micro-ordinateur ou même une calculette programmable. � REMARQUE : la connaissance des orbitales moléculaires nous permettra par la

[2] structure électronique des molécules 5
suite de prévoir la réactivité de l’entité face à un réactif donné, en nous intéressant aux recouvrements entre les orbitales du substrat et celles du réactif. Nous utiliserons pour ce faire l’approximation dite des « orbitales frontières ».
NOTE : la méthode des fragments, qui consiste à découper la molécule en blocs plus simples et, ensuite, à faire interagir les orbitales « de fragment » obtenues, permet souvent de résoudre des problèmes délicats. La seule difficulté réside dans le choix de la fragmentation (elle doit respecter les symétries du problème et donner lieu à des calculs simples) et dans la modélisation desdits fragments. Cette méthode dépasse le cadre de notre programme.
1.4. Principes de la méthode « Hückel simple »
La méthode « Hückel simple » s’applique à l’éthène et aux entités conjuguées, c’est-à-dire à des composés localement plans, où les électrons de N atomes possédant une orbitale atomique de type p, toutes d’axes de révolution parallèles, sont engagés dans la délocalisation électronique. Nous l’utiliserons successivement dans ce cours pour déterminer les orbitales π :
de l’éthène,
des polyènes conjugués – comme le buta-1,3-diène,
des composés aromatiques – comme le benzène,
des composés carbonylés α,β-éthyléniques, comme le propénal,
des énols comme l’éthénol et de leurs bases conjuguées, les énolates.
Il s’agit toujours de construire des orbitales moléculaires monoélectroniques, pouvant donc décrire au maximum deux électrons de spins opposés.
Hypothèses générales � C’est une hypo-thèse essentielle.
Chacun des N atomes engage dans l’orbitale moléculaire une seule � orbitale atomique (OA), normée. Elles sont toutes de même nature, par exemple ici de type
zp2 . � Il faut compren-dre, dans cette écriture, que le résultat de l’application à Ψ de l’opérateur H est égal à E ⋅ Ψ , ce qui est écrit dans le cours de mathéma-tiques sous la forme H( ) EΨ = ⋅ Ψ Les orbitales moléculaires Ψ satisfont à une équation aux valeurs propres, dite
équation de Schrödinger, écrite sous la forme condensée Ψ=Ψ EH �. L’hamiltonien H est un opérateur agissant sur la fonction d’onde, qui ne sera jamais explicité. E est l’énergie d’un électron décrit par la fonction d’onde Ψ. Le but de l’étude est donc de déterminer les valeurs propres E et les fonctions propres Ψ correspondantes de l’opérateur H.
Chaque orbitale moléculaire est une combinaison linéaire d’orbitales atomiques kp : ∑=
=ΨN
kkk pa
1
L’opérateur H est hermitien (voir plus loin) et en particulier linéaire.
Nombre d’électrons mis en jeu dans le système ππππ
À un moment ou à un autre, il nous faut déterminer le nombre d’électrons décrits par le système antisymétrique π. La difficulté n’apparaît en fait que lorsque le squelette contient des hétéroatomes.
O
H
propénal
OH
éthénol

6 LES OUTILS DE LA CHIMIE ORGANIQUE
NOTE : il n’existe en réalité qu’une seule méthode exacte de décompte : elle consiste à déterminer les niveaux d’énergie de toutes les orbitales, y compris celles du système symétrique. Ensuite le décompte est aisé en utilisant les règles classiques de peuplement des niveaux. Comme cette méthode ne nous est pas accessible, nous proposons une technique empirique mais efficace (d’autres moyens analogues peuvent être mis en œuvre).
Il suffit en pratique de compter, dans la (ou les) formule(s) de Lewis du composé étudié, le nombre de doublets d’électrons engagés dans la délocalisation électronique, aussi bien les doublets π que les éventuels doublets non liants, puis de multiplier par deux le nombre obtenu. Illustrons la méthode sur quelques exemples.
L’éthène
Il ne possède qu’une seule double liaison, soit un seul doublet π. Il y a donc deux électrons dans le système π.
Nous pouvons aussi considérer que chaque atome de carbone a contribué pour un seul électron à la liaison π.
Le buta-1,3-diène
Il possède deux doubles liaisons conjuguées soit deux doublets π. Il y a donc quatre électrons à considérer.
CH2H2C
H
H
CC
CH2H2C
H
H
CC
Nous pouvons aussi considérer que chaque atome de carbone a contribué pour un seul électron à l’ensemble du système π : une fois écrite la structure et les liaisons simples assurant le squelette de la molécule, il reste par atome de carbone un seul électron, disponible pour le système π.
Le méthanal
Il possède une double liaison OC = , soit un seul doublet π. Les doublets libres de l’atome d’oxygène ne sont pas engagés dans la conjugaison puisque les directions correspondantes sont situées dans le plan de la molécule. Il y a donc deux électrons dans le système π.
Nous pouvons aussi considérer que l’atome de carbone et l’atome d’oxygène ont apporté chacun un électron au système π.
Le furane
Les propriétés physiques et chimiques du furane permettent de montrer que l’un des doublets de l’atome d’oxygène participe à la délocalisation électronique, ce que nous traduisons sur le schéma ci-après par l’écriture de formules mésomères. Le système π du furane comporte donc deux doublets π et un doublet non liant, soit six électrons délocalisés.
Nous pouvons aussi considérer que chaque atome de carbone a apporté un électron au système π et que l’atome d’oxygène en a apporté deux.
NOTE : ce type de décompte peut sembler étrange mais il est important, dans la méthode de Hückel et notamment dans la mise en place théorique de celle-ci, de connaître le nombre d’électrons apportés au système π par chaque atome. En réalité, il faut bien se rappeler qu’aucune méthode correcte de détermination de ce nombre ne peut être proposée a priori.

[2] structure électronique des molécules 7
1
2
34
5
C C
CO
CH
H H
H
C C
CO
CH
H H
H
C C
CO
CH
H H
H
...
La méthode générale d’obtention des orbitales moléculaires et des niveaux d’énergie correspondant n’est pas au programme. Nous en indiquons seulement le principe et étudions les résultats du calcul. Elle consiste en la minimisation de l’énergie d’un électron par rapport aux coefficients des orbitales atomiques dans l’orbitale moléculaire (méthode des variations).
2. MÉTHODE HÜCKEL SIMPLE APPLIQUÉE AUX NIVEAUX ππππ
DE L’ÉTHÈNE
2.1. Mise en œuvre du calcul
Pour déterminer les niveaux d’énergie des orbitales moléculaires antisymétriques π de l’éthène, nous utilisons les expressions des fonctions d’onde obtenues par l’analyse de la symétrie particulière du problème.
NOTE : cette méthode ne peut être appliquée dans le cas général. La méthode à utiliser (méthode des variations) est explicitée en annexe mais ni sa connaissance ni sa mise en œuvre ne sont au programme des concours.
Chacune des deux fonctions d’onde S S 1 2[ ]a p pΨ = ⋅ + et A A 1 2[ ]a p pΨ = ⋅ − satisfait à l’équation de Schrödinger schématique sous la forme : Ψ=Ψ EH . Ces fonctions sont à valeur réelle.
NOTE : la suite du calcul ne peut pas être exigée au concours. Conformément aux instructions du programme, les résultats doivent être fournis aux étudiants.
Multiplions à droite et à gauche par la fonction Ψ et intégrons sur tout l’espace. Nous obtenons la relation :
2(H ) d despace espace
EΨ ⋅ Ψ τ = ⋅ Ψ τ∫ ∫
Il est donc possible d’en déduire la valeur de l’énergie associée à chaque niveau sous la forme :
( )1 2 1 2
2 21 2
(H ) d ( ) H[ d
d ( ) d
espace espace
espace espace
p p p pE
p p
Ψ ⋅ Ψ τ ± ⋅ ± ] τ= =
Ψ τ ± τ
∫ ∫∫ ∫
Soit tous les signes sont « + » et nous obtenons SE , soit tous les signes sont « – » et nous obtenons AE .
Le développement des termes sous les signes intégrale conduit au résultat suivant :

8 LES OUTILS DE LA CHIMIE ORGANIQUE
1 1 1 2 2 1 2 2
2 21 1 2 2
H d H d H d H d
d 2 d d
esp esp esp esp
esp esp esp
p p p p p p p p
Ep p p p
⋅ τ ± ⋅ τ ± ⋅ τ + ⋅ τ
=τ ± ⋅ τ + τ
∫ ∫ ∫ ∫∫ ∫ ∫
Les deux intégrales 2 di
esp
p τ∫ sont égales à 1 car les fonctions d’onde atomiques sont
normées. Nous définissons successivement :
l’intégrale de recouvrement : 12 1 2 desp
S p p= ⋅ τ∫ ( d 1ii i i
esp
S p p= ⋅ τ =∫ )
l’intégrale coulombienne : dii i i
esp
H p Hp= ⋅ τ∫ notée iα
l’intégrale de résonance : H dij i j
esp
H p p= ⋅ τ∫ , encore appelée intégrale
d’échange et notée ijβ . � REMARQUE : le caractère hermitien de l’opérateur H entraîne, d’après le cours de mathématiques, la relation ij jiH H= soit ij jiα = α .
Ces différentes intégrales vont être traitées comme des paramètres ajustables. Elles dépendent uniquement de la structure géométrique de la molécule et des atomes mis en jeu.
2.2. Valeur des paramètres
La méthode dite Hückel simple repose sur les approximations (brutales) suivantes :
Intégrales de recouvrement
Les atomes sont supposés suffisamment éloignés pour que les intégrales de recouvrement ijS soient prises nulles si i est différent de j. Rappelons que 1iiS =
Nous pouvons utiliser le symbole de Kronecker pour exprimer toutes ces relations sous la forme ijijS δ= où ijδ vaut 1 si ji = et 0 si ji ≠ .
Intégrales coulombiennes
Les intégrales coulombiennes prennent toutes deux une valeur commune notée α, comptée négativement par convention. Cette grandeur représente approximativement l’énergie de l’électron décrit, dans l’atome de carbone isolé, par l’orbitale zp2 considérée.
Intégrales de résonance
L’intégrale de résonance entre atomes connectés (c’est-à-dire reliés par une liaison) est notée β, grandeur négative par convention, qui représente une mesure de la force de la liaison entre les atomes connectés. Elle est souvent prise proportionnelle à

[2] structure électronique des molécules 9
l’intégrale de recouvrement S. Si les atomes ne sont pas reliés, l’intégrale de résonance est nulle.
NOTE : la méthode « Hückel simple » fait l’hypothèse que les intégrales de recouvrement entre atomes sont de valeur très inférieure à 1, à la limite nulle. Le calcul en méthode dite « Hückel étendu », où le recouvrement entre atomes n’est plus négligé, donne qualitativement les mêmes résultats (mêmes signes relatifs, même ordre de grandeur des coefficients) avec des calculs plus laborieux, comme nous pourrons le voir en exercice [exercice 1.1]. Le fait de choisir β proportionnel à S avec β non nul alors que nous prenons 1<<S , même pour deux atomes connectés, est alors un simple passage à la limite, obtenu en faisant tendre S vers 0 dans les expressions obtenues.
2.3. Niveaux énergétiques des électrons
Nous exprimons alors les énergies des niveaux à l’aide des paramètres α et β, sans nous préoccuper des valeurs de ces grandeurs. Il vient alors :
pour l’orbitale S S 1 2[ ]a p pΨ = ⋅ + , symétrique dans la réflexion sur le plan médiateur de la molécule : SE = α + β . Cette orbitale est dite liante car elle ne possède pas de plan nodal (de densité électronique nulle) entre les atomes. Au contraire, la densité électronique est renforcée dans la zone comprise entre les atomes, au détriment de l’extérieur de la molécule.
pour l’orbitale A A 1 2[ ]a p pΨ = ⋅ − , antisymétrique dans la réflexion sur le plan médiateur de la molécule : AE = α −β . Il s’agit de l’orbitale π* antiliante qui possède un plan nodal entre les atomes.
Notons bien la relation S AE E<
Nous obtenons deux niveaux d’énergie non dégénérés. L’un est d’énergie plus basse ( SE = α + β ), l’autre d’énergie plus élevée ( AE = α −β ) que celles des orbitales atomiques initiales (les deux orbitales zp2 d’énergie α sont dégénérées).
Il reste à achever le calcul en normalisant les deux fonctions d’onde moléculaires. Nous obtenons immédiatement, en écrivant que l’intégrale sur l’espace de chacune d’elle est égale à 1, les relations : S A 1 2a a= =
2.4. Diagramme d’énergie des orbitales moléculaire s
Nous pouvons désormais tracer le diagramme d’énergie des orbitales moléculaires (que nous appelons plus simplement le diagramme d’orbitales moléculaires) du système π de l’éthène [ figure 2.4]. Il comporte deux électrons (ce qui correspond à la double liaison dans la formule de Lewis) et le « remplissage électronique » se fait conformément aux règles usuelles. Ainsi, le niveau de plus basse énergie est peuplé par les deux électrons de spins antiparallèles.
L’indice partiel de liaison π, défini comme en première année par la demi-différence du nombre d’électrons sur les niveaux liants et antiliants, vaut 1. La présence de la liaison π stabilise la molécule dans sa conformation plane, d’une énergie valant
β=α−β+α 22)(2 . Nous rendons compte ainsi de la planéité de l’éthène qui n’était pas évidente a priori (dans son premier état excité, la molécule n’est plus plane et il n’y a plus de liaison π).

10 LES OUTILS DE LA CHIMIE ORGANIQUE
BV
HO
2 plans nodaux
1 plan nodal
π*
π
α + β
α − β
α
E
OA OM
p1 p2
Figure 2.4 – Diagramme d’orbitales moléculaires du système π de l’éthène
L’analyse complète de la structure électronique de l’éthène montre que ces deux orbitales constituent les orbitales frontières de la molécule. L’orbitale π correspond au plus niveau peuplé : nous l’appelons HO (plus haute occupée) ou HOMO (highest occupied molecular orbital) en anglais. L’orbitale π* est « la plus basse vacante » : nous l’appelons BV ou LUMO (lowest unoccupied molecular orbital).
NOTE : en toute rigueur, nous devrions parler de niveau peuplé et non d’orbitale occupée. En effet, une orbitale n’est qu’une fonction mathématique monoélectronique. Nous admettrons néanmoins cet usage très courant.
3. SYSTÈME ANTISYMÉTRIQUE ππππ DU BUTA-1,3-DIÈNE
3.1. Présentation
Le buta-1,3-diène de formule brute 64HC est le plus simple des diènes dits « conjugués ». Une telle molécule comporte, en représentation de Lewis, une alternance de simples et de doubles liaisons entre atomes de carbone. L’expérience montre que tous les atomes de la molécule de buta-1,3-diène sont situés dans le même plan.
Cette molécule peut adopter deux conformations privilégiées : cissoïde — ou s-cis — et transsoïde — ou s-trans [ figure 2.5].
s-trans
H C
C C
C H
H
H
H
H
s-cis
H C
C C
C
H
H
H
H
H
1
23
4
d(C C) = 134 pm1 2
d(C C) = 134 pm3 4
d(C C) = 147 pm2 3
Figure 2.5 – Conformations privilégiées du buta-1,3-diène

[2] structure électronique des molécules 11
La conformation privilégiée est en général la conformation s-trans, pour laquelle il y a une moins grande proximité des atomes d’hydrogène en positions 1 et 4. Cependant, dans certains diènes cycliques comme le cyclopenta-1,3-diène, seule la conformation s-cis est possible.
Les diènes conjugués présentent une réactivité et des caractéristiques structurales particulières qu’il n’est pas possible d’interpréter par la seule structure de Lewis à électrons localisés. Ainsi, la barrière de rotation autour de la liaison 2C 3C− est légèrement plus élevée que celle qui est mesurée pour un alcane (de l’ordre de 1molkJ18 −⋅ au lieu d’environ 1molkJ12 −⋅ ).
NOTE : les spécialistes s’accordent pour admettre que la « faible » longueur de liaison observée entre les atomes de carbone 2 et 3 n’est pas un critère de délocalisation électronique.
De même, le spectre d’absorption UV-visible fait apparaître un déplacement vers le rouge de la longueur d’onde correspondant au maximum d’absorption (effet bathochrome) par rapport à l’éthène.
Les diènes conjugués possèdent une réactivité chimique particulière mise en évidence, par exemple, dans la réaction de cycloaddition de Diels-Alder (étudiée au chapitre suivant) :
COOMe
COOMe
‡COOMe
Nous avons, en première année, traduit la délocalisation électronique par l’écriture de formules mésomères. Mais cette méthode est peu satisfaisante car les formules II et III font apparaître des séparations de charge et des lacunes électroniques :
H C
C C
H
H
C H
H
H H C
C C
H
H
C H
H
H H C
C C
H
H
C H
H
H
(II) (III)(I)
3.2. Utilisation des symétries
La méthode de Hückel ne différencie pas les conformations s-cis et s-trans puisqu’elle ne tient compte que des relations de connectivité. Nous étudions par conséquent la conformation s-cis qui possède plus d’éléments de symétrie, notamment le plan orthogonal à la liaison 2C 3C− .
Une orbitale moléculaire du système antisymétrique π se met a priori sous la forme d’une combinaison linéaire des 4 orbitales atomiques 2 zip des 4 atomes de carbone du squelette : 1 1 2 2 3 3 4 4i i i i ia p a p a p a pΨ = ⋅ + ⋅ + ⋅ + ⋅
cyclopentadiène
C
C1
2
3 4
C
C

12 LES OUTILS DE LA CHIMIE ORGANIQUE
Compte tenu de la symétrie de la molécule (elle est transformée en elle-même par la réflexion dans le plan indiqué), la densité électronique est inchangée par cette opération, ce qui entraîne un caractère symétrique ou antisymétrique pour la fonction d’onde.
Ainsi, en notant σ la réflexion dans le plan P nous avons les relations :
S S( )σ Ψ = Ψ ou A A( )σ Ψ = −Ψ
Ces relations se traduisent respectivement par :
S1 4 S2 3 S3 2 S4 1 S1 1 S2 2 S3 3 S4 4a p a p a p a p a p a p a p a p⋅ + ⋅ + ⋅ + ⋅ = ⋅ + ⋅ + ⋅ + ⋅ et
A1 4 A2 3 A3 2 A4 1 A1 1 A2 2 A3 3 A4 4a p a p a p a p a p a p a p a p− ⋅ − ⋅ − ⋅ − ⋅ = ⋅ + ⋅ + ⋅ + ⋅
Nous en déduisons les relations :
dans le cas d’une orbitale symétrique : S2 S3a a= et S4 S1a a=
dans le cas d’une orbitale antisymétrique : A3 A2a a= − et A4 A1a a= −
Les fonctions d’onde ont alors pour expression :
dans le cas d’une orbitale symétrique : ( ) ( )S S1 1 4 2 2 3Sa p p a p pΨ = ⋅ + + ⋅ +
dans le cas d’une orbitale antisymétrique : ( ) ( )A A1 1 4 A2 2 3a p p a p pΨ = ⋅ − + ⋅ −
Il n’est pas possible d’aller plus loin sans utiliser de méthode de calcul. Dans la méthode Hückel simple, implémentée dans de nombreux logiciels et aisée à programmer sur micro-ordinateur (à l’aide d’un logiciel de calcul formel par exemple) ou même une calculette programmable, il est inutile de faire apparaître ces simplifications compte tenu de la rapidité de calcul des outils informatiques d’aujourd’hui. C’est pourquoi nous reprenons l’expression générale de la fonction d’onde.
3.3. Niveaux d’énergie électronique
Principe
NOTE : le principe de ce calcul ne peut être exigé au concours.
L’application de la méthode des variations (voir annexe) conduit à transformer l’équation de Schrödinger Ψ=Ψ EH en une équation matricielle qui prend, nous l’admettons, la forme suivante :
11 14 1 1
41 44 4 4
Idi i
i i
H H a a
E
H H a a
= × …⋮ ⋱ ⋮ ⋮ ⋮⋯
où les coefficients ijH sont définis comme dans la section 3 et où Id représente la matrice identité à 4 lignes et 4 colonnes.
Dans le cas qui nous préoccupe, celui du buta-1,3-diène, nous avons les relations :
α==== 44332211 HHHH car les 4 atomes sont des atomes de carbone

[2] structure électronique des molécules 13
0241413 === HHH car les atomes correspondants ne sont pas connectés
β=== 342312 HHH car les atomes correspondants sont connectés par des
liaisons identiques.
L’équation matricielle s’écrit alors sous la forme détaillée :
1
2
3
4
0 0 0
0 0
0 0
0 0 0
i
i
i
i
aE
aE
aE
aE
α − β β α − β = β α − β β α −
Ce système d’équations n’a de solution non nulle que si le déterminant de la matrice est nul. Les valeurs de l’énergie correspondantes sont données par l’équation déterminantale suivante, appelée « équations séculaire » :
0
00
0
0
00
=
−αββ−αβ
β−αββ−α
E
E
E
E
ou 0
100
110
011
001
=
x
x
x
x
, en posant β−α= E
x
Rechercher les valeurs propres et les vecteurs propres de l’opérateur H revient donc
à déterminer le noyau de l’application dont la matrice est { }ij ijH E− ⋅δ .
Une fois résolue cette équation, les valeurs des coefficients ika pour une valeur iE de
l’énergie s’obtiennent en résolvant le système d’équations précédents, de rang 3. L’indétermination est levée en écrivant que la fonction d’onde est normée
( 2 1ikka =∑ )
Résultats
Le calcul de ce déterminant est possible ici directement. Diverses méthodes plus ou moins élégantes sont utilisables, l’idée étant de réduire le calcul d’un déterminant d’ordre q à un déterminant d’ordre 1−q . Des logiciels du domaine public comme « HMO plus 1.5.1 » (sous MacOS) ou « Hückel 3.0 » (sous Windows™), disponibles sur Internet, donnent très rapidement sur un micro-ordinateur les résultats complets du calcul. Il est aussi possible d’utiliser des logiciels de calcul formel.
En développant le déterminant selon la première colonne, nous obtenons tous calculs faits l’équation séculaire : 013 24 =+− xx .
Les quatre solutions de cette équation bicarrée sont telles que2
532 ±=x . Nous les
exprimons sous la forme : 2
51±±=x
Nous en déduisons les quatre valeurs numériques approchées de l’énergie, par ordre croissant (β est négatif), puis grâce au logiciel (Hückel 3.0 par exemple) les expressions normées des fonctions d’onde moléculaires (ces expressions ne sont pas à connaître).

14 LES OUTILS DE LA CHIMIE ORGANIQUE
β+α= 62,11E )(60,0)(37,0 32411 pppp +++=Ψ
β+α= 62,02E )(37,0)(60,0 32412 pppp −+−=Ψ
β−α= 62,03E )(37,0)(60,0 32413 pppp +−+=Ψ
β−α= 62,14E )(60,0)(37,0 32414 pppp −−−=Ψ
Diagramme d’orbitales moléculaires
Traçons le diagramme d’orbitales moléculaires du système π du buta-1,3-diène [ figure 2.6]. Comme nous l’avons indiqué, nous devons répartir quatre électrons sur les niveaux énergétiques les plus bas. Il est important de repérer la HO ( 2Ψ ), la BV ( 3Ψ ) et en particulier leur symétrie. Ici encore, le calcul complet de la structure électronique montrerait que les orbitales du système π sont les orbitales frontières du butadiène. � REMARQUE : nous observons que le nombre de plans nodaux augmente
régulièrement quand l’énergie de l’orbitale augmente (les orbitales moléculaires présentent toutes au moins un plan nodal, le plan de la molécule). Cette observation permet de retrouver qualitativement la symétrie des orbitales quand on ne s’intéresse pas particulièrement aux valeurs relatives des coefficients.
Ψ1
α − 0,62 β
α
E
Ψ2
Ψ3
Ψ4α − 1,62 β
α + 0,62 β
α + 1,62 β
4 plans nodaux
3 plans nodaux
2 plans nodaux
1 plan nodal
HO
BV
OA OM
pi
Figure 2.6 – Diagramme d’OM du système π du buta-1,3-diène
Énergie de résonance
L’énergie électronique totale du système π est égale à )(2 21 EE +× soit 4 4,48α + β . Si les deux liaisons π étaient indépendantes, analogues à celle de l’éthène, l’énergie électronique vaudrait )(4 β+α× .
Il y a donc une stabilisation de 0,48β par rapport à un système qui aurait pour structure électronique π la formule de Lewis fondamentale. La délocalisation des électrons sur l’ensemble du squelette est bien un phénomène stabilisant.

[2] structure électronique des molécules 15
L’énergie de stabilisation correspond à l’énergie microscopique de résonance, différence entre l’énergie électronique d’une molécule de butadiène et celle d’une molécule du composé hypothétique qui aurait pour structure électronique la formule de Lewis fondamentale.
Indice de liaison ππππ
L’indice partiel de liaison π noté rsl entre deux atomes r et s est calculé selon la formule empirique : ∑=
jjsjrjrs aanl
jn désigne le nombre d’électrons décrits par l’orbitale moléculaire jΨ pour laquelle les coefficients de l’orbitale zrp2 et zsp2 sont respectivement jra et jsa .
Pour le butadiène, seules les deux orbitales 1Ψ et 2Ψ sont utilisées, ce qui donne 89,03412 == ll et 45,023 =l .
L’indice partiel de liaison π est donc beaucoup plus élevé entre les atomes extrêmes qu’entre les atomes centraux, ce qui est compatible avec les résultats issus de l’application de la méthode de la mésomérie.
NOTE : d’autres formules empiriques permettent de calculer les longueurs de liaison π. Nous
trouvons pm1353412 == dd et pm14423 =d , ce qui est ici – un peu par hasard – , tout à fait
compatible avec les données expérimentales.
Charge partielle ππππ et charge nette
La charge partielle π sur l’atome kX , notée πkq , et la charge électronique nette kq
sont données, avec les notations précédentes, par les relations : ∑−=π
jjkjk cneq 2 π+= kkk qemq
km désigne le nombre d’électrons engagés dans la délocalisation par l’atome kX .
Pour le butadiène, nous observons que les charges électroniques π sont égales à e− . Chaque atome porte donc globalement une charge nulle puisque l’atome de carbone engage un seul électron dans le système π.
Ces résultats sont compatibles avec le poids statistique important de la formule de Lewis à deux doubles liaisons : indice de liaison π élevé entre pour les liaisons
C(2)C(1)− et C(4)C(3)− , charge nulle sur chaque atome de carbone.
Nous nous servirons de l’ensemble de ces résultats pour des prévisions de réactivité, dans le modèle des orbitales frontières exposé au chapitre 3. � REMARQUE : dans le cas de l’éthène, nous pourrions aisément vérifier, en
appliquant les formules précédentes, que l’indice partiel de liaison π est égal à 1 et que les charges nettes sont nulles sur chaque atome de carbone, ce qui est en cohérence avec le poids statistique essentiel de la formule classique de Lewis.

16 LES OUTILS DE LA CHIMIE ORGANIQUE
4. ÉTUDE DE L’ÉTHÉNOL ET DU PROPÈNE
4.1. Structure de l’éthénol
Présentation
L’éthénol 2H C CHOH= est le prototype des énols, composés comportant un groupe hydroxyle fixé sur une double liaison C C= . En général ces composés sont particulièrement instables et ne peuvent être isolés, se transformant rapidement et quasi totalement en composé carbonylé (ici l’éthanal H3CCHO). Ils sont néanmoins parfaitement caractérisés en phase gazeuse [figure 2.7] et nous verrons dans le chapitre 4 dédié à l’étude des composés carbonylés qu’ils interviennent comme intermédiaires réactionnels dans de nombreuses réactions de ces derniers.
124 °
126 °121 °
120 °
109 pm
108 pm
133 pm
108 pm
96 pm
C
H
H
H
HO
C
Figure 2.7 – Paramètres géométriques de l’éthénol
La structure de l’énol est localement plane, comme le confirment les caractéristiques géométriques de l’éthénol [ figure 2.7]. La méthode VSEPR permet de rendre compte des valeurs des angles de liaison, voisines de 120 degrés. La longueur de la liaison carbone-carbone est conforme à ce que nous attendons pour une double liaison C C= .
NOTE : les énols sont stables quand ils possèdent certaines caractéristiques structurales, présentes dans les énols de composés β-dicarbonylés, notamment une forte délocalisation électronique et une liaison hydrogène intramoléculaire. Ainsi la cyclohexanone substituée en position 2 par un groupe formyle CHO, n’existe pratiquement que sous forme énolique.
Espèce C
C
C O
O
H
H
C
C
C O
H
O
H
C
C
C O
H
O
H
Pourcentage à l’équilibre
0≈ 24 % 76 %
L’expérience montre que l’éthénol est plus réactif que l’éthène dans les réactions d’addition électrophile et que sa structure ne peut s’interpréter en termes d’électrons localisés, notamment en termes de répartition de charges. La méthode de la mésomérie permet de rendre compte de ces observations, en écrivant deux formules mésomères [figure 2.8].
Ces formules mésomères permettent de rendre compte de la répartition des charges dans la molécule. Une analyse en termes d’électrons décrits par des orbitales délocalisées sur l’ensemble de la structure est néanmoins préférable.

[2] structure électronique des molécules 17
OH OH
Figure 2.8 – délocalisation électronique dans l’éthénol
L’analyse précédente montre que nous pouvons considérer que le système d’orbitales antisymétriques π décrit 4 électrons, l’atome d’oxygène apportant deux électrons au système antisymétrique π tandis que chaque atome de carbone en apporte un.
Orbitales moléculaires antisymétriques ππππ de l’éthénol
En adoptant la numérotation indiquée sur la figure ci-contre, nous obtenons l’équation séculaire sous la forme :
C CC
CC C CO
CO O
0
0
0
E
E
E
α − ββ α − β =
β α −
En effet, en raison des relations de connectivité, nous avons la relation 13 31 0H H= = . Nous posons 12 21 CCH H= = β et 23 32 COH H= = β
Les paramètres Xα et CXβ sont usuellement exprimés à l’aide des paramètres
Cα = α et CCβ = β , à l’aide de coefficients empiriques dont nous adoptons les valeurs sans nous poser d’autre question. L’usage est de poser :
X Xhα = α + ⋅β et CX Xkβ = ⋅β
Dans le cas qui nous intéresse, les valeurs des paramètres sont X 2h = et X 0,8k =
NOTE : la plus basse valeur de α pour l’atome d’oxygène relativement à l’atome de carbone est corrélée à une plus grande électronégativité et à un niveau d’énergie pour les électrons de valence de l’atome d’oxygène plus bas que pour ceux de l’atome de carbone.
Chaque orbitale moléculaire se met sous la forme 1 1 2 2 O Oi i i ia a aΨ = φ + φ + φ . Les résultats du calcul, donnés par le logiciel Hückel 3.0, sont rassemblés dans le tableau ci-après :
Énergie 1C 2C O
2,34α + β 0,16 0,74 0,66
0,77α + β 0,38 0,57 − 0,73
1,11α − β 0,91 − 0,37 0,19
/iQ e − 0,13 0,06 0,07
Les indices partiels de liaison π sont respectivement de 0,96 pour la liaison C C− et 0,27 pour la liaison C O− .
Le diagramme d’orbitales moléculaires est présenté figure 2.9.
OH
1
2
3

18 LES OUTILS DE LA CHIMIE ORGANIQUE
α − 1,11 β
α + 0,77 βα
E
α + 2 βα + 2,33 β
2 C
O
OA OM
Figure 2.9 – Diagramme partiel d’orbitales moléculaires de l’éthénol
L’orbitale de plus basse énergie ne possède aucun plan nodal entre les atomes. Les coefficients des différentes OA sont par conséquent tous de même signe. L’OA la plus proche en énergie étant celle de l’atome d’oxygène, qui est le plus électronégatif des atomes engagés dans ce système π, nous pouvons penser que l’OM totalement liante ressemblera le plus à Op , ce qui est confirmé par le plus grand coefficient sur l’atome d’oxygène.
De même, l’orbitale d’énergie maximale doit posséder un plan nodal entre chaque doublet d’atomes connectés. Les signes des coefficients seront donc alternés. L’OA la plus éloignée en énergie de l’OM étant celle de l’atome d’oxygène, nous pouvons prévoir que le plus petit coefficient, en valeur absolue, sera celui de l’OA Op . � REMARQUE : il est beaucoup plus hasardeux de conclure pour l’OM d’énergie
intermédiaire. Nous pouvons juste prévoir l’existence d’un seul plan nodal entre deux atomes connectés, soit entre C(1) et C(2), soit entre C(2) et O.
Ces résultats sont parfaitement en accord avec les résultats qualitatifs que nous avait donné l’application de la méthode de la mésomérie.
4.2. Le groupe méthyle : un hétéroatome donneur ππππ ?
L’expérience montre que le propène et l’éthène ont des propriétés physiques et des réactivités chimiques différentes, faisant apparaître une participation des électrons du groupe méthyle à la structure électronique de la double liaison.
Il est observé par ailleurs des similitudes dans la réactivité et la structure de l’éthénol et du propène :
existence d’un moment dipolaire, certes très faible, de l’ordre de 0,35 D,
réactivité augmentée vis-à-vis des électrophiles lorsqu’elle est comparée à celle de l’éthène,
régiosélectivité marquée lors de l’addition électrophile de composés dissymétriques.
Une étude complète de la structure électronique du propène peut être menée soit grâce au logiciel de calcul Hyperchem, soit dans la méthode des fragments (voir annexe). Nous leur préférons une analyse simplifiée, analogue à celle menée dans l’étude de l’éthénol par la méthode Hückel simple.

[2] structure électronique des molécules 19
Nous assimilons le groupe méthyle à un hétéroatome X, disposant d’un doublet d’électrons qui peut être délocalisé avec les électrons π de la double liaison CC = . Aussi bizarre que cette affirmation puisse paraître, le propène constitue alors un système à 4 électrons π dispersés sur 3 atomes.
NOTE : la méthode de « l’hyperconjugaison » rend compte de cette description par l’écriture d’une délocalisation électronique partielle entre les électrons π de la double liaison et les électrons de l’une des liaisons HC − , celle qui est située dans le plan orthogonal à la double liaison. On écrit, dans ce modèle, des formules d’hyperconjugaison, analogue aux formules mésomères que nous connaissons [ figure 2.10] :
C C
H
C H
H
H
H
H
C C
H
C H
H
H
H
H
µ = 0,35 D
Figure 2.10 — Formules d’hyperconjugaison pour le propène
Mais il ne faut surtout pas déduire de l’écriture de ces formules une quelconque acidité marquée du propène ! Même si l’anion issu de la déprotonation du propène (anion allyle) possède une certaine stabilité, le propène est loin d’être un acide fort puisque le pKA du couple mis en jeu est de l’ordre de 40… Par ailleurs, la structure géométrique de l’anion allyle (qui est plan) n’a rien à voir avec celle du propène où le groupe méthyle est pyramidal.
Le groupe méthyle est, dans cette description, caractérisé par les valeurs des paramètres β+α=α 2Me et β×=β − 7,0MeC . Ces valeurs permettent de rendre compte d’une petite différence d’électronégativité entre les atomes de carbone 2 et 3, à laquelle est classiquement attribuée l’existence du moment dipolaire de la molécule (sa valeur est de l’ordre de D35,0 ).
1 plan nodal
2 plans nodaux
3 plans nodauxα − 1,08 β
α + 0,81 β
α
E
α + 2 β
α + 2,27 β
2 C
Me
OA OM
Me
Me
Me
Figure 2.11 — Diagramme d’énergie du système antisymétrique du propène

20 LES OUTILS DE LA CHIMIE ORGANIQUE
Les résultats du calcul sont les suivants, fournies par le logiciel Huckel 3.0 :
β−α=β+α=β+α=
08,1
81,0
27,2
3
2
1
E
E
E
Me213
Me212
Me211
16,072,067,0
35,059,073,0
92,035,016,0
ppp
ppp
ppp
+−=Ψ−+=Ψ++=Ψ
Le diagramme d’orbitales moléculaires, ainsi que le peuplement des niveaux, est représenté figure 2.11.
Les résultats du calcul permettent de comprendre l’existence d’une régiosélectivité dans les additions électrophiles sur le propène : en effet, l’orbitale moléculaire correspondant au niveau peuplé de plus haute énergie (HO) est dissymétrique. Nous verrons que si la réaction a lieu « sous contrôle orbitalaire » [cf chapitre suivant] il est possible de rendre compte du lien entre les deux observations, expérimentale et théorique.

[2] structure électronique des molécules 21
Annexes
5. PRINCIPE DE LA MÉTHODE DES VARIATIONS
5.1. Principe du calcul
Nous exprimons l’énergie E à l’aide des différents coefficients introduits et nous recherchons les valeurs des coefficients pour lesquelles E est extrémale. La fonction d’onde est supposée, dans un premier temps, pour plus de généralités, à variables réelles mais à valeurs complexes.
En multipliant à gauche les deux membres de l’équation de Schrödinger par *Ψ , complexe conjugué de Ψ , puis en intégrant sur tout l’espace, nous obtenons la relation : ∫∫ ∫ τΨ⋅=τΨ⋅Ψ⋅=τΨ⋅Ψ
espaceespace espaceEE ddd)(H
2**
Développons la fonction d’onde moléculaire à l’aide des fonctions d’onde atomiques. Nous obtenons l’équation suivante : τ+τ⋅=τ+τ ∑ ∑ ∫∑ ∫∑ ∑ ∫∑ ∫
≠≠ i ijjespace ijji
iespace ii
i ijjespace ijji
iespace iii ppaapaEppaappa
,
*22
,
**2 dddHdH
Comme indiqué dans le cas du butadiène, nous définissons successivement :
l’intégrale de recouvrement : ∫ τ=espace jiij ppS d*
l’intégrale coulombienne : ∫ τ=espace iiii ppH dH*
l’intégrale de résonance : ∫ τ=espace jiij ppH dH*
L’opérateur H est hermitien, ce qui entraîne, d’après le cours de mathématiques, la
relation *jiij HH = . D’autre part, nous avons la relation *
jiij SS = . � REMARQUE : dans la suite du texte, nous choisissons d’utiliser uniquement des fonctions d’onde atomiques ou moléculaires à valeurs réelles. Comme le complexe conjugué d’un nombre réel est égal à lui-même, nous avons les relations ijij HH = et jiij SS =
L’équation la plus générale (1) s’écrit alors sous la forme ci-après : ∑∑∑≠
=−+−i ijj
ijijjii
iii SEHaaEHa,
2 0)()( (1)
ou plus simplement, en se rappelant que 1=iiS : ∑∑ =−i j
ijijji SEHaa 0)(

22 LES OUTILS DE LA CHIMIE ORGANIQUE
5.2. Valeurs des paramètres
Considérons d’abord le cas d’une molécule à squelette purement carboné.
• Intégrales de recouvrement
Dans le modèle « Hückel simple », les atomes sont supposés suffisamment éloignés pour que les intégrales de recouvrement ijS soient prises nulles si i est différent de j. Les intégrales iiS sont par définition égales à 1 puisque les orbitales atomiques sont normées.
Nous pouvons utiliser le symbole de Kronecker pour exprimer toutes ces relations sous la forme ijijS δ= où ijδ vaut 1 si ji = et 0 si ji ≠ .
• Intégrales coulombiennes
Les intégrales coulombiennes prennent toutes une valeur commune notée α, comptée négativement par convention. Cette grandeur représente approximativement l’énergie de l’électron décrit, dans l’atome isolé, par l’orbitale zp2 considérée.
• Intégrales de résonance
Les intégrales de résonance entre atomes connectés (c’est-à-dire reliés par une liaison) prennent une valeur commune notée β, grandeur négative par convention, qui représente une mesure de la force de la liaison entre les atomes connectés. Elle est souvent prise proportionnelle à l’intégrale de recouvrement S. Si les atomes ne sont pas reliés, l’intégrale de résonance est nulle.
NOTE : la méthode « Hückel simple » fait l’hypothèse que les intégrales de recouvrement entre atomes sont de valeur très inférieure à 1, à la limite nulle. Le calcul en méthode dite « Hückel étendu », où le recouvrement entre atomes nest plus négligé, donne qualitativement les mêmes résultats (mêmes signes relatifs, même ordre de grandeur des coefficients) avec des calculs plus laborieux, comme nous pourrons le voir en exercice [exercice 1.1]. Le fait de choisir β proportionnel à S avec β non nul alors que nous prenons 1<<S , même pour deux atomes connectés, est alors un simple passage à la limite, obtenu en faisant tendre S vers 0 dans les expressions obtenues.
Prenons deux exemples :
le butadiène :
1
2
3
4
α==== 44332211 HHHH
β=== 342312 HHH 0241413 === HHH
le cation cyclopropénium : α=== 332211 HHH β=== 312312 HHH
3
2
1C C
C
H
HHC C
C
H
HHC C
C
H
HH
Si le squelette de la molécule comporte des hétéroatomes, les paramètres relatifs aux différents éléments sont exprimés sous la forme β+α=α XX k et β=β CXCX h , où Xk

[2] structure électronique des molécules 23
et CXh sont des coefficients ajustés selon des règles empiriques qui seront précisées dans chaque cas. Ainsi, pour le furane, nous disposons des valeurs suivantes :
1
2
34
5
C C
CO
CH
H H
H
α==== 55443322 HHHH
β+α=α= 2O11H
β=== 453423 HHH
β=β== 8,0CO1512 HH
Déterminant séculaire
Reprenons l’équation (1) reliant les coefficients et l’énergie. En la dérivant par rapport
à chaque coefficient et en exprimant que la dérivée ijjaia
E
≠
∂∂
,
est nulle, nous
obtenons N équations linéaires du premier degré à N inconnues, de la forme : ∑≠
=+−ijj
ijjiii HaEHa,
02)(2 � Rappelons que
jiij HH = que nous rassemblons sous forme matricielle � :
= −α−α
−α−α
−α
−
−
−−−−
−
−
−
0
0
0
0
0
1
3
2
1
1,321
,11,31,21,1
31,32313
21,22312
11,11312 ⋮⋮…… ⋮⋮⋱⋮⋮⋮ ………n
n
nnnnn
nnnnn
nn
nn
nn
a
a
a
a
a
EHHHH
HEHHH
HHEHH
HHHEH
HHHHE
Pour que ce système homogène admette une solution autre que la solution triviale, il faut que le déterminant de la matrice, appelé déterminant séculaire, soit nul. L’équation correspondante est dite équation séculaire. Ensuite, la résolution du système ne pose aucune difficulté théorique et permet l’obtention des solutions associées à chacune des valeurs propres, comme nous allons le montrer sur deux exemples(1).
En pratique, l’écriture du déterminant séculaire repose sur l’observation de quelques règles élémentaires, ne dépendant que des relations de connectivité des atomes dans le squelette de la molécule :
les termes diagonaux sont égaux à E−αX
les termes non diagonaux ijH ne sont non nuls que si les atomes i et j sont
connectés. Dans ce cas, le terme ijH prend la valeur βCXh .
NOTE : dans certains cas, l’étude des symétries du problème permet d’obtenir beaucoup plus rapidement les résultats, en remplaçant la matrice initiale par une « matrice bloc », plus facile à diagonaliser.
(1) Le programme officiel précise : « aucun calcul ne peut être exigé. Les résultats des calculs de Hückel seront donnés ».

24 LES OUTILS DE LA CHIMIE ORGANIQUE
Système ππππ antisymétrique de l’éthène
Nous savons que le système π de l’éthène se compose de 2 orbitales moléculaires (OM), combinaisons linéaires des 2 orbitales atomiques zp2 des atomes de carbone. Nous allons développer la méthode dans son intégralité, même si nous avons vu que les expressions des OM étaient faciles à obtenir.
Les paramètres de Hückel sont : α== 2211 HH , β== 2112 HH . L’équation de Schrödinger s’écrit alors pour une orbitale moléculaire de la forme 2211 papa +=Ψ , pour l’instant non normalisée, et en nous rappelant que l’intégrale 12S est nulle :
)(2 22
2121
22
21 aaEaaaa +⋅=β+α+α
Ici le calcul direct des dérivées partielles de E par rapport à chacun des coefficients est possible, mais nous utilisons dans un premier temps la méthode générale pour bien comprendre son mode d’application.
Niveaux d’énergie
Nous exprimons que l’énergie du système est extrémale : pour cela, nous dérivons l’expression par rapport à chaque coefficient et nous écrivons que chaque dérivée partielle de E est nulle. Nous obtenons successivement :
en dérivant par rapport à 1a : 121 222 aEaa =β+α
en dérivant par rapport à 2a : 221 222 aEaa =α+β
Le système d’équations vérifié par les coefficients 1a et 2a s’écrit sous forme
matricielle : = −αββ−α
0
0
2
1
a
a
E
E
L’équation séculaire est : 0)( 22 =β−−α E . Elle possède deux solutions :
β±α=E
Nous retrouvons donc deux niveaux d’énergie non dégénérés. L’un est d’énergie plus basse ( β+α=1E ), l’autre d’énergie plus élevée ( β−α=2E ) que celles des orbitales atomiques initiales (les deux orbitales zp2 d’énergie α sont dégénérées).
Orbitales moléculaires
Il reste à achever le calcul en résolvant, pour chaque valeur propre, une des équations du système (une seule suffit car le système est de rang 1).
Pour β+α=1E , il vient 21 aa = . Nous obtenons l’orbitale π liante qui ne posséde pas de plan nodal entre les atomes. La normalisation de la fonction permet
d’écrire 211 =a .
Pour β−α=2E , il vient 21 aa −= . Il s’agit de l’orbitale π* antiliante qui possède un plan nodal entre les atomes. La normalisation de la fonction permet d’écrire
211 =a .
[2] structure électronique des molécules 25
Complément
Montrons, sur l’exemple de l’éthène, que l’orbitale liante correspond bien à un minimum d’énergie alors que l’orbitale antiliante correspond à un maximum d’énergie.
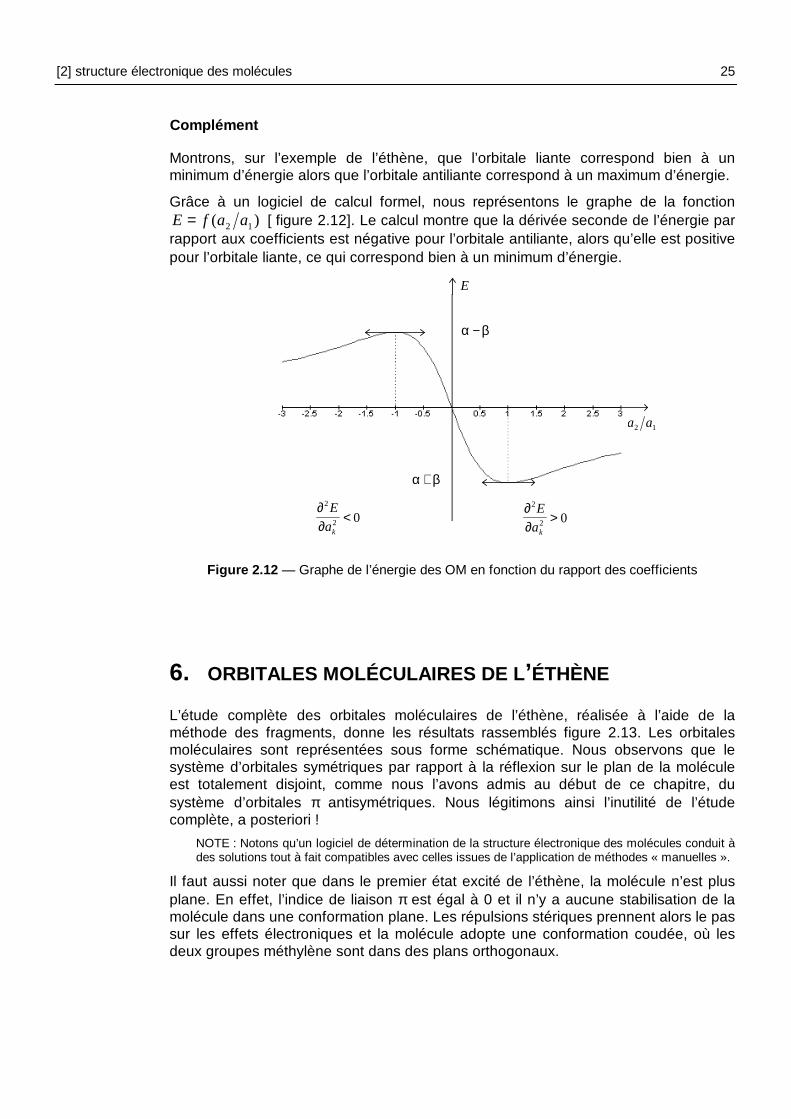
Grâce à un logiciel de calcul formel, nous représentons le graphe de la fonction )( 12 aafE = [ figure 2.12]. Le calcul montre que la dérivée seconde de l’énergie par
rapport aux coefficients est négative pour l’orbitale antiliante, alors qu’elle est positive pour l’orbitale liante, ce qui correspond bien à un minimum d’énergie.
a a2 1
E
α β−
α β+
∂∂
2
2 0E
ak
<∂∂
2
2 0E
ak
>
Figure 2.12 — Graphe de l’énergie des OM en fonction du rapport des coefficients
6. ORBITALES MOLÉCULAIRES DE L ’ÉTHÈNE
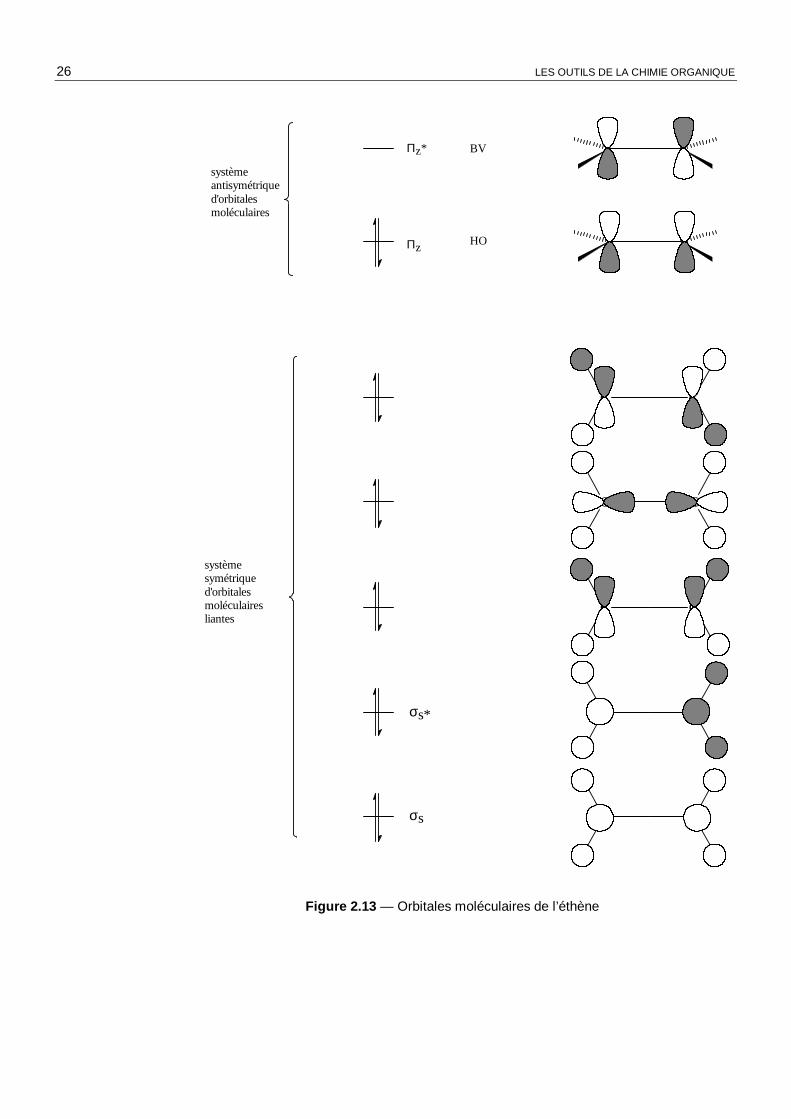
L’étude complète des orbitales moléculaires de l’éthène, réalisée à l’aide de la méthode des fragments, donne les résultats rassemblés figure 2.13. Les orbitales moléculaires sont représentées sous forme schématique. Nous observons que le système d’orbitales symétriques par rapport à la réflexion sur le plan de la molécule est totalement disjoint, comme nous l’avons admis au début de ce chapitre, du système d’orbitales π antisymétriques. Nous légitimons ainsi l’inutilité de l’étude complète, a posteriori !
NOTE : Notons qu’un logiciel de détermination de la structure électronique des molécules conduit à des solutions tout à fait compatibles avec celles issues de l’application de méthodes « manuelles ».
Il faut aussi noter que dans le premier état excité de l’éthène, la molécule n’est plus plane. En effet, l’indice de liaison π est égal à 0 et il n’y a aucune stabilisation de la molécule dans une conformation plane. Les répulsions stériques prennent alors le pas sur les effets électroniques et la molécule adopte une conformation coudée, où les deux groupes méthylène sont dans des plans orthogonaux.
26 LES OUTILS DE LA CHIMIE ORGANIQUE
C C
H
HH
H
C C
H
HH
H
C C
H
HH
H
C C
H
HH
H
C C
H
HH
H
systèmesymétriqued'orbitalesmoléculairesliantes
σs
σs*
systèmeantisymétriqued'orbitalesmoléculaires
Πz
Πz*
HO
BV
Figure 2.13 — Orbitales moléculaires de l’éthène

[2] structure électronique des molécules 27
7. ORBITALES DE FRAGMENT DU GROUPE MÉTHYLE
Le « découpage » en deux fragments du propène conduit à considérer un fragment éthène et un fragment méthyle [ figure 2.14]. L’analyse des orbitales moléculaires du groupe méthyle est présentée sur la figure 2.15. Deux structures géométriques sont envisagées : une conformation plane et une conformation pyramidale. Le remplissage des niveaux montre que le fragment méthyle est plus stable en conformation pyramidale et que l’un des doublets électroniques (de type antisymétrique) peut interagir avec les électrons de la liaison π de l’éthène, justifiant ainsi a posteriori la modélisation précédente du groupe méthyle.
C C
H
C H
H
H
H
H
18 électrons de valence
C C
H
H
H
C
H
H
H
8 électrons de valence 10 électrons de valencedont 2 électrons π
Figure 2.14 – Découpage du propène en fragments
Figure 2.15 – Orbitales moléculaires du fragment méthyle dans deux conformations
28 LES OUTILS DE LA CHIMIE ORGANIQUE
8. ORBITALES MOLÉCULAIRES DU PROPÈNE
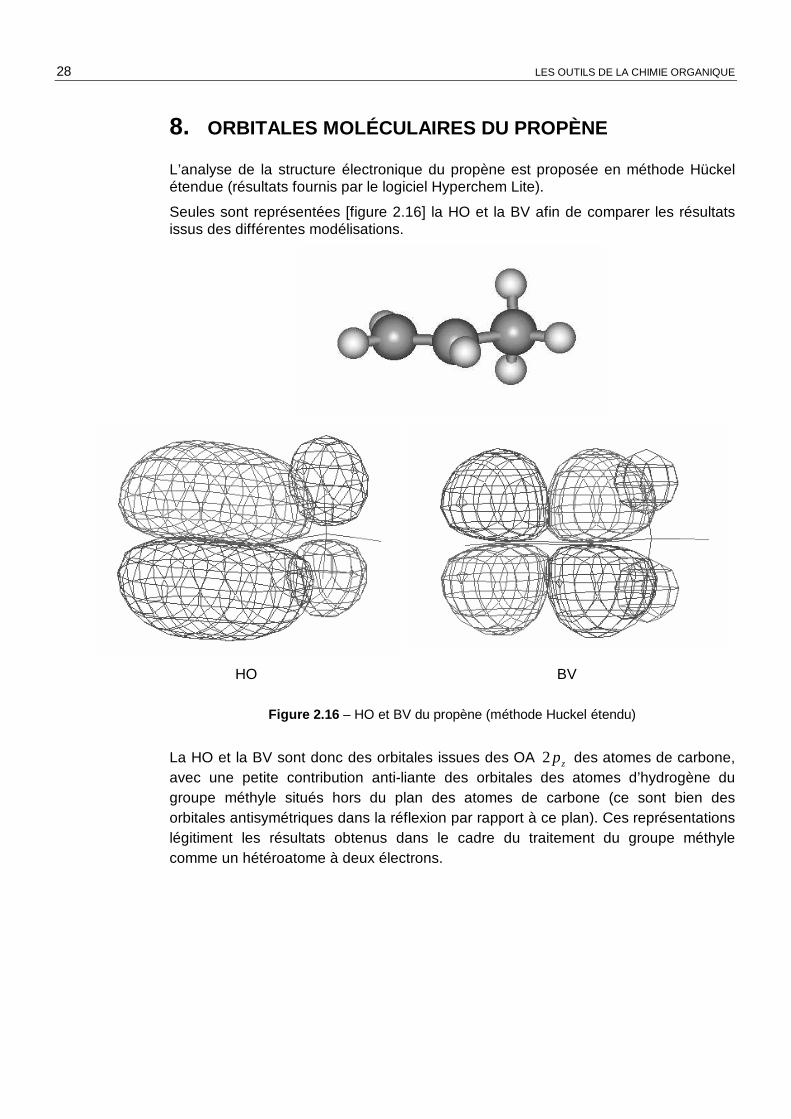
L’analyse de la structure électronique du propène est proposée en méthode Hückel étendue (résultats fournis par le logiciel Hyperchem Lite).
Seules sont représentées [figure 2.16] la HO et la BV afin de comparer les résultats issus des différentes modélisations.
HO BV
Figure 2.16 – HO et BV du propène (méthode Huckel étendu)
La HO et la BV sont donc des orbitales issues des OA zp2 des atomes de carbone, avec une petite contribution anti-liante des orbitales des atomes d’hydrogène du groupe méthyle situés hors du plan des atomes de carbone (ce sont bien des orbitales antisymétriques dans la réflexion par rapport à ce plan). Ces représentations légitiment les résultats obtenus dans le cadre du traitement du groupe méthyle comme un hétéroatome à deux électrons.








