
SMAD6S REGULATES PLASMINOGEN ACTIVATOR INHIBITOR-1 … · inhibitor –1 (PAI-1)1, has been shown...
15
1 SMAD6S REGULATES PLASMINOGEN ACTIVATOR INHIBITOR-1 THROUGH A PKC-b DEPENDENT UP-REGULATION OF TGF-b David T. Berg # , Laura J. Myers # , Mark A. Richardson, George Sandusky and Brian W. Grinnell * From the Division of Biotechnology Discovery Research, Lilly Research Laboratories, Lilly Corporate Center, Indianapolis, IN 46285, USA. Running Title: Smad modulation of PAI-1 # Contributed equally to the study * Address correspondence to: Brian W. Grinnell, Biotechnology Discovery Research, Lilly Research Laboratories, Lilly Corporate Center, Indianapolis, IN 46285-0444, USA. Tel.: 317-276-2293; Fax: 317- 277-2934; E-mail: [email protected] Plasminogen activator inhibitor –1 (PAI- 1) is a serpin class protease inhibitor that plays a central role in the regulation of vascular function and tissue remodeling by modulating thrombosis, inflammation and the extracellular matrix. A central mediator controlling PAI-1 is TGF- b , which induces its expression and promotes fibrosis. We have found that a unique member of the Smad family of signal transduction molecules, Smad6s, modulates the expression of PAI-1. Over expression of Smad6s in endothelial cells increases promoter activity and PAI-1 secretion, and an anti-sense to Smad6s suppresses the induction of PAI-1 by TGF-b . The effect of Smad6s on the PAI-1 promoter appeared to be the result of increase binding of the forkhead winged helix factor FoxD1 to a TGF-b responsive element. Further, the effect of Smad6s on PAI-1 up- regulation and on FoxD1 binding was found to result from up-regulation of TGF-b and could be inhibited by the blocking TGF- b signaling with Smad7. The ability of Smad6s to regulate the TGF- b promoter and subsequent PAI-1 induction was suppressed by a selective PKC- b inhibitor. Consistent with the in vitro data, we found that increased Smad6s in diseased vessels correlated with increased TGF- b and PAI-1 levels. Overall, our results demonstrate that the level of Smad6s can alter the level of TGF-b and the subsequent induction of PAI-1 via a FoxD1 transcription site. Further our data suggest that this process, which is up-regulated in diseased vessels, can be modulated by the inhibition of PKC- b. Vascular injury plays a major role in the pathogenesis of multiple acute and chronic disorders. The response to initial injury triggers activation of coagulation, inflammatory and repair process aimed at maintaining vascular integrity, however, an imbalance in this process can ultimately lead to aberrant fibrosis, structural dysfunction, and ultimately chronic disease. While many factors play a role in the continuum from acute injury to chronic dysfunctional vasculature, the serpin plasminogen activator inhibitor –1 (PAI-1) 1 , has been shown to play a key role in the pathogenesis of both acute and chronic disorders, including cardiovascular, renal, hepatic and pulmonary (reviewed in (1-4). As the physiologic inhibitor of tissue type plasminogen activator, PAI-1 regulates the conversion of plasminogen to plasmin and thus plays a critical role in regulating the balance between coagulation and fibrinolysis. In general, the plasminogen activator (PA)/plasmin pathway has been implicated in a wide variety of physiologic and pathologic processes that require tissue remodeling and cell motility. (5). As such, PAI-1 influences a number of cellular processes (including wound repair, cell migration, angiogenesis, neointima formation, and matrix- dependent cell attachment) largely via interactions with urokinase plasminogen activator receptor, vitronectin and indirectly through plasmin activation of extracellular matrix proteases and latent growth factors (reviewed in (6). While an important component in response to injury, chronically elevated plasma PAI-1 has been shown in increase risk for thrombosis, cancer metastasis, vascular complications of diabetes, and the development of septic shock (7-9) Although a number of cytokines and hormones are known to regulate PAI-1 (reviewed in (10) , a major factor controlling PAI-1 synthesis is transforming growth factor-beta (TFG-β). Both JBC Papers in Press. Published on February 16, 2005 as Manuscript C400579200 Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc. by guest on March 22, 2019 http://www.jbc.org/ Downloaded from
Transcript of SMAD6S REGULATES PLASMINOGEN ACTIVATOR INHIBITOR-1 … · inhibitor –1 (PAI-1)1, has been shown...

1
SMAD6S REGULATES PLASMINOGEN ACTIVATOR INHIBITOR-1 THROUGH A PKC-β DEPENDENT UP-REGULATION OF TGF-β
David T. Berg #, Laura J. Myers #, Mark A. Richardson, George Sandusky and Brian W. Grinnell* From the Division of Biotechnology Discovery Research, Lilly Research Laboratories, Lilly
Corporate Center, Indianapolis, IN 46285, USA. Running Title: Smad modulation of PAI-1
#Contributed equally to the study *Address correspondence to: Brian W. Grinnell, Biotechnology Discovery Research, Lilly Research Laboratories, Lilly Corporate Center, Indianapolis, IN 46285-0444, USA. Tel.: 317-276-2293; Fax: 317-277-2934; E-mail: [email protected]
Plasminogen activator inhibitor –1 (PAI-1) is a serpin class protease inhibitor that plays a central role in the regulation of vascular function and tissue remodeling by modulating thrombosis, inflammation and the extracellular matrix. A central mediator controlling PAI-1 is TGF-β, which induces its expression and promotes fibrosis. We have found that a unique member of the Smad family of signal transduction molecules, Smad6s, modulates the expression of PAI-1. Over expression of Smad6s in endothelial cells increases promoter activity and PAI-1 secretion, and an anti-sense to Smad6s suppresses the induction of PAI-1 by TGF-β. The effect of Smad6s on the PAI-1 promoter appeared to be the result of increase binding of the forkhead winged helix factor FoxD1 to a TGF−β responsive element. Further, the effect of Smad6s on PAI-1 up-regulation and on FoxD1 binding was found to result from up-regulation of TGF-β and could be inhibited by the blocking TGF-β signaling with Smad7. The ability of Smad6s to regulate the TGF-β promoter and subsequent PAI-1 induction was suppressed by a selective PKC-β inhibitor. Consistent with the in vitro data, we found that increased Smad6s in diseased vessels correlated with increased TGF-β and PAI-1 levels. Overall, our results demonstrate that the level of Smad6s can alter the level of TGF-β and the subsequent induction of PAI-1 via a FoxD1 transcription site. Further our data suggest that this process, which is up-regulated in diseased vessels, can be modulated by the inhibition of PKC-β.
Vascular injury plays a major role in the
pathogenesis of multiple acute and chronic
disorders. The response to initial injury triggers activation of coagulation, inflammatory and repair process aimed at maintaining vascular integrity, however, an imbalance in this process can ultimately lead to aberrant fibrosis, structural dysfunction, and ultimately chronic disease. While many factors play a role in the continuum from acute injury to chronic dysfunctional vasculature, the serpin plasminogen activator inhibitor –1 (PAI-1)1, has been shown to play a key role in the pathogenesis of both acute and chronic disorders, including cardiovascular, renal, hepatic and pulmonary (reviewed in (1-4).
As the physiologic inhibitor of tissue type plasminogen activator, PAI-1 regulates the conversion of plasminogen to plasmin and thus plays a critical role in regulating the balance between coagulation and fibrinolysis. In general, the plasminogen activator (PA)/plasmin pathway has been implicated in a wide variety of physiologic and pathologic processes that require tissue remodeling and cell motility. (5). As such, PAI-1 influences a number of cellular processes (including wound repair, cell migration, angiogenesis, neointima formation, and matrix-dependent cell attachment) largely via interactions with urokinase plasminogen activator receptor, vitronectin and indirectly through plasmin activation of extracellular matrix proteases and latent growth factors (reviewed in (6). While an important component in response to injury, chronically elevated plasma PAI-1 has been shown in increase risk for thrombosis, cancer metastasis, vascular complications of diabetes, and the development of septic shock (7-9)
Although a number of cytokines and hormones are known to regulate PAI-1 (reviewed in (10), a major factor controlling PAI-1 synthesis is transforming growth factor-beta (TFG-β). Both
JBC Papers in Press. Published on February 16, 2005 as Manuscript C400579200
Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from

2
TGF- β1 and TGF- β3 have been shown to up-regulate PAI-1 in cultured human endothelial cells at the promoter level (11-14) and although the endothelial regulation in vivo has not be widely studied, Dong et al. (15) have shown that endothelial PAI-1 up-regulation in human aortic allografts is mediated mainly by the TGF-β pathway. TGF- β family signaling is positively modulated by various members of the Smad family of signal transduction proteins (reviewed in (16,17), however, two Smads (Smad6 and 7) inhibit TGF- β signal transduction. The Smad7 protein has been extensively studied and shown to prevent phosphorylation of receptor-activated Smads, thereby inhibiting TGF- β-induced signaling responses. (18,19). Smad6 has been shown to inhibit signaling by the TGF- β superfamily (20,21), and to play a role in the development of the cardiovascular system (22). However, an endothelial splice variant of Smad6, designated Smad6s, recently has been shown to have differential activity relative to the inhibitory Smad6 and Smad7 in a Xenopus model, being an antagonist of the BMP pathway but an agonist of the activin pathways (23), and showing enhanced modulation of TGF-β signaling in human endothelial cells (24).
In this study, we have explored the role of TGF- β signaling in controlling the expression of PAI-1 in human endothelial cells, focusing on the role of the endothelial Smad6s. Using over-expression and antisense modulation, we show that Smad6s stimulates the expression and secretion of PAI-1 by enhancing the binding of nuclear factor FoxD1 to a previously identified TGF-β responsive element in the promoter. Moreover, we demonstrate the up-regulation of PAI-1 synthesis by Smad6s depends on an induction of TGF-β itself, and that this process can be effectively modulated by a selective PKC-β inhibitor. Further, we show that in human diseased vessels over-expressing Smad6s, both TGF-β1 and PAI-1 are over-expressed, consistent with the up-regulation observed in vitro.
EXPERIMENTAL PROCEDURES
Reagents and Cell culture- TGF-β1 and TGF-
β3 were purchased from R&D Systems. SVHA-1 cells, an SV40-transformed human aortic endothelial
cell line, were described previously (24). Human umbilical vein endothelial cells (HUVECs) were obtained from Clonetics (San Diego, CA). The ECV304 cell line was obtained from the (ATCC CRL-1998) and grown as described previously (25). Cells were maintained in DMEM/F-12 (3:1), a medium comprised of a 3:1 v/v mixture of Dulbecco's modified Eagle’s medium and Ham's nutrient mixture F12. DMEM/F-12 (3:1) and fetal bovine serum were purchased from Life Technologies, Inc. The basal medium was supplemented with 10 nM selenium, 50 uM, 2 aminoethanol, 20 mM HEPES, 50 ug/ml gentamycin and 5% fetal bovine serum. The PKC inhibitor LY379196 was from Eli Lilly and Co. All other reagents were of the highest quality available.
DNA transfection, Chloramphenicol acetyltransferace (CAT) ELISA, Luciferase and PAI-1 assays- The vector pOCAT2336 containing the PAI-I promoter and the Smad6s and Smad 7 expression vectors were described previously (23). The TGF-β responsive artificial PAI-1 reporter plasmid p3TP-lux was originally cloned by Dr. J. Massague's laboratory (26). Endothelial cells were seeded in six well plates to 80% confluence. DNA was transfected at a concentration of one microgram for pOCAT2336 and p3TP-lux, and five micrograms for the Smad vectors with Gibco’s lipofectin reagent used accordingly to manufacturer’s instruction. Expressed CAT protein was measured using a CAT ELISA kit from Boehringer Mannheim and performed according to manufacturer’s protocol. The plates were read kinetically and data expressed in mOD/min. The activity of firefly luciferase was measured by a Dynax Microtiter Plate Luminometer. PAI-1 antigen concentration in the conditioned media was measured by a TintElise kit (Biopool International) according to the manufacturer’s instruction.
Antisense oligodeoxynucleotides- The oligonucleotides used in antisense experiments were synthesized with phosphorothioates and C-5 propyne pyrimidines following standard protocols (27). Anti-sense oligodeoxynucleotides (oligos) were designed to hybridize to the region of the Smad6s mRNA encompassing the initial ATG. The sequence of the antisense oligodeoxynucleotide for Smad6s was: 5’-GGTTTGCCCATTCTGGACAT-3’ and the sense strand control was:
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from

3
5’-ATGTCCAGAATGGGCAAACC-3’. Oligos were introduced to cells following the procedure as outlined in (24). After four days each experimental condition was treated with or without TGF- β at a concentration of 1ng/ml in the maintenance medium. The antisense was shown to inhibit the Smad protein levels by Western blot analysis.
TFG-β promoter activation and secretion- TGF-β3 (28) and β1 promoter (29) constructs driving CAT expression (1 µg) were co-transfected with Smad6s vector (5 µg), Smad7 (5µg) or with a control pCIneo vector (Promega, Madison, WI). Transfection with lipofectin and assay for TGF-β1 and TGF-β3 by ELISA were as described previously (24). CAT activity expressed was measured kinetically. Lysates were normalized using a BCA assay measuring total protein concentration. In some experiments, either 1nM staurosporin or 100nM LY379196 were added to the culture 24 hours post transfection.
DNA binding assay – Bioinformatic analysis of the promoter region of PAI-1 was performed using MatInspector release 6.2.1. The gene locus for human PAI-1 was HSPI11, accession number X13323. The match to FoxD1 was 1.0 to the core sequence with an re-value of less than 0.01 matches per 1000 bps. Confirmation of the binding was performed by electrophoretic mobility shift assays as follows. The probe was gamma 32P labeled (NEN) and nuclear extracts were prepared as described previously (30). Reactions containing Buffer D, PolydIC, nuclear extract and labeled oligonucleotide were incubated 15-45 minutes as described previously (31). Samples were run 90 minutes at 110V, 2.0A in 1/2x TBE running buffer. Gels were dried on DE81 paper and developed overnight on Kodak film (Kodak, Rochester, NY), and quantification of was by scanning phosphoimager. In some experiments, detection was using fluorescent labeled oligonucleotides obtained from LI-COR Biosciences (Lincoln, Nebraska USA 68504) 5’ end labeled with IRDye 800 phosphoramidite. Unlabeled single stranded synthetic oligos used for competition studies were obtained from Operon Biotechnologies, Inc. Binding reaction conditions were 5fmol/ul labeled oligonucleotide, 4 units/ul Poly[dIC], and 0.3ug/ul nuclear extract and gels were scanned with the Li-Cor Odyssey Imaging
System. Competition reactions contained from 1X to 100X molar excess unlabeled oligonuleotides. The oligonucleotide containing the FoxD1 sequence was 5’ -CAAGGTTGTTGACACAAGAGAGCCCTCAGG – 3’. A mutant oligonucleotide containing changes to the core FoxD1 binding sequence was 5’ - CAAGGTTGGGGACACAAGAGA GCCCTCAGG – 3’.
Immunohistochemistry- Tissue specimens were retrieved from the tissue bank of Lilly Research Laboratories. These tissues were obtained from the Cooperative Human Tissue Network using an institutional review board-approved protocol. All human samples were derived from surgical specimens obtained during the period extending from 1996-1999. Tissues were fixed overnight in Zn buffered formalin and then transferred to 70% ethanol prior to processing through paraffin. The sectioning and staining with antibodies against Smad6, TGF-β and PAI-1 were as described previously (24). A biotinylated secondary antibody plus streptavidin-horseradish peroxidase kit (Dako LSAB2) was then utilized along with a DAB chromagen and peroxide substrate to detect the bound antibody complexes. The slides were briefly counterstained with hematoxylin, and reviewed by two investigators (GES, MD) using light microscopy to evaluate the intensity and localization of the staining.
RESULTS
TGF-β enhances PAI-1 expression from
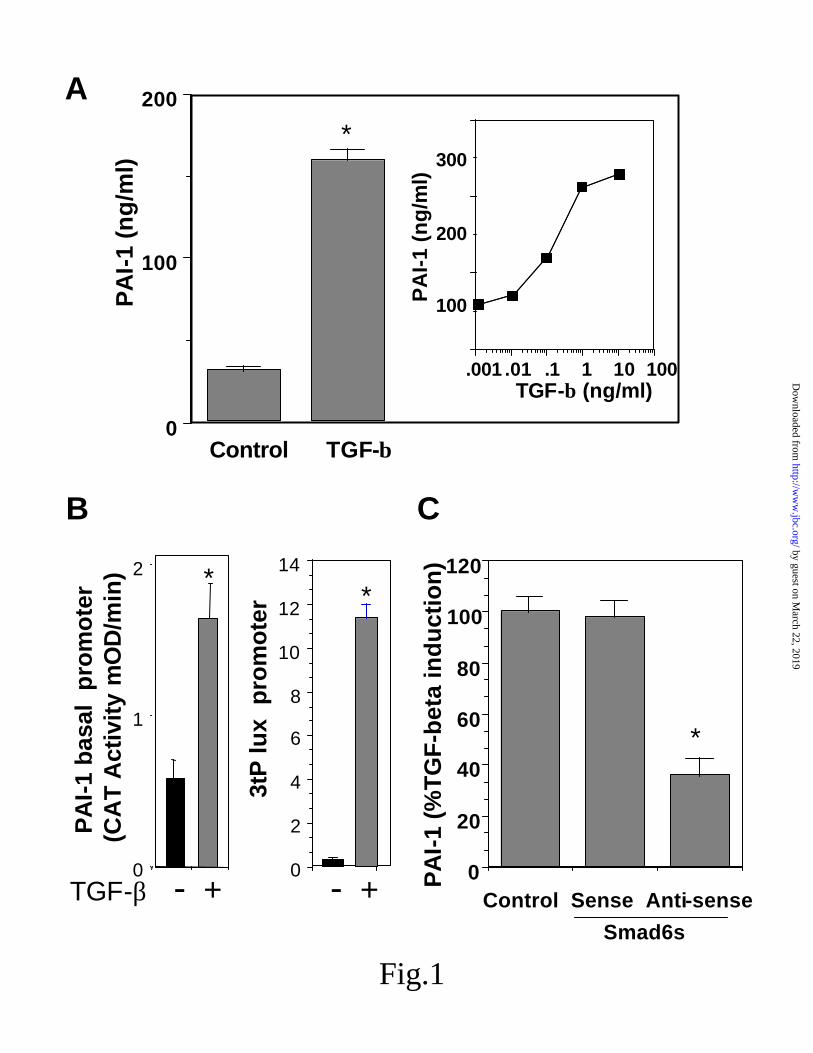
human endothelial cells- As shown in Fig 1A, the level of PAI-1 secreted from human SVHA-1 endothelial line was increased following treatments with TGF- β1 (and TGF-β3, not shown). In both endothelial SVHA-1 cells and in the ECV304 line, the effect was dose dependent with a half maximal response of ~ 25 pM. Moreover, using both the basal PAI-1 promoter and TGF-β sensitive 3TP-lux plasmid (Fig 1B), we observed significant up-regulation of the promoter level by both TGF-β1 and TGF-β3 (not shown) in these cell lines. As indicated above, endothelial Smad6s has been shown to differentially regulate TGF-β family signaling and we sought to determined the role of this regulator on PAI expression. As shown in Fig 1C, an
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from

4
antisense that blocked Smad6s, but not a sense control, significantly reduced the secretion of PAI-1 by TGF-β.
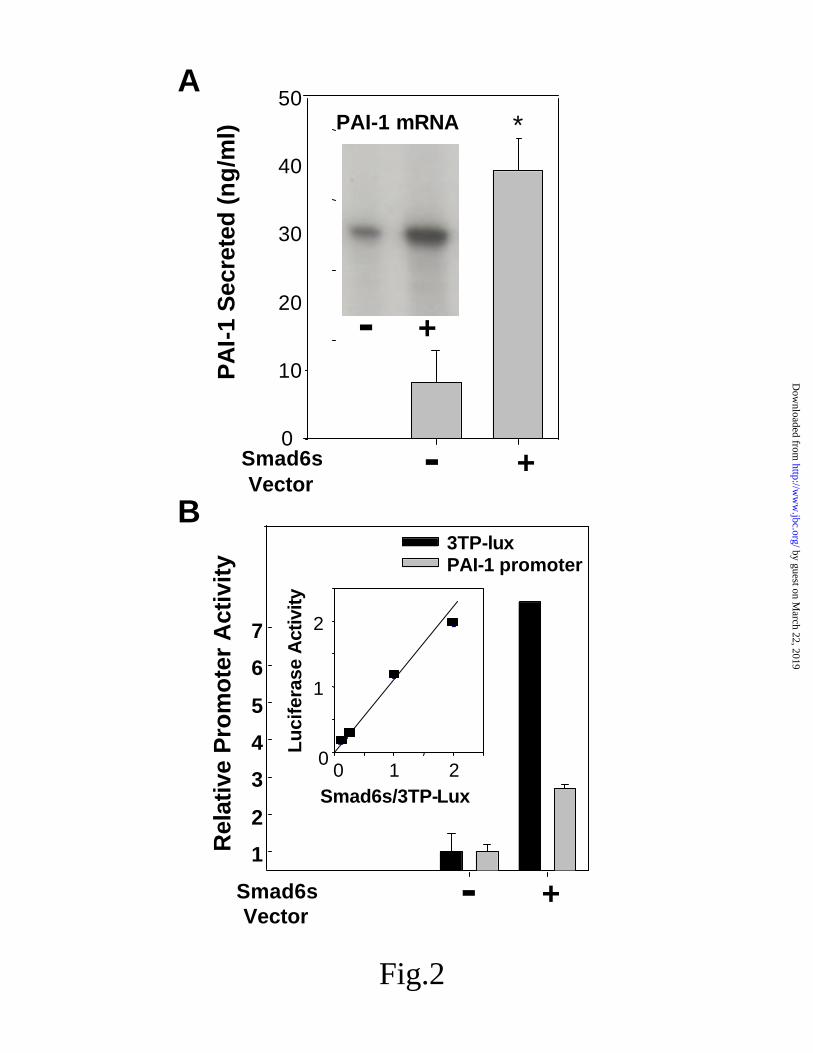
Smad6s enhances TGF-β activation of PAI-1 and nuclear factor FoxD1. The anti-sense data suggested that PAI-1 up-regulation was influenced by the presence of endogenous Smad6s. We confirmed this by over-expressing Smad6s and examining its affect on PAI-1 secretion and promoter activity. As shown in Fig 2A, the over-expression of Smad6s resulted in an increase in both endogenous PAI-1 mRNA and in the secretion of PAI-1. Using both the basal promoter and TGF-β sensitive 3TP-Lux indicator plasmid in co-transfection experiments, over-expression of Smad6s resulted in an increase in PAI-1 promoter activity (Fig 2B), suggesting that the effect on endogenous secretion and mRNA was the result of promoter activation.
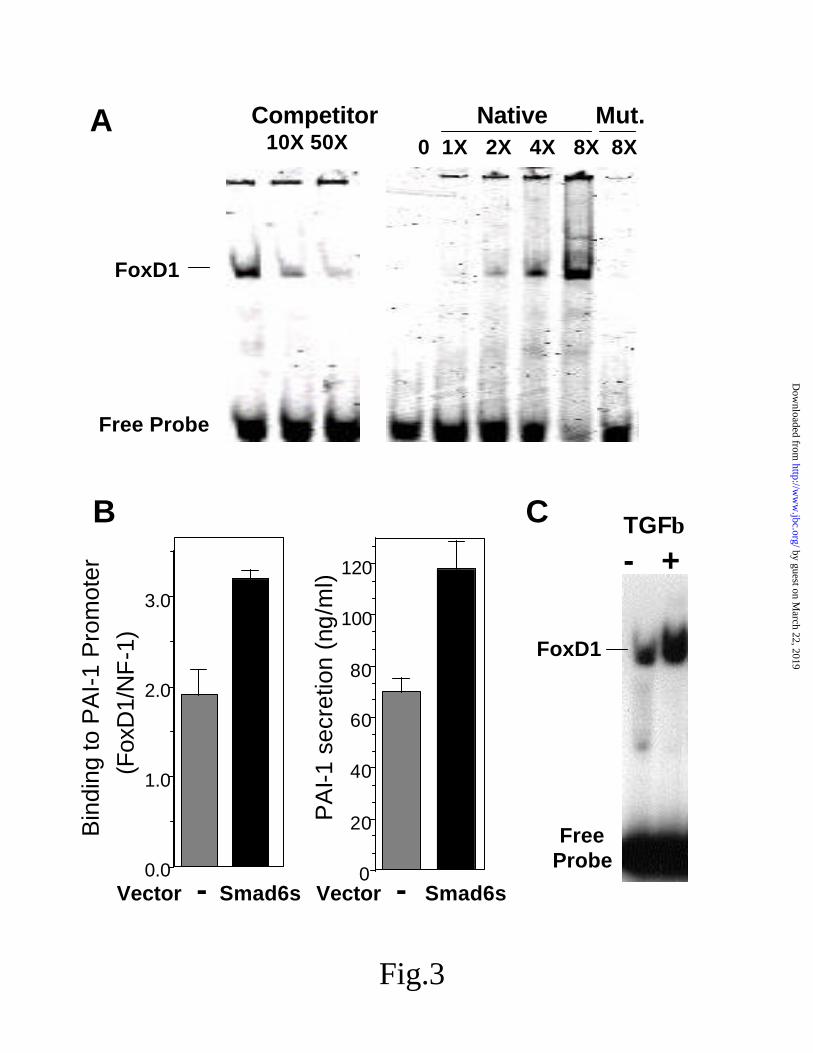
The p3TP-lux promoter contains nucleotides -740 to -636 of the PAI-1 promoter region, and previous studies have suggested several regions important for TGF-β regulation, including the region from –726 to –704 (32). Using an electrophoretic mobility shift assay, we have identified nuclear binding to a site in this region for the forkhead winged helix factor FoxD1 (Fig 3A). The binding was specific and could be competed with cold binding site, and a mutation in the core FoxD1 sequence resulted in poor binding. Moreover, the over-expression of Smad6s was able to enhance the binding of FoxD1 to this region of the PAI-1 promoter, which corresponded with an increase in PAI-1 secretion from the treated cells. (Fig 3B). In addition, the binding of FoxD1 to the promoter region of PAI-1 could be increased by TGF-β alone (Fig 3C).
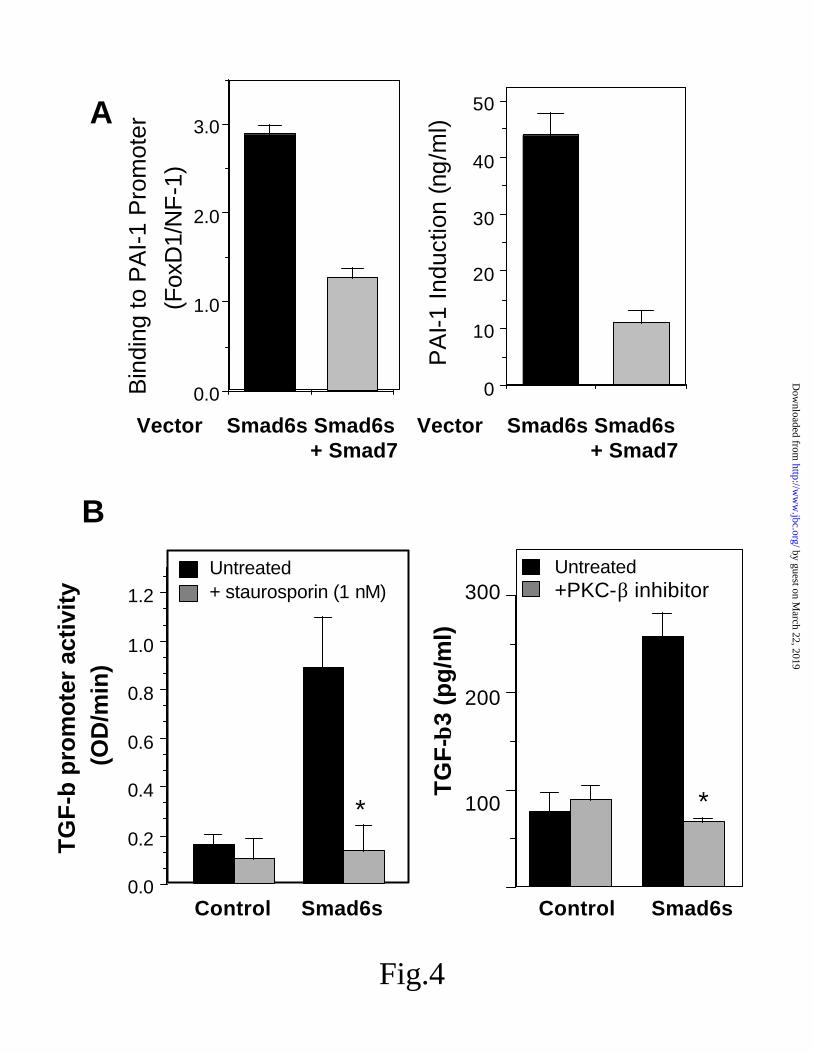
Smad6s induction of TGF-β and suppression by PCK-β inhibition. - As shown above, the over-expression of Smad6s induces PAI-1 and the binding of FoxD1 to the previously identified TGF-responsive site in the PAI-1 promoter, i.e. Smad6s appears to mimic the TGF-β response at the PAI-1 promoter. If the effect of Smad6s on increasing FoxD1 binding were dependent on TGF-β signaling, then we would expect inhibition of TGF-β receptor signaling by Smad7 to inhibit binding. As shown in Fig 4A, both the increase in FoxD1 binding and the subsequent increase in PAI-1 secretion were inhibited by the over-
expression of inhibitory Smad7. In addition, we examined the effect of over-expressing Smad6s on TGF- β levels secreted into the culture medium. As shown, Smad6s over-expression was capable of increasing the amount of TGF-β1 secreted from the cell (Fig 4B). Moreover, the effect on TGF-β appeared to be the result of increased expression as indicated by the ability of Smad6s to induce both the TGF- β1 and TGF- β3 promoters.
Previous studies have suggested that TGF-β mRNA induction can be sensitive to PKC inhibition (33,34). To determine if the Smad6s regulation was kinase dependent, we treated cells with staurosporin at concentrations known to inhibit PKC and found complete inhibition of the Smad6s regulation of TGF-β (Fig 4B). Moreover, using the PKC-β selective inhibitor LY379196 the effect of Smad6s also could be completely eliminated.
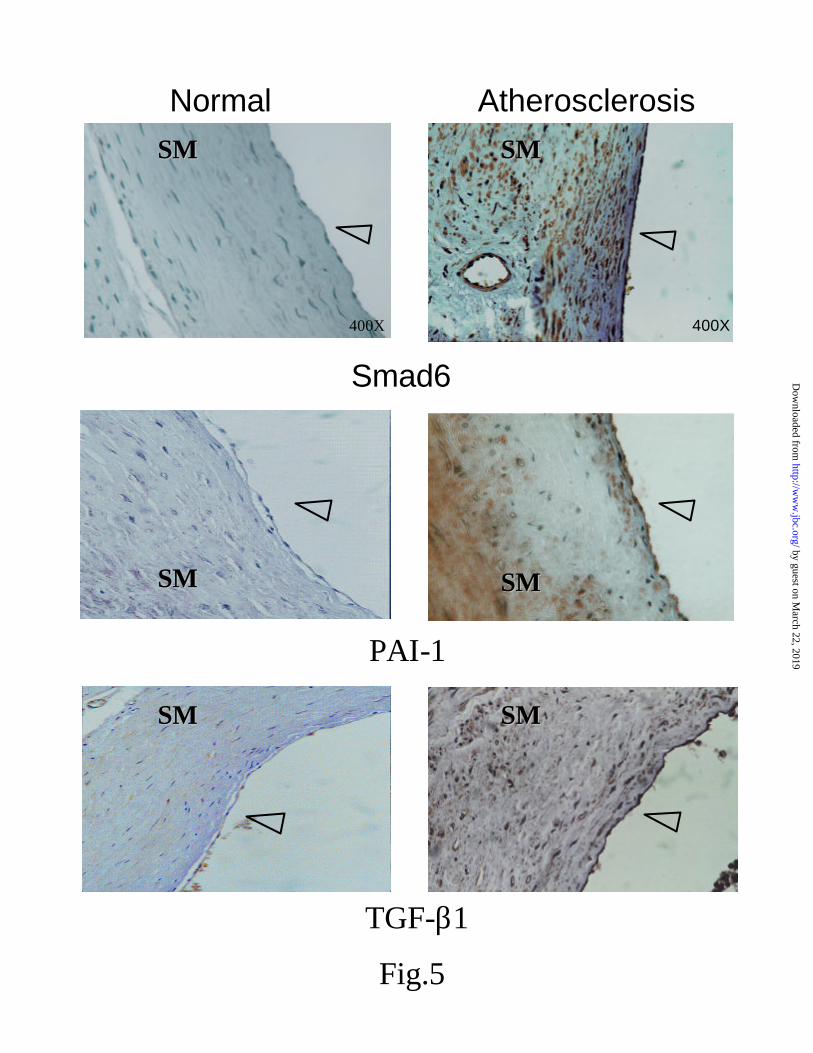
Differential regulation of Smad6s, PAI-1 and TGF-β in vascular disease- The data above suggested that the level of Smad6s in endothelial cells could alter the level of TGF-β and the subsequent induction of PAI-1. To determine if this relationship was observed in endothelial cells in vivo, we examined normal and atherosclerotic human cardiovascular tissues with Smad6s, TGF-β1, and PAI-1 specific antibodies, as studies have previously linked high PAI-1 levels to fibrotic vascular disease, and also have demonstrated TGF- β over-expression in fibroproliferative vascular lesions (35) and lipid-rich aortic intimal lesions (36). In an analysis of 35 CD34 positive vascular samples, Smad6s levels were very low to undetectable in normal vessels, but over-expressed in the endothelium over the plaque, as well as in the plaque vasculature (Fig 5). Whereas little if any staining could be observed in non-diseased vessels with the TGF-β1 and PAI-1 specific antibodies, high levels of expression were observed in the Smad6s positive vessels. These data support the in vitro observations in cultured cells and are consistent with Smad6s playing a role in regulating TFG-β and PAI-1 in vivo.
DISCUSSION
Under normal circumstances, the vascular endothelium displays a number of regulatory mechanisms that modulate coagulation,
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from

5
inflammation and vascular function to maintain homeostatic balance in the local environment (37,38). As this balance is disturbed by either acute or chronic insult, pathological processes ensue that alter vascular function and ultimately organ function. Both PAI-1 and TGF-β play important roles in modulating the response to vascular and tissue injury, but factors that enhance or chronically elevate levels of TGF-β and consequently PAI-1, compromise vascular function and promote pathologic processes. As such, understanding the coordinate regulation of TGF-β and PAI-1 may provide the basis for improved targets for developing therapeutic intervention. In this paper we provide new mechanistic understanding for the control of vascular PAI-1 and TGF-β through the regulatory factor Smad6s, and further demonstrate that the coordinate regulation of PAI-1 by Smad6s induction of TGF-β can be modulated by isozyme specific inhibition of PKC-β.
As reviewed above, the endothelial splice variant Smad6s has been shown to differentially regulate TGF-β family factors in Xenopus (23), and to positively modulate TGF-β suppression of endothelial thrombomodulin (24). As shown in Fig 4, Smad6s functions to increase the expression and subsequent secretion of TGF-β. Interestingly, Smad6 mRNA itself has been reported to be induced by TGF-β1 and BMP-7 (39,40), and BMP-7 has been shown to regulate the expression of PAI-1. While further studies will be required to define these relationships, these observations along with the data presented here suggest that the control of TGF-β / Smad6 related functions form an intricate feedback loop regulating TGF-β levels and the induction of PAI-1.
Several studies have identified the regions in the 5’ flanking region of human PAI-1 gene from -799 and -82 as mediating up-regulation of reporter gene expression by TGF-β1 in endothelial cells, both in vitro and in vivo (41). More specifically the region -791 to -546 upstream of the PAI-1 gene cap site (42) and from -740 to -636 (43) have been shown to confer response to TFG-β. Most notable, a major TGF-β responsive element was identified by DNA footprinting to bind a factor in the region –726 to –703 (32), although at the time this region did not contain consensus sequences for any known transcription factors. Studies by
Kutz et al. (44), recently identified this same general region, from nucleotides -740 and –703, as required for serum response. Our data showed that Smad6s over-expression increased nuclear factor binding to a FoxD1 site centered at –720. The forkhead transcription factor FoxD1, also called FREAC-4 (45) or murine BF-2, appears to play a role in developmental patterning and in kidney morphogeneis/nephrogenesis (46-48). While this study is the first to define a role for FoxD1 in TGF-β response, the xenopus homolog XBF2 has been shown to modulate signaling of another TGF-β family member BMP-4 (49). Of interest, the FoxD1 site is adjacent to a Smad3/Smad4 CAGA box /AGAC site (50,51), a site shown to confer TGF-β stimulation when multimerized in a heterologous reporter construct; future studies will be needed to determine if Smad3/4 interacts with FoxD1 following its induction and binding to this region. Hou et al., (52) recently showed that the upstream regulatory region of PAI-1 is extended up to 15kb, suggesting that future analysis of PAI-1 regulation will need to extend beyond the proximal promoter.
Several studies have shown that both PAI-1 and TGF-β can be regulated by modulating PKC. Using cells deficient in PKC-β, previous studies have shown that PAI synthesis is dependent on PKC-β (53), and that the activation of PKC-β may mediate the production of PAI-1 in endothelial cells (54). In addition, the PKC-β-specific inhibitor LY379196 effectively prevented LDL-induced PAI-1 production (54) as well as PAI-1 mRNA induction via αVβ3 (55). In models of diabetes, studies have shown that TGF-β mRNA expression can be modulated by a PKC-β specific inhibitor (33,34), and the induction of TGF-β1 by cigarette smoke could be significantly suppressed by LY379196, suggesting PKC-β involvement. (56). Our data now supply a possible link to these observations of PKC-β dependence via modulation of Smad6s.
Overall, our data provide new mechanistic understanding for the control of PAI-1 by TGF-β. The results presented here suggest that Smad6s plays a prominent role in controlling the TGF-β dependent expression of PAI-1 and that targeting this Smad signal may provide new opportunities for therapeutic intervention in treating and preventing vascular dysfunction. Specifically, our
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from

6
data give a direct link to previous observations on PKC-β modulation and provide support for this kinase as a target for modulating vascular function where dysregulation of both TGF-β and PAI-1 are important to the pathophysiology of the disorder. While our data give new mechanistic understanding for TGF-b regulation, further studies will be need to determine the relationship
of these finding to other important mediators (57,58) of PAI-1 regulation
Acknowledgements - We thank Rebecca
Fouts of excellent technical help and Dr. Dean Falb (formerly of Millennium Pharmaceuticals Inc.) for supplying Smad vectors.
.
REFERENCES
1. Fay, W. P. (2004) Trends Cardiovasc. Med 14, 196-202 2. Rossini, M., and Fogo, A. B. (2004) Drug Disc. Today 1, 65-72 3. Eddy, A. A. (2002) Am J Physiol 283, F209-220 4. Idell, S. (2003) Crit Care Med 31, S213-220 5. Allan, E. H., and Martin, T. J. (1995) Clinical Orthopaedics & Related Research 313, 54-63 6. Loskutoff, D. J. (1997) Circulation 96(9), 2772-4 7. Margaglione, M., Cappucci, G., Colaizzo, D., Giuliani, N., Vecchione, G., Grandone, E.,
Pennelli, O., and Di Minno, G. (1998) Arteriosclerosis, Thrombosis & Vascular Biology 18(2), 152-6
8. Westendorp, R. G., Hottenga, J. J., and Slagboom, P. E. (1999) Lancet 354(9178), 561-3 9. Chorostowska-Wynimko, J., Skrzypczak-Janjun, E., and Junkun, J. (2004) Int J Mol Med 13,
759-766 10. Chen, Y. Q., Sloan-Lancaster, J., Berg, D. T., Richardson, M. A., Grinnell, B., and
Tseng-Crank, J. (2001) Thrombosis & Haemostasis 86(6), 1563-72 11. Lux, A., Attisano, L., and Marchuk, D. A. (1999) Journal of Biological Chemistry
274(15), 9984-92 12. Sankar, S., Mahooti-Brooks, N., Bensen, L., McCarthy, T. L., Centrella, M., and Madri,
J. A. (1996) Journal of Clinical Investigation 97(6), 1436-46 13. Sawdey, M., Podor, T. J., and Loskutoff, D. J. (1989) Journal of Biological Chemistry
264(18), 10396-401 14. Wileman, S. M., Booth, N. A., Moore, N., Redmill, B., Forrester, J. V., and Knott, R. M.
(2000) British Journal of Ophthalmology 84(4), 417-22 15. Dong, C., Zhu, S., Wang, T., Yoon, W., and Goldschmidt-Clermont, P. J. (1008) Journal
of Heart & Lung Transplantation 21(9), 999-1008 16. Attisano, L., and Wrana, J. (2000) Current Opinion in Cell Biology. 12, 235-43 17. Massague, J., and Wotton, D. (2000) EMBO Journal. 19, 1745-54 18. Hayashi, H., Abdollah, S., Qiu, Y., Cai, J., Xu, Y., Grinnell, B., Richardson, M., Topper,
J., Gimbrone, M. J., Wrana, J., and Falb, D. (1997) Cell 89, 1165-73 19. Nakayama, T., Gardner, H., Berg, L., and Christian, J. (1998) Genes to Cells. 3, 387-94 20. Topper, J., Cai, J., Qiu, Y., Anderson, K., Xu, Y., Deeds, J., Feeley, R., Gimeno, C.,
Woolf, E., Tayber, O., Mays, G., Sampson, B., Schoen, F., Gimbrone, M., and Falb, D. (1997) Proc.Natl Acad. Sci. 93, 14-9
21. Imamura, T., Takase, M., Nishihara, A., Oeda, E., Hanai, J., Kawabata, M., and Miyazono, K. (1997) Nature. 389 (389), 622-6
22. Galvin, K., Donovan, M., Lynch, C., Meyer, R., Paul, R., Lorenz, J., Fairchild-Huntress, V., Dixon, K., Dunmore, J., Gimbrone, M. J., Falb, D., and Huszar, D. (2000) Nature Genetics. 24, 171-4
23. Krishnan, P., King, M. W., Neff, A. W., Sandusky, G. E., Bierman, K. L., Grinnell, B. W., and Smith, R. C. (2001) Develop. Growth and Differ. 43, 115-132
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from

7
24. Sandusky, G., Berg, D. T., Richardson, M. A., Myers, L., and Grinnell, B. W. (2002) Journal of Biological Chemistry 277(51), 49815-9
25. Richardson, M. A., Berg, D. T., Calnek, D. S., A.V.Ciaccia, Joyce, D. E., and Grinnell, B. W. (2000) Endocrinology 141, 3908-3911
26. Wrana, J. L., Attisano, L., Carcamo, J., Zentella, A., Doody, J., Laiho, M., Wang, X. F., and Massague, J. (1992) Cell 71(6), 1003-14
27. Wagner, R. W., Matteucci, M. D., Lewis, J. G., Gutierrez, A. J., Moulds, C., and Froehler, B. C. (1993) Science 260, 1510-1513
28. Lafyatis, R., Lechleider, R., Kim, S. J., Jakowlew, S., Roberts, A. B., and Sporn, M. B. (1990) J. Biol. Chem. 265, 19128-19136
29. Kim, S.-J., Glick, A., Sporn, M. B., and Roberts, A. B. (1989) J. Biol. Chem 264, 402-408
30. Grinnell, B. W., Berg, D. T., and Walls, J. (1986) Mol. Cell. Biol. 6, 3596-3605 31. Reifel-Miller, A. E., Berg, D. T., and Grinnell, B. W. (1991) J. Biol. Chem. 266, 13873-
13882 32. Sandler, M. A., Zhang, J. N., Westerhausen, D. R., Jr., and Billadello, J. J. (1994) Journal
of Biological Chemistry 269(34), 21500-4 33. Koya, D., Jirousek, M. R., Lin, Y. W., Ishii, H., Kuboki, K., and King, G. L. (1997)
Journal of Clinical Investigation 100(1), 115-26 34. Koya, D., Haneda, M., Nakagawa, H., Isshiki, K., Sato, H., Maeda, S., Sugimoto, T.,
Yasuda, H., Kashiwagi, A., Ways, D. K., King, G. L., and Kikkawa, R. (2000) FASEB Journal 14(3), 439-47
35. McCaffrey, T. (2000) Cytokine & Growth Factor Reviews 11, 103-14 36. Bobik, A., Agrotis, A., Kanellakis, P., Dilley, R., Krushinsky, A., Smirnov, V., Tararak,
E., Condron, M., and Kostolias, G. (1999) Circulation. 99 99, 2883-91 37. Rosenberg, R., and Aird, W. (1999) N Engl J Med 340, 1555-1564 38. Vervloet, M., Thijs, L., and Hack, C. (1998) Semin Thromb Hemos 24, 33-44. 39. Takase, M., Imamura, T., Sampath, T., Takeda, K., Ichijo, H., Miyazono, K., and
Kawabata, M. (1998) Biochemical & Biophysical Research Communications. 244, 26-9 40. Afrakhte, M., Moren, A., Jossan, S., Itoh, S., Sampath, K., Westermark, B., Heldin, C.,
Heldin, N., and ten Dijke, P. (1998) Biochemical & Biophysical Research Communications. 249, 505-11
41. Dong, G., Schulick, A. H., DeYoung, M. B., and Dichek, D. A. (1996) Journal of Biological Chemistry 271(47), 29969-77
42. Westerhausen, D. R., Jr., Hopkins, W. E., and Billadello, J. J. (1991) Journal of Biological Chemistry 266(2), 1092-100
43. Keeton, M. R., Curriden, S. A., van Zonneveld, A. J., and Loskutoff, D. J. (1991) Journal of Biological Chemistry 266(34), 23048-52
44. Kutz, S. M., Providence, K. M., and Higgins, P. J. (2001) Cell Motility & the Cytoskeleton 48(3), 163-74
45. Ernstsson, S., Pierrou, S., Hulander, M., Cederberg, A., Hellqvist, M., Carlsson, P., and Enerback, S. (1996) Journal of Biological Chemistry 271(35), 21094-9
46. Cederberg, A., Hulander, M., Carlsson, P., and Enerback, S. (1999) Journal of Biological Chemistry 274(1), 165-9
47. Bard, J. (1996) Bioessays 18(9), 705-7 48. Hatini, V., Huh, S. O., Herzlinger, D., Soares, V. C., and Lai, E. (1996) Genes &
Development 10(12), 1467-78 49. Gomez-Skarmeta, J. L., de la Calle-Mustienes, E., Modolell, J., and Mayor, R. (1999)
Mechanisms of Development 80(1), 15-27 50. Dennler, S., Itoh, S., Vivien, D., ten Dijke, P., Huet, S., and Gauthier, J. M. (1998)
EMBO Journal 17(11), 3091-100
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from

8
51. Song, C. Z., Siok, T. E., and Gelehrter, T. D. (1998) Journal of Biological Chemistry 273(45), 29287-90
52. Hou, B., Eren, M., Painter, C., Covington, J., Dixon, J., Schoenhard, J., and Vaughan, D. (2004) J Biol Chem 279, 18127-18136
53. Lopez, S., Peiretti, F., Morange, P., Laouar, A., Fossat, C., Bonardo, B., Huberman, E., Juhan-Vague, I., and Nalbone, G. (1999) Thrombosis & Haemostasis 81(3), 415-22
54. Ren, S., Shatadal, S., and Shen, G. X. (2000) American Journal of Physiology Endocrinology & Metabolism 278(4)
55. Khatib, A. M., Nip, J., Fallavollita, L., Lehmann, M., Jensen, G., and Brodt, P. (2001) International Journal of Cancer 91(3), 300-8
56. Mur, C., Claria, J., Rodela, S., Lario, S., Campistol, J. M., Titos, E., Inigo, P., Cases, A., Abian, J., and Esmatjes, E. (2004) Life Sciences 75(5), 611-21
57. Brown, N., Vaughan, D., and Fogo, A. (2002) Semin Nephrol 22, 399-406 58. Schoenhard, J., Smith, L., Painter, C., Eren, M., Johnson, C., and Vaughan, D. (2003) J
Mol Cell Cardiol 35(473-81)
FOOTNOTES
1The abbreviations used are: BMP, bone morphogenic protein; CAT, chloramphenicol
acetyltransferase; HUVEC, human umbilical vein endothelial cell; PAI-1, plasminogen activator inhibitor-1; SFM, serum free medium; TGF-β, transforming growth factor-beta; DMEM, Dulbecco’s modified Eagle’s medium; ELISA, enzyme-linked immunosorbent assay; PBS, phosphate-buffered saline; PKC, protein kinase C.
FIGURE LEGENDS
Fig 1. Dependence of Suppression of PAI-1 by TGF-β on Smad6s . A, effect of TGF- β on the level of PAI-1 secretion from human endothelial cells (mean +/- S.D.; n=3) using a PAI-1 ELISA. B, effect of TGF-β on the PAI-1 promoter using pOCAT2336 and on the hyper-responsive 3TP-lux indicator plasmid, measuring the level of CAT activity and luciferase, respectively (mean +/- S.D.; n=3). C, effect of anti-sense and sense oligonucleotides on PAI-1 expression by ELISA (mean +/- S.D.; n=6). * represents p< 0.05.
Fig 2. Smad6s control of PAI-1 secretion, and expression. A, analysis of the effect of Smad6s
over expression on PAI-1 secreted into the media by ELISA and on the level of mRNA by ribonuclease protection assay in cells transfected with the Smad6s expression vector (mean +/- S.D.; n=3). B, determination of the effect of Smad6s over-expression on the PAI-1 promoter following co-transfection with either pOCAT2336 or 3TP-lux. Inset shows the concentration response for promoter activity, measuring luciferase activity from 3TP-lux as a function of increased Smad6s expression plasmid.
Fig 3. Smad6s modulates the binding of nuclear factor FoxD1 to the PAI-1 promoter. A,
demonstration of nuclear factor binding to the region -726 to –704 in the PAI-1 promoter using a gel-shift assay. Gel on the left shows the effect of competition with unlabeled binding site, and on the right the effect of increasing concentration of binding site relative to the binding to an oligonucleotide with a mutation (Mut.) in the FoxD1 core sequence. B, effect of over-expression of Smad6s on the binding activity of FoxD1 to the PAI-1 promoter, and to the subsequent increase in PAI-1 expression in the same experiment (mean +/- S.D.; n=3) . Levels of FoxD1 were made relative to the binding of NF-1 as a control oligonuclotide. C, effect of TGF-β on the binding of FoxD1 to the PAI-1 promoter.
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from

9
Fig 4. Dependence of Smad6s effects on TGF-β induction and kinase activity. A, effect of the TGF-β signaling inhibitor Smad7 on Smad6s induction of FoxD1 binding and PAI-1 expression by ELISA. B, effect of Smad6s over-expression on both TGF-β promoter activity and on the secretion of TGF-β; effect of the inhibition of PKC with staurosporin and of PKC-β with LY379196 on Smad6s induction of TGF-β promoter activity and secretion (mean +/- SE; n=6, p< 0.05 for inhibition of Smad6s effect by staurosporin and LY379196)
Fig 5. Differential expression of Smad6, TGF-β and PAI-1 in normal vs. diseased vessels.
Representative sections from the analysis of 35 samples of normal and diseased vessels stained with antibodies for Smad6s. All samples were viewed at 400X and contained intact endothelium as indicated by CD34 positive staining (data not shown). Also shown are sections stained with either anti-TGF-β1 or anti-PAI-1 antibodies. Arrow indicates the endothelium lining the luminal side of the vessel. SM = smooth muscle.
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from
Control TGF-β0
100
200P
AI-
1 (n
g/m
l)
100101.1.01.001
100
200
300
TGF-β (ng/ml)P
AI-
1 (n
g/m
l)
A
CB
0
2
4
6
8
10
12
14
3tP
lux
pro
mo
ter
- +
PA
I-1
bas
al p
rom
ote
r(C
AT
Act
ivity
mO
D/m
in)
TGF-β - +0
1
2
0
20
40
60
80
100
120
PA
I-1
(%T
GF
-bet
a in
du
ctio
n)
Control Sense Anti-sense
Smad6s
Fig.1
*
*
**
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from
-Smad6sVector
PA
I-1
Sec
rete
d (n
g/m
l)
0
10
20
30
40
50
+
Rel
ativ
e P
rom
ote
r A
ctiv
ity
1
2
3
4
5
6
7
Smad6sVector
3TP-luxPAI-1 promoter
- +
A
B
Smad6s/3TP-Lux
Luci
fera
se A
ctiv
ity
0
1
2
0 1 2
- +
PAI-1 mRNA
Fig.2
*
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from
A
0.0
1.0
2.0
3.0
Bin
ding
to P
AI-
1 P
rom
oter
(Fox
D1/
NF
-1)
0
20
40
60
80
100
120
PA
I-1 s
ecre
tion
(ng/
ml)
Vector - Smad6s Vector - Smad6s
TGFβ
- +
B C
FoxD1
FreeProbe
FoxD1
Free Probe
Fig.3
0 1X 2X 4X 8X 8XCompetitor
10X 50XNative Mut.
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from
0.0
1.0
2.0
3.0
0
10
20
30
40
50
PA
I-1 In
duct
ion
(ng/
ml)
Bin
ding
to P
AI-
1 P
rom
oter
(Fox
D1/
NF
-1)
Vector Smad6s Smad6s + Smad7
Vector Smad6s Smad6s + Smad7
A
B
100
200
300
TG
F-β
3 (p
g/m
l)
Control Smad6s0.0
0.2
0.4
0.6
0.8
1.0
1.2Untreated+ staurosporin (1 nM)
TG
F-b
pro
mo
ter
activ
ity
(OD
/min
)
Control Smad6s
+PKC-β inhibitorUntreated
Fig.4
**
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from
TGF-β1
SMSM
Smad6
PAI-1
SMSM
SMSM
SMSM
SMSM
SMSM
Normal Atherosclerosis
Fig.5
400X400X
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from

GrinnellDavid T. Berg, Laura J. Myers, Mark A. Richardson, George Sandusky and Brian W.
βup-regulation of TGF- dependentβSmad6s regulates plasminogen activator inhibitor-1 through a PKC-
published online February 16, 2005J. Biol. Chem.
10.1074/jbc.C400579200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on March 22, 2019
http://ww
w.jbc.org/
Dow
nloaded from
















![Targeting macrophage checkpoint inhibitor SIRPα for anticancer … · 2020. 6. 18. · lymphocyte–associated protein 4 [CTLA-4] and programmed death 1 [PD-1]), or their ligands](https://static.fdocument.org/doc/165x107/5fd9a9e449b9f25d9f5898e6/targeting-macrophage-checkpoint-inhibitor-sirp-for-anticancer-2020-6-18-lymphocyteaassociated.jpg)

