
Silver Nyambo Department of Chemistry, Marquette University, Wisconsin Resonance enhanced two-photon...
13
Silver Nyambo Department of Chemistry, Marquette University, Wisconsin Resonance enhanced two-photon ionization (R2PI) spectroscopy of halo-aromatic clusters June 2013
-
Upload
koby-sailors -
Category
Documents
-
view
219 -
download
3
Transcript of Silver Nyambo Department of Chemistry, Marquette University, Wisconsin Resonance enhanced two-photon...

Silver NyamboDepartment of Chemistry, Marquette University, Wisconsin
Resonance enhanced two-photon ionization (R2PI) spectroscopy of halo-aromatic clusters
June 2013

Background and motivation
Non covalent interactions such as π-stacking, C-H/π, halogen bonding, hydrogen bond plays a crucial role in chemical and biological processess.
Halobenzenes can be used as prototypes to study these non-covalent interactions.
These interactions may coexist and compete with each other during formation of large molecular assemblies, hence understanding their relative magnitudes becomes important.
1.Ghosh et al Biol. Crystallogr. 2000, 56, 85-1095.2.Kevin , E et al J. Phys. Chem. A 2007, 1111, 1688-16943.Metrangolo, P et al Acc. Chem. Res. 2005, 38, 386-395
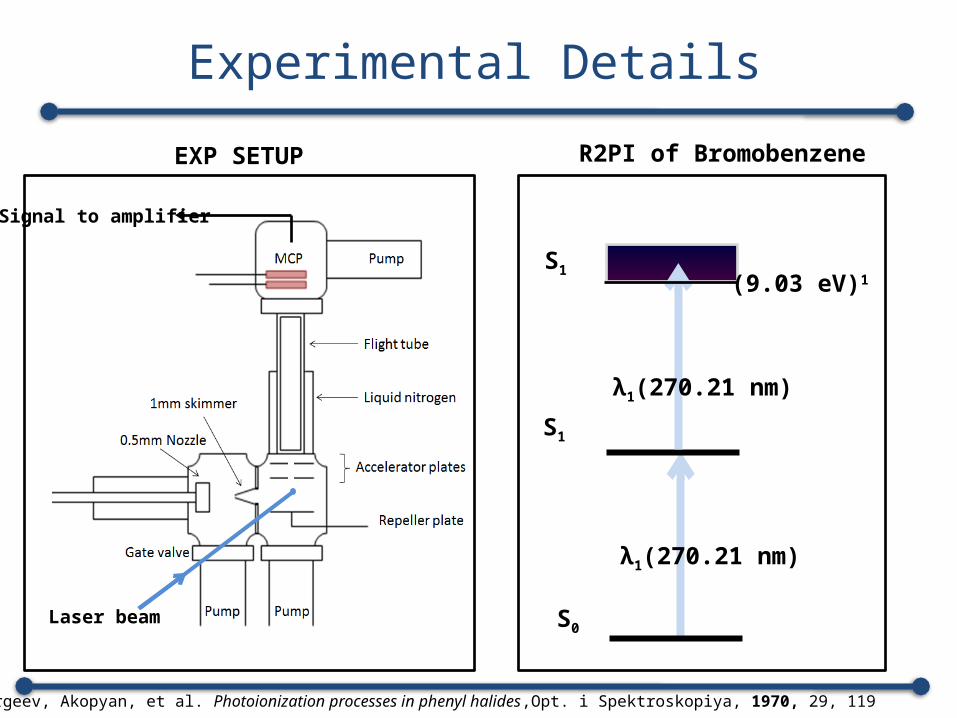
Experimental Details
Laser beam
Signal to amplifier
S1
S0
S1
λ1(270.21 nm)
(9.03 eV)1
R2PI of BromobenzeneEXP SETUP
1. Sergeev, Akopyan, et al. Photoionization processes in phenyl halides,Opt. i Spektroskopiya, 1970, 29, 119
λ1(270.21 nm)
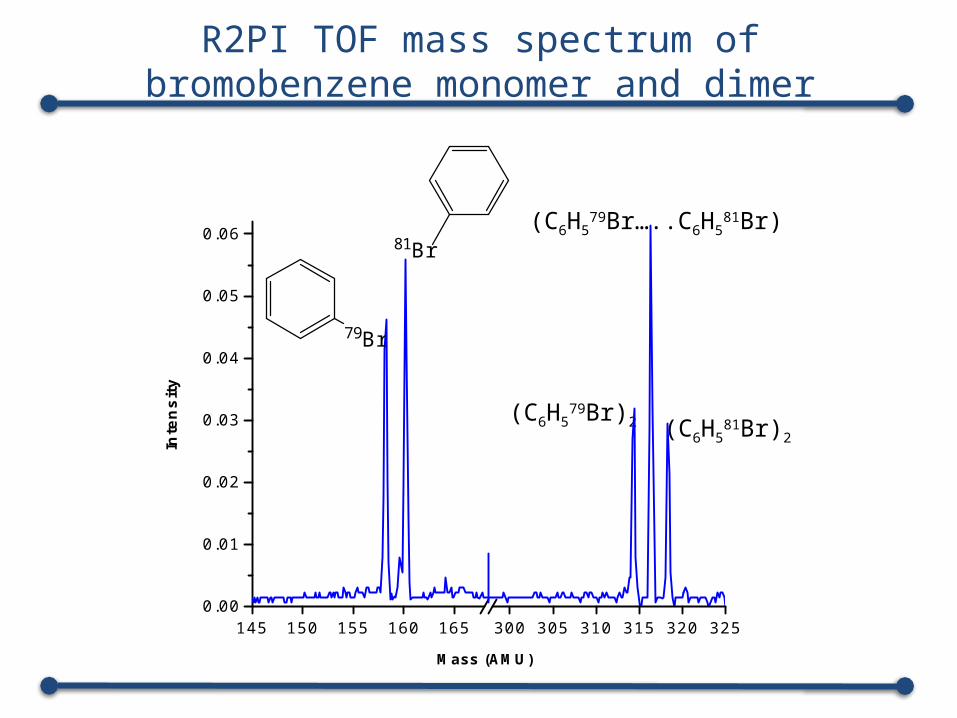
145 150 155 160 165 300 305 310 315 320 3250.00
0.01
0.02
0.03
0.04
0.05
0.06
Inte
nsi
ty
Mass (AMU)
R2PI TOF mass spectrum of bromobenzene monomer and dimer
(C6H579Br)2 (C6H5
81Br)2
(C6H579Br…..C6H5
81Br)
79Br
81Br
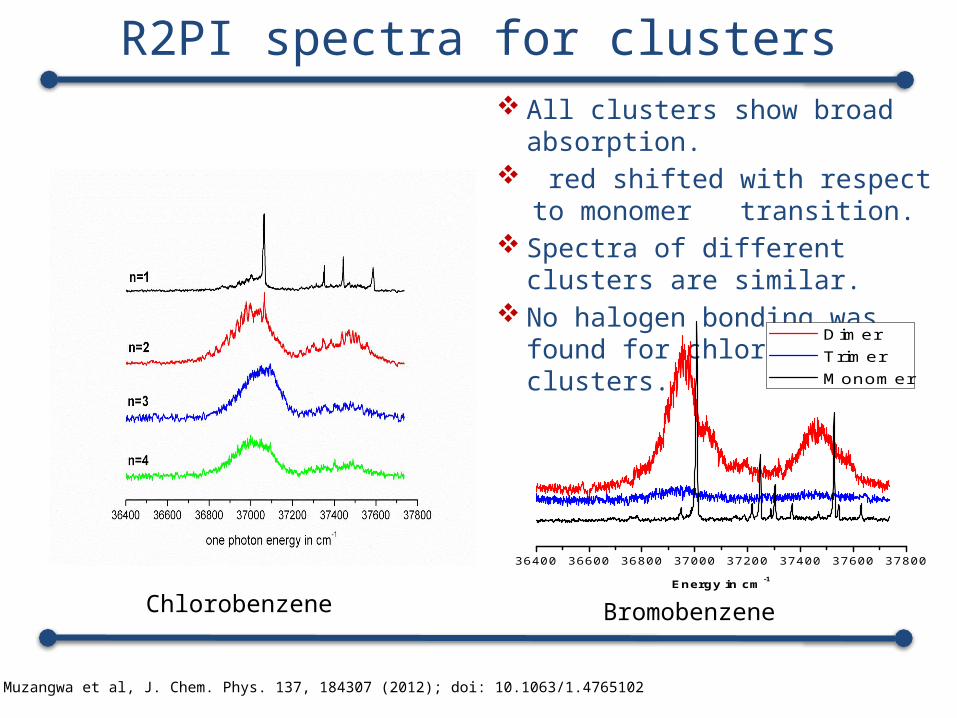
R2PI spectra for clusters
1. Lloyd Muzangwa et al, J. Chem. Phys. 137, 184307 (2012); doi: 10.1063/1.4765102
All clusters show broad absorption.
red shifted with respect to monomer transition.
Spectra of different clusters are similar.
No halogen bonding was found for chlorobenzene clusters.
Chlorobenzene Bromobenzene
36400 36600 36800 37000 37200 37400 37600 37800
Energy in cm-1
Dimer Trimer Monomer

Computational Details
Geometry optimizations and binding energies: M06-2X / aug-cc-pVDZ and corrected for BSSE and ZPE.
Electronic absorption and oscillator strength: TD-DFT (M06-2X/aug-cc-pVDZ)
Sherrill and co-workers benchmarked the performance of DFT methods against high level post-HF ab initio single reference methods and they found out M06-2X was a cost effective method1.
Zhao and Truhlar have shown that M06-2X performs well on describing non covalent interaction energies.2
1.L. A. Burns, A. Vazquez-Mayagoitia, B. G. Sumpter, and C. D. Sherrill, Journal of Chemical Physics 134 084107 (2011).2.Zhao, Y.; Truhlar, D.G. Theor. Chem. Account 2008, 120, 215. DOI 10.1007

Dimer 4 (9.99 KJ/mol) Dimer 5 (6.56 KJ/mol) Dimer 6 (3.47 KJ/mol) Dimer 7 (6.97 KJ/mol)
Optimized dimer conformations: M06-2X/aug-cc-pVDZ
Binding energies were corrected for zero point energy and the counterpoise method was used to correct for basis set superposition error (BSSE).
Dimer 2 (19.43 KJ/mol)Dimer 1 (21.34 KJ/mol) Dimer 3(21.57 KJ/mol)
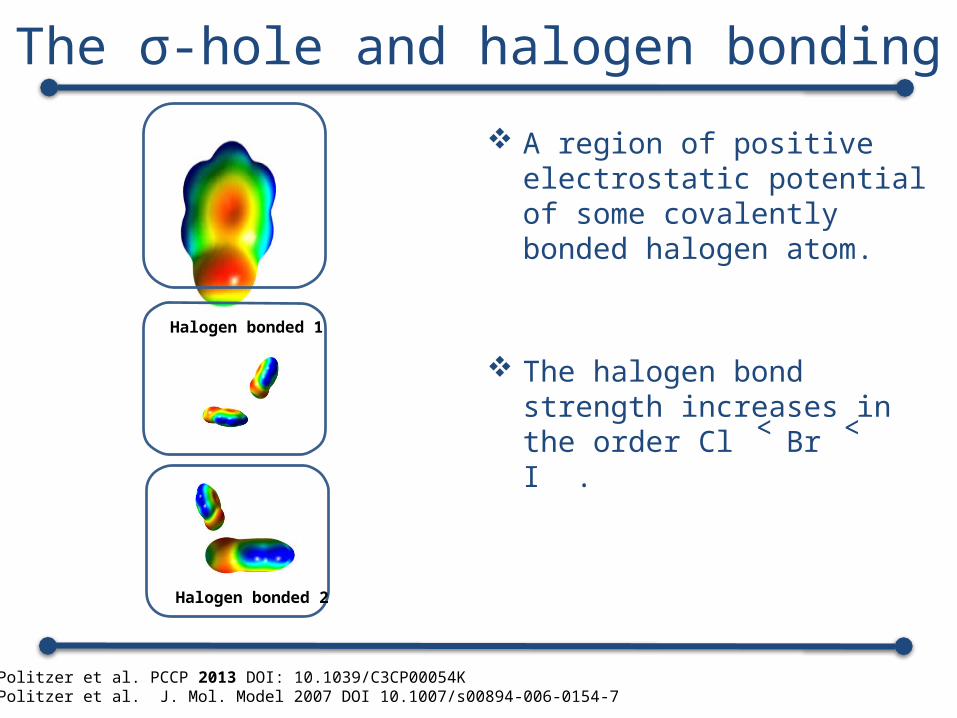
The σ-hole and halogen bonding A region of positive electrostatic
potential of some covalently bonded halogen atom.
The halogen bond strength increases in the order Cl ˂ Br ˂ I .
Peter Politzer et al. PCCP 2013 DOI: 10.1039/C3CP00054KPeter Politzer et al. J. Mol. Model 2007 DOI 10.1007/s00894-006-0154-7
Halogen bonded 1
Halogen bonded 2
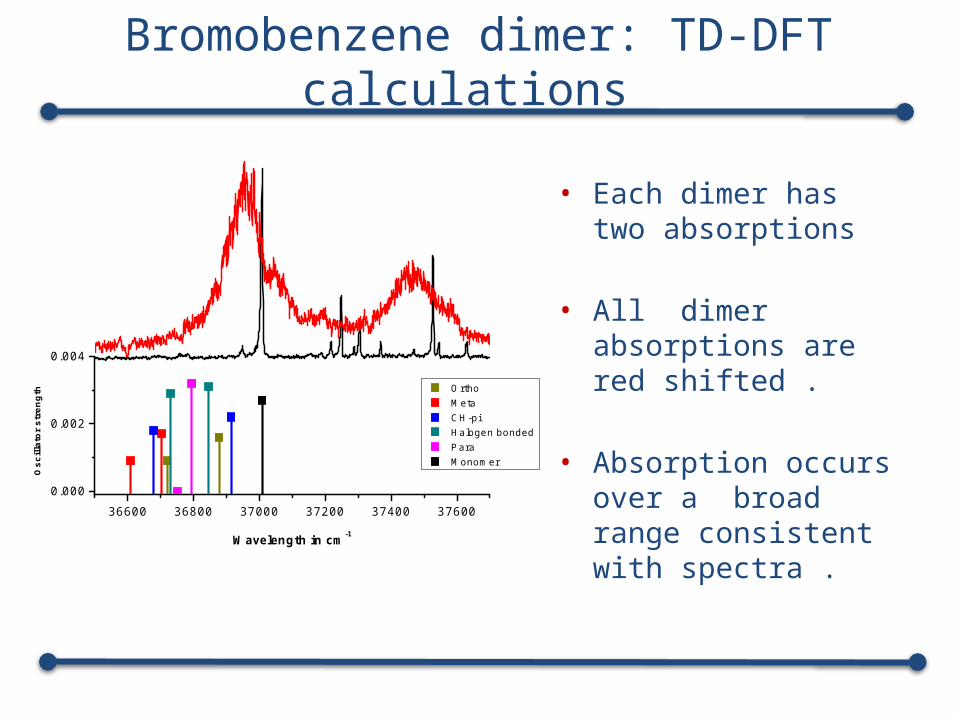
Bromobenzene dimer: TD-DFT calculations
• Each dimer has two absorptions
• All dimer absorptions are red shifted .
• Absorption occurs over a broad range consistent with spectra . 36600 36800 37000 37200 37400 37600
0.000
0.002
0.004
Ortho Meta CH-pi Halogen bonded Para Monomer
Osc
illat
or st
rength
Wavelength in cm-1

3.637
3.015
3.655
3.059
3.629
3.011
2.674
2.480
S1
S0
π …… π Br…….Br and CH……Br CH…….π
TD-DFT: Excited states
• The electronic transition is a π to π* transition• The transition leads to a change in geometry in the S1
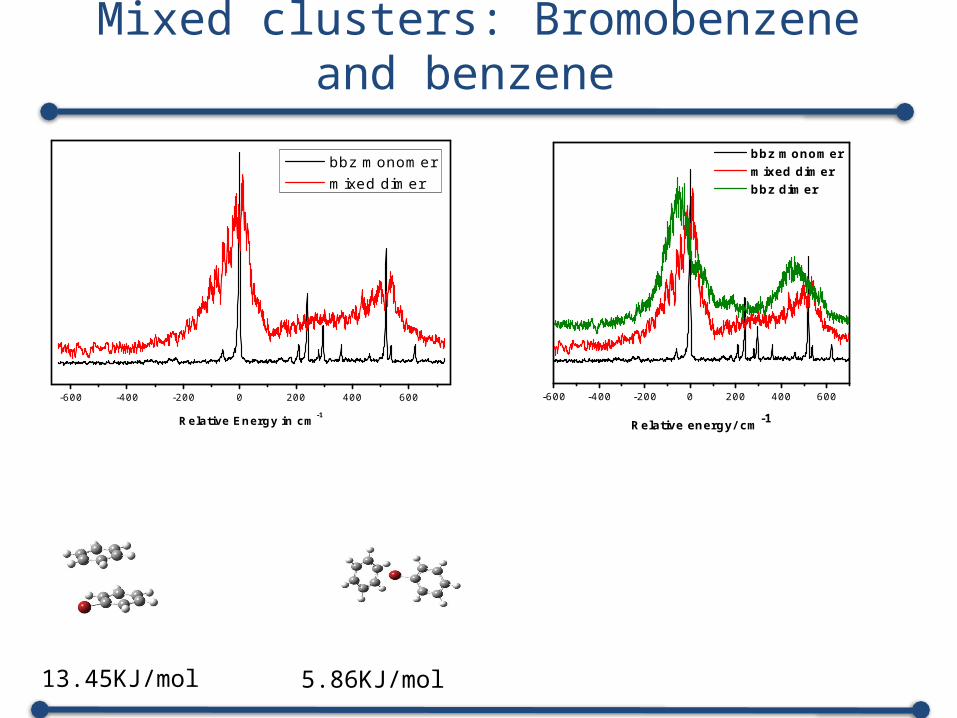
Mixed clusters: Bromobenzene and benzene
-600 -400 -200 0 200 400 600
Relative Energy in cm-1
bbz monomer mixed dimer
13.45KJ/mol 5.86KJ/mol
-600 -400 -200 0 200 400 600
bbz monomer mixed dimer bbz dimer
Relative energy/ cm-1

Summary
Non-covalent interactions in bromobenzene were probed experimentally and computationally.
Computationally, representative minima dimer structures were optimized confirms the existence of multiple conformers.
TD-DFT calculations were used to support our experimental findings.

Acknowledgements
Advisor: Scott A. Reid
Group members:Brandon UrlerLloyd MuzangwaAimable KalumeLisa George
Funding:National Science Foundation


















