
Selective blockade of CaMKII-α inhibits NMDA-induced caspase-3-dependent cell death but does not...
15
Research Report Selective blockade of CaMKII-α inhibits NMDA-induced caspase-3-dependent cell death but does not arrest PARP-1 activation or loss of plasma membrane selectivity in rat retinal neurons Dennis J. Goebel ⁎ Department of Anatomy and Cell Biology, Wayne State University School of Medicine, 540 E. Canfield, Detroit, MI 48201, USA ARTICLE INFO ABSTRACT Article history: Accepted 12 December 2008 Available online 30 December 2008 Calcium/calmodulin-dependent protein kinase II-α (CaMKII-α) has been implicated in a number of receptor mediated events in neurons. Pharmacological blockade of CaMKII-α has been shown to prevent phosphorylation of NMDA-R2A and R2B receptor subunits, suggesting that this enzyme may be linked to receptor trafficking of glutamate receptors and serve as a regulatory protein for neuronal cell death. In the retina, inhibition of CaMKII- α has been reported to be neuroprotective against NMDA-induced cell death by preventing the activation of the caspase-3 dependent pathway. However, the effects of CaMKII-α blockade on the caspase-3 independent, PARP-1 dependent and the non-programmed cell death pathways have not previously been investigated. In the present study, blockade of CaMKII-α with the highly specific antagonist myristoylated autocamtide-2-related inhibitory peptide (AIP) was used in a rat in vivo model of retinal toxicity to compare the effects of on NMDA-induced caspase-3-dependent, PARP-1 dependent and the non- programmed (necrosis) cell death pathways. Results confirmed that AIP fully attenuates caspase-3 activation for at least 8 h following NMDA insult and also significantly improves retinal ganglion cell survival. However, this blockade had little effect on reducing the loss of plasma membrane selectivity (LPMS, e.g. necrosis) in cells located in the ganglion cell and inner nuclear layers and did not alter NMDA-induced PARP-1 hyperactivation, or prevent TUNEL labeling following a moderate NMDA-insult. These findings support a specific role for CaMKII-α in mediating the caspase-3 dependent cell death pathway and provide evidence that it is not directly linked to the signaling of either the PARP-1 dependent or the non-programmed cell death pathways. © 2008 Elsevier B.V. All rights reserved. Keywords: Calcium/calmodulin-dependent kinase II-α blockade Caspase-3 activation PARP-activity TUNEL labeling Ethidium bromide staining 1. Introduction Understanding the mechanisms involved in mediating neuronal cell death linked to stroke/ischemia has been the focus of numerous laboratories over the past four decades. One of the most widely utilized models for initiating cell death in neurons is through the activation of a subset of ionotropic glutamate receptors by the agonist BRAIN RESEARCH 1256 (2009) 190 – 204 ⁎ Fax: +1 313 577 3125. E-mail address: [email protected]. 0006-8993/$ – see front matter © 2008 Elsevier B.V. All rights reserved. doi:10.1016/j.brainres.2008.12.051 available at www.sciencedirect.com www.elsevier.com/locate/brainres
-
Upload
dennis-j-goebel -
Category
Documents
-
view
216 -
download
0
Transcript of Selective blockade of CaMKII-α inhibits NMDA-induced caspase-3-dependent cell death but does not...

B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
ava i l ab l e a t www.sc i enced i rec t . com
www.e l sev i e r. com/ loca te /b ra in res
Research Report
Selective blockade of CaMKII-α inhibits NMDA-inducedcaspase-3-dependent cell death but does not arrest PARP-1activation or loss of plasma membrane selectivity in ratretinal neurons
Dennis J. Goebel⁎
Department of Anatomy and Cell Biology, Wayne State University School of Medicine, 540 E. Canfield, Detroit, MI 48201, USA
A R T I C L E I N F O
⁎ Fax: +1 313 577 3125.E-mail address: [email protected].
0006-8993/$ – see front matter © 2008 Elsevidoi:10.1016/j.brainres.2008.12.051
A B S T R A C T
Article history:Accepted 12 December 2008Available online 30 December 2008
Calcium/calmodulin-dependent protein kinase II-α (CaMKII-α) has been implicated in anumber of receptor mediated events in neurons. Pharmacological blockade of CaMKII-α hasbeen shown to prevent phosphorylation of NMDA-R2A and R2B receptor subunits,suggesting that this enzyme may be linked to receptor trafficking of glutamate receptorsand serve as a regulatory protein for neuronal cell death. In the retina, inhibition of CaMKII-α has been reported to be neuroprotective against NMDA-induced cell death by preventingthe activation of the caspase-3 dependent pathway. However, the effects of CaMKII-αblockade on the caspase-3 independent, PARP-1 dependent and the non-programmed celldeath pathways have not previously been investigated. In the present study, blockade ofCaMKII-α with the highly specific antagonist myristoylated autocamtide-2-relatedinhibitory peptide (AIP) was used in a rat in vivo model of retinal toxicity to compare theeffects of on NMDA-induced caspase-3-dependent, PARP-1 dependent and the non-programmed (necrosis) cell death pathways. Results confirmed that AIP fully attenuatescaspase-3 activation for at least 8 h following NMDA insult and also significantly improvesretinal ganglion cell survival. However, this blockade had little effect on reducing the loss ofplasma membrane selectivity (LPMS, e.g. necrosis) in cells located in the ganglion cell andinner nuclear layers and did not alter NMDA-induced PARP-1 hyperactivation, or preventTUNEL labeling following a moderate NMDA-insult. These findings support a specific rolefor CaMKII-α in mediating the caspase-3 dependent cell death pathway and provideevidence that it is not directly linked to the signaling of either the PARP-1 dependent or thenon-programmed cell death pathways.
© 2008 Elsevier B.V. All rights reserved.
Keywords:Calcium/calmodulin-dependentkinase II-α blockadeCaspase-3 activationPARP-activityTUNEL labelingEthidium bromide staining
1. Introduction
Understanding the mechanisms involved in mediatingneuronal cell death linked to stroke/ischemia has been
er B.V. All rights reserved
the focus of numerous laboratories over the past fourdecades. One of the most widely utilized models forinitiating cell death in neurons is through the activationof a subset of ionotropic glutamate receptors by the agonist
.
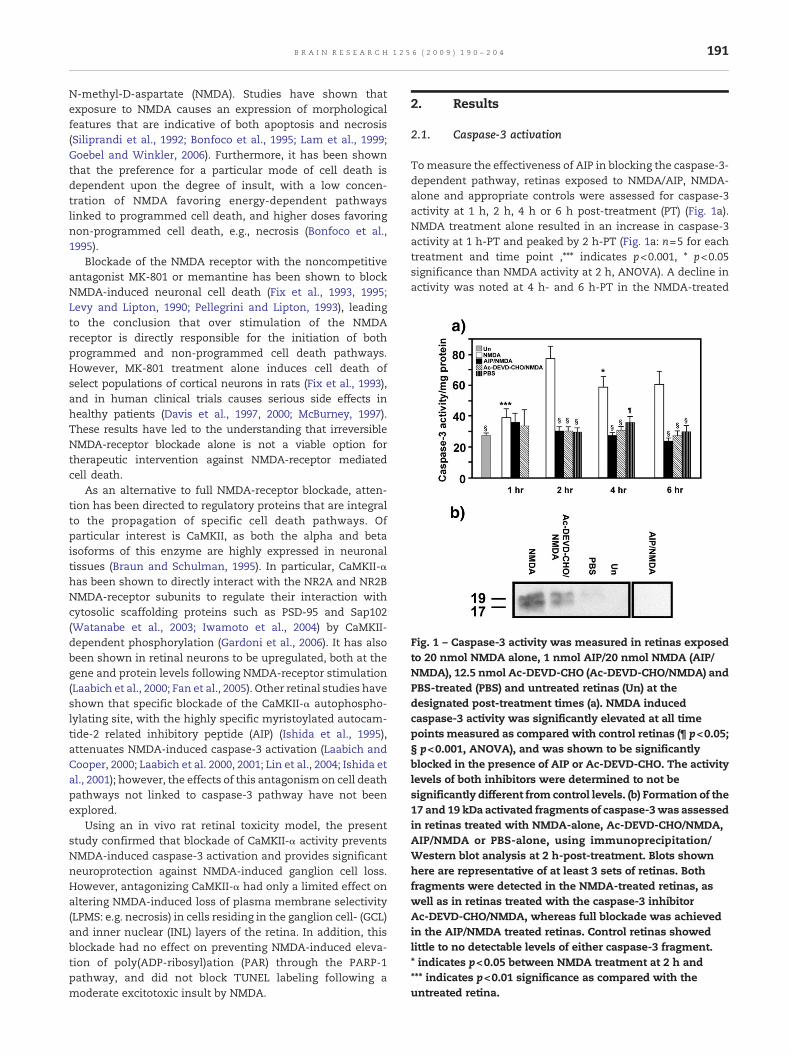
Fig. 1 – Caspase-3 activity was measured in retinas exposedto 20 nmol NMDA alone, 1 nmol AIP/20 nmol NMDA (AIP/NMDA), 12.5 nmol Ac-DEVD-CHO (Ac-DEVD-CHO/NMDA) andPBS-treated (PBS) and untreated retinas (Un) at thedesignated post-treatment times (a). NMDA inducedcaspase-3 activity was significantly elevated at all timepoints measured as compared with control retinas (¶ p<0.05;§ p<0.001, ANOVA), and was shown to be significantlyblocked in the presence of AIP or Ac-DEVD-CHO. The activitylevels of both inhibitors were determined to not besignificantly different from control levels. (b) Formation of the17 and 19 kDa activated fragments of caspase-3was assessedin retinas treated with NMDA-alone, Ac-DEVD-CHO/NMDA,AIP/NMDA or PBS-alone, using immunoprecipitation/Western blot analysis at 2 h-post-treatment. Blots shownhere are representative of at least 3 sets of retinas. Bothfragments were detected in the NMDA-treated retinas, aswell as in retinas treated with the caspase-3 inhibitorAc-DEVD-CHO/NMDA, whereas full blockade was achievedin the AIP/NMDA treated retinas. Control retinas showedlittle to no detectable levels of either caspase-3 fragment.* indicates p<0.05 between NMDA treatment at 2 h and*** indicates p<0.01 significance as compared with theuntreated retina.
191B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
N-methyl-D-aspartate (NMDA). Studies have shown thatexposure to NMDA causes an expression of morphologicalfeatures that are indicative of both apoptosis and necrosis(Siliprandi et al., 1992; Bonfoco et al., 1995; Lam et al., 1999;Goebel and Winkler, 2006). Furthermore, it has been shownthat the preference for a particular mode of cell death isdependent upon the degree of insult, with a low concen-tration of NMDA favoring energy-dependent pathwayslinked to programmed cell death, and higher doses favoringnon-programmed cell death, e.g., necrosis (Bonfoco et al.,1995).
Blockade of the NMDA receptor with the noncompetitiveantagonist MK-801 or memantine has been shown to blockNMDA-induced neuronal cell death (Fix et al., 1993, 1995;Levy and Lipton, 1990; Pellegrini and Lipton, 1993), leadingto the conclusion that over stimulation of the NMDAreceptor is directly responsible for the initiation of bothprogrammed and non-programmed cell death pathways.However, MK-801 treatment alone induces cell death ofselect populations of cortical neurons in rats (Fix et al., 1993),and in human clinical trials causes serious side effects inhealthy patients (Davis et al., 1997, 2000; McBurney, 1997).These results have led to the understanding that irreversibleNMDA-receptor blockade alone is not a viable option fortherapeutic intervention against NMDA-receptor mediatedcell death.
As an alternative to full NMDA-receptor blockade, atten-tion has been directed to regulatory proteins that are integralto the propagation of specific cell death pathways. Ofparticular interest is CaMKII, as both the alpha and betaisoforms of this enzyme are highly expressed in neuronaltissues (Braun and Schulman, 1995). In particular, CaMKII-αhas been shown to directly interact with the NR2A and NR2BNMDA-receptor subunits to regulate their interaction withcytosolic scaffolding proteins such as PSD-95 and Sap102(Watanabe et al., 2003; Iwamoto et al., 2004) by CaMKII-dependent phosphorylation (Gardoni et al., 2006). It has alsobeen shown in retinal neurons to be upregulated, both at thegene and protein levels following NMDA-receptor stimulation(Laabich et al., 2000; Fan et al., 2005). Other retinal studies haveshown that specific blockade of the CaMKII-α autophospho-lylating site, with the highly specific myristoylated autocam-tide-2 related inhibitory peptide (AIP) (Ishida et al., 1995),attenuates NMDA-induced caspase-3 activation (Laabich andCooper, 2000; Laabich et al. 2000, 2001; Lin et al., 2004; Ishida etal., 2001); however, the effects of this antagonism on cell deathpathways not linked to caspase-3 pathway have not beenexplored.
Using an in vivo rat retinal toxicity model, the presentstudy confirmed that blockade of CaMKII-α activity preventsNMDA-induced caspase-3 activation and provides significantneuroprotection against NMDA-induced ganglion cell loss.However, antagonizing CaMKII-α had only a limited effect onaltering NMDA-induced loss of plasma membrane selectivity(LPMS: e.g. necrosis) in cells residing in the ganglion cell- (GCL)and inner nuclear (INL) layers of the retina. In addition, thisblockade had no effect on preventing NMDA-induced eleva-tion of poly(ADP-ribosyl)ation (PAR) through the PARP-1pathway, and did not block TUNEL labeling following amoderate excitotoxic insult by NMDA.
2. Results
2.1. Caspase-3 activation
Tomeasure the effectiveness of AIP in blocking the caspase-3-dependent pathway, retinas exposed to NMDA/AIP, NMDA-alone and appropriate controls were assessed for caspase-3activity at 1 h, 2 h, 4 h or 6 h post-treatment (PT) (Fig. 1a).NMDA treatment alone resulted in an increase in caspase-3activity at 1 h-PT and peaked by 2 h-PT (Fig. 1a: n=5 for eachtreatment and time point ,⁎⁎⁎ indicates p<0.001, ⁎ p<0.05significance than NMDA activity at 2 h, ANOVA). A decline inactivity was noted at 4 h- and 6 h-PT in the NMDA-treated

192 B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
retinas; however, the levels of activity remained significantlyelevated (p<0.05, ANOVA) as compared with time-matchedAIP/NMDA-, Ac-DEVD-CHO/NMDA-, PBS-treated anduntreated retinas (Fig. 1a). Inclusion of the CaMKII-α inhibitorAIP (nN6 for each time point) or the caspase-3 inhibitor Ac-DEVD-CHO (nN5 for each time point) blocked NMDA-induced
Fig. 2 – Rat retinas exposed to 20nmol ofNMDA (a, b)were examinResults from10 retinal sets showed that 20 nmol of NMDAstimulacells in the ganglion cell layer (GCL) and to a lesser extent in the icells increased by 6 h-PT on both the GCL and the INL (b). Treatmlabeling in the INLorGCL (c). Increasing thedoseofAIPhadno furthtreatedwith 4 nmol (d), 8 nmol (e) or 16 nmol AIP (f). Control retinaTUNEL-staining at 6 h-PT. Outer nuclear layer (ONL), Outer plexif
activation of caspase-3 at all time points with no significantdifference in activity noted between these two treatmentconditions, or with untreated retinas, pN0.05 (Fig. 1a). PBStreatment alone did stimulate a slight elevation of caspase 3activity at 4 h following treatment and resulted in a faintdetection of the 19 kDa activated caspase-3 fragment in one
ed for TUNEL-labeling at 4 h (a) and6 h (b) post-treatment (PT).tes TUNEL-labelingwithin 4h-PT, as evidenced by fluorescentnner nuclear layer (INL) (a). The number of TUNEL-stainedent of the retinas with 2 nmol AIP/NMDA did not block TUNELer effect in blockingNMDA-inducedTUNEL labeling in retinass treatedwith 2 nmol AIP/PBS (g) or PBS alone (h) exhibited noorm layer (OPL), inner plexiform layer (IPL).
193B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
set of retinas (Fig. 1b); however, this treatment did not causeany detectable increase in PAR levels (Fig. 6a), LPMS (Figs. 3f, g)or TUNEL-labeling (Figs. 2g, e), nor were there any detectabledifferences in the distribution of surviving ganglion cells ascompared with untreated retina (data not shown). Thevariability and late onset of these events in the PBS treatedeye is likely linked to amild inflammatory response caused bythe injection.
Retinas were also examined for the formation of activatedfragments of caspase-3 (Fig. 1b). Treatment with NMDA alone(n=4 sets) resulted in the visualization of immunoreactive
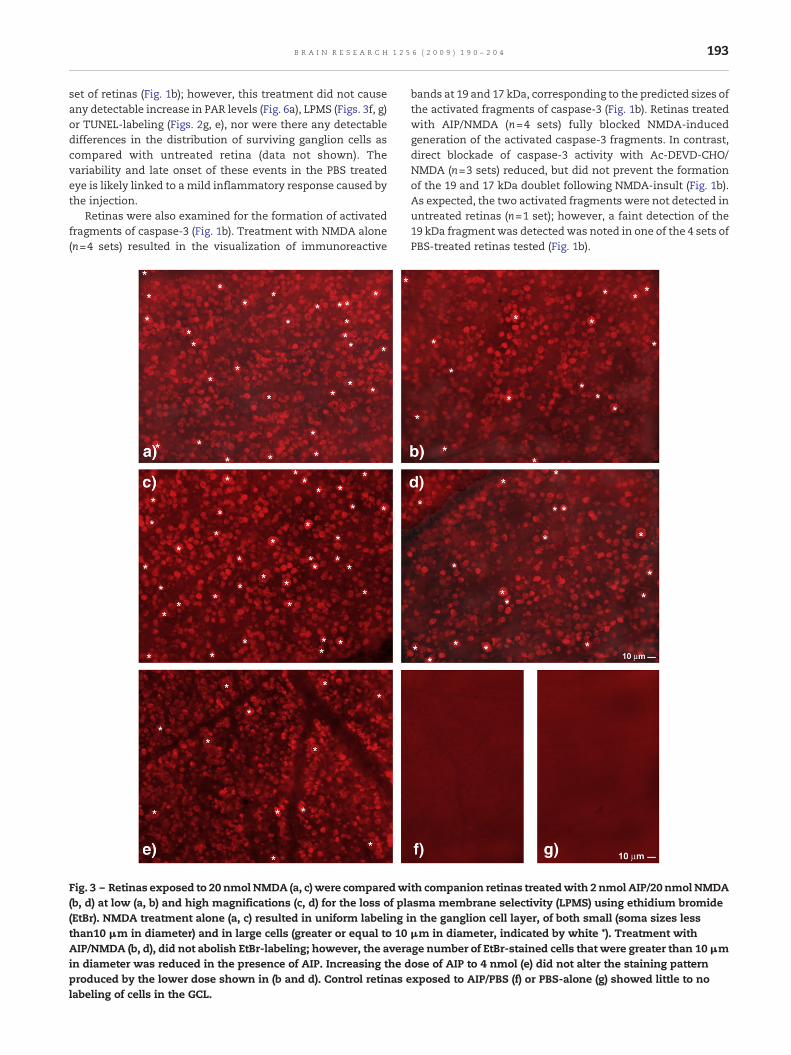
Fig. 3 – Retinas exposed to 20 nmol NMDA (a, c) were comparedw(b, d) at low (a, b) and high magnifications (c, d) for the loss of pla(EtBr). NMDA treatment alone (a, c) resulted in uniform labeling ithan10 μm in diameter) and in large cells (greater or equal to 10AIP/NMDA (b, d), did not abolish EtBr-labeling; however, the averain diameter was reduced in the presence of AIP. Increasing the dproduced by the lower dose shown in (b and d). Control retinas elabeling of cells in the GCL.
bands at 19 and 17 kDa, corresponding to the predicted sizes ofthe activated fragments of caspase-3 (Fig. 1b). Retinas treatedwith AIP/NMDA (n=4 sets) fully blocked NMDA-inducedgeneration of the activated caspase-3 fragments. In contrast,direct blockade of caspase-3 activity with Ac-DEVD-CHO/NMDA (n=3 sets) reduced, but did not prevent the formationof the 19 and 17 kDa doublet following NMDA-insult (Fig. 1b).As expected, the two activated fragments were not detected inuntreated retinas (n=1 set); however, a faint detection of the19 kDa fragment was detectedwas noted in one of the 4 sets ofPBS-treated retinas tested (Fig. 1b).
ith companion retinas treatedwith 2 nmol AIP/20 nmol NMDAsma membrane selectivity (LPMS) using ethidium bromiden the ganglion cell layer, of both small (soma sizes lessμm in diameter, indicated by white *). Treatment withge number of EtBr-stained cells that were greater than 10μmose of AIP to 4 nmol (e) did not alter the staining patternxposed to AIP/PBS (f) or PBS-alone (g) showed little to no
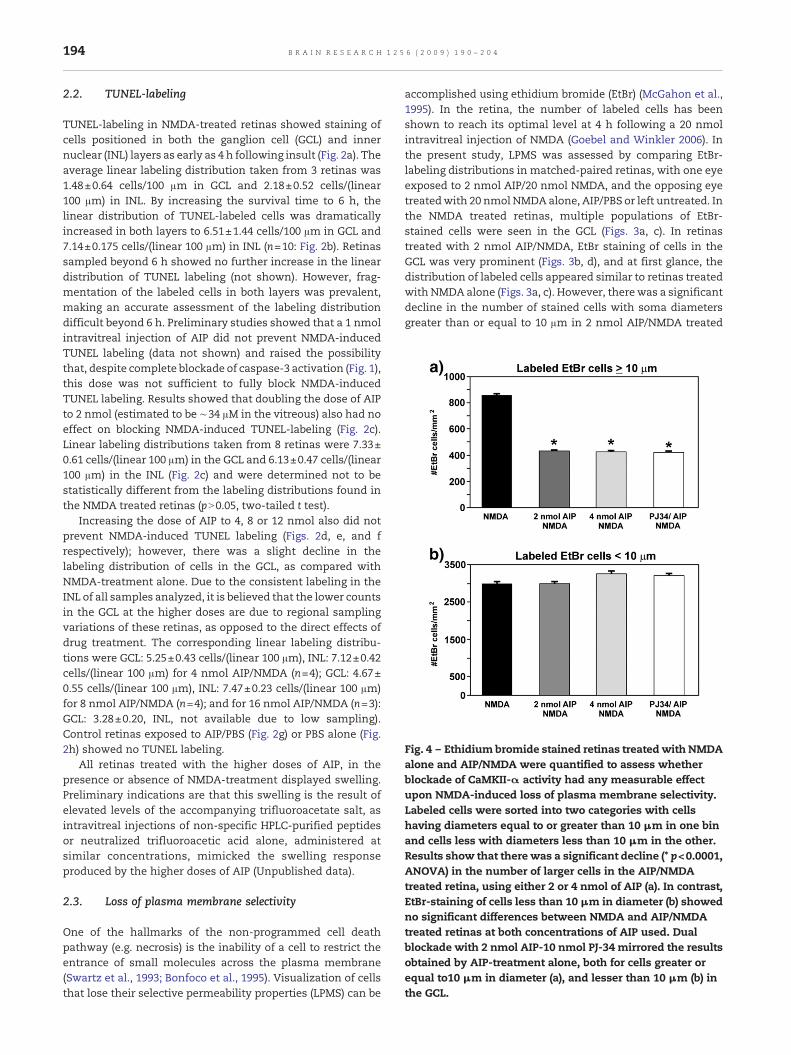
Fig. 4 – Ethidium bromide stained retinas treatedwith NMDAalone and AIP/NMDA were quantified to assess whetherblockade of CaMKII-α activity had any measurable effectupon NMDA-induced loss of plasma membrane selectivity.Labeled cells were sorted into two categories with cellshaving diameters equal to or greater than 10 μm in one binand cells less with diameters less than 10 μm in the other.Results show that there was a significant decline (* p<0.0001,ANOVA) in the number of larger cells in the AIP/NMDAtreated retina, using either 2 or 4 nmol of AIP (a). In contrast,EtBr-staining of cells less than 10 μm in diameter (b) showedno significant differences between NMDA and AIP/NMDAtreated retinas at both concentrations of AIP used. Dualblockade with 2 nmol AIP-10 nmol PJ-34 mirrored the resultsobtained by AIP-treatment alone, both for cells greater orequal to10 μm in diameter (a), and lesser than 10 μm (b) inthe GCL.
194 B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
2.2. TUNEL-labeling
TUNEL-labeling in NMDA-treated retinas showed staining ofcells positioned in both the ganglion cell (GCL) and innernuclear (INL) layers as early as 4 h following insult (Fig. 2a). Theaverage linear labeling distribution taken from 3 retinas was1.48±0.64 cells/100 μm in GCL and 2.18±0.52 cells/(linear100 μm) in INL. By increasing the survival time to 6 h, thelinear distribution of TUNEL-labeled cells was dramaticallyincreased in both layers to 6.51±1.44 cells/100 μm in GCL and7.14±0.175 cells/(linear 100 μm) in INL (n=10: Fig. 2b). Retinassampled beyond 6 h showed no further increase in the lineardistribution of TUNEL labeling (not shown). However, frag-mentation of the labeled cells in both layers was prevalent,making an accurate assessment of the labeling distributiondifficult beyond 6 h. Preliminary studies showed that a 1 nmolintravitreal injection of AIP did not prevent NMDA-inducedTUNEL labeling (data not shown) and raised the possibilitythat, despite complete blockade of caspase-3 activation (Fig. 1),this dose was not sufficient to fully block NMDA-inducedTUNEL labeling. Results showed that doubling the dose of AIPto 2 nmol (estimated to be ∼34 μM in the vitreous) also had noeffect on blocking NMDA-induced TUNEL-labeling (Fig. 2c).Linear labeling distributions taken from 8 retinas were 7.33±0.61 cells/(linear 100 μm) in the GCL and 6.13±0.47 cells/(linear100 μm) in the INL (Fig. 2c) and were determined not to bestatistically different from the labeling distributions found inthe NMDA treated retinas (pN0.05, two-tailed t test).
Increasing the dose of AIP to 4, 8 or 12 nmol also did notprevent NMDA-induced TUNEL labeling (Figs. 2d, e, and frespectively); however, there was a slight decline in thelabeling distribution of cells in the GCL, as compared withNMDA-treatment alone. Due to the consistent labeling in theINL of all samples analyzed, it is believed that the lower countsin the GCL at the higher doses are due to regional samplingvariations of these retinas, as opposed to the direct effects ofdrug treatment. The corresponding linear labeling distribu-tions were GCL: 5.25±0.43 cells/(linear 100 μm), INL: 7.12±0.42cells/(linear 100 μm) for 4 nmol AIP/NMDA (n=4); GCL: 4.67±0.55 cells/(linear 100 μm), INL: 7.47±0.23 cells/(linear 100 μm)for 8 nmol AIP/NMDA (n=4); and for 16 nmol AIP/NMDA (n=3):GCL: 3.28±0.20, INL, not available due to low sampling).Control retinas exposed to AIP/PBS (Fig. 2g) or PBS alone (Fig.2h) showed no TUNEL labeling.
All retinas treated with the higher doses of AIP, in thepresence or absence of NMDA-treatment displayed swelling.Preliminary indications are that this swelling is the result ofelevated levels of the accompanying trifluoroacetate salt, asintravitreal injections of non-specific HPLC-purified peptidesor neutralized trifluoroacetic acid alone, administered atsimilar concentrations, mimicked the swelling responseproduced by the higher doses of AIP (Unpublished data).
2.3. Loss of plasma membrane selectivity
One of the hallmarks of the non-programmed cell deathpathway (e.g. necrosis) is the inability of a cell to restrict theentrance of small molecules across the plasma membrane(Swartz et al., 1993; Bonfoco et al., 1995). Visualization of cellsthat lose their selective permeability properties (LPMS) can be
accomplished using ethidium bromide (EtBr) (McGahon et al.,1995). In the retina, the number of labeled cells has beenshown to reach its optimal level at 4 h following a 20 nmolintravitreal injection of NMDA (Goebel and Winkler 2006). Inthe present study, LPMS was assessed by comparing EtBr-labeling distributions in matched-paired retinas, with one eyeexposed to 2 nmol AIP/20 nmol NMDA, and the opposing eyetreatedwith 20 nmol NMDAalone, AIP/PBS or left untreated. Inthe NMDA treated retinas, multiple populations of EtBr-stained cells were seen in the GCL (Figs. 3a, c). In retinastreated with 2 nmol AIP/NMDA, EtBr staining of cells in theGCL was very prominent (Figs. 3b, d), and at first glance, thedistribution of labeled cells appeared similar to retinas treatedwith NMDA alone (Figs. 3a, c). However, therewas a significantdecline in the number of stained cells with soma diametersgreater than or equal to 10 μm in 2 nmol AIP/NMDA treated

195B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
retinas, as compared with retinas treated with NMDA alone(p<0.0001, ANOVA Tukey–Kramer multiple comparison test,F=136.51, n=3; Fig. 4). Increasing the dose of AIP to 4 nmolproduced an identical effect (Figs. 3e, 4). In contrast, there wasno significant difference in the densities of labeled somaswithdiameters less than 10 μm in the retinas treated with either 2or 4 nmol AIP/NMDA, versus those treated with NMDA alone(pN0.05; ANOVA Tukey–Kramer multiple comparison test,F=2.704). Control retinas treated with 2 nmol AIP/PBS (Fig. 3f)or PBS alone (Fig. 3g) showed only sparse EtBr staining of cellsin the GCL, indicating that loss of plasma membraneselectivity was due to NMDA-insult.
EtBr staining was also present in a large number of cellslocated in the INL in both NMDA- and AIP/NMDA-treatedretinas (Figs. 5a, b respectively), indicating that retinal inter-
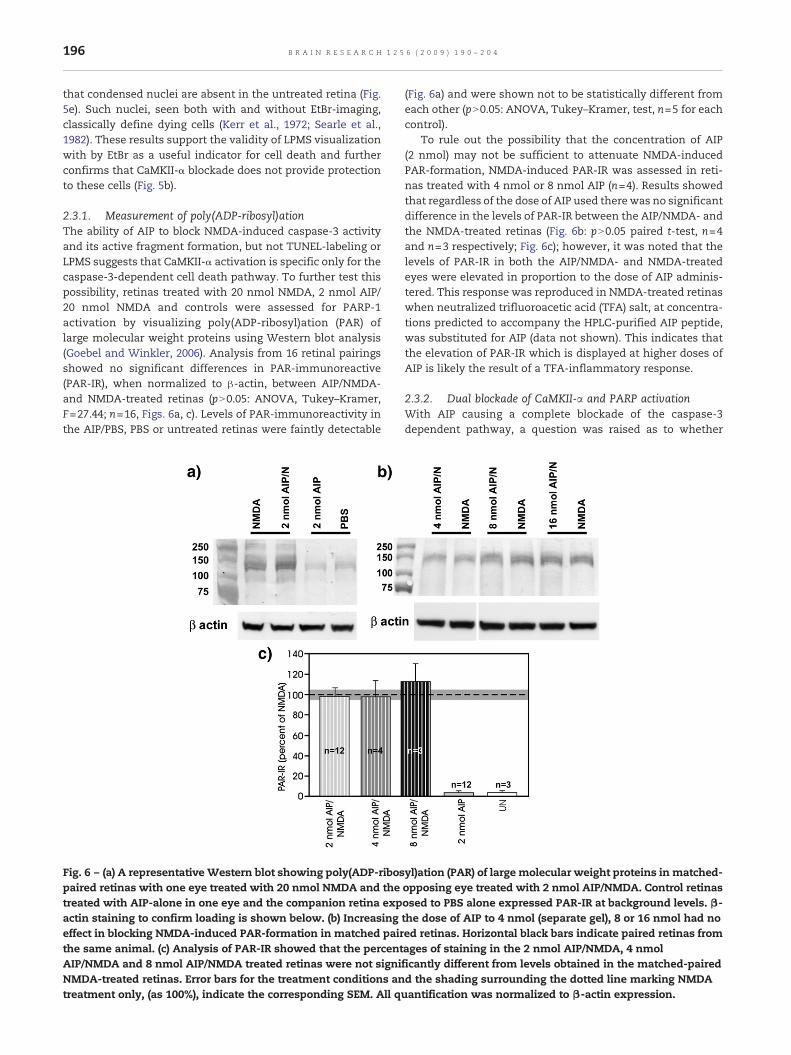
Fig. 5 – Semithin plastic radial sections of EtBr-stained retinas fonumerous labeled cells located in the ganglion cell layer (GCL), aswsame retinal samples were stained with Richardson's stain andAIP/NMDA, d). Both (c and d) show numerous cells with highly codisplay similar distribution patterns to the cells staining positiveNo condensed nuclei were seen in the INL or the GCL of the untrresult of NMDA-insult. Bar in figure b (for a and b), Bar in d (for c
neurons (e.g., amacrine and possibly a subpopulation(s) ofbipolar cell) are also highly susceptible to NMDA-induced lossof plasma membrane selectivity (LPMS).
In order to determine whether the appearance of LPMS isan accurate indication of dying cells, semi-thin radial sectionsof EtBr-stained AIP/NMDA and NMDA-treated retinas werecompared using epifluorescent detection (for LPMS) and brightfield optics (to display morphological characteristics) from thesame retinas. Results show that both the NMDA (Fig. 5a) andAIP/NMDA (Fig. 5b) treated retinas have numerous EtBr-labeled somas in both the GCL and INL. Bight field views ofcorresponding sections stained with Richardson stain (Figs. 5cand d respectively) display shrunken cells with highlycondensed nuclei in the INL, and to a lesser extent in theGCL, that mirrors the pattern produced by EtBr staining. Note
llowing NMDA (a) or 2 nmol AIP/NMDA (b) treatment showell as in the inner nuclear layer (INL). Sections taken from the
are shown at slightly higher magnification (NMDA, c;ndensed nuclei in the INL and in the GCL (white arrows) thatly for EtBr in the corresponding retinas (a and b respectively).eated retina (e), indicating that their appearance is the directand d).
196 B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
that condensed nuclei are absent in the untreated retina (Fig.5e). Such nuclei, seen both with and without EtBr-imaging,classically define dying cells (Kerr et al., 1972; Searle et al.,1982). These results support the validity of LPMS visualizationwith by EtBr as a useful indicator for cell death and furtherconfirms that CaMKII-α blockade does not provide protectionto these cells (Fig. 5b).
2.3.1. Measurement of poly(ADP-ribosyl)ationThe ability of AIP to block NMDA-induced caspase-3 activityand its active fragment formation, but not TUNEL-labeling orLPMS suggests that CaMKII-α activation is specific only for thecaspase-3-dependent cell death pathway. To further test thispossibility, retinas treated with 20 nmol NMDA, 2 nmol AIP/20 nmol NMDA and controls were assessed for PARP-1activation by visualizing poly(ADP-ribosyl)ation (PAR) oflarge molecular weight proteins using Western blot analysis(Goebel and Winkler, 2006). Analysis from 16 retinal pairingsshowed no significant differences in PAR-immunoreactive(PAR-IR), when normalized to β-actin, between AIP/NMDA-and NMDA-treated retinas (pN0.05: ANOVA, Tukey–Kramer,F=27.44; n=16, Figs. 6a, c). Levels of PAR-immunoreactivity inthe AIP/PBS, PBS or untreated retinas were faintly detectable
Fig. 6 – (a) A representativeWestern blot showing poly(ADP-ribospaired retinas with one eye treated with 20 nmol NMDA and thetreated with AIP-alone in one eye and the companion retina expactin staining to confirm loading is shown below. (b) Increasingeffect in blocking NMDA-induced PAR-formation in matched pairthe same animal. (c) Analysis of PAR-IR showed that the percentAIP/NMDA and 8 nmol AIP/NMDA treated retinas were not signiNMDA-treated retinas. Error bars for the treatment conditions antreatment only, (as 100%), indicate the corresponding SEM. All q
(Fig. 6a) and were shown not to be statistically different fromeach other (pN0.05: ANOVA, Tukey–Kramer, test, n=5 for eachcontrol).
To rule out the possibility that the concentration of AIP(2 nmol) may not be sufficient to attenuate NMDA-inducedPAR-formation, NMDA-induced PAR-IR was assessed in reti-nas treated with 4 nmol or 8 nmol AIP (n=4). Results showedthat regardless of the dose of AIP used there was no significantdifference in the levels of PAR-IR between the AIP/NMDA- andthe NMDA-treated retinas (Fig. 6b: pN0.05 paired t-test, n=4and n=3 respectively; Fig. 6c); however, it was noted that thelevels of PAR-IR in both the AIP/NMDA- and NMDA-treatedeyes were elevated in proportion to the dose of AIP adminis-tered. This response was reproduced in NMDA-treated retinaswhen neutralized trifluoroacetic acid (TFA) salt, at concentra-tions predicted to accompany the HPLC-purified AIP peptide,was substituted for AIP (data not shown). This indicates thatthe elevation of PAR-IR which is displayed at higher doses ofAIP is likely the result of a TFA-inflammatory response.
2.3.2. Dual blockade of CaMKII-α and PARP activationWith AIP causing a complete blockade of the caspase-3dependent pathway, a question was raised as to whether
yl)ation (PAR) of large molecular weight proteins in matched-opposing eye treated with 2 nmol AIP/NMDA. Control retinasosed to PBS alone expressed PAR-IR at background levels. β-the dose of AIP to 4 nmol (separate gel), 8 or 16 nmol had noed retinas. Horizontal black bars indicate paired retinas fromages of staining in the 2 nmol AIP/NMDA, 4 nmolficantly different from levels obtained in the matched-pairedd the shading surrounding the dotted line marking NMDAuantification was normalized to β-actin expression.

197B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
dual blockade of CaMKII-α and PARP-1 would result in anydetectable changes in NMDA-induced LPMS. Rats were pre-treated with an intravitreal injection containing 2 nmol AIPand 10 nmol of PJ-34, a dose previously shown to significantlyattenuate NMDA-induced poly(ADP-ribosyl)ation (PAR) in theretina (Goebel and Winkler, 2006), followed by a secondinjection containing the inhibitors and 20 nmol of NMDA.Four hours following insult, these retinas were comparedwithretinas treated with AIP/NMDA for LPMS and PAR-IR.
EtBr staining in the AIP/PJ-34 treated retinas showed nodetectable differences in the staining pattern of labeled cells(Fig. 7a) as compared with AIP/NMDA treated retinas (Fig. 7b).Counts of EtBr-labeled cell distributions confirmed theseobservations, as there were no additive effects in blockingLPMS in the presence of both inhibitors (Figs. 7a, 4a, b) ascompared with retinas pretreated with 2 nmol of AIP alone(ANOVA, pN0.05; Figs. 7b, 4a, b).
PAR-IR from each of the retinal pairings (n=3) was alsomeasured and showed that co-exposure of AIP/PJ-34 (Fig. 7e;lanes 1–3) significantly attenuated NMDA-induced PAR-for-mation by 65%, as compared with NMDA-treatment alone (Fig.
Fig. 7 – Retinas treated with 2 nmol AIP and 10 nmol of the PARP-determine if dual blockade would have any synergistic effect in anot block NMDA-induce loss of plasma membrane selectivity (a)AIP-alone (b). Both (a and b) were obtained from the superior tempnerve head. Assessing PAR-IR in matched paired retinas (c) co-trcorresponding retinas treated with NMDA alone (lanes 4–6) showby 65% (n=3), p<0.019, unpaired t-test) as comparedwith levels excorresponding β-actin signaling (insets).
7c; lanes 4–6; ANOVA, Tukey–Kramer multiple comparisontest, PJ-AIP/NMDA versus NMDA, p<0.05; F=21.212).
2.4. Fluorogold labeling of surviving ganglion cells
The long-term survival of ganglion cells in treated retinas wasexamined through the use of fluorogold retrograde labeling.Retinal pairings (n=3), where the left eyes were treated withAIP/NMDA and the right eyes were treated with NMDA alone,were assessed for surviving ganglion cells 11 days followingNMDA insult. As ameasurement for determining the degree ofganglion cell loss, right eyes of littermates were left untreated(n=5) and compared with the left eyes treated with eitherNMDA alone (n=2), with AIP/NMDA (n=2), or PBS alone (n=1;data not shown) and quantified. Results show that 20 nmol ofNMDA leads to a large depletion in the number of survivingganglion cells in both central (Fig. 8c) and peripheral retina(Fig. 8d), as compared with corresponding regions in theuntreated retina (Figs. 8a, b). Retinas treated with 2 nmol AIP/20 nmol NMDA (Figs. 9e, f) showed a noticeable increase in thenumber of surviving ganglion cells as compared with retinas
1 inhibitor PJ-34 were examined for NMDA-induced LPMS, tottenuating this process. Results show that dual-inhibition did, as these retinas stained identically to retinas treated withoral region of the retina, approximately 1.5mm from the opticeated with 2 nmol AIP/10 nmol PJ-34-NMDA (lanes 1–3) withed significant reductions in PAR-immunoreactivity (reducedpressedwith NMDAalone (d), when normalized against their

Fig. 8 – The number of fluorogold labeled ganglion cells from untreated retinas (a, b) was compared with matched pairs ofretinas treated 11 days previously with 20 nmol NMDA (c, d) or with 2 nmol AIP/20 nmol NMDA (e, f). Representative views (a, cand e) are from central retina, approximately 1.5 mm from the optic nerve head; b, d, and f are from peripheral retinaapproximately 3.2 mm from the optic nerve head. Results show that NMDA induces a dramatic loss in the number of survivingretinal ganglion cells across the full extent of the retina (c, d). Inclusion of AIP improved ganglion cell survival (e, f); however, thisprovided only partial neuroprotection, as the numbers of surviving ganglion cells in these retinas were noticeably less than inthe untreated retinas (a, b).
198 B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
treated with NMDA alone (Figs. 8c, d); however, the level ofrescue provided by AIP-treatment (Figs. 8e, f) was only afraction of the number of cells expressed in the same region ofthe untreated retina (Figs. 8a, b).
Full quantification of the number of surviving ganglioncells in each retina was carried on bins of concentric rings of1 mm each surrounding the optic nerve (see Fig. 9). Theresulting data shows that NMDA-treatment significantlyreduces the number of surviving ganglion cells by an averageof 69.4% in the retina (p<0.001; ANOVA, Bonferroni multiplecomparison test; n=5), as compared with the untreated retina.
AIP significantly improved ganglion cell survival, as comparedwith NMDA treatment alone p<0.01 (ANOVA, Bonferronimultiple comparison test, n=5), at all 4 eccentricities mea-sured (F values from bins 1–4 are 35.065, 62.637, 35.728 and18.681 respectively). Although AIP improved ganglion cellsurvival, full protection was not achieved, as the number ofsurviving cells in bins 1–3 (0–3 mm) of AIP/NMDA treatedretinas was significantly lower (68% of control values; p<0.01,ANOVA) than in the untreated retina. This trend continued ateccentricities beyond 3 mm with AIP/NMDA treated retinashaving a smaller average of surviving cells than the untreated
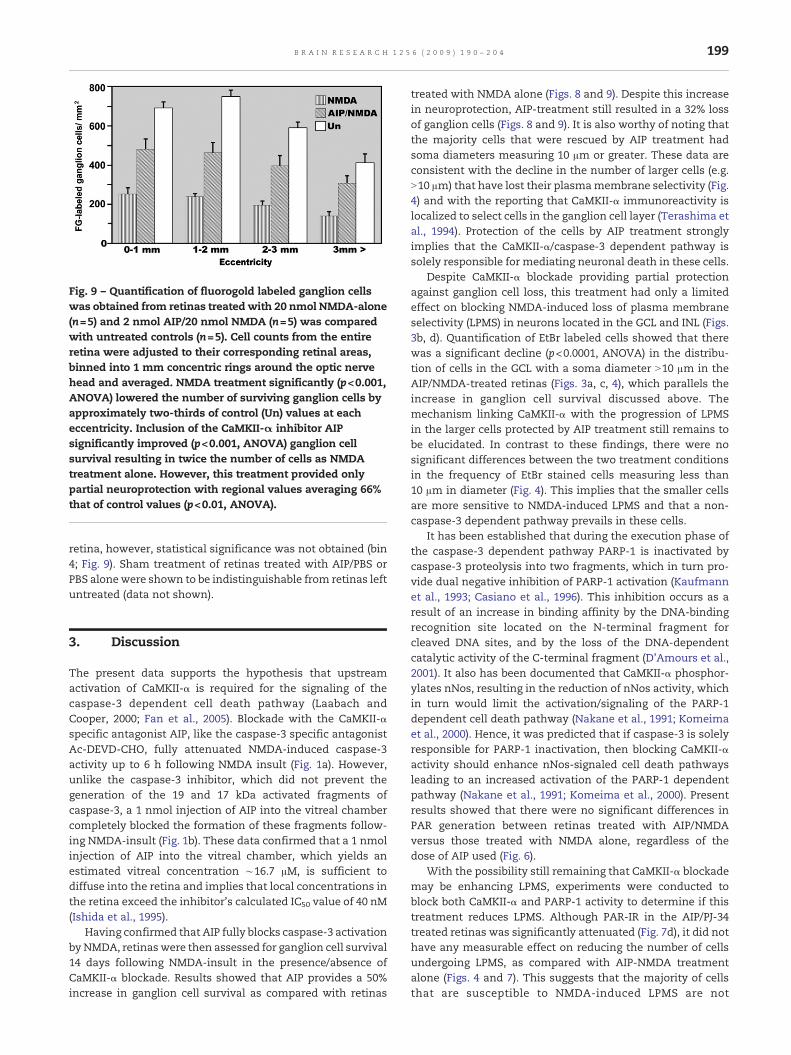
Fig. 9 – Quantification of fluorogold labeled ganglion cellswas obtained from retinas treated with 20 nmol NMDA-alone(n=5) and 2 nmol AIP/20 nmol NMDA (n=5) was comparedwith untreated controls (n=5). Cell counts from the entireretina were adjusted to their corresponding retinal areas,binned into 1 mm concentric rings around the optic nervehead and averaged. NMDA treatment significantly (p<0.001,ANOVA) lowered the number of surviving ganglion cells byapproximately two-thirds of control (Un) values at eacheccentricity. Inclusion of the CaMKII-α inhibitor AIPsignificantly improved (p<0.001, ANOVA) ganglion cellsurvival resulting in twice the number of cells as NMDAtreatment alone. However, this treatment provided onlypartial neuroprotection with regional values averaging 66%that of control values (p<0.01, ANOVA).
199B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
retina, however, statistical significance was not obtained (bin4; Fig. 9). Sham treatment of retinas treated with AIP/PBS orPBS alonewere shown to be indistinguishable from retinas leftuntreated (data not shown).
3. Discussion
The present data supports the hypothesis that upstreamactivation of CaMKII-α is required for the signaling of thecaspase-3 dependent cell death pathway (Laabach andCooper, 2000; Fan et al., 2005). Blockade with the CaMKII-αspecific antagonist AIP, like the caspase-3 specific antagonistAc-DEVD-CHO, fully attenuated NMDA-induced caspase-3activity up to 6 h following NMDA insult (Fig. 1a). However,unlike the caspase-3 inhibitor, which did not prevent thegeneration of the 19 and 17 kDa activated fragments ofcaspase-3, a 1 nmol injection of AIP into the vitreal chambercompletely blocked the formation of these fragments follow-ing NMDA-insult (Fig. 1b). These data confirmed that a 1 nmolinjection of AIP into the vitreal chamber, which yields anestimated vitreal concentration ∼16.7 μM, is sufficient todiffuse into the retina and implies that local concentrations inthe retina exceed the inhibitor's calculated IC50 value of 40 nM(Ishida et al., 1995).
Having confirmed that AIP fully blocks caspase-3 activationby NMDA, retinas were then assessed for ganglion cell survival14 days following NMDA-insult in the presence/absence ofCaMKII-α blockade. Results showed that AIP provides a 50%increase in ganglion cell survival as compared with retinas
treated with NMDA alone (Figs. 8 and 9). Despite this increasein neuroprotection, AIP-treatment still resulted in a 32% lossof ganglion cells (Figs. 8 and 9). It is also worthy of noting thatthe majority cells that were rescued by AIP treatment hadsoma diameters measuring 10 μm or greater. These data areconsistent with the decline in the number of larger cells (e.g.N10 μm) that have lost their plasmamembrane selectivity (Fig.4) and with the reporting that CaMKII-α immunoreactivity islocalized to select cells in the ganglion cell layer (Terashima etal., 1994). Protection of the cells by AIP treatment stronglyimplies that the CaMKII-α/caspase-3 dependent pathway issolely responsible for mediating neuronal death in these cells.
Despite CaMKII-α blockade providing partial protectionagainst ganglion cell loss, this treatment had only a limitedeffect on blocking NMDA-induced loss of plasma membraneselectivity (LPMS) in neurons located in the GCL and INL (Figs.3b, d). Quantification of EtBr labeled cells showed that therewas a significant decline (p<0.0001, ANOVA) in the distribu-tion of cells in the GCL with a soma diameter N10 μm in theAIP/NMDA-treated retinas (Figs. 3a, c, 4), which parallels theincrease in ganglion cell survival discussed above. Themechanism linking CaMKII-α with the progression of LPMSin the larger cells protected by AIP treatment still remains tobe elucidated. In contrast to these findings, there were nosignificant differences between the two treatment conditionsin the frequency of EtBr stained cells measuring less than10 μm in diameter (Fig. 4). This implies that the smaller cellsare more sensitive to NMDA-induced LPMS and that a non-caspase-3 dependent pathway prevails in these cells.
It has been established that during the execution phase ofthe caspase-3 dependent pathway PARP-1 is inactivated bycaspase-3 proteolysis into two fragments, which in turn pro-vide dual negative inhibition of PARP-1 activation (Kaufmannet al., 1993; Casiano et al., 1996). This inhibition occurs as aresult of an increase in binding affinity by the DNA-bindingrecognition site located on the N-terminal fragment forcleaved DNA sites, and by the loss of the DNA-dependentcatalytic activity of the C-terminal fragment (D'Amours et al.,2001). It also has been documented that CaMKII-α phosphor-ylates nNos, resulting in the reduction of nNos activity, whichin turn would limit the activation/signaling of the PARP-1dependent cell death pathway (Nakane et al., 1991; Komeimaet al., 2000). Hence, it was predicted that if caspase-3 is solelyresponsible for PARP-1 inactivation, then blocking CaMKII-αactivity should enhance nNos-signaled cell death pathwaysleading to an increased activation of the PARP-1 dependentpathway (Nakane et al., 1991; Komeima et al., 2000). Presentresults showed that there were no significant differences inPAR generation between retinas treated with AIP/NMDAversus those treated with NMDA alone, regardless of thedose of AIP used (Fig. 6).
With the possibility still remaining that CaMKII-α blockademay be enhancing LPMS, experiments were conducted toblock both CaMKII-α and PARP-1 activity to determine if thistreatment reduces LPMS. Although PAR-IR in the AIP/PJ-34treated retinas was significantly attenuated (Fig. 7d), it did nothave any measurable effect on reducing the number of cellsundergoing LPMS, as compared with AIP-NMDA treatmentalone (Figs. 4 and 7). This suggests that the majority of cellsthat are susceptible to NMDA-induced LPMS are not

200 B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
influenced by either the caspase-3 dependent or the PARP-1dependent cell death pathway.
Despite AIP fully blocking NMDA-induced caspase-3 acti-vation in the retina and providing partial protection againstganglion cell loss, this blockade did not block NMDA-inducedTUNEL labeling (Figs. 2c–f), regardless of the dose of AIPadministered. Cell counts revealed that there were nostatistical differences in the labeling patterns displayed inthe INL or the GCL between 2 nmol AIP/NMDA-treated andNMDA-treatment alone (pN0.05, two-tailed t test, nN8). Thepersistence of TUNEL labeling in the presence of CaMKII-αblockade contradicts Laabach and Cooper's (2000) findings,where they showed AIP to block NMDA-induced TUNEL-labeling at 24 h following treatment. Possible explanationsfor these differences could be the sampling time chosen tomonitor TUNEL-labeling, as we (Goebel andWinkler, 2006) andothers (Manabe and Lipton, 2003; Lam et al., 1999) have shownTUNEL-labeling to be optimal at 6 h-post-treatment followinga 20 nmol NMDA-insult (Fig. 2b). In contrast, Laabach andCooper (2000) used an 8 nmol dose of NMDA, which wouldlikely extend the onset for optimal TUNEL labeling. It is alsopossible that, at this reduced level of insult, the resulting DNAdamage may be mediated solely by the caspase-3 dependentpathway (Bonfoco et al., 1995) and thus would be prevented byCaMKII-α blockade (Laabach and Cooper, 2000). However, thiswould contradict the current thinking that TUNEL-labeling isnot unique to the programmed cell death pathway but canresult from necrosis as well (Nishizaki et al., 1999; Barth et al.,2002). It should be noted here that LPMS still occurs in retinasexposed to a 5 nmol injection of NMDA (Goebel and Winkler,2006). Lastly, we found that the integrity of the AIP-treatedretinas was much more resistant to the diffusion of terminaltransferase into the retinal sections than retinas exposed toNMDA treatment alone. This was evidenced by the need forlonger proteinase K digestion times to optimize TUNELlabeling in the AIP/NMDA-treated retinas. Regardless ofthese technical subtleties, the persistence of TUNEL-labelingin the AIP-treated retinas indicates that DNA fragmentation isoccurring independently of the caspase-3 dependent pathway.The likely sources are either through the activation ofprogrammed cell death by way of a caspase-3 independentpathway (Nicholson et al., 2001; McKernan et al., 2007), and/orby a non-programmed cell death pathway.We cannot rule outthe possibility that one or both pathways are interlinked, orserve as a default when the caspase-3 dependent pathway hasbeen compromised (Lin et al., 2004; Fan et al., 2005; Gillardon etal., 1999; Nakane et al., 1991; Komeima et al., 2000).
Despite the partial protection of retinal ganglion cells byCaMKII-αblockadeorbyblockadeofPARP-1activity (Goebel andWinkler, 2006), it is clear that a significant number of ganglioncells (35%; Figs. 8, 9), as well as smaller inter-neurons in the INLandGCL, are still lost by either a non-caspase-3 dependent and/or a non-programmed cell death pathways. Similar findingshave been reported in the brainwhere only partial neuroprotec-tion of primary cortical neurons following CaMKII-α blockadewas achieved by AIP or KN93 following beta-amyloid insult (Linet al., 2004). Collectively, these findings suggest that themodeofcell death for a given cell type is unique and that, in some cases,it may be multimodal. This is consistent with previousobservations that some dying neurons display ultrastructural
pathologies that appear as hybrids between apoptotic andnecrotic (Baille et al., 2005; Ünal-Çevik et al., 2004) and suggestthat bothpathwaysare concomitantly activated in the samecell(See review, Yaun et al., 2003; Portera-Cailliau et al., 1997).
In conclusion, the present study indicates that blockade ofCaMKII-α provides only partial neuroprotection against retinalganglion cell loss and has a limited effect in attenuating LPMSin the smaller sized cells. These data suggest that yet anotherset(s) of regulatory cell death mediator molecules is beingsimultaneously signaled through NMDA-receptor hyperacti-vation. Finally, this study provides clear evidence that CaMKII-α is not responsible for mediating all modes of NMDA-receptor-induced neuronal cell death.
4. Experimental procedures
4.1. Materials
N-methyl-D-aspartate, alkaline phosphatase-linked goat anti-rabbit-IgG, mouse monoclonal anti-β-actin, PJ-34 (Sigma, StLouis, MO); myristoylated autocamtide-2 related inhibitorypeptide, Ac-DEVD-CHO (Calbiochem-Novabiochem Corp.,LaJolla, CA); Ac-DEVD-fmc (Bachem Bioscience Inc., Torrance,CA); caspase-3 inhibitor I (Ac-Asp-Glu-Val-Asp-CHO, or Ac-DEVD-CHO) (Calbiochem, San Diego, CA); peroxidase-linkedsheep anti-mouse-IgG (Amersham, Piscataway, NJ); In situ celldeath detection kit, rabbit anti-PARP-1, LumilightWestern blotsubstrate, NBT/BCIP in solution (RocheMolecular BiochemicalsInc, Indianapolis, IN); rabbit anti-caspase-3 (Cell SignalingTechnology, Beverly, MA); rabbit anti-PAR (BD Biosciences, SanJose, CA); Fluorogold (Fluorochrome LLC, Denver Colorado);polylysine coated slides (Polyscience Inc. Warrington, PA);Slowfade mounting medium (Molecular Probes, Eugene, OR).
To minimize animal to animal variations (Danias et al.,2002),matched paired retinaswere used throughout this studyto validate changes produced by a given treatment. MaleSprague-Dawley rats weighing 175–225 g were anaesthetizedwith isoflurane and given a 3 μl intravitreal injection contain-ing the CaMKII-α inhibitor autocamtide-2-related inhibitorypeptide (AIP) at 1 or 2, 4, 8, 12 or 16 nanomoles (nmol), thecaspase-3 specific inhibitor Ac-DEVD-CHO (12.5 nmol), or aninhibitor cocktail containing 2 nmol AIP and 10 nmol of thePARP-1 specific inhibitor PJ-34 in to the left eye, while the righteye received either vehicle (0.01 M sterile phosphate bufferedsaline, pH 7.2), inhibitor alone or was left untreated. Assumingthat the vitreal volume of a rat eye is ∼60 μl, it is estimatedthat the vitreal concentrations of AIP ranged from 16.7 μM for1 nmol injected, to 266.7 μM for a 16 nmol injection. Allintravitreal injectionswere administered at the superior-nasalquadrant at the region of the ora serrata using an ultra-fineneedle (32 gauge; Hamilton 701RN that wasmodified to reducethe diameter of the tip and barrel) which was viewed with theaid of a stereo-dissection microscope. The needle tip waspositioned just posterior to the center of the lens and thecontents of the syringe were injected over a 1 min period. Toprevent the loss of the injected material through the injectionsite, the needle remained in place an additional minute beforeit was removed from the eye. Two hours following the initialinjection, a second set of injections containing a mixture of

201B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
20 nmol of NMDA (vitreal concentration estimated to be333.3 μM) and the corresponding inhibitor(s) were adminis-tered to the left eye while the right eye received one of thefollowing treatments: 20 nmol NMDA-alone, PBS, AIP or wasleft untreated. All animals were sacrificed by decapitation attimes previously determined to optimally express the desiredmarker of cell death (Goebel and Winkler, 2006).
4.2. TUNEL-labeling
Retinal pairs were compared at 4 and 6 h following insult forTUNEL-stainingwith one eyebeing treatedwith20nmolNMDAalone and the opposing eye receiving pretreatment of 1, 2, 4, 8or 12 nmol AIP into the vitreous 2 h prior toNMDA-treatment inthepresence ofAIP. Posterior eye cups containing intact retinaswere fixed for 1 h in cold 4% paraformaldehyde, rinsed in 0.1 Mphosphate buffer containing 0.01% sodium azide (PAZ) andthen sequentially cryoprotected in graded sucrose (10, 20 and30%) in PAZ. The individual retinaswere dissected from the eyecups, flat-embedded in Tissue Tec and thenmounted for radialsectioning using a cryostat. Sections (15 μm), cut through thecenter region of the retina,weremounted onpolylysine subbedslides, and stored at −20 °C until use.
For the TUNEL reaction, the slides containing the frozensections were briefly thawed and then rinsed in PBS for 2 minprior to pretreatment with proteinase K (1 mg/ml PK in 10 mMTRIS/HCl, pH 7.5) for 1–2min at room temperature, followed bya rinsed in PBS containing 0.1% Triton X 100. The sectionswerethen incubated for 1 h at 37 °C in terminal transferase mixtureusing fluorescein-dUTP (Roche In situ cell death labeling kit),rinsed several times in PBS and coversliped in Slow fade.Digital photographs were acquired using a Zeiss Apotomemicroscope equipped for epifluorescence.
4.3. Measurement of caspase-3 activity
As an independent measure of programmed cell death(Tenneti and Lipton, 2000), caspase-3 activity was assessedin retinas treatedwithNMDA alone or in the presence of AIP orAc-DEVD-CHO. Treated and control retinas were collected atdesignated time points, individually homogenized in 250ml ofcold lysis buffer (50 mM Tris, 150 mM NaCl, 0.5 nm EDTA and0.5%Nonidet P-40, pH 7.5) and centrifuged to remove insolublematerial. The supernatant was analyzed for total proteincontent (Henkel and Bieger, 1994) and assayed for caspase-3activity following the methods of Rhéaume et al. (1997) andKim et al. (1999). Aliquots of 100 μl of each retinal homogenate(∼200–300 mg total protein) were added to equal volumes ofreaction buffer (20 mM Hepes, 50 mM NaCl, 2.5 mM DTT, pH7.5) containing 40 mM of the caspase-3 specific substrate Ac-DEVD-fmc and incubated at 37 °C for 4 h. Caspase-3 activitywas determined by measuring floromethylcoumarin-fluores-cence (fmc), a byproduct produced by caspase-3 cleavage ofthe DEVD substrate. Measurements were taken at 30 minintervals starting at time zero and ending at 4 h using a 96 wellfluorometer set for 360 nm excitation and 450 nm emission.Caspase-3 activity was corrected to background (time zero)and normalized for protein content. Statistical analysis wasdone using Student t test pair analysis with statisticalsignificance determined at p≤0.05.
4.4. Western blot analysis for the activated fragmentsof caspase-3
Retinas subjected to NMDA, AIP/NMDA, Ac-DEVD-CHO/NMDA,or PBS treatment were pooled (6 retinas per assay) for eachtreatment condition at 2 h post-treatment (PT), homogenized in1 ml of ice-cold lysis buffer (25 mM Tris–HCl (pH 7.5), 100 mMNaCl 1% NP-40, 0.2 mM PMSF. The supernatant was thenimmuno-precipitatedwith anantibody specific for theactivatedfragments of caspase-3 over night at 4 °C. The antigen/antibodycomplexes were captured with the addition of Protein A-Sepharose 4B beads, washed with a series of low and high saltbuffers (50 mM Tris–HCl (pH 7.5), 0.1% NP-40, 10% glycerol,containing either 150 mM NaCl or 500 mM NaCl) and then heatdenatured at 95 °C in an equal volume of 2× gel loading buffer(125 mM Tris–HCl (pH 6.8), 4% SDS, 20% glycerol 0.5% β-mecaptoethanol and50mg/mlbromophenol blue).Thesampleswere separated on a 15% SDS-polyacrylamide gel, electro-transferred to nitrocellulose and then subjected to Westernblot analysis for visualization of the activated caspase-3fragments. The transfer was blocked with 8% dry milk contain-ing 2% whole goat serum and 2% BSA, incubated overnight in a1:1000 dilution of rabbit anti-caspase-3 followed by a 1:10,000dilution of a goat-anti-rabbit IgG-HRP linked secondary anti-body. Immunolabeled bands were visualized using Lumilightchemiluminescence regent and film autoradiography.
4.5. Ethidium bromide staining
We previously have shown that loss of plasma membraneselectivity (LPMS) of retinal neurons, as visualized by ethidiumbromide staining, reaches its peak at 4 h following exposure to20 nmol of NMDA (Goebel and Winkler, 2006). Freshly obtainedposterior eye cups from treated and control eyeswere incubatedfor 1 min in 150 μg/ml ethidium bromide (EtBr) in 0.01 Mphosphate buffer-0.85% saline (PBS), rinsed in cold PBS and thenfixed in 4% paraformaldehyde. Retinas were dissected awayfrom the posterior eye cups, and either flat-mounted, ganglioncell side up on glass slides and coversliped in slowfade, orembedded in Epon-plastic for semi-thin sectioning. The flatmounted retinasweredigitallyphotographed inmontage formatusing a rhodamine filter pack. Quantification of EtBr-labeledcellswasobtained froma750μmthickswatch that includesbothnasal and temporal regions of the retina and passes through theoptic nerve head. Digital images from sampling areas 0.5–3 mmfrom the optic nerve head were normalized to a 256 grayscaleand then manually counted. Previous analysis has shown thatthe number of EtBr labeled cells per unit area in NMDA-treatedretinas is not regionally different between 0.5 mm and 3 mm,allowing for the pooling of the data (unpublished data).Treatment conditions for each retina sampled were thencompared using ANOVA to determine if blockade of CaMKII-αhas any effect in altering the number of cells that undergoNMDA-induced loss of plasma membrane selectivity (LPMS).
4.6. Measurement of poly(ADP-ribose)polymerasedependent polyribosylation of nuclear proteins
Retinas exposed to NMDA, NMDA/AIP or AIP alone wereassessed for poly(ADP-ribose) polymerase-1 activity (PARP-1;

202 B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
EC 2.4.4.30) by measuring the levels of poly(ADP-ribosyl)ation(PAR) of putative large molecular weight proteins obtainedfrom the retinal homogenates (Mandir et al., 2000; Goebel andWinkler, 2006). Previous studies in our lab have determinedthat 20 nmol of NMDA initiates an optimal level of PAR-expression 4 h following insult (Goebel and Winkler, 2006).Following this dose and time frame, individual treated andcontrol retinas were homogenized in cold homogenizingbuffer (62.5 mM Tris–HCl, 6 M Urea, 10% glycerol, 2% sodiumdodecyl sulfate, 5% β-mercaptoethanol and 200 μg/ml benza-mide), as previously described (Goebel and Winkler, 2006).Equal protein loads, ranging from 35–65 μg, of material wereseparated on an 8% polyacrylamide-SDS gel and electro-transferred onto a nitrocellulose coated membrane. Transferswere blocked in 8% driedmilk containing 2%whole goat serum(WGS) in 10 mM Tris-saline-0.5% Tween 20 (TST), incubatedovernight in a 1:2000 dilution of a rabbit generated anti-PARantibody in 1%WGS-TST followed by a 1 h incubation at roomtemperature in a 1:5000 dilution of an alkaline phosphatase-liked goat anti-rabbit-IgG. PAR-immunoreactivity (PAR-IR) wasvisualized using NBT/BCIP and digitally scanned. The result-ing bands were measured for intensity of staining using ScionImage densitometric software (Scion Corporation, Frederick,MD). To account for sample loading, each transfer wasreprobed for β-actin content, as previously described (Goebeland Winkler, 2006).
4.7. Measurement of surviving ganglion cell usingretrograde labeling of the superior colliculus with fluorogold
Ganglion cell distributions for untreated and NMDA- and AIP/NMDA-treated retinas were assessed using fluorogold retro-grade labeling of the superior colliculus. To avoid animal toanimal variations (Danias et al., 2002), matched paired retinaswere used to compare the effects of NMDA induced cell deathin the presence or absence of AIP. Rats were treated as follows,with one group receiving intravitreal injections containing2 nmol AIP/20 nmol NMDA in the left eye and 20 nmol ofNMDA in the right. A second group of animals from the samelitter received 20 nmol of NMDA in the left and either PBS or notreatment in the right eye. One week following intravitrealinjections, the animals were anesthetized using ketamine/xylazine I.P , secured in a stereotaxic holder and prepared forbilateral stereotaxic injections, as previously described (Goe-bel and Winkler 2006). A gelform slurry containing 2%fluorogold (FG: Fluorochrome LLC, Denver CO) was loadedinto a blunt end 18 gauge needle and stereotacticallypositioned just above the superior colliculi. A 20 μl volumewas injected over a 4min period onto each colliculus, followedby a 4min rest period prior to needle retraction. The scalp wassutured closed and the animal was administered lactateringers (10–20 cc I.P.) to maintain hydration. A daily injectionof carprofen (5 mg/kg S.Q.) was given to minimize paindistress. Four days following the stereotaxic procedure therats were euthanized. The matched pairs of eyes were rapidlydissected, fixed in 4% paraformaldehyde and then mountedganglion cell layer side up as described above. Each retina wasmarked for orientation by a single radial incision from themargin to the optic nerve head to the periphery dividing thesuperior-temporal and superior-nasal quadrants. The entire
retina was digitally photographed at 10×, in montage-format,using an epifluorescence Zeiss Apotome system equippedwith a DAPI filter pack. The resulting montage of each retinawas mapped off with a series of 1 mm concentric ringscentered about the optic nerve. The area defining eachconcentric ring was further subdivided into uniform grids, toobtain individual averages of labeled cells per defined area. Toavoid issues involved with regional sampling, FG-labeled cellswere manually counted and pooled from all four retinalquadrants for each concentric area, as previously described(Goebel and Winkler, 2006).
Acknowledgments
I would like to thank Dr. Fei Yu, Dr. Ivanova Elena, and myassistant Eren D. Berberoglu for their advice and technicalexpertise. Also, I wish to acknowledge the data displayed inFig. 6 toMs Tania Borboni and dedicate this work inmemory ofher energetic smile and enthusiasm she brought to our lab.Her tragic departure from this world at such a young age raisesmany unanswerable questions and her absence, will begreatly missed by all that had the opportunity to know her.Supported by NIH grant EY-014430, Vision Core grant P30-EY04068.
R E F E R E N C E S
Baille, V., Clarke, G.H., Brochier, G., Dorandeu, F., Verna, J.-M., Four,E., Lallement, G., Carpentier, P., 2005. Soman-inducedconvulsions: the neuropathology revisited. Toxicology 215, 1–24.
Barth, M., Oulmi, Y., Ehrenreich, H., Schilling, L., 2002.Pre-embedding immunogold labeling of TUNEL stain enablesevaluation of DNA strand breaks and ultrastructural alterationin individual cells of neuronal tissue. Acta Neuropathol. 104,621–636.
Bonfoco, E., Krainc, D., Ankarcrona, M., Nicotera, P., Lipton, S.A.,1995. Apoptosis and necrosis: two distinct events induced,respectively, by mild and intense insults withN-methyl-D-aspartate or nitric oxide/superoxide in corticalcultures. Proc. Natl. Acad. Sci. U. S. A. 92, 7162–7166.
Braun, A.P., Schulman, H., 1995. The multifunctional calcium/calmodulin-dependent protein kinase: From form to function.Annu. Rev. Physiol. 57, 417–445.
Casiano, C.A., Martin, S.J., Green, D.R., Tan, E.M., 1996. Selectivecleavage of nuclear autoantigens during CD95(Fas/APO-1)-mediated T cell apoptosis. J. Exp. Med. 184,765–770.
D'Amours, D., Sallmann, F.R., Dixit, V.M., Poirier, G.G., 2001.Gain-of-function of poly(ADP-ribose) polymerase-1 uponcleavage by apoptotic proteases: implications for apoptosis. J.Cell Sci. 114, 3771–3778.
Danias, J., Shen, F., Goldblum, D., Chen, B., Ramos-Esteban, J.,Podos, S.M., Mittag, T., 2002. Cytoarchitecture of the retinalganglion cells in the rat. Invest. Ophthal. Vis. Sci. 43,587–594.
Davis, S.M., Albers, G.W., Diener, H.C., Lees, K.R., Noris, J., 1997.Termination of acute stroke studies involving selfoteltreatment. ASSIST steering committed. Lancet 349, 32.
Davis, S.M., Lees, K.R., Albers, G.W., Diener, H.C., Markabi, S.,Karlsson, G., Norris, J., 2000. Selfotel in acute ischemic stroke:possible neurotoxic effects of an NMDA antagonist. Stroke 31,347–354.

203B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
Fan, W., Agarwal, N., Kumar, M.D., Cooper, N.G.F., 2005. Retinalganglion cell death and neuroprotection: involvement of theCaMKIIα gene. Mol. Brain Res. 139, 306–316.
Fix, A.S., Horn, J.W., Wrightman, K.A., Johnson, C.A., Long, G.G.,Storts, R.W., Farber, N., Wozniak, D.F., Olney, J.W., 1993.Neuronal vacuolization and necrosis induced by thenoncompetitive N-methyl-D-aspartate (NMDA) antagonist MK(+)801 (Dizocilpine maleate): a light and electron microscopicevaluation of the rat retrosplenial cortex. Exp. Neurol. 123,204–215.
Fix, A.S., Wozniak, D.F., Truex, L.L., McEwen Miller, J.J., Olney, J.W.,1995. Quantitative analysis of factors influencing neuronalnecrosis induced by MK-801 in rat posterior cingulate/retosp-inal cortex. Brain Res. 696, 194–204.
Gardoni, F., Polli, F., Cattabeni, F., Di Luca, M., 2006.Calcium-calmodulin-dependent protein kinase IIphosphorylation modulates PSD-95 binding to NMDAreceptors. Eur. J. Neurosci. 24, 2694–2704.
Gillardon, F., Kiprianova, I., Sandkuhler, J., Hossmann, K.A.,Spranger, M., 1999. Inhibition of caspases prevents cell deathof hippocampal CA1 neurons, but not impairment ofhippocampal long-term potentiation following globalischemia. Neurosci. 93, 1219–1222.
Goebel, D.J., Winkler, B.S., 2006. Blockade of PARP activityattenuates poly(ADP-ribosyl)ation but offers only partialneuroprotection against NMDA-induced cell death in the ratretina. J. Neurochem. 98, 1732–1745.
Henkel, A.W., Bieger, S.C., 1994. Quantification of proteins dissolvein an electrophoresis sample buffer. Anal. Biochem. 223,329–331.
Ishida, A., Kameshita, I., Okuno, S., Kitani, T., Fujisawa, H., 1995. Anovel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem. Biophys. Res. Commun.212, 806–812.
Ishida, A., Shigeri, Y., Tatsu, Y., Wndo, Y., Kameshita, I., Okuno, S.,Kitani, T., Takeuchi, M., Yumoto, N., Fujisawa, H., 2001.Substrate specificity of Ca(2+)/calmodulin-dependent proteinkinase phosphatase: kinetic studies using syntheticphosphopeptides as model substrates. J. Biochem. (Tokyo) 129,745–753.
Iwamoto, T., Yamada, Y., Hori, K., Wantanabe, Y., Sobue, K., Inui,M., 2004. Differential modulation of NR1-R2A and NR1-R2Bsubtypes of NMDA receptor by PDZ domain-containingproteins. J. Neurochem. 89, 100–108.
Kaufmann, S.H., Desnoyers, S., Ottaviano, Y., Davidson, N.E.,Poirier, G.G., 1993. Specific proteolytic cleavage ofPoly(ADP-ribose) polymerase: an early marker ofchemotherapy-induced apoptosis. Cancer Res. 53, 3976–3985.
Kerr, J.F., Wyllie, A.H., Currie, A.R., 1972. Apoptosis: a basicbiological phenomenon with wide-ranging implications intissue kinetics. Br. J. Cancer 4, 239–257.
Kim, H.-H.C., Luo, Y., Li, G., Kessel, D., 1999. Enhanced apoptoticresponse to photodynamic therapy after bcl-2 transfection.Cancer Res. 59, 3429–3432.
Komeima, K., Hayashi, Y., Naito, Y., Watanabe, Y., 2000. Inhibitionof neuronal nitric-oxide synthase by calcium/calmodulin-dependent protein kinase IIα through Ser847
phosphorylation in NG108-15 neuronal cells. J. Biochem. 275,28139–28143.
Laabich, A., Cooper, N.G.F., 2000. Neuroprotective effect of AIP onN-methyl-D-aspartate-induced cell death in retinal neurons.Mol. Brain Res. 85, 32–40.
Laabich, A., Li, G., Cooper, N.G.F., 2000. Calcium/calmodulin-dependent protein kinase II containing a nuclearlocalizing signal is altered in retinal neurons exposed toN-methyl-D-aspartate. Mol. Brain Res. 76, 253–265.
Laabich, A., Li, G., Cooper, N.G.F., 2001. Characterization ofapoptosis-genes associated with NMDA mediated cell death inthe adult retina. Mol. Brain Res. 91, 34–42.
Lam, T.T., Abler, A.S., Kwong, J.M., Tso, M.O., 1999.N-methyl-D-aspartate (NMDA)-induced apoptosis in rat retina.Investig. Ophthalmol. Vis. Sci. 40, 2391–2397.
Levy, D.I., Lipton, S.A., 1990. Comparison of delayedadministration of competitive and uncompetitive antagonistin preventing NMDA receptor-mediated neuronal death.Neurology 40, 852–855.
Lin, K.-F., Chang, R.C.C., Suen, K.-A., So, K.-F., Hugon, J., 2004.Modulation of calcium/calmodulin kinase-II provides partialneuroprotection against beta-amyloid peptide toxicity. Eur. J.Neurosci. 19, 2047–2055.
Manabe, S., Lipton, S.A., 2003. Divergent NMDA signals leading toproapoptotic and antiapoptotic pathways in the rat retina.Investig. Ophthalmol. Vis. Sci. 44, 385–392.
Mandir, A.S., Poitras, M.F., Berliner, A.R., Herring,W.J., Guastella, D.B., Feldman, A., Poirier, G.G., Wang, Z.Q., Dawson, T.M.,Dawson, V.L., 2000. NMDA but not non-NMDA excitotoxicity ismediated by Poly(ADP-ribose) polymerase. J. Neurosci. 20,8005–8011.
McBurney, R.N., 1997. Development of the NMDA ion-channelblocker, aptiganel hydrochloride, as a neuroprotective agentfor acute CNS injury. Int. Rev. Neurobiol. 40, 173–195.
McGahon, A.J., Martin, S.J., Bissonnette, R.P., Mahboubi, A., Shi, Y.,Mogil, R.J., Nishioka, W.K., Green, D.R., 1995. The end of the(cell) line: methods for the study of apoptosis in vitro. MethodsCell Biol. 46, 153–185.
McKernan, D.P., Guerin, M.B., O'Brian, C.J., Cotter, T.G., 2007. A keyrole for calpains in retinal ganglion cell death. Investig.Ophthalmol. Vis. Sci. 48, 5420–5430.
Nakane, M., Mitchell, J., Forstermann, U., Murad, F., 1991.Phosphorylation by calcium calmodulin-dependent proteinkinase, protein kinase C modulates the activity of nitricoxide synthase. Biochem. Biophys. Res. Commun. 180,1396–1402.
Nicholson, D.W., All, A., Thornberry, N.A., Vaillancourt, J.P., Ding,C.K., Gallant, M., Gareau, Y., Griffin, P.R., Labelle, M., Lazebnik,Y.A., Munday, N.A., Raju, S.M., Oppenheim, R.W., Flavell, R.A.,Vinsant, S., Prevette, D., Kuan, C.-Y., Rakic, P., 2001.Programmed cell death of developing mammalian neuronsafter genetic depletion of caspases. J. Neurosci. 21, 4752–7560.
Nishizaki, K., Yoshino, T., Orita, Y., Nomiya, S., Masuda, Y., 1999.TUNEL staining of inner ear structures may reflect autolysis,not apoptosis. Hear. Res. 130, 131–136.
Pellegrini, J.W., Lipton, S.A., 1993. Delayed administration ofmemantine prevents N-methyl-D-aspartatereceptor-mediated neurotoxicity. Ann. Neurol. 33, 403–435.
Portera-Cailliau, C., Price, D.L., Martin, L.J., 1997. Non-NMDA andNMDA receptor-mediated excitotoxic neuronal deaths in adultbrain are morphologically distinct: further evidence forapoptosis-necrosis continuum. J. Comp. Neurol. 378, 88–104.
Rhéaume, E., Cohen, L.Y., Uhlmann, F., Lazure, C., Alam, A.,Hurwitz, J., Sekaly, R.P., Denis, F., 1997. The large subunit ofreplication factor C is a substrate for caspase-3 in vitro and iscleaved by a caspase-3-like protease during Fas-mediatedapoptosis. EMBO J. 16, 6346–6354.
Searle, J., Kerr, J.F., Bishop, C.J., 1982. Necrosis and apoptosis:distinct modes of cell death with fundamentally differentsignificance. Pathol. Annu. 17 (2), 229–259.
Siliprandi, R., Canella, R., Carmignoto, G., Schiavo, N., Zanellato,A., Zanoni, R., Vantini, G., 1992. N-methyl-D-aspartate-inducedneurotoxicity in the adult rat retina. Vis. Neurosci. 8, 567–573.
Swartz, L.M., Smith, S.W., Jones, M.E.E., Osborne, B.A., 1993. Do allprogrammed cell deaths occur via apoptosis? Proc. Natl. Acad.Sci. U. S. A. 90, 980–984.
Tenneti, L., Lipton, S.A., 2000. Involvement of activatedcaspase-3-like proteases in N-methyl-D-aspartate-inducedapoptosis in cerebrocortical neurons. J. Neurochem. 74, 134–142.
Terashima, T., Ochiishi, T., Yamauchi, T., 1994.Immunocytochemical localization of calcium/

204 B R A I N R E S E A R C H 1 2 5 6 ( 2 0 0 9 ) 1 9 0 – 2 0 4
calmodulin-dependent protein kinase II isoforms in theganglion cells of the rat retina: immunofluorescencehistochemistry combined with a fluorescent retrograde tracer.Brain Res. 650, 133–139.
Ünal-Çevik, I., Kilinç, M., Can, A., Gürsoy-Özdemir, Y., Dalkara, T.,2004. Apoptotic and necrotic death mechanisms areconcomitantly activated in the same cell after cerebralischemia. Stroke 35, 2189–2194.
Watanabe, Y., Song, T., Sugimoto, K., Horii, M., Araki, N.,Tokumitsu, H., Tezuka, T., Yamamoto, T., Tokuda, M., 2003.Post-synaptic density-95 promotes calcium/calmodulin-dependent protein kinase II-mediated Ser847phosphorylation of neuronal nitric oxide synthase. Biochem. J.372, 465–471.
Yaun, J., Lipinski, M., Degterev, A., 2003. Diversity in themechanisms of neuronal cell death. Neuron 40, 401–413.


![Ivyspring International Publisher Theranostics · response to acute or chronic retinal injury including inflammation, ischemia and neurodegeneration [1-4]. Fibrosis alters the retinal](https://static.fdocument.org/doc/165x107/600a05c5fd5be725da7f0a44/ivyspring-international-publisher-theranostics-response-to-acute-or-chronic-retinal.jpg)













