
Schrödinger Equation in Born-Oppenheimer Approximation (1) · Schrödinger Equation in...
22
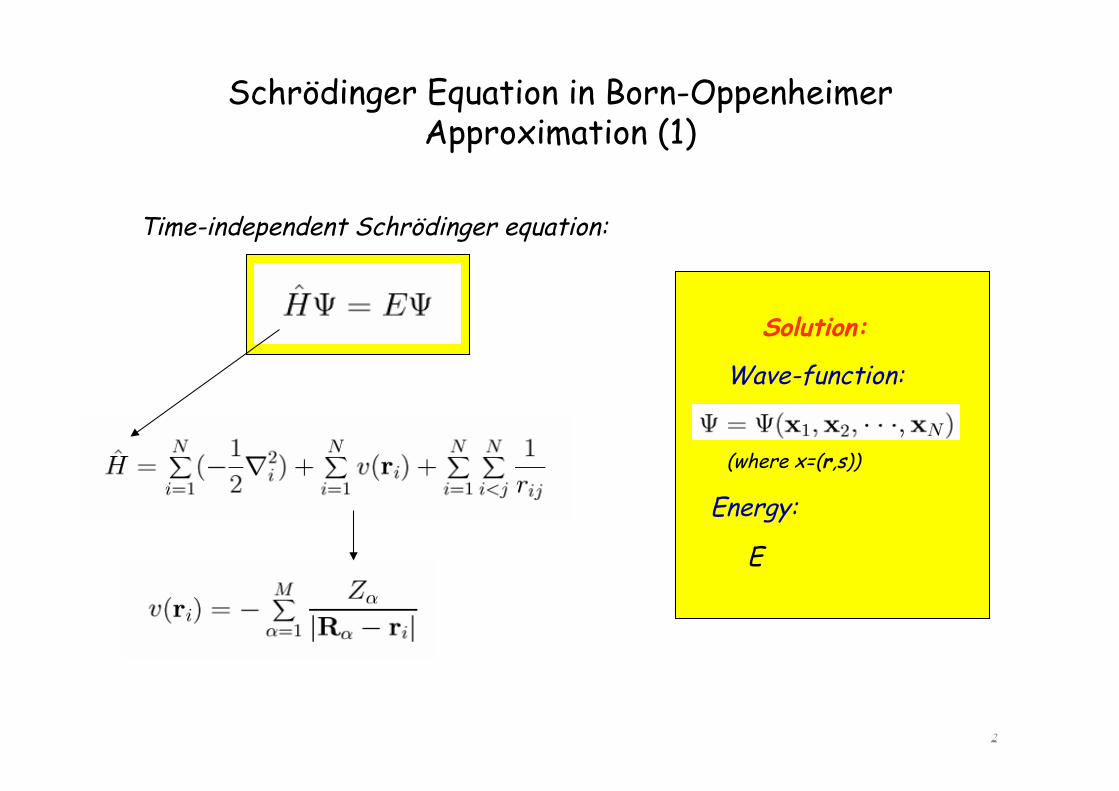
Schrödinger Equation in Born-Oppenheimer Approximation (1) Time-independent Schrödinger equation: Wave-function: (where x=(r,s)) Solution: Energy: E 2
Transcript of Schrödinger Equation in Born-Oppenheimer Approximation (1) · Schrödinger Equation in...
Schrödinger Equation in Born-Oppenheimer Approximation (1)
Time-independent Schrödinger equation:
Wave-function:
(where x=(r,s))
Solution:
Energy:
E
2

Schrödinger Equation in Born-Oppenheimer Approximation (2)
Knowledge of the wavefunction Ψ(x1,x2,..,xN) is sufficient to obtain all observable quantities, electron density ρ(r) in particular:
ρ(r)= ∫dx1dx2..dxN ∑iN δ(r-ri) |Ψ(x1,x2,..,xN)|2
3

Variational principle:
E0=min<ΨN|H |ΨN> ΨN
where, E0 is ground-state energy and ΨN is a trial N-electron wavefunction
Variational principle, is the basis for a large group of methods to obtain an approximate solution of Schrödinger equation (HF, MCSCF, CI, for instance).
Schrödinger Equation in Born-Oppenheimer Approximation (3)
3
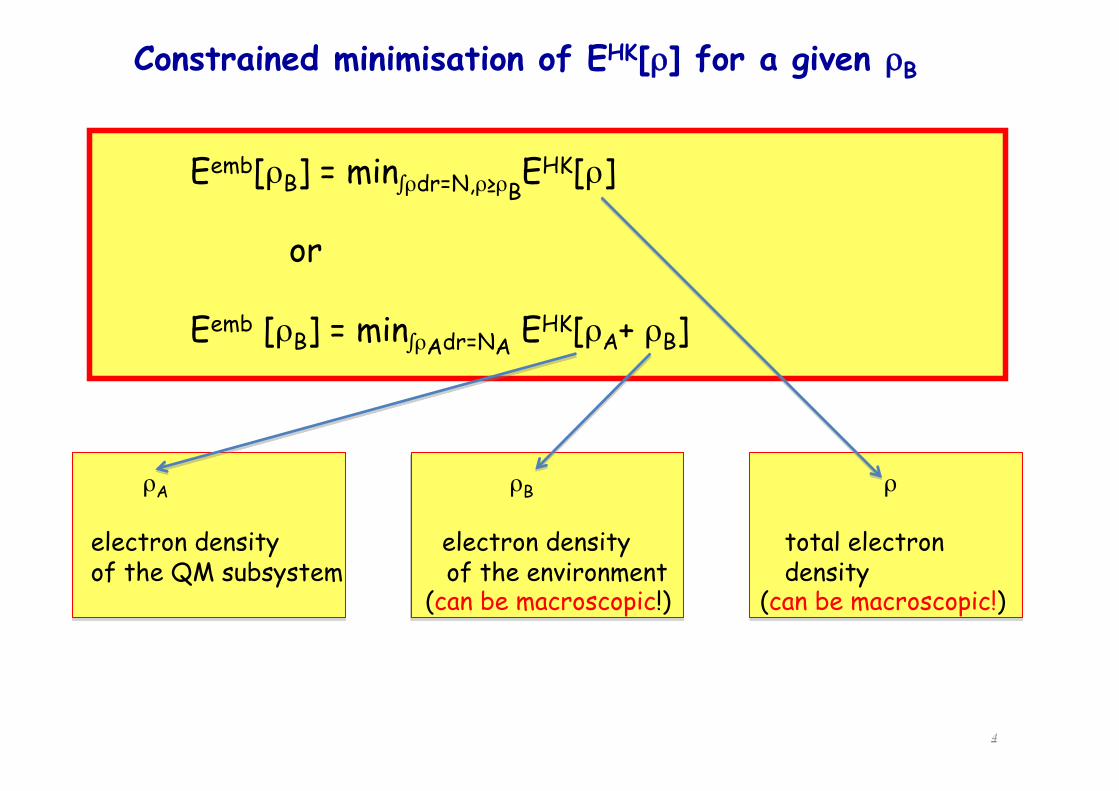
Constrained minimisation of EHK[ρ] for a given ρB
Eemb[ρB] = min∫ρdr=N,ρ≥ρBEHK[ρ]
or
Eemb [ρB] = min∫ρAdr=NA EHK[ρA+ ρB]
ρA ρB ρ
electron density electron density total electron of the QM subsystem of the environment density
(can be macroscopic!) (can be macroscopic!)
4

Definitions of density functionals through constrained search
The minimiser (Ψo) is used to define the following density functionals
“By a functional, we mean a correspondence which assigns a definite (real) number to each function (or a curve) belonging to some class.” [I.M. Gelfand and S.V. Fomin, "Calculus of Variations" Prentice-Hall, Inc. 1963, page 1] 5

The non-additive bi-functionals:
For a given functional F[ρ], the corresponding non-additive bi-functional is: Fnad[ρA,ρB] = F[ρA+ρB]- F[ρA] – F[ρB]
A given functional F[ρ] is additive if:
Fnad[ρA,ρB] = F[ρA+ρB]- F[ρA] – FρB] =0.
Additive functionals: N[ρ], Vext[ρ]
Non-additive functionals: J[ρ] ,T[ρ], Eex[ρ], Ts[ρ], EHK[ρ]
6

Embedding potentials for variational QC methods
Q1: Can conventional methods of Quantum Chemistry be modified by adding some local potential (embedding potential) to perform the constrained minimisation Eemb = min∫ρdr=N,ρ≥ρB
EHK[ρ]? Q2: Can such local potential be obtained as universal functional of ρA and ρB?
[H0 + Vemb(r)]Ψemb = Eemb Ψemb
7

Embedding potentials for variational QC methods
Q1: Can conventional methods of Quantum Chemistry be modified by adding some local potential (embedding potential) to perform the constrained minimisation Eemb = min∫ρdr=N,ρ≥ρB
EHK[ρ]? Q2: Can such local potential be obtained as universal functional of ρA and ρB?
Answers:
α) Φsemb embedded non-interacting
wavefunction (Kohn-Sham system) [Wesolowski & Warshel, J. Phys. Chem. 97 (1993) 8050]
β) Ψsemb embedded interacting wavefunction
[Wesolowski, Phys. Rev.A. 77 (2008) 012504]
γ) Γsemb embedded one-matrix
[Pernal & Wesolowski, Intl. J. Quant. Chem .109 (2009) 2520]
Φsemb, Ψemb, or Γemb
Auxiliary quantities used to access:
- the total energy (- energy of the frozen part)
- properties directly related to the electronic structure
[H0 + Vemb(r)]Ψemb = Eemb Ψemb
8
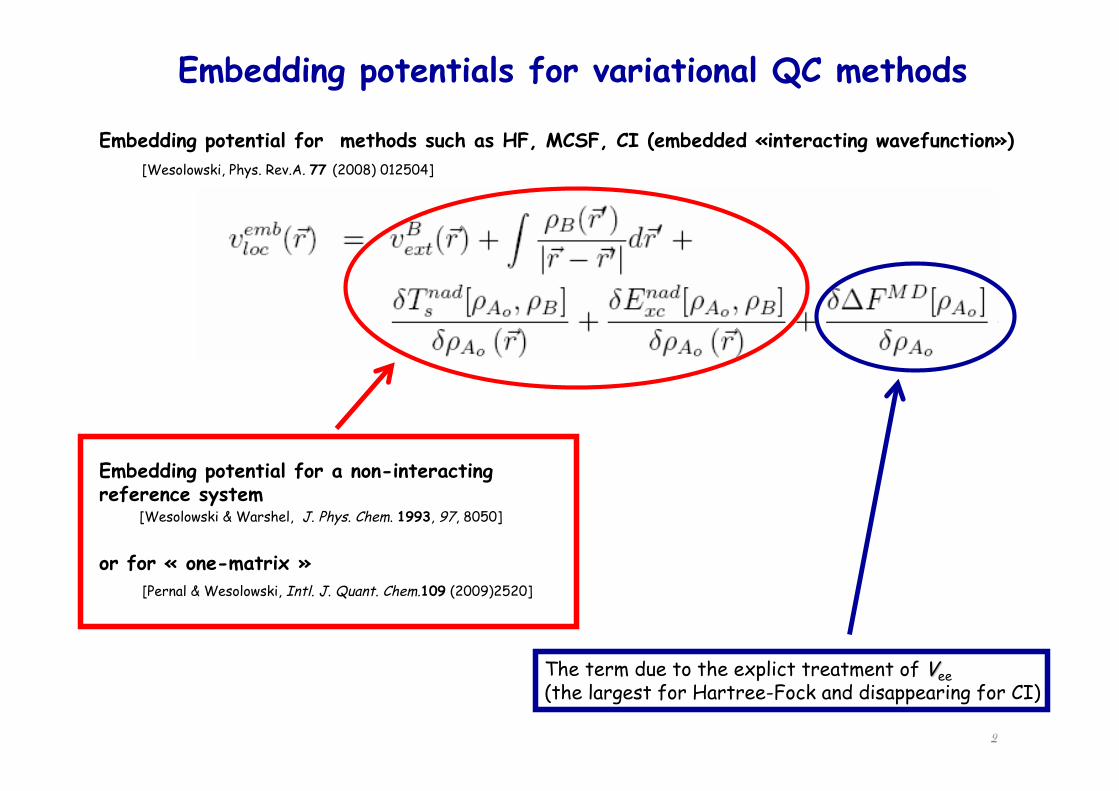
Embedding potentials for variational QC methods
Embedding potential for a non-interacting reference system
or for « one-matrix »
Embedding potential for methods such as HF, MCSF, CI (embedded «interacting wavefunction») [Wesolowski, Phys. Rev.A. 77 (2008) 012504]
[Pernal & Wesolowski, Intl. J. Quant. Chem.109 (2009)2520]
[Wesolowski & Warshel, J. Phys. Chem. 1993, 97, 8050]
9

Embedding potentials for variational QC methods
Embedding potential for methods such as HF, MCSF, CI (embedded «interacting wavefunction»)
10
question: “What local correlation potential do you have toadd to the Hartree-Fock equation so that the density is ex-act.” In this work, we consider not only the Hartree-Fockmethod but any method which treats the electron-electroninteractions exactly and uses a wave function comprising notonly one but any arbitrarily chosen number of determinants.Moreover, the target electron density is not the exact ground-state electron density, but the one which minimizes theHohenberg-Kohn energy functional keeping one componentof the total electron density constant in the present consider-ations. The potential analyzed in this work is obtained usingthe following steps:
!i" In all considerations, the environment of an embeddedsystem comprising 2NA electrons is represented by means ofa given electron density (!B!r!") and the Coulomb potentialgenerated by nuclei of the environment (vext
B !r!").!ii" Two formal frameworks for obtaining the ground-state
electron density are considered. In the first one, the embed-ded electron density is represented by means of embeddedKohn-Sham orbitals, which are obtained from one-electronequations #Eqs. !20" and !21" of Ref. #7$$. In these equations,the total effective potential and its embedding component inparticular is expressed by means of explicit density function-als. For the exact effective potential, these equations lead tothe density !Ao
1, which minimizes the Hohenberg-Kohn en-ergy functional for a fixed !B !i.e., at "!B=0". The secondframework uses a multideterminantal wave function to rep-resent the embedded electron density and treats the electron-electron interactions exactly whereas the presence of envi-ronment is accounted for by means of the operator V̂emb:
#T̂2NA+ V̂2NA
ee + V̂extA + V̂emb$#A = EA#A, !1"
where the first three operators define the isolated subsystem.!iii" The embedding operator is postulated to take the
form of a local potential,
V̂emb = %i
2NA
vlocemb!r!i" . !2"
The electron density !!Ao2" obtained as an approximate solu-
tion of Eq. !1" by means of variational calculations usingtrial wave functions of the multideterminantal form with afixed number of determinants, such as multiconfigurationalself consistent-field calculations !MCSCF" for instance, de-pends on vloc
emb!r!i". By imposing that
!Ao1 = !Ao
2 = !Ao, !3"
vlocemb!r!i" is constructed leading to the principal result of this
work—the relation between vlocemb!r!i" and universal density
functionals.Note that we take a particular perspective on the relation
between the wave-function-based methods and density-functional theory. A multideterminantal wave function isconsidered in this work as an auxiliary quantity used to ob-tain the approximate solution of Eq. !1" and the correspond-ing electron density by means of variational calculations,whereas the relevant density functionals are considered to be
exact in the derivation of the basic relation. Approximatefunctionals are considered only in the discussion part in viewof the prospects for practical calculations.
B. Key definitions and notation
The functionals are denoted with capital letters and thequantities, on which they depend explicitly, are given withinsquare brackets !as in F#y$". Unless indicated by tildes, theconsidered functionals are assumed to be exact. The formu-las are given in atomic units for spin-compensated electrondensities.
For 2N electrons in an external potential vext!r!", theHohenberg-Kohn energy functional #1$ is defined as
EHK#!$ = FHK#!$ +& vext!r!" !!r!" dr! , !4"
where the constrained search definition of FHK#!$ #18$ reads
FHK#!$ = min#!!
'#(T̂2N + V̂2Nee (#) . !5"
The electron densities considered in the search for theminimum of EHK#!$ are required to be N representable #16$.In finite Coulomb systems, N representability can be easilyassured #16,17$. In this work, a stronger condition—the re-quirement that ! is pure-state noninteracting v representable#17$ !v representable in short" is also relevant. For electrondensities, which belong to this category !obtained from theKohn-Sham equations #2$ or other one-electron equationswith a multiplicative potential, for instance", additional ex-plicit density functionals can be defined using the con-strained search procedure #18$
Ts#!$ = min*$i+!!
,2%i
N -$i.!12
!2.$i/0= 2%
i
N -$io.!
12
!2.$io/ , !6"
where the search procedure is performed among the orbitals*$i+ preserving the normalization !2%i
N ($i(2=!" and ortho-normality !'$i ($ j)="ij" conditions.
Exc#!$ is subsequently defined in the following decompo-sition of FHK#!$:
FHK#!$ = Ts#!$ + J#!$ + Exc#!$ , !7"
where J#!$ is the Coulomb repulsion integral
J#!$ =12 & & !!r!"!!r!!"
(r! ! r!!(dr!!dr! . !8"
In this work, functionals depending on other quantities thanelectron density are considered. In the Kohn-Sham frame-work, which is based on a reference system of noninteractingelectrons, the total energy functional !%KS" depends on a setof one-electron functions !*$i+"—the Kohn-Sham orbitals
TOMASZ A. WESO"OWSKI PHYSICAL REVIEW A 77, 012504 !2008"
012504-2question: “What local correlation potential do you have toadd to the Hartree-Fock equation so that the density is ex-act.” In this work, we consider not only the Hartree-Fockmethod but any method which treats the electron-electroninteractions exactly and uses a wave function comprising notonly one but any arbitrarily chosen number of determinants.Moreover, the target electron density is not the exact ground-state electron density, but the one which minimizes theHohenberg-Kohn energy functional keeping one componentof the total electron density constant in the present consider-ations. The potential analyzed in this work is obtained usingthe following steps:
!i" In all considerations, the environment of an embeddedsystem comprising 2NA electrons is represented by means ofa given electron density (!B!r!") and the Coulomb potentialgenerated by nuclei of the environment (vext
B !r!").!ii" Two formal frameworks for obtaining the ground-state
electron density are considered. In the first one, the embed-ded electron density is represented by means of embeddedKohn-Sham orbitals, which are obtained from one-electronequations #Eqs. !20" and !21" of Ref. #7$$. In these equations,the total effective potential and its embedding component inparticular is expressed by means of explicit density function-als. For the exact effective potential, these equations lead tothe density !Ao
1, which minimizes the Hohenberg-Kohn en-ergy functional for a fixed !B !i.e., at "!B=0". The secondframework uses a multideterminantal wave function to rep-resent the embedded electron density and treats the electron-electron interactions exactly whereas the presence of envi-ronment is accounted for by means of the operator V̂emb:
#T̂2NA+ V̂2NA
ee + V̂extA + V̂emb$#A = EA#A, !1"
where the first three operators define the isolated subsystem.!iii" The embedding operator is postulated to take the
form of a local potential,
V̂emb = %i
2NA
vlocemb!r!i" . !2"
The electron density !!Ao2" obtained as an approximate solu-
tion of Eq. !1" by means of variational calculations usingtrial wave functions of the multideterminantal form with afixed number of determinants, such as multiconfigurationalself consistent-field calculations !MCSCF" for instance, de-pends on vloc
emb!r!i". By imposing that
!Ao1 = !Ao
2 = !Ao, !3"
vlocemb!r!i" is constructed leading to the principal result of this
work—the relation between vlocemb!r!i" and universal density
functionals.Note that we take a particular perspective on the relation
between the wave-function-based methods and density-functional theory. A multideterminantal wave function isconsidered in this work as an auxiliary quantity used to ob-tain the approximate solution of Eq. !1" and the correspond-ing electron density by means of variational calculations,whereas the relevant density functionals are considered to be
exact in the derivation of the basic relation. Approximatefunctionals are considered only in the discussion part in viewof the prospects for practical calculations.
B. Key definitions and notation
The functionals are denoted with capital letters and thequantities, on which they depend explicitly, are given withinsquare brackets !as in F#y$". Unless indicated by tildes, theconsidered functionals are assumed to be exact. The formu-las are given in atomic units for spin-compensated electrondensities.
For 2N electrons in an external potential vext!r!", theHohenberg-Kohn energy functional #1$ is defined as
EHK#!$ = FHK#!$ +& vext!r!" !!r!" dr! , !4"
where the constrained search definition of FHK#!$ #18$ reads
FHK#!$ = min#!!
'#(T̂2N + V̂2Nee (#) . !5"
The electron densities considered in the search for theminimum of EHK#!$ are required to be N representable #16$.In finite Coulomb systems, N representability can be easilyassured #16,17$. In this work, a stronger condition—the re-quirement that ! is pure-state noninteracting v representable#17$ !v representable in short" is also relevant. For electrondensities, which belong to this category !obtained from theKohn-Sham equations #2$ or other one-electron equationswith a multiplicative potential, for instance", additional ex-plicit density functionals can be defined using the con-strained search procedure #18$
Ts#!$ = min*$i+!!
,2%i
N -$i.!12
!2.$i/0= 2%
i
N -$io.!
12
!2.$io/ , !6"
where the search procedure is performed among the orbitals*$i+ preserving the normalization !2%i
N ($i(2=!" and ortho-normality !'$i ($ j)="ij" conditions.
Exc#!$ is subsequently defined in the following decompo-sition of FHK#!$:
FHK#!$ = Ts#!$ + J#!$ + Exc#!$ , !7"
where J#!$ is the Coulomb repulsion integral
J#!$ =12 & & !!r!"!!r!!"
(r! ! r!!(dr!!dr! . !8"
In this work, functionals depending on other quantities thanelectron density are considered. In the Kohn-Sham frame-work, which is based on a reference system of noninteractingelectrons, the total energy functional !%KS" depends on a setof one-electron functions !*$i+"—the Kohn-Sham orbitals
TOMASZ A. WESO"OWSKI PHYSICAL REVIEW A 77, 012504 !2008"
012504-2
Embedding a multideterminantal wave function in an orbital-free environment
Tomasz A. Weso!owskiDépartement de Chimie Physique, 30 quai Ernest-Ansermet, Université de Genève, CH-1211 Genève 4, Switzerland
!Received 3 October 2006; revised manuscript received 15 October 2007; published 11 January 2008"
Variational methods to treat a many-electron system embedded in the environment, which is represented bymeans of only its electron density, are considered. It is shown that the embedding operator is a local potentialin the case where the electron-electron repulsion is treated exactly and the trial embedded wave function takesthe multideterminantal form with a fixed number of determinants. The local embedding potential is constructedby imposing that it leads to the same electron density as the one which minimizes the Hohenberg-Kohnfunctional. For the limiting cases of single-determinant and configuration interaction forms of the embeddedwave function, the expressions for the local embedding potential using commonly known density functionalsare given. The relation between the derived local embedding potential and the effective embedding potential inthe case of the embedded Kohn-Sham system #T. A. Weso!owski and A. Warshel, J. Phys. Chem. 97, 8050!1993"$ is discussed in detail.
DOI: 10.1103/PhysRevA.77.012504 PACS number!s": 31.15.E!, 31.70.Dk
I. INTRODUCTION
Numerical simulations of condensed-matter systems at thequantum-mechanics level face inevitably a dilemma of trad-ing accuracy of the applied approximate method for the pos-sibility of including a large number of atoms in the model.The Hohenberg-Kohn-Sham formulation of density-functional theory #1,2$ proved essential in moving the sizelimits upwards. Indeed, the commonly used approximationsto the exchange-correlation !Exc#"$" energy are sufficientlyadequate because the errors due to the fact that they are notexact are acceptable for a large number of problems andsystems. Nevertheless, some well-defined systems cannot bedescribed satisfactorily using common simple approxima-tions to Exc#"$. Such cases include systems for which single-determinantal wave function provides a qualitatively wrongapproximation of the exact one #3$. Development of suchbeyond-Kohn-Sham formalisms, which are based onHohenberg-Kohn theorems but use other reference systemsthan the noninteracting electrons, is currently the area of in-tensive research #3,4$ motivated by these flaws. The wave-function-based methods are free from such deficiencies butare applicable only to systems of relatively small size. For aparticular type of problems, where the primary interests con-cern details of the electronic structure which are well-localized in real space, the size limits can be pushed furtherby means of applying the embedding strategy. In this strat-egy, the wave function or the Kohn-Sham orbitals are con-structed for the selected subsystem, whereas the effects ofthe environment are accounted for by means of a specialoperator !embedding operator". In the simplest case, the em-bedding operator takes into account only the electrostaticcontributions. In more refined approaches, effects ofquantum-mechanical origin are also represented using suchdescriptors of the environment as pseudopotentials or orbit-als #5,6$.
If the embedded system is described by means of embed-ded Kohn-Sham orbitals, the exact effective potential can beexpressed using universal density functionals #7,8$. The partof the whole effective potential taking into account the pres-
ence of the environment !orbital-free effective embeddingpotential", does not require any information about the envi-ronment besides its electron density and the electrostatic po-tential generated by other electric charges !nuclei". It is ap-pealing, therefore, to combine the density-functional-theoryderived orbital-free effective embedding potential of Ref. #7$with a multideterminantal representation of the embeddedsubsystem. Such a combination has a potential to overcometwo types of limitations of the Kohn-Sham-based methodsby pushing the size limit of amenable model systems up-wards !when applicable" and the possibility of treating thesystems of multideterminantal character. Indeed, these ad-vantages have been recognized by Carter, Wang, and col-laborators who applied such an combination in numericalsimulations #9,10$. A straightforward application of such acombination leads, however, to the risk of inconsistent treat-ment of some energy components of the total energy andeven conceptual difficulties #11$. In this work, we addressthese issues by identifying the assumptions and approxima-tions involved. To this end, we derive the relation betweenthe exact local embedding operator and the quantities ex-pressed by means of exact density functionals.
Finally, we stress that the common element in the presentconsiderations is the requirement that only electron density isused as a descriptor of electrons in the environment. Possibleformal frameworks, in which the environment is describedusing other quantities—pseudopotentials or orbitals for in-stance #5,6$—lie outside of the scope of this work. Theorbital-free representation of the environment is of key im-portance for applications of the resulting computational ap-proach in the domain of nonempirical multiscale computersimulations #8$ because the electron density is a well-definedquantity at both microscopic and macroscopic scales.
II. EMBEDDING A MULTIDETERMINANTAL WAVEFUNCTION IN AN ORBITAL-FREE ENVIRONMENT
A. Construction of the local embedding operator—outline
The strategy applied in this work follows the same gen-eral lines as the ones used by others #12–15$ to answer the
PHYSICAL REVIEW A 77, 012504 !2008"
1050-2947/2008/77!1"/012504!9" ©2008 The American Physical Society012504-1

!! "AWF(r)
Embedded interacting electrons (1) construction of the embedding operator Vemb
11

We are looking for a local potential Vemb(r), such that
EHK[!AWF[Vemb] +!B] = min"!Adr=NA EHK[!A+!B]
for some arbitrarily chosen !B!
1) The definition of the functional #EWF[$A,!B]!
! where $A is of the multi-determinant form ($AMD). !
2) showing that
3) !extracting the form of Vemb from the condition: !AMD=!WF[Vemb] !
Embedded interacting electrons (2) construction of the embedding operator Vemb
12

Depending on the number of determinants (MD) the following relation holds:
where
Embedded interacting electrons (3) construction of the embedding operator Vemb
13

Only the first term depends explicitly on !AMD.!
For !AOMD obtained from Euler-Lagrange equations,
the sum of the first and the last term is equal to FHK["A]=Ts["A]+J["A]+Exc["A].!
Therefore:
#EWF[!A0MD,"B]= FHK["A+"B]+$vext
AB(r)("A+"B)dr= EHK["A+"B]!
Embedded interacting electrons (4) construction of the embedding operator Vemb
14
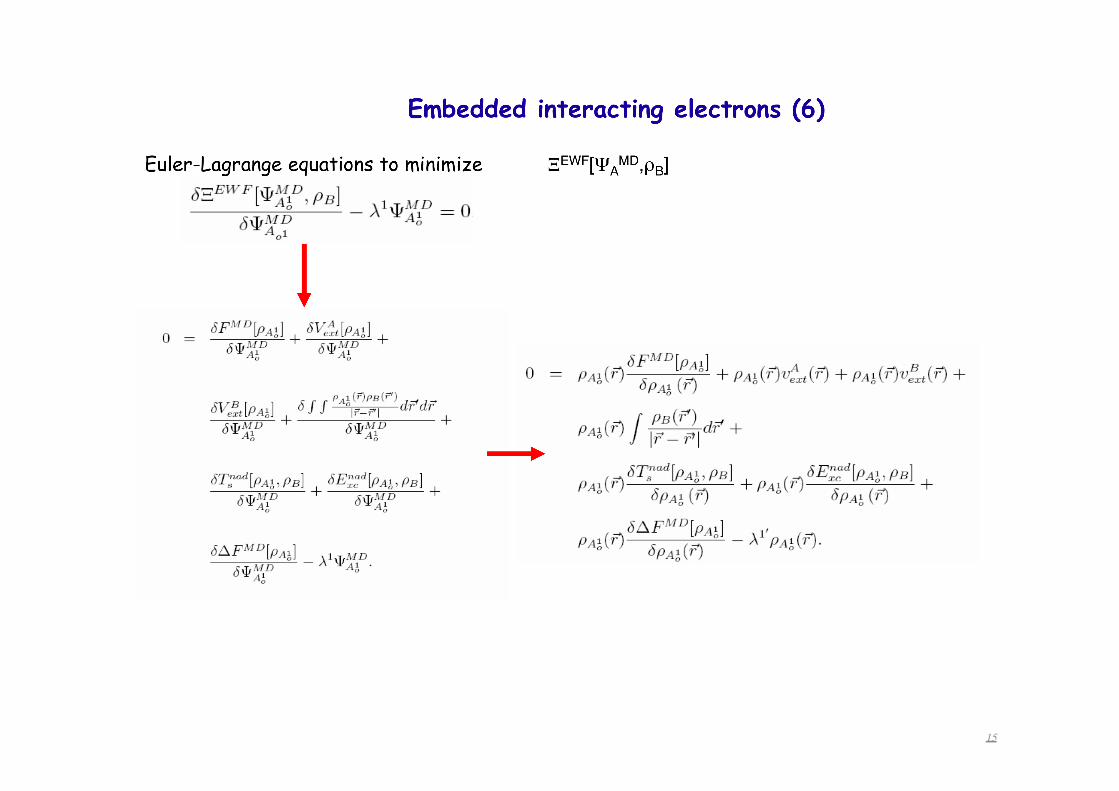
15

Euler-Lagrange equations to minimize
Embedded interacting electrons (6) construction of the embedding operator Vemb
16
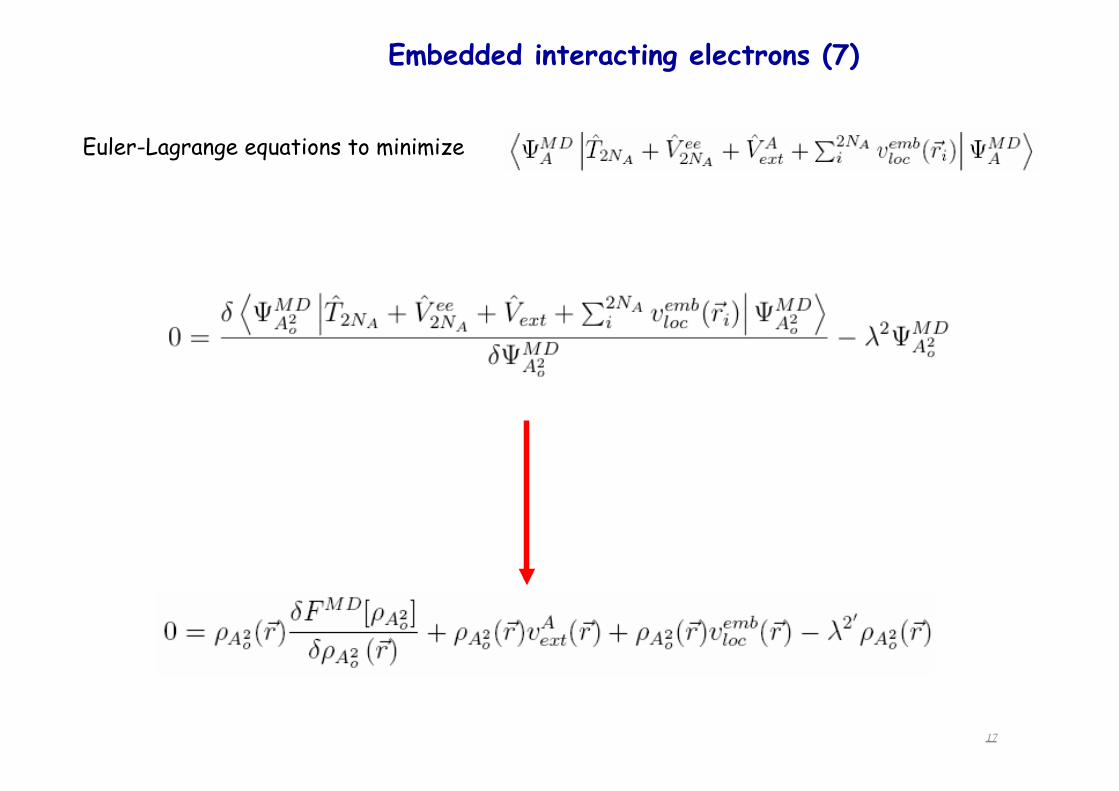
Euler-Lagrange equations to minimize
Embedded interacting electrons (7)!
17

18
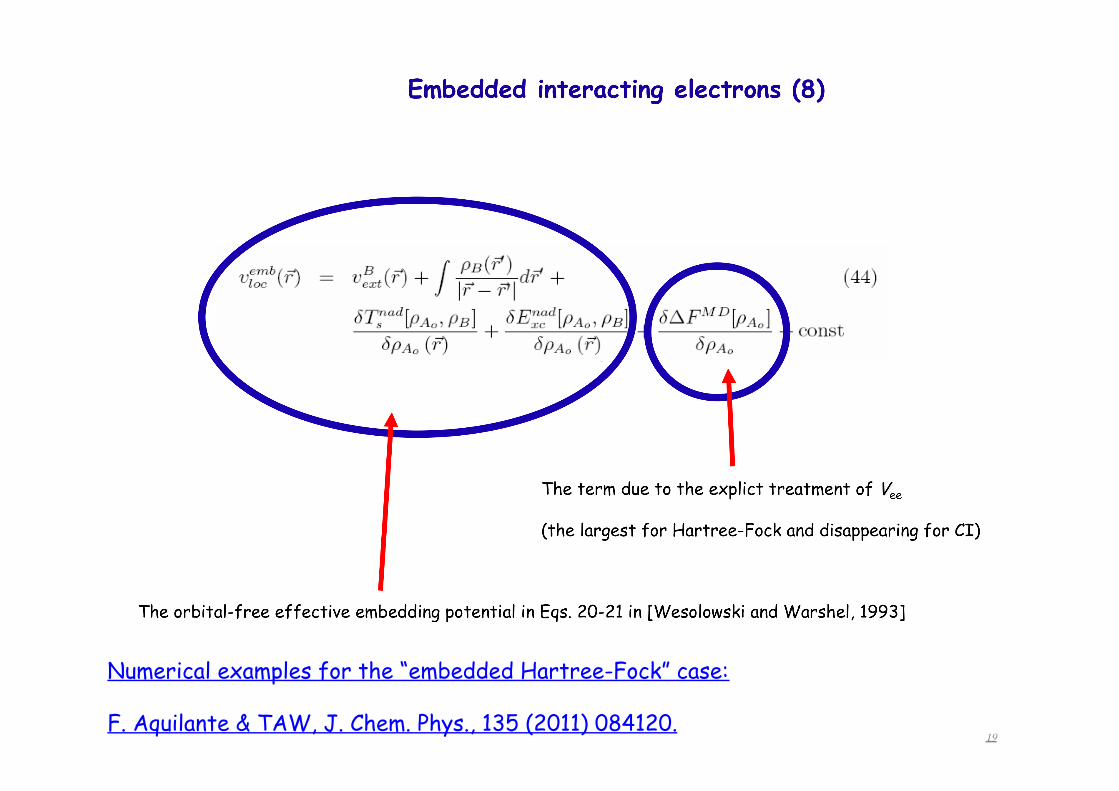
19
Numerical examples for the “embedded Hartree-Fock” case:
F. Aquilante & TAW, J. Chem. Phys., 135 (2011) 084120.

Embedding an « interacting wavefunction »
• The embedding operator is a local potential Vemb(r)
• For a given external potential Vemb(r) is a bi-functional of ρA and ρB (it is orbital-free therefore).
• The embedded wavefunction is an auxiliary quantity. Its relation to the exact wavefunction for the whole wavefunction is not direct.
• Depending on the form of the embedded wavefunction, the functional dependence of the potential Vemb(r) on ρA and ρB might be different.
• Vemb(r) and the potential Veffemb(KS)[ρA,ρB,r] for embedding a non-
interacting system (Wesolowski-Warshel, 1993) are closely related. They are equal if the embedded wavefunction has the full CI form. The difference is bound from below by (-Ec[ρ]) and from above (it is non-positive).
20
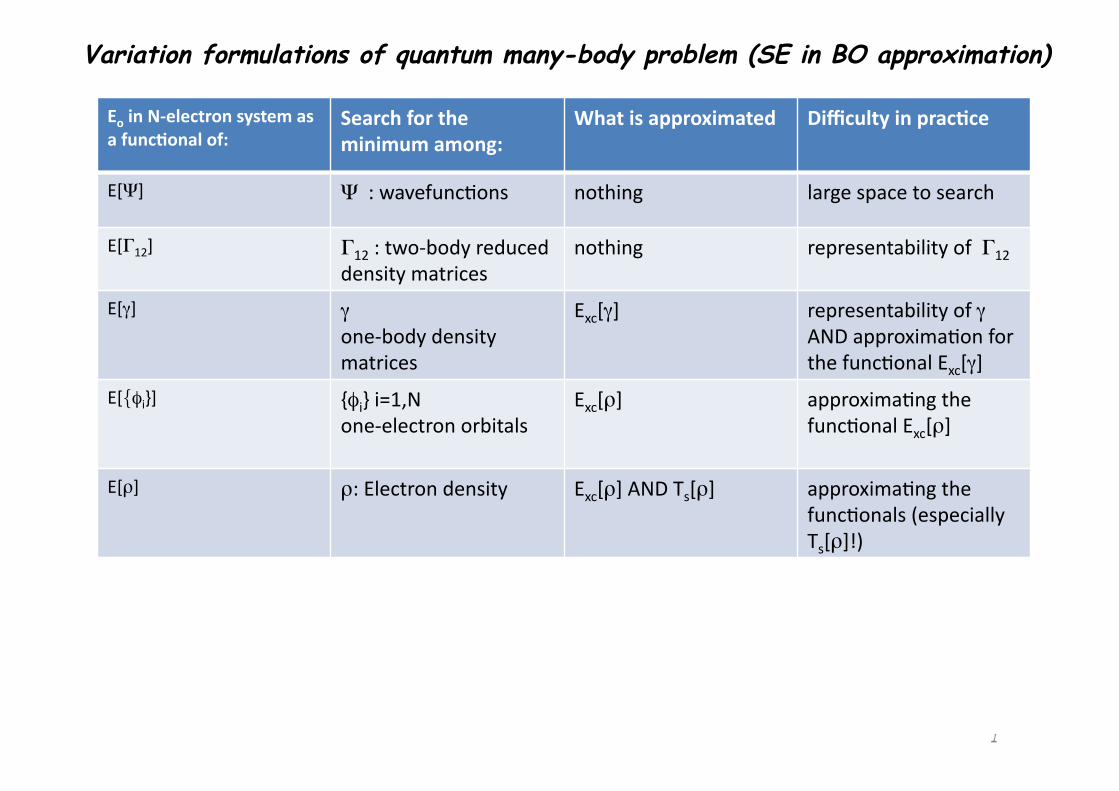
1
Eo in N-‐electron system as a func3onal of:
Search for the minimum among:
What is approximated Difficulty in prac3ce
E[Ψ] Ψ : wavefunc1ons nothing large space to search
E[Γ12] Γ12 : two-‐body reduced density matrices
nothing representability of Γ12
E[γ] γ one-‐body density matrices
Exc[γ] representability of γ AND approxima1on for the func1onal Exc[γ]
E[{φi}] {φi} i=1,N one-‐electron orbitals
Exc[ρ] approxima1ng the func1onal Exc[ρ]
E[ρ] ρ: Electron density Exc[ρ] AND Ts[ρ] approxima1ng the func1onals (especially Ts[ρ]!)
Variation formulations of quantum many-body problem (SE in BO approximation)
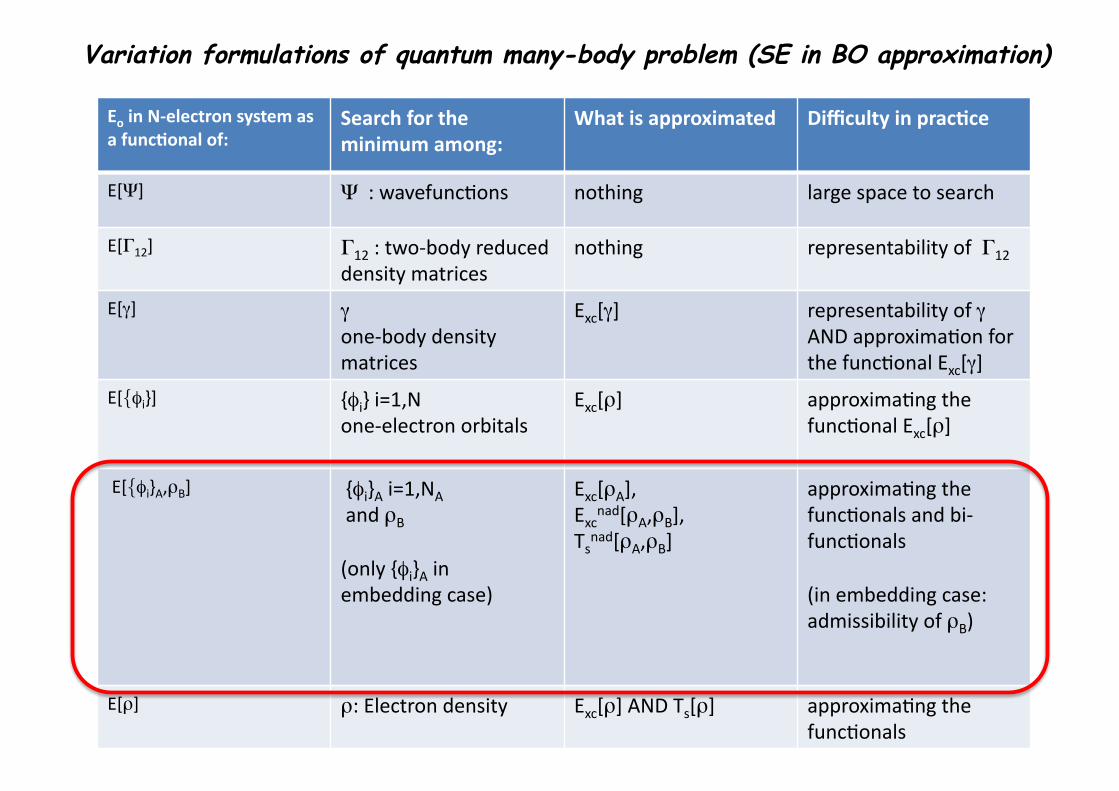
1
Eo in N-‐electron system as a func3onal of:
Search for the minimum among:
What is approximated Difficulty in prac3ce
E[Ψ] Ψ : wavefunc1ons nothing large space to search
E[Γ12] Γ12 : two-‐body reduced density matrices
nothing representability of Γ12
E[γ] γ one-‐body density matrices
Exc[γ] representability of γ AND approxima1on for the func1onal Exc[γ]
E[{φi}] {φi} i=1,N one-‐electron orbitals
Exc[ρ] approxima1ng the func1onal Exc[ρ]
E[{φi}A,ρB] {φi}A i=1,NA and ρB
(only {φi}A in embedding case)
Exc[ρA], Excnad[ρA,ρB], Tsnad[ρA,ρB]
approxima1ng the func1onals and bi-‐func1onals
(in embedding case: admissibility of ρB)
E[ρ] ρ: Electron density Exc[ρ] AND Ts[ρ] approxima1ng the func1onals
Variation formulations of quantum many-body problem (SE in BO approximation)









![THE HVZ THEOREM FOR -PARTICLE SCHRODINGER ...in the Born-Oppenheimer approximation was obtained in [19, 20, 21] in terms of two-cluster decompositions. In [22], the HVZ theorem is](https://static.fdocument.org/doc/165x107/613facd2f0f55d448e4cf2bc/the-hvz-theorem-for-particle-schrodinger-in-the-born-oppenheimer-approximation.jpg)








