
Review of Laboratory 3 Spectrophotometric determination of DNA quantity, purity Abs 260 nmAbs 280...
31
Review of Laboratory 3 Review of Laboratory 3 ectrophotometric determination of DNA quantity, pur Abs 260 nm Abs 280 nm Abs 320 nm Abs 260/Abs 280 0.05241 0.03110 0.00261 1.75 Restriction digest, agarose gel electrophoresis pM AB58 7577 bps 1000 2000 3000 4000 5000 6000 7000 S w aI P puM I A hdI AlwN I A sp718I K pnI ApaI Bsp120I StyI Bsm I P acI BsrG I B saB I N spV R srII Eco47III B seR I S exA I MluI E coR I X baI Bsm I BsrG I Sm aI B saB I E coR I NotI MluI BsrG I A atII ++ SacII Bam H I D raIII B su36I X baI BspEI S naB I AR S4/C en6 Am p ori pAD H N LS AD attB1 ccdB attB2 T er A D H pT 7 F1 TRP1 Expected results:
-
date post
21-Dec-2015 -
Category
Documents
-
view
217 -
download
4
Transcript of Review of Laboratory 3 Spectrophotometric determination of DNA quantity, purity Abs 260 nmAbs 280...

Review of Laboratory 3Review of Laboratory 3
• Spectrophotometric determination of DNA quantity, purity
Abs 260 nm Abs 280 nm Abs 320 nm Abs 260/Abs 280
0.05241 0.03110 0.00261 1.75
• Restriction digest, agarose gel electrophoresis
pMAB58
7577 bps
1000
2000
3000
4000
5000
6000
7000
SwaIPpuMI
AhdI
AlwNI
Asp718IKpnIApaIBsp120I
StyIBsmI
PacIBsrGI
BsaBINspV
RsrIIEco47III
BseRISexAIMluI
EcoRIXbaI
BsmIBsrGISmaI
BsaBIEcoRI
NotIMluI
BsrGIAatII++
SacII
BamHI
DraIII
Bsu36IXbaI
BspEISnaBI
ARS4/Cen6
Amp
ori
pADHNLS
ADattB1
ccdB
attB2
Ter ADH
pT7
F1
TRP1
Expected results:

Genomic DNA Uncut Plasmid DNA
Lab 3 electrophoresis resultsLab 3 electrophoresis resultsW R
• problems with ladder (λ HindIII)• incomplete digestion• what do these banding patterns represent?

Our good friend Our good friend PhaeodactylumPhaeodactylumtricornutumtricornutum

Cycle Sequencing andCycle Sequencing andBioinformatics ToolsBioinformatics Tools

Broad and Long Term ObjectiveBroad and Long Term Objective
To characterize a single clone from an To characterize a single clone from an Emiliania huxleyiEmiliania huxleyi cDNA library using cDNA library using sequence analysissequence analysis
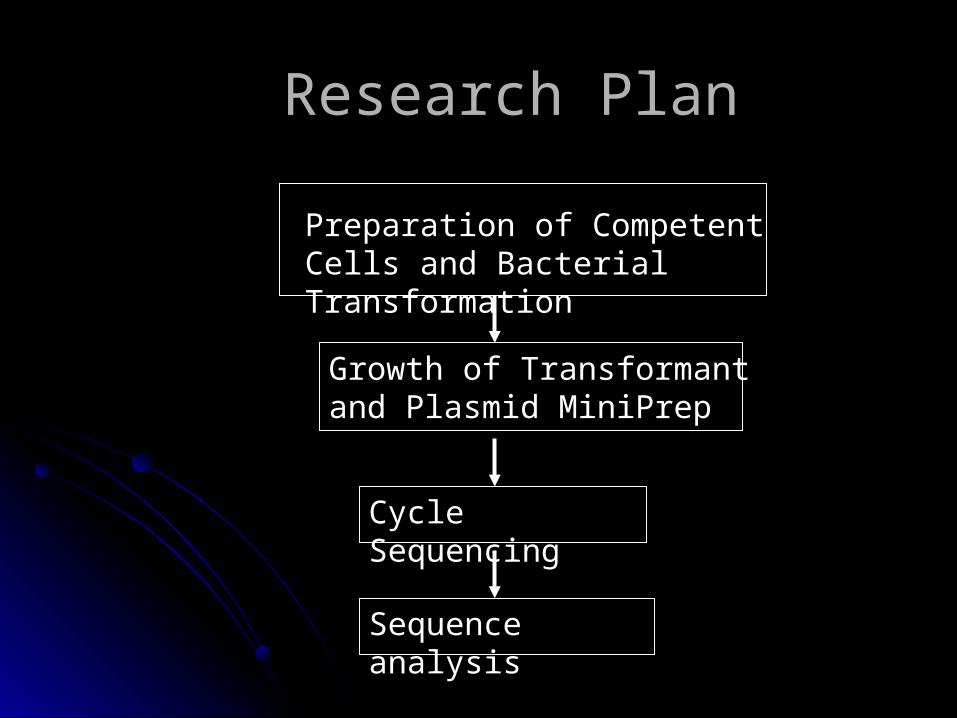
Research PlanResearch Plan
Preparation of Competent Cells and Bacterial Transformation
Growth of Transformant and Plasmid MiniPrep
Cycle Sequencing
Sequence analysis

Today’s Laboratory Objectives Today’s Laboratory Objectives
1. To understand the theoretical basis of Sanger-type DNA sequencing
2. To learn how to analyze your E. huxleyi cDNA sequence using various web-based bioinformatics tools

What is Cycle Sequencing?What is Cycle Sequencing?
Based on the Sanger Dideoxy chain termination methodBased on the Sanger Dideoxy chain termination method
(1974) (1974)
DNA synthesis reaction whereby fluorescently labeled DNA synthesis reaction whereby fluorescently labeled dideoxynucleotides are incorportated into the newly dideoxynucleotides are incorportated into the newly replicated DNA by DNA polymerase in a primer replicated DNA by DNA polymerase in a primer extension reactionextension reaction
Separation of labeled fragments by polyacrylamide gel Separation of labeled fragments by polyacrylamide gel electrophoresiselectrophoresis
*
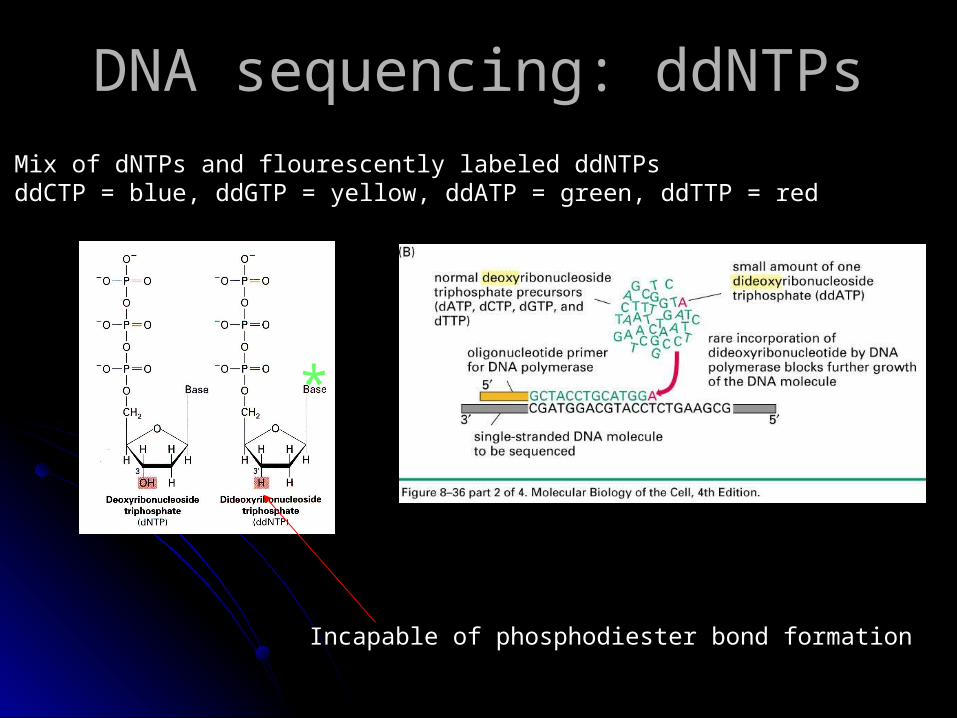
• Mix of dNTPs and flourescently labeled ddNTPs• ddCTP = blue, ddGTP = yellow, ddATP = green, ddTTP = red
DNA sequencing: ddNTPsDNA sequencing: ddNTPs
Incapable of phosphodiester bond formation
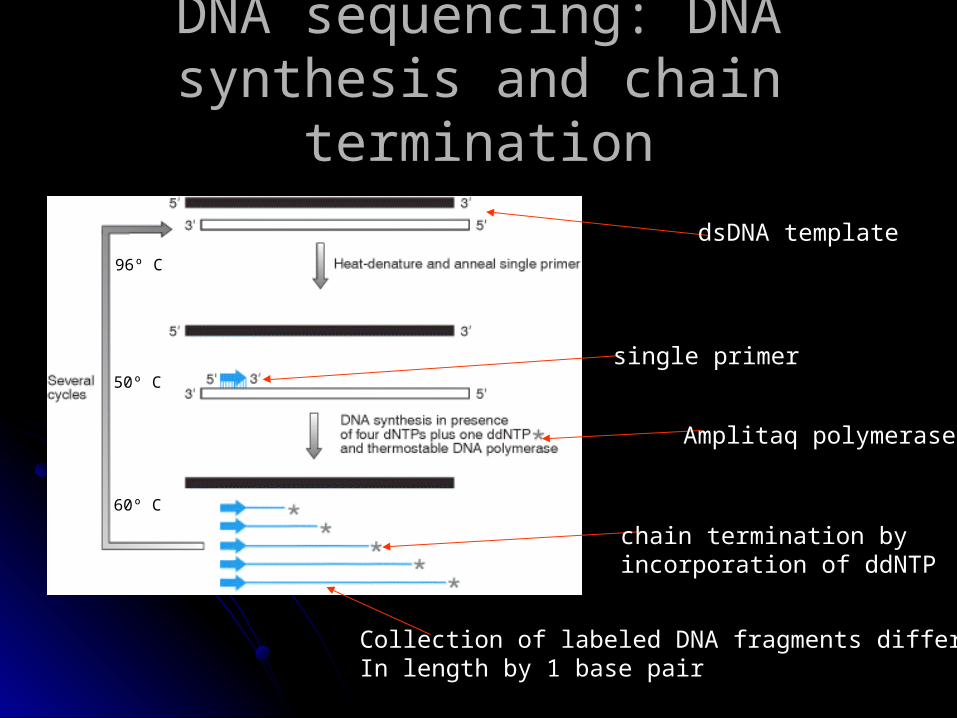
DNA sequencing: DNA synthesis DNA sequencing: DNA synthesis and chain terminationand chain termination
dsDNA template
single primer
chain termination byincorporation of ddNTP
Collection of labeled DNA fragments differing In length by 1 base pair
Amplitaq polymerase
96º C
50º C
60º C

Labeled DNA fragmentsLabeled DNA fragments
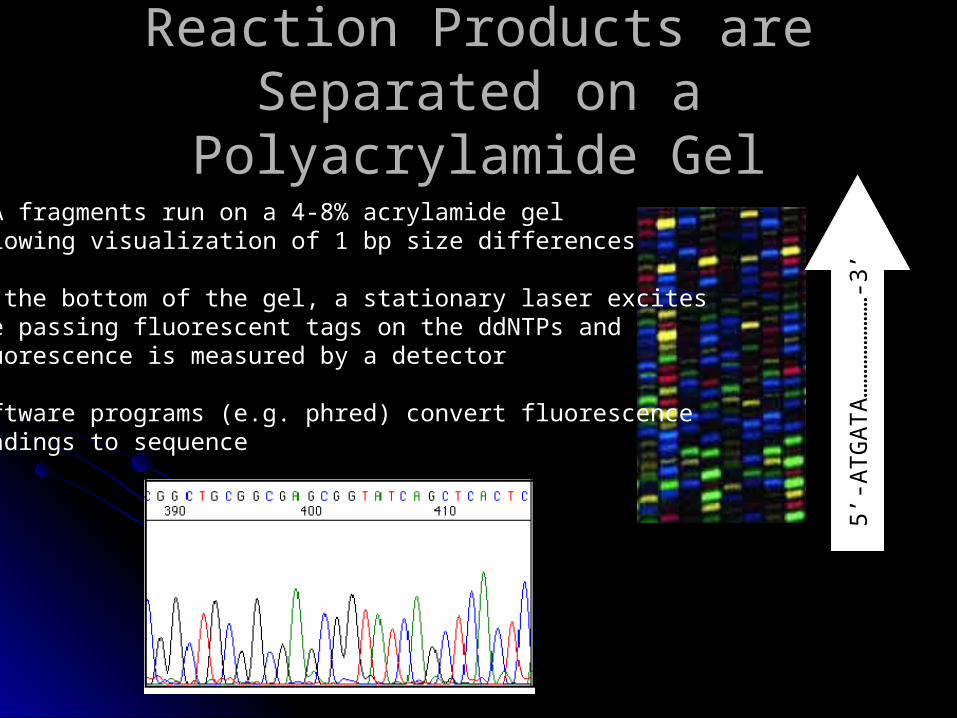
Reaction Products are Separated Reaction Products are Separated on a Polyacrylamide Gelon a Polyacrylamide Gel
5’-A
TG
AT
A…
……
……
……
-3’
• DNA fragments run on a 4-8% acrylamide gel allowing visualization of 1 bp size differences
• At the bottom of the gel, a stationary laser excites the passing fluorescent tags on the ddNTPs and fluorescence is measured by a detector
• software programs (e.g. phred) convert fluorescence readings to sequence

DNA sequence analysisDNA sequence analysis
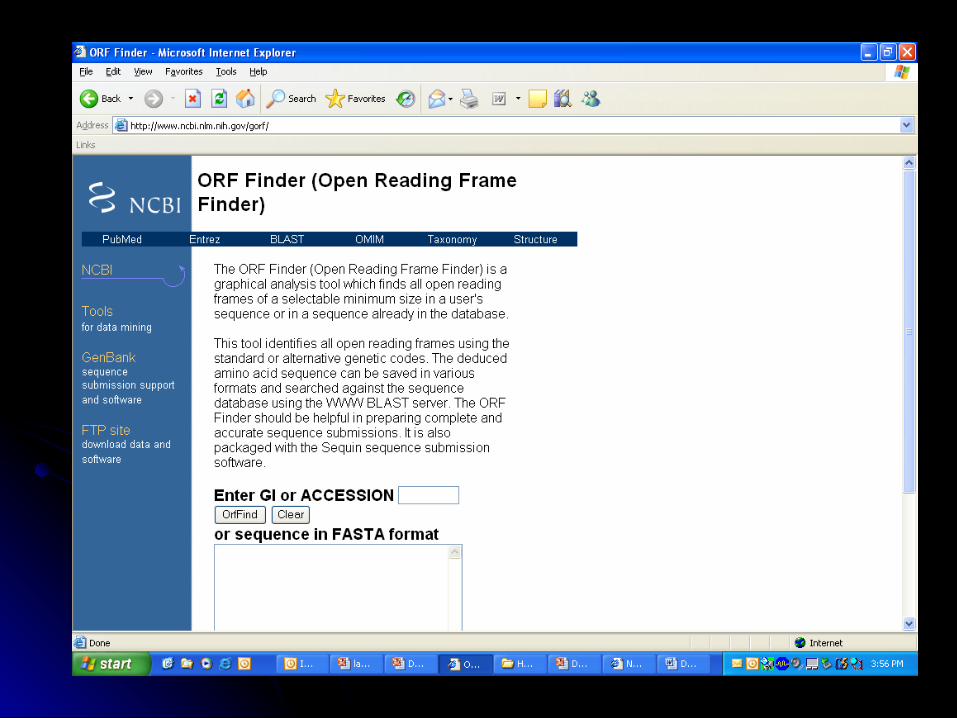
ORF Finder- http://www.ncbi.nlm.nih.gov/gorf/*
BLASTN- http://www.ncbi.nlm.nih.gov/BLAST/*
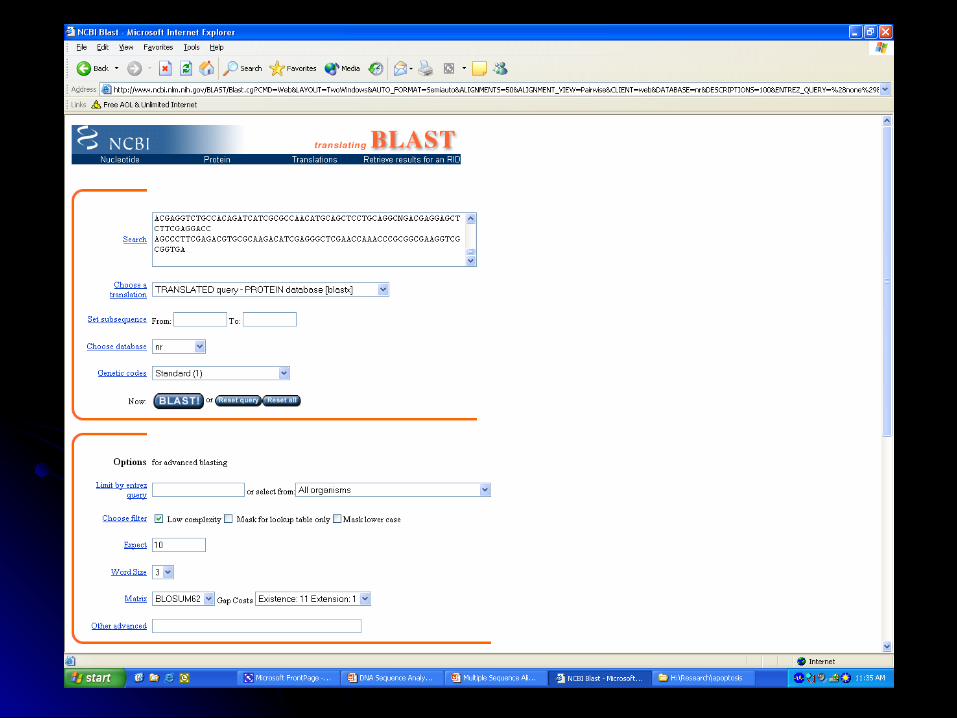
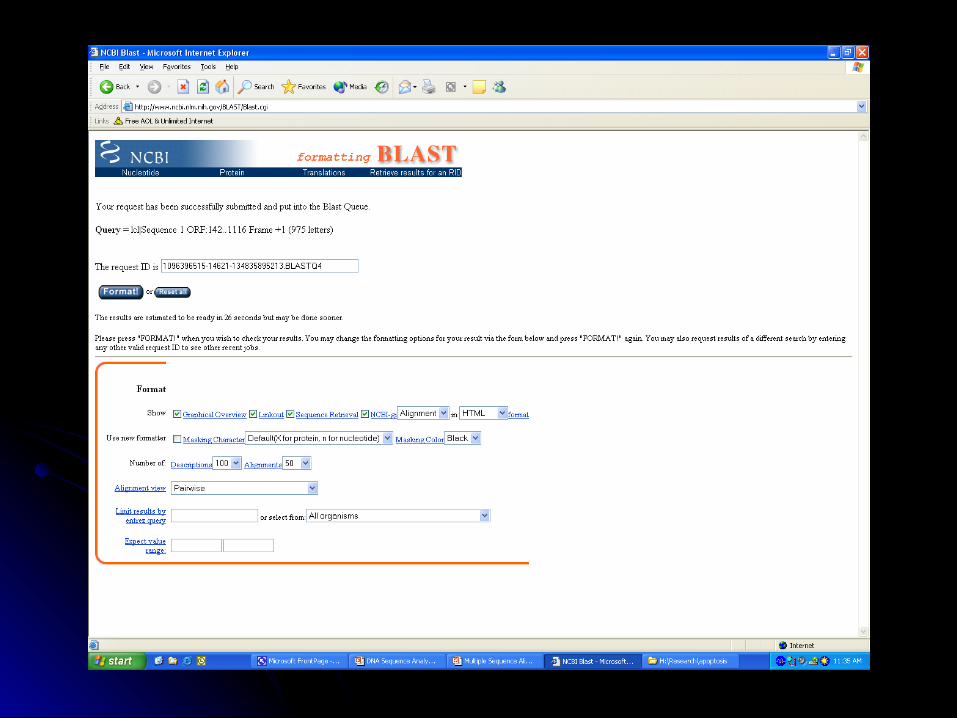
BLASTX- http://www.ncbi.nlm.nih.gov/BLAST/*
Clustal W- http://www.ebi.ac.uk/clustalw/*
For more information- http://www.ncbi.nlm.nih.gov/Education/index.html
ORF finderORF finder
• ORF (Open reading frame) finder: http://www.ncbi.nlm.nih.gov/gorf/
• Why would I use it?Is there a potential protein-coding region in this sequence?Is the ORF complete (with defined start and stop codons)?Identifies ORFs in DNA sequences
• How does it work?Scans all six possible reading frames (+1, +2, +3, -1, -2, -3) for potential start codons (ATG) followed by in-frame stop codons (TAA, TGA, TAG)
Assumption: larger ORFs more likely to code for proteins
• Useful for prokaryotic genomic DNA/cDNA, eukaryotic cDNA
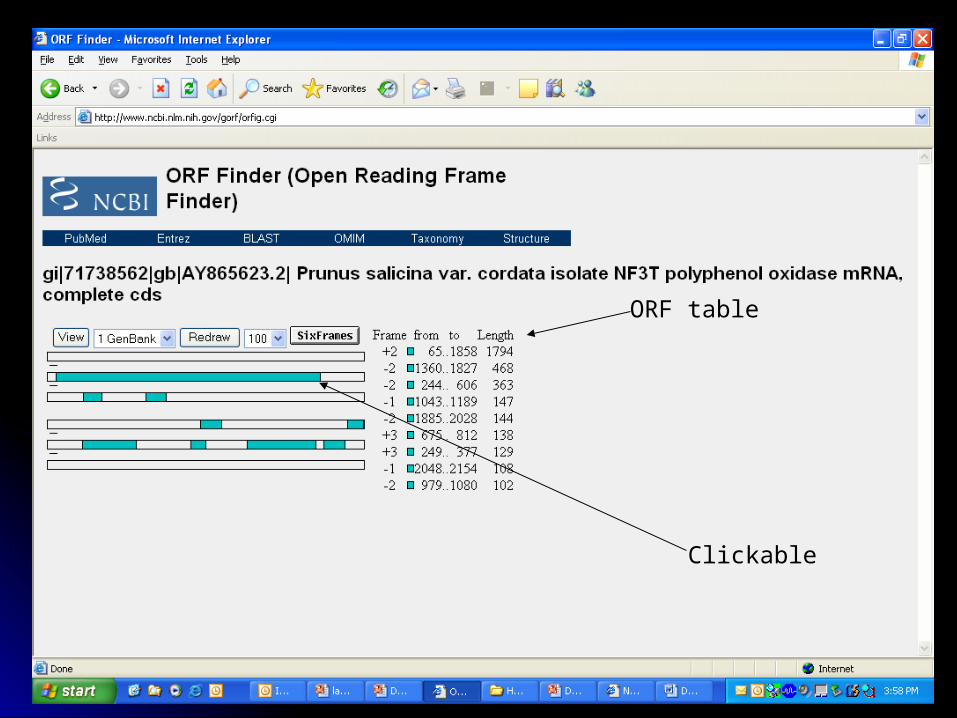
ORF table
Clickable
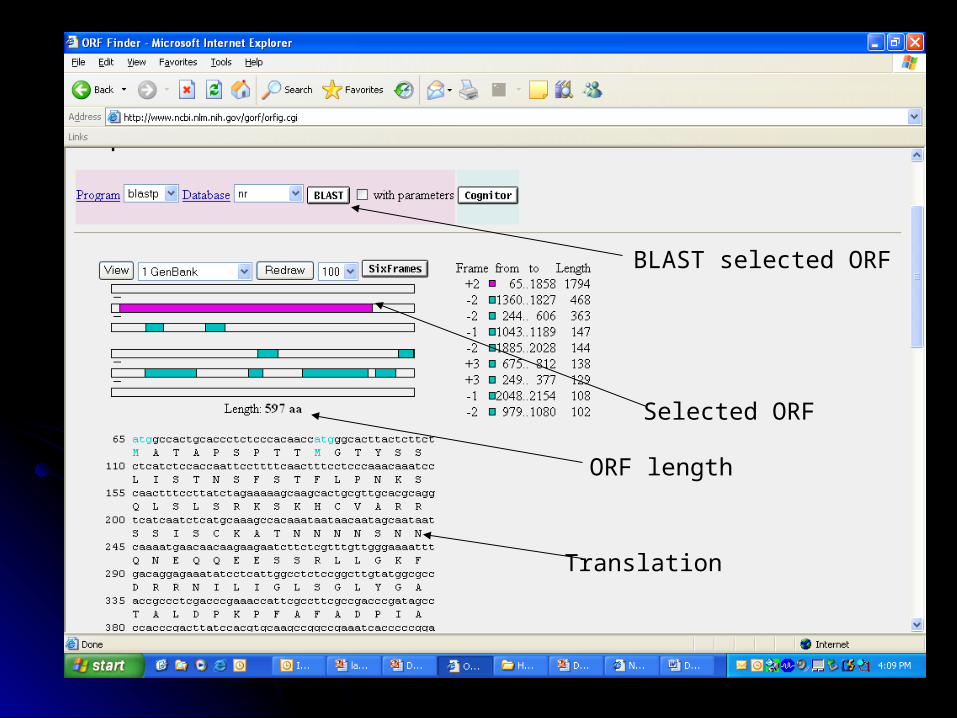
Selected ORF
BLAST selected ORF
ORF length
Translation
BLAST Database Search ToolBLAST Database Search Tool
• BLAST- Basic Local Alignment Search Tool: http://www.ncbi.nlm.nih.gov/BLAST/
• Why would I use it?Find similarity between your nucleotide/protein sequence and otherseqeuences deposited in very large (updated daily, >25 million
entry) public databases
Assumption 1: Similar DNA/protein sequences indicate descent from a common ancestral sequence (i.e. homology)
Assumption 2: Similar protein sequences indicate a similar structure and function

Program Query Sequence Database Target
BLASTN Nucleotide (both strands)
Optimized for speed not accuracy
Not good for distant homologs
Dust Filter Option (low complexity)
Nucleotide Database
BLASTX Nucleotide translated 6 frames
Less sensitive to sequence errors and mismatches
Useful for preliminary data/EST
Dust Filter Option
Protein Database
BLASTP Protein Protein Database
The BLAST FamilyThe BLAST Family

How does it work? How does it work? The BLAST AlgorithmThe BLAST Algorithm
Performs similarity search between your query sequence and aPerforms similarity search between your query sequence and anucleotide/protein database.nucleotide/protein database.
Pairwise alignment (two sequences compared)Pairwise alignment (two sequences compared)
Query sequence is split into small fragments (words) of defaultQuery sequence is split into small fragments (words) of defaultsizes of 11 bp or 3 amino acidssizes of 11 bp or 3 amino acids
Words are compared to every sequence in the target databaseWords are compared to every sequence in the target database(sliding window)(sliding window)
ATTGTACCGTA…GCGAT-ATACAGTTTTA…

How does it work?How does it work?The BLAST AlgorithmThe BLAST Algorithm
• Every window is scored using a scoring matrix, e.g. Every window is scored using a scoring matrix, e.g. BLOSUM62 or PAMBLOSUM62 or PAM
Nucleotide matrix- Match = +3, Substitution = -1, Gap = -5Nucleotide matrix- Match = +3, Substitution = -1, Gap = -5 Amino acid matrix- more complex relationships betweenAmino acid matrix- more complex relationships between amino acid substitutionsamino acid substitutions
• Goal to identify HSP’s (High Scoring Segment Pairs), Goal to identify HSP’s (High Scoring Segment Pairs), examine adjacent regions of sequence (Local alignment)examine adjacent regions of sequence (Local alignment)
• total alignment score is subjected to statistical analysis to total alignment score is subjected to statistical analysis to calculate the significance vs. random chance of the scorecalculate the significance vs. random chance of the score
ATTGTACCGTA…GCGAT-ATACAGTTTTA…
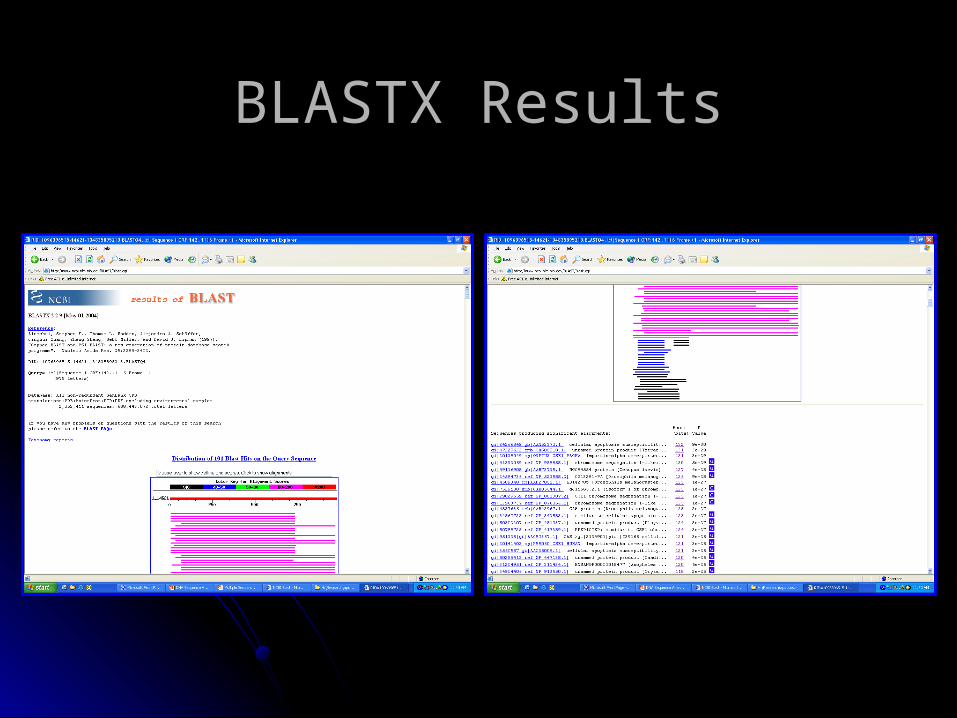
BLASTX ResultsBLASTX Results
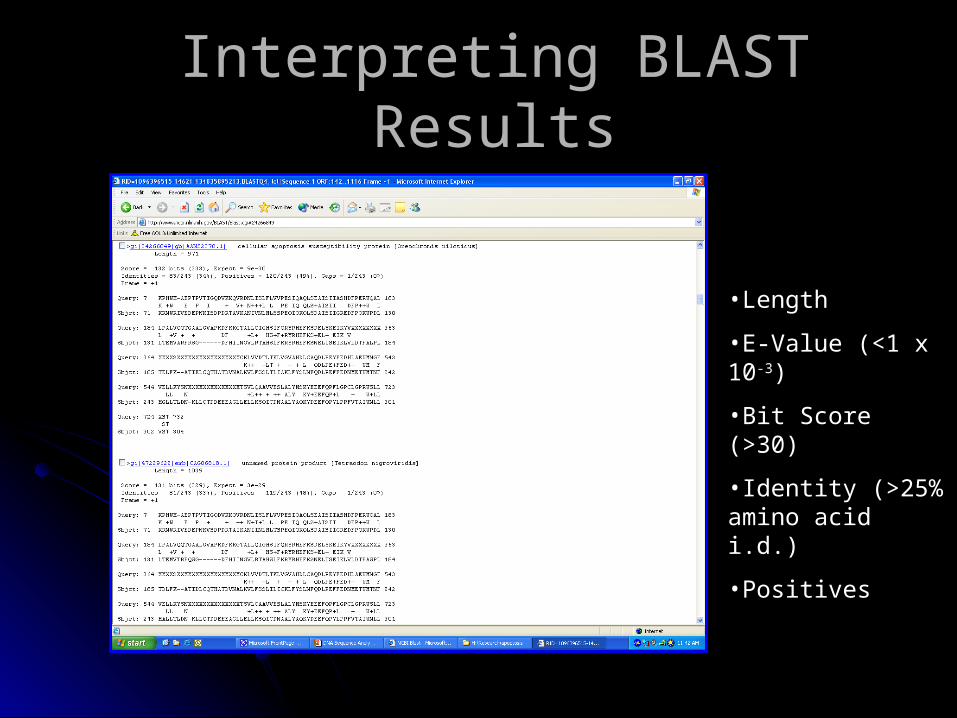
Interpreting BLAST ResultsInterpreting BLAST Results
•Length
•E-Value (<1 x 10-3)
•Bit Score (>30)
•Identity (>25% amino acid i.d.)
•Positives
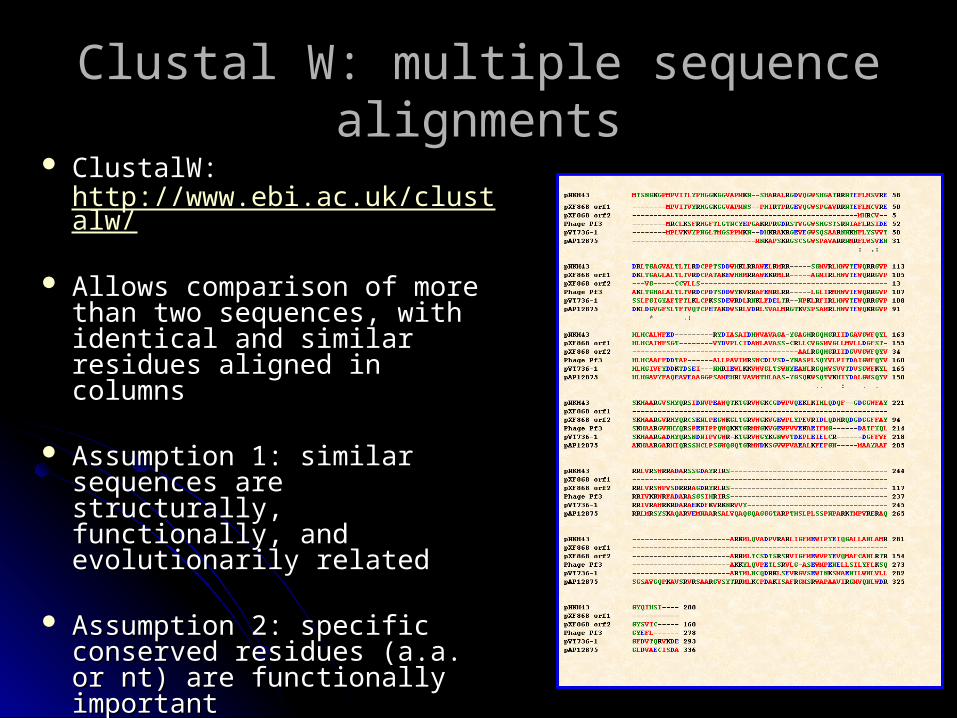
Clustal W: multiple sequence Clustal W: multiple sequence alignmentsalignments
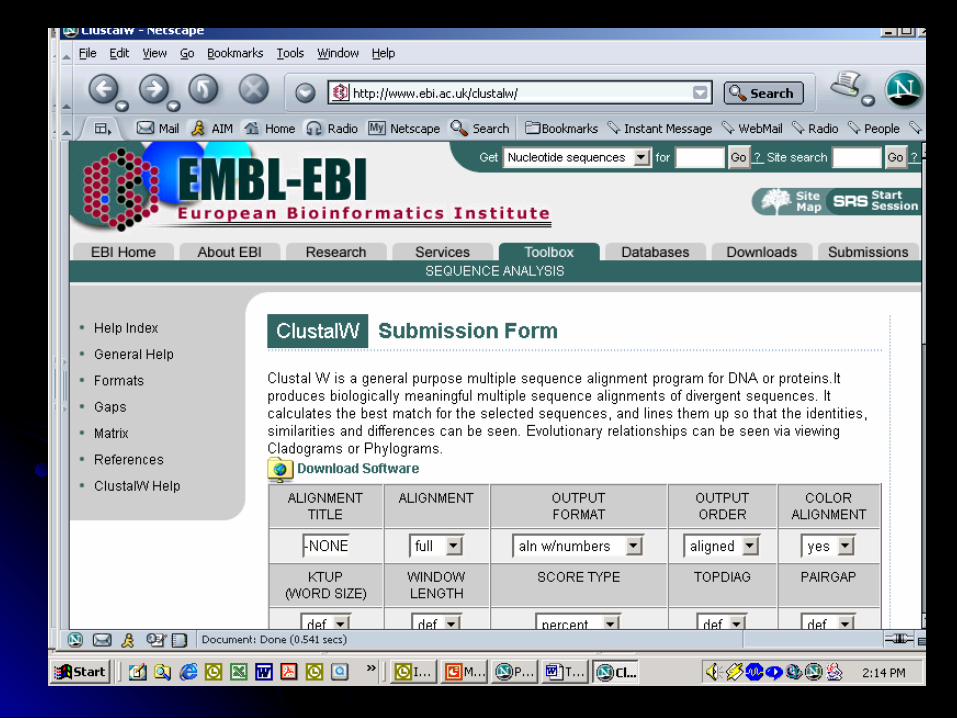
ClustalW: http://www.ebi.ac.uk/clustalw/
Allows comparison of more Allows comparison of more than two sequences, with than two sequences, with identical and similar residues identical and similar residues aligned in columnsaligned in columns
Assumption 1: similar Assumption 1: similar sequences are structurally, sequences are structurally, functionally, and evolutionarily functionally, and evolutionarily relatedrelated
Assumption 2: specific Assumption 2: specific conserved residues (a.a. or conserved residues (a.a. or nt) are functionally importantnt) are functionally important

Why would I do a multiple Why would I do a multiple sequence alignment?sequence alignment?
Phylogenetic comparisons (more similar Phylogenetic comparisons (more similar sequences are more closely related)sequences are more closely related)
Identify highly conserved amino Identify highly conserved amino acid/nucleotide sequences (may be critical acid/nucleotide sequences (may be critical for function)for function)
Characterize gene familyCharacterize gene family Identify conserved regions for the design Identify conserved regions for the design
of PCR primersof PCR primers

How does Clustal W work?How does Clustal W work? True multiple sequence alignment is extremely True multiple sequence alignment is extremely
computationally taxingcomputationally taxing Clustal W strategy: progressive pairwise Clustal W strategy: progressive pairwise
alignmentsalignments
A+B C+D E+F
Consensus1
Consensus2
Consensus3
+
Consensus4 + Final alignment
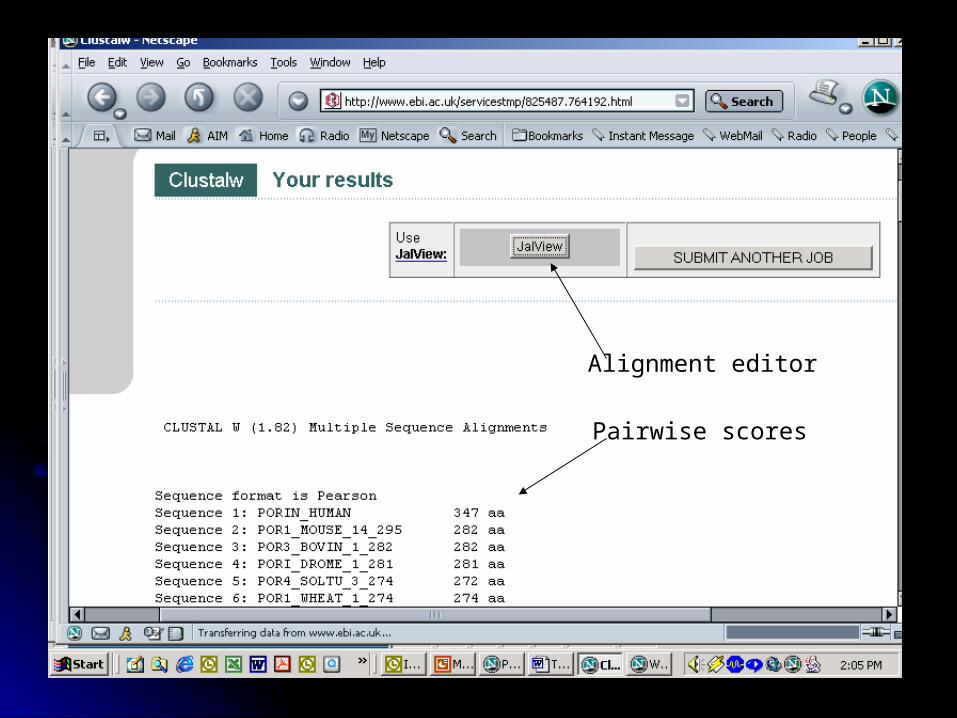
Pairwise scores
Alignment editor
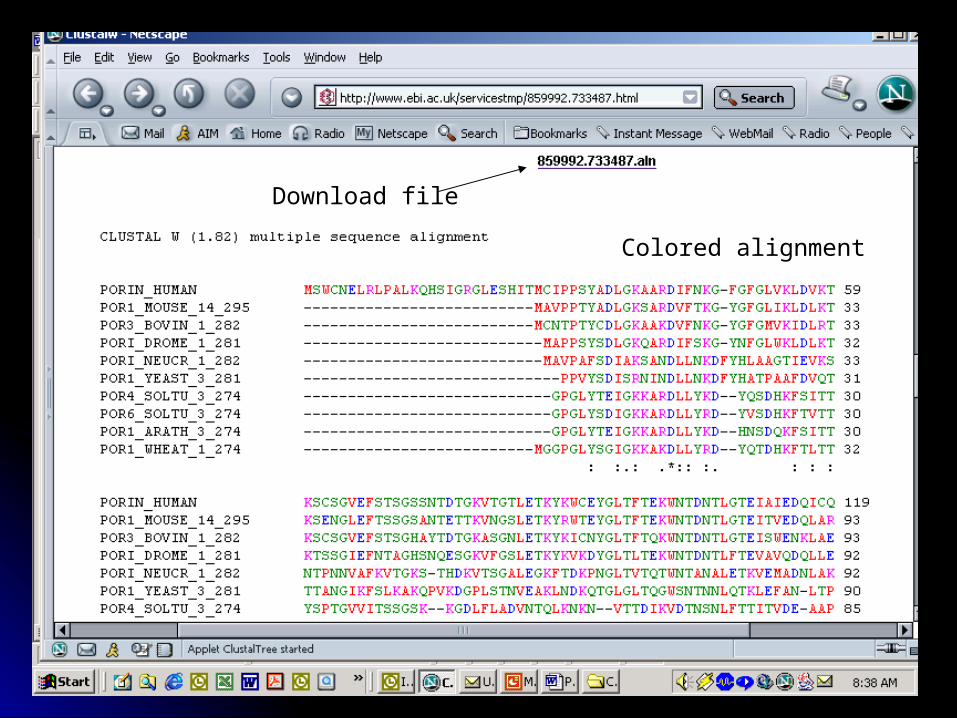
Colored alignment
Download file

![4-Bromo-a-PVP - SWGDRUG · 2016. 4. 20. · EI4-Bromo-α-PVP HCl; Lot# Mass RM-160316-01 Spectrum: 40 60 80 100 120 140 160 180 200 220 240 260 280 300 m/z [x 10 6] Intensity 2 4](https://static.fdocument.org/doc/165x107/6112b50edc449d558f354d04/4-bromo-a-pvp-swgdrug-2016-4-20-ei4-bromo-pvp-hcl-lot-mass-rm-160316-01.jpg)
















