
QXFOHLQLQ Neurodegenerative Diseases
122
New Insight into Mechanisms of Transcellular Propagation of Tau and α-Synuclein in Neurodegenerative Diseases XU YAN DISSERTATIONES SCHOLAE DOCTORALIS AD SANITATEM INVESTIGANDAM UNIVERSITATIS HELSINKIENSIS 7/2017 NEUROSCIENCE CENTER AND DEPARTMENT OF BIOSCIENCES FACULTY OF BIOLOGICAL AND ENVIRONMENTAL SCIENCES DOCTORAL PROGRAMME BRAIN & MIND UNIVERSITY OF HELSINKI
Transcript of QXFOHLQLQ Neurodegenerative Diseases

New Insight into Mechanisms of Transcellular Propagation of Tau and α-Synuclein in Neurodegenerative Diseases
XU YAN
dissertationes scholae doctoralis ad sanitatem investigandam universitatis helsinkiensis 7/2017
7/2017Helsinki 2017 ISSN 2342-3161 ISBN 978-951-51-2865-2
XU
YA
N N
ew In
sight into M
echanism
s of Transcellular P
ropagation of Tau an
d α-Synuclein
in N
eurodegenerative D
iseases
Recent Publications in this Series
77/2016 Aino SalonsalmiAlcohol Drinking, Health-Related Functioning and Work Disability78/2016 Heini WennmanPhysical Activity, Sleep and Cardiovascular Diseases: Person-oriented and Longitudinal Perspectives79/2016 Andres LõhmusHelper Component-Proteinase and Coat Protein are Involved in the Molecular Processes of Potato Virus A Translation and Replication80/2016 Li MaBrain Immune Gene Network in Inbred Mouse Models of Anxiety- and Sociability-related Neuropsychiatric Disorders81/2016 Finny S. VargheseCracking the Code of Chikungunya Virus: Inhibitors as Tools to Explore Alphavirus Biology82/2016 Vera ShirokovaTranscription Factor Foxi3 in Hair Follicle Development and Homeostasis83/2016 Daria BulanovaNovel Genetic Determinants of Breast Cancer Progression84/2016 Hugh ChapmanThe hERG1 (KV11.1) Potassium Channel: Its Modulation and the Functional Characterisation of Genetic Variants85/2016 Katja RostiExpression and Characterization of Neuronal Membrane Receptor Proteins86/2016 Irepan Salvador-MartínezEstimating Complexity and Adaptation in the Embryo: A Statistical Developmental Biology Approach87/2016 Vigneshwari SubramanianField-based Proteochemometric Models Derived from 3D Protein Structures: A Novel Approach to Visualize Affinity and Selectivity Features88/2016 Anita LampinenSignalling and Expression of the Ang-Tie Pathway in Tumor Vasculature1/2017 Essi HavulaTranscriptional Control of Dietary Sugar Metabolism and Homeostasis by Mondo-Mlx Transcription Factors2/2017 Satu MassinenSpecific Reading Disorder: Cellular and Neurodevelopmental Functions of Susceptibility Genes3/2017 Margarita AndreevskayaEcological Fitness and Interspecies Interactions of Food-Spoilage-Associated Lactic Acid Bacteria: Insights from the Genome Analyses and Transcriptome Profiles4/2017 Mikko SiuralaImproving Adenovirus-Based Immunotherapies for Treatment of Solid Tumors5/2017 Inken KörberMicroglial Dysfunction in Cstb-/- Mice, a Model for the Neurodegenerative Disorder Progressive Myoclonus Epilepsy of Unverricht-Lundborg Type, EPM16/2017 Shrikanth KulashekharThe Role of Cortical Oscillations in the Estimation and Maintenance of Sensory and Duration Information in Working Memory
NEUROSCIENCE CENTER AND DEPARTMENT OF BIOSCIENCESFACULTY OF BIOLOGICAL AND ENVIRONMENTAL SCIENCESDOCTORAL PROGRAMME BRAIN & MINDUNIVERSITY OF HELSINKI

Neuroscience CenterAnd
Department of BiosciencesFaculty of Biological and Environmental Sciences
Doctoral programme in Brain & MindUniversity of Helsinki
NEW INSIGHT INTO MECHANISMS OF TRANSCELLULAR PROPAGATION OF TAU
AND -SYNUCLEIN IN NEURODEGENERATIVE DISEASES
Xu Yan
ACADEMIC DISSERTATION
To be presented, with the permission of the Faculty of Biological and Environmental Sciences, University of Helsinki,
for public examination in lecture room B105, Cultivator II, Viikki,on 10th February 2017, at 12 noon.
Helsinki 2017

II
Supervisor Docent Henri Huttunen, PhDNeuroscience Center,University of Helsinki, Finland
Thesis committee Docent Mikko Airavaara, PhDInstitute of Biotechnology,University of Helsinki, Finland
Docent Tomi Rantamäki, PhD, Department of Biosciences,University of Helsinki, Finland
Pre-examiners Docent Mikko Airavaara, PhDInstitute of Biotechnology, University of Helsinki, Finland
Docent Katja Kanninen, PhD, Institute for Molecular Sciences,University of Eastern Finland, Finland
Opponent Assistant Professor Kelvin C Luk, PhDDepartment of Pathology and Laboratory Medicine, Perelman School of Medicine,University of Pennsylvania, U.S.A.
Custos Professor Juha Voipio, PhDDepartment of Biosciences,University of Helsinki, Finland
Dissertationes Scholae Doctoralis Ad Sanitatem Investigandam Universitatis Helsinkiensis
ISSN 2342-3161 (print)ISSN 2342-317X (online)ISBN 978-951-51-2865-2 (paperback)ISBN 978-951-51-2866-9 (PDF)Unigrafia and Hansaprint Helsinki 2017

III
TABLE OF CONTENTS List Of Original Publications .................................................................................................. V Abbreviations ......................................................................................................................... VI Abstract ................................................................................................................................ VIII 1 Introduction .......................................................................................................................... 1 2 Review of literature ............................................................................................................. 3
Protein misfolding and aggregation ............................................................................ 3 2.1 α-synuclein ..................................................................................................................... 5 2.2
2.2.1 Structure and biological functions ........................................................................ 5 2.2.2 α-synuclein aggregation ....................................................................................... 7 2.2.3 Clearance of aggregation .................................................................................... 10 2.2.4 α-synuclein toxic species .................................................................................... 11 2.2.5 Prolyl oligopeptidase .......................................................................................... 12
Tau ............................................................................................................................ 13 2.32.3.1 Structure and biological functions ...................................................................... 13 2.3.2 Phosphorylation .................................................................................................. 14 2.3.3 Kinases and phosphatases .................................................................................. 15 2.3.4 Other post-translational modifications ............................................................... 16 2.3.5 Tau aggregation .................................................................................................. 17 2.3.6 Genetic risk factors of Alzheimer’s disease ....................................................... 20 2.3.7 Tau ubiquitination and clearance ....................................................................... 23 2.3.8 Toxic species of tau ............................................................................................ 24 2.3.9 Interconnection between aSyn and tau ............................................................... 25 2.3.10 Therapeutic strategies against tauopathies and synucleinopathies ..................... 27
Misfolded protein propagation in pathological conditions ..................................... 28 2.42.4.1 Prion-like seeding ............................................................................................... 29 2.4.2 Cell-to-cell transmission of α-synuclein and tau ................................................ 32 2.4.3 Spread of pathology ............................................................................................ 37 2.4.4 The spread of pathology- what is missing from the story? ................................. 38
3 Aims of the study ................................................................................................................ 40 4 Materials and methods ...................................................................................................... 41
Plasmid constructs and recombinant proteins ......................................................... 41 4.1 Chemicals .................................................................................................................... 41 4.2 Antibodies .................................................................................................................... 42 4.3 Cell culture and transfections .................................................................................... 42 4.4 Protein-fragment complementation assay ................................................................ 42 4.5
4.5.1 Tau and aSyn secretion assay ............................................................................. 43 4.5.2 Assessment of tau and aSyn uptake .................................................................... 43
Electrophoretic techniques ........................................................................................ 43 4.64.6.1 Western blotting ................................................................................................. 43 4.6.2 Native polyacrylamide gel electrophoresis......................................................... 44
Fractionation techniques ............................................................................................ 44 4.74.7.1 Protein fractionation ........................................................................................... 44 4.7.2 Media fractionation ............................................................................................ 44
Protein crosslinking .................................................................................................... 45 4.8 Microscale Thermophoresis ....................................................................................... 45 4.9

IV
Immunofluorescence microscopy ............................................................................ 45 4.10 Cell viability............................................................................................................... 46 4.11 Statistical analyses .................................................................................................... 46 4.12
5 Results ................................................................................................................................. 47 Clarifying molecular mechanisms of prolyl oligopeptidase modulating α-5.1
Synuclein intracellular dimerization (I) ................................................................... 47 5.1.1 Prolyl oligopeptidase promotes α-Synuclein dimerization................................. 47 5.1.2 Direct protein-protein interaction between prolyl oligopeptidase and α-
Synuclein ............................................................................................................ 47 5.1.3 KYP-2047 alters the conformation of PREP...................................................... 48
Assay development for studying secretion and uptake of α-Synuclein and tau (I, 5.2II).................................................................................................................................. 49 5.2.1 Development of an assay for studying α-Synuclein release and uptake............. 49 5.2.2 Tau secretion and uptake assay development..................................................... 51
Functional association of LOAD susceptibility genes with tau secretion and 5.3uptake mechanisms (II).............................................................................................. 53 5.3.1 RNAi screen of LOAD risk genes using the tau secretion and uptake assays ... 53 5.3.2 CD2AP overexpression does not alter tau secretion .......................................... 54 5.3.3 FRMD4A-cytohesin signaling pathway regulates tau secretion ........................ 54
The impact of transcellular propagation of tau on cellular stress (III) ................. 56 5.45.4.1 Hyperphosphorylated tau is recruited to stress granules after internalization .... 56 5.4.2 Tau uptake sensitizes cells to stress.................................................................... 57
6 Discussion ........................................................................................................................... 59 Assay development for studying cell-to-cell propagation of α-synuclein and tau. 59 6.1 The effect of prolyl oligopeptidase on α-synuclein oligomerization ....................... 61 6.2 The effect of LOAD genes on cell-to-cell transmission of tau................................. 62 6.3 Pathophysiological consequences triggered by tau internalization........................ 66 6.4
6.4.1 Role of TIA-1 in mediating internalized tau-induced stress and toxicity through stress granules..................................................................................................... 66
6.4.2 The role of tau hyperphosphorylation in cell-to-cell transmission of tau........... 67 Understanding cell-to-cell transfer of disease-associated protein as a whole........ 68 6.5
7 Concluding remarks and future prospects ...................................................................... 71 Acknowledgements ................................................................................................................. 72References................................................................................................................................ 73

V
List of Publications included: I Mari Savolainen*, Xu Yan*, Timo Myöhänen and Henri J. Huttunen
(2015). Prolyl oligopeptidase enhances α-synuclein dimerization via direct protein-protein interaction. J. Biol. Chem. 290(8):5117-26. * = equal contribution.
II Xu Yan, Niko-Petteri Nykänen, Cecilia A. Brunello, Annakaisa
Haapasalo, Mikko Hiltunen, Riikka-Liisa E. Uronen and Henri J. Huttunen (2016). FRMD4A-cytohesin signaling modulates cellular release of Tau. J Cell Sci. 129(10):2003-15.
III Cecilia A. Brunello, Xu Yan and Henri J. Huttunen (2016). Internalized
Tau sensitizes cells to stress by promoting formation and stability of stress granules. Sci Rep. 6:30498.
Author’s contribution to the original publications included in the Thesis:
I: The author participated in experimental design, data analysis and writing the manuscript. The author conducted most of the experiments.
II: The author participated in experimental design, data analysis and writing the manuscript. The author conducted most of the experiments.
III: The author participated in experimental design, data analysis and writing the manuscript. The author conducted assay development and some of the experiments.

VI
Abbreviations
α-Synuclein (aSyn)Alzheimer’s disease (AD)Amyloid Precursor Protein (APP)Amyotrophic lateral sclerosis (ALS)Annular protofibrils (APFs)Apolipoprotein E (APOE4)Argyrophilic grain disease (AGD)Autophagy-lysosomal pathway (ALP)Bridging integrator 1(BIN1)Calmodulin-dependent protein kinase II (CaMKII)Casein kinase 1(CK1)CD2-associated protein (CD2AP)Cerebrospinal fluid (CSF)Central nervous system (CNS)Clusterin (CLU)Chaperone-mediated autophagy (CMA)Complement receptor 1 (CR1)Corticobasal degeneration (CBD)Cyclin-dependent kinase 5 (CDK5)Cyclic AMP-dependent protein kinase (PKA)Dementia with Lewy bodies (DLB)Dopamine (DA)Down syndrome (DS)Dual-specificity tyrosine-phosphorylation-related kinase 1A (DYRK1A)Electron microscopy (EM)Endoplasmic reticulum (ER)Extracellular signal–regulated kinases (ERK)FERM containing domain 4A (FRMD4A)Gaussia princeps luciferase (GLuc)Genome-wide association studies (GWAS)Glycogen synthase kinase-3 (GSK-3)Immunofluorescence microscopy (IF)Jun NH2-terminal kinase (JNK)Lactate dehydrogenase (LDH)Late-onset Alzheimer’s disease (LOAD)Lewy body variant of AD (LBVAD)Membrane-spanning 4-domains subfamily A (MS4A)Metalloproteinases 9 (MMP9)Microtubule-binding protein (MAP)MicroRNA (miRNA)Misfolding-associated protein secretion (MAPS)Mitogen-activated protein kinase family (MAPKs)Microscale Thermophoresis (MST)Native Polyacrylamide Gel Electrophoresis (PAGE)Neurofibrillary tangles (NFTs)

VII
Nuclear magnetic resonance (NMR)Parkinson’s disease (PD)Paired-helical filaments (PHFs)Phosphatidylinoaitol binding clathrin assembly protein (PICALM)Pick’s disease (PiD)Protein-fragment complementation assay (PCA)Preformed fibrils (Pffs)Progressive supranuclear palsy (PSP)Prolyl oligopeptidase (PREP)Protein-protein interactions (PPIs)Protein kinase C (PKC)Reactive oxygen species (ROS)Small hairpin RNA (shRNA)Small interfering RNA (siRNA)Sortilin-related receptor LDLR class A repeats containing (SOR1)Stress Granules (SGs)Superoxide dismutase 1 (SOD1)RNA-binding Protein (RBP)TAR DNA-binding protein 43 (TDP-43)Triggering receptor expressed on myeloid cells 2 (TREM2)Tyrosine hydroxylase (TH)Ubiquitin-proteasome system (UPS)

VIII
Abstract
Progressive development of pathology in neuroanatomically connected brain regions is a common feature of many neurodegenerative diseases. The spread of disease pathology is suggested to be dependent on the transmissibility of disease-associated proteins, particularly soluble aggregates of misfolded proteins. Emerging evidence suggests that many disease-associated proteins such as α-synuclein (aSyn) and tau, in certain misfolded and aggregated states convert from physiologically normal proteins into forms that lead to progression of disease pathology in a template-dependent manner, which is also known as seeding. The propagation and the proteinopathy have been suggested to occur via cell-to-cell transmission. The exact mechanisms involved in the seeding and spreading process are incompletely understood. In this thesis work, three critical steps of the seeding pathway (a process involves multiple steps), the intracellular aggregation, cellular release and uptake of aSyn and tau, were carefully studied primarily via a newly developed platform based on protein-fragmentcomplementation assay. The main findings of this thesis are:
a) Prolyl oligopeptidase (PREP) is a serine peptidase that was previously known to accelerate the process of aSyn aggregation and suppress autophagy clearance in cells and transgenic aSyn mice. The results of this thesis show that PREP directly interacts with aSyn in neuro2A cells and cell-free environment, and enhances aSyn dimerization, which is an early event in aSyn aggregation pathway. In addition, the PREP-mediated aSyn dimerization can be antagonized by KYP-2047, a small-molecule PREP inhibitor.
b) Late-onset Alzheimer’s disease (LOAD) susceptibility genes affect the individual risk of developing Alzheimer’s disease, which is one of the common tauopathies. In this work, the functional connection between selected LOAD susceptibility genes and cell-to-cell transmission of tau was studied in vitro. We observed that RNAi knockdown ofCD2AP and FRMD4A reduced tau secretion, and knockdown of APOE reduced tau uptake in HEK293T cells. Further mechanistic studies revealed that FRMD4A modulates tau secretion via the FRMD4A-cytohesin-Arf6 signalling pathway and the Par6/aPKC polarity signalling complex. This data, for the first time, demonstrates a functional connection between LOAD risk genes and cell-to-cell propagation of tau.
c) Following internalization, extracellular, hyperphosphorylated tau was found to be recruited to stress granules, transient non-membraneous cytosolic structures composed of RNA and self-aggregating RNA-binding proteins. Tau recruitment was dependent on TIA-1, an RNA-binding stress granule protein. Importantly, the stress granules induced by and containing internalized tau were resistant to normal clearance and associated with increased sensitivity of cells to other stresses. This data describe a previously unrecognized mechanism and pathological consequence of cell-to-cell propagation of tau-mediated by stress granules, which have previously been associated with the pathophysiology of various neurodegenerative diseases.
Overall, the work described in this thesis provides several novel findings that improve our understanding of cellular mechanisms underlying the development and spreading of aSyn and tau-related neurodegenerative pathologies. These pieces of knowledge may be potential avenues towards the development of crucial therapeutics against aSyn and tau-relatedneurodegenerative diseases.

1
1 Introduction
Dynamic protein-protein interactions (PPIs) that constitute multi-protein complexes and networks predominantly determine the cellular functionalities. Impairments in these complexes and networks could result in various pathological disorders. For example, protein misfolding and subsequent aggregation in certain brain regions is a pathological characteristic shared by many neurodegenerative diseases (Lashuel et al., 2013, Lee et al., 2001).
Cerebral accumulation and aggregation of microtubule-associated protein tau is a common feature of diseases associated with tauopathy such as Alzheimer’s disease (AD), which is the most common neurodegenerative disease with age-related dementia (Lee et al., 2001). The accumulation and aggregation of α-Synuclein (aSyn) in various brain regions such as substantia nigra is a hallmark of Parkinson’s disease (PD), which is the most common neurodegenerative movement disorder (Fahn, 2003, Spillantini et al., 1997). Despite the fact that tau and aSyn are distinct proteins, and have been extensively studied in distinct pathological contexts, the mechanisms involved in the aggregation process and propagation of pathology of these two proteins are proposed to be highly converged and overlapping based on existing evidence (Moussaud et al., 2014). For example, both aSyn and tau aggregation exhibits an inducible nucleation-elongation mechanism (Wang and Mandelkow, 2016, Lashuel et al., 2013). Both aSyn and tau are present in cerebrospinal fluid (CSF) of human patients, and are transmissible between cells and animal models (Blennow et al., 1995, Borghi et al., 2000, Guo and Lee, 2014b). In the theories of Braak staging, Lewy body pathology and tauopathies develop sequentially in neuroanatomically connected brain regions in a time-dependent manner (Braak and Braak, 1991, Bancher et al., 1993, Braak et al., 2003). This thus implicates that pathological forms of aSyn and tau could get access to the extracellular space, and moreover spread from one region to another during the pathogenesis of neurodegenerative diseases.
Emerging evidence on the spread of various disease-associated proteins in a "prion-like" manner in vitro and in vivo have implicated the existence of a common mechanism in the spread of pathology of neurodegenerative diseases as reviewed in Guo and Lee (2014a). In the “prion-like” paradigm, it was suggested that many amyloidogenic proteins, including aSyn, tau and as well as other known pathology-related proteins such as amyloid-β (Aβ), TDP-43, superoxidase dismutase 1 and huntingtin, might transmit between cells and spread pathology into distinct but connected brain regions with a mechanism similar to prion proteins. In prion diseases, such as Creutzfeldt-Jacob disease (CJD), normal prion proteins sporadically convert to pathological species that have altered conformations and act as infectious agents that further convert normal prion proteins into pathological species in a template-directed manner, and thus spread the pathology rapidly (Bolton et al., 1982, Aguzzi, 2009). The amyloidogenic non-prion proteins mentioned above were also proposed to undergo this feedforward loop by seeding aggregation into neighbouring cells (Guo and Lee, 2014b, Brettschneider et al., 2015). The exact mechanisms of cell-to-cell transfer and seeding of the disease-associated proteins are poorly understood, but the mechanisms are implicated in involving gain of seeding property, release from donor cells and uptake by recipient cells. Hence understanding the molecular mechanisms, protein pathways and risk factors related to the transcellular propagation of disease-associated proteins would grant us not only knowledge of our physiological and pathological conditions but also avenues to develop crucial therapeutics.
The improved well-being and success of modern medicinal therapeutics have expanded our life expectancy. However, longer life also results in various age-related diseases that add a high cost to our society. Among all, neurodegenerative diseases lay a particular burden. For

2
example, in 2015 there were 47 million patients with neurodegenerative dementia and the global economic cost of this was $818 billion (Wimo et al., 2016). Hence, it grows urgent for us to put a vast investigation effort into this field. The development of therapeutics and early diagnosis of neurodegenerative diseases can be expensive and time-consuming, and thus the fundamental research should focus more on the early events in disease pathogenesis such as abnormal PPIs. Furthermore, in synucleinopathies and tauopathies, neurodegeneration is caused by multiple cascade-events, and thus the study should investigate the mechanisms involved in the multiple steps of cell-to-cell propagation of pathological form of aSyn and tau.In this thesis, aggregation, cellular release and uptake of aSyn and tau, which are the three critical steps involved in the cell-to-cell propagation, are thoroughly investigated. Understanding how protein interaction partners and genetic risk factors contribute to these steps could potentially reveal novel therapeutic targets and grant us knowledge of our physiological and pathological conditions.

3
2 Review of literature
Protein misfolding and aggregation 2.1
Protein misfolding is a cascade of events, starting from the natively unfolded protein and culminating in the mature fibril formation that is collectively termed aggregation. Protein misfolding associated with cellular dysfunction and cell death is a common molecular event in many neurodegenerative diseases. In physiological conditions, protein folds into its native state, which is the lowest energy state with hydrophobic residues inside the folded protein structure, after translation (Jahn and Radford, 2005). aSyn and tau are primarily translated in the cytosol, whereas secretory and membrane proteins are translated into the lumen of endoplasmic reticulum (ER). Many factors could impair this process, such as genetic mutations, environmental factors, etc. When misfolding occurs in certain proteins, the hydrophobic residues may become exposed forming unspecific interactions with other proteins, causing clustering and aggregation. In this process, a “healthy” protein may be trapped, or sequestered into a largely irreversible complex, known as aggregates, which may severely impair cellular functions (Ogen-Shtern et al., 2016).
To maintain cellular homeostasis during these situations as illustrated in Figure 1, molecular chaperones such as heat shock family proteins, can bind to the hydrophobic motifs of misfolded proteins in the cytosol, and prevent them from further interactions and aggregation (Chaari et al., 2013). The bound misfolded proteins may be conjugated with ubiquitin, which diverts them for proteasomal degradation (Balch et al., 2008), or direct to autosomal-lysosomal degradation pathway (Rubinsztein, 2006). However, when the formation of misfolded or aggregated proteins overruns the capacity of the clearance machinery, as the final attempt these proteins could be packed into β-sheet-like structures (also known as amyloids), which can further be assembled into amyloid fibrils reducing toxic interactions to the minimum (Eisenberg and Jucker, 2012). During pathogenesis process, certain protein monomers that may be natively unfolded oligomerize and gradually form large irreversible aggregates and fibrils. The solubility has been shown to decrease as the degree of aggregation increased in cells or animal models (Kothawala et al., 2012, Hirata-Fukae et al., 2009). However, the exact mechanisms involved are poorly understood as not all the intermediates involved in the aggregation pathway have been fully characterized(Kothawala et al., 2012, Hirata-Fukae et al., 2009). The misfolded secretory and membrane protein in ER may active unfold protein response, and are refolded by ER chaperones such as binding immunoglobulin protein and ER-localized J-protein (Schroder and Kaufman, 2005). Some of the misfolded proteins could re-translocate through ER membrane into the cytoplasm, and become ubiquitinated and are delivered for proteasomal degradation in a process called ER-associated degradation (Bernales et al., 2012). (Bernales et al., 2012) It has been suggested that the misfolding of cytosolic proteins such as aSyn and tau could enhance ER stress, elevate the rate of unfold protein response and disrupt the protein homeostasis in ER (Matus et al., 2011).
These insoluble protein deposits can take many forms and are reported in many neurodegenerative diseases. For example, in PD and other synucleinopathies, cytoplasmic inclusions occur in many regions of central nervous system (CNS) and autonomic system. These inclusions are also known as Lewy bodies, which are plaques primarily composed of misfolded aSyn and a variety of other components such as ubiquitin, cytochrome C, MAP-family proteins, tyrosine hydroxylase, superoxide dismutase, lipids, etc. ((Lashuel et al., 2013, Dennis Dickson, 2011, Braak and Del Tredici, 2008). In AD, plaques mainly composed of Aβ aggregates (also known as amyloid) and plaques primarily composed of hyperphosphorylated
4
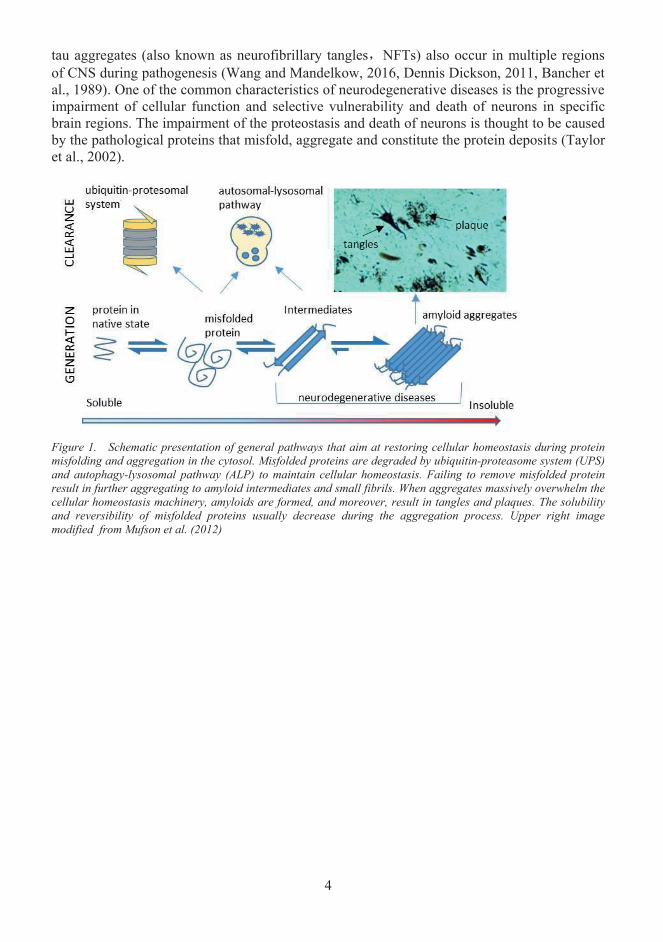
tau aggregates (also known as neurofibrillary tangles NFTs) also occur in multiple regions of CNS during pathogenesis (Wang and Mandelkow, 2016, Dennis Dickson, 2011, Bancher et al., 1989). One of the common characteristics of neurodegenerative diseases is the progressive impairment of cellular function and selective vulnerability and death of neurons in specificbrain regions. The impairment of the proteostasis and death of neurons is thought to be caused by the pathological proteins that misfold, aggregate and constitute the protein deposits (Taylor et al., 2002).
Figure 1. Schematic presentation of general pathways that aim at restoring cellular homeostasis during protein misfolding and aggregation in the cytosol. Misfolded proteins are degraded by ubiquitin-proteasome system (UPS) and autophagy-lysosomal pathway (ALP) to maintain cellular homeostasis. Failing to remove misfolded protein result in further aggregating to amyloid intermediates and small fibrils. When aggregates massively overwhelm the cellular homeostasis machinery, amyloids are formed, and moreover, result in tangles and plaques. The solubility and reversibility of misfolded proteins usually decrease during the aggregation process. Upper right image modified from Mufson et al. (2012)
5
α-synuclein2.2
2.2.1 Structure and biological functions
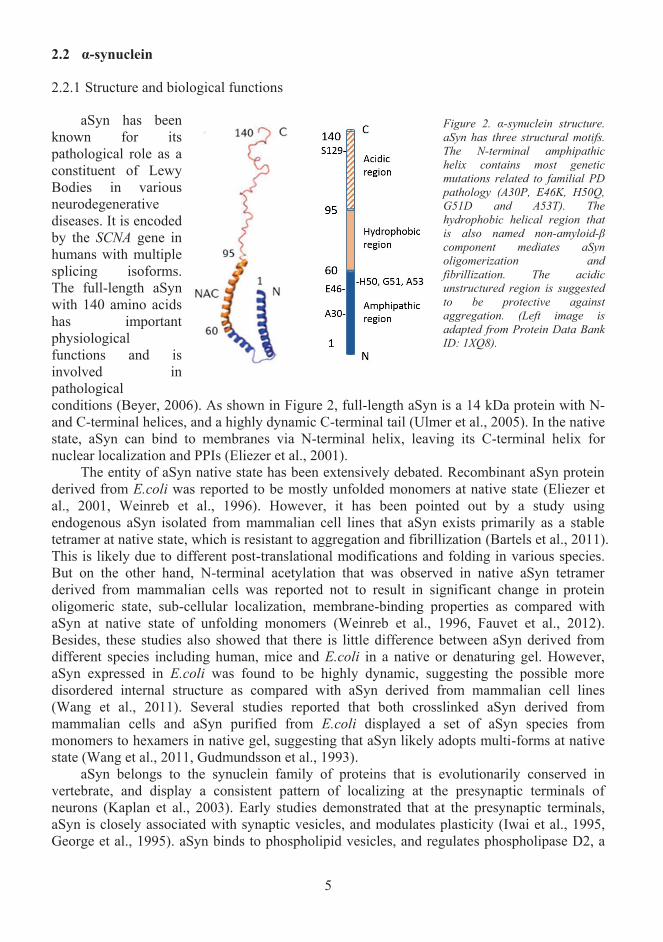
aSyn has been known for its pathological role as aconstituent of LewyBodies in various neurodegenerative diseases. It is encoded by the SCNA gene in humans with multiple splicing isoforms. The full-length aSyn with 140 amino acidshas important physiological functions and is involved in pathological conditions (Beyer, 2006). As shown in Figure 2, full-length aSyn is a 14 kDa protein with N-and C-terminal helices, and a highly dynamic C-terminal tail (Ulmer et al., 2005). In the native state, aSyn can bind to membranes via N-terminal helix, leaving its C-terminal helix for nuclear localization and PPIs (Eliezer et al., 2001).
The entity of aSyn native state has been extensively debated. Recombinant aSyn protein derived from E.coli was reported to be mostly unfolded monomers at native state (Eliezer et al., 2001, Weinreb et al., 1996). However, it has been pointed out by a study using endogenous aSyn isolated from mammalian cell lines that aSyn exists primarily as a stable tetramer at native state, which is resistant to aggregation and fibrillization (Bartels et al., 2011).This is likely due to different post-translational modifications and folding in various species. But on the other hand, N-terminal acetylation that was observed in native aSyn tetramer derived from mammalian cells was reported not to result in significant change in protein oligomeric state, sub-cellular localization, membrane-binding properties as compared with aSyn at native state of unfolding monomers (Weinreb et al., 1996, Fauvet et al., 2012).Besides, these studies also showed that there is little difference between aSyn derived fromdifferent species including human, mice and E.coli in a native or denaturing gel. However, aSyn expressed in E.coli was found to be highly dynamic, suggesting the possible more disordered internal structure as compared with aSyn derived from mammalian cell lines(Wang et al., 2011). Several studies reported that both crosslinked aSyn derived from mammalian cells and aSyn purified from E.coli displayed a set of aSyn species from monomers to hexamers in native gel, suggesting that aSyn likely adopts multi-forms at native state (Wang et al., 2011, Gudmundsson et al., 1993).
aSyn belongs to the synuclein family of proteins that is evolutionarily conserved in vertebrate, and display a consistent pattern of localizing at the presynaptic terminals of neurons (Kaplan et al., 2003). Early studies demonstrated that at the presynaptic terminals,aSyn is closely associated with synaptic vesicles, and modulates plasticity (Iwai et al., 1995, George et al., 1995). aSyn binds to phospholipid vesicles, and regulates phospholipase D2, a
Figure 2. α-synuclein structure. aSyn has three structural motifs.The N-terminal amphipathic helix contains most genetic mutations related to familial PD pathology (A30P, E46K, H50Q, G51D and A53T). The hydrophobic helical region that is also named non-amyloid-β component mediates aSyn oligomerization and fibrillization. The acidic unstructured region is suggested to be protective against aggregation. (Left image is adapted from Protein Data Bank ID: 1XQ8).
6
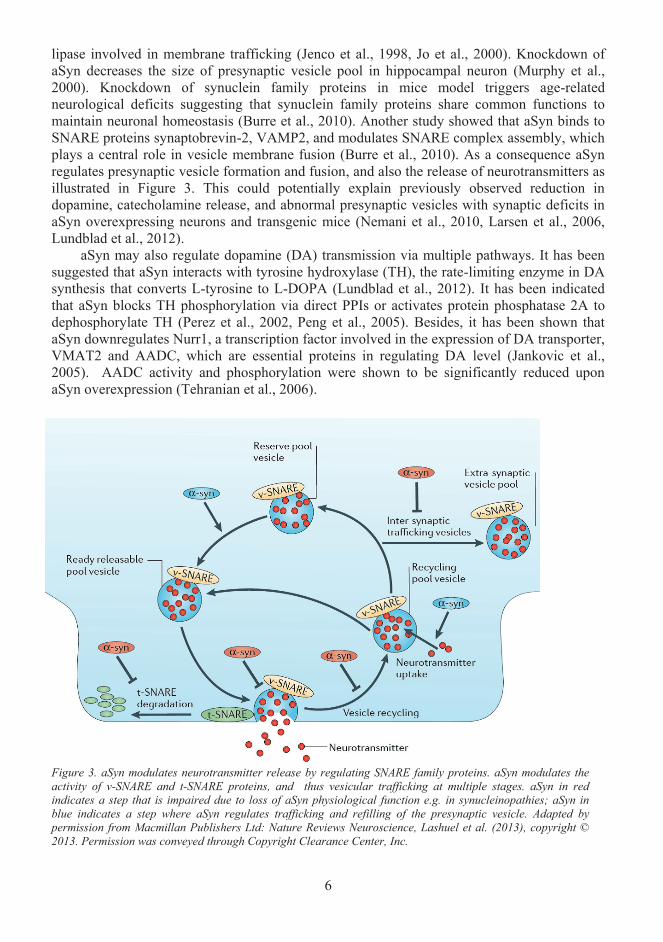
lipase involved in membrane trafficking (Jenco et al., 1998, Jo et al., 2000). Knockdown of aSyn decreases the size of presynaptic vesicle pool in hippocampal neuron (Murphy et al., 2000). Knockdown of synuclein family proteins in mice model triggers age-related neurological deficits suggesting that synuclein family proteins share common functions to maintain neuronal homeostasis (Burre et al., 2010). Another study showed that aSyn binds toSNARE proteins synaptobrevin-2, VAMP2, and modulates SNARE complex assembly, which plays a central role in vesicle membrane fusion (Burre et al., 2010). As a consequence aSyn regulates presynaptic vesicle formation and fusion, and also the release of neurotransmitters as illustrated in Figure 3. This could potentially explain previously observed reduction in dopamine, catecholamine release, and abnormal presynaptic vesicles with synaptic deficits inaSyn overexpressing neurons and transgenic mice (Nemani et al., 2010, Larsen et al., 2006, Lundblad et al., 2012).
aSyn may also regulate dopamine (DA) transmission via multiple pathways. It has been suggested that aSyn interacts with tyrosine hydroxylase (TH), the rate-limiting enzyme in DA synthesis that converts L-tyrosine to L-DOPA (Lundblad et al., 2012). It has been indicated that aSyn blocks TH phosphorylation via direct PPIs or activates protein phosphatase 2A todephosphorylate TH (Perez et al., 2002, Peng et al., 2005). Besides, it has been shown that aSyn downregulates Nurr1, a transcription factor involved in the expression of DA transporter, VMAT2 and AADC, which are essential proteins in regulating DA level (Jankovic et al., 2005). AADC activity and phosphorylation were shown to be significantly reduced upon aSyn overexpression (Tehranian et al., 2006).
Figure 3. aSyn modulates neurotransmitter release by regulating SNARE family proteins. aSyn modulates the activity of v-SNARE and t-SNARE proteins, and thus vesicular trafficking at multiple stages. aSyn in red indicates a step that is impaired due to loss of aSyn physiological function e.g. in synucleinopathies; aSyn in blue indicates a step where aSyn regulates trafficking and refilling of the presynaptic vesicle. Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Neuroscience, Lashuel et al. (2013), copyright ©2013. Permission was conveyed through Copyright Clearance Center, Inc.

7
2.2.2 α-synuclein aggregation
Misfolding of a protein is a consequence of failing to achieve or maintain the correct structure, which is generally considered as the prerequisite of aggregation. Misfolded aSynoligomerizes and aggregates following the general pattern explained in section 2.1(Eliezer et al., 2001, Weinreb et al., 1996, Bartels et al., 2011). A few studies including electronmicroscopy and immunoblot have demonstrated that aSyn (expressed in E.coli) can take different forms that exist in equilibrium with each other (Weinreb et al., 1996, Pountney et al., 2004, Volles and Lansbury, 2002, Horvath et al., 2012). These primarily include unfolded andfolded membrane-bound monomers, dimers or trimers, ring- or pore-shaped oligomers, and beta-sheet intermediates as shown in Figure 4. Various factors promote aSyn to adopt certain conformations, e.g. aSyn favour α-helical structure upon binding to phospholipid membrane(Davidson et al., 1998). However, it has been shown that aSyn derived from mammalian cells acquires the helical structure without need to bind to phospholipid membrane, suggesting a more stable internal structure as compared with E.coli derived aSyn (Bartels et al., 2011).Many factors contributing to aSyn aggregation are directly linked with aSyn structure. Some genetic mutations, gene duplication or truncation can alter aSyn structure to favour aggregation (Lashuel et al., 2013). Some external factors, such as phosphorylation, oxidative stress, fatty acid, proteolysis failure, can also modulate aSyn or aSyn-related pathways that enhance aSyn aggregation, accumulation or reduce aSyn clearance. Some of these morphologies of aSyn were suggested to be linked with its physiological or pathological roles that will be reviewed later in the thesis.
Figure 4 A schematic representation of aSyn species and aggregation pathways. aSyn takes forms of unfolded,folded or membrane-bound monomers, which have the potency to form higher order oligomers. Some membrane-bound aSyn monomers with α-helical structures could also form β-sheet intermediate oligomers. These oligomers including some amorphous ones can also transform into different forms, e.g. ring-shaped or membrane-embedded pore-like structures. At high monomer concentrations, they have a slow tendency to form fibril-like structuresirreversibly. During some pathological conditions, small fibrils further aggregate into amyloid fibrils, which gather together to form visible protein deposits in cells known as Lewy bodies. Modified from Lashuel et al. (2013).
2.2.2.1 α-synuclein structure and aggregation
As shown in Figure 2, the non-amyloid component fragment (NAC) of aSyn composed of 35 amino acid residues (61-95) composes central hydrophobic region, and it plays a major role in aSyn oligomerization and aggregation (Hashimoto et al., 2000). As explained in section 2.1, this hydrophobic motif may get exposed during misfolding, and initiate aggregation.

8
Mutation or deletion of this region significantly reduces aSyn filament assembly (Giasson et al., 2001). The C-terminal tail of aSyn was suggested to have a protective function against aggregation, and the residues 104, 105, 114 and 115 on the C-terminal tail are needed for this function (Murray et al., 2003). Even though the mechanism is not fully understood, the truncation of the C-terminal tail leads to increased aSyn aggregation in both experimental and pathological conditions (Li et al., 2005).
2.2.2.2 Genetic factors
The mutations that are associated with familial form of PD are mostly reported at the N-terminal helix of aSyn. A30P, E46K, H50Q, G51D and A53T mutation at the N-terminus have been proposed to likely associate with the acceleration of aSyn fibrillization and possibly elevate aSyn-mediated toxicity (Choi et al., 2004, Beyer, 2006, Lesage et al., 2013, Rutherford et al., 2014). These mutation-mediated effects were suggested to be a result of altered aSynsecondary structure (A30P, A53T), enhanced aSyn binding to phospholipids (E46K) or aSyn fibril formation that are more prone to activate caspase-3 related apoptotic pathways (G51D).In rare cases, duplication and triplication of SNCA gene lead to the autosomal dominant form of PD (Ibanez et al., 2004, Singleton et al., 2003, Chartier-Harlin et al., 2004). The mechanism of how multiplication of SNCA gene contributes to PD pathogenesis is poorly understood, but this is a strong indication that aSyn plays an important role in the development of the disease.
2.2.2.3 Post-translational modification
An early in vitro study demonstrated that aSyn S87 and S129 residues are constitutively phosphorylated (Okochi et al., 2000). However, it was later discovered that both pS87 (phosphorylation of S87) and pS129 are significantly increased in Lewy bodies from both patient samples and animal models (Paleologou et al., 2010, Fujiwara et al., 2002).Phosphorylation of S87 was found to increase aSyn conformational flexibility, which leads to reduced binding affinity to lipid membranes, and reduced potential to form fibrils (Oueslati et al., 2012). The same study also showed that hyperphosphorylation or mutation mimicking hyperphosphorylation of S87 (S87E) reduced aSyn aggregation and toxicity in a rodent model.
aSyn with pS129 is found in a minor population of total aSyn at physiological conditions but increase to over 90% of total aSyn in Lewy bodies context (Fujiwara et al., 2002, Anderson et al., 2006). Also, aSyn with pS129 hyperphosphorylation is also exclusively ubiquitinated in pathological conditions. As certain patterns of poly-ubiquitination of proteinsdirect them to proteasomal degradation, pS129 was thought to play a critical role in mediating aSyn proteasomal degradation (Anderson et al., 2006). For example, S129A mutation blocking S129 phosphorylation was found to reduce the rate of aSyn proteasomal and autophagy degradation (Tenreiro et al., 2014).
C-terminal truncation of aSyn is another common post-translational modification, and the amount of aSyn with C-terminal truncation was found to be significantly enriched in Lewy bodies (Li et al., 2005). The mechanism is poorly understood, but in vitro experiments demonstrated that C-terminally truncated aSyn induced significant wild-type aSyn aggregation upon co-expression in a template-directed manner, suggesting a prion-like behaviour of truncated aSyn species (Liu et al., 2005a). The site-directed expression of C-terminal truncated aSyn at nigral dopamine neurons was shown to markedly reduce dopamine levels in a transgenic mouse model, suggesting a failure to maintain aSyn regulated DA homeostasis as explained in section 2.2.1 (Daher et al., 2009).
Oxidative damage can happen to any protein typically resulting in oxidative modification of cysteine, tyrosine and methionine residues. aSyn has four tyrosine and methionine residues,

9
make it susceptible to oxidative stress. Nitration of tyrosine and oxidation of methionine are common oxidative modifications of aSyn found in Lewy bodies (Chavarria and Souza, 2013). Reactive oxygen species (ROS) and reactive nitrogen species from mitochondrial metabolism during cellular stress conditions were suggested to promote aSyn aggregation (Andersen, 2004). Application of rotenone, a drug that generates ROS by inhibiting mitochondrial complex I, induces aSyn aggregation in neurons (Chaves et al., 2010).
It was previously found that nitrated aSyn species are enriched in PD brain lysate (Giasson et al., 2000). The mechanism and significance of this phenomenon are poorly understood. Application of a nitrating agent to cell culture was reported to promote aSyn aggregation (Paxinou et al., 2001). However, controversial shreds of evidence showed that nitration reduced aSyn fibrillization by nitrating aSyn at tyrosine residues, and stabilized aSyn oligomers with improved folded secondary structure (Uversky et al., 2005).
Methionine oxidation of aSyn is thought to be primarily metal-catalysed (Chavarria and Souza, 2013). aSyn is most susceptible to copper oxidation because it has two copper binding sites at N- and C-termini, which can simultaneously accommodate Cu (I) and (II), and facilitate copper reduction (Binolfi et al., 2008, Jiang et al., 2007). The compound of aSyn-Cu (I) can further generate oxidative damage, and causing more aSyn to aggregate (Camponeschi et al., 2013). Other metal ions like iron were also shown to induce aSyn oxidation and aggregation when co-incubated with peroxide in vitro (Hashimoto et al., 1999a).
Di-tyrosine crosslink is another common oxidative modification found in aSyn structure both in vitro and in vivo (Souza et al., 2000, Pennathur et al., 1999). Cytochrome was shown to be important in the induction of aSyn di-tyrosine formation in the presence of peroxide (Ruf et al., 2008, Bayir et al., 2009). This cytochrome peroxidation system is thought to be a good model for studying aSyn aggregation in PD, as cytochrome is also found in Lewy bodies (Hashimoto et al., 1999b).
2.2.2.4 Lipids
There have been numerous studies showing that lipid binding promotes aSyn fibrillization, and aSyn aggregation is significantly enhanced in the presence of lipids both in vitro and in vivo (Rivers et al., 2008, Cole et al., 2002, Zhu et al., 2003, Lee et al., 2002, Sharon et al., 2003a). Lipid composition and level is associated with synucleinopathy progression in mouse brain, suggesting a relationship between aSyn aggregation and lipids during pathological conditions (Sharon et al., 2003b). Higher lipid concentrations protect aSyn from aggregation in dopaminergic neurons, whereas low lipid concentrations promote aSyn aggregation, suggesting that the ratio between aSyn and lipids may be the determinant of the aggregation (Zhu and Fink, 2003). It was proposed that lipid membrane may serve as a 2D scaffold, binding to membrane increases aSyn local concentration thus promotes aggregation (Aisenbrey et al., 2008). This is in agreement with the observation that even at low nanomolar range concentration, spontaneous aSyn aggregation can still be induced by the presence of lipid (Rabe et al., 2013). A recent study explained this phenomenon by demonstrating that local aSyn concentration is boosted by at least 1000-fold upon binding to lipid vesicle membrane (Galvagnion et al., 2015).
Several pathological mutations of aSyn are associated with changing its binding affinity to lipids. E46K is close to residue 39-45, which is the section of aSyn that penetrates to membranes the most. Hence the E46K mutant shows increased binding affinity to membranes, resulting in forming a higher local aSyn concentration that can lead to increased aggregation (Stockl et al., 2008). NRM data show that after binding to the phospholipid membrane, A30P and G51D mutated aSyn purified from E.coli adapt a membrane-bound conformation with the

10
hydrophobic core exposed instead of burying it in the membrane, causing membrane-induced aggregation with a mechanism explained in section 2.2.2.1 (Jensen et al., 1998, Ysselstein et al., 2015).
2.2.3 Clearance of aggregation
2.2.3.1 Ubiquitin-Proteasomal degradation
As mentioned in section 2.1, misfolded or aggregated proteins are generally degraded through the ubiquitin-proteasome system (UPS) and autophagy-lysosomal pathway (ALP) (Figure 1)(Rubinsztein, 2006). In UPS, protein is first conjugated with ubiquitin by a chain of reactions via enzyme E1, E2 and E3, then recognized by 26S proteasome, and is degraded by subunits 19S and 20S via proteolytic activity (Pickart and VanDemark, 2000, Jariel-Encontre et al., 2008). aSyn can be degraded via UPS and ubiquitinated aSyn and components from UPS are frequently found in Lewy bodies (Kuzuhara et al., 1988, Zhou et al., 2004, Bennett et al., 1999). In addition, the reduction of UPS subunit expression and UPS activity are seen throughout the development of synucleinopathies (Bukhatwa et al., 2010). Mutation of two subunits of UPS, UCH-L1 and parkin, are associated with the development of familial PD (Shimura et al., 2001, Leroy et al., 1998). Inhibition of UPS promotes accumulation aSyn thus aggregation in vitro and in vivo, suggesting that malfunction of UPS can be responsible for aSyn aggregation (McNaught and Olanow, 2006). Mutated forms of aSyn such as A30P and A53T, exhibit a slower turnover rate by UPS, and in some cases reduced UPS activity, suggesting that some forms of aSyn may be partially resistant to proteolytic degradation, and thus impair the UPS pathway (Bennett et al., 1999, Tanaka et al., 2001, Smith et al., 2005).Because protein needs to be unfolded by the 19S domain into a peptide chain to translocate into the active site of 20S catalytic domain, it is possible that high degree of aggregation could prohibit the unfolding of aSyn, thus impair the normal function of 19S domain (Shabek et al., 2012). It is currently thought that aSyn aggregation and UPS may exhibit a “Chicken or the egg” relationship: severe aSyn aggregation blocks and impairs UPS; and dysfunction of UPS promotes further aSyn aggregation (Ebrahimi-Fakhari et al., 2012).
2.2.3.2 Autosomal-lysosomal pathway
ALP consists of multiple pathways including macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy, all of which mediate proteins for lysosomal degradation. In CMA, proteins, including aSyn, with a KFERQ peptide sequence are recognized by chaperones like Hsp70 and are directed to a lysosomal receptor, such as LAMP-2A, which mediate protein translocation across the lysosomal membrane for degradation (Cuervo and Wong, 2014). Lack of this motif in aSyn results in reduced cellular turnover rate (Vogiatzi et al., 2008). A reduction of ALP activity and expression of ALP-components are seen throughout the development of synucleinopathies (Ebrahimi-Fakhari et al., 2012). Mice with a deficiency of cathepsin D, a lysosomal enzyme, showed significant accumulation of endogenous aSyn but not aSyn mRNA in brain neurons (Qiao et al., 2008). Knockdown of LAMP-2A was also reported to induce aSyn deposits in cells (Vogiatzi et al., 2008). These pieces of evidence suggest that the impairment of ALP results in a failure of aSyn clearance. On the other hand, both dopamine-modified and mutated aSyn such as A30P and A53T, display a higher affinity to LAMP-2A. These mutations interrupt LAMP-2A mediated translocation of aSyn by blocking the receptor, hence impairing CMA activity and increase aSyn accumulation (Cuervo et al., 2004, Xilouri et al., 2009, Martinez-Vicente et al., 2008). In these studies, some post-translational modifications in aSyn, such as pS129 and certain

11
nitrated forms, also showed to reduce lysosomal translocation of aSyn, suggesting that certain forms of aSyn could impair and block CMA. Hence the "Chicken or the egg" relationship described for UPS and aSyn could also apply to the case of ALP: certain aSyn aggregationmay overrun and impair ALP, and dysfunction of ALP promotes further aSyn aggregation.
In addition to CMA, macroautophagy is also involved in aSyn degradation. Unlike CMA that is highly selective and constantly active, macroautophagy is only effective under certain circumstances like cellular starvation or stress. In conditions like these, cellular components are engulfed via autophagosome for bulk degradation (Ebrahimi-Fakhari et al., 2012).Inhibition of autophagy by bafilomycin A1 mildly increases aSyn aggregation under an unstressed condition in cells, whereas induction of autophagy by rapamycin significantly boosts aSyn clearance (Webb et al., 2003). The expression of Beclin-1, a protein important for autophagy protein sorting, and LH3-II, an autophagosome membrane protein, were found to be increased in PD patients and mice with Lewy bodies, whereas mTOR, a negative regulator of autophagy, was reduced, showing an altered autophagy activity during synucleinopathies (Yu et al., 2009, Crews et al., 2010).
2.2.4 α-synuclein toxic species
aSyn aggregates frequently remain in Lewy bodies after the death of neuron. Hence it is commonly deduced that the aSyn aggregates are the toxic species that cause neuronal degeneration. It has been reported that S87E aSyn mutant aggregated less and exhibits less toxicity on DA neurons, whereas S87A showed the opposite (Oueslati et al., 2012). Direct application of aSyn fibrils to cells exhibits significantly higher toxicity than oligomers likely due to disruption of membrane permeability, suggesting that aSyn aggregates may exhibit a higher level of toxicity than soluble oligomers (Pieri et al., 2012). On the other hand, there is also controversial evidence. A wildly recognized view suggests that aSyn aggregation may be a protective process responding to the overwhelming pre-fibrillar aSyn oligomer that is toxic to the cells (Wan and Chung, 2012). For example, cellular toxicity of aSyn was shown to be reduced by directly or indirectly speeding up aSyn aggregation process in pharmacological approaches (Bodner et al., 2006, Outeiro et al., 2007). An in vivo study confirmed that aSyn species that aggregated faster were less toxic, whereas aSyn species that stayed as soluble oligomers caused the most damage to neurons (Winner et al., 2011). To make this more puzzling, recent study points to a direction that the toxicity of aSyn is highly dependent on various properties of different aSyn strains, which could result in distinct inclusion structure and pathological phenotypes (Peelaerts et al., 2015). In this study, a structurally defined aSynoligomer, ribbon and fibril assemblies were injected to mouse. aSyn fibrils caused the most toxicity, oligomers the least, and ribbons resulted in a phenotype resemble the pathological condition of PD the most. Another study on oligomer related toxicity demonstrated that by using different oligomerization protocol, different strains of aSyn oligomers were induced, likely associated with distinct mechanisms of disrupting cellular homeostasis and aggregation in as strain-specific manner (Danzer et al., 2007). It was revealed by an electron microscopy (EM) study that even the well-characterised genetic mutations such as A30P and A53T, tend to form different protofibrils strains under different conditions (Lashuel et al., 2002b). Like other neurodegenerative proteins, such as Tau, prion protein and Aβ, the strain properties might play a critical role in modulating the process of aSyn aggregation, intermediate structures, final aggregates and the toxicity. Currently little is known about the underlying molecular mechanisms. Details of how strains of aSyn play a role in the cell-to-cell propagation of synucleinopathy will be reviewed under a different section below.

12
Despite the ongoing debate on aSyn toxic species, the intracellular locations of abnormal aSyn species may potentially implicate the mechanisms of aSyn to exhibit toxicity. aSyn annular protofibrils were shown to bind to cell membranes, obtained an octameric structure that shares homology to bacterial pore-forming toxins, permeabilized membranes causing an influx of calcium (Ding et al., 2002, Tsigelny et al., 2012, Kim et al., 2009a). aSyn was also reported to bind and permeabilize the outer membrane of mitochondria, and thus triggered the release of cytochrome c, which is an apoptotic factor, and promoted oxidative stress and apoptosis (Hashimoto et al., 2004, Parihar et al., 2008). The A53T mutant of aSyn was shown to interact with ER chaperones, and sensitized cells to ER stress and disrupted ER-Golgi transport upon overexpression (Heller et al., 2012, Cooper et al., 2006). Apart from these gain of toxic functions, loss of aSyn physiological function could also result in toxicity. As reviewed in section 2.2.1, the loss of physiological function of aSyn in pathological conditions would result in the impairment of DA homeostasis and pre-synaptic vesicular trafficking. For example, the abnormal pre-synaptic vesicles morphology and impaired neurotransmitter release, which lead to the synaptic deficit, have been reported in aSyn overexpression transgenic mice (Scott et al., 2010).
2.2.5 Prolyl oligopeptidase
Prolyl oligopeptidase (PREP) is one of the evolutionarily conserved serine peptidases, which cleaves peptides with less than 30 amino acids at the C-terminal side of proline residues(Rawlings and Barrett, 1994). PREP consists of a hydrolytic and a β-propeller domain, arranged in a “PacMan” shape with the active site located between the two domains (Fulop et al., 2000). PREP is widely expressed in the body tissues such as brain, liver, lung and spleen,and is likely involved in the hydrolysis of substance P, angiotensin, thyrotropin and arginine-vasopressin (Myohanen et al., 2012b, Garcia-Horsman et al., 2007, Mannisto et al., 2007). A Recent study suggested that PREP also regulates pancreatic insulin and glucagon secretion in mice (Kim et al., 2014).
PREP has been implicated in neuronal function(s). For example, PREP has been suggested to modulate the function of GABAergic and cholinergic neurotransmitter release, and thus is involved in excitatory and inhibitory signalling pathway in the brain (Peltonen et al., 2011). PREP inhibition benefits the spatial memory in mice (Jalkanen et al., 2007). PREP deficiency impairs synaptic plasticity in mice (Hofling et al., 2016). PREP regulates inositol metabolism that serves as an important drug target in bipolar affective disorder (Williams et al., 2002). Also, PREP activity in brain changes during normal aging, but also in AD and PD (Myohanen et al., 2009). For example, hippocampal PREP activity is increased in the brains of aged mice and mice with AD as compared with young or wild-type ones (Rossner et al., 2005).PREP was found to co-localize with aSyn and tau in AD and PD patient brain samples (Hannula et al., 2013). PREP was reported to enhance aSyn aggregation in vitro, which can be blocked by PREP inhibitors (Brandt et al., 2008). Some studies demonstrated that PREP inhibitor benefited learning and memory by modulating neuropeptide level, whereas some other studies showed the opposite (Shishido et al., 1998, Toide et al., 1997, Morain et al., 2002, Jalkanen et al., 2007). KYP-2047, which is a small-molecule inhibitor of PREP, was shown to reduce aSyn-mediated cytotoxicity in responding to oxidative stress and aSyn oligomerization in in vitro and in vivo studies (Dokleja et al., 2014, Myohanen et al., 2012a). KYP-2047 treatment in transgenic mice and cells of PD model was also shown to increase cell and neuron viability with reduced high-molecular-weight oligomeric aSyn (Savolainen et al.,2014). In the same study, it was also shown that KYP-2047-mediated inhibition of PREP also

13
significantly increased autophagy activity, and thus suggesting that PREP inhibition could elevate autophagy activity and enhance protein clearance. It is currently not known if PREP has a direct effect on aSyn even though they have been reported to co-localize in PD-relatedinclusions (Brandt et al., 2008). In the same study, aSyn seemed not to be a substrate of PREP enzyme activity as co-incubation of aSyn and PREP in vitro did not result in truncation of full-length aSyn, which contains five Proline residues that may serve as potential hydrolytic sites for PREP.
Tau2.3
2.3.1 Structure and biological functions
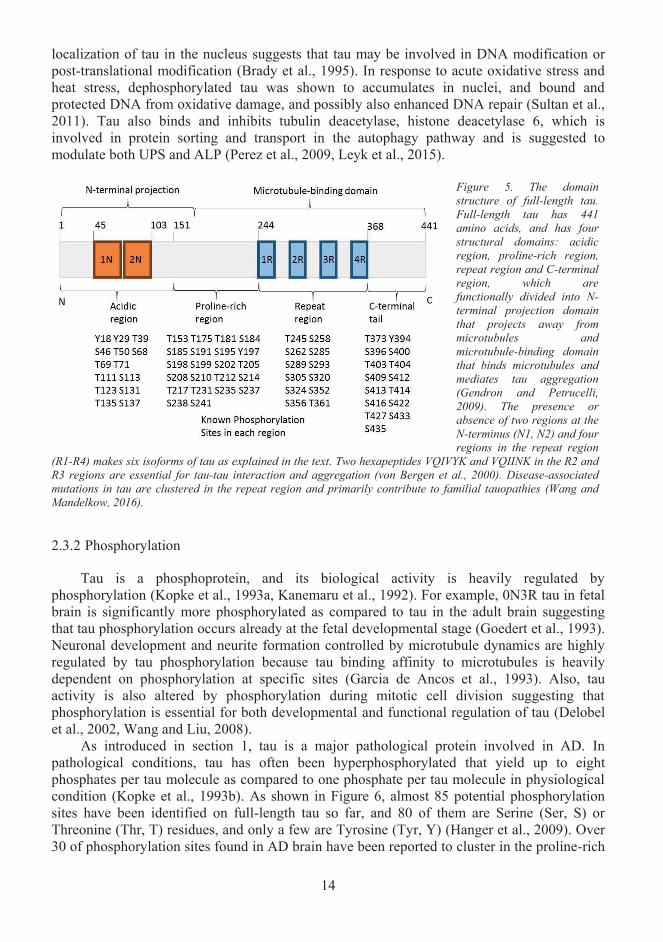
Tau was first discovered and known for its physiological role in regulating microtubule assembly and stability (Weingarten et al., 1975). It belongs to microtubule-binding protein (MAP) family and is encoded by 16 exons of MAPT gene on chromosome locus 17q21 (Neveet al., 1986). As shown in Figure 5, the full-length human tau protein has 441 amino acids.Tau protein is structurally divided into the acidic region, the proline-rich region, themicrotubule-binding repeat region and C-terminal region (Mandelkow et al., 1996). The four regions constitute two functional domains: the N-terminal projection domain (N-domain) and the microtubule-binding domain (R-domain). Depending on the presence or absence of 1N, 2N or 2R regions, there are six tau isoforms found in man: 0N3R (1R, 3R and 4R only), 1N3R (1N, 1R, 3R, 4R), 2N3R, 0N4R, 1N4R (1N, 1R-4R) and 2N4R (Gendron and Petrucelli, 2009).N-terminal projection domain is mostly acidic, hence negatively charged, bends and projects from the surface of microtubule as shown in Figure 7 (Hirokawa et al., 1988). This domain interacts with many known proteins, and was suggested to modulate cellular signalling by interacting with several Src-family kinases, phospholipase C-γ, growth factor receptor-bound protein 2, phosphatidylinositol bisphosphate, Pin1. (Reynolds et al., 2008, Surridge and Burns, 1994, Flanagan et al., 1997, Morris et al., 2011, Lu and Kosik, 2001). The microtubule-binding domain functions primarily through the four highly conserved repetitive motives (R1-R4) that regulate microtubule polymerization (Simic et al., 2016). The adult tau isoforms with R1-R4 repeats show significantly higher efficacy in promoting microtubule assembly than fetal tau isoform 0N3R, which lacks R2 region, highlighting the importance of R2 region inregulating microtubule polymerization (Simic et al., 2003). In vitro study further demonstrated 4R tau has a significantly higher binding affinity to microtubules than 3R tau (Lu and Kosik, 2001).
Tau is widely expressed such as in submandibular gland, sigmoid colon, liver, scalp, and abdominal skin(Dugger et al., 2016), and is most abundant in brain, especially in neuron axonsand somatodendritic compartment (Gu et al., 1996, Dugger et al., 2016). The primary function of tau, regulating microtubule assembly and stability, is highly compensated by other MAP family proteins. Silencing tau in neuronal culture does not trigger neurodegeneration or prevent axon growth (Qiang et al., 2006). It is suggested that MAP1B rather than tau is more crucial in regulating microtubule stability (Takei et al., 2000). In neurons, tau was shown to interact with many proteins including Fyn kinase, post-synaptic density 95 and NMDA receptor, which are highly expressed in synaptic terminals (Klein et al., 2002, Lee et al., 1998, Mondragon-Rodriguez et al., 2012). Silencing tau in mice impairs long-term potentiation suggesting a role of tau in NMDA receptor-dependent memory (Ahmed et al., 2014).Overexpression of tau in cells induces tau secretion in vesicles-bound form suggesting certain homeostatic cell mechanisms regulate the intracellular level of tau (Simon et al., 2012a). The
14
localization of tau in the nucleus suggests that tau may be involved in DNA modification or post-translational modification (Brady et al., 1995). In response to acute oxidative stress and heat stress, dephosphorylated tau was shown to accumulates in nuclei, and bound and protected DNA from oxidative damage, and possibly also enhanced DNA repair (Sultan et al., 2011). Tau also binds and inhibits tubulin deacetylase, histone deacetylase 6, which is involved in protein sorting and transport in the autophagy pathway and is suggested to modulate both UPS and ALP (Perez et al., 2009, Leyk et al., 2015).
Figure 5. The domain structure of full-length tau. Full-length tau has 441 amino acids, and has four structural domains: acidic region, proline-rich region, repeat region and C-terminal region, which are functionally divided into N-terminal projection domain that projects away from microtubules andmicrotubule-binding domain that binds microtubules and mediates tau aggregation (Gendron and Petrucelli, 2009). The presence or absence of two regions at theN-terminus (N1, N2) and four regions in the repeat region
(R1-R4) makes six isoforms of tau as explained in the text. Two hexapeptides VQIVYK and VQIINK in the R2 and R3 regions are essential for tau-tau interaction and aggregation (von Bergen et al., 2000). Disease-associatedmutations in tau are clustered in the repeat region and primarily contribute to familial tauopathies (Wang and Mandelkow, 2016).
2.3.2 Phosphorylation
Tau is a phosphoprotein, and its biological activity is heavily regulated by phosphorylation (Kopke et al., 1993a, Kanemaru et al., 1992). For example, 0N3R tau in fetal brain is significantly more phosphorylated as compared to tau in the adult brain suggesting that tau phosphorylation occurs already at the fetal developmental stage (Goedert et al., 1993).Neuronal development and neurite formation controlled by microtubule dynamics are highly regulated by tau phosphorylation because tau binding affinity to microtubules is heavily dependent on phosphorylation at specific sites (Garcia de Ancos et al., 1993). Also, tau activity is also altered by phosphorylation during mitotic cell division suggesting that phosphorylation is essential for both developmental and functional regulation of tau (Delobel et al., 2002, Wang and Liu, 2008).
As introduced in section 1, tau is a major pathological protein involved in AD. Inpathological conditions, tau has often been hyperphosphorylated that yield up to eight phosphates per tau molecule as compared to one phosphate per tau molecule in physiological condition (Kopke et al., 1993b). As shown in Figure 6, almost 85 potential phosphorylation sites have been identified on full-length tau so far, and 80 of them are Serine (Ser, S) or Threonine (Thr, T) residues, and only a few are Tyrosine (Tyr, Y) (Hanger et al., 2009). Over 30 of phosphorylation sites found in AD brain have been reported to cluster in the proline-rich

15
and C-terminal regions (Wang and Liu, 2008). Among them, 17 Thr or Ser sites prior to Proline (Pro) are found abnormally hyperphosphorylated in AD brain and are considered most attractive regarding pathology (Wang and Mandelkow, 2016). A few widely used antibodies such as AT8, AT100, AT180, 12E8 and PHF-1 have been developed to specifically recognize them (the identified epitopes are as shown in Figure 6).
2.3.3 Kinases and phosphatases
Tau phosphorylation homeostasis is regulated by multiple protein kinases and phosphatases (Liu et al., 2007). Depending on motif-specificity, kinases are divided intoproline-directed (phosphorylate Thr or Ser sites prior to Pro) and non-proline-directed protein kinases (Meraz-Rios et al., 2010). The major proline-directed protein kinases related to tau include glycogen synthase kinase-3 (GSK-3), cyclin-dependent kinase 5 (CDK5), dual-specificity tyrosine-phosphorylation-related kinase 1A (DYRK1A) and mitogen-activated protein kinase family (MAPKs) (Wang and Liu, 2008, Martin et al., 2013). All of them, especially GSK-3β (one of the two GSK-3 proteins), are strongly associated with tau phosphorylation in both physiological and pathological conditions(Wang and Liu, 2008, Martin et al., 2013).
GSK-3, in general, regulates multiple cellular signalling, metabolic and structural proteins functions (Grimes and Jope, 2001). GSK-3, especially the β form, is one of the most important tau kinases that phosphorylate tau at over 30 sites known to regulate tau physiological functions (Cho and Johnson, 2003, Ishiguro et al., 1993, Grimes and Jope, 2001).In addition, GSK-3β also impairs tau binding affinity to microtubule by hyperphosphorylating tau in pathological conditions. CDK5 is involved in maintaining cellular function and development of CNS (Lew et al., 1994). Prolonged activity of CDK5 under stress conditions
Figure 6. Tau phosphorylation sites. Tau phosphorylation sites showed in the figure are derived from experiments, theories and hypothesis. Tau phosphorylation sites in physiological and pathological conditions are indicated by different colours. The commonly used antibodies recognize pathologically relevant phosphoepitopes are shown in purple. Adopted from Simic et al. (2016) with permission under Creative Commons Attribution License, copyright (2017).

16
lead to tau hyperphosphorylation, cytoskeletal abnormality and neuron degeneration (Patrick et al., 1999). DYRK1A was suggested to play a major role in neuronal growth and development (Duchon and Herault, 2016). In addition, DYRK1A encoding gene is known to localize on chromosome 21, trisomy of which causes Down syndrome (DS) (Wiseman et al., 2009). As it was reported that most DS patient developed AD-like dementia by the age of 40, the fact the DYRK1A regulates tau phosphorylation become interesting in term of connected pathologies (Park et al., 2009). MAPKs are a family of kinases including MAPK, MAPK2/3, extracellular signal–regulated kinases (ERK), Jun NH2-terminal kinase (JNK), p38 etc, and are primarily involved in signalling transduction and several cellular functions such as cell growth, apoptosis and proliferation (Schaeffer and Weber, 1999, Munoz and Ammit, 2010).MAPKs-mediated pathways have been suggested to be involved in several types of cancers, and also AD, PD and amyotrophic lateral sclerosis (ALS) (Kim and Choi, 2010). Several members of MAPKs such as JNK, ERK1/2 and p38 were reported to be related to abnormal tau phosphorylation in pathological conditions (Churcher, 2006).
Non-proline-directed protein kinases that phosphorylate tau include casein kinase 1(CK1),protein kinase C (PKC), calmodulin-dependent protein kinase II (CaMKII), Fyn, and cyclic AMP-dependent protein kinase (PKA), all of which apart from Fyn phosphorylate tau at Ser/Thr sites (Meraz-Rios et al., 2010, Wang and Liu, 2008). All of these kinases have beenshown to be related to abnormal phosphorylation of tau in pathological conditions (Kuret et al., 1997, Yamamoto et al., 2002, Liu et al., 2003, Zhang et al., 2006, Lee et al., 2004). PKA isprimarily involved in cAMP-mediated cellular signalling pathways and was also shown tophosphorylate GSK-3 and CDK5 in addition to tau (Wang et al., 2007a). Fyn is one of the very few kinases phosphorylate tau at Tyr residues, and is involved in cell signalling and several neuronal functions (Resh, 1998). In addition, Fyn has been reported to hyperphosphorylate tau in an Aβ-dependent manner (Williamson et al., 2002).
In mammalian cells, the protein phosphatase (PP) 1, PP5, PP2A, PP2B and PP2C are the major phosphatases that dephosphorylate tau at specific Ser/Thr residues (Liu et al., 2005b). It has been reported that PP2A activity is decreased in AD brains while the endogenous inhibitor of PP2A, I1
PP2A and I2PP2A level are increased (Tanimukai et al., 2005). In addition, PP2A,
I1PP2A and I2
PP2A colocalize with tau aggregates in pathological inclusions, suggesting that PP2A is closely linked with abnormal tau phosphorylation in pathological conditions, and thusmay serve as a drug target for AD (Liu et al., 2005b, Tian and Wang, 2002).
2.3.4 Other post-translational modifications
Apart from phosphorylation, other important tau post-translational modifications includetruncation, glycosylation, acetylation, nitration, methylation and ubiquitinations (Gong et al., 2005). In glycosylation, proteins are covalently linked with oligosaccharides in N- or O-glycosidic bond. Paired helical filaments (PHF) has been reported to consist of tau with N-glycosylation modification, which was suggested to affect tau phosphorylation via kinases-and phosphatases-mediated pathways (Liu et al., 2002). O-glycosylation of tau was reported to take place at Ser or Thr residues that are important residues for tau phosphorylation during pathogenesis of AD as mentioned above, and thus was suggested to be protective inpathological conditions, possibly by competing Thr/Ser sites with proline-directed kinases against hyperphosphorylation (Morris et al., 2015, Liu et al., 2004). Tau poly-ubiquitination is linked with tau clearance and is discussed in a different section below. Hyper-acetylation of tau has been reported to inhibit ubiquitin-mediated tau degradation, thus enhances accumulation of hyperphosphorylated tau, and moreover induces toxicity in pathological conditions (Min et al., 2010). Nitrations of tau were reported to occur on tyrosine residues 18,

17
29, 197 and 394 in NFTs, and thus were suggested to have a link to tau fibrillization(Reynolds et al., 2005). Methylation of tau has been observed on lysine residues, but the exact functional relevance is yet not fully understood (Thomas et al., 2012). Tau truncations, especially C-terminal truncations, were suggested to contribute to tau aggregation pathways, and are discussed below.
2.3.5 Tau aggregation
It is widely recognized that hyperphosphorylation is a significant contributor and regulator to tau aggregation pathway. In physiological condition, tau binds and stabilizes microtubules, and its phosphorylation homeostasis is maintained by transient reversible phosphorylation via kinases and phosphatases as shown in Figure 7. However, abnormal phosphorylation of tau occurs when the phosphorylation/dephosphorylation cycle is disturbed resulting in accumulation of hyperphosphorylate tau in cells. Certain patterns of hyperphosphorylation lead to reduced tau binding affinity to microtubules and destabilize microtubules. Free tau molecules could further oligomerize, forming fibrils known as paired helical filaments (PHF). Additional aggregation of PHFs forms NTFs that are commonly observed after the death of neurons in tauopathies.
Tau aggregation follows a similar pattern of nucleation-elongation mechanism as aSyn. Several factors contributing to the aggregation process are shared between aSyn and tau, such as structural characteristics favouring aggregation, truncations, post-translational modifications and genetic mutations (Wang and Mandelkow, 2016).
2.3.5.1 β-sheet intermediate
Despite the fact that natively unfolded tau monomers are structurally highly dynamic, tau aggregates are generally packed into well-ordered β-sheet-like structures (Simic et al., 2016).Two hexapeptides VQIINK and VQIVYK on R2 and R3 motives of tau (Figure 5) are
Figure 7. A schematic representation of tau aggregation pathway. In physiological conditions, tau promotes microtubule assembly and stabilization by binding to microtubules via the microtubule-binding domain. Homeostasis of tau phosphorylation is maintained by kinases and phosphatases. In pathological conditions, tau can be hyperphosphorylated due to increased kinase activity or decreased phosphatase activity, and detaches from microtubules. This causes microtubules to disassemble and loss physiological functions. Free soluble tau with hyperphosphorylation and other post-translational modifications such as truncations tend to aggregate to paired-helical filaments (PHF), which further aggregate into insoluble neurofibrillary tangles (NFT) that are commonly observed in the clinical situations of tauopathies. Right top image is modified from Clavaguera et al. (2009).

18
essential for the formation of “zipper-like” interdigitated β-sheet structures, and thus play a major role in mediating tau oligomerization and aggregation (von Bergen et al., 2000). Certain mutations, e.g. mutations at K280 and P301, which disrupt the structure of paired β-sheet, tend to have a significant impact on tau aggregation. For example addition of proline after K280 significantly reduces the tendency of tau aggregation, whereas certain mutations like deletionof K280 or P301L strongly promote tau aggregation in vitro (Khlistunova et al., 2006). Most of these mutations were originally identified in patients who have familial dementia (Rizzu et al., 1999, Iijima et al., 1999, Hutton et al., 1998). It was suggested that the aggregation in the manner of “steric zipper” is a characteristic shared between over 30 different fibril-forming-prone proteins including amyloid-β and aSyn, suggesting that amyloid diseases share common mechanistic features at the molecular level during aggregation (Sawaya et al., 2007).
2.3.5.2 Truncation
Truncation is one of the post-translational modifications that are closely linked with the aggregation property of tau. There are multiple potential cleavage sites on full-length tau as shown in Figure 8. Truncations of tau lead to many fragments containing the microtubule-binding region that are highly prone to aggregation (Wang and Mandelkow, 2016).Inoculation of tau151-391 that was isolated from PHFs via pronase was shown to induce wildtype tau aggregation and neurofibrillary pathology in a transgenic mouse model (Wischiket al., 1988b, Zilka et al., 2006). Inoculation of tau1-421 that is cleaved by caspase 3 was also shown to form NTFs in a transgenic mouse model (de Calignon et al., 2010). Lysosomal asparagine endopeptidases cleave tau at Asn255 or Asn368, and the resulting tau1-368 is not only prone to aggregation but also has compromised ability to bind and to stabilize microtubules (Zhang et al., 2014). In the cellular model of tauopathy, deletion of P280 triggers a stepwise proteolysis of tau: starting from N-terminal cleavage by thrombin-like protease preceding to C-terminal cleavage by cathepsin I, resulting in fragments F1 (Tau257-441), F2 (Tau257-363), F3 (Tau257-360) (Wang et al., 2007b). The F3 fragment was found to aggregate rapidly and can form the core of AD-related PHFs (Wang et al., 2009b, Simic et al., 2016).
2.3.5.3 The role of phosphorylation in tau aggregation
In 1990 M. Goedert and colleagues found all isoforms of tau are hyperphosphorylated within PHFs, and after that, it has been widely accepted that PHFs consist of mostly hyperphosphorylated tau species (Goedert and Jakes, 1990). In addition, since tau hyperphosphorylation has long been observed in tau aggregates from patients or animal models, it has been reasoned that phosphorylation is responsible for driving tau aggregation (Braak et al., 1994b). There is evidence support this theory. For example, hyperphosphorylated tau isolated from AD patients’ brain self-assembles into PHFs (Alonso et al., 2001), but it is not known if other co-factors or modifications are involved. On the other hand, AD-like tau hyperphosphorylation without aggregation is observed in hibernating animals or anaesthesia-induced hypothermia indicating that hyperphosphorylation may not be sufficient to trigger tau aggregation alone (Arendt et al., 2003, Planel et al., 2007). Some phosphorylation events, e.g. Ser214/262 even show protection against aggregation (Schneider et al., 1999). No evidence has yet proved that phosphorylation alone accounts for tau aggregation. Many contradictory conclusions are derived probably due to the complexity and a significant number of tau phosphorylation sites (Simic et al., 2016). But indeed tau hyperphosphorylation is linked to aggregation, for example, it has been proposed that hyperphosphorylation of tau could induce tau detachment from microtubules that may potentially accelerate and feedforward the aggregation process (Wang and Mandelkow, 2016).
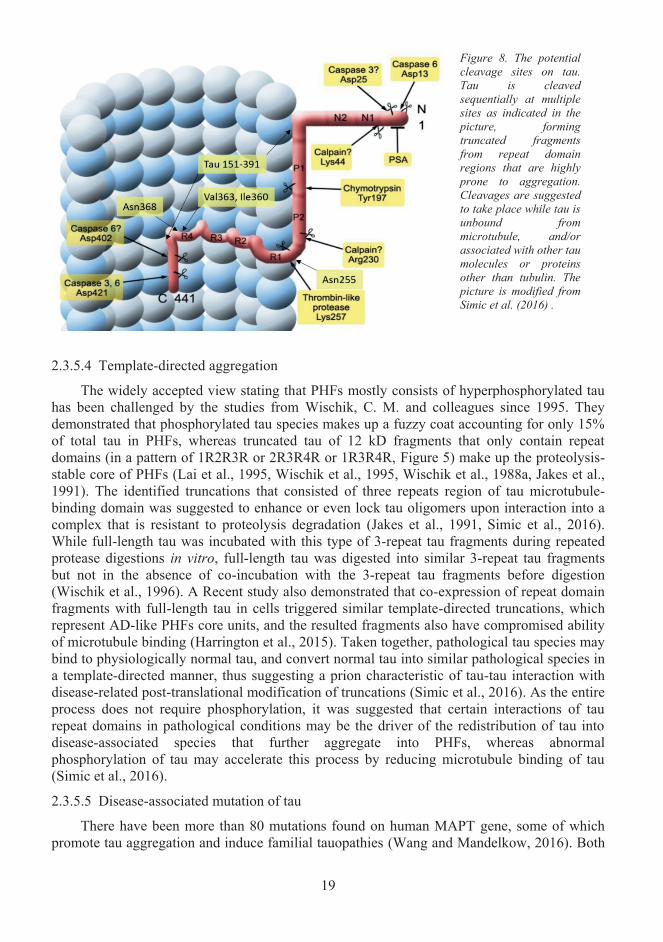
19
2.3.5.4 Template-directed aggregation
The widely accepted view stating that PHFs mostly consists of hyperphosphorylated tau has been challenged by the studies from Wischik, C. M. and colleagues since 1995. They demonstrated that phosphorylated tau species makes up a fuzzy coat accounting for only 15% of total tau in PHFs, whereas truncated tau of 12 kD fragments that only contain repeat domains (in a pattern of 1R2R3R or 2R3R4R or 1R3R4R, Figure 5) make up the proteolysis-stable core of PHFs (Lai et al., 1995, Wischik et al., 1995, Wischik et al., 1988a, Jakes et al., 1991). The identified truncations that consisted of three repeats region of tau microtubule-binding domain was suggested to enhance or even lock tau oligomers upon interaction into acomplex that is resistant to proteolysis degradation (Jakes et al., 1991, Simic et al., 2016).While full-length tau was incubated with this type of 3-repeat tau fragments during repeatedprotease digestions in vitro, full-length tau was digested into similar 3-repeat tau fragmentsbut not in the absence of co-incubation with the 3-repeat tau fragments before digestion (Wischik et al., 1996). A Recent study also demonstrated that co-expression of repeat domain fragments with full-length tau in cells triggered similar template-directed truncations, which represent AD-like PHFs core units, and the resulted fragments also have compromised ability of microtubule binding (Harrington et al., 2015). Taken together, pathological tau species may bind to physiologically normal tau, and convert normal tau into similar pathological species in a template-directed manner, thus suggesting a prion characteristic of tau-tau interaction with disease-related post-translational modification of truncations (Simic et al., 2016). As the entire process does not require phosphorylation, it was suggested that certain interactions of tau repeat domains in pathological conditions may be the driver of the redistribution of tau into disease-associated species that further aggregate into PHFs, whereas abnormal phosphorylation of tau may accelerate this process by reducing microtubule binding of tau (Simic et al., 2016).
2.3.5.5 Disease-associated mutation of tau
There have been more than 80 mutations found on human MAPT gene, some of which promote tau aggregation and induce familial tauopathies (Wang and Mandelkow, 2016). Both
Figure 8. The potential cleavage sites on tau. Tau is cleaved sequentially at multiple sites as indicated in the picture, formingtruncated fragmentsfrom repeat domainregions that are highly prone to aggregation.Cleavages are suggested to take place while tau is unbound from microtubule, and/or associated with other tau molecules or proteins other than tubulin. The picture is modified fromSimic et al. (2016) .

20
missense and splicing mutations have been reported previously. As shown in Figure 5, most of the missense mutations are clustered in or near the repeat domains and have a tendency to reduce tau binding affinity to microtubules and to increase tau aggregation (Barghorn et al., 2000, Hong et al., 1998). There are also mutations far from the repeat domain regions exhibit a similar effect on tau. For example, it was reported that progressive supranuclear palsy (PSP)-related mutation A152T located far from the repeat domains reduced tau binding to microtubule and tau-mediated microtubule assembly (Coppola et al., 2012). R5H and R5L mutations, which disrupt tau binding to p150 that is a dynactin subunit acts as a cofactor of microtubule motor dynein, reduced microtubule-mediated axonal transport (Magnani et al., 2007). Splicing mutations are mostly clustered in or near intron 10 (N279K, L284L, ∆N296,N296N, N296H, S305N, S305S, etc.), and result in increased inclusion of exon 10, which encodes for the R2 region, thus alter the 1:1 physiological ratio of R4:R3 tau isoforms (Wang and Mandelkow, 2016, Ballatore et al., 2007). 0N4R tau was shown to have significantly higher efficacy in promoting microtubule assembly than 0N3R tau that lacks R2 region(seeFigure 5), highlighting the importance of R2 region in regulating tau-mediate microtubule assembly and stability (Simic et al., 2003). In addition, R2 region of tau contains the VQIINKhexapeptide (Figure 5) that is essential for tau oligomerization and aggregation (von Bergen et al., 2000). Taken together, this evidence could potentially offer an explanation of the increased R4: R3 ratio as a characteristic of tauopathies in frontotemporal dementias (Hong et al., 1998).
2.3.6 Genetic risk factors of Alzheimer’s disease
AD is one of age-related dementia characterized by progressive neurodegeneration in certain connected brain regions, which correlates with the gradual decline of cognitive function (Dennis Dickson, 2011). Study of AD brain at autopsy revealed numerous senile plaques and deposits in blood vessels, which primarily consist of Aβ and NFTs (Braak and Braak, 1991). The etiology of AD is currently incompletely understood, but many studies have made the consensus that the disease progression and onset are closely linked with genetic factors. The misfolding and aggregation of tau play essential roles in the pathogenic pathway of AD. There are many genetic risk factors are potentially associated with the increased chance of developing AD. Currently, there lacks direct connection between some of these genetic risk factors and tauopathies observed in AD. Part of this thesis work demonstrates a previously unrecognized mechanism of a protein that is encoded by a late-onset Alzheimer’s disease (LOAD) susceptibility gene FRMD4A, modulating the cellular propagation of tau.
Occasionally before the age of 65, patients with rare mutations in APP and PSEN1/2genes (also known as early-onset AD risk genes) develop familial AD (Cacace et al., 2016).However, over 95% AD cases are the late-onset (LOAD) type. A large number (>700) of risk genes have been identified for LOAD using genome-wide association studies (GWAS) and candidate gene approaches, which correlate the frequency of the risk variants of certain alleles with diseases in defined groups (Bertram et al., 2010, Chouraki and Seshadri, 2014). GWAScovers entire genome with known polymorphisms, and screens potential association of SNPs to phenotypes in a manner of hypothesis-free. Thus GWAS is statistically powerful and generates massive data. In GWAS discovery stage, a p-value threshold of 5 x 10-8 is usually set to avoid false-positive (McCarthy et al., 2008). In addition, during replication stage, selected variants are often re-studied by de novo genotyping multiple independent populations to further filter out false-positives (Chouraki and Seshadri, 2014). Since 2007 when APOEwas the first LOAD risk gene reported by GWAS, multiple genes with SNPs or variants associated with AD had been identified: ABCA7, BIN1, CASS4, CD33, CD2AP, CELF1, CLU,CR1, DSG2, EPHA1, FERMT2, HLA-DRB5/DRB1, INPP5D, MS4A, MEF2C, NME8,

21
PICALM, PTK2B, SLC24H-RIN3, SORL1 and ZCWPW1 (Medway and Morgan, 2014, Calero et al., 2015, Karch and Goate, 2015). Using other similar approaches such as genome-wide haplotype association study or alternative methods such as exome sequencing, FRMD4A,TRIP4, TREM2 and PLD3 were also reported as novel loci related to AD susceptibility (Lambert et al., 2013a, Ruiz et al., 2014, Jonsson et al., 2013, Cruchaga et al., 2014, Guerreiro et al., 2013a)
Among all the LOAD susceptibility genes, APOE, especially APOE ε4 allele, is by far the most important and frequently reported genetic risk factor related to the pathogenesis of non-familial, sporadic late-onset AD (Strittmatter et al., 1993, Corder et al., 1993). APOEgene locates on chromosome 19q13.2, and encodes for apolipoprotein E (APOE) that physiologically functions as lipoprotein ligand, which regulates lipoprotein internalization, cholesterol and other lipids transport and metabolisms in brain and periphery (Pericak-Vance et al., 1991, Siest et al., 1995, Mahley, 1988, Kim et al., 2009b). APOE has three isoforms depending on two variations in amino acid sequence. APOE4 (R112, R158) corresponds to APOE ε4, APOE2 (C112, C158) corresponds to APOE ε2, and APOE3 (C112, R158) corresponds to APOE ε3 (Zannis et al., 1982, Weisgraber et al., 1981). As schematically illustrated in Figure 9, APOE ε4 could elevate the AD risk from 3-15 folds depending on whether the allele is in homozygosis or heterozygosis (Kim et al., 2009b). But unlike PSEN1/2and APP mutations, APOE ε4 alone is not sufficient to induce AD (Bertram et al., 2007, Farrer et al., 1997). Many LOAD risk genes including APOE may not significantly contribute to AD by itself, but the presence of multiple LOAD risk gene with disease-associated SNPs or variants at the same time would remarkably increase the risk of developing AD (Bertram et al., 2007). Over half of AD patients were not APOE ε4 carriers, but the presence of APOE ε4 was suggested to affect the age of onset of familial AD (Corder et al., 1993). By far the exact role and molecular mechanisms of APOE in AD pathogenesis are not fully understood (Chouraki and Seshadri, 2014). Evidence suggested that APOE could modulate Aβ aggregation and metabolism through direct or indirect routes such as binding or regulating low-density lipoprotein receptor-related protein 1 (LRP1)(Verghese et al., 2013, Castellano et al., 2011). In addition, APOE was also shown to bind to tau, but the functional relevance of this binding is yet unknown (Strittmatter et al., 1994). Fortunately, APOE ε3 is the most common genotype, and together with APOE ε2 show protective role against the development of AD (Weisgraber et al., 1981, Corder et al., 1994).
Table 1 and Figure 9 summarize some of the top susceptibility risk genes with associated cellular functions. These genes are functionally grouped into vesicular trafficking [bridging integrator 1(BIN1), CD2-associated protein (CD2AP), FERM containing domain 4A (FRMD4A), Phosphatidylinoaitol binding clathrin assembly protein (PICALM) and sortilin-related receptor LDLR class A repeats containing (SOR1)], cholesterol metabolism [ATP-binding cassette transporte A7 (ABCA7), ADAM10, apolipoprotein E (APOE4), clusterin (CLU) and SOR1] and immune response [ABCA7, CLU, complement receptor 1 (CR1),CD33, triggering receptor expressed on myeloid cells 2 (TREM2) and membrane-spanning 4-domains subfamily A (MS4A)] (Karch and Goate, 2015, Jones et al., 2010, Medway and Morgan, 2014). It is not fully understood that how these LOAD risk genes are connected to AD or tau, but BIN1, CD2AP, and APOE were previously reported to influence CSF tau level and tau-mediated neurotoxicity (Chapuis et al., 2013b, Cruchaga et al., 2013b, Shulman et al., 2011). The exact function(s) of other newly identified LOAD risk genes are yet to be elucidated. Functional studies to explore the connections between LOAD risk genes and tau propagation pathway are particularly interesting in term of pathology and may identify newdrug targets.
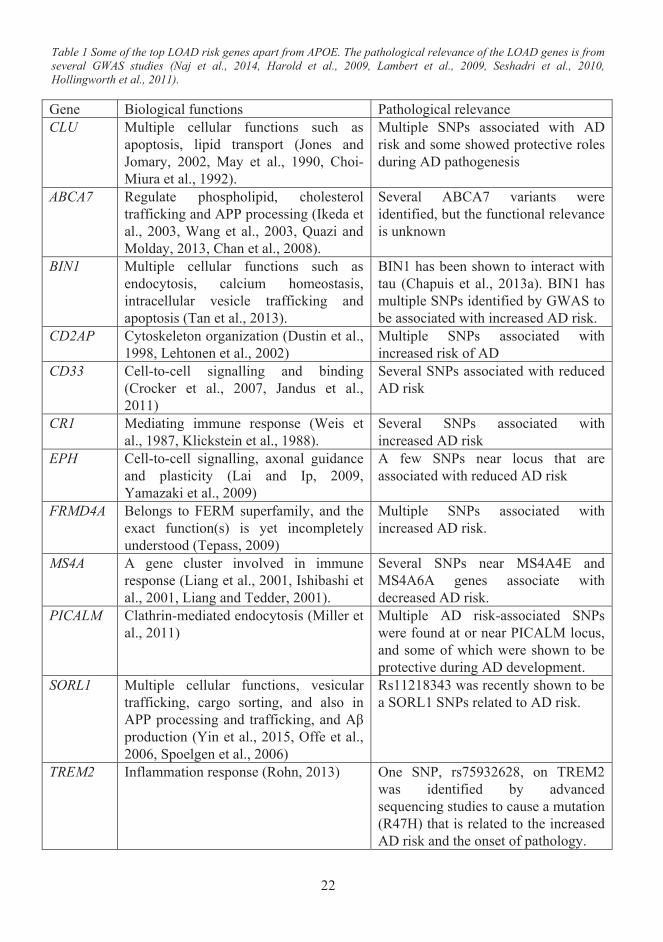
22
Table 1 Some of the top LOAD risk genes apart from APOE. The pathological relevance of the LOAD genes is from several GWAS studies (Naj et al., 2014, Harold et al., 2009, Lambert et al., 2009, Seshadri et al., 2010, Hollingworth et al., 2011).
Gene Biological functions Pathological relevanceCLU Multiple cellular functions such as
apoptosis, lipid transport (Jones and Jomary, 2002, May et al., 1990, Choi-Miura et al., 1992).
Multiple SNPs associated with AD risk and some showed protective roles during AD pathogenesis
ABCA7 Regulate phospholipid, cholesterol trafficking and APP processing (Ikeda et al., 2003, Wang et al., 2003, Quazi and Molday, 2013, Chan et al., 2008).
Several ABCA7 variants were identified, but the functional relevance is unknown
BIN1 Multiple cellular functions such as endocytosis, calcium homeostasis, intracellular vesicle trafficking and apoptosis (Tan et al., 2013).
BIN1 has been shown to interact with tau (Chapuis et al., 2013a). BIN1 has multiple SNPs identified by GWAS to be associated with increased AD risk.
CD2AP Cytoskeleton organization (Dustin et al., 1998, Lehtonen et al., 2002)
Multiple SNPs associated with increased risk of AD
CD33 Cell-to-cell signalling and binding (Crocker et al., 2007, Jandus et al., 2011)
Several SNPs associated with reduced AD risk
CR1 Mediating immune response (Weis et al., 1987, Klickstein et al., 1988).
Several SNPs associated with increased AD risk
EPH Cell-to-cell signalling, axonal guidance and plasticity (Lai and Ip, 2009, Yamazaki et al., 2009)
A few SNPs near locus that are associated with reduced AD risk
FRMD4A Belongs to FERM superfamily, and the exact function(s) is yet incompletely understood (Tepass, 2009)
Multiple SNPs associated with increased AD risk.
MS4A A gene cluster involved in immune response (Liang et al., 2001, Ishibashi et al., 2001, Liang and Tedder, 2001).
Several SNPs near MS4A4E and MS4A6A genes associate with decreased AD risk.
PICALM Clathrin-mediated endocytosis (Miller et al., 2011)
Multiple AD risk-associated SNPs were found at or near PICALM locus, and some of which were shown to be protective during AD development.
SORL1 Multiple cellular functions, vesicular trafficking, cargo sorting, and also in APP processing and trafficking, and Aβ production (Yin et al., 2015, Offe et al., 2006, Spoelgen et al., 2006)
Rs11218343 was recently shown to be a SORL1 SNPs related to AD risk.
TREM2 Inflammation response (Rohn, 2013) One SNP, rs75932628, on TREM2 was identified by advanced sequencing studies to cause a mutation (R47H) that is related to the increasedAD risk and the onset of pathology.

23
2.3.7 Tau ubiquitination and clearance
Tau isolated from AD brains was shown to carry ubiquitin-modifications at multiple sites,such as Lys254/257/311/317/353, which are clustered in or near the repeat domains and influence microtubule binding affinity of tau (Mori et al., 1987, Morishima-Kawashima et al., 1993, Cripps et al., 2006, Matenia and Mandelkow, 2009). Some of these poly-ubiquitin-modifications are chained through Lys48/63, which are previously described signalling pathways for proteasomal degradation (Pickart, 2001). The C-terminus of heat-shock cognate 70-interacting protein (CHIP) and tumour-necrosis-factor-receptor-associated factor 6(TRAF6) are the two known ubiquitin E3 subunits that ubiquitinate tau and colocalize with tau in NFTs (Shimura et al., 2004, Hatakeyama et al., 2004, Babu et al., 2005). Specific proline-directed Ser/Thr phosphorylation of tau is needed for CHIP recognition, whereas KXGSphosphorylation motif in the repeat domain reduces tau ubiquitination (Dickey et al., 2007, Shimura et al., 2004). Overexpression of CHIP or promoting formation polyubiquitin chainedat Lys48/63 can trigger tau aggregation suggesting ubiquitination plays an important role regulating tau aggregation (Petrucelli et al., 2004, Tan et al., 2008).
Despite the fact that tau is polyubiquitinated, it is currently debated if tau ispredominantly degraded by UPS (Wang and Mandelkow, 2012). Tau appears to be the substrate of UPS in some studies but is hardly influenced by UPS in other studies (Shimura et al., 2004, Dickey et al., 2007, David et al., 2002, Wang et al., 2009b, Brown et al., 2005, Feuillette et al., 2005). Cell lines, tau species, microenvironments, etc. differences between studies are considered as the cause of the contradictory results suggesting tau may be degraded by distinct mechanisms under different circumstances (Wang and Mandelkow, 2012). Incancer cell lines such as SH-SY5Y, tau is degraded by UPS, whereas in primary neurons, tau is actively degraded by ALP (David et al., 2002, Kruger et al., 2012). While using the same immortalized mouse cortical neuron CN1.4, full-length tau is degraded by UPS whereas Asp421 truncated tau that represents an AD pathological species is predominantly degraded via the ALP (Dolan and Johnson, 2010). In rat primary neurons, after inhibition of heat-shock protein Hsp90, Tau P301L, an aggregation-prone mutant, is actively degraded via UPS whileWT tau is not (Luo et al., 2007). Natively unfolded tau can be degraded via 20S proteasome in
Figure 9. Genetic landscape of the topsusceptibility genes related to late-onset Alzheimer’sdisease. Genome-wide association and related studies on LOAD genes generated an enormous amount of data that provides a statistic view on the chance of a gene variant to trigger diseases, and the distribution in the population. Compared with autosomal dominant disease causative genes
such as APP, PSEN1/2, the LOAD risk genes have much lower chance to trigger disease alone. APOE4 homo-and heterozygote have different contribution to disease. The primary cellular functions of the proteins encoded by these genes are indicated as colour in the boxes, and the additional functions are indicated as the edge lines. Modified from Karch et al. (2015).

24
vitro, and this can be inhibited by GSK3- phosphorylation of tau (Poppek et al., 2006, David et al., 2002). Additionally, like aSyn (reviewed in section 2.2.3.1), higher order oligomers and aggregates of tau are precluded from UPS as the narrow channel of UPS only allow unfolded and linearized peptide chain to pass through (Rubinsztein, 2006). In many cases, these tau fibrils impair UPS activity (Keck et al., 2003).
Just like aSyn, tau also has the KFERQ motif that is recognized by Hsp70 and is used to direct tau for ALP degradation (Wang et al., 2009b). Activation of autophagy by rapamycin reduces tau level in cell and Drosophila models, whereas inhibition of autophagy promotes tauaggregation (Hamano et al., 2008, Berger et al., 2006). ALP degrades most forms of tau, including both soluble and insoluble tau, including tau with KXGS motif phosphorylated that is not degraded by UPS (Wang et al., 2009b). Like aSyn, certain tau aggregates or mutantssuch as P301L, G272V, R406W, can impair ALP-mediated degradation (Shemesh and Spira, 2010, Lin et al., 2003, Lim et al., 2001).
2.3.8 Toxic species of tau
The pathogenesis of neurodegenerative diseases with tauopathies is not fully understood (Wang and Mandelkow, 2016). It is currently debated if NFTs is associated with the cognitive decline seen in AD patients due to limited data from human brains and lack of understanding of disease mechanisms (DeFelipe, 2016). Several studies carried out in transgenic mice indicated that NFTs may not strongly relate to cognitive impairment (Gomez-Isla et al., 1997, Andorfer et al., 2005, Spires-Jones et al., 2008). However, tau aggregation is a common characteristic of several neurodegenerative diseases and is also observed in aged brains from cognitively normal subjects (Crary et al., 2014). Like aSyn, the tau aggregates such as NFTs were at first considered to be toxic due to the correlation with diseases progression (Wang and Mandelkow, 2016). But later a lot of evidence has suggested that insoluble tau aggregates maynot be the most toxic species. For example, switching off tau P301L expression (an aggregation-prone mutant) improved memory dysfunction in transgenic mouse regardless of the presence of NFTs suggesting that synapse loss or cognitive deficit is not dependent on the presence of tau aggregates (Santacruz et al., 2005). In several other animal models, neuronaldeath is not correlated with NFTs, and neurons can live surrounded by NFTs for years (Andorfer et al., 2005, Spires-Jones et al., 2008). This agrees with the observations in human. In superior temporal sulcus region of 37 AD patients’ brains, the ratio between the neuronal loss to NFT population can be 7:1, suggesting a lot more neurons dead in the absence of NFTs(Gomez-Isla et al., 1997). With computer modelling, neurons in man were shown to surviveover 20 years with NFTs (Morsch et al., 1999).
There have been views suggesting that the filamental assembly of pathological tau species might be neuron-protective, possibly by “packaging up” pathological tau species into PHFs and NFTs to prevent further cellular damage (Alonso Adel et al., 2006). As mentioned previously, certain misfolded tau species have their hydrophobic residues exposed and could recruit normal proteins with low complexity including tau itself upon interaction, and form a further aggregation. Hence by packaging into fibrils, the hydrophobic residues could be buried inside, and thus prevents pathological interaction that could cause other normal proteins and tau to gain toxic function or to lose their physiological functions.
There are also views suggesting that the most toxic tau species are certain oligomericintermediates (Lasagna-Reeves et al., 2012c, Maeda et al., 2007). However, due to the heterogeneity of tau oligomers, there has not been a clearly defined toxic property assigned to different oligomers species of tau (Wang and Mandelkow, 2016). For example, exposing primary neuron to hyperphosphorylated full-length tau oligomers trigger degeneration of

25
dendritic spines (Tepper et al., 2014). Treating SH-SY5Y cells with tau monomers, oligomersor fibrils demonstrated that tau oligomers are the most toxic species, which caused significant neuronal death possibly by disrupting membrane integrity (Flach et al., 2012). Incubating human neuroblastoma cells with tau monomers, dimers and trimers showed that trimer was the most toxic species even at a low nanomolar concentration (Tian et al., 2013). Some studies have tested both pro-aggregant and anti-aggregant tau mutants in mouse models, showing that only pro-aggregant tau induced the development of AD-like pathology in mice suggesting that tau aggregation potential may be needed for toxicity and brain pathology (Eckermann et al., 2007, Mocanu et al., 2008). Previous studies suggested that tau strains/species, especially preformed fibrils (Pffs), affect the rate of cell-to-cell transmission, possibly also the propagation of pathology (Sanders et al., 2014a). The contribution of tau and aSyn to the spread of pathology will be discussed in a different section below.
Apart from tau oligomer related-toxicity, hyperphosphorylation could also induce taugain of toxic functions. For example, hyperphosphorylation promotes tau interaction with JIP1, and impair kinesin complex while normal tau does not (Bhaskar et al., 2005). In a Drosophilamodel, hyperphosphorylation of tau impairs the actin network and causes neurodegeneration(Fulga et al., 2007). It was suggested that this gain of toxic function could be a result of tau re-localization to postsynaptic spines after hyperphosphorylation or caused by mutations and impairs synaptic functions (Tai et al., 2014, Hoover et al., 2010, Thies and Mandelkow, 2007).It has been reported that missorted dendritic tau mediates A toxicity by recruiting tyrosine ligase-like enzyme 6 into dendrites and triggering Spastin to sever microtubules, causing dendritic spine degeneration (Zempel et al., 2013).
Neurotoxicity could also be due to loss of tau’s physiological functions. Loss of tau function is generally considered as a loss of microtubule binding that results in microtubule disassembly (Wang and Mandelkow, 2016). As reviewed in section 2.3.1, the function of tau can be compensated by other proteins like MAP1, but acute silencing of tau in 12-month old mice showed impaired cognitive behaviour suggesting that tau is also needed for normal neuronal functions (Lei et al., 2014). Considering that tau is also involved in DNA protection, neurogenesis, etc. (reviewed in section 2.3.1), loss of these functions may also contribute to neurodegeneration (Wang and Mandelkow, 2016).
2.3.9 Interconnection between aSyn and tau
It has been widely recognized that tauopathies are not restricted to AD, nor synucleinopathies restricted to PD (Galpern and Lang, 2006). Many familial cases with mutations in MAPT or SNCA genes can have both Parkinsonism and dementia at the same time (Polymeropoulos et al., 1997, Zarranz et al., 2004, Fujioka et al., 2014, Spillantini et al., 1998, Dumanchin et al., 1998). As schematically illustrated in Figure 10, aSyn and tau inclusion could co-occur in multiple diseases such as Lewy body variant of AD (LBVAD), Parkinson’s disease with dementia (PDD), dementia with Lewy bodies (DLB), Guam-Parkinson-ALS dementia complex, and even Down’s syndrome (Lippa et al., 1998, Lippa et al., 1999, Forman et al., 2002, Moussaud et al., 2014). PD patients have increased risk of developing dementia, and frequently developed NFTs at autopsy, while many AD patients developed Lewy bodies at autopsy, suggesting remarkable crosstalk between synucleinopathies and tauopathies (Aarsland et al., 2005, Leverenz et al., 2009, Leverenz et al., 2008, Hamilton, 2000, Galpern and Lang, 2006, Bancher et al., 1993).
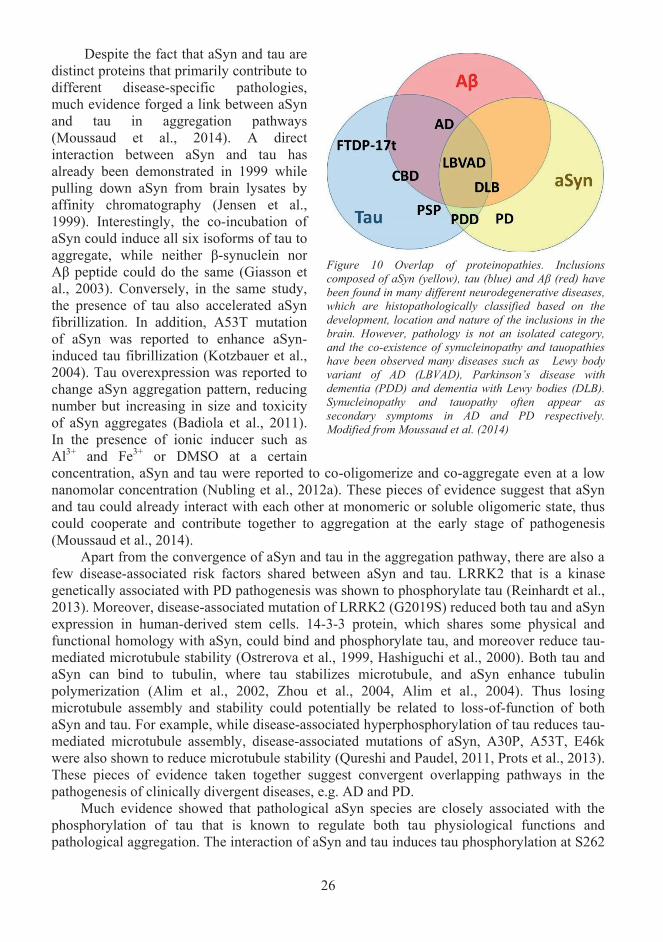
26
Despite the fact that aSyn and tau are distinct proteins that primarily contribute to different disease-specific pathologies, much evidence forged a link between aSyn and tau in aggregation pathways(Moussaud et al., 2014). A direct interaction between aSyn and tau has already been demonstrated in 1999 while pulling down aSyn from brain lysates by affinity chromatography (Jensen et al., 1999). Interestingly, the co-incubation of aSyn could induce all six isoforms of tau to aggregate, while neither β-synuclein norAβ peptide could do the same (Giasson et al., 2003). Conversely, in the same study, the presence of tau also accelerated aSyn fibrillization. In addition, A53T mutation of aSyn was reported to enhance aSyn-induced tau fibrillization (Kotzbauer et al., 2004). Tau overexpression was reported to change aSyn aggregation pattern, reducing number but increasing in size and toxicity of aSyn aggregates (Badiola et al., 2011).In the presence of ionic inducer such as Al3+ and Fe3+ or DMSO at a certain concentration, aSyn and tau were reported to co-oligomerize and co-aggregate even at a low nanomolar concentration (Nubling et al., 2012a). These pieces of evidence suggest that aSyn and tau could already interact with each other at monomeric or soluble oligomeric state, thus could cooperate and contribute together to aggregation at the early stage of pathogenesis (Moussaud et al., 2014).
Apart from the convergence of aSyn and tau in the aggregation pathway, there are also a few disease-associated risk factors shared between aSyn and tau. LRRK2 that is a kinase genetically associated with PD pathogenesis was shown to phosphorylate tau (Reinhardt et al., 2013). Moreover, disease-associated mutation of LRRK2 (G2019S) reduced both tau and aSyn expression in human-derived stem cells. 14-3-3 protein, which shares some physical and functional homology with aSyn, could bind and phosphorylate tau, and moreover reduce tau-mediated microtubule stability (Ostrerova et al., 1999, Hashiguchi et al., 2000). Both tau and aSyn can bind to tubulin, where tau stabilizes microtubule, and aSyn enhance tubulin polymerization (Alim et al., 2002, Zhou et al., 2004, Alim et al., 2004). Thus losing microtubule assembly and stability could potentially be related to loss-of-function of both aSyn and tau. For example, while disease-associated hyperphosphorylation of tau reduces tau-mediated microtubule assembly, disease-associated mutations of aSyn, A30P, A53T, E46k were also shown to reduce microtubule stability (Qureshi and Paudel, 2011, Prots et al., 2013).These pieces of evidence taken together suggest convergent overlapping pathways in the pathogenesis of clinically divergent diseases, e.g. AD and PD.
Much evidence showed that pathological aSyn species are closely associated with the phosphorylation of tau that is known to regulate both tau physiological functions and pathological aggregation. The interaction of aSyn and tau induces tau phosphorylation at S262
Figure 10 Overlap of proteinopathies. Inclusions composed of aSyn (yellow), tau (blue) and Aβ (red) have been found in many different neurodegenerative diseases,which are histopathologically classified based on the development, location and nature of the inclusions in the brain. However, pathology is not an isolated category, and the co-existence of synucleinopathy and tauopathies have been observed many diseases such as Lewy body variant of AD (LBVAD), Parkinson’s disease with dementia (PDD) and dementia with Lewy bodies (DLB). Synucleinopathy and tauopathy often appear as secondary symptoms in AD and PD respectively. Modified from Moussaud et al. (2014)

27
in vitro, which is a phosphorylation associated with loss of tau-mediated microtubule stability (Qureshi and Paudel, 2011). In the same study, disease-associated aSyn mutations, A30P, A53T and E46K were shown to enhance aSyn interaction with tau, and thus enhance loss-of-function of tau. Multiple evidence suggested that aSyn forms a complex with tau and GSK-3β via NAC and acidic region, and simulate GSK-3β-mediated phosphorylation of tau at AT270 (T181, an established biomarker for AD) and PHF-1/AD2 (S396 and S404, AD-associated phosphoepitope) (Duka et al., 2006, Duka et al., 2009). The activity of ERK and JNK, which are two kinases of tau, and tau phosphorylation at PHF-1/AD2 were increased in transgenic mice overexpressing aSyn or aSyn mutant A30P or A53T (Kaul et al., 2011, Oaks et al., 2013, Frasier et al., 2005). The interaction of tau with aSyn and phosphorylation at AT270 and PHF-1/AD2 can be suppressed by Hsp70 overexpression or be enhanced by aSyn A53T mutation (Ciaccioli et al., 2013, Kawakami et al., 2011). In cell model of PD, GSK-3β inhibition reduced tau phosphorylation but increased aSyn accumulation suggesting a role of tau in aSyn aggregation (Duka et al., 2009). It was reported that exogenous aSyn of certain strains such as pre-formed fibrils could induce tau hyperphosphorylation and aggregation followed by internalization into neurons overexpressing tau (Guo et al., 2013, Waxman and Giasson, 2011). These observations could potentially provide mechanisms of how the co-existence of synucleinopathies and tauopathies could worsen the clinical outcome during diseases such as Lewy body variant of AD (LBVAD) and dementia with Lewy bodies (DLB).
Taken together, aSyn and tau are interconnected and may feed-forward each other during the development and progression of neurodegenerative diseases. Understanding the connection between aSyn and tau pathogenic pathway could provide novel insights into the pathogenesis mechanism(s) and new drug targets (Moussaud et al., 2014).
2.3.10 Therapeutic strategies against tauopathies and synucleinopathies
Although the roles and species of tau and aSyn causing neurotoxicity in tauopathies and synucleinopathies remain debatable, it is widely accepted that these disease-associated proteins are likely building blocks of these diseases (Wang and Mandelkow, 2016). In general, tau and aSyn-directed strategies against tauopathies and synucleinopathies focus on reducing the neurotoxic gain of functions of tau and aSyn. This may be achieved by reducing tau and aSyn accumulation, aggregation and transmission as shown in Figure 11. MicroRNA (miRNA) or small interfering RNA (siRNA) may be used to silence MAPT and SNCA gene or to suppress the correspondent promoters, and thus reduce the synthesis and intracellular accumulation of tau and aSyn. Some preclinical studies in mice have shown some benefit of knockdown these genes, but target delivery and genetic manipulation in human have always been obstacles (DeVos et al., 2013, Cooper et al., 2014). An alternative way could involve promoting the clearance of tau and aSyn. Activation of ALS and UPS pathways would elevate the overall rate of proteins and macromolecules degradation. The proteolytic degradation of tau and aSyn may also be enhanced by activating various related-proteases such as cathepsin D, neurosin, and metalloproteinases 9 (MMP9), etc. (Nubling et al., 2012b, Iwata et al., 2003, Malik et al., 2011). Apart from modulating tau and aSyn accumulation, reducing the aggregation of tau and aSyn was another traditional approach to alleviate the correspondent neurotoxicity. Many post-translational modifications are associated with the aggregation property of tau and aSyn, such as phosphorylation, C-terminal cleavage, oxidation and nitration, etc. as mentioned previously. Reduction of these aggregation-prone factors was thought to alleviate related-pathology. For example, tideglusib, an inhibitor of GSK-3β that heavily phosphorylates tau, has entered Phase II trial in individual with mild AD (Lovestone et al., 2015). On the other hand, dithiocarbamates, a small molecule activator of GSK-3β that is

28
upstream of nuclear factor erythroid 2-related factor 2-mediated neuroprotective pathway, was also suggested to exert therapeutic potentials, revealing the fact that the choice of therapeutic targets can be complicated even contradictory in these diseases (Kanninen et al., 2011). Direct intervention of the aggregation processes was also interesting in term of therapeutics. For example, entacapone and tolcapone that inhibit the aggregation of aSyn and Aβ by stabilizing high-molecular-weight oligomers, are protective against extracellular neurotoxicity from aSyn and Aβ aggregation (Di Giovanni et al., 2010). Methylene blue treatment, which inhibits tau aggregation, was shown to preserve cognition in AD mice model, and one of its derivatives further showed benefit in Phase II trial in individual with mild AD (Wischik et al., 2015, Hochgrafe et al., 2015). Anti-oxidants, such as Omega-3, have been interesting to public not only because of its role in reducing protein oxidation including tau and aSyn, but also its effect in neuroprotection (Green et al., 2007). Other non-tau and aSyn-directed approaches against tauopathies and synucleinopathies involve growth factors such as GDNF, and anti-inflammatories, which are mainly for neuroprotection during tauopathies and synucleinopathies (Lashuel et al., 2013). Immunotherapy has emerged to be an important therapeutic strategy against tauopathies and synucleinopathies. Specific antibodies were thought to bind to tau and aSyn, and reduce the cell-to-cell transmission (Wang and Mandelkow, 2016, Lashuel et al., 2013). For example, one of the aSyn antibodies could bind to preformed fibrils of aSyn (Pffs), and inhibit its uptake of by cells (Tran et al., 2014). One of the tau antibody blocks cell-to-cell transmission of tau, and improved cognition of AD mice (Yanamandra et al., 2013). The antibodies were also reported enter cells via Fcγ-receptors, and inhibited intracellular aggregation of tau and aSyn (Sahin et al., 2016). For example, an antibody of cis-tau reduces tau aggregation in Fcγ-receptor-mediated manner, and decrease the development and spread of tauopathies in mice with traumatic brain injury (Kondo et al., 2015).
Figure 11 Possible therapeutic strategies against tauopathies and synucleinopathies. Possible strategies focus on reducing tau and aSyn accumulation, aggregation and transmission. siRNA and miRNA may be used to reduced tau and aSyn synthesis. Activation of mechanisms or protein involved in clearance would reduce aberrant protein accumulation. Approaches that involve anti-aggregating, antioxidant or post-translational modification could be used to
reduce the aggregation of tau and aSyn. Immunotherapy may be used to reduce tau and aSyn transmission. Modified from Lashuel (2013).
Misfolded protein propagation in pathological conditions 2.4
Intraneuronal accumulation of protein aggregates is a common pathological feature shared by many neurodegenerative diseases. Increasing evidence indicates that transcellular transmission of these aggregates may be a common mechanism of the propagation of pathologies. Many disease-associated proteins that were previously not known to transfer between cells have been shown to seed aggregation during cell-to-cell transmission in a “prion-like” manner.

29
2.4.1 Prion-like seeding
The term and theory of “prion-like” come from the molecular mechanisms of the propagation of prion diseases, which are also known as spongiform encephalopathies. In prion diseases, normal prion proteins such as PrPC, sporadically convert to misfolded pathological species, e.g. PrPSc, which have altered conformations and act as infectious agents that spread the pathology rapidly by interacting and converting normal prion proteins into pathological species in a template-directed manner, and thus is also called “seeding” (Bolton et al., 1982, Aguzzi and Rajendran, 2009). The molecular mechanism of “prion-like” seeding was originally proposed as seeded polymerization of amyloid-proteins that may be involved in AD and scrapie (Jarrett and Lansbury, 1993). Many neurodegenerative disease-associated proteins that were originally thought to lack seeding properties, such as aSyn and tau, also exhibit characteristics of “prion-like” self-propagation (Guo and Lee, 2014b). As summarized inTable 2, various species of A and tau, aSyn, superoxide dismutase 1 (SOD1) and TDP-43(TAR DNA-binding protein 43, related to Amyotrophic Lateral Sclerosis, ALS), Polyglutamine repeat proteins, such as mutant Huntingtin (mtHH, related to Huntington’sDisease) have been experimentally shown to seed aggregation in different model systems(Guo and Lee, 2014b). Most of these studies have confirmed that the transfer of lysates from human or mouse brain containing disease-associated protein aggregates to healthy cells or animal branch can seed the corresponding pathology suggesting that these disease-associated proteins can transmit pathology in a “prion-like” manner, and this characteristic can bepathologically specific to each disease.
Studies of tau propagation revealed that Pffs of tau proteins are highly efficient in sequestering soluble tau into aggregates, causing tau loss-of-function toxicity in a time-dependent manner (Guo and Lee, 2011a, Iba et al., 2013a). From the same study, theobservation that tau Pffs can be spontaneously uptaken by cells suggests that the uptake of tau could involve pinocytosis. Tau seeding was reported to occur more predominantly in cells overexpressing 4R tau but less in 3R tau overexpressing cells, and also when using synthetic fibrils derived from 4R tau as compared to 3R tau or fibrils composed of other proteins such as aSyn, suggesting that tau isoforms, especially ones with R2 domain, could play a dictateseeding (Iba et al., 2013a, Nonaka et al., 2010b). Besides, tau monomer was reported not be able to seed aggregation suggesting that the oligomeric state of tau is required for seeding (Frost et al., 2009). These shreds of evidence indicate that tau propagation is highly dependent on different physiochemical and oligomeric properties of various strains and species (Sanders et al., 2014a).
A few aSyn studies have demonstrated that both synthetic aSyn fibrils and lysates from PD mice brains induce synucleinopathies sufficiently with endogenous aSyn, unlike tau that requires overexpression of tau in recipient cells or tissue (Volpicelli-Daley et al., 2011, Luk et al., 2012a). This suggests that aSyn may be more aggregation prone under physiological conditions than tau. In cells overexpressing aSyn, recombinant aSyn fibrils were reported to recruit soluble aSyn, and converting them into hyperphosphorylated (S129) and ubiquitinated aggregates (Luk et al., 2009). Similar post-translational modification of aSyn aggregates was also induced by introducing recombinant nucleation seeds via transfection in SH-SY5Y cells (Nonaka et al., 2010b). Interestingly, in the same study, applying a series of chemicals such asexifone and gossypetin, which block amyloid filament formation, significantly reduced the toxicity of aSyn inclusions in recipient cells, suggesting a potential therapeutic approach by preventing protein aggregation at the step of cell-to-cell transmission. Inoculation of aSyn synthetic fibrils induces Lewy bodies-like pathology in mice, whereas soluble aSyn does not,
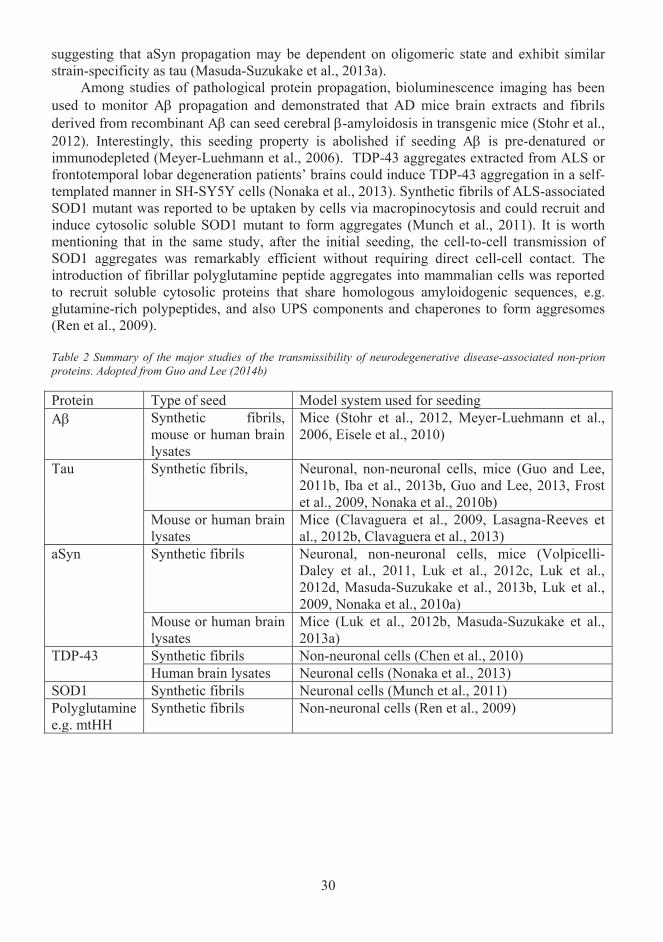
30
suggesting that aSyn propagation may be dependent on oligomeric state and exhibit similar strain-specificity as tau (Masuda-Suzukake et al., 2013a).
Among studies of pathological protein propagation, bioluminescence imaging has been used to monitor A propagation and demonstrated that AD mice brain extracts and fibrils derived from recombinant A can seed cerebral -amyloidosis in transgenic mice (Stohr et al., 2012). Interestingly, this seeding property is abolished if seeding A is pre-denatured or immunodepleted (Meyer-Luehmann et al., 2006). TDP-43 aggregates extracted from ALS or frontotemporal lobar degeneration patients’ brains could induce TDP-43 aggregation in a self-templated manner in SH-SY5Y cells (Nonaka et al., 2013). Synthetic fibrils of ALS-associated SOD1 mutant was reported to be uptaken by cells via macropinocytosis and could recruit and induce cytosolic soluble SOD1 mutant to form aggregates (Munch et al., 2011). It is worth mentioning that in the same study, after the initial seeding, the cell-to-cell transmission of SOD1 aggregates was remarkably efficient without requiring direct cell-cell contact. The introduction of fibrillar polyglutamine peptide aggregates into mammalian cells was reported to recruit soluble cytosolic proteins that share homologous amyloidogenic sequences, e.g. glutamine-rich polypeptides, and also UPS components and chaperones to form aggresomes (Ren et al., 2009).
Table 2 Summary of the major studies of the transmissibility of neurodegenerative disease-associated non-prion proteins. Adopted from Guo and Lee (2014b)
Protein Type of seed Model system used for seedingA Synthetic fibrils,
mouse or human brain lysates
Mice (Stohr et al., 2012, Meyer-Luehmann et al., 2006, Eisele et al., 2010)
Tau Synthetic fibrils, Neuronal, non-neuronal cells, mice (Guo and Lee, 2011b, Iba et al., 2013b, Guo and Lee, 2013, Frost et al., 2009, Nonaka et al., 2010b)
Mouse or human brain lysates
Mice (Clavaguera et al., 2009, Lasagna-Reeves et al., 2012b, Clavaguera et al., 2013)
aSyn Synthetic fibrils Neuronal, non-neuronal cells, mice (Volpicelli-Daley et al., 2011, Luk et al., 2012c, Luk et al., 2012d, Masuda-Suzukake et al., 2013b, Luk et al., 2009, Nonaka et al., 2010a)
Mouse or human brain lysates
Mice (Luk et al., 2012b, Masuda-Suzukake et al., 2013a)
TDP-43 Synthetic fibrils Non-neuronal cells (Chen et al., 2010)Human brain lysates Neuronal cells (Nonaka et al., 2013)
SOD1 Synthetic fibrils Neuronal cells (Munch et al., 2011)Polyglutaminee.g. mtHH
Synthetic fibrils Non-neuronal cells (Ren et al., 2009)
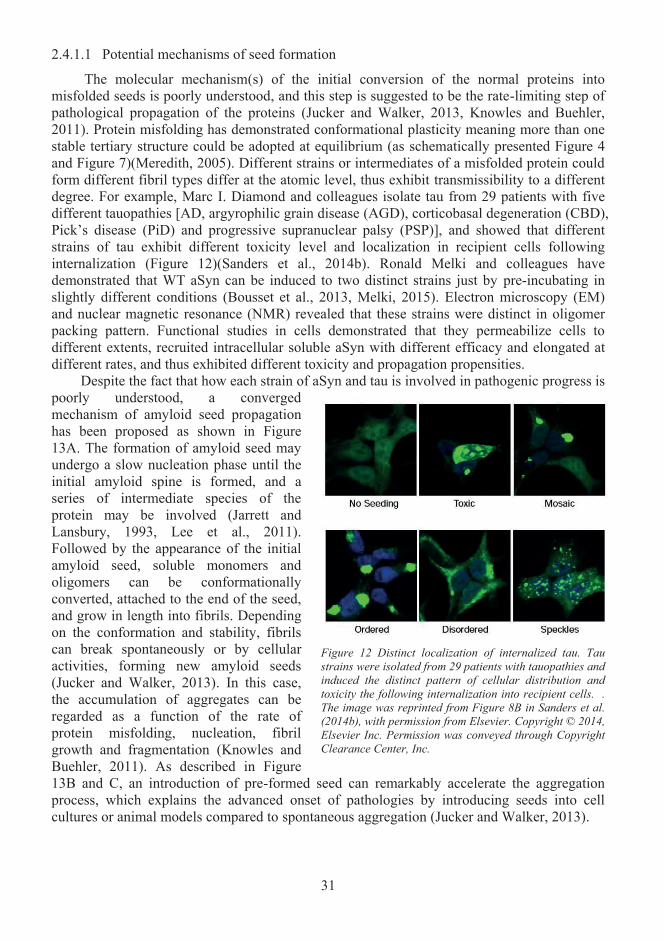
31
2.4.1.1 Potential mechanisms of seed formation
The molecular mechanism(s) of the initial conversion of the normal proteins into misfolded seeds is poorly understood, and this step is suggested to be the rate-limiting step of pathological propagation of the proteins (Jucker and Walker, 2013, Knowles and Buehler, 2011). Protein misfolding has demonstrated conformational plasticity meaning more than one stable tertiary structure could be adopted at equilibrium (as schematically presented Figure 4and Figure 7)(Meredith, 2005). Different strains or intermediates of a misfolded protein could form different fibril types differ at the atomic level, thus exhibit transmissibility to a different degree. For example, Marc I. Diamond and colleagues isolate tau from 29 patients with fivedifferent tauopathies [AD, argyrophilic grain disease (AGD), corticobasal degeneration (CBD), Pick’s disease (PiD) and progressive supranuclear palsy (PSP)], and showed that differentstrains of tau exhibit different toxicity level and localization in recipient cells following internalization (Figure 12)(Sanders et al., 2014b). Ronald Melki and colleagues have demonstrated that WT aSyn can be induced to two distinct strains just by pre-incubating inslightly different conditions (Bousset et al., 2013, Melki, 2015). Electron microscopy (EM)and nuclear magnetic resonance (NMR) revealed that these strains were distinct in oligomer packing pattern. Functional studies in cells demonstrated that they permeabilize cells to different extents, recruited intracellular soluble aSyn with different efficacy and elongated at different rates, and thus exhibited different toxicity and propagation propensities.
Despite the fact that how each strain of aSyn and tau is involved in pathogenic progress ispoorly understood, a converged mechanism of amyloid seed propagation has been proposed as shown in Figure13A. The formation of amyloid seed may undergo a slow nucleation phase until the initial amyloid spine is formed, and aseries of intermediate species of the protein may be involved (Jarrett and Lansbury, 1993, Lee et al., 2011).Followed by the appearance of the initial amyloid seed, soluble monomers and oligomers can be conformationallyconverted, attached to the end of the seed, and grow in length into fibrils. Dependingon the conformation and stability, fibrils can break spontaneously or by cellular activities, forming new amyloid seeds (Jucker and Walker, 2013). In this case, the accumulation of aggregates can be regarded as a function of the rate of protein misfolding, nucleation, fibril growth and fragmentation (Knowles and Buehler, 2011). As described in Figure 13B and C, an introduction of pre-formed seed can remarkably accelerate the aggregation process, which explains the advanced onset of pathologies by introducing seeds into cell cultures or animal models compared to spontaneous aggregation (Jucker and Walker, 2013).
Figure 12 Distinct localization of internalized tau. Tau strains were isolated from 29 patients with tauopathies and induced the distinct pattern of cellular distribution and toxicity the following internalization into recipient cells. .The image was reprinted from Figure 8B in Sanders et al. (2014b), with permission from Elsevier. Copyright © 2014,Elsevier Inc. Permission was conveyed through Copyright Clearance Center, Inc.
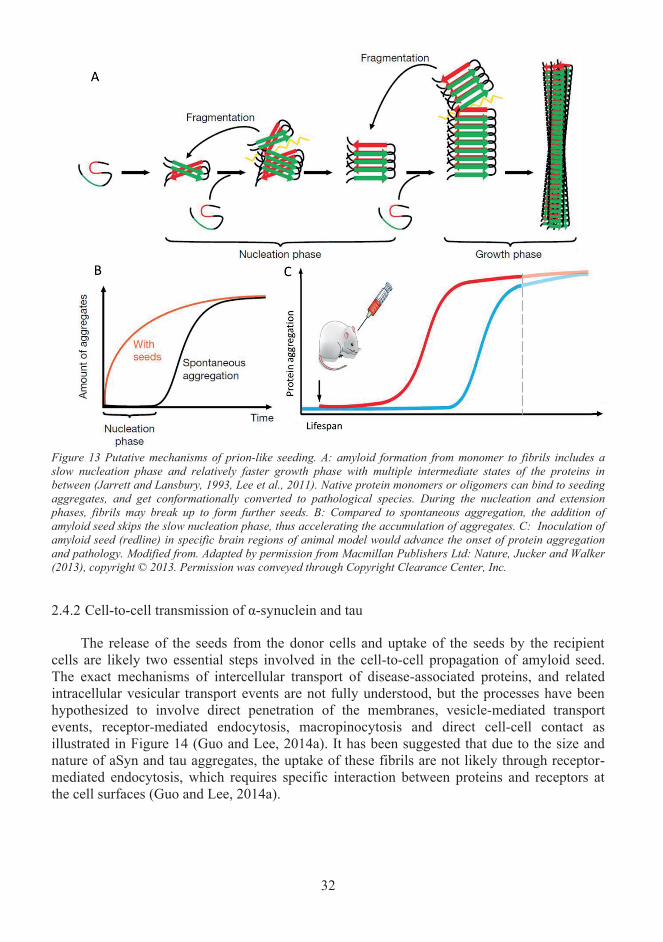
32
Figure 13 Putative mechanisms of prion-like seeding. A: amyloid formation from monomer to fibrils includes a slow nucleation phase and relatively faster growth phase with multiple intermediate states of the proteins in between (Jarrett and Lansbury, 1993, Lee et al., 2011). Native protein monomers or oligomers can bind to seeding aggregates, and get conformationally converted to pathological species. During the nucleation and extensionphases, fibrils may break up to form further seeds. B: Compared to spontaneous aggregation, the addition of amyloid seed skips the slow nucleation phase, thus accelerating the accumulation of aggregates. C: Inoculation of amyloid seed (redline) in specific brain regions of animal model would advance the onset of protein aggregation and pathology. Modified from. Adapted by permission from Macmillan Publishers Ltd: Nature, Jucker and Walker (2013), copyright © 2013. Permission was conveyed through Copyright Clearance Center, Inc.
2.4.2 Cell-to-cell transmission of α-synuclein and tau
The release of the seeds from the donor cells and uptake of the seeds by the recipient cells are likely two essential steps involved in the cell-to-cell propagation of amyloid seed. The exact mechanisms of intercellular transport of disease-associated proteins, and related intracellular vesicular transport events are not fully understood, but the processes have been hypothesized to involve direct penetration of the membranes, vesicle-mediated transport events, receptor-mediated endocytosis, macropinocytosis and direct cell-cell contact as illustrated in Figure 14 (Guo and Lee, 2014a). It has been suggested that due to the size and nature of aSyn and tau aggregates, the uptake of these fibrils are not likely through receptor-mediated endocytosis, which requires specific interaction between proteins and receptors at the cell surfaces (Guo and Lee, 2014a).

33
Figure 14 Putative pathways of cell-to-cell transmission of aggregated proteins at the synaptic cleft. Protein or protein aggregates can be released into extracellular space as (a) free monomers or oligomers or protofibrils, and enter plasma membrane ofrecipient cells by 1) direct penetration, 2) fluid-phaseendocytosis or macropinocytosis, 3) receptor-mediated endocytosis. The protein may also be released in (b) membrane-bound vesicles like exosomes or
ectosomes, and uptake by recipient cells by 4) membrane fusion. In some cases, direct cell-cell contact or 5) nanotubes connecting two cells provide direct routes for protein transfer. Modified from Guo and Lee (2014a).
2.4.2.1 Vesicular release of α-synuclein
It has been proposed that the release of vesicle-bound aSyn and tau may be through the unconventional secretory pathway, in another word not via classical ER-Golgi pathway (Chai et al., 2012b, Malm et al., 2016). The release of vesicles through unconventional secretary pathway primarily included secretory lysosome and endoplasmic vesicles release, and microvesicles shedding (Nickel and Rabouille, 2009). Exosomes can be released with the last two ways, whereas ectosomes are generally released by membrane budding. The released proteins are surrounded by vesicles, which can be recognized and purified based on their size, e.g. exosome (50-200 nm) and ectosome (0.1–1 μm). The largely unexplored secretary pathway(s) is also called misfolding-associated protein secretion (MAPS), which is not likely specific to synapses and could be studied in non-neuronal cells with certain limitations.
aSyn has been identified in the CSF and plasma of patients with synucleinopathy,suggesting that aSyn is secreted into the extracellular space in human (Borghi et al., 2000) (El-Agnaf et al., 2003). Despite the fact that the mechanisms of aSyn release are incompletely understood, aSyn has been identified in the exosome-bound and free form in conditioned media from SH-SY5Y cells (Emmanouilidou et al., 2010). Many groups have reported that free aSyn is the predominantly secreted species by neuron and non-neuronal cells with exosomal fraction representing a smaller population (Pan-Montojo et al., 2012, Vella et al., 2016, Paillusson et al., 2013). A study using protein-fragment complementation assay has demonstrated that exosomal aSyn is internalized much more efficiently than free aSyn suggesting that vesicle might offer a more efficient route for aSyn to get access into cells(Danzer et al., 2012). In vivo studies have demonstrated that exosomal aSyn level in plasmaand CSF of PD patients is associated with severity of the pathology (Shi et al., 2014).
Accumulating evidence suggest that exosomal release of aSyn may be modulated by the ALP (Vella et al., 2016). It is not fully understood if this is specific to the aSyn release, because ALP induction and stimulation have been shown to modulate the amount of exosome released, and ALP impairment causes intracellular accumulation of misfolded aSyn (Fader et al., 2008, Baixauli et al., 2014). As reviewed in section 2.2.3.2, ALP dysfunction plays animportant role in PD pathology, hence the exosomal release of aSyn may be altered in PD. Indeed, inhibition of fusion of autophagosome to lysosome via bafilomycin has been shown to increase the exosomal release of aSyn (Poehler et al., 2014, Alvarez-Erviti et al., 2011, Danzer

34
et al., 2012). Impairment of mitochondrial, lysosomal and autophagic function via treatment of rotenone also enhanced neuronal release of aSyn in exosomes (Pan-Montojo et al., 2012).Also, a few PD-related genes, such as LRRK2, VPS35 and PARK9 are linked to autophagic and endocytic pathways, and were also shown to influence exosome release (Fraser et al., 2013, Follett et al., 2014, Kong et al., 2014).
2.4.2.2 Vesicular release of tau
Tau can be detected in CSF of both healthy individuals and AD patients, but the amount is increased in pathological conditions (Blennow et al., 1995, Vigo-Pelfrey et al., 1995). The mechanisms of tau secretion have not been extensively studied like aSyn and Aβ, because tau release was originally thought to be consequences of neuronal death or exocytic process (Frost et al., 2009). However, increasing evidence reveal that tau can be released both passively and by exosomes, and free tau represents the major species (Saman et al., 2012b, Chai et al., 2012b, Simon et al., 2012b). Recent evidence shows that vesicular tau is predominantly released in ectosomes, which are similar but larger vesicles compared with exosomes (up to1000nm), and are formed directly from plasma membrane budding (Dujardin et al., 2014a, Fevrier and Raposo, 2004). It has been proposed that vesicular release of tau could be induced by overexpressing tau in cell models and is likely to be one route for cells to eliminate excess proteins to reduce toxicity (Simon et al., 2012b). Microvesicles-associated tau has been reported in human CSF sample and was shown to be phosphorylated at T181, which is also known as AT270, an established biomarker for AD (Saman et al., 2012b). This suggests that vesicle-bound tau species could be pathologically relevant.
Some studies indicated that the presence of N-terminus, C-terminal truncation, tau isoforms, synaptic activities, several MAPT mutations (P301L/S, R406W) might have a link to tau secretion, but the mechanisms are poorly understood (Pooler et al., 2013, Yamada et al., 2014, Karch et al., 2012, Kanmert et al., 2015). For example, activation of AMPA receptor contributes to tau release in cortical neurons in a calcium-dependent manner that can be reduced by tetanus toxin or tetrodotoxin, which block pre-synaptic vesicle release or neuronal activity (Pooler et al., 2013). Another in vivo study also showed similar observation that extracellular tau level is rapidly increased when neuronal activity is enhanced (Yamada et al., 2014). In the same study, tau was released within a few hours, but the extracellular tau had ahalf-life of more than 11 days, suggesting a much less sufficient clearance of tau outside cells.Karch CM. and colleagues have explored the effect of various factors on tau release (Karch et al., 2012). Apart from confirming the previous findings that both phosphorylated and unphosphorylated tau can be secreted via unconventional pathways in a calcium-dependent manner, they showed that there was little difference between the level of secreted 0N3R or 0N4R tau (Figure 5). However, extracellular 2N3R level was significantly higher than 2N4R, suggesting that various isoforms of tau may have different propagation propensities (how prone they are to propagate). They also showed that several disease-associated mutations (P301L, P301S and R406W) reduced tau secretion, suggesting a role of mutations in tau secretion pathway. In the studies conducted by Kanmert D. and colleagues, C-terminal truncated tau was shown to be the predominantly secreted species from three neuronal models, while intracellular tau was mostly full-length (Kanmert et al., 2015). Among these C-terminal truncated tau species in the extracellular space, a minor population had microtubule-binding domains that are required for tau aggregation as mentioned previously. Interestingly, the microtubule-binding domains containing tau fragments were primarily released passively during cell death rather than active secretion, suggesting that the factors inducing cell death may be needed to seed tau aggregation.

35
2.4.2.3 Annular protofibrils-mediated membrane permeabilization
Free aSyn and tau have been proposed as the major extracellular species, but the exact mechanisms of how they are secreted by cells are poorly understood (Vella et al., 2016, Dujardin et al., 2014a). Both aSyn and tau have been reported to form intermediate annular protofibrils (APFs) that adopt ring/pore-like structures share homology with some bacterial toxins (Lasagna-Reeves et al., 2014, Ding et al., 2002). It was previously shown that many bacterial toxins such as α-hemolysin assemble into pore-like oligomers and permeabilize cell membrane, implicating that aSyn and tau APFs might also form pore in plasma membrane and induce release of free aSyn and tau, and thus provide an explanation for the observed membrane permeabilization induced by aSyn and tau in several in vitro and in vivo studies (Flach et al., 2012, Jones et al., 2012, Stefanovic et al., 2014, Parker and Feil, 2005, Montoya and Gouaux, 2003).
APFs of several disease-associated proteins including aSyn, tau and Aβ have been identified from human brain of pathological conditions (Lasagna-Reeves et al., 2011, Pountney et al., 2004, Lasagna-Reeves et al., 2014). Disease-associated mutations have been reported to accelerate the formation of aSyn and Aβ APFs (Lashuel et al., 2002a). Besides, both phosphorylation and disease-associated mutation (P301L) have been implicated to affect tau APFs formation (Lasagna-Reeves et al., 2014). A Recent study showed that aSyn amyloid fibrils could permeabilize membranes, and the treatment of flavonoid epigallocatechin gallate (EGCG), which is small molecule previously described to remodel aSyn aggregation pattern and reduced tau mutant aggregation, could inhibit this process (Wobst et al., 2015, Lorenzen et al., 2014). In this study, aSyn fibrils did not only permeabilize cell membrane but also vesicle membranes, suggesting that vesicle post-secretion release may also contribute to the excess of extracellular free aSyn and tau.
2.4.2.4 Macropinocytosis
Macropinocytosis is a subtype of bulk endocytosis and is defined by its function to uptake large volumes of extracellular liquid and dissolved molecules (Bloomfield and Kay, 2016). Macropinocytosis is highly conserved in eukaryotic cells and has different functions in various cell types, e.g. ingestion of extracellular material in leukocytes, bulk endocytosis in neuron during synaptic activity (Amyere and A. and Courtoy, 2001, Clayton and Cousin, 2009). However, it is worth mentioning that macropinocytosis involves extensive membrane turnover, and thus is not highly active in mature neurons. Macropinocytosis has been proposedas an important route for cells to uptake large aggregates during cell-to-cell transmission of protein aggregates (Zeineddine and Yerbury, 2015). Extracellular SOD1, TDP-43 and aSyn aggregates were recently reported to bind to the cell surface, followed by internalization viaRac1-mediated membrane ruffling and macropinocytosis (Zeineddine et al., 2015). The same study also demonstrated that these disease-associated proteins enter the cytosol by rupturing macropinosomes membrane. Low molecular weight recombinant tau aggregates and short fibrils were shown to be uptake by macropinocytosis at synaptic terminals, and transportedalong axons (Wu et al., 2013). It has also been reported that tau and aSyn fibrils bind to heparan sulphate proteoglycans, and stimulate macropinocytotic uptake of fluid andaggregates (Holmes et al., 2013). Amyloid Precursor Protein (APP) can also be internalized from cell surface via macropinocytosis that bypasses early and late endosome, and thus directsAPP to lysosomes in an Arf6-dependent manner (Tang et al., 2015). APP is degraded to A in lysosomes, suggesting a role of macropinocytosis in A accumulation that could contribute to AD pathogenesis (Tang et al., 2015).

36
2.4.2.5 Other routes for internalization of aSyn and tau
Some studies have proposed that aSyn may bind to dynamin, and enter cells via receptor-mediated endocytosis (Hansen et al., 2011, Desplats et al., 2009). Some other studiessuggested that there might a link between the uptake of aSyn and tau and adsorptive endocytosis, which exhibit the characteristic of both receptor-mediated endocytosis and non-specific bulk endocytosis (Volpicelli-Daley et al., 2011, Guo and Lee, 2011b). In these studies, the induction of cytoplasmic aggregates via seeding did not give rise to vesicle-bound species of aSyn or tau inside cells, suggesting that either receptor-mediated endocytosis is not the major route for aSyn and tau internalization or these proteins escape rapidly from vesicles into the cytosol.
Tunnelling nanotubes (TNTs) that serve a role in cell-to-cell communication have been proposed to participate in mediating prion proteins propagation (Gousset et al., 2009). A Recent study has demonstrated that TNTs also mediate cell-to-cell transmission of aSyn by providing a shortcut between two cells for transferring lysosomal vesicles that contain aSyn aggregates (Abounit et al., 2016). It is recently reported that impairment of UPS lead to the accumulation of misfolded proteins in the cytoplasm, and the excess misfolded proteins are deubiquitylated and packaged by ER-resident deubiquitylase USP19 into late endosomes, which then fuse with plasma membrane and release the misfolded proteins into extracellular space (Lee et al., 2016, Volkmar et al., 2016). Interestingly, apart from the donor cells and the recipient cells, a "third party," microglial cells were also suggested to participate in mediating cell-to-cell transmission of misfolded proteins. Microglial cells may collect and package misfolded proteins that are secreted by neurons with pathology from extracellular space into vesicles via phagocytic endocytosis, and deliver them back to neurons via vesicle-mediated transport events, e.g. exosomes (Asai et al., 2015a). It was previously noted that microglial cells internalize microvesicles, which contain Aβ and tau, from other neurons and neuroglia (Fruhbeis et al., 2013, Yuyama et al., 2012, Asai et al., 2015a). This may have a connection to the fact that the activation of microglia in in vivo AD models benefits amyloid pathology possibly by promoting A clearance while worsening tau pathology (Lee et al., 2013, Malm et al., 2016). Hence the participation of microglia to the propagation of pathology may be specific to tau, but further studies are needed to clarify these findings.
2.4.2.6 Stress granules
Stress granules (SGs) are transiently formed non-membrane-bound RNA granules nucleated by various RNA-binding proteins (RBPs) under stress conditions to reduce translation (Anderson and Kedersha, 2008, Gilks et al., 2004). Many of these RBPs, such as TDP-43, fused in sarcoma proteins and heterogeneous nuclear ribonucleoproteins, have been shown to be related to neurodegenerative diseases (Kim et al., 2013, Liu-Yesucevitz et al., 2010, Sreedharan et al., 2008, Vance et al., 2009). Increasing evidence support the hypothesis that stress granules (SGs) may provide a location for misfolded and normal proteins to interact, and possibly converting normal protein to pathological species upon interaction, hence seeding aggregation (Wolozin and Apicco, 2015). Mutation in valosin-containing protein gene that mediates autophagosomal clearance of SGs was reported to cause ALS (Johnson et al., 2010). SG makers have also been reported in several disease-associated inclusions (Bentmann et al., 2013). One SGs maker, TIA-1 has been reported to co-localize with NFTs in AD, and increase in the amount proportionally with increased diseases severity (Vanderweyde et al., 2012). It is yet unclear the relation between tau and SGs. Part of this thesis work partially provides a mechanistic explanation of how tauopathies propagate in an SGs mediated-manner.
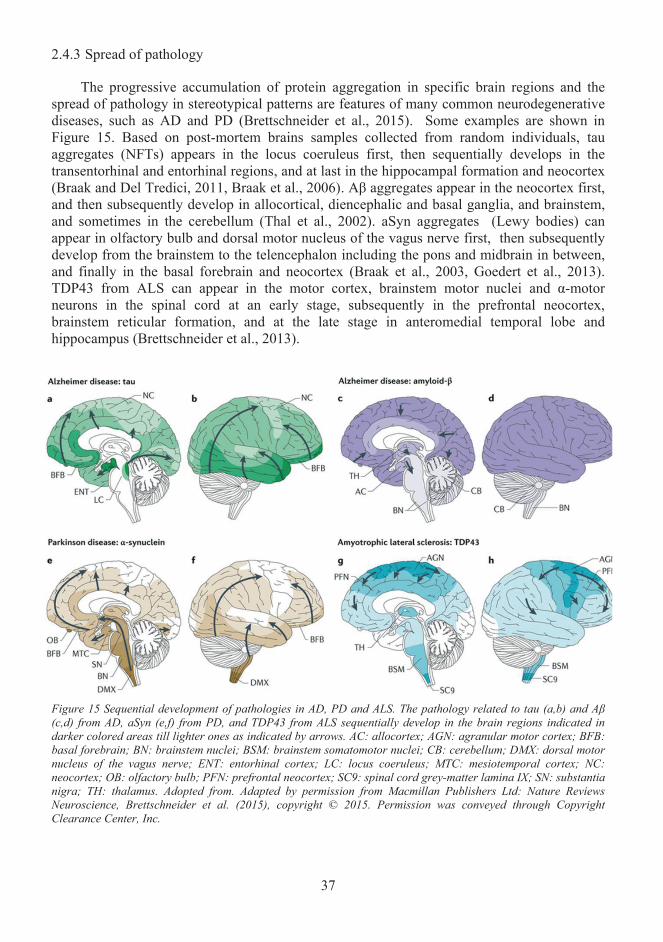
37
2.4.3 Spread of pathology
The progressive accumulation of protein aggregation in specific brain regions and the spread of pathology in stereotypical patterns are features of many common neurodegenerativediseases, such as AD and PD (Brettschneider et al., 2015). Some examples are shown inFigure 15. Based on post-mortem brains samples collected from random individuals, tauaggregates (NFTs) appears in the locus coeruleus first, then sequentially develops in the transentorhinal and entorhinal regions, and at last in the hippocampal formation and neocortex(Braak and Del Tredici, 2011, Braak et al., 2006). Aβ aggregates appear in the neocortex first,and then subsequently develop in allocortical, diencephalic and basal ganglia, and brainstem, and sometimes in the cerebellum (Thal et al., 2002). aSyn aggregates (Lewy bodies) canappear in olfactory bulb and dorsal motor nucleus of the vagus nerve first, then subsequently develop from the brainstem to the telencephalon including the pons and midbrain in between,and finally in the basal forebrain and neocortex (Braak et al., 2003, Goedert et al., 2013).TDP43 from ALS can appear in the motor cortex, brainstem motor nuclei and α-motor neurons in the spinal cord at an early stage, subsequently in the prefrontal neocortex, brainstem reticular formation, and at the late stage in anteromedial temporal lobe andhippocampus (Brettschneider et al., 2013).
Figure 15 Sequential development of pathologies in AD, PD and ALS. The pathology related to tau (a,b) and Aβ(c,d) from AD, aSyn (e,f) from PD, and TDP43 from ALS sequentially develop in the brain regions indicated in darker colored areas till lighter ones as indicated by arrows. AC: allocortex; AGN: agranular motor cortex; BFB:basal forebrain; BN: brainstem nuclei; BSM: brainstem somatomotor nuclei; CB: cerebellum; DMX: dorsal motor nucleus of the vagus nerve; ENT: entorhinal cortex; LC: locus coeruleus; MTC: mesiotemporal cortex; NC:neocortex; OB: olfactory bulb; PFN: prefrontal neocortex; SC9: spinal cord grey-matter lamina IX; SN: substantia nigra; TH: thalamus. Adopted from. Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Neuroscience, Brettschneider et al. (2015), copyright © 2015. Permission was conveyed through Copyright Clearance Center, Inc.

38
2.4.4 The spread of pathology- what is missing from the story?
The sequential appearances of the accumulation of NFTs in AD and Lewy bodies in PD in different brain regions characterized by Braak and colleagues have been widely cited to support the hypothesis of the spread of pathologies that are via certain neuronal networks and pathways (Brettschneider et al., 2015). However these elegant studies were based on post-mortem brains from uncontrolled subjects at various age and various causes, and thus wereargued not to fully explain the progression of the pathology (Walsh and Selkoe, 2016). Also, it has been argued that the clinical severity of diseases is not always associated with amyloidogenesis, e.g. synucleinopathy often occurs without neurological sign in aged-human autopsy brain and animal models (Burke et al., 2008). Alternatively to the hypothesis of thespread of pathology, the sequential appearance of the pathologies in different brain regionswas proposed to be a result of selective neuronal vulnerability (Walsh and Selkoe, 2016). The concept of selective neuronal vulnerability suggests that certain neurons are intrinsically more vulnerable than others during pathogenic processes (Jackson, 2014). What determines “intrinsic vulnerability” is poorly understood, and it may refer to gene expression profiles that make some neuron more and earlier dysfunctional and structurally abnormal than others.Apart from neurons, microglial cells also exhibit changes in number, density and gene expression during AD pathology, e.g. several genes including TREM2, CD33 mainly expressed in microglia are associated with AD risk, and thus suggesting that the intrinsic vulnerability of neurons may be influenced by neighboring microglial cells (Grubman et al., 2016, Wes et al., 2016, Bachstetter et al., 2015). Interestingly, in autopsy-based studies, early aSyn aggregates can be detected in the neurons of gut, submandibular gland and olfactory bulb,and this corresponds to the fact that some patients with early PD developed symptoms such as dysphagia and constipation, thus suggesting that the free transfer of disease-associated protein is not only restricted in CNS but also in peripheral nervous system (Del Tredici et al., 2010, Edwards et al., 1992, Hawkes et al., 1999, Holmqvist et al., 2014, Wakabayashi et al., 1990).
Currently, the exact mechanisms of cell-to-cell transmission of disease-associatedproteins are poorly understood. Thus there lacks a bridge connecting the emerging evidence of seeded propagation of pathology from in vitro studies and the observed stereotypical patterns of pathology in vivo. In animal experiments, certain groups of neurons are sequentially affected depending on the location of the initial injection of pre-formed fibrils (Pffs). For example, injection of tau Pffs into the striatum of transgenic mice induced tau aggregation in a time-dependent manner in substantia nigra and thalamus, which are both connected to striatum, whereas injection of tau Pffs into the hippocampus induced sequential aggregation of tau in entorhinal cortex and contralateral hippocampus (Iba et al., 2013a). Injection of brain lysates from the PD-mice model or aSyn Pffs into the neocortex and striatum of aSyn transgenic mice induced Lewy body-like inclusions in the interconnected area such as frontal cortex, thalamus, hypothalamus, brainstem nuclei, and major white-matter tracts (Luk et al., 2012d). Intrastriatal inoculation of aSyn Pffs in wild-type mice induced Lewy body-like inclusions in the ventral striatum, thalamus, and occipital cortex in a time-dependent manner from 30-180 days post injection (Luk et al., 2012c). Different groups of neurons are affected depending on the location of Pffs inoculated but not neuron types, suggesting that gene profile of an individual group of neurons may not be the key determinant of the propagation of pathology. On the other hand, the human brain is so complex, based on countless neuronal connectivity it is not fully understood why the propagation of observed lesions only follow certain neuroanatomical pathways (Walsh and Selkoe, 2016). Thus neither selective neuronal vulnerability nor spread of pathology via a certain neuronal network would be a perfect model to explain the

39
pathogenic pathways of AD and PD (Brettschneider et al., 2015). Alternatively, selective vulnerability of certain neuronal network may also be an explanation. Lastly, it is worth questioning that to what extend mouse model represent human, e.g. some transgenic mouse with NFT showed no disruption in neuronal functions (Santacruz et al., 2005, Berger et al., 2007).
In conclusion, despite the missing knowledge in the field, such as the initiation of aSyn and tau proteins misfolding, what are the species of aSyn and tau that are transmissible, andhow misfolded aSyn and tau transfer between cells, it has been suggested that therapeutics should focus on preventing the cellular release and uptake of the misfolded proteins(Brettschneider et al., 2015, Walsh and Selkoe, 2016). Understanding the pathway(s) of cell-to-cell transmission of misfolded aSyn and tau could provide novel insights into the etiologyof pathogenesis and new drug targets.

40
3 Aims of the study
The principal aim of this thesis was to develop a novel live-cell platform method basedon the principle of protein-fragment complementation assay, to investigate the hence oligomerization and cell-to-cell transmission of aSyn and tau, and further explore cellularprocesses regulating secretion and uptake of aSyn and tau.
The specific aims of the thesis were to:
1. Set up and functionally validate an assay platform to study the effect of prolyl oligopeptidase on intracellular dimerization, cellular secretion and uptake of aSyn.
2. Set up and functionally validate an assay platform to study the effect of late-onset Alzheimer’s disease susceptibility gene silencing on intracellular dimerization, cellular secretion and uptake of tau.
3. Conduct mechanistic studies to further understand the fate of tau after internalization from the extracellular space, and to explore potential mechanisms involved in tau uptake-induced cellular stress.

41
4 Materials and methods
The methodology is briefly described here, for more detailed pleases refer to materials and methods/ experimental procedures sections of adjoining original publications (I-III).
Plasmid constructs and recombinant proteins4.1
The split Gaussia princeps luciferase (GLuc) plasmids were originally provided by prof. Stephen Michnick (Université de Montréal, Montreal, Canada), and are expressed as previously described (Michnick, 2001). Human aSyn cDNA was obtained from ORFeome library (accession number BC013293, Version 3.1, Genome Biology Unit, Institute of Biotechnology, University of Helsinki), and was used to clone aSyn(Δ118–140) and aSyn(Δ98–140) (with amino acid 118-140 or 980-140 removed respectively), and were PCR-cloned into GLuc vector with HA-tag at KpnI-XhoI restriction sites (Publication I/Figure 2A and 4A). The human PREP and PREP(S554A) plasmids were as previously described,and were cloned into PREP-GLuc2 and PREP(S554A)-GLuc2 (publication I/Figure3A)(Savolainen et al., 2014). The human tau cDNAs was purchased from Thermo (isoform 0N4R, BC114948), and was cloned to GLuc as previously described (Nykanen et al., 2012).Human CD2AP cDNAs were purchased from Thermo (BC019744). The FRMD4A–GFP plasmid was kindly provided by Junichi Ikenouchi (Kyushu University, Fukuoka, Japan).pcDNA3/HA-Arf6 (Addgene plasmid # 10834) was kindly provided by Thomas Roberts (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA). pCMV5B-Flag-Par6 wt (Addgene plasmid # 11748) and pMEP5-Flag Par6 S345A (Addgene plasmid # 24648) were kindly provided by Jeff Wrana (Department of Molecular Genetics, University of Toronto, Toronto, Canada). aPKCζ cDNA was from ORFeome library (University of Helsinki)and was used to clone C20ζ (amino acids 405–592) into the pcDNA6-V5/His expression plasmid (Invitrogen). TIA-1 cDNA was purchased from Thermo (BC046812.1). TauE14plasmid was kindly provided by Mel B. Feany (Harvard Medical School, Boston, MA, USA). Tau(P301L) plasmid (Addgene plasmid #46908) was kindly provided by Karen Ashe (University of Minnesota, Minneapolis, MN, USA). Split GFP plasmids (1–214 and 215–230amino acid) were obtained from Sandia Biotech Inc., USA. Human TIA-1, TREM2 and CD33shRNA, and mouse FRMD4A shRNA (in pLKO.1 vector backbone) were obtained from the TRC1.0 library (Functional Genomics Unit Biomedicum Helsinki). siRNAs for silencing LOAD risk genes follow-up from Invitrogen (Ambion Silencer Select Predesigned siRNA). Real-time quantitative polymerase chain reaction (qPCR) was used to determine the knockdown efficacy of the siRNAs with primers as previously described (Martiskainen et al., 2015a). Primers for qPCR of TIA-1 were: (forward) 5′-ACAGCAGAACAAAGGAACCC-3′, (reversed) 5′-TGTCTGTTTCCTTGCTGGTT-3′. PREP(S554A) protein was kindly provided by Anne-Marie Lambeir (University of Antwerp, Belgium). All plasmids were sequenced to confirm their identity. Recombinant porcine PREP was purified as previously described (Venalainen et al., 2006).
Chemicals4.2
Dimethyl sulfoxide (DMSO), sodium arsenite, rotenone, salubrinal, DBeQ, GW4869 and recombinant human aSyn protein were purchased from Sigma-Aldrich. aSyn protein was expressed in E. coli, and thus may not be correctly folded. SecinH3 was purchased from R&D Systems. TAT-TAMRA peptide was purchased from Anaspec. KYP-2047 was synthesized by

42
the School of Pharmacy, University of Eastern Finland, as previously described (Jarho et al., 2004). Hoechst 33342 was purchased from Invitrogen.
Antibodies4.3
The following primary antibodies were used: anti-HA (Sigma), Tau-5 (Invitrogen), AT8 (Thermo), anti-PREP (R&D Systems), anti-PREP serum (collected from a rabbit immunized against DPDSEQTKAFVEAQNK peptide, and validated by ELISA from Thermo), anti-Flotillin-1 (BD Transduction Labs) and anti-GAPDH (Millipore). The following secondary antibodies were used: Alexa-Fluor-568-conjugated anti-mouse-IgG secondary antibody (Invitrogen) and horseradish peroxidase-conjugated secondary antibodies (Thermo). The dilutions of antibodies were as recommended by manufacturers and providers, e.g. 1:500-1:1000 for Western blotting and IF.
Cell culture and transfections4.4
Mouse Neuro-2A (N2A) neuroblastoma cells, human embryonic kidney 293T cells (HEK293T) and human microglial CHME-5 cells (kindly provided by Dr. Mikko Airavarra, University of Helsinki) were cultured in full Dulbecco’s Modified Eagle Medium [DMEM with 1% (v/v) L-Glutamine-Penicillin-Streptomycin solution (Lonza), 10% (v/v) FBS (Gibco, Invitrogen)]. Mouse primary cortical neurons were prepared as previously described (Nykanen et al., 2012). Primary neurons were cultured in neurobasal medium with 2% B27 supplement, 1% penicillin/streptomycin, and 1% glutamine. 50% of the medium Ultracentrifuge every three days. All cells were cultured at 37°C, 5% CO2 and water saturated air.
JetPei and Jetprime (Polyplus) were used to transfect N2A, HEK293T and CHME5-cellswith plasmid DNA and/or siRNA-DNA according to manufacturer’s instructions. Neurons were transduced at 21 DIV with the lentiviral vector that was prepared and operated as previously described (Kysenius et al., 2012). Media was conditioned for three days post-transduction. Real-time quantitative (qPCR, SYBR Green Maxima kit, Thermo, and RT-PCR cycler, Bio-Rad CFX95) was used to study the knockdown efficacy of siNRAs and shRNAs. Quantitation of qPCR data used comparative Ct method and was normalized to GAPDH.
Protein-fragment complementation assay4.5
Protein-fragment complementation assay (PCA) is a live-cell method to study protein-protein interactions. In PCA, proteins of interest are tagged with fragments of the reporter protein. The reporter protein fragment reconstitutes enzymatic activity if they are brought to proximity less than 10Å during the interaction of the protein of interest, and generate bioluminescence when the substrate is present (Figure 17A)(Michnick et al., 2007).Humanized Gaussia princeps luciferase was used as a reporter (GLuc, 19.9 kDa, 185 aa), and native coelenterazine (nCol, NanoLight Technology) as substrate.
Cells were transiently transfected with proteins tagged or untagged to reporters and/or cotransfected with siRNA (100 ng of DNA and 5 nM of siRNA per well) 24 h post-plating on 96-well plates (9,000-10,000 cells per well, poly-L-lysine-coated) as previously described (Martiskainen et al., 2015b). PCA signal is measured from cells 48 h post-transfection. 30min before signal measurement, media was changed to phenol red-free DMEM (Gibco, Invitrogen) without serum (PRF-DMEM) after gently washing cells with PBS. Victor3 1420 Multilabel Counter (PerkinElmer) or Varioskan Flash multiplate reader (Thermo) are used to titrate nCol

43
(final concentration of 20 M) and monitor signal from 4 replicates wells of each condition. Chemicals were pre-diluted into PRF-DMEM media and applied to cells.
4.5.1 Tau and aSyn secretion assay
Cells were plated and transfected as described above. 16 h before signal measurement,cells were gently washed once with warm PBS and changed to PRF-DMEM (140 μl per well). 30 minutes before signal measurement, cells were spin down at 200-300 g for 3-5 minutes. Conditioned cell-free PRF-DMEM was replaced with fresh PRF-DMEM, and transferred to different 96-well plates. PCA signal from 75 μl of this conditioned media and cells were read separately. 50 μl of the conditioned media was analyzed with lactate dehydrogenase (LDH) release assay according to manufacturer’s instructions (Promega CytoTox 96® Non-Radioactive Cytotoxicity Assay, G1781).
4.5.2 Assessment of tau and aSyn uptake
4.5.2.1 Conditioned media production
HEK293T or N2A cells were plated into poly-L-lysine-coated 10-cm plates at a density of 2-3 million cells per plate and transiently transfected with 9 mg of tau-GLuc1/2 or aSyn1/2 plasmids 24 h post-plating. Cells were washed once with PBS to remove remainingtransfection reagent 24 h post-transfection. Then media was changed to PRF-DMEM. Media was conditioned for 24 hours. Next, media was collected, and centrifuged at 3,000 g for 30 min to remove cell debris. The level of tau-GLuc1/2 and aSyn-GLuc1/2 dimers/oligomers in the media was determined by PCA. Total human Tau ELISA kit was used to determine tau level in the conditioned media according to manufacturer’s instructions (#KHB0041, Novex/Life Technologies). Media (full neurobasal media) was conditioned from mouse primary cortical neurons for 3 d post-transduction, and tau in the conditioned media was determined by mouse-specific tau ELISA kit (KMB7011, Novex, ThermoFisher).
4.5.2.2 Tau uptake assay
Naïve N2A cells or HEK293T cells were plated and transfected as described above on 96-well plates. 24 h post-transfection, cells were washed once with PBS and changed toconditioned media that contained reporter proteins and were produced as described above.Cells were exposed to conditioned media for 4 h, and washed with PBS, then incubated with 20 μg/ml heparin (Sigma) diluted in PRF-DMEM for 5 min [remove cell surface-boundproteins (Holmes et al., 2013)]. Next, cells were washed with PBS and changed to 75 μl PRF-DMEM, and intracellular PCA signal was read as described above. All wash processes were done gently to prevent disturbing cells.
Electrophoretic techniques4.6
4.6.1 Western blotting
Western blotting (WB) were done as previously described (Nykanen et al., 2012). Cells were plated and transfected as mentioned above. 48 h post-transfection, cells were washed twice with ice-cold PBS, and extracted with extracting buffer [10 mM Tris-HCl, pH 6.8, 150mM NaCl, 1 μM NaF, 1% Triton X-100, 0.25% Nonidet P-40, 1 mM EDTA, Protease and Phosphatase Inhibitor cocktail tablets (Roche)], and incubated on ice for 30 min. Extracts were

44
centrifuged at 16,000 g for 10 min to remove debris, and BCA protein assay kit (Thermo)was used to determine protein concentration according to manufacturer’s instruction. Equal amounts of total proteins from cell lysates were loaded per well, and ran on 4–12% gradient Bis-Tris gels according to manufacturer’s instruction (Novex, Invitrogen). Proteins were then transferred from the gel to PVDF membranes (GE Healthcare) via semidry blotting (Bio-Rad). Blots are stained with primary antibodies as mentioned in the chemical section. Horseradish-conjugated secondary antibodies and ECL Western blotting detection reagent (Thermo) were used to generate chemiluminescence. QuantityOne software (Bio-Rad) was used to analyse the blots semi-quantitatively.
4.6.2 Native polyacrylamide gel electrophoresis
Native Polyacrylamide Gel Electrophoresis (PAGE) in publication I was prepared and performed as previously described (Szeltner et al., 2013). 6 μg of recombinant porcine PREP or PREP(S554A) proteins were pre-incubated with KYP-2047 or DMSO for 30 min at room temperature. An equal amount of sample per lane was run on 10% native gel containing a stacking gel. The gel was fixed using destaining buffer (45% methanol, 10% acetic acid). 30 min post-fixation, the gel was stained in 0.1% Coomassie Brilliant Blue R-250 for 2 h with additional washes.
In publication II, conditioned media was prepared as described above, and concentrated using Amicon Ultra-15 Centrifugal Filters (Merck Millipore, UFC903008). Native-PAGE Novex™ Tris-Glycine Native Sample Buffer (LC2673), Novex™ Tris-Glycine Native Running Buffer (LC2672) and NuPAGE™ Novex™ 3-8% Tris-Acetate Protein Gels (EA0375BOX; Novex, ThermoFisher Scientific) were used to prepare and ran samples according to manufacturer's instruction. An equal volume of sample was loaded per well. Gels of Native-PAGE from both publication I and II was then blotted and analysed as described in Western blotting.
Fractionation techniques4.7
4.7.1 Protein fractionation
Protein solubility was studied by protein fractionation. Protein samples were extracted from cells as described above. After incubation on ice, an equal volume of samples was loaded into polycarbonate centrifugation tubes (Beckman Coulter), and centrifuged at 100,000 x g for 30 min using TLA-100 rotor and a tabletop ultracentrifuge (OptimaTM ultracentrifuge, Beckman Coulter). After centrifugation, soluble fraction was collected into a different tube. Insoluble pellets were washed once with milliQ-water and resolved in 50 μl of Laemmli buffer (75 mm Tris-HCl with pH 6.8, 3% SDS, 15% glycerol, 3.75 mm EDTA, pH 7.4) with the aid of a rod sonicator (Labsonic® M.B. Braun Biotech International). Both samples from soluble fractions and insoluble fractions were then analysed by Western blotting as described above.
4.7.2 Media fractionation
Conditioned media was produced as described above, and was fractionated into vesicle-free, larger microvesicles or ectosomes and exosomes fractions with a previously described method (Théry et al., 2006). Briefly, media was centrifuged in a sequence of 3000 x g for 30min (to clear debris), 20,000 x g for 60 min (to pellet the ectosomal fraction), and 100,000 gfor 70 min (to pellet the exosomal fraction) using ultra-centrifuges (SW41 Ti rotor, Sorvall

45
WX Floor Ultra centrifuge). The supernatants from each centrifuge step were gently collected without disturbing the pellets. The pellets were resolved in PBS first, and re-centrifuged to pellet again, and were resuspended either in PRF DMEM for PCA measurement, or 1.5x Laemmli buffer for Western blotting analysis. Flotillin-1 (BD Transduction Labs #F65020, 1:500) was used as a marker for ectosomal vesicles.
Protein crosslinking 4.8
Proteins in cell lysates and conditioned media were crosslinked to study their oligomeric state. Samples from cell lysates and conditioned media were prepared as described above. Conditioned media was concentrated by Amicon Ultra-15 Centrifugal Filters (Merck Millipore, UFC903008). All samples were crosslinked by BS3 (final concentration of 5 mM; Pierce, Thermo) according to the manufacturer's instructions. Tris-HCl (final concentration of 50 mM, pH 7.5) was used to quench the crosslink reaction buffer. Samples of equal volume were analysed by Western blotting.
Microscale Thermophoresis 4.9
Microscale Thermophoresis (MST) is a novel cell-free method studies interaction between molecules such as proteins and chemicals (NanoTemper Technologies). Recombinant porcine PREP and PREP(S554A) proteins (activity was validated as previously described) were fluorescently labelled with the Monolith NT.115 protein labelling kit (NT-647 N-hydroxysuccinimide, NanoTemper Technologies) according to the manufacturer's instructions (Savolainen et al., 2014). Labelled PREP and PREP(S554A) were eluted in PBS with 0.05% Tween-20. Monolith NT.115 Microscale Thermophoresis device and standard treated capillaries (NanoTemper Technologies) were used to study the interaction between aSyn, PREP/PREP(S54A) and KYP-2047 (final concentration of 0.05% Tween-20, 0.5 mg/ml BSA, and 2.5 mm DTT in PBS). Equal amounts of labelled PREP or PREP(S554A) proteins were titrated by recombinant human aSyn (starting from 35 μM; Sigma, #S7820) or KYP-2047 (starting from 10μM) in series dilution. Curve-fitting was done by NTanalysis software (NanoTemper Technologies). GraphPad Prism software was used to calculate the Kd values using non-linear regression and one site-specific binding with Hill slope.
Immunofluorescence microscopy 4.10
Immunofluorescence microscopy (IF) was performed as previously described (Kysenius et al., 2012). Antibodies used are listed in the Antibodies section. Zeiss AxioImager M1 epifluorescence microscope was used to take images. Adobe Photoshop was used compile raw images. Stress granule-positive cells per total number of cells were manually quantified in randomly selected fields. Each of selected field contained at least 50 cells.
TAT-TAMRA peptide (Anaspec) requires live cell activity. In live cell immunofluorescence imaging, naïve HEK293T cells were exposed to tau-BiFC conditioned media for 4 h, and 10 μM of TAT-TAMRA peptide was added to media in the last hour. Then media was changed to PRF DEME with 15 mM Hepes buffer (Life Technologies) before taking the image. The image was taken with Zeiss LSM 710 upright confocal and analysed with ImageJ software.

46
Cell viability4.11
Two cell viability assays were run side-by-side to monitor cell viability and membrane integrity according to manufacturers' instructions. In resazurin assay, cells were exposed to PRF DEME with resazurin sodium salt (final concentration of 100 μM; Sigma) at +37 °C with 5% CO2 for 2 hours. Fluorescence was measured at 530 nm excitation and 590 nm emission filters by Victor as previously described (Asai et al., 2015b).
Cellular release of LDH was measured by CitoTox 96 Non-Radio Cytotoxicity Assay (Promega). LDH level from cell and media was determined by measuring absorbance at 490 nm with Victor plate reader 30 min after applying reagent. Relative LDH release is calculated as a ratio of LDH (media): LDH (total). Three independent experiments with 4-8replicate wells per condition were analysed.
Statistical analyses4.12
A minimum of three independent replications were used for each experiment, and four independent wells per condition were calculated in average as one datapoint in PCA (I, II, III).Microsoft Excel software and GraphPad Prism were used to make statistical analyses and graphs. Statistical significance was evaluated with Student's t-test (two groups) or one-wayANOVA with Bonferroni's post-tests (three or more groups), with the significance threshold set at p < 0.05 (*), and ** = p < 0.01 and *** = p < 0.001 (I, II, III).

47
5 Results
Clarifying molecular mechanisms of prolyl oligopeptidase modulating α-Synuclein 5.1intracellular dimerization (I)
Dimerization of aSyn is the first event towards aSyn oligomerization and aggregation. To study the molecular mechanisms of aSyn dimerization, a live-cell platform method based onprotein-fragment complementation assay (PCA) using split humanized Gaussia princeps luciferase (GLuc) was adopted to monitor aSyn dimerization in an in vitro model quantitatively. PCA detects direct protein-protein interaction by the reconstitution of complementary reporter protein fragments tagged to proteins of interest as described in Figure 17A (Remy and Michnick, 2006a). Compared with other PCA methods such as split GFP, reconstitution of complementary GLuc-reporter protein fragments is reversible. Thus GLuc-based PCA in live-cell platform also monitors the dynamics of the protein-protein interactions in their native cellular environment (Chun et al., 2007a).
5.1.1 Prolyl oligopeptidase promotes α-Synuclein dimerization
Prolyl oligopeptidase (PREP) was previously shown to modulate aSyn aggregation in vitro and in vivo as reviewed in section 2.2.5. We chose to co-express PREP and aSyn-GLuc (aSyn tagged with GLuc-reporters) in N2A neuroblastoma cells, which express a low level ofendogenous aSyn and PREP (I/Figure 1). PREP(S554A) is an enzymatically inactive PREPmutant and was used as a control. Interestingly, both PREP and PREP(S554A) overexpression mildly increased aSyn level (I/Figure 1). A previously developed and functionally validated PCA assay for monitoring aSyn dimerization was used to study if aSyn dimerization was increased by PREP (Yan, 2013). We overexpressed aSyn-GLuc1 and aSyn-GLuc2 (hereafter aSyn-GLuc1/2, with constructs indicated in I/Figure 2A) at a constant level while co-expressing PREP and PREP(S554A) at increasing level. As shown in I/Figure 2B, aSyn dimerization was increased in a dose-dependent manner with increasing PREP and PREP(S554A) overexpression, suggesting that overexpression of PREP promotes aSyn dimerization, and this is independent of its enzyme activity.
Inhibition of PREP was shown to reduce the amount of aggregated aSyn in cells and transgenic mice overexpressing aSyn (Brandt et al., 2008, Myohanen et al., 2012a). We treated cells co-expressing PREP or PREP(S554A) and aSyn-GLuc1/2 with KYP-2047, a small-molecule inhibitor of PREP enzyme activity, and observed a reduction in PREP-induced aSyn dimerization in cell overexpressing wild-type PREP but not PREP(S554A) mutant (I/Figure 2C). This effect was specific to PREP as the KYP-2047 treatment had no effect on controlcells overexpressing aSyn only (I/Figure 2D). Besides, overexpression of PREP or treatmentof KYP had no effect on aSyn solubility (I/Figure 2E) suggesting that we are studying an early event of aSyn aggregation pathway, where aSyn stayed mostly as soluble dimer or oligomers.
5.1.2 Direct protein-protein interaction between prolyl oligopeptidase and α-Synuclein
PREP was reported to co-localize with aSyn in Lewy bodies in PD patients brain (Hannula et al., 2013). Hence to explore possible mechanisms of PREP-induced aSyn dimerization, we decided to address the potential interaction between aSyn and PREP by generating PREP-GLuc2 and PREP(S554A)-GLuc2 constructs (I/Figure 3A). We validated the expression level and activity of PREP-GLuc2 and confirmed that PREP-GLuc2 and

48
PREP(S554A)-GLuc2 are expressed in N2A cells but at a lower level than untagged PREP (I/Figure 3B), and that PREP-GLuc2 maintained hydrolytic activity (I/Figure 3C).
In PCA study, we observed a significant amount of PCA signal from co-expression of aSyn-GLuc1 with both PREP-GLuc2 or PREP(S554A)-GLuc2 (I/Figure 3D), where aSyn-PREP(S554A) PCA signal was slightly higher than aSyn-PREP. This suggests that there is an interaction between aSyn and PREP and PREP(S554A), and the aSyn-PREP interaction could be independent of PREP enzyme activity. We further tested the interaction of aSyn-PREP and aSyn-PREP(S554A) combination using Microscale Thermophoresis (MST), a cell-free system that quantitatively monitors direct protein-protein interactions in solution (Wienken et al., 2010). In the MST study, recombinant aSyn and PREP interacted with an affinity of Kd=2.96μM, whereas recombinant aSyn and PREP(S554A) interaction had a Kd of 1.41 μM (I/Figure 5A, B). Surface plasmon resonance was used to confirm the direct interaction and showed aKd of 3.61 μM of aSyn-PREP interaction (I/Figure 5C). These results are in line with PCA data suggesting that aSyn-PREP(S554A) interaction had a slightly higher affinity than aSyn-PREP, and thus confirmed the direct interaction between aSyn and PREP.
PREP was previously reported to cleave at proline residues at the C-termini of its substrates, and aSyn contains five proline residues at its C-terminus (Walter et al., 1971). To confirm that aSyn-PREP interaction is independent of PREP enzyme activity, we generated two aSyn mutants, aSyn(Δ118–140)-GLuc1 (lacking three proline residues) and aSyn(Δ98–140)-GLuc1 (lacking all proline residues) as illustrated in I/Figure 4A. Both aSyn mutants are expressed in N2A cells as confirmed by WB (I/Figure 4B). Expression of all three pairs of aSyn (mutants)-GLuc1 with PREP-GLuc2 showed a comparable level of PCA signal (I/Figure 4C), suggesting that the C-terminal proline residues of aSyn were not needed for PREP binding. This is in line with a previous study, which showed that aSyn is not cleaved by PREP hydrolytic activity (Brandt et al., 2008). The signal from aSyn(Δ98–140)-GLuc1 and PREP-GLuc2 was about 30% lower as compared to the other two pairs, but this may be explained by the slightly lower expression level of aSyn(Δ98–140)-GLuc1 (I/Figure 4B).
5.1.3 KYP-2047 alters the conformation of PREP
As a previous study indicated that inhibition of PREP by KYP-2047 reduced PREP-dependent aSyn dimerization, we studied the effect of KYP-2047 on aSyn-PREP interaction using PCA. We treated N2A cells with KYP-2047 and observed roughly a doubled PCA signal with aSyn-PREP interaction but an insignificant change in aSyn-PREP(S554A), suggesting that KYP-2047 binding enhances PREP interaction with aSyn (I/Figure 3E). KYP-2047 was previously shown to bind and modify the conformation of PREP (Szeltner et al., 2013). We confirmed KYP-2047 binding to both recombinant porcine PREP and PREP(S554A) by using MST as described in Figure 16, where the Kd values are within physiological range. We preincubated recombinant porcine PREP and PREP(S554A) protein with KYP-2047, and resolved on naive PAGE gel. With Western blotting analysis, weobserved three bands of PREP and PREP(S554A) on the native gel (I/Figure 6). This could indicate the three different conformations of PREP, monomeric compact, oligomeric and monomeric open form, as previously described (Szeltner et al., 2013). However, after pre-incubation with KYP-2047, PREP adopted only one faster-migrating band (I/Figure 6), which represents the monomeric compact form of PREP. This KYP-2047-induced conformational change of PREP was not observed with PREP(S554A), although KYP-2047 also binds to PREP(S554A). This could provide an explanation about the insensitivity of PREP(S554A) to KYP-2047 treatment observed in PCA assays.

49
Figure 16 KYP-2047 binding to PREP (A) and PREP (S554) (B) was studied by MST. An equal amount of fluorescence labeled, purified recombinant PREP or PREP(S554A) was titrated by KYP-2047 at various concentrations. KYP-2047 binds to PREP and PREP(S554A) at Kd of ~5.8 nM and ~2.7 nM, respectively. The curve and Kd were calculated by averaging Kd curves assimilated using NTanalysis software from independent experiments (mean ± SEM; N=3 independent experiments).
Assay development for studying secretion and uptake of α-Synuclein and tau (I, II)5.2
We utilized highly sensitive PCA-based platform as a basis (Figure 17A), and further developed the platform to monitor cellular release and uptake of tau and aSyn dimers. As shown in II/Figure 1, the concept of monitoring release of protein dimers via PCA is to measure both PCA signal from cells and corresponding media at the end of the experiment, hence quantitatively analysing the amount of dimers that have been released into media from the cells. In uptake assay, recipient cells that are not expressing GLuc-tagged proteins are incubated with the conditioned media that contain reporter protein dimers. By measuring the PCA signal in recipient cells at the end of the experiment after thorough washing, we can quantitatively analyse the amount of dimers that have been internalized.
5.2.1 Development of an assay for studying α-Synuclein release and uptake
PREP has been previously suggested to bind to the microtubule, and serve a role in intracellular transport and protein secretion by regulating axonal transport in neuroblastoma cells (Morawski et al., 2011). miRNA silencing and chemically inhibition of PREP were also reported to increase the overall protein secretion in glioma cells (Schulz et al., 2005). As afollow-up study of the aSyn-PREP project, we wanted to investigate if PREP modulates aSyn dimer release and uptake. We first co-expressed aSyn–GLuc1/2 constructs in increasing amount (Figure 17B). The PCA signal in washed cell monolayers and media increased in a dose-dependent manner, suggesting both intracellular and secreted aSyn levels correlated with the level of aSyn–GLuc1/2 reporter gene dose. To confirm the observed signal in the medium is specific to extracellular aSyn dimer and is not due to passive release related to cell death, we expressed aSyn–GLuc1 and aSyn–GLuc2 separately. Expression of aSyn reporters alone did not generate luminescence signal in either cells or media (Figure 17C). aSyn dimer PCAsignal from the medium was independent of changes in lactate dehydrogenase (LDH) release, suggesting that the release of aSyn dimers into the media was not a result of passive release or membrane leakage. During an 18-h observation period, aSyn–GLuc1/2 reporters accumulated in serum-free culture medium with nearly linear kinetics for up to 12 hours, as shown by an LDH-normalized aSyn secretion PCA (Pearson r2=0. 0.9337)(Figure 17D). Using the aSyn secretion assay, we co-expressed aSyn with PREP or PREP(S554A). Both PCA signals from
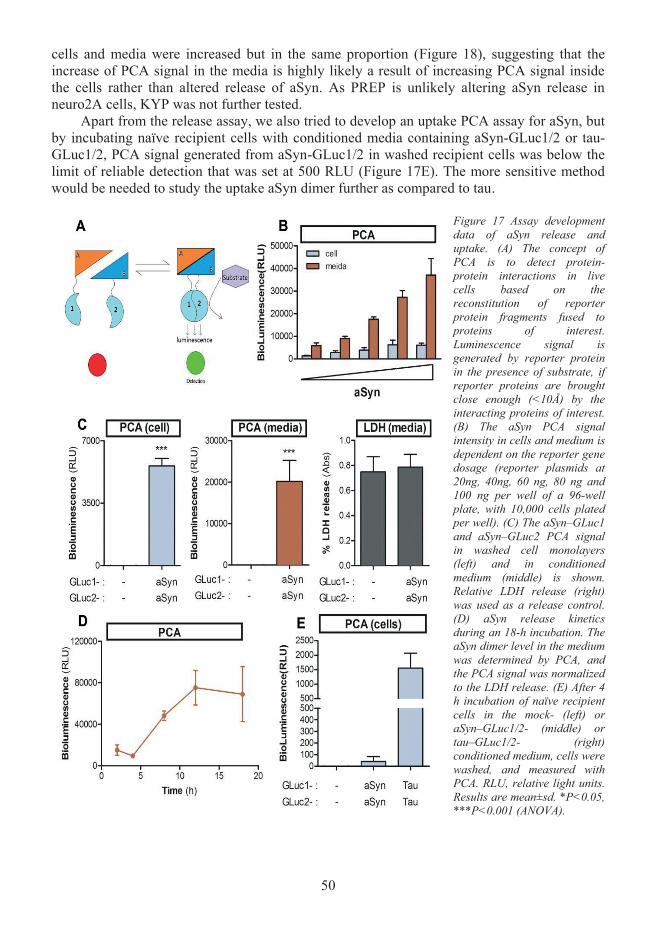
50
cells and media were increased but in the same proportion (Figure 18), suggesting that the increase of PCA signal in the media is highly likely a result of increasing PCA signal inside the cells rather than altered release of aSyn. As PREP is unlikely altering aSyn release in neuro2A cells, KYP was not further tested.
Apart from the release assay, we also tried to develop an uptake PCA assay for aSyn, but by incubating naïve recipient cells with conditioned media containing aSyn-GLuc1/2 or tau-GLuc1/2, PCA signal generated from aSyn-GLuc1/2 in washed recipient cells was below the limit of reliable detection that was set at 500 RLU (Figure 17E). The more sensitive method would be needed to study the uptake aSyn dimer further as compared to tau.
Figure 17 Assay development data of aSyn release and uptake. (A) The concept of PCA is to detect protein-protein interactions in live cells based on the reconstitution of reporter protein fragments fused to proteins of interest.Luminescence signal is generated by reporter protein in the presence of substrate, if reporter proteins are brought close enough (<10Å) by the interacting proteins of interest.(B) The aSyn PCA signal intensity in cells and medium is dependent on the reporter gene dosage (reporter plasmids at 20ng, 40ng, 60 ng, 80 ng and 100 ng per well of a 96-wellplate, with 10,000 cells plated per well). (C) The aSyn–GLuc1 and aSyn–GLuc2 PCA signal in washed cell monolayers (left) and in conditioned medium (middle) is shown. Relative LDH release (right) was used as a release control.(D) aSyn release kinetics during an 18-h incubation. The aSyn dimer level in the medium was determined by PCA, and the PCA signal was normalized to the LDH release. (E) After 4 h incubation of naïve recipient cells in the mock- (left) oraSyn–GLuc1/2- (middle) or tau–GLuc1/2- (right) conditioned medium, cells were washed, and measured with PCA. RLU, relative light units. Results are mean±sd. *P<0.05, ***P<0.001 (ANOVA).
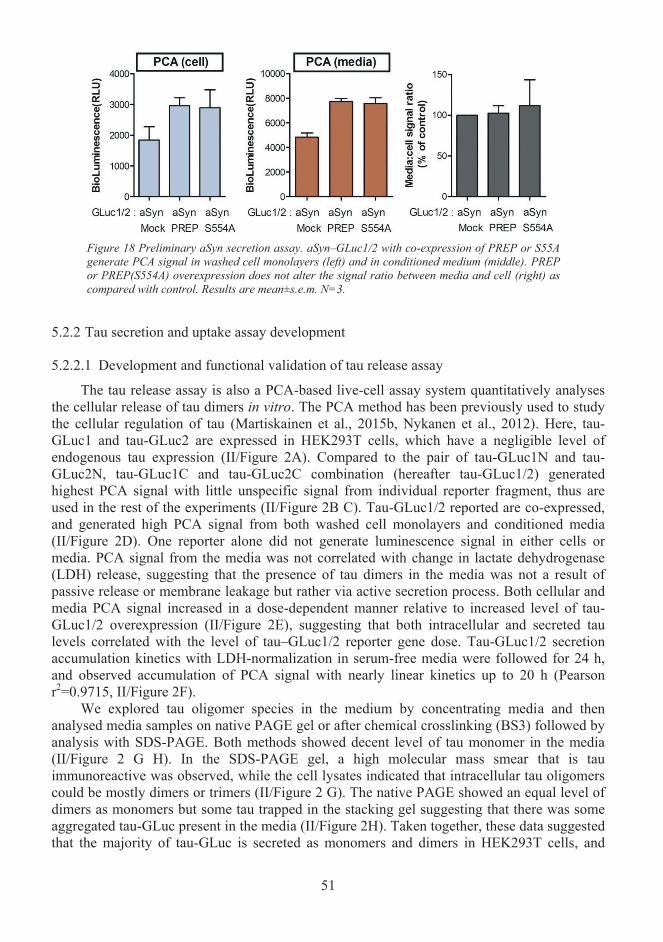
51
Figure 18 Preliminary aSyn secretion assay. aSyn–GLuc1/2 with co-expression of PREP or S55A generate PCA signal in washed cell monolayers (left) and in conditioned medium (middle). PREP or PREP(S554A) overexpression does not alter the signal ratio between media and cell (right) ascompared with control. Results are mean±s.e.m. N=3.
5.2.2 Tau secretion and uptake assay development
5.2.2.1 Development and functional validation of tau release assay
The tau release assay is also a PCA-based live-cell assay system quantitatively analysesthe cellular release of tau dimers in vitro. The PCA method has been previously used to study the cellular regulation of tau (Martiskainen et al., 2015b, Nykanen et al., 2012). Here, tau-GLuc1 and tau-GLuc2 are expressed in HEK293T cells, which have a negligible level of endogenous tau expression (II/Figure 2A). Compared to the pair of tau-GLuc1N and tau-GLuc2N, tau-GLuc1C and tau-GLuc2C combination (hereafter tau-GLuc1/2) generated highest PCA signal with little unspecific signal from individual reporter fragment, thus are used in the rest of the experiments (II/Figure 2B C). Tau-GLuc1/2 reported are co-expressed, and generated high PCA signal from both washed cell monolayers and conditioned media(II/Figure 2D). One reporter alone did not generate luminescence signal in either cells ormedia. PCA signal from the media was not correlated with change in lactate dehydrogenase (LDH) release, suggesting that the presence of tau dimers in the media was not a result of passive release or membrane leakage but rather via active secretion process. Both cellular and media PCA signal increased in a dose-dependent manner relative to increased level of tau-GLuc1/2 overexpression (II/Figure 2E), suggesting that both intracellular and secreted taulevels correlated with the level of tau–GLuc1/2 reporter gene dose. Tau-GLuc1/2 secretion accumulation kinetics with LDH-normalization in serum-free media were followed for 24 h, and observed accumulation of PCA signal with nearly linear kinetics up to 20 h (Pearsonr2=0.9715, II/Figure 2F).
We explored tau oligomer species in the medium by concentrating media and then analysed media samples on native PAGE gel or after chemical crosslinking (BS3) followed by analysis with SDS-PAGE. Both methods showed decent level of tau monomer in the media (II/Figure 2 G H). In the SDS-PAGE gel, a high molecular mass smear that is tau immunoreactive was observed, while the cell lysates indicated that intracellular tau oligomers could be mostly dimers or trimers (II/Figure 2 G). The native PAGE showed an equal level of dimers as monomers but some tau trapped in the stacking gel suggesting that there was some aggregated tau-GLuc present in the media (II/Figure 2H). Taken together, these data suggestedthat the majority of tau-GLuc is secreted as monomers and dimers in HEK293T cells, and

52
could also include soluble pre-aggregates or fibrils, indicating that GLuc-tag did not interfere with known tau oligomerization and aggregation properties.
It was previously reported that tau dimerizes irreversibly in the split GFP system that is another form of PCA (Chun et al., 2007b). Hence we tested the reversibility of tau-GLuc1/2 dimers by titrating conditioned medium with several detergents. Titration of the mediasamples with SDS abolished luminescence signals generated from full-length GLuc or tau-GLuc1/2, because reporter proteins are denatured (II/Figure 2I). Triton X-100, a mild non-ionic detergent, and Saponin, another mild detergent derived from glucoside, were capable of dissociating the protein complexes without denaturing them at relatively low concentration.Titration of the media samples with either one of the detergents reduced luminescence generated by tau-GLuc1/2 complex but not full-length GLuc enzyme (II/Figure 2I), suggesting that most tau-GLuc1/2 dimers are reversibly associated. This further indicates that tau-GLuc PCA is reversible, and HEK293T cells secreted tau species are not predominantly in pre-aggregation or fibril forms, in which case PCA signal will be diminished.
To show that tau secretion is specific to tau dimer, we demonstrated that the complex of tau-GLuc2 and Pin1-GLuc1, a peptidyl-prolyl cis-trans-isomerase that was previously shown to interact with tau by PCA, was not secreted as efficiently as tau-GLuc1/2 (II/Figure 2J)(Nykanen et al., 2012). Treatment of cells with GW4869, a neutral sphingomyelinase (nSMase) inhibitor, reduced tau-GLuc1/2 secretion by 32% (II/Figure 2K). nSMase generates ceramide that regulates exocytosis and exosomal secretion (Rohrbough et al., 2004, Trajkovic et al., 2008). Hence this result suggests that tau could be secreted in small vesicles as has been previously reported (Saman et al., 2012a, Dujardin et al., 2014a).
Total tau level in cell and media samples were analysed by ELISA, and the data was normalized to total protein content. As shown in II/Figure 3A, there is 16,700±1300 pg of tau per μg of protein in average in cell lysates, and 77.6±7.3 pg of tau per μg of total protein onaverage in media, suggesting that less than 0.5% of cellular tau has been released to medium by HEK293T cells in our system. This is in line with a previous observation where only 0.1-0.3% of cellular tau was released from transfected HEK293 T-Rex cells and human neurons induced from pluripotent stem cells (Chai et al., 2012a). To explore the 0.5% secreted tau that is associated with vesicles, a previously described method for characterising vesicle-boundaSyn was adopted (Danzer et al., 2012). Conditioned media was treated with 0.005% (v/v) trypsin, and noticed that 99.8% of luminescence signals was depleted (II/Figure 3B), whiletreatment of the media sample with trypsin plus 0.005% (v/v) saponin, which permeabilizesvesicles without rupturing the vesicles, removed 99.9% signal. These results suggest that tau is primarily secreted as vesicle-free dimers, and that only roughly 0.1% of all secreted tau wasinside vesicles. We further fractionated conditioned media into ectosomal, exosomal and vesicle-free fractions with a previously described method (II/Figure 3C), and analysed via Western blotting, where flotillin-1 was used as a maker for specifying ectosomes as previously described (II/Figure 3D) (Dujardin et al., 2014a, Kowal et al., 2016). Semi-quantitative analysis of the Western blots confirmed that 99.7% of tau was secreted in vesicle-free form by HEK293T cells, while 0.22% of tau in ectosomes and 0.05% in exosomes (II/Figure 3E). This was also confirmed by PCA analysis of the media fractions, showed that majority of tau dimers were in the vesicle-free fraction (II/Figure 3F).
5.2.2.2 Development and functional validation of tau uptake assay
In the tau uptake assay, media was conditioned for 24 h with HEK293T cells overexpressing tau-GLuc1/2. Naïve recipient cells were incubated with the tau-GLuc1/2 conditioned medium for 4 h, and then washed with PBS and immunostained for tau. We
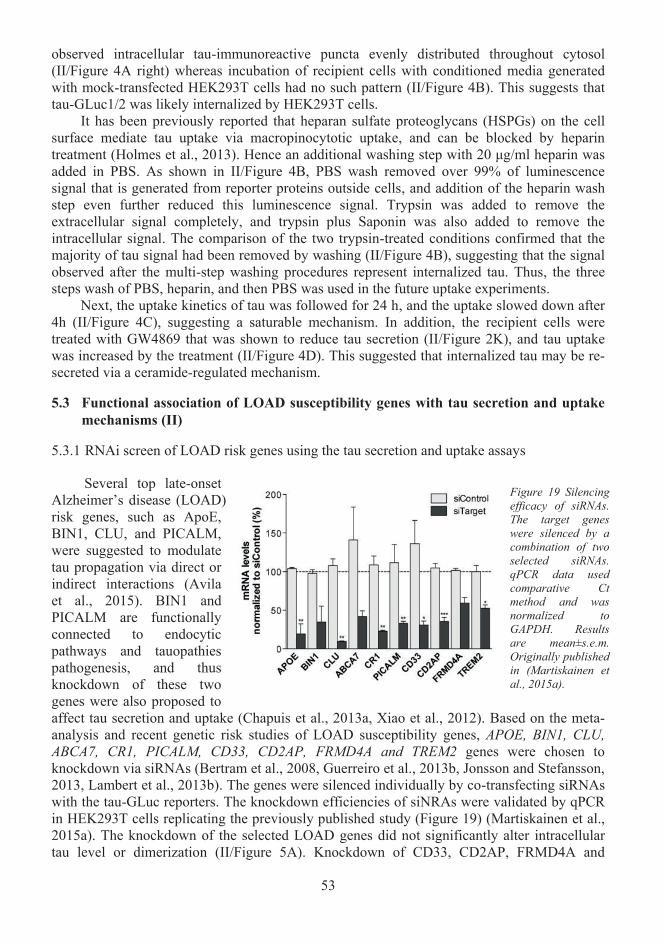
53
observed intracellular tau-immunoreactive puncta evenly distributed throughout cytosol(II/Figure 4A right) whereas incubation of recipient cells with conditioned media generated with mock-transfected HEK293T cells had no such pattern (II/Figure 4B). This suggests thattau-GLuc1/2 was likely internalized by HEK293T cells.
It has been previously reported that heparan sulfate proteoglycans (HSPGs) on the cellsurface mediate tau uptake via macropinocytotic uptake, and can be blocked by heparin treatment (Holmes et al., 2013). Hence an additional washing step with 20 μg/ml heparin was added in PBS. As shown in II/Figure 4B, PBS wash removed over 99% of luminescencesignal that is generated from reporter proteins outside cells, and addition of the heparin wash step even further reduced this luminescence signal. Trypsin was added to remove theextracellular signal completely, and trypsin plus Saponin was also added to remove theintracellular signal. The comparison of the two trypsin-treated conditions confirmed that the majority of tau signal had been removed by washing (II/Figure 4B), suggesting that the signal observed after the multi-step washing procedures represent internalized tau. Thus, the three steps wash of PBS, heparin, and then PBS was used in the future uptake experiments.
Next, the uptake kinetics of tau was followed for 24 h, and the uptake slowed down after 4h (II/Figure 4C), suggesting a saturable mechanism. In addition, the recipient cells weretreated with GW4869 that was shown to reduce tau secretion (II/Figure 2K), and tau uptake was increased by the treatment (II/Figure 4D). This suggested that internalized tau may be re-secreted via a ceramide-regulated mechanism.
Functional association of LOAD susceptibility genes with tau secretion and uptake 5.3mechanisms (II)
5.3.1 RNAi screen of LOAD risk genes using the tau secretion and uptake assays
Several top late-onset Alzheimer’s disease (LOAD) risk genes, such as ApoE, BIN1, CLU, and PICALM,were suggested to modulate tau propagation via direct or indirect interactions (Avila et al., 2015). BIN1 and PICALM are functionally connected to endocytic pathways and tauopathies pathogenesis, and thusknockdown of these two genes were also proposed toaffect tau secretion and uptake (Chapuis et al., 2013a, Xiao et al., 2012). Based on the meta-analysis and recent genetic risk studies of LOAD susceptibility genes, APOE, BIN1, CLU, ABCA7, CR1, PICALM, CD33, CD2AP, FRMD4A and TREM2 genes were chosen to knockdown via siRNAs (Bertram et al., 2008, Guerreiro et al., 2013b, Jonsson and Stefansson, 2013, Lambert et al., 2013b). The genes were silenced individually by co-transfecting siRNAs with the tau-GLuc reporters. The knockdown efficiencies of siNRAs were validated by qPCR in HEK293T cells replicating the previously published study (Figure 19) (Martiskainen et al., 2015a). The knockdown of the selected LOAD genes did not significantly alter intracellular tau level or dimerization (II/Figure 5A). Knockdown of CD33, CD2AP, FRMD4A and
Figure 19 Silencing efficacy of siRNAs. The target genes were silenced by a combination of two selected siRNAs. qPCR data used comparative Ct method and was normalized to GAPDH. Results are mean±s.e.m.Originally published in (Martiskainen et al., 2015a).

54
TREM2 decreased tau secretion (CD33 by -27%, CD2AP by -23%, FRMD4A by -19% and TREM2 by -55%) as shown by LDH-normalized PCA signal (II/Figure 5B). In the tau uptake assay, only the knockdown of ApoE in recipient cells increased tau uptake (by +29%,II/Figure 5C).
As the endogenous level of TREM2 and CD33 are very low in HEK293T cells, the findings in HEK293T cells were validated in CHME-5 cell line, a human fetal microglia-derived cell line that expresses relatively higher levels of endogenous TREM2 and CD33 as verified by qPCR (data not shown). However, knockdown of TREM2 and CD33 in CHME-5cells did not alter tau secretion in any way (II/Figure 5D) suggesting that the effect of TREM2 and CD33 on tau secretion in HEK293T cells might be a false-positive. Therefore, CD2AP and FRMD4A were explored in further tau secretion studies.
5.3.2 CD2AP overexpression does not alter tau secretion
CD2-associated protein (CD2AP) is encoded by gene CD2AP on chromosome 6p12, which has two SNPs, rs9296559 and rs934907 associated with increased AD risk (Hollingworth et al., 2011). CD2AP is known as a scaffolding protein involved inactin cytoskeleton organization and polarity (Dustin et al., 1998, Lehtonen et al., 2002).CD2AP was also reported to regulate synapse formation, cell-to-cell interaction and endocytosis (Wolf and Stahl, 2003, Cormont et al., 2003). Knockdown of CD2AP in Drosophila model of AD was shown to increase tau-mediated toxicity (Shulman et al., 2014). Here, CD2AP was overexpressed in HEK293T cells, and only very subtle changesin tau PCA signal was observed in cells and media (Figure 20), suggesting that endogenousCD2AP may be sufficient to modulate tau secretion, thus unlike knockdown, the overexpression of CD2AP would not alter tau secretion. The tau toxicity-modulating effect of CD2AP may thus be related to other mechanisms other than tau secretion.
5.3.3 FRMD4A-cytohesin signaling pathway regulates tau secretion
FRMD4A belongs to the FERM superfamily and is encoded by FRMD4A gene on human chromosome 10p13 (Tepass, 2009). The FRMD4A gene has multiple SNPs associated with increased AD risk (Lambert et al., 2013a). The exact function(s) of FRMD4A are poorly understood. A recent study showed reduced expression of FRMD4A in AD patients’ brains, and knockdown FRMD4A in vitro showed increased tau phosphorylation and AD-related NFT pathology (Martiskainen et al., 2015a). Here, the intracellular localization of FRMD4A was first verified by immunofluorescent study. Both HA-tagged and GFP-tagged FRMD4A showed similar punctate intracellular distribution (II/Figure 6A). This punctate distribution of FRMD4A did not fully colocalize with tau that is overexpressed, but some FRMD4A-positive vesicles at or near the plasma membrane were closely associated with tau (II/Figure 6B). We also used FRMD4A-HA to confirm sufficient knockdown efficacy of siRNA, and analysis with Western blotting showed a reduction of FRMD4A level by 39% (II/Figure 6C). Next,FRMD4A was overexpressed in increasing amount in HEK293T cells, and a significant increase in tau PCA signal was observed in the media in an FRMD4A gene dose-dependent
Figure 20 The tauPCA signal intensity in cells and media is notaltered by CD2APgene dosage (CD2AP plasmids at 0ng, 10 ng, 30ng, 50 ng per well). Results are mean±s.e.m, N=3

55
manner (up to +146% increase of secreted tau) while no change in cell-derived tau PCA signal was observed (II/Figure 6D). This suggested that FRMD4A regulates tau secretion.
FRMD4A has been reported to serve as a scaffold protein that mediates the binding of cytohesins to the polarity signalling complex Par3/Par6/aPKCζ (Partitioning defective 3/6, atypical Protein Kinase C ζ), resulting in ADP ribosylation factor 6 (Arf6) activation (Ikenouchi and Umeda, 2010). Arf6 is a small GTPase localized in intracellular membranes,and plays a key role in regulating actin cytoskeleton dynamics and membrane trafficking (D'Souza-Schorey and Chavrier, 2006). Like other small GTPases, Arf6 activity can beregulated by many guanine nucleotide exchange factors (GEF), GTPase activating proteins (GAPs) and guanosine nucleotide dissociation inhibitors (GDIs) (Hongu and Kanaho, 2014).Cytohesins (also known as Sec7 in yeast) are GEFs of Arf6 (Ashery et al., 1999). Here, cellswere first treated with SecinH3, a small-molecule inhibitor of cytohesin GEF activity, and observed a reduction in FRMD4A-induced tau secretion, and increased intracellular tau dimer content (II/Figure 6E), suggesting an accumulation of intracellular tau. Similar but milder effect was also observed when cells expressing endogenous FRMD4A were treated with secinH3 (II/Figure 6F). Next, Arf6 was overexpressed, and a strong increase in tau secretion was observed (II/Figure 6G). Overexpression of constitutively active Arf6(Q67L) mutantsignificantly enhanced this effect (II/Figure 6H), whereas siRNA knockdown of Arf6 or alternatively expressing Arf6 dominant-negative mutant Arf6(T27N) significantly suppressed this effect (II/Figure 6G, H).
Polarity signalling complex Par3/Par6/aPKCζ is connected to the FRMD4A-cytohesin pathway as mentioned above. The primary function of this complex is to regulate cellularpolarization, which is a key event involved in cell proliferation, migration, differentiation, etc.(Suzuki and Ohno, 2006, Goldstein and Macara, 2007). Here, aPKCζ, a ceramide-binding protein previously reported to be involved in several membrane trafficking events and exocytosis, and C20ζ, the C-terminal ceramide-binding region of aPKCζ (amino acid 405–592) were chosen to be overexpressed (Horikoshi et al., 2009, Joberty et al., 2000, Wang et al., 2009a). Overexpression of aPKCζ significantly increased tau secretion whereas overexpression of C20ζ had no effect (II/Figure 7A). Co-expression of aPKCζ with C20ζsuppressed aPKCζ-induced tau secretion, suggesting that both may compete to bind to ceramide that appears to be involved in tau secretion. In the polarity complex that regulates Arf6 activation in an FRMD4A-dependent manner, Par6 was shown to be an important adaptor connects Par3 and aPKCζ in order to carry out normal function at tight junction during epithelial cell polarization (Ikenouchi and Umeda, 2010, Joberty et al., 2000). While Par6 wasoverexpressed, tau secretion was significantly increased (II/Figure 7C), which was likelydependent on the Park6 kinase activity as expression of Par6 inactive mutant, Par6(S345A)did not have this effect. The addition of SecinH3 did not alter aPKCζ- or Par6- induced tau secretion (II/Figure 7B, D), suggesting that the polarity complex induced tau secretion does not require cytohesin GEF activity. Taken together, the data suggest that FRMD4A–cytohesin–Arf6 pathway and the polarity complex with Par6/aPKCζ are involved in the regulation of tau secretion.
Arf6 regulates synaptic vesicle endocytosis and recycling, and also cytohesins have been implicated in presynaptic vesicle transport (Ashery et al., 1999, Krauss et al., 2003, Tagliatti et al., 2016). Hence neuronal culture expressing endogenous tau was used to validate the findings further. Mouse cortical neuron cultures (21 DIV) were transduced with FRMD4A shRNA using lentiviral vectors, which was shown to result in 99% knockdown of endogenous FRMD4A mRNA (II/Figure 8A). Media was conditioned with transduced neurons for 3 d, and analysed by mouse-specific tau ELISA. Compared to control and GFP-transduced conditions,

56
silencing of FRDM4A expression significantly increased tau secretion (+390%, II/Figure 8B). Cortical neurons were treated with SecinH3 in increasing doses, and an increase in tau secretion was also observed (up to +204% at 20 μM of SecinH3) as analysed by ELISA (II/Figure 8C). Taken together, these results confirm that the FRMD4A-cytohesin signalling pathway modulates endogenous tau secretion.
The impact of transcellular propagation of tau on cellular stress (III)5.4
5.4.1 Hyperphosphorylated tau is recruited to stress granules after internalization
During tau uptake assay development, the punctate distribution of tau in naïve cells exposed to tau conditioned media was particularly interesting, and possibly indicated intracellular localization of internalized tau in endosomal or lysosomal compartments. Henceintracellular localization of tau was followed after uptake using immunofluorescence microscopy and compartmental markers. Interestingly, tau was not localized to early endosomes (as stained by Rab5, III/Figure D, left), late endosomes (as stained by Rab7,III/Figure 2D, middle) or lysosomes (as stained by Lamp2, III/Figure 2D, right) in recipient cells. Instead, some tau localized to macropinosomes (as stained by fluorescently labelledTAT-TAMRA peptide in live cell imaging, III/Figure 2E), which is in line with a previous study (Holmes et al., 2013). Since (1) there was no co-localization with endo/lysosomal markers, (2) only some punctate tau-positive structures co-stained with TAT-TAMRA, and (3) some of the structures varied significantly in size and morphology, we hypothesized that these tau-containing structures could be non-vesicular cytosolic bodies. The number, size and shape of the tau-positive structures indicated stress granules as the most likely candidate. Indeed, co-staining with a stress granule marker TIA-1 showed significant co-localization of internalized tau-GLuc with TIA-1 (III/Figure 2G). Interestingly, TIA-1 had just been reported to co-localize with tau in brains of AD mouse models, including JNPL3 and rTg4510, as well as postmortem human AD patient brain tissue (Vanderweyde et al., 2012).
Stress granules (SGs) are transiently formed non-membrane bound RNA granules nucleated by various RNA-binding proteins (RBPs) under stress conditions in order to suppress translation (Anderson and Kedersha, 2008, Gilks et al., 2004). SG markers have been found in several disease-associated inclusions (Bentmann et al., 2013). Both untagged tau (III/Figure 2F) and tau with GLuc-tag (III/Figure 2G) were recruited to SGs by co-stain tau with several SG markers, TIA1 and eIF3η (III/Figure 2F, 2G top and middle panels), but TTP did not colocalize with tau (III/Figure 2G bottom). On the other hand, exposing cells toconditioned media containing tau or tau-GLuc significantly increased the amount of SG-positive cells (+80%, III/Figure 2I), suggesting that tau uptake enhanced SGs formation.
TIA-1 knockdown was shown to reduce tau misfolding and toxicity in hippocampal neuronal culture (Vanderweyde et al., 2016). Knockdown of TIA-1 significantly reduced colocalization of internalized tau with both SGs markers, TIA-1 and eIF3η (III/Figure 5B with quantitative analysis in 5C), suggesting that TIA-1 is required for recruitment of tau to SGs after being uptake. Interestingly, knockdown of TIA-1 also significantly reduced tau-induced toxicity after internalization (III/Figure 5D). This is in line with a recent study where TIA1 knockdown reduced tau misfolding and toxicity in hippocampal neuronal culture (Vanderweyde et al., 2016).
It was recently proposed that tau hyperphosphorylation promotes tau internalization (Wauters et al., 2016). Tau, tau-GLuc and tauE14-GLuc, which is apseudohyperphosphorylated tau mutant carrying 14 phosphomimetic mutations, were overexpressed and cells co-stained with Tau-5 and TIA-1 antibodies (Khurana et al., 2006).

57
Interestingly, compared to tau or tau-GLuc that did not co-localize with TIA-1 (III/Figure 1B top), tauE14-GLuc overexpression promoted the formation of SGs, into which tauE14-GLuc was recruited (III/Figure 1B bottom left, C), suggesting that hyperphosphorylation of tau isinvolved in tau recruitment into SGs.
TIA-1 was previously reported to interact with tau (Vanderweyde et al., 2012). We generated a TIA-1-GLuc1 construct and observed PCA signal from TIA-1-GLuc1 and tau-GLuc2 (III/Figure 1F), and 6-fold higher PCA signal from TIA-1-GLuc1 and tauE14-GLuc2, further suggesting hyperphosphorylation could promote tau binding to TIA-1 or possibly to SGs. To further study if tau localization to SGs is a consequence of cell-to-cell transmission, cells were first transfected individually with one of the four reporter proteins, TIA-1-GLuc1,tau-GLuc2, tauE14-GLuc2 and GSK3β-GLuc2 and then co-plated into same plates. 24 h post co-plating, strong PCA signal was observed from both combinations of TIA-1-GLuc1/tau-GLuc2 and TIA-1-GLuc1/tauE14-GLuc2 (III/Figure 1G), confirming the cell-to-cell transmission of tau. Notably, wild-type tau-GLuc2 showed a comparable level of interaction with TIA-1-GLuc1 in the neighboring cells to tauE14-GLuc2, suggesting that during the secretion/uptake process wild-type tau-GLuc2 had acquired a property that enhances its TIA-1interaction and SG recruitment in the recipient cells.
Several lines of evidence indicated that hyperphosphorylation might play an important role in tau uptake. We analysed conditioned media and corresponding cell lysates by Western blotting. Despite the fact that tau is mostly located intracellularly (III/Figure J bottom), AT8,an AD-associated phosphoepitope of tau, staining showed a remarkable enrichment in tau oligomers in the media while in cells phosphorylated tau was mostly monomeric (III/Figure J top). Immunofluorescence microscopy also confirmed that the internalized and SGs-associated tau was also phosphorylated at the AT8 epitope (III/Figure I).
Altogether, this data, for the first time, indicates that SGs are a critical cellular structure in the cell-to-cell transmission process that underlies the spread of tau pathology in the brain. This suggests that TIA-1 and possibly also other SG proteins, which have an intrinsic self-aggregating property needed for formation of SGs, play a key role in the seeding and transmission of pathological, misfolded and hyperphosphorylated tau to healthy cells.
5.4.2 Tau uptake sensitizes cells to stress
RNA-binding proteins and SGs have been implicated to be involved in the pathogenesis of neurodegenerative diseases (Maniecka and Polymenidou, 2015, Wolozin and Apicco, 2015).For example, in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD),mutations in Tar DNA-binding protein 43 (TDP-43), fused in sarcoma protein (FUS), and heterogeneous nuclear ribonucleoproteins (hnRNPA1/hnRNPA2B1) alter their intracellular localization and promote aggregation, and thus have been linked to the pathogenic pathway (Kim et al., 2013, Liu-Yesucevitz et al., 2010, Vance et al., 2009, Sreedharan et al., 2008, Tradewell et al., 2012).
A 4 h incubation with salubrinal induced SG formation, which disappeared during a 4 hwashout period (III/Figure 6A, top panels, with quantitative analysis in 6B). With the addition of DBeQ, an inhibitor that blocks valosin-containing protein-mediated SG clearance,numerous SGs remained in cells after the 4 h washout (III/Figure 6A, right, 6B). Interestingly, when cells were exposed with to conditioned media containing tau-GLuc for 4 h, SGs wereinduced but could not be resolved by even a 20 h of washout period using fresh media without tau (III/Figure 6A, bottom panels, 6B). In another experiment, cells were exposed to tau conditioned media or salubrinal for 4 hours, followed by 4 h washout with fresh media, and then cells were treated with rotenone, a mitochondrial complex I inhibitor that induces

58
oxidative stress. A significant reduction in cell viability was observed in the tau media pre-exposed condition (III/Figure 6E) but not in the salubrinal pre-treatment (III/Figure 6D). Thus, SG-association of tau impairs normal clearance dynamics of SGs and sensitizes cells to secondary stress. Taken together, these data showed that tau recruitment to SGs afterinternalization significantly alters the cells’ normal stress-coping mechanisms and increases their sensitivity to other types of stress. This mechanism provides a previously unrecognized contributor to tau toxicity that may have particular importance in post-mitotic neurons that are exposed to various stresses in the aging or injured brain, as occurs in tauopathies and chronic traumatic encephalopathy (CTE)(McKee et al., 2009).

59
6 Discussion
Assay development for studying cell-to-cell propagation of α-synuclein and tau6.1
Both aSyn and tau have been identified in healthy human plasma and cerebrospinal fluid,and their level are altered during the pathogenic processes (Borghi et al., 2000, El-Agnaf et al., 2003, Blennow et al., 1995, Vigo-Pelfrey et al., 1995). This indicates that aSyn and tau are constitutively released, but the mechanisms might be altered during pathogenesis. Braak staging proposed by Dr. Heiko Braak and colleagues described the sequential stage of development of synucleinopathies and tauopathies that are primarily composed aggregated aSyn and tau, respectively, in connected brain regions in a time-dependent manner (Braak and Braak, 1991, Bancher et al., 1993, Braak et al., 2003). This may offer a potential clinical consequence of aSyn and tau misfolding and transmission in specific brain areas with neurological manifestation. Despite the debate between the theories of “the spread of pathology”, which suggests that the transmission of disease-associated proteins occurs through specific neuronal network and between specific groups of cells, and the theory of “selective neuronal vulnerability”, which suggests that the transmission of disease-associated proteins are ubiquitous but certain neuronal populations are more affected than others, disease-associated proteins need first to adapt the conformations that could seed pathology, and then spread from the donor cells to the recipient cells (Brettschneider et al., 2015, Walsh and Selkoe, 2016). In the past two decades, the studies of aSyn and tau aggregation pathways have been intensively investigated, because these studies can offer potential mechanistic explanations for the initial and final events leading to the development of synucleinopathies and tauopathies. The missing gaps in between are potentially filled and explained by the cell-to-cell propagation of aSyn and tau. The seeding of aggregates has been widely demonstrated in a series of in vitro and in vivo studies. These recent findings on the cell-to-cell transmission of aSyn and tau have received a lot of attention, and may offer hope for better understanding of disease mechanisms and identify novel therapeutic targets for many incurable neurodegenerative diseases such as AD, PD and ALS.
In this thesis work, a novel system was developed for monitoring cellular oligomerization, secretion and uptake of tau and aSyn. The assays were built based on PCA system that is widely utilized for studying protein-protein interactions in live cells (Remy and Michnick, 2006b, Michnick et al., 2007). Although the assay was designed to detect protein-protein interactions in their native environment with minimum invasive impact, and thus has many benefits, this system also has its own weaknesses. The use of GLuc-reporter fragments fused to the protein of interest at N- or C- termini and the requirement of overexpression of reporter proteins are the two important caveats. The C-terminal fusion of GLuc reporter fragments to tau did not significantly alter cellular tau expression level nor the localization and oligomerization/aggregation behaviour in HEK293T cells. N-terminal GLuc fusion of aSyn did not interfere with the expression of aSyn nor oligomerization behaviour at the early stage of aggregation. A previous immunofluorescence study of aSyn-GLuc showed diffuse cytosolic signals, and staining on or near the cell surface and in protrusions, which was similar to endogenous aSyn expression (Yan, 2013, Lashuel et al., 2013). In transient transfection-based studies (publications I and II), overexpression of aSyn and tau reporter proteins did not causesignificant cytotoxicity or unspecific release of aSyn and tau from N2A and HEK293T cells, respectively, as confirmed by the measurement of LDH release.
In the assay system for studying cell-to-cell transmission of tau, the PCA signal from intracellular and secreted tau appears to be largely derived from tau dimers, which were previously described as the essential building blocks of tau aggregates and PHFs (Friedhoff et

60
al., 1998). Through validation via native gel and Western blotting analysis, tau-GLuc proteins were also shown to assemble into dimers, and to some extend into soluble aggregates. These observations indicate that tau generated in our system largely represents pathological conditions.
It was previously demonstrated that the association of GLuc-reporter protein fragments used in PCA is reversible (Remy and Michnick, 2006b). However, in bimolecular fluorescence complementation assay, the complementary fragments of fluorescent reporter proteins form irreversible dimers (Kodama, 2012). In our GLuc PCA system, tau-GLuc1/2 in conditioned media dissociated with low concentrations of two mild detergents, Triton X-100 and Saponin, suggesting that our GLuc PCA system replicates the previously described reversibility. At 0.1% Triton X-100 that would not be able to dissolve tau aggregates or fibrils, PCA signal was lost, suggesting that aggregates were not generating signals. This could be in line with a previous observation that tau signal was reduced upon aggregation in the bimolecular fluorescence complementation system (Chun et al., 2007a).
In conditioned media, over 99% tau-GLuc was secreted in vesicle-free form, while a minor population represented vesicle-bound tau, in which ectosomes represented the major species. Even though this is in line with previous observations, there is still a possibility that post-secretion of tau from microvesicles in the media could overemphasize the vesicle-free fraction of tau (Dujardin et al., 2014a, Saman et al., 2012b). On the other hand, post-release/escape of tau from microvesicles may also represent a physiological consequence of tau binding to the vesicle membranes in plasma or CSF. Interestingly, the internalization of vesicle-associated tau is significantly more efficient than vesicle-free fractions. This observation could be explained by vesicle offering a more efficient internalization route into cells. However, as tau internalization may take multiple routes that are incompletely understood, further studies are needed to address specific mechanisms involved.
Macropinocytosis represents a subtype of bulk endocytosis, and has been described as a major route for cells to uptake macrosolutes such as extracellular tau fibrils (Holmes et al., 2013). An immunofluorescence study in study III confirmed that macropinocytosis is involved in uptake of tau dimers. The mechanisms of how tau escapes macropinosomes and gets recruited to other intracellular compartments such as stress granules remain unknown. Disruption of vesicle membranes by misfolding and formation of annular protofibrils as previously described could be one potential explanation (Jones et al., 2012, Zeineddine et al., 2015). However, depending on the overall rate of endocytic and exocytic events in cells, macropinosomes could undergo rapid recycling through exocytosis (Falcone et al., 2006). This offers a potential explanation for the low uptake rate as compared to the higher level of secretion. Overall, it could be implicated that most tau is internalized to macropinosomes and is recycled back via active secretion, while a smaller pool of vesicle-associated tau gets access to the cytosol. It is worth mentioning that ceramide-regulated mechanism may also be involved, as GW4869 that is a neutral sphingomyelinase (nSMase) inhibitor increased intracellular accumulation of tau following internalization.
In our system developed to study cell-to-cell transmission of aSyn, aSyn dimers were clearly present in the media. The ratio between intracellular and extracellular aSyn dimers is similar to tau. The release kinetics of aSyn observed within 18 hours also showed similar saturable mechanisms as observed with tau. These preliminary data implicate that the release mechanisms of aSyn dimer could be similar to tau. Despite the fact that we have not fully characterized the extracellular aSyn oligomer species, intracellular aSyn is predominantly soluble with minor SDS-insoluble fraction present possibly indicating a relatively low level of aggregation, and suggesting that the assay reports an early event in the aSyn aggregation

61
pathway. The level of internalization of aSyn-GLuc from conditioned media is lower than tau, possibly indicating the different routes of aSyn internalization. Further studies are needed to address aSyn uptake mechanisms and require more sensitive means to detect internalized aSyn dimers/oligomer/aggregates.
The effect of prolyl oligopeptidase on α-synuclein oligomerization6.2
Prolyl oligopeptidase (PREP) has been previously shown to enhance aSyn aggregation (Brandt et al., 2008). Immunohistochemical assay also suggests that PREP co-localizes with aSyn in Lewy bodies in PD patients’ brain (Hannula et al., 2013). A small-molecule inhibitor of PREP, KYP-2047 reduces the amount of aggregated aSyn in cells and transgenic mice overexpressing wild-type aSyn and aggregation-prone aSyn mutant as verified by immunohistochemical assay and Western blotting, and in addition promotes the clearance of aSyn aggregates in transgenic mice models (Myohanen et al., 2012a). Previously, there has been interests in KYP-2047 in term of pathology inhibition effect, but the molecular mechanisms of PREP-mediated aSyn aggregation and the role of KYP-2047 in this process were incompletely understood. It has been suggested that PREP may serve as a nucleation point for aSyn aggregation (Brandt et al., 2008, Lambeir, 2011). Many proteins and small molecules such as tyrosine hydroxylase, reactive oxygen species, metal ions and lipids, have been shown to interact with aSyn, some of which also enhance aSyn aggregation in vitro and in vivo (Chavarria and Souza, 2013, Binolfi et al., 2008, Rivers et al., 2008, Lundblad et al., 2012). It has been proposed that aSyn aggregation exhibits an inducible nucleation-elongationmechanism, e.g. in the case of aSyn binding to lipids vesicles (Rabe et al., 2013). Hence it is possible that some of the interaction partners of aSyn also serve as nucleation points seeding aSyn oligomerization and aggregation (Galvagnion et al., 2015, Uversky, 2007).
In our study, PCA system for studying intracellular dimerization of aSyn is complemented with microscale thermophoresis (MST), a novel cell-free method to demonstrate side-by-side that PREP interacts with aSyn in both live-cell and cell-freeenvironments, and that this interaction enhances aSyn dimerization. In addition, this effect canbe inhibited by KYP-2047 via modulating PREP conformation but not its enzyme activity. Apart from PREP-induced aSyn dimerization, PREP expression also elevated the level of intracellular aSyn, which could also contribute to enhanced aSyn dimerization. It was previously reported that KYP-2047 inhibition of PREP in aSyn transgenic mice accelerated aSyn clearance, and the overexpression of PREP in cell culture reduced autophagy activity (Savolainen et al., 2014). Hence the enhanced aSyn level observed in our study may also be related to reduced aSyn clearance(Savolainen et al., 2014).
It was previously reported that aSyn colocalizes with PREP in aSyn inclusions that were formed under oxidative stress conditions in SH-SY5Y human neuroblastoma cells overexpressing aSyn, and this colocalization in inclusions could be reduced by KYP-2047 treatment (Myohanen et al., 2012a). This led to a hypothesis that KYP-2047 treatment interferes with the interaction between aSyn and PREP, which may be responsible for the reduced aSyn aggregation. Based on the data from two independent methods in cell and cell-free systems, we showed that KYP-2047 inhibition of PREP leads to enhanced PREP-aSyninteraction. We further explored potential PREP binding sites by generating different C-terminal truncated aSyn mutants, because the C-terminal of full length aSyn contains proline residues that are known to be important for substrate specificity of PREP (Walter et al., 1971).Our results indicate that the C-terminal proline residues of aSyn are not needed for PREP-aSyn interaction. This also suggests that PREP-aSyn interaction is likely independent of PREP enzyme activity.

62
It has been reported that PREP adopts three different conformations, open and compact monomeric forms and an oligomeric form that exist in equilibrium (Szeltner et al., 2013, Szeltner et al., 2010, Tarrago et al., 2009). In one of the studies, KYP-2047 induced a large portion of PREP shift toward the compact monomeric form (Szeltner et al., 2013). Our data from native PAGE confirmed that KYP-2047 induces PREP to adopt the compact monomeric form predominantly in our system. Based on previous data, the compact monomers of PREP might have a higher binding affinity to aSyn but lower ability in promoting aSyn dimerization. These observations are in agreement with previous studies that demonstrated the conformations and the higher order oligomer structure of PREP are closely associated with PREP-induced aSyn aggregation and fibrillization in cells (Brandt et al., 2008, Szeltner et al., 2013). Interestingly, PREP(S554A) was also observed to induce aSyn dimerization via a direct interaction similarly to wild-type PREP. However, KYP-2047 had no effect on PREP(S554A)-induced aSyn dimerization nor on PREP(S554A)-aSyn interaction. We showed that PREP(S554A) adopted three oligomeric states similar to wild type PREP, but KYP-2047treatment did not induce a conformational shift of PREP(S554A) towards the compact monomers. This inability for conformational shifting may explain the observation that PREP(S554A) is insensitive to KYP-2047 treatment.
The binding sites on PREP for proteins and inhibitors remain incompletely understood, but the access loop structures have been suggested to play key roles in mediating ligand binding and inhibitor-induced conformational shift (Kaszuba et al., 2012). PREP has three loops at 189-209 (loop A), 577-608 (loop B), and 636-646 (loop C), and its two domains are held together by a stable network of hydrogen bonds. A molecular dynamics simulationshowed that PREP inhibition breaks hydrogen bonds holding loop B and A, which gates the active site of PREP, and this results in an overall conformational shift where the active sites are no longer accessible (Kaszuba et al., 2012). Our data suggests that although PREP(S554A) is enzymatically inactive, the mutation may have stabilized the monomeric open conformation and oligomeric conformation. This may implicate that PREP oligomers and open monomers would be responsible for inducing aSyn oligomerization independent of enzymatic activity, whereas the compact monomers form strong interaction with aSyn but do no promote aSyn nucleation. We tried to isolate these PREP oligomeric and monomeric pools from gels, but because PREP activity and structure is very environment-sensitive and does not withstand harsh conditions, these attempts failed. In the follow-up studies, we also tried to investigate if PREP overexpression alters aSyn dimer secretion, and we observed an elevated level of intracellular and extracellular aSyn dimer in the same proportion, possibly indicating that aSyn secretion mechanisms were not altered by PREP.
In conclusion, our study for the first time provides solid evidence and mechanistic explanation for the direct effect of PREP on aSyn. Together with previous studies showing that PREP enhances aSyn aggregation and modulates aSyn autophagy clearance in vivo, the study on PREP in this thesis fills an important missing gap in the molecular mechanisms of PREP-induced synucleinopathies (Brandt et al., 2008, Savolainen et al., 2014). PREP has long been interested as a drug target, and there has been numerous of patents and clinical trials of PREP inhibitors (Lopez et al., 2011, Morain et al., 2002). Understanding the molecular mechanisms of how KYP-2047 reduces synucleinopathies shed light and confidence in developing new therapeutics targeting the PREP-aSyn pathway.
The effect of LOAD genes on cell-to-cell transmission of tau6.3
A siRNA-based screen was conducted to study the functional connection betweencellular secretion and uptake of tau and 10 LOAD susceptibility genes. It has been previously

63
shown that several late-onset Alzheimer’s disease (LOAD) risk genes, such as BIN1, CLU,and PICALM, might modulate tau propagation or toxicity via direct or indirect interactions with tau (Avila et al., 2015). We hypothesized that two of these selected LOAD susceptibility genes, BIN1 and PICALM may regulate tau secretion and uptake, as both genes are functionally connected to endocytic pathways (Chapuis et al., 2013a, Xiao et al., 2012).However, in the tau uptake study, only the knockdown of APOE mildly affected tau uptake, which could be related to the previously described interaction between tau and ApoE proteins (Fleming et al., 1996, Strittmatter et al., 1994). It was somewhat surprising that the knockdown of both BIN1 and PICALM expression had little effect on tau uptake, suggesting that tau internalization is not heavily dependent on receptor-mediated endocytosis, such as clathrin-mediated endocytosis. On the other hand, a recent study showed that tau uptake can occur via a bulk endocytic mechanism, such as macropinocytosis (Holmes et al., 2013). The macropinocytotic process involves extensive membrane turnover and is not very active inmature neurons. Thus cell types and relevant physiological context should be taken into consideration in the future studies.
In the tau secretion study, siRNA knockdown of CD2AP, CD33, FRMD4A and TREM2significantly reduced tau secretion in HEK293T cells. The knockdown efficiency of TREM2gene was relatively low as compared to other siRNAs, but the effect on tau secretion was remarkably high. TREM2 gene cluster was previously reported to contain SNPs associated with CSF tau and tau phosphorylation in a genome-wide association study, which used CSFtau phosphorylation at Thr181 as an endophenotype of LOAD (Cruchaga et al., 2013a).However, both TREM2 and CD33 are functionally associated with immune responses, and are mainly expressed in immune cells. Moreover, their proximal signalling partners such as DAP12 are expressed at very low level in HEK293T cells, and thus their function may be reduced or altered in the HEK293T cells system, which was used in our siRNA screen (Rohn, 2013, Crocker et al., 2007, Jandus et al., 2011). Knockdown of TREM2 and CD33 in a microglial CHME-5 cell line, which expresses a high endogenous level of both genes, showed no effects on tau secretion. It is thus possible that the reduced tau secretion in HEK293T cells have been caused by off-target effects of TREM2 and CD33 siRNAs. The false-positive hits reflect the fact that although in vitro functional screening of genes is efficient, just like genome-wide association studies, functional verification of gene-screen hits in different independent population group sample sets should always be included to filter out false-positive hits. In addition, microglia has been reported to accelerate the cell-to-cell transmission of tau by internalizing and packing extracellular free tau into exosomes, and delivering them to neighbouring neurons (Asai et al., 2015a). This not only indicates a new role of microglia in the propagation of tau pathology but also highlights the importance of non-transsynaptic tau propagation mechanisms that need to be studied in the right model system.
Both FRMD4A and CD2AP have been described as scaffolding proteins involved in actin cytoskeleton organization and polarity (Dustin et al., 1998, Lehtonen et al., 2002, Ikenouchi and Umeda, 2010). In addition, CD2AP was also reported to regulate synapse formation and endocytosis (Wolf and Stahl, 2003, Cormont et al., 2003). A recent study in CD2AP double-knockout mice revealed a novel function of CD2AP in maintaining blood-brain barrier (BBB) integrity, which was previously shown to be impaired during the pathogenesis of tauopathy in a mouse model (Cochran et al., 2015, Blair et al., 2015). In addition, CD2AP deficiency also promotes tau-mediated toxicity in a Drosophila model of AD (Shulman et al., 2014). Taken together, impairment of CD2AP function(s) might alter the physiological cerebrovascular role of CD2AP for maintaining BBB integrity, and lead to AD-

64
related vascular changes that may promote tau toxicity and propagation in vivo. The reduction of tau secretion induced by knockdown of CD2AP expression level could indicate a potential connection between cell-to-cell transmission of tau and CD2AP-mediated BBB dysregulation in pathological conditions, which can be a subject of future studies.
A recent functional screen showed that in vitro knockdown of FRMD4A altered phosphorylation of tau (Martiskainen et al., 2015b). Moreover, FRMD4A expression in human patients’ brain samples was reported to decrease during progressive development of AD pathology. In this thesis work, cellular level of FRMD4A was shown to be closely associated with tau secretion. In HEK293T cells, tau secretion level is increased with FRMD4A overexpression, and decreased with siRNA knockdown. It is worth mentioning that knockdown efficacy of FRMD4A was relatively low as compared to other siRNA (Figure 19), suggesting a robust functional effect of partial knockdown. It has been previously suggested that reduction in tau secretion might lead to intracellular accumulation of tau with elevated toxicity (Gendreau and Hall, 2013, Hall and Saman, 2012). In our study, reduction in tau secretion level only increased intracellular tau level in HEK293T cells, and the overexpression of FRMD4A only enhanced extracellular tau level without significantly inducing toxicity. As there could be an equilibrium between intracellular and secreted tau, overexpression may not fully represent physiological conditions. For example, overexpression of tau has been previously shown to induce secretion of tau associated with membrane vesicles (Simon et al., 2012a). However, tau in the condition media was observed to contain hyperphosphorylated tau species, a large quantity of dimers and soluble aggregates, suggesting that this is rather a model of pathological tau secretion than physiological.
In mature mouse cortical neurons, which represent a more physiological model than HEK293T cells, reduced FRMD4A level and inhibition of cytohesin activity remarkably enhanced endogenous tau secretion, which is opposite to the response observed in HEK293T cells. The different response between neuronal and non-neuronal cell lines may be explained by the fact that neurons have more specialized and an intense network of secretion machinery, and moreover, tau is mostly expressed endogenously in neurons (Sudhof, 2013, Feng and Arnold, 2016). This is also in line with previous observations in AD patients’ brain that FRMD4A level is progressively reduced during the development of AD pathology (Martiskainen et al., 2015b).
As mentioned above, FRMD4A is associated with altered tau phosphorylation (Martiskainen et al., 2015b). Interestingly, in study III, extracellular tau hyperphosphorylation at AD-associated epitope AT8 (Ser199, Ser202 and Thr205) was shown to impair stress-coping mechanisms of cells after tau was internalized. The AT8 epitope of tau is one of the most extensively studied tau phosphorylation epitopes, and is suggested to be a central mediator of various cascades regulating tau phospho-priming and other related pathways (Bertrand et al., 2010). It has been proposed that increased tau level and phosphorylation (Thr181, also known as AT270) in CSF are associated with cognitive decline and can be used as a biomarker for AD prediction (Kester et al., 2009, Wallin et al., 2010). Phosphorylation of tau at Thr231 has also been reported to correlate with memory deficits in AD and mild cognitive impairment (MCI) (Hampel et al., 2010). The exact molecular pathway of how these abnormal phosphorylation events contribute to AD pathogenesis in human patients are incompletely understood, but some of the AD-related tau phosphoepitopes (Figure 6), such as Ser202, Thr205 and Thr231 were shown to associate with aggregation and tau-mediated toxicity in vitro (Alonso Adel et al., 2004). One potential mechanism was demonstrated by a GWAS study using CSF phospho-tau (Thr181) as an endophenotype of AD showing that an SNP on PPP3R1 gene that is associated with reduced expression of protein phosphatase B

65
(calcineurin, a known tau phosphatase) in the parietal lobes, could be responsible for elevated phospho-tau level in CSF and accelerated AD pathogenesis (Cruchaga et al., 2010). These pieces of evidence and examples taken together suggest that FRMD4A is possibly involved in modulating both the mechanism of tau phosphorylation and cell-to-cell transmission of tau that might contribute to AD- and MCI-related cognitive impairment, memory deficit and neuronal death owning to tau-mediated cellular stress and toxicity. In the future studies, it would be important to investigate the change in other disease-associated phosphorylation sites of tau both in cell and conditioned media that might have been altered by FRMD4A knockdown or overexpression.
It has been reported that by binding to cytohesins, FRMD4A serves as a scaffold protein that promotes the interaction between Arf6 and the polarity signalling complex Par3/Par6/aPKCζ (Ikenouchi and Umeda, 2010). This polarity complex has been suggested to regulate endocytic trafficking including vesicle recycling and exocytotic secretion (Balklava et al., 2007). Cytohesin-1, which is one of the cytohesin family proteins, was reported to promote basal synaptic transmission at Xenopus neuromuscular junctions by activating and promoting Arf6 translocation to the cell membrane, and moreover, by increasing the amount of vesicle priming at the presynaptic terminal (Ashery et al., 1999). In addition, cytohesin-1was also shown to interact with Munc13-1, which is a key protein for vesicle priming at the membrane, thus offering another potential mechanistic explanation for the cytohesin-1-induced increase in presynaptic vesicle pools (Neeb et al., 1999). As presynaptic vesicle release was proposed as one of the routes for tau secretion, both cytohesin-Arf6 and cytohesin-Munc13 pathway might be involved in modulating tau secretion as described in Figure 21(Yamada et al., 2014). Arf6 silencing or cytohesin inhibition in hippocampal neuron culture was recently reported to increase the amount of synaptic vesicle priming, and also synaptic release (Tagliatti et al., 2016). This is in line with our observation that cytohesin inhibition and siRNA silencing of FRMD4A significantly boosted tau secretion levels in neurons. This implicates that FRMD4A might facilitate synaptic transmission by forming direct interaction with cytohesin that further regulates the activity of Arf6 and Munc13-1 to increase the reserve of presynaptic vesicles ready to fuse at the synaptic terminals, and thus modulate tau secretionthrough presynaptic vesicle release. Interestingly, a recent in vitro study showed that cytohesin is also involved in ALS (Zhai et al., 2015). It would be interesting to investigate possible connections of this study with our findings in the future studies.
In addition to cytohesins and Arf6, the data indicate that two major components of the polarity signalling complex, Par6 and aPKCζ are also involved in the tau secretion pathway. The Par polarity signalling complex plays a role in regulating cellular polarization, which is a key event involved in many cellular functions such as cell proliferation, development, differentiation, and more importantly polarized delivery of proteins and lipids to specific sites of the plasma membrane (Suzuki and Ohno, 2006, Goldstein and Macara, 2007). The FRMD4A-cytohesin-Arf6 pathway was previously reported to be involved in cell polarization events in epithelial cells where the Par complex was shown to activate Arf6 in an FRMD4A-dependent manner at primordial adherent junctions (Ikenouchi and Umeda, 2010). One potential explanation is that the Par polarity signalling complex might indirectly influence tau secretion by delivering microtubule-bound tau and vesicles to the presynaptic terminals or generally to the polarized plasma membrane in non-neuronal cells, such as HEK293T. Nevertheless, addressing this novel molecular pathway of Par6-aPKCζ-FRMD4A-cytohesin-Arf6 and candidates as illustrated in Figure 21 would benefit the understanding of tauopathy propagation, and possibly provide new therapeutic targets.

66
Pathophysiological consequences triggered by tau internalization6.4
6.4.1 Role of TIA-1 in mediating internalized tau-induced stress and toxicity through stress granules
In AD pathogenesis, tau pathology appears in connected brain regions in a time-dependent manner suggesting that pathological tau species might spread from one cell to another and induce toxicity and neurodegeneration (Braak et al., 1994a, Lasagna-Reeves et al., 2012a, Dujardin et al., 2014b). Little is known about the mechanism of tau uptake and the pathophysiological consequences of tau internalization. In this thesis work, extracellular tau was for the first time shown to be recruited to stress granules following internalization, which resulted in altered stress-coping capability in cells, and increased their sensitivity to othertypes of stress. In addition, knockdown of TIA-1 halted the recruitment of tau to stress granules, and reduced internalized tau-mediated toxicity. This is in line with a recent study that demonstrated the knockdown of TIA-1 reduced tau misfolding and toxicity in neurons (Vanderweyde et al., 2016).
Previous literature has described an association of stress granules (SGs) with a number of neurodegenerative diseases. As mentioned previously, SGs are transiently formed non-membrane bound RNA granules, which are nucleated by various RNA-binding proteins (RBPs) under stress conditions in order to suppress translation and promote cell survival (Anderson and Kedersha, 2008, Gilks et al., 2004). The abnormality of SGs has been implicated to be involved in the pathogenesis of neurodegenerative diseases (Maniecka and Polymenidou, 2015, Wolozin and Apicco, 2015). Many of the RBPs, particularly TDP-43, fused in sarcoma (FUS) proteins and certain heterogeneous nuclear ribonucleoproteins(hnRNPs), have been shown to be genetically associated with frontotemporal dementia (FTD)and amyotrophic lateral sclerosis (ALS) (Kim et al., 2013, Liu-Yesucevitz et al., 2010, Sreedharan et al., 2008, Vance et al., 2009). SG markers such as TIA-1, which is a key protein
Figure 21 The proposed role of FRMD4A-cytohesin-Arf6 pathway in presynaptic tau secretion. FRMD4A mediates the activation and translocation of Arf6 to the membrane by recruiting cytohesin and polarity signaling complex Par3/Par6/aPKCζ. Other than Arf6, Munc13 that also interact with the cytohesins are involved in synaptic vesicle pool filling and priming. It is yet not fully understood how cytohesins modulate Munc13 activity in neuronal and non-neuronal cells. Cellular secretion of tau is enhanced by synaptic activity. Hence this pathway is closely associated with presynaptic tau release. LOAD risk variants disrupting physiological function of FRMD4A or SecinH3 inhibition of cytohesin activity increase tau release. Image modified from Uronen RL and HJ (2016)

67
in the assembly of certain types of SGs, were reported to localize to NFTs in AD (Ash et al., 2014, Vanderweyde et al., 2012).
Together, it was somewhat surprising to see that internalized tau is recruited to SGs in a TIA-1 dependent manner. This novel mechanism described a previously unrecognized role of TIA-1-containing stress granules in mediating internalized tau-induced cellular stress and toxicity that may be an important contributor in tauopathies and chronic traumatic encephalopathy, where post-mitotic neurons are exposed to various stresses in post brain injury (McKee et al., 2009). In the future studies, it will be interesting to see if knockdown of other disease-associated genes/proteins such as TDP-43, FUS and hnRNPs could have an effect on cell-to-cell transmission of tau or others and related toxicity.
Lastly, it is worth mentioning that there could be various SG compositions. For example, TTP is another SG marker previously reported to interact with tau in vivo (Vanderweyde et al., 2012). However TTP did not co-localize with SGs that are tau-positive in N2A and HEK293T cells, suggesting that there might be a variety of SGs composed with different components, and TTP is not recruited to the SGs induced by tau internalization, or alternatively TTP may not interact with tau in in vitro models (Vanderweyde et al., 2012). Thus, the nature of SG components should also be taken into account in the future studies.
6.4.2 The role of tau hyperphosphorylation in cell-to-cell transmission of tau
Tau strains have been previously reported to affect the rate of cell-to-cell transmission and the ability to promote seeded aggregation (Sanders et al., 2014a). The internalization of the hyperphosphorylated tau species, but not with cytosolic overexpression of tau, was observed to induce formation of stress granules. Together with the evidence showing that pseudo-hyperphosphorylated tau formed significantly stronger interaction with TIA-1 than normal tau and promoted formation of SGs, it can be concluded that either certain tau species are more prone for cell-to-cell transmission or tau composition and structures are modified during secretion and uptake, e.g. by phosphorylation and aggregation during these processes. This is in line with the observation that secreted, SG-promoting tau is predominantly in hyperphosphorylated form, at least at the AT8 epitope. Both Ser202 and Thr205 sites of tau protein are phosphorylated by AD-associated kinase CDK5 and GSK3-β, and hyperphosphorylation at Ser202 and Thr205 was shown to be linked with a reduction in the ability of tau to nucleate microtubule assembly (Wada et al., 1998). As mentioned above, CSF tau level and phosphorylation are closely associated with AD pathology, particularly memory deficits and cognitive impairment (Hampel et al., 2010, Kester et al., 2009, Wallin et al., 2010, Cruchaga et al., 2010). This implicates that the emerging hyperphosphorylation at the AT8 epitope of extracellular tau may be a result of lost physiological function that impairs microtubule functionality and contributes to worsening clinical outcome in memory and cognitive functions during AD pathogenesis. Under this hypothesis, it will be highly important and interesting to fully characterize the tau phosphorylation state in the conditioned media in order to address further hyperphosphorylated epitopes that could be relevant regarding pathogenesis.
Increasing evidence support the hypothesis that stress granules (SGs) may provide a location for misfolded and normal low complexity proteins to interact, and upon interaction to convert normal, misfolding-prone proteins to pathological species, hence seeding aggregation (Wolozin, 2012, Wolozin and Apicco, 2015, Maniecka and Polymenidou, 2015).Hyperphosphorylated tau-induced SGs were shown to be hard to resolve. This may be explained by the fact that tau and TIA-1 have low complexity sequences and flexible secondary structures, and the initial tau/TIA-1 complexes might lead to the formation of a

68
nucleation core that cannot be degraded or resolved as compared with SGs clearance under physiological conditions (Calabretta and Richard, 2015, Wolozin and Apicco, 2015). The role of hyperphosphorylation in cell-to-cell transmission and seeding of tau has not been fully explored. It was previously demonstrated that certain strains of tau aggregates such as preformed fibrils strongly promote tau seeding (Sanders et al., 2014a). On the other hand, hyperphosphorylation of tau has been widely recognized as the major factor contributing to tau aggregation, dysfunction and related neurofibrillary degeneration in AD (Iqbal et al., 2009, Alonso et al., 1994). Taken together, the unresolvable nucleation core appears to consist of hyperphosphorylated tau and TIA-1, which may serve as a seeding point for further aggregation of normal tau, and thus act as an important mediator of seeded aggregation. This proposes a previously unrecognized connection between disease-associated hyperphosphorylation and cell-to-cell seeding property of tau. Further studies are needed to confirm this hypothesis, for example by testing the nucleation behavior of TIA-1 and hyperphosphorylated tau in a cell-free system such as Microscale Thermophoresis.
Understanding cell-to-cell transfer of disease-associated protein as a whole6.5
The pathogenic pathways of several neurodegenerative diseases such as Parkinson’s disease and Alzheimer’s disease remain incompletely understood. As illustrated in Figure 22,the development of synucleinopathies and tauopathies start at the sporadic aggregation of disease-specific proteins. Despite the fact that previous studies have proposed many factors,such as genetic risk factors, post-translational modifications, metal ion-related oxidative stresses, lipid abnormalities, impaired cellular waste management and altered energy homeostasis, could modulate the aggregation pathway(s) of aSyn and tau, we still do not have enough knowledge to confidently declare the initial triggers of the pathological protein aggregation in human patients. Most likely this process involves a complex interplay of genetic and environmental factors, including aging-related deterioration of cellular defence mechanisms. One thing that has repeatedly been observed is the progressive development of synucleinopathies and tauopathies and related degeneration of neurons in neuroanatomically connected brain regions in a time-dependent manner (Braak et al., 2003, Braak and DelTredici, 2011). In addition, both aSyn and tau are present in CSF of human patients (Blennow et al., 1995, Borghi et al., 2000). Taken together, it seems that aSyn and tau get access to the extracellular space and spread from one region to another during the progression of neurodegenerative diseases. Thus, for therapeutic purposes, it may be critical to halting the pathogenic spreading by interrupting these processes. However, there are many missing gaps in our knowledge about the mechanisms of the pathology spreading pathways, as summarized in Figure 22, and this hinders us from selecting the right biomarkers for diagnostics and right protein candidates for drug targeting.
In the literature, the term “prion-like” is often used to describe the transmissibility and seeded aggregation of aSyn and tau. One recent publication even claimed that aSyn is a prion, and aSyn-mediated human multiple system atrophy (MSA) is a prion disease simply because inoculation of lysate from human MSA post-mortem brain induced a synucleinopathy in transgenic mice and cells (Prusiner et al., 2015). To be “prion-like” would mean pathological species of aSyn and tau acting as infectious agents that spread the pathology rapidly by interacting and converting normal aSyn and tau proteins into pathological species in a template-directed manner, and that this phenomenon could also be transmitted from one individual to another (Bolton et al., 1982, Aguzzi and Rajendran, 2009). This is indeed true for some species or strains of aSyn and tau as demonstrated in cells and transgenic mice models. This thus creates an impression that when aSyn and tau are called "prion-like," the puzzle of

69
how aSyn and tau contribute to the spread of pathology is solved. However, it is not as simple as that. First of all, it may be questioned that to what extend mouse models represent human, e.g. some NFTs-bearing transgenic mice showed no impairment in neuronal functions (Santacruz et al., 2005, Berger et al., 2007). Due to the ethical concerns, a few studies of pathological protein propagation have been carried out in human neurons induced from pluripotent stem cells (Chai et al., 2012a, Wu et al., 2016). These neurons derived from differentiated human stem cells provide a better model for studying tau and aSyn transmission in the context of "human," but it is still far away from representing the complex and systemic network of the human brain. Apart from the limitation of the model system, the term "prion-like" only provides an existing model to explain how aSyn and tau might seed the aggregation at the molecular level. Except two studies implicated that contaminated surgical grafts of dura mater and growth hormones prepared from cadavers of Creutzfeldt–Jakob disease (CJD) might be related to Aβ plaques observed in the brain of patients who received the transplantation, there has not been any indication that AD and PD are transmissible in man (Zane Jaunmuktane and Simon Mead, 2015, Frontzek et al., 2016). Apart from the fact that not all the strains and species of aSyn and tau have been fully characterized by their property of seeding, the mechanism of cellular release and uptake, potential post-release events in extracellular space or human plasma and CSF, and the pathological consequences of internalization of these disease-associated proteins are currently poorly understood (Figure 22). In addition, many factors such as genetic risk factors, post-translational modifications, chemicals and protein modulators can readily modify the propagation pathway(s) of aSyn and tau in pathological conditions. Thus, it is important to dissect the entire propagation pathway into sub-processes, and to study the mechanisms and modulators involved in each step of the pathway in order to understand the propagation of disease-associated proteins as a whole.
In this thesis, the mechanisms of cell-to-cell transmission of disease-associated proteins were studied at steps of dimerization/oligomerization, cellular release and uptake, and a post-internalization event. Both tau and aSyn were used as models, although side-by-side comparisons of the two proteins were not directly performed. It may be criticized that the study should have just focused on one protein rather than two proteins that are structurally different and primarily contribute to distinct disease-specific pathologies. However, the aSyn and tau pathologies are not strictly isolated, and a lot of evidence supports significant co-occurrence and overlap of aSyn and tau pathologies in individual patients in many diseases such as Lewy body variant of AD (LBVAD), Parkinson’s disease with dementia (PDD),dementia with Lewy bodies (DLB), Guam-Parkinson-ALS dementia complex, and even Down’s syndrome etc (Lippa et al., 1998, Lippa et al., 1999, Forman et al., 2002, Moussaud et al., 2014). In addition, synucleinopathy and tauopathy are not hermetically restricted in PD or AD, but often contributes to secondary symptoms. As mentioned previously, there is a lot of similarity and overlap in the aggregation pathways and properties of transmissibility between aSyn and tau, suggesting convergent pathways of aSyn and tau aggregation and propagation in the pathogenesis of clinically divergent neurodegenerative diseases such as AD and PD.Therefore, further studies of the two remarkably interconnected disease-associated proteins could generate complementary knowledge that would benefit our understanding of the poorly understood pathogenic pathways as a whole.
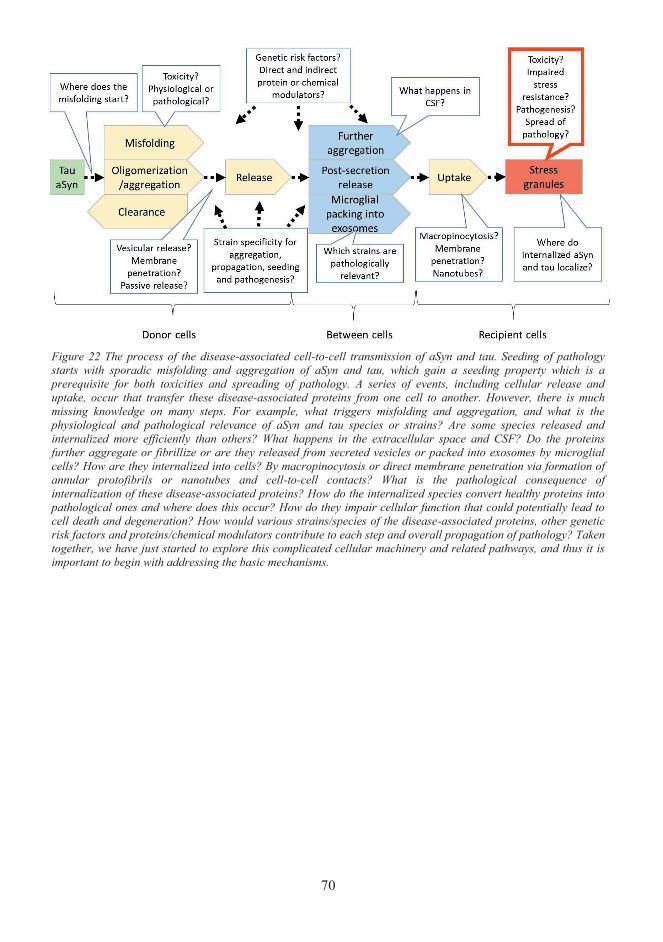
70
Figure 22 The process of the disease-associated cell-to-cell transmission of aSyn and tau. Seeding of pathology starts with sporadic misfolding and aggregation of aSyn and tau, which gain a seeding property which is a prerequisite for both toxicities and spreading of pathology. A series of events, including cellular release and uptake, occur that transfer these disease-associated proteins from one cell to another. However, there is much missing knowledge on many steps. For example, what triggers misfolding and aggregation, and what is the physiological and pathological relevance of aSyn and tau species or strains? Are some species released and internalized more efficiently than others? What happens in the extracellular space and CSF? Do the proteins further aggregate or fibrillize or are they released from secreted vesicles or packed into exosomes by microglial cells? How are they internalized into cells? By macropinocytosis or direct membrane penetration via formation of annular protofibrils or nanotubes and cell-to-cell contacts? What is the pathological consequence of internalization of these disease-associated proteins? How do the internalized species convert healthy proteins into pathological ones and where does this occur? How do they impair cellular function that could potentially lead to cell death and degeneration? How would various strains/species of the disease-associated proteins, other genetic risk factors and proteins/chemical modulators contribute to each step and overall propagation of pathology? Taken together, we have just started to explore this complicated cellular machinery and related pathways, and thus it is important to begin with addressing the basic mechanisms.

71
7 Concluding remarks and future prospects
The aggregation, cellular secretion and uptake of aSyn and tau are fundamental processes involved in the propagation of synucleinopathies and tauopathies that contribute to many neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. The studies presented in this thesis provide an important novel insight into the mechanisms of aSyn aggregation, and cellular secretion and uptake of tau.
First, we have developed and validated a novel method based on protein-fragment complementation assay to study protein-protein interactions of aSyn and tau in live cells and conditioned media (I, II, III). Moreover, the protein-fragment complementation assay (PCA)-based platform was used to study dimerization change of aSyn and tau upon pharmacological and genetic modulation in cells (I, II). Additionally, we have further optimized and validated these assays to investigate the mechanisms and consequences of cellular secretion and internalization of tau in live cell environment (II, III). The main findings of this thesis are:
Study I: Prolyl-oligo peptidase (PREP) interacts with aSyn. The direct protein-protein interaction between PREP and aSyn promote aSyn dimerization independent of PREP enzymatic activity. KYP-2047, a small molecule inhibitor of PREP, reduces PREP-induced aSyn dimerization, possibly by inducing PREP conformational shift to compact monomeric form.
Study II: The expression level of FRMD4A, a late-onset Alzheimer’s disease susceptibility gene, modulates cellular secretion of tau, likely through cell polarity signalling complex Par6/aPKC -FRMD4A-cytohesin-Arf6 signalling pathway.
Study III: Extracellular tau is internalized by cells and recruited to stress granules in a TIA-1 dependent manner. Macropinocytosis appears as the main route of entry mediating tau internalization. Hyperphosphorylation of tau at the phosphoepitope AT8 likely contributes to induction of the abnormal stress granules that are hard to resolve and sensitize cells tosecondary stress.
Due to the increases in demographic aging, age-related diseases such as neurodegenerative diseases have emerged as a heavy burden to the society. There is yet no effective curable therapeutics or ever ways to delay the disease progression in most of these diseases, and most clinical trials for Alzheimer’s disease and Parkinson’s disease have been disappointing. There thus is an urgent need for a better understanding of the etiology of these diseases in order to forge effective strategies for development of therapeutics. This thesis provides novel data in identifying the modulators and risk factors of the diseases and identifying novel molecular mechanisms, which give the opportunity for novel drug targets and development of disease-modifying treatments and strategies to halt the progression of disease-specific pathology in the future.

72
Acknowledgements
The work included in my thesis was carried out in the laboratory of Docent Henri Huttunen at the Viikki campus of Neuroscience Centre, Helsinki University. The work was funded by the Neuroscience center, Academy of Finland, Brain and Mind Doctoral Programme.
I wish to express my deepest gratitude to my supervisor Docent Henri Huttunen. I am very appreciated that Henri has chosen me to be his Ph.D. student and taught me tremendously about neuroscience and how to be a scientific researcher. Thank you for your valuable instructions and advices on my research. Your patience, trust and encouragement have accompanied me in my journey of obtaining the degree. I can honestly say that you have been the best supervisor and colleague I could expect. Thank you for everything.
I am deeply thankful to Dr. Kelvin Luk for kindly accepting the invitation being my opponent, and to Prof. Juha Voipio for kindly accepting being custos in my Ph.D. defence. I would also like to thank Dr. Mikko Airavaara, who has been both my pre-examiner and thesis committee member, Dr. Katja Kanninen, who has been my pre-examiner, and Dr. Tomi Rantamäki, who has been my thesis committee member. Your critical and delightful comments have been a great help in improving my thesis. Also, I wish to express my gratitude to my collaborators Dr. Timo Myöhänen, Dr. Mari Savolainen and Prof. Mikko Hiltunen. Your contribution has made this work possible.
I would like to thank the former and present colleagues in the Huttunen lab (in alphabetic order): Ana, Emmi, Cecillia, Kai, Liang, Maria, Niko, Pranuti, Prasanna, Riikka, and Wei. I appreciate a lot of your friendship, positive environment, help and scientific discussion over the years. In addition, I would like to thank all other colleagues and staff members at the Neuroscience Centre. Special thanks to Dr. Li Ma, Dr. Liang Zhou and Dr. Zhilin Li (in alphabetic order) for all the friendship, interesting discussion and outdoor activities.
I would like to thank my relatives in China for their caring, support and all the good time spent together. I would also like to thank my friend in Finland and abroad for the fun time and friendship.
For my parents, Mei Liu and Zhizhong Yan, I am very grateful for all the support, caring and love. Thank you for letting me chose my own path and career. Your guidance and helphave been fundamental elements for pursuing my scientific career. Thank you for your understanding and comforts when I am depressed. In addition, I would also like to thank my girlfriend Siyi Fu. I appreciate your love and support. Your accompany has been delightful and encouraging as always. You all mean the world to me.
Helsinki, February 2017
Xu Yan

73
References
AARSLAND, D., ZACCAI, J. & BRAYNE, C. 2005. A systematic review of prevalence studies of dementia in Parkinson's disease. Mov Disord, 20, 1255-63.AGUZZI, A. 2009. Cell biology: Beyond the prion principle. Nature, 459, 924-5.AGUZZI, A. & RAJENDRAN, L. 2009. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron, 64, 783-90.AHMED, T., VAN DER JEUGD, A., BLUM, D., GALAS, M. C., D'HOOGE, R., BUEE, L. & BALSCHUN, D. 2014. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol Aging, 35, 2474-8.AISENBREY, C., BOROWIK, T., BYSTROM, R., BOKVIST, M., LINDSTROM, F., MISIAK, H., SANI, M. A. & GROBNER, G. 2008. How is protein aggregation in amyloidogenic diseases modulated by biological membranes? Eur Biophys J, 37, 247-55.ALIM, M. A., HOSSAIN, M. S., ARIMA, K., TAKEDA, K., IZUMIYAMA, Y., NAKAMURA, M., KAJI, H., SHINODA, T., HISANAGA, S. & UEDA, K. 2002. Tubulin seeds alpha-synuclein fibril formation. J Biol Chem, 277, 2112-7.ALIM, M. A., MA, Q. L., TAKEDA, K., AIZAWA, T., MATSUBARA, M., NAKAMURA, M., ASADA, A., SAITO, T., KAJI, H., YOSHII, M., HISANAGA, S. & UEDA, K. 2004. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J Alzheimers Dis, 6, 435-42; discussion 443-9.ALONSO, A., ZAIDI, T., GRUNDKE-IQBAL, I. & IQBAL, K. 1994. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A,91, 5562-6.ALONSO, A., ZAIDI, T., NOVAK, M., GRUNDKE-IQBAL, I. & IQBAL, K. 2001. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A, 98, 6923-8.ALONSO ADEL, C., LI, B., GRUNDKE-IQBAL, I. & IQBAL, K. 2006. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A, 103,8864-9.ALONSO ADEL, C., MEDERLYOVA, A., NOVAK, M., GRUNDKE-IQBAL, I. & IQBAL, K. 2004.Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem, 279,34873-81.ALVAREZ-ERVITI, L., SEOW, Y., SCHAPIRA, A. H., GARDINER, C., SARGENT, I. L., WOOD, M. J. & COOPER, J. M. 2011. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis, 42, 360-7.AMYERE, M., METTLEN, M., VAN DER SMISSEN, P., PLATEK, A., PAYRASTRE, B., VEITHEN, & A. AND COURTOY, P. J. 2001. Origin, originality, functions, subversions andmolecular signalling of macropinocytosis. Int. J. Med. Microbiol., 487-494.ANDERSEN, J. K. 2004. Oxidative stress in neurodegeneration: cause or consequence? Nat Med, 10Suppl, S18-25.ANDERSON, J. P., WALKER, D. E., GOLDSTEIN, J. M., DE LAAT, R., BANDUCCI, K., CACCAVELLO, R. J., BARBOUR, R., HUANG, J., KLING, K., LEE, M., DIEP, L., KEIM, P. S., SHEN, X., CHATAWAY, T., SCHLOSSMACHER, M. G., SEUBERT, P., SCHENK, D., SINHA, S., GAI, W. P. & CHILCOTE, T. J. 2006. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem, 281, 29739-52.ANDERSON, P. & KEDERSHA, N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem Sci,33, 141-50.ANDORFER, C., ACKER, C. M., KRESS, Y., HOF, P. R., DUFF, K. & DAVIES, P. 2005. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci, 25,5446-54.

74
ARENDT, T., STIELER, J., STRIJKSTRA, A. M., HUT, R. A., RUDIGER, J., VAN DER ZEE, E. A., HARKANY, T., HOLZER, M. & HARTIG, W. 2003. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci, 23, 6972-81.ASAI, H., IKEZU, S., TSUNODA, S., MEDALLA, M., LUEBKE, J., HAYDAR, T., WOLOZIN, B., BUTOVSKY, O., KUGLER, S. & IKEZU, T. 2015a. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci, 18, 1584-93.ASAI, H., IKEZU, S., TSUNODA, S., MEDALLA, M., LUEBKE, J., HAYDAR, T., WOLOZIN, B., BUTOVSKY, O., KUGLER, S. & IKEZU, T. 2015b. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci.ASH, P. E., VANDERWEYDE, T. E., YOUMANS, K. L., APICCO, D. J. & WOLOZIN, B. 2014. Pathological stress granules in Alzheimer's disease. Brain Res, 1584, 52-8.ASHERY, U., KOCH, H., SCHEUSS, V., BROSE, N. & RETTIG, J. 1999. A presynaptic role for the ADP ribosylation factor (ARF)-specific GDP/GTP exchange factor msec7-1. Proc Natl Acad Sci U S A,96, 1094-9.AVILA, J., GOMEZ-RAMOS, A. & BOLOS, M. 2015. AD genetic risk factors and tau spreading. Front Aging Neurosci, 7, 99.BABU, J. R., GEETHA, T. & WOOTEN, M. W. 2005. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem, 94, 192-203.BACHSTETTER, A. D., VAN ELDIK, L. J., SCHMITT, F. A., NELTNER, J. H., IGHODARO, E. T., WEBSTER, S. J., PATEL, E., ABNER, E. L., KRYSCIO, R. J. & NELSON, P. T. 2015. Disease-related microglia heterogeneity in the hippocampus of Alzheimer's disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun, 3, 32.BADIOLA, N., DE OLIVEIRA, R. M., HERRERA, F., GUARDIA-LAGUARTA, C., GONCALVES, S. A., PERA, M., SUAREZ-CALVET, M., CLARIMON, J., OUTEIRO, T. F. & LLEO, A. 2011. Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS One, 6,e26609.BAIXAULI, F., LOPEZ-OTIN, C. & MITTELBRUNN, M. 2014. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol, 5, 403.BALCH, W. E., MORIMOTO, R. I., DILLIN, A. & KELLY, J. W. 2008. Adapting proteostasis for disease intervention. Science, 319, 916-9.BALKLAVA, Z., PANT, S., FARES, H. & GRANT, B. 2007. Genome-wide analysis identifies a general requirement for polarity proteins in endocytic traffic. Nat Cell Biol, 9, 1066-73.BALLATORE, C., LEE, V. M. & TROJANOWSKI, J. Q. 2007. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci, 8, 663-72.BANCHER, C., BRAAK, H., FISCHER, P. & JELLINGER, K. A. 1993. Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer's and Parkinson's disease patients. Neurosci Lett,162, 179-82.BANCHER, C., BRUNNER, C., LASSMANN, H., BUDKA, H., JELLINGER, K., WICHE, G., SEITELBERGER, F., GRUNDKE-IQBAL, I., IQBAL, K. & WISNIEWSKI, H. 1989. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res, 477, 90-9.BARGHORN, S., ZHENG-FISCHHOFER, Q., ACKMANN, M., BIERNAT, J., VON BERGEN, M., MANDELKOW, E. M. & MANDELKOW, E. 2000. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry, 39, 11714-21.BARTELS, T., CHOI, J. G. & SELKOE, D. J. 2011. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature, 477, 107-10.BAYIR, H., KAPRALOV, A. A., JIANG, J., HUANG, Z., TYURINA, Y. Y., TYURIN, V. A., ZHAO, Q., BELIKOVA, N. A., VLASOVA, II, MAEDA, A., ZHU, J., NA, H. M., MASTROBERARDINO, P. G., SPARVERO, L. J., AMOSCATO, A. A., CHU, C. T., GREENAMYRE, J. T. & KAGAN, V. E. 2009. Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome C:

75
protection against apoptosis versus delayed oxidative stress in Parkinson disease. J Biol Chem, 284,15951-69.BENNETT, M. C., BISHOP, J. F., LENG, Y., CHOCK, P. B., CHASE, T. N. & MOURADIAN, M. M. 1999. Degradation of alpha-synuclein by proteasome. J Biol Chem, 274, 33855-8.BENTMANN, E., HAASS, C. & DORMANN, D. 2013. Stress granules in neurodegeneration--lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J, 280, 4348-70.BERGER, Z., RAVIKUMAR, B., MENZIES, F. M., OROZ, L. G., UNDERWOOD, B. R., PANGALOS, M. N., SCHMITT, I., WULLNER, U., EVERT, B. O., O'KANE, C. J. & RUBINSZTEIN, D. C. 2006. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet, 15,433-42.BERGER, Z., RODER, H., HANNA, A., CARLSON, A., RANGACHARI, V., YUE, M., WSZOLEK,Z., ASHE, K., KNIGHT, J., DICKSON, D., ANDORFER, C., ROSENBERRY, T. L., LEWIS, J., HUTTON, M. & JANUS, C. 2007. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci, 27, 3650-62.BERNALES, S., SOTO, M. M. & MCCULLAGH, E. 2012. Unfolded protein stress in the endoplasmic reticulum and mitochondria: a role in neurodegeneration. Front Aging Neurosci, 4, 5.BERTRAM, L., LANGE, C., MULLIN, K., PARKINSON, M., HSIAO, M., HOGAN, M., SCHJEIDE, B., HOOLI, B., DIVITO, J., IONITA, I., JIANG, H., LAIRD, N., MOSCARILLO, T., OHLSEN, K., ELLIOTT, K., WANG, X., HU-LINCE, D., RYDER, M., MURPHY, A., WAGNER, S., BLACKER, D., BECKER, K. & TANZI, R. 2008. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet, 83, 623-32.BERTRAM, L., LILL, C. & TANZI, R. 2010. The genetics of Alzheimer disease: back to the future. Neuron, 68, 270-81.BERTRAM, L., MCQUEEN, M., MULLIN, K., BLACKER, D. & TANZI, R. 2007. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet, 39, 17-23.BERTRAND, J., PLOUFFE, V., SENECHAL, P. & LECLERC, N. 2010. The pattern of human tau phosphorylation is the result of priming and feedback events in primary hippocampal neurons. Neuroscience, 168, 323-34.BEYER, K. 2006. Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol, 112, 237-51.BHASKAR, K., YEN, S. H. & LEE, G. 2005. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem, 280, 35119-25.BINOLFI, A., LAMBERTO, G. R., DURAN, R., QUINTANAR, L., BERTONCINI, C. W., SOUZA, J. M., CERVENANSKY, C., ZWECKSTETTER, M., GRIESINGER, C. & FERNANDEZ, C. O. 2008. Site-specific interactions of Cu(II) with alpha and beta-synuclein: bridging the molecular gap between metal binding and aggregation. J Am Chem Soc, 130, 11801-12.BLAIR, L., FRAUEN, H., ZHANG, B., NORDHUES, B., BIJAN, S., LIN, Y., ZAMUDIO, F., HERNANDEZ, L., SABBAGH, J., SELENICA, M. & DICKEY, C. 2015. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol Commun, 3,8.BLENNOW, K., WALLIN, A., AGREN, H., SPENGER, C., SIEGFRIED, J. & VANMECHELEN, E. 1995. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol, 26, 231-45.BLOOMFIELD, G. & KAY, R. R. 2016. Uses and abuses of macropinocytosis. J Cell Sci, 129, 2697-705.BODNER, R. A., OUTEIRO, T. F., ALTMANN, S., MAXWELL, M. M., CHO, S. H., HYMAN, B. T., MCLEAN, P. J., YOUNG, A. B., HOUSMAN, D. E. & KAZANTSEV, A. G. 2006. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington's and Parkinson's diseases. Proc Natl Acad Sci U S A, 103, 4246-51.BOLTON, D., MCKINLEY, M. & PRUSINER, S. 1982. Identification of a protein that purifies with the scrapie prion. Science, 218, 1309-11.

76
BORGHI, R., MARCHESE, R., NEGRO, A., MARINELLI, L., FORLONI, G., ZACCHEO, D., ABBRUZZESE, G. & TABATON, M. 2000. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson's disease and normal subjects. Neurosci Lett, 287, 65-7.BOUSSET, L., PIERI, L., RUIZ-ARLANDIS, G., GATH, J., JENSEN, P. H., HABENSTEIN, B., MADIONA, K., OLIERIC, V., BOCKMANN, A., MEIER, B. H. & MELKI, R. 2013. Structural and functional characterization of two alpha-synuclein strains. Nat Commun, 4, 2575.BRAAK, E., BRAAK, H. & MANDELKOW, E. 1994a. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol, 87, 554-67.BRAAK, H., ALAFUZOFF, I., ARZBERGER, T., KRETZSCHMAR, H. & DEL TREDICI, K. 2006. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol, 112, 389-404.BRAAK, H. & BRAAK, E. 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol, 82, 239-59.BRAAK, H., BRAAK, E. & STROTHJOHANN, M. 1994b. Abnormally phosphorylated tau protein related to the formation of neurofibrillary tangles and neuropil threads in the cerebral cortex of sheep and goat. Neurosci Lett, 171, 1-4.BRAAK, H. & DEL TREDICI, K. 2008. Invited Article: Nervous system pathology in sporadic Parkinson disease. Neurology, 70, 1916-25.BRAAK, H. & DEL TREDICI, K. 2011. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol, 121, 171-81.BRAAK, H., DEL TREDICI, K., RUB, U., DE VOS, R. A., JANSEN STEUR, E. N. & BRAAK, E. 2003. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging, 24, 197-211.BRADY, R., ZINKOWSKI, R. & BINDER, L. 1995. Presence of tau in isolated nuclei from human brain. Neurobiol Aging, 16, 479-86.BRANDT, I., GERARD, M., SERGEANT, K., DEVREESE, B., BAEKELANDT, V., AUGUSTYNS, K., SCHARPE, S., ENGELBORGHS, Y. & LAMBEIR, A. M. 2008. Prolyl oligopeptidase stimulates the aggregation of alpha-synuclein. Peptides, 29, 1472-8.BRETTSCHNEIDER, J., DEL TREDICI, K., LEE, V. M. & TROJANOWSKI, J. Q. 2015. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci, 16, 109-20.BRETTSCHNEIDER, J., DEL TREDICI, K., TOLEDO, J. B., ROBINSON, J. L., IRWIN, D. J., GROSSMAN, M., SUH, E., VAN DEERLIN, V. M., WOOD, E. M., BAEK, Y., KWONG, L., LEE, E. B., ELMAN, L., MCCLUSKEY, L., FANG, L., FELDENGUT, S., LUDOLPH, A. C., LEE, V. M., BRAAK, H. & TROJANOWSKI, J. Q. 2013. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol, 74, 20-38.BROWN, M. R., BONDADA, V., KELLER, J. N., THORPE, J. & GEDDES, J. W. 2005. Proteasome or calpain inhibition does not alter cellular tau levels in neuroblastoma cells or primary neurons. JAlzheimers Dis, 7, 15-24.BUKHATWA, S., ZENG, B. Y., ROSE, S. & JENNER, P. 2010. A comparison of changes in proteasomal subunit expression in the substantia nigra in Parkinson's disease, multiple system atrophy and progressive supranuclear palsy. Brain Res, 1326, 174-83.BURKE, R. E., DAUER, W. T. & VONSATTEL, J. P. 2008. A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol, 64, 485-91.BURRE, J., SHARMA, M., TSETSENIS, T., BUCHMAN, V., ETHERTON, M. R. & SUDHOF, T. C. 2010. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science, 329, 1663-7.CACACE, R., SLEEGERS, K. & VAN BROECKHOVEN, C. 2016. Molecular genetics of early-onset Alzheimer's disease revisited. Alzheimers Dement, 12, 733-48.CALABRETTA, S. & RICHARD, S. 2015. Emerging Roles of Disordered Sequences in RNA-Binding Proteins. Trends Biochem Sci, 40, 662-72.CALERO, M., GOMEZ-RAMOS, A., CALERO, O., SORIANO, E., AVILA, J. & MEDINA, M. 2015. Additional mechanisms conferring genetic susceptibility to Alzheimer's disease. Front Cell Neurosci, 9,138.

77
CAMPONESCHI, F., VALENSIN, D., TESSARI, I., BUBACCO, L., DELL'ACQUA, S., CASELLA, L., MONZANI, E., GAGGELLI, E. & VALENSIN, G. 2013. Copper(I)-alpha-synuclein interaction: structural description of two independent and competing metal binding sites. Inorg Chem, 52, 1358-67.CASTELLANO, J., KIM, J., STEWART, F., JIANG, H., DEMATTOS, R., PATTERSON, B., FAGAN, A., MORRIS, J., MAWUENYEGA, K., CRUCHAGA, C., GOATE, A., BALES, K., PAUL, S., BATEMAN, R. & HOLTZMAN, D. 2011. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med, 3, 89ra57.CHAARI, A., HOARAU-VECHOT, J. & LADJIMI, M. 2013. Applying chaperones to protein-misfolding disorders: molecular chaperones against alpha-synuclein in Parkinson's disease. Int J Biol Macromol, 60, 196-205.CHAI, X., DAGE, J. & CITRON, M. 2012a. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis, 48, 356-66.CHAI, X., DAGE, J. L. & CITRON, M. 2012b. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis, 48, 356-66.CHAPUIS, J., HANSMANNEL, F., GISTELINCK, M., MOUNIER, A., VAN CAUWENBERGHE, C., KOLEN, K., GELLER, F., SOTTEJEAU, Y., HAROLD, D., DOURLEN, P., GRENIER-BOLEY, B., KAMATANI, Y., DELEPINE, B., DEMIAUTTE, F., ZELENIKA, D., ZOMMER, N., HAMDANE, M., BELLENGUEZ, C., DARTIGUES, J., HAUW, J., LETRONNE, F., AYRAL, A., SLEEGERS, K., SCHELLENS, A., BROECK, L., ENGELBORGHS, S., DE DEYN, P., VANDENBERGHE, R., O'DONOVAN, M., OWEN, M., EPELBAUM, J., MERCKEN, M., KARRAN, E., BANTSCHEFF, M., DREWES, G., JOBERTY, G., CAMPION, D., OCTAVE, J., BERR, C., LATHROP, M., CALLAERTS, P., MANN, D., WILLIAMS, J., BUEE, L., DEWACHTER, I., VAN BROECKHOVEN, C., AMOUYEL, P., MOECHARS, D., DERMAUT, B. & LAMBERT, J. 2013a. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry, 18, 1225-34.CHAPUIS, J., HANSMANNEL, F., GISTELINCK, M., MOUNIER, A., VAN CAUWENBERGHE, C., KOLEN, K. V., GELLER, F., SOTTEJEAU, Y., HAROLD, D., DOURLEN, P., GRENIER-BOLEY, B., KAMATANI, Y., DELEPINE, B., DEMIAUTTE, F., ZELENIKA, D., ZOMMER, N., HAMDANE, M., BELLENGUEZ, C., DARTIGUES, J. F., HAUW, J. J., LETRONNE, F., AYRAL, A. M., SLEEGERS, K., SCHELLENS, A., BROECK, L. V., ENGELBORGHS, S., DE DEYN, P. P., VANDENBERGHE, R., O'DONOVAN, M., OWEN, M., EPELBAUM, J., MERCKEN, M., KARRAN, E., BANTSCHEFF, M., DREWES, G., JOBERTY, G., CAMPION, D., OCTAVE, J. N., BERR, C., LATHROP, M., CALLAERTS, P., MANN, D., WILLIAMS, J., BUEE, L., DEWACHTER, I., VAN BROECKHOVEN, C., AMOUYEL, P., MOECHARS, D., DERMAUT, B., LAMBERT, J. C. & CONSORTIUM, G. 2013b. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry, 18, 1225-34.CHARTIER-HARLIN, M. C., KACHERGUS, J., ROUMIER, C., MOUROUX, V., DOUAY, X., LINCOLN, S., LEVECQUE, C., LARVOR, L., ANDRIEUX, J., HULIHAN, M., WAUCQUIER, N., DEFEBVRE, L., AMOUYEL, P., FARRER, M. & DESTEE, A. 2004. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet, 364, 1167-9.CHAVARRIA, C. & SOUZA, J. M. 2013. Oxidation and nitration of alpha-synuclein and their implications in neurodegenerative diseases. Arch Biochem Biophys, 533, 25-32.CHAVES, R. S., MELO, T. Q., MARTINS, S. A. & FERRARI, M. F. 2010. Protein aggregation containing beta-amyloid, alpha-synuclein and hyperphosphorylated tau in cultured cells of hippocampus, substantia nigra and locus coeruleus after rotenone exposure. BMC Neurosci, 11, 144.CHEN, A. K., LIN, R. Y., HSIEH, E. Z., TU, P. H., CHEN, R. P., LIAO, T. Y., CHEN, W., WANG, C. H. & HUANG, J. J. 2010. Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J Am Chem Soc, 132, 1186-7.CHO, J. & JOHNSON, G. 2003. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J Biol Chem, 278, 187-93.

78
CHOI, W., ZIBAEE, S., JAKES, R., SERPELL, L. C., DAVLETOV, B., CROWTHER, R. A. & GOEDERT, M. 2004. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett, 576, 363-8.CHOURAKI, V. & SESHADRI, S. 2014. Genetics of Alzheimer's disease. Adv Genet, 87, 245-94.CHUN, W., WALDO, G. & JOHNSON, G. 2007a. Split GFP complementation assay: a novel approach to quantitatively measure aggregation of tau in situ: effects of GSK3beta activation and caspase 3 cleavage. J Neurochem, 103, 2529-39.CHUN, W., WALDO, G. S. & JOHNSON, G. V. 2007b. Split GFP complementation assay: a novel approach to quantitatively measure aggregation of tau in situ: effects of GSK3beta activation and caspase 3 cleavage. J Neurochem, 103, 2529-39.CHURCHER, I. 2006. Tau therapeutic strategies for the treatment of Alzheimer's disease. Curr Top Med Chem, 6, 579-95.CIACCIOLI, G., MARTINS, A., RODRIGUES, C., VIEIRA, H. & CALADO, P. 2013. A powerful yeast model to investigate the synergistic interaction of alpha-synuclein and tau in neurodegeneration. PLoS One, 8, e55848.CLAVAGUERA, F., AKATSU, H., FRASER, G., CROWTHER, R. A., FRANK, S., HENCH, J., PROBST, A., WINKLER, D. T., REICHWALD, J., STAUFENBIEL, M., GHETTI, B., GOEDERT, M. & TOLNAY, M. 2013. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A, 110, 9535-40.CLAVAGUERA, F., BOLMONT, T., CROWTHER, R. A., ABRAMOWSKI, D., FRANK, S., PROBST, A., FRASER, G., STALDER, A. K., BEIBEL, M., STAUFENBIEL, M., JUCKER, M., GOEDERT, M. & TOLNAY, M. 2009. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol, 11, 909-13.CLAYTON, E. L. & COUSIN, M. A. 2009. The molecular physiology of activity-dependent bulk endocytosis of synaptic vesicles. J Neurochem, 111, 901-14.COCHRAN, J., RUSH, T., BUCKINGHAM, S. & ROBERSON, E. 2015. The Alzheimer's disease risk factor CD2AP maintains blood-brain barrier integrity. Hum Mol Genet.COLE, N. B., MURPHY, D. D., GRIDER, T., RUETER, S., BRASAEMLE, D. & NUSSBAUM, R. L. 2002. Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha-synuclein. J Biol Chem, 277, 6344-52.COOPER, A. A., GITLER, A. D., CASHIKAR, A., HAYNES, C. M., HILL, K. J., BHULLAR, B., LIU, K., XU, K., STRATHEARN, K. E., LIU, F., CAO, S., CALDWELL, K. A., CALDWELL, G. A., MARSISCHKY, G., KOLODNER, R. D., LABAER, J., ROCHET, J. C., BONINI, N. M. & LINDQUIST, S. 2006. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science, 313, 324-8.COOPER, J. M., WIKLANDER, P. B., NORDIN, J. Z., AL-SHAWI, R., WOOD, M. J., VITHLANI, M., SCHAPIRA, A. H., SIMONS, J. P., EL-ANDALOUSSI, S. & ALVAREZ-ERVITI, L. 2014. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov Disord, 29, 1476-85.COPPOLA, G., CHINNATHAMBI, S., LEE, J. J., DOMBROSKI, B. A., BAKER, M. C., SOTO-ORTOLAZA, A. I., LEE, S. E., KLEIN, E., HUANG, A. Y., SEARS, R., LANE, J. R., KARYDAS, A. M., KENET, R. O., BIERNAT, J., WANG, L. S., COTMAN, C. W., DECARLI, C. S., LEVEY, A. I., RINGMAN, J. M., MENDEZ, M. F., CHUI, H. C., LE BER, I., BRICE, A., LUPTON, M. K., PREZA, E., LOVESTONE, S., POWELL, J., GRAFF-RADFORD, N., PETERSEN, R. C., BOEVE, B. F., LIPPA, C. F., BIGIO, E. H., MACKENZIE, I., FINGER, E., KERTESZ, A., CASELLI, R. J., GEARING, M., JUNCOS, J. L., GHETTI, B., SPINA, S., BORDELON, Y. M., TOURTELLOTTE, W. W., FROSCH, M. P., VONSATTEL, J. P., ZAROW, C., BEACH, T. G., ALBIN, R. L., LIEBERMAN, A. P., LEE, V. M., TROJANOWSKI, J. Q., VAN DEERLIN, V. M., BIRD, T. D., GALASKO, D. R., MASLIAH, E., WHITE, C. L., TRONCOSO, J. C., HANNEQUIN, D., BOXER, A. L., GESCHWIND, M. D., KUMAR, S., MANDELKOW, E. M., WSZOLEK, Z. K., UITTI, R. J., DICKSON, D. W., HAINES, J. L., MAYEUX, R., PERICAK-VANCE, M. A., FARRER, L. A., ALZHEIMER'S

79
DISEASE GENETICS, C., ROSS, O. A., RADEMAKERS, R., SCHELLENBERG, G. D., MILLER, B. L., MANDELKOW, E. & GESCHWIND, D. H. 2012. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer's diseases. Hum Mol Genet, 21, 3500-12. CORDER, E., SAUNDERS, A., RISCH, N., STRITTMATTER, W., SCHMECHEL, D., GASKELL, P. J., RIMMLER, J., LOCKE, P., CONNEALLY, P., SCHMADER, K. & ET, A. 1994. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet, 7, 180-4. CORDER, E., SAUNDERS, A., STRITTMATTER, W., SCHMECHEL, D., GASKELL, P., SMALL, G., ROSES, A., HAINES, J. & PERICAK-VANCE, M. 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science, 261, 921-3. CORMONT, M., METON, I., MARI, M., MONZO, P., KESLAIR, F., GASKIN, C., MCGRAW, T. & LE MARCHAND-BRUSTEL, Y. 2003. CD2AP/CMS regulates endosome morphology and traffic to the degradative pathway through its interaction with Rab4 and c-Cbl. Traffic, 4, 97-112. CRARY, J. F., TROJANOWSKI, J. Q., SCHNEIDER, J. A., ABISAMBRA, J. F., ABNER, E. L., ALAFUZOFF, I., ARNOLD, S. E., ATTEMS, J., BEACH, T. G., BIGIO, E. H., CAIRNS, N. J., DICKSON, D. W., GEARING, M., GRINBERG, L. T., HOF, P. R., HYMAN, B. T., JELLINGER, K., JICHA, G. A., KOVACS, G. G., KNOPMAN, D. S., KOFLER, J., KUKULL, W. A., MACKENZIE, I. R., MASLIAH, E., MCKEE, A., MONTINE, T. J., MURRAY, M. E., NELTNER, J. H., SANTA-MARIA, I., SEELEY, W. W., SERRANO-POZO, A., SHELANSKI, M. L., STEIN, T., TAKAO, M., THAL, D. R., TOLEDO, J. B., TRONCOSO, J. C., VONSATTEL, J. P., WHITE, C. L., 3RD, WISNIEWSKI, T., WOLTJER, R. L., YAMADA, M. & NELSON, P. T. 2014. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol, 128, 755-66. CREWS, L., SPENCER, B., DESPLATS, P., PATRICK, C., PAULINO, A., ROCKENSTEIN, E., HANSEN, L., ADAME, A., GALASKO, D. & MASLIAH, E. 2010. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One, 5, e9313. CRIPPS, D., THOMAS, S. N., JENG, Y., YANG, F., DAVIES, P. & YANG, A. J. 2006. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem, 281, 10825-38. CROCKER, P., PAULSON, J. & VARKI, A. 2007. Siglecs and their roles in the immune system. Nat Rev Immunol, 7, 255-66. CRUCHAGA, C., KARCH, C., JIN, S., BENITEZ, B., CAI, Y., GUERREIRO, R., HARARI, O., NORTON, J., BUDDE, J., BERTELSEN, S., JENG, A., COOPER, B., SKORUPA, T., CARRELL, D., LEVITCH, D., HSU, S., CHOI, J., RYTEN, M., HARDY, J., RYTEN, M., TRABZUNI, D., WEALE, M., RAMASAMY, A., SMITH, C., SASSI, C., BRAS, J., GIBBS, J., HERNANDEZ, D., LUPTON, M., POWELL, J., FORABOSCO, P., RIDGE, P., CORCORAN, C., TSCHANZ, J., NORTON, M., MUNGER, R., SCHMUTZ, C., LEARY, M., DEMIRCI, F., BAMNE, M., WANG, X., LOPEZ, O., GANGULI, M., MEDWAY, C., TURTON, J., LORD, J., BRAAE, A., BARBER, I., BROWN, K., PASSMORE, P., CRAIG, D., JOHNSTON, J., MCGUINNESS, B., TODD, S., HEUN, R., KOLSCH, H., KEHOE, P., HOOPER, N., VARDY, E., MANN, D., PICKERING-BROWN, S., BROWN, K., KALSHEKER, N., LOWE, J., MORGAN, K., DAVID SMITH, A., WILCOCK, G., WARDEN, D., HOLMES, C., PASTOR, P., LORENZO-BETANCOR, O., BRKANAC, Z., SCOTT, E., TOPOL, E., MORGAN, K., ROGAEVA, E., SINGLETON, A., HARDY, J., KAMBOH, M., ST GEORGE-HYSLOP, P., CAIRNS, N., MORRIS, J., KAUWE, J. & GOATE, A. 2014. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature, 505, 550-4. CRUCHAGA, C., KAUWE, J., HARARI, O., JIN, S., CAI, Y., KARCH, C., BENITEZ, B., JENG, A., SKORUPA, T., CARRELL, D., BERTELSEN, S., BAILEY, M., MCKEAN, D., SHULMAN, J., DE JAGER, P., CHIBNIK, L., BENNETT, D., ARNOLD, S., HAROLD, D., SIMS, R., GERRISH, A., WILLIAMS, J., VAN DEERLIN, V., LEE, V., SHAW, L., TROJANOWSKI, J., HAINES, J., MAYEUX, R., PERICAK-VANCE, M., FARRER, L., SCHELLENBERG, G., PESKIND, E.,

80
GALASKO, D., FAGAN, A., HOLTZMAN, D., MORRIS, J. & GOATE, A. 2013a. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron, 78, 256-68.CRUCHAGA, C., KAUWE, J., MAYO, K., SPIEGEL, N., BERTELSEN, S., NOWOTNY, P., SHAH, A., ABRAHAM, R., HOLLINGWORTH, P., HAROLD, D., OWEN, M., WILLIAMS, J., LOVESTONE, S., PESKIND, E., LI, G., LEVERENZ, J., GALASKO, D., MORRIS, J., FAGAN, A., HOLTZMAN, D. & GOATE, A. 2010. SNPs associated with cerebrospinal fluid phospho-tau levels influence rate of decline in Alzheimer's disease. PLoS Genet, 6.CRUCHAGA, C., KAUWE, J. S., HARARI, O., JIN, S. C., CAI, Y., KARCH, C. M., BENITEZ, B. A., JENG, A. T., SKORUPA, T., CARRELL, D., BERTELSEN, S., BAILEY, M., MCKEAN, D., SHULMAN, J. M., DE JAGER, P. L., CHIBNIK, L., BENNETT, D. A., ARNOLD, S. E., HAROLD, D., SIMS, R., GERRISH, A., WILLIAMS, J., VAN DEERLIN, V. M., LEE, V. M., SHAW, L. M., TROJANOWSKI, J. Q., HAINES, J. L., MAYEUX, R., PERICAK-VANCE, M. A., FARRER, L. A., SCHELLENBERG, G. D., PESKIND, E. R., GALASKO, D., FAGAN, A. M., HOLTZMAN, D. M., MORRIS, J. C., CONSORTIUM, G., ALZHEIMER'S DISEASE NEUROIMAGING, I., ALZHEIMER DISEASE GENETIC, C. & GOATE, A. M. 2013b. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron, 78, 256-68.CUERVO, A. M., STEFANIS, L., FREDENBURG, R., LANSBURY, P. T. & SULZER, D. 2004. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science, 305, 1292-5.CUERVO, A. M. & WONG, E. 2014. Chaperone-mediated autophagy: roles in disease and aging. Cell Res, 24, 92-104.D'SOUZA-SCHOREY, C. & CHAVRIER, P. 2006. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol, 7, 347-58.DAHER, J. P., YING, M., BANERJEE, R., MCDONALD, R. S., HAHN, M. D., YANG, L., FLINT BEAL, M., THOMAS, B., DAWSON, V. L., DAWSON, T. M. & MOORE, D. J. 2009. Conditional transgenic mice expressing C-terminally truncated human alpha-synuclein (alphaSyn119) exhibit reduced striatal dopamine without loss of nigrostriatal pathway dopaminergic neurons. Mol Neurodegener, 4, 34.DANZER, K. M., HAASEN, D., KAROW, A. R., MOUSSAUD, S., HABECK, M., GIESE, A., KRETZSCHMAR, H., HENGERER, B. & KOSTKA, M. 2007. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci, 27, 9220-32.DANZER, K. M., KRANICH, L. R., RUF, W. P., CAGSAL-GETKIN, O., WINSLOW, A. R., ZHU, L., VANDERBURG, C. R. & MCLEAN, P. J. 2012. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener, 7, 42.DAVID, D. C., LAYFIELD, R., SERPELL, L., NARAIN, Y., GOEDERT, M. & SPILLANTINI, M. G. 2002. Proteasomal degradation of tau protein. J Neurochem, 83, 176-85.DAVIDSON, W. S., JONAS, A., CLAYTON, D. F. & GEORGE, J. M. 1998. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem, 273, 9443-9.DE CALIGNON, A., FOX, L. M., PITSTICK, R., CARLSON, G. A., BACSKAI, B. J., SPIRES-JONES, T. L. & HYMAN, B. T. 2010. Caspase activation precedes and leads to tangles. Nature, 464,1201-4.DEFELIPE, J. 2016. Phospho-Tau and Cognitive Decline in Alzheimer's Disease. Commentary: Tau in physiology and pathology. Front Neuroanat, 10, 44.DEL TREDICI, K., HAWKES, C. H., GHEBREMEDHIN, E. & BRAAK, H. 2010. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson's disease. Acta Neuropathol, 119, 703-13.DELOBEL, P., FLAMENT, S., HAMDANE, M., MAILLIOT, C., SAMBO, A., BEGARD, S., SERGEANT, N., DELACOURTE, A., VILAIN, J. & BUEE, L. 2002. Abnormal Tau phosphorylation of the Alzheimer-type also occurs during mitosis. J Neurochem, 83, 412-20.

81
DENNIS DICKSON, C. B., JEAN-JACQUES HAUW, KURT A. JELLINGER, PETER L. LANTOS, HIDEHIRO MIZUSAWA 2011. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. John Wiley & Sons, 2011.DESPLATS, P., LEE, H. J., BAE, E. J., PATRICK, C., ROCKENSTEIN, E., CREWS, L., SPENCER, B., MASLIAH, E. & LEE, S. J. 2009. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A, 106, 13010-5.DEVOS, S. L., GONCHAROFF, D. K., CHEN, G., KEBODEAUX, C. S., YAMADA, K., STEWART, F. R., SCHULER, D. R., MALONEY, S. E., WOZNIAK, D. F., RIGO, F., BENNETT, C. F., CIRRITO, J. R., HOLTZMAN, D. M. & MILLER, T. M. 2013. Antisense reduction of tau in adult mice protects against seizures. J Neurosci, 33, 12887-97.DI GIOVANNI, S., ELEUTERI, S., PALEOLOGOU, K. E., YIN, G., ZWECKSTETTER, M., CARRUPT, P. A. & LASHUEL, H. A. 2010. Entacapone and tolcapone, two catechol O-methyltransferase inhibitors, block fibril formation of alpha-synuclein and beta-amyloid and protect against amyloid-induced toxicity. J Biol Chem, 285, 14941-54.DICKEY, C. A., KAMAL, A., LUNDGREN, K., KLOSAK, N., BAILEY, R. M., DUNMORE, J., ASH, P., SHORAKA, S., ZLATKOVIC, J., ECKMAN, C. B., PATTERSON, C., DICKSON, D. W., NAHMAN, N. S., JR., HUTTON, M., BURROWS, F. & PETRUCELLI, L. 2007. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest, 117, 648-58.DING, T. T., LEE, S. J., ROCHET, J. C. & LANSBURY, P. T., JR. 2002. Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry, 41, 10209-17.DOKLEJA, L., HANNULA, M. J. & MYOHANEN, T. T. 2014. Inhibition of prolyl oligopeptidase increases the survival of alpha-synuclein overexpressing cells after rotenone exposure by reducing alpha-synuclein oligomers. Neurosci Lett, 583, 37-42.DOLAN, P. J. & JOHNSON, G. V. 2010. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem, 285, 21978-87.DUCHON, A. & HERAULT, Y. 2016. DYRK1A, a Dosage-Sensitive Gene Involved in Neurodevelopmental Disorders, Is a Target for Drug Development in Down Syndrome. Front Behav Neurosci, 10, 104.DUGGER, B. N., WHITESIDE, C. M., MAAROUF, C. L., WALKER, D. G., BEACH, T. G., SUE, L. I., GARCIA, A., DUNCKLEY, T., MEECHOOVET, B., REIMAN, E. M. & ROHER, A. E. 2016. The Presence of Select Tau Species in Human Peripheral Tissues and Their Relation to Alzheimer's Disease. J Alzheimers Dis, 51, 345-56.DUJARDIN, S., BEGARD, S., CAILLIEREZ, R., LACHAUD, C., DELATTRE, L., CARRIER, S., LOYENS, A., GALAS, M., BOUSSET, L., MELKI, R., AUREGAN, G., HANTRAYE, P., BROUILLET, E., BUEE, L. & COLIN, M. 2014a. Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One, 9, e100760.DUJARDIN, S., LECOLLE, K., CAILLIEREZ, R., BEGARD, S., ZOMMER, N., LACHAUD, C., CARRIER, S., DUFOUR, N., AUREGAN, G., WINDERICKX, J., HANTRAYE, P., DEGLON, N., COLIN, M. & BUEE, L. 2014b. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol Commun, 2, 14.DUKA, T., DUKA, V., JOYCE, J. N. & SIDHU, A. 2009. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson's disease models. FASEB J, 23, 2820-30.DUKA, T., RUSNAK, M., DROLET, R. E., DUKA, V., WERSINGER, C., GOUDREAU, J. L. & SIDHU, A. 2006. Alpha-synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. FASEB J, 20, 2302-12.DUMANCHIN, C., CAMUZAT, A., CAMPION, D., VERPILLAT, P., HANNEQUIN, D., DUBOIS, B., SAUGIER-VEBER, P., MARTIN, C., PENET, C., CHARBONNIER, F., AGID, Y., FREBOURG, T. & BRICE, A. 1998. Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet, 7, 1825-9.

82
DUSTIN, M., OLSZOWY, M., HOLDORF, A., LI, J., BROMLEY, S., DESAI, N., WIDDER, P., ROSENBERGER, F., VAN DER MERWE, P., ALLEN, P. & SHAW, A. 1998. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell, 94, 667-77.EBRAHIMI-FAKHARI, D., WAHLSTER, L. & MCLEAN, P. J. 2012. Protein degradation pathways in Parkinson's disease: curse or blessing. Acta Neuropathol, 124, 153-72.ECKERMANN, K., MOCANU, M. M., KHLISTUNOVA, I., BIERNAT, J., NISSEN, A., HOFMANN, A., SCHONIG, K., BUJARD, H., HAEMISCH, A., MANDELKOW, E., ZHOU, L., RUNE, G. & MANDELKOW, E. M. 2007. The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem, 282, 31755-65.EDWARDS, L. L., QUIGLEY, E. M. & PFEIFFER, R. F. 1992. Gastrointestinal dysfunction in Parkinson's disease: frequency and pathophysiology. Neurology, 42, 726-32.EISELE, Y. S., OBERMULLER, U., HEILBRONNER, G., BAUMANN, F., KAESER, S. A., WOLBURG, H., WALKER, L. C., STAUFENBIEL, M., HEIKENWALDER, M. & JUCKER, M. 2010. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science, 330,980-2.EISENBERG, D. & JUCKER, M. 2012. The amyloid state of proteins in human diseases. Cell, 148,1188-203.EL-AGNAF, O. M., SALEM, S. A., PALEOLOGOU, K. E., COOPER, L. J., FULLWOOD, N. J., GIBSON, M. J., CURRAN, M. D., COURT, J. A., MANN, D. M., IKEDA, S., COOKSON, M. R., HARDY, J. & ALLSOP, D. 2003. Alpha-synuclein implicated in Parkinson's disease is present in extracellular biological fluids, including human plasma. FASEB J, 17, 1945-7.ELIEZER, D., KUTLUAY, E., BUSSELL, R., JR. & BROWNE, G. 2001. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol, 307, 1061-73.EMMANOUILIDOU, E., MELACHROINOU, K., ROUMELIOTIS, T., GARBIS, S. D., NTZOUNI, M., MARGARITIS, L. H., STEFANIS, L. & VEKRELLIS, K. 2010. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci, 30,6838-51.FADER, C. M., SANCHEZ, D., FURLAN, M. & COLOMBO, M. I. 2008. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic, 9, 230-50.FAHN, S. 2003. Description of Parkinson's disease as a clinical syndrome. Ann N Y Acad Sci, 991, 1-14.FALCONE, S., COCUCCI, E., PODINI, P., KIRCHHAUSEN, T., CLEMENTI, E. & MELDOLESI, J. 2006. Macropinocytosis: regulated coordination of endocytic and exocytic membrane traffic events. JCell Sci, 119, 4758-69.FARRER, L., CUPPLES, L., HAINES, J., HYMAN, B., KUKULL, W., MAYEUX, R., MYERS, R., PERICAK-VANCE, M., RISCH, N. & VAN DUIJN, C. 1997. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA, 278, 1349-56.FAUVET, B., MBEFO, M. K., FARES, M. B., DESOBRY, C., MICHAEL, S., ARDAH, M. T., TSIKA, E., COUNE, P., PRUDENT, M., LION, N., ELIEZER, D., MOORE, D. J., SCHNEIDER, B., AEBISCHER, P., EL-AGNAF, O. M., MASLIAH, E. & LASHUEL, H. A. 2012. alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem, 287, 15345-64.FENG, S. & ARNOLD, D. B. 2016. Techniques for studying protein trafficking and molecular motors in neurons. Cytoskeleton (Hoboken), 73, 508-15.FEUILLETTE, S., BLARD, O., LECOURTOIS, M., FREBOURG, T., CAMPION, D. & DUMANCHIN, C. 2005. Tau is not normally degraded by the proteasome. J Neurosci Res, 80, 400-5.FEVRIER, B. & RAPOSO, G. 2004. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol, 16, 415-21.FLACH, K., HILBRICH, I., SCHIFFMANN, A., GARTNER, U., KRUGER, M., LEONHARDT, M., WASCHIPKY, H., WICK, L., ARENDT, T. & HOLZER, M. 2012. Tau oligomers impair artificial membrane integrity and cellular viability. J Biol Chem, 287, 43223-33.

83
FLANAGAN, L. A., CUNNINGHAM, C. C., CHEN, J., PRESTWICH, G. D., KOSIK, K. S. & JANMEY, P. A. 1997. The structure of divalent cation-induced aggregates of PIP2 and their alteration by gelsolin and tau. Biophys J, 73, 1440-7.FLEMING, L., WEISGRABER, K., STRITTMATTER, W., TRONCOSO, J. & JOHNSON, G. 1996. Differential binding of apolipoprotein E isoforms to tau and other cytoskeletal proteins. Exp Neurol,138, 252-60.FOLLETT, J., NORWOOD, S. J., HAMILTON, N. A., MOHAN, M., KOVTUN, O., TAY, S., ZHE, Y., WOOD, S. A., MELLICK, G. D., SILBURN, P. A., COLLINS, B. M., BUGARCIC, A. & TEASDALE, R. D. 2014. The Vps35 D620N mutation linked to Parkinson's disease disrupts the cargo sorting function of retromer. Traffic, 15, 230-44.FORMAN, M. S., SCHMIDT, M. L., KASTURI, S., PERL, D. P., LEE, V. M. & TROJANOWSKI, J. Q. 2002. Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol, 160, 1725-31.FRASER, K. B., MOEHLE, M. S., DAHER, J. P., WEBBER, P. J., WILLIAMS, J. Y., STEWART, C. A., YACOUBIAN, T. A., COWELL, R. M., DOKLAND, T., YE, T., CHEN, D., SIEGAL, G. P.,GALEMMO, R. A., TSIKA, E., MOORE, D. J., STANDAERT, D. G., KOJIMA, K., MOBLEY, J. A. & WEST, A. B. 2013. LRRK2 secretion in exosomes is regulated by 14-3-3. Hum Mol Genet, 22, 4988-5000.FRASIER, M., WALZER, M., MCCARTHY, L., MAGNUSON, D., LEE, J. M., HAAS, C., KAHLE, P. & WOLOZIN, B. 2005. Tau phosphorylation increases in symptomatic mice overexpressing A30P alpha-synuclein. Exp Neurol, 192, 274-87.FRIEDHOFF, P., VON BERGEN, M., MANDELKOW, E., DAVIES, P. & MANDELKOW, E. 1998. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci U S A, 95,15712-7.FRONTZEK, K., LUTZ, M. I., AGUZZI, A., KOVACS, G. G. & BUDKA, H. 2016. Amyloid-beta pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt-Jakob disease after dural grafting. Swiss Med Wkly, 146, w14287.FROST, B., JACKS, R. L. & DIAMOND, M. I. 2009. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem, 284, 12845-52.FRUHBEIS, C., FROHLICH, D., KUO, W. P., AMPHORNRAT, J., THILEMANN, S., SAAB, A. S., KIRCHHOFF, F., MOBIUS, W., GOEBBELS, S., NAVE, K. A., SCHNEIDER, A., SIMONS, M., KLUGMANN, M., TROTTER, J. & KRAMER-ALBERS, E. M. 2013. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol, 11, e1001604.FUJIOKA, S., OGAKI, K., TACIK, P. M., UITTI, R. J., ROSS, O. A. & WSZOLEK, Z. K. 2014. Update on novel familial forms of Parkinson's disease and multiple system atrophy. Parkinsonism Relat Disord, 20 Suppl 1, S29-34.FUJIWARA, H., HASEGAWA, M., DOHMAE, N., KAWASHIMA, A., MASLIAH, E., GOLDBERG, M. S., SHEN, J., TAKIO, K. & IWATSUBO, T. 2002. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol, 4, 160-4.FULGA, T. A., ELSON-SCHWAB, I., KHURANA, V., STEINHILB, M. L., SPIRES, T. L., HYMAN, B. T. & FEANY, M. B. 2007. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol, 9, 139-48.FULOP, V., SZELTNER, Z. & POLGAR, L. 2000. Catalysis of serine oligopeptidases is controlled by a gating filter mechanism. EMBO Rep, 1, 277-81.GALPERN, W. R. & LANG, A. E. 2006. Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol, 59, 449-58.GALVAGNION, C., BUELL, A. K., MEISL, G., MICHAELS, T. C., VENDRUSCOLO, M., KNOWLES, T. P. & DOBSON, C. M. 2015. Lipid vesicles trigger alpha-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol, 11, 229-34.GARCIA-HORSMAN, J. A., MANNISTO, P. T. & VENALAINEN, J. I. 2007. On the role of prolyl oligopeptidase in health and disease. Neuropeptides, 41, 1-24.

84
GARCIA DE ANCOS, J., CORREAS, I. & AVILA, J. 1993. Differences in microtubule binding and self-association abilities of bovine brain tau isoforms. J Biol Chem, 268, 7976-82.GENDREAU, K. & HALL, G. 2013. Tangles, Toxicity, and Tau Secretion in AD - New Approaches to a Vexing Problem. Front Neurol, 4, 160.GENDRON, T. & PETRUCELLI, L. 2009. The role of tau in neurodegeneration. Mol Neurodegener, 4,13.GEORGE, J. M., JIN, H., WOODS, W. S. & CLAYTON, D. F. 1995. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron, 15, 361-72.GIASSON, B. I., DUDA, J. E., MURRAY, I. V., CHEN, Q., SOUZA, J. M., HURTIG, H. I., ISCHIROPOULOS, H., TROJANOWSKI, J. Q. & LEE, V. M. 2000. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science, 290, 985-9.GIASSON, B. I., FORMAN, M. S., HIGUCHI, M., GOLBE, L. I., GRAVES, C. L., KOTZBAUER, P. T., TROJANOWSKI, J. Q. & LEE, V. M. 2003. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science, 300, 636-40.GIASSON, B. I., MURRAY, I. V., TROJANOWSKI, J. Q. & LEE, V. M. 2001. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem, 276, 2380-6.GILKS, N., KEDERSHA, N., AYODELE, M., SHEN, L., STOECKLIN, G., DEMBER, L. M. & ANDERSON, P. 2004. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell, 15, 5383-98.GOEDERT, M. & JAKES, R. 1990. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J, 9, 4225-30.GOEDERT, M., JAKES, R., CROWTHER, R., SIX, J., LUBKE, U., VANDERMEEREN, M., CRAS, P., TROJANOWSKI, J. & LEE, V. 1993. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci U S A, 90,5066-70.GOEDERT, M., SPILLANTINI, M. G., DEL TREDICI, K. & BRAAK, H. 2013. 100 years of Lewy pathology. Nat Rev Neurol, 9, 13-24.GOLDSTEIN, B. & MACARA, I. G. 2007. The PAR proteins: fundamental players in animal cell polarization. Dev Cell, 13, 609-22.GOMEZ-ISLA, T., HOLLISTER, R., WEST, H., MUI, S., GROWDON, J. H., PETERSEN, R. C., PARISI, J. E. & HYMAN, B. T. 1997. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol, 41, 17-24.GONG, C., LIU, F., GRUNDKE-IQBAL, I. & IQBAL, K. 2005. Post-translational modifications of tau protein in Alzheimer's disease. J Neural Transm, 112, 813-38.GREEN, K. N., MARTINEZ-CORIA, H., KHASHWJI, H., HALL, E. B., YURKO-MAURO, K. A., ELLIS, L. & LAFERLA, F. M. 2007. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-beta and tau pathology via a mechanism involving presenilin 1 levels. J Neurosci,27, 4385-95.GRIMES, C. & JOPE, R. 2001. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol, 65, 391-426.GRUBMAN, A., KANNINEN, K. M. & MALM, T. 2016. Multitasking Microglia and Alzheimer's Disease: Diversity, Tools and Therapeutic Targets. J Mol Neurosci, 60, 390-404.GU, Y., OYAMA, F. & IHARA, Y. 1996. Tau is widely expressed in rat tissues. J Neurochem, 67,1235-44.GUDMUNDSSON, S., EINARSSON, S., ERLENDSDOTTIR, H., MOFFAT, J., BAYER, W. & CRAIG, W. A. 1993. The post-antibiotic effect of antimicrobial combinations in a neutropenic murine thigh infection model. J Antimicrob Chemother, 31 Suppl D, 177-91.GUERREIRO, R., LOHMANN, E., BRAS, J., GIBBS, J., ROHRER, J., GURUNLIAN, N., DURSUN, B., BILGIC, B., HANAGASI, H., GURVIT, H., EMRE, M., SINGLETON, A. & HARDY, J. 2013a.

85
Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol, 70, 78-84.GUERREIRO, R., WOJTAS, A., BRAS, J., CARRASQUILLO, M., ROGAEVA, E., MAJOUNIE, E., CRUCHAGA, C., SASSI, C., KAUWE, J., YOUNKIN, S., HAZRATI, L., COLLINGE, J., POCOCK, J., LASHLEY, T., WILLIAMS, J., LAMBERT, J., AMOUYEL, P., GOATE, A., RADEMAKERS, R., MORGAN, K., POWELL, J., ST GEORGE-HYSLOP, P., SINGLETON, A. & HARDY, J. 2013b. TREM2 variants in Alzheimer's disease. N Engl J Med, 368, 117-27.GUO, J. & LEE, V. 2011a. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem, 286, 15317-31.GUO, J. & LEE, V. 2014a. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med, 20, 130-8.GUO, J. L., COVELL, D. J., DANIELS, J. P., IBA, M., STIEBER, A., ZHANG, B., RIDDLE, D. M., KWONG, L. K., XU, Y., TROJANOWSKI, J. Q. & LEE, V. M. 2013. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell, 154, 103-17.GUO, J. L. & LEE, V. M. 2011b. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem, 286, 15317-31.GUO, J. L. & LEE, V. M. 2013. Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett, 587, 717-23.GUO, J. L. & LEE, V. M. 2014b. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med, 20, 130-8.HALL, G. F. & SAMAN, S. 2012. Death or secretion? The demise of a plausible assumption about CSF-tau in Alzheimer Disease? Commun Integr Biol, 5, 623-6.HAMANO, T., GENDRON, T. F., CAUSEVIC, E., YEN, S. H., LIN, W. L., ISIDORO, C., DETURE, M. & KO, L. W. 2008. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci, 27, 1119-30.HAMILTON, R. L. 2000. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol, 10, 378-84.HAMPEL, H., BLENNOW, K., SHAW, L., HOESSLER, Y., ZETTERBERG, H. & TROJANOWSKI, J. 2010. Total and phosphorylated tau protein as biological markers of Alzheimer's disease. Exp Gerontol, 45, 30-40.HANGER, D. P., ANDERTON, B. H. & NOBLE, W. 2009. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med, 15, 112-9.HANNULA, M. J., MYOHANEN, T. T., TENORIO-LARANGA, J., MANNISTO, P. T. & GARCIA-HORSMAN, J. A. 2013. Prolyl oligopeptidase colocalizes with alpha-synuclein, beta-amyloid, tau protein and astroglia in the post-mortem brain samples with Parkinson's and Alzheimer's diseases. Neuroscience, 242, 140-50.HANSEN, C., ANGOT, E., BERGSTROM, A. L., STEINER, J. A., PIERI, L., PAUL, G., OUTEIRO, T. F., MELKI, R., KALLUNKI, P., FOG, K., LI, J. Y. & BRUNDIN, P. 2011. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest, 121, 715-25.HAROLD, D., ABRAHAM, R., HOLLINGWORTH, P., SIMS, R., GERRISH, A., HAMSHERE, M., PAHWA, J., MOSKVINA, V., DOWZELL, K., WILLIAMS, A., JONES, N., THOMAS, C., STRETTON, A., MORGAN, A., LOVESTONE, S., POWELL, J., PROITSI, P., LUPTON, M., BRAYNE, C., RUBINSZTEIN, D., GILL, M., LAWLOR, B., LYNCH, A., MORGAN, K., BROWN, K., PASSMORE, P., CRAIG, D., MCGUINNESS, B., TODD, S., HOLMES, C., MANN, D., SMITH, A., LOVE, S., KEHOE, P., HARDY, J., MEAD, S., FOX, N., ROSSOR, M., COLLINGE, J., MAIER, W., JESSEN, F., SCHURMANN, B., HEUN, R., VAN DEN BUSSCHE, H., HEUSER, I., KORNHUBER, J., WILTFANG, J., DICHGANS, M., FROLICH, L., HAMPEL, H., HULL, M., RUJESCU, D., GOATE, A., KAUWE, J., CRUCHAGA, C., NOWOTNY, P., MORRIS, J., MAYO, K., SLEEGERS, K., BETTENS, K., ENGELBORGHS, S., DE DEYN, P., VAN BROECKHOVEN, C., LIVINGSTON, G., BASS, N., GURLING, H., MCQUILLIN, A., GWILLIAM, R., DELOUKAS, P.,

86
AL-CHALABI, A., SHAW, C., TSOLAKI, M., SINGLETON, A., GUERREIRO, R., MUHLEISEN, T., NOTHEN, M., MOEBUS, S., JOCKEL, K., KLOPP, N., WICHMANN, H., CARRASQUILLO, M., PANKRATZ, V., YOUNKIN, S., HOLMANS, P., O'DONOVAN, M., OWEN, M. & WILLIAMS, J. 2009. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet, 41, 1088-93.HARRINGTON, C. R., STOREY, J. M., CLUNAS, S., HARRINGTON, K. A., HORSLEY, D., ISHAQ, A., KEMP, S. J., LARCH, C. P., MARSHALL, C., NICOLL, S. L., RICKARD, J. E., SIMPSON, M., SINCLAIR, J. P., STOREY, L. J. & WISCHIK, C. M. 2015. Cellular Models of Aggregation-dependent Template-directed Proteolysis to Characterize Tau Aggregation Inhibitors for Treatment of Alzheimer Disease. J Biol Chem, 290, 10862-75.HASHIGUCHI, M., SOBUE, K. & PAUDEL, H. K. 2000. 14-3-3zeta is an effector of tau protein phosphorylation. J Biol Chem, 275, 25247-54.HASHIMOTO, M., HSU, L. J., XIA, Y., TAKEDA, A., SISK, A., SUNDSMO, M. & MASLIAH, E. 1999a. Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport, 10, 717-21.HASHIMOTO, M., KAWAHARA, K., BAR-ON, P., ROCKENSTEIN, E., CREWS, L. & MASLIAH, E. 2004. The Role of alpha-synuclein assembly and metabolism in the pathogenesis of Lewy body disease. J Mol Neurosci, 24, 343-52.HASHIMOTO, M., TAKEDA, A., HSU, L. J., TAKENOUCHI, T. & MASLIAH, E. 1999b. Role of cytochrome c as a stimulator of alpha-synuclein aggregation in Lewy body disease. J Biol Chem, 274,28849-52.HASHIMOTO, M., TAKENOUCHI, T., MALLORY, M., MASLIAH, E. & TAKEDA, A. 2000. The role of NAC in amyloidogenesis in Alzheimer's disease. Am J Pathol, 156, 734-6.HATAKEYAMA, S., MATSUMOTO, M., KAMURA, T., MURAYAMA, M., CHUI, D. H., PLANEL, E., TAKAHASHI, R., NAKAYAMA, K. I. & TAKASHIMA, A. 2004. U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat Tau and is involved in neurodegeneration of tauopathy. J Neurochem, 91, 299-307.HAWKES, C. H., SHEPHARD, B. C. & DANIEL, S. E. 1999. Is Parkinson's disease a primary olfactory disorder? QJM, 92, 473-80.HELLER, J., RUHNKE, N., ESPINO, J. J., MASSAROLI, M., COLLADO, I. G. & TUDZYNSKI, P. 2012. The mitogen-activated protein kinase BcSak1 of Botrytis cinerea is required for pathogenic development and has broad regulatory functions beyond stress response. Mol Plant Microbe Interact,25, 802-16.HIRATA-FUKAE, C., LI, H. F., MA, L., HOE, H. S., REBECK, G. W., AISEN, P. S. & MATSUOKA, Y. 2009. Levels of soluble and insoluble tau reflect overall status of tau phosphorylation in vivo. Neurosci Lett, 450, 51-5.HIROKAWA, N., SHIOMURA, Y. & OKABE, S. 1988. Tau proteins: the molecular structure and mode of binding on microtubules. J Cell Biol, 107, 1449-59.HOCHGRAFE, K., SYDOW, A., MATENIA, D., CADINU, D., KONEN, S., PETROVA, O., PICKHARDT, M., GOLL, P., MORELLINI, F., MANDELKOW, E. & MANDELKOW, E. M. 2015. Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta Neuropathol Commun, 3, 25.HOFLING, C., KULESSKAYA, N., JAAKO, K., PELTONEN, I., MANNISTO, P. T., NURMI, A., VARTIAINEN, N., MORAWSKI, M., ZHARKOVSKY, A., VOIKAR, V., ROSSNER, S. & GARCIA-HORSMAN, J. A. 2016. Deficiency of prolyl oligopeptidase in mice disturbs synaptic plasticity and reduces anxiety-like behaviour, body weight, and brain volume. EurNeuropsychopharmacol, 26, 1048-61.HOLLINGWORTH, P., HAROLD, D., SIMS, R., GERRISH, A., LAMBERT, J., CARRASQUILLO, M., ABRAHAM, R., HAMSHERE, M., PAHWA, J., MOSKVINA, V., DOWZELL, K., JONES, N., STRETTON, A., THOMAS, C., RICHARDS, A., IVANOV, D., WIDDOWSON, C., CHAPMAN, J., LOVESTONE, S., POWELL, J., PROITSI, P., LUPTON, M., BRAYNE, C., RUBINSZTEIN, D.,

87
GILL, M., LAWLOR, B., LYNCH, A., BROWN, K., PASSMORE, P., CRAIG, D., MCGUINNESS, B., TODD, S., HOLMES, C., MANN, D., SMITH, A., BEAUMONT, H., WARDEN, D., WILCOCK, G., LOVE, S., KEHOE, P., HOOPER, N., VARDY, E., HARDY, J., MEAD, S., FOX, N., ROSSOR, M., COLLINGE, J., MAIER, W., JESSEN, F., RUTHER, E., SCHURMANN, B., HEUN, R., KOLSCH, H., VAN DEN BUSSCHE, H., HEUSER, I., KORNHUBER, J., WILTFANG, J., DICHGANS, M., FROLICH, L., HAMPEL, H., GALLACHER, J., HULL, M., RUJESCU, D., GIEGLING, I., GOATE, A., KAUWE, J., CRUCHAGA, C., NOWOTNY, P., MORRIS, J., MAYO, K., SLEEGERS, K., BETTENS, K., ENGELBORGHS, S., DE DEYN, P., VAN BROECKHOVEN, C., LIVINGSTON, G., BASS, N., GURLING, H., MCQUILLIN, A., GWILLIAM, R., DELOUKAS, P., AL-CHALABI, A., SHAW, C., TSOLAKI, M., SINGLETON, A., GUERREIRO, R., MUHLEISEN, T., NOTHEN, M., MOEBUS, S., JOCKEL, K., KLOPP, N., WICHMANN, H., PANKRATZ, V., SANDO, S., AASLY, J., BARCIKOWSKA, M., WSZOLEK, Z., DICKSON, D., GRAFF-RADFORD, N., PETERSEN, R., et al. 2011. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet, 43, 429-35.HOLMES, B. B., DEVOS, S. L., KFOURY, N., LI, M., JACKS, R., YANAMANDRA, K., OUIDJA, M. O., BRODSKY, F. M., MARASA, J., BAGCHI, D. P., KOTZBAUER, P. T., MILLER, T. M., PAPY-GARCIA, D. & DIAMOND, M. I. 2013. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A, 110, E3138-47.HOLMQVIST, S., CHUTNA, O., BOUSSET, L., ALDRIN-KIRK, P., LI, W., BJORKLUND, T., WANG, Z. Y., ROYBON, L., MELKI, R. & LI, J. Y. 2014. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol, 128, 805-20.HONG, M., ZHUKAREVA, V., VOGELSBERG-RAGAGLIA, V., WSZOLEK, Z., REED, L., MILLER, B. I., GESCHWIND, D. H., BIRD, T. D., MCKEEL, D., GOATE, A., MORRIS, J. C., WILHELMSEN, K. C., SCHELLENBERG, G. D., TROJANOWSKI, J. Q. & LEE, V. M. 1998. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science, 282,1914-7.HONGU, T. & KANAHO, Y. 2014. Activation machinery of the small GTPase Arf6. Adv Biol Regul,54, 59-66.HOOVER, B. R., REED, M. N., SU, J., PENROD, R. D., KOTILINEK, L. A., GRANT, M. K., PITSTICK, R., CARLSON, G. A., LANIER, L. M., YUAN, L. L., ASHE, K. H. & LIAO, D. 2010. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron, 68, 1067-81.HORIKOSHI, Y., SUZUKI, A., YAMANAKA, T., SASAKI, K., MIZUNO, K., SAWADA, H., YONEMURA, S. & OHNO, S. 2009. Interaction between PAR-3 and the aPKC-PAR-6 complex is indispensable for apical domain development of epithelial cells. J Cell Sci, 122, 1595-606.HORVATH, I., WEISE, C. F., ANDERSSON, E. K., CHORELL, E., SELLSTEDT, M., BENGTSSON, C., OLOFSSON, A., HULTGREN, S. J., CHAPMAN, M., WOLF-WATZ, M., ALMQVIST, F. & WITTUNG-STAFSHEDE, P. 2012. Mechanisms of protein oligomerization: inhibitor of functional amyloids templates alpha-synuclein fibrillation. J Am Chem Soc, 134, 3439-44.HUTTON, M., LENDON, C. L., RIZZU, P., BAKER, M., FROELICH, S., HOULDEN, H., PICKERING-BROWN, S., CHAKRAVERTY, S., ISAACS, A., GROVER, A., HACKETT, J., ADAMSON, J., LINCOLN, S., DICKSON, D., DAVIES, P., PETERSEN, R. C., STEVENS, M., DE GRAAFF, E., WAUTERS, E., VAN BAREN, J., HILLEBRAND, M., JOOSSE, M., KWON, J. M., NOWOTNY, P., CHE, L. K., NORTON, J., MORRIS, J. C., REED, L. A., TROJANOWSKI, J., BASUN, H., LANNFELT, L., NEYSTAT, M., FAHN, S., DARK, F., TANNENBERG, T., DODD, P. R., HAYWARD, N., KWOK, J. B., SCHOFIELD, P. R., ANDREADIS, A., SNOWDEN, J., CRAUFURD, D., NEARY, D., OWEN, F., OOSTRA, B. A., HARDY, J., GOATE, A., VAN SWIETEN, J., MANN, D., LYNCH, T. & HEUTINK, P. 1998. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature, 393, 702-5.

88
IBA, M., GUO, J., MCBRIDE, J., ZHANG, B., TROJANOWSKI, J. & LEE, V. 2013a. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci, 33, 1024-37.IBA, M., GUO, J. L., MCBRIDE, J. D., ZHANG, B., TROJANOWSKI, J. Q. & LEE, V. M. 2013b. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci, 33, 1024-37.IBANEZ, P., BONNET, A. M., DEBARGES, B., LOHMANN, E., TISON, F., POLLAK, P., AGID, Y., DURR, A. & BRICE, A. 2004. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet, 364, 1169-71.IIJIMA, M., TABIRA, T., POORKAJ, P., SCHELLENBERG, G. D., TROJANOWSKI, J. Q., LEE, V. M., SCHMIDT, M. L., TAKAHASHI, K., NABIKA, T., MATSUMOTO, T., YAMASHITA, Y., YOSHIOKA, S. & ISHINO, H. 1999. A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport, 10, 497-501.IKENOUCHI, J. & UMEDA, M. 2010. FRMD4A regulates epithelial polarity by connecting Arf6 activation with the PAR complex. Proc Natl Acad Sci U S A, 107, 748-53.IQBAL, K., LIU, F., GONG, C., ALONSO ADEL, C. & GRUNDKE-IQBAL, I. 2009. Mechanisms oftau-induced neurodegeneration. Acta Neuropathol, 118, 53-69.ISHIGURO, K., SHIRATSUCHI, A., SATO, S., OMORI, A., ARIOKA, M., KOBAYASHI, S., UCHIDA, T. & IMAHORI, K. 1993. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett, 325, 167-72.IWAI, A., MASLIAH, E., YOSHIMOTO, M., GE, N., FLANAGAN, L., DE SILVA, H. A., KITTEL, A. & SAITOH, T. 1995. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron, 14, 467-75.IWATA, A., MARUYAMA, M., AKAGI, T., HASHIKAWA, T., KANAZAWA, I., TSUJI, S. & NUKINA, N. 2003. Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum Mol Genet, 12, 2625-35.JACKSON, W. S. 2014. Selective vulnerability to neurodegenerative disease: the curious case of Prion Protein. Dis Model Mech, 7, 21-9.JAHN, T. R. & RADFORD, S. E. 2005. The Yin and Yang of protein folding. FEBS J, 272, 5962-70.JAKES, R., NOVAK, M., DAVISON, M. & WISCHIK, C. M. 1991. Identification of 3- and 4-repeat tau isoforms within the PHF in Alzheimer's disease. EMBO J, 10, 2725-9.JALKANEN, A. J., PUTTONEN, K. A., VENALAINEN, J. I., SINERVA, V., MANNILA, A., RUOTSALAINEN, S., JARHO, E. M., WALLEN, E. A. & MANNISTO, P. T. 2007. Beneficial effect of prolyl oligopeptidase inhibition on spatial memory in young but not in old scopolamine-treated rats. Basic Clin Pharmacol Toxicol, 100, 132-8.JANDUS, C., SIMON, H. & VON GUNTEN, S. 2011. Targeting siglecs--a novel pharmacological strategy for immuno- and glycotherapy. Biochem Pharmacol, 82, 323-32.JANKOVIC, J., CHEN, S. & LE, W. D. 2005. The role of Nurr1 in the development of dopaminergic neurons and Parkinson's disease. Prog Neurobiol, 77, 128-38.JARHO, E. M., VENALAINEN, J. I., HUUSKONEN, J., CHRISTIAANS, J. A., GARCIA-HORSMAN, J. A., FORSBERG, M. M., JARVINEN, T., GYNTHER, J., MANNISTO, P. T. & WALLEN, E. A. 2004. A cyclopent-2-enecarbonyl group mimics proline at the P2 position of prolyl oligopeptidase inhibitors. J Med Chem, 47, 5605-7.JARIEL-ENCONTRE, I., BOSSIS, G. & PIECHACZYK, M. 2008. Ubiquitin-independent degradation of proteins by the proteasome. Biochim Biophys Acta, 1786, 153-77.JARRETT, J. T. & LANSBURY, P. T., JR. 1993. Seeding "one-dimensional crystallization" of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell, 73, 1055-8.JENCO, J. M., RAWLINGSON, A., DANIELS, B. & MORRIS, A. J. 1998. Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry, 37, 4901-9.

89
JENSEN, P. H., HAGER, H., NIELSEN, M. S., HOJRUP, P., GLIEMANN, J. & JAKES, R. 1999. alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem, 274, 25481-9.JENSEN, P. H., NIELSEN, M. S., JAKES, R., DOTTI, C. G. & GOEDERT, M. 1998. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson's disease mutation. J Biol Chem,273, 26292-4.JIANG, D., MEN, L., WANG, J., ZHANG, Y., CHICKENYEN, S., WANG, Y. & ZHOU, F. 2007. Redox reactions of copper complexes formed with different beta-amyloid peptides and their neuropathological [correction of neuropathalogical] relevance. Biochemistry, 46, 9270-82.JO, E., MCLAURIN, J., YIP, C. M., ST GEORGE-HYSLOP, P. & FRASER, P. E. 2000. alpha-Synuclein membrane interactions and lipid specificity. J Biol Chem, 275, 34328-34.JOBERTY, G., PETERSEN, C., GAO, L. & MACARA, I. 2000. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol, 2, 531-9.JOHNSON, J. O., MANDRIOLI, J., BENATAR, M., ABRAMZON, Y., VAN DEERLIN, V. M., TROJANOWSKI, J. Q., GIBBS, J. R., BRUNETTI, M., GRONKA, S., WUU, J., DING, J., MCCLUSKEY, L., MARTINEZ-LAGE, M., FALCONE, D., HERNANDEZ, D. G., AREPALLI, S., CHONG, S., SCHYMICK, J. C., ROTHSTEIN, J., LANDI, F., WANG, Y. D., CALVO, A., MORA, G., SABATELLI, M., MONSURRO, M. R., BATTISTINI, S., SALVI, F., SPATARO, R., SOLA, P., BORGHERO, G., CONSORTIUM, I., GALASSI, G., SCHOLZ, S. W., TAYLOR, J. P., RESTAGNO, G., CHIO, A. & TRAYNOR, B. J. 2010. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron, 68, 857-64.JONES, E. M., DUBEY, M., CAMP, P. J., VERNON, B. C., BIERNAT, J., MANDELKOW, E., MAJEWSKI, J. & CHI, E. Y. 2012. Interaction of tau protein with model lipid membranes induces tau structural compaction and membrane disruption. Biochemistry, 51, 2539-50.JONES, L., HOLMANS, P., HAMSHERE, M., HAROLD, D., MOSKVINA, V., IVANOV, D., POCKLINGTON, A., ABRAHAM, R., HOLLINGWORTH, P., SIMS, R., GERRISH, A., PAHWA, J., JONES, N., STRETTON, A., MORGAN, A., LOVESTONE, S., POWELL, J., PROITSI, P., LUPTON, M., BRAYNE, C., RUBINSZTEIN, D., GILL, M., LAWLOR, B., LYNCH, A., MORGAN, K., BROWN, K., PASSMORE, P., CRAIG, D., MCGUINNESS, B., TODD, S., HOLMES, C., MANN, D., SMITH, A., LOVE, S., KEHOE, P., MEAD, S., FOX, N., ROSSOR, M., COLLINGE, J., MAIER, W., JESSEN, F., SCHURMANN, B., HEUN, R., KOLSCH, H., VAN DEN BUSSCHE, H., HEUSER, I., PETERS, O., KORNHUBER, J., WILTFANG, J., DICHGANS, M., FROLICH, L., HAMPEL, H., HULL, M., RUJESCU, D., GOATE, A., KAUWE, J., CRUCHAGA, C., NOWOTNY, P., MORRIS, J., MAYO, K., LIVINGSTON, G., BASS, N., GURLING, H., MCQUILLIN, A., GWILLIAM, R., DELOUKAS, P., AL-CHALABI, A., SHAW, C., SINGLETON, A., GUERREIRO, R., MUHLEISEN, T., NOTHEN, M., MOEBUS, S., JOCKEL, K., KLOPP, N., WICHMANN, H., RUTHER, E., CARRASQUILLO, M., PANKRATZ, V., YOUNKIN, S., HARDY, J., O'DONOVAN, M., OWEN, M. & WILLIAMS, J. 2010. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS One, 5, e13950.JONSSON, T., STEFANSSON, H., STEINBERG, S., JONSDOTTIR, I., JONSSON, P., SNAEDAL, J., BJORNSSON, S., HUTTENLOCHER, J., LEVEY, A., LAH, J., RUJESCU, D., HAMPEL, H., GIEGLING, I., ANDREASSEN, O., ENGEDAL, K., ULSTEIN, I., DJUROVIC, S., IBRAHIM-VERBAAS, C., HOFMAN, A., IKRAM, M., VAN DUIJN, C., THORSTEINSDOTTIR, U., KONG, A. & STEFANSSON, K. 2013. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med, 368, 107-16.JONSSON, T. & STEFANSSON, K. 2013. TREM2 and neurodegenerative disease. N Engl J Med, 369,1568-9.JUCKER, M. & WALKER, L. C. 2013. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature, 501, 45-51.KANEMARU, K., TAKIO, K., MIURA, R., TITANI, K. & IHARA, Y. 1992. Fetal-type phosphorylation of the tau in paired helical filaments. J Neurochem, 58, 1667-75.

90
KANMERT, D., CANTLON, A., MURATORE, C., JIN, M., O'MALLEY, T., LEE, G., YOUNG-PEARSE, T., SELKOE, D. & WALSH, D. 2015. C-Terminally Truncated Forms of Tau, But Not Full-Length Tau or Its C-Terminal Fragments, Are Released from Neurons Independently of Cell Death. JNeurosci, 35, 10851-65.KANNINEN, K., WHITE, A. R., KOISTINAHO, J. & MALM, T. 2011. Targeting Glycogen Synthase Kinase-3beta for Therapeutic Benefit against Oxidative Stress in Alzheimer's Disease: Involvement of the Nrf2-ARE Pathway. Int J Alzheimers Dis, 2011, 985085.KAPLAN, B., RATNER, V. & HAAS, E. 2003. Alpha-synuclein: its biological function and role in neurodegenerative diseases. J Mol Neurosci, 20, 83-92.KARCH, C. & GOATE, A. 2015. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry, 77, 43-51.KARCH, C., JENG, A. & GOATE, A. 2012. Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies. J Biol Chem, 287, 42751-62.KASZUBA, K., ROG, T., DANNE, R., CANNING, P., FULOP, V., JUHASZ, T., SZELTNER, Z., ST PIERRE, J. F., GARCIA-HORSMAN, A., MANNISTO, P. T., KARTTUNEN, M., HOKKANEN, J. & BUNKER, A. 2012. Molecular dynamics, crystallography and mutagenesis studies on the substrate gating mechanism of prolyl oligopeptidase. Biochimie, 94, 1398-411.KAUL, T., CREDLE, J., HAGGERTY, T., OAKS, A. W., MASLIAH, E. & SIDHU, A. 2011. Region-specific tauopathy and synucleinopathy in brain of the alpha-synuclein overexpressing mouse model of Parkinson's disease. BMC Neurosci, 12, 79.KAWAKAMI, F., SUZUKI, M., SHIMADA, N., KAGIYA, G., OHTA, E., TAMURA, K., MARUYAMA, H. & ICHIKAWA, T. 2011. Stimulatory effect of alpha-synuclein on the tau-phosphorylation by GSK-3beta. FEBS J, 278, 4895-904.KECK, S., NITSCH, R., GRUNE, T. & ULLRICH, O. 2003. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J Neurochem, 85, 115-22.KESTER, M. I., VAN DER VLIES, A. E., BLANKENSTEIN, M. A., PIJNENBURG, Y. A., VAN ELK, E. J., SCHELTENS, P. & VAN DER FLIER, W. M. 2009. CSF biomarkers predict rate ofcognitive decline in Alzheimer disease. Neurology, 73, 1353-8.KHLISTUNOVA, I., BIERNAT, J., WANG, Y., PICKHARDT, M., VON BERGEN, M., GAZOVA, Z., MANDELKOW, E. & MANDELKOW, E. M. 2006. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem, 281, 1205-14.KHURANA, V., LU, Y., STEINHILB, M. L., OLDHAM, S., SHULMAN, J. M. & FEANY, M. B. 2006. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol, 16, 230-41.KIM, E. & CHOI, E. 2010. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta, 1802, 396-405.KIM, H. J., KIM, N. C., WANG, Y. D., SCARBOROUGH, E. A., MOORE, J., DIAZ, Z., MACLEA,K. S., FREIBAUM, B., LI, S., MOLLIEX, A., KANAGARAJ, A. P., CARTER, R., BOYLAN, K. B., WOJTAS, A. M., RADEMAKERS, R., PINKUS, J. L., GREENBERG, S. A., TROJANOWSKI, J. Q., TRAYNOR, B. J., SMITH, B. N., TOPP, S., GKAZI, A. S., MILLER, J., SHAW, C. E., KOTTLORS, M., KIRSCHNER, J., PESTRONK, A., LI, Y. R., FORD, A. F., GITLER, A. D., BENATAR, M., KING, O. D., KIMONIS, V. E., ROSS, E. D., WEIHL, C. C., SHORTER, J. & TAYLOR, J. P. 2013. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature, 495, 467-73.KIM, H. Y., CHO, M. K., KUMAR, A., MAIER, E., SIEBENHAAR, C., BECKER, S., FERNANDEZ, C. O., LASHUEL, H. A., BENZ, R., LANGE, A. & ZWECKSTETTER, M. 2009a. Structural properties of pore-forming oligomers of alpha-synuclein. J Am Chem Soc, 131, 17482-9.KIM, J., BASAK, J. & HOLTZMAN, D. 2009b. The role of apolipoprotein E in Alzheimer's disease. Neuron, 63, 287-303.

91
KIM, J. D., TODA, C., D'AGOSTINO, G., ZEISS, C. J., DILEONE, R. J., ELSWORTH, J. D., KIBBEY, R. G., CHAN, O., HARVEY, B. K., RICHIE, C. T., SAVOLAINEN, M., MYOHANEN, T., JEONG, J. K. & DIANO, S. 2014. Hypothalamic prolyl endopeptidase (PREP) regulates pancreatic insulin and glucagon secretion in mice. Proc Natl Acad Sci U S A, 111, 11876-81.KLEIN, C., KRAMER, E. M., CARDINE, A. M., SCHRAVEN, B., BRANDT, R. & TROTTER, J. 2002. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci, 22, 698-707.KNOWLES, T. P. & BUEHLER, M. J. 2011. Nanomechanics of functional and pathological amyloid materials. Nat Nanotechnol, 6, 469-79.KODAMA, Y. A. H., C.-D. 2012. Bimolecular fluorescence complementation (BiFC): a 5-year update and future perspectives. Biotechniques 285-298.KONDO, A., SHAHPASAND, K., MANNIX, R., QIU, J., MONCASTER, J., CHEN, C. H., YAO, Y., LIN, Y. M., DRIVER, J. A., SUN, Y., WEI, S., LUO, M. L., ALBAYRAM, O., HUANG, P., ROTENBERG, A., RYO, A., GOLDSTEIN, L. E., PASCUAL-LEONE, A., MCKEE, A. C., MEEHAN, W., ZHOU, X. Z. & LU, K. P. 2015. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature, 523, 431-6.KONG, S. M., CHAN, B. K., PARK, J. S., HILL, K. J., AITKEN, J. B., COTTLE, L., FARGHAIAN, H., COLE, A. R., LAY, P. A., SUE, C. M. & COOPER, A. A. 2014. Parkinson's disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum Mol Genet, 23, 2816-33.KOPKE, E., TUNG, Y., SHAIKH, S., ALONSO, A., IQBAL, K. & GRUNDKE-IQBAL, I. 1993a.Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem, 268, 24374-84.KOPKE, E., TUNG, Y. C., SHAIKH, S., ALONSO, A. C., IQBAL, K. & GRUNDKE-IQBAL, I. 1993b. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem, 268, 24374-84.KOTHAWALA, A., KILPATRICK, K., NOVOA, J. A. & SEGATORI, L. 2012. Quantitative analysis of alpha-synuclein solubility in living cells using split GFP complementation. PLoS One, 7, e43505.KOTZBAUER, P. T., GIASSON, B. I., KRAVITZ, A. V., GOLBE, L. I., MARK, M. H., TROJANOWSKI, J. Q. & LEE, V. M. 2004. Fibrillization of alpha-synuclein and tau in familial Parkinson's disease caused by the A53T alpha-synuclein mutation. Exp Neurol, 187, 279-88.KOWAL, J., ARRAS, G., COLOMBO, M., JOUVE, M., MORATH, J. P., PRIMDAL-BENGTSON, B., DINGLI, F., LOEW, D., TKACH, M. & THERY, C. 2016. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A, 113, E968-77.KRAUSS, M., KINUTA, M., WENK, M. R., DE CAMILLI, P., TAKEI, K. & HAUCKE, V. 2003. ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type Igamma. J Cell Biol, 162, 113-24.KRUGER, U., WANG, Y., KUMAR, S. & MANDELKOW, E. M. 2012. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging, 33, 2291-305.KURET, J., JOHNSON, G., CHA, D., CHRISTENSON, E., DEMAGGIO, A. & HOEKSTRA, M. 1997. Casein kinase 1 is tightly associated with paired-helical filaments isolated from Alzheimer's disease brain. J Neurochem, 69, 2506-15.KUZUHARA, S., MORI, H., IZUMIYAMA, N., YOSHIMURA, M. & IHARA, Y. 1988. Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol, 75,345-53.KYSENIUS, K., MUGGALLA, P., MATLIK, K., ARUMAE, U. & HUTTUNEN, H. J. 2012. PCSK9 regulates neuronal apoptosis by adjusting ApoER2 levels and signaling. Cell Mol Life Sci, 69, 1903-16.LAI, R. Y., GERTZ, H. N., WISCHIK, D. J., XUEREB, J. H., MUKAETOVA-LADINSKA, E. B., HARRINGTON, C. R., EDWARDS, P. C., MENA, R., PAYKEL, E. S., BRAYNE, C. & AL., E. 1995.

92
Examination of phosphorylated tau protein as a PHF-precursor at early stage Alzheimer’s disease. . Neurobiol. Aging, 433-445.LAMBEIR, A. M. 2011. Interaction of prolyl oligopeptidase with alpha-synuclein. CNS Neurol Disord Drug Targets, 10, 349-54.LAMBERT, J., GRENIER-BOLEY, B., HAROLD, D., ZELENIKA, D., CHOURAKI, V., KAMATANI, Y., SLEEGERS, K., IKRAM, M., HILTUNEN, M., REITZ, C., MATEO, I., FEULNER, T., BULLIDO, M., GALIMBERTI, D., CONCARI, L., ALVAREZ, V., SIMS, R., GERRISH, A., CHAPMAN, J., DENIZ-NARANJO, C., SOLFRIZZI, V., SORBI, S., AROSIO, B., SPALLETTA, G., SICILIANO, G., EPELBAUM, J., HANNEQUIN, D., DARTIGUES, J., TZOURIO, C., BERR, C., SCHRIJVERS, E., ROGERS, R., TOSTO, G., PASQUIER, F., BETTENS, K., VAN CAUWENBERGHE, C., FRATIGLIONI, L., GRAFF, C., DELEPINE, M., FERRI, R., REYNOLDS, C., LANNFELT, L., INGELSSON, M., PRINCE, J., CHILLOTTI, C., PILOTTO, A., SERIPA, D., BOLAND, A., MANCUSO, M., BOSSU, P., ANNONI, G., NACMIAS, B., BOSCO, P., PANZA, F., SANCHEZ-GARCIA, F., DEL ZOMPO, M., COTO, E., OWEN, M., O'DONOVAN, M., VALDIVIESO, F., CAFFARRA, P., SCARPINI, E., COMBARROS, O., BUEE, L., CAMPION, D., SOININEN, H., BRETELER, M., RIEMENSCHNEIDER, M., VAN BROECKHOVEN, C., ALPEROVITCH, A., LATHROP, M., TREGOUET, D., WILLIAMS, J. & AMOUYEL, P. 2013a. Genome-wide haplotype association study identifies the FRMD4A gene as a risk locus for Alzheimer's disease. Mol Psychiatry, 18, 461-70.LAMBERT, J., HEATH, S., EVEN, G., CAMPION, D., SLEEGERS, K., HILTUNEN, M., COMBARROS, O., ZELENIKA, D., BULLIDO, M., TAVERNIER, B., LETENNEUR, L., BETTENS, K., BERR, C., PASQUIER, F., FIEVET, N., BARBERGER-GATEAU, P., ENGELBORGHS, S., DE DEYN, P., MATEO, I., FRANCK, A., HELISALMI, S., PORCELLINI, E., HANON, O., DE PANCORBO, M., LENDON, C., DUFOUIL, C., JAILLARD, C., LEVEILLARD, T., ALVAREZ, V., BOSCO, P., MANCUSO, M., PANZA, F., NACMIAS, B., BOSSU, P., PICCARDI, P., ANNONI, G., SERIPA, D., GALIMBERTI, D., HANNEQUIN, D., LICASTRO, F., SOININEN, H., RITCHIE, K., BLANCHE, H., DARTIGUES, J., TZOURIO, C., GUT, I., VAN BROECKHOVEN, C., ALPEROVITCH, A., LATHROP, M. & AMOUYEL, P. 2009. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet, 41, 1094-9.LAMBERT, J., IBRAHIM-VERBAAS, C., HAROLD, D., NAJ, A., SIMS, R., BELLENGUEZ, C., JUN, G., DESTEFANO, A., BIS, J., BEECHAM, G., GRENIER-BOLEY, B., RUSSO, G., THORNTON-WELLS, T., JONES, N., SMITH, A., CHOURAKI, V., THOMAS, C., IKRAM, M., ZELENIKA, D., VARDARAJAN, B., KAMATANI, Y., LIN, C., GERRISH, A., SCHMIDT, H., KUNKLE, B., DUNSTAN, M., RUIZ, A., BIHOREAU, M., CHOI, S., REITZ, C., PASQUIER, F., HOLLINGWORTH, P., RAMIREZ, A., HANON, O., FITZPATRICK, A., BUXBAUM, J., CAMPION, D., CRANE, P., BALDWIN, C., BECKER, T., GUDNASON, V., CRUCHAGA, C., CRAIG, D., AMIN, N., BERR, C., LOPEZ, O., DE JAGER, P., DERAMECOURT, V., JOHNSTON, J., EVANS, D., LOVESTONE, S., LETENNEUR, L., MORON, F., RUBINSZTEIN, D., EIRIKSDOTTIR, G., SLEEGERS, K., GOATE, A., FIEVET, N., HUENTELMAN, M., GILL, M., BROWN, K., KAMBOH, M., KELLER, L., BARBERGER-GATEAU, P., MCGUINNESS, B., LARSON, E., GREEN, R., MYERS, A., DUFOUIL, C., TODD, S., WALLON, D., LOVE, S., ROGAEVA, E., GALLACHER, J., ST GEORGE-HYSLOP, P., CLARIMON, J., LLEO, A., BAYER, A., TSUANG, D., YU, L., TSOLAKI, M., BOSSU, P., SPALLETTA, G., PROITSI, P., COLLINGE, J., SORBI, S., SANCHEZ-GARCIA, F., FOX, N., HARDY, J., NARANJO, M., BOSCO, P., CLARKE, R., BRAYNE, C., GALIMBERTI, D., MANCUSO, M., MATTHEWS, F., MOEBUS, S., MECOCCI, P., DEL ZOMPO, M., MAIER, W., et al. 2013b. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet.LARSEN, K. E., SCHMITZ, Y., TROYER, M. D., MOSHAROV, E., DIETRICH, P., QUAZI, A. Z., SAVALLE, M., NEMANI, V., CHAUDHRY, F. A., EDWARDS, R. H., STEFANIS, L. & SULZER, D. 2006. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci, 26, 11915-22.

93
LASAGNA-REEVES, C., CASTILLO-CARRANZA, D., SENGUPTA, U., GUERRERO-MUNOZ, M., KIRITOSHI, T., NEUGEBAUER, V., JACKSON, G. & KAYED, R. 2012a. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep, 2, 700.LASAGNA-REEVES, C. A., CASTILLO-CARRANZA, D. L., SENGUPTA, U., GUERRERO-MUNOZ, M. J., KIRITOSHI, T., NEUGEBAUER, V., JACKSON, G. R. & KAYED, R. 2012b. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep, 2, 700.LASAGNA-REEVES, C. A., CASTILLO-CARRANZA, D. L., SENGUPTA, U., SARMIENTO, J., TRONCOSO, J., JACKSON, G. R. & KAYED, R. 2012c. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J, 26, 1946-59.LASAGNA-REEVES, C. A., GLABE, C. G. & KAYED, R. 2011. Amyloid-beta annular protofibrils evade fibrillar fate in Alzheimer disease brain. J Biol Chem, 286, 22122-30.LASAGNA-REEVES, C. A., SENGUPTA, U., CASTILLO-CARRANZA, D., GERSON, J. E., GUERRERO-MUNOZ, M., TRONCOSO, J. C., JACKSON, G. R. & KAYED, R. 2014. The formation of tau pore-like structures is prevalent and cell specific: possible implications for the disease phenotypes. Acta Neuropathol Commun, 2, 56.LASHUEL, H. A., HARTLEY, D., PETRE, B. M., WALZ, T. & LANSBURY, P. T., JR. 2002a. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature, 418, 291.LASHUEL, H. A., OVERK, C. R., OUESLATI, A. & MASLIAH, E. 2013. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci, 14, 38-48.LASHUEL, H. A., PETRE, B. M., WALL, J., SIMON, M., NOWAK, R. J., WALZ, T. & LANSBURY, P. T., JR. 2002b. Alpha-synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol, 322, 1089-102.LEE, D. C., RIZER, J., HUNT, J. B., SELENICA, M. L., GORDON, M. N. & MORGAN, D. 2013. Review: experimental manipulations of microglia in mouse models of Alzheimer's pathology: activation reduces amyloid but hastens tau pathology. Neuropathol Appl Neurobiol, 39, 69-85.LEE, G., NEWMAN, S. T., GARD, D. L., BAND, H. & PANCHAMOORTHY, G. 1998. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci, 111 ( Pt 21), 3167-77.LEE, G., THANGAVEL, R., SHARMA, V., LITERSKY, J., BHASKAR, K., FANG, S., DO, L., ANDREADIS, A., VAN HOESEN, G. & KSIEZAK-REDING, H. 2004. Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci, 24, 2304-12.LEE, H. J., CHOI, C. & LEE, S. J. 2002. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem, 277, 671-8.LEE, J., CULYBA, E. K., POWERS, E. T. & KELLY, J. W. 2011. Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat Chem Biol, 7, 602-9.LEE, V., GOEDERT, M. & TROJANOWSKI, J. 2001. Neurodegenerative tauopathies. Annu Rev Neurosci, 24, 1121-59.LEHTONEN, S., ZHAO, F. & LEHTONEN, E. 2002. CD2-associated protein directly interacts with the actin cytoskeleton. Am J Physiol Renal Physiol, 283, F734-43.LEI, P., AYTON, S., MOON, S., ZHANG, Q., VOLITAKIS, I., FINKELSTEIN, D. I. & BUSH, A. I. 2014. Motor and cognitive deficits in aged tau knockout mice in two background strains. MolNeurodegener, 9, 29.LEROY, E., BOYER, R., AUBURGER, G., LEUBE, B., ULM, G., MEZEY, E., HARTA, G., BROWNSTEIN, M. J., JONNALAGADA, S., CHERNOVA, T., DEHEJIA, A., LAVEDAN, C., GASSER, T., STEINBACH, P. J., WILKINSON, K. D. & POLYMEROPOULOS, M. H. 1998. The ubiquitin pathway in Parkinson's disease. Nature, 395, 451-2.LESAGE, S., ANHEIM, M., LETOURNEL, F., BOUSSET, L., HONORE, A., ROZAS, N., PIERI, L., MADIONA, K., DURR, A., MELKI, R., VERNY, C., BRICE, A. & FRENCH PARKINSON'S DISEASE GENETICS STUDY, G. 2013. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol, 73, 459-71.LEVERENZ, J. B., HAMILTON, R., TSUANG, D. W., SCHANTZ, A., VAVREK, D., LARSON, E. B., KUKULL, W. A., LOPEZ, O., GALASKO, D., MASLIAH, E., KAYE, J., WOLTJER, R., CLARK,

94
C., TROJANOWSKI, J. Q. & MONTINE, T. J. 2008. Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol, 18, 220-4.LEVERENZ, J. B., QUINN, J. F., ZABETIAN, C., ZHANG, J., MONTINE, K. S. & MONTINE, T. J. 2009. Cognitive impairment and dementia in patients with Parkinson disease. Curr Top Med Chem, 9,903-12.LEW, J., HUANG, Q., QI, Z., WINKFEIN, R., AEBERSOLD, R., HUNT, T. & WANG, J. 1994. A brain-specific activator of cyclin-dependent kinase 5. Nature, 371, 423-6.LEYK, J., GOLDBAUM, O., NOACK, M. & RICHTER-LANDSBERG, C. 2015. Inhibition of HDAC6 modifies tau inclusion body formation and impairs autophagic clearance. J Mol Neurosci, 55,1031-46.LI, W., WEST, N., COLLA, E., PLETNIKOVA, O., TRONCOSO, J. C., MARSH, L., DAWSON, T. M., JAKALA, P., HARTMANN, T., PRICE, D. L. & LEE, M. K. 2005. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc Natl Acad Sci U S A, 102, 2162-7.LIM, F., HERNANDEZ, F., LUCAS, J. J., GOMEZ-RAMOS, P., MORAN, M. A. & AVILA, J. 2001. FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol Cell Neurosci, 18, 702-14.LIN, W. L., LEWIS, J., YEN, S. H., HUTTON, M. & DICKSON, D. W. 2003. Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol, 32, 1091-105.LIPPA, C. F., FUJIWARA, H., MANN, D. M., GIASSON, B., BABA, M., SCHMIDT, M. L., NEE, L. E., O'CONNELL, B., POLLEN, D. A., ST GEORGE-HYSLOP, P., GHETTI, B., NOCHLIN, D., BIRD, T. D., CAIRNS, N. J., LEE, V. M., IWATSUBO, T. & TROJANOWSKI, J. Q. 1998. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol, 153, 1365-70.LIPPA, C. F., SCHMIDT, M. L., LEE, V. M. & TROJANOWSKI, J. Q. 1999. Antibodies to alpha-synuclein detect Lewy bodies in many Down's syndrome brains with Alzheimer's disease. Ann Neurol,45, 353-7.LIU-YESUCEVITZ, L., BILGUTAY, A., ZHANG, Y. J., VANDERWEYDE, T., CITRO, A., MEHTA, T., ZAARUR, N., MCKEE, A., BOWSER, R., SHERMAN, M., PETRUCELLI, L. & WOLOZIN, B. 2010. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One, 5, e13250.LIU, C. W., GIASSON, B. I., LEWIS, K. A., LEE, V. M., DEMARTINO, G. N. & THOMAS, P. J. 2005a. A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J Biol Chem, 280, 22670-8.LIU, F., GRUNDKE-IQBAL, I., IQBAL, K. & GONG, C. 2005b. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci, 22,1942-50.LIU, F., IQBAL, K., GRUNDKE-IQBAL, I., HART, G. & GONG, C. 2004. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci U S A, 101, 10804-9.LIU, F., LI, B., TUNG, E., GRUNDKE-IQBAL, I., IQBAL, K. & GONG, C. 2007. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci, 26,3429-36.LIU, F., ZAIDI, T., IQBAL, K., GRUNDKE-IQBAL, I. & GONG, C. 2002. Aberrant glycosylation modulates phosphorylation of tau by protein kinase A and dephosphorylation of tau by protein phosphatase 2A and 5. Neuroscience, 115, 829-37.LIU, S. J., ZHANG, A. H., LI, H. L., WANG, Q., DENG, H. M., NETZER, W. J., XU, H. & WANG, J. Z. 2003. Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. Journal of Neurochemistry, 87, 1333-1344.

95
LOPEZ, A., TARRAGO, T. & GIRALT, E. 2011. Low molecular weight inhibitors of Prolyl Oligopeptidase: a review of compounds patented from 2003 to 2010. Expert Opin Ther Pat, 21, 1023-44.LORENZEN, N., NIELSEN, S. B., YOSHIMURA, Y., VAD, B. S., ANDERSEN, C. B., BETZER, C., KASPERSEN, J. D., CHRISTIANSEN, G., PEDERSEN, J. S., JENSEN, P. H., MULDER, F. A. & OTZEN, D. E. 2014. How epigallocatechin gallate can inhibit alpha-synuclein oligomer toxicity in vitro. J Biol Chem, 289, 21299-310.LOVESTONE, S., BOADA, M., DUBOIS, B., HULL, M., RINNE, J. O., HUPPERTZ, H. J., CALERO, M., ANDRES, M. V., GOMEZ-CARRILLO, B., LEON, T., DEL SER, T. & INVESTIGATORS, A. 2015. A phase II trial of tideglusib in Alzheimer's disease. J Alzheimers Dis, 45, 75-88.LU, M. & KOSIK, K. 2001. Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Mol Biol Cell, 12, 171-84.LUK, K., KEHM, V., CARROLL, J., ZHANG, B., O'BRIEN, P., TROJANOWSKI, J. & LEE, V. 2012a. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science, 338, 949-53.LUK, K., KEHM, V., ZHANG, B., O'BRIEN, P., TROJANOWSKI, J. & LEE, V. 2012b. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med, 209, 975-86.LUK, K. C., KEHM, V., CARROLL, J., ZHANG, B., O'BRIEN, P., TROJANOWSKI, J. Q. & LEE, V. M. 2012c. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science, 338, 949-53.LUK, K. C., KEHM, V. M., ZHANG, B., O'BRIEN, P., TROJANOWSKI, J. Q. & LEE, V. M. 2012d. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med, 209, 975-86.LUK, K. C., SONG, C., O'BRIEN, P., STIEBER, A., BRANCH, J. R., BRUNDEN, K. R., TROJANOWSKI, J. Q. & LEE, V. M. 2009. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A, 106, 20051-6.LUNDBLAD, M., DECRESSAC, M., MATTSSON, B. & BJORKLUND, A. 2012. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A, 109, 3213-9.LUO, W., DOU, F., RODINA, A., CHIP, S., KIM, J., ZHAO, Q., MOULICK, K., AGUIRRE, J., WU, N., GREENGARD, P. & CHIOSIS, G. 2007. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc Natl Acad Sci U S A, 104, 9511-6.MAEDA, S., SAHARA, N., SAITO, Y., MURAYAMA, M., YOSHIIKE, Y., KIM, H., MIYASAKA, T., MURAYAMA, S., IKAI, A. & TAKASHIMA, A. 2007. Granular tau oligomers as intermediates of tau filaments. Biochemistry, 46, 3856-61.MAGNANI, E., FAN, J., GASPARINI, L., GOLDING, M., WILLIAMS, M., SCHIAVO, G., GOEDERT, M., AMOS, L. A. & SPILLANTINI, M. G. 2007. Interaction of tau protein with the dynactin complex. EMBO J, 26, 4546-54.MAHLEY, R. 1988. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science, 240, 622-30.MALIK, M., FENKO, M. D., SHEIKH, A. M., WEN, G. & LI, X. 2011. A novel approach for characterization of cathepsin D protease and its effect on tau and beta-amyloid proteins. Neurochem Res,36, 754-60.MALM, T., LOPPI, S. & KANNINEN, K. M. 2016. Exosomes in Alzheimer's disease. Neurochem Int,97, 193-9.MANDELKOW, E. M., SCHWEERS, O., DREWES, G., BIERNAT, J., GUSTKE, N., TRINCZEK, B. & MANDELKOW, E. 1996. Structure, microtubule interactions, and phosphorylation of tau protein. Ann N Y Acad Sci, 777, 96-106.MANIECKA, Z. & POLYMENIDOU, M. 2015. From nucleation to widespread propagation: A prion-like concept for ALS. Virus Res, 207, 94-105.

96
MANNISTO, P. T., VENALAINEN, J., JALKANEN, A. & GARCIA-HORSMAN, J. A. 2007. Prolyl oligopeptidase: a potential target for the treatment of cognitive disorders. Drug News Perspect, 20, 293-305.MARTIN, L., LATYPOVA, X., WILSON, C. M., MAGNAUDEIX, A., PERRIN, M. L., YARDIN, C. & TERRO, F. 2013. Tau protein kinases: involvement in Alzheimer's disease. Ageing Res Rev, 12, 289-309.MARTINEZ-VICENTE, M., TALLOCZY, Z., KAUSHIK, S., MASSEY, A. C., MAZZULLI, J., MOSHAROV, E. V., HODARA, R., FREDENBURG, R., WU, D. C., FOLLENZI, A., DAUER, W., PRZEDBORSKI, S., ISCHIROPOULOS, H., LANSBURY, P. T., SULZER, D. & CUERVO, A. M. 2008. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest, 118,777-88.MARTISKAINEN, H., VISWANATHAN, J., NYKANEN, N., KURKI, M., HELISALMI, S., NATUNEN, T., SARAJARVI, T., KURKINEN, K., PURSIHEIMO, J., RAURAMAA, T., ALAFUZOFF, I., JAASKELAINEN, J., LEINONEN, V., SOININEN, H., HAAPASALO, A., HUTTUNEN, H. & HILTUNEN, M. 2015a. Transcriptomics and mechanistic elucidation of Alzheimer's disease risk genes in the brain and in vitro models. Neurobiol Aging, 36, 1221.e15-28.MARTISKAINEN, H., VISWANATHAN, J., NYKANEN, N. P., KURKI, M., HELISALMI, S., NATUNEN, T., SARAJARVI, T., KURKINEN, K. M., PURSIHEIMO, J. P., RAURAMAA, T., ALAFUZOFF, I., JAASKELAINEN, J. E., LEINONEN, V., SOININEN, H., HAAPASALO, A., HUTTUNEN, H. J. & HILTUNEN, M. 2015b. Transcriptomics and mechanistic elucidation of Alzheimer's disease risk genes in the brain and in vitro models. Neurobiol Aging, 36, 1221 e15-28.MASUDA-SUZUKAKE, M., NONAKA, T., HOSOKAWA, M., OIKAWA, T., ARAI, T., AKIYAMA, H., MANN, D. & HASEGAWA, M. 2013a. Prion-like spreading of pathological alpha-synuclein in brain. Brain, 136, 1128-38.MASUDA-SUZUKAKE, M., NONAKA, T., HOSOKAWA, M., OIKAWA, T., ARAI, T., AKIYAMA, H., MANN, D. M. & HASEGAWA, M. 2013b. Prion-like spreading of pathological alpha-synuclein in brain. Brain, 136, 1128-38.MATENIA, D. & MANDELKOW, E. M. 2009. The tau of MARK: a polarized view of the cytoskeleton. Trends Biochem Sci, 34, 332-42.MATUS, S., GLIMCHER, L. H. & HETZ, C. 2011. Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr Opin Cell Biol, 23, 239-52.MCCARTHY, M., ABECASIS, G., CARDON, L., GOLDSTEIN, D., LITTLE, J., IOANNIDIS, J. & HIRSCHHORN, J. 2008. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet, 9, 356-69.MCKEE, A. C., CANTU, R. C., NOWINSKI, C. J., HEDLEY-WHYTE, E. T., GAVETT, B. E., BUDSON, A. E., SANTINI, V. E., LEE, H. S., KUBILUS, C. A. & STERN, R. A. 2009. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol, 68, 709-35.MCNAUGHT, K. S. & OLANOW, C. W. 2006. Proteasome inhibitor-induced model of Parkinson's disease. Ann Neurol, 60, 243-7.MEDWAY, C. & MORGAN, K. 2014. Review: The genetics of Alzheimer's disease; putting flesh on the bones. Neuropathol Appl Neurobiol, 40, 97-105.MELKI, R. 2015. Role of Different Alpha-Synuclein Strains in Synucleinopathies, Similarities with other Neurodegenerative Diseases. J Parkinsons Dis, 5, 217-27.MERAZ-RIOS, M., LIRA-DE LEON, K., CAMPOS-PENA, V., DE ANDA-HERNANDEZ, M. & MENA-LOPEZ, R. 2010. Tau oligomers and aggregation in Alzheimer's disease. J Neurochem, 112,1353-67.MEREDITH, S. C. 2005. Protein denaturation and aggregation: Cellular responses to denatured and aggregated proteins. Ann N Y Acad Sci, 1066, 181-221.MEYER-LUEHMANN, M., COOMARASWAMY, J., BOLMONT, T., KAESER, S., SCHAEFER, C., KILGER, E., NEUENSCHWANDER, A., ABRAMOWSKI, D., FREY, P., JATON, A. L.,

97
VIGOURET, J. M., PAGANETTI, P., WALSH, D. M., MATHEWS, P. M., GHISO, J., STAUFENBIEL, M., WALKER, L. C. & JUCKER, M. 2006. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science, 313, 1781-4.MICHNICK, S. 2001. Exploring protein interactions by interaction-induced folding of proteins from complementary peptide fragments. Curr Opin Struct Biol, 11, 472-7.MICHNICK, S., EAR, P., MANDERSON, E., REMY, I. & STEFAN, E. 2007. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov,6, 569-82.MIN, S., CHO, S., ZHOU, Y., SCHROEDER, S., HAROUTUNIAN, V., SEELEY, W., HUANG, E., SHEN, Y., MASLIAH, E., MUKHERJEE, C., MEYERS, D., COLE, P., OTT, M. & GAN, L. 2010. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron, 67, 953-66.MOCANU, M. M., NISSEN, A., ECKERMANN, K., KHLISTUNOVA, I., BIERNAT, J., DREXLER, D., PETROVA, O., SCHONIG, K., BUJARD, H., MANDELKOW, E., ZHOU, L., RUNE, G. & MANDELKOW, E. M. 2008. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci, 28, 737-48.MONDRAGON-RODRIGUEZ, S., TRILLAUD-DOPPIA, E., DUDILOT, A., BOURGEOIS, C., LAUZON, M., LECLERC, N. & BOEHM, J. 2012. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem,287, 32040-53.MONTOYA, M. & GOUAUX, E. 2003. Beta-barrel membrane protein folding and structure viewed through the lens of alpha-hemolysin. Biochim Biophys Acta, 1609, 19-27.MORAIN, P., LESTAGE, P., DE NANTEUIL, G., JOCHEMSEN, R., ROBIN, J. L., GUEZ, D. & BOYER, P. A. 2002. S 17092: a prolyl endopeptidase inhibitor as a potential therapeutic drug for memory impairment. Preclinical and clinical studies. CNS Drug Rev, 8, 31-52.MORAWSKI, M., SCHULZ, I., ZEITSCHEL, U., BLOSA, M., SEEGER, G. & ROSSNER, S. 2011. Role of prolyl endopeptidase in intracellular transport and protein secretion. CNS Neurol Disord Drug Targets, 10, 327-32.MORI, H., KONDO, J. & IHARA, Y. 1987. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science, 235, 1641-4.MORISHIMA-KAWASHIMA, M., HASEGAWA, M., TAKIO, K., SUZUKI, M., TITANI, K. & IHARA, Y. 1993. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron, 10, 1151-60.MORRIS, M., KNUDSEN, G., MAEDA, S., TRINIDAD, J., IOANOVICIU, A., BURLINGAME, A. & MUCKE, L. 2015. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci.MORRIS, M., MAEDA, S., VOSSEL, K. & MUCKE, L. 2011. The many faces of tau. Neuron, 70,410-26.MORSCH, R., SIMON, W. & COLEMAN, P. D. 1999. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol, 58, 188-97.MOUSSAUD, S., JONES, D. R., MOUSSAUD-LAMODIERE, E. L., DELENCLOS, M., ROSS, O. A. & MCLEAN, P. J. 2014. Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegener, 9, 43.MUFSON, E. J., BINDER, L., COUNTS, S. E., DEKOSKY, S. T., DE TOLEDO-MORRELL, L., GINSBERG, S. D., IKONOMOVIC, M. D., PEREZ, S. E. & SCHEFF, S. W. 2012. Mild cognitive impairment: pathology and mechanisms. Acta Neuropathol, 123, 13-30.MUNCH, C., O'BRIEN, J. & BERTOLOTTI, A. 2011. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci U S A, 108, 3548-53.MUNOZ, L. & AMMIT, A. 2010. Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology, 58, 561-8.

98
MURPHY, D. D., RUETER, S. M., TROJANOWSKI, J. Q. & LEE, V. M. 2000. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci, 20, 3214-20.MURRAY, I. V., GIASSON, B. I., QUINN, S. M., KOPPAKA, V., AXELSEN, P. H., ISCHIROPOULOS, H., TROJANOWSKI, J. Q. & LEE, V. M. 2003. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry, 42, 8530-40.MYOHANEN, T. T., GARCIA-HORSMAN, J. A., TENORIO-LARANGA, J. & MANNISTO, P. T. 2009. Issues about the physiological functions of prolyl oligopeptidase based on its discordant spatial association with substrates and inconsistencies among mRNA, protein levels, and enzymatic activity. JHistochem Cytochem, 57, 831-48.MYOHANEN, T. T., HANNULA, M. J., VAN ELZEN, R., GERARD, M., VAN DER VEKEN, P.,GARCIA-HORSMAN, J. A., BAEKELANDT, V., MANNISTO, P. T. & LAMBEIR, A. M. 2012a. A prolyl oligopeptidase inhibitor, KYP-2047, reduces alpha-synuclein protein levels and aggregates in cellular and animal models of Parkinson's disease. Br J Pharmacol, 166, 1097-113.MYOHANEN, T. T., PYYKKO, E., MANNISTO, P. T. & CARPEN, O. 2012b. Distribution of prolyl oligopeptidase in human peripheral tissues and in ovarian and colorectal tumors. J Histochem Cytochem,60, 706-15.NAJ, A., JUN, G., REITZ, C., KUNKLE, B., PERRY, W., PARK, Y., BEECHAM, G., RAJBHANDARY, R., HAMILTON-NELSON, K., WANG, L., KAUWE, J., HUENTELMAN, M., MYERS, A., BIRD, T., BOEVE, B., BALDWIN, C., JARVIK, G., CRANE, P., ROGAEVA, E., BARMADA, M., DEMIRCI, F., CRUCHAGA, C., KRAMER, P., ERTEKIN-TANER, N., HARDY, J., GRAFF-RADFORD, N., GREEN, R., LARSON, E., ST GEORGE-HYSLOP, P., BUXBAUM, J., EVANS, D., SCHNEIDER, J., LUNETTA, K., KAMBOH, M., SAYKIN, A., REIMAN, E., DE JAGER, P., BENNETT, D., MORRIS, J., MONTINE, T., GOATE, A., BLACKER, D., TSUANG, D., HAKONARSON, H., KUKULL, W., FOROUD, T., MARTIN, E., HAINES, J., MAYEUX, R., FARRER, L., SCHELLENBERG, G., PERICAK-VANCE, M., ALBERT, M., ALBIN, R., APOSTOLOVA, L., ARNOLD, S., BARBER, R., BARNES, L., BEACH, T., BECKER, J., BEEKLY, D., BIGIO, E., BOWEN, J., BOXER, A., BURKE, J., CAIRNS, N., CANTWELL, L., CAO, C., CARLSON, C., CARNEY, R., CARRASQUILLO, M., CARROLL, S., CHUI, H., CLARK, D., CORNEVEAUX, J., CRIBBS, D., CROCCO, E., DECARLI, C., DEKOSKY, S., DICK, M., DICKSON, D., DUARA, R., FABER, K., FALLON, K., FARLOW, M., FERRIS, S., FROSCH, M., GALASKO, D., GANGULI, M., GEARING, M., GESCHWIND, D., GHETTI, B., GILBERT, J., GLASS, J., GROWDON, J., HAMILTON, R., HARRELL, L., HEAD, E., HONIG, L., HULETTE, C., et al. 2014. Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: a genome-wide association study. JAMA Neurol, 71, 1394-404.NEEB, A., KOCH, H., SCHURMANN, A. & BROSE, N. 1999. Direct interaction between the ARF-specific guanine nucleotide exchange factor msec7-1 and presynaptic Munc13-1. Eur J Cell Biol, 78,533-8.NEMANI, V. M., LU, W., BERGE, V., NAKAMURA, K., ONOA, B., LEE, M. K., CHAUDHRY, F. A., NICOLL, R. A. & EDWARDS, R. H. 2010. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron, 65, 66-79.NEVE, R., HARRIS, P., KOSIK, K., KURNIT, D. & DONLON, T. 1986. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res, 387, 271-80.NICKEL, W. & RABOUILLE, C. 2009. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol, 10, 148-55.NONAKA, T., MASUDA-SUZUKAKE, M., ARAI, T., HASEGAWA, Y., AKATSU, H., OBI, T., YOSHIDA, M., MURAYAMA, S., MANN, D. M., AKIYAMA, H. & HASEGAWA, M. 2013. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep, 4, 124-34.

99
NONAKA, T., WATANABE, S., IWATSUBO, T. & HASEGAWA, M. 2010a. Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem, 285,34885-98.NONAKA, T., WATANABE, S. T., IWATSUBO, T. & HASEGAWA, M. 2010b. Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem,285, 34885-98.NUBLING, G., BADER, B., LEVIN, J., HILDEBRANDT, J., KRETZSCHMAR, H. & GIESE, A. 2012a. Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with alpha-synuclein at the single molecule level. Mol Neurodegener, 7, 35.NUBLING, G., LEVIN, J., BADER, B., ISRAEL, L., BOTZEL, K., LORENZL, S. & GIESE, A. 2012b. Limited cleavage of tau with matrix-metalloproteinase MMP-9, but not MMP-3, enhances tau oligomer formation. Exp Neurol, 237, 470-6.NYKANEN, N. P., KYSENIUS, K., SAKHA, P., TAMMELA, P. & HUTTUNEN, H. J. 2012. gamma-Aminobutyric acid type A (GABAA) receptor activation modulates tau phosphorylation. J Biol Chem,287, 6743-52.OAKS, A. W., FRANKFURT, M., FINKELSTEIN, D. I. & SIDHU, A. 2013. Age-dependent effects of A53T alpha-synuclein on behavior and dopaminergic function. PLoS One, 8, e60378.OGEN-SHTERN, N., BEN DAVID, T. & LEDERKREMER, G. Z. 2016. Protein aggregation and ER stress. Brain Res.OKOCHI, M., WALTER, J., KOYAMA, A., NAKAJO, S., BABA, M., IWATSUBO, T., MEIJER, L., KAHLE, P. J. & HAASS, C. 2000. Constitutive phosphorylation of the Parkinson's disease associated alpha-synuclein. J Biol Chem, 275, 390-7.OSTREROVA, N., PETRUCELLI, L., FARRER, M., MEHTA, N., CHOI, P., HARDY, J. & WOLOZIN, B. 1999. alpha-Synuclein shares physical and functional homology with 14-3-3 proteins. JNeurosci, 19, 5782-91.OUESLATI, A., PALEOLOGOU, K. E., SCHNEIDER, B. L., AEBISCHER, P. & LASHUEL, H. A. 2012. Mimicking phosphorylation at serine 87 inhibits the aggregation of human alpha-synuclein and protects against its toxicity in a rat model of Parkinson's disease. J Neurosci, 32, 1536-44.OUTEIRO, T. F., KONTOPOULOS, E., ALTMANN, S. M., KUFAREVA, I., STRATHEARN, K. E., AMORE, A. M., VOLK, C. B., MAXWELL, M. M., ROCHET, J. C., MCLEAN, P. J., YOUNG, A. B., ABAGYAN, R., FEANY, M. B., HYMAN, B. T. & KAZANTSEV, A. G. 2007. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science, 317, 516-9.PAILLUSSON, S., CLAIREMBAULT, T., BIRAUD, M., NEUNLIST, M. & DERKINDEREN, P. 2013. Activity-dependent secretion of alpha-synuclein by enteric neurons. J Neurochem, 125, 512-7.PALEOLOGOU, K. E., OUESLATI, A., SHAKKED, G., ROSPIGLIOSI, C. C., KIM, H. Y., LAMBERTO, G. R., FERNANDEZ, C. O., SCHMID, A., CHEGINI, F., GAI, W. P., CHIAPPE, D., MONIATTE, M., SCHNEIDER, B. L., AEBISCHER, P., ELIEZER, D., ZWECKSTETTER, M., MASLIAH, E. & LASHUEL, H. A. 2010. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci,30, 3184-98.PAN-MONTOJO, F., SCHWARZ, M., WINKLER, C., ARNHOLD, M., O'SULLIVAN, G. A., PAL, A., SAID, J., MARSICO, G., VERBAVATZ, J. M., RODRIGO-ANGULO, M., GILLE, G., FUNK, R. H. & REICHMANN, H. 2012. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep, 2, 898.PARIHAR, M. S., PARIHAR, A., FUJITA, M., HASHIMOTO, M. & GHAFOURIFAR, P. 2008. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci, 65, 1272-84.PARK, J., SONG, W. & CHUNG, K. 2009. Function and regulation of Dyrk1A: towards understanding Down syndrome. Cell Mol Life Sci, 66, 3235-40.PARKER, M. W. & FEIL, S. C. 2005. Pore-forming protein toxins: from structure to function. Prog Biophys Mol Biol, 88, 91-142.

100
PATRICK, G., ZUKERBERG, L., NIKOLIC, M., DE LA MONTE, S., DIKKES, P. & TSAI, L. 1999. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature, 402, 615-22.PAXINOU, E., CHEN, Q., WEISSE, M., GIASSON, B. I., NORRIS, E. H., RUETER, S. M., TROJANOWSKI, J. Q., LEE, V. M. & ISCHIROPOULOS, H. 2001. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci, 21, 8053-61.PEELAERTS, W., BOUSSET, L., VAN DER PERREN, A., MOSKALYUK, A., PULIZZI, R., GIUGLIANO, M., VAN DEN HAUTE, C., MELKI, R. & BAEKELANDT, V. 2015. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature, 522, 340-4.PELTONEN, I., MYOHANEN, T. T. & MANNISTO, P. T. 2011. Association of prolyl oligopeptidase with conventional neurotransmitters in the brain. CNS Neurol Disord Drug Targets, 10, 311-8.PENG, X., TEHRANIAN, R., DIETRICH, P., STEFANIS, L. & PEREZ, R. G. 2005. Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J Cell Sci, 118, 3523-30.PENNATHUR, S., JACKSON-LEWIS, V., PRZEDBORSKI, S. & HEINECKE, J. W. 1999. Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and o,o'-dityrosine in brain tissue of 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson's disease. J Biol Chem, 274, 34621-8.PEREZ, M., SANTA-MARIA, I., GOMEZ DE BARREDA, E., ZHU, X., CUADROS, R., CABRERO, J. R., SANCHEZ-MADRID, F., DAWSON, H. N., VITEK, M. P., PERRY, G., SMITH, M. A. & AVILA, J. 2009. Tau--an inhibitor of deacetylase HDAC6 function. J Neurochem, 109, 1756-66.PEREZ, R. G., WAYMIRE, J. C., LIN, E., LIU, J. J., GUO, F. & ZIGMOND, M. J. 2002. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci, 22, 3090-9.PERICAK-VANCE, M., BEBOUT, J., GASKELL, P. J., YAMAOKA, L., HUNG, W., ALBERTS, M., WALKER, A., BARTLETT, R., HAYNES, C., WELSH, K. & ET, A. 1991. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet, 48, 1034-50.PETRUCELLI, L., DICKSON, D., KEHOE, K., TAYLOR, J., SNYDER, H., GROVER, A., DE LUCIA, M., MCGOWAN, E., LEWIS, J., PRIHAR, G., KIM, J., DILLMANN, W. H., BROWNE, S. E., HALL, A., VOELLMY, R., TSUBOI, Y., DAWSON, T. M., WOLOZIN, B., HARDY, J. & HUTTON, M. 2004. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet, 13, 703-14.PICKART, C. M. 2001. Ubiquitin enters the new millennium. Mol Cell, 8, 499-504.PICKART, C. M. & VANDEMARK, A. P. 2000. Opening doors into the proteasome. Nat Struct Biol, 7,999-1001.PIERI, L., MADIONA, K., BOUSSET, L. & MELKI, R. 2012. Fibrillar alpha-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys J, 102, 2894-905.PLANEL, E., RICHTER, K. E., NOLAN, C. E., FINLEY, J. E., LIU, L., WEN, Y., KRISHNAMURTHY, P., HERMAN, M., WANG, L., SCHACHTER, J. B., NELSON, R. B., LAU, L. F. & DUFF, K. E. 2007. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci, 27, 3090-7.POEHLER, A. M., XIANG, W., SPITZER, P., MAY, V. E., MEIXNER, H., ROCKENSTEIN, E., CHUTNA, O., OUTEIRO, T. F., WINKLER, J., MASLIAH, E. & KLUCKEN, J. 2014. Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy,10, 2171-92.POLYMEROPOULOS, M. H., LAVEDAN, C., LEROY, E., IDE, S. E., DEHEJIA, A., DUTRA, A., PIKE, B., ROOT, H., RUBENSTEIN, J., BOYER, R., STENROOS, E. S., CHANDRASEKHARAPPA, S., ATHANASSIADOU, A., PAPAPETROPOULOS, T., JOHNSON, W. G., LAZZARINI, A. M., DUVOISIN, R. C., DI IORIO, G., GOLBE, L. I. & NUSSBAUM, R. L. 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science, 276, 2045-7.POOLER, A., PHILLIPS, E., LAU, D., NOBLE, W. & HANGER, D. 2013. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep, 14, 389-94.

101
POPPEK, D., KECK, S., ERMAK, G., JUNG, T., STOLZING, A., ULLRICH, O., DAVIES, K. J. & GRUNE, T. 2006. Phosphorylation inhibits turnover of the tau protein by the proteasome: influence of RCAN1 and oxidative stress. Biochem J, 400, 511-20.POUNTNEY, D. L., LOWE, R., QUILTY, M., VICKERS, J. C., VOELCKER, N. H. & GAI, W. P. 2004. Annular alpha-synuclein species from purified multiple system atrophy inclusions. J Neurochem,90, 502-12.PROTS, I., VEBER, V., BREY, S., CAMPIONI, S., BUDER, K., RIEK, R., BOHM, K. J. & WINNER, B. 2013. alpha-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J Biol Chem, 288,21742-54.PRUSINER, S. B., WOERMAN, A. L., MORDES, D. A., WATTS, J. C., RAMPERSAUD, R., BERRY, D. B., PATEL, S., OEHLER, A., LOWE, J. K., KRAVITZ, S. N., GESCHWIND, D. H., GLIDDEN, D. V., HALLIDAY, G. M., MIDDLETON, L. T., GENTLEMAN, S. M., GRINBERG, L. T. & GILES, K. 2015. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A, 112, E5308-17.QIANG, L., YU, W., ANDREADIS, A., LUO, M. & BAAS, P. W. 2006. Tau protects microtubules in the axon from severing by katanin. J Neurosci, 26, 3120-9.QIAO, L., HAMAMICHI, S., CALDWELL, K. A., CALDWELL, G. A., YACOUBIAN, T. A.,WILSON, S., XIE, Z. L., SPEAKE, L. D., PARKS, R., CRABTREE, D., LIANG, Q., CRIMMINS, S., SCHNEIDER, L., UCHIYAMA, Y., IWATSUBO, T., ZHOU, Y., PENG, L., LU, Y., STANDAERT, D. G., WALLS, K. C., SHACKA, J. J., ROTH, K. A. & ZHANG, J. 2008. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain, 1, 17.QURESHI, H. Y. & PAUDEL, H. K. 2011. Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and alpha-synuclein mutations promote Tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. J Biol Chem, 286, 5055-68.RABE, M., SORAGNI, A., REYNOLDS, N. P., VERDES, D., LIVERANI, E., RIEK, R. & SEEGER, S. 2013. On-surface aggregation of alpha-synuclein at nanomolar concentrations results in two distinct growth mechanisms. ACS Chem Neurosci, 4, 408-17.RAWLINGS, N. D. & BARRETT, A. J. 1994. Families of serine peptidases. Methods Enzymol, 244,19-61.REINHARDT, P., SCHMID, B., BURBULLA, L. F., SCHONDORF, D. C., WAGNER, L., GLATZA, M., HOING, S., HARGUS, G., HECK, S. A., DHINGRA, A., WU, G., MULLER, S., BROCKMANN, K., KLUBA, T., MAISEL, M., KRUGER, R., BERG, D., TSYTSYURA, Y., THIEL, C. S., PSATHAKI, O. E., KLINGAUF, J., KUHLMANN, T., KLEWIN, M., MULLER, H., GASSER, T., SCHOLER, H. R. & STERNECKERT, J. 2013. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell, 12, 354-67.REMY, I. & MICHNICK, S. 2006a. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods, 3, 977-9.REMY, I. & MICHNICK, S. W. 2006b. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods, 3, 977-9.REN, P. H., LAUCKNER, J. E., KACHIRSKAIA, I., HEUSER, J. E., MELKI, R. & KOPITO, R. R. 2009. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol, 11, 219-25.RESH, M. 1998. Fyn, a Src family tyrosine kinase. Int J Biochem Cell Biol, 30, 1159-62.REYNOLDS, C. H., GARWOOD, C. J., WRAY, S., PRICE, C., KELLIE, S., PERERA, T., ZVELEBIL, M., YANG, A., SHEPPARD, P. W., VARNDELL, I. M., HANGER, D. P. & ANDERTON, B. H. 2008. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J Biol Chem,283, 18177-86.

102
REYNOLDS, M., BERRY, R. & BINDER, L. 2005. Site-specific nitration and oxidative dityrosine bridging of the tau protein by peroxynitrite: implications for Alzheimer's disease. Biochemistry, 44,1690-700.RIVERS, R. C., KUMITA, J. R., TARTAGLIA, G. G., DEDMON, M. M., PAWAR, A., VENDRUSCOLO, M., DOBSON, C. M. & CHRISTODOULOU, J. 2008. Molecular determinants of the aggregation behavior of alpha- and beta-synuclein. Protein Sci, 17, 887-98.RIZZU, P., VAN SWIETEN, J. C., JOOSSE, M., HASEGAWA, M., STEVENS, M., TIBBEN, A., NIERMEIJER, M. F., HILLEBRAND, M., RAVID, R., OOSTRA, B. A., GOEDERT, M., VAN DUIJN, C. M. & HEUTINK, P. 1999. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet, 64,414-21.ROHN, T. 2013. The triggering receptor expressed on myeloid cells 2: "TREM-ming" the inflammatory component associated with Alzheimer's disease. Oxid Med Cell Longev, 2013, 860959.ROHRBOUGH, J., RUSHTON, E., PALANKER, L., WOODRUFF, E., MATTHIES, H., ACHARYA, U., ACHARYA, J. & BROADIE, K. 2004. Ceramidase regulates synaptic vesicle exocytosis and trafficking. J Neurosci, 24, 7789-803.ROSSNER, S., SCHULZ, I., ZEITSCHEL, U., SCHLIEBS, R., BIGL, V. & DEMUTH, H. U. 2005. Brain prolyl endopeptidase expression in aging, APP transgenic mice and Alzheimer's disease. Neurochem Res, 30, 695-702.RUBINSZTEIN, D. C. 2006. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature, 443, 780-6.RUF, R. A., LUTZ, E. A., ZIGONEANU, I. G. & PIELAK, G. J. 2008. Alpha-Synuclein conformation affects its tyrosine-dependent oxidative aggregation. Biochemistry, 47, 13604-9.RUIZ, A., HEILMANN, S., BECKER, T., HERNANDEZ, I., WAGNER, H., THELEN, M., MAULEON, A., ROSENDE-ROCA, M., BELLENGUEZ, C., BIS, J., HAROLD, D., GERRISH, A., SIMS, R., SOTOLONGO-GRAU, O., ESPINOSA, A., ALEGRET, M., ARRIETA, J., LACOUR, A., LEBER, M., BECKER, J., LAFUENTE, A., RUIZ, S., VARGAS, L., RODRIGUEZ, O., ORTEGA, G., DOMINGUEZ, M., MAYEUX, R., HAINES, J., PERICAK-VANCE, M., FARRER, L., SCHELLENBERG, G., CHOURAKI, V., LAUNER, L., VAN DUIJN, C., SESHADRI, S., ANTUNEZ, C., BRETELER, M., SERRANO-RIOS, M., JESSEN, F., TARRAGA, L., NOTHEN, M., MAIER, W., BOADA, M. & RAMIREZ, A. 2014. Follow-up of loci from the International Genomics of Alzheimer's Disease Project identifies TRIP4 as a novel susceptibility gene. Transl Psychiatry, 4, e358.RUTHERFORD, N. J., MOORE, B. D., GOLDE, T. E. & GIASSON, B. I. 2014. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of alpha-synuclein. J Neurochem, 131, 859-67.SAHIN, C., LORENZEN, N., LEMMINGER, L., CHRISTIANSEN, G., MOLLER, I. M., VESTERAGER, L. B., PEDERSEN, L. O., FOG, K., KALLUNKI, P. & OTZEN, D. E. 2016. Antibodies against the C-terminus of alpha-synuclein modulate its fibrillation. Biophys Chem.SAMAN, S., KIM, W., RAYA, M., VISNICK, Y., MIRO, S., SAMAN, S., JACKSON, B., MCKEE, A., ALVAREZ, V., LEE, N. & HALL, G. 2012a. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem,287, 3842-9.SAMAN, S., KIM, W., RAYA, M., VISNICK, Y., MIRO, S., SAMAN, S., JACKSON, B., MCKEE, A. C., ALVAREZ, V. E., LEE, N. C. & HALL, G. F. 2012b. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. JBiol Chem, 287, 3842-9.SANDERS, D., KAUFMAN, S., DEVOS, S., SHARMA, A., MIRBAHA, H., LI, A., BARKER, S., FOLEY, A., THORPE, J., SERPELL, L., MILLER, T., GRINBERG, L., SEELEY, W. & DIAMOND, M. 2014a. Distinct Tau Prion Strains Propagate in Cells and Mice and Define Different Tauopathies. Neuron.

103
SANDERS, D. W., KAUFMAN, S. K., DEVOS, S. L., SHARMA, A. M., MIRBAHA, H., LI, A., BARKER, S. J., FOLEY, A. C., THORPE, J. R., SERPELL, L. C., MILLER, T. M., GRINBERG, L. T., SEELEY, W. W. & DIAMOND, M. I. 2014b. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron, 82, 1271-88.SANTACRUZ, K., LEWIS, J., SPIRES, T., PAULSON, J., KOTILINEK, L., INGELSSON, M., GUIMARAES, A., DETURE, M., RAMSDEN, M., MCGOWAN, E., FORSTER, C., YUE, M., ORNE, J., JANUS, C., MARIASH, A., KUSKOWSKI, M., HYMAN, B., HUTTON, M. & ASHE, K. H. 2005. Tau suppression in a neurodegenerative mouse model improves memory function. Science, 309, 476-81.SAVOLAINEN, M. H., RICHIE, C. T., HARVEY, B. K., MANNISTO, P. T., MAGUIRE-ZEISS, K.A. & MYOHANEN, T. T. 2014. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol Dis, 68, 1-15.SAWAYA, M. R., SAMBASHIVAN, S., NELSON, R., IVANOVA, M. I., SIEVERS, S. A., APOSTOL, M. I., THOMPSON, M. J., BALBIRNIE, M., WILTZIUS, J. J., MCFARLANE, H. T., MADSEN, A. O., RIEKEL, C. & EISENBERG, D. 2007. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature, 447, 453-7.SCHAEFFER, H. & WEBER, M. 1999. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol, 19, 2435-44.SCHNEIDER, A., BIERNAT, J., VON BERGEN, M., MANDELKOW, E. & MANDELKOW, E. M. 1999. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry, 38, 3549-58.SCHRODER, M. & KAUFMAN, R. J. 2005. The mammalian unfolded protein response. Annu Rev Biochem, 74, 739-89.SCHULZ, I., ZEITSCHEL, U., RUDOLPH, T., RUIZ-CARRILLO, D., RAHFELD, J. U., GERHARTZ, B., BIGL, V., DEMUTH, H. U. & ROSSNER, S. 2005. Subcellular localization suggests novel functions for prolyl endopeptidase in protein secretion. J Neurochem, 94, 970-9.SCOTT, D. A., TABAREAN, I., TANG, Y., CARTIER, A., MASLIAH, E. & ROY, S. 2010. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. JNeurosci, 30, 8083-95.SESHADRI, S., FITZPATRICK, A., IKRAM, M., DESTEFANO, A., GUDNASON, V., BOADA, M., BIS, J., SMITH, A., CARASSQUILLO, M., LAMBERT, J., HAROLD, D., SCHRIJVERS, E., RAMIREZ-LORCA, R., DEBETTE, S., LONGSTRETH, W. J., JANSSENS, A., PANKRATZ, V., DARTIGUES, J., HOLLINGWORTH, P., ASPELUND, T., HERNANDEZ, I., BEISER, A., KULLER, L., KOUDSTAAL, P., DICKSON, D., TZOURIO, C., ABRAHAM, R., ANTUNEZ, C., DU, Y., ROTTER, J., AULCHENKO, Y., HARRIS, T., PETERSEN, R., BERR, C., OWEN, M., LOPEZ-ARRIETA, J., VARADARAJAN, B., BECKER, J., RIVADENEIRA, F., NALLS, M., GRAFF-RADFORD, N., CAMPION, D., AUERBACH, S., RICE, K., HOFMAN, A., JONSSON, P., SCHMIDT, H., LATHROP, M., MOSLEY, T., AU, R., PSATY, B., UITTERLINDEN, A., FARRER, L., LUMLEY, T., RUIZ, A., WILLIAMS, J., AMOUYEL, P., YOUNKIN, S., WOLF, P., LAUNER, L., LOPEZ, O., VAN DUIJN, C. & BRETELER, M. 2010. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA, 303, 1832-40.SHABEK, N., HERMAN-BACHINSKY, Y., BUCHSBAUM, S., LEWINSON, O., HAJ-YAHYA, M., HEJJAOUI, M., LASHUEL, H. A., SOMMER, T., BRIK, A. & CIECHANOVER, A. 2012. The size of the proteasomal substrate determines whether its degradation will be mediated by mono- orpolyubiquitylation. Mol Cell, 48, 87-97.SHARON, R., BAR-JOSEPH, I., FROSCH, M. P., WALSH, D. M., HAMILTON, J. A. & SELKOE, D. J. 2003a. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron, 37, 583-95.SHARON, R., BAR-JOSEPH, I., MIRICK, G. E., SERHAN, C. N. & SELKOE, D. J. 2003b. Altered fatty acid composition of dopaminergic neurons expressing alpha-synuclein and human brains with alpha-synucleinopathies. J Biol Chem, 278, 49874-81.

104
SHEMESH, O. A. & SPIRA, M. E. 2010. Hallmark cellular pathology of Alzheimer's disease induced by mutant human tau expression in cultured Aplysia neurons. Acta Neuropathol, 120, 209-22.SHI, M., LIU, C., COOK, T. J., BULLOCK, K. M., ZHAO, Y., GINGHINA, C., LI, Y., ARO, P., DATOR, R., HE, C., HIPP, M. J., ZABETIAN, C. P., PESKIND, E. R., HU, S. C., QUINN, J. F., GALASKO, D. R., BANKS, W. A. & ZHANG, J. 2014. Plasma exosomal alpha-synuclein is likely CNS-derived and increased in Parkinson's disease. Acta Neuropathol, 128, 639-50.SHIMURA, H., SCHLOSSMACHER, M. G., HATTORI, N., FROSCH, M. P., TROCKENBACHER, A., SCHNEIDER, R., MIZUNO, Y., KOSIK, K. S. & SELKOE, D. J. 2001. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science, 293,263-9.SHIMURA, H., SCHWARTZ, D., GYGI, S. P. & KOSIK, K. S. 2004. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem, 279, 4869-76.SHISHIDO, Y., FURUSHIRO, M., TANABE, S., TANIGUCHI, A., HASHIMOTO, S., YOKOKURA, T., SHIBATA, S., YAMAMOTO, T. & WATANABE, S. 1998. Effect of ZTTA, a prolyl endopeptidase inhibitor, on memory impairment in a passive avoidance test of rats with basal forebrain lesions. Pharm Res, 15, 1907-10.SHULMAN, J., IMBOYWA, S., GIAGTZOGLOU, N., POWERS, M., HU, Y., DEVENPORT, D., CHIPENDO, P., CHIBNIK, L., DIAMOND, A., PERRIMON, N., BROWN, N., DE JAGER, P. & FEANY, M. 2014. Functional screening in Drosophila identifies Alzheimer's disease susceptibility genes and implicates Tau-mediated mechanisms. Hum Mol Genet, 23, 870-7.SHULMAN, J. M., CHIPENDO, P., CHIBNIK, L. B., AUBIN, C., TRAN, D., KEENAN, B. T., KRAMER, P. L., SCHNEIDER, J. A., BENNETT, D. A., FEANY, M. B. & DE JAGER, P. L. 2011. Functional screening of Alzheimer pathology genome-wide association signals in Drosophila. Am J Hum Genet, 88, 232-8.SIEST, G., PILLOT, T., REGIS-BAILLY, A., LEININGER-MULLER, B., STEINMETZ, J., GALTEAU, M. & VISVIKIS, S. 1995. Apolipoprotein E: an important gene and protein to follow in laboratory medicine. Clin Chem, 41, 1068-86.SIMIC, G., BABIC LEKO, M., WRAY, S., HARRINGTON, C., DELALLE, I., JOVANOV-MILOSEVIC, N., BAZADONA, D., BUEE, L., DE SILVA, R., DI GIOVANNI, G., WISCHIK, C. & HOF, P. R. 2016. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer's Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules, 6, 6.SIMIC, G., DIANA, A. & HOF, P. R. 2003. Phosphorylation pattern of tau associated with distinct changes of the growth cone cytoskeleton. Prog Mol Subcell Biol, 32, 33-48.SIMON, D., GARCIA-GARCIA, E., GOMEZ-RAMOS, A., FALCON-PEREZ, J. M., DIAZ-HERNANDEZ, M., HERNANDEZ, F. & AVILA, J. 2012a. Tau overexpression results in its secretion via membrane vesicles. Neurodegener Dis, 10, 73-5.SIMON, D., GARCIA-GARCIA, E., ROYO, F., FALCON-PEREZ, J. M. & AVILA, J. 2012b. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett, 586,47-54.SINGLETON, A. B., FARRER, M., JOHNSON, J., SINGLETON, A., HAGUE, S., KACHERGUS, J., HULIHAN, M., PEURALINNA, T., DUTRA, A., NUSSBAUM, R., LINCOLN, S., CRAWLEY, A., HANSON, M., MARAGANORE, D., ADLER, C., COOKSON, M. R., MUENTER, M., BAPTISTA, M., MILLER, D., BLANCATO, J., HARDY, J. & GWINN-HARDY, K. 2003. alpha-Synuclein locus triplication causes Parkinson's disease. Science, 302, 841.SMITH, W. W., JIANG, H., PEI, Z., TANAKA, Y., MORITA, H., SAWA, A., DAWSON, V. L., DAWSON, T. M. & ROSS, C. A. 2005. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet, 14, 3801-11.SOUZA, J. M., GIASSON, B. I., CHEN, Q., LEE, V. M. & ISCHIROPOULOS, H. 2000. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem, 275, 18344-9.

105
SPILLANTINI, M. G., MURRELL, J. R., GOEDERT, M., FARLOW, M. R., KLUG, A. & GHETTI, B. 1998. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A, 95, 7737-41.SPILLANTINI, M. G., SCHMIDT, M. L., LEE, V. M., TROJANOWSKI, J. Q., JAKES, R. & GOEDERT, M. 1997. Alpha-synuclein in Lewy bodies. Nature, 388, 839-40.SPIRES-JONES, T. L., DE CALIGNON, A., MATSUI, T., ZEHR, C., PITSTICK, R., WU, H. Y., OSETEK, J. D., JONES, P. B., BACSKAI, B. J., FEANY, M. B., CARLSON, G. A., ASHE, K. H.,LEWIS, J. & HYMAN, B. T. 2008. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci, 28, 862-7.SREEDHARAN, J., BLAIR, I. P., TRIPATHI, V. B., HU, X., VANCE, C., ROGELJ, B., ACKERLEY, S., DURNALL, J. C., WILLIAMS, K. L., BURATTI, E., BARALLE, F., DE BELLEROCHE, J., MITCHELL, J. D., LEIGH, P. N., AL-CHALABI, A., MILLER, C. C., NICHOLSON, G. & SHAW, C. E. 2008. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science, 319, 1668-72.STEFANOVIC, A. N., STOCKL, M. T., CLAESSENS, M. M. & SUBRAMANIAM, V. 2014. alpha-Synuclein oligomers distinctively permeabilize complex model membranes. FEBS J, 281, 2838-50.STOCKL, M., FISCHER, P., WANKER, E. & HERRMANN, A. 2008. Alpha-synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J Mol Biol, 375, 1394-404.STOHR, J., WATTS, J. C., MENSINGER, Z. L., OEHLER, A., GRILLO, S. K., DEARMOND, S. J., PRUSINER, S. B. & GILES, K. 2012. Purified and synthetic Alzheimer's amyloid beta (Abeta) prions. Proc Natl Acad Sci U S A, 109, 11025-30.STRITTMATTER, W., SAUNDERS, A., GOEDERT, M., WEISGRABER, K., DONG, L., JAKES, R., HUANG, D., PERICAK-VANCE, M., SCHMECHEL, D. & ROSES, A. 1994. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A, 91, 11183-6.STRITTMATTER, W., SAUNDERS, A., SCHMECHEL, D., PERICAK-VANCE, M., ENGHILD, J., SALVESEN, G. & ROSES, A. 1993. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A,90, 1977-81.SUDHOF, T. C. 2013. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron, 80, 675-90.SULTAN, A., NESSLANY, F., VIOLET, M., BEGARD, S., LOYENS, A., TALAHARI, S., MANSUROGLU, Z., MARZIN, D., SERGEANT, N., HUMEZ, S., COLIN, M., BONNEFOY, E., BUEE, L. & GALAS, M. C. 2011. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem,286, 4566-75.SURRIDGE, C. D. & BURNS, R. G. 1994. The difference in the binding of phosphatidylinositol distinguishes MAP2 from MAP2C and Tau. Biochemistry, 33, 8051-7.SUZUKI, A. & OHNO, S. 2006. The PAR-aPKC system: lessons in polarity. J Cell Sci, 119, 979-87.SZELTNER, Z., JUHASZ, T., SZAMOSI, I., REA, D., FULOP, V., MODOS, K., JULIANO, L. & POLGAR, L. 2013. The loops facing the active site of prolyl oligopeptidase are crucial components in substrate gating and specificity. Biochim Biophys Acta, 1834, 98-111.SZELTNER, Z., MORAWSKI, M., JUHASZ, T., SZAMOSI, I., LILIOM, K., CSIZMOK, V., TOLGYESI, F. & POLGAR, L. 2010. GAP43 shows partial co-localisation but no strong physical interaction with prolyl oligopeptidase. Biochim Biophys Acta, 1804, 2162-76.TAGLIATTI, E., FADDA, M., FALACE, A., BENFENATI, F. & FASSIO, A. 2016. Arf6 regulates the cycling and the readily releasable pool of synaptic vesicles at hippocampal synapse. Elife, 5.TAI, H. C., WANG, B. Y., SERRANO-POZO, A., FROSCH, M. P., SPIRES-JONES, T. L. & HYMAN, B. T. 2014. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer's disease. Acta Neuropathol Commun, 2, 146.TAKEI, Y., TENG, J., HARADA, A. & HIROKAWA, N. 2000. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol, 150, 989-1000.

106
TAN, J. M., WONG, E. S., KIRKPATRICK, D. S., PLETNIKOVA, O., KO, H. S., TAY, S. P., HO, M. W., TRONCOSO, J., GYGI, S. P., LEE, M. K., DAWSON, V. L., DAWSON, T. M. & LIM, K. L. 2008. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet, 17, 431-9.TANAKA, Y., ENGELENDER, S., IGARASHI, S., RAO, R. K., WANNER, T., TANZI, R. E., SAWA, A., V, L. D., DAWSON, T. M. & ROSS, C. A. 2001. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet, 10, 919-26.TANG, W., TAM, J. H., SEAH, C., CHIU, J., TYRER, A., CREGAN, S. P., MEAKIN, S. O. & PASTERNAK, S. H. 2015. Arf6 controls beta-amyloid production by regulating macropinocytosis of the Amyloid Precursor Protein to lysosomes. Mol Brain, 8, 41.TANIMUKAI, H., GRUNDKE-IQBAL, I. & IQBAL, K. 2005. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol, 166, 1761-71.TARRAGO, T., MARTIN-BENITO, J., SABIDO, E., CLAASEN, B., MADURGA, S., GAIRI, M., VALPUESTA, J. M. & GIRALT, E. 2009. A new side opening on prolyl oligopeptidase revealed by electron microscopy. FEBS Lett, 583, 3344-8.TAYLOR, J. P., HARDY, J. & FISCHBECK, K. H. 2002. Toxic proteins in neurodegenerative disease. Science, 296, 1991-5.TEHRANIAN, R., MONTOYA, S. E., VAN LAAR, A. D., HASTINGS, T. G. & PEREZ, R. G. 2006. Alpha-synuclein inhibits aromatic amino acid decarboxylase activity in dopaminergic cells. JNeurochem, 99, 1188-96.TENREIRO, S., REIMAO-PINTO, M. M., ANTAS, P., RINO, J., WAWRZYCKA, D., MACEDO, D., ROSADO-RAMOS, R., AMEN, T., WAISS, M., MAGALHAES, F., GOMES, A., SANTOS, C. N., KAGANOVICH, D. & OUTEIRO, T. F. 2014. Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson's disease. PLoS Genet, 10, e1004302.TEPASS, U. 2009. FERM proteins in animal morphogenesis. Curr Opin Genet Dev, 19, 357-67.TEPPER, K., BIERNAT, J., KUMAR, S., WEGMANN, S., TIMM, T., HUBSCHMANN, S., REDECKE, L., MANDELKOW, E. M., MULLER, D. J. & MANDELKOW, E. 2014. Oligomer formation of tau protein hyperphosphorylated in cells. J Biol Chem, 289, 34389-407.THAL, D. R., RUB, U., ORANTES, M. & BRAAK, H. 2002. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology, 58, 1791-800.THIES, E. & MANDELKOW, E. M. 2007. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci, 27, 2896-907.THOMAS, S., FUNK, K., WAN, Y., LIAO, Z., DAVIES, P., KURET, J. & YANG, A. 2012. Dualmodification of Alzheimer's disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol, 123, 105-17.TIAN, H., DAVIDOWITZ, E., LOPEZ, P., EMADI, S., MOE, J. & SIERKS, M. 2013. Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int J Cell Biol, 2013, 260787.TIAN, Q. & WANG, J. 2002. Role of Serine/Threonine Protein Phosphatase in Alzheimer’s Disease. Neurosignals, 11, 262-269.TOIDE, K., SHINODA, M., FUJIWARA, T. & IWAMOTO, Y. 1997. Effect of a novel prolyl endopeptidase inhibitor, JTP-4819, on spatial memory and central cholinergic neurons in aged rats. Pharmacol Biochem Behav, 56, 427-34.TRADEWELL, M. L., YU, Z., TIBSHIRANI, M., BOULANGER, M. C., DURHAM, H. D. & RICHARD, S. 2012. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum Mol Genet, 21, 136-49.TRAJKOVIC, K., HSU, C., CHIANTIA, S., RAJENDRAN, L., WENZEL, D., WIELAND, F., SCHWILLE, P., BRUGGER, B. & SIMONS, M. 2008. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science, 319, 1244-7.

107
TRAN, H. T., CHUNG, C. H., IBA, M., ZHANG, B., TROJANOWSKI, J. Q., LUK, K. C. & LEE, V. M. 2014. Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell Rep, 7, 2054-65.TSIGELNY, I. F., SHARIKOV, Y., WRASIDLO, W., GONZALEZ, T., DESPLATS, P. A., CREWS, L., SPENCER, B. & MASLIAH, E. 2012. Role of alpha-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J, 279, 1000-13.ULMER, T. S., BAX, A., COLE, N. B. & NUSSBAUM, R. L. 2005. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem, 280, 9595-603.URONEN RL & HJ, H. 2016. Genetic risk factors of Alzheimer’s disease and cell-to-cell transmission of Tau J Neurol Neuromed, 1, 17-22.UVERSKY, V. N. 2007. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J Neurochem, 103, 17-37.UVERSKY, V. N., YAMIN, G., MUNISHKINA, L. A., KARYMOV, M. A., MILLETT, I. S., DONIACH, S., LYUBCHENKO, Y. L. & FINK, A. L. 2005. Effects of nitration on the structure and aggregation of alpha-synuclein. Brain Res Mol Brain Res, 134, 84-102.VANCE, C., ROGELJ, B., HORTOBAGYI, T., DE VOS, K. J., NISHIMURA, A. L., SREEDHARAN, J., HU, X., SMITH, B., RUDDY, D., WRIGHT, P., GANESALINGAM, J., WILLIAMS, K. L., TRIPATHI, V., AL-SARAJ, S., AL-CHALABI, A., LEIGH, P. N., BLAIR, I. P., NICHOLSON, G., DE BELLEROCHE, J., GALLO, J. M., MILLER, C. C. & SHAW, C. E. 2009. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science, 323, 1208-11.VANDERWEYDE, T., APICCO, D. J., YOUMANS-KIDDER, K., ASH, P. E., COOK, C., LUMMERTZ DA ROCHA, E., JANSEN-WEST, K., FRAME, A. A., CITRO, A., LESZYK, J. D., IVANOV, P., ABISAMBRA, J. F., STEFFEN, M., LI, H., PETRUCELLI, L. & WOLOZIN, B. 2016. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep, 15, 1455-66.VANDERWEYDE, T., YU, H., VARNUM, M., LIU-YESUCEVITZ, L., CITRO, A., IKEZU, T., DUFF, K. & WOLOZIN, B. 2012. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci, 32, 8270-83.VELLA, L. J., HILL, A. F. & CHENG, L. 2016. Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Biomarker Potential in Alzheimer's and Parkinson's Disease. Int J Mol Sci, 17, 173.VENALAINEN, J. I., GARCIA-HORSMAN, J. A., FORSBERG, M. M., JALKANEN, A., WALLEN, E. A., JARHO, E. M., CHRISTIAANS, J. A., GYNTHER, J. & MANNISTO, P. T. 2006. Binding kinetics and duration of in vivo action of novel prolyl oligopeptidase inhibitors. Biochem Pharmacol,71, 683-92.VERGHESE, P., CASTELLANO, J., GARAI, K., WANG, Y., JIANG, H., SHAH, A., BU, G., FRIEDEN, C. & HOLTZMAN, D. 2013. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci U S A, 110, E1807-16.VIGO-PELFREY, C., SEUBERT, P., BARBOUR, R., BLOMQUIST, C., LEE, M., LEE, D., CORIA, F., CHANG, L., MILLER, B., LIEBERBURG, I. & ET, A. 1995. Elevation of microtubule-associated protein tau in the cerebrospinal fluid of patients with Alzheimer's disease. Neurology, 45, 788-93.VOGIATZI, T., XILOURI, M., VEKRELLIS, K. & STEFANIS, L. 2008. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem, 283,23542-56.VOLLES, M. J. & LANSBURY, P. T., JR. 2002. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry, 41, 4595-602.VOLPICELLI-DALEY, L. A., LUK, K. C., PATEL, T. P., TANIK, S. A., RIDDLE, D. M., STIEBER, A., MEANEY, D. F., TROJANOWSKI, J. Q. & LEE, V. M. 2011. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron, 72, 57-71.

108
VON BERGEN, M., FRIEDHOFF, P., BIERNAT, J., HEBERLE, J., MANDELKOW, E. M. & MANDELKOW, E. 2000. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci U S A, 97,5129-34.WADA, Y., ISHIGURO, K., ITOH, T. J., UCHIDA, T., HOTANI, H., SAITO, T., KISHIMOTO, T. & HISANAGA, S. 1998. Microtubule-stimulated phosphorylation of tau at Ser202 and Thr205 by cdk5 decreases its microtubule nucleation activity. J Biochem, 124, 738-46.WAKABAYASHI, K., TAKAHASHI, H., OHAMA, E. & IKUTA, F. 1990. Parkinson's disease: an immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol, 79, 581-3.WALLIN, A. K., BLENNOW, K., ZETTERBERG, H., LONDOS, E., MINTHON, L. & HANSSON, O. 2010. CSF biomarkers predict a more malignant outcome in Alzheimer disease. Neurology, 74, 1531-7.WALSH, D. & SELKOE, D. 2016. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci, 17, 251-60.WALTER, R., SHLANK, H., GLASS, J. D., SCHWARTZ, I. L. & KERENYI, T. D. 1971. Leucylglycinamide released from oxytocin by human uterine enzyme. Science, 173, 827-9.WAN, O. W. & CHUNG, K. K. 2012. The role of alpha-synuclein oligomerization and aggregation in cellular and animal models of Parkinson's disease. PLoS One, 7, e38545.WANG, G., KRISHNAMURTHY, K., UMAPATHY, N., VERIN, A. & BIEBERICH, E. 2009a. The carboxyl-terminal domain of atypical protein kinase Czeta binds to ceramide and regulates junction formation in epithelial cells. J Biol Chem, 284, 14469-75.WANG, J., GRUNDKE-IQBAL, I. & IQBAL, K. 2007a. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci, 25, 59-68.WANG, J. & LIU, F. 2008. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol, 85, 148-75.WANG, W., PEROVIC, I., CHITTULURU, J., KAGANOVICH, A., NGUYEN, L. T., LIAO, J., AUCLAIR, J. R., JOHNSON, D., LANDERU, A., SIMORELLIS, A. K., JU, S., COOKSON, M. R., ASTURIAS, F. J., AGAR, J. N., WEBB, B. N., KANG, C., RINGE, D., PETSKO, G. A., POCHAPSKY, T. C. & HOANG, Q. Q. 2011. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A, 108, 17797-802.WANG, Y. & MANDELKOW, E. 2012. Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans, 40, 644-52.WANG, Y. & MANDELKOW, E. 2016. Tau in physiology and pathology. Nat Rev Neurosci, 17, 5-21.WANG, Y., MARTINEZ-VICENTE, M., KRUGER, U., KAUSHIK, S., WONG, E., MANDELKOW, E. M., CUERVO, A. M. & MANDELKOW, E. 2009b. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet, 18, 4153-70.WANG, Y. P., BIERNAT, J., PICKHARDT, M., MANDELKOW, E. & MANDELKOW, E. M. 2007b. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc Natl Acad Sci U S A, 104, 10252-7.WAUTERS, M., WATTIEZ, R. & RIS, L. 2016. Internalization of the Extracellular Full-Length Tau Inside Neuro2A and Cortical Cells Is Enhanced by Phosphorylation. Biomolecules, 6.WAXMAN, E. A. & GIASSON, B. I. 2011. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci, 31,7604-18.WEBB, J. L., RAVIKUMAR, B., ATKINS, J., SKEPPER, J. N. & RUBINSZTEIN, D. C. 2003. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem, 278, 25009-13.WEINGARTEN, M., LOCKWOOD, A., HWO, S. & KIRSCHNER, M. 1975. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A, 72, 1858-62.WEINREB, P. H., ZHEN, W., POON, A. W., CONWAY, K. A. & LANSBURY, P. T., JR. 1996. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry, 35,13709-15.

109
WEISGRABER, K., RALL, S. J. & MAHLEY, R. 1981. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J Biol Chem, 256, 9077-83.WES, P. D., SAYED, F. A., BARD, F. & GAN, L. 2016. Targeting microglia for the treatment of Alzheimer's Disease. Glia, 64, 1710-32.WIENKEN, C. J., BAASKE, P., ROTHBAUER, U., BRAUN, D. & DUHR, S. 2010. Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun, 1, 100.WILLIAMS, R. S., CHENG, L., MUDGE, A. W. & HARWOOD, A. J. 2002. A common mechanism of action for three mood-stabilizing drugs. Nature, 417, 292-5.WILLIAMSON, R., SCALES, T., CLARK, B., GIBB, G., REYNOLDS, C., KELLIE, S., BIRD, I., VARNDELL, I., SHEPPARD, P., EVERALL, I. & ANDERTON, B. 2002. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: involvement of Src family protein kinases. J Neurosci, 22, 10-20.WIMO, A., GUERCHET, M., ALI, G. C., WU, Y. T., PRINA, A. M., WINBLAD, B., JONSSON, L., LIU, Z. & PRINCE, M. 2016. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimers Dement.WINNER, B., JAPPELLI, R., MAJI, S. K., DESPLATS, P. A., BOYER, L., AIGNER, S., HETZER, C., LOHER, T., VILAR, M., CAMPIONI, S., TZITZILONIS, C., SORAGNI, A., JESSBERGER, S., MIRA, H., CONSIGLIO, A., PHAM, E., MASLIAH, E., GAGE, F. H. & RIEK, R. 2011. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A, 108, 4194-9.WISCHIK, C. M., EDWARDS, P. C., LAI, R. Y., GERTZ, H. N., XUEREB, J. H., PAYKEL, E. S., BRAYNE, C., HUPPERT, F. A., MUKAETOVA-LADINSKA, E. B. & MENA, R. 1995. Quantitative analysis of tau protein in paired helical filament preparations: Implications for the role of tau protein phosphorylation in PHF assembly in Alzheimer’s disease. . Neurobiol. Aging, 409-417, discussion 418–431.WISCHIK, C. M., EDWARDS, P. C., LAI, R. Y., ROTH, M. & HARRINGTON, C. R. 1996. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A, 93,11213-8.WISCHIK, C. M., NOVAK, M., EDWARDS, P. C., KLUG, A., TICHELAAR, W. & CROWTHER, R. A. 1988a. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A, 85, 4884-8.WISCHIK, C. M., NOVAK, M., THOGERSEN, H. C., EDWARDS, P. C., RUNSWICK, M. J., JAKES, R., WALKER, J. E., MILSTEIN, C., ROTH, M. & KLUG, A. 1988b. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A, 85,4506-10.WISCHIK, C. M., STAFF, R. T., WISCHIK, D. J., BENTHAM, P., MURRAY, A. D., STOREY, J. M., KOOK, K. A. & HARRINGTON, C. R. 2015. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer's disease. J Alzheimers Dis, 44, 705-20.WISEMAN, F., ALFORD, K., TYBULEWICZ, V. & FISHER, E. 2009. Down syndrome--recent progress and future prospects. Hum Mol Genet, 18, R75-83.WOBST, H. J., SHARMA, A., DIAMOND, M. I., WANKER, E. E. & BIESCHKE, J. 2015. The green tea polyphenol (-)-epigallocatechin gallate prevents the aggregation of tau protein into toxic oligomers at substoichiometric ratios. FEBS Lett, 589, 77-83.WOLF, G. & STAHL, R. 2003. CD2-associated protein and glomerular disease. Lancet, 362, 1746-8.WOLOZIN, B. 2012. Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener, 7, 56.WOLOZIN, B. & APICCO, D. 2015. RNA binding proteins and the genesis of neurodegenerative diseases. Adv Exp Med Biol, 822, 11-5.WU, J. W., HERMAN, M., LIU, L., SIMOES, S., ACKER, C. M., FIGUEROA, H., STEINBERG, J. I., MARGITTAI, M., KAYED, R., ZURZOLO, C., DI PAOLO, G. & DUFF, K. E. 2013. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem, 288, 1856-70.

110
WU, J. W., HUSSAINI, S. A., BASTILLE, I. M., RODRIGUEZ, G. A., MREJERU, A., RILETT, K., SANDERS, D. W., COOK, C., FU, H., BOONEN, R. A., HERMAN, M., NAHMANI, E., EMRANI, S., FIGUEROA, Y. H., DIAMOND, M. I., CLELLAND, C. L., WRAY, S. & DUFF, K. E. 2016. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci, 19, 1085-92.XIAO, Q., GIL, S., YAN, P., WANG, Y., HAN, S., GONZALES, E., PEREZ, R., CIRRITO, J. & LEE, J. 2012. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J Biol Chem,287, 21279-89.XILOURI, M., VOGIATZI, T., VEKRELLIS, K., PARK, D. & STEFANIS, L. 2009. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One, 4, e5515.YAMADA, K., HOLTH, J., LIAO, F., STEWART, F., MAHAN, T., JIANG, H., CIRRITO, J., PATEL, T., HOCHGRAFE, K., MANDELKOW, E. & HOLTZMAN, D. 2014. Neuronal activity regulates extracellular tau in vivo. J Exp Med.YAMAMOTO, H., YAMAUCHI, E., TANIGUCHI, H., ONO, T. & MIYAMOTO, E. 2002. Phosphorylation of microtubule-associated protein tau by Ca2+/calmodulin-dependent protein kinase II in its tubulin binding sites. Arch Biochem Biophys, 408, 255-62.YAN, X. 2013. A novel approach for studying α- synuclein oligomerization in living cell. Master Thesis, https://1drv.ms/b/s!AtkIVetzEE3gsHkD7KoegcwM6P64.YANAMANDRA, K., KFOURY, N., JIANG, H., MAHAN, T. E., MA, S., MALONEY, S. E., WOZNIAK, D. F., DIAMOND, M. I. & HOLTZMAN, D. M. 2013. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron, 80,402-14.YSSELSTEIN, D., JOSHI, M., MISHRA, V., GRIGGS, A. M., ASIAGO, J. M., MCCABE, G. P., STANCIU, L. A., POST, C. B. & ROCHET, J. C. 2015. Effects of impaired membrane interactions on alpha-synuclein aggregation and neurotoxicity. Neurobiol Dis, 79, 150-63.YU, W. H., DORADO, B., FIGUEROA, H. Y., WANG, L., PLANEL, E., COOKSON, M. R., CLARK, L. N. & DUFF, K. E. 2009. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol, 175, 736-47.YUYAMA, K., SUN, H., MITSUTAKE, S. & IGARASHI, Y. 2012. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J Biol Chem, 287, 10977-89.ZANE JAUNMUKTANE & SIMON MEAD, M. E., JONATHAN D. F. WADSWORTH, ANDREW J. NICOLL, JOANNA KENNY, FRANCESCA LAUNCHBURY, JACQUELINE LINEHAN, ANGELA RICHARD-LOENDT, A. SARAH WALKER, PETER RUDGE, JOHN COLLINGE & SEBASTIAN BRANDNER 2015. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature, 525
247-250.ZANNIS, V., BRESLOW, J., UTERMANN, G., MAHLEY, R., WEISGRABER, K., HAVEL, R., GOLDSTEIN, J., BROWN, M., SCHONFELD, G., HAZZARD, W. & BLUM, C. 1982. Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes. J Lipid Res, 23, 911-4.ZARRANZ, J. J., ALEGRE, J., GOMEZ-ESTEBAN, J. C., LEZCANO, E., ROS, R., AMPUERO, I., VIDAL, L., HOENICKA, J., RODRIGUEZ, O., ATARES, B., LLORENS, V., GOMEZ TORTOSA, E., DEL SER, T., MUNOZ, D. G. & DE YEBENES, J. G. 2004. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol, 55, 164-73.ZEINEDDINE, R., PUNDAVELA, J. F., CORCORAN, L., STEWART, E. M., DO-HA, D., BAX, M., GUILLEMIN, G., VINE, K. L., HATTERS, D. M., ECROYD, H., DOBSON, C. M., TURNER, B. J., OOI, L., WILSON, M. R., CASHMAN, N. R. & YERBURY, J. J. 2015. SOD1 protein aggregates stimulate macropinocytosis in neurons to facilitate their propagation. Mol Neurodegener, 10, 57.ZEINEDDINE, R. & YERBURY, J. J. 2015. The role of macropinocytosis in the propagation of protein aggregation associated with neurodegenerative diseases. Front Physiol, 6, 277.

111
ZEMPEL, H., LUEDTKE, J., KUMAR, Y., BIERNAT, J., DAWSON, H., MANDELKOW, E. & MANDELKOW, E. M. 2013. Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J, 32, 2920-37.ZHAI, J., ZHANG, L., MOJSILOVIC-PETROVIC, J., JIAN, X., THOMAS, J., HOMMA, K., SCHMITZ, A., FAMULOK, M., ICHIJO, H., ARGON, Y., RANDAZZO, P. A. & KALB, R. G. 2015. Inhibition of Cytohesins Protects against Genetic Models of Motor Neuron Disease. J Neurosci, 35,9088-105.ZHANG, Y., LI, H., WANG, D., LIU, S. & WANG, J. 2006. A transitory activation of protein kinase-Ainduces a sustained tau hyperphosphorylation at multiple sites in N2a cells-imply a new mechanism in Alzheimer pathology. J Neural Transm, 113, 1487-97.ZHANG, Z., SONG, M., LIU, X., KANG, S. S., KWON, I. S., DUONG, D. M., SEYFRIED, N. T., HU, W. T., LIU, Z., WANG, J. Z., CHENG, L., SUN, Y. E., YU, S. P., LEVEY, A. I. & YE, K. 2014. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer's disease. Nat Med, 20, 1254-62.ZHOU, Y., GU, G., GOODLETT, D. R., ZHANG, T., PAN, C., MONTINE, T. J., MONTINE, K. S., AEBERSOLD, R. H. & ZHANG, J. 2004. Analysis of alpha-synuclein-associated proteins by quantitative proteomics. J Biol Chem, 279, 39155-64.ZHU, M. & FINK, A. L. 2003. Lipid binding inhibits alpha-synuclein fibril formation. J Biol Chem,278, 16873-7.ZHU, M., LI, J. & FINK, A. L. 2003. The association of alpha-synuclein with membranes affects bilayer structure, stability, and fibril formation. J Biol Chem, 278, 40186-97.ZILKA, N., FILIPCIK, P., KOSON, P., FIALOVA, L., SKRABANA, R., ZILKOVA, M., ROLKOVA, G., KONTSEKOVA, E. & NOVAK, M. 2006. Truncated tau from sporadic Alzheimer's disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett, 580, 3582-8.


New Insight into Mechanisms of Transcellular Propagation of Tau and α-Synuclein in Neurodegenerative Diseases
XU YAN
dissertationes scholae doctoralis ad sanitatem investigandam universitatis helsinkiensis 7/2017
7/2017Helsinki 2017 ISSN 2342-3161 ISBN 978-951-51-2865-2
XU
YA
N N
ew In
sight into M
echanism
s of Transcellular P
ropagation of Tau an
d α-Synuclein
in N
eurodegenerative D
iseases
Recent Publications in this Series
77/2016 Aino SalonsalmiAlcohol Drinking, Health-Related Functioning and Work Disability78/2016 Heini WennmanPhysical Activity, Sleep and Cardiovascular Diseases: Person-oriented and Longitudinal Perspectives79/2016 Andres LõhmusHelper Component-Proteinase and Coat Protein are Involved in the Molecular Processes of Potato Virus A Translation and Replication80/2016 Li MaBrain Immune Gene Network in Inbred Mouse Models of Anxiety- and Sociability-related Neuropsychiatric Disorders81/2016 Finny S. VargheseCracking the Code of Chikungunya Virus: Inhibitors as Tools to Explore Alphavirus Biology82/2016 Vera ShirokovaTranscription Factor Foxi3 in Hair Follicle Development and Homeostasis83/2016 Daria BulanovaNovel Genetic Determinants of Breast Cancer Progression84/2016 Hugh ChapmanThe hERG1 (KV11.1) Potassium Channel: Its Modulation and the Functional Characterisation of Genetic Variants85/2016 Katja RostiExpression and Characterization of Neuronal Membrane Receptor Proteins86/2016 Irepan Salvador-MartínezEstimating Complexity and Adaptation in the Embryo: A Statistical Developmental Biology Approach87/2016 Vigneshwari SubramanianField-based Proteochemometric Models Derived from 3D Protein Structures: A Novel Approach to Visualize Affinity and Selectivity Features88/2016 Anita LampinenSignalling and Expression of the Ang-Tie Pathway in Tumor Vasculature1/2017 Essi HavulaTranscriptional Control of Dietary Sugar Metabolism and Homeostasis by Mondo-Mlx Transcription Factors2/2017 Satu MassinenSpecific Reading Disorder: Cellular and Neurodevelopmental Functions of Susceptibility Genes3/2017 Margarita AndreevskayaEcological Fitness and Interspecies Interactions of Food-Spoilage-Associated Lactic Acid Bacteria: Insights from the Genome Analyses and Transcriptome Profiles4/2017 Mikko SiuralaImproving Adenovirus-Based Immunotherapies for Treatment of Solid Tumors5/2017 Inken KörberMicroglial Dysfunction in Cstb-/- Mice, a Model for the Neurodegenerative Disorder Progressive Myoclonus Epilepsy of Unverricht-Lundborg Type, EPM16/2017 Shrikanth KulashekharThe Role of Cortical Oscillations in the Estimation and Maintenance of Sensory and Duration Information in Working Memory
NEUROSCIENCE CENTER AND DEPARTMENT OF BIOSCIENCESFACULTY OF BIOLOGICAL AND ENVIRONMENTAL SCIENCESDOCTORAL PROGRAMME BRAIN & MINDUNIVERSITY OF HELSINKI





![Skin Diseases Expert System using Dempster- Shafer … · Skin Diseases Expert System using Dempster-Shafer Theory ... was coined by J. A. Barnett [8] ... {Θ} = 1 - 0.3 = 0.7 TABEL](https://static.fdocument.org/doc/165x107/5afc38da7f8b9a44659153ed/skin-diseases-expert-system-using-dempster-shafer-diseases-expert-system-using.jpg)












