PPARγγγ antagonist Gleevec improves insulin...
40
PPARγ antagonist Gleevec improves insulin sensitivity and promotes the browning of white adipose tissue Running title : Gleevec blocks PPARγ phosphorylation at Ser273 Sunsil Choi a , Eun-Sun Kim a , Ji-Eun Jung a , David P. Marciano b , Ala Jo c , Ja Young Koo c , Soo Youn Choi a , Yong Ryoul Yang a , Hyun-Jun Jang a , Eung-Kyun Kim a , Jiyoung Park a , Hyug Moo Kwon a , In Hee Lee d , Seung Bum Park c, e , Kyung-Jae Myung f , Pann-Ghill Suh a , Patrick R. Griffin b , Jang Hyun Choi a,* a Department of Biological Sciences, Ulsan National Institute of Science and Technology (UNIST), Ulsan, 689-798, Korea b Department of Molecular Therapeutics, The Scripps Research Institute, Scripps Florida, Jupiter FL, USA c Department of Chemistry, Seoul National University, Seoul, 151-747, Korea d Department of Medical Chemistry, Hyundai Pharm Co., Ltd., Suwon, Gyonggi 443-270, Korea e Department of Biophysics and Chemical Biology/N-Bio institute, Seoul National University, Seoul, 151-747, Korea f Center for Genomic Integrity (CGI), Institute for Basic Science (IBS), Department of Biological Sciences, Ulsan National Institute of Science and Technology (UNIST), Ulsan, 689-798, Korea Page 2 of 40 Diabetes Diabetes Publish Ahead of Print, published online January 6, 2016
Transcript of PPARγγγ antagonist Gleevec improves insulin...

PPARγγγγ antagonist Gleevec improves insulin sensitivity and promotes
the browning of white adipose tissue
Running title : Gleevec blocks PPARγγγγ phosphorylation at Ser273
Sunsil Choia, Eun-Sun Kim
a, Ji-Eun Jung
a, David P. Marciano
b, Ala Jo
c, Ja Young Koo
c,
Soo Youn Choia, Yong Ryoul Yang
a, Hyun-Jun Jang
a, Eung-Kyun Kim
a, Jiyoung Park
a,
Hyug Moo Kwona, In Hee Lee
d, Seung Bum Park
c, e, Kyung-Jae Myung
f, Pann-Ghill
Suha, Patrick R. Griffin
b, Jang Hyun Choi
a,*
a Department of Biological Sciences, Ulsan National Institute of Science and Technology
(UNIST), Ulsan, 689-798, Korea
b Department of Molecular Therapeutics, The Scripps Research Institute, Scripps Florida,
Jupiter FL, USA
c Department of Chemistry, Seoul National University, Seoul, 151-747, Korea
d Department of Medical Chemistry, Hyundai Pharm Co., Ltd., Suwon, Gyonggi 443-270,
Korea
e Department of Biophysics and Chemical Biology/N-Bio institute, Seoul National University,
Seoul, 151-747, Korea
f Center for Genomic Integrity (CGI), Institute for Basic Science (IBS), Department of
Biological Sciences, Ulsan National Institute of Science and Technology (UNIST), Ulsan,
689-798, Korea
Page 2 of 40Diabetes
Diabetes Publish Ahead of Print, published online January 6, 2016

* Corresponding author
Prof. Jang Hyun Choi
Department of Biological Sciences, Ulsan National Institute of Science and Technology
(UNIST), Ulsan, 689-798, Korea
Phone : 82-52-217-2543
Fax : 82-52-217-5219
e-mail : [email protected]
Words: 3999; Figure: 6; Supplementary Figure: 9
Page 3 of 40 Diabetes

Abstract
Blocking phosphorylation of peroxisome proliferator-activated receptor γ (PPARγ)
at Ser273 is one of the key mechanisms for anti-diabetic drugs to target PPARγ. Using high
throughput phosphorylation screening, we here describe that Gleevec blocks CDK5-mediated
PPARγ phosphorylation devoid of classical agonism as a PPARγ antagonist ligand. In high
fat-fed mice, Gleevec improved insulin sensitivity without causing severe side effects
associated with other PPARγ-targeting drugs. Furthermore, Gleevec reduces lipogenic and
gluconeogenic gene expression in liver and ameliorates inflammation in adipose tissues.
Interestingly, Gleevec increases browning of white adipose tissue (WAT) and energy
expenditure. Taken together, Gleevec exhibits greater beneficial effects on both glucose/lipid
metabolism and energy homeostasis by blocking PPARγ phosphorylation. These data
illustrate that Gleevec could be a novel therapeutic agent for use in insulin resistance and type
2 diabetes.
Page 4 of 40Diabetes

As the prevalence of obesity has exploded over the last several decades, associated
metabolic disorders, including type 2 diabetes, dyslipidemia, hypertension, and
cardiovascular diseases, have also increased dramatically. As PPARγ agonists,
thiazolidinediones (TZDs), which include pioglitazone, represent synthetic insulin-sensitizing
drugs that have been widely prescribed for the treatment of type 2 diabetes (1, 2). However,
the use of TZDs is associated with unwanted side effects, including weight gain, fluid
retention, bone fracture, cardiovascular disease, and bladder cancer (3-7). Thus, the United
States Food and Drug Administration (FDA) recently restricted the use of one TZD,
rosiglitazone, for the treatment of type 2 diabetes.
PPARγ is a master regulator of adipocyte differentiation, glucose and lipid
metabolism, and inflammation (8-10). Recently, we demonstrated that phosphorylation of
PPARγ at Ser273 (pS273) is linked to obesity and insulin resistance (11). Phosphorylation
does not globally alter its transcriptional activity, but dysregulates a specific set of genes with
roles in obesity and diabetes (11, 12). Moreover, both TZDs and SPPARMs inhibit cyclin-
dependent kinase 5 (CDK5)-mediated PPARγ phosphorylation at Ser273 (11-13). More
specifically, non-agonist PPARγ ligands (SR1664 or UHC1) are anti-diabetic and have
reduced signals of undesirable side effects caused by TZDs (12, 13). These observations
indicate that blocking pS273 without classical agonism is an important mechanism to
consider in the development of novel anti-diabetic drugs targeting PPARγ.
In the present study, we screened a chemical library for compounds that inhibit
pS273 in vitro, and found that Gleevec, a well-known anti-cancer drug, blocked PPARγ
phosphorylation as a PPARγ ligand without classical agonism. Gleevec improved insulin
sensitivity without the commonly observed side effects of TZDs in mice fed a high-fat diet
(HFD). Furthermore, it negatively regulated pro-inflammatory responses and glucose
Page 5 of 40 Diabetes

production in white adipose tissue (WAT) and liver, respectively. Importantly, it increased
energy expenditure by regulating a thermogenic program in subcutaneous WAT (sWAT),
resulting in anti-obesity effects. Our results demonstrate that Gleevec is a potent therapeutic
agent for both diabetes and obesity.
Research Design and Methods
Cell Culture
3T3-L1, HEK-293 and Raw264.7 cells were obtained from ATCC (VA, USA) and
cultured in Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum.
FLAG-PPARγWT
and FLAG-PPARγS273A
were subcloned into pMSCV-puro retroviral vector
(Agilent Tech., CA, USA). Adipocyte differentiation of 3T3-L1 or mouse embryonic
fibroblast (MEFs) expressing PPARγWT
or PPARγS273A
were induced differentiation as
previously described (11). Fully differentiated 3T3-L1 and MEFs or Raw264.7 cells were
preincubated with Gleevec for 24 h and treated with TNF-α (50 ng/ml) for 3 h or LPS (10
ng/ml) for 6 h, respectively. All chemicals for cell culture were obtained from Sigma (MO,
USA) unless otherwise indicated.
In vitro kinase assay
Active Cdk5/p35 or ERK was purchased from Millipore. In vitro kinase assay was
performed as previously described (11). Briefly, 0.5 µg of recombinant PPARγ (Cayman
Chem., MI, USA) were incubated with active CDK or ERK kinases in kinase assay buffer (25
mM Tris-HCl pH 7.5, 5 mM beta-glycerophosphate, 2 mM dithiothreitol (DTT), 0.1 mM
Na3VO4, 10 mM MgCl2) containing 10 µM ATP for 15 min at 30°C. Rb (Cell Signaling
Technology, MA, USA) was used as a positive control.
Page 6 of 40Diabetes

Immunoprecipitation and immunoblotting
HEK-293 cells expressing PPARγ were treated with TNF-α (50 ng/ml) and total cell
lysates were incubated with FLAG M2 agarose (Sigma-Aldrich, MO, USA) at 4°C.
Immunoprecipitates and total cell lysates were analyzed with phospho-specific antibody
against ser273 (11) or anti-PPARγ antibody (Santa Cruz, TX, USA).
Primary hepatocytes isolation and glucose production assay
Primary mouse hepatocytes were isolated by the two-step collagenase perfusion
method from male C57BL/6 after HFD as previously described (14). Primary hepatocytes
were plated and treated with Gleevec for 24 h following to treat forskolin (10 µM) for 6 h.
The glucose concentration in the media were measured by glucose assay kit (Sigma-Aldrich,
MO, USA)
Gene expression analysis
Total RNA was isolated from cells or tissues using Trizol reagents (Invitrogen, CA,
USA). The RNA was reverse-transcribed using ABI reverse transcription kit. Quantitative
PCR reactions were performed with SYBR green fluorescent dye using an ABI9300 PCR
machine. Relative mRNA expression was determined by the ∆∆-Ct method normalized to
TATA-binding protein (TBP) levels.
Reporter gene assay
Page 7 of 40 Diabetes

HEK-293 cells were transfected with pDR-1 luciferase reporter plasmid, PPARγ,
PPARα or PPARδ, RXRα and pRL-renillia, respectively (Invitrogen, CA, USA). Reporter
gene assay was performed as previous described (11).
Animals
All animal experiments were performed according to procedures approved by Ulsan
National Institute of Science and Technology’s Institutional Animal Care and Use Committee.
5-week-old male C57BL/6J mice (DBL, Korea) were fed a high fat diet (60% kcal fat,
D12492, Research Diets Inc., NJ, USA) for 10 weeks. After 7-day-intraperitoneal (i.p.)
injection with Gleevec (25 mg/kg; the human equivalent dose would be around1500mg/daily)
or vehicle mice were injected with D-glucose (1.5 g/kg body weight) after overnight
starvation or human insulin (0.75 U/kg body weight) after 6 h starvation for glucose tolerant
tests (GTTs) or insulin tolerance tests (ITTs), respectively. To determine the energy
expenditure and inflammation, mice were injected daily 20 mg/kg of Gleevec or vehicle for 3
weeks. Oxygen consumption, carbon dioxide production, and food intake were measured by
the Comprehensive Laboratory Animal Monitoring System (CLAMS™; Columbus
Instruments, OH, USA) and body temperatures were measured rectally using digital
thermometer. Blood glucose levels were determined using tail blood and glucometer. Serum
insulin (Crystal Chem., IL, USA) and serum cholesterol, TG and FFAs were determined by
ELISA (Cayman Chem., MI, USA & Millipore, MA, USA).
Statistical analysis
Data are presented as means ± standard errors of the means (SEMs) as indicated in
the figure legends. Comparisons between two groups were made by unpaired two-tailed
Page 8 of 40Diabetes

Student's t tests. P values of <0.05 were considered statistically significant. Microsoft Excel
was used for statistical calculations.
Results
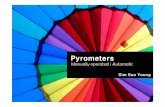
Gleevec blocks CDK5-mediated PPARγ phosphorylation at Ser273
To identify alternative non-agonist PPARγ ligands that block CDK5-mediated
PPARγ phosphorylation at Ser273, chemical screening was performed using an in vitro
kinase assay with 780 FDA-approved drugs. Among the positive candidates, we noted with
interest that Gleevec, a potent-anti-cancer drug (Fig. 1A), blocked pS273. Although the
beneficial effects of Gleevec on glucose metabolism have been demonstrated previously, the
molecular and cellular mechanisms remain unclear. Therefore, we focused on the molecular
mechanisms how Gleevec regulates glucose/fat metabolism. As shown in Fig. 1B, Gleevec
inhibited PPARγ phosphorylation in a dose-dependent manner, and its inhibitory effect was
similar to that of rosiglitazone at 10 µM. However, it does not inhibit the CDK5-mediated
phosphorylation of retinoblastoma (Rb), another CDK5 substrate (Fig. 1C) (11). Furthermore,
Gleevec blocked ERK-mediated PPARγ phosphorylation, indicating that Gleevec directly
targets PPARγ and inhibits pS273 regardless of the kinases (15) (Supplementary Fig. 1). In
human embryonic kidney 293 (HEK293) cells expressing PPARγ, Gleevec significantly
inhibited TNF-α-mediated PPARγ phosphorylation (Fig. 1D). Because Gleevec has been
widely used for the treatment of chronic myelogenous leukemia (CML) by specifically
targeting BCR-Abl tyrosine kinase, we tested whether other BCR-Abl tyrosine kinase
inhibitors block PPARγ phosphorylation. As shown in Fig. 1E, while nilotinib, dasatinib, and
sunitinib (Fig. 1A) inhibited pS273 in a similar manner as that of Gleevec, they also blocked
Page 9 of 40 Diabetes

Rb phosphorylation by CDK5, indicating that only Gleevec targets PPARγ. These results
suggest that Gleevec specifically blocks pS273, independent of its BCR-Abl targeting.
Gleevec is a PPARγ ligand that lacks classical agonism
Next, a LanthaScreen TR-FRET competitive binding assay was performed to assess
whether Gleevec directly binds PPARγ. The half-maximum inhibitory concentration (IC50) of
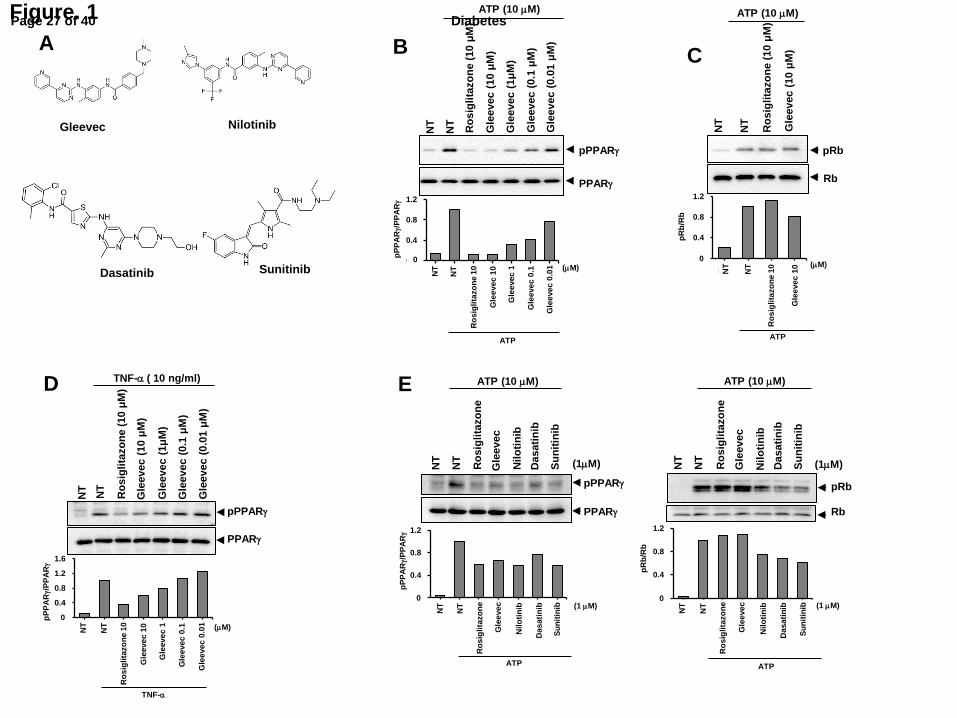
Gleevec (1-3 µM) was higher than that of rosiglitazone (0.1µM) (Fig. 2A). Gleevec did not
induce transcriptional activity of PPARγ at any concentration tested (Fig. 2B). A coactivator
recruitment assay showed that Gleevec impaired recruitment of the CBP coactivator to
PPARγ, which was recruited to PPARγ in the presence of rosiglitazone (Fig. 2D).
Furthermore, Gleevec did not transcriptionally activate either PPARα or PPARδ
(Supplementary Fig. 2).
It has been reported that full agonist ligands form a hydrogen bond with tyrosine 473
on PPARγ H12; this interaction stabilizes the agonist conformation and allows H12 to serve
as the coactivator binding site (AF-2 surface) (16). Therefore, the stability of the AF2 surface
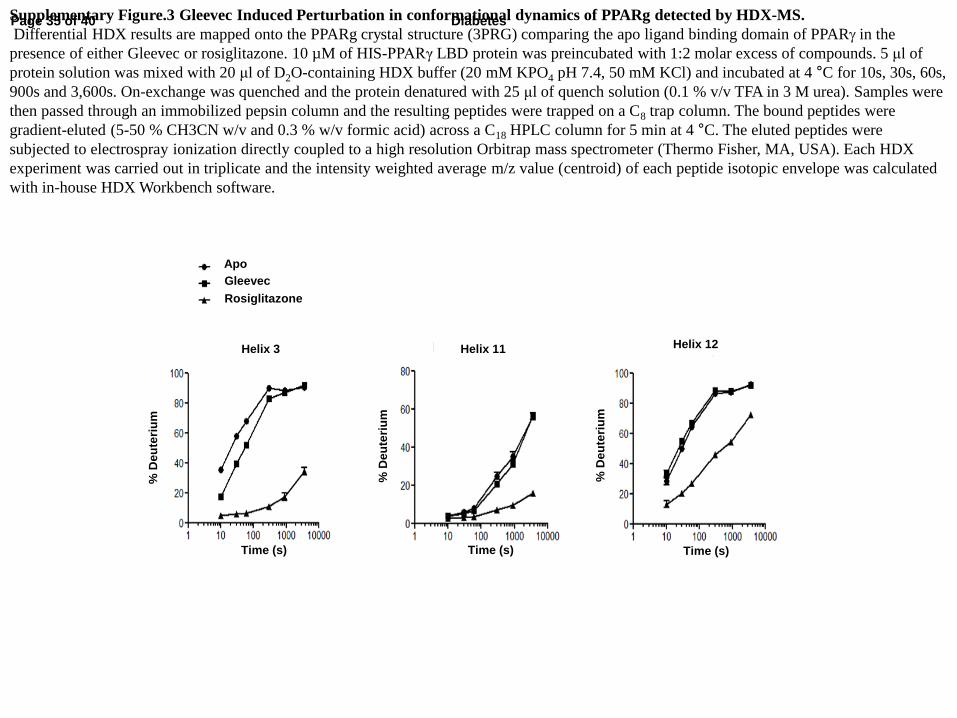
is an important determinant for the transactivation of PPARγ. In hydrogen/deuterium
exchange (HDX) analyses of the PPARG LBD, Gleevec binding induced no change in H12
conformational dynamics, in contrast to rosiglitazone that strongly stabilized the same region
(Fig. 2C and Supplementary Fig. 3). This suggests that the conformational stability of H12,
unaffected by Gleevec is consistent with the observed absence of transcriptional activity (Fig.
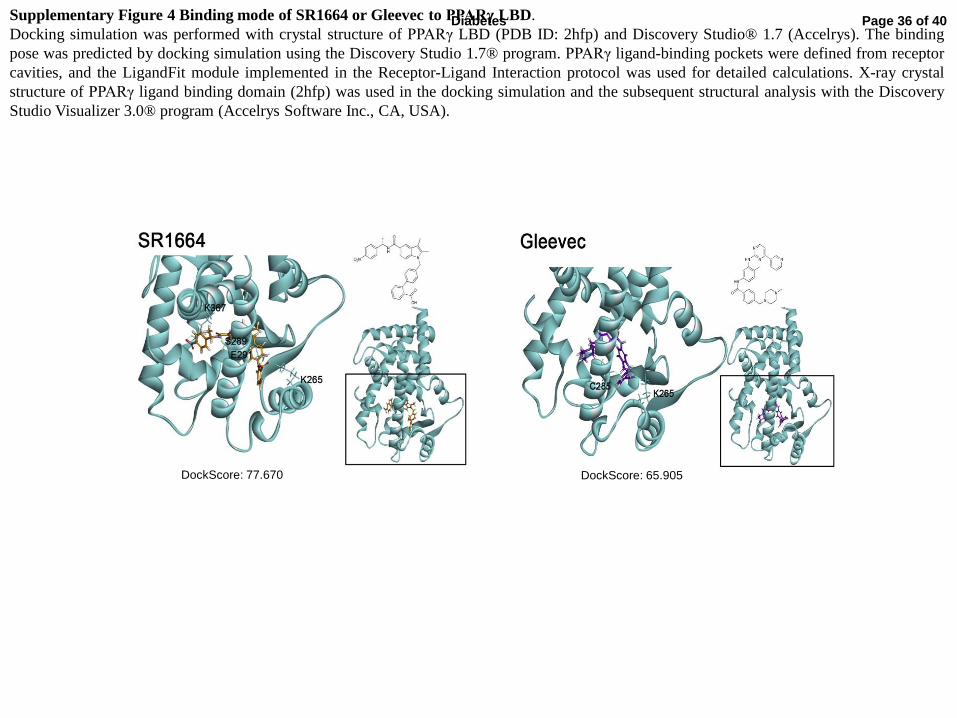
2B) and coactivator recruitment (Fig. 2D). Next, we compared the in silico docking
simulations of Gleevec and SR1664 (Supplementary Fig. 4). The docking score for the LBD
of PPARγ revealed that Gleevec fits in a manner similar to that of SR1664 in the proper
binding mode.
Page 10 of 40Diabetes

To further determine whether Gleevec lacks classical agonism, we tested its effects on
adipocyte differentiation (8, 9). As shown in Fig. 2E, rosiglitazone dramatically stimulated
adipocyte differentiation of fibroblasts (pre-adipocytes), whereas Gleevec did not increase
lipid accumulation. Moreover, the expression of adipogenic markers was also increased by
rosiglitazone, but not by Gleevec (Fig. 2F). Then, we tested the ability of Gleevec to regulate
gene expression in fully differentiated adipocytes (Fig. 2G). As shown in Fig. 2G, Gleevec
upregulated the expression of 11/17 (64%) diabetic genes dysregulated by pS273 in fully
differentiated adipocytes (Fig. 2G) (11). These data suggest that Gleevec is not a classical
transcriptional agonist of PPARγ, but specifically regulates pS273.
Gleevec improves insulin sensitivity in HFD-induced obese mice
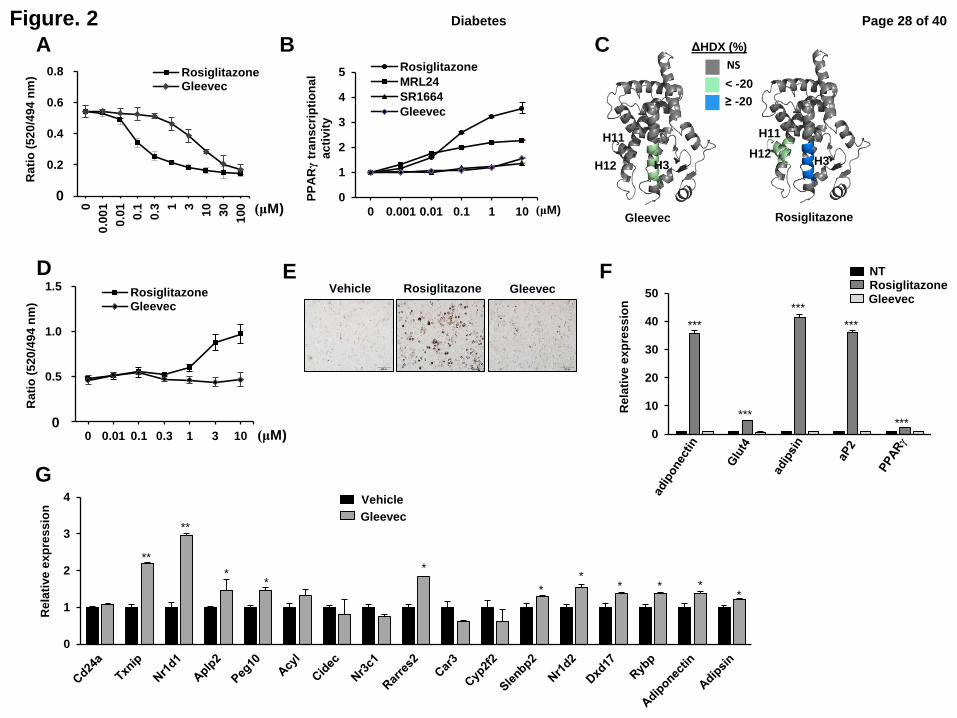
Next, we determined whether Gleevec exerts anti-diabetic properties in vivo. After
wild type C57BL/6 mice were fed an HFD for 10 weeks, glucose tolerance tests (GTTs) and
insulin tolerance tests (ITTs) were performed following treatment with 25 mg/kg/day
Gleevec for 7 days. Both GTTs and ITTs were markedly improved without affecting body
weight (Fig. 3A, B, E). Treatment with Gleevec in HFD-fed mice significantly decreased
pS273 (Fig. 3C). Furthermore, the potency of phosphorylation inhibition was positively
correlated with improved glucose tolerance (Fig. 3D). Control mice that received vehicle only
remained hyperinsulinemic, but Gleevec substantially reduced insulin levels in these mice
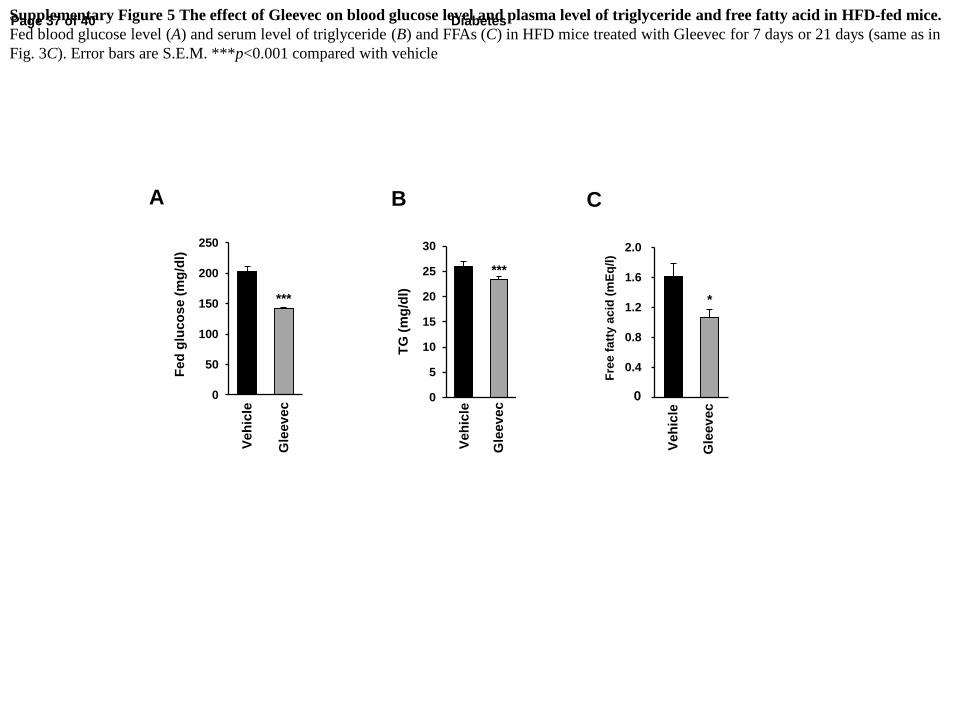
(Fig. 3F), and the level of fed blood glucose was significantly reduced (Supplementary Fig.
5A). Insulin resistance, as computed by HOMA-IR, showed a clear improvement following
treatment with Gleevec (Fig. 3G). In addition, serum triglyceride (TG) and free fatty acid
(FFA) levels significantly decreased in Gleevec-treated mice (Supplementary Fig. 5B-C).
Treatment with Gleevec also altered the expression of 9/17 (52.9%) diabetic genes
dysregulated by pS273, all in the direction predicted for inhibition of pS273 (Fig. 3H) (11).
Page 11 of 40 Diabetes

The expression of adiponectin seemed to be unaffected by treatment with Gleevec for 1 week,
but we observed the expressions of both adiponectin and adipsin were significantly increased
by treatment with Gleevec for 3 weeks in WAT (data not shown). This difference could be
caused by other environmental factors such as the communications with immune cells
because other diabetic genes dysregulated by phosphorylation of PPARγ did not exactly
matched in between adipocytes and adipose tissue after treating with Gleevec. Taken together,
these results indicate that Gleevec has a potent insulin-sensitizing effect with preferential
regulation of diabetic genes sensitive to pS273.
Gleevec results in significantly reduced common TZDs’ side effects
TZDs such as rosiglitazone can cause weight gain and fluid retention, all of which
contribute to increased cardiac dysfunction (3). They also increase the risk for bone fracture
by reducing bone formation and bone mineral density (4). As shown in Fig. 3I, treatment
human mesenchymal stem cells (hMSCs) with rosiglitazone reduced the expression of genes
involved in bone formation, including bone sialoprotein (Bsp), osteocalcin (Ocn), and osterix
(Osx). Importantly, Gleevec did not affect the expression of these gene sets (Fig. 3I). An
increase in body fat was also observed following treatment with rosiglitazone, but Gleevec
treatment did not cause any changes in either body fat or lean mass percentage (Fig. 3K, L).
As indicated in Fig. 3J, Gleevec had no detectable effect on hemodilution compared to either
vehicle or rosiglitazone treatment.
Next, we analyzed the expression of cardiac genes associated with heart failure or
hypertrophy. Cardiac hypertrophy is characterized by increased protein synthesis and
enlarged cardiomyocytes, leading to increased cardiac muscle mass (17). As shown in Fig.
3N, the expression of natriuretic peptide type B (Bnp), myosin heavy chain β (β-Mhc), and
nandrolone phenylpropionate (Npp) were increased in rosiglitazone-treated mice. However,
Page 12 of 40Diabetes

Gleevec did not alter their expression. Consistent with these results, HFD-fed mice treated
with rosiglitazone, but not Gleevec, showed increased heart weight (Fig. 3M). These results
strongly suggest that Gleevec greatly improves insulin sensitivity without the associated
adverse effects observed following treatment with most TZDs in vivo.
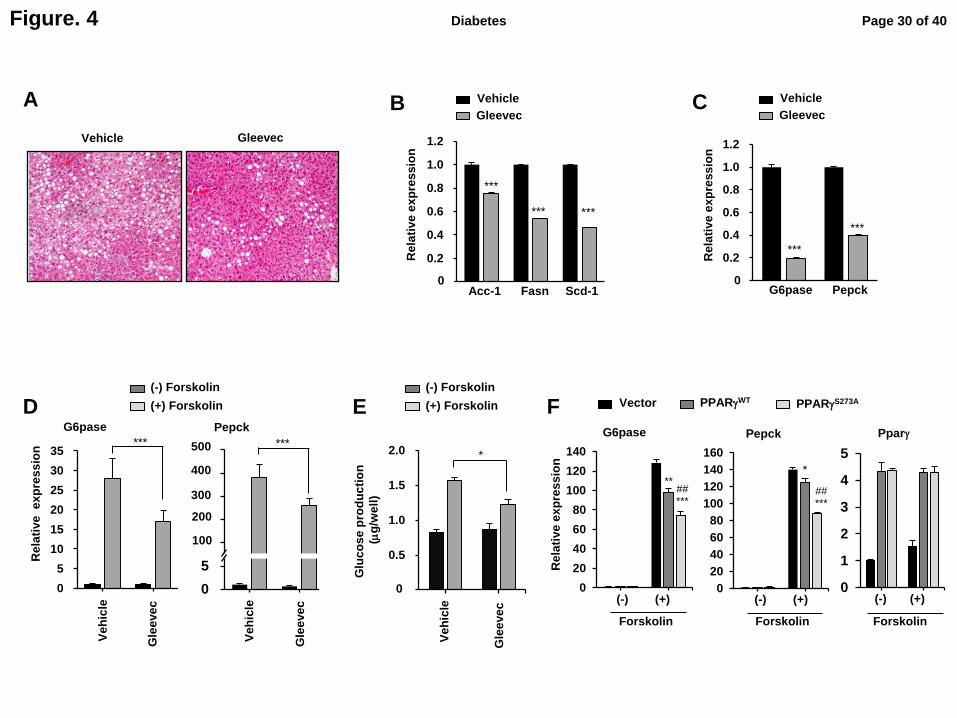
Gleevec ameliorates hepatic steatosis and reduces hepatic glucose production.
Obesity-induced insulin resistance is associated with fatty liver disease, and
dysregulation of hepatic glucose output greatly contributes to hyperglycemia in both humans
and mice. Histological observations revealed that Gleevec remarkably reduced hepatic
steatosis in HFD-induced obese mice (Fig. 4A). Consistent with hematoxylin and eosin (H&E)
staining, it also decreased the expression of hepatic lipogenic genes, including acetyl-CoA
carboxylase 1 (Acc-1), fatty acid synthase (Fasn), and stearoyl-CoA desaturase-1 (Scd-1) (Fig.
4B). Furthermore, Gleevec reduced the expression of gluconeogenic genes, such as glucose-
6-phosphatase (G6pase) and phosphoenolpyruvate carboxykinase (Pepck) in the livers of
HFD-fed mice (Fig. 4B, C).
Next, we examined whether the reduced expression of gluconeogenic genes by
Gleevec in vivo is a direct cell-autonomous effect. As shown in Fig. 4D and E, Gleevec
directly decreased the expression of G6pase and Pepck (Fig. 4D) and glucose production (Fig.
4E) in primary hepatocytes. We also determined whether pS273 plays a role hepatic
gluconeogenesis. When wild-type PPARγ (PPARγWT
) or a phosphorylation-deficient PPARγ
mutant (PPARγS273A
) was expressed in primary hepatocytes, PPARγS273A
suppressed the
expression of G6pase and Pepck genes more efficiently than PPARγWT
without altering the
expression of PPARγ itself (Fig. 4F). These results strongly suggest that Gleevec improves
hepatic steatosis and directly regulates hepatic glucose production in a pS273-dependent
manner.
Page 13 of 40 Diabetes

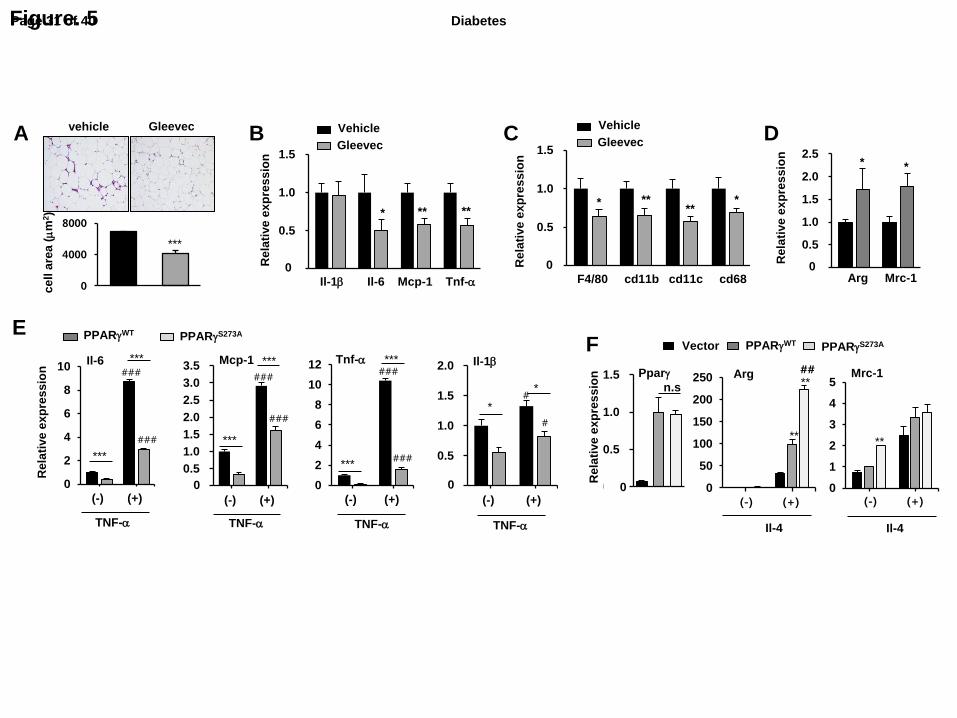
Gleevec ameliorates adipose tissue inflammation.
Many studies have reported that obesity is associated with a state of chronic low-
grade inflammation that facilitates the development of insulin resistance (18, 19). Therefore,
suppression of inflammation in adipose tissue may have therapeutic potential for the
treatment of enhanced inflammatory responses in obesity. Therefore, we determined whether
Gleevec suppresses chronic inflammation in the obese state. As shown in Fig. 5A,
histological sections of WAT in HFD-fed mice treated with Gleevec for 21 days had smaller
adipocytes than those of vehicle-treated mice; they also had fewer crown-like structures
formed by aggregated macrophages in adipose tissue (Fig. 5A) (20). To further investigate the
effects of Gleevec on inflammation in WAT, we assessed the expression of pro-inflammatory
and macrophage marker genes. As shown in Fig. 5B and C, pro-inflammatory genes such as
interleukin-6 (Il-6), monocyte-chemoattractant protein-1 (Mcp-1), and tumor necrosis factor-
α (Tnf-α) were significantly reduced in WAT following treatment with Gleevec whereas IL-
1β was not changed. Moreover, the expression of macrophage marker genes (F4/80, cd68,
and cd11b) and the M1 macrophage marker gene, cd11c, were also downregulated by
Gleevec. Interestingly, Gleevec-treated HFD-fed mice showed significantly increased
expression of M2 macrophage markers, including arginase (Arg) and mannose receptor (Mrc-
1) in WAT (Fig. 5D).
In previous study, we have reported that pS273 of PPARγ directly effects on
suppressing M1 macrophage activation (13). Thus we next investigated whether pS273 of
PPARγ is directly involved in anti-inflammatory activity in adipocytes and M2 polarization
in macrophages. Overexpression of PPARγS273A
in PPARγ-deficient mouse embryonic
fibroblast (MEF) cells significantly blocked pro-inflammatory gene expression compared to
that of PPARγWT
(Fig. 5E). In macrophages, overexpression of PPARγS273A
promoted
Page 14 of 40Diabetes

interleukin 4 (IL-4)-mediated M2 macrophage activation (Fig. 5F). Consistent with these
results, Gleevec significantly suppressed TNF-α-induced pro-inflammatory gene expression
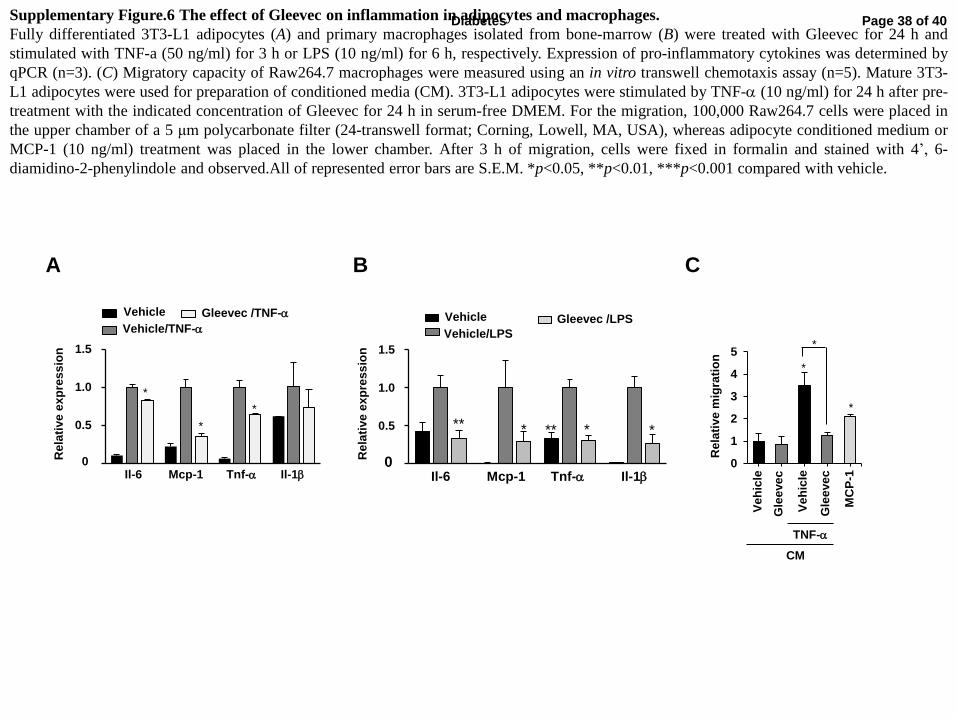
in both 3T3-L1 adipocytes and primary macrophages (Supplementary Fig. 6A, B).
Furthermore, in vitro transwell chemotaxis assay showed that macrophages had lower
chemotactic capacity toward conditioned media (CM) from Gleevec-treated 3T3-L1 cells
compared to vehicle-treated CM (Supplementary Fig. 6C). Taken together, these data indicate
that blocking pSer273 by Gleevec causes decreased macrophage inflammation and
infiltration to WAT by substantially suppressing the pro-inflammatory the pro-inflammatory
responses in adipocytes, thus ameliorating adipose inflammation.
Gleevec increases energy expenditure and adaptive thermogenesis.
Recent studies have shown that PPARγ ligands affect energy balance by promoting
browning of WAT (21). Therefore, we investigated whether Gleevec regulates energy
expenditure using comprehensive lab animal monitoring system (CLAMS). As shown in Fig.
6A and B, we observed a highly significant increase in energy expenditure in Gleevec-treated
mice compared to vehicle-treated controls. Importantly, there was no change in respiratory
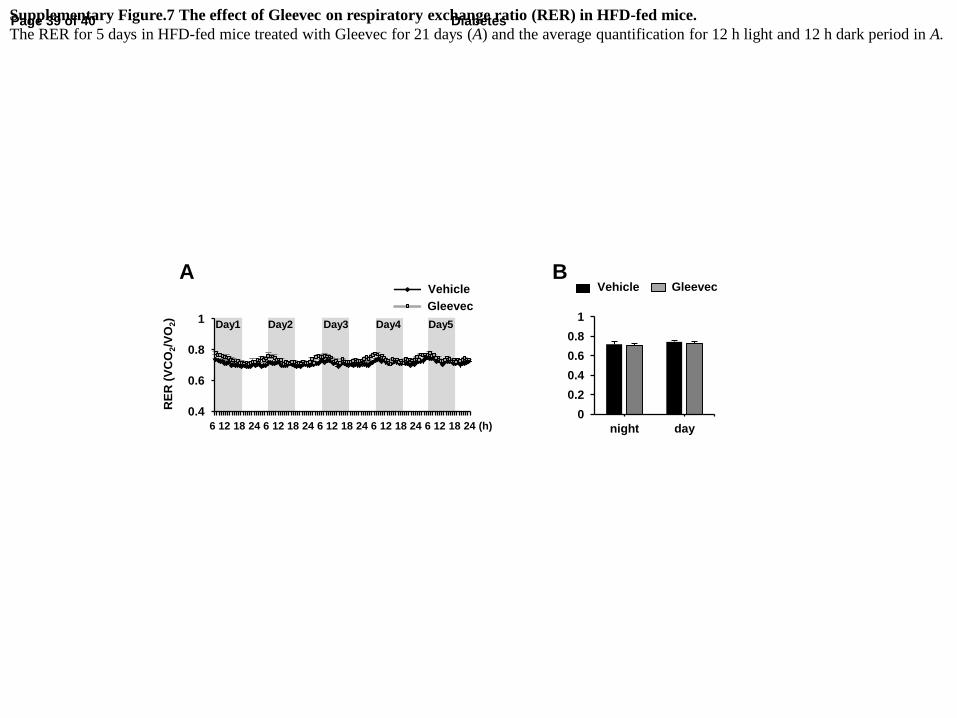
exchange ratio (RER), indicating that Gleevec did not stimulate any substantial shift from
carbohydrate to fat-based fuels (Supplementary Fig. 7). Importantly, these changes in energy
expenditure in Gleevec-treated mice were not dependent on food intake or physical activity
(Fig. 6B). At the molecular level, Gleevec produced a significant increase in broad
brown/beige fat thermogenic and mitochondrial genes, including Ucp-1, Pgc-1α, and cox-5b
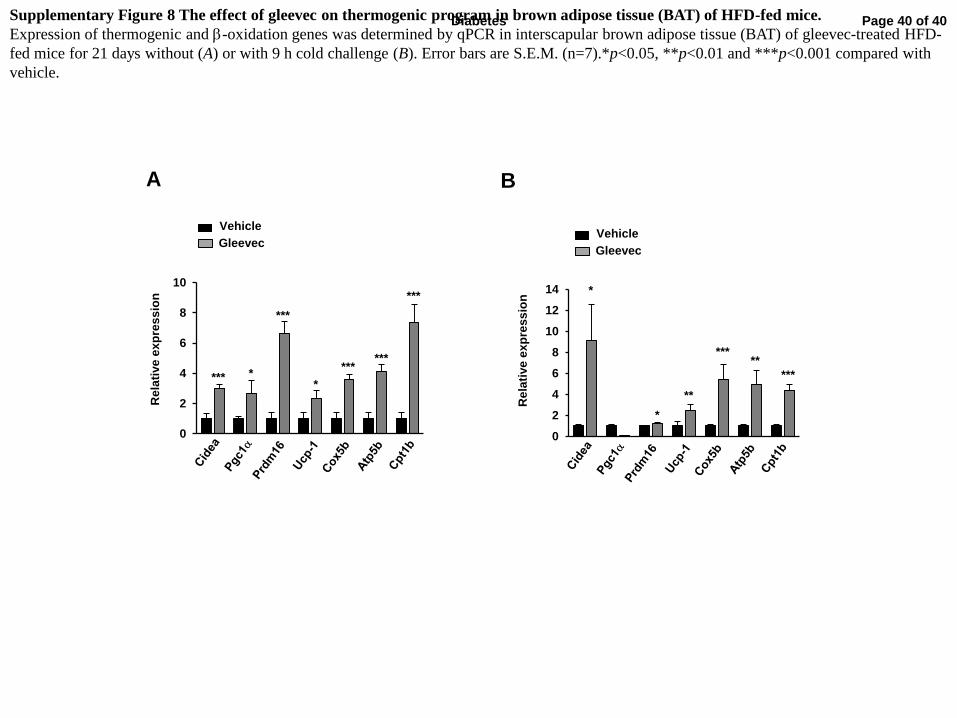
in WAT and interscapular brown adipose tissue (BAT) (Fig. 6C, Supplementary Fig. 8A). In
addition, Gleevec stimulated the expression of beige adipocyte marker (Cd137 and Tmem26)
and β-oxidation genes (carnitine palmitoyltransferase 1b, Cpt1b) (Fig. 6C).
Page 15 of 40 Diabetes

To further examine the effects of Gleevec on cold-induced thermogenesis in vivo, mice
were challenged with cold exposure at 4°C. Acute exposure (9 h) to cold significantly
dropped rectal temperatures (Fig. 6D). As expected, Gleevec-treated mice showed a more
resistant phenotype (decreased rectal temperature) against cold (Fig. 6D). WAT morphology
was also analyzed under these conditions by H&E staining. As shown in Fig. 6E, multilocular
adipocytes were observed in the sWAT of Gleevec-treated mice. Furthermore, the expression
of thermogenic and mitochondrial genes, including Ucp-1, Cidea, Cox5b, Atp5b and Cpt1b
were significantly increased in scWAT and BAT of Gleevec-treated mice following cold
exposure (Fig. 6F and Supplementary Fig. 8B).
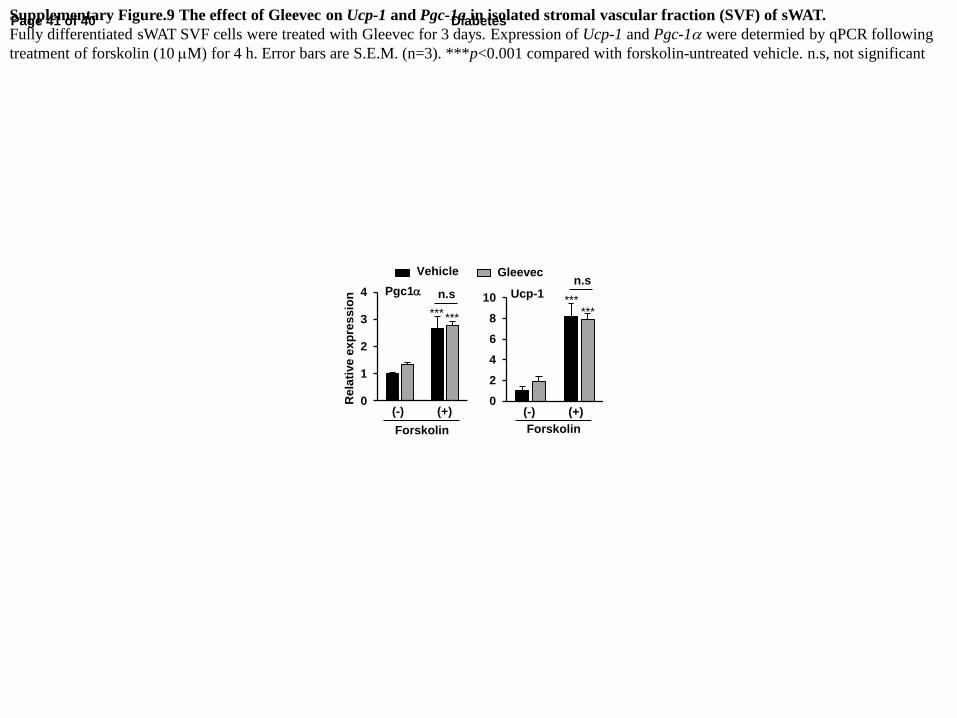
To determine whether the browning effect of Gleevec is cell-autonomous, Gleevec was
administered to the stromal vascular fraction (SVF) of inguinal WAT during differentiation in
vitro. There were no detectable differences in the expression of Ucp-1 or Pgc-1α in fully
differentiated adipocytes (Supplementary Fig. 9), indicating that Gleevec may induce adipose
tissue thermogenesis in vivo independent of its direct action on adipocytes.
Recent studies have demonstrated that IL-4 activates M2 macrophage polarization and
alternatively activated macrophage produces catecholamine to be important for induction of
thermogesis in WAT and BAT (22). As Gleevec promoted the expression of M2 marker
genes, including Arg and Mrc-1 in adipose tissue (Fig. 5C), we next examined the expression
of IL-4, IL-13, and thyrosine hydroxylase (Th), the rate-limiting enzyme to synthesize
catecholamines in WAT. As shown in Fig. 6G and H, treatment with Gleevec resulted in
significantly increased expression of IL-4, IL-13 and Th either during cold exposure (Fig. 6G)
or in normal housing temperature (22°C) (Fig. 6H). Taken together, these results indicate that
Gleevec increases energy expenditure by upregulating beige/brown fat thermogenic genes in
vivo. And these effects were induced by a phenotypic switch in adipose tissue macrophages,
Page 16 of 40Diabetes

along with increase expression of Th possibly via inducing expression of the IL-4 and IL-13.
Discussion
Full agonist PPARγ ligands, including TZDs, have been widely used to treat Type 2
diabetes (8, 9, 2). However, patients treated with these drugs have exhibited higher incidence
of serious adverse effects, such as weight gain, bone fracture, and congestive heart failure
compared with other oral hypoglycemic agent. Recent studies have shown that the insulin-
sensitizing effects of PPARγ ligands are not dependent on classical agonism, but rather are a
consequence of ligand-dependent inhibition of pS273 by CDK5/ERK kinases (11, 12, 15).
More specifically, non-agonist PPARγ ligands, such as SR1664 and UHC1, have illustrated
similar glucose-lowering effects as TZDs while lacking the commonly observed side effects.
These studies have allowed us the opportunity to find a potential compound for the treatment
of type 2 diabetes. Thus, we aimed to discover compounds that block pS273 and lack
classical agonism with high binding affinity to PPARγ. Drug repositioning is the application
of known drugs and compounds to new indications, which can save time and costs because
they have already been tested in humans and detailed information is available on their
pharmacology, formulation, and potential toxicity (23). Thus, we screened PPARγ ligands
that block pS273 with an FDA-approved drug library and Gleevec was determined to fit these
criteria.
Gleevec, a specific BCR-Abl kinase inhibitor, is a well-known anti-cancer drug that
exhibits dramatic effects for the treatment of CML and gastrointestinal stromal tumors
(GISTs) (24, 25). In addition to its anti-neoplastic activity, several studies have reported that
Gleevec has a blood glucose-lowering effect in patients suffering from both CML and type 2
Page 17 of 40 Diabetes

diabetes (26, 27) or GISTs (28). Restoration of insulin sensitivity or amelioration of
hyperlipidemia has also been noted in non-diabetic CML patients who show significant
insulin resistance at diagnosis (29, 30). Although the beneficial effects of Gleevec on glucose
and lipid metabolism have been demonstrated previously, the molecular and cellular
mechanisms remain unclear. Previous studies have demonstrated that Gleevec controls
hyperglycemia by preserving pancreatic β-cell mass or reducing endoplasmic reticulum (ER)
stress in diet-induced obese rats or diabetic db/db mice (31-34), while the present study
clearly demonstrates that the potent insulin-sensitizing action of Gleevec is derived from a
distinct molecular mechanism. Gleevec acts as a PPARγ antagonist ligand that directly blocks
pS273 while lacking classical agonism (Fig. 2 and Supplementary Fig. 1-4). Several lines of
evidence have shown improved insulin sensitivity through blocking pS273 by Gleevec:
Gleevec ameliorates hepatic steatosis and glucose production in liver (Fig. 4), attenuates pro-
inflammatory responses in WAT (Fig. 5), and promotes browning and energy expenditure in
WAT (Fig. 6), all of which contribute to increased insulin sensitivity (Fig. 3).
In our previous study, we demonstrated that blocking pS273 inhibits
lipopolysaccharide (LPS)-induced inflammation in macrophages (13). Furthermore, Gleevec
reportedly suppresses LPS- or TNF-α-induced inflammatory cytokine production in
macrophages by blocking IκB phosphorylation and subsequent DNA binding of nuclear
factor kappa B (NF-κB), thus inhibiting NF-κB activation (36). Thus, we speculate that
inhibition of pS273 by Gleevec regulates adipose tissue inflammation in the obese state.
Indeed, we observed that Gleevec redirects ATMs from an M1 to an M2 polarization state
(Fig. 5), suggesting that Gleevec reduces macrophage infiltration by suppressing the pro-
inflammatory response in adipocytes, as well as regulating M1/M2 polarization in
macrophages, thus ameliorating adipose tissue inflammation.
Page 18 of 40Diabetes

In the present results, we observed Gleevec induces adipose tissue thermogenesis in
vivo independent of its direct action on adipocytes. How does Gleevec promote browning of
WAT? According to recent evidence, an increase in M2 macrophages produces
catecholamine in the induction of thermogenic and β-oxidation gene expression in WAT and
BAT in cold-exposed mice (22). Furthermore, this effect is accompanied by increased
eosinophil-derived IL-4 in WAT (37). Accordingly, genetic deletion of Il4ra or Th in
myeloid cells significantly impairs the development of thermogenic beige fat in mice (37, 38).
Recently, Lee et al. reported that IL-4, derived from type 2 innate lymphoid (ILC2s),
stimulated eosinophils and directly promoted the proliferation and commitment of platelet-
derived growth factor receptor α+ (Pdgfrα
+) adipocyte precursors (APs), which are
bipotential cells that differentiate into either beige or white adipocytes within sWAT and
enhances differentiation into beige adipocytes (39). Interestingly, we have shown that
Gleevec treatment increased the proportion of M2 macrophages (Fig. 5) and the expression of
beige maker genes in sWAT (Fig. 6). Furthermore, we observed that PPARγS273A
mutant
increased IL-4-induced M2 polarization of macrophages (Fig. 5). Together, induction of IL-
4/IL-13 expression and alternative macrophage activation by inhibition of PPARγ
phosphorylation might promote regulation of expression of genes associated with
brown/beige adipose thermogenesis.
In conclusion, we found that Gleevec has potent beneficial effects on insulin
sensitivity to relieve metabolic disorders by regulating glucose and lipid metabolism without
causing several adverse effects, including body weight gain, fatty liver, fluid retention, bone
fracture, and cardiac hypertrophy, which have been observed following treatment with most
TZDs. Notably, Gleevec regulates energy expenditure by promoting the development of
beige fat in WAT. These results establish an important role for Gleevec in regulating adipose
Page 19 of 40 Diabetes

tissue thermogenesis and white adipose plasticity towards BAT, and shed the light on the
development of novel therapeutic drugs for the treatment of obesity and type 2 diabetes.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant
funded by the Korea government (MSIP) (No. 2014M3A9D8034456)(S.S.C.), the Korean
Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea.
(HI14C2518)(J.H.C), the National Research Foundation of Korea (NRF-
2013R1A1A2060283, NRF-2011-0020163)(J.H.C), and the Institute for Basic Science (IBS-
R022-D1-2015)(J.H.C, K.J.M).
E.S.K., J.E.J., D.P.M., A.J., J.Y.K., S.Y.C., Y.R.Y., H.J.J., E.K.K., J.P., H.M.K.,
I.H.L., S.B.P., K.J.M., P.G.S., P.R.G. researched data and assisted with data interpretation.
S.S.C. and J.H.C. concived the hypothesis, designed and researched data, and wrote the
manuscript. J.H.C. is the guarantor of this work and, as such, has full access to all the data in
the study and takes responsibility for the integrity of the data and the accuracy of the data
analysis.
No potential conflicts of interest relevant to this article were reported.
References
1. Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta
12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell
1995;83:803–812
2. Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An
antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated
receptor gamma (PPAR gamma). J Biol Chem 1995;270:12953–12956
3. Nesto RW, Bell D, Bonow RO, Fonseca V, Grundy SM, Horton ES, Le Winter M, Porte D,
Semenkovich CF, Smith S, Young LH, Kahn R. Thiazolidinedione use, fluid retention, and
congestive heart failure: a consensus statement from the American Heart Association and
American Diabetes Association. Diabetes Care 2004;27:256–263
Page 20 of 40Diabetes

4. Kahn SE, Zinman B, Lachin JM, Haffner SM, Herman WH, Holman RR, Kravitz BG, Yu
D, Heise MA, Aftring RP, Viberti G; Diabetes Outcome Progression Trial (ADOPT) Study
Group. Rosiglitazone-associated fractures in type 2 diabetes: an Analysis from A Diabetes
Outcome Progression Trial (ADOPT). Diabetes Care 2008;31:845–851
5. Grey A, Bolland M, Gamble G, Wattie D, Horne A, Davidson J, Reid IR. The peroxisome
proliferator-activated receptor-gamma agonist rosiglitazone decreases bone formation and
bone mineral density in healthy postmenopausal women: a randomized, controlled trial. J
Clin Endocrinol Metab 2007;92:1305–1310
6. Kostapanos MS, Elisaf MS, Mikhailidis DP. Pioglitazone and cancer: angel or demon?
Curr Pharm Des 2013;19:4973–4929
7. Ferwana M, Firwana B, Hasan R, Al-Mallah MH, Kim S, Montori VM, Murad MH.
Pioglitazone and risk of bladder cancer: a meta-analysis of controlled studies. Diabet Med
2013;30:1026–1032
8. Evans RM, Barish GD,Wang YX. PPARs and the complex journey to obesity. Nat Med
2004;10:355–361
9. Tontonoz P,Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Ann
Rev Biochem 2008;77:289–312
10. Willson TM, Lambert MH,Kliewer SA. Peroxisome proliferator-activated receptor
gamma and metabolic disease. Ann Rev Biochem 2001;70:341–367
11. Choi JH, Banks AS, Estall JL, Kajimura S, Boström P, Laznik D, Ruas JL, Chalmers MJ,
Kamenecka TM, Blüher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-
linked phosphorylation of PPARgamma by Cdk5. Nature 2010;466:451–456
12. Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS,
Shin Y, He Y, Bruning JB, Marciano DP, Cameron MD, Laznik D, Jurczak MJ, Schürer SC,
Vidović D, Shulman GI, Spiegelman BM, Griffin PR. Antidiabetic actions of a non-agonist
PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature 2011;477:477–481
13. Choi SS, Kim ES, Koh M, Lee SJ, Lim D, Yang YR, Jang HJ, Seo KA, Min SH, Lee IH,
Park SB, Suh PG, Choi JH. A novel non-agonist peroxisome proliferator-activated receptor γ
(PPARγ) ligand UHC1 blocks PPARγ phosphorylation by cyclin-dependent kinase 5 (CDK5)
and improves insulin sensitivity. J Biol Chem 2014;289:26618–26629
14. Liu S, Hatano B, Zhao M, Yen CC, Kang K, Reilly SM, Gangl MR, Gorgun C, Balschi
JA, Ntambi JM, Lee CH. Role of peroxisome proliferator-activated receptor {delta}/{beta} in
hepatic metabolic regulation. J Biol Chem 2011;286:1237–1247
15. Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D,
Shulman GI, Gygi SP, Spiegelman BM. An ERK/Cdk5 axis controls the diabetogenic actions
of PPARγ. Nature 2015;517:391–395
16. Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG,
Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the
peroxisome proliferator-activated receptor-gamma. Nature 1998;395:137-143
17. Hannan RD, Jenkins A, Jenkins AK,Brandenburger Y. Cardiac hypertrophy: a matter of
translation. Clin Exp Pharmacol Physiol 2003;30:517-527.
18. Matsuzawa Y, Funahashi T, Nakamura T. Molecular mechanism of metabolic syndrome
X: contribution of adipocytokines adipocyte-derived bioactive substances. Ann N Y Acad Sci
1999;892:146–154
19. Dandona P, Aljada A,Bandyopadhyay A. Inflammation: the link between insulin
resistance, obesity and diabetes. Trends Immunol 2004;25:4–7
20. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M,
Greenberg AS, Obin MS. (Adipocyte death defines macrophage localization and function in
adipose tissue of obese mice and humans. J Lipid Res 2005;46:2347–2355
Page 21 of 40 Diabetes

21. Ohno H, Shinoda K, Spiegelman BM, Kajimura S. PPARγ agonists induce a white-to-
brown fat conversion through stabilization of PRDM16 protein. Cell Metab 2012;15:395-404
22. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F,
Locksley RM, Chawla A. Alternatively activated macrophages produce catecholamines to
sustain adaptive thermogenesis. Nature 2011;480:104–108
23. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for
existing drugs. Nat Rev Drug Discov 2004;3:678–683
24. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J,
Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-
Abl positive cells. Nat Med 1996;2:561–566
25. Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, Fletcher CD,
Demetri GD, Fletcher JA. Mechanisms of oncogenic KIT signal transduction in primary
gastrointestinal stromal tumors (GISTs). Oncogene 2004;23:3999–4006
26. Veneri D, Franchini M, Bonora E. Imatinib and regression of type 2 diabetes. N Engl J
Med 2005;352:1049–1050
27. Breccia M, Muscaritoli M, Aversa Z, Mandelli F, Alimena G. Imatinib mesylate may
improve fasting blood glucose in diabetic Ph+chronic myelogenous leukemia patients
responsive to treatment. J Clin Oncol 2004;22:4653–4655
28. Hamberg P, de Jong FA, Boonstra JG, van Doorn J, Verweij J, Sleijfer S. Non-islet-cell
tumor induced hypoglycemia in patients with advanced gastrointestinal stromal tumor
possibly worsened by imatinib. J Clin Oncol 2006;24:e30–e31
29. Tsapas A, Vlachaki E, Sarigianni M, Klonizakis F, Paletas K. Restoration of insulin
sensitivity following treatment with imatinib mesylate (Gleevec) in non-diabetic patients with
chronicmyelogenic leukemia (CML). Leuk Res 2008;32:674–675
30. Gologan R, Constantinescu G, Georgescu D, Ostroveanu D, Vasilache D, Dobrea C,
Iancu D, Popov V. Hypolipemiant besides antileukemic effect of imatinib mesylate. Leuk Res
2009;33:1285–1287
31. Hagerkvist R, Jansson L, Welsh N. Imatinib mesylate improves insulin sensitivity and
glucose disposal rates in high-fat diet fed rats. Clin Sci 2007;114:65–71
32. Han MS, Chung KW, Cheon HG, Rhee SD, Yoon CH, Lee MK, Kim KW, Lee MS.
Imatinib mesylate reduces endoplasmic reticulum stress and induces remission of diabetes in
db/db mice. Diabetes doi: 10.2337/db08-0080
33. H¨agerkvist R, Sandler S, Mokhtari D, Welsh N. Amelioration of diabetes by imatinib
mesylate (Gleevec): role of β-cell NF-κB activation and anti-apoptotic preconditioning.
FASEB J 2007;21:618–628
34. H¨agerkvist R, Makeeva N, Elliman S, Welsh N. Imatinib mesylate (Gleevec) protects
against streptozotocin-induced diabetes and islet cell death in vitro. Cell Biol Int
2006;30:1013–1017
35. Donath MY,Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev
Immunology 2011;11:98–107
36. Wolf AM, Wolf D, Rumpold H, Ludwiczek S, Enrich B, Gastl G, Weiss G, Tilg H. The
kinase inhibitor imatinib mesylate inhibits TNF-{alpha} production in vitro and prevents
TNF-α dependent acute hepatic inflammation. Proc Natl Acad Sci U S A 2005;102:13622–
13627
37. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla
A, Locksley RM. Eosinophils sustain adipose alternatively activated macrophages associated
with glucose homeostasis. Science 2011;332:243–247
Page 22 of 40Diabetes

38. Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, Palmiter RD, Chawla A.
Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of
functional beige fat. Cell 2014;157:1292–1308
39. Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, Yun K,
Locksley RM, Chawla A. Activated type 2 innate lymphoid cells regulate beige fat
biogenesis. Cell 2015;160:74–87
Figure Legends
Fig. 1. Gleevec blocks CDK5-mediated PPARγ phosphorylation. (A) Chemical structure
of compounds. (B) In vitro CDK5 assay on full-length PPARγ incubated with rosiglitazone or
Gleevec. (C) Phosphorylation of Rb after treating with rosiglitazone or Gleevec. (D) TNF-α-
induced phosphorylation of PPARγ in HEK-293 cells expressing PPARγ treated with
rosiglitazone or Gleevec. (E) In vitro CDK5 assay on full-length PPARγ or Rb incubated
with rosiglitazone, Gleevec, Nilotinib, Dasatinib, and Sunitinib. NT, not treated
Fig. 2. Gleevec is a PPARγ ligand that lacks classical agonism. (A) Binding affinity of
Gleevec or rosiglitazone to the LBD of PPARγ by LanthaScreen assay. (B) Transcriptional
activity of a PPAR-derived reporter gene in HEK-293 cells following treatment with
rosiglitazone, MRL24, SR1664 or Gleevec. (C) Differential HDX results are mapped onto the
PPARγ crystal structure (3PRG) comparing the LBD of PPARγ in the presence of Gleevec
(left) and rosiglitazone (right). (D) Recruitment of coactivator to PPARγ in the presence of
rosiglitazone or Gleevec by LanthaScreen assay. (E) Lipid accumulation in differentiated
3T3-L1 adipocytes treated with rosiglitazone or Gleevec following Oil-red O staining.
Expression of adipogenic marker genes (F) and gene sets regulated by PPARγ
phosphorylation (G) in these cells. All of error bars represented are S.E.M. (n=3). *p<0.05,
**p<0.01, ***p<0.001 compared with vehicle.
Page 23 of 40 Diabetes

Fig. 3. Gleevec has potent insulin-sensitizing effects in high fat-induced obese mice
without severe side effects that TZDs have. Intraperitoneal glucose tolerant test (IPGTT) (A)
and intraperitoneal insulin tolerance test (IPITT) (B) after 7 days of treatment with vehicle or
Gleevec (25 mg/kg) in HFD-fed mice treated (n=5). Inlet, AUC. (C) Phosphorylation of
PPARγ in WAT. Quantification of PPARγ phosphorylation compared to total PPARγ was
performed (n=5). (D) Correlation between the levels of PPARγ phosphorylation normalized
to the total PPARγ protein and the changes of AUC by GTT. Pearson correlation coefficient
and p value are shown. Fasting body weight (E), fasting insulin (F), HOMA-IR (G) were
determined in these mice (n=5). (H) Expression of gene sets regulated by PPARγ
phosphorylation in WAT (n=5). (I) hMSCs were differentiated with 10 mM β-
glycerophosphate, 50 µM ascorbate-2-phosphate, 100 nM DEX treated with Gleevec or
rosiglitazone for 2 weeks. The expression of osteoblast marker genes were determined by
qPCR (n=3). Packed cell volume (PVC) in whole blood (J), the percent of body fat mass (K),
the percent of lean mass (L), heart weight (M) were measured and the expression of marker
genes for heart failure and cardiac hypertrophy in heart (N) was determined in HFD-fed mice
treated with rosiglitazone or Gleevec for 14 days (n=6). All of represented error bars are
S.E.M. (n=5). *p<0.05, **p<0.01, ***p<0.001 compared with vehicle.
Fig. 4. Gleevec reduces hepatic steatosis and glucose production in liver. (A) Histological
analysis by hematolxylin-eosin (H&E) staining of liver in HFD-fed mice treated with
Gleevec for 7 days. Expression of lipogenic (B) and gluconeogenic (C) genes in liver (n=5).
(D) Expression of gluconeogenic genes in primary hepatocytes isolated from HFD-fed mice
for 10 weeks following treatment with Gleevec (n=3). (E) Glucose concentration in the
primary hepatocyte media (n=3). (F) Expression of gluconeogenic genes and PPARγ in the
Page 24 of 40Diabetes

primary hepatocyes expressing PPARγWT
or PPARγS273A
(n=3). All of represented error bars
are S.E.M. *p<0.05, **p<0.01, ***p<0.001 compared with vector; ##
p<0.01 compared
between PPARγWT
and PPARγS273A
Fig. 5. Gleevec ameliorates adipose tissue inflammation. (A) Histological analysis by H&E
staining of WAT in HFD-fed mice treated with Gleevec for 21 days. Adipocyte size was
calculated on histological sections of WAT (n=6). Expression of marker genes for M1
macrophage (B), total macrophage (C) and M2 macrophage (D) (n=6). All of represented
error bars are S.E.M. *p<0.05, **p<0.01, ***p<0.001 compared with vehicle. (E) Expression
of pro-inflammatory genes in mouse embryonic fibroblast (MEFs) expressing PPARγWT
or
PPARγS273A
treated with TNF-α (50 ng/ml) (n=3). All of represented error bars are S.E.M.
*p<0.05, **p<0.01, ***p<0.001 compared with PPARγWT
; #p<0.05,
##p<0.01,
###p<0.001
compared with TNF-α treated cells; n.s, not significant. (F) Expression of pro-imflammatory
genes in Raw264.7 macrophages expressing PPARγWT
or PPARγS273A
treated with LPS (10
ng/ml) (n=3). All of represented error bars are S.E.M. **p<0.01 compared with vector and
##p<0.01 compared with PPARγ
WT ; n.s, not significant
Fig. 6. Gleevec increases energy expenditure and adaptive thermogenesis. (A-B) O2
consumption rate (VO2), CO2 production rate (VCO2), food intake, and locomotors activity
(XTOT) were measured by CLAMS in HFD-fed mice treated with Gleevec for 21 days (n=4).
(C) Expression of thermogenic, β-oxidation and beige marker genes in sWAT (n=6). (D)
Rectal temperature were measured at the indicated time points for mice placed in a cold room
(n=4). (E) H&E staining of sWAT in these mice. (F) Expression of genes for thermogenesis
and β-oxidation (n=4). Expression of Il-4, Il-13 and Th in subcutaneous WAT were
Page 25 of 40 Diabetes

determined by qPCR with (G) or without cold exposure (H) (n=4-6). All of represented error
bars are S.E.M. *p<0.05, **p<0.01, ***p<0.001 compared with vehicle.
Page 26 of 40Diabetes
pPPARg
PPARg
NT
Gle
evec (
10 μ
M)
ATP (10 mM)
NT
Ro
sig
lita
zo
ne
(10 μ
M)
Gle
evec (
1μ
M)
Gle
evec (
0.1
μM
)
Gle
evec (
0.0
1 μ
M)
TNF-a ( 10 ng/ml)
NT
Gle
evec (
10 μ
M)
NT
Ro
sig
lita
zo
ne
(10 μ
M)
Gle
evec (
1μ
M)
Gle
evec (
0.1
μM
)
Gle
evec (
0.0
1 μ
M)
pPPARg
PPARg
Figure. 1
C
D
Gleevec
Dasatinib
Nilotinib
Sunitinib
Nil
oti
nib
Su
nit
inib
Dasati
nib
Gle
evec
Ro
sig
lita
zo
ne
NT
NT
pPPARg
PPARg
(1mM)
A B
pRb
Rb
NT
NT
Gle
evec (
10 μ
M)
Ro
sig
lita
zo
ne
(10 μ
M)
E
pRb
Rb
Nil
oti
nib
Su
nit
inib
Dasati
nib
Gle
evec
Ro
sig
lita
zo
ne
NT
NT
(1mM)
0.0
0.4
0.8
1.2
pP
PA
Rg/
PP
AR
g
NT
Ro
sig
lita
zo
ne
10
NT
Gle
eve
c 1
0
Gle
eve
c 1
Gle
eve
c 0
.1
Gle
eve
c 0
.01
(mM)
ATP
ATP (10 mM)
ATP (10 mM) ATP (10 mM)
0.0
0.4
0.8
1.2
NT NT R10 G10
pR
b/R
b
(mM)
NT
NT
Ro
sig
lita
zo
ne
10
Gle
eve
c 1
0
ATP
0 0
0.0
0.4
0.8
1.2
1.6
pP
PA
Rg/
PP
AR
g
TNF-a
0
NT
Ro
sig
lita
zo
ne
10
NT
Gle
eve
c 1
0
Gle
eve
c 1
Gle
eve
c 0
.1
Gle
eve
c 0
.01
0.0
0.4
0.8
1.2
NT NT R G N D S
pP
PA
Rg/
PP
AR
g
ATP
0
Nilo
tin
ib
Su
nit
inib
Das
ati
nib
Gle
eve
c
Ro
sig
lita
zo
ne
NT
NT
0.0
0.4
0.8
1.2
NT NT R G N D S
pR
b/R
b
ATP
Nilo
tin
ib
Su
nit
inib
Das
ati
nib
Gle
eve
c
Ro
sig
lita
zo
ne
NT
NT
0
(mM)
(1 mM) (1 mM)
Page 27 of 40 Diabetes
0.0
0.2
0.4
0.6
0.8
0
0.0
01
0.0
1
0.1
0.3 1 3
10
30
100
Rati
o (
52
0/4
94
nm
) RosiglitazoneGleevec
(μM) 0
1
2
3
4
5
0 0.001 0.01 0.1 1 10
Rosiglitazone
MRL24
SR1664
Gleevec
(μM)
PP
ARγ t
ran
sc
rip
tio
na
l
ac
tivit
y
Figure. 2
A B C
0
1
2
3
4
Rela
tive
ex
pre
ss
ion
**
**
* *
*
* *
* * * *
Vehicle
D E Rosiglitazone
G
Gleevec
H12
H11
H3
≥ -20
< -20
ΔHDX (%)
NS
H12
H11
H3
Rosiglitazone
F
0
Vehicle
Gleevec
0.0
0.5
1.0
1.5
0 0.01 0.1 0.3 1 3 10
Rati
o (
52
0/4
94
nm
)
RosiglitazoneGleevec
(μM) 0
0
10
20
30
40
50
Rela
tive
ex
pre
ss
ion
***
NT Rosiglitazone
Gleevec
***
***
***
***
Gleevec
Page 28 of 40Diabetes
0
10
20
30
40
50
Bo
dy w
eig
ht
(g)
Figure. 3
Vehicle Gleevec
pPPARg
PPARg
(0.0)
0.3
0.6
0.9
1.2
pP
PA
Rg/
PP
AR
g
***
A B C D
E F G H
0
Vehicle Gleevec
R² = 0.7633
0.0
0.5
1.0
1.5
2.0
0.0 0.1 0.2 0.3 0.4 0.5
pPPARg/PPARg
Rela
tive
AU
C o
f G
TT
0
0
n.s
0
1
2
3
4
5
Fa
sti
ng
in
su
lin
(n
g/m
l)
***
0
1
2
3
HO
MA
-IR
***
0
1
2
3
Rela
tive
ex
pre
ss
ion
** ** *
** **
**
*** ***
**
Vehicle
Gleevec
0
1
2
BSP OCN ostrix
**
** **
**
Re
lati
ve
ex
pre
ss
ion
**
Bsp Ocn Osx
*
*
0
10
20
30
40
50
60
PV
C (
%)
32
34
36
38
40
42
44
Bo
dy f
at
(%)
*
54
55
56
57
58
59
60
Le
an
(%
)
***
I J K L M N
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Atp1a2 Bnp Myhcβ Npp
***
* *
6.0
7.0
0 100
110
120
130
140
150
Hea
rt w
eig
ht
(mg
) **
Re
lati
ve
ex
pre
ss
ion
Vehicle
Rosiglitazone
Gleevec
Vehicle
Gleevec
0
100
200
300
400
500
600
0 20 40 60 90 120
Vehicle
Gleevec 25 mg/kg
Glu
co
se
(m
g/d
l)
***
*** ***
0.0
0.4
0.8
1.2
Re
lati
ve
AU
C
** Vehicle
Gleevec
(min)
Vehicle
Gleevec
Vehicle
Gleevec Vehicle
Gleevec
Vehicle
Rosiglitazone
Gleevec
Vehicle
Rosiglitazone
Gleevec
Vehicle
Rosiglitazone
Gleevec
Vehicle
Rosiglitazone
Gleevec
Vehicle
Rosiglitazone
Gleevec
0
20
40
60
80
100
120
0 20 40 60 90 120
Glu
co
se
(m
g/d
l)
VehicleGleevec
**
(min)
Re
lati
ve
AU
C
Vehicle
Gleevec
0
0.4
0.8
1.2
**
Page 29 of 40 Diabetes
Vehicle Gleevec
A B C
0
5
10
15
20
25
30
35
Rela
tive
e
xp
res
sio
n
ee
Gle
eve
c
Ve
hic
le
*
0
5
10
15
20
25
30500
400
300
200
100
Gle
eve
c
Ve
hic
le
0.0
0.5
1.0
1.5
2.0
Glu
co
se
pro
du
cti
on
(mg
/we
ll)
Gle
eve
c
Ve
hic
le
D
*** *** *
** ## ***
0
20
40
60
80
100
120
140
G6pase Pepck G6pase Pepck
F
Rela
tive
ex
pre
ss
ion
*
## ***
0
20
40
60
80
100
120
140
160
0
1
2
3
4
5
Pparg
(-) (+) (-) (+) (-) (+)
Forskolin Forskolin Forskolin
Vector PPARgWT PPARgS273A
Figure. 4
E (-) Forskolin
(+) Forskolin
(-) Forskolin
(+) Forskolin
Vehicle
Gleevec
0
0.0
0.2
0.4
0.6
0.8
1.0
1.2
G6Pase PEPCK
Rela
tive
ex
pre
ss
ion
***
***
0 G6pase Pepck
0.0
0.2
0.4
0.6
0.8
1.0
1.2
Acc-1 Fasn Scd-1
Vehicle
Gleevec
***
*** ***
Rela
tive
ex
pre
ss
ion
Acc-1 Fasn Scd-1 0
Page 30 of 40Diabetes
vehicle Gleevec
0
4000
8000
***
A B C D
E
0
2
4
6
8
10
(-) (+)
Il-6
TNF-a
***
***
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
(-) (+)
Mcp-1
TNF-a
***
0
2
4
6
8
10
12
(-) (+)
Tnf-a
TNF-a
***
***
0.0
0.5
1.0
1.5
2.0
(-) (+)
Il-1b
TNF-a
*
*
Rela
tive
ex
pre
ss
ion
c
ell
are
a (
mm
2) *
0.0
0.5
1.0
1.5
Il-1b Il-6 Mcp-1 Tnf-a
** **
Rela
tive
ex
pre
ss
ion
Vehicle
Gleevec
0 0.0
0.5
1.0
1.5
F4/80 cd11b cd11c cd68
Rela
tive
ex
pre
ss
ion
* * ** **
F4/80 cd11b cd11c cd68
Vehicle
Gleevec
0
PPARgWT PPARgS273A
0 0
Rela
tive
ex
pre
ss
ion
0.0
0.5
1.0
1.5
2.0
2.5
0 Arg Mrc-1
* *
Figure. 5
F
0
1
2
3
4
5
0.0
0.5
1.0
1.5
0
Rela
tive
ex
pre
ss
ion
Vector PPARgWT PPARgS273A
Pparg
n.s
Il-4 Il-4
(-) (+) (-) (+)
Arg Mrc-1
**
##
0
50
100
150
200
250 **
**
*** ###
###
###
###
###
###
#
#
Page 31 of 40 Diabetes
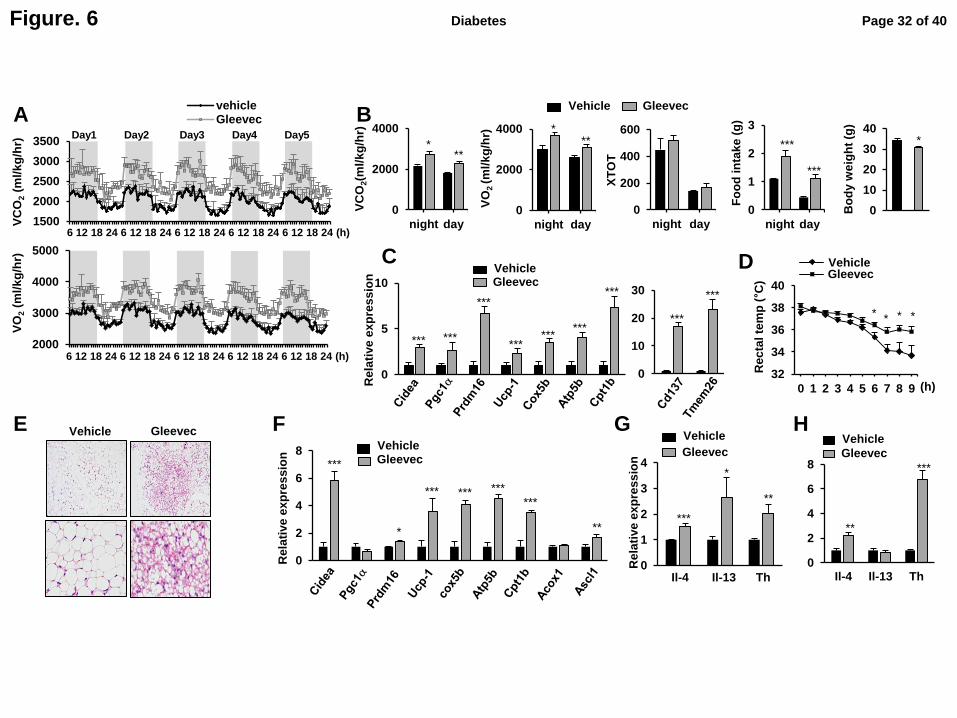
Figure. 6
A B
2000
3000
4000
5000
6 12 6 12 6 12 6 12 6 12 6 12 6 12 6 12 6 12 6 12
VO
2 (
ml/
kg
/hr)
6 12 18 24 6 12 18 24 6 12 18 24 6 12 18 24 6 12 18 24 (h)
* **
0
2000
4000
night day
VC
O2(m
l/k
g/h
r)
0
200
400
600
night day
XT
OT
0
1
2
3
night day
***
***
Fo
od
in
tak
e (
g)
0
10
20
30
40
Bo
dy w
eig
ht
(g)
*
0
2000
4000
night day
VO
2 (m
l/k
g/h
r) *
**
Vehicle Gleevec
Vehicle E F G H
C
Gleevec
0
5
10
Cidea PGC-1a PRDM16 UCP-1 cox5b ATP5B CPT-1bRe
lati
ve
ex
pre
ss
ion
*** ***
***
*** ***
***
***
Vehicle Gleevec
0
10
20
30
***
***
0
2
4
6
8
cidea PGC-1α PRDM16 UCP-1 cox5a ATP5b CPT-1a Acox1 Ascl1
***
*** *** ***
***
* **
Vehicle Gleevec
Rela
tive
ex
pre
ss
ion
0 1 2 3 4 5 6 7 8 9
32
34
36
38
40
vehicle Gleevec
* * * *
Gleevec Vehicle
(h)
Rec
tal te
mp
(°C
)
1500
2000
2500
3000
3500
6 12 6 12 6 12 6 12 6 12 6 12 6 12 6 12 6 12 6 12
vehicleGleevec 20 mg/kg
6 12 18 24 6 12 18 24 6 12 18 24 6 12 18 24 6 12 18 24 (h)
VC
O2 (
ml/
kg
/hr)
Day1 Day2 Day3 Day4 Day5
D
***
**
*
Rela
tive
ex
pre
ss
ion
Vehicle
Gleevec
0
1
2
3
4
IL-4 IL-13 THIl-4 Il-13 Th
Vehicle
Gleevec
0
2
4
6
8
IL-4 IL-13 TH
**
***
Il-4 Il-13 Th
Page 32 of 40Diabetes

Supplementary Figure 1 Gleevec blocks ERK-mediated PPARγ phosphorylation.
In vitro ERK kinase assay on full-length PPARγ incubated with rosiglitazone or Gleevec.
pPPARg
PPARg
NT
Gle
eve
c (
10
μM
)
ATP
NT
Ro
sig
lita
zo
ne
(1
0 μ
M)
Gle
eve
c (
1μ
M)
Gle
eve
c (
0.1
μM
)
Gle
eve
c (
0.0
1 μ
M)
Page 33 of 40 Diabetes

Supplymentary Figure. 2 Gleevec does not activate PPARa and PPARd.
Transcriptional activity of a PPAR-derived reporter gene in HEK-293 cells following treatment with GW7647, L165041 or Gleevec.
A B P
PA
Ra
tra
ns
cri
pti
on
al a
cti
vit
y
(μM) (μM)
PP
AR
d t
ran
sc
rip
tio
na
l a
cti
vit
y
0
1
2
3
0.01 0.1 1 10
GW7647
Gleevec
L16540
Gleevec
0
1
2
3
4
5
0.01 0.1 1 10
GW7647
Gleevec
Page 34 of 40Diabetes
Apo
Gleevec
Rosiglitazone
Helix 3
% D
eu
teri
um
Time (s)
Helix 11
% D
eu
teri
um
Time (s)
Helix 12
% D
eu
teri
um
Time (s)
Supplementary Figure.3 Gleevec Induced Perturbation in conformational dynamics of PPARg detected by HDX-MS.
Differential HDX results are mapped onto the PPARg crystal structure (3PRG) comparing the apo ligand binding domain of PPARg in the
presence of either Gleevec or rosiglitazone. 10 µM of HIS-PPARγ LBD protein was preincubated with 1:2 molar excess of compounds. 5 μl of
protein solution was mixed with 20 μl of D2O-containing HDX buffer (20 mM KPO4 pH 7.4, 50 mM KCl) and incubated at 4 °C for 10s, 30s, 60s,
900s and 3,600s. On-exchange was quenched and the protein denatured with 25 μl of quench solution (0.1 % v/v TFA in 3 M urea). Samples were
then passed through an immobilized pepsin column and the resulting peptides were trapped on a C8 trap column. The bound peptides were
gradient-eluted (5-50 % CH3CN w/v and 0.3 % w/v formic acid) across a C18 HPLC column for 5 min at 4 °C. The eluted peptides were
subjected to electrospray ionization directly coupled to a high resolution Orbitrap mass spectrometer (Thermo Fisher, MA, USA). Each HDX
experiment was carried out in triplicate and the intensity weighted average m/z value (centroid) of each peptide isotopic envelope was calculated
with in-house HDX Workbench software.
Page 35 of 40 Diabetes
DockScore: 77.670
DockScore: 65.905
Supplementary Figure 4 Binding mode of SR1664 or Gleevec to PPARγ LBD.
Docking simulation was performed with crystal structure of PPARγ LBD (PDB ID: 2hfp) and Discovery Studio® 1.7 (Accelrys). The binding
pose was predicted by docking simulation using the Discovery Studio 1.7® program. PPARγ ligand-binding pockets were defined from receptor
cavities, and the LigandFit module implemented in the Receptor-Ligand Interaction protocol was used for detailed calculations. X-ray crystal
structure of PPARγ ligand binding domain (2hfp) was used in the docking simulation and the subsequent structural analysis with the Discovery
Studio Visualizer 3.0® program (Accelrys Software Inc., CA, USA).
Page 36 of 40Diabetes
0
5
10
15
20
25
30
TG
(m
g/d
l)
***
Veh
icle
Gle
ev
ec
0
50
100
150
200
250
***
Fed
glu
co
se (
mg
/dl)
Gle
ev
ec
Veh
icle
A B
Supplementary Figure 5 The effect of Gleevec on blood glucose level and plasma level of triglyceride and free fatty acid in HFD-fed mice.
Fed blood glucose level (A) and serum level of triglyceride (B) and FFAs (C) in HFD mice treated with Gleevec for 7 days or 21 days (same as in
Fig. 3C). Error bars are S.E.M. ***p<0.001 compared with vehicle
C
0.0
0.4
0.8
1.2
1.6
2.0
Fre
e f
att
y a
cid
(m
Eq
/l)
Veh
icle
Gle
ev
ec
*
0
Page 37 of 40 Diabetes
A B C
0.0
0.5
1.0
1.5
IL-6 TNF-α MCP-1 IL-1β
Rela
tive
ex
pre
ss
ion
*
*
*
Il-6 Mcp-1 Tnf-a Il-1b
Vehicle
Vehicle/TNF-a
0
Gleevec /TNF-a
0
1
2
3
4
5
Rela
tive
mig
rati
on
Ve
hic
le
Gle
eve
c
Ve
hic
le
Gle
eve
c
MC
P-1
TNF-a
CM
*
*
*
0.0
0.5
1.0
1.5
IL-1β IL-6 MCP-1 TNF-α
Rela
tive
ex
pre
ss
ion
0
** ** * * *
Il-6 Mcp-1 Tnf-a Il-1b
Gleevec /LPS
Vehicle
Vehicle/LPS
Supplementary Figure.6 The effect of Gleevec on inflammation in adipocytes and macrophages.
Fully differentiated 3T3-L1 adipocytes (A) and primary macrophages isolated from bone-marrow (B) were treated with Gleevec for 24 h and
stimulated with TNF-a (50 ng/ml) for 3 h or LPS (10 ng/ml) for 6 h, respectively. Expression of pro-inflammatory cytokines was determined by
qPCR (n=3). (C) Migratory capacity of Raw264.7 macrophages were measured using an in vitro transwell chemotaxis assay (n=5). Mature 3T3-
L1 adipocytes were used for preparation of conditioned media (CM). 3T3-L1 adipocytes were stimulated by TNF-a (10 ng/ml) for 24 h after pre-
treatment with the indicated concentration of Gleevec for 24 h in serum-free DMEM. For the migration, 100,000 Raw264.7 cells were placed in
the upper chamber of a 5 μm polycarbonate filter (24-transwell format; Corning, Lowell, MA, USA), whereas adipocyte conditioned medium or
MCP-1 (10 ng/ml) treatment was placed in the lower chamber. After 3 h of migration, cells were fixed in formalin and stained with 4’, 6-
diamidino-2-phenylindole and observed.All of represented error bars are S.E.M. *p<0.05, **p<0.01, ***p<0.001 compared with vehicle.
Page 38 of 40Diabetes
0.4
0.6
0.8
1
6 1 8 3 10 5 12 7 2 9 4 11 6 1 8 3 10
RE
R (
VC
O2/V
O2)
vehicle
gleevec
Supplementary Figure.7 The effect of Gleevec on respiratory exchange ratio (RER) in HFD-fed mice.
The RER for 5 days in HFD-fed mice treated with Gleevec for 21 days (A) and the average quantification for 12 h light and 12 h dark period in A.
Vehicle
Gleevec
6 12 18 24 6 12 18 24 6 12 18 24 6 12 18 24 6 12 18 24 (h)
Day1 Day2 Day3 Day4 Day5
0
0.2
0.4
0.6
0.8
1
night day
Vehicle Gleevec A B
Page 39 of 40 Diabetes
Vehicle
Gleevec Vehicle
Gleevec
A B
0
2
4
6
8
10
12
14
Rela
tive
ex
pre
ss
ion
***
*** **
**
*
*
0
2
4
6
8
10
Rela
tive
ex
pre
ss
ion
***
***
*** ***
***
* *
Supplementary Figure 8 The effect of gleevec on thermogenic program in brown adipose tissue (BAT) of HFD-fed mice.
Expression of thermogenic and b-oxidation genes was determined by qPCR in interscapular brown adipose tissue (BAT) of gleevec-treated HFD-
fed mice for 21 days without (A) or with 9 h cold challenge (B). Error bars are S.E.M. (n=7).*p<0.05, **p<0.01 and ***p<0.001 compared with
vehicle.
Page 40 of 40Diabetes
Rela
tive
ex
pre
ss
ion
Vehicle Gleevec
*** Ucp-1
0
2
4
6
8
10
***
(-) (+)
Forskolin
n.s Pgc1a
0
1
2
3
4
*** ***
(-) (+)
Forskolin
n.s
Supplementary Figure.9 The effect of Gleevec on Ucp-1 and Pgc-1a in isolated stromal vascular fraction (SVF) of sWAT.
Fully differentiated sWAT SVF cells were treated with Gleevec for 3 days. Expression of Ucp-1 and Pgc-1a were determied by qPCR following
treatment of forskolin (10 mM) for 4 h. Error bars are S.E.M. (n=3). ***p<0.001 compared with forskolin-untreated vehicle. n.s, not significant
Page 41 of 40 Diabetes










![arXiv:1312.7826v2 [hep-ex] 1 Aug 2014 · 51University of Science and Technology of China, Hefei 230026 52Seoul National University, Seoul 151-742 53Soongsil University, Seoul 156-743](https://static.fdocument.org/doc/165x107/5fdbad0cd4fd056cbc36c199/arxiv13127826v2-hep-ex-1-aug-2014-51university-of-science-and-technology-of.jpg)