Platelet storage pool disorders
36
Platelet Storage Pool Disease by: amirhossein heydarian
-
Upload
amirhossein-heydarian -
Category
Health & Medicine
-
view
34 -
download
5
Transcript of Platelet storage pool disorders

Platelet Storage Pool Disease
by: amirhossein heydarian

Introduction
Platelets contain α-granules, dense granules, and lysosomes.
Platelet secretion or exocytosis releases molecules at sites of vascular injury to activate other cells or to facilitate cellular adhesion
These secretory organelles have unique molecular contents, and genetic disorders

Platelet Storage Pool Disease
The term SPD refers to patients with deficiencies in platelet content:
• dense granules (δ-SPD)
• α-granules (α-SPD)
• both types of granules (αδSPD)
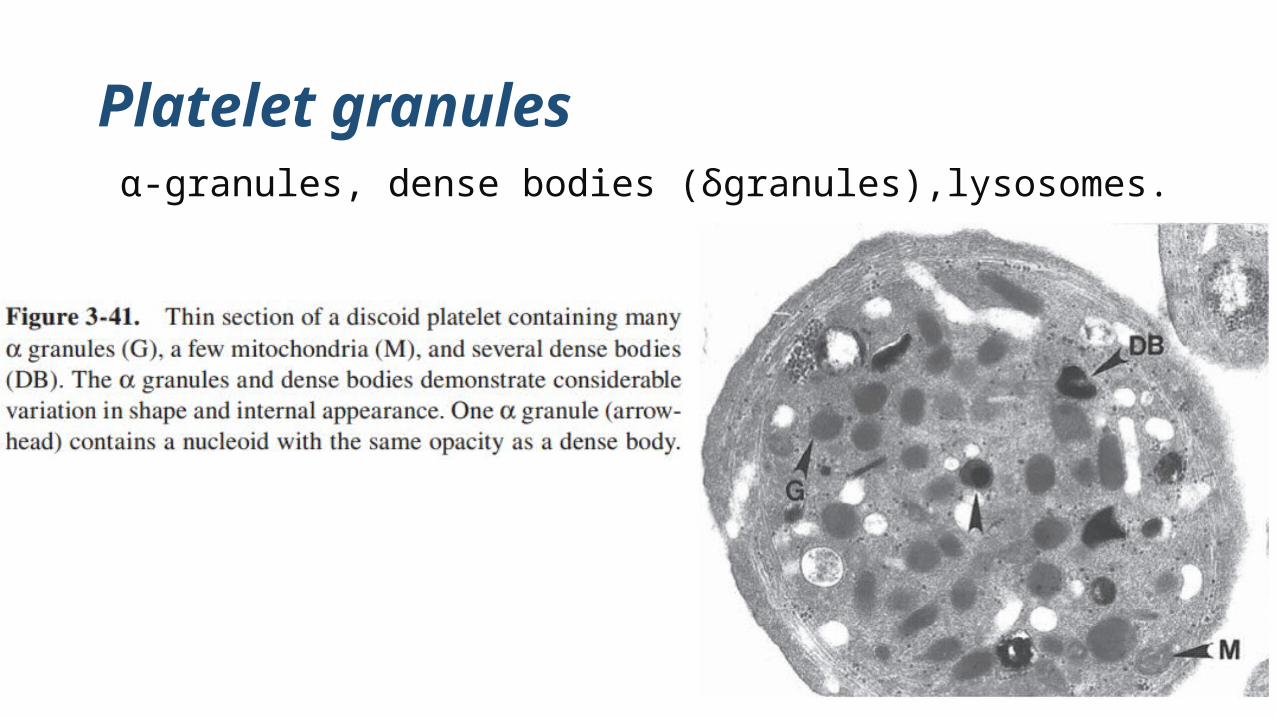
Platelet granules α-granules, dense bodies (δgranules),lysosomes.

Platelet granules con..
the term "alpha granules" is used to describe granules containing several growth factors
Dense granules (also known as dense bodies or delta granules) are specialized secretory organelles

α-granules
α-granules are the most numerous of the platelet organelles
There are usually 40 to 80 α-granules per platelet, but large and giant
cells may have well over 100
α-granules in resting platelets remain separated from each other
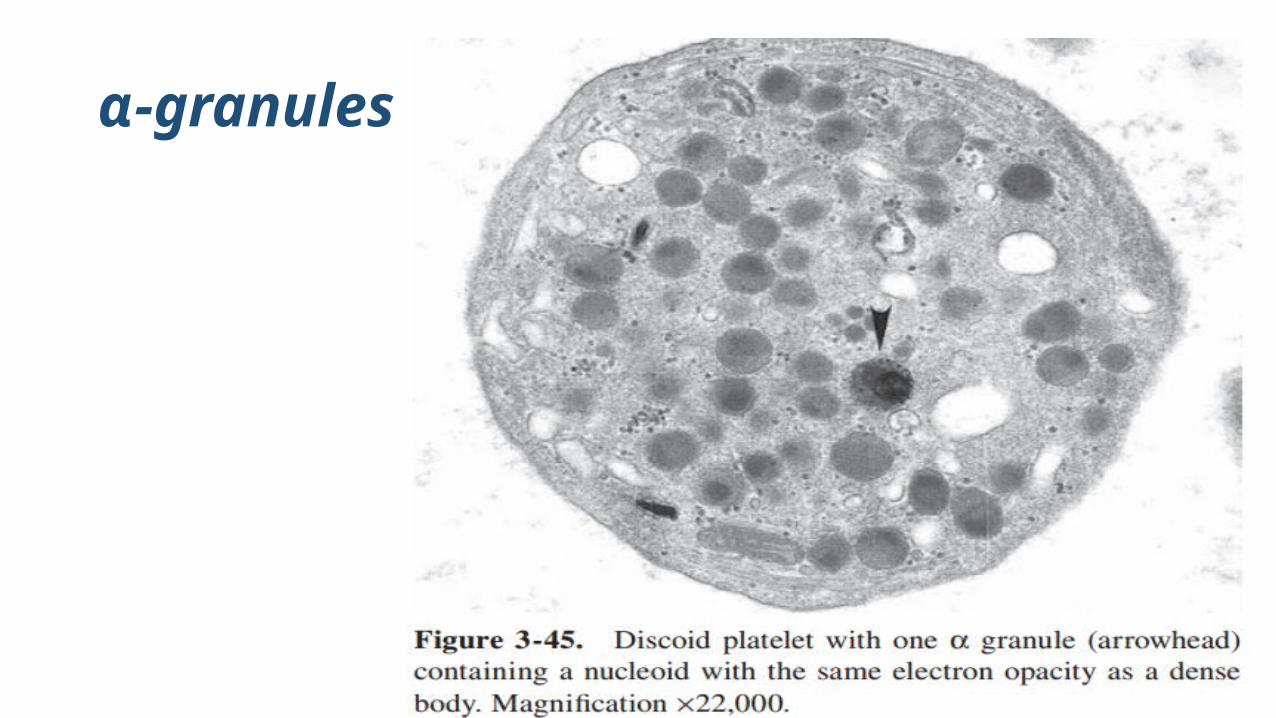
α-granules

Contents of alpha-granules
Contents include:
• insulin-like growth factor 1
• platelet-derived growth factors (PDGF)
• platelet factor 4 (PF4)
• thrombospondin
• fibronectin
• factor V
• von Willebrand factor (vWF)
• P-selectin
• fibrinogen, albumin, immunoglobulin

Contents of alpha-granules con..
proteins that are either synthesized in megakaryocytes:
platelet factor 4 [PF4], β-thromboglobulin [βTG], VWF, platelet-derived growth factor [PDGF]
endocytosed from plasma:
fibrinogen, albumin, immunoglobulin

a-Granule Disorders
I. Gray Platelet Syndrome (GPS)
II. Quebec Platelet Disorder

Gray Platelet Syndrome
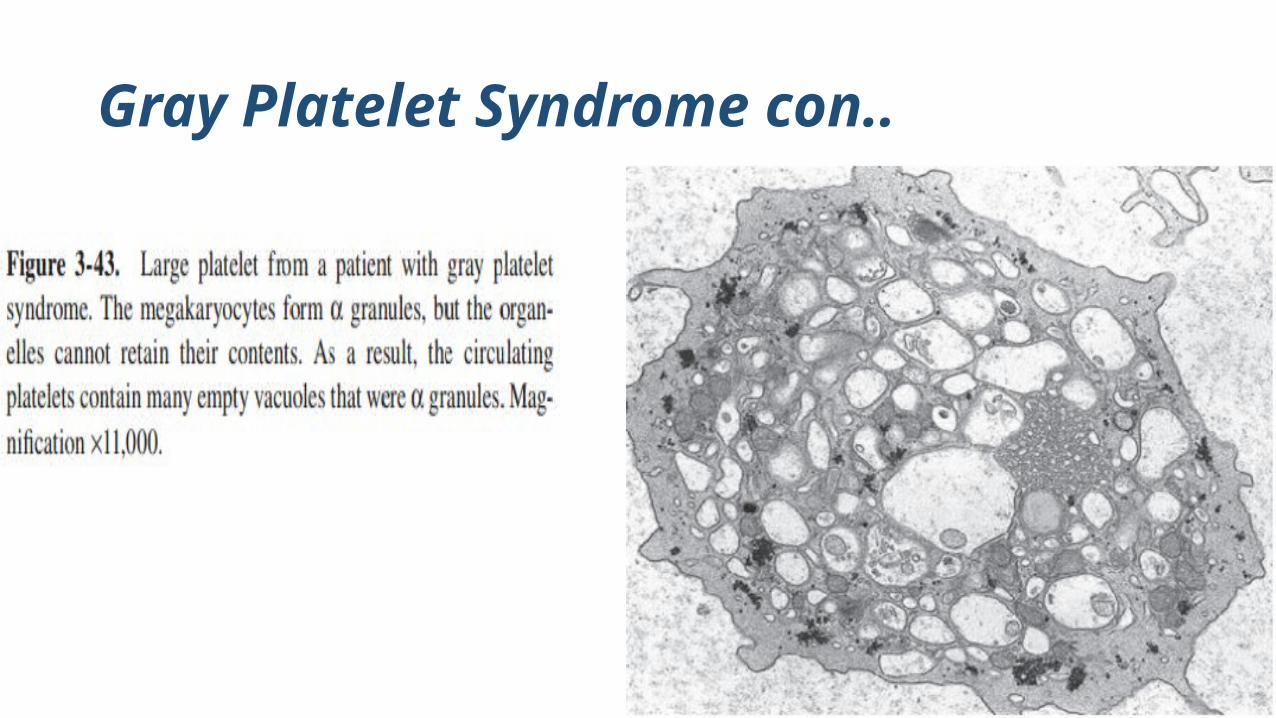
GPS is a heterogeneous disorder associated with abnormalities in α-granule formation and maturation
autosomal recessive inheritancemild thrombocytopeniaA mild bleeding disorder (prolonged bleeding Time)
characterized by:
platelet’s inability to store α-granule proteins
platelets appearing gray on stained blood smears

Gray Platelet Syndrome con..
Under the electron microscope, platelets and megakaryocytes reveal absent or markedly decreased α-granules or small and almost empty
Platelets are severely and selectively deficient in α-granule
proteins
Platelet aggregation responses have been variable:
Responses to ADP and epinephrine were normal in most patients; however, in some patients, aggregation responses to thrombin, collagen, and ADP have been impaired.

Gray Platelet Syndrome con..
Inability of megakaryocytes to store newly synthesized growth factors
and cytokines such as PDGF, and TGF-β1
Plasma levels of PF4 and βTG have been found to be raised in some patients, suggesting that the defect is not in their synthesis by megakaryocytes but in their packaging into granules

Gray Platelet Syndrome con..
The neutrophils from these patients also had decreased granules.
Reticulin is increased in the bone marrow from GPS patients
which can be attributed to elevated plasma PDGF levels
Gray Platelet Syndrome con..

Quebec Platelet Disorderautosomal dominant bleeding disorder
was first described in two FrenchCanadian families
normal to reduced platelet counts
associated with delayed bleeding and abnormal proteolysis of α-granule proteins caused by increased amounts of platelet urokinase-type plasminogen activator (u-PA)

Quebec Platelet Disorder con..
proteolytic degradation of soluble and membrane proteins of α-granules
Platelet factor V but not plasma factor V is degraded along with other α-granule proteins, fibrinogen, vWF, thrombospondin, osteonectin, fibronectin, and P-selectin

Quebec Platelet Disorder con..
platelets are also severely deficient in multimerin.
“multimerin is a high molecular mass protein that is stored as a complex with factor Factor V in α-granules”
platelets of these patients show protease related degradation of many α-granule proteins (including P-selectin) even though α-granule ultrastructure is preserved
Thrombocytopenia is sometimes observed

Quebec Platelet Disorder con..
The platelet aggregation deficiency is, surprisingly, most striking with epinephrine (selective defective aggregation with epinephrine)
the bleeding syndrome responds to fibrinolytic inhibitors rather than platelet transfusions
Originally thought to involve factor V

Quebec Platelet Disorder con..
Patients with QPD suffer from mucocutaneous bleeding, which often is delayed by 12–24 hours following injury
This bleeding is unresponsive to platelet transfusions but responsive to fibrinolytic inhibitors

δ-granules
Human platelet dense bodies are smaller than the αgranules, fewer in number, and have high morphological variability
Normal platelets possess 3-8 dense granules/platelet
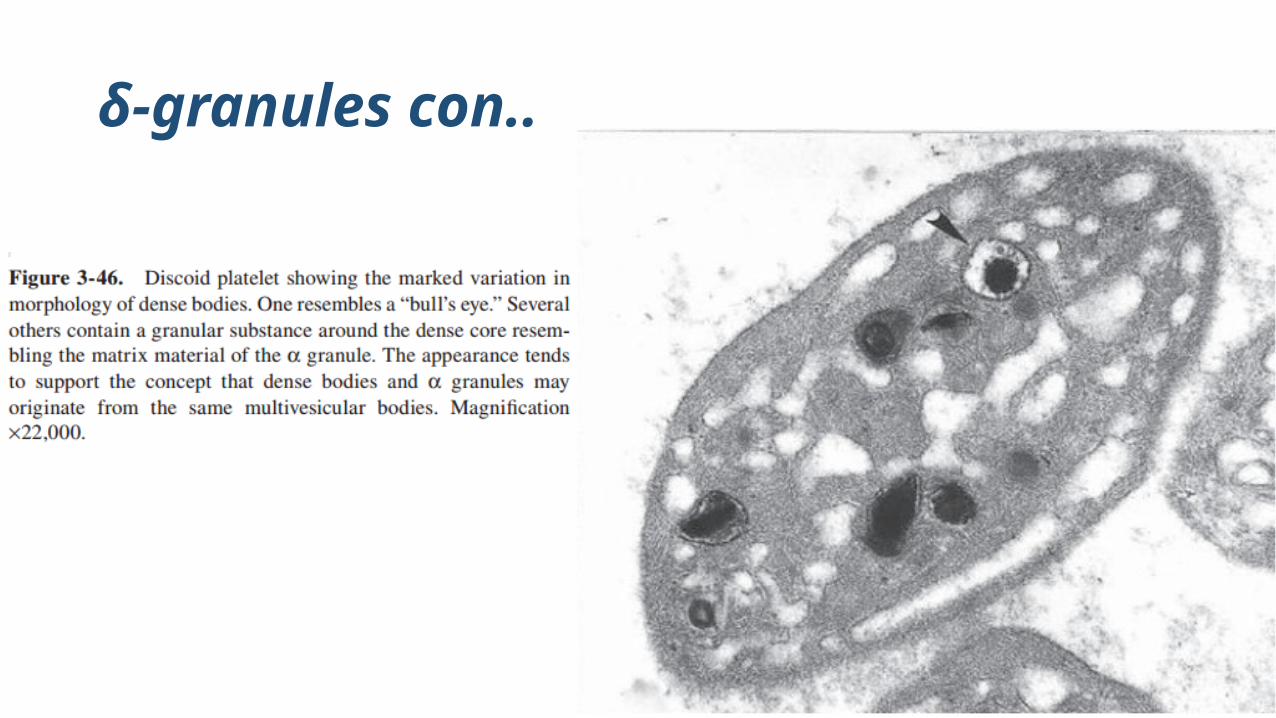
δ-granules con..

Contents of dense granules
Contents include:
• adenosine diphosphate (ADP) (more than ATP)
• adenosine triphosphate (ATP)
• ionized calcium
• magnesium
• histamine
• serotonin
• pyrophosphate

Dense (δ) Granule Disorders
Patients with δ-SPD have a mild to moderate bleeding diathesis associated with prolonged bleeding time
In platelet studies the second wave of aggregation :
• Response to ADP and epinephrine is absent or blunted
• The collagen response is markedly Impaired
• Both impaired and normal aggregation responses to arachidonic acid have been noted

Dense (δ) Granule Disorders con..
Under the electron microscope, dense granules are decreased in δ-SPD platelets
in δ-SPD platelets, the ratio of total ATP to ADP is increased (>2.5) compared with that in normal platelets.

Dense (δ) Granule Disorders con..
δ-SPD has been reported in association with other inherited disorders such as the:
• Hermansky-Pudlak syndrome (HPS)
• Chédiak-Higashi syndrome (CHS)
• Wiskott–Aldrich Syndrome (WAS)
• Thrombocytopenia with absent radii syndrome (TAR)
• Griscelli syndrome

Hermansky-Pudlak syndrome
• Is a rare autosomal recessive disorder
• There are eight classic forms of the disorder, based on the genetic mutation from which the disorder stems
• HPS can be caused by mutations in several genes: HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6 and HPS7(dysbindin)

HPS Symptoms
• Albinism (decrease amounts of skin pigment (melanin))
• light sensitivity (photophobia)
• strabismus (crossed eyes)
• Bleeding disorders (platelet dysfunction)
• Cellular storage disorders (wax-like substance (ceroid) to accumulate in the body tissues and cause damage, especially in the lungs and kidneys)

Chediak–Higashi syndrome
• is a rare autosomal recessive disorder
• mutation of a lysosomal trafficking regulator protein (also called LYST)
• leads to a decrease in phagocytosis
• The disease is characterised by large lysosome vesicles in phagocytes (neutrophils), which thus have poor bactericidal function, abnormalities in nuclear structure of leukocytes, anemia, and hepatomegaly

CHS Symptoms
• light skin and silvery hair (partial albinism)
• complain of solar sensitivity and photophobia
• frequent infections are common
• Neuropathy often begins in the teenage years
• organ failure
• Most children ultimately reach a stage known as the accelerated phase, also known as the lymphoma-like-syndrome
• Bone marrow transplants appear to have been successful in several patients

Wiskott–Aldrich Syndrome (WAS)
• is an X-linked recessive disease
• mutations in a gene on the short arm of the X chromosome, which was termed Wiskott-Aldrich syndrome protein gene (WASp).
• The WASp gene codes for the protein WASp, which is 502 amino acids long and is mainly expressed in hematopoietic cells
• WAS platelets show a decreased aggregation response to ADP, epinephrine, and collagen and have a reduced dense granule number

WAS Symptoms
• most patients are male
• petechiae and bruising, resulting from a low platelet count
• Epistaxis and bloody diarrhea are common
• Eczema develops within the first month of life
• Recurrent bacterial infections
• The majority of WAS children develop at least one autoimmune disorder, and cancers (mainly lymphoma and leukemia)
• (IgM) levels are reduced, IgA and IgE are elevated, and IgG levels can be normal, reduced, or elevated

TAR syndrome
• This syndrome may occur as a part of the 1q21.1 deletion syndrome
• is a rare genetic disorder that is characterized by the absence of the radius bone
• dramatically reduced platelet count
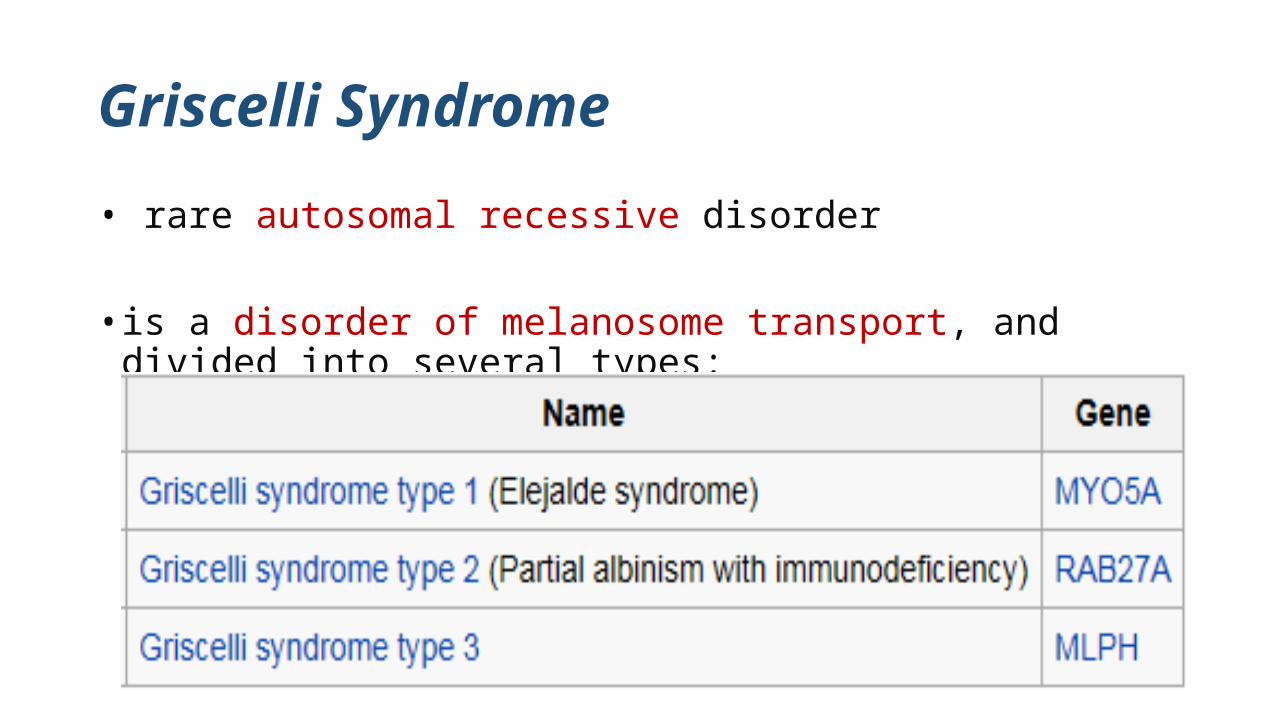
Griscelli Syndrome
• rare autosomal recessive disorder
• is a disorder of melanosome transport, and divided into several types:

Griscelli Syndrome Symptoms
• Hypopigmentation
• Frequent pyogenic infection
• Hepatosplenomegaly
• Neutropenia
• Thrombocytopenia
• Impaired NK cell activity