Nuclear analogs of β-lactam antibiotics. X. Synthesis of 2-substituted desthiocephalosporins
5
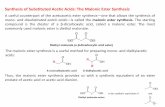
Nuclear analogs of p-lactam antibiotics. X. Synthesis of 2-substituted desthiocephalosporins TERRE~CE WILLIAM DoYLE,*'~ TERRY THOMAS COUWAY.~ MICHAEL CASEY, AND GARY LIM Brrstol Luborarorie5 of Carradn, 100 Ind~lsrrial Boule~iird, Cundinc, P.Q., Cunucici J5R 1J1 Recclvcd Apr 11 20, 1978 TERRENCE WILLIAM DOYLE, TERRY THOMAS CO~WAY, MICHAEL CASEY, and GARY LIM. Can. J. Chem. 57.222 (1979) The syntheses of the desthiocephem system from the en01 2 is described. Thus conversion of 2 to its tr~flate 3 followed by treatment with the sodium salts of malonate esters gave the cyclized products 4a-c. Azide reduction followed by coupling of phenoxyacetic acid and hydrogenolysis gave the acids 7 and 8. Compound 15 was also prepared. TFRRE~CE WILL~AM DOYLE, TERRY THOMAS COYWAY, MICHAEL CASEY et GARY LIM. Can. J. Chem. 57. 222 (1979). On decrit les syntheses du systeme desthiocephem a partir de l'enol 2. Par conversion de 2 en son triflate 3, puis un traitement avec les sels de sodium des esters malonates, on a obtenu les produits cyclises 40-c. La reduction de l'azide, puis le couplage avec l'acide phenoxyacetique et I'hydrogenolyse ont conduit aux acides 7 et 8. On a egaleme~lt prepare le compose 15. [Traduit par le journal] Over the past several years a number of leports on +OCH~CONHJ~N:^+ the preparation of nuclear analogs of the cephalo- x = 0, NR', S, SO, SO,, C spor~ns have appeared ~nthe Ilterature." Previously we 0 R \R" have reported (2)5 the syntheses of a nunlber of C02H ailalogs (Flg. 1) in ~vhlch the sulfur atom at 1 has t been replaced by a inethyle~le (or subst~tutedcar- Frc 1 Cephalosporln analog, bon) and a hetiro atom ;ntrod;ced in position 2. The syntheses of these analogs proceeded via the of in the presence triethyl- common intermediate 2, ~i~~~~ base treatment of amine with trifluoromethane sulfonic anhydride gave the eno] 2 gave the 0-2-isocephem series whereas the h i g h yield of the correspondi~lg triflate 3 as a mix- preparation of the 2-isocephem (X = S) and N-2- ture of isomers (1). It proved possible to obtain one isocephem systems proceeded nucleophilic attack of the isomers as a crystalline solid at the expense of on the triflate 3 (or the corresponding mesylate). ~t I o ~ ~ E considerable amoullts of product. he crude occurred to us that treatment of 3 with aDDroDriate triflate was generally sufficiently pure to be used as LA carbon nucleoph~les might well provlde access to the desth~ocephalospor~ns At the tlme no ~eports of such analogs had appeared In the literature but about the tlrne Me uele completing our own work a report appeared by Chrlstensen and co-uorkers (3) of the synthes~s of the I-oxacephalosporln and 1 -car bacephalosporln ( = desth~ocephalospor~~~) sys- tems These authors reported that the carbon analog exhlblted activity approx~mately equal to that In the natural serles In this and the accompanying papers we should 11ke to report our own results In thls area. 'For Part IX of this series see ref. 5. 'To whom correspondence concerning this paper may be addressed. 3Present address: Bristol Laboratories, P.O. Box 657, Syracuse, NY 13201, U.S.A. 4For a brief review of some of these papers see ref. 2a. 5For a detailed argument concerding the conformations of these and similar analogs in solution see ref. 26. such. As we had anticipated, treatment of 3 with sodium dimethylmalonate in tetrahydrofuran gave the desired cyclized product 40 in 68;; yield after chro- matography. Similarly replacing the dimethyl mal- onate in the reaction with dibenzyl malonate or di-trrt-butyl malonate gave the products 4b and 4c in 60 and 52% yields, respectively. The structures assigned to 4a-c were made on the basis of their elemental analyses and by con~parison of their uv, ir, and nrnr spectra with those of the earlier nuclear analogs synthesized in these laboratories. Examina- tion of the proton nmr (Table 1) enables us to assign the conformation illustrated in Fig. 2 to these molecules. Reduction of the azido function in 4a using triethylamine - hydrogen sulfide gave the amine 50 in 91:5 yield. Conversion of the amine 5ci to the amide 6a proceeded in near quantitative yield using 0008-4042/79/020222-05m .W/0 a1979 National Research Council of CanadalConseil national de recherches du Canada Can. J. Chem. Downloaded from www.nrcresearchpress.com by CASE WESTERN RESERVE UNIV on 11/09/14 For personal use only.
Transcript of Nuclear analogs of β-lactam antibiotics. X. Synthesis of 2-substituted desthiocephalosporins

Nuclear analogs of p-lactam antibiotics. X. Synthesis of 2-substituted desthiocephalosporins
T E R R E ~ C E WILLIAM DoYLE,*'~ TERRY THOMAS C O U W A Y . ~ MICHAEL CASEY, AND GARY LIM Brrstol Luborarorie5 of Carradn, 100 Ind~lsrrial Boule~iird, Cundinc, P.Q., Cunucici J5R 1J1
Recclvcd Apr 11 20, 1978
TERRENCE WILLIAM DOYLE, TERRY THOMAS CO~WAY, MICHAEL CASEY, and GARY LIM. Can. J. Chem. 57.222 (1979)
The syntheses of the desthiocephem system from the en01 2 is described. Thus conversion of 2 to its tr~flate 3 followed by treatment with the sodium salts of malonate esters gave the cyclized products 4a-c. Azide reduction followed by coupling of phenoxyacetic acid and hydrogenolysis gave the acids 7 and 8. Compound 15 was also prepared.
T F R R E ~ C E WILL~AM DOYLE, TERRY THOMAS COYWAY, MICHAEL CASEY et GARY LIM. Can. J. Chem. 57. 222 (1979).
On decrit les syntheses du systeme desthiocephem a partir de l'enol 2. Par conversion de 2 en son triflate 3, puis un traitement avec les sels de sodium des esters malonates, on a obtenu les produits cyclises 40-c. La reduction de l'azide, puis le couplage avec l'acide phenoxyacetique et I'hydrogenolyse ont conduit aux acides 7 et 8. On a egaleme~lt prepare le compose 15.
[Traduit par le journal]
Over the past several years a number of leports on +OCH~CONHJ~N:^+ the preparation of nuclear analogs of the cephalo- x = 0, NR' , S , SO, SO,, C
spor~ns have appeared ~n the Ilterature." Previously we 0 R \R"
have reported (2)5 the syntheses of a nunlber of C 0 2 H
ailalogs (Flg. 1) in ~vhlch the sulfur atom at 1 has t
been replaced by a inethyle~le (or subst~tuted car- Frc 1 Cephalosporln analog,
bon) and a hetiro atom ;ntrod;ced in position 2. The syntheses of these analogs proceeded via the of in the presence triethyl- common intermediate 2, ~i~~~~ base treatment of amine with trifluoromethane sulfonic anhydride gave the eno] 2 gave the 0-2-isocephem series whereas the h i g h yield of the correspondi~lg triflate 3 as a mix- preparation of the 2-isocephem (X = S) and N-2- ture of isomers (1). I t proved possible to obtain one
isocephem systems proceeded nucleophilic attack of the isomers as a crystalline solid at the expense of
on the triflate 3 (or the corresponding mesylate). ~t I o ~ ~ E considerable amoullts of product. he crude occurred to us that treatment of 3 with aDDroDriate triflate was generally sufficiently pure to be used as
L A
carbon nucleoph~les might well provlde access to the desth~ocephalospor~ns At the tlme no ~ e p o r t s of such analogs had appeared In the literature but about the tlrne Me uele completing our own work a report appeared by Chrlstensen and co-uorkers (3) of the synthes~s of the I-oxacephalosporln and 1 -car bacephalosporln ( = des th~ocepha lospor~~~) sys- tems These authors reported that the carbon analog exhlblted activity approx~mately equal to that In the natural serles In this and the accompanying papers we should 11ke to report our own results In thls area.
'For Part IX of this series see ref. 5. 'To whom correspondence concerning this paper may be
addressed. 3Present address: Bristol Laboratories, P.O. Box 657,
Syracuse, NY 13201, U.S.A. 4For a brief review of some of these papers see ref. 2a. 5For a detailed argument concerding the conformations of
these and similar analogs in solution see ref. 26.
such. As we had anticipated, treatment of 3 with sodium
dimethylmalonate in tetrahydrofuran gave the desired cyclized product 40 in 68;; yield after chro- matography. Similarly replacing the dimethyl mal- onate in the reaction with dibenzyl malonate or di-trrt-butyl malonate gave the products 4b and 4c in 60 and 52% yields, respectively. The structures assigned to 4a-c were made on the basis of their elemental analyses and by con~parison of their uv, ir, and nrnr spectra with those of the earlier nuclear analogs synthesized in these laboratories. Examina- tion of the proton nmr (Table 1) enables us to assign the conformation illustrated in Fig. 2 to these molecules.
Reduction of the azido function in 4a using triethylamine - hydrogen sulfide gave the amine 50 in 91:5 yield. Conversion of the amine 5ci t o the amide 6a proceeded in near quantitative yield using
0008-4042/79/020222-05m .W/0 a1979 National Research Council of CanadalConseil national de recherches du Canada
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CA
SE W
EST
ER
N R
ESE
RV
E U
NIV
on
11/0
9/14
For
pers
onal
use
onl
y.

DOYLE ET AL. I 223
TABLE 1. Nuclear magnetic resonance spectraa
Compound Aromatic protons ArCH, CH 3 Other
4.45 (m, 3H, CHCH,OMes)
2.65(1H, dd, Jl = 13 J2 = 3.5)
2.14 (IH, m) 2.66 (dd, J1 = 13
J, = 3 .5 , lH) 2.14 (m, 1H)
2.59 (dd, lH , J 1 = 13, J2 = 3.5)
1 .92(dd, I H, Jl = 12, J2 = 13)
2.68 (dd, 1H, J1 = 13 J2 = 3.5)
1.94 (dd, 1H, J1 = 13 J, = 12)
1.68 (bs, 2H, NH,) 1.92 (dd, Jl = 13,
J, = 12) 2.68 (dd, J1 = 13,
J, = 3.5) 1 .50 (bs, 2H, NH,) 4.46 (s, 2H, 40CH2) 1.84 (dd, J l = 13
J2 = 12) 2.51 (dd, J 1 = 13
J* = 3.5) 2.53 (m, 1H) 1.84 (m, lH) 4.38 (s, 2H, +OCH2) 4.50 (s, 2H, +OCH2) 2.30 (m, 2H)
1-3 (bs, 2H)
3.30 (bd, lH, CHCOZH)
1.70-2.6 (m 2H) 4.73 (bs, lH,
CHCO~BZ) 2.60 (bd, J - 8 Hz) 3.35 m(1H) 1 .7-2.4(m, 2H) 1 .60 (bs, NH,) 1.7-2.4 (m, 2H) 3.33 m (lH, bd,
J - 6) 4.48 (s, 2H, +OCH2) 1 .5-2.3 (m, OH) 3.22 (m, lH , bd
J = -6) 4.55 ( s , 2H, (bOCH2) 3.32 (m, IN) 1 .5-2.3 (m, 2H)
2.08(s) 3.70(s, OMe) 2 . 1 0(s) 3.73(s)
"Recorded at 60 MHz in CDCI, unless otherwise noted. The coupling constants are recorded in DArms of AB quartet. CMinor isomer. QDMSO-d,.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CA
SE W
EST
ER
N R
ESE
RV
E U
NIV
on
11/0
9/14
For
pers
onal
use
onl
y.

C.4N. J . CHEM. VOL. 57. 1979
c ~ f ~ o ~ R " FIG. 2. Conformation of desthiocephalosporins
2 - ethoxy - N - ethoxycarbonyl - 1,2 - dihydroquinoline (EEDQ) and phenoxyacetic acid (4). Similarly 46 was converted to 56 and subsequently to 6b.
Hydrogenation of 60 over 10% palladium-on- carbon gave 3-methyl-2,2-biscarbomethoxy-7-P-phe- noxyacetamido-A3-desthiocephem-4-carboxyic acid 7 in 70% yield. Reduction of 66 over 1 0 s palladium- on-carbon in acetic acid resulted in loss of all three benzyl functions and decarboxylation to give 8 in 44% yield.
To explore the synthesis of various 2-mono- substituted analogs the deesterification of 4c was studied next (Scheme 2). Treatment of 4c with tri- fluoroacetic acid a t 60cC gave a foam the nmr spec- trum of which was compatible with the diacid 9. A solution of the foam in toluene ivas boiled at reflux for 10 min and evaporated to yield a mixture of the
two conlpounds 10 and 11, in a 3 : 1 ratio. Partial resolution of the mixture of acidc was achieved by colunln chromatography on silica gel and the struc- tural assigilmellts made on the basis of their nmr s ~ e c t r a . The acid mixture was used as such in the next step. Thus treatment of the mixture with methyl chloroformate and tricthylamine gave the methyl ester 12 in - 5 5 3 i e l d . The crude ester was reduced to the amine 13 using triethylamine - hydrogen sul- fide and 13 was coupled to phenoxyacctic acid using EEDQ to give 14. Hydrogenolysis of 14 gave the desired acid 15. The stereochernistry of the car- bomethoxy group in 14 and 15 was not determined owing to poor resolution of the nmr spectrum.
The biological activities of compounds 1, 8, and 15 were disappointingly low and will be discussed in full in a forthcoming structure-activity relationship paper.
Experimental The infrared spectra were recorded on a Unicam Sp-200 G
grating ir spectrophotometer. The nmr spectra were deter- ~nined on a Varian A60-A spcctronieter using tetramethylsilane as an internal standard. Melting points are uncorrected except where noted and were determined on a Gallenkan~p melting point apparatus. The analyses were performed by Micro- Tech Laboratories, Skokie, IL.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CA
SE W
EST
ER
N R
ESE
RV
E U
NIV
on
11/0
9/14
For
pers
onal
use
onl
y.

DOYLE ET AL. I 225
Preparation of Vinyl TriJlhte 3 The en01 2 (48.0 g, 0.117 mol) was dissolved in 500 ml
CaC12-dried CH,CI, and then cooled in an ice bath under N,. Triflic anhydride (33.0 g, 19.5 nil, 0.1 17 niol) was added in one portion. A solution o f triethylarnine (11.8 g, 16.3 nsl, 0.117 mol) in 80 ml CH,CI, was added dropwise over a period o f 40 min. When the addition was complete, the ice bath was removed and the mixture stirred at 25-C for a further 45 min.
The inixture was poured into 300 ml ice-H,G and washed with cold H,O until the pH was - 6 (five or six washings were necessary). The CH2CI, solution was dried (Na2SO4) and evaporated to give a dark red oil (54.0 g) which was dissolved in 400 ml benzene and passed through a If-in. pad o f silica gel (15% water, w!w) (,-- 150g); the pad was washed with 1 f o f benzene. Evaporation o f the benzene gave a yellow oil (38.3 g) which contained -20% unreacted enol. Eluting the pad with 500 mI CH2CI, gave 5.42 g o f a 40:60 mixture o f triflate and enol. Further elution with 500 ml E t20 gave 5.34 g of unreacted enol.
The yellow oil which contained the two isomeric triflates was triturated with 50 ml o f absolute EtOH until there were no more large gummy particles. After cooling to 0°C for a few hours, the suspension was filtered to afford 19.5 g o f a white powder consisting o f one triflate isomer, mp 57-59°C; ir (neat): 2110, 1790, 1730, 1656 cm-' ; uv (EtOH) 1..,,,: 242 (E
6360). A~ml . calcd. for C1,H,,F,N,09S2: C 37.67, H 3.14, N 10.33, S 11.82; found: C 37.40, N 3.12, N 10.43, S 11.73.
Preparation of Compozrnds 4a-c Conlpo~trld 4a T o a suspension o f 225 mg (5.34 mmol) o f sodium hydride
(57% mineral oil dispersion washed three times with petroleum ether) in 2 ml dry tetrahydrofuran, under nitrogen, and cooled to 0-5"C, was added 1.32 g (2.43 mmol) o f 3 and 282 mg (2.43 mol) o f dimethylmalonate in 3 ml o f dry tetrahydrofuran over 10 min. A red color developed and hydrogen was evolved. The reaction was stirred at 25'C for I .5 h at the end o f which time the reaction was complete as determined by thin-layer chromatography.
Five percent HCl was added t o the reaction (pH 3.4) and was then distributed between ether and water. The organic phase was washed with water, dried (Na,S04), and concen- trated in cncuo to give 4a (1.02 g, 98%) as a yellow oil. The oil was chromatographed on silica gel (15% H,O), developed with 1 volume hexane, and eluted with benzene. Pure 40 was ob- tained (688 mg, 66%); ir (neat): 2110, 1780, 1740, 1730, 1630 c m - ' ; uv (EtOH) A,,,,: 270 (E 1 1 200).
Co~npound 4b Similarly treatment o f 504 mg (1 1.95 mmol) o f sodium
hydride with 2.95 g (5.45 mmol) o f 3 and 1.54 g (5.45 mmol) o f dibenzyl malonate in THF gave 1.88 g (60%) o f 4b as an oil
after chromatography; ir (neat): 21 10, 1780, 1740, 1730 cnl-' ; uv (EtOH) i,,,,: 270 ( E 10 967). Anal. calcd. for C 3 2 H 2 S N 4 0 7 : C 66.12, H 4.86, N 9.65; found: C 66.68, H 4.99, N 9.31.
Con?polmd 4c Treatment o f 2.59 g (12 mmol) o f di-terr-butyl nialonate and
6.65 g (12 mmol) o f 3 with 1.05 g (24 n ~ n ~ o l ) sodium hydride in tetrahydrofuran as in the previous examples gave, after the usual work-up, 4c as a light brown oil. Trituration o f this oil with a small volume o f ethanol induced crystallization to give 3.2 g (52%) o f 4c as a colorless solid, nsp 146-147°C after recrystallization from ether; ir (CHCI,): 21 10, 1780, 1725(s 1738) c m - ' ; uv (EtOH) F.,,,,: 271 ( E 1 1 724). Anal. calcd. for C Z ~ H ~ Z N ~ O ~ : C 60.92, H 6.29, N 10.93; foulld: C 61.03, H 6.38, N 11.03.
Preparation of Compornlds 5a and b Compound 5a The azide 4n (588 mg, 1.37 mmol) was dissolved in 10 rnl
lnethylene chloride and triethylamine (700 mg, 0.96 ml, 6.93 mmol) was added. Hydrogen sulphide was bubbled in slowly producing an orange-red color and complete reaction to a niore polar (tlc) material after 45 min. The solution was con- centrated in uacuo and the yellow residue distributed between ether and 10% HCI. The organic phase was washed once with 10% HCI which was then combined with the previous washings and extracted once with ether. The acidic solution was brought to pH 8 with solid NaHCO, (foams!) and saturated with NaCl precipitating the amine 5u as a thick oil which was extracted into CH,CI,. The combined extracts were washed once with brine, dried (Na,SO,), and evaporated in caclro leaving 5a (500 nig, 9 1 z ) , which was used as such in the following reac- tion. A small amount o f this material was converted to the hydrochloride salt (EtOH-HCI; Et,O), mp 178-180°C; ir (free base, neat): 3410, 3350, 1770, 1740, 1725 cm-I . Ai2al. calcd. for C,,H22N207~HC1: C 54.73, H 5.28, N 6.38; found: C 54.10, H 5.41, N 6.26.
Coinpound 5b Treatment o f 1.88 g (3.08 mmol) o f 4b with triethylamine
(6.16 mmol) and hydrogen sulfide as in the above example gave 1.07 g (100%) o f 5b as an oil on work-up. The amine 5b was used as such in the subsequent reaction.
Preparation of Comnpounds 6a und b Compound 6a Amlne 5n (350 mg, 0.87 mmol) was st~rred in methylene
chloride (20 ml) with phenoxyacetic acid (132 mg, 0.86 mmol) and EEDQ (220 mg, 0.89 mmol) until CO, evolution had ceased (80 min). The clear solutlon was washed with 1% NaHCO, (2 :: 15 ml), 10% HC1 (2 x 10 ml), brine (2 x 20 ml), dried (Na,SQ4), and concentrated in cacuo t o give 6n (480 mg, 103%) as a white foam. The material was chroma-
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CA
SE W
EST
ER
N R
ESE
RV
E U
NIV
on
11/0
9/14
For
pers
onal
use
onl
y.

226 CAN. .I. CHEM. VOL. 57. 1979
tographed on 10 g silica gel (15% H,O) in CHC1,-Et20 (9: 1). Pure 6a was lsolated (480 mg, 103%) as a glas5y solld; ir (sn~eal): 3400, 1780, 1740, 1685, 1600, 1530, 1495 cnl-'; uv (EtOH) i.,,,: 269 (E 6778). Anal. calcd. for C28H2sN209: C 61.64, H 5.35, N 5.13; found: C 61.37, H 5.26, N 5.02.
Compound 6b Anline Sb (1.8 g, 2.13 rnmol) was stirred in CH,C12 (100
ml) with phenoxyacetic acid (471 mg, 3.1 mmol) and EEDQ (765 rng, 3.1 mmol) until CO, evolution had ceased (1.5 h). The solution was washed with 1% NaHCO, (2 x 15 ml), 10% HC1 (2 x 10 ml), brine (2 x 20 ml), dried (Na2S04), and con- centrated in cncuo to give 66 (2.05 g) as a red oil. This material was chromatographed on SiO, (35 g, 15% H,O) eluting with CHCI, to give white crystals (1.2 g) mp 134-135'C (corrected); ir (Nujol mull): 3320, 1770, 1725, 1680, 1550, 1495 cm- ' ; uv (EtOH) ).,,,: 269 (E 14 556). Anal. calcd. for C4,H,,N,0,: C 69.85, H 5.12, N 4.07; found: C 69.51, H 5.33, N 3.94.
3-1Methyl-2,2-biscm.bot~~ethoxy-7-[3-(phe1zo.xyaceramido) -A3- cle~thiocephern-4-carbosjlic Acid (7)
Amide 60 (410 mg, 0.76 mniol) was dissolved in 60 mI EtOH - 8 ml T H F and hydrogenated at 1 atrn and 25°C over 10% Pd/C (410 mg); hydrogen uptake was complete at 10 min (27 nil, 145%). The catalyst was filtered off and washed with EtOH (50 1111) and evaporation of the filtrate left 7 (306 mg, 90%) as a clear oil. It was crystallized from acetone-pentane to give 237mg (70x1 of a white powder, mp 186-188'C (corrected); ir (Nujol mull): 3315, 3340, 2500-3600, 1785, 1755, 1725(1745s), 1665, 1600, 1550, 1500 cm-' ; uv (EtOH)
267 (E 9580). Atznl. calcd. for C, ,H2,N20a: C 56.50, H 4.96, N 6.27; found: C 56.47, H 4.97, N 6.24.
3-:Methyl-7-[3-(pheno.xynceiur~1icio) -A3-&.c-tl~iocephenz-2,4- dicarho.uylic Acid ( 8 )
A mixture of amide 60 (270 ~ n g , 0.39 mmol) and 10% Pd/C (270 mg) in glacial AcOH (10 ml) was hydrogenated at 1 atni and 25'C until H, uptake ceased. (More than the theoretical amount of H Z was absorbed in all cases.) The catalyst was filtered off and the AcOH was evaporated at t 1 Torr, 40-50'C water bath. The glassy residue was covered with ben- zene and reevaporated to remove the last traces of AcOH. The residue was crystallized by warming in water. The resulting white powdery crystals were dried in cacuo over P2O5 for 18 h (64 mg, 43.5%), mp 158-159'C (corrected); ir (Nujol mull): 3410, 3370, 2500-3600, 1775, 1750, 1725, 1715, 1685, 1650, 1600, 1525, 1500 C I ~ I - ' ; uv (EtOH) h,,,,: 268 (E 5180). Arral. calcd. for C,8H,8N207.+H20: C 56.39, H 4.99, N 7.30; found: C 56.70, H 4.83, N 7.35.
Ester Cleauage of4c A solution of 555 rng (1.085 mmol) of 4c in 15 ml of trifluo-
roacetic acid was placed on a rotary evaporator at 62'C and the solvent evaporated over a period of 5-10 min to give 425 mg of a foam the nmr spectrum of which indicated the com- plete loss of the tert-butyl signals. The foam was taken up in toluene and refluxed 10 min following which the toluene was evaporated to yield 380 mg (98.5%) of a white foam. The nmr spectrum of the crude material showed two methyl signals and two benzyl signals in a ratio of 3: 1 indicating that decarboxyla- tion of the diacid had occurred. The foam was chromato- graphed on 10 g silica gel (15z water w/w) using chloroforn~ as eluent. The eluting solvent was gradually changed to ether. The earlier fractions gave material greatly enriched in com- pound 10, the major isomer, while the colun~n tailings were mostly compound 11. P~epuration of Conlpound I 4
To a solution of 500 mg (1.4 mmol) of crude acid (3 : 1 mix-
ture of 10 and 11) in 25 ml methylene chloride was added 150 Ing (1.5 mmol) of triethylamine. The solution was cooled to - 10-C (ice-methanol) and 150 mg (1 .6 mmol) methyl chloroformate in 2 ml CH,C12 was added dropwise. The solu- tion was stirred 1.5 h. The solution was evaporated to dryncss at reduced pressure and the residue partitioned between water and ether. The ethereal extracts were washed with 10% hydro- chloric acid and then saturated sodium bicarbonate solution. Evaporation of the organic layer gave 300 mg crude methyl ester 12 which was used as such in the subsequent step. From the basic extracts, 120 mg of starting acid was recovered (mixture).
Reduction of 300 mg of c r ~ ~ d e ester using triethylamine (100 mg) - hydrogen sulfide gave, upon work-up (see prepara- tion of Srr), 200 rng of 13 as an oil; ir (film): 3420, 3360, 1770, 1750, 1740, 1640 cm- ' .
Treatment of 172 mg (0.5 mmol) of the amine 13 with 76 mg (0.5 mmol) phenoxyacetic acid and 124 mg (0.5 mmol) EEDQ in 10 ml methylene chloride gave 220 mg (92%) of 14 as a white foam. Chromatography of this material on silica gel (1 5% water, w/w) using ether as eluent gave 200 mg of pure 14, mp 57-58°C; ir (CHCI,): 3430, 1770, 1740, 1695, 1600, 1590, 1525, 1495 cm- ' ; uv (EtOH) i,,,,: 268 (E 12 900). Anal. calcd. for C2,H,,N,0,~)H20: C 64.06, H 5.58, N 5.75; found: C 64.31, H 5.52, N 5.60.
3-Methyl-2-carbornethoxy-7-a- (phenoxyacetnnlido) -A3- desthiocephern-4-curho.xj~lic Acid ( I S )
Reduction of I00 mg (0.208 mmol) of 14 with I00 mg 10% Pd/C in 5 1111 T H F and 10 ml ethanol at 1 atm and 22'C for 3 h gave, after work-up (see preparation of 7), 75 mg of a white powder, lnp 8 4 T (dec.); ir (CHCI,): 3420, 2500-3600, 1773, 1740, 1695, 1600, 1530, 1495 cnl-'; uv (EtOH) I,,,: 265 (E 8150). Anal. calcd. for C19HZONZ02.+H20: C 57.43, H 5.33, N 7.04; found: C 57.87, H 5.60, N 6.61.
Acknowledgements
Partial financial support of this work by the National Research Council of Canada through its Industrial Research Assistance Plan is gratefully acknowledged. We also thank Dr. Bernard Belleau of McGill University for helpful discussions during the course of this work
1. T. T. CONWAY. G. LIM, J . L. DOUGL.AS. M. MENARD. T. W. DOYLE; P. KIVEST. D. HORNING, L. K. MORRIS, and D. CIMON. Can. J . Chem. 56, 1335 (1978).
2. ((1) T. W. DOYLE, B. BELLEAU. B. LUH. C. F . FERRARI, and M. P. CUNNINGHAM. Can. J . Chem. 55. 468 (1977); (h) T. W. DOYLE, B. BELLEAU, B. LVH, T. T. CONWAY, M. MENARD, J . L . DOUGLAS, D. T. CHU, G. LIM, L. R. MOR- RIS, P. RIVEST. and M. CASEY. Can. J . Chem. 55, 2719 (1977); (c) T. W. DOYLE, B. Y. LUH, D. T. CHU, and B. BELLEAU. Can. J . Chem. 55,484 (1977): (d) T. W. DOYLE, J . L . DOUGLAS, B. BELLEAU, J . MEUNIER, and B. LUH. Can. J . Chem. 55,2873 (1977).
3. L. D. CAMA and B. G. CHRISTENSEN. J . Am. Chem. Soc. 96, 7582 (1974); R. N. GUTHIKONDA. L. D. CAIVIA, and B. G. CHRISTEUSEN. J. Am. Chern. Soc. 96,7584 (1974).
4. B. BELLEAU and G. MALEK. J . Am. Chem. Soc. 90. 1651 (1968).
5. T. T. CONWAY, J . L . DOUGLAS, and D. E. HORNING. Can. J . Chem. 56,2879 (1978).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CA
SE W
EST
ER
N R
ESE
RV
E U
NIV
on
11/0
9/14
For
pers
onal
use
onl
y.