
Nickel promoted olefin dimerizations - Harvard...
35
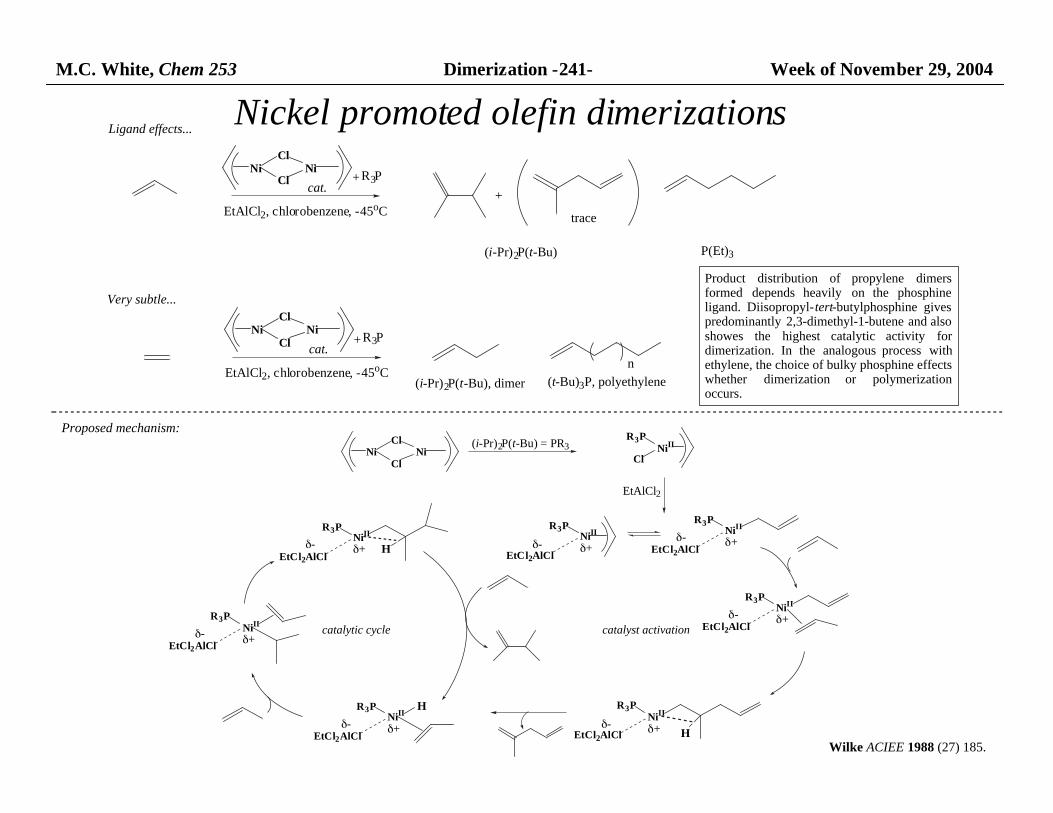
M.C. White, Chem 253 Dimerization -241- Week of November 29, 2004 Nickel promoted olefin dimerizations Ni Cl Cl Ni (i-Pr) 2 P(t-Bu) = PR 3 Ni II R 3 P Cl EtAlCl 2 Ni II R 3 P EtCl 2 AlCl δ+ Ni II R 3 P EtCl 2 AlCl δ+ Ni II R 3 P EtCl 2 AlCl δ+ δ- δ- δ- Ni II R 3 P EtCl 2 AlCl δ+ δ- H Ni II R 3 P EtCl 2 AlCl δ+ H δ- Ni II R 3 P EtCl 2 AlCl δ+ δ- Ni II R 3 P EtCl 2 AlCl δ+ δ- H catalyst activation catalytic cycle Proposed mechanism: Wilke ACIEE 1988 (27) 185. Ni Cl Cl Ni cat. EtAlCl 2 , chlorobenzene, -45 o C R 3 P + (i-Pr) 2 P(t-Bu) P(Et) 3 Product distribution of propylene dimers formed depends heavily on the phosphine ligand. Diisopropyl- tert -butylphosphine gives predominantly 2,3-dimethyl-1-butene and also showes the highest catalytic activity for dimerization. In the analogous process with ethylene, the choice of bulky phosphine effects whether dimerization or polymerization occurs. Ligand effects... Very subtle... Ni Cl Cl Ni cat. EtAlCl 2 , chlorobenzene, -45 o C R 3 P + (i-Pr) 2 P(t -Bu), dimer n (t-Bu) 3 P, polyethylene trace +
Transcript of Nickel promoted olefin dimerizations - Harvard...
M.C. White, Chem 253 Dimerization -241- Week of November 29, 2004
Nickel promoted olefin dimerizations
NiCl
ClNi
(i-Pr)2P(t-Bu) = PR3 NiIIR3P
Cl
EtAlCl2
NiIIR3P
EtCl2AlClδ+
NiIIR3P
EtCl2AlClδ+
NiIIR3P
EtCl2AlClδ+
δ-δ-
δ-
NiIIR3P
EtCl2AlClδ+δ-
H
NiIIR3P
EtCl2AlClδ+
H
δ-
NiIIR3P
EtCl2AlClδ+δ-
NiIIR3P
EtCl2AlClδ+δ- H
catalyst activationcatalytic cycle
Proposed mechanism:
Wilke ACIEE 1988 (27) 185.
NiCl
ClNi
cat.
EtAlCl2, chlorobenzene, -45oC
R3P+
(i-Pr)2P(t-Bu) P(Et)3
Product distribution of propylene dimersformed depends heavily on the phosphineligand. Diisopropyl-tert-butylphosphine givespredominantly 2,3-dimethyl-1-butene and alsoshowes the highest catalytic activity fordimerization. In the analogous process withethylene, the choice of bulky phosphine effects whether dimerization or polymerizationoccurs.
Ligand effects...
Very subtle...
NiCl
ClNi
cat.
EtAlCl2, chlorobenzene, -45oC
R3P+
(i-Pr)2P(t-Bu), dimer
n
(t-Bu)3P, polyethylene
trace
+
M.C. White, Q. Chen Chem 253 Dimerization -242- Week of November 29, 2004
ZrI V
Cl
Cl MAO ZrIV
ClAl(MAO)
Me
δ-
?
EtZrI V
ClAl(MAO)
Meδ-
Et
ZrI V
ClAl(MAO)δ-
Et
δ+ δ+
ZrI V
ClAl(MAO)
Hδ- Et
ZrIV
ClAl(MAO)
Hδ-
Et
δ+
ZrIV
ClAl(MAO)δ-δ+
Et
ZrIV
ClAl(MAO)δ-
Et
δ+
Et
ZrI V
ClAl(MAO)δ-δ+
EtEt
H
Et
Et
Bergman JACS 1996 (118) 4715.
Et
Et
Et
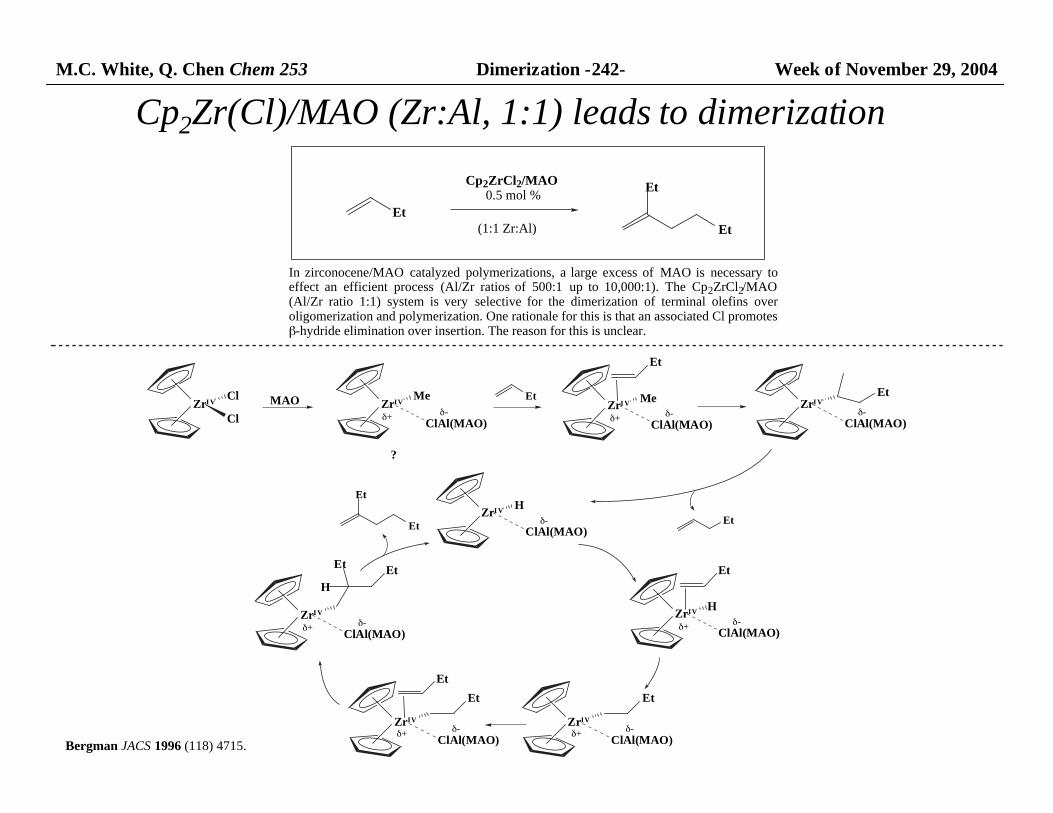
In zirconocene/MAO catalyzed polymerizations, a large excess of MAO is necessary toeffect an efficient process (Al/Zr ratios of 500:1 up to 10,000:1). The Cp2ZrCl2/MAO(Al/Zr ratio 1:1) system is very selective for the dimerization of terminal olefins overoligomerization and polymerization. One rationale for this is that an associated Cl promotes β-hydride elimination over insertion. The reason for this is unclear.
Cp2ZrCl2/MAO 0.5 mol %
(1:1 Zr:Al)
Cp2Zr(Cl)/MAO (Zr:Al, 1:1) leads to dimerization
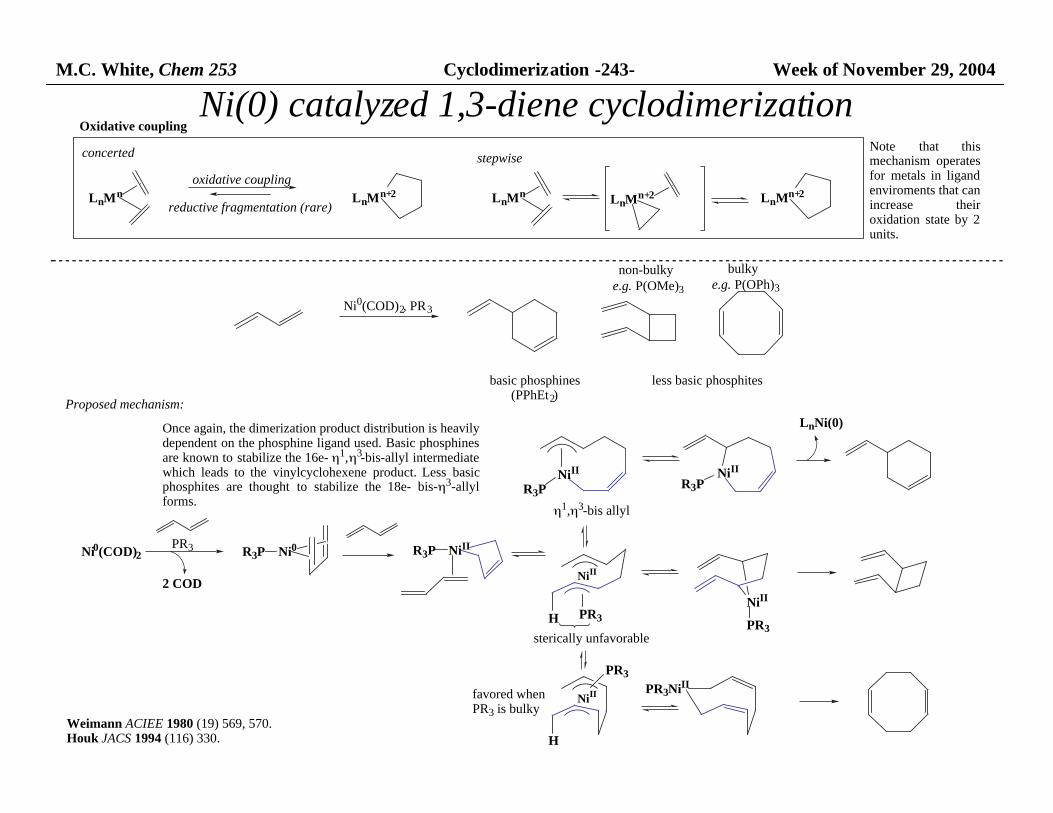
M.C. White, Chem 253 Cyclodimerization -243- Week of November 29, 2004
Ni(0) catalyzed 1,3-diene cyclodimerization
Ni0R3P NiIIR3P
NiII
R3PNiII
R3P
NiII
PR3HNiII
PR3
NiII
H
PR3
PR3NiII
Ni0(COD)2PR3
2 COD
η1,η3-bis allyl
LnNi(0)
sterically unfavorable
favored when PR3 is bulky
Proposed mechanism:
Weimann ACIEE 1980 (19) 569, 570.Houk JACS 1994 (116) 330.
Once again, the dimerization product distribution is heavily dependent on the phosphine ligand used. Basic phosphinesare known to stabilize the 16e- η1,η3-bis-allyl intermediatewhich leads to the vinylcyclohexene product. Less basicphosphites are thought to stabilize the 18e- bis-η3-allylforms.
LnMn
oxidative coupling
reductive fragmentation (rare)LnMn+2
Note that thismechanism operatesfor metals in ligandenviroments that can increase theiroxidation state by 2units.
LnMn+2
concerted stepwise
LnMn LnMn+2
Oxidative coupling
Ni0(COD)2, PR3
basic phosphines(PPhEt2)
less basic phosphites
bulky e.g. P(OPh)3
non-bulky e.g. P(OMe)3
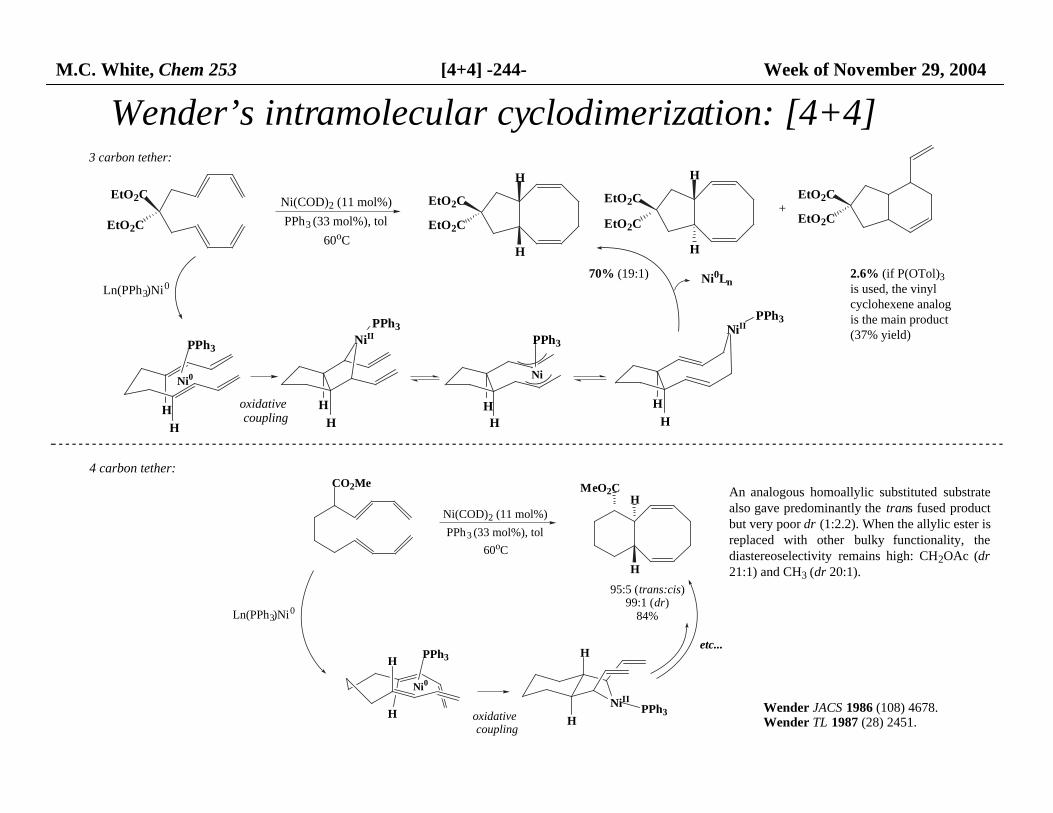
M.C. White, Chem 253 [4+4] -244- Week of November 29, 2004
Wender’s intramolecular cyclodimerization: [4+4]
EtO2C
EtO2C
Ni(COD)2 (11 mol%)
PPh3 (33 mol%), tol
60oC
H
H
EtO2C
EtO2C
H
H
EtO2C
EtO2C
H
H
70% (19:1)
+EtO2C
EtO2C
2.6% (if P(OTol)3 is used, the vinyl cyclohexene analog is the main product (37% yield)
3 carbon tether:
H
H
Ni0
PPh3NiII
PPh3
H
H
Ni
PPh3
H
H
NiIIPPh3
Ni0Ln
oxidative coupling
Ln(PPh3)Ni0
CO2Me
Ni(COD)2 (11 mol%)
PPh3 (33 mol%), tol
60oC
H
H
MeO2C
95:5 (trans:cis)99:1 (dr)
84%
H
H
Ni0
PPh3 H
H
NiIIPPh3
Ln(PPh3)Ni0
oxidative coupling
etc...
An analogous homoallylic substituted substratealso gave predominantly the trans fused productbut very poor dr (1:2.2). When the allylic ester is replaced with other bulky functionality, thediastereoselectivity remains high: CH2OAc (dr21:1) and CH3 (dr 20:1).
Wender JACS 1986 (108) 4678.Wender TL 1987 (28) 2451.
4 carbon tether:
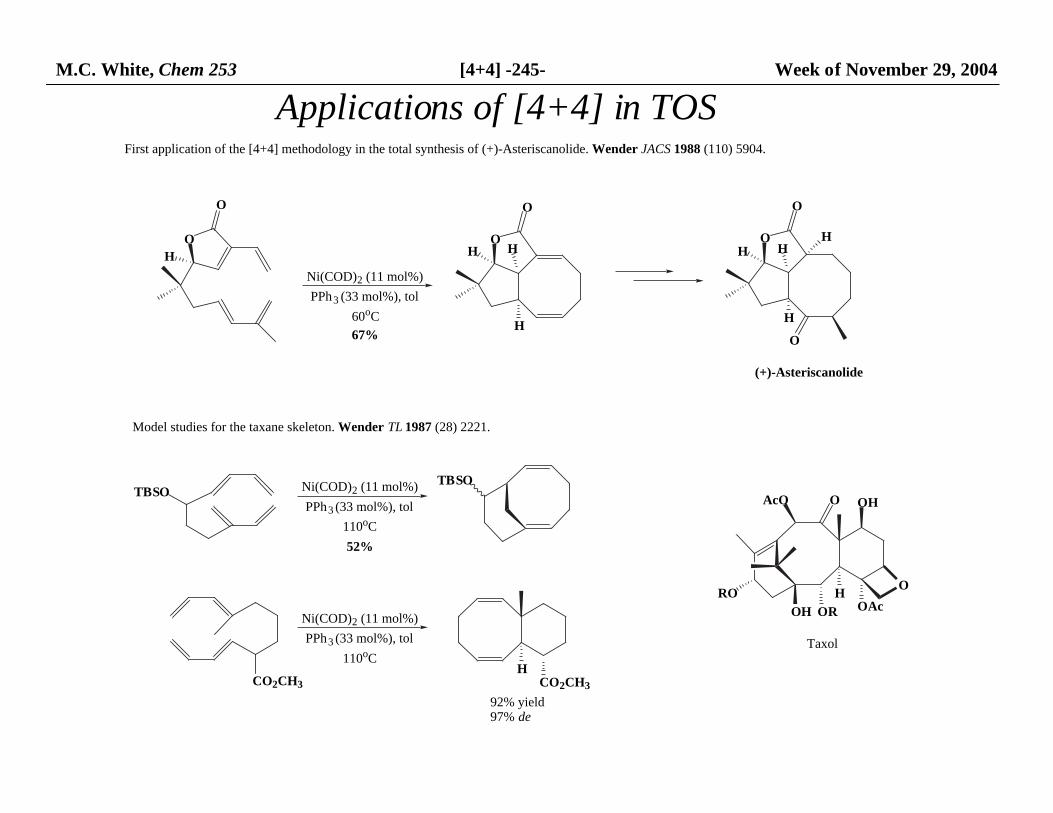
M.C. White, Chem 253 [4+4] -245- Week of November 29, 2004
Applications of [4+4] in TOS
Ni(COD)2 (11 mol%)
PPh3 (33 mol%), tol
60oC
O
O
H
67%H
O
O
H H
H
O
O
H HH
O
(+)-Asteriscanolide
First application of the [4+4] methodology in the total synthesis of (+)-Asteriscanolide. Wender JACS 1988 (110) 5904.
TBSO Ni(COD)2 (11 mol%)
PPh3 (33 mol%), tol
110oC
52%
TBSO
CO2CH3
Ni(COD)2 (11 mol%)
PPh3 (33 mol%), tol
110oCH
CO2CH3
92% yield97% de
AcO O
RO
OH OR
H
OH
O
OAc
Taxol
Model studies for the taxane skeleton. Wender TL 1987 (28) 2221.
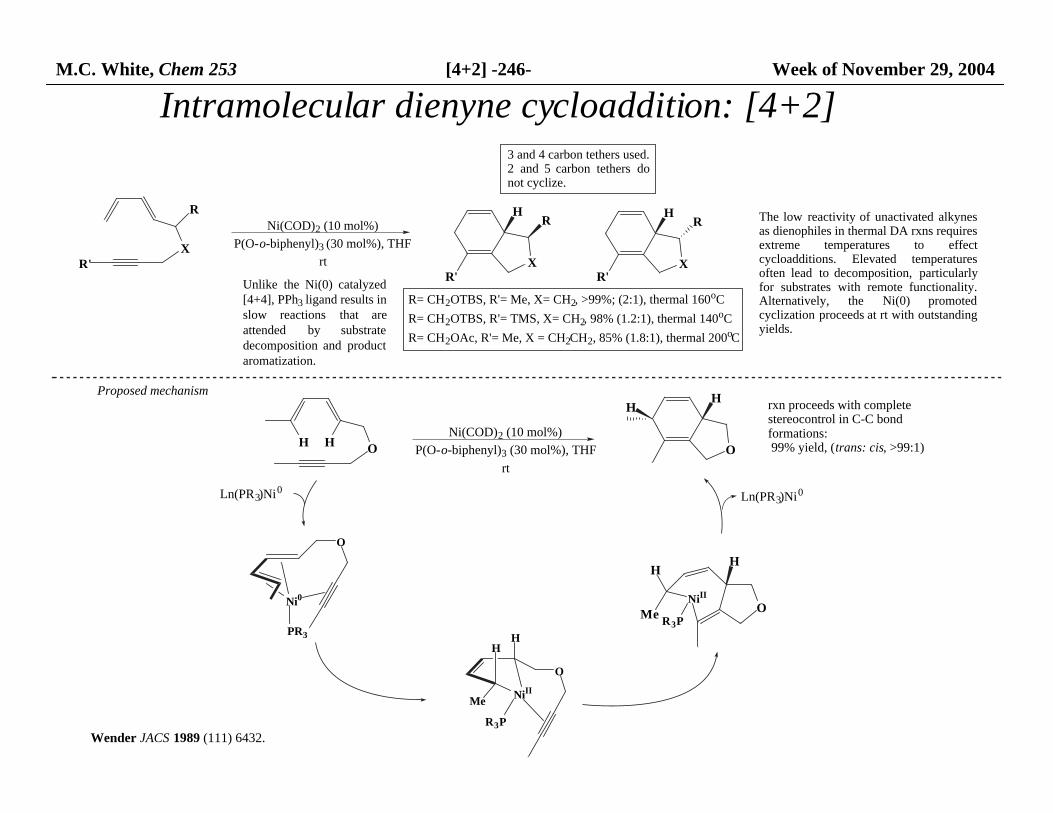
M.C. White, Chem 253 [4+2] -246- Week of November 29, 2004
Intramolecular dienyne cycloaddition: [4+2]
O
Ni(COD)2 (10 mol%)
P(O-o-biphenyl)3 (30 mol%), THF
rt
O
H
H H
rxn proceeds with complete stereocontrol in C-C bond formations: 99% yield, (trans: cis, >99:1)
H
O
Ni0
PR3
O
NiII
H
Me
H
R3P
ONiII
H
Me
H
R3P
Ln(PR3)Ni0Ln(PR3)Ni0
Proposed mechanism
Wender JACS 1989 (111) 6432.
Ni(COD)2 (10 mol%)
P(O-o-biphenyl)3 (30 mol%), THF
rtX
R'
R
R'X
HR
R'X
HR
R= CH2OTBS, R'= Me, X= CH2, >99%; (2:1), thermal 160oC
R= CH2OTBS, R'= TMS, X= CH2, 98% (1.2:1), thermal 140oC
R= CH2OAc, R'= Me, X = CH2CH2, 85% (1.8:1), thermal 200oC
The low reactivity of unactivated alkynesas dienophiles in thermal DA rxns requires extreme temperatures to effectcycloadditions. Elevated temperaturesoften lead to decomposition, particularlyfor substrates with remote functionality.Alternatively, the Ni(0) promotedcyclization proceeds at rt with outstandingyields.
3 and 4 carbon tethers used. 2 and 5 carbon tethers donot cyclize.
Unlike the Ni(0) catalyzed[4+4], PPh3 ligand results in slow reactions that areattended by substratedecomposition and productaromatization.
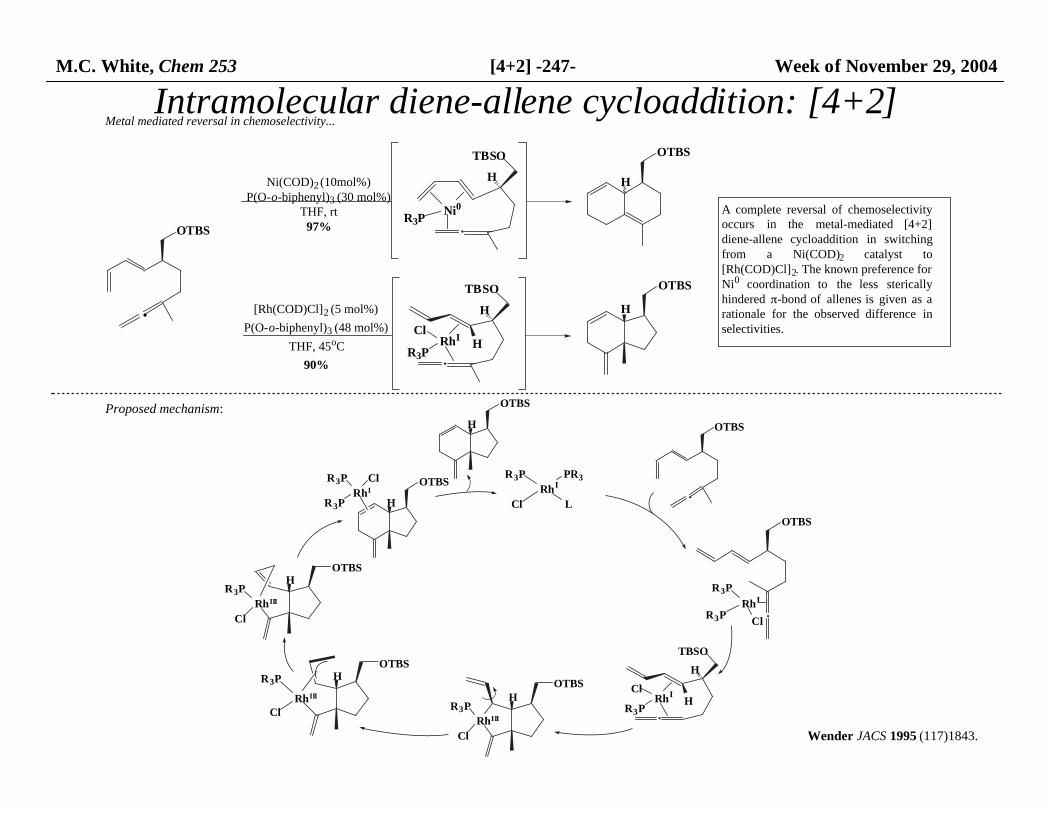
M.C. White, Chem 253 [4+2] -247- Week of November 29, 2004
Intramolecular diene-allene cycloaddition: [4+2]
·
OTBS
RhIR3P
Cl
PR3
L
H
TBSO
RhICl
R3P
H
OTBS
RhI
R3P
R3PCl
RhIII
OTBS
R3P
Cl
HRhIII
OTBS
R3P
Cl
H
RhIII
OTBS
R3P
Cl
H
OTBS
HRhI
R3P
R3P Cl
OTBS
H
Proposed mechanism:
Wender JACS 1995 (117)1843.
Ni(COD)2 (10mol%)P(O-o-biphenyl)3 (30 mol%)
THF, rt97%
[Rh(COD)Cl]2 (5 mol%)
P(O-o-biphenyl)3 (48 mol%)
THF, 45oC
90%
OTBS
OTBS
H
OTBS
H
H
TBSO
RhICl
R3P
H
TBSO
H
Ni0R3P
Metal mediated reversal in chemoselectivity...
A complete reversal of chemoselectivityoccurs in the metal-mediated [4+2]diene-allene cycloaddition in switchingfrom a Ni(COD)2 catalyst to[Rh(COD)Cl]2. The known preference for Ni0 coordination to the less stericallyhindered π-bond of allenes is given as arationale for the observed difference inselectivities.
·
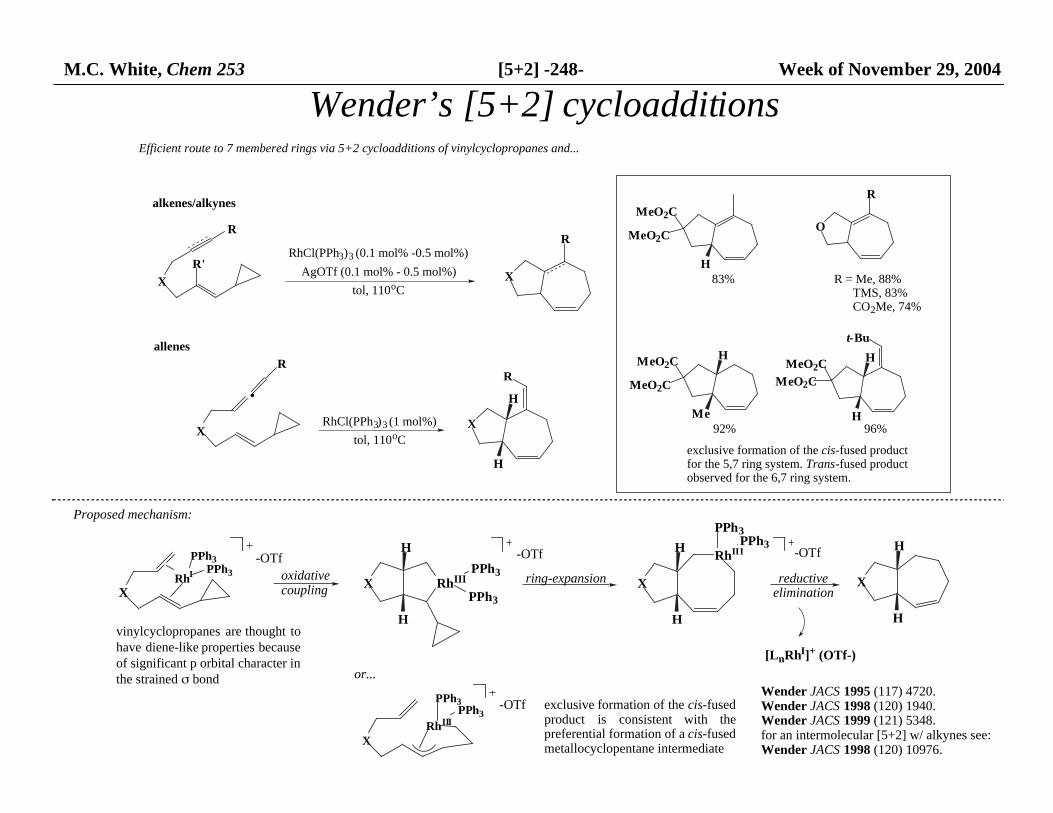
M.C. White, Chem 253 [5+2] -248- Week of November 29, 2004
X
R
RhCl(PPh3)3 (0.1 mol% -0.5 mol%)
AgOTf (0.1 mol% - 0.5 mol%)
tol, 110oCX
R
Efficient route to 7 membered rings via 5+2 cycloadditions of vinylcyclopropanes and...
MeO2C
MeO2C
H
O
R
R'
MeO2C
MeO2C
Me
H
alkenes/alkynes
allenes
X
·
R
RhCl(PPh3)3 (1 mol%)
tol, 110oC
X
R
H
H
t-Bu
H
H
MeO2C
MeO2C
83% R = Me, 88% TMS, 83% CO2Me, 74%
92%
exclusive formation of the cis-fused product for the 5,7 ring system. Trans-fused product observed for the 6,7 ring system.
96%
XRhI PPh3
PPh3
oxidativecoupling
X RhIII
H
H
exclusive formation of the cis-fusedproduct is consistent with thepreferential formation of a cis-fused metallocyclopentane intermediate
PPh3
PPh3
ring-expansion X
H
Hvinylcyclopropanes are thought tohave diene-like properties becauseof significant p orbital character in the strained σ bond
RhIII
PPh3PPh3
reductiveelimination
X
H
H
-OTf -OTf -OTf
[LnRhI]+ (OTf-)
XRhIII
PPh3
PPh3 -OTf
or...
Wender JACS 1995 (117) 4720.Wender JACS 1998 (120) 1940.Wender JACS 1999 (121) 5348.for an intermolecular [5+2] w/ alkynes see: Wender JACS 1998 (120) 10976.
Proposed mechanism:
Wender’s [5+2] cycloadditions
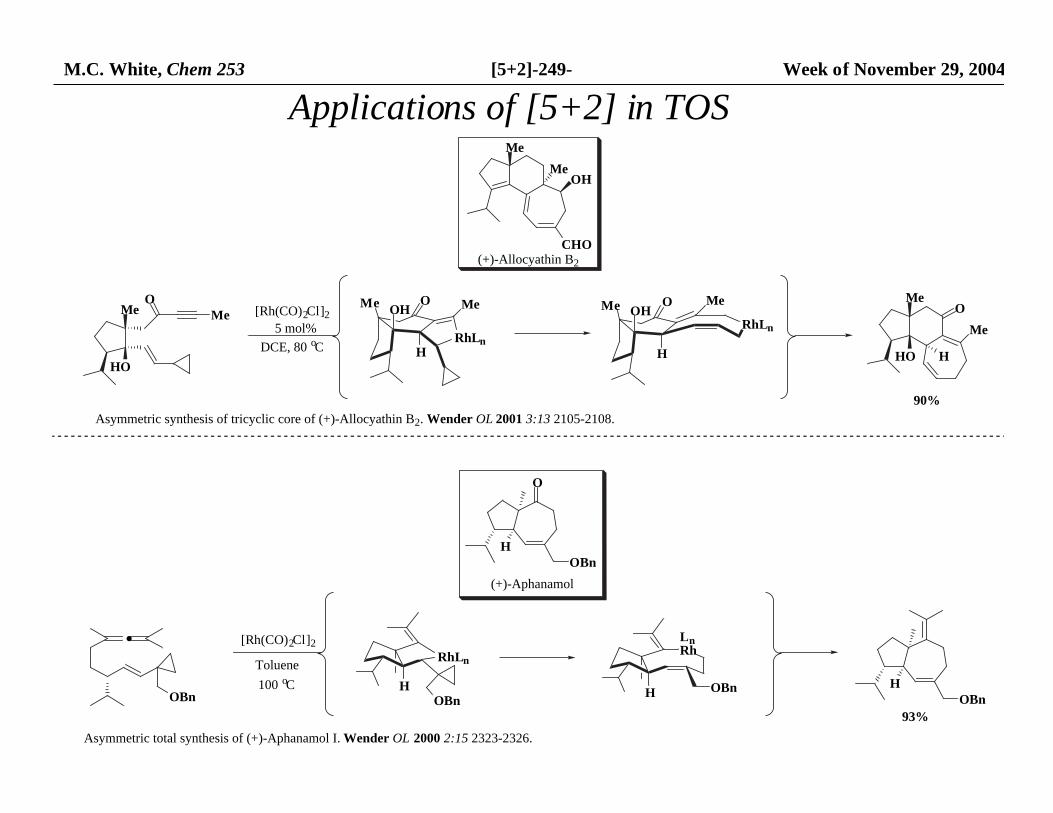
M.C. White, Chem 253 [5+2]-249- Week of November 29, 2004
Applications of [5+2] in TOS Me
OHMe
CHO(+)-Allocyathin B2
·
OBn
[Rh(CO)2Cl]2
Toluene
100 oC
RhLn
OBnH
H
LnRh
OBn HOBn
Asymmetric total synthesis of (+)-Aphanamol I. Wender OL 2000 2:15 2323-2326.
93%
H
O
OBn
(+)-Aphanamol
Asymmetric synthesis of tricyclic core of (+)-Allocyathin B2. Wender OL 2001 3:13 2105-2108.
Me
HO
MeO O
OHMe
RhLn
Me
H
OOHMe Me
H
RhLn
Me
HO
O
Me
H
[Rh(CO)2Cl]2
DCE, 80 oC
5 mol%
90%
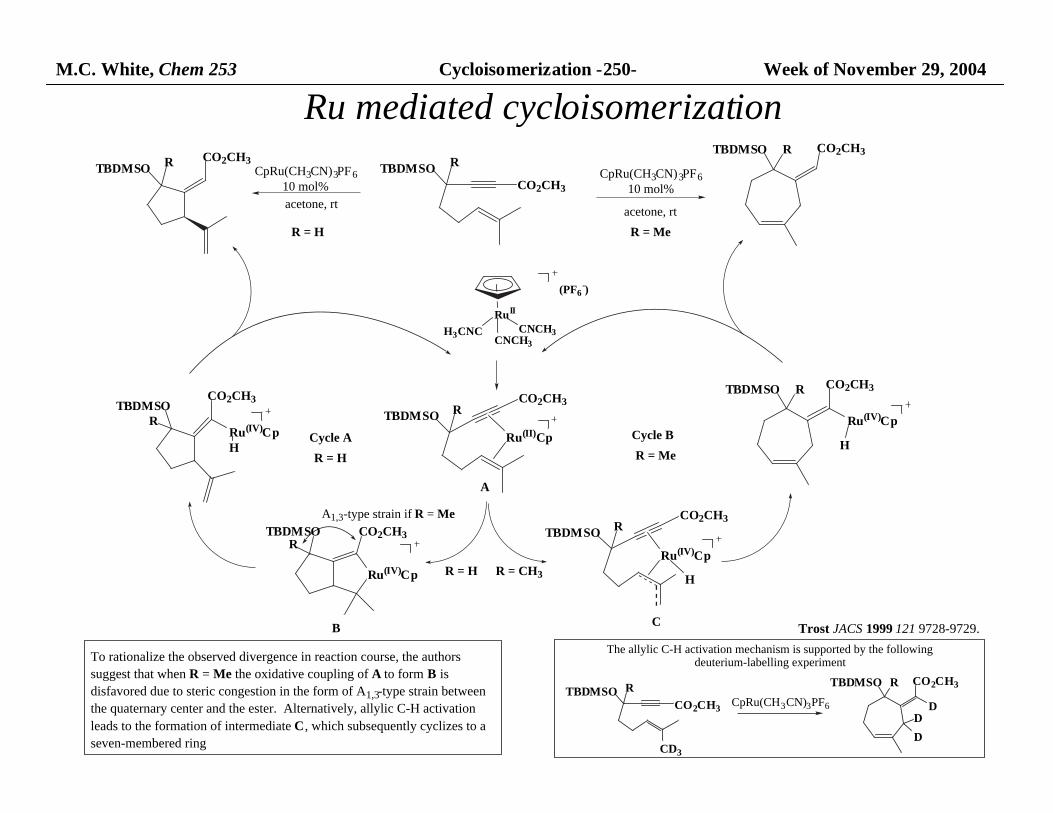
M.C. White, Chem 253 Cycloisomerization -250- Week of November 29, 2004
CO2CH3RTBDMSO
CO2CH3
RTBDMSO
CO2CH3RTBDMSO
Ru(II)Cp
Ru(IV)Cp
CO2CH3TBDMSOR
CO2CH3R
TBDMSO
Ru(IV)Cp
H
TBDMSOR
CO2CH3
Ru(IV)CpH
A
BC
TBDMSO R
CO2CH3
Ru(IV)Cp
TBDMSO R
H
R = CH3R = H
Cycle A Cycle B
CpRu(CH3CN)3PF6
10 mol%
acetone, rt
CpRu(CH3CN)3PF6
10 mol%
acetone, rt
R = H
R = Me
A1,3-type strain if R = Me
RuII
H3CNCCNCH3
CNCH3
(PF6-)
CO2CH3
R = H
R = Me
CO2CH3
CD3
RTBDMSOD
TBDMSO R
D
D
CO2CH3
The allylic C-H activation mechanism is supported by the following deuterium-labelling experiment
CpRu(CH3CN)3PF6
To rationalize the observed divergence in reaction course, the authors
suggest that when R = Me the oxidative coupling of A to form B is
disfavored due to steric congestion in the form of A1,3-type strain between
the quaternary center and the ester. Alternatively, allylic C-H activation
leads to the formation of intermediate C, which subsequently cyclizes to a
seven-membered ring
Trost JACS 1999 121 9728-9729.
Ru mediated cycloisomerization
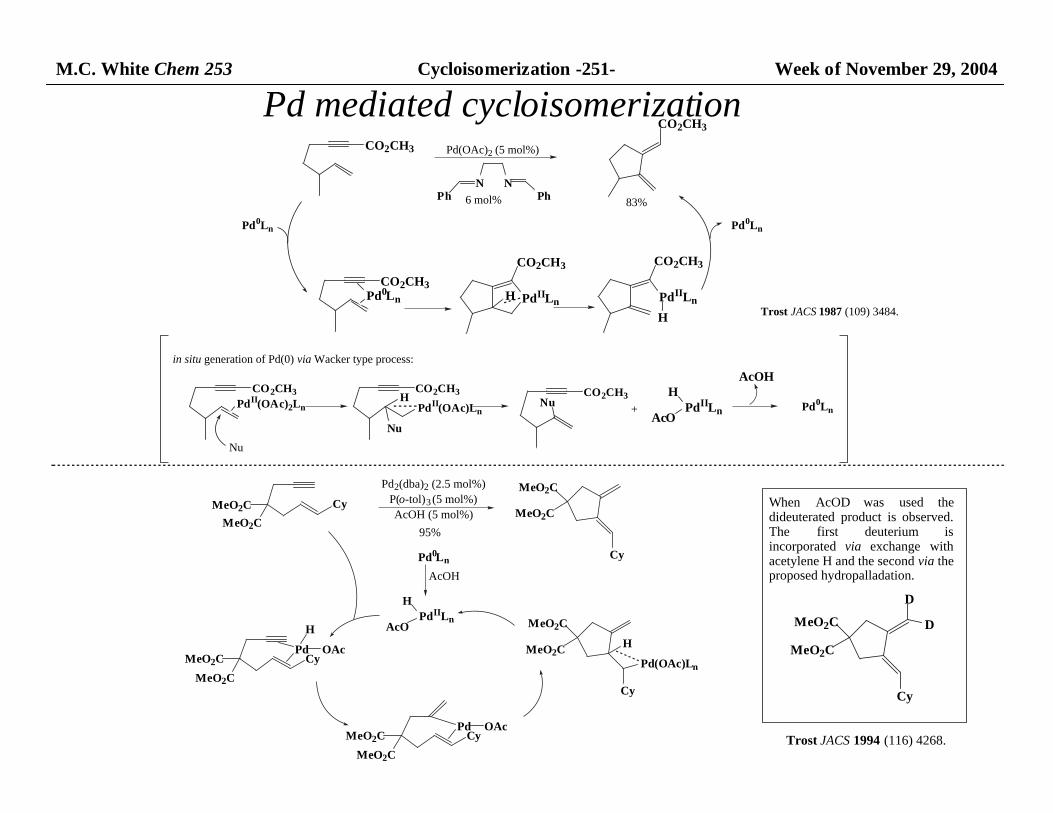
M.C. White Chem 253 Cycloisomerization -251- Week of November 29, 2004
CO2CH3
N NPh Ph
CO2CH3
CO2CH3
PdII(OAc)2Ln
Nu
CO2CH3
PdII(OAc)Ln
Nu
H CO2CH3Nu PdIILn
AcO
HAcOH
CO2CH3Pd0Ln PdIILn
CO2CH3
H PdIILn
CO2CH3
H
Pd(OAc)2 (5 mol%)
6 mol% 83%
Trost JACS 1987 (109) 3484.
in situ generation of Pd(0) via Wacker type process:
+ Pd0Ln
Pd0Ln Pd0Ln
Pd mediated cycloisomerization
Pd2(dba)2 (2.5 mol%)
P(o-tol)3 (5 mol%)
AcOH (5 mol%)MeO2C
MeO2C
Cy
Cy
95%
MeO2C
MeO2C
Pd0Ln
AcOH
PdIILnAcO
H
MeO2C
MeO2C
CyPd OAc
H
MeO2C
MeO2C
CyPd OAc
Cy
MeO2C
MeO2CPd(OAc)Ln
H
When AcOD was used thedideuterated product is observed.The first deuterium isincorporated via exchange withacetylene H and the second via the proposed hydropalladation.
Cy
MeO2C
MeO2C
D
D
Trost JACS 1994 (116) 4268.
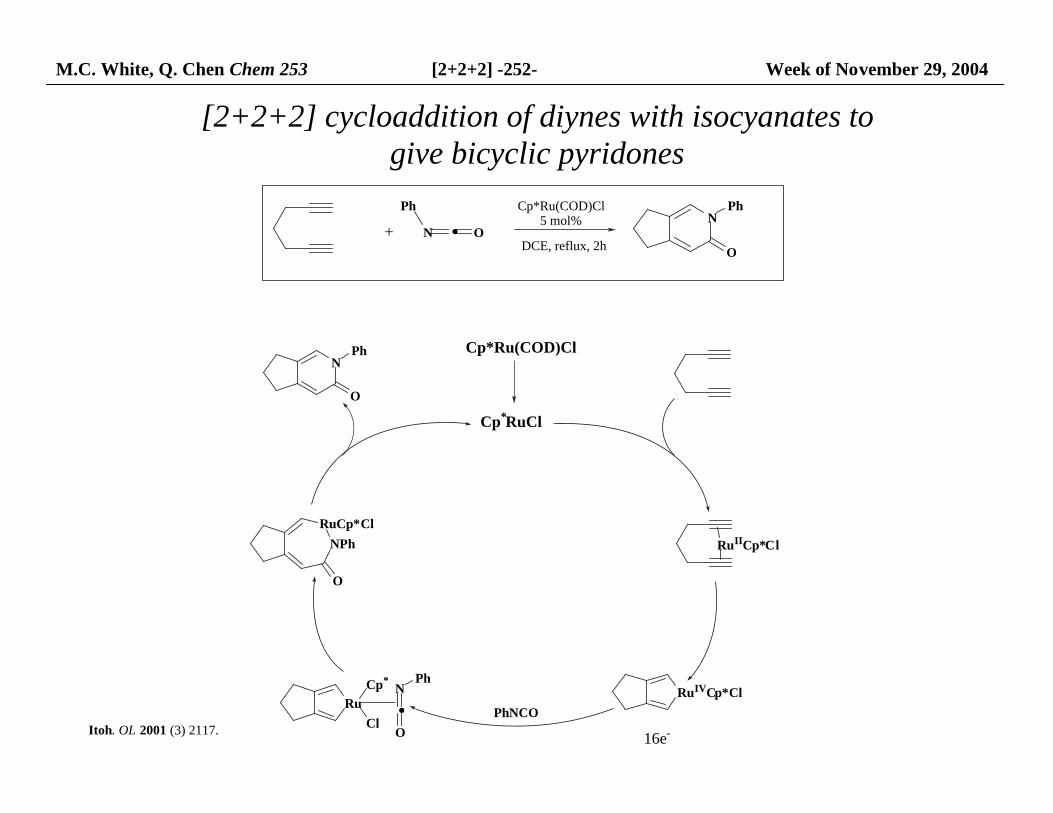
M.C. White, Q. Chen Chem 253 [2+2+2] -252- Week of November 29, 2004
[2+2+2] cycloaddition of diynes with isocyanates to give bicyclic pyridones
NPh
O
RuIICp*Cl
RuIVCp*Cl
PhNCORu
N
·O
Ph
NPh
RuCp*Cl
O
NPh
O
+
Cp*Ru(COD)Cl5 mol%
DCE, reflux, 2h
16e-
Cp*RuCl
Cp*Ru(COD)Cl
Cp*
Cl
· ON
Ph
Itoh. OL 2001 (3) 2117.
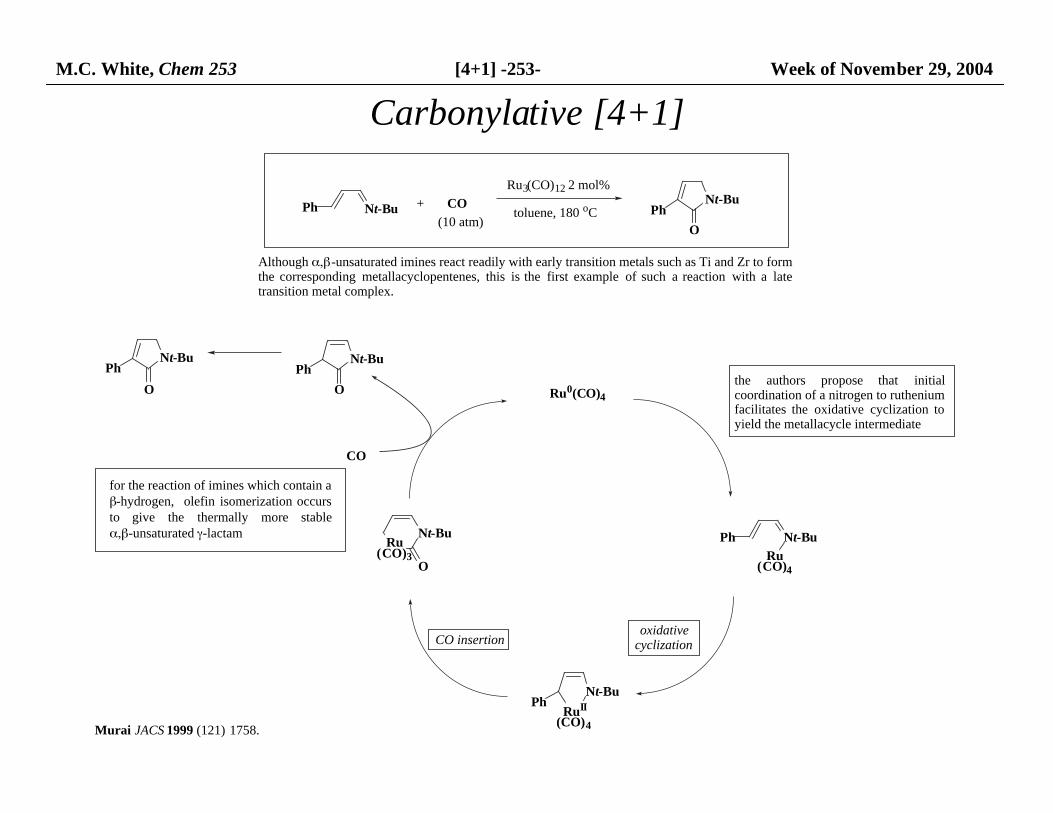
M.C. White, Chem 253 [4+1] -253- Week of November 29, 2004
Ph Nt-Bu + CO
Ru3(CO)12 2 mol%
toluene, 180 oCNt-Bu
O
Ph
Although α,β-unsaturated imines react readily with early transition metals such as Ti and Zr to form the corresponding metallacyclopentenes, this is the first example of such a reaction with a latetransition metal complex.
(10 atm)
the authors propose that initialcoordination of a nitrogen to ruthenium facilitates the oxidative cyclization toyield the metallacycle intermediate
for the reaction of imines which contain a β-hydrogen, olefin isomerization occursto give the thermally more stableα,β-unsaturated γ-lactam
Ru0(CO)4
Ph Nt-Bu
Ru (CO)4
RuII (CO)4
Nt-Bu
Ru (CO)3
Nt-Bu
O
Nt-Bu
O
Ph
PhNt-Bu
O
Ph
oxidativecyclizationCO insertion
CO
Murai JACS 1999 (121) 1758.
Carbonylative [4+1]
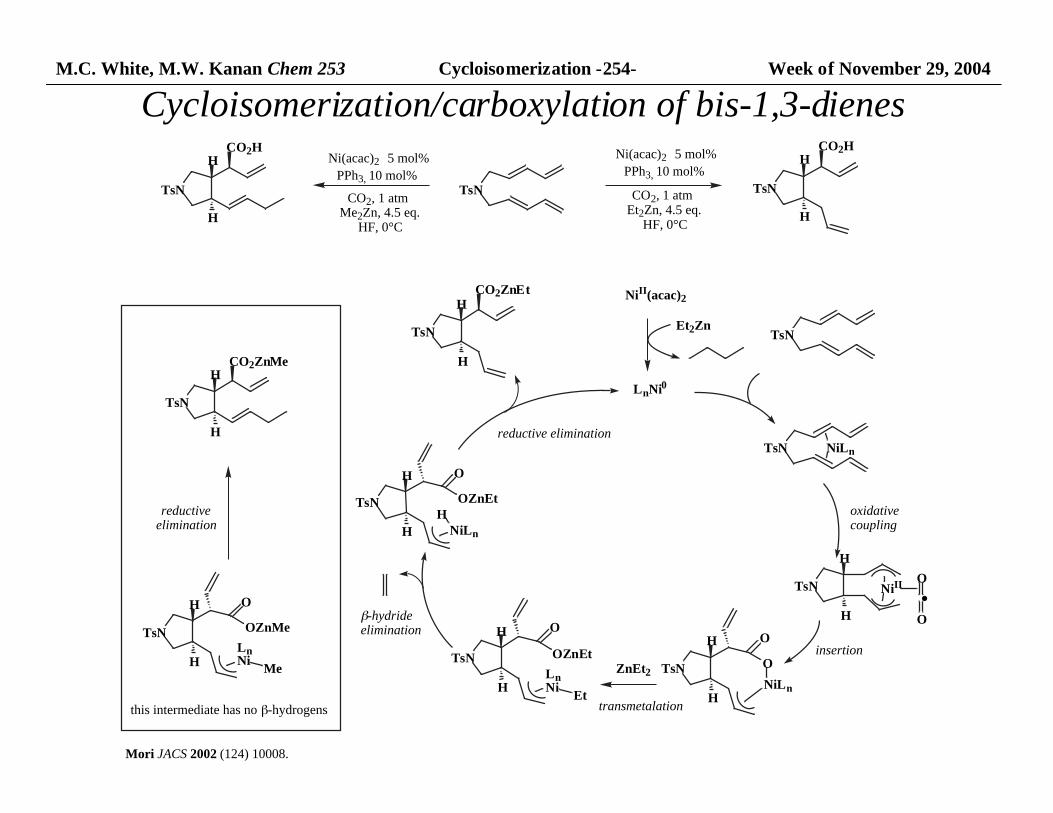
M.C. White, M.W. Kanan Chem 253 Cycloisomerization -254- Week of November 29, 2004
TsN
H
H
CO2H
TsN
TsN NiLn
TsN
H
H
NiII
TsN
H
H
O
O
NiLn
TsN
H
H
O
OZnEt
LnNi
Et
TsN
H
H
O
OZnEt
NiLn
H
TsN
H
H
CO2ZnEt
Et2Zn
ZnEt2
TsN
H
H
CO2H
LnNi
Me
TsN
H
H
O
OZnMe
TsN
H
H
CO2ZnMe
NiII(acac)2
LnNi0
Mori JACS 2002 (124) 10008.
Ni(acac)2 5 mol%
PPh3, 10 mol%
CO2, 1 atm Me2Zn, 4.5 eq.
HF, 0°C
Ni(acac)2 5 mol%
PPh3, 10 mol%
CO2, 1 atm Et2Zn, 4.5 eq.
HF, 0°C
reductiveelimination
β-hydride elimination
reductive elimination
this intermediate has no β-hydrogens
·O
O
TsN
oxidativecoupling
insertion
transmetalation
Cycloisomerization/carboxylation of bis-1,3-dienes
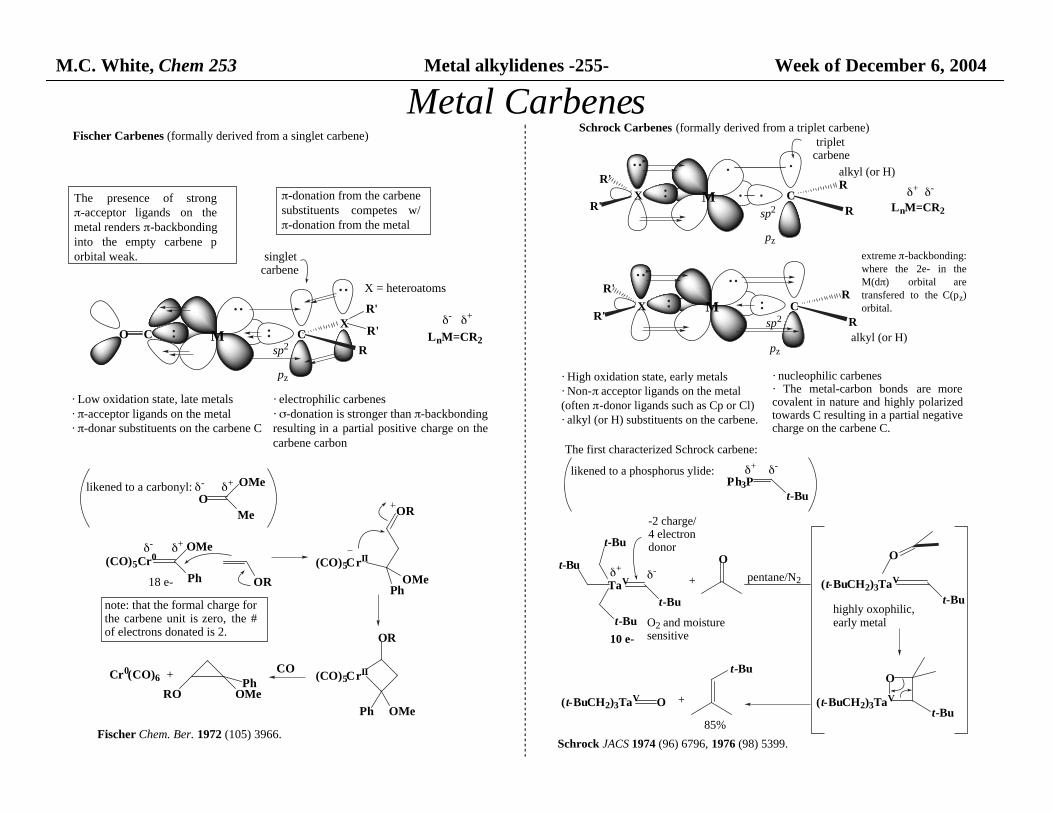
M.C. White, Chem 253 Metal alkylidenes -255- Week of December 6, 2004
Metal Carbenes
singletcarbene
M CX
sp2
pz
R'
R
O C LnM=CR2
δ+δ-
Fischer Carbenes (formally derived from a singlet carbene)
(CO)5Cr0OMe
Ph OR
(CO)5CrII
PhOMe
OR
(CO)5CrII
Ph OMe
OR
COPh
OMeRO
Cr0(CO)6
Fischer Chem. Ber. 1972 (105) 3966.
· Low oxidation state, late metals· π-acceptor ligands on the metal· π-donar substituents on the carbene C
δ+δ-
+
· electrophilic carbenes· σ-donation is stronger than π-backbonding resulting in a partial positive charge on thecarbene carbon
note: that the formal charge for the carbene unit is zero, the #of electrons donated is 2.
π-donation from the carbene substituents competes w/π-donation from the metal
The presence of strongπ-acceptor ligands on themetal renders π-backbonding into the empty carbene porbital weak.
R'
X = heteroatoms
likened to a carbonyl:O
OMe
Me
δ+δ-
18 e-
triplet carbene
Schrock Carbenes (formally derived from a triplet carbene)
M CR
sp2
pz
R
· The metal-carbon bonds are morecovalent in nature and highly polarizedtowards C resulting in a partial negative charge on the carbene C.
alkyl (or H)
XR'
R'
LnM=CR2
δ-δ+
· High oxidation state, early metals· Non-π acceptor ligands on the metal (often π-donor ligands such as Cp or Cl)· alkyl (or H) substituents on the carbene.
· nucleophilic carbenes
The first characterized Schrock carbene:
TaV
t-Bu
t-Bu
t-Bu
t-Bu
δ-δ+
+
likened to a phosphorus ylide:Ph3P
t-Bu
δ-δ+
O
pentane/N2
O2 and moisture sensitive
-2 charge/4 electron donor
10 e-
(t-BuCH2)3TaV
t-Bu
O
(t-BuCH2)3TaV
O
t-Bu
highly oxophilic,early metal
(t-BuCH2)3TaV O
t-Bu
+
85%
Schrock JACS 1974 (96) 6796, 1976 (98) 5399.
M CR
sp2
pz
R
alkyl (or H)
XR'
R'
extreme π-backbonding: where the 2e- in theM(dπ) orbital aretransfered to the C(pz)orbital.
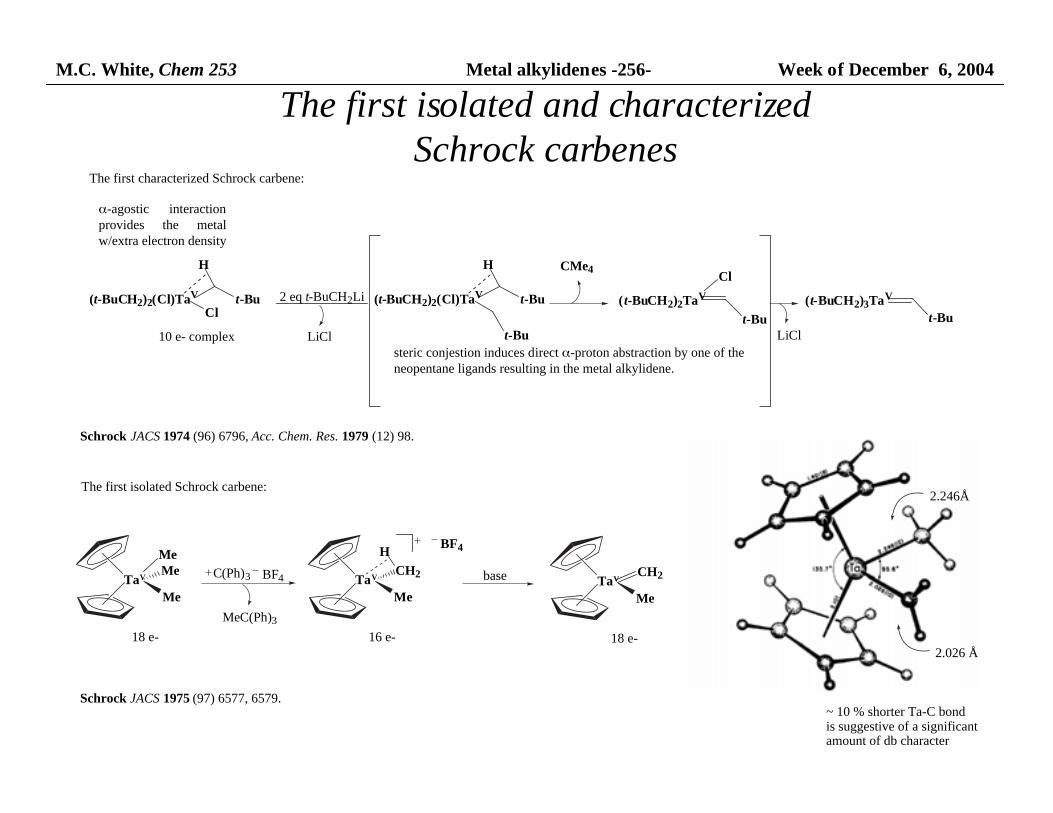
M.C. White, Chem 253 Metal alkylidenes -256- Week of December 6, 2004
The first isolated and characterizedSchrock carbenes
The first isolated Schrock carbene:
TaV
Me
MeMe
18 e-
C(Ph)3 BF4
MeC(Ph)3
TaV
Me
CH2
16 e-
BF4H
base TaV
Me
CH2
18 e-
Schrock JACS 1975 (97) 6577, 6579.
2.026 Å
2.246Å
~ 10 % shorter Ta-C bondis suggestive of a significant amount of db character
The first characterized Schrock carbene:
(t-BuCH2)2(Cl)TaV t-BuCl
H
10 e- complex
α-agostic interactionprovides the metalw/extra electron density
2 eq t-BuCH2Li (t-BuCH2)2(Cl)TaV t-Bu
H
t-Bu
steric conjestion induces direct α-proton abstraction by one of the neopentane ligands resulting in the metal alkylidene.
(t-BuCH2)2TaV
t-Bu
ClCMe4
(t-BuCH2)3TaV
t-Bu
LiCl LiCl
Schrock JACS 1974 (96) 6796, Acc. Chem. Res. 1979 (12) 98.
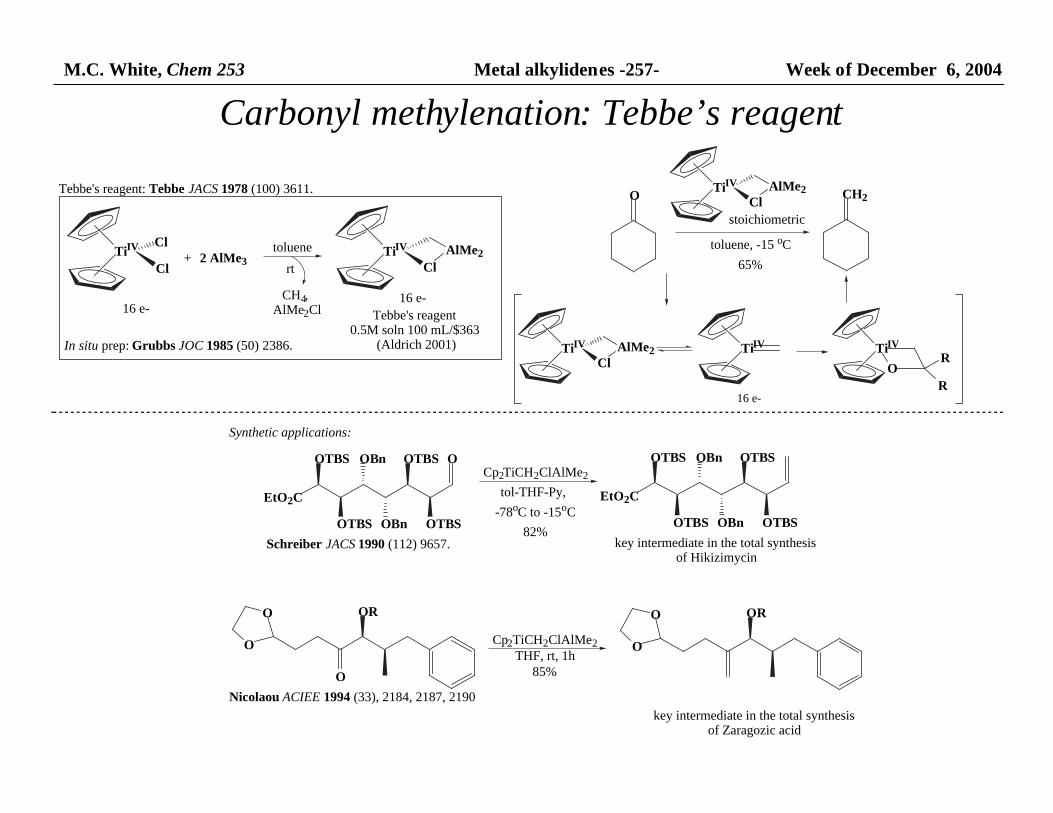
M.C. White, Chem 253 Metal alkylidenes -257- Week of December 6, 2004
Carbonyl methylenation: Tebbe’s reagent
TiIV
Cl
Cl
16 e-
+ 2 AlMe3
toluene
rt
TiIV
Cl
16 e-
AlMe2
CH4, AlMe2Cl
Tebbe's reagent: Tebbe JACS 1978 (100) 3611.
Tebbe's reagent 0.5M soln 100 mL/$363
(Aldrich 2001) TiIV
ClAlMe2 TiIV
O
TiIV
OR
R
CH2
16 e-
TiIV
ClAlMe2
stoichiometric
toluene, -15 oC
65%
Synthetic applications:
EtO2C
O
OTBS
OTBS
OBn
OBn
OTBS
OTBSCp2TiCH2ClAlMe2
tol-THF-Py,
-78oC to -15oC
82%
EtO2C
OTBS
OTBS
OBn
OBn
OTBS
OTBS
key intermediate in the total synthesisof Hikizimycin
Schreiber JACS 1990 (112) 9657.
OR
O
O
O Cp2TiCH2ClAlMe2
THF, rt, 1h85%
ORO
O
Nicolaou ACIEE 1994 (33), 2184, 2187, 2190
key intermediate in the total synthesisof Zaragozic acid
In situ prep: Grubbs JOC 1985 (50) 2386.
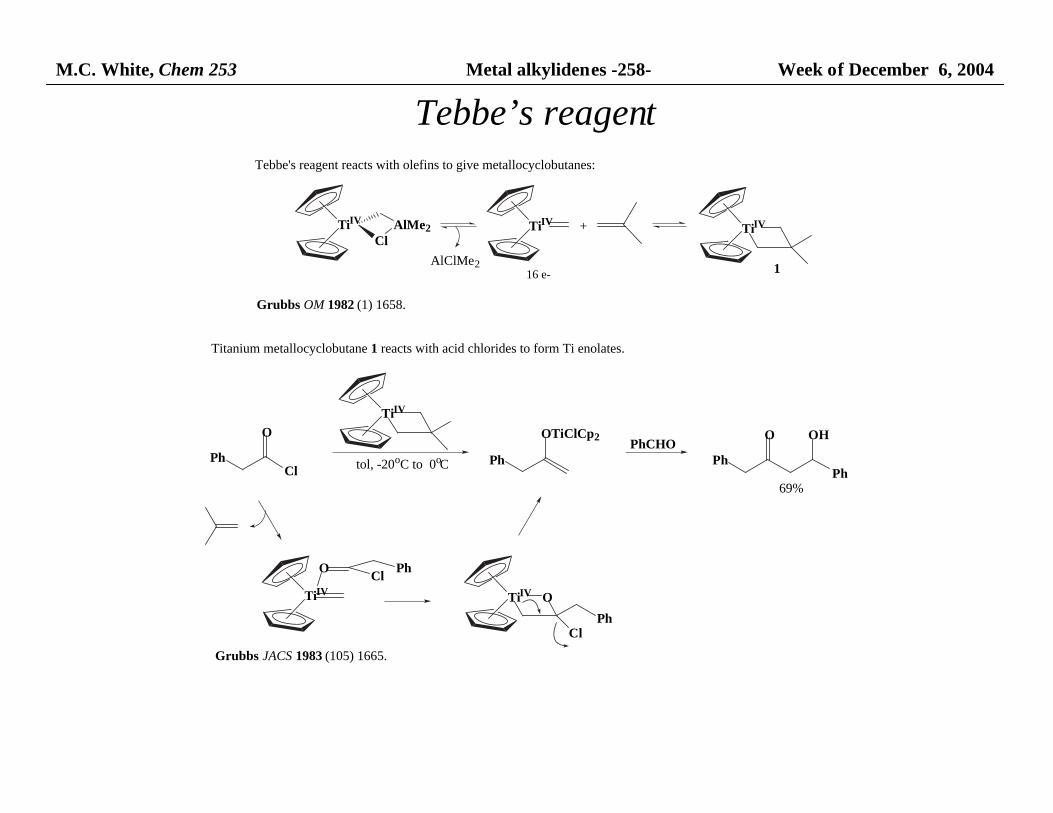
M.C. White, Chem 253 Metal alkylidenes -258- Week of December 6, 2004
Tebbe’s reagent
TiIV
ClAlMe2 TiIV
16 e-
+
AlClMe2
TiIV
TiIV
Tebbe's reagent reacts with olefins to give metallocyclobutanes:
Titanium metallocyclobutane 1 reacts with acid chlorides to form Ti enolates.
1
Grubbs OM 1982 (1) 1658.
O
ClPh
tol, -20oC to 0oC
OTiClCp2
PhPhCHO
O
Ph
OH
Ph69%
TiIVTiIV O
ClPh
OCl
Ph
Grubbs JACS 1983 (105) 1665.
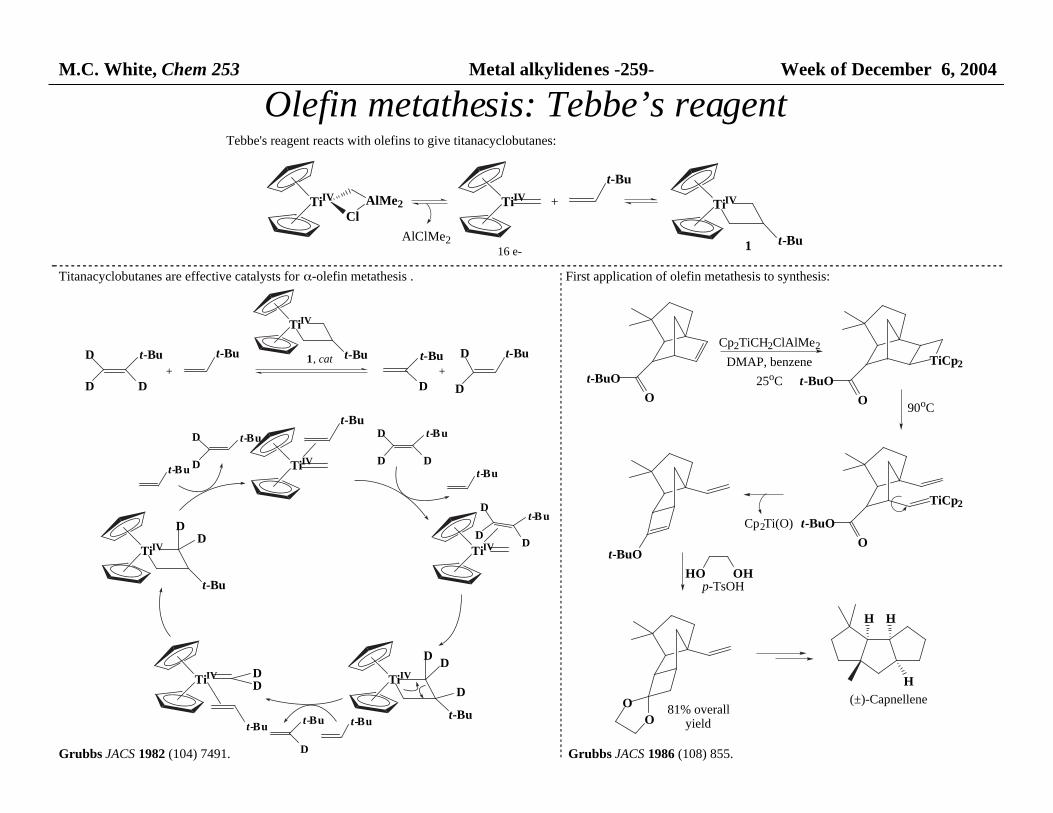
M.C. White, Chem 253 Metal alkylidenes -259- Week of December 6, 2004
Olefin metathesis: Tebbe’s reagent
TiIV
ClAlMe2 TiIV
16 e-
+
t-Bu
AlClMe2
TiIV
t-Bu
Tebbe's reagent reacts with olefins to give titanacyclobutanes:
1
D
D D
t-Bu t-Bu
TiIV
t-Bu
D
t-Bu t-BuD
D
TiIV
D
D D
t-But-Bu
TiIV
D
DD
t-Bu
TiIV
t-Bu
D
DD
TiIV
t-Bu
DD
TiIV
t-Bu
DD
t-Bu
t-Bu
D
t-Bu
t-Bu
t-BuD
D
+1, cat
+
Titanacyclobutanes are effective catalysts for α-olefin metathesis .
Grubbs JACS 1982 (104) 7491.
O
t-BuO
O
t-BuO
TiCp2
O
t-BuO
TiCp2
t-BuO
HO OH
O
O
H H
H
Cp2TiCH2ClAlMe2
DMAP, benzene
25oC
90oC
p-TsOH
81% overallyield
Cp2Ti(O)
(±)-Capnellene
First application of olefin metathesis to synthesis:
Grubbs JACS 1986 (108) 855.
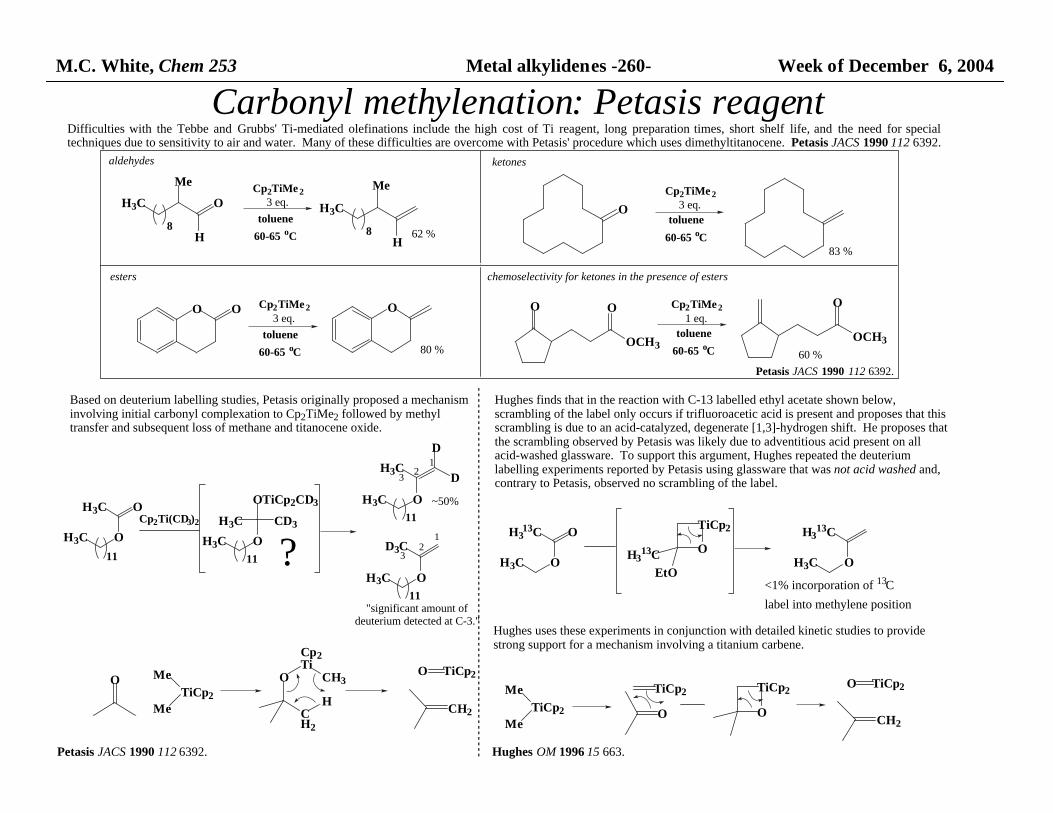
M.C. White, Chem 253 Metal alkylidenes -260- Week of December 6, 2004
Carbonyl methylenation: Petasis reagent
OO
H
H3C
Me
O O
H3C O
OH3C OH3C
OTiCp2CD3
H3C CD3
H
H3C
Me
O
D3C
OH3C
H3C
OH3C
D
D
O
OCH3
O
OCH3
O
8 8 62 %
83 %
aldehydes ketones
esters chemoselectivity for ketones in the presence of esters
80 % 60 %
Cp2TiMe 2
3 eq.
toluene
60-65 oC
Cp2TiMe 2
3 eq.
toluene
60-65 oC
Cp2TiMe 2
3 eq.
toluene
60-65 oC
Cp2TiMe 2
1 eq.
toluene
60-65 oC
Based on deuterium labelling studies, Petasis originally proposed a mechanism involving initial carbonyl complexation to Cp2TiMe2 followed by methyl transfer and subsequent loss of methane and titanocene oxide.
Petasis JACS 1990 112 6392.
Difficulties with the Tebbe and Grubbs' Ti-mediated olefinations include the high cost of Ti reagent, long preparation times, short shelf life, and the need for specialtechniques due to sensitivity to air and water. Many of these difficulties are overcome with Petasis' procedure which uses dimethyltitanocene. Petasis JACS 1990 112 6392.
11
Cp2Ti(CD3)2
11 ?11
11
~50%
"significant amount of deuterium detected at C-3."
H313C O
OH3CO
TiCp2
H313C
EtO
H313C
OH3C
<1% incorporation of 13C
label into methylene position
Hughes finds that in the reaction with C-13 labelled ethyl acetate shown below, scrambling of the label only occurs if trifluoroacetic acid is present and proposes that this scrambling is due to an acid-catalyzed, degenerate [1,3]-hydrogen shift. He proposes that the scrambling observed by Petasis was likely due to adventitious acid present on all acid-washed glassware. To support this argument, Hughes repeated the deuterium labelling experiments reported by Petasis using glassware that was not acid washed and, contrary to Petasis, observed no scrambling of the label.
O Me
TiCp2
Me
O
Cp2Ti
CH2
H
CH3
CH2
O TiCp2
Petasis JACS 1990 112 6392.
Me
TiCp2
MeO
TiCp2
O
TiCp2
CH2
O TiCp2
Hughes uses these experiments in conjunction with detailed kinetic studies to provide strong support for a mechanism involving a titanium carbene.
Hughes OM 1996 15 663.
12
3
12
3
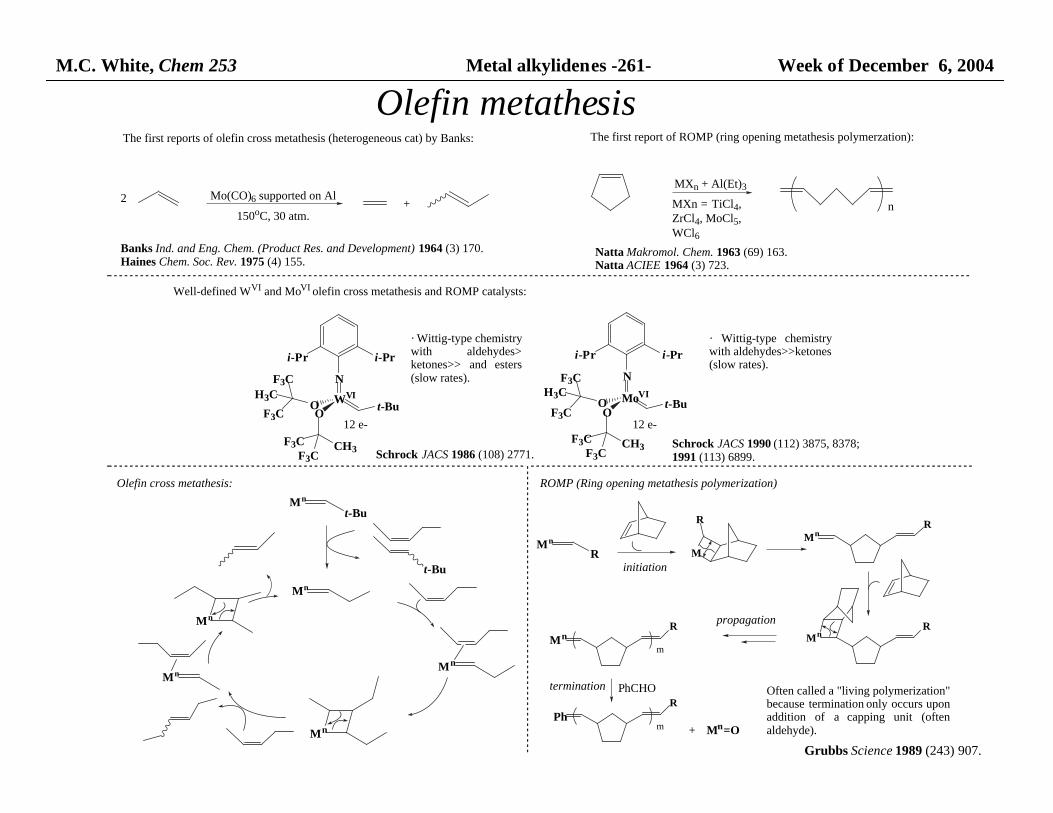
M.C. White, Chem 253 Metal alkylidenes -261- Week of December 6, 2004
Olefin metathesisThe first reports of olefin cross metathesis (heterogeneous cat) by Banks:
2 Mo(CO)6 supported on Al
150oC, 30 atm.+
Banks Ind. and Eng. Chem. (Product Res. and Development) 1964 (3) 170.Haines Chem. Soc. Rev. 1975 (4) 155.
The first report of ROMP (ring opening metathesis polymerzation):
MXn + Al(Et)3
nMXn = TiCl4,ZrCl4, MoCl5, WCl6
Natta Makromol. Chem. 1963 (69) 163.Natta ACIEE 1964 (3) 723.
Well-defined WVI and MoVI olefin cross metathesis and ROMP catalysts:
WVI
N
t-BuO
O
F3C
H3C
F3C
F3CF3C
CH3
i-Pr i-Pr
MoVI
N
t-BuO
O
F3CH3C
F3C
F3CF3C
CH3
i-Pr i-Pr
· Wittig-type chemistry with aldehydes>ketones>> and esters(slow rates).
· Wittig-type chemistrywith aldehydes>>ketones (slow rates).
Schrock JACS 1986 (108) 2771.Schrock JACS 1990 (112) 3875, 8378;1991 (113) 6899.
Olefin cross metathesis:
Mn
t-Bu
12 e- 12 e-
Mn
t-Bu
Mn
Mn
Mn
Mn
ROMP (Ring opening metathesis polymerization)
Mn
R M
R
MnR
MnR
MnR
m
propagation
PhCHO
PhR
m
termination
+ Mn=O
initiation
Often called a "living polymerization" because termination only occurs uponaddition of a capping unit (oftenaldehyde).
Grubbs Science 1989 (243) 907.
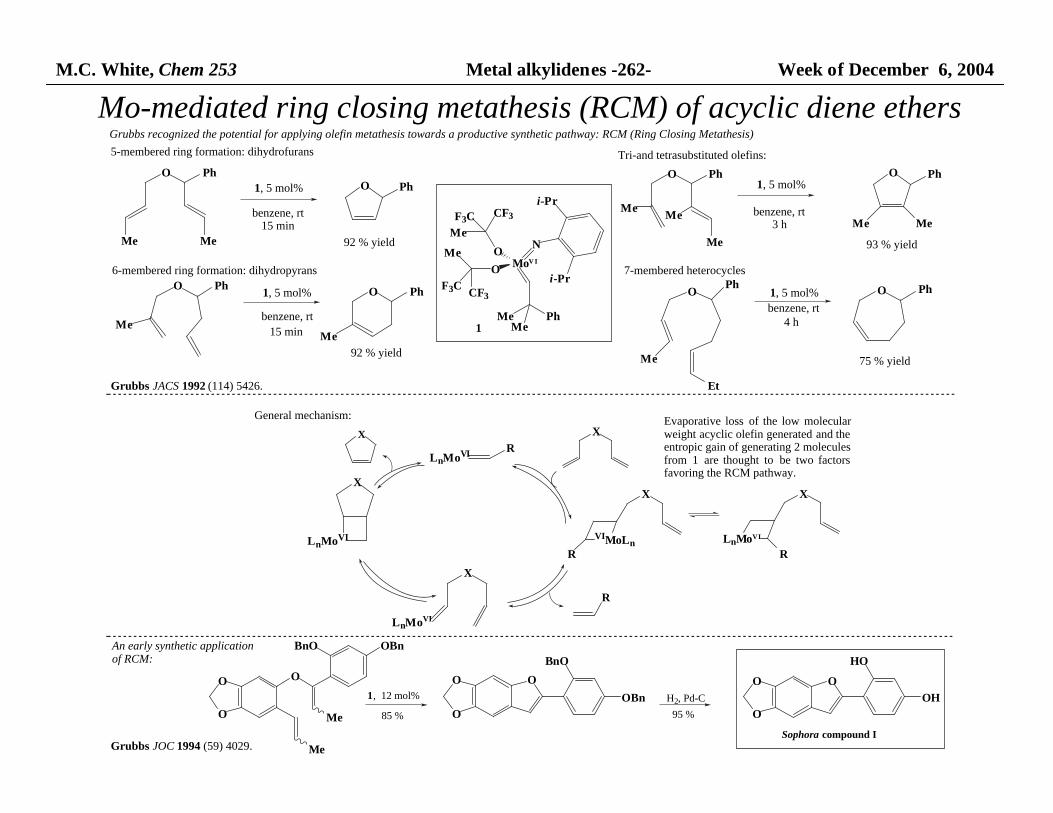
M.C. White, Chem 253 Metal alkylidenes -262- Week of December 6, 2004
Mo-mediated ring closing metathesis (RCM) of acyclic diene ethers
O PhO Ph
Me
benzene, rt15 min
92 % yield
O Ph
Me
O
Me
Ph
92 % yield
benzene, rt
15 min
O
O
Me
O
BnO OBn
Me
O
O
O
BnO
OBn
O
O
O
HO
OH
Me
O
Me
Ph
Me
O Ph
MeMebenzene, rt
3 h
93 % yieldMe
OPh
Et
Me
O Ph
benzene, rt4 h
75 % yield
1, 5 mol% 1, 5 mol%
1, 5 mol% 1, 5 mol%
MoV I
NO
O
i-Pr
i-Pr
MeMe
Ph
CF3F3C
Me
Me
F3C CF3
1
Grubbs JACS 1992 (114) 5426.
1, 12 mol%
85 % 95 %
Sophora compound IGrubbs JOC 1994 (59) 4029.
H2, Pd-C
Grubbs recognized the potential for applying olefin metathesis towards a productive synthetic pathway: RCM (Ring Closing Metathesis)
General mechanism:
X
LnMoVIR
LnMoVI
X
VIMoLn
X
RR
X
LnMoVI
X
LnMoVI
X
5-membered ring formation: dihydrofurans Tri-and tetrasubstituted olefins:
6-membered ring formation: dihydropyrans 7-membered heterocycles
R
Evaporative loss of the low molecularweight acyclic olefin generated and theentropic gain of generating 2 molecules from 1 are thought to be two factorsfavoring the RCM pathway.
An early synthetic application of RCM:
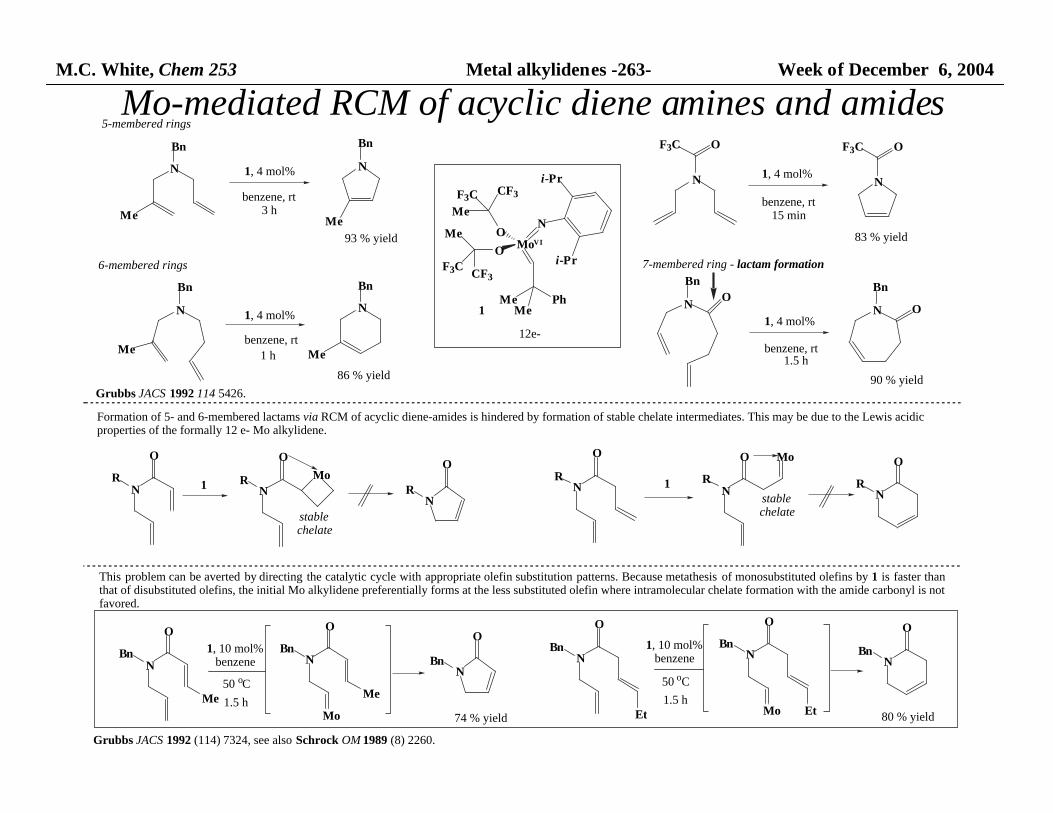
M.C. White, Chem 253 Metal alkylidenes -263- Week of December 6, 2004
Mo-mediated RCM of acyclic diene amines and amides
N
Me
Bn
N
Me
Bn
N
O
R
N
Me
Bn
N
O
R Mo
N
Bn
Me
MoVI
NO
O
i-Pr
i-Pr
MeMe
Ph
CF3F3C
Me
Me
F3C CF3
N
O
R
N
OF3C
N
Bn
O
N
OF3C
N
Bn
O
benzene, rt3 h
93 % yield
86 % yield
benzene, rt
1 h
benzene, rt15 min
83 % yield
benzene, rt1.5 h
90 % yield
1, 4 mol% 1, 4 mol%
1, 4 mol% 1, 4 mol%1
Grubbs JACS 1992 114 5426.
Formation of 5- and 6-membered lactams via RCM of acyclic diene-amides is hindered by formation of stable chelate intermediates. This may be due to the Lewis acidic properties of the formally 12 e- Mo alkylidene.
1 N
O
R1
N
O
R
Mo
N
O
R
N
O
Bn
Me
N
O
Bn
Mo
Me
N
O
Bn
stablechelate
stablechelate
N
O
BnN
O
Bn
MoEt
N
O
Bn1, 10 mol%benzene
50 oC
1.5 h
74 % yieldEt 80 % yield
1, 10 mol%benzene
50 oC
1.5 h
This problem can be averted by directing the catalytic cycle with appropriate olefin substitution patterns. Because metathesis of monosubstituted olefins by 1 is faster thanthat of disubstituted olefins, the initial Mo alkylidene preferentially forms at the less substituted olefin where intramolecular chelate formation with the amide carbonyl is not favored.
Grubbs JACS 1992 (114) 7324, see also Schrock OM 1989 (8) 2260.
5-membered rings
6-membered rings 7-membered ring - lactam formation
12e-
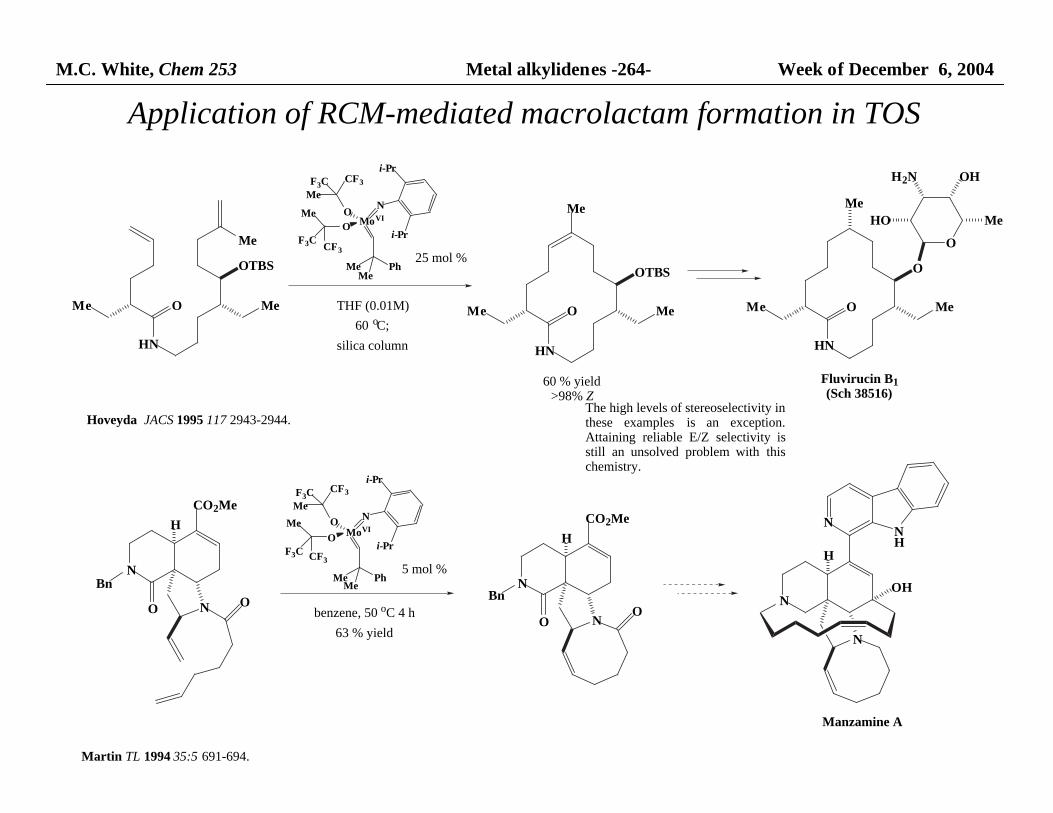
M.C. White, Chem 253 Metal alkylidenes -264- Week of December 6, 2004
Application of RCM-mediated macrolactam formation in TOS
MoVI
NO
O
i-Pr
i-Pr
MeMe
Ph
CF3F3C
Me
Me
F3C CF3
HN
Me
O
MeMe O
O
HO Me
OHH2N
Fluvirucin B1(Sch 38516)
HN
Me
OTBS
MeMe O
HN
OTBS
MeMe O
Me
25 mol %
THF (0.01M)
60 oC;
silica column
Hoveyda JACS 1995 117 2943-2944.
60 % yield>98% Z
N
O
Bn
CO2Me
H
N O
MoVI
NO
O
i-Pr
i-Pr
MeMe
Ph
CF3F3C
Me
Me
F3C CF3
5 mol %
benzene, 50 oC 4 h
63 % yield
N
O
Bn
CO2Me
H
NO
N
H
N
NNH
OH
Manzamine A
Martin TL 1994 35:5 691-694.
The high levels of stereoselectivity in these examples is an exception.Attaining reliable E/Z selectivity isstill an unsolved problem with thischemistry.
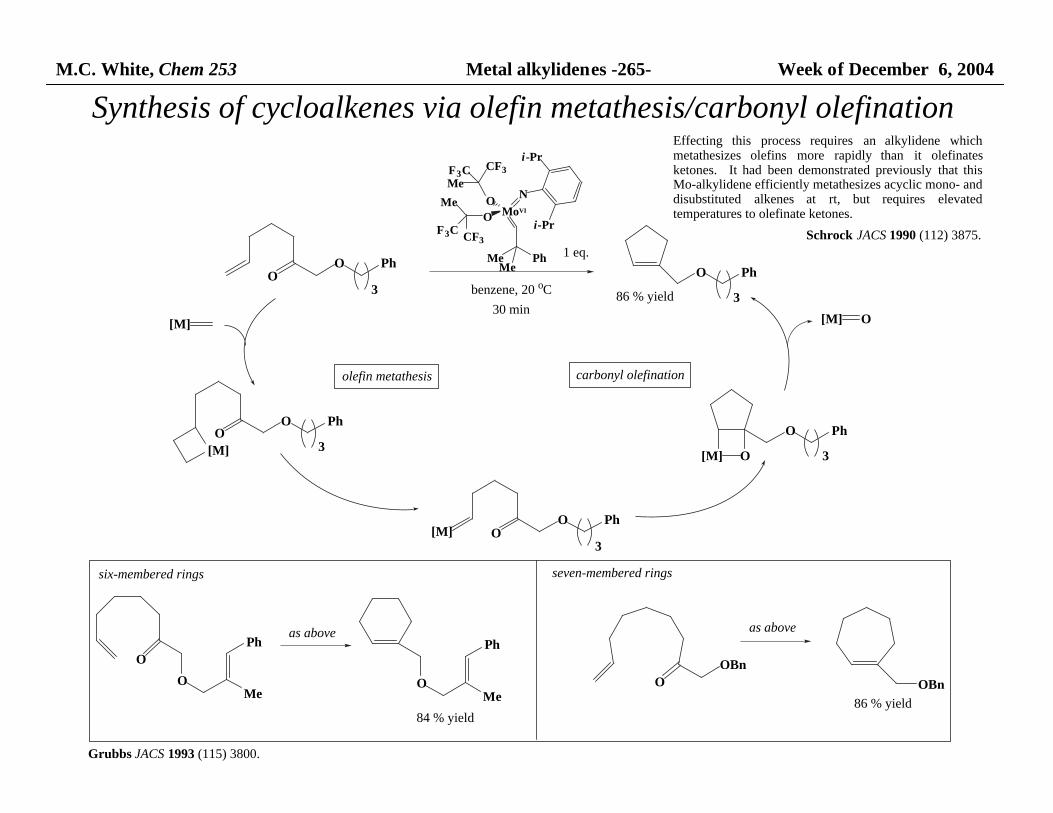
M.C. White, Chem 253 Metal alkylidenes -265- Week of December 6, 2004
Synthesis of cycloalkenes via olefin metathesis/carbonyl olefination
OO Ph
3
MoVI
NO
O
i-Pr
i-Pr
MeMe
Ph
CF3F3C
Me
MeF3C CF3
O Ph
3
1 eq.
benzene, 20 oC
30 min[M]
OO Ph
3[M]
[M] OO Ph
3
[M] O
O Ph
3
[M] O
86 % yield
olefin metathesis carbonyl olefination
Effecting this process requires an alkylidene whichmetathesizes olefins more rapidly than it olefinatesketones. It had been demonstrated previously that thisMo-alkylidene efficiently metathesizes acyclic mono- and disubstituted alkenes at rt, but requires elevatedtemperatures to olefinate ketones.
six-membered rings seven-membered rings
O
O
Ph
MeO
Ph
Me
as above
84 % yield
O
OBn
OBn
86 % yield
as above
Grubbs JACS 1993 (115) 3800.
Schrock JACS 1990 (112) 3875.
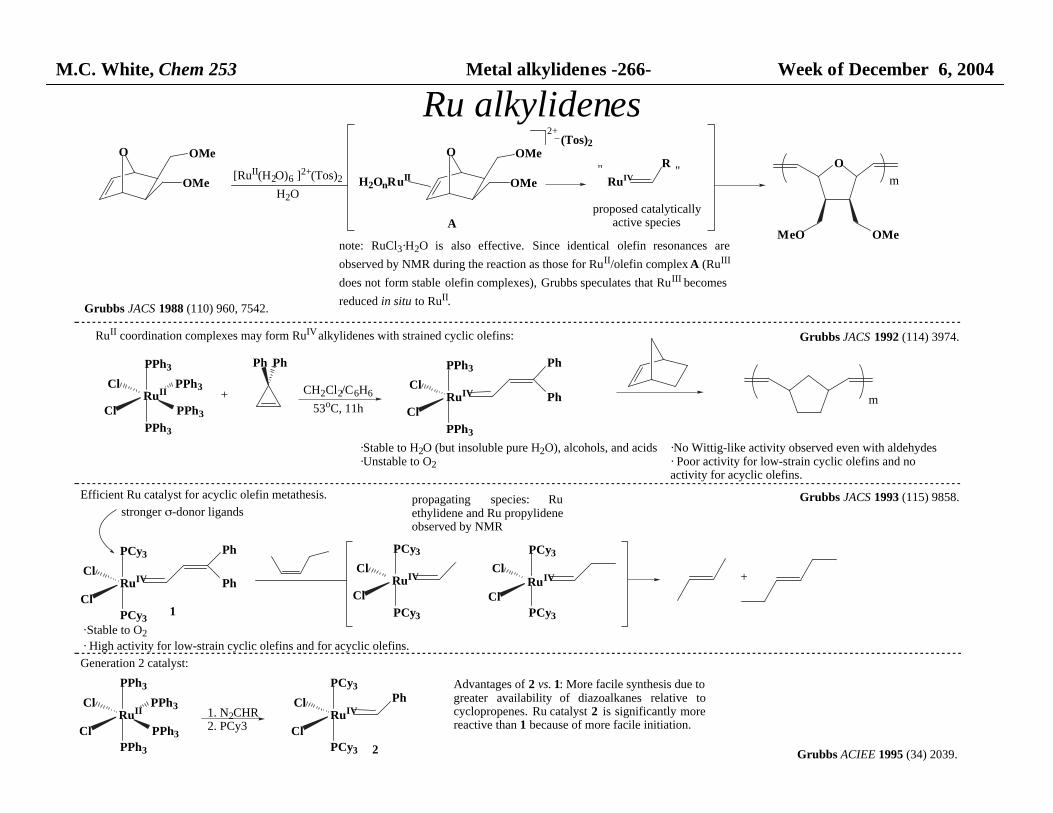
M.C. White, Chem 253 Metal alkylidenes -266- Week of December 6, 2004
Ru alkylidenesO OMe
OMe[RuII(H2O)6 ]2+(Tos)2
H2O
O OMe
OMeH2OnRuII RuIV
2+(Tos)2
R" "
proposed catalytically active species
O
m
MeO OMenote: RuCl3·H2O is also effective. Since identical olefin resonances are
observed by NMR during the reaction as those for RuII/olefin complex A (RuIII
does not form stable olefin complexes), Grubbs speculates that RuIII becomes
reduced in situ to RuII.
A
Grubbs JACS 1988 (110) 960, 7542.
RuII coordination complexes may form RuIV alkylidenes with strained cyclic olefins:
ClRuII
Cl PPh3
PPh3
PPh3
PPh3
+
Ph Ph
CH2Cl2/C6H6
53oC, 11h ClRuIV
Cl
PPh3
PPh3
Ph
Ph
·Stable to H2O (but insoluble pure H2O), alcohols, and acids·Unstable to O2
m
Grubbs JACS 1992 (114) 3974.
Efficient Ru catalyst for acyclic olefin metathesis.
Cl
RuIVCl
PCy3
PCy3
Ph
Ph
stronger σ-donor ligands
·Stable to O2
· High activity for low-strain cyclic olefins and for acyclic olefins.
ClRuIV
Cl
PCy3
PCy3
ClRuIV
Cl
PCy3
PCy3
propagating species: Ruethylidene and Ru propylidene observed by NMR
+
Grubbs JACS 1993 (115) 9858.
·No Wittig-like activity observed even with aldehydes· Poor activity for low-strain cyclic olefins and no activity for acyclic olefins.
Cl
RuIVCl
PCy3
PCy3
Ph
Generation 2 catalyst:
Cl
RuIICl PPh3
PPh3
PPh3
PPh3
1. N2CHR2. PCy3
Advantages of 2 vs. 1: More facile synthesis due to greater availability of diazoalkanes relative tocyclopropenes. Ru catalyst 2 is significantly morereactive than 1 because of more facile initiation.
1
2 Grubbs ACIEE 1995 (34) 2039.
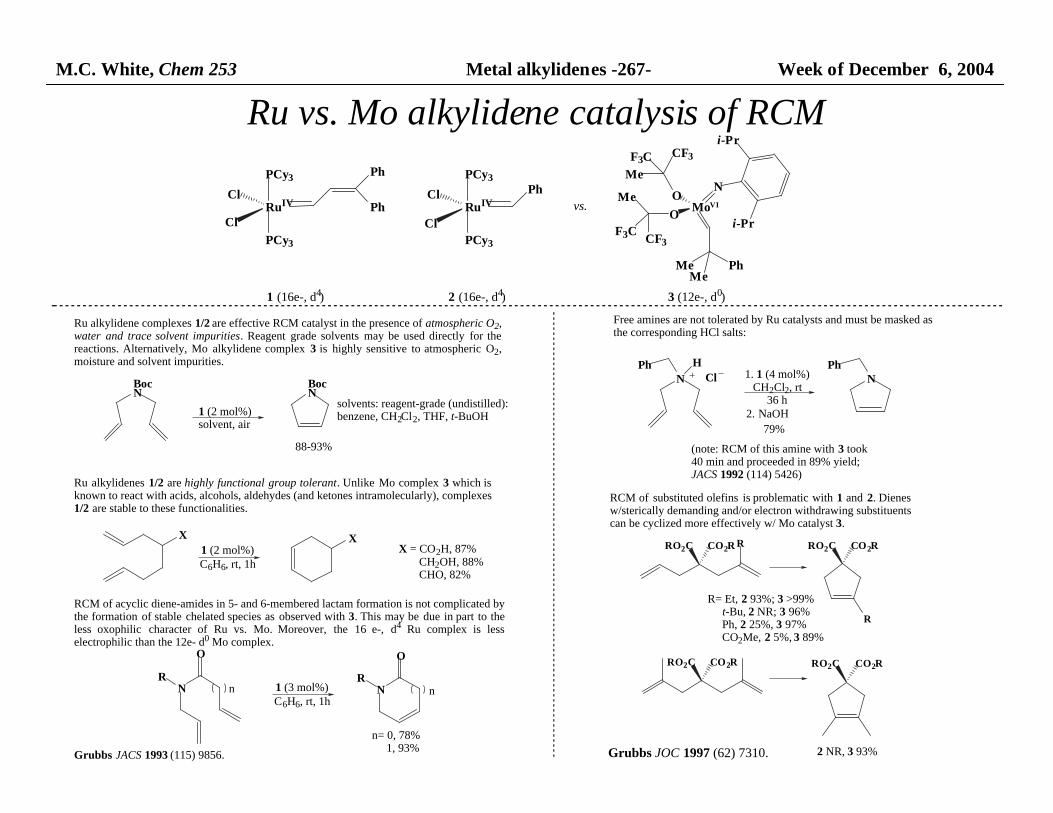
M.C. White, Chem 253 Metal alkylidenes -267- Week of December 6, 2004
Ru vs. Mo alkylidene catalysis of RCM
ClRuIV
Cl
PCy3
PCy3
Ph
Ph
1 (16e-, d4)
vs. MoVI
NO
O
i-Pr
i-Pr
MeMe
Ph
CF3F3C
Me
Me
F3C CF3
3 (12e-, d0)
Cl
RuIVCl
PCy3
PCy3
Ph
2 (16e-, d4)
Ru alkylidene complexes 1/2 are effective RCM catalyst in the presence of atmospheric O2, water and trace solvent impurities. Reagent grade solvents may be used directly for thereactions. Alternatively, Mo alkylidene complex 3 is highly sensitive to atmospheric O2,moisture and solvent impurities.
BocN
1 (2 mol%)solvent, air
BocN
88-93%
solvents: reagent-grade (undistilled): benzene, CH2Cl2, THF, t-BuOH
Ru alkylidenes 1/2 are highly functional group tolerant. Unlike Mo complex 3 which isknown to react with acids, alcohols, aldehydes (and ketones intramolecularly), complexes 1/2 are stable to these functionalities.
X
1 (2 mol%)C6H6, rt, 1h
XX = CO2H, 87% CH2OH, 88% CHO, 82%
RCM of acyclic diene-amides in 5- and 6-membered lactam formation is not complicated by the formation of stable chelated species as observed with 3. This may be due in part to theless oxophilic character of Ru vs. Mo. Moreover, the 16 e-, d4 Ru complex is lesselectrophilic than the 12e- d0 Mo complex.
N
O
Rn 1 (3 mol%)
C6H6, rt, 1hN
O
Rn
n= 0, 78% 1, 93%
Grubbs JACS 1993 (115) 9856.
Free amines are not tolerated by Ru catalysts and must be masked as the corresponding HCl salts:
NPh H
Cl 1. 1 (4 mol%)CH2Cl2, rt
36 h2. NaOH
NPh
(note: RCM of this amine with 3 took 40 min and proceeded in 89% yield; JACS 1992 (114) 5426)
79%
RCM of substituted olefins is problematic with 1 and 2. Dienesw/sterically demanding and/or electron withdrawing substituents can be cyclized more effectively w/ Mo catalyst 3.
RRO2C CO2R
R
RO2C CO2R
R= Et, 2 93%; 3 >99% t-Bu, 2 NR; 3 96% Ph, 2 25%, 3 97% CO2Me, 2 5%, 3 89%
RO2C CO2R RO2C CO2R
2 NR, 3 93%Grubbs JOC 1997 (62) 7310.
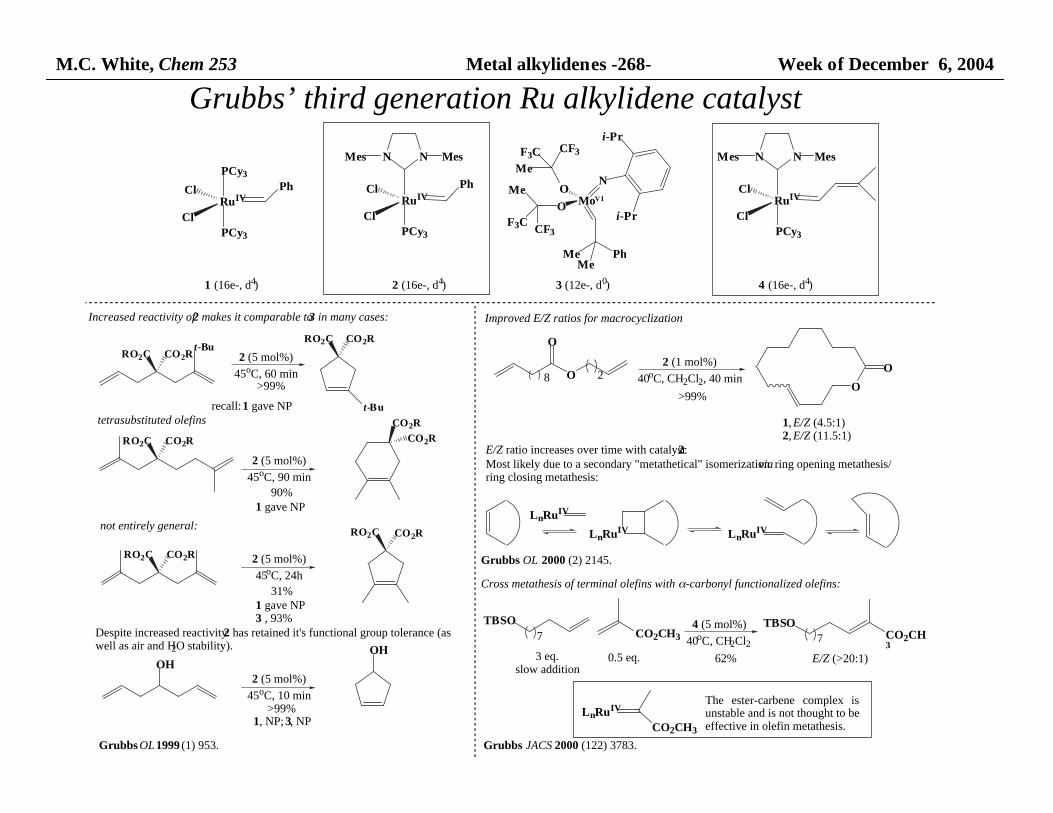
M.C. White, Chem 253 Metal alkylidenes -268- Week of December 6, 2004
Grubbs’ third generation Ru alkylidene catalyst
Cl
RuIVCl
PCy3
PCy3
Ph
1 (16e-, d4)
Cl
RuIVCl
PCy3
Ph
NN MesMes
2 (16e-, d4)
MoV I
NO
O
i-Pr
i-Pr
MeMe
Ph
CF3F3C
Me
Me
F3C CF3
3 (12e-, d0)
t-BuRO2C CO2R
t-Bu
RO2C CO2R
RO2C CO2R
CO2R
CO2R
RO2C CO2R
RO2C CO2R
Improved E/Z ratios for macrocyclization
O
OO
O
LnRuIV
LnRuIV LnRuIV
Grubbs OL 2000 (2) 2145.
OHOH
Increased reactivity of 2 makes it comparable to 3 in many cases:
2 (5 mol%)
45oC, 60 min
recall: 1 gave NP
2 (5 mol%)
45oC, 90 min
>99%
90%
tetrasubstituted olefins
1 gave NP
not entirely general:
2 (5 mol%)
45oC, 24h
31%1 gave NP3 , 93%
Despite increased reactivity 2 has retained it's functional group tolerance (as well as air and H2O stability).
2 (5 mol%)
45oC, 10 min>99%
1, NP; 3, NP
Grubbs OL 1999 (1) 953.
Cross metathesis of terminal olefins with α-carbonyl functionalized olefins:
TBSO
7 CO2CH3
Cl
RuIVCl
PCy3
NN MesMes
4 (16e-, d4)
3 eq.slow addition
0.5 eq.
4 (5 mol%)
40oC, CH2Cl2
62%
TBSO
7
E/Z (>20:1)
CO2CH3
8 22 (1 mol%)
40oC, CH2Cl2, 40 min
>99%
1, E/Z (4.5:1)2, E/Z (11.5:1)
E/Z ratio increases over time with catalyst 2:Most likely due to a secondary "metathetical" isomerization via ring opening metathesis/ ring closing metathesis:
LnRuIV
CO2CH3
The ester-carbene complex isunstable and is not thought to be effective in olefin metathesis.
Grubbs JACS 2000 (122) 3783.
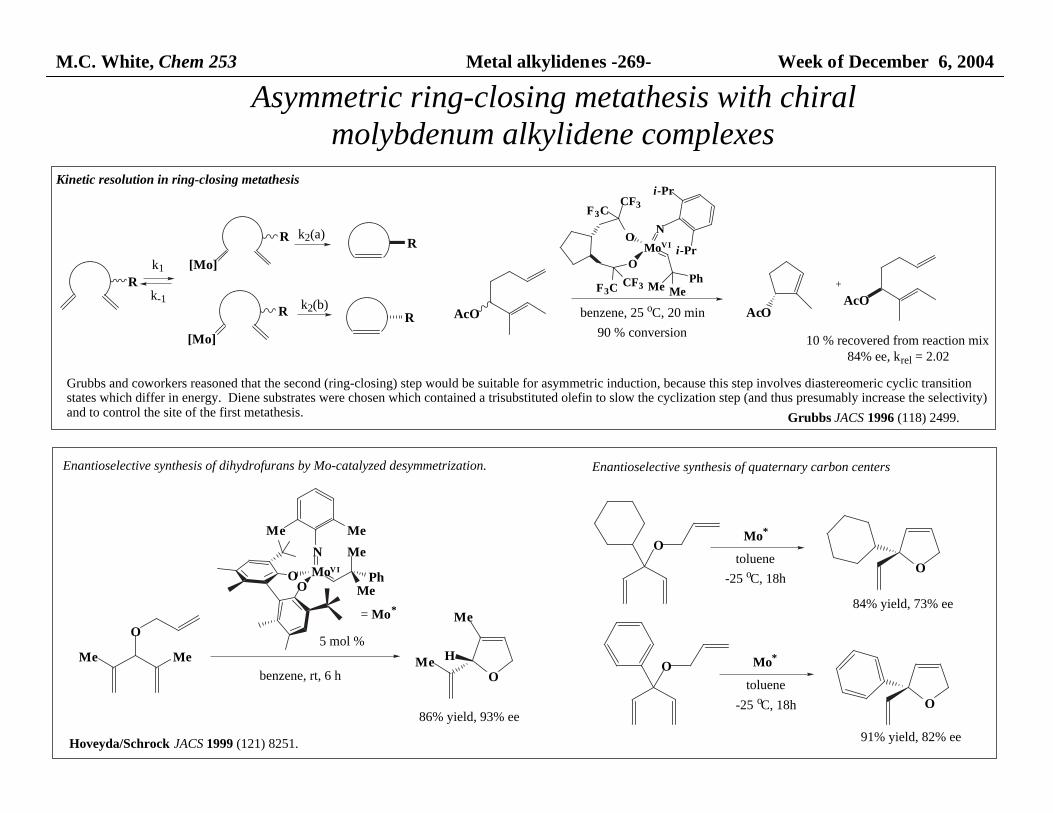
M.C. White, Chem 253 Metal alkylidenes -269- Week of December 6, 2004
Asymmetric ring-closing metathesis with chiral molybdenum alkylidene complexes
R
MeMe
O
[Mo]
[Mo]
R
R
MoVI
N
OO
Me Me
Me
PhMe
R
R
O
Me
HMe
AcO
MoVI
NO
O
i-Pr
i-Pr
Me MePhCF3F3C
F3CCF3
O
O
AcO
O
AcO
O
benzene, 25 oC, 20 min
90 % conversion
+
Kinetic resolution in ring-closing metathesis
10 % recovered from reaction mix84% ee, krel = 2.02
k2(a)
k2(b)
k1
k-1
Grubbs and coworkers reasoned that the second (ring-closing) step would be suitable for asymmetric induction, because this step involves diastereomeric cyclic transition states which differ in energy. Diene substrates were chosen which contained a trisubstituted olefin to slow the cyclization step (and thus presumably increase the selectivity) and to control the site of the first metathesis.
Enantioselective synthesis of dihydrofurans by Mo-catalyzed desymmetrization.
Grubbs JACS 1996 (118) 2499.
Hoveyda/Schrock JACS 1999 (121) 8251.
5 mol %
benzene, rt, 6 h
86% yield, 93% ee
Mo*
toluene
-25 oC, 18h
Mo*
toluene
-25 oC, 18h
84% yield, 73% ee
91% yield, 82% ee
= Mo*
Enantioselective synthesis of quaternary carbon centers
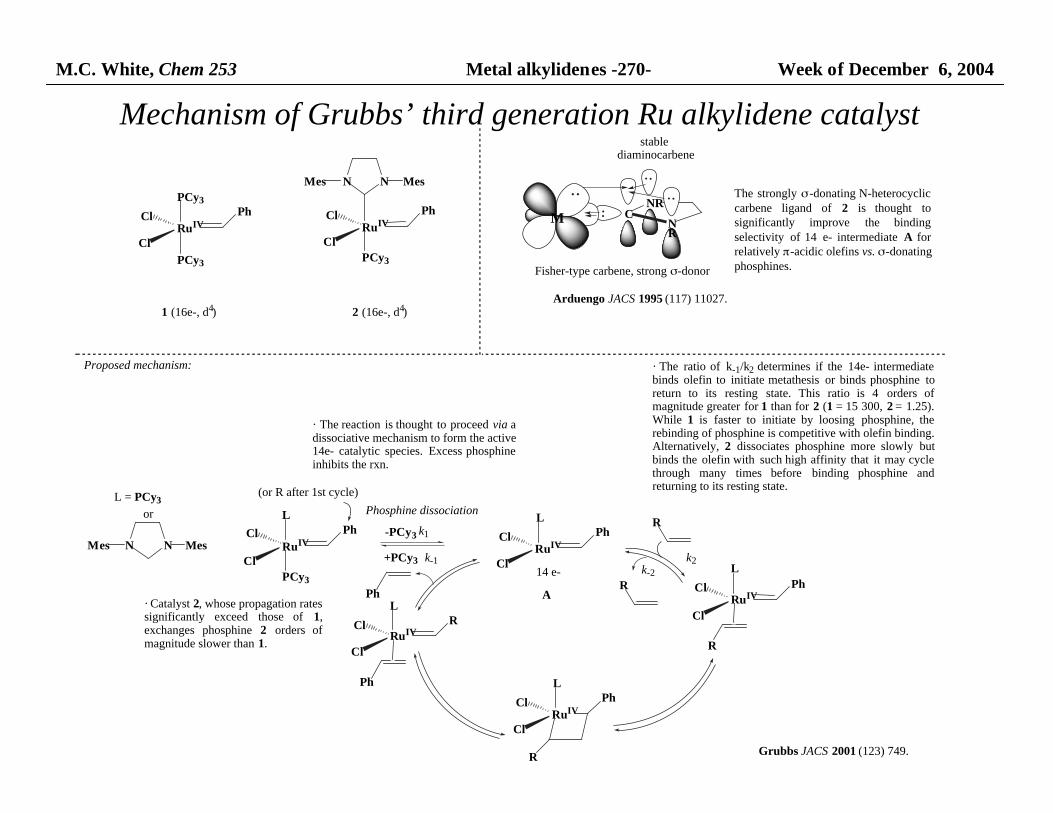
M.C. White, Chem 253 Metal alkylidenes -270- Week of December 6, 2004
Mechanism of Grubbs’ third generation Ru alkylidene catalyst
ClRuIV
Cl
PCy3
PCy3
Ph
1 (16e-, d4)
ClRuIV
Cl
PCy3
Ph
NN MesMes
2 (16e-, d4)
CNR
NR
stable diaminocarbene
Arduengo JACS 1995 (117) 11027.
M
Fisher-type carbene, strong σ-donor
Proposed mechanism:
ClRuIV
Cl
L
PCy3
Ph
L = PCy3
NN MesMes
or Phosphine dissociation
-PCy3
+PCy3 ClRuIV
Cl
LPhk1
k-1
14 e-
Cl
RuIVCl
L
Ph
R
R
k2k-2
R
ClRuIV
Cl
LPh
R
(or R after 1st cycle)
Cl
RuIVCl
LR
Ph
Ph
· The reaction is thought to proceed via adissociative mechanism to form the active 14e- catalytic species. Excess phosphineinhibits the rxn.
· Catalyst 2, whose propagation rates significantly exceed those of 1,exchanges phosphine 2 orders ofmagnitude slower than 1.
· The ratio of k-1/k2 determines if the 14e- intermediatebinds olefin to initiate metathesis or binds phosphine toreturn to its resting state. This ratio is 4 orders ofmagnitude greater for 1 than for 2 (1 = 15 300, 2 = 1.25).While 1 is faster to initiate by loosing phosphine, therebinding of phosphine is competitive with olefin binding. Alternatively, 2 dissociates phosphine more slowly butbinds the olefin with such high affinity that it may cyclethrough many times before binding phosphine andreturning to its resting state.
The strongly σ-donating N-heterocycliccarbene ligand of 2 is thought tosignificantly improve the bindingselectivity of 14 e- intermediate A forrelatively π-acidic olefins vs. σ-donating phosphines.
A
Grubbs JACS 2001 (123) 749.
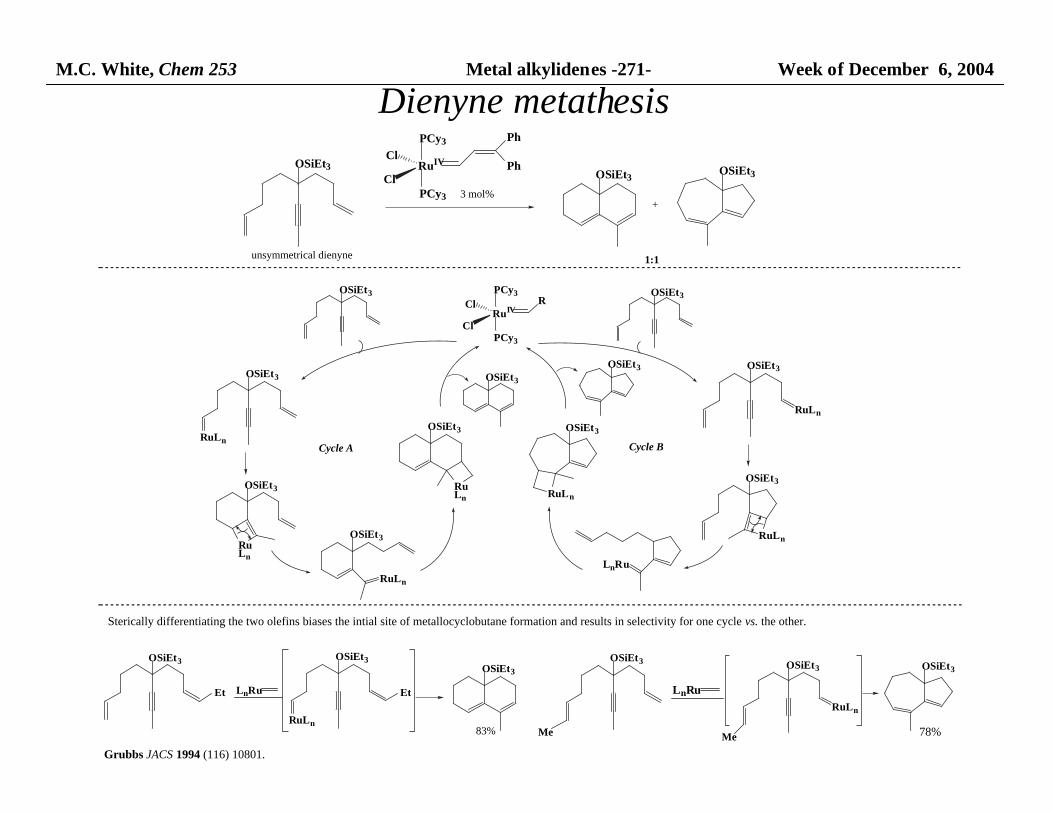
M.C. White, Chem 253 Metal alkylidenes -271- Week of December 6, 2004
OSiEt3
ClRuIV
Cl
PCy3
PCy3
Ph
PhOSiEt3
OSiEt3
OSiEt3
RuLn
OSiEt3
RuLn
OSiEt3
OSiEt3
RuLn
OSiEt3
RuLn
ClRuIV
Cl
PCy3
PCy3
ROSiEt3
OSiEt3
RuLn
LnRu
OSiEt3
RuLn
OSiEt3
RuLn
OSiEt3
OSiEt3
3 mol%+
1:1unsymmetrical dienyne
Cycle A Cycle B
OSiEt3
Et
RuLn
OSiEt3
EtLnRu
OSiEt3
OSiEt3
LnRu
Me
RuLn
OSiEt3
Me
OSiEt3
83%
Sterically differentiating the two olefins biases the intial site of metallocyclobutane formation and results in selectivity for one cycle vs. the other.
78%
Grubbs JACS 1994 (116) 10801.
Dienyne metathesis
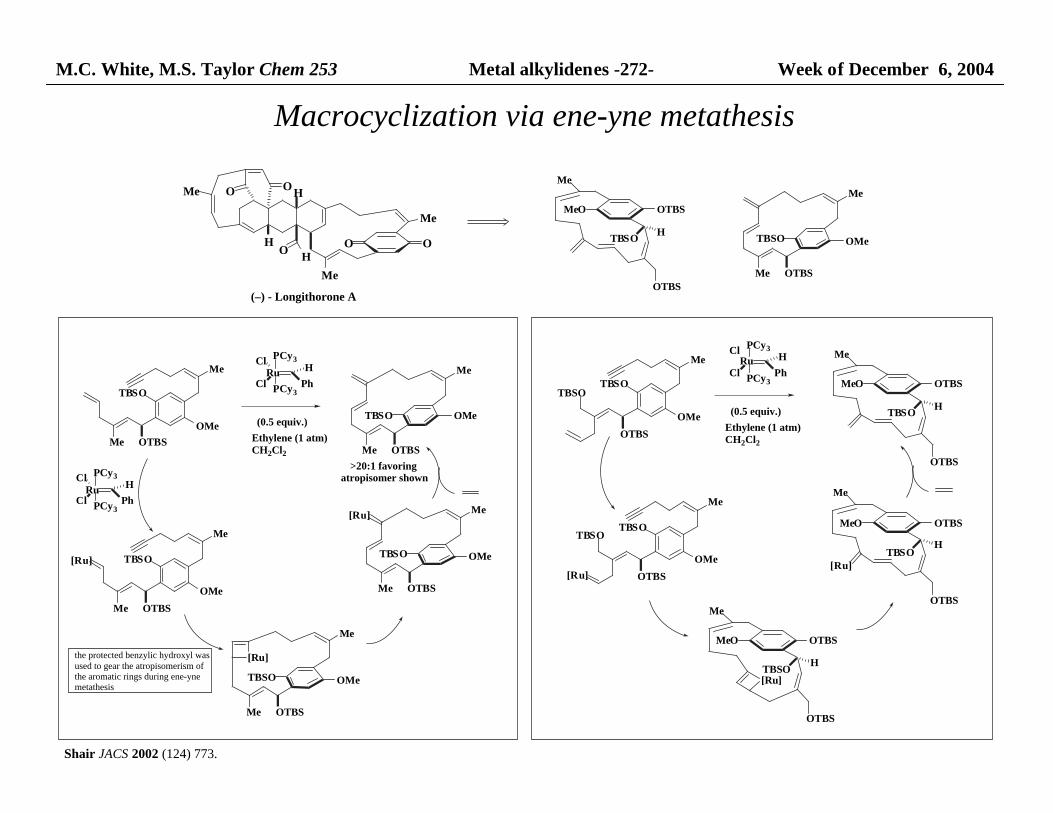
M.C. White, M.S. Taylor Chem 253 Metal alkylidenes -272- Week of December 6, 2004
O O
Me
Me
H
H
Me OO
OH
(–) - Longithorone A
Me
MeO OTBS
TBSOH
OTBS
OTBS
TBSO OMe
Me
Me
Macrocyclization via ene-yne metathesis
Me
TBSO
OMe
Me OTBS
Ru
PCy3
PCy3Ph
HCl
Cl
OTBS
TBSO OMe
Me
Me
[Ru]
Me
OMe
Me
[Ru]
OTBS
TBSO
Ru
PCy3
PCy3Ph
HCl
Cl
OTBS
TBSO OMe
Me
Me
[Ru]
OTBS
TBSO OMe
Me
Me
(0.5 equiv.)
Ethylene (1 atm)CH2Cl2
>20:1 favoring atropisomer shown
Shair JACS 2002 (124) 773.
the protected benzylic hydroxyl was used to gear the atropisomerism of the aromatic rings during ene-yne metathesis
Me
TBSO
OMe
OTBS
TBSO
Ru
PCy3
PCy3Ph
HCl
Cl
Me
[Ru]
MeO OTBS
TBSOH
OTBS
Me
TBSO
OMe
OTBS[Ru]
TBSO
Me
[Ru]
MeO OTBS
TBSOH
OTBS
Me
MeO OTBS
TBSOH
OTBS
(0.5 equiv.)
Ethylene (1 atm)CH2Cl2
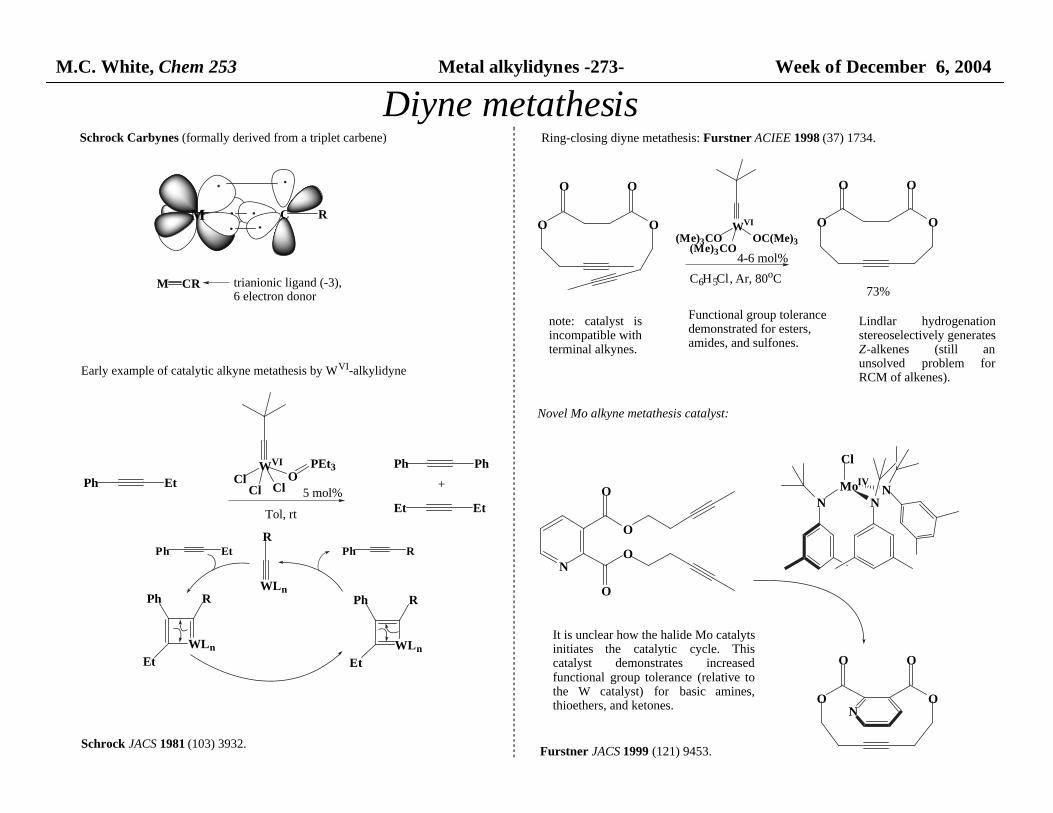
M.C. White, Chem 253 Metal alkylidynes -273- Week of December 6, 2004
Schrock Carbynes (formally derived from a triplet carbene)
M C R
M CR trianionic ligand (-3), 6 electron donor
Early example of catalytic alkyne metathesis by WVI-alkylidyne
Ph Et
WVI
ClCl Cl
OPEt3
Tol, rt
5 mol%
Ph Ph
+
Et Et
WLn
R
WLn
RPh
Et
WLn
RPh
Et
Ph Et Ph R
Schrock JACS 1981 (103) 3932.
Ring-closing diyne metathesis: Furstner ACIEE 1998 (37) 1734.
O
OO
O
WVI
(Me)3CO(Me)3CO
OC(Me)3
O
OO
O
4-6 mol%
C6H5Cl, Ar, 80oC73%
Functional group tolerance demonstrated for esters, amides, and sulfones.
note: catalyst isincompatible with terminal alkynes.
Lindlar hydrogenationstereoselectively generates Z-alkenes (still anunsolved problem forRCM of alkenes).
Novel Mo alkyne metathesis catalyst:
MoIV
Cl
N NN
N
O
O
O
O
O
OO
O
N
It is unclear how the halide Mo catalyts initiates the catalytic cycle. Thiscatalyst demonstrates increasedfunctional group tolerance (relative tothe W catalyst) for basic amines,thioethers, and ketones.
Furstner JACS 1999 (121) 9453.
Diyne metathesis
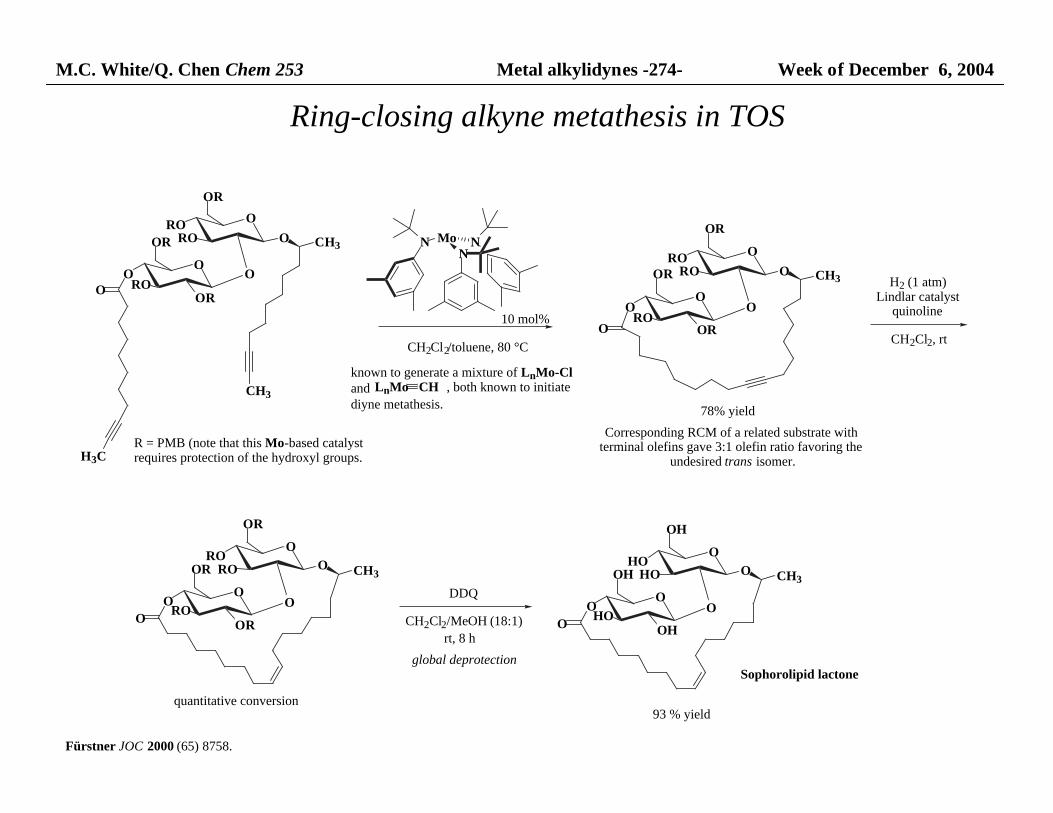
M.C. White/Q. Chen Chem 253 Metal alkylidynes -274- Week of December 6, 2004
OO
ROOR
O
OR
ORO
RO O
OR
CH3
CH3
O
H3C
OO
ROOR
O
OR
ORO
RO O
OR
CH3
O
N MoN
N
DDQ
R = PMB (note that this Mo-based catalystrequires protection of the hydroxyl groups.
Sophorolipid lactone
CH2Cl2/toluene, 80 °C
OO
ROOR
O
OR
ORO
RO O
OR
CH3
O
78% yield
H2 (1 atm)Lindlar catalyst
quinoline
CH2Cl2, rt
Corresponding RCM of a related substrate with terminal olefins gave 3:1 olefin ratio favoring the
undesired trans isomer.
OO
HOOH
O
OH
OHO
HO O
OH
CH3
OCH2Cl2/MeOH (18:1)
global deprotection
quantitative conversion
rt, 8 h
93 % yield
10 mol%
known to generate a mixture of LnMo-Cland LnMo CH , both known to initiate
diyne metathesis.
Ring-closing alkyne metathesis in TOS
Fürstner JOC 2000 (65) 8758.
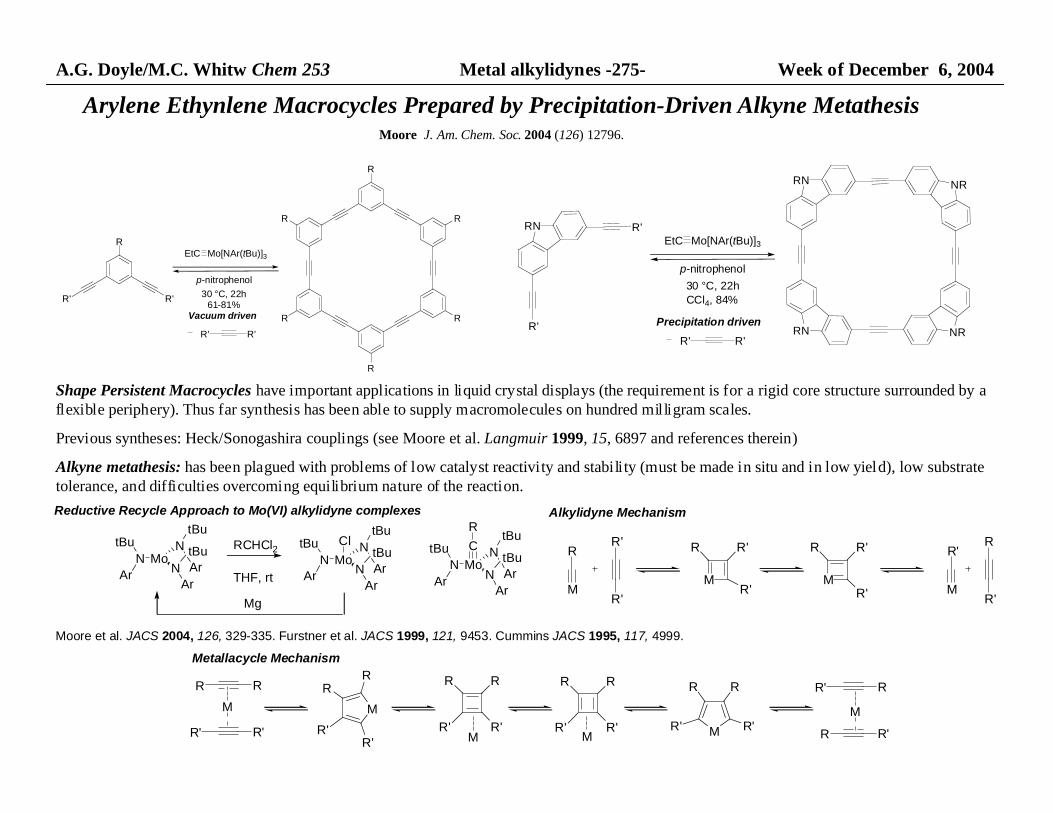
A.G. Doyle/M.C. Whitw Chem 253 Metal alkylidynes -275- Week of December 6, 2004
Arylene Ethynlene Macrocycles Prepared by Precipitation-Driven Alkyne MetathesisMoore J. Am. Chem. Soc. 2004 (126) 12796.
R
R'R'
R
R R
R R
R
R'R'
EtC Mo[NAr(tBu)]3
p-nitrophenol
30 °C, 22h61-81%
Vacuum driven
RN
R'
RN
RN
NR
NR
R'EtC Mo[NAr(tBu)]3
p-nitrophenol
30 °C, 22hCCl4, 84%
R'R'
Precipitation driven
Shape Persistent Macrocycles have important applications in liquid crystal displays (the requirement is for a rigid core structure surrounded by aflexible periphery). Thus far synthesis has been able to supply macromolecules on hundred milligram scales.
Previous syntheses: Heck/Sonogashira couplings (see Moore et al. Langmuir 1999, 15, 6897 and references therein)
Alkyne metathesis: has been plagued with problems of low catalyst reactivity and stability (must be made in situ and in low yield), low substratetolerance, and difficulties overcoming equilibrium nature of the reaction.
N
ArMoN
tBu
ArN
tBu
Ar
tBuRCHCl2
THF, rt
N
ArMoN
tBu
ArN
tBu
Ar
tBuCl
N
ArMoN
tBu
ArN
tBu
Ar
tBuC
R
Mg
Reductive Recycle Approach to Mo(VI) alkylidyne complexes
Moore et al. JACS 2004, 126, 329-335. Furstner et al. JACS 1999, 121, 9453. Cummins JACS 1995, 117, 4999.
R'
R'
R
MM
R'
R'
R
M
R'
R'
R R
R'
R'
M
Alkylidyne Mechanism
R' R'
R R
M
R'R'
RR
R
R'
R
R'
R
R'
R
R' M
R
R' R'
R
R R'
R' R
M
M M
M
Metallacycle Mechanism













![Room-temperature polymerization of ββββ-pinene by niobium ......polymerization [4,5]. Lewis acid-promoted cationic polymerization represents the most efficient method in the commercial](https://static.fdocument.org/doc/165x107/61290b395072b0244f019799/room-temperature-polymerization-of-pinene-by-niobium-polymerization.jpg)
![Synthesis ].Cl and [Ni(en) ] - Shodhgangashodhganga.inflibnet.ac.in/bitstream/10603/21087/8/12_chapter 3.pdf · NiSn 2 Cl 6] displayed a π→π* transitions bands at 272 and 274](https://static.fdocument.org/doc/165x107/5cdd36ca88c993b1358da484/synthesis-cl-and-nien-3pdf-nisn-2-cl-6-displayed-a-transitions.jpg)


