
NF-kB1 Inhibits TLR-Induced IFN-b Production in ... · unclear. In this study, we analyzed the gene...
9
of September 20, 2018. This information is current as Dependent ERK Activation - Production in Macrophages through TPL-2 β B1 Inhibits TLR-Induced IFN- κ NF- Fox, Steven C. Ley and Bruce H. Horwitz O'Garra, Michal F. Tomczak, Susan E. Erdman, James G. Demissie, Stamatia Papoutsopoulou, Agnes Mambole, Anne Huei-Ting Yang, Yanyan Wang, Xixing Zhao, Ezana http://www.jimmunol.org/content/186/4/1989 doi: 10.4049/jimmunol.1001003 January 2011; 2011; 186:1989-1996; Prepublished online 7 J Immunol Material Supplementary 3.DC1 http://www.jimmunol.org/content/suppl/2011/01/07/jimmunol.100100 References http://www.jimmunol.org/content/186/4/1989.full#ref-list-1 , 22 of which you can access for free at: cites 38 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2011 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on September 20, 2018 http://www.jimmunol.org/ Downloaded from by guest on September 20, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of NF-kB1 Inhibits TLR-Induced IFN-b Production in ... · unclear. In this study, we analyzed the gene...

of September 20, 2018.This information is current as
Dependent ERK Activation−Production in Macrophages through TPL-2
βB1 Inhibits TLR-Induced IFN-κNF-
Fox, Steven C. Ley and Bruce H. HorwitzO'Garra, Michal F. Tomczak, Susan E. Erdman, James G.Demissie, Stamatia Papoutsopoulou, Agnes Mambole, Anne Huei-Ting Yang, Yanyan Wang, Xixing Zhao, Ezana
http://www.jimmunol.org/content/186/4/1989doi: 10.4049/jimmunol.1001003January 2011;
2011; 186:1989-1996; Prepublished online 7J Immunol
MaterialSupplementary
3.DC1http://www.jimmunol.org/content/suppl/2011/01/07/jimmunol.100100
Referenceshttp://www.jimmunol.org/content/186/4/1989.full#ref-list-1
, 22 of which you can access for free at: cites 38 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2011 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on September 20, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on September 20, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
NF-kB1 Inhibits TLR-Induced IFN-b Production inMacrophages through TPL-2–Dependent ERK Activation
Huei-Ting Yang,*,1 Yanyan Wang,†,1 Xixing Zhao,† Ezana Demissie,†
Stamatia Papoutsopoulou,* Agnes Mambole,* Anne O’Garra,‡ Michal F. Tomczak,†
Susan E. Erdman,x James G. Fox,x Steven C. Ley,* and Bruce H. Horwitz†,{
Although NF-kB1 p50/p105 has critical roles in immunity, the mechanism by which NF-kB1 regulates inflammatory responses is
unclear. In this study, we analyzed the gene expression profile of LPS-stimulated Nfkb12/2 macrophages that lack both p50 and
p105. Deficiency of p50/p105 selectively increased the expression of IFN-responsive genes, which correlated with increased IFN-b
expression and STAT1 phosphorylation. IFN Ab-blocking experiments indicated that increased STAT1 phosphorylation and
expression of IFN-responsive genes observed in the absence of p50/p105 depended upon autocrine IFN-b production. Markedly
higher serum levels of IFN-b were observed in Nfkb12/2 mice than in wild-type mice following LPS injection, demonstrating that
Nfkb1 inhibits IFN-b production under physiological conditions. TPL-2, a mitogen-activated protein kinase kinase kinase stabi-
lized by association with the C-terminal ankyrin repeat domain of p105, negatively regulates LPS-induced IFN-b production by
macrophages via activation of ERK MAPK. Retroviral expression of TPL-2 in Nfkb12/2 macrophages, which are deficient in
endogenous TPL-2, reduced LPS-induced IFN-b secretion. Expression of the C-terminal ankyrin repeat domain of p105 in
Nfkb12/2 macrophages, which rescued LPS activation of ERK, also inhibited IFN-b expression. These data indicate that p50/
p105 negatively regulates LPS-induced IFN signaling in macrophages by stabilizing TPL-2, thereby facilitating activation of
ERK. The Journal of Immunology, 2011, 186: 1989–1996.
The NF-kB family of transcription factors, which share anN-terminal Rel-homology domain, includes five subunits,NF-kB1 p50, p65 (RelA), c-Rel, RelB, and NF-kB2 p52
(1). These proteins form homo- and heterodimers that are retainedin the cytoplasm of unstimulated cells by members of the IkBfamily of ankyrin repeat-containing proteins (IkB-a, -b, and -ε)(2). Inflammatory stimuli activate the IkB kinase complex (IKK),which phosphorylates two serines found within the N termini ofthe IkBs, directing proteosomal degradation (3). IkB degradationallows nuclear translocation of liberated NF-kB dimers, which thenmodulate transcription by binding to kB sites found within the
promoter and enhancer regions of many genes involved in immuneand inflammatory responses (1, 4). p65, c-Rel, and RelB, whicheach have a C-terminal trans-activation domain, are synthesized intheir mature form. In contrast, p50 and p52 are produced from the Ntermini of larger precursor proteins NF-kB1 p105 and NF-kB2p100, respectively, by proteolytic removal of their C termini by theproteasome, and lack trans-activation domains (2).NF-kB1 p50 induces transcription in heterodimeric complexes
with trans-activating Rel subunits. However, p50 homodimers canrepress transcription by interfering with the binding of other activeNF-kB dimers and recruiting histone deacetylases to the promo-ter regions of kB-target genes (5, 6). It has been suggested thatp50 homodimers are responsible for inhibiting TNF expressionin LPS-tolerized macrophages (7–9), and maintaining an anti-inflammatory phenotype in M2-polarized tumor-associated mac-rophages (9, 10). p105 also has NF-kB inhibitory activity, due toits C-terminal ankyrin repeats. These are homologous to thosefound in the classical IkB proteins, and retain NF-kB dimers inthe cytoplasm of unstimulated cells. IKK phosphorylation of twoserines in the C-terminal region of p105 after agonist stimulationleads to proteosomal-dependent complete degradation of p105(11–13), releasing associated p50, p65, and c-Rel to translocateinto the nucleus (14). The C-terminal portion of p105 additionallybinds to TPL-2 (also known as MAP3K8 and Cot) (11, 15), amitogen-activated protein kinase kinase kinase that is essentialfor ERK activation following LPS stimulation of macrophages(16). Binding of p105 prevents TPL-2 from phosphorylating itsdownstream targets, MEK-1/2, and is also required to maintainTPL-2 protein stability. Consequently, LPS-induced activationof ERK-1/2 is blocked in bone marrow-derived macrophages(BMDMs) that lack p50/p105, due to substantially decreasedamounts of TPL-2 protein (17).The multiple biochemical functions of NF-kB1 p50/p105 sug-
gest that its role in immune responses is complex. This is consistent
*Division of Immune Cell Biology, National Institute for Medical Research, London,United Kingdom; ‡Division of Immunoregulation, National Institute for MedicalResearch, London, United Kingdom; †Department of Pathology, Brigham and Wom-en’s Hospital, Boston, MA 02115; xDivision of Comparative Medicine, Cambridge,MA 02139; and {Division of Emergency Medicine, Children’s Hospital, Boston, MA02115
1H.-T.Y. and Y.W. contributed equally to this work.
Received for publication March 29, 2010. Accepted for publication December 8,2010.
This work was supported by National Institutes of Health Grants AI52267 (to B.H.H.and S.E.E.), CA108854 (to S.E.E.), and CA67529 (to J.G.F.). H.-T.Y., S.P., andS.C.L. were supported by the United Kingdom Medical Research Council.
The microarray data presented in this article have been submitted to the NationalCenter for Biotechnology Information’s Gene Expression Omnibus under accessionnumber GSE19941.
Address correspondence and reprint requests to Dr. Bruce H. Horwitz or Dr. StevenC. Ley, Department of Pathology, Brigham and Women’s Hospital, HNRB 630E,77 Avenue Louis Pasteur, Boston, MA 02115 (B.H.H.), or Division of ImmuneCell Biology, National Institute for Medical Research, Mill Hill, London NW71AA, United Kingdom (S.C.L.). E-mail addresses: [email protected] (B.H.H.) and [email protected] (S.C.L.)
The online version of this article contains supplemental material.
Abbreviations used in this article: BMDM, bone marrow-derived macrophage; IKK,IkB kinase complex; WT, wild-type.
Copyright� 2011 by The American Association of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1001003
by guest on September 20, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

with observations that mice lacking p50/p105 exhibit both multi-focal immune defects and increased sensitivity to inflammation (18,19). To further define the function of p50/p105 during TLR-inducedinflammatory responses, we profiled gene expression followingLPS stimulation of Nfkb12/2 macrophages. This analysis revealedthat macrophages lacking p50/p105 demonstrated augmented in-duction of a type I IFN response. We show that the ability of NF-kB1 p50/p105 to inhibit this response depends on its ability tostabilize TPL-2 and facilitate TLR-induced ERK activation. Thus,p50/p105-dependent ERK activation plays an important role inregulating the nature of TLR-induced inflammatory responses.
Materials and MethodsExperimental animals
Il102/2, Nfkb12/2Il102/2, and Il102/2Rag22/2 mice were maintained onthe 129S6/SvEvTac background, as previously described (20, 21).Nfkb12/2
Il102/2Rag2/2 mice were generated by crossing Nfkb12/2Il102/2 andIl102/2Rag22/2 mice. Map3k82/2 mice (provided by Professor PhilipTsichlis, Tufts University, Boston and Thomas Jefferson University, Phil-adelphia, PA) were on the C57BL/6 background. In experiments compar-ing Map3k82/2 mice with wild-type (WT) and Nfkb12/2 mice, all strainswere on the C57BL/6 background.
In vitro stimulation of BMDM
BMDM were grown as previously described (20) and stimulated with LPSfrom Escherichia coli 0127:B8 (Sigma-Aldrich, St. Louis, MO) at 1 ng/ml(unless stated otherwise) or mouse IFN-b (PBL Biomedical Laboratories,New Brunswick, NJ) at 10 U/ml. rIL-10 (BD Biosciences, San Diego, CA)was used at 0.3 ng/ml. IFN-b depletion assays were performed by includinganti–IFN-b Ab (75-D3; Yamasa Soya, Tokyo, Japan) or control IgG ata concentration of 6.7 mg/ml. PD184352 (provided by Professor Sir PhilipCohen, University of Dundee, U.K.) was used at a concentration of 2 mM.
Preparation of RNA
RNA was isolated in TRIzol (Invitrogen, Carlsbad, CA) or using RNeasymini kit (Qiagen, Valencia, CA) following the manufacturer’s instructions.
Gene expression profiling
Probes were prepared from isolated RNA, and hybridized to Affymetrix430A2.0 arrays. Two independent sampleswere analyzed for each condition.Expression data were read from CEL files, and background correction,
normalization, and summarization of the probe-level data into probe setexpression values were accomplished using Robust Multi-Array Analysis.Differential expression between samples was analyzed using the LIMMApackage of the afflylmGUIwithin theBioconductorR environment (22). Themicroarray data discussed in this publication are accessible throughNationalCenter for Biotechnology Information’s Gene Expression Omnibus, ac-cession number GSE19941 (http://www.ncbi.nlm.nih.gov/geo).
RT-PCR
RT-PCR analysis was performed using probes from Applied Biosystems(Foster City, CA), as per the manufacturer’s instructions. Expression foreach sample was analyzed in duplicate and normalized between samplesusing the DD cycle threshold method. Fold-change is reported as relative touninduced levels in control BMDM. Each data point represents the average6 SEM for two or three independent macrophage pools. Experiments arerepresentative of two or three independent experiments with consistentresults.
ELISA
IFN-b was analyzed in culture supernatants and serum using a commer-cially available IFN-b–specific ELISA kit (PBL Biomedical Laboratories),according to the manufacturer’s instructions.
Immunoblotting
ERK (p44/42 MAPK), phospho-ERK (p44/42 MAPK) (Thr202/Tyr204), andphospho-STAT1 (Y701) Abs were purchased from Cell Signaling Tech-nology (Beverly, MA). STAT1 p84/p91 (E-23), TFIID (Sl-1), c-Fos (sc-52), and TPL-2 (sc-726) Abs were purchased from Santa Cruz Bio-technology (Santa Cruz, CA).
In vivo LPS challenge
Adult male mice (10-12 wk old) were injected i.p. with LPS (200 mg) andsacrificed 4 h later. Serum was collected, and IFN-b was measured byELISA.
Retrovirally mediated gene expression
For expression of TPL-2 and c-Fos, BMDM were infected with ecotropicretroviruses expressing the gene of interest and GFP, or GFP alone, asdescribed (23). Following infection, GFP-positive BMDMwere isolated byFACS. Cells were cultured at 23 105 cells/well in 200 ml medium in a 96-well plate and stimulated with 10 ng/ml LPS for 24 h. The culture mediumwas used for IFN-b–specific ELISA, and cell pellets were lysed in 50 mllysis buffer (24) for measurement of total protein by Bradford assay (Bio-Rad). For expression of p105DN, BMDM were infected with retrovirusesexpressing p105DN (murine p105 aa 434-971) and the gene for puromycin
Table I. Top 20 genes expressed at higher levels in Nfkb12/2Il102/2 BMDM than in Il102/2 BMDMfollowing LPS stimulation
Symbol Name Fold-Change p Value
Gbp1 Guanylate nucleotide-binding protein 1 182 0.00Iigp1 IFN-inducible GTPase 1 19.0 0.01Rsad2 Radical S-adenosyl methionine domain containing 2 14.2 0.00Mx1 Myxovirus (influenza virus) resistance 1 14.0 0.01Oasl1 29-59 Oligoadenylate synthetase-like 1 13.7 0.00Tyki Thymidylate kinase family LPS-inducible member 12.6 0.01Tgtp T cell-specific GTPase 12.2 0.01Slfn1 Schlafen 1 12.2 0.02Ifit2 IFN-induced protein with tetratricopeptide repeats 2 10.6 0.01Mpa2l Macrophage activation 2-like 10.3 0.00Il12b IL-12b 10.2 0.00Usp18 Ubiquitin-specific peptidase 18 9.96 0.01Gfi1 Growth factor independent 1 8.50 0.00Il15 IL-15 7.24 0.00Nt5c3 59-Nucleotidase, cytosolic III 7.01 0.00Mx2 Myxovirus (influenza virus) resistance 2 6.89 0.00Iigp2 IFN-inducible GTPase 2 6.29 0.00Cxcl10 Chemokine (C-X-C motif) ligand 10 6.06 0.00Chi3l1 Chitinase 3-like 1 5.82 0.00Cxcl9 Chemokine (C-X-C motif) ligand 9 5.75 0.01
Fold-change refers to expression in Nfkb12/2Il102/2 BMDM divided by expression in Il102/2 BMDM following stim-ulation with LPS in the presence of exogenous IL-10. p refers to the p value between genotypes calculated using theBenjamini-Hochberg correction for multiple comparison testing. All listed genes were induced at least 4-fold by LPS ineither Il102/2 or Nfkb12/2Il102/2 BMDM. Duplicates, expressed sequence tags, and hypothetical genes have been removed.
1990 NF-kB1 INHIBITS TLR-INDUCED IFN SIGNALING
by guest on September 20, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
resistance, or the gene for puromycin resistance alone. Puromcyin-resistantcells were isolated by selection for 72 h in media containing puromycin.After selection, cells were replated at 53 105 cells/well in 1 ml medium ina 24-well plate for total protein extraction, and at 5 3 104 cells/well in 200ml media for analysis of cytokine secretion. Cells were stimulated thefollowing day with LPS. Culture supernatants were assayed for IFN-b byELISA.
p50 DNA-binding ELISA
p50 DNA-binding activity in nuclear extracts derived from LPS-stimulatedWT and Map3k82/2 BMDM was measured with the p50 DNA-bindingELISA kit from Active Motif (Carlsbad, CA), as per the manufacturer’sinstructions. Data are normalized to total protein in the nuclear extractsdetermined by Bradford assay (Bio-Rad).
Statistical analysis
All data analysis was performed using GraphPad Prism software (GraphPadSoftware, San Diego, CA). Data generated by RT-PCR or ELISA werecompared using t tests for parametric data; *p , 0.05, **p , 0.01, and***p , 0.001. Error bars represent SEM.
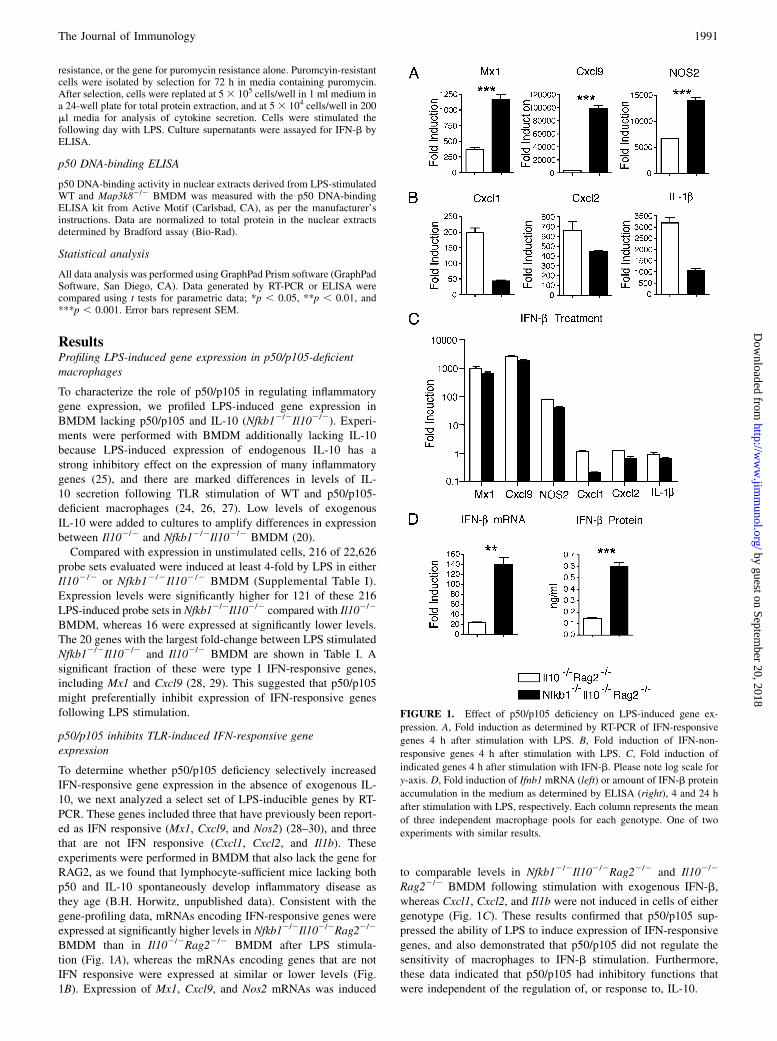
ResultsProfiling LPS-induced gene expression in p50/p105-deficientmacrophages
To characterize the role of p50/p105 in regulating inflammatorygene expression, we profiled LPS-induced gene expression inBMDM lacking p50/p105 and IL-10 (Nfkb12/2Il102/2). Experi-ments were performed with BMDM additionally lacking IL-10because LPS-induced expression of endogenous IL-10 has astrong inhibitory effect on the expression of many inflammatorygenes (25), and there are marked differences in levels of IL-10 secretion following TLR stimulation of WT and p50/p105-deficient macrophages (24, 26, 27). Low levels of exogenousIL-10 were added to cultures to amplify differences in expressionbetween Il102/2 and Nfkb12/2Il102/2 BMDM (20).Compared with expression in unstimulated cells, 216 of 22,626
probe sets evaluated were induced at least 4-fold by LPS in eitherIl102/2 or Nfkb12/2Il102/2 BMDM (Supplemental Table I).Expression levels were significantly higher for 121 of these 216LPS-induced probe sets in Nfkb12/2Il102/2 compared with Il102/2
BMDM, whereas 16 were expressed at significantly lower levels.The 20 genes with the largest fold-change between LPS stimulatedNfkb12/2Il102/2 and Il102/2 BMDM are shown in Table I. Asignificant fraction of these were type I IFN-responsive genes,including Mx1 and Cxcl9 (28, 29). This suggested that p50/p105might preferentially inhibit expression of IFN-responsive genesfollowing LPS stimulation.
p50/p105 inhibits TLR-induced IFN-responsive geneexpression
To determine whether p50/p105 deficiency selectively increasedIFN-responsive gene expression in the absence of exogenous IL-10, we next analyzed a select set of LPS-inducible genes by RT-PCR. These genes included three that have previously been report-ed as IFN responsive (Mx1, Cxcl9, and Nos2) (28–30), and threethat are not IFN responsive (Cxcl1, Cxcl2, and Il1b). Theseexperiments were performed in BMDM that also lack the gene forRAG2, as we found that lymphocyte-sufficient mice lacking bothp50 and IL-10 spontaneously develop inflammatory disease asthey age (B.H. Horwitz, unpublished data). Consistent with thegene-profiling data, mRNAs encoding IFN-responsive genes wereexpressed at significantly higher levels in Nfkb12/2Il102/2Rag22/2
BMDM than in Il102/2Rag22/2 BMDM after LPS stimula-tion (Fig. 1A), whereas the mRNAs encoding genes that are notIFN responsive were expressed at similar or lower levels (Fig.1B). Expression of Mx1, Cxcl9, and Nos2 mRNAs was induced
to comparable levels in Nfkb12/2Il102/2Rag22/2 and Il102/2
Rag22/2 BMDM following stimulation with exogenous IFN-b,whereas Cxcl1, Cxcl2, and Il1b were not induced in cells of eithergenotype (Fig. 1C). These results confirmed that p50/p105 sup-pressed the ability of LPS to induce expression of IFN-responsivegenes, and also demonstrated that p50/p105 did not regulate thesensitivity of macrophages to IFN-b stimulation. Furthermore,these data indicated that p50/p105 had inhibitory functions thatwere independent of the regulation of, or response to, IL-10.
FIGURE 1. Effect of p50/p105 deficiency on LPS-induced gene ex-
pression. A, Fold induction as determined by RT-PCR of IFN-responsive
genes 4 h after stimulation with LPS. B, Fold induction of IFN-non-
responsive genes 4 h after stimulation with LPS. C, Fold induction of
indicated genes 4 h after stimulation with IFN-b. Please note log scale for
y-axis. D, Fold induction of Ifnb1 mRNA (left) or amount of IFN-b protein
accumulation in the medium as determined by ELISA (right), 4 and 24 h
after stimulation with LPS, respectively. Each column represents the mean
of three independent macrophage pools for each genotype. One of two
experiments with similar results.
The Journal of Immunology 1991
by guest on September 20, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
We next determined whether the increase in IFN-responsive geneexpression observed following LPS stimulation of BMDM lackingp50/p105 could result from increased production of IFN-b itself.Consistent with this hypothesis, we found that LPS induced sig-nificantly higher expression of Ifnb1 mRNA, as well as higherlevels of secreted IFN-b protein, in Nfkb12/2Il102/2Rag22/2
BMDM than in Il102/2Rag22/2 BMDM (Fig. 1D).
Increased IFN signaling observed in the absence of p50/p105depends on expression of IFN-b
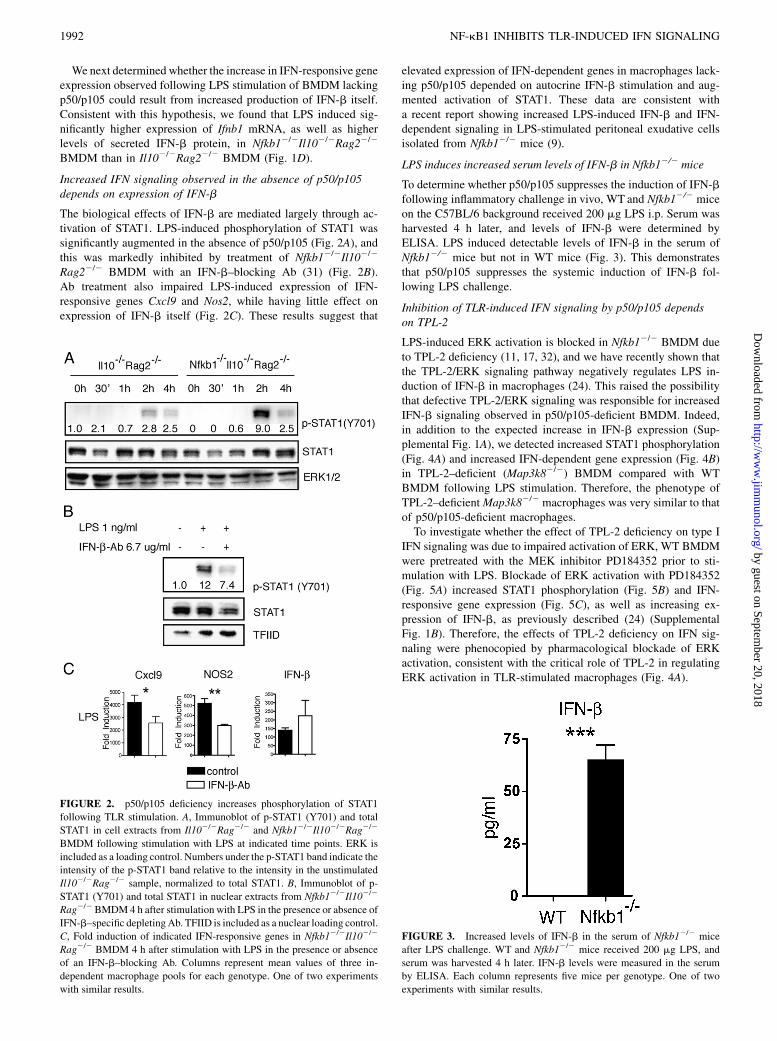
The biological effects of IFN-b are mediated largely through ac-tivation of STAT1. LPS-induced phosphorylation of STAT1 wassignificantly augmented in the absence of p50/p105 (Fig. 2A), andthis was markedly inhibited by treatment of Nfkb12/2Il102/2
Rag22/2 BMDM with an IFN-b–blocking Ab (31) (Fig. 2B).Ab treatment also impaired LPS-induced expression of IFN-responsive genes Cxcl9 and Nos2, while having little effect onexpression of IFN-b itself (Fig. 2C). These results suggest that
elevated expression of IFN-dependent genes in macrophages lack-ing p50/p105 depended on autocrine IFN-b stimulation and aug-mented activation of STAT1. These data are consistent witha recent report showing increased LPS-induced IFN-b and IFN-dependent signaling in LPS-stimulated peritoneal exudative cellsisolated from Nfkb12/2 mice (9).
LPS induces increased serum levels of IFN-b in Nfkb12/2 mice
To determine whether p50/p105 suppresses the induction of IFN-bfollowing inflammatory challenge in vivo, WT and Nfkb12/2 miceon the C57BL/6 background received 200 mg LPS i.p. Serum washarvested 4 h later, and levels of IFN-b were determined byELISA. LPS induced detectable levels of IFN-b in the serum ofNfkb12/2 mice but not in WT mice (Fig. 3). This demonstratesthat p50/p105 suppresses the systemic induction of IFN-b fol-lowing LPS challenge.
Inhibition of TLR-induced IFN signaling by p50/p105 dependson TPL-2
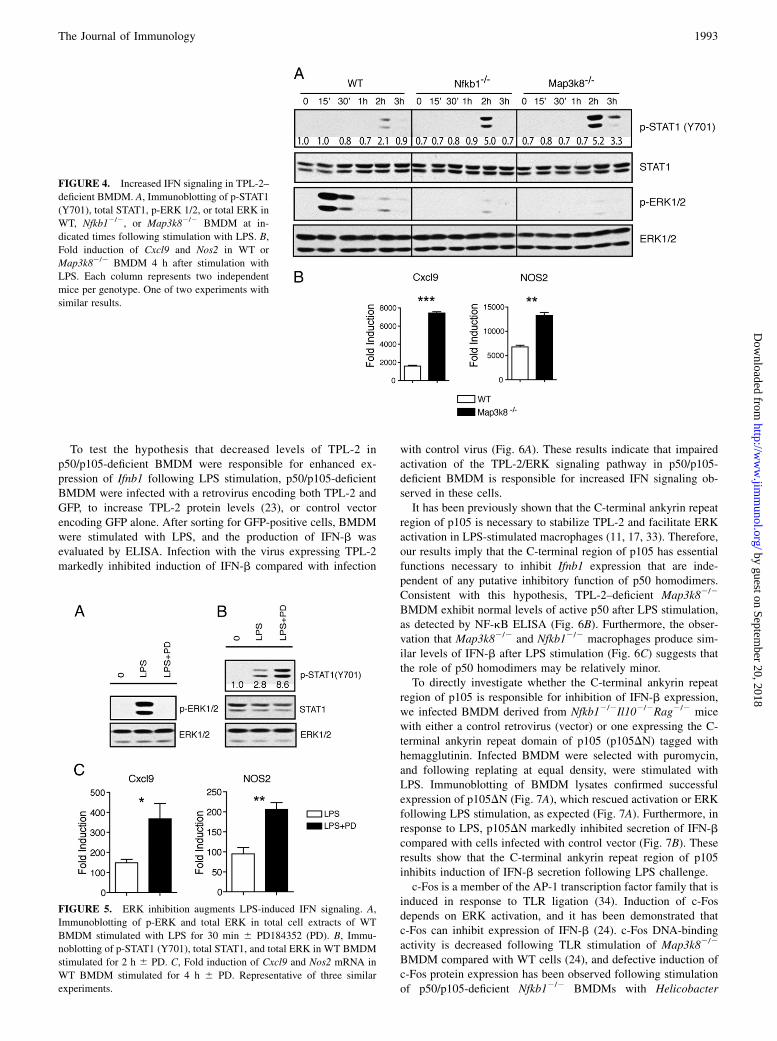
LPS-induced ERK activation is blocked in Nfkb12/2 BMDM dueto TPL-2 deficiency (11, 17, 32), and we have recently shown thatthe TPL-2/ERK signaling pathway negatively regulates LPS in-duction of IFN-b in macrophages (24). This raised the possibilitythat defective TPL-2/ERK signaling was responsible for increasedIFN-b signaling observed in p50/p105-deficient BMDM. Indeed,in addition to the expected increase in IFN-b expression (Sup-plemental Fig. 1A), we detected increased STAT1 phosphorylation(Fig. 4A) and increased IFN-dependent gene expression (Fig. 4B)in TPL-2–deficient (Map3k82/2) BMDM compared with WTBMDM following LPS stimulation. Therefore, the phenotype ofTPL-2–deficientMap3k82/2 macrophages was very similar to thatof p50/p105-deficient macrophages.To investigate whether the effect of TPL-2 deficiency on type I
IFN signaling was due to impaired activation of ERK, WT BMDMwere pretreated with the MEK inhibitor PD184352 prior to sti-mulation with LPS. Blockade of ERK activation with PD184352(Fig. 5A) increased STAT1 phosphorylation (Fig. 5B) and IFN-responsive gene expression (Fig. 5C), as well as increasing ex-pression of IFN-b, as previously described (24) (SupplementalFig. 1B). Therefore, the effects of TPL-2 deficiency on IFN sig-naling were phenocopied by pharmacological blockade of ERKactivation, consistent with the critical role of TPL-2 in regulatingERK activation in TLR-stimulated macrophages (Fig. 4A).
FIGURE 2. p50/p105 deficiency increases phosphorylation of STAT1
following TLR stimulation. A, Immunoblot of p-STAT1 (Y701) and total
STAT1 in cell extracts from Il102/2Rag2/2 and Nfkb12/2Il102/2Rag2/2
BMDM following stimulation with LPS at indicated time points. ERK is
included as a loading control. Numbers under the p-STAT1 band indicate the
intensity of the p-STAT1 band relative to the intensity in the unstimulated
Il102/2Rag2/2 sample, normalized to total STAT1. B, Immunoblot of p-
STAT1 (Y701) and total STAT1 in nuclear extracts from Nfkb12/2Il102/2
Rag2/2 BMDM4 h after stimulation with LPS in the presence or absence of
IFN-b–specific depletingAb. TFIID is included as a nuclear loading control.
C, Fold induction of indicated IFN-responsive genes in Nfkb12/2Il102/2
Rag2/2 BMDM 4 h after stimulation with LPS in the presence or absence
of an IFN-b–blocking Ab. Columns represent mean values of three in-
dependent macrophage pools for each genotype. One of two experiments
with similar results.
FIGURE 3. Increased levels of IFN-b in the serum of Nfkb12/2 mice
after LPS challenge. WT and Nfkb12/2 mice received 200 mg LPS, and
serum was harvested 4 h later. IFN-b levels were measured in the serum
by ELISA. Each column represents five mice per genotype. One of two
experiments with similar results.
1992 NF-kB1 INHIBITS TLR-INDUCED IFN SIGNALING
by guest on September 20, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
To test the hypothesis that decreased levels of TPL-2 inp50/p105-deficient BMDM were responsible for enhanced ex-pression of Ifnb1 following LPS stimulation, p50/p105-deficientBMDM were infected with a retrovirus encoding both TPL-2 andGFP, to increase TPL-2 protein levels (23), or control vectorencoding GFP alone. After sorting for GFP-positive cells, BMDMwere stimulated with LPS, and the production of IFN-b wasevaluated by ELISA. Infection with the virus expressing TPL-2markedly inhibited induction of IFN-b compared with infection
with control virus (Fig. 6A). These results indicate that impairedactivation of the TPL-2/ERK signaling pathway in p50/p105-deficient BMDM is responsible for increased IFN signaling ob-served in these cells.It has been previously shown that the C-terminal ankyrin repeat
region of p105 is necessary to stabilize TPL-2 and facilitate ERKactivation in LPS-stimulated macrophages (11, 17, 33). Therefore,our results imply that the C-terminal region of p105 has essentialfunctions necessary to inhibit Ifnb1 expression that are inde-pendent of any putative inhibitory function of p50 homodimers.Consistent with this hypothesis, TPL-2–deficient Map3k82/2
BMDM exhibit normal levels of active p50 after LPS stimulation,as detected by NF-kB ELISA (Fig. 6B). Furthermore, the obser-vation that Map3k82/2 and Nfkb12/2 macrophages produce sim-ilar levels of IFN-b after LPS stimulation (Fig. 6C) suggests thatthe role of p50 homodimers may be relatively minor.To directly investigate whether the C-terminal ankyrin repeat
region of p105 is responsible for inhibition of IFN-b expression,we infected BMDM derived from Nfkb12/2Il102/2Rag2/2 micewith either a control retrovirus (vector) or one expressing the C-terminal ankyrin repeat domain of p105 (p105DN) tagged withhemagglutinin. Infected BMDM were selected with puromycin,and following replating at equal density, were stimulated withLPS. Immunoblotting of BMDM lysates confirmed successfulexpression of p105DN (Fig. 7A), which rescued activation or ERKfollowing LPS stimulation, as expected (Fig. 7A). Furthermore, inresponse to LPS, p105DN markedly inhibited secretion of IFN-bcompared with cells infected with control vector (Fig. 7B). Theseresults show that the C-terminal ankyrin repeat region of p105inhibits induction of IFN-b secretion following LPS challenge.c-Fos is a member of the AP-1 transcription factor family that is
induced in response to TLR ligation (34). Induction of c-Fosdepends on ERK activation, and it has been demonstrated thatc-Fos can inhibit expression of IFN-b (24). c-Fos DNA-bindingactivity is decreased following TLR stimulation of Map3k82/2
BMDM compared with WT cells (24), and defective induction ofc-Fos protein expression has been observed following stimulationof p50/p105-deficient Nfkb12/2 BMDMs with Helicobacter
FIGURE 4. Increased IFN signaling in TPL-2–
deficient BMDM. A, Immunoblotting of p-STAT1
(Y701), total STAT1, p-ERK 1/2, or total ERK in
WT, Nfkb12/2, or Map3k82/2 BMDM at in-
dicated times following stimulation with LPS. B,
Fold induction of Cxcl9 and Nos2 in WT or
Map3k82/2 BMDM 4 h after stimulation with
LPS. Each column represents two independent
mice per genotype. One of two experiments with
similar results.
FIGURE 5. ERK inhibition augments LPS-induced IFN signaling. A,
Immunoblotting of p-ERK and total ERK in total cell extracts of WT
BMDM stimulated with LPS for 30 min 6 PD184352 (PD). B, Immu-
noblotting of p-STAT1 (Y701), total STAT1, and total ERK in WT BMDM
stimulated for 2 h 6 PD. C, Fold induction of Cxcl9 and Nos2 mRNA in
WT BMDM stimulated for 4 h 6 PD. Representative of three similar
experiments.
The Journal of Immunology 1993
by guest on September 20, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
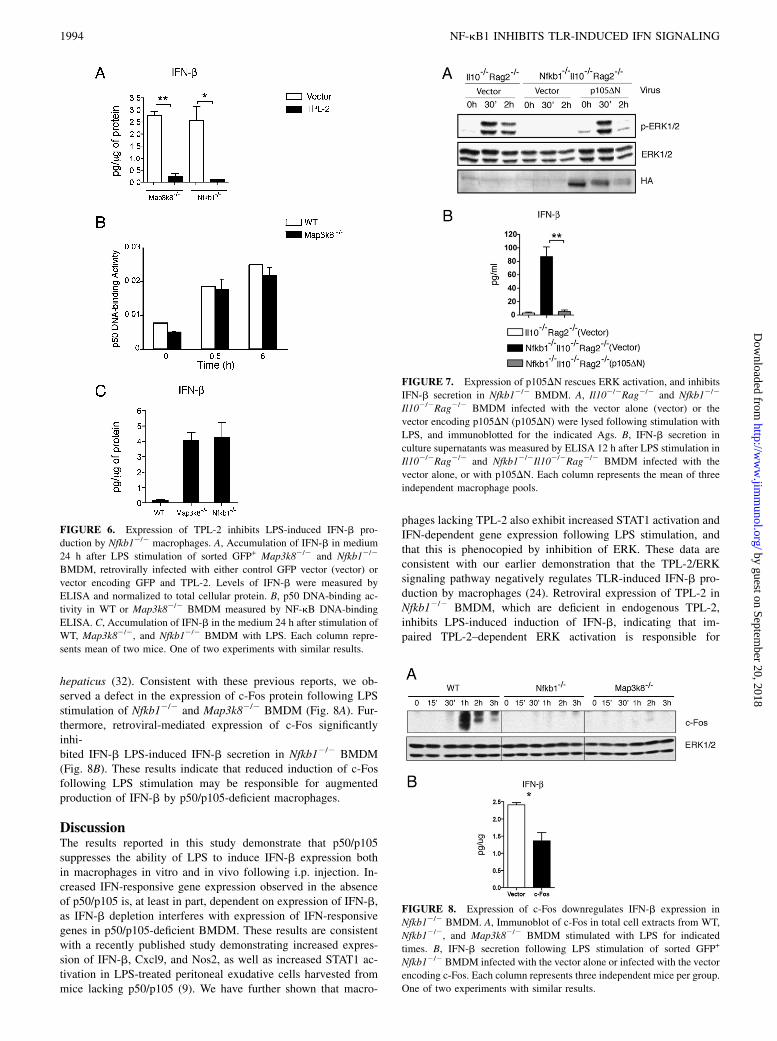
hepaticus (32). Consistent with these previous reports, we ob-served a defect in the expression of c-Fos protein following LPSstimulation of Nfkb12/2 and Map3k82/2 BMDM (Fig. 8A). Fur-thermore, retroviral-mediated expression of c-Fos significantlyinhi-bited IFN-b LPS-induced IFN-b secretion in Nfkb12/2 BMDM(Fig. 8B). These results indicate that reduced induction of c-Fosfollowing LPS stimulation may be responsible for augmentedproduction of IFN-b by p50/p105-deficient macrophages.
DiscussionThe results reported in this study demonstrate that p50/p105suppresses the ability of LPS to induce IFN-b expression bothin macrophages in vitro and in vivo following i.p. injection. In-creased IFN-responsive gene expression observed in the absenceof p50/p105 is, at least in part, dependent on expression of IFN-b,as IFN-b depletion interferes with expression of IFN-responsivegenes in p50/p105-deficient BMDM. These results are consistentwith a recently published study demonstrating increased expres-sion of IFN-b, Cxcl9, and Nos2, as well as increased STAT1 ac-tivation in LPS-treated peritoneal exudative cells harvested frommice lacking p50/p105 (9). We have further shown that macro-
phages lacking TPL-2 also exhibit increased STAT1 activation andIFN-dependent gene expression following LPS stimulation, andthat this is phenocopied by inhibition of ERK. These data areconsistent with our earlier demonstration that the TPL-2/ERKsignaling pathway negatively regulates TLR-induced IFN-b pro-duction by macrophages (24). Retroviral expression of TPL-2 inNfkb12/2 BMDM, which are deficient in endogenous TPL-2,inhibits LPS-induced induction of IFN-b, indicating that im-paired TPL-2–dependent ERK activation is responsible for
FIGURE 6. Expression of TPL-2 inhibits LPS-induced IFN-b pro-
duction by Nfkb12/2 macrophages. A, Accumulation of IFN-b in medium
24 h after LPS stimulation of sorted GFP+ Map3k82/2 and Nfkb12/2
BMDM, retrovirally infected with either control GFP vector (vector) or
vector encoding GFP and TPL-2. Levels of IFN-b were measured by
ELISA and normalized to total cellular protein. B, p50 DNA-binding ac-
tivity in WT or Map3k82/2 BMDM measured by NF-kB DNA-binding
ELISA. C, Accumulation of IFN-b in the medium 24 h after stimulation of
WT, Map3k82/2, and Nfkb12/2 BMDM with LPS. Each column repre-
sents mean of two mice. One of two experiments with similar results.
FIGURE 7. Expression of p105DN rescues ERK activation, and inhibits
IFN-b secretion in Nfkb12/2 BMDM. A, Il102/2Rag2/2 and Nfkb12/2
Il102/2Rag2/2 BMDM infected with the vector alone (vector) or the
vector encoding p105DN (p105DN) were lysed following stimulation with
LPS, and immunoblotted for the indicated Ags. B, IFN-b secretion in
culture supernatants was measured by ELISA 12 h after LPS stimulation in
Il102/2Rag2/2 and Nfkb12/2Il102/2Rag2/2 BMDM infected with the
vector alone, or with p105DN. Each column represents the mean of three
independent macrophage pools.
FIGURE 8. Expression of c-Fos downregulates IFN-b expression in
Nfkb12/2 BMDM. A, Immunoblot of c-Fos in total cell extracts from WT,
Nfkb12/2, and Map3k82/2 BMDM stimulated with LPS for indicated
times. B, IFN-b secretion following LPS stimulation of sorted GFP+
Nfkb12/2 BMDM infected with the vector alone or infected with the vector
encoding c-Fos. Each column represents three independent mice per group.
One of two experiments with similar results.
1994 NF-kB1 INHIBITS TLR-INDUCED IFN SIGNALING
by guest on September 20, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

increased IFN expression and IFN signaling observed in BMDMlacking p50/p105. Expression of the C-terminal ankyrin repeatdomain of p105 in Nfkb12/2 macrophages, which reconstitutesLPS-induced ERK activation, also suppresses IFN-b secretion,consistent with the role of the C-terminal domain in stabilizing thesteady-state expression of TPL-2. Finally, expression of c-Fosin Nfkb12/2 BMDM inhibits IFN-b secretion, suggesting thatthe reduction in ERK-dependent c-Fos expression observed inNfkb12/2 BMDM may be an important factor leading to elevatedproduction of IFN-b.IL-10 expression is reduced in macrophages lacking p50/p105
compared with WT cells after TLR stimulation. Because IL-10is a potent inhibitor of many TLR-inducible genes, includingIFN-b and CXCL9 (35), it is possible that increased expressionof IFN-responsive genes observed following LPS stimulation ofNfkb12/2 BMDM could in part result from decreased productionof IL-10. However, as our initial experiments were performedusing macrophages that also lack the ability to secrete IL-10,differences in gene expression observed in this study could notbe solely due to altered endogenous production of IL-10. p50/p105 therefore has inhibitory functions that are independent ofIL-10. Consistent with this conclusion, we previously demon-strated that TPL-2/ERK signaling negatively regulates IFN-bproduction in the absence of IL-10 (24).It has been reported that macrophages lacking IKK-b exhibited
augmented activation of STAT1 following stimulation with heat-killed group B Streptococcus (36). Because IKK-b regulates ac-tivation of the TPL-2/ERK signaling pathway via phosphorylationof p105 (11, 13), it might be expected that increased STAT1 ac-tivation in IKK-b–deficient macrophages resulted from aug-mented IFN-b production, as in Nfkb12/2 macrophages. Surpri-singly, however, IFN-b production was reported to be reducedin the absence of IKK-b (36). These data suggest that IKK-bmost likely has roles in both the induction, and p105-dependentinhibition, of IFN-b expression following LPS stimulation. Theobservation that LPS-induced IFN-b expression is lower in mac-rophages lacking IKK-b suggests that any decrease in activationof the p105-dependent inhibitory pathway does not overcome thereduction in the primary IKK/NF-kB–dependent induction ofIFN-b transcription. The mechanism for the negative regulation ofSTAT1 activation by IKK-b, which is apparently independentof effects on IFN-b production, to our knowledge, remains un-known.Macrophages can exhibit divergent functional programs de-
pending on the nature of the activating stimuli (37). Classic orM1 activation is induced by microbial products and/or IFN-g, which induce a proinflammatory response (increased IL-12,NOS2, MHC class II, and IFN-b expression) required to kill in-tracellular pathogens. In contrast, M2 alternative activation byIL-4 and IL-13 following parasite infection is characterized bydecreased expression of NOS2 and MHC class II, and increasedexpression of anti-inflammatory cytokines such as IL-10. Basedon analyses of Nfkb12/2 macrophages, Porta et al. (9) have pro-posed that p50 inhibits M1 polarization of LPS-stimulated mac-rophages. As overexpression of p50 in RAW 267.4 macrophagessuppressed LPS-induced IFN-b promoter activity, it was sug-gested that p50 homodimers might directly inhibit IFN-b tran-scription (9). Our study indicates that the C-terminal ankyrinrepeat domain of p105, which is known to interact with and sta-bilize TPL-2 (17), inhibits IFN-b expression by facilitating LPS-induced ERK activation. These results are consistent with thehypothesis that defective TPL-2/ERK signaling is the predominantfactor leading to elevated expression of IFN-b in p50/p105-deficient macrophages, rather than the absence of p50. Our ex-
periments further suggest that LPS-induced expression of c-Fos,which depends on the p105/TPL-2/ERK pathway, inhibits IFN-bexpression, providing a mechanism for the increase in IFN-bexpression observed in Nfkb12/2 macrophages.In conclusion, this study shows that a major function for NF-kB1
p50/p105 in regulating gene expression in TLR-stimulated mac-rophages is to facilitate activation of the TPL-2/ERK signalingpathway by maintaining steady-state TPL-2 protein levels. Thesedata raise the possibility that in vivo phenotypes of Nfkb12/2 micethat have been attributed to altered NF-kB function might actuallybe due, at least in part, to impaired activation of ERK MAPK ininnate immune responses. This is supported by the observationthat mice lacking only p105 are susceptible to colitis (38), similarto the colitis-susceptible phenotype previously observed in micelacking both p50 and p105 (18, 20, 27). Further studies will benecessary to define the relative importance of impaired activationof the TPL-2/ERK pathway versus altered NF-kB activation forthe phenotype of Nfkb12/2 mice in models of infectious diseaseand autoimmunity.
DisclosuresThe authors have no financial conflicts of interest.
References1. Grilli, M., J. J. Chiu, and M. J. Lenardo. 1993. NF-kappa B and Rel: participants
in a multiform transcriptional regulatory system. Int. Rev. Cytol. 143: 1–62.2. Ghosh, S., M. J. May, and E. B. Kopp. 1998. NF-kappa B and Rel proteins:
evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol.16: 225–260.
3. Rothwarf, D. M., and M. Karin. 1999. The NF-kappa B activation pathway:a paradigm in information transfer from membrane to nucleus. Sci. STKE 1999:RE1.
4. Li, Q., and I. M. Verma. 2002. NF-kappaB regulation in the immune system. Nat.Rev. Immunol. 2: 725–734.
5. Kang, S. M., A. C. Tran, M. Grilli, and M. J. Lenardo. 1992. NF-kappa B subunitregulation in nontransformed CD4+ T lymphocytes. Science 256: 1452–1456.
6. Zhong, H., M. J. May, E. Jimi, and S. Ghosh. 2002. The phosphorylation statusof nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1.Mol. Cell 9: 625–636.
7. Bohuslav, J., V. V. Kravchenko, G. C. Parry, J. H. Erlich, S. Gerondakis,N. Mackman, and R. J. Ulevitch. 1998. Regulation of an essential innate immuneresponse by the p50 subunit of NF-kappaB. J. Clin. Invest. 102: 1645–1652.
8. Kastenbauer, S., and H. W. Ziegler-Heitbrock. 1999. NF-kappaB1 (p50) is up-regulated in lipopolysaccharide tolerance and can block tumor necrosis factorgene expression. Infect. Immun. 67: 1553–1559.
9. Porta, C., M. Rimoldi, G. Raes, L. Brys, P. Ghezzi, D. Di Liberto, F. Dieli,S. Ghisletti, G. Natoli, P. De Baetselier, et al. 2009. Tolerance and M2 (alter-native) macrophage polarization are related processes orchestrated by p50 nu-clear factor kappaB. Proc. Natl. Acad. Sci. USA 106: 14978–14983.
10. Saccani, A., T. Schioppa, C. Porta, S. K. Biswas, M. Nebuloni, L. Vago,B. Bottazzi, M. P. Colombo, A. Mantovani, and A. Sica. 2006. p50 nuclearfactor-kappaB overexpression in tumor-associated macrophages inhibits M1inflammatory responses and antitumor resistance. Cancer Res. 66: 11432–11440.
11. Beinke, S., M. J. Robinson, M. Hugunin, and S. C. Ley. 2004. Lipopolysac-charide activation of the TPL-2/MEK/extracellular signal-regulated kinasemitogen-activated protein kinase cascade is regulated by IkappaB kinase-induced proteolysis of NF-kappaB1 p105. Mol. Cell. Biol. 24: 9658–9667.
12. Lang, V., J. Janzen, G. Z. Fischer, Y. Soneji, S. Beinke, A. Salmeron, H. Allen,R. T. Hay, Y. Ben-Neriah, and S. C. Ley. 2003. betaTrCP-mediated proteolysis ofNF-kappaB1 p105 requires phosphorylation of p105 serines 927 and 932. Mol.Cell. Biol. 23: 402–413.
13. Waterfield, M., W. Jin, W. Reiley, M. Zhang, and S. C. Sun. 2004. IkappaBkinase is an essential component of the Tpl2 signaling pathway. Mol. Cell. Biol.24: 6040–6048.
14. Sriskantharajah, S., M. P. Belich, S. Papoutsopoulou, J. Janzen, V. Tybulewicz,B. Seddon, and S. C. Ley. 2009. Proteolysis of NF-kappaB1 p105 is essential forT cell antigen receptor-induced proliferation. Nat. Immunol. 10: 38–47.
15. Belich, M. P., A. Salmeron, L. H. Johnston, and S. C. Ley. 1999. TPL-2 kinaseregulates the proteolysis of the NF-kappaB-inhibitory protein NF-kappaB1 p105.Nature 397: 363–368.
16. Dumitru, C. D., J. D. Ceci, C. Tsatsanis, D. Kontoyiannis, K. Stamatakis,J. H. Lin, C. Patriotis, N. A. Jenkins, N. G. Copeland, G. Kollias, andP. N. Tsichlis. 2000. TNF-alpha induction by LPS is regulated posttranscrip-tionally via a Tpl2/ERK-dependent pathway. Cell 103: 1071–1083.
17. Waterfield, M. R., M. Zhang, L. P. Norman, and S. C. Sun. 2003. NF-kappaB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by gov-erning the stability and function of the Tpl2 kinase. Mol. Cell 11: 685–694.
The Journal of Immunology 1995
by guest on September 20, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

18. Erdman, S., J. G. Fox, C. A. Dangler, D. Feldman, and B. H. Horwitz. 2001.Typhlocolitis in NF-kappa B-deficient mice. J. Immunol. 166: 1443–1447.
19. Sha, W. C., H. C. Liou, E. I. Tuomanen, and D. Baltimore. 1995. Targeteddisruption of the p50 subunit of NF-kappa B leads to multifocal defects in im-mune responses. Cell 80: 321–330.
20. Tomczak, M. F., S. E. Erdman, A. Davidson, Y. Y. Wang, P. R. Nambiar,A. B. Rogers, B. Rickman, D. Luchetti, J. G. Fox, and B. H. Horwitz. 2006.Inhibition of Helicobacter hepaticus-induced colitis by IL-10 requires the p50/p105 subunit of NF-kappa B. J. Immunol. 177: 7332–7339.
21. Wang, Y., B. H. Rickman, T. Poutahidis, K. Schlieper, E. A. Jackson,S. E. Erdman, J. G. Fox, and B. H. Horwitz. 2008. c-Rel is essential for thedevelopment of innate and T cell-induced colitis. J. Immunol. 180: 8118–8125.
22. Wettenhall, J. M., K. M. Simpson, K. Satterley, and G. K. Smyth. 2006.affylmGUI: a graphical user interface for linear modeling of single channelmicroarray data. Bioinformatics 22: 897–899.
23. Robinson, M. J., S. Beinke, A. Kouroumalis, P. N. Tsichlis, and S. C. Ley. 2007.Phosphorylation of TPL-2 on serine 400 is essential for lipopolysaccharide ac-tivation of extracellular signal-regulated kinase in macrophages. Mol. Cell. Biol.27: 7355–7364.
24. Kaiser, F., D. Cook, S. Papoutsopoulou, R. Rajsbaum, X. Wu, H. T. Yang,S. Grant, P. Ricciardi-Castagnoli, P. N. Tsichlis, S. C. Ley, and A. O’Garra. 2009.TPL-2 negatively regulates interferon-beta production in macrophages andmyeloid dendritic cells. J. Exp. Med. 206: 1863–1871.
25. Lang, R., D. Patel, J. J. Morris, R. L. Rutschman, and P. J. Murray. 2002.Shaping gene expression in activated and resting primary macrophages by IL-10.J. Immunol. 169: 2253–2263.
26. Cao, S., X. Zhang, J. P. Edwards, and D. M. Mosser. 2006. NF-kappaB1 (p50)homodimers differentially regulate pro- and anti-inflammatory cytokines inmacrophages. J. Biol. Chem. 281: 26041–26050.
27. Tomczak, M. F., S. E. Erdman, T. Poutahidis, A. B. Rogers, H. Holcombe,B. Plank, J. G. Fox, and B. H. Horwitz. 2003. NF-kappa B is required within theinnate immune system to inhibit microflora-induced colitis and expression of IL-12 p40. J. Immunol. 171: 1484–1492.
28. Hug, H., M. Costas, P. Staeheli, M. Aebi, and C. Weissmann. 1988. Organizationof the murine Mx gene and characterization of its interferon- and virus-induciblepromoter. Mol. Cell. Biol. 8: 3065–3079.
29. Amichay, D., R. T. Gazzinelli, G. Karupiah, T. R. Moench, A. Sher, andJ. M. Farber. 1996. Genes for chemokines MuMig and Crg-2 are induced inprotozoan and viral infections in response to IFN-gamma with patterns of tissueexpression that suggest nonredundant roles in vivo. J. Immunol. 157: 4511–4520.
30. Vadiveloo, P. K., G. Vairo, P. Hertzog, I. Kola, and J. A. Hamilton. 2000. Role oftype I interferons during macrophage activation by lipopolysaccharide. Cytokine12: 1639–1646.
31. Toshchakov, V., B. W. Jones, P. Y. Perera, K. Thomas, M. J. Cody, S. Zhang,B. R. Williams, J. Major, T. A. Hamilton, M. J. Fenton, and S. N. Vogel. 2002.TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependentgene expression in macrophages. Nat. Immunol. 3: 392–398.
32. Tomczak, M. F., M. Gadjeva, Y. Y. Wang, K. Brown, I. Maroulakou,P. N. Tsichlis, S. E. Erdman, J. G. Fox, and B. H. Horwitz. 2006. Defectiveactivation of ERK in macrophages lacking the p50/p105 subunit of NF-kappaBis responsible for elevated expression of IL-12 p40 observed after challenge withHelicobacter hepaticus. J. Immunol. 176: 1244–1251.
33. Beinke, S., J. Deka, V. Lang, M. P. Belich, P. A. Walker, S. Howell,S. J. Smerdon, S. J. Gamblin, and S. C. Ley. 2003. NF-kappaB1 p105 negativelyregulates TPL-2 MEK kinase activity. Mol. Cell. Biol. 23: 4739–4752.
34. Introna, M., T. A. Hamilton, R. E. Kaufman, D. O. Adams, and R. C. Bast, Jr.1986. Treatment of murine peritoneal macrophages with bacterial lipopolysac-charide alters expression of c-fos and c-myc oncogenes. J. Immunol. 137: 2711–2715.
35. Moore, K. W., R. de Waal Malefyt, R. L. Coffman, and A. O’Garra. 2001.Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19: 683–765.
36. Fong, C. H., M. Bebien, A. Didierlaurent, R. Nebauer, T. Hussell, D. Broide,M. Karin, and T. Lawrence. 2008. An antiinflammatory role for IKKbeta throughthe inhibition of “classical” macrophage activation. J. Exp. Med. 205: 1269–1276.
37. Mosser, D. M., and J. P. Edwards. 2008. Exploring the full spectrum of mac-rophage activation. Nat. Rev. Immunol. 8: 958–969.
38. Chang, M., A. J. Lee, L. Fitzpatrick, M. Zhang, and S. C. Sun. 2009. NF-kappaB1 p105 regulates T cell homeostasis and prevents chronic inflammation. J.Immunol. 182: 3131–3138.
1996 NF-kB1 INHIBITS TLR-INDUCED IFN SIGNALING
by guest on September 20, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from


















