
Modern Heterocyclic Chemistry (BARLUENGA:HETEROCYC. 4VOL O-BK) || The Chemistry of 2-Azetidinones...
57
24 The Chemistry of 2-Azetidinones (b-Lactams) Benito Alcaide, Pedro Almendros, and Amparo Luna 24.1 Monocyclic Derivatives 24.1.1 Introduction The large number of recent reports on b-lactam chemistry demonstrates the increasing interest in this important class of compounds. Monocyclic b-lactams frequently serve as precursors for the synthesis of classical bicyclic b-lactam anti- biotics. The cyclic 2-azetidinone skeleton has been extensively used as a template on which to build the heterocyclic structure fused to the four-membered ring, using the chirality and functionalization of the b-lactam nucleus as a stereocontrolling element. The discovery of nonclassical b-lactam antibiotics, such as monobactams and nocardicins, coupled with ever-growing new applications such as enzyme inhibition has triggered a renewed interest in the building of new monocyclic b-lactam derivatives. Besides the utility of b-lactams as biologically active agents, they are used as intermediates in a- and b-amino acid synthesis, as well as building blocks for alkaloids, heterocycles, taxoids and other types of compounds of biological and medicinal interest. 24.1.2 Physicochemical Data 24.1.2.1 Computational Chemistry Theoretical studies show that b-lactams are weaker bases, in the gas phase, than acyclic amides [1]. The attenuation of basicity upon cyclization is stronger than that found for cyclic ketones of similar size due to the existence of a negative hypercon- jugation effect that is present in b-lactams but not in cyclic ketones. Ab initio calculations indicate that both b-lactams and acyclic amides are oxygen bases, but the gap between the oxygen and nitrogen intrinsic basicities is much smaller in the former due to the aforementioned cyclization effects. This decrease of the oxygen Modern Heterocyclic Chemistry, First Edition. Edited by Julio Alvarez-Builla, Juan Jose Vaquero, and José Barluenga. Ó 2011 Wiley-VCH Verlag GmbH & Co. KGaA. Published 2011 by Wiley-VCH Verlag GmbH & Co. KGaA. j2117
Transcript of Modern Heterocyclic Chemistry (BARLUENGA:HETEROCYC. 4VOL O-BK) || The Chemistry of 2-Azetidinones...

24The Chemistry of 2-Azetidinones (b-Lactams)Benito Alcaide, Pedro Almendros, and Amparo Luna
24.1Monocyclic Derivatives
24.1.1Introduction
The large number of recent reports on b-lactam chemistry demonstrates theincreasing interest in this important class of compounds. Monocyclic b-lactamsfrequently serve as precursors for the synthesis of classical bicyclic b-lactam anti-biotics. The cyclic 2-azetidinone skeleton has been extensively used as a template onwhich to build the heterocyclic structure fused to the four-membered ring, using thechirality and functionalization of theb-lactamnucleus as a stereocontrolling element.The discovery of nonclassical b-lactam antibiotics, such as monobactams andnocardicins, coupled with ever-growing new applications such as enzyme inhibitionhas triggered a renewed interest in the building of new monocyclic b-lactamderivatives. Besides the utility of b-lactams as biologically active agents, they areused as intermediates in a- and b-amino acid synthesis, as well as building blocksfor alkaloids, heterocycles, taxoids and other types of compounds of biological andmedicinal interest.
24.1.2Physicochemical Data
24.1.2.1 Computational ChemistryTheoretical studies show that b-lactams are weaker bases, in the gas phase, thanacyclic amides [1]. The attenuation of basicity upon cyclization is stronger than thatfound for cyclic ketones of similar size due to the existence of a negative hypercon-jugation effect that is present in b-lactams but not in cyclic ketones. Ab initiocalculations indicate that both b-lactams and acyclic amides are oxygen bases, butthe gap between the oxygen and nitrogen intrinsic basicities is much smaller in theformer due to the aforementioned cyclization effects. This decrease of the oxygen
Modern Heterocyclic Chemistry, First Edition.Edited by Julio Alvarez-Builla, Juan Jose Vaquero, and José Barluenga.� 2011 Wiley-VCH Verlag GmbH & Co. KGaA. Published 2011 by Wiley-VCH Verlag GmbH & Co. KGaA.
j2117

intrinsic basicity of b-lactams with respect to the aliphatic amides of the same size isa direct consequence of the hybridization changes undergone by the carbonyl carbonand is very well described by a topological analysis of the corresponding electroniccharge densities. The topological analysis of bond activations upon protonationreveals that for 2-azetidinones these effects are not dramatic when protonation takesplace at the oxygen atom, whereas they are quite significant if protonation takes placeat the ring nitrogen.
Model chiral b-lactam molecules, (3S)- and (4R)-fluoro-2-azetidinone, have beencalculated at the B3PW91/aug-cc-pVTZ level to be hydrogen bonded with achiral HXmolecules (X¼F,Cl, Br) [2]. Two stable structures of the complex are possible: a cyclicor a bentH-bond, inwhich theHXmolecule interacts with theCOgroup and is eitherclose to NH or CH2 (CHF) moiety, respectively. The VCD effect of these two formsdiffers in several respects; however, the main difference is the sign of the VCDrotational strength of the n(HX) stretching vibrations, revealing the geometry of thehydrogen bond complex. A related report on halogenoazetidinones has considered inthe influence of the halogen atom, at the C4 position of the 2-azetidinone ring, on thegeometry, IR, Raman and vibrational circular dichroism spectra [3]. The vibrationalspectra were calculated for the chiral (4R)-X-2-azetidinone (X¼F,Cl or Br)moleculesat the B3PW91/aug-cc-pVTZ level. It was shown that the geometry of the moleculesstudied do not change much upon changing the halogen atom. In case of thevibrational spectra, the position and, evenmore so, the intensities depend strongly onthe kind of halogen substituent.
Ab initioMP2/6-31G(d,p) and 6-31 þ þG(d,p) calculations have been performedto investigate the intramolecular hydrogen-bonding in two model monocyclicb-lactams: monobactams and nocardicins [4]. It was found that the intramolecularC¼O � � �H�O¼S� hydrogen bond stabilizes a monobactam, while a nocardicin isdestabilized by C¼O � � �H�O�C¼O� hydrogen bond formation. This observationsuggests that monobactams could block themselves by the intramolecular bond and,therefore, could be less active towards a receptor active site than nocardicins.
The effect of an ancillary water molecule on the neutral and alkaline hydrolysismechanisms of a simple b-lactammolecule (N-methylazetidinone) has been studiedat the Hartree–Fock and MP2 levels using the 6-31G� and 6-31 þ G� basis sets [5].Solvent effects have been also considered by means of a polarizable continuummodel. In neutral hydrolysis, the additional water molecule diminishes the free-energy barriers only when correlation energy is taken into account, Concerted andstepwise mechanisms have been described. The corresponding barriers are close,and the actual mechanism could be conditioned by the molecular environment,solution, protein, and so on. Using the results of amolecular dynamics simulation ofN-methylazetidinone in aqueous solution, it has been shown that the stepwiseprocess is more likely to occur in such conditions. In alkaline hydrolysis, the firstreaction step consists of the formation of a tetrahedral intermediate that requiresa desolvation of the hydroxyl anion, which is difficult to reproduce by calculation.Afterward, the hydrolysis reaction proceeds through either concerted or stepwisemechanisms for ring opening and proton transfer. The concerted channel presentsa very low energy barrier, and the species involved are dependent on the calculation
2118j 24 The Chemistry of 2-Azetidinones (b-Lactams)

level. The stepwise mechanism is virtually the same as that previously reported forthe unassisted hydrolysis, the relative energy of all the points along the path beingdiminished and the energy barriers remaining essentially unaltered.
Kinetic experiments have been performed to characterize the reactivity ofaztreonam against amine nucleophiles relative to that of penicillin compounds(6-APA) [6]. The magnitude of the experimentally determined kinetic constants (k1,k2 and k3) shows that aztreonam is slightly more reactive than 6-APA, despitecommon assumptions that the amide bond should be less activated in mono-bactams. Interestingly, these kinetic results are consistent with the experimentallydetermined rate for aztreonam covalent linkage to the e-amino groups of lysineresidues in HSA plasma proteins (70% of the initial aztreonam fixed to HSA after24 h of reaction), which is higher than that reported for benzylpenicillins (3% after48 h). Furthermore, the kinetic influence of substitution on the attacking nucle-ophile was also investigated. Most remarkably, for ethanolamine in reaction witheither aztreonam or 6-APA, the corresponding rate law has a kinetic termproportional to [RNH2][RNH3
þ ], in contrast with previous reports on the reactionbetween benzylpenicillin and ethanolamine. To gain a better understanding of thevarious effects controlling the rates of the reactions between b-lactams and amines,the molecular details of the reactive processes have been investigated by quantumchemical calculations. The APA and MONO model systems were considered tocompute the rate-determining DGsolution barriers corresponding to various reactionmechanisms, all involving bifunctional catalysis by water, a second amine moleculeor the N-sulfonate groups of monobactams. The theoretical results confirm theability of the water-assisted (k1) and amine-assisted (k2) mechanisms to explainexperimental data on the aminolysis of b-lactams. Thus, the computed DGsolution
barriers have moderate values ranging from about 26 to about 34 kcalmol�1. Forthe aminolysis of monobactams, the previously proposed N-SO3
�-assisted mech-anism turns out to be 5.2 kcalmol�1 less stable than the water-assisted route.Moreover, the theoretical calculations undertaken in this study satisfactorilyreproduce several experimentally observed kinetic trends: the prevalence of theamine-assisted mechanism (k2 term in the rate law) over the water-assisted route(k1) and the higher reactivity exhibited by the monobactam. Nevertheless, the mostinteresting prediction made by these calculations is that the kinetic term in theexperimental rate law proportional to [CH2OHCH2NH2]�[CH2OHCH2NH3
þ ] canbe interpreted in terms of the bifunctional catalysis performed by the hydroxygroup of the protonated amine molecule. Finally, from comparison betweenexperimental and theoretical data, it was concluded that a combination of standardDFT gas-phase calculations with SCRF solvation methodologies can yield relativeDGsolution barriers with semiquantitative accuracy and give valuable insights intothe various factors controlling the rate of chemical processes in the condensedphase.
24.1.2.2 Experimental Structural MethodsThe analysis of b-lactams by X-ray diffraction indicates that the four-membered ringis planar. Several 2-azetidinone derivatives, for example,b-lactampseudopeptides [7],
24.1 Monocyclic Derivatives j2119

4-aryl-substituted b-lactams [8], 3,3-dichloro-N-p-methoxyphenyl-4-(2-phenylstyryl)-2-azetidinone [9], 4-(2-oxoethylidene)azetidin-2-ones [10], isoxazolidinyl- andpyrrolidinil-b-lactams [11], 4-(1-hydroxy-3-oxobutyl)-b-lactams [12] and an oxiranyl-b-lactam [13], have been recently studied by X-ray crystallography.
The method most useful for the determination of the relative stereochemistry ofb-lactams is 1H NMR spectroscopy. The assignment of the cis-stereochemistry tob-lactams is based on the observed coupling constants of about 5.0Hz for methineprotons H3 and H4, whereas trans-stereochemistry is consistent with methinecoupling constants of about 2.0Hz in their 1H NMR spectra [14]. The 13C NMRspectra of 2-azetidinones show the carbonyl resonance between 160 and 167 ppm.Interestingly, the carbonyl resonances of c- and larger-membered lactams appearbetween 170 and 180 ppm. The infrared C¼O absorption frequency for the mono-cyclic 2-azetidinone ring is about 1745 cm�1.
24.1.3Biologically Relevant Monocyclic b-Lactams
The word �antibiotic� refers to a chemical agent that either kills or prevents thegrowth ofmicroorganisms and is itself derived from amicroorganism. Although theterm �antimicrobial� is better and more precise because it includes the syntheticagents that have been commonly employed for several decades to treat infections, forease of use the prevalent term antibiotic will be kept herein.
Theminimum structural features believed to be essential for antimicrobial activityin the b-lactam antibiotics have undergone considerable revision. Since in recentyears several natural monocyclic b-lactams have been shown to exhibit high anti-bacterial activity, it now appears that theminimumrequirement for biological activityis a suitably substituted monocyclic 2-azetidinone ring. The most representativeexamples of these monocyclic b-lactams exhibiting antibiotic activities are thenaturally occurring nocardicins 1 [15], and monobactams 2 [16].
NO
HN OH
H CO2H
NO SO3H
R2R
O
HNR1
O
21
The common structural feature of these monocyclic b-lactams is the absenceof substitution at the C4 carbon of the 2-azetidinone ring. The antibiotic activityof this type of b-lactams has stimulated considerable activity in this area. As aconsequence, aztreonam (3) [17] and carumonam (4) [18], both with a monobactamstructure but bearing substituents at C4, have been synthesized. The relevantfeature of these compounds is the b-lactam nucleus, but the nature as well as thesterical arrangement of the substituents also play an important role in the antibioticactivity.
2120j 24 The Chemistry of 2-Azetidinones (b-Lactams)

NO
H H
SO3
MeHN
O
N
N
SH3N
O
CO2H
NO
H H
SO3
HN
O
N
N
SH3N
O
CO2H
OCONH2
43
Recent discoveries have shown other biological properties of these compoundsapart from their antibacterial action. Some of themore notable advances concern theuse in gene activation as well as the development of mechanism-based serineprotease inhibitors of elastase, cytomegalovirus protease, thrombin, prostate specificantigen, and cell metastasis and as inhibitors of acyl-CoA cholesterol acyl transfer-ase [19]. The cholesterol-lowering agent ezetimibe (5) [20], as well as the irreversibleinhibitor of glutamine synthetase tabtoxinine-b-lactam (6) [21], are representativeexample of these trends.
5
NO
F
OHOHF
N
OH
H
HOOC
NH2
6
O
24.1.42-Azetidinone Nucleus Synthesis
The vast number of syntheses of b-lactams amply illustrates the ongoing vitality of2-azetidinone chemistry. Obviously, this chapter cannot offer a comprehensivedescription of all the aspects of the various types of b-lactam syntheses emanatingfrom research groups active in this area, and so we have concentrated our efforts onthe more relevant aspects. Major synthetic routes, for example, the cycloaddition ofketenes and imines, cannot be covered completely and readers are advised to consultreviews on this topic for more details.
24.1.4.1 Ketene-Imine Cycloaddition (Staudinger Reaction)[2 þ 2] Cycloaddition reactions between ketenes, bearing amino-, oxy-, or halo-groups, and imines are recognized as being among the most important and directroutes to b-lactams [22]. Alkyl-substituted ketenes also furnish the correspondingb-lactams upon reaction with activated imines (iminoesters). In general, ketenes aregenerated from the appropriate acid chloride and a tertiary amine. The major or soleproduct of the cycloaddition is usually the cis-b-lactam, although a few exceptionsshowing trans selectivity are known. In this way b-lactams with a widely varying
24.1 Monocyclic Derivatives j2121

substitution pattern at the C3 and C4 positions of the ring are constructed stereo-selectively. The diastereoselection of the cycloaddition process can be controlled withvariable success from chiral groups attached to either the ketene or the iminecomponent, or alternatively to both. More recently, chiral catalysts have been usedin the asymmetric Staudinger reaction.
Staudinger-like cycloaddition between proline-derived formaldehyde hydrazonesand functionalized ketenes constitutes an efficient methodology for the stereose-lective construction of 4-unsubstituted b-lactams 7 (yield: 80–96%, dr up to 99 : 1)(Scheme 24.1) [23]. Enantiopure N,N-dialkylhydrazones react with N-benzyloxycar-bonyl-N-benzyl glycine as an aminoketene precursor to afford trans-3-amino-4-alkylazetidin-2-ones 8 as single diastereomers [24a]. Oxidative N�N bond cleavageof cycloadducts 7 and 8 afforded free N-H-azetidinones in high yields [24b].
Lectka and colleagues have reacted achiral ketenes with achiral imines to achieveasymmetric induction in the synthesis of cis-b-lactams through the use of a bifunc-tional catalyst system consisting of a chiral nucleophile (benzoylquinine) and anachiral Lewis acid [25], while a catalytic, highly diastereoselective process for thesynthesis of trans-b-lactams has been reported based on a phosphonium fluorideprecatalyst that both activates the nucleophile and directs the reaction process forhigh yield and diastereoselectivity [26a]. It has been demonstrated as well that aplanar-chiral azaferrocene derivative of 4-(pyrrolidino)pyridine is an excellent catalystfor the enantioselective Staudinger reaction, providing b-lactams 9 with very goodstereoselection and yield (Scheme 24.2) [26b].
More recently, azolium salts that belong to the extraordinary class ofN-heterocycliccarbenes (NHCs) have been found to be efficient catalysts for the enantioselective
NO
BnO
N
H H
N
Cl
O+
N
7
R
R
OMe
RROMe N
O
CbzBnN
N
8
R
OBn
Et3 tolueneN,
reflux
Scheme 24.1
NO
R1
Ts9
R2
R3
R3
R2
C OH
R1
N Ts +catalyst10% N
RR
RR
R
Fe
R2N
Scheme 24.2
2122j 24 The Chemistry of 2-Azetidinones (b-Lactams)

synthesis of b-lactams through Staudinger reaction of ketenes with N-tosyl, N-ben-zyloxycarbonyl, N-tert-butoxycarbonyl, or N-(4-nitrobenzenesulfonyl) imines [27].
The rapid development of solid-supported combinatorial chemistry has increasedin a spectacular way the complexity and the diversity of reactions by using solidsupport. Among others, the Staudinger reaction has found various uses in polymer-assisted synthesis [28]. Sasrin, preloaded with an Fmoc-protected (Fmoc¼ 9-fluor-enylmethoxycarbonyl) amino acid, has beenused byGallop as the startingmaterial 10(Scheme 24.3) [29]. After deprotection, the resin yields a free primary amine, whichcan be reacted with aldehydes, in the presence of trimethyl orthoformate as adesiccant, to afford the desired polymer-bound imines 11. These, in turn, are treatedwith an acid chloride in the presence of triethylamine to produce the polymer-supported b-lactams 12, which are liberated from the resin to give in good yields 2-azetidinones 13 by treatment with CF3COOH. The polymer-bound lactams can befurther derivatized by Suzuki and Heck coupling reactions, upon selection ofproperly functionalized aldehydes, to form the imines. The Staudinger reaction ona solid phase has also been accomplished using imines obtained from commerciallyavailable fluorinated a-amino acids. Thus, the b-lactam formation on a solid phasecan be monitored by 19F NMR spectroscopy [30].
The stereochemical selectivity of this procedure, through the effect of chiralketenes and chiral imines, has also been investigated [31]. In both cases only cis-diastereomers are observed. The diastereoselectivities of the b-lactams producedwere in a range of 8 : 1 to greater than 25 : 1 when using a ketene bearing a chiraloxazolidinone moiety, and a range of 2 : 1 to greater than 25 : 1 when using chiralaldehydes to form the imines.
In the previous examples, the imine intermediate is generated from a polymer-bound amine, but it may also be generated from a polymer-bound aldehyde [32]. Theuse acetoxyketene [33] allows further modifications to give carbamate products. This
O
R1
NHFmoc
O
O
R1
N
OR2
O
R1
N
OR3 R2
OOH
R1
N
OR3 R2
O
11
1213
10–100%=d.e.58–97%;
10
NMPinPiperidine30%i)
Rii) 2 HC(OMe)CHO, 3 CH, 2Cl2
R3CH2 EtCOCl, 3 CHN, 2Cl2
CF3% 3 CHinCOOH 2Cl2
N-methylpyrrolidone=NMP
R1 Ralkyl;= 2 aryl;alkyl,=
R3 alkyl,= O-alkyl, O-aryl,
N-phthalimidoyl
Scheme 24.3
24.1 Monocyclic Derivatives j2123

chemistry has been extrapolated to the synthesis of enantiomerically enrichedb-lactams starting from a polymer-bound version of Garner�s aldehyde [34]. Apolymer-supported Mukaiyama-type reagent has been used for the preparation ofb-lactams, using the Staudinger reaction. The products were obtained by generatingthe ketene from a carboxylic acid under sonicationwith the resin followed by reactionwith the imine [35]. These approaches also exclusively produce the cis-diastereomersof the lactams. The solid-phase synthesis of trans-3-alkyl-b-lactams from non-acti-vated acid chlorides has been reported recently [36].
Lectka has devised a method in which a polymer-bound base, used as a packingmaterial for a jacketed column cooled to�78 �C, effects the dehydrohalogenation ofacyl halides to generate ketenes [37]. When a solution of the acid chloride is added tothe top of the column, a solution of the ketene percolates at the bottom and can beeither trapped by another reagent or eluted through another column packed witha different polymer-bound reagent/scavenger for further transformations. The use ofa polymer-bound cinchona alkaloid as both the nucleophilic catalyst and the baseeffecting the dehydrohalogenation has been reported [38]. This polymeric reagentwas regenerated in situ with K2CO3 or sodium hydride in a rather unusual solid-gelshuttle deprotonation between a solid and a gel. Although this b-lactam formationinvolves a single step, the presence of a regenerating base seems to induce somescrambling in its stereoselectivity. All of the polymers can be recycled by simplyelutingwashing solutions through the columns,which seems to have only amarginaleffect on the reaction results.
24.1.4.2 Metalloester Enolate-Imine CondensationThe metalloester enolate-imine condensation represents one of the most popularentries to b-lactams [39]. Various ester types and imine types can be utilized in thisone-pot reaction between imines and metal ester enolates (or their syntheticequivalents, the silylketene acetals). The reaction can be promoted by variousmetals,including aluminium, boron, indium, lithium, titanium, zinc and zirconium.
The Reformatsky addition reaction to imines has been employed as a method tosynthesize b-lactams. For example, in the presence of Zn/Cp2TiCl2 (cat.), a-bro-moacetates, or c-bromocrotonates, react with imines in one-pot to form b-lactams, atroom temperature without the need for pretreatment of the solvent and Zn [40].Reformatsky reactions between enolizable and non-enolizable aldimines and a-bro-moesters of differing steric demands, in the presence of zinc dust and a catalyticamount of iodine in dioxaneunder high intensity ultrasound (HIU) irradiation affordshort reaction times and high yields of b-lactams 14 (Scheme 24.4) [41]. In a similarway, indium can mediate the synthesis of 3-unsubstituted b-lactams [42].
NO
N
R1 H
R2
OEt
O+
R2
14
Br
R3
R3
R1R3
R3IZn, 2 (cat.)
20HIU,dioxane, oC
72–94%
Scheme 24.4
2124j 24 The Chemistry of 2-Azetidinones (b-Lactams)

The use of silyl enolates or S-thioester, instead of carboxylic ester, metal enolates inthe condensation with imines provides a mild route to 2-azetidinones. 2,20-Diben-zothiazolyl disulfide is a versatile reagent that provides a convenient and efficientroute for the synthesis of b-lactams from Schiff�s bases and alkoxy/aryloxy aceticacids. The process involves the formation of thioester of the corresponding acid.Finally, condensation of titanium enolates, derived from these esters, with iminescompletes the synthesis of 2-azetidinones [43]. Highly substituted b-lactams havebeen synthesized by addition of air-stable ethyl(trimethylsilyl)acetate derivatives toN-(2-hydroxyphenyl)aldimine sodium salts [44].
The asymmetric version of the metalloester enolate-imine condensation routehas been explored using a chiral enolate. The diastereoselectivity of the reaction ofthe lithium enolate of menthyl isobutyrate with imines has been improved by theaddition of a catalytic amount (5mol%) of a chiral tridentate aminodiether ligand togive the corresponding b-lactams in high enantioselectivities [45a]. Matchingand mismatching phenomena were observed by the reaction of L- and D-menthylisobutyrates. The asymmetric Reformatsky-type reaction of (–)-menthylbromodifluoroacetate with imines in the presence of RhCl(PPh3)3 affords (S)-difluoro-b-lactams inmoderate to good yields and high diastereoselectivities throughspontaneous removal of the auxiliary [45b]. A systematic investigation of chiral ligandmediated addition of imines to lithium ester enolates to give b-lactams has beencarried out to study the effects of the variation of the alkoxy group in the latterreagent [46]. A maximum of 93% ee was obtained.
The ester enolate-imine condensation route to b-lactams via an immobilized esterenolate has been achieved [47]. The key reaction in the synthesis is the cyclization ofthe resin-bound ester dianion and an imine. Traceless cleavage from the T1-triazenelinker system yields the desired b-lactams (Scheme 24.5).
24.1.4.3 Isocyanate-Alkene CyclocondensationThe reaction of isocyanates with alkenes to give b-lactams requires activation of theisocyanate moiety by electron-withdrawing substituents or activation of the alkenepartner by electron-donating groups. 4-Aryl-2-azetidinones have been prepared byreacting N-chlorosulfonyl isocyanate with styrene and 4-methylstyrene [48]. Thereaction between the same isocyanate with an enantiopure (E)-vinyl sulfide givesa 2.5 : 1 diastereomeric mixture of phenylthioazetidinones [49]. The facial selectivity
N
N
N
NH
Me
O
O
OMe 1550–96%=d.e.53–71%;
NO
RMeHN
O
THF,LHMDS,equiv.2.2i) – °C78Requiv.3ii) 2CH=NPh, – RTto°C78
CF5%iii) 3 CHinCOOH 2Cl2°C60(5/2),THF/DMFiv)
Scheme 24.5
24.1 Monocyclic Derivatives j2125

in the cycloaddition is explained by the conformational preference of the allylicgroups in the transition structure. The [2 þ 2] cycloaddition of chlorosulfonylisocyanate (CSI) to alkoxyallenes derived fromethylidene and benzylidene erythritolsand threitols proceeds with a moderate asymmetric induction in the case ofthe erythritols (Scheme 24.6) and with a very low induction in the case of threitols.This indicates that the erythritol derivatives may exist in solution in onepredominant conformation while the threitol derivatives behave as a conformationalensemble [50].
24.1.4.4 Chromium Carbene-Imine CyclizationThe preparation of 2-azetidinones through the photochemical reaction of chromiumcarbene complexes with imines is a convenient method [51]. A vast array of imines,including simple imines, a-iminoketones, a-diimines, iminodithiocarbonates, andferrocene imines [52], can be used. The asymmetric version of this route can beaccomplished on using enantiopure chromium carbenes, such as the (R)-phenyl-glycine derivative 17, which allowed the preparation of optically active b-lactams 18(Scheme 24.7) [53]. A theoretical-experimental approach to the mechanism of thephotocarbonylation of chromium(0) (Fischer)-carbene complexes and their reactionwith imines to give b-lactams has been published [54].
24.1.4.5 Cyclization of b-Amino Acids and DerivativesThe cyclization of b-amino acids to give b-lactams can be achieved through the use ofa numerous reagents and conditions [55]. Interesting examples include the prepa-ration of C3 unsubstituted b-lactams by using tert-butylmagnesium chloride [56], thesynthesis of 2-azetidinones bearing a C4 quaternary stereocenter by using 4-pyrro-lidinopyridine [57], the preparation of the key b-lactam precursor in the totalsynthesis of lankacidin C [58], the preparation of the chartelline framework bysimple heating [59], the LHMDS-promoted cyclization of an aspartic acid derivative to
O
O O
R2O
R1O
O O
R2O
R1
HNO
H
16
NaCSI,i) 2CO3 toluene,, – °C78
Reii) – toluene,Al, – °C7850–65%=d.e.66–87%;
Scheme 24.6
NO
N
R1 R2
R3
+
R3
O
N
HPh(OC)5Cr
18
R1R2
HN
OPh
17
hυ CH, 2Cl2 RT,
70–97%=d.e.20–91%;
Scheme 24.7
2126j 24 The Chemistry of 2-Azetidinones (b-Lactams)

provide a carbapenem PS-6 precursor [60] and the preparation of the spirocyclicb-lactam 19 by sequential base and acid treatment (Scheme 24.8) [61].
24.1.4.6 Hydroxamate CyclizationThe cyclization of a b-hydroxyhydroxamate derived from an amino acid isa straightforward approach to 2-azetidinones [62]. The stereoselective synthesis of3,4-substituted b-lactams by bromine-induced oxidative cyclization of O-acylb,c-unsaturated hydroxamic acid derivatives is a classical example [63]. The intra-molecular Mitsunobu reaction of a b-hydroxy hydroxamic acid derivative has beenused for the preparation of the b-lactam precursors in the preparation of cobactinanalogs and in the total syntheses of pateamine A and sitagliptin [64]. A relatedcontribution is the cyclization of b-hydroxy-a-thioalkylhydroxamates in the presenceof AgClO4 [65]. The hydroxamate synthesis of b-lactams carried out on solid phasehas been reported [66]. The strategy chosen was to link the amino acid derivative toa polystyrene-supported hydroxylamine, and finally carry out the cyclization underMitsunobu conditions. This approach is particularly suitable for solid-phase syn-thesis as the supported b-lactam can be easily separated from the by-products of theMitsunobu reaction. The linker employed was a polystyrene resin carrying aO-trityl-hydroxylamine linker. The cyclization occurred in THFusing freshly distilled DEADand PPh3. The resin was treated with a commercially available solution of SmI2 inTHF, and free 2-azetidinones 20 were recovered from the solution after hydrolyticworkup and passage through a short silica gel column (Scheme 24.9).
24.1.4.7 Metal-Catalyzed Insertions of Diazo CompoundsIn recent years, metal-catalyzed intramolecular C�H insertion has emerged asa general strategy for the construction of numerous cyclic and heterocyclic
H
ON
EtO2C H
PMP+
NO PMP
19
CO2Et
HNO
CO2Et
PMP
HL-Proline
RTDMSO,
NaClOi) 2 NaH, 2PO4,
t-BuOH-H 2 RT(5:1),O
(1M),NaOHequiv.1ii)
RTEtOAc,
(1N),HClequiv.1.1iii)
RTEtOAc,
80%
98%=e.e.94%;
Scheme 24.8
24.1 Monocyclic Derivatives j2127

compounds, among which b-lactams are especially noteworthy [67]. The success ofthis approach is related to the level of regio- and stereocontrol and, in some cases, tothe high enantioselectivity of the C�H insertion process. It has been recentlydemonstrated that water is an efficient solvent for the Rh2(OAc)4-catalyzed intra-molecular C�H insertion of a range of diazo substrates to yield 2-azetidinones 21without competitive water insertion (Scheme 24.10) [68]. Owing to the high solubilityand stability of the catalyst in water, the catalyst can be efficiently reused.
An operationally simple catalytic system based on [RuCl2(p-cymene)]2 has beendeveloped for the stereoselective cyclization of a-diazoacetamides by intramolecularcarbenoid C�H insertion, and b-lactams 22 have been produced in excellent yieldsand>99% cis-stereoselectivity (Scheme 24.11) [69a]. The Ru-catalyzed reactions canbe performed without the need for slow addition of diazo compounds and inertatmosphere. The stereoselectivity of the related polymer-supported ruthenium-catalyzed intramolecular carbenoid C�H insertion of a-diazoacetamides to yieldb-lactams has been shown to be similar to the analogous reactions with thehomogeneus [RuCl2(p-cymene)]2 catalyst [69b].
Aryl tosylhydrazones are converted into b-lactams in good yields and remarkablecis selectivity (up to 99%) using a ruthenium porphyrin-catalyzed stereoselectiveintramolecular carbenoid C�H insertion [70].
20
NHO
RCbzHN
O
HN
O
NHCbz
RHO PPhequiv.10DEAD,equiv.5i) 3,
h24RT,THF,
SmIii) 2 h4RT,THF,inM0.1
45–52%
Scheme 24.9
21
NON2
N
O
XX
Rhmol%1 2(OAc)4 H, 2 °C80O,
73–75%
Scheme 24.10
22
NON2 R2
N
O
R1
R2
CO2EtR1O
EtO
[RuClmol%2.5 2(p-cymene)2],
°C70toluene,
12–99%
Scheme 24.11
2128j 24 The Chemistry of 2-Azetidinones (b-Lactams)

24.1.4.8 Multicomponent ReactionsMulticomponent reactions (MCRs) have recently emerged as a highly valuablesynthetic tool in the context of modern drug discovery. The atom economical andconvergent character, the simplicity of a one-pot procedure, the possible structuralvariations, the accessible complexity of the molecules, as well as the very largenumber of accessible compounds are among the described advantages of MCRs.Thus, for example, the reactivity of transition-metal catalysts can be exploited todesign one-step methods to convert readily available building blocks directly intoa diverse array of products, including b-lactams [71]. A multicomponent reaction ofb-aminothiocarboxylic acids, aldehydes and 3-dimethylamino-2-isocyanoacylate hasbeen used for the preparation of b-lactams 23 (Scheme 24.12) [72]. During thisreaction two heterocyclic moieties, a thiazole and a 2-azetidinone ring, are formedsimultaneously and under mild conditions. The increase in molecular complexityhere is dramatic as twoC�N, twoC�S and oneC�Cbonds are formed in a new �one-pot� multicomponent reaction.
A tandem Petasis–Ugi multicomponent condensation strategy and 1,3-diisopro-pylcarbodiimide condensation reaction can be used to prepare aza-b-lactams contain-ing two to four elements of diversity [73a]. Although the yields are onlymoderate, themethods provide rapid entry into this interesting structural class of molecules. Thecreation of the b-lactam ring by Ugi reaction with b-keto-acids is unknown in organicsolvents, as exemplified by the complete failure of the reaction in MeOH, THF orCH2Cl2. However, this reaction proceeds well in water to give 2-azetidinones 24(Scheme 24.13) [73b]. A library of 32 b-lactams has been created by Ugi reaction inwater. TheHPLCpurity of the crude reactions productswas 70–99%, and the yields ofthese products were 71–89%.
N
R3
O
R1S
NR2
H2NCOSH
R3
NC
N
R2++
23
R1CHOMgSO4 RTMeOH,,
36–69%
Scheme 24.12
HO2C
NH2
R2R1
NO
24
R1 R2
O
R3NH
R4++ R3 RCHO 4NCRTM),2.5(aqLiCl
71–89%
Scheme 24.13
24.1 Monocyclic Derivatives j2129

24.1.4.9 Coupling of Terminal Alkynes and Nitrones (Kinugasa Reaction)The Kinugasa reaction is a convergent route to b-lactams through the reaction ofa copper acetylide with a nitrone [74]. Appealing features of this process include theready availability of the startingmaterials and its high functional group tolerance. TheKinugasa reaction has been used for the asymmetric synthesis of b-lactams 25 viacycloaddition between chiral oxazolidinyl propynes (or related chiral ynamides) andnitrones, in the presence of cuprous iodide (Scheme 24.14) [75].
Recently, asymmetric Kinugasa reactions have been accomplished using enantio-selective catalysis. Thus, 3-exomethylene b-lactams have been obtained via Cu(I)-mediated cycloaddition between propargyl alcohol and nitrones in the presence of L-proline [76a], while 3,4-diaryl b-lactams have been observed for the asymmetricintermolecular Kinugasa reaction using P,N-ligands [76b]. A chiral bis(oxazoline)/Cu(OTf)2 derivative, a chiral tris(oxazoline)/Cu(II) system, and a chiral i-Pr-trisoxazo-line/Cu(ClO4)2�6H2O complex under air atmosphere catalyzed the coupling ofterminal alkynes and nitrones to afford b-lactams with reasonable enantioselectiv-ities [77]. A versatile system for the copper-catalyzed asymmetric coupling of alkyneswith nitrones to form cis-b-lactams has been developed using a bis(azaferrocene)ligand [78].
24.1.4.10 Photochemical and Radical Methodsa-Oxoamides 26 undergo c-hydrogen (with respect to the benzylic carbonyl) abstrac-tion under photochemical treatment in the crystalline state, leading tob-lactams 27 inwhich a new chiral center is generated at the benzylic carbon (Scheme 24.15) [79]. Ithas been shown that the crystal lattice preorganizes the reactant molecules towardsa single diastereomer of the b-lactam and prevents large motions of the 1,4-diradicalintermediate that would result in the loss of stereochemical memory.
ON
O
R1
N
HR2
PhO++
NO
R2
Ph25
NO
O
R1
EtCuI, 3 RTDMF,N,
20–50%=d.e.62–70%;
Scheme 24.14
O
N
O
NO
OH
27
O
R*
O
R*
26
hυ
Scheme 24.15
2130j 24 The Chemistry of 2-Azetidinones (b-Lactams)

Photosensitized decomposition of oxime oxalate amides and a-oxoamides isa useful new route to carbamoyl radicals that may cyclize to afford b-lactams [80].The kinetics of the 4-exo cyclizations of these carbamoyl radicals onto C¼C andC¼NO bonds, leading to b-lactam-containing species, have been studied by EPRspectroscopy [81]. Similarly, b-lactams have been prepared via ring closures ofunsaturated carbamoyl radicals derived from 1-carbamoyl-1-methylcyclohexa-2,5-dienes [82]. The free-radical mediated stannylcarbonylation of azaenynes providesa 4-exo annulation approach leading to a-stannylmethylene b-lactams 28(Scheme 24.16) [83]. The stereochemical results of b-lactam formation with respectto newly formed C�C double bonds depend strongly on the substitution pattern atthe propargylic position. Thus, if the substituent is anything other than hydrogen, thetributyltin group tends to be disposed syn to the carbonyl group to avoid strain.
24.1.4.11 Synthesis from Carbo- or HeterocyclesThe regioselectivity and efficiency of the ring opening of aziridines has beenexploited for the synthesis of b-lactams through carbonylation of the aziridinenucleus in the presence of a catalytic amount of [Rh(CO)2Cl]2 [84a], or using afour-component reaction [84b]. The carbonyl insertion is regio- and stereospecific,occurring at the most substituted carbon–nitrogen bond in the aziridine ring, andproceeding with retention of stereochemistry of the substituents linked to theaziridinic carbon atoms. The four-component reaction for the rapid synthesis of1,3,4,4-tetrasubstituted b-lactams from methyleneaziridines consists of a sequencethat involves aziridine opening, C-alkylation, and Staudinger [2p þ 2p] cycloaddi-tion. cis-Aziridines 29 have been employed in the carbonylation reaction by treatmentwith Co2(CO)8, giving rise to trans-b-lactams 30, which were obtained as singlediastereo- and regioisomers in good yields (Scheme 24.17) [85a]. Nucleophilic ringopening of the cis startingmaterial results in inversion of configuration, thus leadingto the trans-b-lactam. The exclusive formation of the 2-azetidinones 30 is a conse-quence of the completely regioselective ring opening of the aziridine [85b].
N
R
NO
28
Bu3SnRBu3 CO,AIBN,SnH,
C6H6 90, oC
E/Z 0–32:100–68=
40–84%
Scheme 24.16
N
R3 R2
R1
NO
R3
30
R2
R1
29
Co2(CO)8 CO (500psi).,
100DME, oC
42–95%
Scheme 24.17
24.1 Monocyclic Derivatives j2131

A theoretical investigation of the related Co2(CO)4�-catalyzed carbonylative ring
expansion of N-benzoyl-2-methylaziridine to b-lactams has been performed [85c].The synthesis of 3-unsubstituted 4,4-disubstituted b-lactams by silver-induced
ring expansion of the corresponding 2,2-disubstituted N-chloro-1-hydroxycyclopro-pylamines is, according to theoretical calculations, a very efficient process thatyields a regio- and stereoselective product [86]. This process presents a two-stepmechanism proceeding through a nitrenium intermediate. The rate-determiningstep corresponds to the extrusion of AgCl. This pathway could be an interesting newsynthetic route for obtaining the useful 3-unsubstituted 4-alkoxycarbonyl-4-alkyl-2-azetidinones.
A single-pot, mild conversion of b-lactones into N-benzyloxy-b-lactams has beenaccomplished by a ring opening/Mitsunobu sequence [87]. The transformationproceeded with high stereochemical fidelity, with the Mitsunobu reactions proceed-ing as expected with inversion of configuration at the b-carbon. In contrast to whatwas expected, the reaction of 1,3-thiazolium-4-olates (thioisom€unchones) 31 witharomatic aldehydes yielded b-lactams 32 bearing a sulfur-containing side chain(Scheme 24.18) [88]. In every case, b-lactams 32 were formed as a mixture of cis andtrans isomers (with respect to the orientation of aryl substituents at C–3 and C–4).Individual diastereomers were separated either by fractional crystallization orpreparative chromatography. A plausible rationale to account for the formation ofb-lactams involves first a [3 þ 2] cycloaddition in which thioisom€unchones play therole of the dipole to produce a transient cycloadduct that undergoes a spontaneousC�N bond cleavage, followed by a rearrangement under the reaction conditions.
1,3-Dipolar cycloaddition of nitrones to bicyclopropylidene or fluoroalkenes givesthe corresponding cycloadducts [89]. Catalytic hydrogenolysis of theN�Obondof thefluorinated isoxazolidine derivatives leads to a-trifluoromethylated b-lactams, whiletreatment of the bis-spirocyclopropanated isoxazolidines 33 with trifluoroacetic acidin acetonitrile furnishes the corresponding 3-spirocyclopropanated b-lactams 34 ingood yields. Thus, this newmethod affords compounds with a 5-azaspiro[2.3]hexan-4-one skeleton in 68–94% overall yield in two simple steps (Scheme 24.19).
1-(o-Nitrobenzyl)-2-acylpyrazolidin-3-ones upon irradiation through Pyrex andthen through Vycor yield 1-(acylamino)azetidin-2-ones. Removal of the acyl residuefrom the extraannular nitrogen produces 1-aminoazetidin-2-ones. The suggestedmechanism for this tandem photochemical synthesis of b-lactams involves initial
S
N
Ph
O
NMe
BnAr1
+ –Ar2CHO
NAr1O
cis-32 (29–54%)
SPh
N
O
MeBn
HAr2+
NAr1O
trans-32 (20–34%)
SPh
N
O
MeBn
Ar2H+
31
C6H6
reflux
Scheme 24.18
2132j 24 The Chemistry of 2-Azetidinones (b-Lactams)

removal of the N1 o-nitrobenzyl substituent, followed by ring contraction viaa diazabicyclo[2.1.0]pentane intermediate [90]. Highly enantioselective photocycliza-tion in the solid state of 1-alkyl-2-pyridones has been achieved in inclusion crystalswith optically active host compounds to give cyclobutene fused b-lactams, which aftersequential treatment with ozone and sodium borohydride afford the correspondingmonocyclic b-lactams [91].
24.1.4.12 MiscellaneousThe asymmetric synthesis of 4-alkyl-4-carboxy-2-azetidinones 35 has been achievedthrough base-mediated intramolecular cyclization of the corresponding N-a-chlor-oacetyl derivatives bearing (þ )- or (�)-10-(N,N-dicyclohexylsulfamoyl)isoborneol aschiral auxiliary (ee up to 82%) [92]. More recently, it has been noted that theasymmetric induction observed during cyclization of N-alkyl-N-chloroacetyl aminoacid derivatives to b-lactams 35 may be ascribed to chirality memory, being depen-dent on the substituents on the starting material, and can be controlled by theappropriate choice of the base and solvent (Scheme 24.20) [93].
Cycloaddition of lithium ynolates toN-sulfonyl imines has been reported to afford2-azetidinones [94]. Unactivated imines such as N-4-methoxyphenyl imines are,however, much less reactive in this reaction. The benzylic lithiation of substitutedacrylamides bearing a b-electron-withdrawing group, followed by 4-exo-trig cycliza-tion, has yielded b-lactams in modest yields [95]. Iridium-catalyzed reductivecoupling of acrylates and imines provides trans b-lactams 36 with high diastereo-selection (Scheme 24.21) [96]. The optimal catalyst allows for the synthesis of transb-lactams bearing aromatic, alkenyl and alkynyl side chains. This reaction appears toproceed through a reductive Mannich addition–cyclization mechanism. Examina-tion of substituent effects reveals a linear Hammett correlation for both the N-arylgroup on the imine and the aryloxy group on the acrylate, thereby pointing to rate-determining cyclization in the reaction mechanism.
NO R1
34
R2
NO
R2
R1O
N
R2
R1–++
33
20–60 oC
C6H6
71–100%
MeCNTFA,
70 oC75–96%
Scheme 24.19
NR2O
35
N
R3
Cl
O
R2
CO2R1
CO2R1
R3
(Csbase 2CO3 NaOH),BTPP,,
CHDMF,(MeCN,solvent 2Cl2)
RT
Scheme 24.20
24.1 Monocyclic Derivatives j2133

Allyl halides of different structures, under CO pressure, undergo a [2 þ 2]cycloaddition with imines in the presence of Pd(OAc)2, PPh3, and Et3N to afford2-azetidinones [97]. The PdCl2-catalyzed cyclocarbonylation of propargylic amineswith CuCl2 and benzoquinone affords (E)-a-chloroalkylidene-b-lactams 37 in mod-erate to good yields (Scheme24.22) [98]. Formation of the corresponding (Z)-isomersor five-membered products was not observed. The stereoselectivity in this reaction isdifferent from that observed with propargylic alcohols.
The electrochemically induced synthesis of b-lactams by C3�C4 bond formationhas been accomplished [99]. 4-Alkylidene gem-difluoro b-lactams have been synthe-sized through intramolecular hydroamination reaction of difluoropropargyl amidesvia a Baldwin disfavored 4-exo-digonal cyclization using palladium acetate as thecatalyst [100a], while 4-alkylidene b-lactams have been obtained by Cu(I)-catalyzedintramolecular C�Ncoupling of amides with vinyl bromides, revealing that the 4-exoring closure is preferred over othermodes (5-exo, 6-exo, and 6-endo) [100b]. A copper-catalyzed skeletal rearrangement of O-propargyl arylaldoximes has produced thecorresponding 4-arylidene-2-azetidinones in good yields [101]. It was found thatthe thermal rearrangement of aminocyclobutenones in the presence of an appro-priate amine produced either cis- or trans-b-lactams with high selectivities [102].
24.1.5Reactivity of the 2-Azetidinone Ring
24.1.5.1 Nucleophilic Attack at CarbonThe functionalization of 4-acetoxy-b-lactams at the C4 position is a key step in thesynthesis of 1-b-methylcarbapenems. Most of these efforts have been devoted tothe stereoselective introduction of different moieties on the commercially available
NO
R
PMP
Me
NRPMP OC6F5
O
+
36
[(cod)IrCl]%mol2.5 2,
P(OPh)%mol10 3,
Et2 60MeSiH, oC
58–80%
Scheme 24.21
NO
R1 R2
R1
Cl
NHR3
R2
R3
37
PdCl%mol5 2,
CuClequiv.2 2,benzoquinone,equiv.1psi),(300CO
40THF, oC
32–80%
Scheme 24.22
2134j 24 The Chemistry of 2-Azetidinones (b-Lactams)

4-acetoxy-b-lactam 38, including a highly diastereoselective condensation betweenthe titanium enolate of 20-hydroxypropiophenone with 2-azetidinone 38 followed byozonolysis of the resulting ketone to the carboxylic acid [103], the synthesis of 4-(2-oxoethylidene)azetidin-2-ones by a Lewis acid mediated reaction of acyldiazo com-pounds with 4-acetoxy derivative 38 [104], the reaction of 38 with organoindiumreagents generated in situ from indium powder and c-substituted propargylbromides in the presence of KI in DMF to selectively produce 4-allenyl-2-azetidi-nones 39 in good to excellent yields, the reaction of 4-acetoxy-2-azetidinones withorganoindium reagents generated in situ from indium and 1,4-dibromo-2-butyne inthe presence of LiCl in DMF to selectively produce 2-azetidinones that contain a 1,3-butadienyl-2-yl group at theC4-position in good yields [105], the reaction of 4-acetoxy-b-lactams with organoindium reagent generated in situ from indium and 1,6-dibromo-2,4-hexadiyne in the presence of LiCl in DMF to selectively produce2-azetidinones possessing 1,2,4,5-hexatetraen-3-yl group on the C4-position [106],as well as the coupling with a-substituted propargyl bromides to give 4-propargyl-2-azetidinones 40 selectively (Scheme 24.23) [107].
The stereoselective anti SN20 attack of NaN3 to 3-alkenyl-3-bromo-azetidin-2-onesgives a mixture of diastereomeric azides in rapid equilibrium. The [3,3]-sigmatropicrearrangement of allylic azides occurs with complete stereocontrol, allowing theequilibrium to be directed preferentially toward the (E)- or (Z)-isomer, which areuseful precursors of 3(20-amino)-b-lactams [108]. Azetidine-2,3-diones 41 and var-ious stabilized organic halides undergo coupling under Barbier-type conditions inthe presence of different metals (indium, tin, zinc) and additives [ammoniumchloride, hydrobromic acid, bismuth(III) chloride, hafnium(IV) chloride]. Theregiochemistry of the processes (carbonyl-allylation [109], bromoallylation [110],1,3-butadien-2-ylation [111], propargylation [112] or allenylation [112] reactions) aregenerally excellent. Similarly, the reaction of various activated vinyl systems, includ-ing 2-cyclopenten-1-one, with enantiopure azetidine-2,3-diones 41 has been pro-moted byDABCO to afford the corresponding optically pure Baylis–Hillman adductswithout detectable epimerization [110]. In addition, the reactions of enantiopureazetidine-2,3-diones with unmodified ketones or nitromethane were catalyzedby proline and N-methylephedrine, respectively, to give the corresponding aldoland nitroaldol adducts [113]. On this basis, simple and fast protocols for the synthesisof the bioactive 3-substituted 3-hydroxy-b-lactam moiety have been developed(Scheme 24.24).
NHO
OAcHHTBSOBr Br
NHO
HHTBSO
++ NHO
HHTBSOR2
R1
R2
R1
3839 40
In,equiv.2
KI,equiv.3
RTDMF,
81–93%
In,equiv.2
KI,equiv.3
RTDMF,
69–95%
Scheme 24.23
24.1 Monocyclic Derivatives j2135

24.1.5.2 Electrophilic Attack at CarbonUsing a halogen–lithium exchange reaction on 4-aryl-3,3-dichloro-2-azetidinones 42,followed by treatment with alkyl halides as electrophiles, the synthesis of cis-3-alkyl-3-chloro-4-arylazetidin-2-ones 43 has been accomplished (Scheme 24.25) [114].
It has been reported that the achiral bis(trimethylsilyl)methyl group acts as anefficient stereochemical determinant of the a-alkylation reaction in b-brancheda-phenyloxazolidinyl-b-lactams 44 andprovides stereocontrolled access to syn-a-ami-no-a,b-dialkyl(aryl)-b-lactams 45 (Scheme 24.26) [115], which are readily trans-formed into type II b-turn mimetic surrogates [116]. In situ generated organozincreagents of 3-alkenyl-3-bromoazetidin-2-ones react with aromatic and aliphaticaldehydes to give the corresponding alcohol derivatives, which could be of intereston account of their structural similarity with known cholesterol adsorptioninhibitors [117].
24.1.5.3 Electrophilic Attack at NitrogenConventionally, an alkyl or acyl side chain is introduced at the N1 position by base-mediated N-alkylation or N-acylation of the nitrogen atom with the appropriate alkyl
NO
HO
EWGR2
R1N
O
R2
R1N
O
R2
R1
HOOH
X
X
41
+EWG
+
In,equiv.2
THF–H2 (1:1),O
RT
38–62%
DABCO,mol%50
–20MeCN, oC
40–90%
Scheme 24.24
NO
HCl
43
R1
R2
R3
NO
Cl
42
R1
R2
Cli) n-BuLi, –78THF, oC
ii) R3 –78THF,X, o RTtoC23–57%
Scheme 24.25
NO
N
SiMe3
SiMe3
R1O
O
Ph
R2
NO
N
44SiMe3
SiMe3
R1O
O
Ph
H
45
–78THF,LDA,i) oC
Rii) 2 –78THF,X, o RTtoC58–92%
syn:anti 98:2toup=
Scheme 24.26
2136j 24 The Chemistry of 2-Azetidinones (b-Lactams)

or acyl halide [118]. A representative example is shown in Scheme 24.27 for thepreparation of 2-azetidinone 46 [119]. However, some unexpected results have beenreported. For example, in a tentative acylation reaction of the b-lactam nitrogen atomof (E)- and (Z)-4-alkylidene-b-lactamswith acetic anhydride under basic conditions itwas found that the (E) isomer is readily acylated, whereas the (Z)-isomer reactedsluggishly, rearranging to the corresponding oxazin-6-one. The N-acylation of (Z)-isomers has been successful, though, with oxalyl- or malonyl chlorides in benzene atreflux [120]. The treatment ofNH-b-lactams with aldehydes under heat or sonicationformed the corresponding N-hydroxyalkyl-2-azetidinones [121].
The copper-catalyzed couplings of NH-b-lactams with aryl and vinyl halides havebeen developed as an efficient procedure for the preparation of unsubstituted N-aryland N-vinyl-2-azetidinones [122]. This protocol has been fruitful for the synthesis ofthe spiro-b-lactam 47, which contains the enamide moiety of natural chartellines(Scheme 24.28) [123].
24.1.5.4 Radical TransformationsThe treatment of 4-thiophenyl-2-azetidinoneswith tributyltin hydride in the presenceof AIBN initiator yields the corresponding C4-desulfenylated b-lactam [124]. Thegeneration of radicals at C4 has been used for the synthesis of C4-unsubstitutedb-lactams 48, which are conveniently prepared using as the key step a radicalreductive decarbonylation of 4-carboxy derivatives through their phenyl selenoesters(Scheme 24.29) [125]. 3,3-Dibromosubstituted b-lactams can be dehalogenated orC3-functionalizated by treatment withmethyl acrylate under radical conditions [126].Upon using triethylborane as the radical initiator, b-lactamido N-sulfonyl radicalscould be allylated and added onto electron-rich olefins [127]. The radicals do notundergo desulfonylation and are electrophilic in nature.
NO H
46
NO
OBOMOBOM
CO2Me
BrCH2CO2Et,
0THF,KOH, oC
84%
Scheme 24.27
N
N
R1
O
O
47
N
N
Me
O
O
H I
Ph
H
Cl+
H
Cl
PhCuI,mol%5(CH2NHMe)2 K, 2CO3
refluxtoluene,76%
Scheme 24.28
24.1 Monocyclic Derivatives j2137

24.1.5.5 Reduction ReactionsThe application of metal hydrides in the search for general and efficient methods forthe one-step conversion of b-lactams into different building blocks has beenexamined. The reaction of different b-lactams with diborane gives rise to c-aminoalcohols [128]. Lithium aluminium hydride (2 molar equiv) in diethyl ether underreflux for 7–16 h converted 1,4,4-trisubstituted b-lactams into azetidines 49 in goodyields 63–82% yield (Scheme 24.30) [129]. By contrast, treatment of 4-(1-chloroethyl)-b-lactams with two molar equivalents of lithium aluminium hydride in diethyl etherat 0 �C for two hours afforded 3-chloropyrrolidines [130], while various novel syn-2-alkoxy-3-amino-3-arylpropan-1-ols, easily converted into antimalarial cis-5-alkoxy-4-aryl-1,3-oxazinanes, have been prepared through LiAlH4-promoted reductivering-opening of cis-3-alkoxy-4-aryl-b-lactams in Et2O [131].
Attempted reduction by BH3.THF (22 h in refluxing dioxane) and NaBH4–AlCl3(3.5 h in refluxing ether) resulted in a complete recovery of the starting 2-azetidinone.The reduction with LiAlH4, LiBEt3H or LiB-sec-Bu3H in THF at room temperaturegave exclusively the corresponding c-amino alcohol through 1,2-bond fission. It wasfound that the reduction of various 2-azetidinones with DIBAL-H in THFaffords thecorresponding azetidines in reasonable yields, although a small amount of c-aminoalcohols was also produced. The use of alane (AlH3) for the reduction of theazetidinone nucleus results in the formation of a mixture of compounds, with thefour-membered heterocycle and the c-amino alcohol being the minor and majorcomponent, respectively [132]. Examination of the reactivities of monochloroalane(AlH2Cl) and dichloroalane (AlHCl2) toward b-lactams has revealed that AlH2Cl andAlHCl2 prepared in situ from LiAlH4 and AlCl3 in ether converts 2-azetidinones 50into azetidines 51 in quite high yields (50–97%) without being accompanied byc-amino alcohols (Scheme 24.31) [133]. The reduction of 2-azetidinones by metalhydrides to afford azetidines is not compatible with the presence of ester groups.Reduction with diphenylsilane and catalytic amounts of tris(triphenylphosphine)
NO R1
R2
SePh
O
NO R1
R2
48
Bu3 AIBN,SnH,
C6H6 reflux,
45–85%
Scheme 24.29
NO
R3
R1
R2
N
R3
R1
R2
49
LiAlH4 Et, 2 refluxO,
63–82%
Scheme 24.30
2138j 24 The Chemistry of 2-Azetidinones (b-Lactams)

rhodium(I) carbonyl hydrides is a chemoselective method for the transformation ofb-lactams into the corresponding azetidines [134].
24.1.5.6 Cis/Trans IsomerizationRegarding the stereochemical outcome of the routes to prepare b-lactams, a verystrong preference for cis-b-lactam formation, a kinetic control product, is observed.Consequently, the development of different strategies to access to trans-2-azetidi-nones is of interest. Isomerization of cis-b-lactams to trans-b-lactams usuallyrequires as starting materials 2-azetidinones bearing acid or basic sensitivemoieties (e.g., aldehyde, ketone, ester, amine, amide) at the position susceptibleto epimerization. Epimerization at C3 and/or C4 is effected by different reagents,such as CF3COOH [135], Me3SiOTf [136], DBN [137], DBU [138] and NaOH/BuLi [139]. A more recent report involves Na2CO3-promoted regiospecific C4-epimerization of cis-4-formyl-b-lactams 52 into trans-4-formyl-b-lactams 53(Scheme 24.32) [14b].
A thermal conversion method for switching the stereochemistry of the 4-aryl-b-lactam ring from cis to trans involving a homolytic cleavage of the C3�C4 bond hasbeen reported (Scheme 24.33) [140]. These results are the only available examples ofisomerization in b-lactams induced by heat.
NO R1
R3R2
NR1
R3R2
5150
LiAlH4, AlCl3,
Et2 refluxO,
50–97%
Scheme 24.31
NO
R2 CHOH H
R1N
O
R2 CHOH
R1
H
5352
Na2CO3 RTMeCN,,
58–92%100:0toupr.d.
Scheme 24.32
NO R1
R2 HH
NO R1
R2 HH
54
R3 R3230Toluene, oC
44–67%
Scheme 24.33
24.1 Monocyclic Derivatives j2139

24.1.5.7 Ring-Opening and Rearrangement ReactionsIn recent years many 2-azetidinone-based methods for the synthesis of nitrogen-containing compounds of biological relevance have appeared [141]. b-Lactams areprecursors to a- and b-amino acids. They have been used to introduce the C13 side-chain of the anticancer compound paclitaxel (taxol) and related analogues [142]. Thering expansion of a-hydroxy b-lactams 55 through a regioselective Baeyer–Villigerrearrangement of an in situ generated azetidine-2,3-dione by means of sodiumhypochlorite and a catalytic amount of TEMPO, affords N-carboxy anhydrides(NCAs) 56, which after coupling with amines or alcohols produces a-amino acidderivatives 57 (Scheme 24.34) [143]. A related one-pot procedure starting fromazetidine-2,3-diones has been documented [144].
Palladium-catalyzed hydrogenolysis of 4-aryl-b-lactams 58 proceeds exclusivelyto give a-amino acid derivatives 59 (Scheme 24.35). The ring strain of the 2-azetidinone nucleus greatly accelerates the cleavage of the N1�C4 bond, ratherthan the more usual N1�C2 bond breakage, when an aryl substituent is attached tothe C4 position [145]. Addition reaction of 2-(trimethylsilyl)thiazole to cis- or trans-4-formyl-b-lactams has been reported to give enantiopure a-alkoxy-c-keto acidderivatives [146]. Also, the first organocatalytic N1�C4 bond breakage of theb-lactam skeleton has been uncovered, providing a direct method for the prepa-ration of enantiopure 5-arylimino-pyrrolidin-2-ones as well as pyrrolidin-2,5-diones(succinimides) from 4-(arylimino)-methyl-azetidin-2-ones [147]. In addition, asingle-step catalytic ring expansion approach from 4-oxoazetidine-2-carbaldehydesto enantiopure succinimides has been achieved by the use of a base (DBU) anda thiazolium salt precatalyst [148], and its mechanism has been studied usingDFT methods [149].
55
ON
O
O
H
Bn
R2
OR1
57
R3X
NHBn
R2
OR1OH
NO Bn
HOP
HHO
R2
56
KNaOCl, 2HPO4,
(cat),TEMPO
CH2Cl2–H2 0O, oC
91–96%
R3 CHXH, 2Cl2,
RT
70–80%
ONH,=X
Scheme 24.34
NO Ph
58
X Ar
Ar
Y
NHPh
O
59
Pd–H2,
RTAcOEt,
62–99%
N=X 3 OBn,
NH=Y 2 OH,
Scheme 24.35
2140j 24 The Chemistry of 2-Azetidinones (b-Lactams)
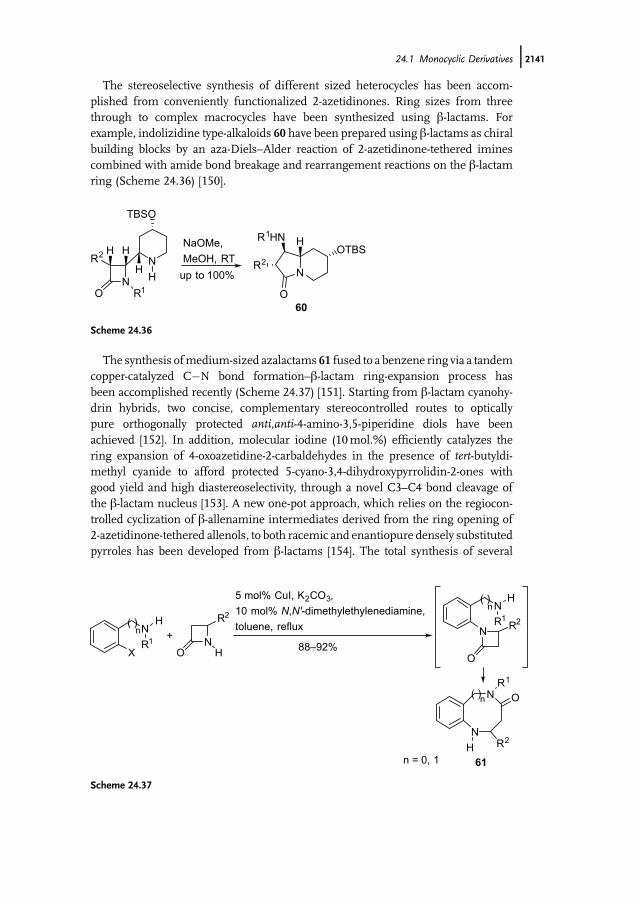
The stereoselective synthesis of different sized heterocycles has been accom-plished from conveniently functionalized 2-azetidinones. Ring sizes from threethrough to complex macrocycles have been synthesized using b-lactams. Forexample, indolizidine type-alkaloids 60 have been prepared using b-lactams as chiralbuilding blocks by an aza-Diels–Alder reaction of 2-azetidinone-tethered iminescombined with amide bond breakage and rearrangement reactions on the b-lactamring (Scheme 24.36) [150].
The synthesis ofmedium-sized azalactams 61 fused to a benzene ring via a tandemcopper-catalyzed C�N bond formation–b-lactam ring-expansion process hasbeen accomplished recently (Scheme 24.37) [151]. Starting from b-lactam cyanohy-drin hybrids, two concise, complementary stereocontrolled routes to opticallypure orthogonally protected anti,anti-4-amino-3,5-piperidine diols have beenachieved [152]. In addition, molecular iodine (10mol.%) efficiently catalyzes thering expansion of 4-oxoazetidine-2-carbaldehydes in the presence of tert-butyldi-methyl cyanide to afford protected 5-cyano-3,4-dihydroxypyrrolidin-2-ones withgood yield and high diastereoselectivity, through a novel C3–C4 bond cleavage ofthe b-lactam nucleus [153]. A new one-pot approach, which relies on the regiocon-trolled cyclization of b-allenamine intermediates derived from the ring opening of2-azetidinone-tethered allenols, to both racemic and enantiopure densely substitutedpyrroles has been developed from b-lactams [154]. The total synthesis of several
NO R1
H HR2
N
H
TBSO
H N
O
R1HNOTBS
R2
H
60
NaOMe,
RTMeOH,
100%toup
Scheme 24.36
X
N)(n
NO H
R2
R1
H+
)(
N
Nn
R1
O
H
N)(n
R1
H
N
O
R2
R2
61
KCuI,mol%5 2CO3,
mol%10 N,N'-dimethylethylenediamine,
refluxtoluene,
88–92%
10,=n
Scheme 24.37
24.1 Monocyclic Derivatives j2141

natural products such as biotin [155], cribrostatin 4 [156], and himandrine [157]has also been carried out using 2-azetidinones as important building blocks. Thesyntheses of pyrrolizidines [158], fused prolines [159], oxazinones [160], aminoglycals [161], aminocyclobutanes [162], bicyclic c-lactams [163], medium-sizedheterocycles [164], and complex macrocycles [165] deserve to be mentioned as well.
24.1.5.8 Reactions of Substituents Attached to Carbon AtomsThe oxidation of a-ethylidene b-lactams is a useful method to prepare azetidine-2,3-diones [166]. A general and efficient synthesis of cis and trans b-lactams bearinga quinone moiety at N1, C3 or C4 positions, which can be regarded as hybrids of thepharmacologically relevant subunits of b-lactam and quinone, has been developed.The target molecules 62 are smoothly prepared via oxidative demethylation of theappropriate 2,5-dimethoxyphenyl substituted-b-lactams using ceric ammoniumnitrate (CAN) in aqueous acetonitrile (Scheme 24.38) [167].
A stereoselective synthesis of 1,2,3-trisubstituted b-lactam-1,3-dienes 63 has beendeveloped from 2-azetidinone-tethered a-allenols just by treatment with a metha-nesulfonyl chloride/tertiary amine system. This transformation might be tentativelyexplained through a migration of the methanesulfonyl group in the initially formeda-allenic methanesulfonate to give the corresponding mesyloxy-diene via [3,3]-sigmatropic rearrangement (Scheme 24.39) [168]. Mesylates of 2-azetidinone-teth-ered homoallylic alcohols by gentle heating in benzene or toluene in the presenceof DBU have been used for the stereoselective preparation of 4-butadienyl-2-azetidinones [169].
The benzylidene moiety of b-lactam 64 is cleaved by ozonolysis and reductivetreatment with NaBH4 to afford the primary alcohol, which is then activated as thetosylate. After replacement of the benzyl group with the TBS group, a four-stepsequence including SN2 reaction of caesium thioacetate, methanolysis of the
NR2
O
R1
CH3O OCH3
NR2
O
R1
O O
62
MeCN–HCAN, 2 (3:1),O
RT
44–98%
Scheme 24.38
OHR3
R3
MsO
NO R1
R2
NO R1
R2
63
CH3SO2 EtNCl, 3,
CH2Cl2 RT,
46–96%
Scheme 24.39
2142j 24 The Chemistry of 2-Azetidinones (b-Lactams)

thioacetate, chloromethylation of the thiol, and SN2 reaction of N-hydroxyphthali-mide sodiumsalt, gives the protectedN-alkoxyamine 65 (Scheme 24.40), which bearsthe side chain required for the construction of the oxathiazepin ring of naturaleudistomins [170]. N-Terminal chain elongation on amido substituents at the C3-position of the b-lactam moiety has been achieved using conventional peptidesynthesis (saponification, activation as pentafluorophenyl esters and subsequentcleavage of theN-terminal Boc-protecting group) [171]. The olefin crossmetathesis ofa-methylene-b-lactams [172] and the selective Diels-Alder reaction of a-dienyl-b-lactams [173] have been developed. Vinylic halogenation and halodecarboxylationreactions of 4-alkylidene-b-lactams have been performed [174]. It has been reportedthat, by adopting Pd(II)-catalyzed conditions, the cycloisomerization/dimerizationratio of 4-buta-2,3-dienoyl-azetidin-2-ones is controlled by the substitution of theallene moiety: unsubstituted allenones mainly afford dimerization, whereas alle-nones bearing an internal substituent favor the formation of cycloisomerizationproducts [175].
24.1.5.9 Reactions of Substituents Attached to Nitrogen AtomA three-step synthesis of N-vinyl-2-azetidinones starting from a- or b-amino esterimines has been developed [176]. Enolate formation on the amino estermoiety of the2-azetidinone 66, selenylation and finally MCPBA treatment affords N-vinyl-2-azetidinones 67 in good yields (Scheme 24.41).
NO
BnO
65 (34%)64
OMOM
PhS
NO
TBSO
OMOM
H H H H
ONPhth
Oi) 3 CH, 2Cl2 –78–MeOH, oC,
NaBHthen 4 –78, o RTtoC
CHpyridine,TsCl,ii) 2Cl2 RT,
HCOPd/C,iii) 2NH4 refluxMeOH,,
RTDMF,imidazole,TBSCl,iv)
CsAcSH,v) 2CO3 60DMF,, oC
0MeOH,NaOMe,vi) oC
ClCHvii) 2 BnEtKOH,Br, 3 (cat.),NCl
CH2Cl2–H2 RTO,
NaH,viii) N-hydroxyphthalimide,
RTDMF,
Scheme 24.40
NO
R3
67
R4
R1R2
NO
R3 R4
R1R2
66
–78THF,LHMDS,i) oC
–78THF,PhSeBr,ii) oC
CHMCPBA,iii) 2Cl2 –78, oC
55–91%
Scheme 24.41
24.1 Monocyclic Derivatives j2143

The selective N-oxidation of the most nucleophilic amino nitrogen atom of thehydrazidemoiety in 1-dialkylamino azetidin-2-ones is central for the cleavage of theirN�N bonds under oxidative conditions by treatment with peracids such as magne-sium monoperoxyphthalate hexahydrate (MMPP�6H2O) or meta-chloroperbenzoicacid (MCPBA) [177]. A Grubbs� carbene catalyzed isomerization ofN-allyl b-lactamsintoN-vinyl b-lactams affords, after RuCl3–NaIO4 treatment, the correspondingNH-b-lactams [178]. It has been shown thatN-(4-methoxy or 4-ethoxyphenyl) groups canbe oxidatively removed by silica gel supported ceric ammonium nitrate under mildconditions in solution and on column [179]. The cis-2-azetidinones 68 have beenreactedwith PhI(CF3CO2)2 andNaHCO3 and the crude reaction products purified bycolumn chromatography on silica gel to give a diastereomericmixture of hemiketals,which was quantitatively converted into the lactones 69 by oxidation with PDC(Scheme 24.42) [180].
24.2Penicillins and Cephalosporins
24.2.1Introduction
The antibacterial effect of b-lactam antibiotics such as penicillins (70) and cepha-losporins (71) is due to their capacity to disrupt bacterial cell wall biosynthesis [181].This is achieved by the antibiotics acting as inhibitors of penicillin binding proteins(PBPs), which are membrane bound serine peptidases. PBPs recognize D-alanyl-D-alanine peptide termini and the structural and conformational similarity of theb-lactam antibiotics to these natural peptide substrates for PBPs is believed to ensuretheir acceptance by the target proteins. The high reactivity of the fused b-lactam ringtowards nucleophiles then results in the formation of a covalent PBP–antibioticcomplex that prevents the PBPs from taking further part in bacterial cell wallsynthesis.
NO
R1H H
O
Ph
R2
O
69
NO
R1H H
Ph
68
CH(SEt)2
OO
R2
PhI(CFi) 3CO2)2 NaHCO, 3,
CH3CN–H2 RT(85:15),O
CHPDC,ii) 2Cl2 RT,
54–75%
Scheme 24.42
2144j 24 The Chemistry of 2-Azetidinones (b-Lactams)

NO
HN
NO
R
O
HNR
O
1707
S
CO2HH
Me
MeS
CO2H
CH2OAc
b-Lactamase hydrolytic enzymes are the most common, and growing, form ofbacterial resistance to their normally lethal action [182]. b-Lactamases catalyze thehydrolysis of the b-lactam antibiotic to give the ring opened and bacterially inertb-amino acid (Scheme 24.43). The problem of b-lactamases became critical in 1960when widespread use of penicillin G led to an alarming increase of Staphylococcusaureus infections. These problem strains had gained the lactamase enzyme and hadthus gained resistance to the drug. At one point, 80% of all Staphylococcus aureusinfections in hospitals were due to virulent, penicillin-resistant strains. Alarmingly,these strains were also resistant to all other available antibiotics.
Two main therapeutic strategies have been adopted to counteract bacterial resis-tance to b-lactam antibiotics. One strategy consists of modifying the structure of theb-lactam antibiotic, aiming to render it insensitive to the b-lactamase attack. Recently,trinems antibiotics (Figure 24.1) have been the subject of considerable study owing totheir broad spectrum of antibacterial activity, resistance to b-lactamases and stabilityto renal dehydropeptidases [183]. As a result of their impressive biological activity,tricyclic b-lactams have become interesting targets for synthesis. A second approachuses a reagent, typically a b-lactam derivative, which incapacitates the b-lactamase, insynergy with the b-lactam antibiotic. Clavulanic acid (Figure 24.1) is the archetype ofb-lactamase inhibitors [184]: in synergistic mixture with amoxicillin (Figure 24.1),under the name �augmentin,� it arrived at the practice some years ago. Both
NO
HNR
O
S NH
HO2C
O
R β-lactamase
CO2H
HN
S
CO2H
Scheme 24.43
NO
O
NO
HOH
CO2H
acidclavulanic
NO
S
CO2H
amoxicillin
HN
OHO
NH2
CO2H
OMe
HO H
trinems
Me
Me
Figure 24.1 New generation b-lactam antimicrobials: trinems and �augmentin.�
24.2 Penicillins and Cephalosporins j2145

approaches have produced results and a new generation of antibiotics has beendeveloped.
24.2.2Physicochemical Data
24.2.2.1 Computational ChemistryThe calculated STO-3G energy of formamide in a penicillin-like geometry is only2.8 kcalmol�1 higher than the planar geometry [185]. In addition, the geometricalparameters associated with the 2-azetidinone nucleus generally suffer a slightvariation with changes in the hybridization at nitrogen. However, the C�N bondlength becomes longer as the nitrogen atom becomes pyramidal.
Two penicillin derivatives, the active penamecillin and the inactive penamecillin-1b-sulfoxide, have been used to study the relationship between their charge densityand their activity [186]. Single crystals of both compounds have beenmeasured at thesynchrotron beamline F1 at the HASYLAB/DESY, at 100K and up to resolutions ofaround 0.4A
�. Experimental charge densities have been obtained by using the
Hansen–Coppens multipole formalism. The cleavage of the amide bond in theb-lactam ring is of paramount importance in the mechanism of action of penicillins.Topological analysis of this bond in terms of Bader�s AIM theory showed that itsstrength is equal in both compounds; therefore, a direct influence of bond strengthon the activity can be ruled out. However, the two derivatives differ significantly intheir experimental electrostatic potentials. These differences provide further insightinto the chemistry and activity of penicillins.
Theoretical results have been reported on the conformational properties ofbenzylpenicillin, which are characterized by means of quantum chemical calcula-tions (MP2/6-31G� and B3LYP/6-31G�) and classical molecular dynamics simula-tions (5 ns) both in the gas phase and in aqueous solution [187]. In the gas phase, thebenzylpenicillin conformer in which the thiazolidine ring has the carboxylate grouporiented axially is the most favored one. Both intramolecular CH � � � O and disper-sion interactions contribute to stabilize the axial conformer with respect to theequatorial one. In aqueous solution, a molecular dynamics simulation predictsa relative population of the axial : equatorial conformers of 0.70 : 0.30 in consonancewith NMR experimental data. Overall, the quantum chemical calculations as well asthe simulations give insight into substituent effects, the conformational dynamics ofbenzylpenicillin, the frequency of ring-puckeringmotions, and the correlation of sidechain and ring-puckering motions.
The mechanisms of antibiotic resistance have been studied using a combinedquantum mechanical and molecular mechanical (QM/MM) modeling of the acyl-ation reaction of a class A b-lactamase with benzylpenicillin [188]. Hybrid Car–Parrinello QM/MM calculations have been used to investigate the reaction mech-anism of hydrolysis of a common cephalosporin-type substrate (cefotaxime) by themonozinc b-lactamase from Bacillus cereus [189]. Theoretical studies on the confor-mational similarity of penicillins and cephalosporins to X-D-alanyl-D-alanine andcorrelation of their structure with activity has been examined by stereochemical
2146j 24 The Chemistry of 2-Azetidinones (b-Lactams)

criteria, concluding that the conformation of these b-lactam antibiotics is similar toX-D-alanyl-D-alanine due to the presence of the lactam ring [190].
24.2.2.2 Experimental Structural MethodsThe degree of coplanarity of the b-lactamnitrogen atom inb-lactam antibiotics can beexpressed either by the perpendicular distance, h, of the nitrogen from the plane ofits three substituents or by the sum of the bond angles about nitrogen. The former iseasier to visualize and the nitrogen ranges from being essentially in the plane of itsthree substituents in monocyclic b-lactams to being 0.5A
�out of the plane in bicyclic
systems [191]. There is no direct correlation between h values and chemical reactivity.In non-planar penicillins and cephalosporins there is a general trend for the C�Nbond length to increase as the C¼O bond length decreases. However, this trend is byno means linear. Bond lengths for C¼O vary from 1.17 to 1.24A
�and for C�N from
1.33 to 1.46A�. There is also a tendency for the C�N bond length to increase with h. It
is difficult to discern reasons and reactivity consequences of these differences inbond length. Penicillin V (70 R¼PhOCH2) shows the longest C–N bond length of1.46A
�and yet the C¼O bond length (1.21A
�) is identical to that commonly found in
planar monocyclic b-lactams. In monocyclic b-lactams the nitrogen is coplanar withits three substituents and yet the bond length differences are also in the directionpredictedby inhibitionof amide resonance.Thedegree ofnon-planarity inpenicillinVand ampicillin [70 R¼PhCH(NH2)] is similar (h¼ 0.40 and 0.38A
�, respectively) and
yet the C�N bond length in the former is 0.10A�longer than in the latter. Structural
data have also been used to support the suggestion that enamine resonance isimportant in cephalosporins and that this also reduces amide resonance [192].However, there is no significant difference in the C�O and C�N bond lengths incephalosporins from the general trend exhibited by penicillins. It became apparentthat variations in bond lengths within penicillins and cephalosporins are due to thenature of substituents and the minimization of unfavorable strain energies caused bythe geometry of the molecule rather than to the inhibition of the amide resonance.
The 13C NMRspectra of bicyclicb-lactamantibiotic show the carbonyl resonance atabout 165 ppm. The 2-azetidinone carbonyl carbon in penicillins resonates around10 ppm to lower field than that in cephalosporins. The chemical shifts for thebiologically active D3- and the inactive D2-cephalosporins are similar. The 15N NMRspectra show an upfield shift of 30 ppm in the b-lactam nitrogen on going from non-planar penicillins to planar 2-cephems [193]. The infraredC¼Oabsorption frequencyfor the bicyclic b-lactam antibiotics is in the 1760–1780 cm�1 range. The frequency incephalosporins increases by about 5 cm�1 when the ring sulfur is substituted byoxygen but decreases by a similar amount when the 7-a-hydrogen is replaced byamethoxy group. Structural studies confirm that N-fused b-lactam systems generallyhave a higher C¼O stretching frequency than the C-fused structures, indicating agreater amount of ring strain and chemical reactivity toward nucleophiles [194].N-Fused lactams are highly respondent to the geometric constraints imposed by thesecond ring to which it is fused, as evidenced from the increase in the infraredabsorption frequency for the lactam carbonyl as the size of the second ring isdecreased. 13C NMR spectroscopic data for third-generation cephalosporins, such as
24.2 Penicillins and Cephalosporins j2147
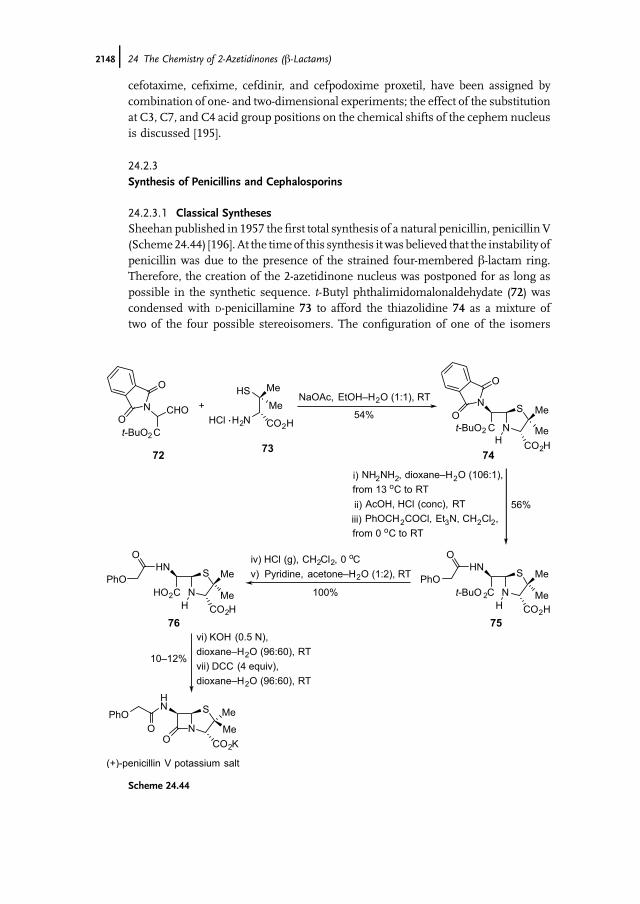
cefotaxime, cefixime, cefdinir, and cefpodoxime proxetil, have been assigned bycombination of one- and two-dimensional experiments; the effect of the substitutionat C3, C7, and C4 acid group positions on the chemical shifts of the cephem nucleusis discussed [195].
24.2.3Synthesis of Penicillins and Cephalosporins
24.2.3.1 Classical SynthesesSheehan published in 1957 thefirst total synthesis of a natural penicillin, penicillin V(Scheme24.44) [196]. At the timeof this synthesis it was believed that the instability ofpenicillin was due to the presence of the strained four-membered b-lactam ring.Therefore, the creation of the 2-azetidinone nucleus was postponed for as long aspossible in the synthetic sequence. t-Butyl phthalimidomalonaldehydate (72) wascondensed with D-penicillamine 73 to afford the thiazolidine 74 as a mixture oftwo of the four possible stereoisomers. The configuration of one of the isomers
HS
H2N
Me
Me
CO2HHCl
N
O
Ot-BuO2 C N
S Me
Me
N
O
Ot-BuO2 C
CHO
CO2H
+
HN
t-BuO 2C N
S Me
MeCO2H
7273
H
74
H
O
PhO
75
HN
HO2C N
S Me
MeCO2H
H
O
PhO
76
NO
HN
O
S
CO2K
Me
MePhO
saltpotassiumV(+)-penicillin
EtOH–HNaOAc, 2 RT(1:1),O
54%
NHi) 2NH2 dioxane–H, 2 (106:1),O
13from o RTtoC
RT(conc),HClAcOH,ii)PhOCHiii) 2 EtCOCl, 3 CHN, 2Cl2,
0from o RTtoC
56%
CH(g),iv) HCl 2Cl2 0, oC
acetone–HPyridine,v) 2 RT(1:2),O
100%
N),(0.5KOHvi)
dioxane–H2 RT(96:60),O
equiv),(4DCCvii)
dioxane–H2 RT(96:60),O
10–12%
Scheme 24.44
2148j 24 The Chemistry of 2-Azetidinones (b-Lactams)
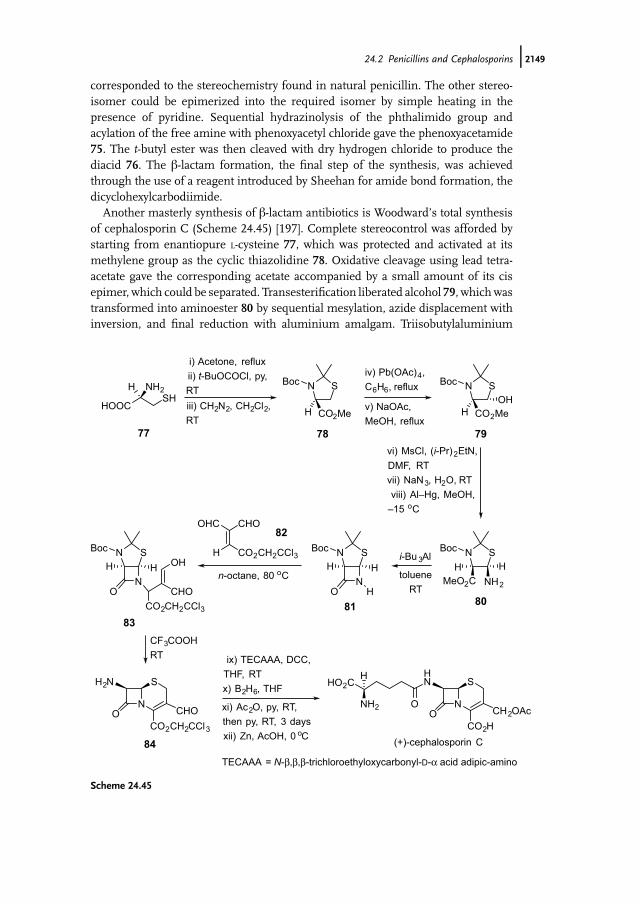
corresponded to the stereochemistry found in natural penicillin. The other stereo-isomer could be epimerized into the required isomer by simple heating in thepresence of pyridine. Sequential hydrazinolysis of the phthalimido group andacylation of the free amine with phenoxyacetyl chloride gave the phenoxyacetamide75. The t-butyl ester was then cleaved with dry hydrogen chloride to produce thediacid 76. The b-lactam formation, the final step of the synthesis, was achievedthrough the use of a reagent introduced by Sheehan for amide bond formation, thedicyclohexylcarbodiimide.
Another masterly synthesis of b-lactam antibiotics is Woodward�s total synthesisof cephalosporin C (Scheme 24.45) [197]. Complete stereocontrol was afforded bystarting from enantiopure L-cysteine 77, which was protected and activated at itsmethylene group as the cyclic thiazolidine 78. Oxidative cleavage using lead tetra-acetate gave the corresponding acetate accompanied by a small amount of its cisepimer, which could be separated. Transesterification liberated alcohol 79, whichwastransformed into aminoester 80 by sequential mesylation, azide displacement withinversion, and final reduction with aluminium amalgam. Triisobutylaluminium
NO
HN
O
S
CO2H
CH2OAc
HOOCSH
H NH2 SN
CO2MeH
Boc SN
CO2MeH
Boc
OH
SN
MeO2CH
Boc
NH2
77 7978
80
SNBoc
81
NO H
H H
SNBoc
83
NO
H Hn-octane, 80 oC
H
OHC
CO2CH2CCl3
CHO
CO2CH2CCl3
CHO
OH
NO
H2N S
CO2CH2CCl3
CHO
CF3COOH
RT
82
84 C(+)-cephalosporin
HO2C
NH2
H
H
refluxAcetone,i)
ii) t-BuOCOCl, py,
RT
CHiii) 2N2 CH, 2Cl2,
RT
Pb(OAc)iv) 4,
C6H6 reflux,
NaOAc,v)
refluxMeOH,
(i-Pr)MsCl,vi) 2EtN,
RTDMF,
NaNvii) 3 H, 2 RTO,
MeOH,Al–Hg,viii)
–15 oC
i-Bu 3Al
toluene
RT
DCC,TECAAA,ix)
RTTHF,
Bx) 2H6 THF,
Acxi) 2 RT,py,O,
days3RT,py,then
0AcOH,Zn,xii) oC
=TECAAA N-β,β,β-trichloroethyloxycarbonyl-D-α acid adipic-amino
Scheme 24.45
24.2 Penicillins and Cephalosporins j2149

effected smooth conversion of aminoester 80 into the bicyclic 2-azetidinone 81,which is a key intermediate containing the basic structural features common to bothpenicillins and cephalosporins. Conjugate addition of the b-lactam nitrogen atom toester 82, which was obtained through the condensation between malondialdehydeand trichloroethyl glyoxylate, generated compound 83. All the functionalitiesrequired for the cephem skeleton formation are present in fused 2-azetidinone 83.Treatment of 83 with trifluoroacetic acid induced cyclization with concomitantdeprotection of both the amino and mercapto groups to give bicycle 84. The totalsynthesis of cephalosporin C was completed after acylation with a protected N-b,b,b-trichloroethyloxycarbonyl-D-a-amino adipic acid, followed by aldehyde reduc-tion with diborane, acetylation, isomerization of the olefin under basic conditions,and treatment with zinc in acetic acid. This last step was the first use of thetrichloroethyl moiety as protecting group in synthesis.
24.2.3.2 Industrial Production of b-Lactam AntibioticsThe industrial production of b-lactam antibiotics by fermentation over the past 50years is one of the outstanding examples of biotechnology [198]. Today, b-lactamantibiotics, particularly penicillins and cephalosporins, are the world�s major bio-technology products with worldwide dosage form sales of�US$ 15 billion or�65%of the total world market for antibiotics. Over the past five decades, major improve-ments in the productivity of the producer organisms, Penicillium chrysogenum andAcremonium chrysogenum (syn. Cephalosporium acremonium) and improved fermen-tation technology have culminated in enhanced productivity and substantial costreduction. Major fermentation producers are now estimated to record harvest titersof 40–50 g L�1 for penicillin and 20–25 g L�1 for cephalosporin C. Recovery yields forpenicillin G or penicillin V are now >90%. Chemical and enzymatic hydrolysisprocess technology for 6-aminopenicillanic acid (6-APA) 85 or 7-aminocephalos-poranic acid (7-ACA) 86 is also highly efficient (�80–90%), with new enzymetechnology leading to major cost reductions over the past decade.
NO
H2N
NO
H2N
6858
S
CO2HH
Me
MeS
CO2H
CH2OAc
24.2.3.2.1 Commercial Production of Penicillins The fermentation production ofpenicillin-G or -V is a fed-batch process carried out aseptically in stainless steel tankreactors of 30 000–100 000 gallon capacity. The fermentation usually involves two tothree initial seed growth phases followed by a fermentation production phase havinga time cycle ranging from 120 to 200 h. High dissolved oxygen levels are critical,especially during peak growth periods that often occur at the 40–50 h time-period ofthe cycle. The fermentation mode is fed-batch and crude sugar and precursor arefed throughout the cycle. Current penicillin fermentations are highly computerizedand automated. Temperature, pH, dissolved oxygen, carbon dioxide, sugar,
2150j 24 The Chemistry of 2-Azetidinones (b-Lactams)

precursor, ammonia, and so on are closely monitored and controlled for optimalantibiotic production [199]. Various carbon sources have been adopted for thefermentation, including glucose, sucrose and other crude sugars. About 65% of thecarbon is metabolized for cellular maintenance, 20–25% for growth and 10–12% forpenicillin production [200]. Sugar and precursor are fed continuously and the sugar isalso used to help regulate the pH of the fermentation to between 6.4–6.8 during theactive penicillin production phase. Corn steep liquor and cottonseed or soybeanmeal,ammonia and ammonium sulfate represent major nitrogen sources. The essentialprecursor substances are phenylacetic acid (for penicillin G) or phenoxyacetic acid(for penicillin V) that are either fed or batched.
Mini-harvest protocols are often used in penicillin fermentations. This �batch-filland withdraw� system involves the removal of 20–40% of the fermentor contentswith replacement with fresh sterile medium. This procedure can be repeated severaltimes during the fermentation without yield reduction and, in reality, can enhancethe total penicillin yield per fermentor. Penicillin is excreted into the medium and isrecovered at the end of the fermentation.Whole broth extraction is usually performedat acidic pH by most manufacturers and has resulted in a 2–5% improvement inoverall extraction efficiency by the elimination of the rotary vacuum filtration step.Solvent extraction of chilled acidified broth is carried out with amyl, butyl or isobutylacetate. Multiple back-extractions into buffer and solvent at varying pH usingcountercurrent contactors has led to considerable penicillin concentration in theearly recovery stages of the purification process. Pigments and other broth impuritiesare removed by the use of activated charcoal. The penicillin is crystallized upon theaddition of potassium acetate and is isolated as a crystalline potassium salt. Addi-tional carbon treatments and solvent washes result in a highly purified final product.
Approximately 75% of the total bulk penicillin volume produced in 1995,�33 000tons, was used for the production of semi-synthetic penicillins and cephalosporins.The penicillin nucleus (6-APA) has enabled researchers to develop many excellentsemi-synthetic penicillins. 6-APAcan also be chemically ring-expanded to 7-ADCA togenerate several important orally-active cephalosporins (cephalexin, cephradine,cefadroxyl, etc.). 6-APA has now grown to be the world�s largest selling b-lactambulk intermediate.
24.2.3.2.2 Commercial Production of Cephalosporin C High-yielding strains of A.chrysogenum are used in large-scale, fed-batch fermentations. Major fermentationproducers of cephalosporin C obtain harvest titers in the range of 20–25 g L�1.Production-scale fermentations are fed-batch with carbon supplied as simple orcomplex carbohydrate feeds during the growth phase of the fermentation. As thefermentation progresses, sugar feeds are reduced and are usually replaced by higherenergy oils such as soybean oil or peanut oil. Energy conservation from oil asa substrate is considerably less efficient and leads to slower growth, with thevegetative mycelium becoming largely transformed into multicellular arthrospores.The arthrospore stage leads to greater oxygen availability to the organism and resultsin rapid cephalosporin production. DL-Methionine addition, which also results in theonset of arthrospore formation, is often added to themediumduring the early growth
24.2 Penicillins and Cephalosporins j2151

phase of the fermentation. The formation of arthrospores is also correlated withimproved dissolved oxygen concentration in the broth and is critical for maximalexpression of the important biosynthetic cyclase and expandase enzymes. Organicnitrogen is often supplied as a combination of soybean and cottonseed mealssupplemented with ammonium sulfate and ammonia that is also used to helpcontrol the pH throughout the fermentation. Corn steep liquor is also supplied as acheap nitrogen source and is rich in amino acids, vitamins, organic acids and traceelements. The pH of the fermentation is maintained between 6.2 and 7.0 and thetemperature range is controlled between 24 and 28 �C.
A major problem associated with cephalosporin C fermentation is the inherentchemical instability of the cephalosporin C molecule. This is probably one ofthe major reasons why long-cycle cephalosporin C fermentations often result inreduced cephalosporin production compared to typical long-cycle penicillin fer-mentations. Cephalosporin C is readily degraded to 2-(D-4-amino-4-carboxybutyl)-thiazole-4-carboxylic acid, which can account for as much as a �40% loss of thecephalosporin C produced [201]. The biosynthetic precursor molecules of cepha-losporin C, deacetylcephalosporin C and DAOC have much more chemical stability.Strains of the yeast Rhodosporidium toruloides possess a potent acetyl esterase and,when the organism is added to active cephalosporin C fermentations, result inincreased levels of deacetylcephalosporin C with an increase in total cephalosporinnucleus levels of �40%. Over the past decade, the cloning of many of the genesinvolved in the biosynthetic pathway of cephalosporins has resulted in moreproductive strains.
The purification and recovery of harvest cephalosporin C broth begins with therapid chilling of the active broth to 3–5 �C followed by removal of the mycelial solidseither by filtration or by centrifugation. The active broth contains not only the desiredcephalosporin C component but also small quantities of the biosynthetic precursorspenicillin N, DAOC, deacetylcephalosporin C and the degraded cephalosporin Cproduct, 2-(D-4-amino-4-carboxybutyl)-thiazole-4-carboxylic acid. Two major strate-gies can be used for the recovery and purification of cephalosporin C. One strategyinvolves the use of activated carbon or the use of a non-ionic resin. Because of thehigh selectivity of the resin, cephalosporinC is preferentially adsorbed over penicillinN or the contaminating biosynthetic precursor molecules. Most of the penicillin N isremoved in the pH 2.0 acidification step. An additional anion- and cation-exchangestep usually results in high quality cephalosporin C. A large fraction of the ceph-alosporin C is converted into 7-ACA and derivatized to semi-synthetic cephalospor-ins. A second purification strategy involves the substitution of the amine moiety onthe a-aminoadipyl side-chain at C7. Two substituted derivatives, N-2,4-dichloroben-zoyl cephalosporin C and tetrabromocarboxybenzoyl cephalosporin C, can be crys-tallized from acidic aqueous solution. Alternatively, salts can be formed between theN-substituted derivatives, and an organic base such as dicyclohexylamine ordimethylbenzylamine results in cephalosporin salts that are solvent extractable.Bristol-Myers Squibb uses a solvent-extractable process resulting in the isochlor-obutylformate (ICBF) ester of cephalosporin C, termed cephalosporin D.Several extraction steps are usually necessary to achieve the final desired purity.
2152j 24 The Chemistry of 2-Azetidinones (b-Lactams)

N-Substituted cephalosporin C salts containing small amounts of contaminants canbe effectively converted into 7-ACA. Efficient enzymatic processes are now utilizedfor the conversion of cephalosporins into 7-ACA, which has resulted in a dramaticcost reduction for this important bulk intermediate. Two key genetically engineeredenzymes are involved. The initial step is reaction of the a-aminoadipyl group withD-amino acid oxidase to produce glutaryl-7-ACA. This reaction proceeds througha keto-7-ACA intermediate that undergoes an oxidative decarboxylation in thepresence of hydrogen peroxide. A glutaryl acylase is used to remove the glutarylside-chain to produce 7-ACA. Two-thirds of the commercial cephalosporins arederived from 7-ACA that is produced from cephalosporin C by either chemical orenzymatic deacylation. In the chemical process, after protection of the amino andcarboxyl groups, reactionwith potassiumpentachloride in the presence of base formsan iminochloride derivative. The iminoether is formed on the addition of alcohol.The iminoether is hydrolyzed to form 7-ACA.
24.2.4Reactivity of Penicillins and Cephalosporins
24.2.4.1 Basicity of b-Lactam NitrogenIf amide resonance in penicillins is inhibited because of the pyramidal nature of theb-lactamnitrogen, penicillins should show enhanced basicity comparedwith normalamides. By contrast, penicillins appear to show reduced basicity and cannot bedetectably protonated even in 12M hydrochloric acid [202]. Another indication ofincreased nitrogen basicity would be a large binding constant of penicillin to metalions. The equilibrium constant for metal-ion coordination between the carboxylgroup and the b-lactam nitrogen in penicillins is about 100–200M�1 for variousmetal ions [203], which is the same order of magnitude expected for coordinationbetween a normal amide and a carboxyl group. Therefore, it became evident thatthere is not a substantial enhancement for the electron pair donating ability either toa proton or to a metal in penicillins.
24.2.4.2 HydrolysisThe bicyclic system in penicillin consists of a four-membered ring and a five-membered ring. As a result, penicillin suffers large angle and torsional strains.Ring opening relieves these strains by cleavage of the more highly strainedfour-membered lactam ring (Scheme 24.46).
NO
HNR
O
S
CO2H
Me
Me
NO
HNR
O
S
CO2H
Me
Me
HN
HN S
CO2H
Me
Me
H2O
–H+
HO–
H+
O
RHO2C
Scheme 24.46
24.2 Penicillins and Cephalosporins j2153

The carbonyl group in the b-lactam ring is highly susceptible to nucleophiles andas such does not behave like normal tertiary amides, which are usually quite resistantto nucleophilic attack. This difference in reactivity is due mainly to the fact thatstabilization of the carbonyl is possible in the tertiary amide but impossible in theb-lactam nucleus. The b-lactam nitrogen is unable to feed its lone pair of electronsinto the carbonyl group since this would require the bicyclic rings to adopt animpossibly strained flat system. As a result, the lone pair is localized on the nitrogenatom and the carbonyl group is far more electrophilic than one would expect fora tertiary amide.
The acyl side chain can actively participate (neighboring group participation) ina mechanism to open up the 2-azetidinonemoiety (Scheme 24.47). Thus, penicillinshave a built-in self-destruct mechanism. However, if a good electron-withdrawinggroup is attached to the carbonyl group, then the inductive pulling effect shoulddraw electrons away from the carbonyl oxygen and reduce its tendency to act asa nucleophile.
Enzyme-catalyzed hydrolysis of the b-lactam ring uncovers the thiazolidine-ringnitrogen as a nucleophile that drives a rapid intramolecular displacement on the sidechain. Attachment of 7-hydroxy-4-methylcoumarin as the releasable group of thisside chain generates a penicillin structure that can function as a fluorescence-basedreporter substance/diagnostic for the presence of low levels ofb-lactamase enzyme insolution [204].
The major structural differences between penicillins and cephalosporins are thatthe five-membered thiazolidine ring of penicillins is replaced by a six-membereddihydrothiazine ring in cephalosporins and that the degree of pyramidalization of theb-lactam nitrogen is generally smaller in cephalosporins. In addition, many cepha-losporins bear a potential leaving group at theC30 position (pyridine, acetate, or thiol),which is expelled during the hydrolysis of the 2-azetidinone nucleus to give anexo-methylene cyclic imine (Scheme 24.48). Experimental observations have led tothe conclusion that b-lactamC�Nbond fission is not concerted with the departure ofthe leaving group, and that the tetrahedral intermediate breaks down by proton
NO
HNR
O
S
CO2H
Me
Me
N
S
CO2H
Me
Me
–
H+
–H+ N
OR
O
HN
S
CO2H
Me
MeN
OR
O
N
S
CO2H
Me
MeN
HO2C
R
steps
OO
NH
R
CO2H
MeHS
Me
+
acidspenillicacidspenicillenic
Scheme 24.47
2154j 24 The Chemistry of 2-Azetidinones (b-Lactams)

transfer to generate an intermediate enamine,which subsequently, in a separate step,expels the leaving group [205]. The similarity in the second-order rate constants forthe hydroxide-promoted hydrolysis of penicillins and cephalosporins points to anabsence of influence of the leaving group at C30 in cephalosporins.
The hydrolysis of an acetoxy ester side chain at C3 in cephalosporins is competitivewith the hydrolysis of the b-lactam ring. It may be due to the comparable reactivity ofthe 2-azetidinone nucleus of cephalosporins and a simple ester such as ethyl acetate.No significant spontaneous hydrolysis is observed in the pH–rate profile for thehydrolysis of penicillins, but the b-lactam moiety does undergo an acid-catalyzeddegradation. By contrast, the hydrolysis of cephalosporins shows a spontaneous pH-independent hydrolysis and is less reactive by a factor of about 104 towards acid thanare penicillins [206]. Penicillins undergo an acid- and a base-catalyzed hydrolysis, butthere is no significant uncatalyzed reaction, the pH minimum being around 7 forspontaneous or water-induced degradation. However, cephalosporins often exhibita significant pH-independent reaction in the pH range 3–7. The evaluation ofdifferent glutaryl acylase mutants to improve the hydolysis of cephalosporin C inthe absence of hydrogen peroxide has been reported [207].
24.2.4.3 Alcoholysis, Thiolysis, and AminolysisThe reactions of b-lactam antibiotics and their derivatives with nucleophiles havebeen studied extensively. For example, nucleophilic substitution at the b-lactamcarbonyl center of penicillins occurs, in water, with amines [208], alcohols [209], andthiols [210] in competition with that by hydroxide ion. These are acyl transferprocesses involving covalent bond formation between the carbonyl carbon and thenucleophile and C�N bond fission of the b-lactam. In general, covalent bondformation to the incoming nucleophile occurs before b-lactam C�N bond fission,resulting in the reversible formation of a tetrahedral intermediate. The rate-limitingstep in these reactions is thus commonly ring opening and breakdown of thetetrahedral intermediate. Formation of the tetrahedral intermediate also changesthe basicity of the leaving group amine, as amide resonance in the b-lactam is lost and
HNO
RHO2C HN
S
CO2H
CH2X
HNO
RHO2C N
S
CO2H
CH2
X–
X– –
+
NO
HNR
O NO
HNR
O
H2OHO
–
H+
–H+S
CO2H
CH2X
S
CO2H
CH2X
Scheme 24.48
24.2 Penicillins and Cephalosporins j2155

proton transfer to nitrogen changes from an unfavorable to a thermodynamicallyfavorable process. Thus, many of these reactions require general acid catalysis andprotonation of the amine nitrogen leaving group to facilitate C�N bond fission andavoid amine anion expulsion. Although the release of strain energy, which accom-panies ring opening, could possibly decrease the need for protonation, C�N fissionin penicillins appears to require some form of catalysis. For example, the alcoholysisand thiolysis of penicillins occur with rate-limiting breakdown of the tetrahedralintermediate facilitated by proton transfer from solvent water to the departing amine.Exceptionally, the thiolysis of some cephalosporins appears to involve the breakdownof the tetrahedral intermediate by the expulsion of an enamine anion [211].
Unlike the strongly base catalyzed aminolysis of b-lactam antiobiotics, such aspenicillins and cephaloridines, the rate law for the aminolysis ofN-aroyl b-lactams isdominated by a termwith afirst-order dependence on amine concentration in its freebase form, which is indicative of an uncatalyzed aminolysis reaction that proceededby a concertedmechanism [212]. The relative sequence of bondmaking and breakingbetween heavy atoms in the aminolysis of b-lactam antibiotics is a result of subtleeffects that often involve proton transfer. A step-wise process for aminolysis occursthrough the formation of a tetrahedral intermediate, resulting from the attack on thecarbonyl center by an amine,which gives rise to a large change in the pKa of the amineNH as a result of covalent bond formation. Proton transfer from the aminenucleophile to a base catalyst thus occurs after full covalent bond formation, as itchanges from a thermodynamically unfavorable to a favorable process. Henceaminolysis usually requires general base catalysis to remove a proton from theattacking amine and this is the dominant term in the rate law – in fact it isexperimentally difficult to determine the rate constant for any uncatalyzed reaction.With penicillins and cephalosporins this proton transfer occurs after initial C�Nbond formation in a rate-limiting step that is diffusion controlled. The aminolysis ofb-lactam antibiotics also requires b-lactam C�N bond fission and expulsion of anamine. The rate of aminolysis of benzylpenicillin and cephaloridine by hydroxyl-amine, unlike other amines, shows only a first order dependence on amineconcentration [213]. The rate enhancement compared with that predicted from aBrønsted plot for other primary amineswith benzylpenicillin is greater than 106. Thisis much more than an a-effect and is compatible with rate-limiting formation of thetetrahedral intermediate due to a rapid intramolecular general acid catalyzedbreakdown of the intermediate. For cephaloridine, the rate enhancement is greaterthan 104, which demonstrates that b-lactam C�N bond fission and expulsion of theleaving group at C30 are not concerted.
24.2.4.4 Destruction of b-Lactam Antibiotics by b-Lactamasesb-Lactamases hydrolyze the four-membered b-lactam ring in both penicillin andcephalosporin classes of antibiotics (Scheme 24.49). They thereby destroy theantibacterial activity by deactivating the chemical warhead in the molecule [182],the strained b-lactam that is the chemically reactive acylating group formodifying theactive-site serine side chains in the penicillin-binding proteins (PBPs) (the trans-peptidases and carboxypeptidases in peptidoglycan [PG] crosslinking).
2156j 24 The Chemistry of 2-Azetidinones (b-Lactams)
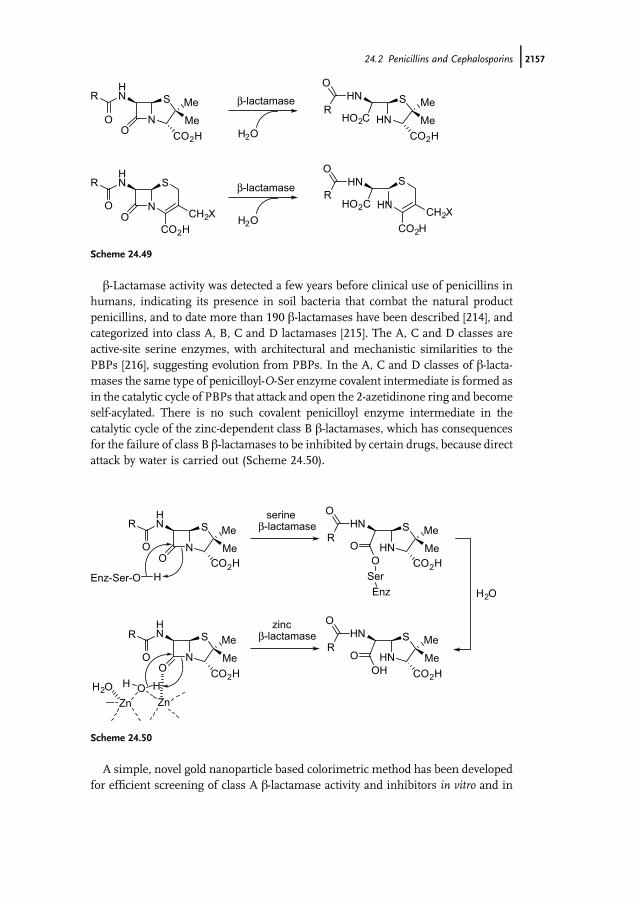
b-Lactamase activity was detected a few years before clinical use of penicillins inhumans, indicating its presence in soil bacteria that combat the natural productpenicillins, and to date more than 190 b-lactamases have been described [214], andcategorized into class A, B, C and D lactamases [215]. The A, C and D classes areactive-site serine enzymes, with architectural and mechanistic similarities to thePBPs [216], suggesting evolution from PBPs. In the A, C and D classes of b-lacta-mases the same type of penicilloyl-O-Ser enzyme covalent intermediate is formed asin the catalytic cycle of PBPs that attack and open the 2-azetidinone ring and becomeself-acylated. There is no such covalent penicilloyl enzyme intermediate in thecatalytic cycle of the zinc-dependent class B b-lactamases, which has consequencesfor the failure of class B b-lactamases to be inhibited by certain drugs, because directattack by water is carried out (Scheme 24.50).
A simple, novel gold nanoparticle based colorimetric method has been developedfor efficient screening of class A b-lactamase activity and inhibitors in vitro and in
NO
HNR
O
S
CO2H
Me
Me
HN
HN S
CO2H
Me
Me
H2O
O
RHO2C
HNO
RHO2C HN
S
CO2H
CH2XN
O
HNR
O
S
CO2H
CH2X
β-lactamase
H2O
β-lactamase
Scheme 24.49
NO
HNR
O
S
CO2HMe
Me
HN
HN S
CO2HMe
Me
O
R
serine β-lactamase
Enz-Ser-O H
OO
Ser
Enz
NO
HNR
O
S
CO2H
Me
Me
HN
HN S
CO2H
Me
Me
O
R
zinc β-lactamase
OOH
O H
ZnZn
H2O
HH2O
Scheme 24.50
24.2 Penicillins and Cephalosporins j2157

bacterial strains [217]. A novel protein labeling system that combines a geneticallymodified, noncatalytic b-lactamase variant and specific mechanism-based fluores-cent probes has also been developed [218].
BcII is a B1 metallo-b-lactamase that is found in both mononuclear and dinuclearforms. Despite very elegant studies, there is still controversy over the nature of theactive BcII species. Anon-steady-state study of the hydrolysis of penicillinG catalyzedby Co(II)-substituted BcII has been carried out, and the modifications occurring atthe active site of the enzyme have been followed.Working at different metal/enzymeratios it has been demonstrated that both mono-Co(II) and di-Co(II) BcII are activemetallo-b-lactamases. In addition, evidence has been presented that during penicillinGhydrolysis catalyzed bymono-Co(II) BcII themetal is localized in theDCHsite (theZn2 site in B1 enzymes) [219].
To probe the role of the Zn(II) sites in metallo-b-lactamase L1, mononuclearmetal ion containing and heterobimetallic analogues of the enzyme have beengenerated and characterized using kinetic and spectroscopic studies. MononuclearZn(II)-containing L1, which binds Zn(II) in the consensus Zn1 site, was shown tobe slightly active; however, this enzyme did not stabilize a nitrocefin-derivedreaction intermediate that had been previously detected. Mononuclear Co(II)- andFe(III)-containing L1 were essentially inactive, and NMR and EPR studies suggestthat these metal ions bind to the consensus Zn2 site in L1. Heterobimetallicanalogues (ZnCo and ZnFe) analogues of L1 have been generated, and stopped-flow kinetic studies revealed that these enzymes rapidly hydrolyze nitrocefin andthat there are large amounts of the reaction intermediate formed during thereaction. These studies demonstrate that the metal ion in the Zn1 site is essentialfor catalysis in L1 and that the metal ion in the Zn2 site is crucial for stabilization ofthe nitrocefin-derived reaction intermediate [220].
24.2.4.5 Conversion of Penicillins into CephalosporinsThe chemical relationship of the penicillin (thiazolidine) and the cephalosporin(dihydrothiazine) skeletons was established in the early 1960s, when it was dem-onstrated that penicillin sulfoxides could be rearranged to form 3-methylatedcephalosporins [221]. Treatment of phenoxymethylpenicillin sulfoxide methyl ester87, which can be obtained from phenoxymethyl penicillin via periodate oxidationfollowed by esterificationwith diazomethane, with a trace of p-toluenesulfonic acid inxylene at reflux temperature gave the cephalosporin derivative 88. A plausiblepathway for this acid-catalyzed ring-expansion involves a sulfoxide elimination tointermediate 89, subsequent addition of the sulfenic acid to the double bondwith thesulfur becoming attached to the primary carbon and the loss of a proton to yield 88(Scheme 24.51).
Eventually, amore efficient processwas developed using silyl protection during thering expansion rearrangement. Silyl protection chemistry has led to efficient chem-ical production of 7-ADCA and has led to highly efficient production of the oralcephalosporins, cephalexin and cephradine. Cephadroxyl is synthesized after silyla-tion of 7-aminocephalosporanic acid (7-ACA) followed by acylation with a mixedanhydride prepared from a salt of p-hydroxyphenylglycine and ethylchloroformate.
2158j 24 The Chemistry of 2-Azetidinones (b-Lactams)

Amoxicillin is synthesized using a similar process. An important Lilly product,cefaclor, involves a ring enlargement of a penicillin V ester to an expanded ceph-alosporin-S oxidewith an exocyclic double bond. The product is a useful intermediatein that it can be converted into 3-substituted cephalosporins and into cefaclor, a highlyprescribed oral cephalosporin with chlorine on the C3 position.
24.2.4.6 Reactions for the Transformation of Functional Groups in Side ChainsThis chapter cannot give a complete overview of all the possible transformations forpenicillins and cephalosporins at a specific position, because of the enormous varietyof reactions involved. We focus instead on some of the most relevant reactions [181,222]. The most important transformation for the amino function in penicillins andcephalosporins is the protection, which is introduced in general as amide by acylatingthe 6-APA or 7-ACA [223]. In a recent contribution, 6-aminopenicillanates 90 havebeen N-acylated in a three-component reaction with an aldehyde and Ph3PCCO togive the corresponding 6-[(E)-20-alkenoyl]amides 91 via a domino addition–Wittigalkenation sequence (Scheme 24.52) [224]. In addition to the amide group, severaldifferent protective groups are used for the transformation of the amino moiety ofpenicillins and cephalosporins. The amine can be converted into a carbamate group,to give the corresponding Boc [225], Cbz [226] or Teoc [227] derivatives. The Daneprotecting group is very suitable for penicillins and cephalosporins, because the
NO
HNR
O
S
CO2Me
Me
Me
O–
+
PhOCH=R 2
87
NO
HNR
O
S
CO2Me
CH3
NO
HNR
O
S
CO2Me
CH3
NO
HNR
O
S
CO2Me
Me
CH2
OH+ H
OH
+
89
NO
HNR
O
S
CO2Me
CH3
88
H2O–H+
NO
HNR
O
S
CO2Me
CH3
H+
H H
OH–H2O
(cat.)PTSA
xylene,
reflux
Scheme 24.51
NO
H2N S
CO2R1Me
Me
90
NO
HN
O
S
CO2R1
Me
Me
91
R2Ph3P=C=C=O,
R2 60THF,CHO, oC
50–77%
Scheme 24.52
24.2 Penicillins and Cephalosporins j2159

proton remaining on the nitrogen is stabilized by hydrogen bonding with the estercarbonyl group, which allows reactions that are normally possible only in doublyprotected derivatives [228]. Double protection of the amino group is desirable inmany operations, particularly with strongly basic reagents, and imines are oftenused [229].
The amino moiety of penicillins and cephalosporins can be transformed intodifferent heteroatomic groups, as shown in Scheme 24.53. 6-Hydroxypenicillanicacid (92) has been synthesized by treatment of a solution of 6-APA in aqueousperchloric acid with sodium nitrite [230], while 6,6-dibromopenicillanic acid 93 hasbeen prepared on reacting 6-APA with bromine [231]. A regiospecific methodologyfor the preparation of penicillate derivatives 6a-(1R-hydroxyoctyl)penicillanic acidand 6b-(1R-hydroxyoctyl)penicillanic acid – which will be valuable tools in theinvestigation of mechanistic and structural details of class D b-lactamases – hasbeen described from 6,6-dibromopenicillanic acid 93 [232]. Esterification of 7-ACAfollowed by sequential treatment with excess triethylamine and trifluoromethane-sulfonic anhydride gives an imine, which can then be hydrolyzed to generate the7-oxo-cephalosporanate 94 [233]. A carbenechromium(0)-containing tether maybe incorporated into penicillin G or cephalotin by using a (bromopropylamino)carbenechromium(0) complex to give metalla-penicillin and-cephalosporin deriva-tives, stable compounds which can be transformed into antibiotic derivatives bearingtripeptide side-chains [234].
6-Aminopenicillanic acid and two of its derivatives, 6-APA benzyl ester andpenicillin G, have been evaluated as catalysts for use in direct cross-aldol reactionsin different solvents and mixtures [235]. A thermal decarbonylation of penamb-lactams has been reported [236]. With the exception of monobactams, a carboxylicacid functiona to theb-lactamnitrogen is essential for good antibacterial activity, andit is nearly always necessary to protect this carboxylic acid function during thepreparation of derivatives. The first esterification of the carboxylic acid of penicillinswas achieved on preparing penicillin G benzhydryl ester [237]. For penicillins and
NO
H2N S
CO2H
Me
Me
NO
Br S
CO2H
Me
Me
93
NO
H2N S
CO2H
NO
S
CO2CHPh2
94
OAc OAc
O
NO
HO S
CO2H
Me
Me
92
BrHClO4 (aq),
NaNO2,
H2 0O, oC
32%
Br2 NaNO, 2,
HN2.5 2SO4
CH2Cl2 5, oC
80%
Phi) 2CN2 CHMeOH,, 2Cl2 RT,
Etii) 3 TfN, 2 CHO, 2Cl2,–78 oC
RT(aq),iii) HCl
40%
Scheme 24.53
2160j 24 The Chemistry of 2-Azetidinones (b-Lactams)

cephalosporins, the most often used protective groups are functionalities that canbe removed under acidic conditions, such as benzhydryl [238], tert-butyl [239] andp-methoxybenzyl [240]. A procedure that has been employed for some time toimprove the absorption of penicillins and cephalosporins after oral administrationis the use of special esters that readily undergo enzymatic hydrolysis in vivo, withliberation of the active drug. These esters, which are mostly bis-acyl derivatives offormaldehyde or acetaldehyde, can also provide protection for carboxylic acidfunctions during derivatization and synthetic transformations [222].
Transformations of 3-acetoxymethyl cephalosporins are of central importance. Thecleavage of the acetyl residue can be carried out both enzymatically [241] andchemically [242] under mild conditions. Oxidation of alcohols obtained in this wayallows preparation of the corresponding 3-formylcephalosporins, which can sufferfurther transformations such as the Wittig olefination [243] or Barbier-type allyla-tion [244]. 3-Halomethyl cephalosporins canbe synthesized via substitution reactionsin 3-acetoxy(hydroxy)methyl cephalosporins [245]. These 3-halo derivatives usuallyact as building blocks for more complex derivatives. For example, the cephalosporinderivative 96, which has been confirmed as a novel fluorogenic substrate for imagingb-lactamase gene expression, has been prepared from the 3-chloromethyl cephalo-sporin 95 (Scheme 24.54) [246]. Apractical route to the preparation of nitrocefin froma 3-chloromethyl cephalosporin related to 95 has been reported using a similarsynthetic sequence [247]. Employing a multivalent approach to drug discovery,vancomycin and cephalosporin synthons have been chemically linked to yieldheterodimer antibiotics, which simultaneously target the principal cellular targetsof both glycopeptides and b-lactams [248].
The sulfur in penicillins and cephalosporins can be oxidized, leading to sulfoxidesor sulfones [224, 231, 246, 249]. This oxidation is usually accomplished for one ofthree reasons: (i) sulfoxide formation to obtain reactive intermediates for furthertransformations; (ii) sulfoxide formation with subsequent reduction in cephemsto shift the double bond from position 2 to position 3; (iii) preparation of sulfones asb-lactamase or elastase inhibitors.
NO
HS
O –
+
OCHPh2O
O O OH
N
H
Bn
ONO
H
96
S
O –
+
OHO
O O OH
N
H
Bn
O
NO
HS
OCHPh2O
PPh3I
HN
H
Bn
ONO
HS
OCHPh2O
Cl
HN
H
Bn
O
95
OHC
O O Oacetone,NaI,i)
RT
PPhii) 3 EtOAc,,
RTNaOH,1Miii)
CH2Cl2 RT,
MCPBA,iv)
CH2Cl2 RT,
anisole,TFA,
CH2Cl2 0, oC
17%
Scheme 24.54
24.2 Penicillins and Cephalosporins j2161

Abbreviations
AIBN a,a0-azoisobutyronitrile7-ACA 7-aminocephalosporanic acid6-APA 6-aminopenicillanic acid (6-APA)Bn benzylBoc tert-butyloxycarbonylBOM benzyloxymethylBTPP phosphazene base P1-t-Bu-tris(tetramethylene)CAN ceric ammonium nitrateCbz benzyloxycarbonylCp cyclopentadienylCSI chlorosulfonyl isocyanateDABCO 1,4-diazabicyclo[2.2.2]octaneDBN 1,5-diazabicyclo[4.3.0]non-5-eneDBU 1,8-diazabicyclo[5.4.0]undec-7-eneDCC N,N0-dicyclohexylcarbodiimideDEAD diethyl azodicarboxylateDIBAL-H (diisobutyl)aluminium hydrideDMAP 4-dimethylaminopyridineDMF dimethylformamideDMSO dimethyl sulfoxidede diastereomeric excessdr diastereomeric ratioee enantiomeric excessFmoc 9-fluorenylmethoxycarbonylHIU high intensity ultrasoundHMDS hexamethyldisilazaneHPLC high-performance liquid chromatographyIR infraredLHMDS lithium hexamethyldisilazaneLDA lithium diisopropylamideMCPBA meta-chloroperoxybenzoic acidMCR multicomponent reactionMOM methoxymethylMM molecular mechanicalMMPP magnesium monoperoxyphthalateMS mass spectrometryNCA N-carboxy anhydrideNMP N-methylpyrrolidoneNMR nuclear magnetic resonancePBP penicillin binding proteinPDC pyridinium dichromatePhth phthalimidoylPMB p-methoxybenzyl
2162j 24 The Chemistry of 2-Azetidinones (b-Lactams)

PMP p-methoxyphenylPPTS pyridinium p-toluenesulfonatePTSA p-toluenesulfonic acidQM quantum mechanicalRCM ring closing metathesisRT room temperatureTBDPS tert-butyldiphenylsilylTBS tert-butyldimethylsilylTES triethylsilylTFA trifluoroacetic acidTHF tetrahydrofuranTIPS triisopropylsilylTEMPO 2,2,6,6-tetramethylpiperidine 1-oxylTs tosyl
References
1 Abboud, J.L.M., Ca~nada, T., Homan, H.,et al. (1992) Journal of the AmericanChemical Society, 114, 4728.
2 Rode, J.E. and Dobrowolski, J.Cz. (2003)Journal of Molecular Structure-THEOCHEM, 637, 81.
3 Rode, J.E. and Dobrowolski, J.Cz. (2003)Journal of Molecular Structure-THEOCHEM, 651, 705.
4 Dobrowolski, J.C., Sadlej, J., andMazurek, A.P. (2003) Journal ofMolecularStructure-THEOCHEM, 638, 229.
5 Pitarch, J., Ruiz-Lopez, M.F., Silla, E.,et al. (1998) Journal of the AmericanChemical Society, 120, 2146.
6 D�ıaz, N., Su�arez, D., Sordo, T.L., et al.(2003) European Journal of OrganicChemistry, 4161.
7 Palomo, C., Aizpurua, J.M., Benito, A.,et al. (2003) Journal of the AmericanChemical Society, 125, 16243.
8 Carr, J.A., Al-Azemi, T.F., Long, T.E., et al.(2003) Tetrahedron, 59, 9147. Bhargava,G., Mahajan, M.P., Saito, T., et al. (2005)European Journal of Organic Chemistry,2397.
9 Kabak, M., Guner, V., Elerman, Y.I., andDurlu, T.N. (2003)Analytical Sciences, 19,969.
10 Cainelli, G., Giacomini, D., Galletti, P.,and Quintavalla, A. (2003)
European Journal of Organic Chemistry,1765.
11 Grigg, R., Thornton-Pett, M., Xu, J., andXu, L.-H. (1999) Tetrahedron, 55, 13841.Alcaide, B., Almendros, P., Alonso, J.M.,et al. (2002) The Journal of OrganicChemistry, 67, 7004. Subramaniyan, G.,Raghunathan, R., and Castro, A.M.M.(2003) Tetrahedron, 59, 335.
12 Alcaide, B., Almendros, P., Luna, A., andTorres,M.R. (2006)The Journal of OrganicChemistry, 71, 4818.
13 Alcaide, B., Almendros, P., Luna, A., andTorres, M. R. (2008) Organic andBiomolecular Chemistry, 6, 1635.
14 Alcaide, B., Almendros, P., Salgado,N.R.,and Rodr�ıguez-Vicente, A. (2000) TheJournal of Organic Chemistry, 65, 4453.Alcaide, B., Aly, M., Rodr�ıguez, C., andRodr�ıguez-Vicente, A. (2000) The Journalof Organic Chemistry, 65, 3453. Ren, X.F.,Turos, E., Lake,C.H., andChurchill,M.R.(1995) The Journal of Organic Chemistry,60, 6468. Wagle, D.R., Garai, C., Chiang,J., et al. (1988) The Journal of OrganicChemistry, 53, 4227.
15 Aoki, H., Sakai, H., Kohsaka, M., et al.(1976) Journal of Antibiotics, 29, 890.Hashimoto, M., Komori, T., and Kamiya,T. (1976) Journal of the American ChemicalSociety, 98, 3023. Gerardin-Charbonnier,
References j2163

C., Auberger, S., Molina, L., et al. (1999)Preparative Biochemistry & Biotechnology,29, 257. Kelly, W.L. and Townsend, C.A.(2005) Journal of Bacteriology, 187, 739.
16 Imada, A., Kitano, K., Kintaka, K., et al.(1981) Nature (London), 289, 590. Sykes,R.B., Cimarusti, C.M., Boner, D.P., et al.(1981) Nature (London), 291, 489. Jones,R.N., Rhomberg, P.R., Varnam, D.J., andMathai, D. (2002) International Journal ofAntimicrobial Agents, 20, 426.Ortega, E.,Escobar, A., Gaforio, J.J., and deCienfuegos, G.A. (2003) In: NewApproaches in the Use of Antibiotics,Research Signpost, Kerala, India,123–147. Ball, A.P., Bartlett, J.G., Craig,W.A., et al. (2004) Journal ofChemotherapy, 16, 419.
17 Sykes, R.B. and Phillips, J. (1981) TheJournal of Antimicrobial Chemotherapy, 8,(Suppl E), 1–141. Muratani, T., Akasaka,S., Kobayashi, T., et al. (2001)Antimicrobial Agents and Chemotherapy,45, 3603. Murray, C.K. and Hospenthal,D.R. (2004) Antimicrobial Agents andChemotherapy, 48, 4002.
18 Kishimoto, S., Sendai, M., Hashiguchi,S., et al. (1983) Journal of Antibiotics, 36,1421. Guanti, G., Riva, R., Cascio, G.,et al. (1998) Farmaco (Societa ChimicaItaliana: 1989), 53, 173.
19 For reviews, see: Veinberg, G., Vorona,M., Shestakova, I., et al. (2003) CurrentMedicinal Chemistry, 10, 1741. Burnett,D.A. (2004) Current Medicinal Chemistry,11, 1873. For a leading reference, see:Rothstein, J.D., Patel, S., Regan, M.R.,et al. (2005) Nature, 433, 73.
20 For a review, see: Clader, J.W. (2004)Journal of Medicinal Chemistry, 47, 1. Fora leading reference, see: Kuaerno, L.,Ritter, T., Werder, M., et al. (2004)AngewandteChemie, International Edition,43, 4653.
21 Stewart, W.W. (1971) Nature, 229, 174.Meek, T.D. and Villafranca, J.V. (1980)Biochemistry, 19, 5513. Unkefer, C.J.,London, R.E., Durbin, R.D., et al. (1987)The Journal of Biological Chemistry, 262,4993. Dolle, R.E., Li, C.S., Novelli, R.,et al. (1992) The Journal of OrganicChemistry, 57, 128. Kiyota, H., Takai, T.,Saitoh, M., et al. (2004) Tetrahedron
Letters, 45, 8191. Kiyota, H., Takai, T.,Shimasaki, Y., Sitoh, M., Nakayama, O.,Takada, T., and Kuwahara, S. (2007)Synthesis, 2471.
22 For reviews, see: Palomo, C., Aizpurua,J.M., Ganboa, I., and Oiarbide, M. (1999)European Journal of Organic Chemistry,3223. Palomo, C., Aizpurua, J.M.,Ganboa, I., and Oiarbide, M. (2004)Current Medicinal Chemistry, 11, 1837.Cossío, F.P., Arrieta, A., and Sierra, M.A.(2008) Accounts of Chemical Research, 41,925; Tidwell, T.T. (2008) AngewandteChemie International Edition, 47, 1016;Fu, N., and Tidwell, T.T. (2008)Tetrahedron, 64, 10465; Xu, J. (2009)Arkivoc, 21; For a recent example, see:(g) Jarrahpor, A., and Zarei, M. (2009)Tetrahedron Letters, 50, 1568.
23 Mart�ın-Zamora, E., Ferrete, A., Llera,J.M., et al. (2004) Chemistry – A EuropeanJournal, 10, 6111.
24 D�ıez, E., Fern�andez, R., Marqu�es-L�opez,E., et al. (2004) Organic Letters, 6, 2749.Fern�andez, R., Ferrete, A., Llera, J.M.,et al. (2004) Chemistry – A EuropeanJournal, 10, 737.
25 For reviews, see: France, S., Weatherwax,A., Taggi, A.E., and Lectka, T. (2004)Accounts of Chemical Research, 37, 592.Paull, D. H., Weatherwax, A., and Lectka,T. (2009)Tetrahedron, 65, 6771. France, S.,Shah, M.H., and Weatherwax, A., et al.(2005) Journal of the American ChemicalSociety, 124, 1578.
26 Zhang, Y.-R., He, L., Wu, X., Shao, P.-L.,and Ye, S. (2008) Organic Letters, 10, 277;Duguet, N., Campbell, C. D., Slawin, A.M. Z., and Smith, A. D. (2008) Organicand Biomolecular Chemistry, 6, 1108;Sereda, O., Blanrue, A., and Wilhelm, R.(2009) Chemical Communications, 1040.
27 Abraham,C.J., Paull,D.H.,Dogo-Isonagie,C., and Lectka, T. (2009) Synlett, 1651;Hodous, B.L., and Fu, G.C. (2002) Journalof the American Chemical Society, 127, 1206.
28 For an overview on solid-phase synthesis,including b-lactam formation, see: Far,A.R. (2003) Angewandte Chemie,International Edition, 42, 2340. Thepolymer-supported and combinatorialsynthesis of b-lactams has been recentlyreviewed: Laborde, M.A. and Mata, E.G.
2164j 24 The Chemistry of 2-Azetidinones (b-Lactams)

(2006) Mini-Reviews in MedicinalChemistry, 6, 109.
29 Ruhland, B., Bhandari, A., Gordon, E.M.,and Gallop, M.A. (1996) Journal of theAmerican Chemical Society, 118, 253.Ruhland, B., Bombrun, A., and Gallop,M.A. (1997) The Journal of OrganicChemistry, 62, 7820.
30 LeRoy, I.,Mouysset,D.,Mignani, S., et al.(2003) Tetrahedron, 59, 3719.
31 Delpiccolo, C.M.L. and Mata, E.G. (2002)Tetrahedron: Asymmetry, 13, 905.
32 Shou, W.-G., Yang, Y.-Y., and Wang, Y.-G.(2005) Synthesis, 530.
33 Singh, R. and Nuss, J.M. (1999)Tetrahedron Letters, 40, 1249.
34 Gordon, K., Bolger, M., Khan, N., andBalasubramanian, S. (2000) TetrahedronLetters, 41, 8621.
35 Donati, D., Morelli, C., Porcheddu, A.,and Taddei, M. (2004) The Journal ofOrganic Chemistry, 69, 9316.
36 Delpiccolo, C.M.L., andMata, E.G. (2004)Tetrahedron Letters, 45, 4085.
37 Hafez, A.M., Taggi, A.E., and Lectka, T.(2002) Chemistry – A European Journal, 8,4115.
38 Hafez, A.M., Taggi, A.E., Drury, W.J., III,and Lectka, T. (2001) Journal of theAmerican Chemical Society, 123, 10853.Taggi, A.E., Hafez, A.M., Wack, H., et al.(2002) Journal of the American ChemicalSociety, 124, 6626.
39 For reviews, see: Georg, G.I. (1989) inNatural Products Chemistry (ed. Atta-ur-Rahman), Elsevier, pp. 431.Hart,D.J. andHa, D.C. (1989) Chemical Reviews, 89,1447.Cainelli, G., Panunzio, M.,Giacomini, D., et al., (1996) in ChemicalSynthesis, Gnosis to Prognosis (eds C.Chatgilialoglu, and V. Snieckus), KluwerAcademic, Amsterdam, pp. 25. Fujisawa,T. and Shimizu, M. (1996) Reviews onHeteroatomChemistry, 15, 203; Brandi, A.,Cicchi, S., and Cordero, F. (2008)Chemical Reviews, 108, 3988.
40 Chen, L., Zhao, G., and Ding, Y. (2003)Tetrahedron Letters, 44, 2611.
41 Ross, N.A.,MacGregor, R.R., and Bartsch,R.A. (2004) Tetrahedron, 60, 2035.
42 Banik, B.K., Ghatak, A., and Becker, F.F.(2000) Journal of the Chemical Society,Perkin Transactions 1, 2179.
43 For a review, see: Benaglia, M.,Cinquini, M., and Cozzi, F. (2000)European Journal of Organic Chemistry,563; Sharma, S.D. and Kanwar, S. (2004)Synlett, 2824.
44 Gembus, V., Poisson, T., Oudeyer, S.,Marsais, F., and Levacher, V. (2009)Synlett, 2437.
45 Hata, S., Iwasawa, T., Iguchi, M., et al.(2004) Synthesis, 1471; Tarui, A., Ozaki,D., Nakajima, N., Yokota, Y., Sokeirik,Y.S., Sato, K., Omote, M., Kumadaki, I.,and Ando, A. (2008) Tetrahedron Letters,49, 3839.
46 Kambara, T., Hussein, M.A., Fujieda,H., et al. (1998) Tetrahedron Letters, 39,9055.
47 Schunk, S. and Enders, D. (2000)OrganicLetters, 2, 907. Schunk, S. and Enders, D.(2002) The Journal of Organic Chemistry,67, 8034.
48 Forro, E. and Fulop, F. (2000)Tetrahedron:Asymmetry, 12, 2351.
49 Lee, K.-I., Yoon, T.-H., Shim, Y.-K., andKim, W.-J. (2000) Bulletin of the ChemicalSociety of Korea, 22, 935.
50 Danh, T.T., Bocian, W., Kozerski, L., et al.(2005) European Journal of OrganicChemistry, 429.
51 For a review, see: Hegedus, L.S. (1997)Tetrahedron, 53, 4105.
52 Sierra, M.A., Manche~no, M.J., Vicente,R., and G�omez-Gallego, M. (2001) TheJournal of Organic Chemistry, 66, 8920;Lage, M.L., Fern�andez, I., Manche~no,M.J., G�omez-Gallego, M., and Sierra,M.A. (2009) Chemistry–A EuropeanJournal, 15, 593.
53 Hegedus, L. S., Imwinkelried, R., Alarid-Sargent, M., et al. (1990) Journal of theAmerican Chemical Society, 112, 1109.Narukawa, Y., Juneau, K.N., Snustad, D.,et al. (1992) The Journal of OrganicChemistry, 57, 5453.
54 Fern�andez, I., Sierra, M.A., Manche~no,M.J., G�omez-Gallego, M., and Coss�ıo,F.P. (2008) Journal of the AmericanChemical Society, 130, 13892.
55 For a discussion on b-amino acids,including b-lactam formation, see:Juaristi, E. (ed.) (1997) EnantioselectiveSynthesis of b-Amino Acids, Wiley–VCHVerlag GmbH, Weinheim. Palomo, C.,
References j2165

Aizpurua, J.M., Ganboa, I., and Oiarbide,M. (2005) Synthesis of b-Amino Acidsand Their Derivatives from b-Lactams:Update in Enantioselective Synthesis ofb-Amino Acids, 2nd edn (eds E. Juaristiand V.A. Soloshonok), John Wiley &Sons, Inc,, Hoboken, NJ, Ch. 20, pp, 477.
56 Lacroix, S., Cheguillaume, A., G�erard, S.,and Marchand-Brynaert, J. (2003)Synthesis, 2483.
57 Vassiliou, S., Dimitropoulos, C., andMagriotis, P.A. (2003) Synlett, 2398.
58 Kende, A.S., Liu, K., Kaldor, I., et al.(1995) Journal of the American ChemicalSociety, 117, 8258. Chen, A., Nelson, A.,Tanikkul, N., and Thomas, E.J. (2001)Tetrahedron Letters, 42, 1251. Brain, C.T.,Chen, A., Nelson, A., et al. (2001)Tetrahedron Letters, 42, 1247.
59 Nishikawa, T., Kajii, S., and Isobe, M.(2004) Chemistry Letters, 33, 440.
60 C�ordova, A., Watanabe, S., Tanaka, F.,et al. (2002) Journal of the AmericanChemical Society, 124, 1866.
61 Chowdari, N.S., Suri, J.F., and Barbas,C.F., III (2004) Organic Letters, 6, 2507.
62 For a review, see: Miller, M.J. (1986)Accounts of Chemical Research, 19, 49.
63 Rajendra, G. and Miller, M.J. (1987) TheJournal of Organic Chemistry, 52, 4471.
64 Romo, D., Rzasa, R.M., Shea, H.A., et al.(1998) Journal of the American ChemicalSociety, 120, 12237; Romo, D., Choi, N.S.,Li, S., et al. (2004) Journal of the AmericanChemical Society, 126, 10582; Walz, A.J.,andMiller,M.J. (2007)Tetrahedron Letters,48, 5103; (d)Hansen, K.B., Hsiao, Y., Xu,F., Rivera, N., Clausen, A., Kubryk, M.,Krska, S., Rosner, T., Simmons, B.,Balsells, J., Ikemoto,N., Sun, Y., Spindler,F., Malan, C., Grabowski, E.J.J., andArmstrongIII, J.D. (2009) Journal of theAmerican Chemical Society, 131, 8798.
65 Kamimura, A., Morita, R., Matsuura, K.,et al. (2003) Tetrahedron, 59, 9931.
66 Meloni, M.M. and Taddei, M. (2001)Organic Letters, 3, 337.
67 For a review, see: Gois, P.M.P. andAfonso, C.A.M. (2004) European Journalof Organic Chemistry, 3773. For recentcontributions, see: Grohmann, M., Buck,S., Sch€affler, L., and Maas, G. (2006)Advanced Synthesis and Catalysis, 348,
2203. Gois, P.M.P. and Afonso, C.A.M.(2003) Tetrahedron Letters, 44, 6571. Gois,P.M.P. and Afonso, C.A.M. (2003)European Journal of Organic Chemistry,3798.
68 Candeias, N.R., Gois, P.M.P., andAfonso, C.A.M. (2005) ChemicalCommunications, 391. Candeias, N.R.,Gois, P.M.P., and Afonso, C.A.M. (2006)The Journal of Organic Chemistry, 71,5489.
69 Choi, M.K.-W., Yu, W.-Y., and Che, C.-M.(2005) Organic Letters, 7, 1081; Choi,M.K.-W., Yu, W.-Y., So, M.-H., Zhou, C.-Y., Deng, Q.-H., and Che, C.-M. (2008)Chemistry–An Asian Journal, 3, 1256.
70 Cheung, W.-H., Zheng, S.-L., Yu, W.-Y.,et al. (2003) Organic Letters, 5, 2535.
71 Arndtsen, B.A. (2009) Chemistry–AEuropean Journal, 15, 302.
72 Kolb, J., Beck, B., and D€omling, A. (2002)Tetrahedron Letters, 43, 6897.
73 Naskar, D., Roy, A., Seibel, W.L., et al.(2003) Tetrahedron Letters, 44, 6297;Pirrung, M.C. and Sarma, K.D. (2004)Journal of the American Chemical Society,126, 444.
74 For reviews, see: Marco-Contelles, J.(2004) Angewandte Chemie, InternationalEdition, 43, 2198; Pal, R., Ghosh, S.C.,Chandra, K., and Basak, A. (2007)Synlett, 2321; For recent contributions,see: Zhao, L. and Li, C.-J. (2006)Asian Journal of Chemistry, 1, 203;Basak, A., Chandra, K., Pal, R., andGhosh, S.C. (2007) Synlett, 1585;McKay, C.S., Kennedy, D.C., andPezacki, J.P. (2009) Tetrahedron Letters,50, 1893.
75 Basak, A., Ghosh, S.C., Bhowmick, T.,et al. (2002) Tetrahedron Letters, 43, 6259;Zhang, X., Hsung, R.P., Li, H., Zhang, Y.,Johnson, W.L., and Figueroa, R. (2008)Organic Letters, 10, 3477.
76 Basak, A. and Ghosh, S.C. (2004) Synlett,1637; Coyne, A.G., M€uller-Bunz, H., andGuiry, P.J. (2007)Tetrahedron: Asymmetry,18, 199.
77 Ye, M.-C., Zhou, J., Huang, Z.-Z., andTang, Y. (2003) ChemicalCommunications, 2554. Ye, M.-C., Zhou,J., and Tang, Y. (2006) The Journal ofOrganic Chemistry, 71, 3576; Saito, T.,
2166j 24 The Chemistry of 2-Azetidinones (b-Lactams)

Kikuchi, T., Tanabe, H., Yahiro, J.,and Otani, T. (2009) Tetrahedron Letters,50, 4969.
78 Lo,M.M.-C. andFu,G.C. (2002) Journal ofthe American Chemical Society, 124, 4572.
79 Natarajan, A.,Wang, K., Ramamurthy, V.,et al. (2002) Organic Letters, 4, 1443.Natarajan, A., Mague, J.T., andRamamurthy, V. (2005) Journal of theAmerican Chemical Society, 127, 3568.
80 Scanian, E.M. and Walton, J.C. (2002)Chemical Communications, 2086.Scanlan, E.M., Slawin, A.M.Z., andWalton, J.C. (2004) Organic andBiomolecular Chemistry, 2, 716. Natarajan,A. and Ramamurthy, V. (2006) Organicand Biomolecular Chemistry, 4, 4533.
81 DiLabio, G.A., Scanlan, E.M., andWalton, J.C. (2005)Organic Letters, 7, 155.
82 Bella, A.F., Jackson, L.V., andWalton, J.C.(2004) Organic and BiomolecularChemistry, 2, 421.
83 Ryu, I., Miyazato, H., Kuriyama, H., et al.(2003) Journal of the American ChemicalSociety, 125, 5632.
84 Lu, S.-M. andAlper,H. (2004)The Journalof Organic Chemistry, 69, 3558; Cariou,C.C., Clarkson, G.J., and Shipman, M.(2008) Journal of Organic Chemistry, 73,9762.
85 Aggarwal, V.K., Alonso, E., Ferrara, M.,and Spey, S.E. (2002) The Journal ofOrganic Chemistry, 67, 2335; Piotti, M.E.and Alper, H. (1996) Journal of theAmerican Chemical Society, 118, 111;Ardura, D., and L�opez, R. (2007) Journalof Organic Chemistry, 72, 3259.
86 Campomames, P., Men�endez, M.I., andSordo, T.L. (2003) The Journal of OrganicChemistry, 68, 6685.
87 Yang, H.W. and Romo, D. (1999) TheJournal of Organic Chemistry, 64, 7657.
88 Avalos, M., Babiano, R., Cintas, P., et al.(1999) Chemical Communications, 1589.Avalos, M., Babiano, R., Cintas, P., et al.(2003) The Journal of Organic Chemistry,68, 6338.
89 Cordero, F.M., Salvati,M., Pisaneschi, F.,and Brandi, A. (2004) European Journalof Organic Chemistry, 2205. Zanobini, A.,Gensini, M., Magull, J., et al. (2004)European Journal of Organic Chemistry,4158; Zanobini, A., Brandi, A., and de
Meijere, A. (2006) European Journal ofOrganic Chemistry, 1251; Jakowiecki, J.,Loska,R., andMakosza,M. (2008) Journalof Organic Chemistry, 73, 5436.
90 Perri, S.T., Slater, S.C., Toske, S.G., andWhite, J.D. (1990) The Journal of OrganicChemistry, 55, 6037.
91 Ashcroft, C.J., Brennan, J., Newall, C.E.,and Roberts, S.M. (1984) TetrahedronLetters, 25, 877. Tanaka, K., Fujiwara, T.,and Urbanczyk-Lipkowska, Z. (2002)Organic Letters, 4, 3255.
92 Gerona-Navarro, G., Garc�ıa-L�opez, M.T.,and Gonz�alez-Mu~niz, R. (2002) TheJournal of Organic Chemistry, 67, 3953.Gerona-Navarro, G., Bonache, M.A.,Reyero, N., et al. (2002) Heterocycles, 56,501.
93 Bonache, M.A., Gerona-Navarro, G.,Garc�ıa-Aparicio, C., et al. (2003)Tetrahedron: Asymmetry, 14, 2161.Bonache, M.A., Gerona-Navarro, G.,Mart�ın-Mart�ınez, M., et al. (2003) Synlett,1007.
94 Shindo, M., Oya, S., Murakami, R., et al.(2000) Tetrahedron Letters, 41, 5943.
95 Clayden, J., Watson, D.W., Helliwell, M.,and Chambers, M. (2003) ChemicalCommunications, 2582.
96 Townes, J.A., Evans, M.A., Queffelec, J.,et al. (2002) Organic Letters, 4, 2537.
97 Troisi, L., De Vitis, L., Granito, C., andEpifani, E. (2004) European Journal ofOrganic Chemistry, 1357; Troisi, L., DeVitis, L., Granito, C., et al. (2004)Tetrahedron, 60, 6895; Troisi, L.,Pindinelli, E., Strusi, V., and Trinchera, P.(2009) Tetrahedron: Asymmetry, 20, 368.
98 Ma, S., Wu, B., and Jiang, X. (2005) TheJournal of Organic Chemistry, 70, 2588.Ma, S., Jiang, X., Cheng, X., and Hou, H.(2006) Advanced Synthesis and Catalysis,348, 2114.
99 Feroci, M. (2007) Advanced Synthesis andCatalysis, 349, 2177; Feroci, M.,Chiarotto, I., Orsini, M., Sotgiu, G., andInesi, A. (2008) Advanced Synthesis andCatalysis, 350, 1355.
100 Fustero, S., Fern�andez, B., Bello, P., delPozo, C., Arimitsu, S., andHammond,G.B. (2007)Organic Letters, 9,4251; Zhao, Q., and Li, C. (2008) OrganicLetters, 10, 4037.
References j2167

101 Nakamura, I., Araki, T., and Terada, M.(2009) Journal of the American ChemicalSociety, 131, 2804.
102 Hachiya, I., Yoshitomi, T., Yamaguchi, Y.,and Shimizu, M. (2009) Organic Letters,11, 3266.
103 Lee, Y.-S., Choung,W.-K., Kim,K.H., et al.(2004) Tetrahedron, 60, 867.
104 Cainelli, G., Giacomini, D., Galletti, P.,and Quintavalla, A. (2003) EuropeanJournal of Organic Chemistry, 1765.
105 Lee, K., and Lee P.H. (2007) Chemistry–AEuropean Journal, 13, 8877.
106 Yu, H., and Lee P.H. (2008) Journal ofOrganic Chemistry, 73, 5183.
107 Lee, P.H., Kim, H., Lee, K., et al. (2005)Angewandte Chemie, International Edition,44, 1840.
108 Cardillo, G., Fabbroni, S., Gentilucci, L.,et al. (2005) Organic Letters, 7, 533.
109 Jayaraman, M., Manhas, M.S., and Bose,A.K. (1997) Tetrahedron Letters, 38, 709.Paquette, L.A., Rothhaar, R.R., Isaac, M.,et al. (1998) The Journal of OrganicChemistry, 63, 5463; Alcaide, B.,Almendros, P., Aragoncillo, C., Cabrero,G., Callejo, and Ruiz, M.P. (2008)European Journal of Organic Chemistry,4434.
110 Alcaide, B., Almendros, P., Aragoncillo,C., and Rodr�ıguez-Acebes, R. (2004) TheJournal of Organic Chemistry, 69, 826.
111 Alcaide, B., Almendros, P., andRodr�ıguez-Acebes, R. (2002) The Journalof Organic Chemistry, 67, 1925.
112 Alcaide, B., Almendros, P., andAragoncillo, C. (2000) Organic Letters, 2,1411. Alcaide, B., Almendros, P.,Aragoncillo, C., and Rodr�ıguez-Acebes,R. (2001)The Journal ofOrganic Chemistry,66, 5208.
113 Alcaide, B., Almendros, P., and Luna, A.(2009) Tetrahedron, 63, 3102.
114 Dejaegher, Y., Denolf, B., Stevens, C.V.,and De Kimpe, N. (2005) Synthesis, 193.
115 Palomo, C., Aizpurua, J.M., Ganboa, I.,et al. (2004) Organic Letters, 6, 4443.
116 Palomo, C., Aizpurua, J.M., Benito, A.,et al. (2003) Journal of the AmericanChemical Society, 125, 16243; Aizpurua,J.M., Palomo, C., Fratila, R.M., Ferr�on, P.,Benito, A., G�omez-Bengoa, E., Miranda,
J.I., and Santos, J.I. (2009) Journal ofOrganic Chemistry, 74, 6691.
117 Benfatti, F., Cardillo, G., Fabbroni, S.,et al. (2005) Synthesis, 61.
118 Barrett, A.G.M., Ahmed, M., Baker, S.P.,et al. (2000) The Journal of OrganicChemistry, 65, 3716; Clemente, A.,Domingos, A., Grancho, A.P., et al. (2001)Bioorganic & Medicinal Chemistry Letters,11, 1065; Ojima, I., Geng, X., Lin, S., et al.(2002) Bioorganic & Medicinal ChemistryLetters, 12, 349. Hitchcock, P.B.,Papadopoulos, K., and Young, D.W.(2003) Organic and BiomolecularChemistry, 1, 2670.
119 Banfi, L., Guanti, G., and Rasparini, M.(2003) European Journal of OrganicChemistry, 1319.
120 Cainelli, G., Giacomini, D., Gazzano, M.,et al. (2003) Tetrahedron Letters, 44, 6269.
121 Forro, E. and Fulop, F. (2000)Tetrahedron:Asymmetry, 12, 2351. Penfold, D.J., Pike,K., Genge, A., et al. (2000) TetrahedronLetters, 41, 10347.
122 Klapars, A., Huang, X., and Buchwald,S.L. (2002) Journal of the AmericanChemical Society, 124, 7421; Jiang, L., Job,G.E., Klapars, A., and Buchwald, S.L.(2003) Organic Letters, 5, 3667. Sun, C.,Camp, J.E., and Weinreb, S.M. (2006)Organic Letters, 8, 1779.
123 Nishikawa, T., Kajii, S., and Isobe, M.(2004) Chemistry Letters, 33, 440.
124 Buynak, J.D., Rao, M.N., Pajouhesh, H.,et al. (1985) The Journal of OrganicChemistry, 50, 4245.
125 Alcaide, B., Rodr�ıguez-Vicente, A., andSierra,M.A. (1998)Tetrahedron Letters, 39,163.
126 Manhas, M.S., Khayavi, M.S., Bari, S.S.,and Bose, A.K. (1983) Tetrahedron Letters,24, 2323. Sacripante, G. and Just, G.(1987) The Journal of Organic Chemistry,52, 3659.
127 Montermini, F., Lacote, E., and Malacria,M. (2004) Organic Letters, 6, 921.
128 Sammes, P. and Smith, S. (1984) Journalof the Chemical Society, Perkin Transactions1, 2415.
129 De Kimpe, N., Tehrani, K.A., and Fonck,G. (1996) The Journal of OrganicChemistry, 61, 6500.
2168j 24 The Chemistry of 2-Azetidinones (b-Lactams)

130 D�hooghe, M., Van Brabandt, W.,Dekeukeleire, S., Dejaegher, Y., andDe Kimpe, N. (2008) Chemistry–AEuropean Journal, 14, 6336.
131 D�hooghe, M., Dekeukeleire, S., Mollet,K., Lategan, C., Smith, P.J., Chibale, K.,and De Kimpe, N. (2008) Journal ofMedicinal Chemistry, 52, 4058.
132 Yamashita, M. and Ojima, I. (1983)Journal of the American Chemical Society,105, 6339.
133 Ojima, I., Zhao, M., Yamato, T., et al.(1991) The Journal of Organic Chemistry,56, 5263. Alcaide, B., Almendros, P.,Aragoncillo, C., and Salgado, N.R. (1999)The Journal ofOrganicChemistry, 64, 9596.
134 Gerona-Navarro, G., Bonache, M.A.,Al�ıas, M., et al. (2004) Tetrahedron Letters,45, 2193.
135 Kawabata, T., Itoh, K., and Hiyama, T.(1989) Tetrahedron Letters, 30, 4837.
136 Chiba, T. and Nakai, T. (1985) TetrahedronLetters, 26, 4647.
137 Bose, A.K., Narayanan,C.S., andManhas,M.S. (1970) Journal of theChemical Society,Chemical Communications, 975. Wagle,D.R.,Garai, C., Chiang, J., et al. (1988)TheJournal of Organic Chemistry, 53, 4227.
138 Hart, D.J. and Ha, D.-Ch. (1985)Tetrahedron Letters, 26, 5493.
139 Alcaide, B., Dom�ınguez, G., Escobar, G.,et al. (1986) Heterocycles, 24, 1579.
140 Alcaide, B., Almendros, P., Salgado, N.R.,and Rodr�ıguez-Vicente, A. (2000) TheJournal of Organic Chemistry, 65, 4453.
141 For selected recent reviews, see: Alcaide,B., Almendros, P., and Aragoncillo, C.(2009) Chemical Reviews, 107, 4437;Alcaide, B., and Almendros, P. (2004)Current Medicinal Chemistry, 11, 1921;Deshmukh, A.R.A.S., Bhawal, B.M.,Krishnaswamy, D., Govande, V.V.,Shinkre, B.A., and Jayanthi, A. (2004)Current Medicinal Chemistry, 11, 1889.
142 Suffness, M. (1995) Taxol Science andApplications, CRC Press, Boca Raton, FL,USA; For a recent example, see: Ge, H.,Spletstoser, J.T., Yang, Y., Kayser, M., andGeorg, G.I. (2007) Journal of OrganicChemistry, 72, 756.
143 For reviews, see: Palomo, C., Aizpurua,J.M., Ganboa, I., and Oiarbide, M. (2001)Synlett, 1813. Palomo, C., Aizpurua, J.M.,
Ganboa, I., and Oiarbide, M. (1999)Amino Acids, 16, 321.
144 Alcaide, B., Almendros, P., andAragoncillo, C. (2000) ChemicalCommunications, 757. Alcaide, B.,Almendros, P., andAragoncillo, C. (2002)Chemistry – A European Journal, 8, 3646.
145 For reviews, see: Ojima, I. and Delaloge,F. (1997) Chemical Society Reviews, 26,377. Ojima, I. (1995) Advances inAsymmetric Syntheses, 1, 95.
146 Alcaide, B., Almendros, P., and Redondo,M.C. (2004) Organic Letters, 6, 1765;Alcaide, B., Almendros, P., andRedondo,M.C. (2007)European Journal ofOrganic Chemistry, 3707.
147 Alcaide, B., Almendros, P., Cabrero, G.,and Ruiz, M.P. (2005) Organic Letters, 7,3981.
148 Alcaide, B., Almendros, P., Cabrero, G.,and Ruiz, M.P. (2007) ChemicalCommunications, 4788.
149 Domingo, L.R., Aurell,M.J., andArn�o,M.(2009) Tetrahedron, 65, 3432.
150 Alcaide, B., Almendros, P., Alonso, J.M.,and Aly, M.F. (2003) Chemistry – AEuropean Journal, 9, 3415. Alcaide, B.,Almendros, P., and Alonso, J.M.(2003) Chemistry – A European Journal,9, 5793.
151 Klapars, A., Parris, S., Anderson, K.W.,and Buchwald, S.L. (2004) Journal of theAmerican Chemical Society, 126, 3529.
152 Alcaide, B., Almendros, P., Cabrero, G.,and Ruiz, M.P. (2007) Journal of OrganicChemistry, 72, 7980.
153 Alcaide, B., Almendros, P., Cabrero, G.,and Ruiz, M.P. (2008) ChemicalCommunications, 615.
154 Alcaide, B., Almendros, P., and Redondo,M.C. (2006) Chemical Communications,2616; Alcaide, B., Almendros, P.,Carrascosa, R., andRedondo,M.C. (2008)Chemistry–A European Journal, 14, 637.
155 Kale, A.S., Puranik, V. G., andDeshmukh, A.R.A.S. (2007) Synthesis,1159.
156 Williams, R.M., and Vincent, G. (2007)Angewandte Chemie International Edition,46, 1517.
157 Movassaghi, M., Tjandra, M., and Qi, J.(2009) Journal of the American ChemicalSociety, 131, 9648.
References j2169

158 Cavagna, F., Linkies, A., Pietsch, H., andReuschling, D. (1980) AngewandteChemie, International Edition, 19, 129.Palomo, C., Aizpurua, J.M., Cuevas, C.,et al. (1996) Anales de QuimicaInternational Edition, 92, 134. Alcaide, B.,Almendros, P., Alonso, J.M., and Aly,M.F. (2000) Chemical Communications,485. Alcaide, B., Almendros, P., Alonso,J.M., and Aly, M.F. (2001) The Journalof Organic Chemistry, 66, 1351.
159 Jacobsen, M.F., Turks, M., Hazell, R.,and Skrydstrup, T. (2002) The Journalof Organic Chemistry, 67, 2411.
160 Alajar�ın, M., Vidal, A., S�anchez-Andrada,S., et al. (2000) Organic Letters, 2, 965;Alajar�ın, M., S�anchez-Andrada, S.,Coss�ıo, F.P., et al. (2001) The Journal ofOrganic Chemistry, 68, 8470; Cainelli, G.,Giacomini, D., Gazzano, M., et al. (2003)Tetrahedron Letters, 44, 6269;
161 Cutchins, W.W. and McDonald, F.E.(2002) Organic Letters, 4, 749.
162 Cheung, L.L.W., and Yudin, A.K. (2009)Organic Letters, 11, 1281.
163 Dekeukeleire, S., D�hooghe, M., and DeKimpe, N. (2009) Journal of OrganicChemistry, 74, 11644.
164 Crombie, L., Jones, R.C.F.,Mat-Zin, A.R.,and Osborne, S. (1983) Journal of theChemical Society. ChemicalCommunications, 960; Alcaide, B.,Rodr�ıguez-Ranera, C., and Rodr�ıguez-Vicente, A. (2001) Tetrahedron Letters, 42,3081; Vasudevan, A., Villamil, C.I., andDjuric, S.W. (2004) Organic Letters, 6,3361.
165 Betschart, C. and Hegedus, L.S. (1992)Journal of the American Chemical Society,114, 5010; Ojima, I., Lin, S., Inoue, T.,Miller, M.L., et al. (2000) Journal of theAmerican Chemical Society, 122, 5343;Chen, A., Nelson, A., Tanikkul, N., andThomas, E.J. (2001) Tetrahedron Letters,42, 1251; Wasserman, H.H., Matsuyama,H., and Robinson, R.P. (2002)Tetrahedron, 58, 7177; Vidya, R., Eggen,M.J., Nair, S.V., et al. (2003) The Journalof Organic Chemistry, 68, 9687; Geng, X.,Miller, M.L., Lin, S., and Ojima, I. (2003)Organic Letters, 5, 3733; Romo, D., Choi,N.S., Li, S., et al. (2004) Journal of theAmerican Chemical Society, 126, 10582.
166 Tufariello, J.J., Pinto, D.J.P., Milowsky,A.S., and Reinhardt, D.V. (1987)Tetrahedron Letters, 28, 5481; Alcaide, B.,Esteban, G., Mart�ın-Cantalejo, Y., et al.(1994) The Journal of Organic Chemistry,59, 7994.
167 Alcaide, B., Almendros, P., andRodr�ıguez-Salgado, N. (2001) TetrahedronLetters, 42, 1503.
168 Alcaide, B., Almendros, P., Aragoncillo,C., and Redondo, M.C. (2002) ChemicalCommunications, 1472; Alcaide, B.,Almendros, P., Aragoncillo, C., andRedondo, M.C. (2005) European Journalof Organic Chemistry, 98.
169 Alcaide, B. and Almendros, P. (1999)Tetrahedron Letters, 40, 1015; Alcaide, B.,Almendros, P., and Rodr�ıguez-Salgado,N. (2000) The Journal of OrganicChemistry, 65, 3310.
170 Yamashita, T., Tokuyama, H., andFukuyama, T. (2003) Synlett, 738.
171 Maier, T.C. and Podlech, J. (2004)European Journal of Organic Chemistry,4379.
172 Liang, Y., Raju, R., Le, T., Taylor, C.D., andHowell, A.R. (2009) Tetrahedron Letters,50, 1020.
173 Bhargava, G., Anand, A., Mahajan, M.P.,Saito, T., Sakai, K., and Medhi, C. (2008)Tetrahedron, 64, 6801.
174 Cainelli, G., Galletti, P., Giacomini, D.,Licciulli, S., and Quintavalla, A. (2007)European Journal of Organic Chemistry,256; Galletti, P., Quintavalla, A.,Ventrici, C., and Giacomini, D. (2009)European Journal of Organic Chemistry,4541.
175 Alcaide, B., Almendros, P., and Mart�ınezdel Campo, T. (2007) European Journal ofOrganic Chemistry, 2844.
176 Alcaide, B., Polanco, C., and Sierra,M. (1998) Synlett, 416; Alcaide, B.,Polanco, C., and Sierra, M.A. (1998)European Journal of OrganicChemistry, 416.
177 Fern�andez, R., Ferrete, A., Llera, J.M.,et al. (2004) Chemistry – A EuropeanJournal, 10, 737.
178 Alcaide, B., Almendros, P., and Alonso,J.M. (2003) Tetrahedron Letters, 44, 8693.
179 Jarrahpour, A., and Zarei, M. (2008)Synlett, 381.
2170j 24 The Chemistry of 2-Azetidinones (b-Lactams)

180 Ruano, G., Grande, M., and Anaya, J.(2002) The Journal of Organic Chemistry,67, 8243.
181 For recent comprehensive reviews onpenicillins and cefalosphorins, see:Marchand-Brynaert, J., Brul�e, C. (2008) inComprehensive Heterocyclic Chemistry IIIVol. 2, (eds A.R. Katritzky, C.A.Ramsden, E.F.V., Scriven, and R. Taylor),Elsevier, Oxford, pp 173–238; Alcaide, B.,Almendros, P., andAragoncillo, C. (2008)in Comprehensive Heterocyclic ChemistryIII Vol. 2, (eds A.R. Katritzky, C.A.Ramsden, E.F.V., Scriven, and R. Taylor),Elsevier, Oxford, pp 111–172.
182 Fisher, J.F., Meroueh, S.O., andMobashery, S. (2005) Chemical Reviews,105, 395; Birck, C., Cha, J.Y., Cross, J.,et al. (2004) Journal of the AmericanChemical Society, 126, 13945;Walsh, C.(2003) Antibiotics: Actions, Origins,Resistance, ASM Press; Mascaretti, O.A.(2003)Bacteria versus Antibacterial Agents,ASM Press, Washington, DC;Sandanayaka, V.P. and Prashad, A.S.(2002) Current Medicinal Chemistry, 9,1145; Walsh, C. (2000) Nature, 406, 775;Page,M.I. and Laws, A.P. (1998)ChemicalCommunications, 1609; Niccolai, D.,Tarsi, L., and Thomas, R.J. (1997)Chemical Communications, 2333; Hook,V. (1997) Chemistry in Britain, 33, 34;Spratt, B.G. (1994) Science, 264, 388;Davies, J. (1994) Science, 264, 375;Neuhaus, F.C. and Georgeopapadakou,N.H. (1992) in Emerging Targets inAntibacterial and Antifungal Chemoterapy(eds J. Sutcliffe and N.H.Georgeopapadakou), Chapman & Hall,New York.
183 Cabri, W. and Di Fabio, R. (2000)Sanfetrinems in From Bench to Market: TheEvolution of Chemical Synthesis, OxfordUniversity Press, pp. 72–96; Kanno, O.and Kawamoto, I. (2000) Tetrahedron, 56,5639; Hanessian, S. and Reddy, B. (1999)Tetrahedron, 55, 3427; Biondi, S.,Pecunioso, A., Busi, F., et al. (2000)Tetrahedron, 56, 5649;Ghiron, C. andRossi, T. (1997) The chemistry of trinemsin Targets in Heterocyclic Systems-Chemistry and Properties, Vol. 1 (eds O.A.Attanasi and D. Spinelli), Societa
Chimica Italiana, Rome, pp. 161–186;Ngo, J. and Casta~ner, J. (1996) Drugs ofthe Future, 21, 1238.
184 Brown, A.G., Butterworth, D., Cole, M.,et al. (1976) Journal of Antibiotics, 29, 668.
185 Neu, H.C. (1983) Reviews of InfectiousDiseases, 5 (Suppl 2), 319.
186 Wagner, A., Flaig, R., Dittrich, B., et al.(2004)Chemistry–AEuropean Journal, 10,2977.
187 D�ıaz, N., Su�arez, D., and Sordo, T.L.(2003) Journal of ComputationalChemistry, 24, 1864.
188 Hermann, J.C., Hensen, C., Ridder, L.,et al. (2005) Journal of the AmericanChemical Society, 127, 4454.
189 Dal Peraro, M., Llarrull, L.I.,Rothlisberger, U., et al. (2004) Journalof the American Chemical Society, 126,12661.
190 Virudachalam, R., and Rao, V.S.R. (2009)International Journal of Peptide and ProteinResearch, 10, 51.
191 Page, M.I. (1992) in The Chemistry ofb-Lactams (ed. M.I. Page), BlackieAcademic & Professional, London,pp. 79–100.
192 Doyle, F.P., Long, A.A.W., Naylor, J.H.C.,and Stove, E.R. (1961) Nature, 192, 1183.
193 Acred, P., Brown, D.M., Knudsen, E.T.,et al. (1967) Nature, 215, 66. 166.
194 Sweet, R.M. (1972) Cephalosporins andPenicillins, Chemistry and Biology, (ed.E.H. Flynn), Academic Press, New York,p. 280; Dunn, G.L., (1984) inComprehensive Heterocyclic Chemistry(ed. W. Lwowski), Pergamon Press,New York, Vol. 7, pp. 341–363.
195 L�opez, M.A., Rodr�ıguez, Z., Gonz�alez,M., Tol�on, B., Avila, R., Rodr�ıguez, J.C.,V�elez-Castro, H., and Fininueva, A.(2007) Magnetic Resonance in Chemistry,45, 236.
196 Sheehan, J.C. and Henery-Logan, K.R.(1957) Journal of the American ChemicalSociety, 79, 1262. Sheehan, J.C. andHenery-Logan, K.R. (1959) Journal ofthe American Chemical Society, 81, 3089.
197 Woodward, R.B., Heusler, K., Gosteli, J.,et al. (1966) Journal of the AmericanChemical Society, 88, 852.
198 Elander, R.P. (2003) Applied Microbiologyand Biotechnology, 61, 385.
References j2171

199 Waites, M.J., Morgan, N.L., Rockey, J.S.,and Higton, G. (2001) IndustrialMicrobiology: An Introduction, Blackwell,Oxford, UK.
200 Van Nistelrooij, M., Krijgsman, J.,DeVroom, E., and Oldenhof, C. (1998)Penicillin update in Penicillin: a Paradigmfor Biotechnology (ed. R.I. Mateles),Candida, Chicago, pp. 85–91.
201 Usher, J.J., Lewis, M.A., Hughes, D.W.,and Compton, B.J. (1988) BiotechnologyLetters, 10, 543.
202 Lund, F. and Tybring, L. (1977) Nature,236, 135.
203 Lund, F.J. (1977) in Recent Advances inthe Chemistry of b-Lactam Antibiotics (ed.J. Elks), Royal Society of Chemistry,London, pp. 24–45.
204 Ruddle, C.C., and Smyth, T.P. (2007)OrganicandBiomolecularChemistry,5, 160.
205 Faraci, W.S. and Pratt, R.F. (1986) Journalof the American Chemical Society, 108,5328. Buckwell, S.C., Page, M.I.,Longridge, J.L., and Waley, S.G. (1988)Journal of the Chemical Society-PerkinTransactions 2, 1823.
206 Proctor, P., Gensmantel, N.P., and Page,M.I. (1982) Journal of the Chemical Society-Perkin Transactions 2, 1185.
207 L�opez-Gallego, F., Betancor, L., Sio, C.F.,Reis, C.R., Jim�enez, P.N., Guisan, J.M.,Quax, W.J., and Fern�andez-Lafuente, R.(2008) Advanced Synthesis and Catalysis,350, 343.
208 Gensmantel, N.P. and Page, M.I. (1978)Journal of the Chemical Society. ChemicalCommunications, 374; Martin, A.F.,Morris, J.J., and Page, M.I. (1979) Journalof the Chemical Society. ChemicalCommunications, 298; Morris, J.J. andPage, M.I. (1980) Journal of the ChemicalSociety-Perkin Transactions 2, 212; Morris,J.J. and Page, M.I. (1980) Journal of theChemical Society-Perkin Transactions 2,220; Gensmantel, N.P. and Page, M.I.(1982) Journal of the Chemical Society-Perkin Transactions 2, 137.
209 Davis, A.M., Proctor, P., and Page, M.I.(1991) Journal of the Chemical Society-Perkin Transactions 2, 1213.
210 Llin�as, A., Donoso, J., Vilanova, B., et al.(2000) Journal of the Chemical Society-Perkin Transactions 2, 1521.
211 Llin�as, A., Vilanova, B., and Page, M.I.(2004) Journal of Physical OrganicChemistry, 17, 521.
212 Tsang, W.Y., Ahmed, N., and Page, M.I.(2007) Organic and BiomolecularChemistry, 5, 485.
213 Llin�as, A. and Page, M.I. (2004) Organicand Biomolecular Chemistry, 2, 651.
214 Thomson, K.S. and Moland, E.S. (2000)Microbes and Infection/Institut Pasteur, 2,1225.
215 Jacoby, G.A. (2006) Antimicrobial Agentsand Chemotherapy, 50, 1123. Bush, K. andMobashery, S. (1998) Advances inExperimental Medicine and Biology,456, 71.
216 Knox, J.R. (1995)Antimicrobial Agents andChemotherapy, 39, 2593. Knox, J.R.,Moews, P.C., and Frere, J.M. (1996)Chemistry & Biology, 3, 937.
217 Jiang, T., Liu, R., Huang, X., Feng, H.,Teo, W., and Xing, B. (2009) ChemicalCommunications, 1972.
218 Mizukami, S., Watanabe, S., Hori, Y., andKikuchi, K. (2009) Journal of the AmericanChemical Society, 131, 5016.
219 Llarrull, L.I., Tioni, M.F., and Vila, A.J.(2008) Journal of the American ChemicalSociety, 130, 15842.
220 Hu, Z., Periyannan, G., Bennett, B., andCrowder, M.W. (2008) Journal of theAmerican Chemical Society, 130, 14207.
221 Morin, R.B., Jackson, B.G., Mueller, R.A.,et al. (1963) Journal of the AmericanChemical Society, 85, 1896.
222 For more details, see: Georg, G.I. (ed.)(1992)The Organic Chemistry of b-lactams,VCH Publishers. Zhang, T.Y. andHatfield, L.D., (1996) in ComprehensiveHeterocyclic Chemistry, 2nd edn, Vol. 1B(eds A.R. Katritzky, C.W. Rees, and E.F.V.Scriven) Elsevier, Oxford, pp. 591.
223 Hatfield, L.D., Lunn, W.H.W., Jackson,B.G., et al. (1981) inRecent Advances in theChemistry of b-Lactam Antibiotics (ed. G.I.Gregory), Special Publication No. 38,Royal Society of Chemistry, pp. 109.Newall, C.E.in (1985) Recent Advances inthe Chemistry of b-Lactam Antibiotics (edsA.G. Brown and S.M. Roberts), SpecialPublication No. 52, Royal Society ofChemistry, London, p. 1; Huang, H.T.,English, A.R., Seto, T.A., et al. (1960)
2172j 24 The Chemistry of 2-Azetidinones (b-Lactams)

Journal of the American Chemical Society,82, 3790.
224 Schobert, R. and Stangl, A. (2005)Tetrahedron Letters, 46, 1127.
225 Sheehan, J.C., Buku, A., Chacko, E., et al.(1977) The Journal of Organic Chemistry,42, 4045. Tsuji, T., Kataoka, T., Yoshioka,M., et al. (1979) Tetrahedron Letters, 2793.
226 Kurita, A., Atarashi, S., Hattori, K., andTakano, T. (1966)The Journal of AntibioticsSeries A, 19, 243.
227 Corbett, D.F., Frydrych, C.H., Southgate,R., and Basker, M.J. (1990) Journal ofAntibiotics, 43, 1042.
228 Dane, E. and Dockner, T. (1965)Chemische Berichte, 98, 789; Manhas,M.S., Gala, K., Bari, S.A., and Bose, A.K.(1983) Synthesis, 649.
229 Cama, L.D. and Christensen, B.G. (1973)Tetrahedron Letters, 3505. Heuser, L.J.,Anderson, C.F., Applegate, H.E., et al.(1974) The Journal of Organic Chemistry,39, 3929. Grant, J.W. and Smyth, T.P.(2004) The Journal of Organic Chemistry,69, 7965.
230 Buynak, J.D., Borate, H.B., Lamb, G.W.,et al. (1993) The Journal of OrganicChemistry, 58, 1325.
231 Volkmann, R.A., Carroll, R.D., Drolet,R.B., et al. (1982) The Journal of OrganicChemistry, 47, 3344; Buynak, J.D.,Ghadachanda, V.R., Vogeti, L., et al.(2005) The Journal of Organic Chemistry,70, 4510.
232 Testero, S.A., O�Daniel, P.I., Shi, Q., Lee,M., Hesek, D., Ishiwata, A., Noll, B.C.,and Mobashery, S. (2009) Organic Letters,11, 2515.
233 Buynak, J.D., Khasnis, D., Bachmann, B.,et al. (1994) Journal of the AmericanChemical Society, 116, 10955.
234 Sierra, M.A., Rodr�ıguez-Fern�andez, M.,Casarrubios, L., G�omez-Gallego, M., andManche~no, M.J. (2009) European Journalof Organic Chemistry, 2998.
235 Emer, E., Galletti, P., and Giacomini, D.(2009) European Journal of OrganicChemistry, 3155.
236 Wiitala, K.W., Tian, Z., Cramer, C.J., andHoye, T.R. (2008) Journal of OrganicChemistry, 73, 3024.
237 Meyer, K., Hobby, G.L., and Chaffee, E.(1943) Science, 97, 205.
238 Kametani, T., Sekine, H., and Honda, T.(1982) Chemical & PharmaceuticalBulletin, 30, 4545; Yokoo, C., Goi, M.,Onodera, A., et al. (1988) Journal ofAntibiotics, 41, 181; Torii, S., Tanaka, H.,Taniguchi, M., et al. (1991) The Journalof Organic Chemistry, 56, 3633.
239 Takasugi, H., Kochi, H., Masugi, T., et al.(1983) Journal of Antibiotics, 36, 846.Guest, A.W., Branch, C.L., Finch, S.C.,et al. (1987) Journal of the Chemical Society,Perkin Transactions 1, 45.
240 Iwamatsu, K., Atsumi, K., Sakagami,K., et al. (1990) Journal of Antibiotics, 43,1450.
241 Heyninge, E.V. (1965) Journal ofMedicinal Chemistry, 8, 22; Peter, H. andBickel, H. (1974)Helvetica Chimica Acta,57, 2044.
242 Mobashery, S. and Johnston, M. (1986)Tetrahedron Letters, 27, 3333; Murakami,M., Aoki, T., and Nagata, W. (1990)Heterocycles, 30, 567; Koyama, Y., Huang,S.-P., Ikeda, D., and Kondo, S. (1990)Synlett, 389.
243 Naito, T., Hoshi, H., Aburaki, S., et al.(1987) Journal of Antibiotics, 40, 991;Kamachi, H., Oka, M., Narita, Y., et al.(1990) Journal of Antibiotics, 43, 533.
244 Keltjens, R., Vadivel, S.K., de Gelder, R.,et al. (2003) European Journal of OrganicChemistry, 1749.
245 Yamanaka, H., Chiba, T., Kawabata, K.,et al. (1985) Journal of Antibiotics, 38, 1738;Bonjouklian, R. and Philips, M.L. (1981)Tetrahedron Letters, 3915.
246 Gao, W., Xing, B., Tsien, R.Y., and Rao, J.(2003) Journal of the American ChemicalSociety, 125, 11146.
247 Lee, M., Hesek, D., and Mobashery, S.(2005) The Journal of Organic Chemistry,70, 367.
248 Long, D.D., Aggen, J.B., Christensen,B.G., Judice, J.K., Hegde, S.S., Kaniga, K.,Krause, K.M., Linsell, M.S., Moran, E.J.,and Pace, J.L. (2008) Journal of Antibiotics,61, 595.
249 Micetich, R.G., Singh, R., and Maiti, S.N.(1984) Heterocycles, 22, 531. Micetich,R.G., Singh, R., and Shaw, C. (1986)The Journal ofOrganicChemistry, 51, 1811.Tanaka,M., Konoike, T., and Yoshioka,M.(1989) Synthesis, 197.
References j2173










![Supporting Information - Royal Society of Chemistry · Supporting Information N-Heterocyclic Carbene-Catalyzed [3+2] Annulation of Bromoenals with 3-Aminooxindoles: Highly Enantioselective](https://static.fdocument.org/doc/165x107/5f0dee5b7e708231d43cc95a/supporting-information-royal-society-of-supporting-information-n-heterocyclic.jpg)







