
Misfolding proteico2015
99
Ruolo del misfolding proteico nelle patologie neurodegenerative Ruolo del misfolding proteico nelle patologie neurodegenerative
Transcript of Misfolding proteico2015

Ruolo del misfoldingproteico nelle patologie
neurodegenerative
Ruolo del misfoldingproteico nelle patologie
neurodegenerative
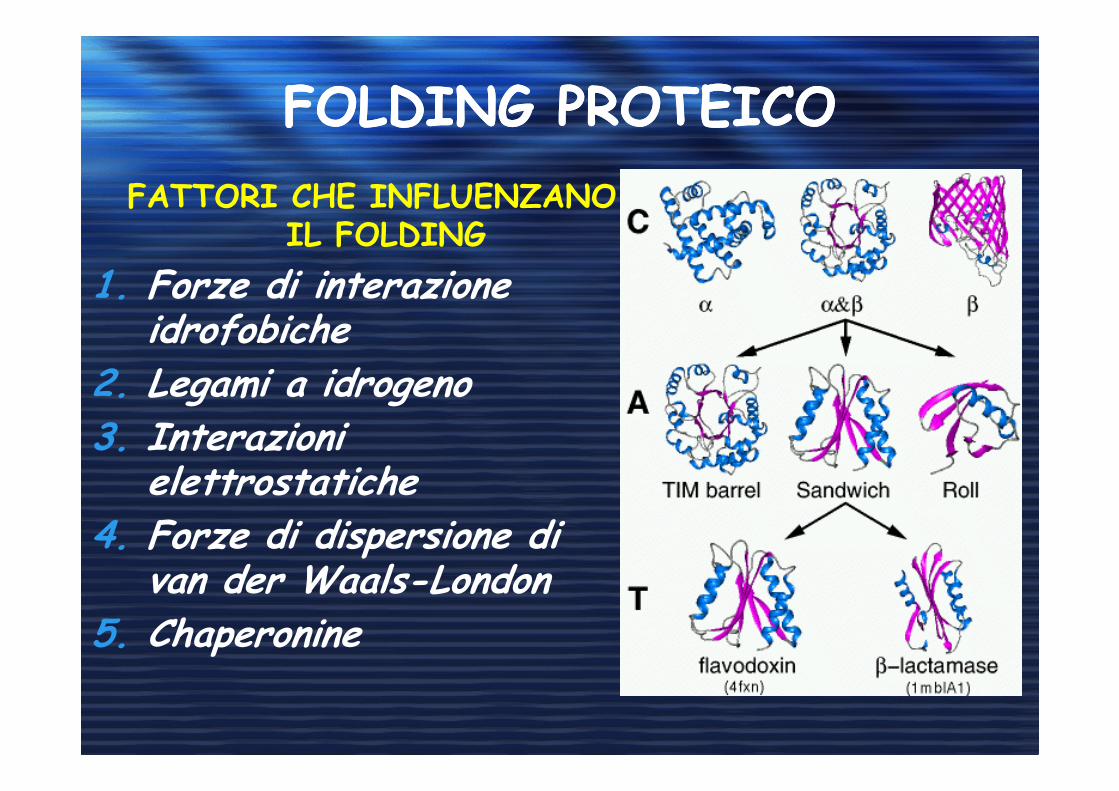
FOLDING PROTEICOFOLDING PROTEICO
FATTORI CHE INFLUENZANO IL FOLDING
1. Forze di interazione idrofobiche
2. Legami a idrogeno3. Interazioni
elettrostatiche4. Forze di dispersione di
van der Waals-London5. Chaperonine
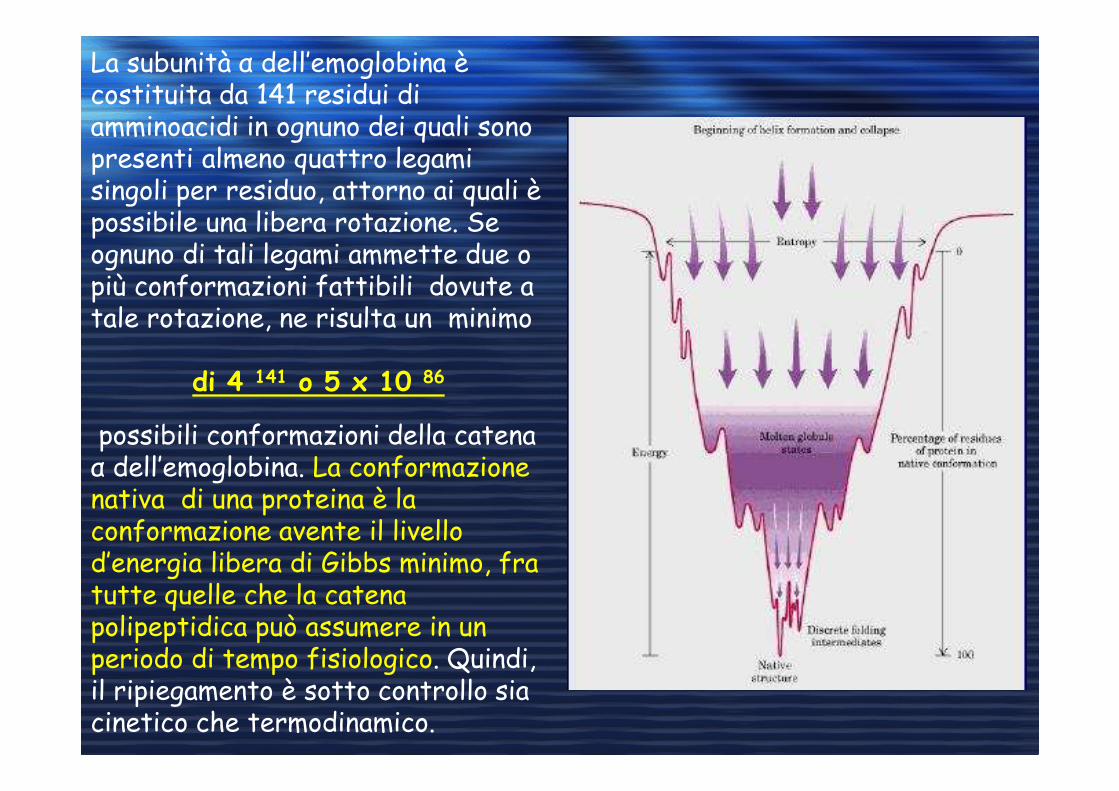
La subunità α dell’emoglobina è costituita da 141 residui di amminoacidi in ognuno dei quali sono presenti almeno quattro legami singoli per residuo, attorno ai quali è possibile una libera rotazione. Se ognuno di tali legami ammette due o più conformazioni fattibili dovute a tale rotazione, ne risulta un minimo
di 4 141 o 5 x 10 86
possibili conformazioni della catena α dell’emoglobina. La conformazione nativa di una proteina è la conformazione avente il livello d’energia libera di Gibbs minimo, fra tutte quelle che la catena polipeptidica può assumere in un periodo di tempo fisiologico. Quindi, il ripiegamento è sotto controllo sia cinetico che termodinamico.

FOLDING PROTEICOFOLDING PROTEICO
http://www.biocomp.chem.uw.edu.pl/movies.php
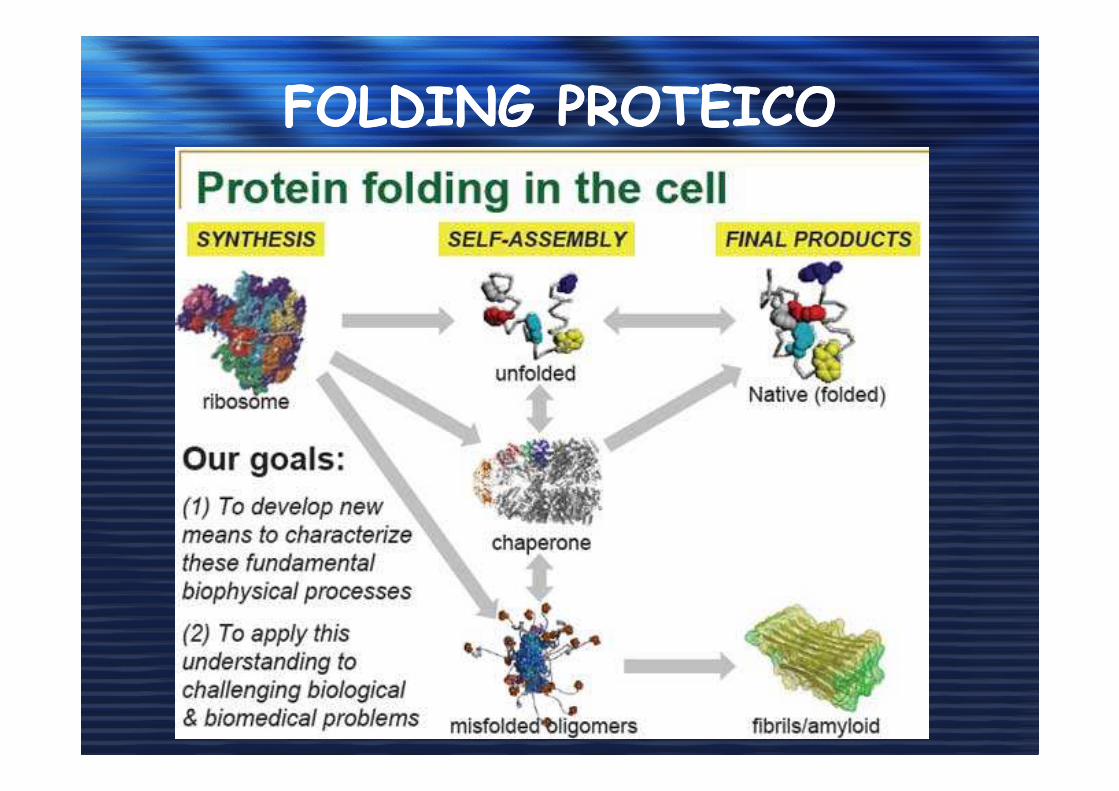
FOLDING PROTEICOFOLDING PROTEICO
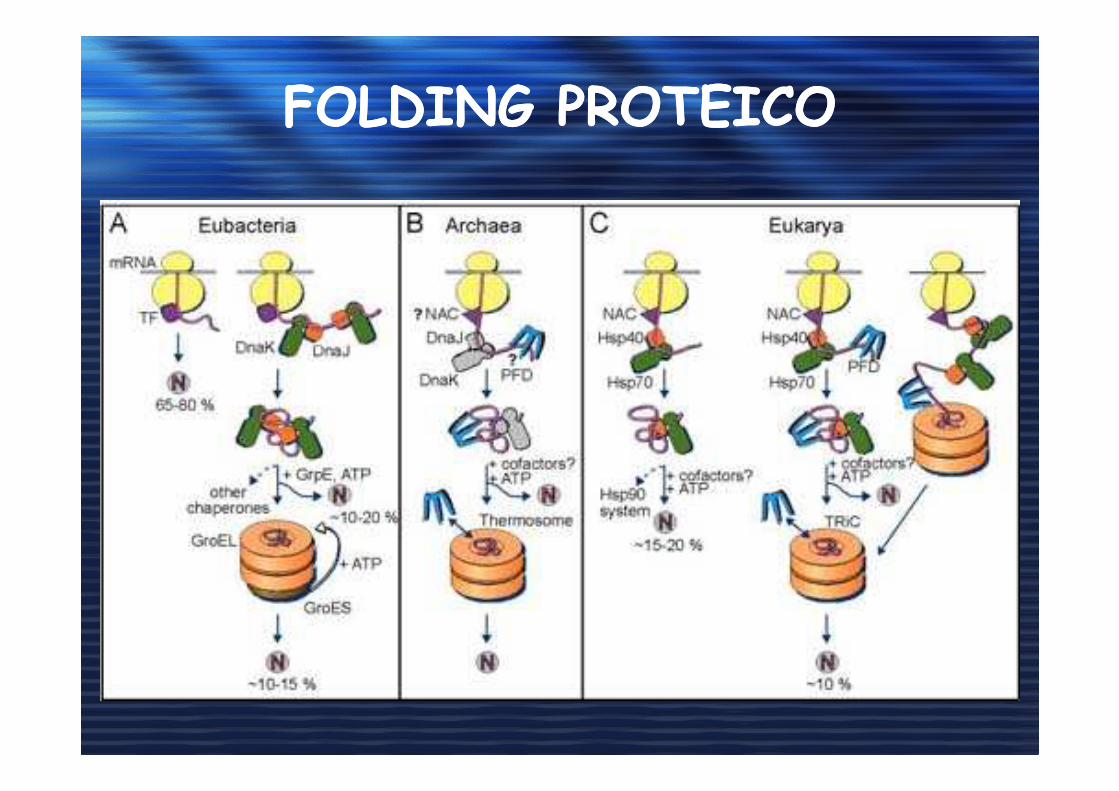
FOLDING PROTEICOFOLDING PROTEICO
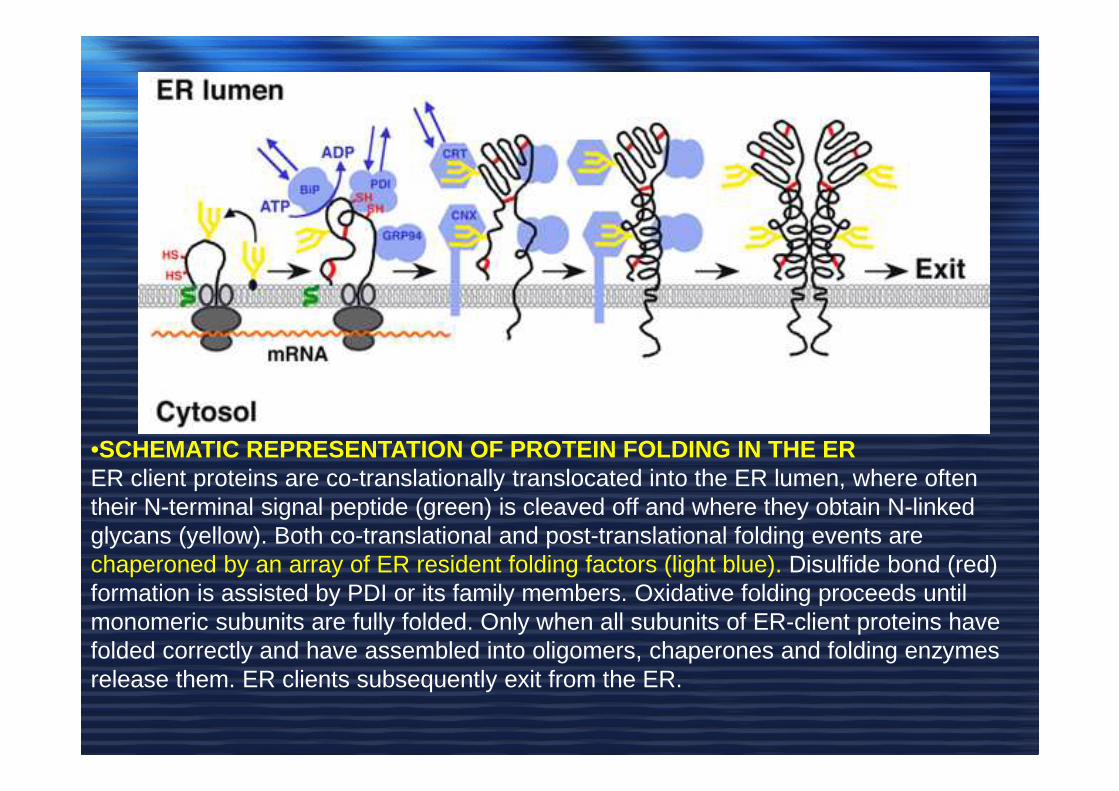
•SCHEMATIC REPRESENTATION OF PROTEIN FOLDING IN THE ERER client proteins are co-translationally translocated into the ER lumen, where often their N-terminal signal peptide (green) is cleaved off and where they obtain N-linked glycans (yellow). Both co-translational and post-translational folding events are chaperoned by an array of ER resident folding factors (light blue). Disulfide bond (red) formation is assisted by PDI or its family members. Oxidative folding proceeds until monomeric subunits are fully folded. Only when all subunits of ER-client proteins have folded correctly and have assembled into oligomers, chaperones and folding enzymes release them. ER clients subsequently exit from the ER.
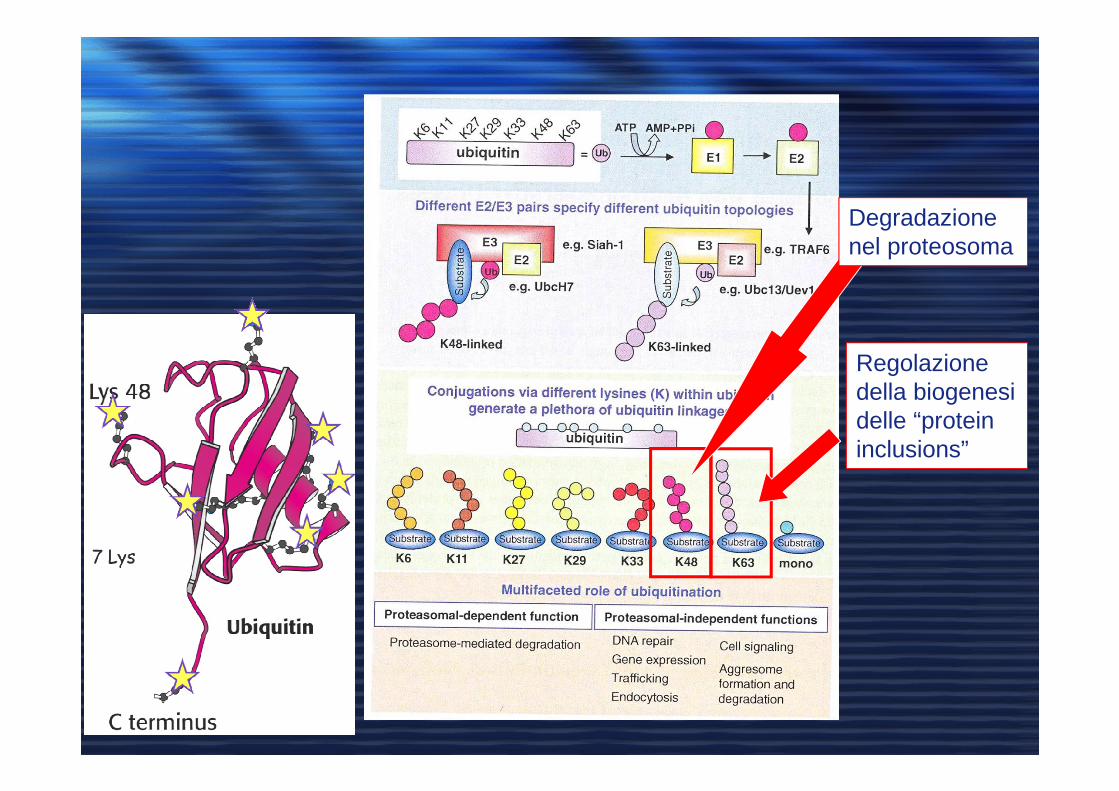
Regolazione della biogenesi delle “protein inclusions”
Degradazione nel proteosoma
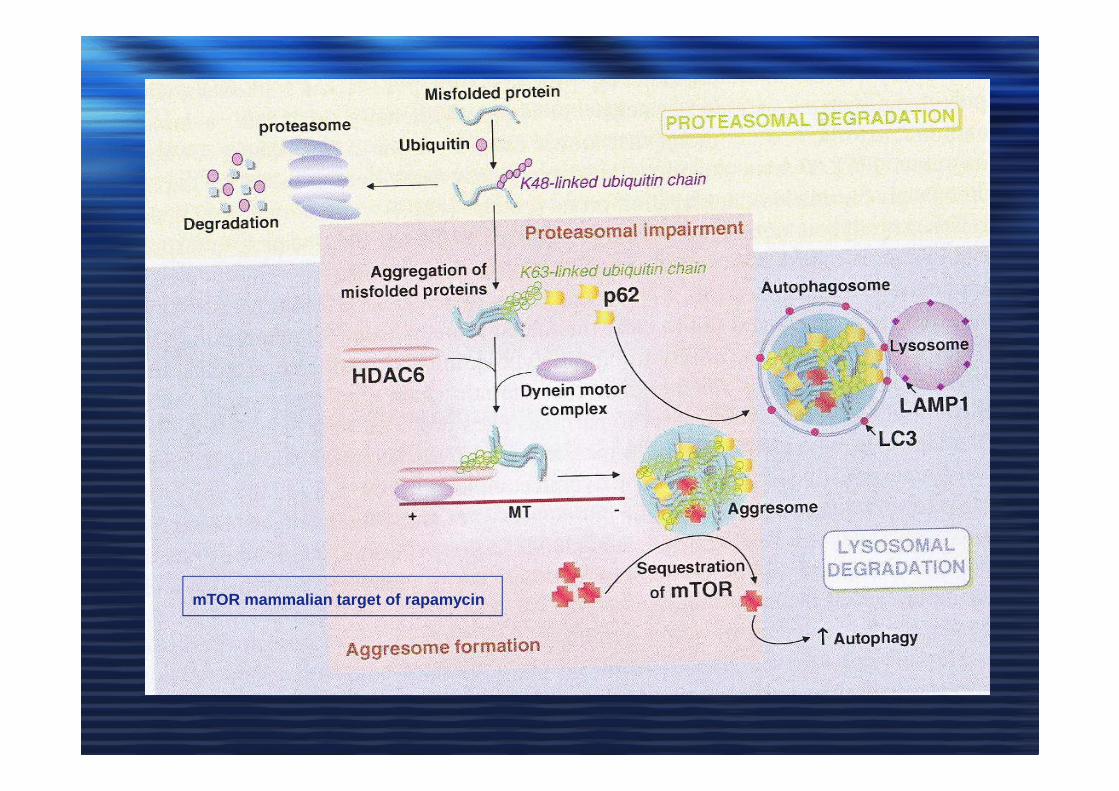
mTOR mammalian target of rapamycin
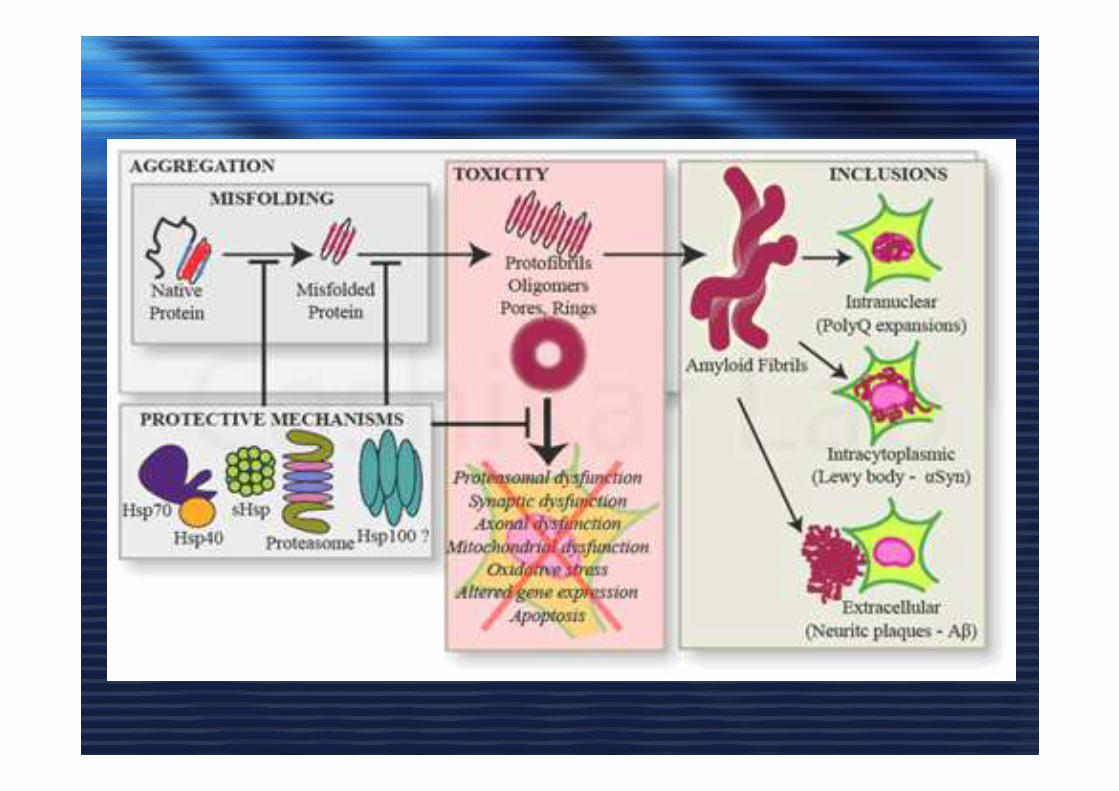
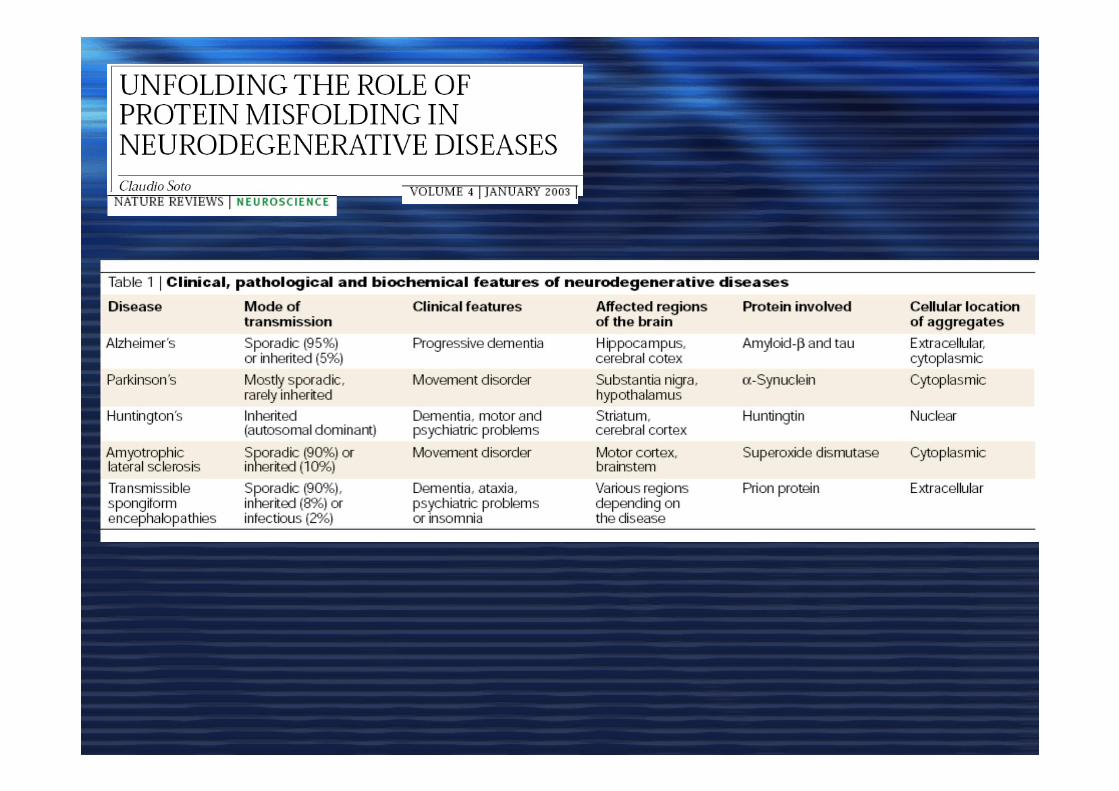
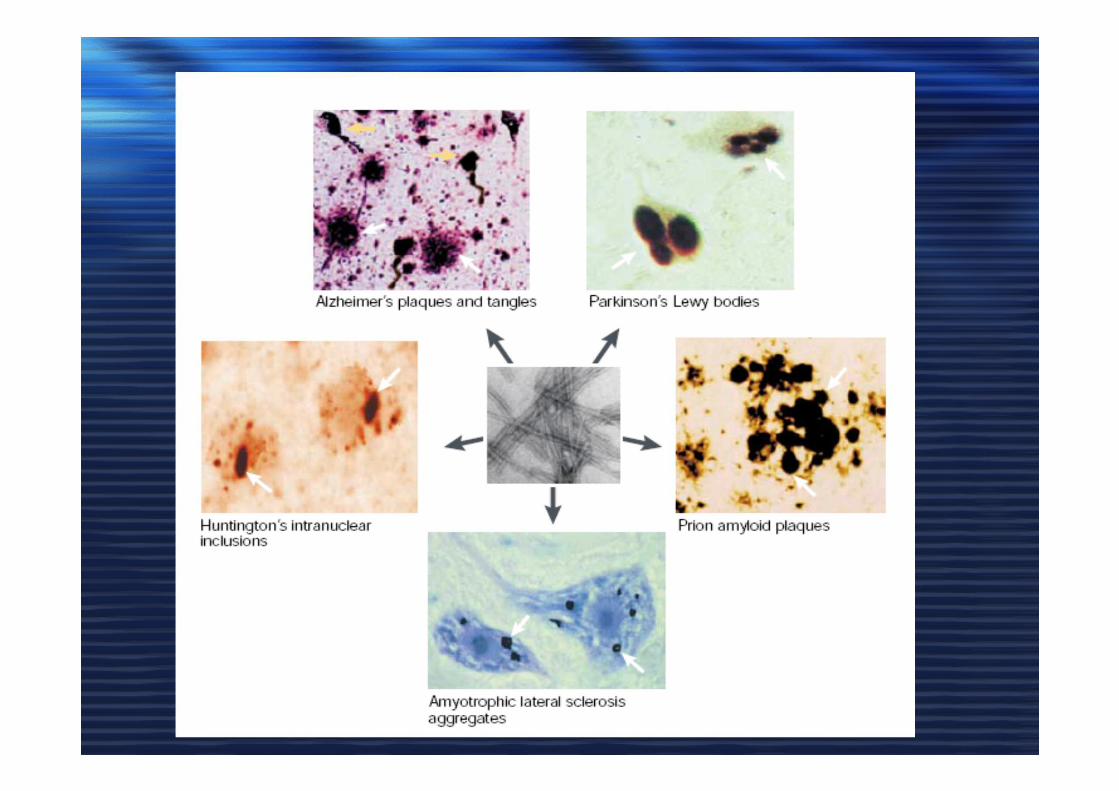

AMILOIDEAMILOIDE• Aggregati organizzati in strutture beta
• Reattivi ad alcuni coloranti (Congo Red)
• Alta resistenza alle proteasi
LA CAUSA (CONSEGUENZA ???)
di QUESTE MALATTIE NEURODEGENERATIVE è
L’ACCUMULO DEGLI AGGREGATI di PROTEINE MISFOLDING
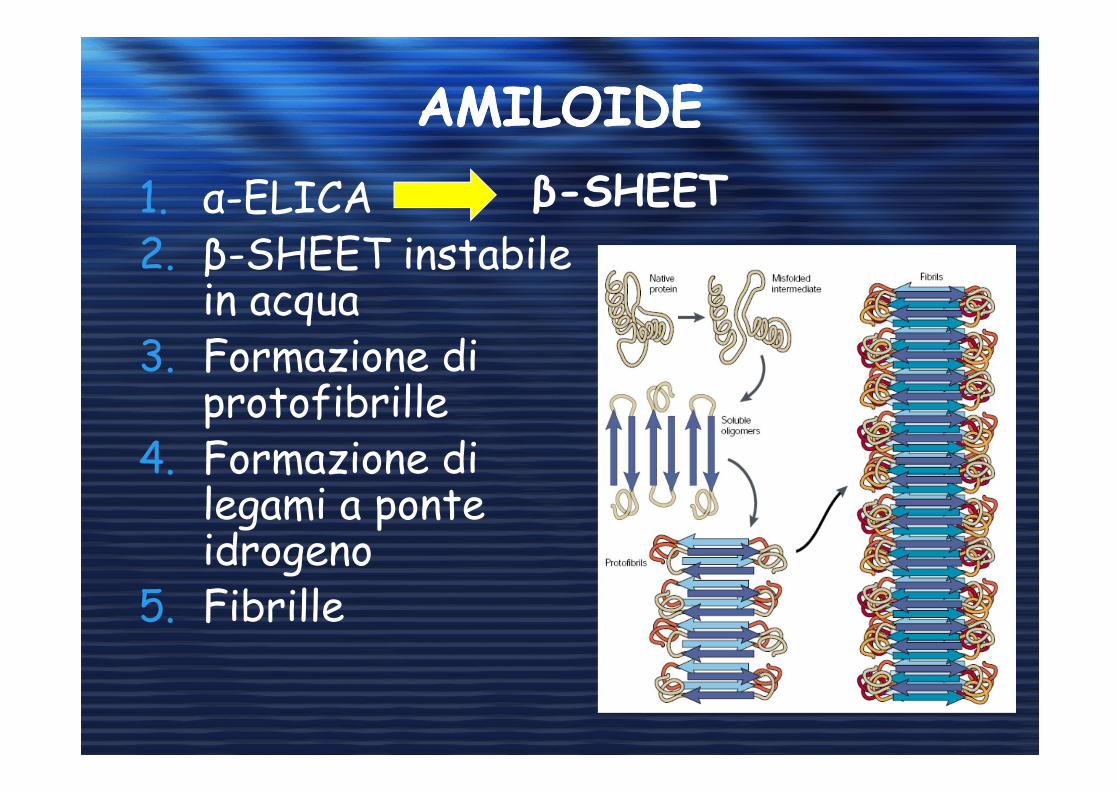
AMILOIDEAMILOIDE
1. α-ELICA 2. β-SHEET instabile
in acqua3. Formazione di
protofibrille4. Formazione di
legami a ponte idrogeno
5. Fibrille
β-SHEET

AMILOIDEAMILOIDE
Alterazioni concentrazione di ioni metallici
Proteine chaperone alterate
Alterazioni del pH
Stress ossidativo –ROS-
Aumento localizzato di macromolecole
Stress da Reticolo
INVECCHIAMENTO

LA MALATTIA DI ALZHEIMER
50% di tutte le forme di demenza
In Italia: 70.000 casi/anno
cognitive
Forma sporadica (>60 y): 90%
Forma familiare (40-50 y): 10%
sociali
emotive
1906
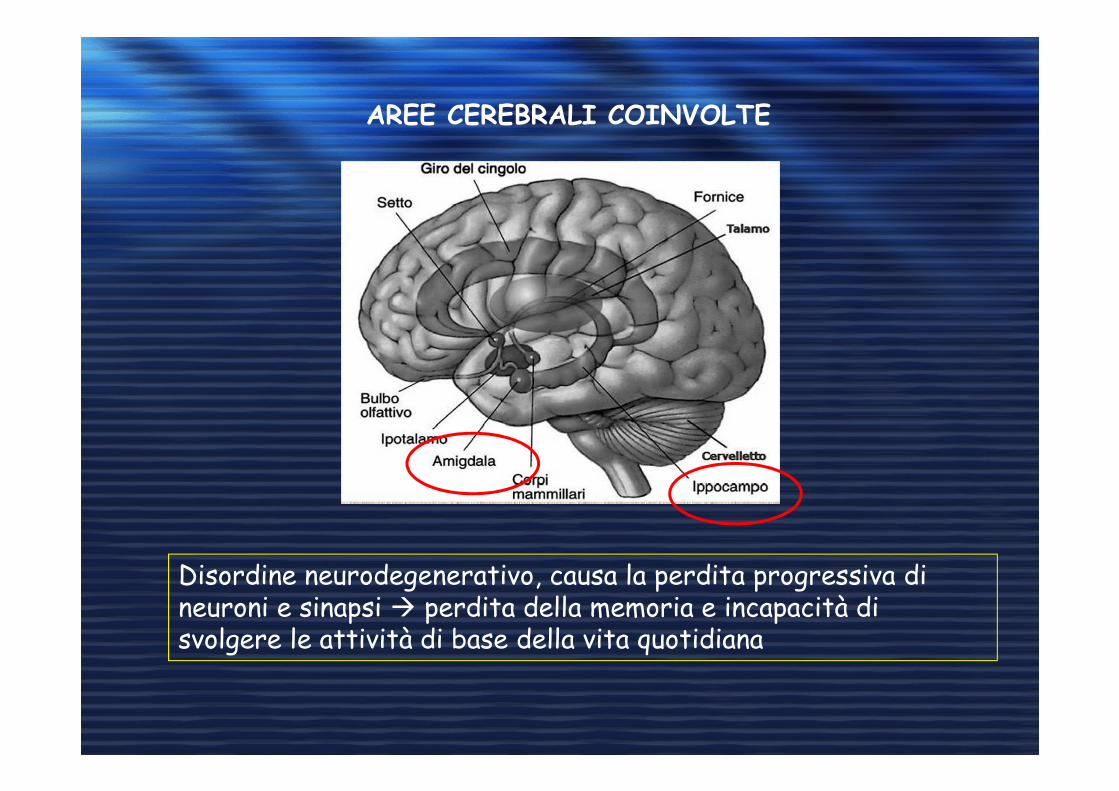
AREE CEREBRALI COINVOLTE
Disordine neurodegenerativo, causa la perdita progressiva di neuroni e sinapsi � perdita della memoria e incapacità di svolgere le attività di base della vita quotidiana
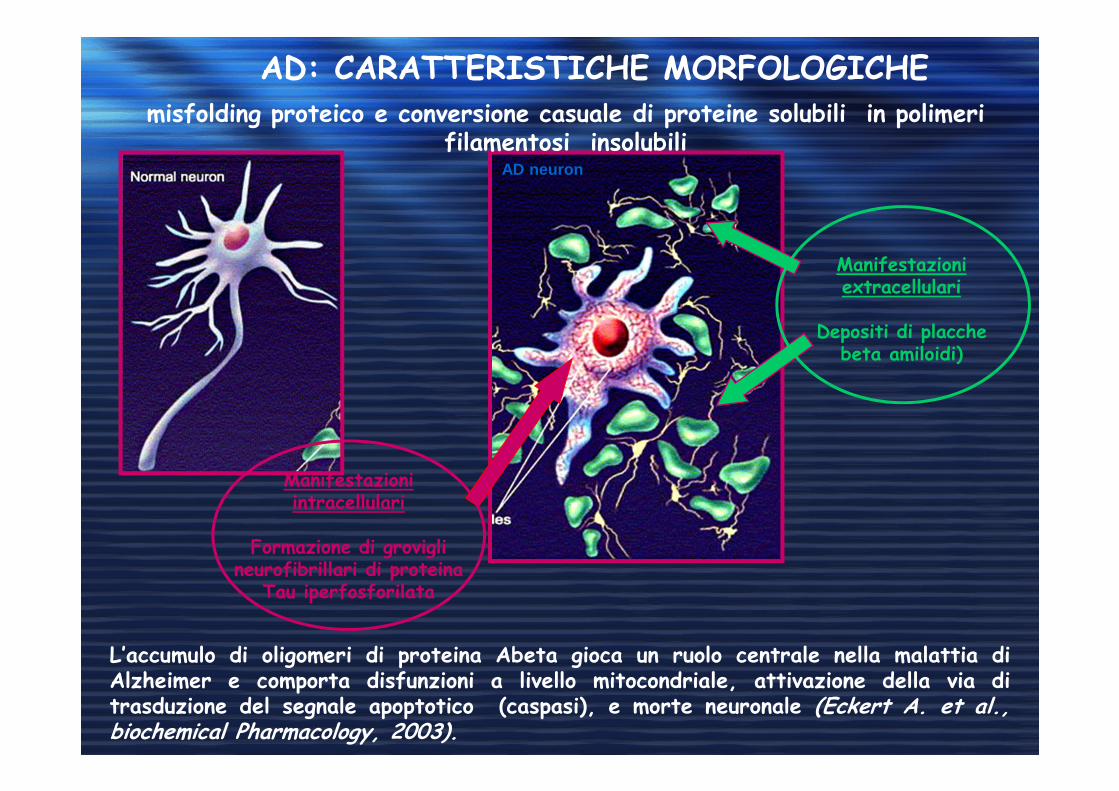
AD: CARATTERISTICHE MORFOLOGICHEmisfolding proteico e conversione casuale di proteine solubili in polimeri
filamentosi insolubili
Manifestazioni intracellulari
Formazione di grovigli neurofibrillari di proteina
Tau iperfosforilata
Manifestazioni extracellulari
Depositi di placche beta amiloidi)
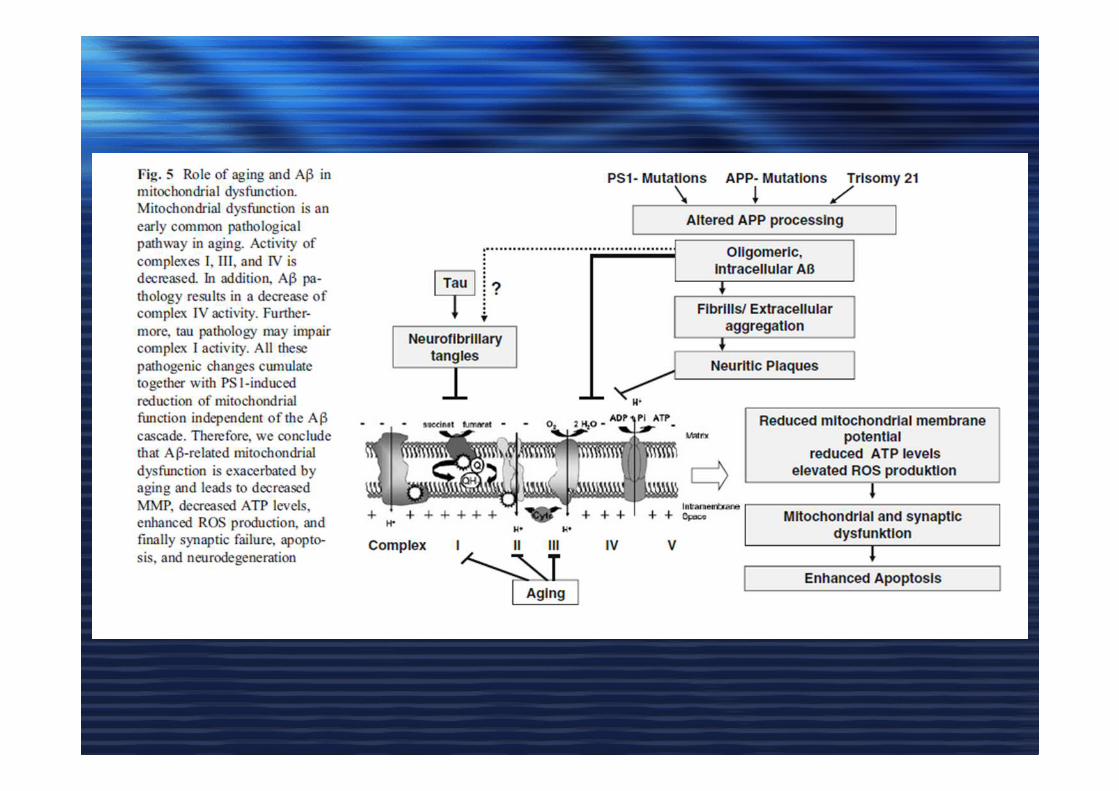
L’accumulo di oligomeri di proteina Abeta gioca un ruolo centrale nella malattia diAlzheimer e comporta disfunzioni a livello mitocondriale, attivazione della via ditrasduzione del segnale apoptotico (caspasi), e morte neuronale (Eckert A. et al.,biochemical Pharmacology, 2003).
AD neuron
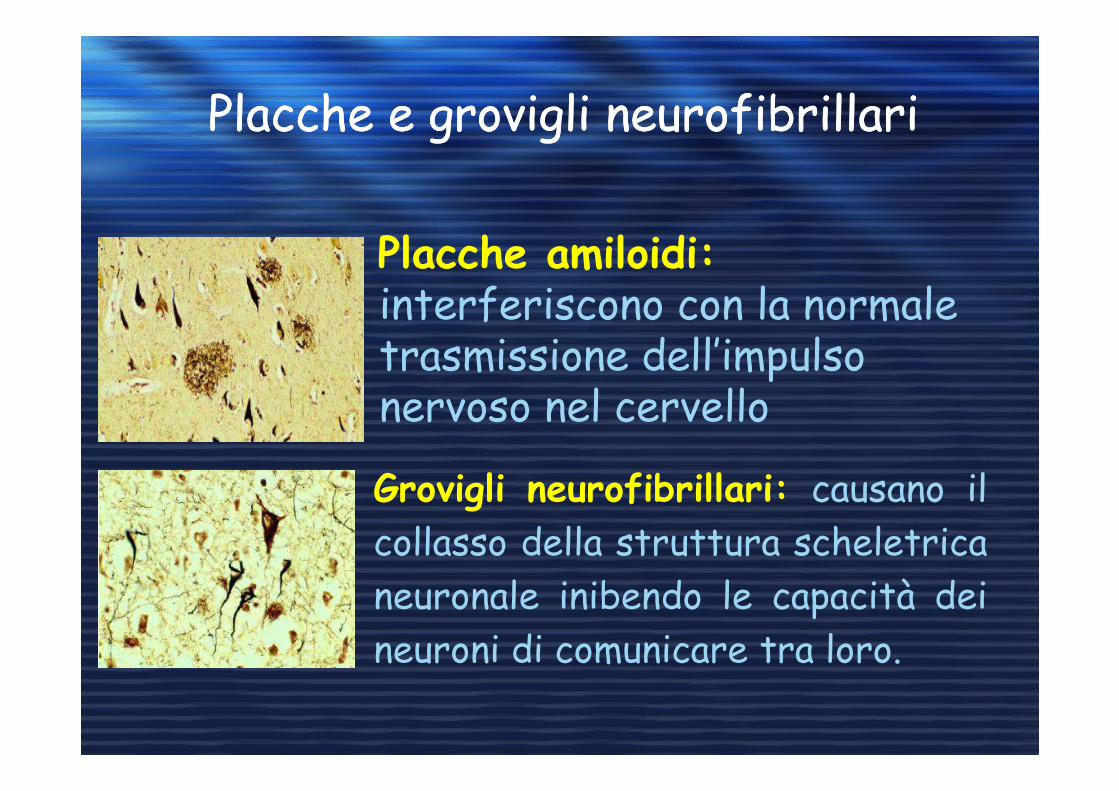
Placche e grovigli neurofibrillariPlacche e grovigli neurofibrillari
Placche amiloidi:interferiscono con la normale trasmissione dell’impulso nervoso nel cervello
Grovigli neurofibrillari: causano il
collasso della struttura scheletrica
neuronale inibendo le capacità dei
neuroni di comunicare tra loro.
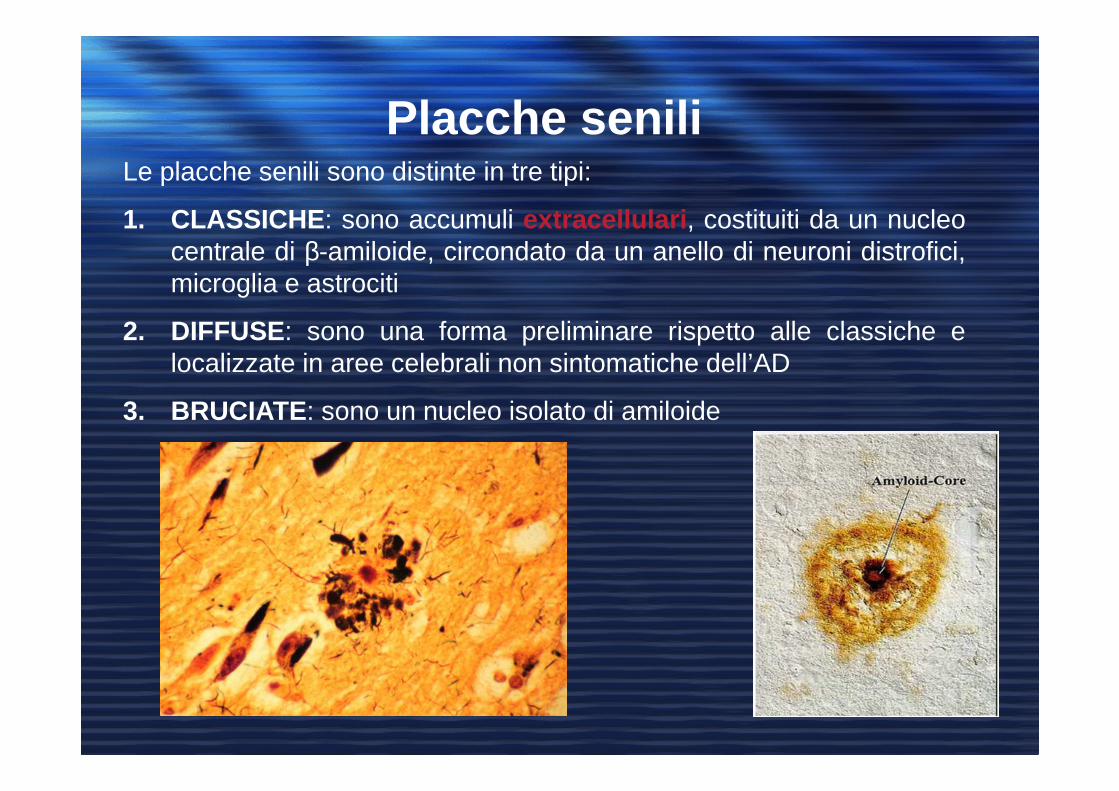
Le placche senili sono distinte in tre tipi:
1. CLASSICHE : sono accumuli extracellulari , costituiti da un nucleocentrale di β-amiloide, circondato da un anello di neuroni distrofici,microglia e astrociti
2. DIFFUSE: sono una forma preliminare rispetto alle classiche elocalizzate in aree celebrali non sintomatiche dell’AD
3. BRUCIATE : sono un nucleo isolato di amiloide
Placche senili
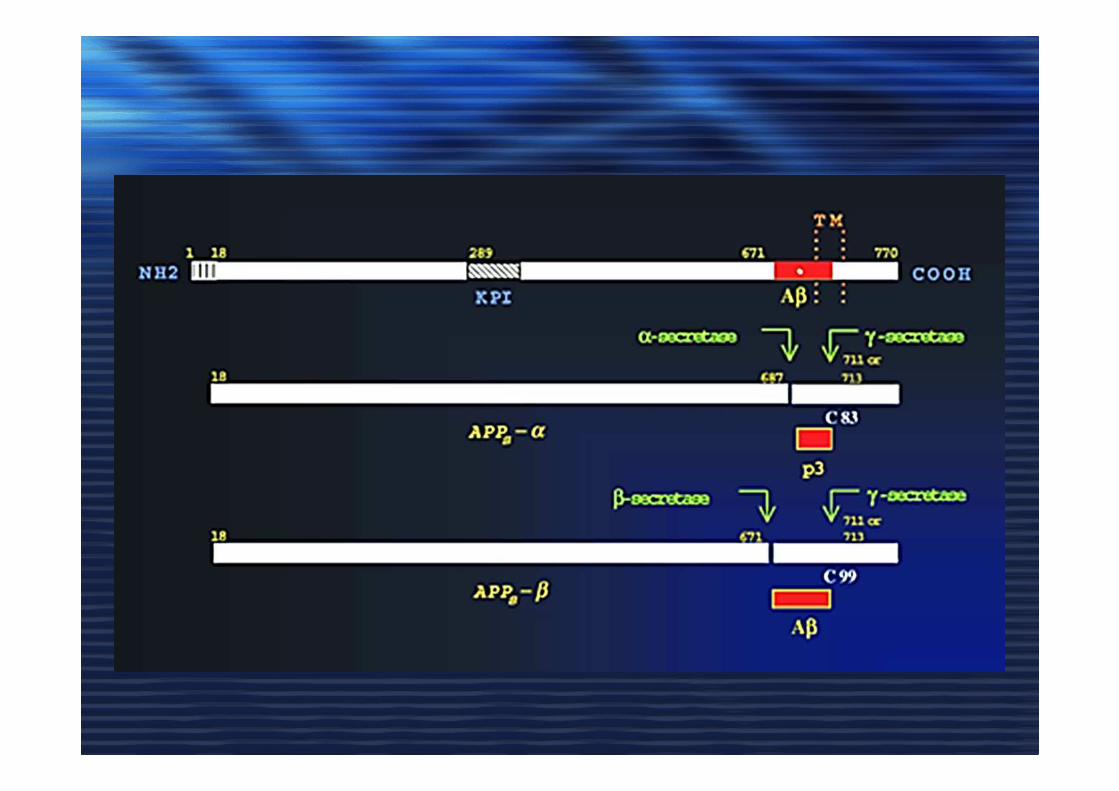
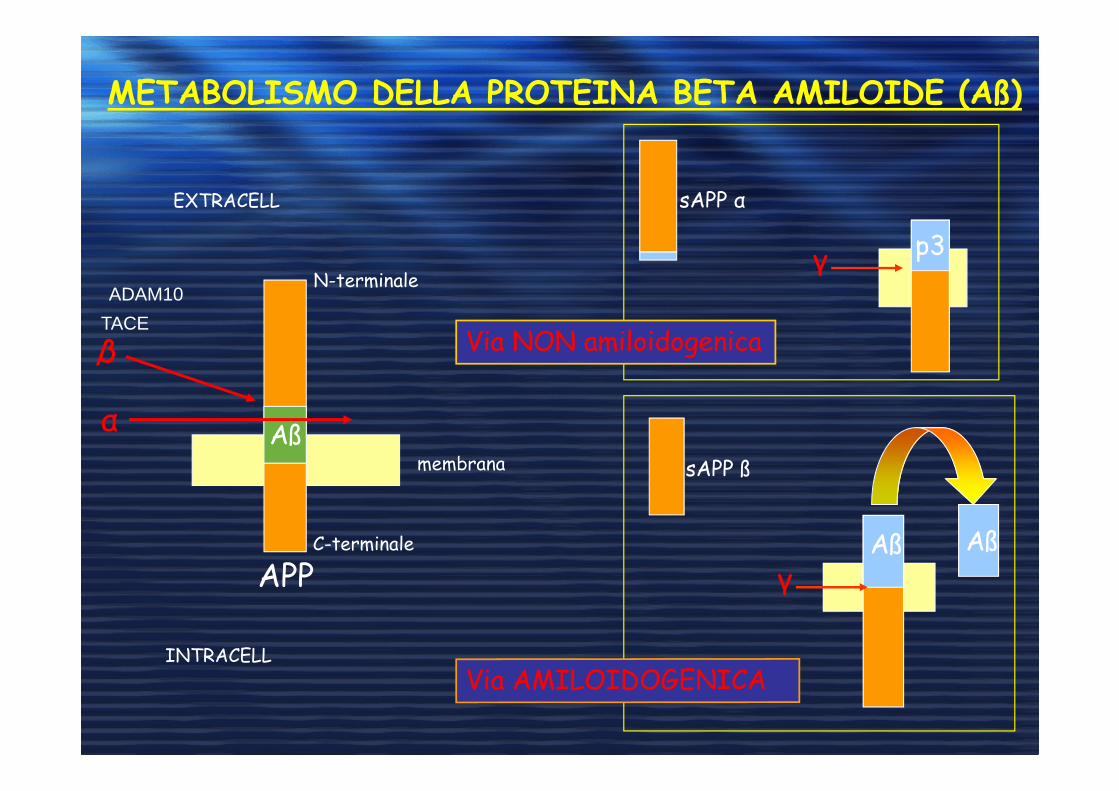
METABOLISMO DELLA PROTEINA BETA AMILOIDE (Aß)
p3
membrana
APPC-terminale
N-terminale
Aß
INTRACELL
EXTRACELL
α
Aß Aß
sAPP ß
sAPP α
γ
γ
Via NON amiloidogenica
ADAM10
TACE
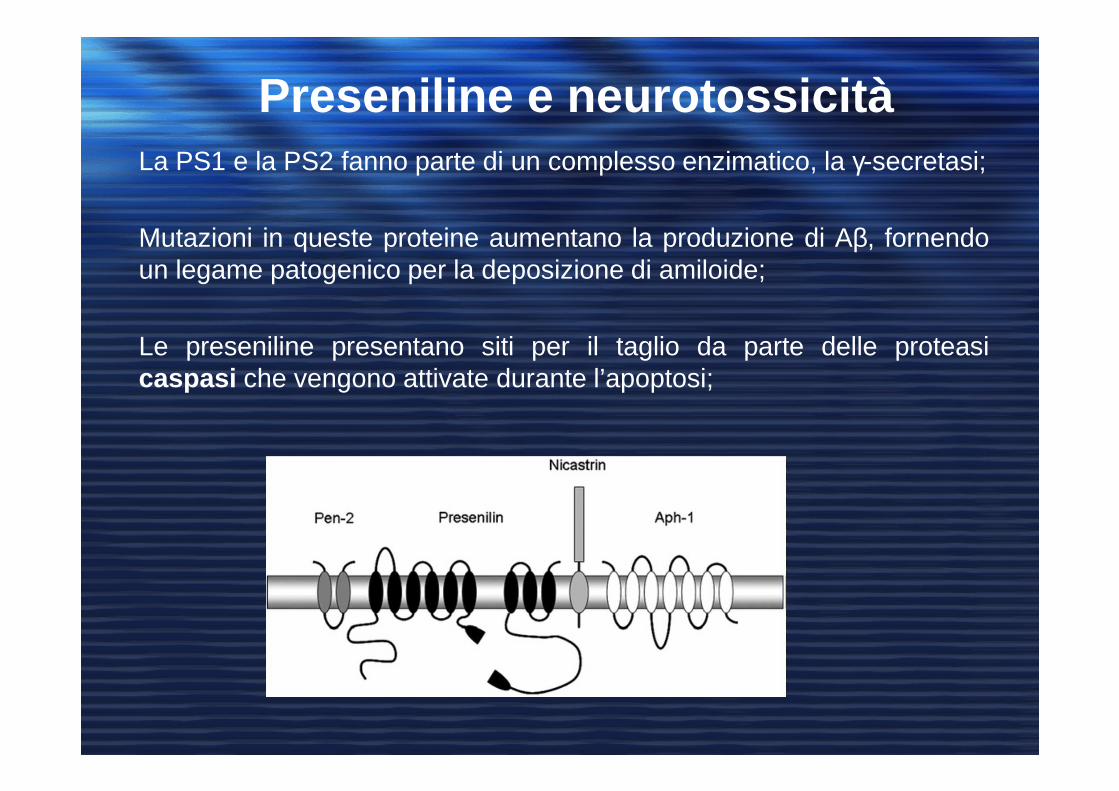
La PS1 e la PS2 fanno parte di un complesso enzimatico, la γ-secretasi;
Mutazioni in queste proteine aumentano la produzione di Aβ, fornendoun legame patogenico per la deposizione di amiloide;
Le preseniline presentano siti per il taglio da parte delle proteasicaspasi che vengono attivate durante l’apoptosi;
Preseniline e neurotossicità
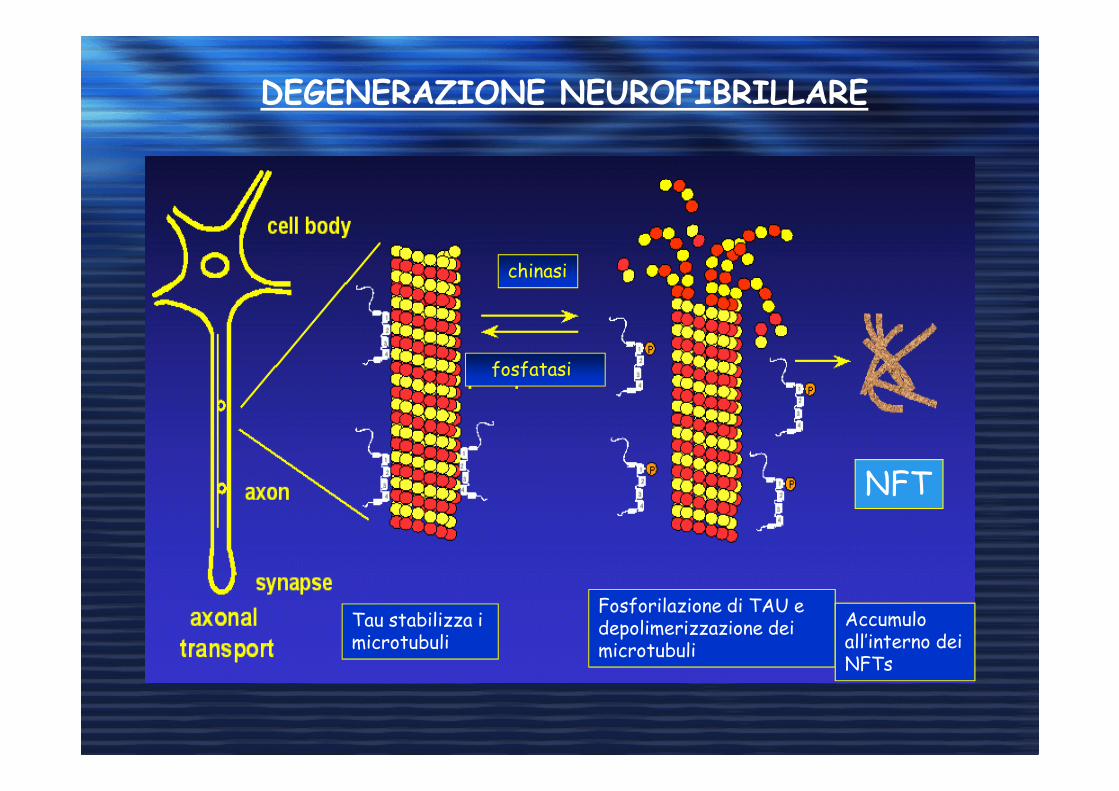
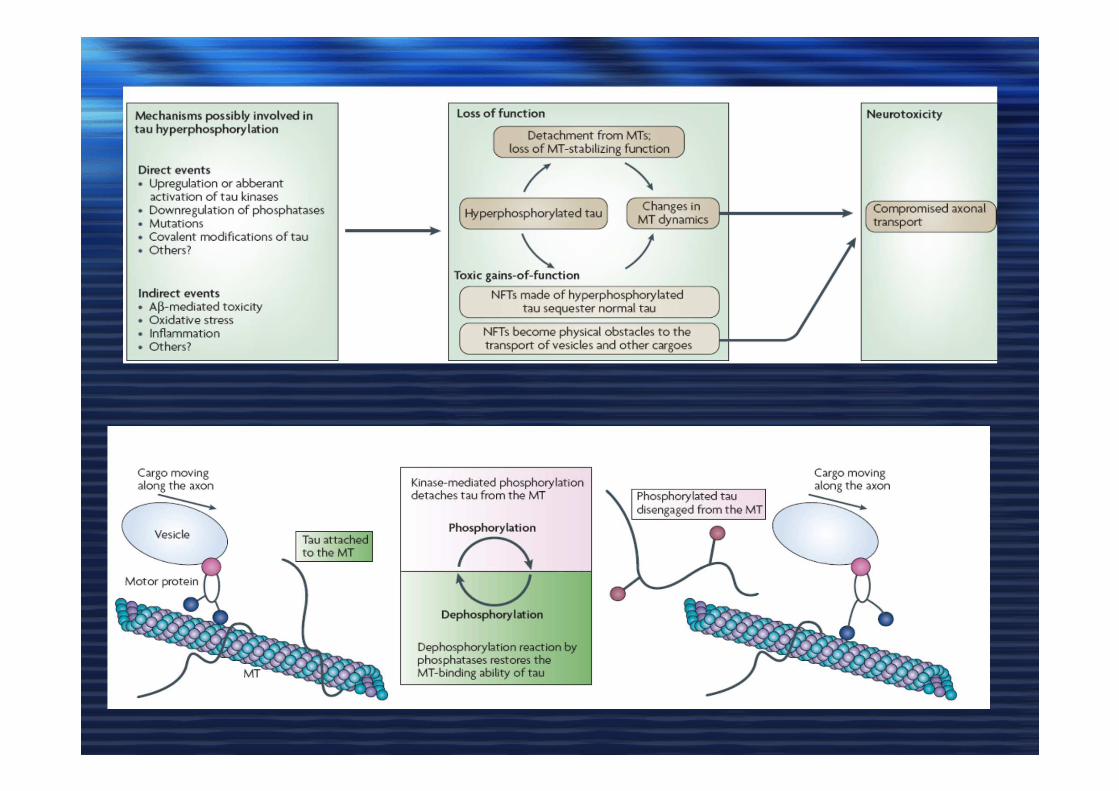
DEGENERAZIONE NEUROFIBRILLARE
chinasi
fosfatasi
Tau stabilizza i microtubuli
Fosforilazione di TAU e depolimerizzazione dei microtubuli
Accumulo all’interno dei NFTs
NFT
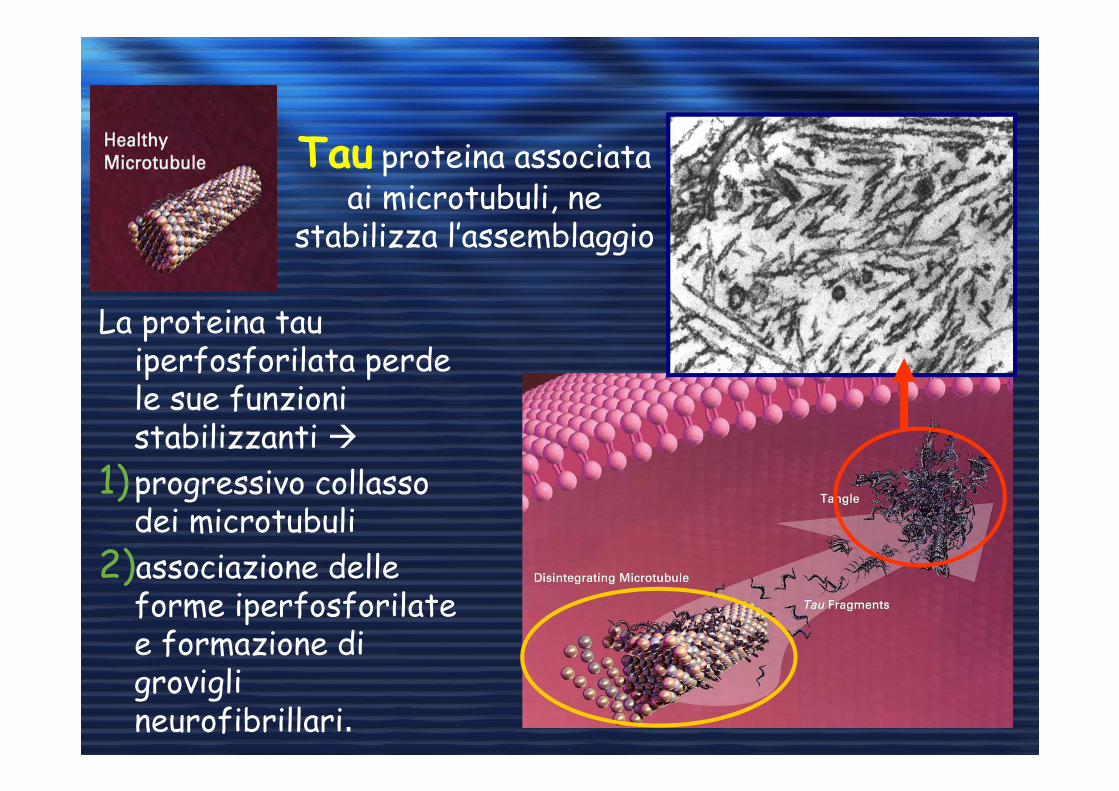
Tau proteina associata ai microtubuli, ne
stabilizza l’assemblaggio
La proteina tau iperfosforilata perde le sue funzioni stabilizzanti �
1)progressivo collasso dei microtubuli
2)associazione delle forme iperfosforilate e formazione di grovigli neurofibrillari.

Early Onset Alzheimer’s

Late Onset Alzheimer’s
• In late onset Alzheimer’s, there are no specific gene mutations that are associated with the inheritance of the disease
• Sorlin 1??

Polimorfismo ApoE e ALZHEIMERPolimorfismo ApoE e ALZHEIMER
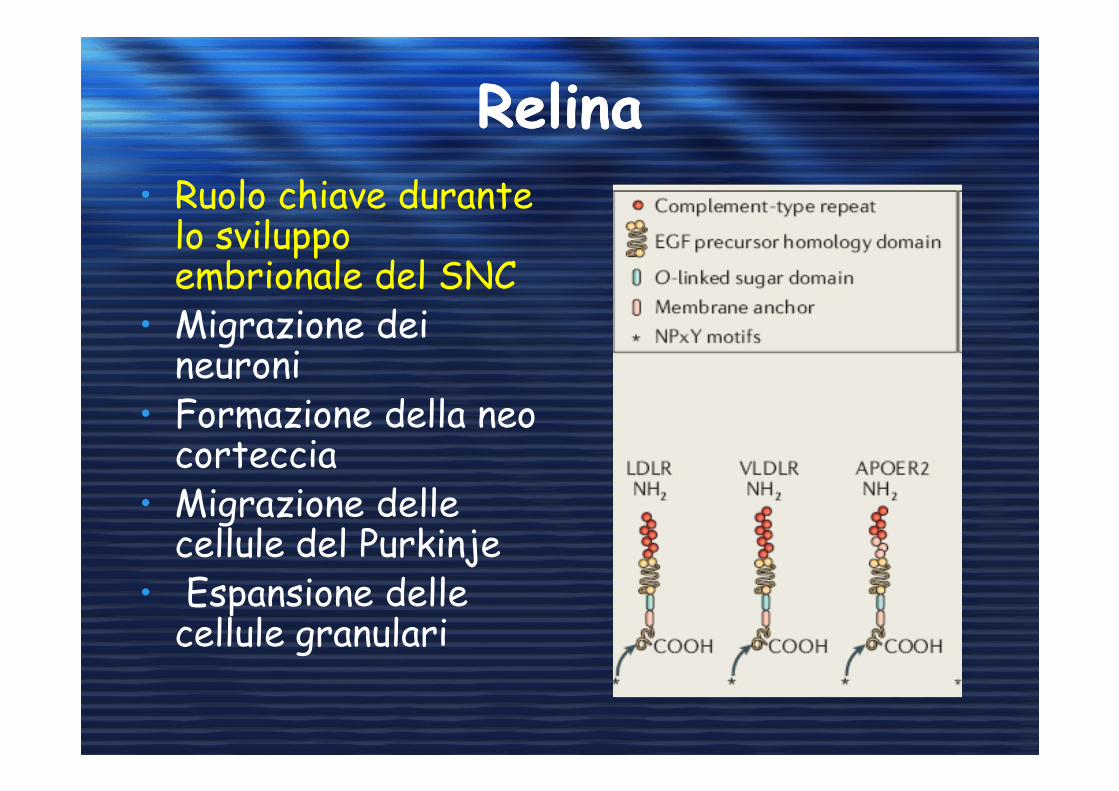
RelinaRelina
• Ruolo chiave durante lo sviluppo embrionale del SNC
• Migrazione dei neuroni
• Formazione della neo corteccia
• Migrazione delle cellule del Purkinje
• Espansione delle cellule granulari
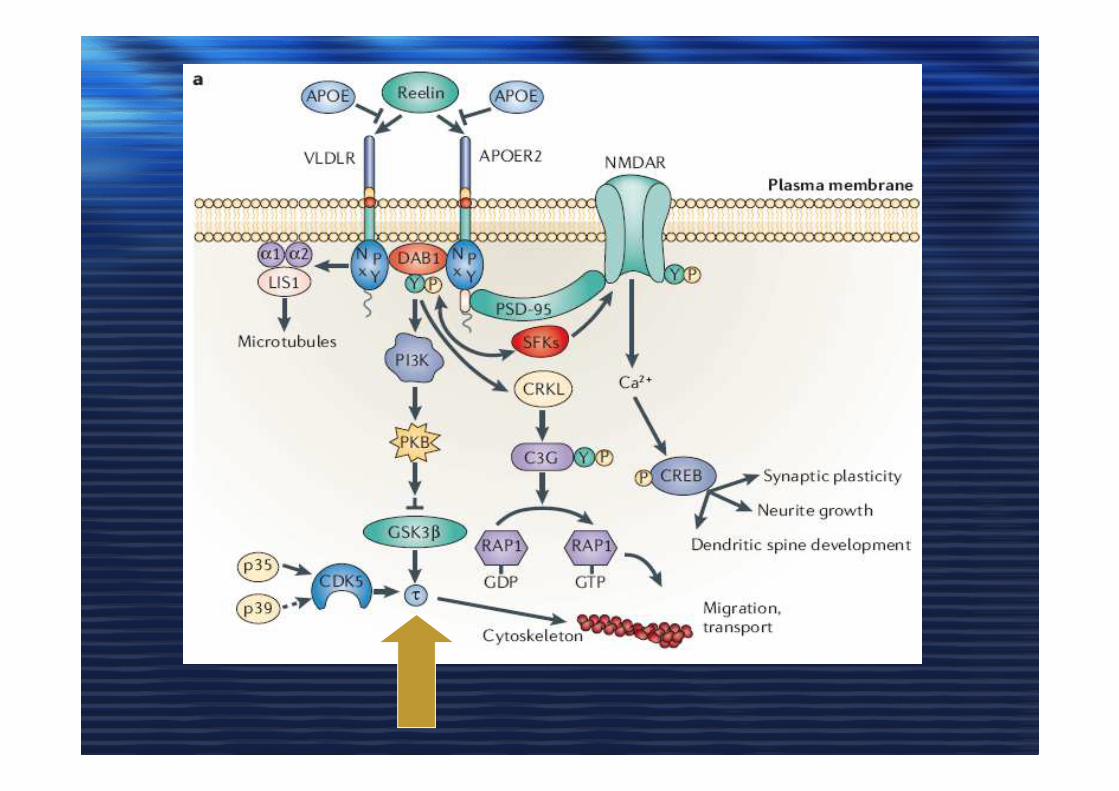
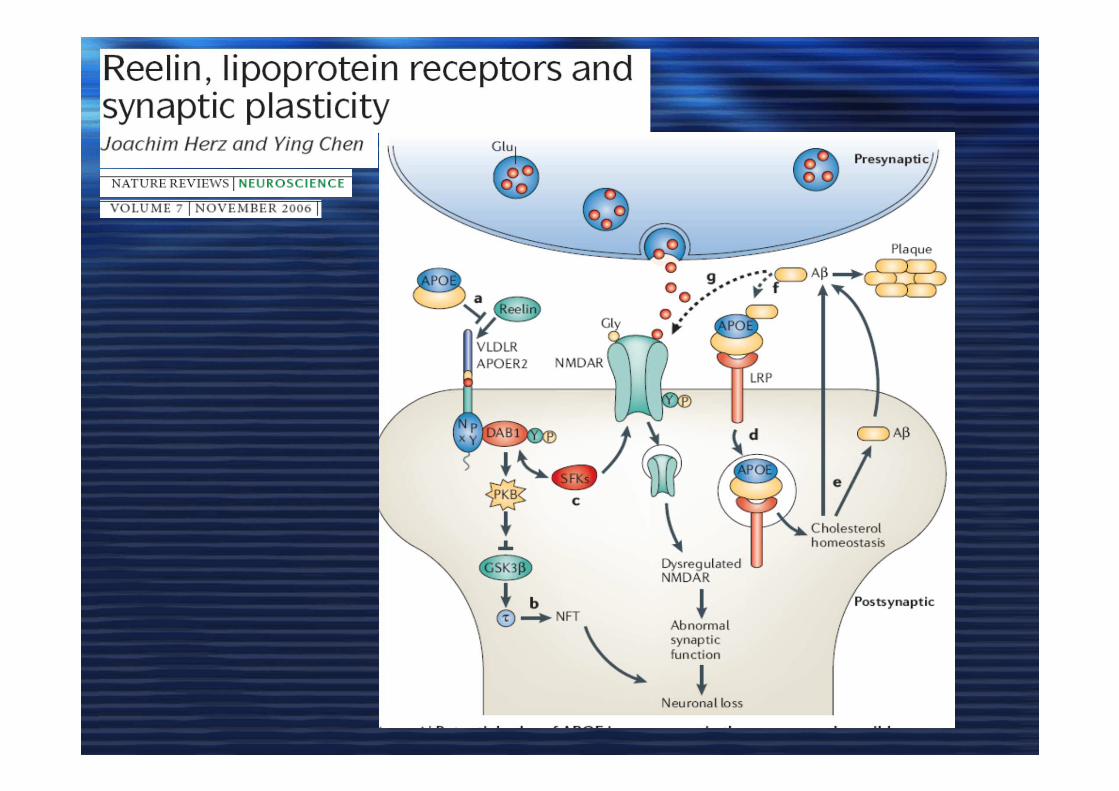

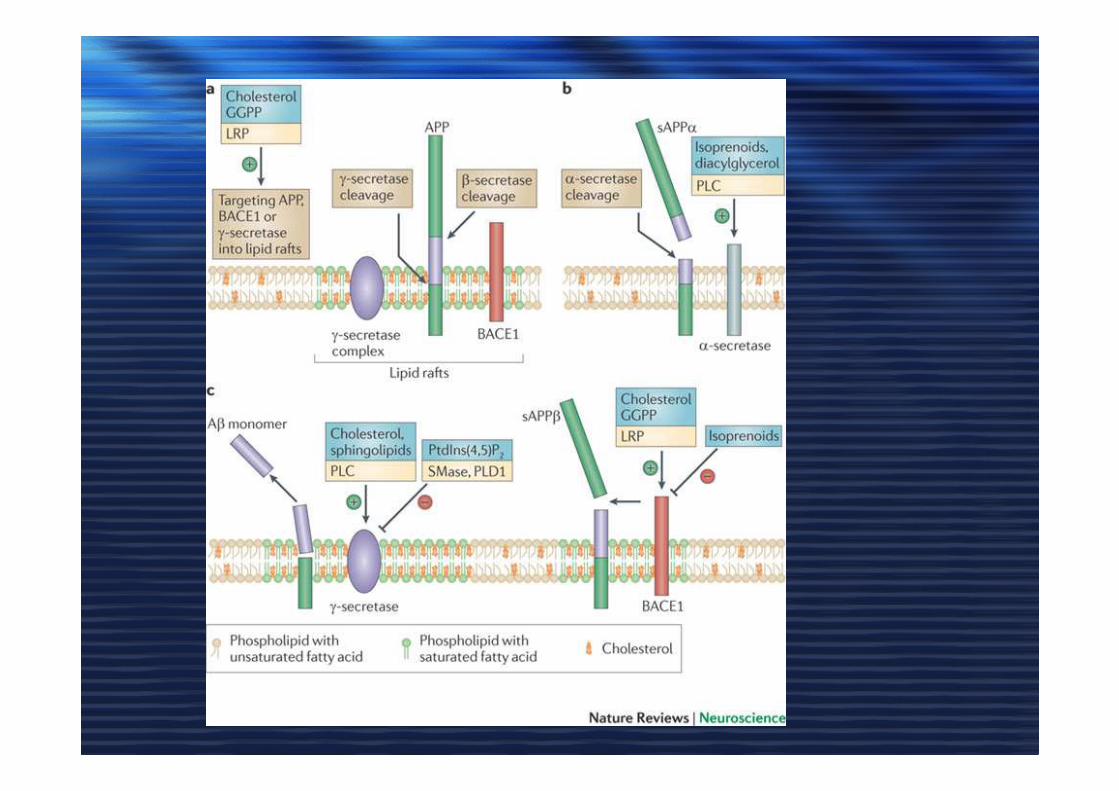
ApoE, COLESTEROLO e ADApoE, COLESTEROLO e AD
• Astrociti producono apoE /HDL
• Colesterolo indispensabile nella plasticità sinaptica e in LTP
• apoE4 e apoE3
• Deplezione di colesterolo Abeta
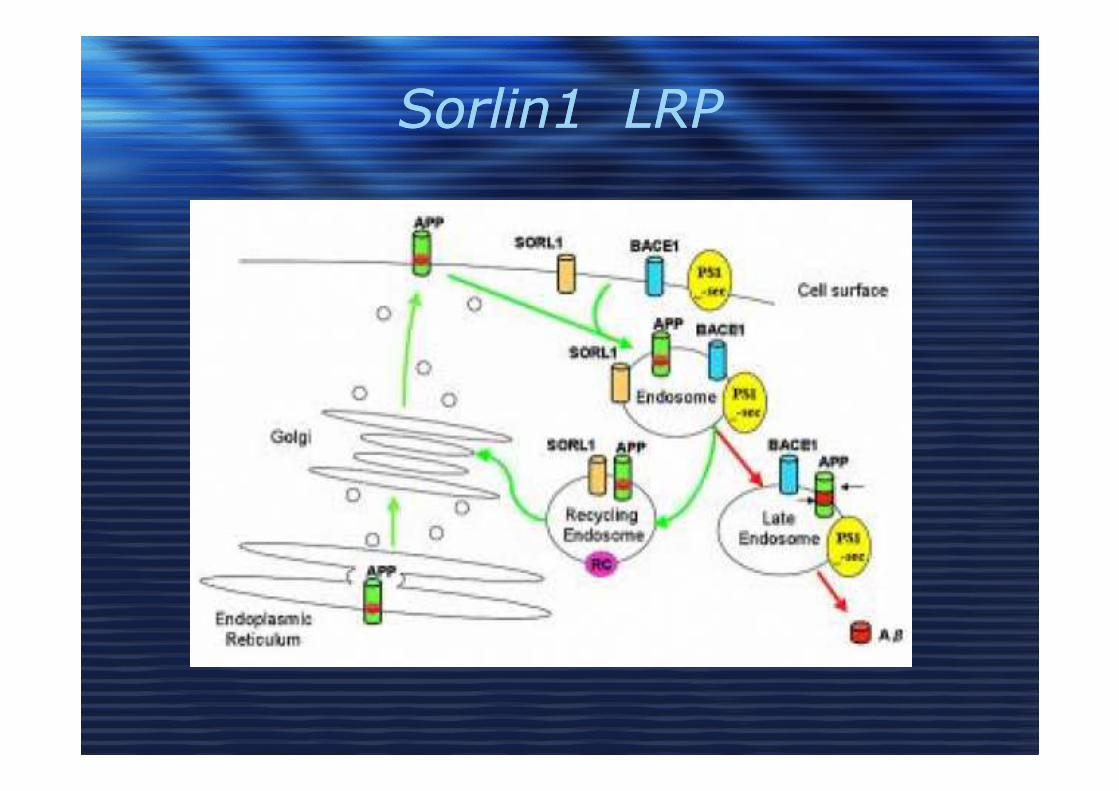
Sorlin1 LRPSorlin1 LRP

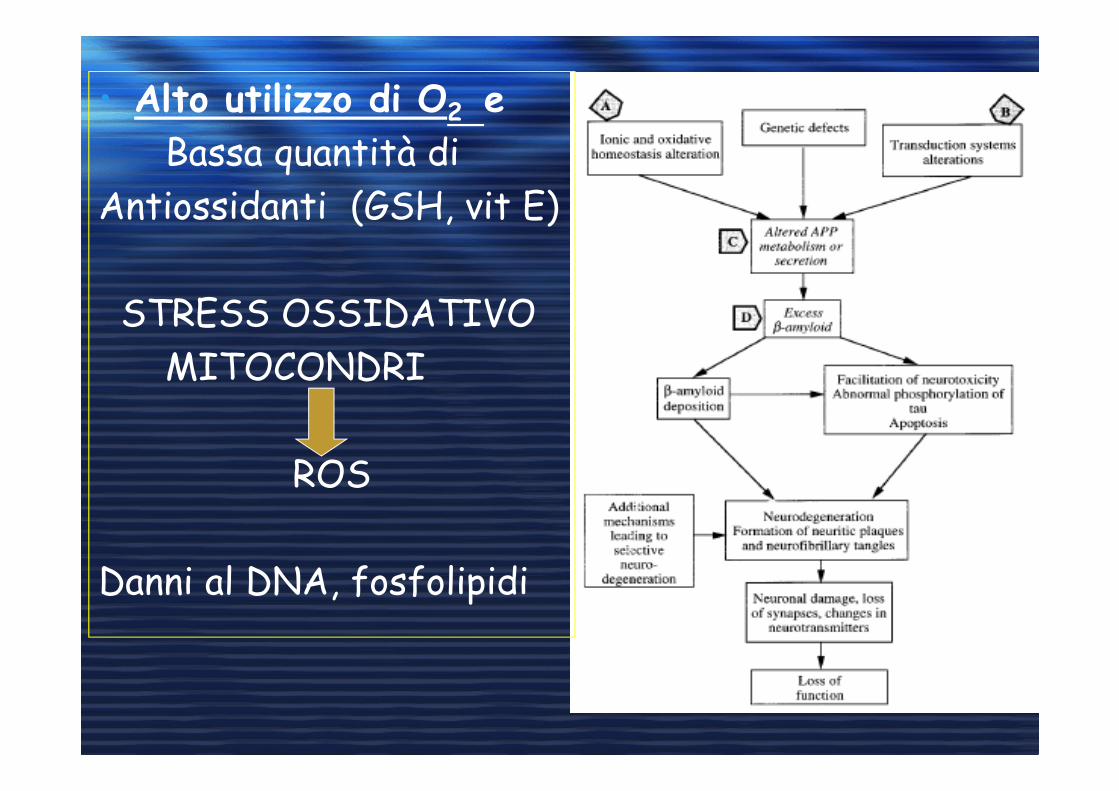
• Alto utilizzo di O2 e
Bassa quantità di
Antiossidanti (GSH, vit E)
STRESS OSSIDATIVO
MITOCONDRI
ROS
Danni al DNA, fosfolipidi
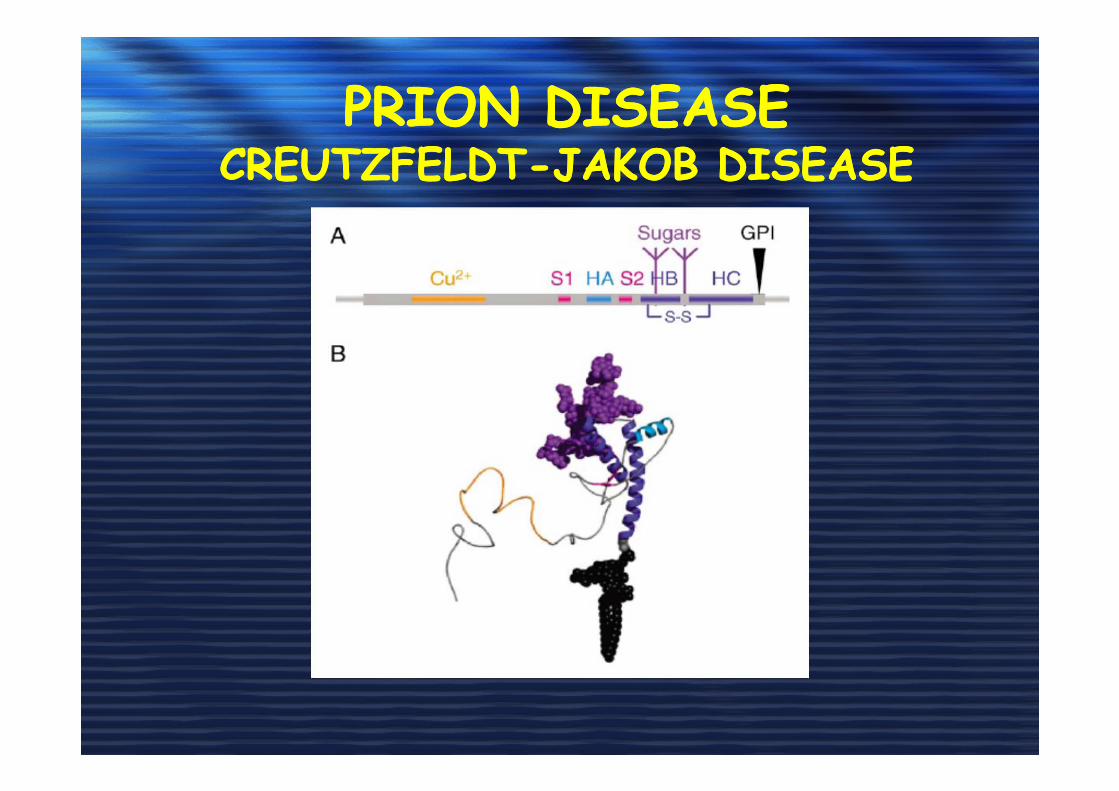
PRION DISEASE CREUTZFELDT-JAKOB DISEASE
PRION DISEASE CREUTZFELDT-JAKOB DISEASE
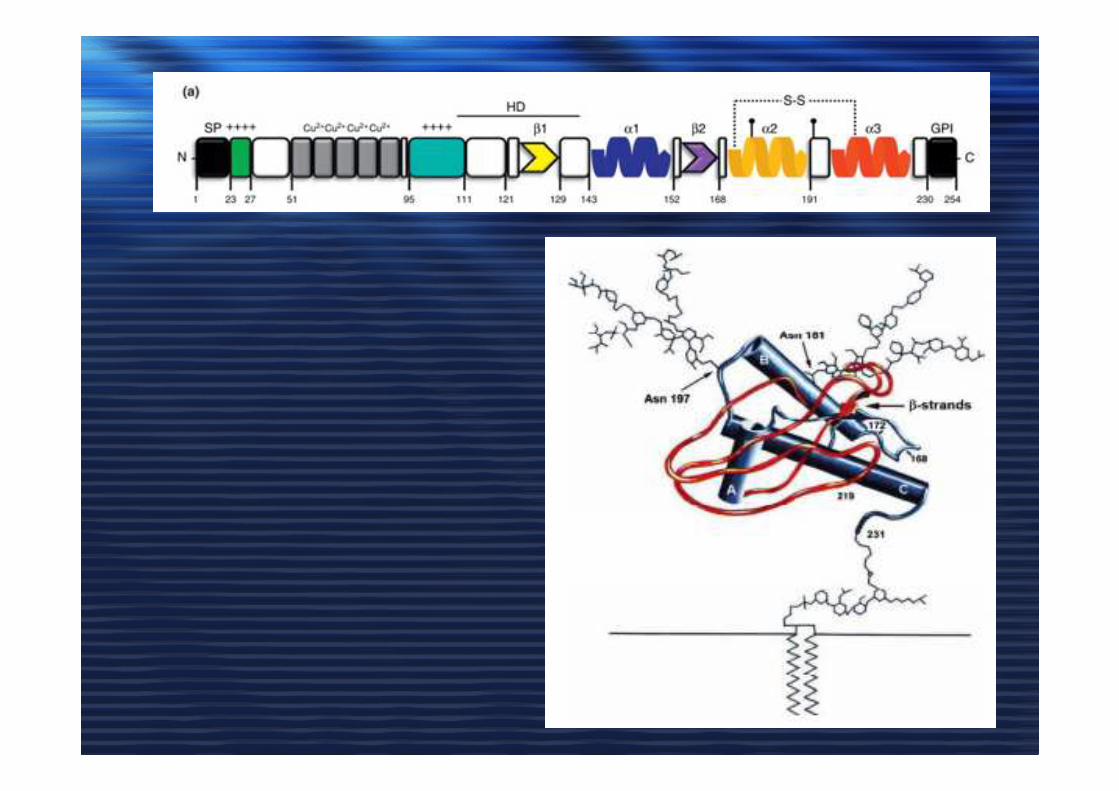

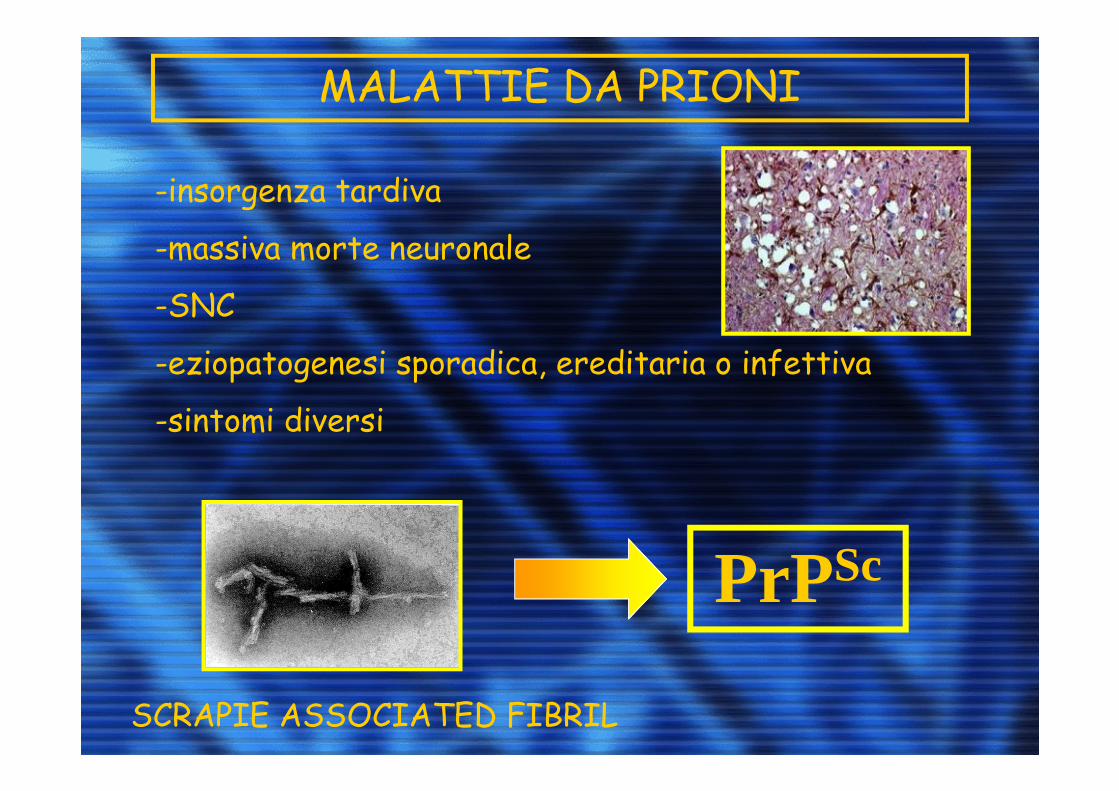
MALATTIE DA PRIONI
-insorgenza tardiva
-massiva morte neuronale
-SNC
-eziopatogenesi sporadica, ereditaria o infettiva
-sintomi diversi
SCRAPIE ASSOCIATED FIBRIL
PrPSc

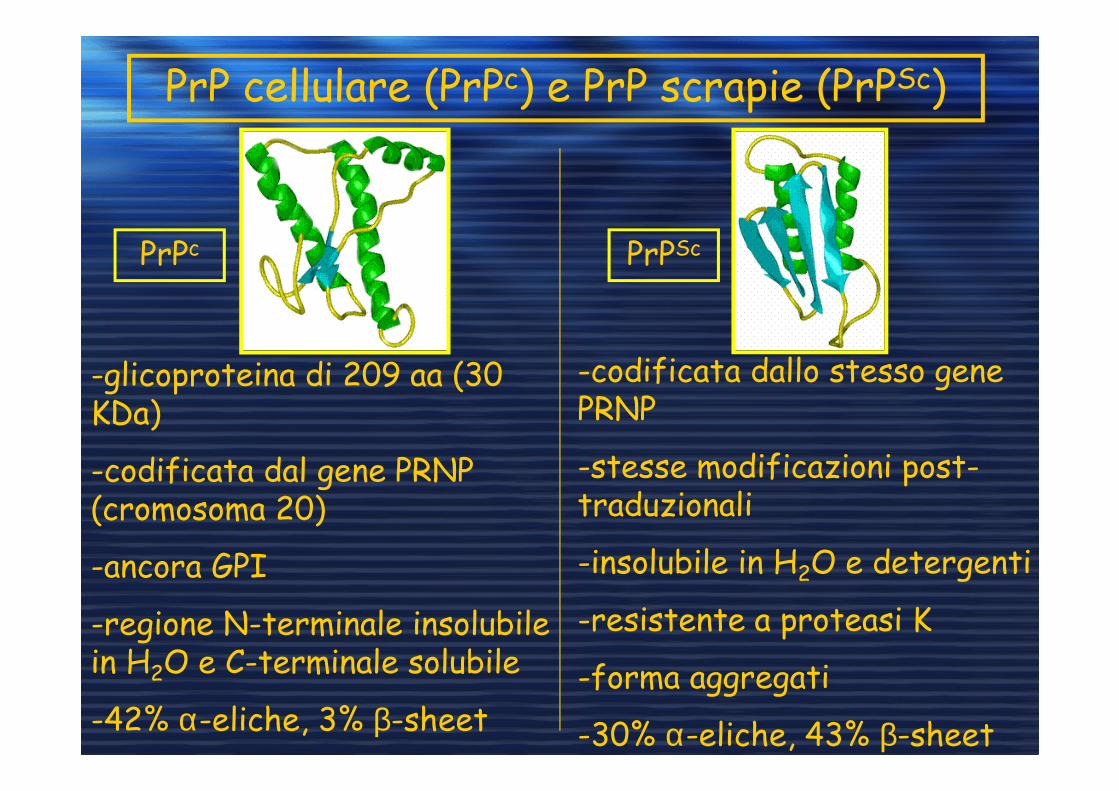
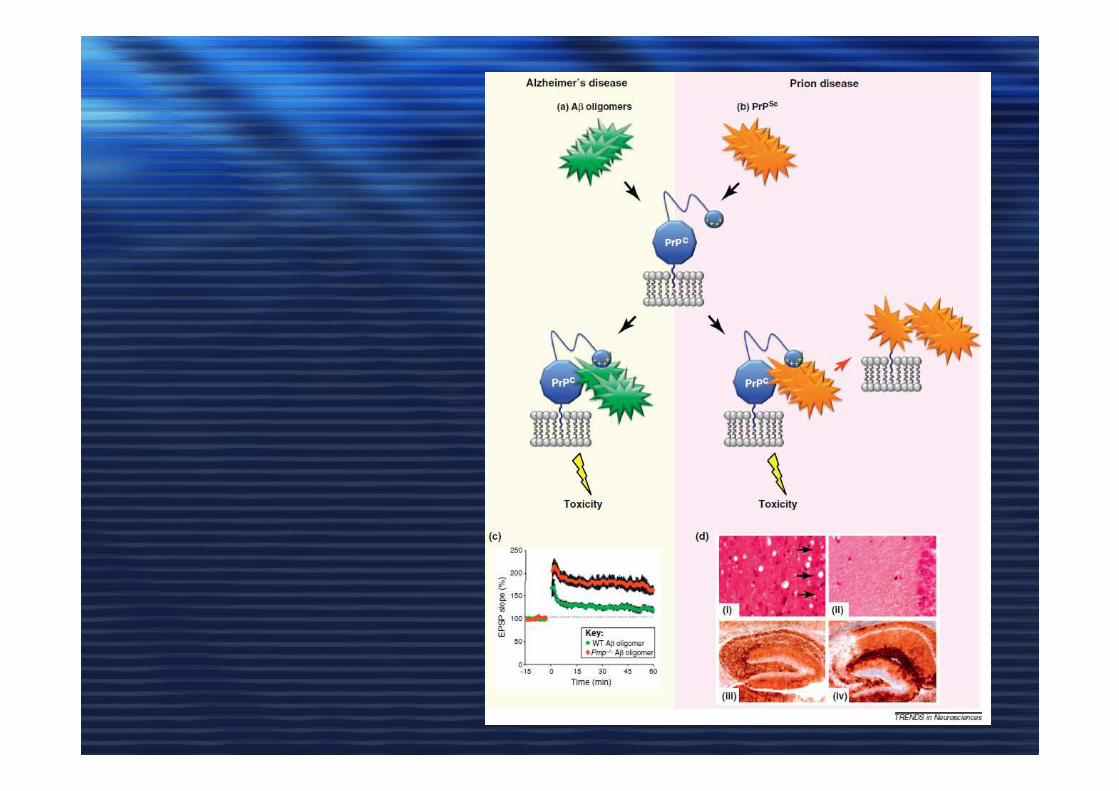
PrP cellulare (PrPc) e PrP scrapie (PrPSc)
-glicoproteina di 209 aa (30 KDa)
-codificata dal gene PRNP (cromosoma 20)
-ancora GPI
-regione N-terminale insolubile in H2O e C-terminale solubile
-42% α-eliche, 3% β-sheet
PrPc PrPSc
-codificata dallo stesso gene PRNP
-stesse modificazioni post-traduzionali
-insolubile in H2O e detergenti
-resistente a proteasi K
-forma aggregati
-30% α-eliche, 43% β-sheet
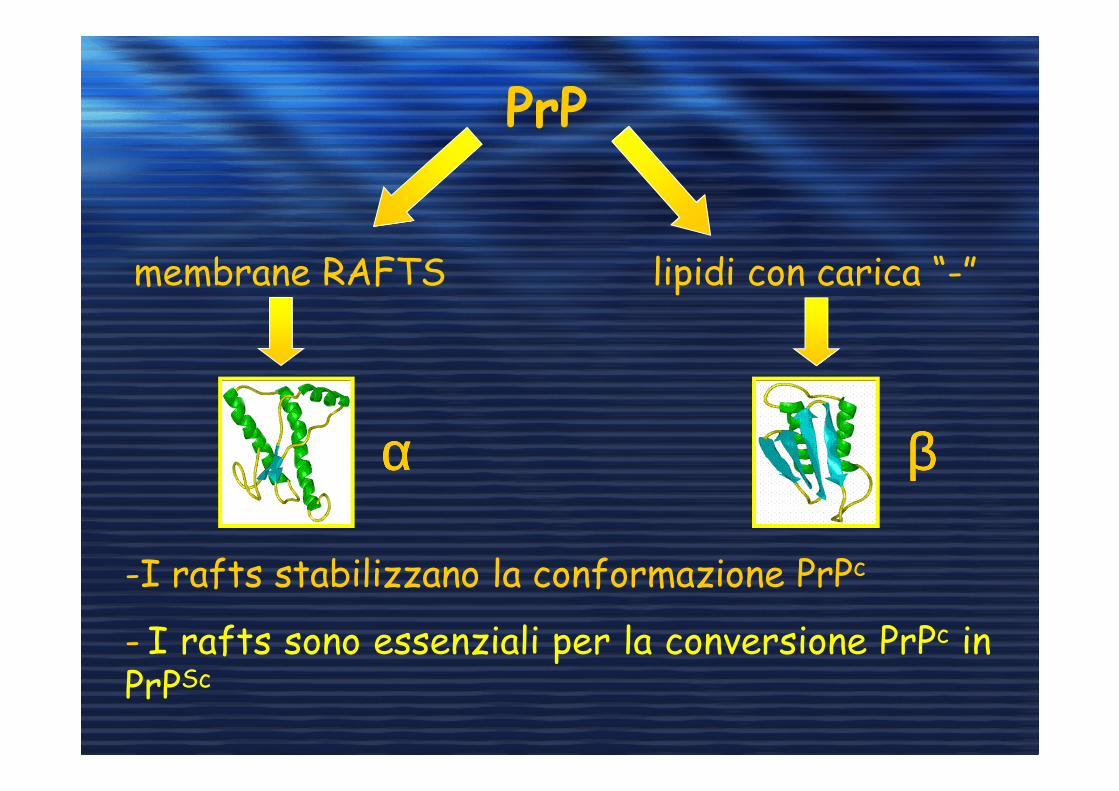
PrP
membrane RAFTS
αααα ββββ
lipidi con carica “-”
-I rafts stabilizzano la conformazione PrPc
- I rafts sono essenziali per la conversione PrPc inPrPSc
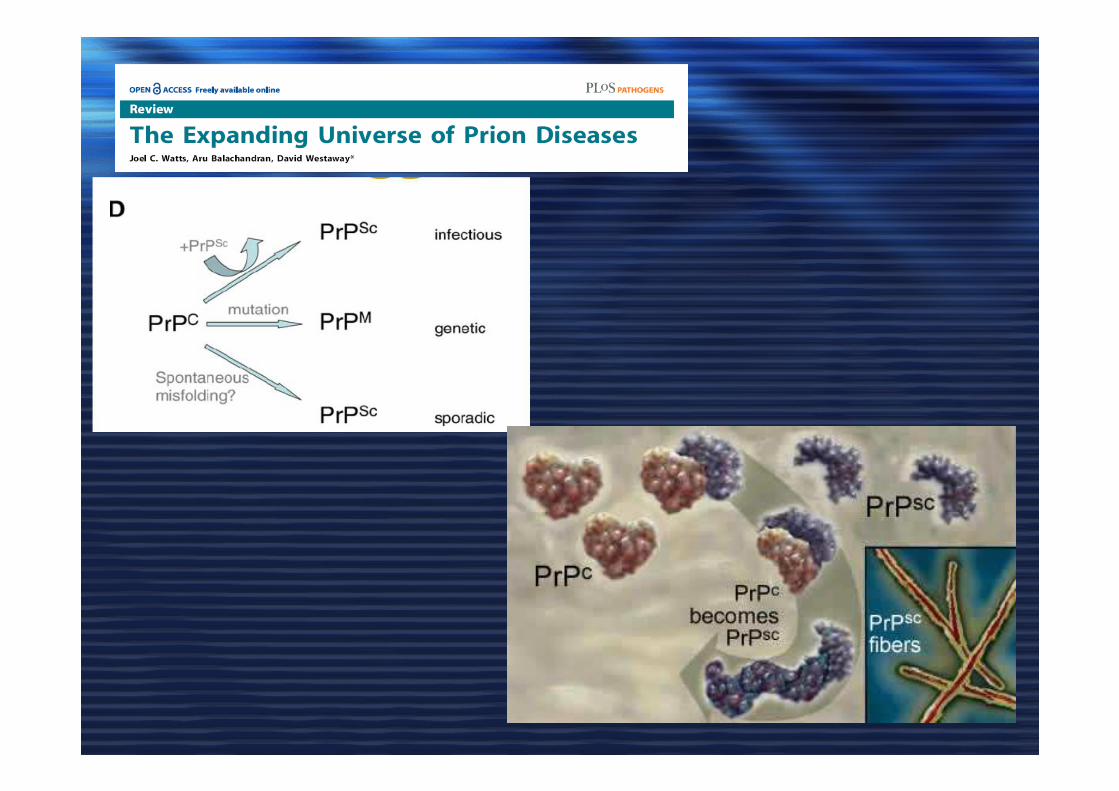
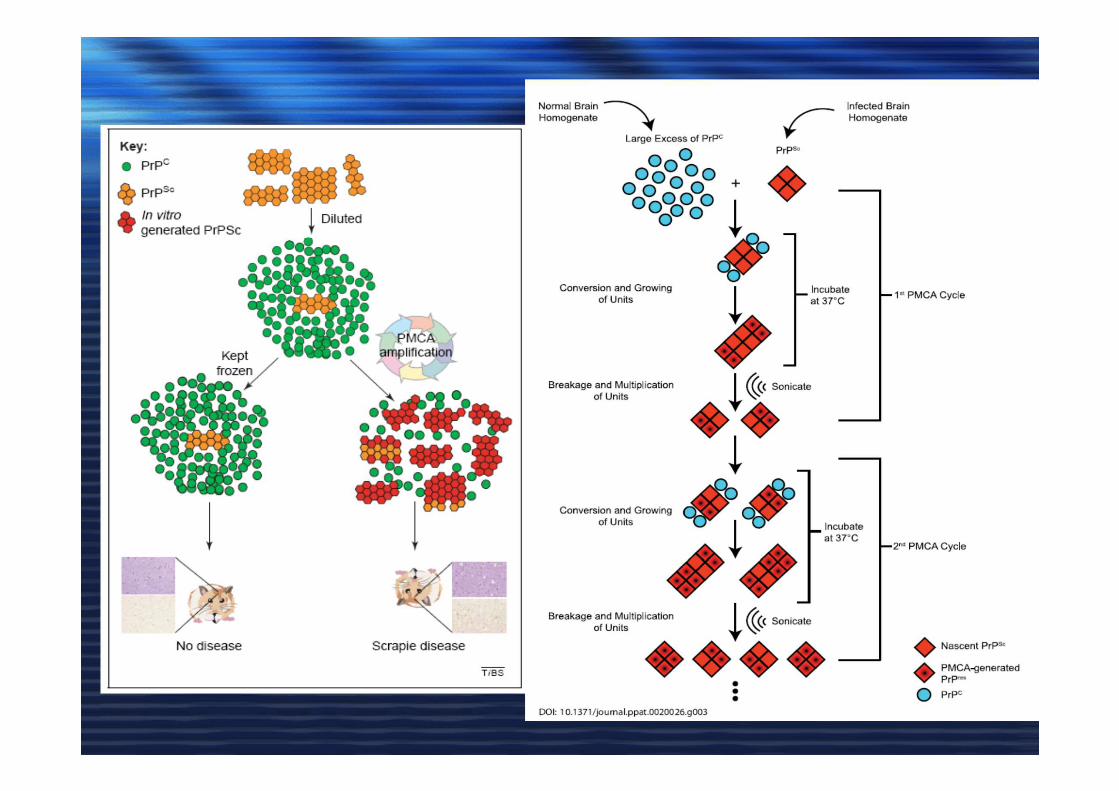
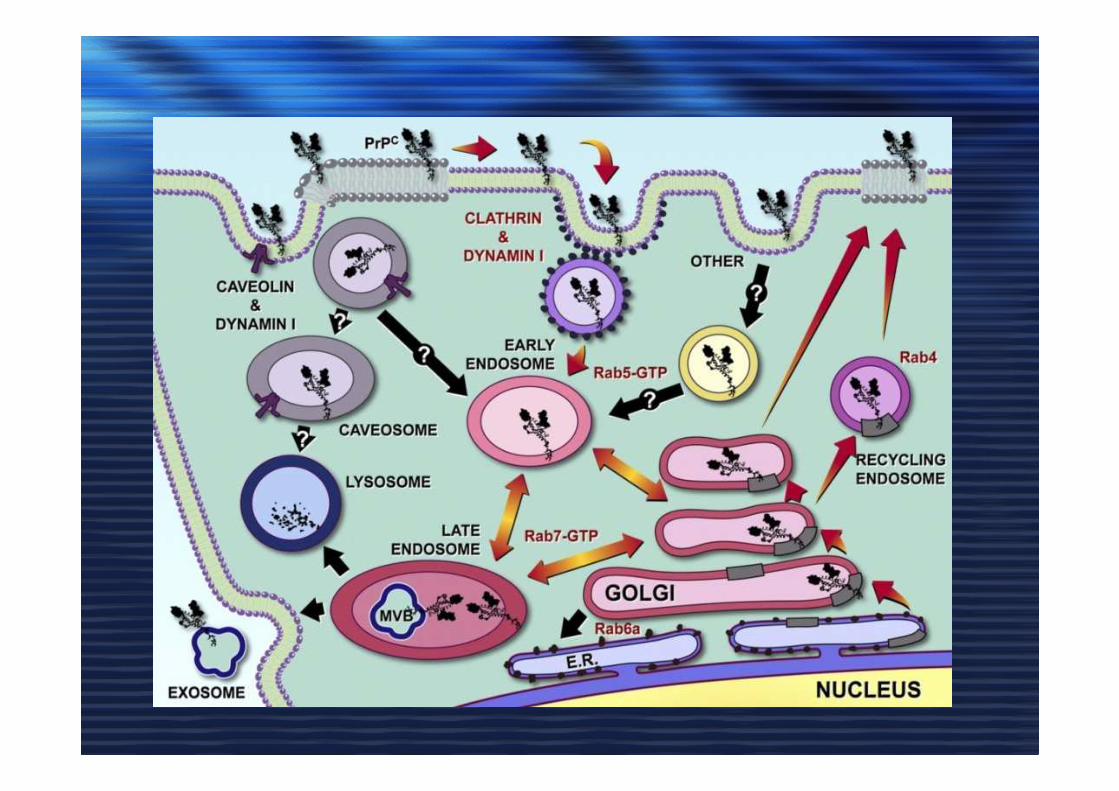

MisfoldingMisfolding
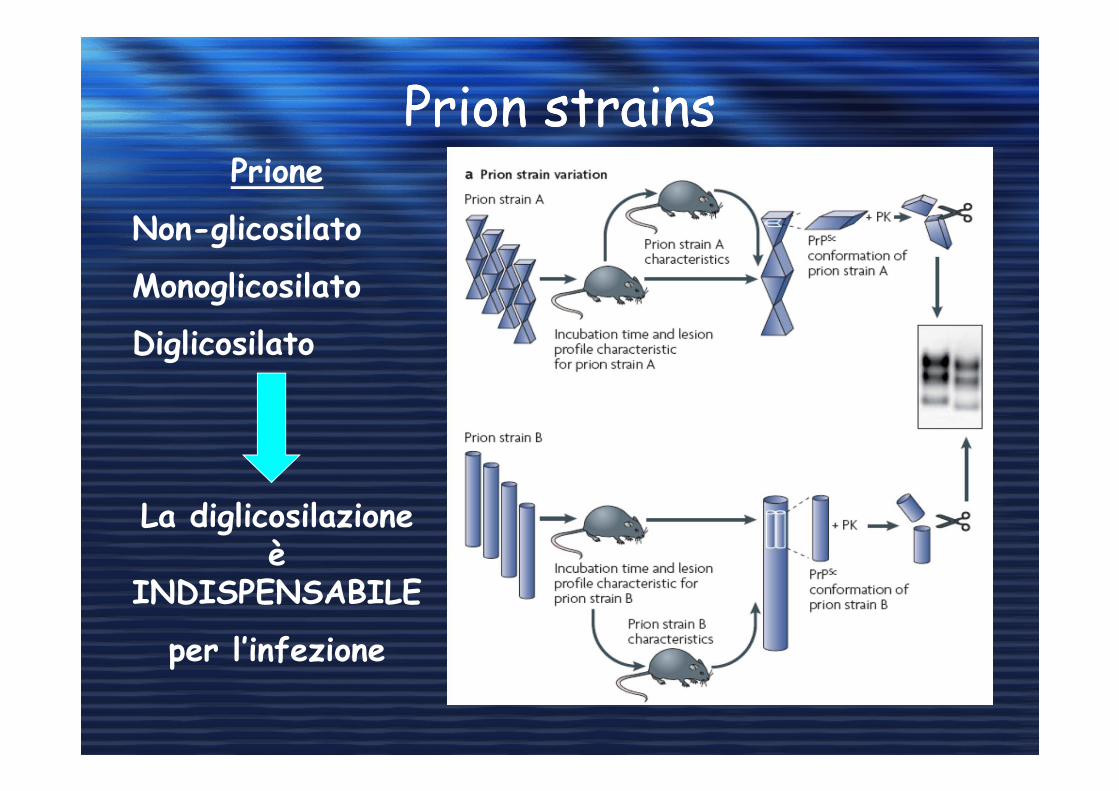
Prion strainsPrion strainsPrione
Non-glicosilato
Monoglicosilato
Diglicosilato
La diglicosilazione è
INDISPENSABILE
per l’infezione
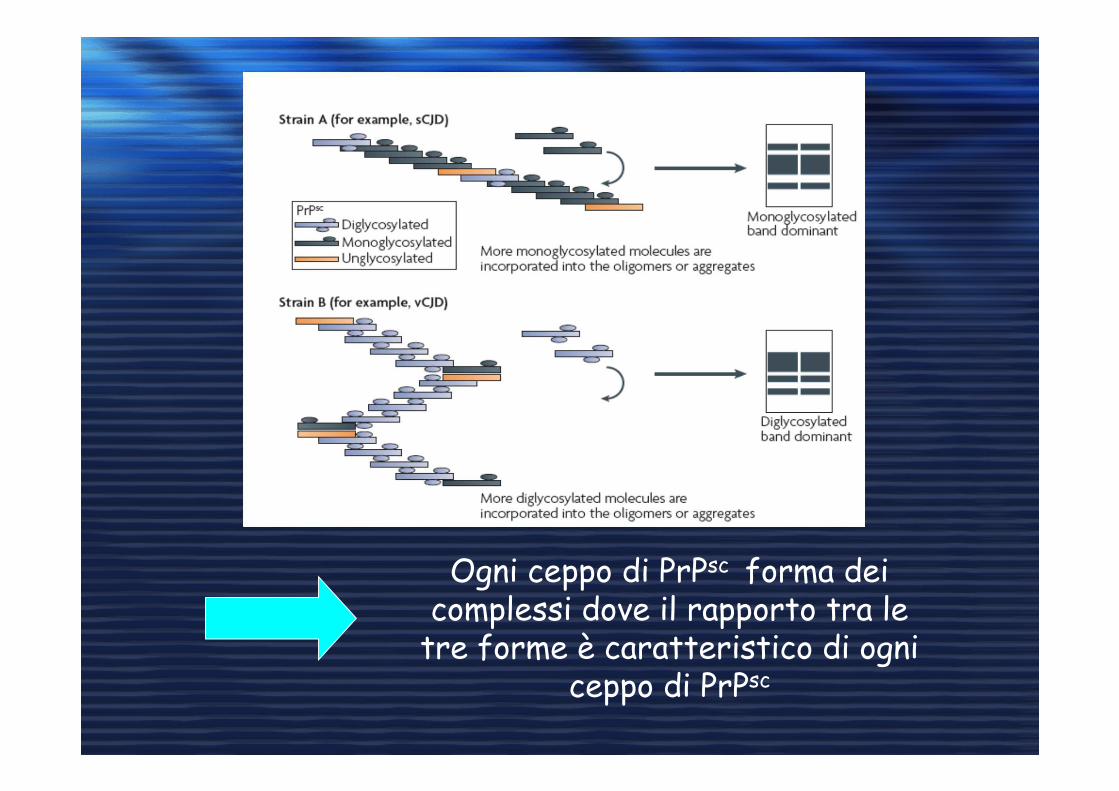
Ogni ceppo di PrPsc forma dei complessi dove il rapporto tra le
tre forme è caratteristico di ogni ceppo di PrPsc
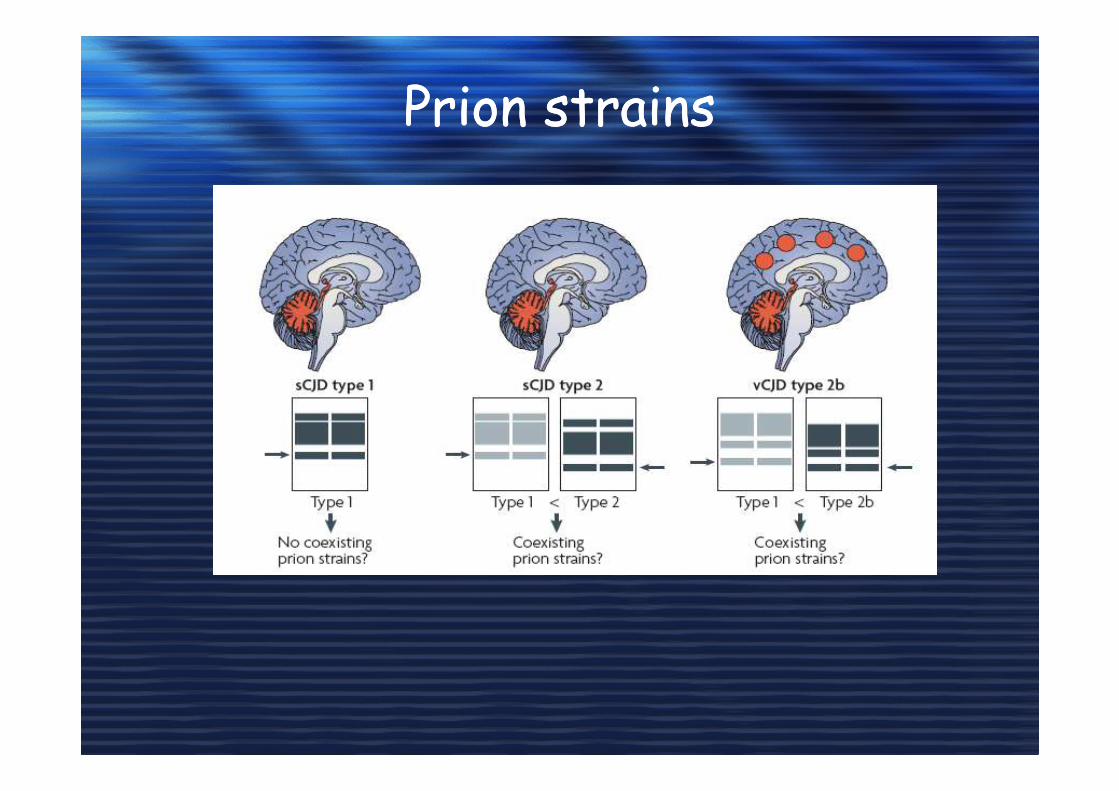
Prion strainsPrion strains
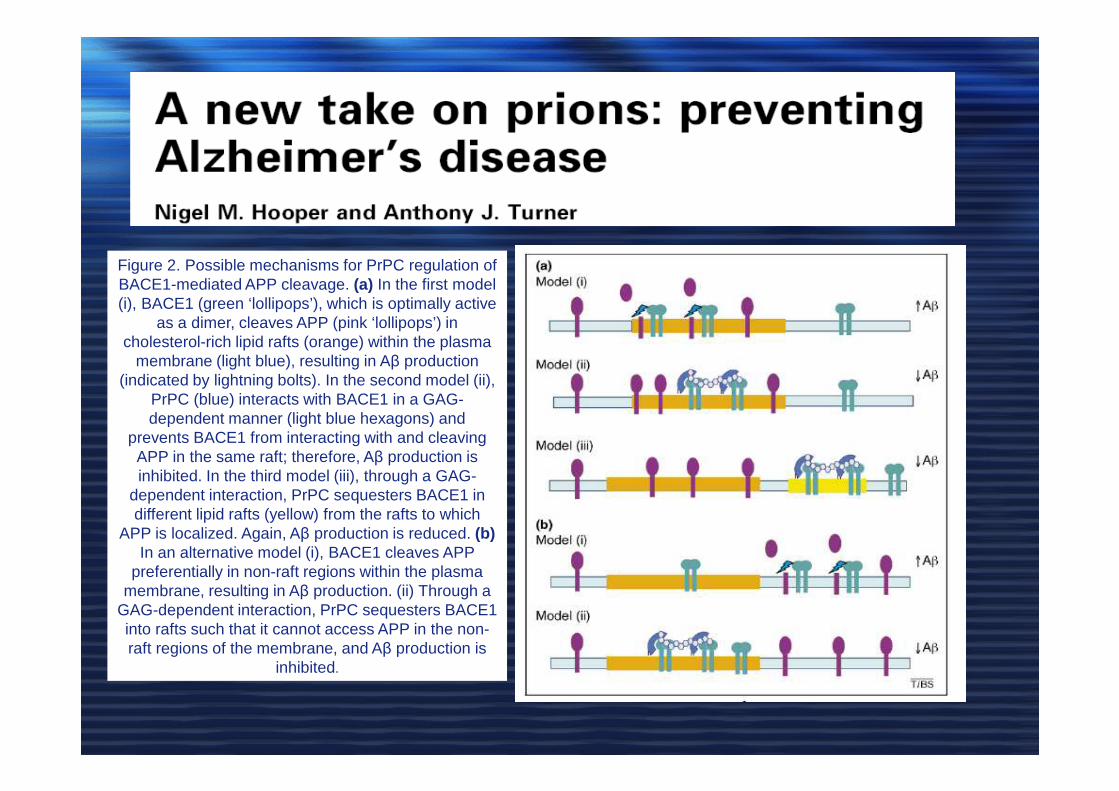
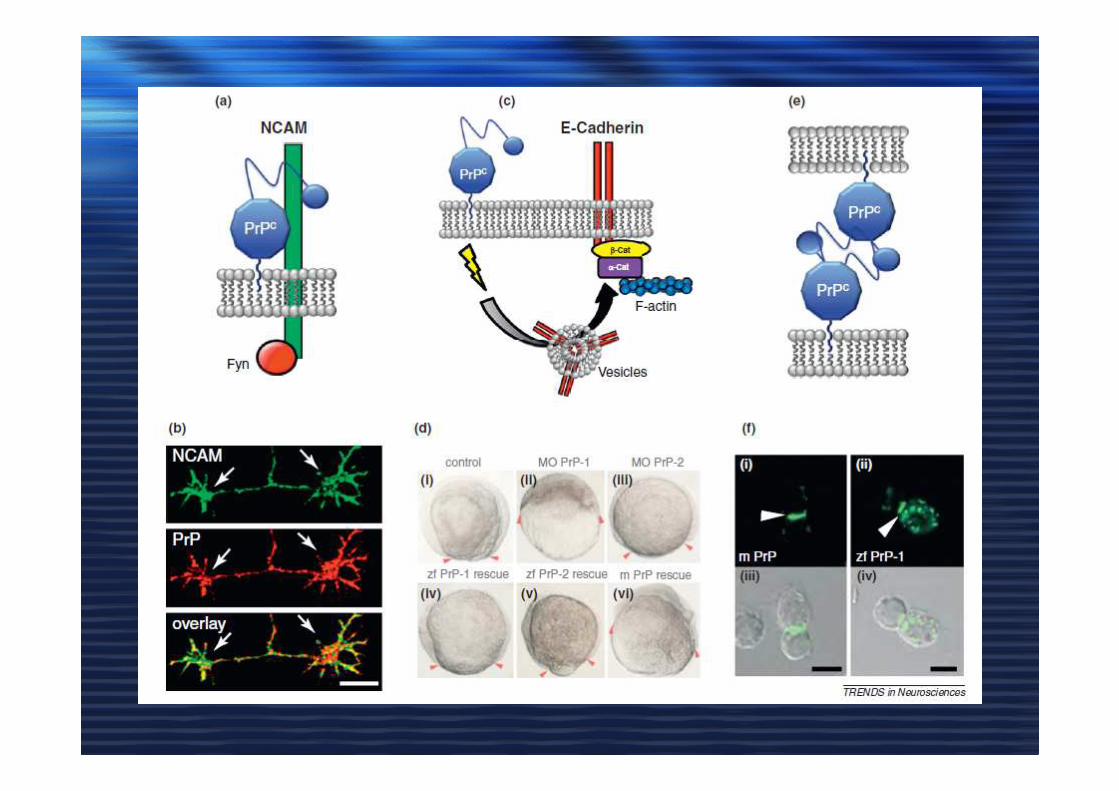
Figure 2. Possible mechanisms for PrPC regulation of BACE1-mediated APP cleavage. (a) In the first model (i), BACE1 (green ‘lollipops’), which is optimally active
as a dimer, cleaves APP (pink ‘lollipops’) in cholesterol-rich lipid rafts (orange) within the plasma
membrane (light blue), resulting in Aβ production (indicated by lightning bolts). In the second model (ii),
PrPC (blue) interacts with BACE1 in a GAG-dependent manner (light blue hexagons) and
prevents BACE1 from interacting with and cleaving APP in the same raft; therefore, Aβ production is inhibited. In the third model (iii), through a GAG-
dependent interaction, PrPC sequesters BACE1 in different lipid rafts (yellow) from the rafts to which
APP is localized. Again, Aβ production is reduced. (b)In an alternative model (i), BACE1 cleaves APP
preferentially in non-raft regions within the plasma membrane, resulting in Aβ production. (ii) Through a
GAG-dependent interaction, PrPC sequesters BACE1 into rafts such that it cannot access APP in the non-raft regions of the membrane, and Aβ production is
inhibited.
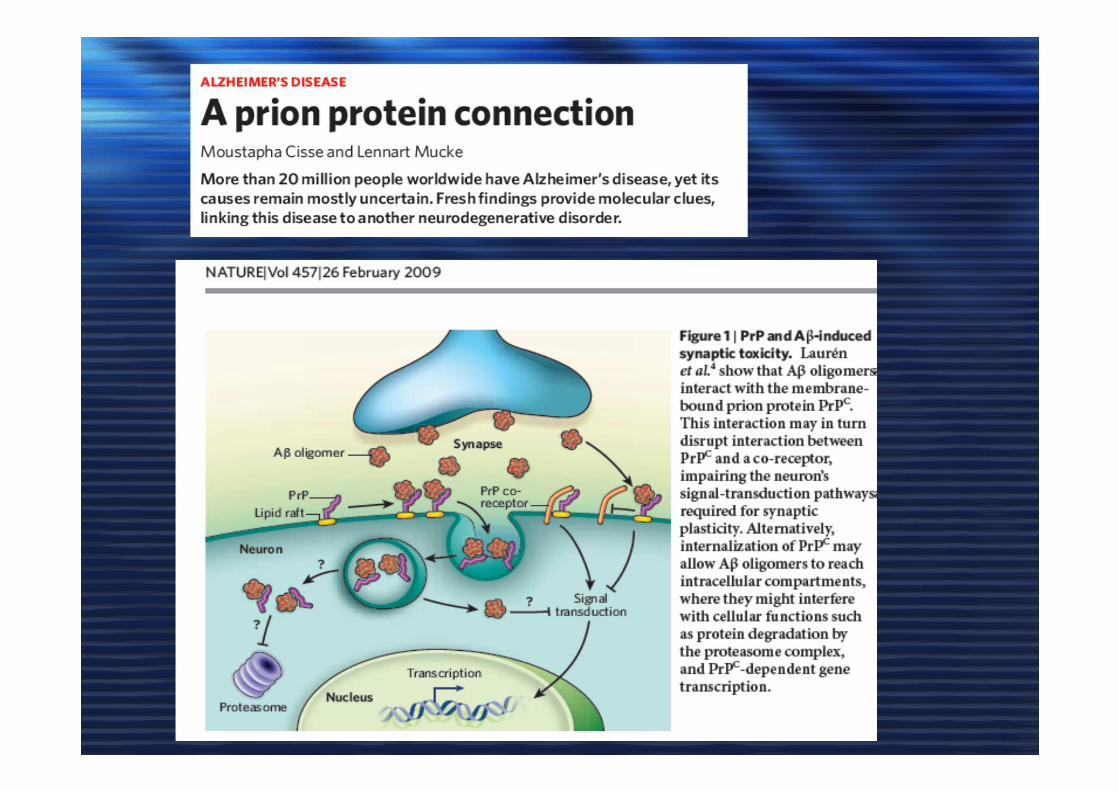
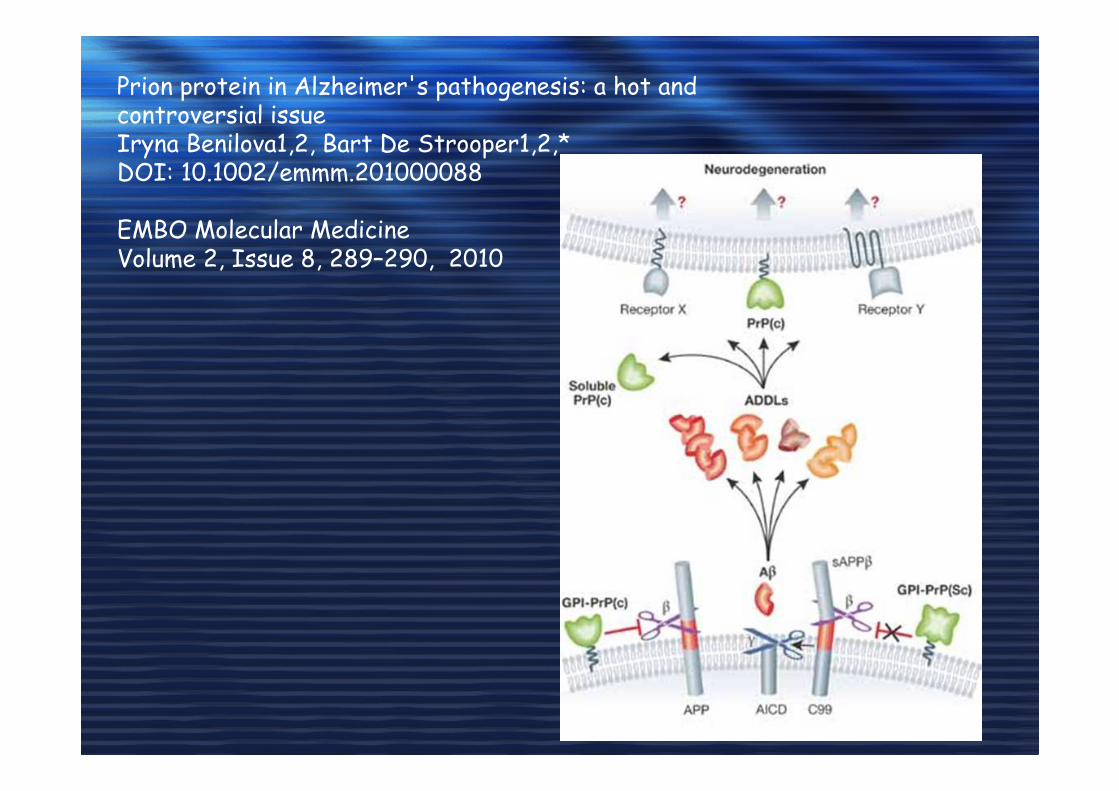
Prion protein in Alzheimer's pathogenesis: a hot and controversial issueIryna Benilova1,2, Bart De Strooper1,2,*DOI: 10.1002/emmm.201000088
EMBO Molecular MedicineVolume 2, Issue 8, 289–290, 2010

Malattie da poliglutammine PoliQMalattie da poliglutammine PoliQ
• Atassia spinocerebellare SCA• Sei diversi tipi - Ataxina 1,2,3,7 proteine solubili
• espansione della tripletta CAG > 36-45
• Huntington (40-60 anni) autosomica dominante
• Cromosoma 4 (espansione della tripletta CAG > 36)
• Disturbi del movimento (corea e bradicinesia)
• Disturbi cognitivi psichiatrici (demenza)
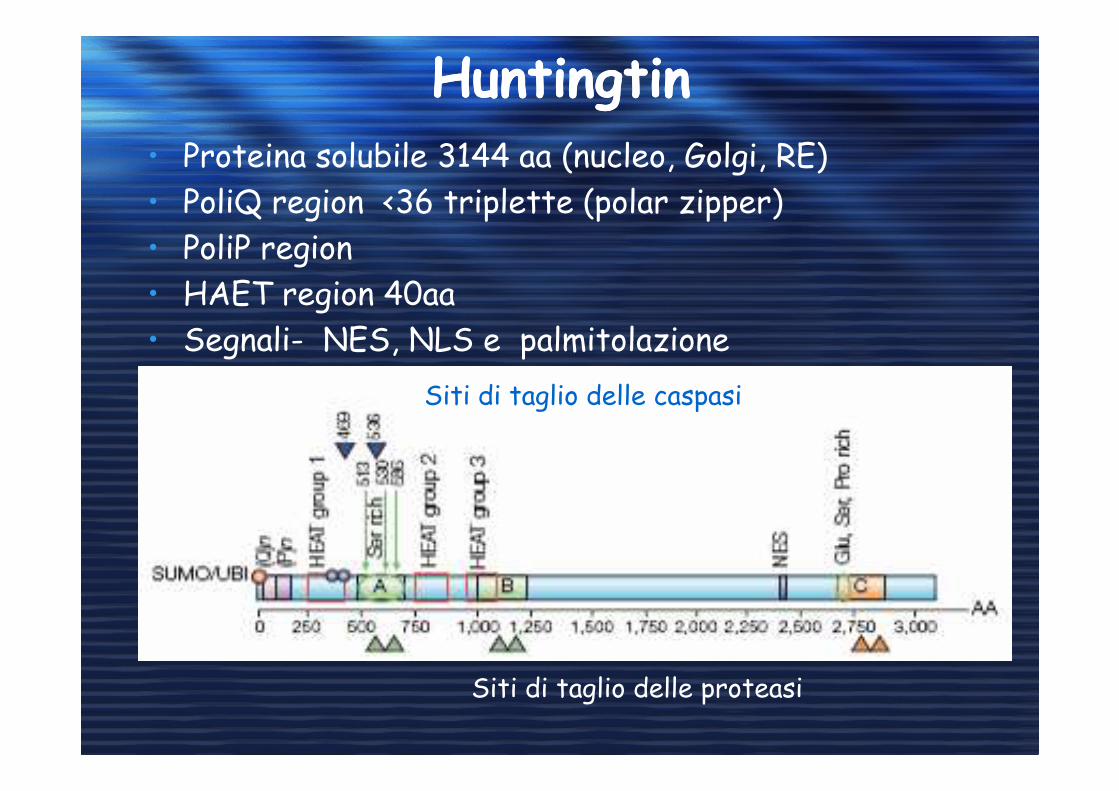
HuntingtinHuntingtin• Proteina solubile 3144 aa (nucleo, Golgi, RE)
• PoliQ region <36 triplette (polar zipper)
• PoliP region
• HAET region 40aa
• Segnali- NES, NLS e palmitolazione
Siti di taglio delle proteasi
Siti di taglio delle caspasi

Huntingtin wild-tipeHuntingtin wild-tipe
• Sviluppo embrionale (non alterato nelle poliQ expansion)
• Sviluppo del SNC
• Neuroprotettiva agli stimoli apoptotici (siero deprivazione,
ROS)
• Trasporto veloce assonale
• BDNF; incrementa la trascrizione e la quantità totale della
proteina ; stimola il trasporto vescicolare
• Implicata nella trasmissione sinaptica
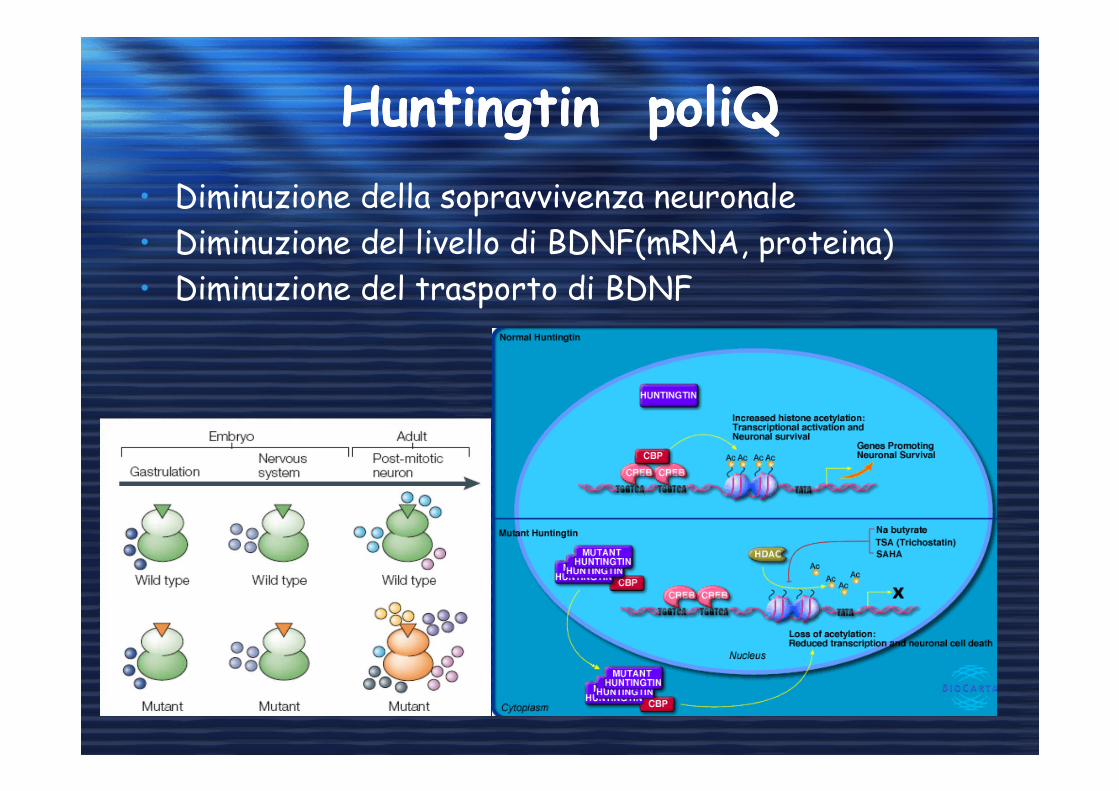
Huntingtin poliQHuntingtin poliQ
• Diminuzione della sopravvivenza neuronale
• Diminuzione del livello di BDNF(mRNA, proteina)
• Diminuzione del trasporto di BDNF
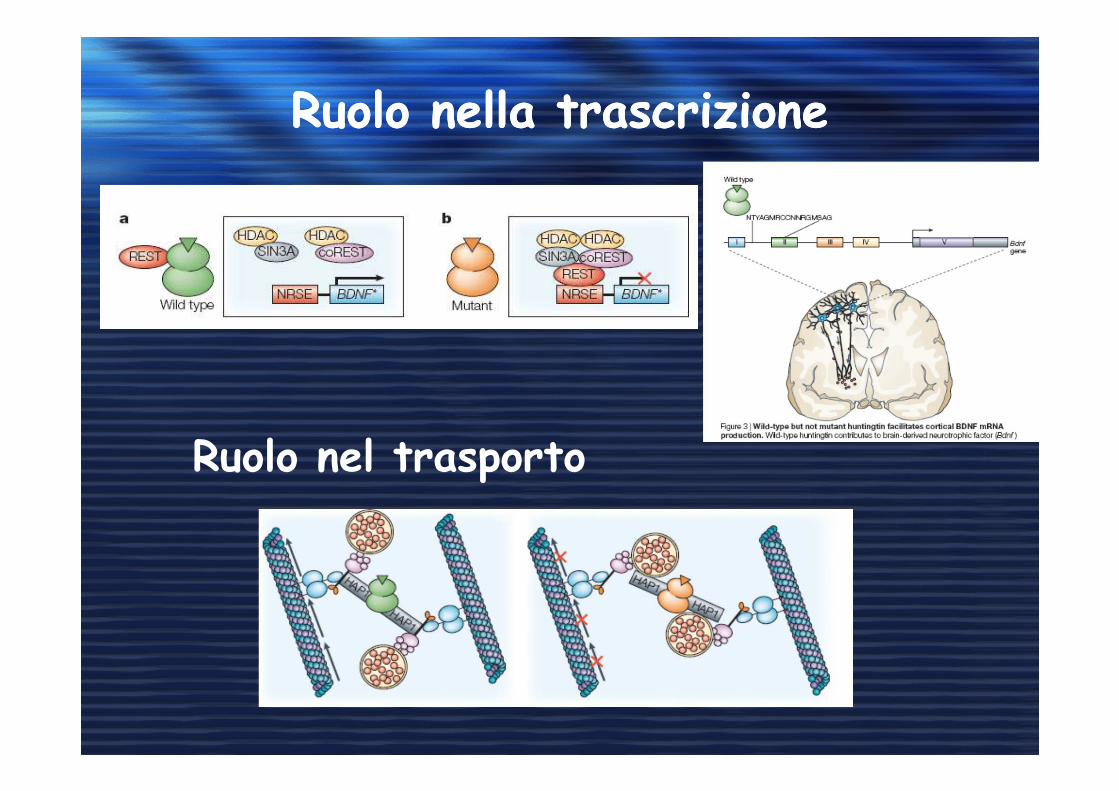
Ruolo nella trascrizioneRuolo nella trascrizione
Ruolo nel trasporto


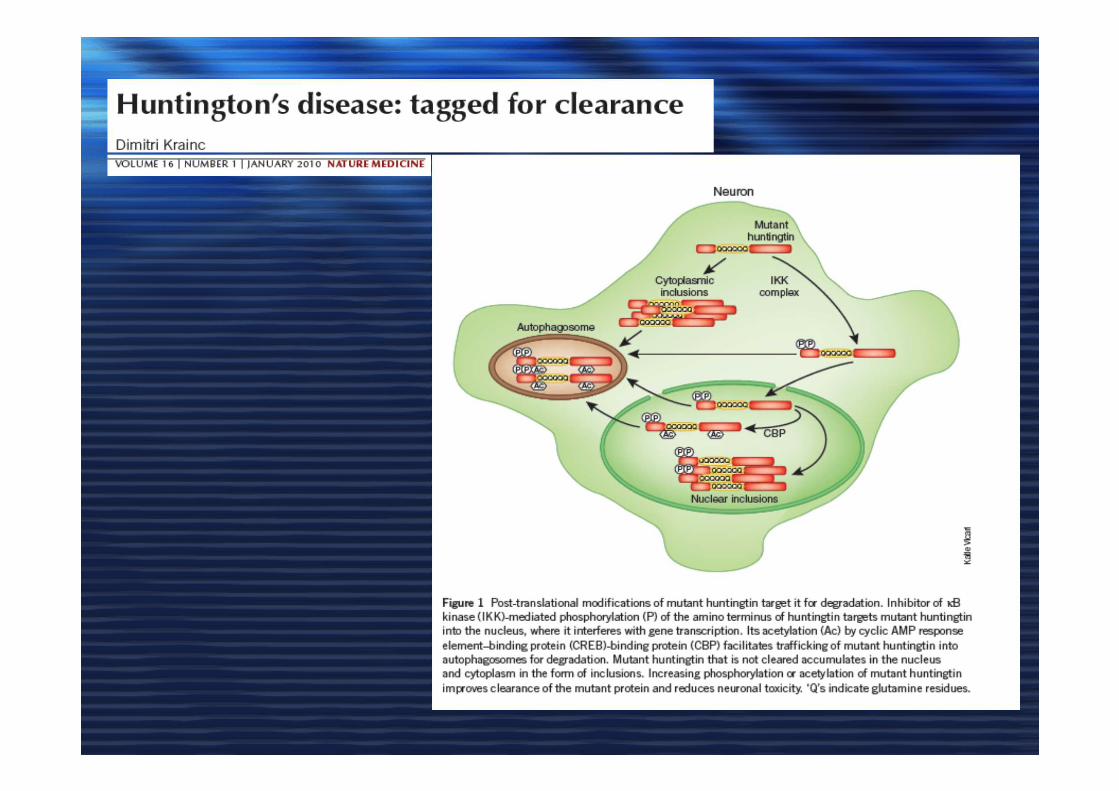
MECCANISMI IMPLICATI NELLA MORTE NEURONALE
MECCANISMI IMPLICATI NELLA MORTE NEURONALE
• AD ippocampo, amigdala, corteccia e neocorteccia
• HD neostriato, corteccia cerebrale
• TSE degenerazione spongiforme estesa e varia
APOPTOSI NEURONALE

1. IPOTESI PERDITA’ FUNZIONALE1. IPOTESI PERDITA’ FUNZIONALE
• Neurodegenerazione causata dalla perdita di
attività della proteina durante il misfolding
• HD - substrato delle caspasi-
• PrP -segnali neuroprotettivi-
• APP e α-sinucleina -regolazione apoptosi-
• PrP, Aβ - incapacità di legare metalli -
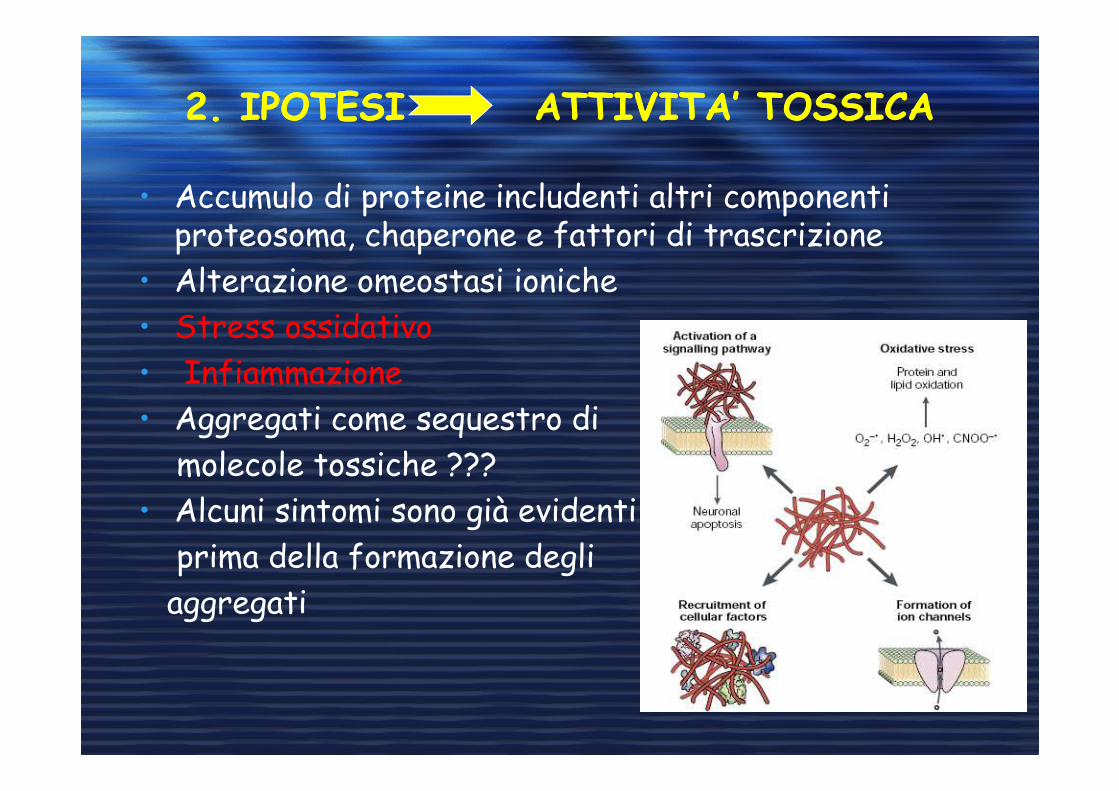
2. IPOTESI ATTIVITA’ TOSSICA2. IPOTESI ATTIVITA’ TOSSICA
• Accumulo di proteine includenti altri componenti proteosoma, chaperone e fattori di trascrizione
• Alterazione omeostasi ioniche
• Stress ossidativo
• Infiammazione
• Aggregati come sequestro di
molecole tossiche ???
• Alcuni sintomi sono già evidenti
prima della formazione degli
aggregati

3. IPOTESI SNC INFIAMMAZIONE3. IPOTESI SNC INFIAMMAZIONE
• Gli aggregati inducono una infiammazione cronica che conduce al danno neuronale
• Attivazione degli astrociti e della microglia
• Accumulo di proteine infiammatorie (citokine)
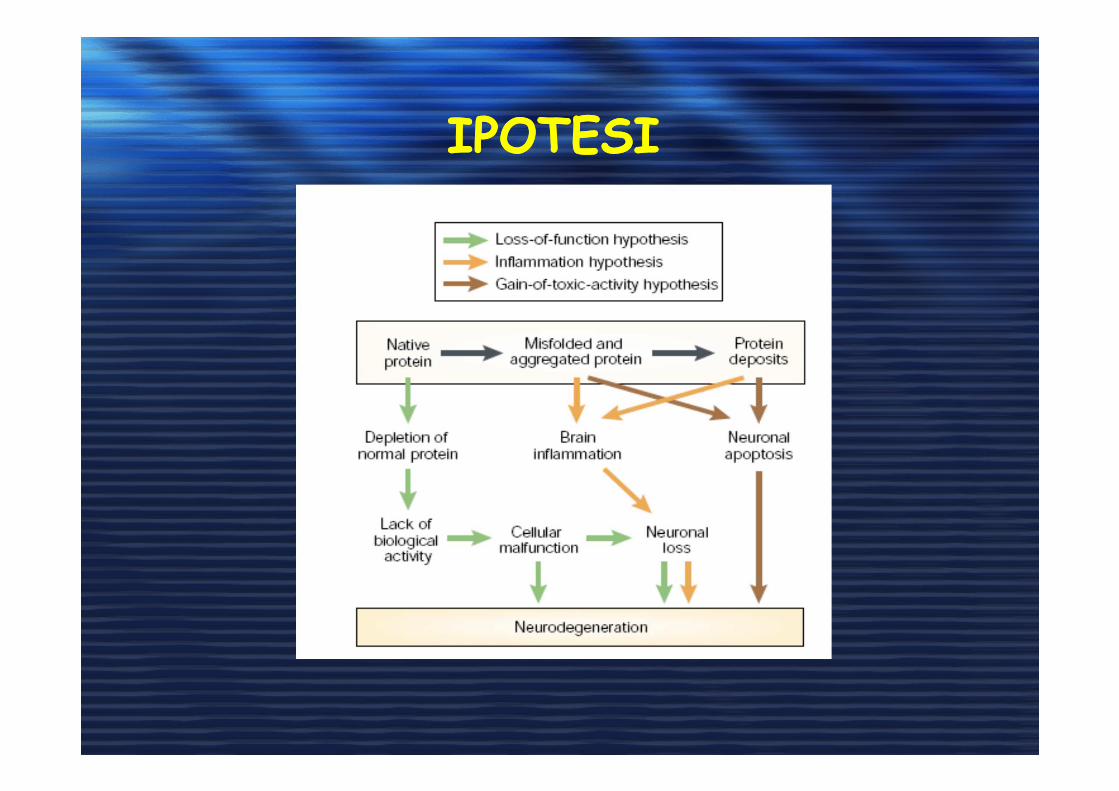
IPOTESIIPOTESI
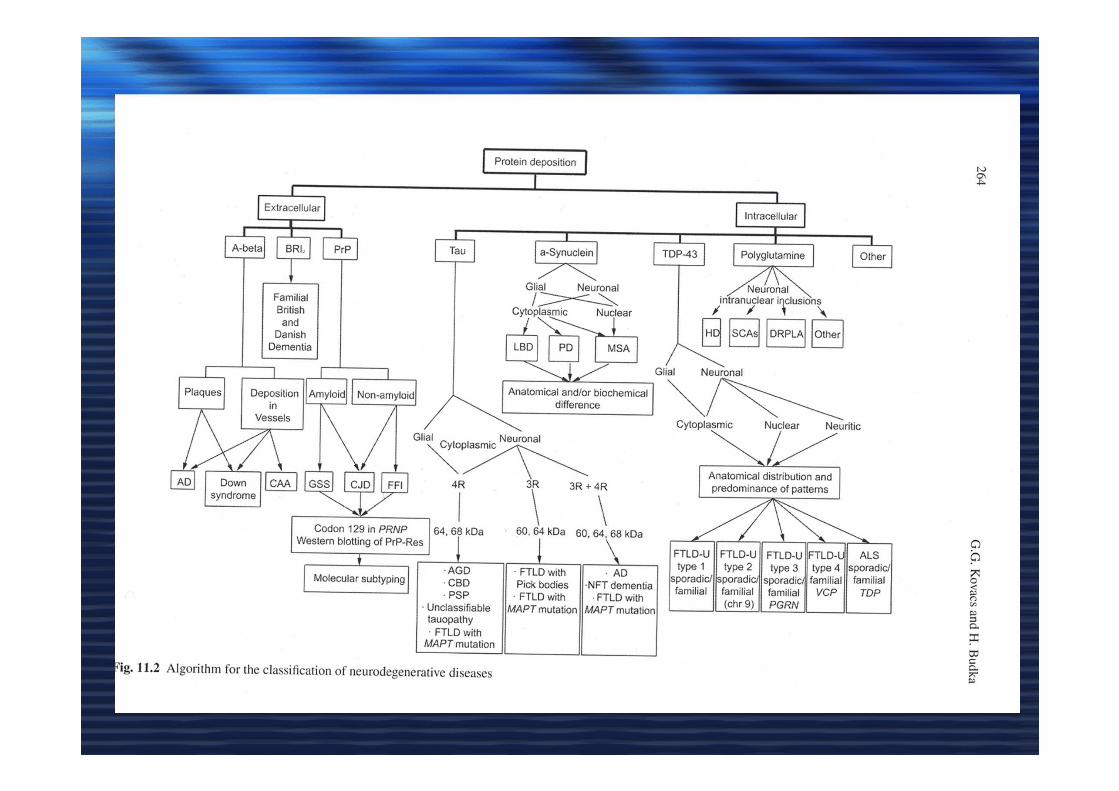

DOMANDE DOMANDE
• Le patologie neurodegenerative possono
trasmettersi mediante infezione (prion-like) ??
• La morte neuronale è la causa iniziale ????
• Gli aggregati sono stabili ??? frammentazione
• Gli aggregati sono tossici ???

A-beta, SOD1, TDP-43 Tau alfa-synuclein, poli Q Huntingtin
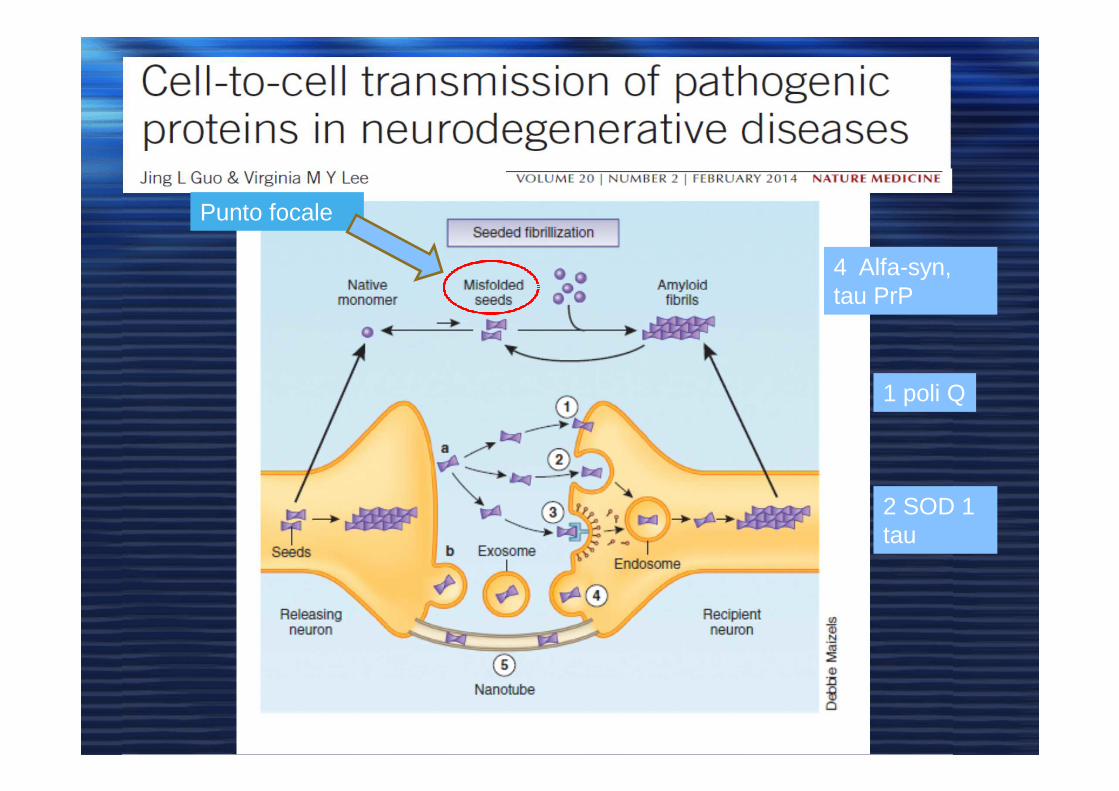
Punto focale
4 Alfa-syn, tau PrP
1 poli Q
2 SOD 1 tau

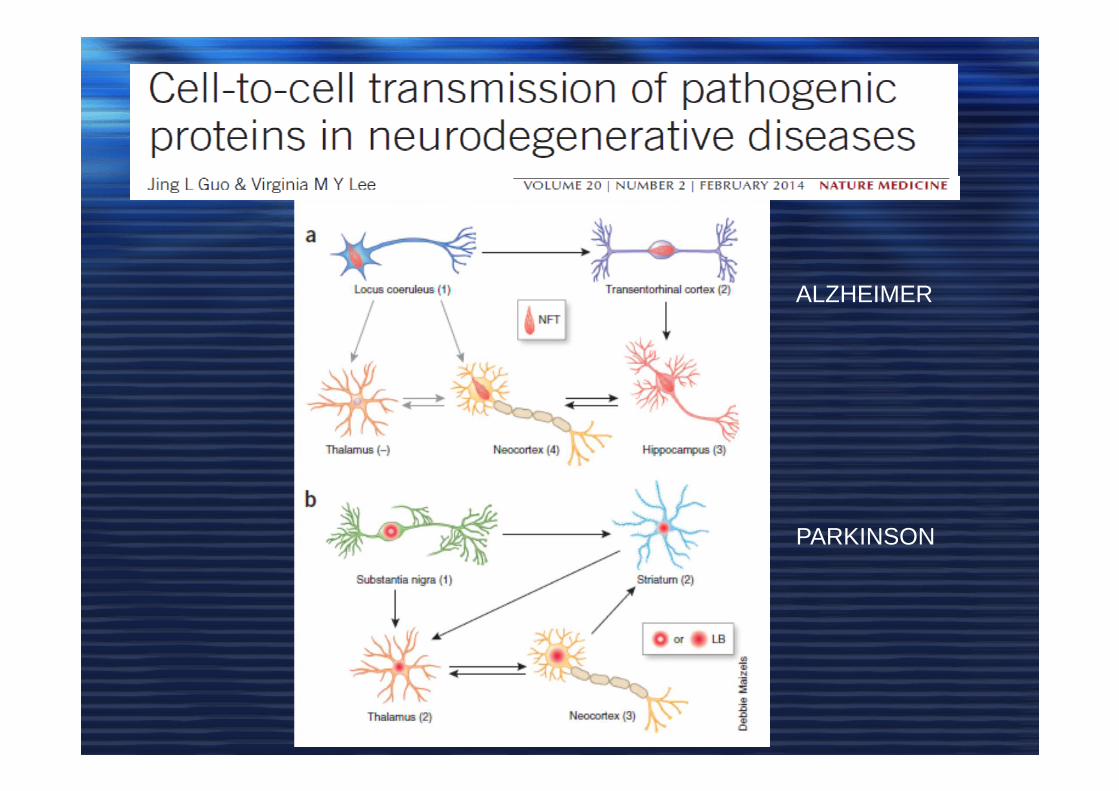
ALZHEIMER
PARKINSON
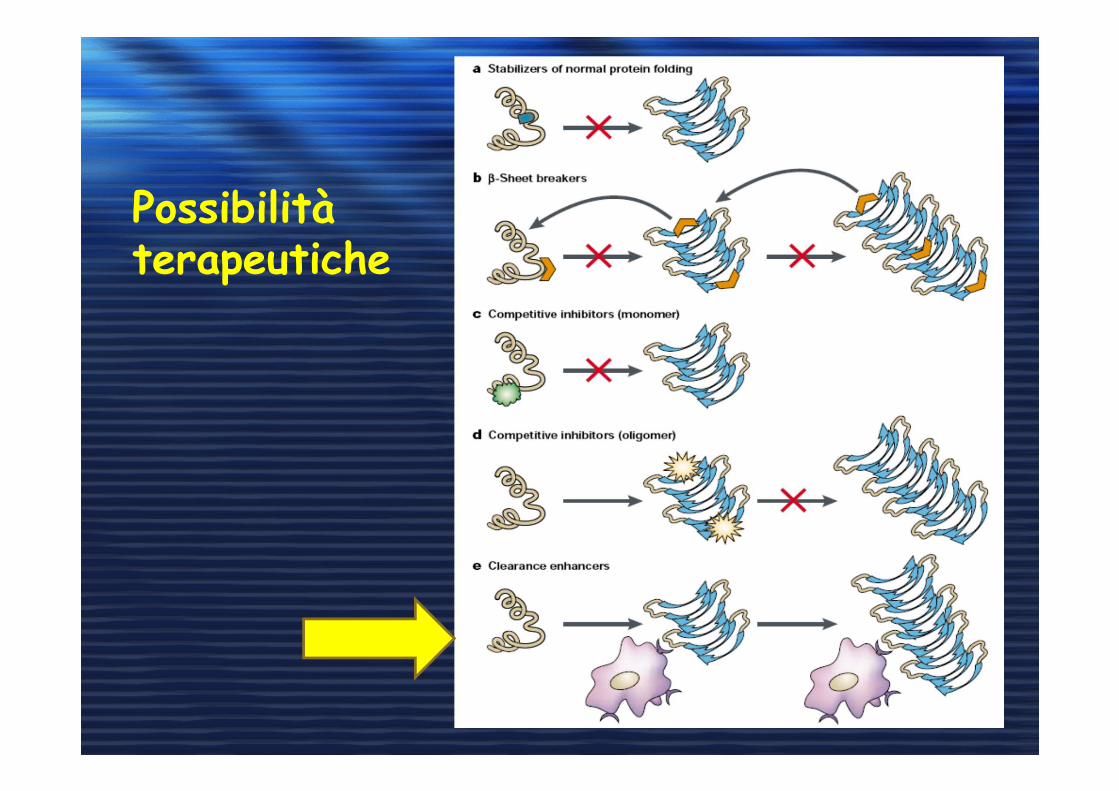
Possibilità terapeutiche

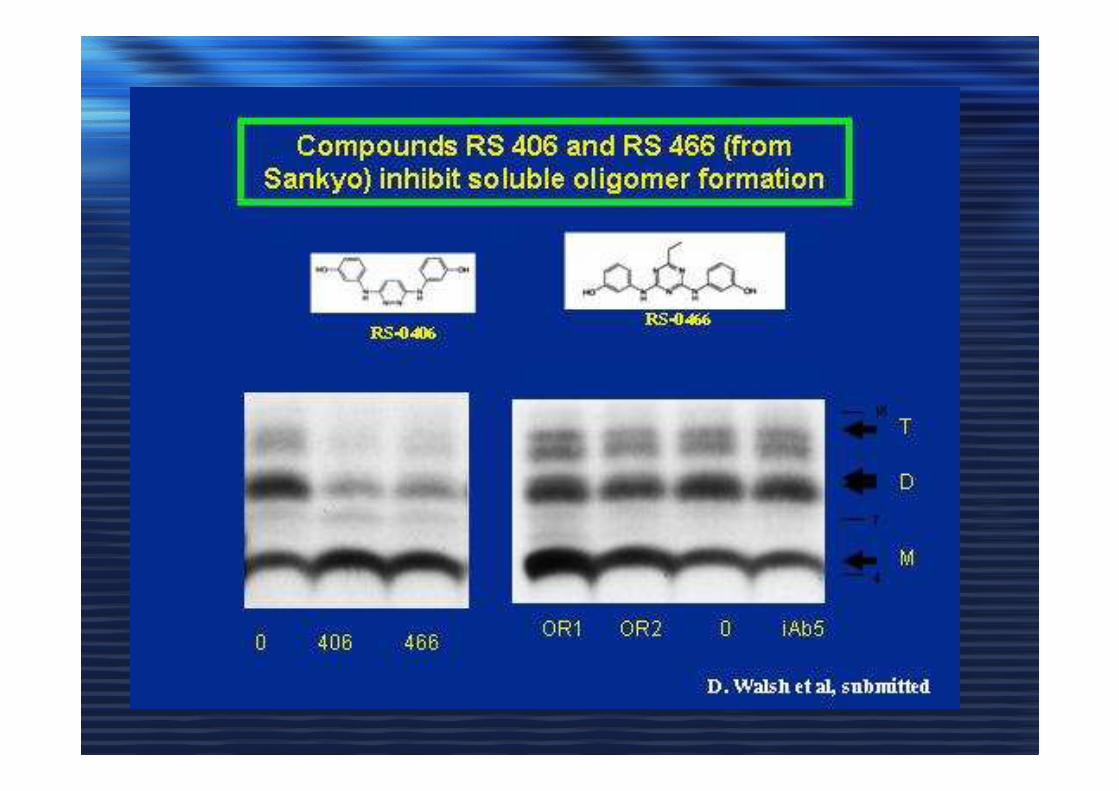
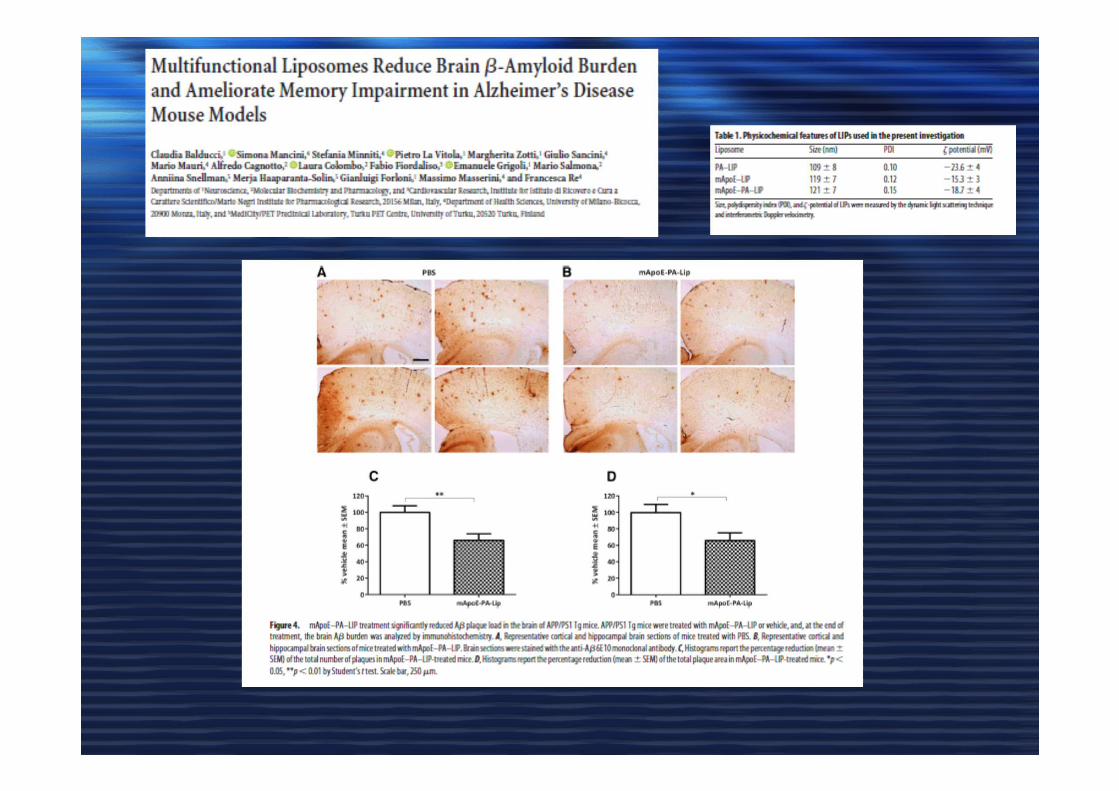


Microdomini di membranaMicrodomini di membrana
Caveolae and lipid rafts
http://web.medicina.unimib.it/palestini/

1972
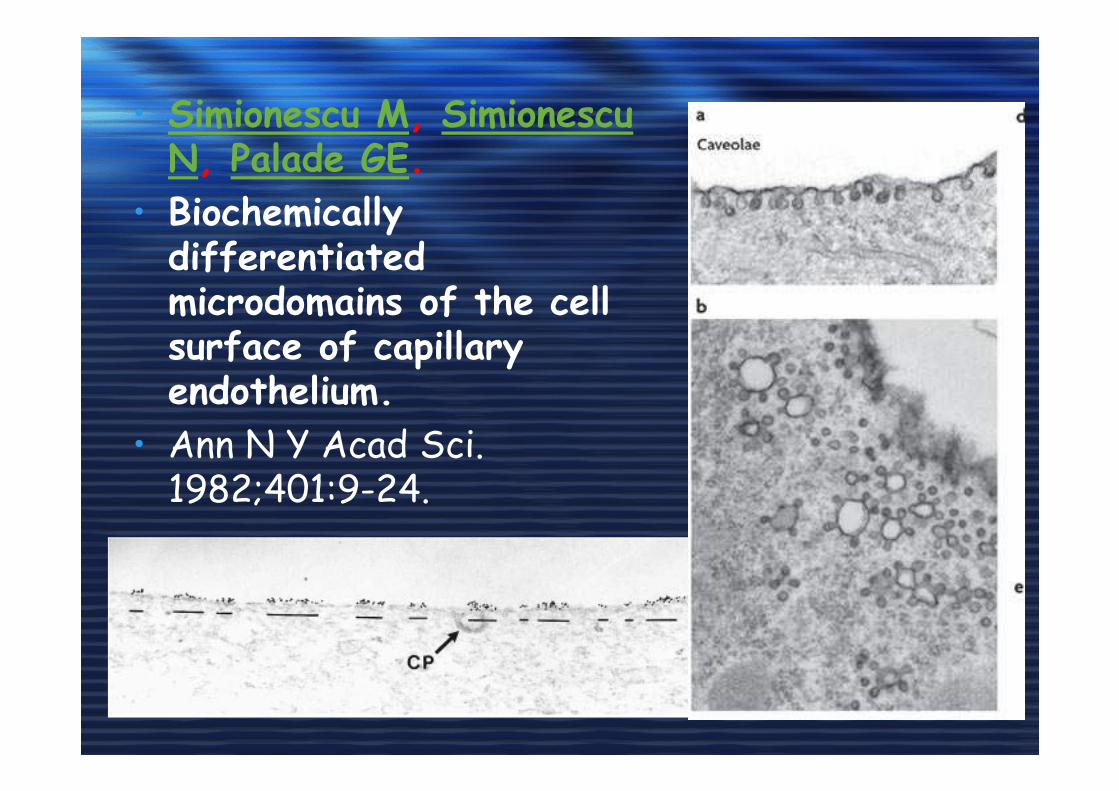
• Simionescu M, Simionescu N, Palade GE.
• Biochemically differentiated microdomains of the cell surface of capillary endothelium.
• Ann N Y Acad Sci. 1982;401:9-24.
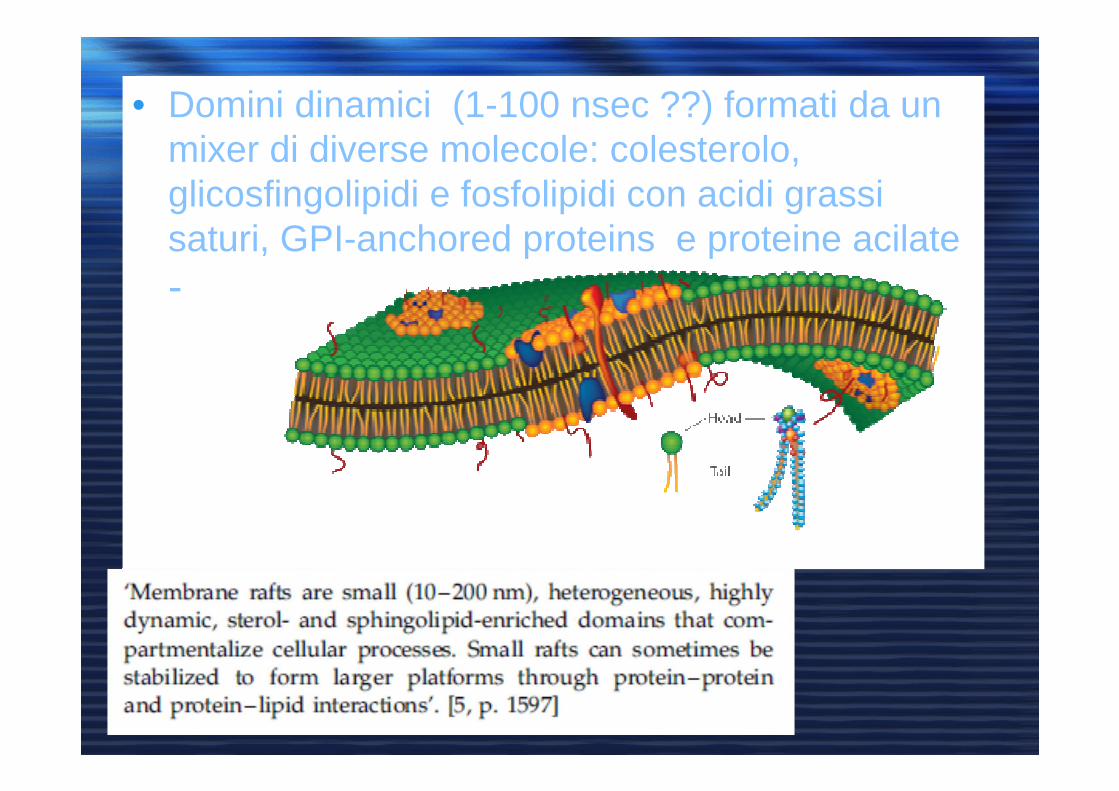
• Domini dinamici (1-100 nsec ??) formati da un mixer di diverse molecole: colesterolo, glicosfingolipidi e fosfolipidi con acidi grassi saturi, GPI-anchored proteins e proteine acilate-
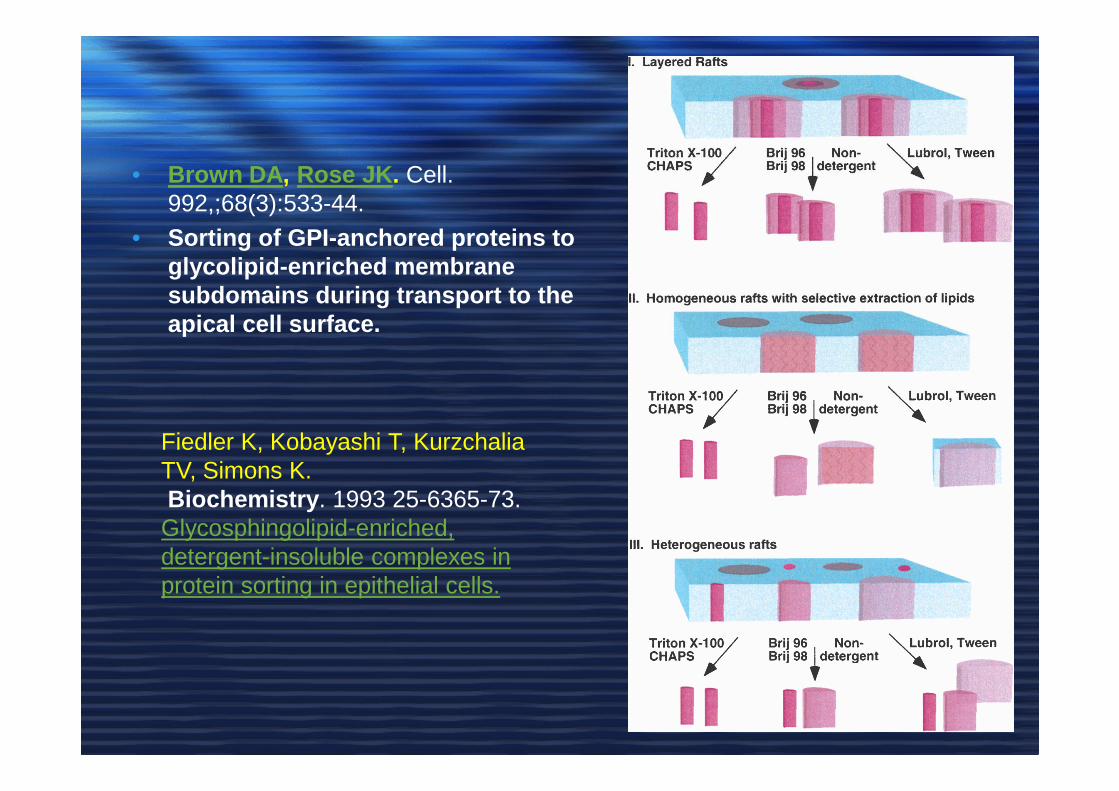
• Brown DA , Rose JK . Cell. 992,;68(3):533-44.
• Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface.
Fiedler K, Kobayashi T, Kurzchalia TV, Simons K.Biochemistry . 1993 25-6365-73.
Glycosphingolipid-enriched, detergent-insoluble complexes in protein sorting in epithelial cells.

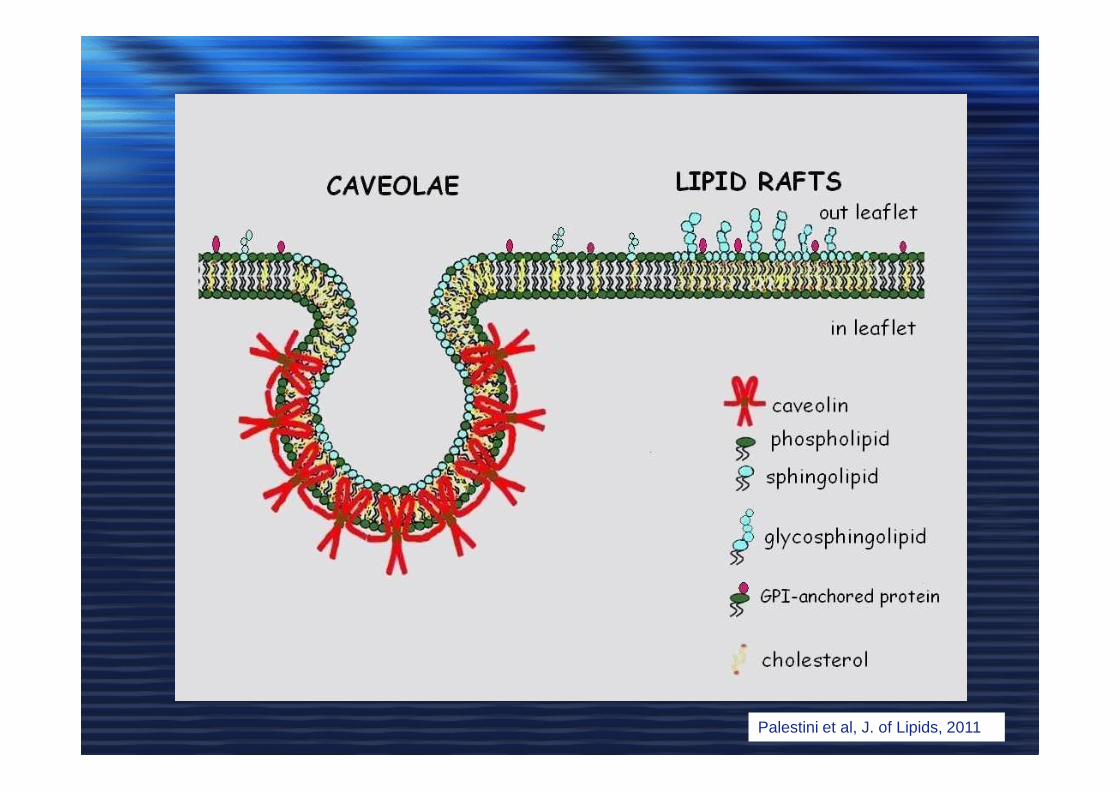
Palestini et al, J. of Lipids, 2011
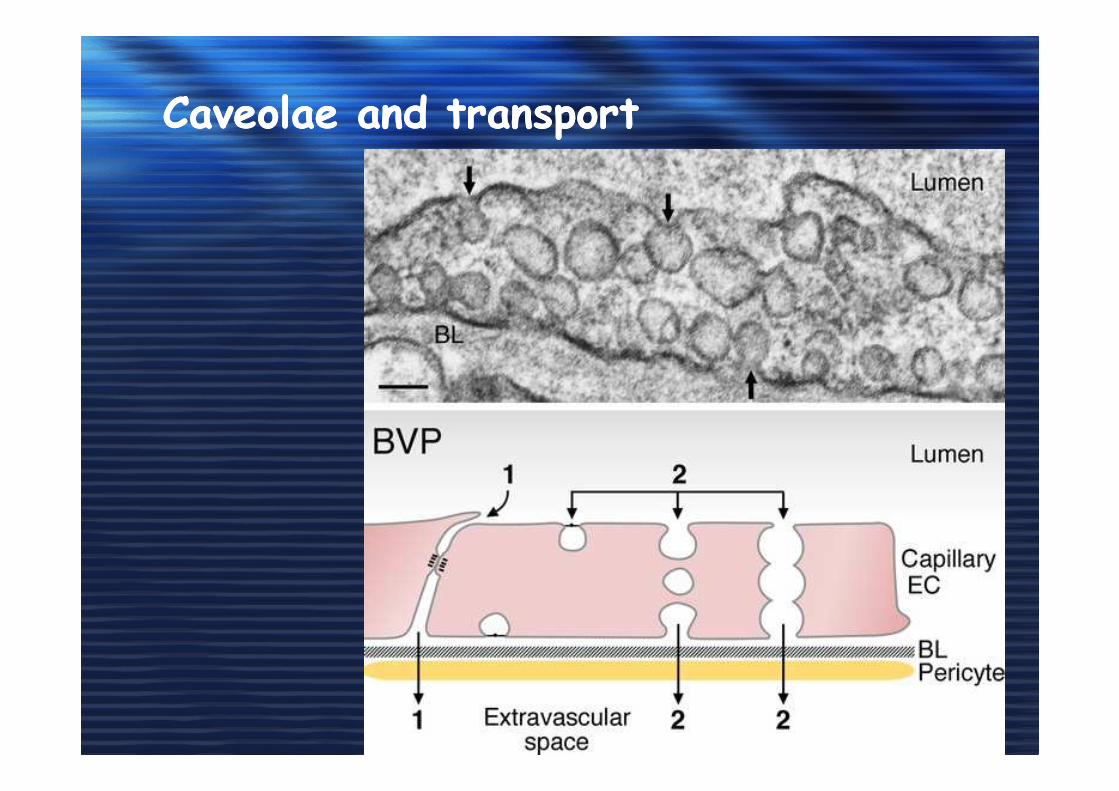
Caveolae and transportCaveolae and transport
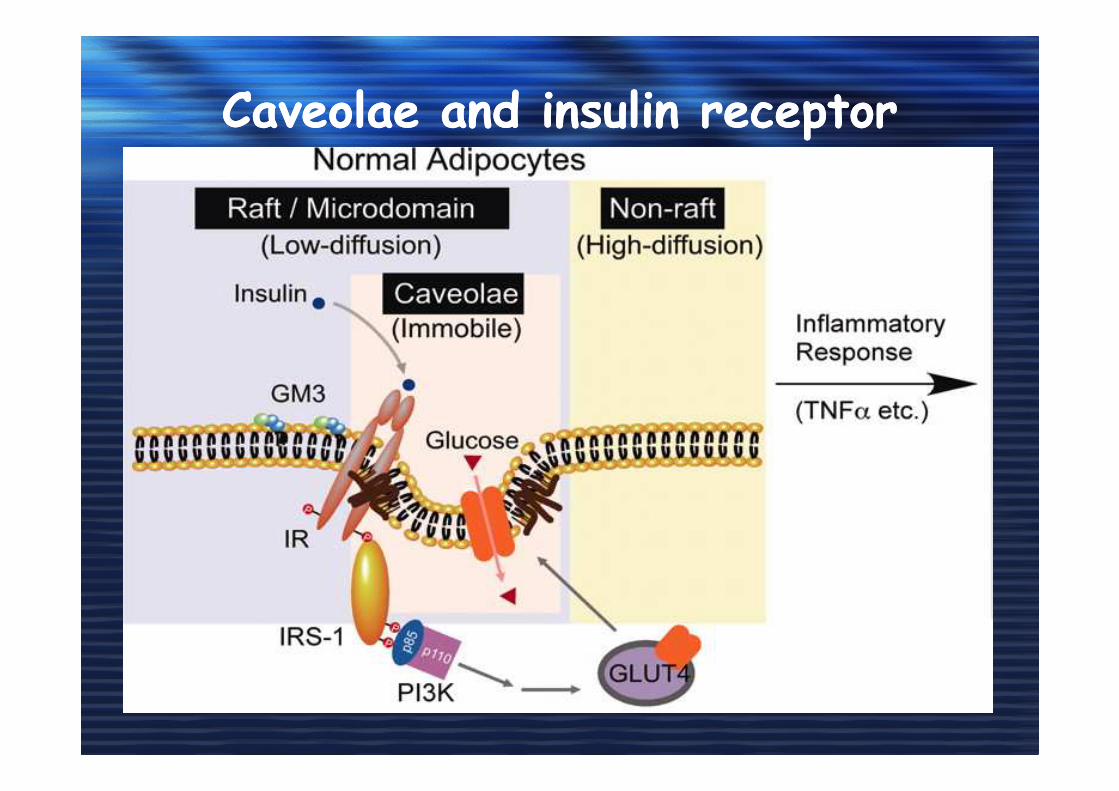
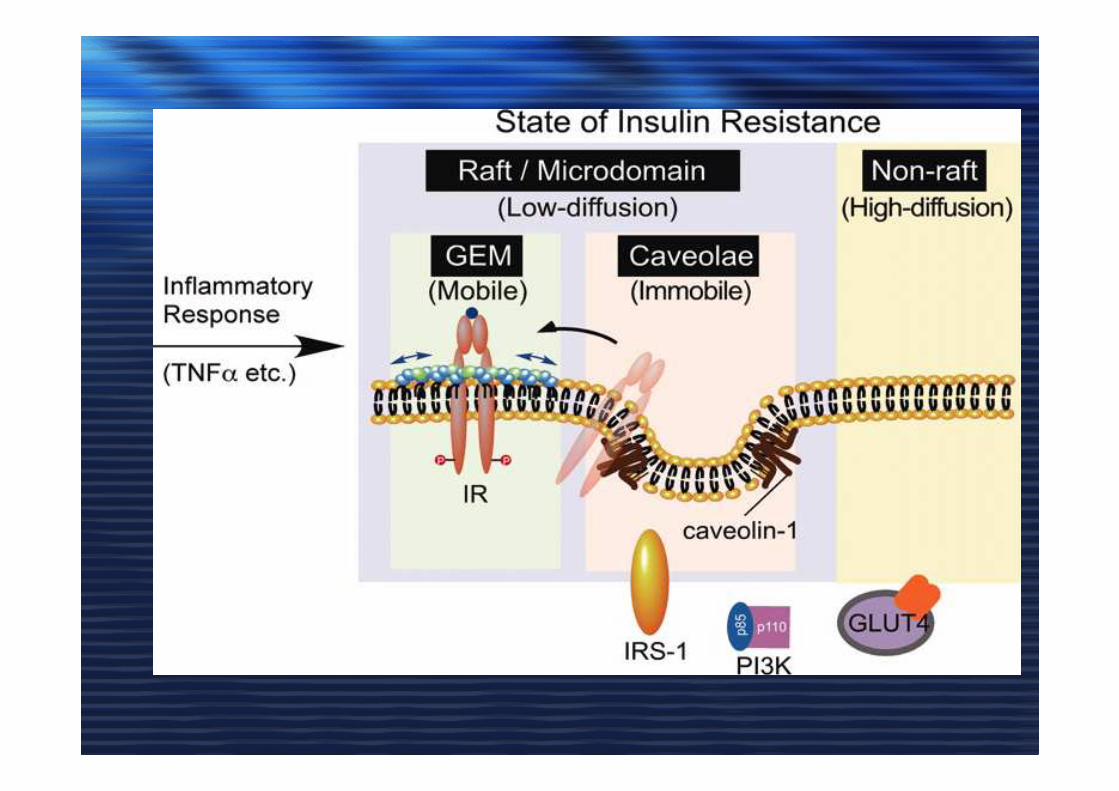
Caveolae and insulin receptorCaveolae and insulin receptor
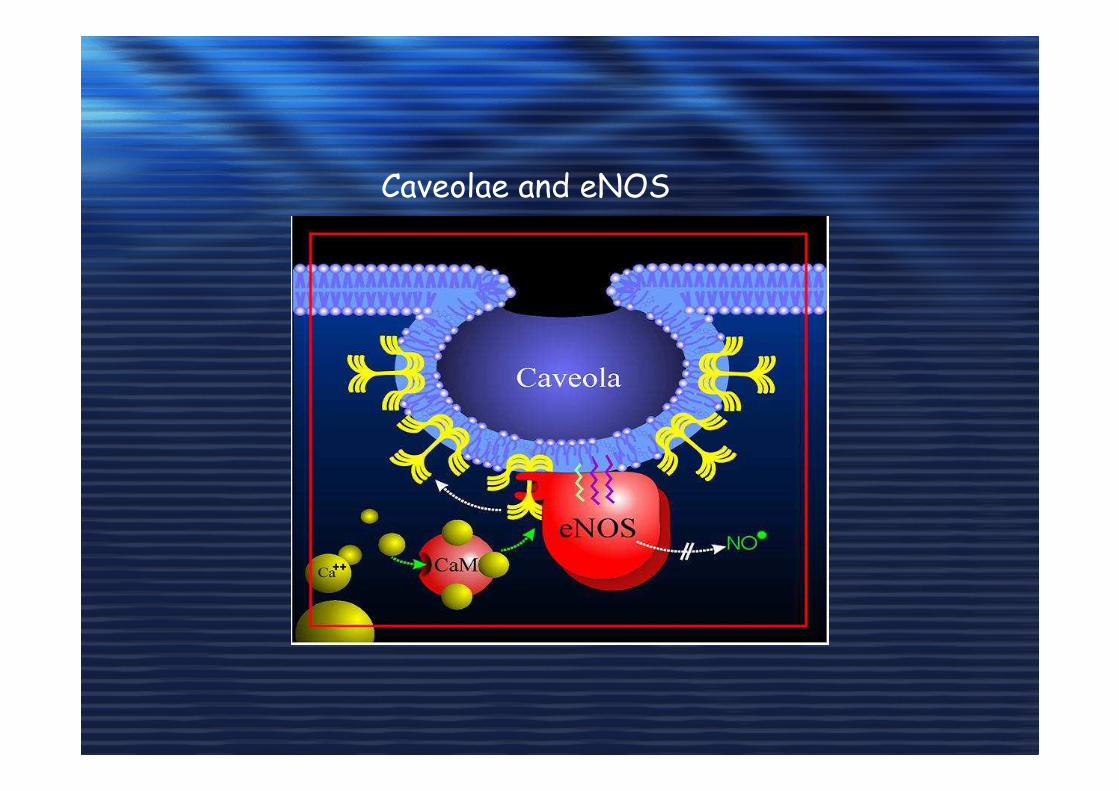
Caveolae and eNOS
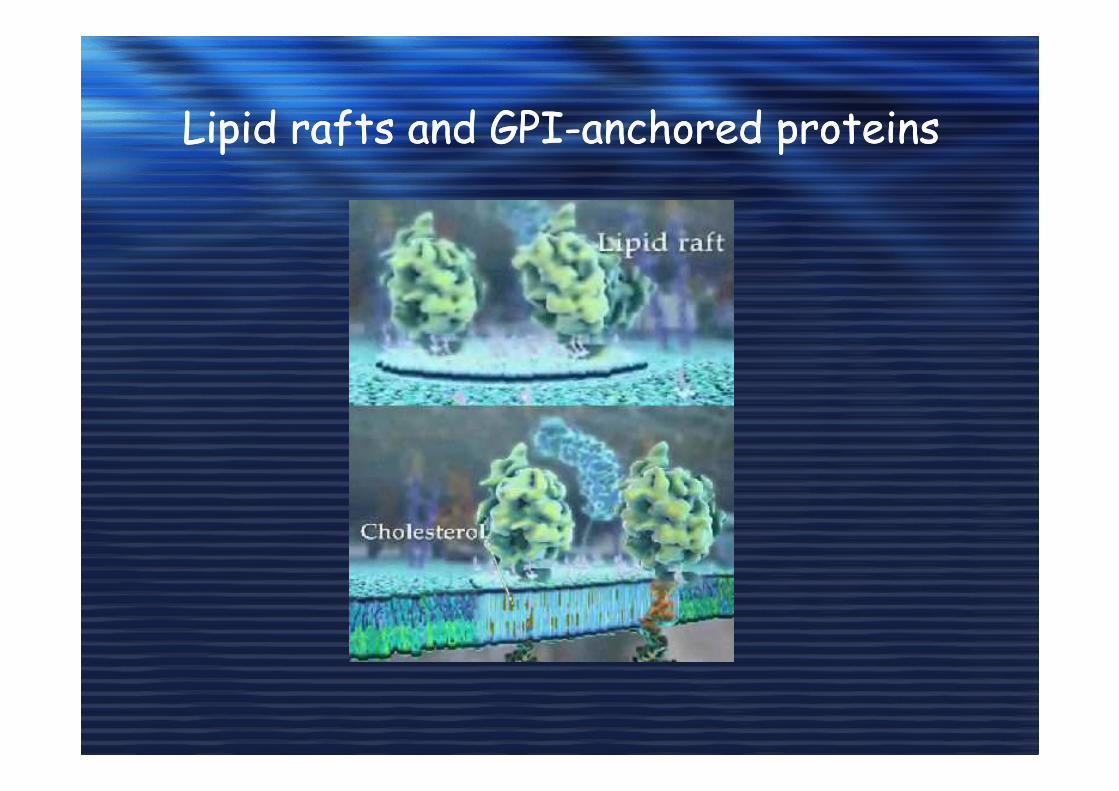
Lipid rafts and GPI-anchored proteins Lipid rafts and GPI-anchored proteins




