
In Copyright - Non-Commercial Use Permitted Rights / …30395/... · ANGELO STORNI dipl....
74
Research Collection Doctoral Thesis Stereochemische Untersuchungen an α-Ambrinol und γ-Iron Author(s): Storni, Angelo Publication Date: 1962 Permanent Link: https://doi.org/10.3929/ethz-a-000089173 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection . For more information please consult the Terms of use . ETH Library
Transcript of In Copyright - Non-Commercial Use Permitted Rights / …30395/... · ANGELO STORNI dipl....
Research Collection
Doctoral Thesis
Stereochemische Untersuchungen an α-Ambrinol und γ-Iron
Author(s): Storni, Angelo
Publication Date: 1962
Permanent Link: https://doi.org/10.3929/ethz-a-000089173
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 3203
Stereochemische Untersuchungen
an a -Ambrinol und ^ -Iron
Von der
EIDGENÖSSISCHEN TECHNISCHEN
HOCHSCHULE IN ZÜRICH
zur Erlangung
der Würde eines Doktors der Naturwissenschaften
genehmigte
PROMOTIONSARBEIT
vorgelegt von
ANGELO STORNI
dipl. Naturwissenschaftei E. T. H.
von Lugaggia (Tessin)
Referent: Herr Prof. Dr. A. Eschenmoser
Korreferent: Herr Prof. Dr. E. Heilbrunner
Juris-Verlag Zürich
1962

Leer - Vide - Empty

Ai miei cari genitori
con tanto affetto

Leer - Vide - Empty

Meinem hochverehrten Lehrer,
Herrn Prof. Dr. V.Prelog,
bin ich für sein stetes Wohlwollen zu grossem Dank verpflichtet.
Herrn Prof. Dr. A. Eschenmoser
unter dessen Anleitung ich die vorliegende Promotionsarbeit ausführen konnte, möchte
ich besonders herzlich danken für wertvolle Anregungen und die grosszügige Hilfe, die
er mir jederzeit entgegenbrachte.

Leer - Vide - Empty

- 7 -
INHALTSVERZEICHNIS
THEORETISCHER TEIL 9
Einleitung 9
I. Kapitel: STEREOCHEMIE DER SAEUREKATALYSIERTEN
CYCLISATION VON DIHYDRO- Jf-JONON 10
Literaturbesprechung 10
Eigene Arbeiten 14
1. Problemstellung 14
2. Die relative Konfiguration der isomeren ot-Ambrinole 21
3. Relative Cyclisationsgeschwindigkeit von (±)-Dihydro- y-jonon und
(+)-cis(2,6)-Dihydro- f-iron 27
II. Kapitel: DIE KONFIGURATION DES NATÜRLICHEN ^-IRONS 31
Literaturbesprechung und Problem Stellung 31
Eigene Arbeiten 37
1. Die relative Konfiguration des natürlichen (+)- #"-Irons 37
2. Diskussion der Auwers-Skita'schen Regel bei den Tetrahydro-ironen 42
EXPERIMENTELLER TEIL 47
1. Stereochemie der säurekatalysierten Cyclisation von Dihydro- y--jonon 47
2. Die Konfiguration des natürlichen (+)-Y" -Irons 54
Zusammenfassung 65
Literaturverzeichnis 66

Leer - Vide - Empty

- 9 -
THEORETISCHER TEIL
Einleitung
Im Laufe der letzten Jahre wurden eingehende Untersuchungen auf dem Gebiet
der säurekatalysierten Cyclisation'von aliphatischen, terpenoiden Polyenen ausge¬
führt, die zum Ziel hatten, eine möglichst umfassende Kenntnis jener Faktoren zu er¬
langen, welche den sterischen Verlauf dieser Reaktion bestimmen. Den Anstoss dazu
gab die Vorstellung, dass dieser Reaktionstypus ein chemisches Analogon der struk¬
turell weitgehend ähnlich verlaufenden biologischen Ringschlussreaktion darstellt, wel¬
che bei der Biogenese der Steroide und cyclischen Terpene eine zentrale Stellung ein-2)
nimmt. Im Falle der Uebergänge von Squalen zu Lanosterin ' sowie von markiertem
3)Mevalolacton zu den Sojasapogenolen ' ist diese Hypothese unlängst bewiesen worden.
Einer der Hauptunterschiede zwischen der Reaktion in vitro und der Cyclisation
in vivo wird heute darin vermutet, dass die säurekatalysierte Cyclisation von einem
Konstellationsgleichgewicht aus erfolge, während die enzymatische Cyclisation von fi¬
xierten, zum Ringschluss befähigten Konstellationen ausgehe. Es wäre deshalb von
theoretischem Interesse, bei einer Cyclisation in vitro den Einfluss der konstellativen
Faktoren zu untersuchen.
Das 1. Kapitel der vorliegenden Arbeit befasst sich mit einer derartigen Unter-4) 51
suchung am Beispiel der säurekatalysierten Cyclisation von Dihydro- y-jonon' '. Die
von dieser Reaktion bevorzugte Ringschlusskonstellation könnte auf Grund der Stereo¬
chemie des Cyclisationshauptproduktes (ot-Ambrinol) und mit Hilfe eines Vergleiches
der Cyclisationsgeschwindigkeiten von Dihydro- V--jonon und Dihydro- Jf-iron bestimmt
werden.
Im Hinblick auf diese kinetischen Untersuchungen stellte sich im weiteren die Fra¬
ge nach der relativen Konfiguration des aus natürlichem (+)-y"-Iron gewonnenen (+)-Di-
hydro-V--irons. Mit diesem Problem befasst sich das 2. Kapitel dieser Arbeit.
la)*) Vgl. P. A.Stadler
, A.Nechvatal, A. J .
F r ey und A.E schenmoser
und die darin zitierte Literatur, sowie A .Eschenmoser
, D.Felix, M.Gut
J.Meier und P .A . Stadle rlb).
- 10 -
1. Kapitel
STEREOCHEMIE DER SAEUREKATALYSIERTEN CYCLEATION VON
DIHYDRO-^f-JONON
Literaturbesprechung
4)5)
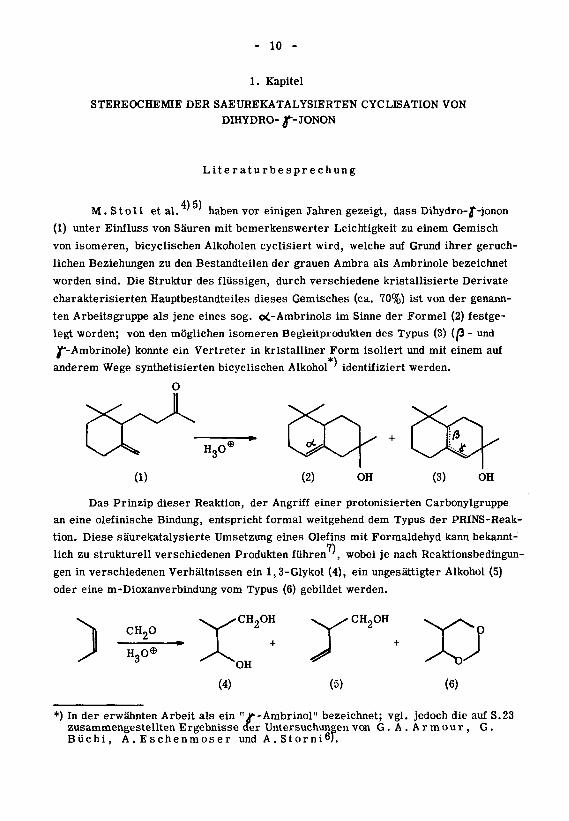
M. S toll et al. ' ' haben vor einigen Jahren gezeigt, dass Dihydro-f-jonon
(1) unter Einfluss von Säuren mit bemerkenswerter Leichtigkeit zu einem Gemisch
von isomeren, bicyclischen Alkoholen cyclisiert wird, welche auf Grund ihrer geruch¬
lichen Beziehungen zu den Bestandteilen der grauen Ambra als Ambrinole bezeichnet
worden sind. Die Struktur des flüssigen, durch verschiedene kristallisierte Derivate
charakterisierten Hauptbestandteiles dieses Gemisches (ca. 70%) ist von der genann¬
ten Arbeitsgruppe als jene eines sog. oi-Ambrinols im Sinne der Formel (2) festge¬
legt worden; von den möglichen isomeren Begleitprodukten des Typus (3) (0- und
/"-Ambrinole) konnte ein Vertreter in kristalliner Form isoliert und mit einem auf
anderem Wege synthetisierten bicyclischen Alkohol ' identifiziert werden.
O
H3°
(1)
Das Prinzip dieser Reaktion, der Angriff einer protonisierten Carbonylgruppe
an eine olefinische Bindung, entspricht formal weitgehend dem Typus der PRINS-Reak-
tion. Diese säurekatalysierte Umsetzung eines Olefins mit Formaldehyd kann bekannt-
7)lieh zu strukturell verschiedenen Produkten führen ', wobei je nach Reaktionsbedingun¬
gen in verschiedenen Verhältnissen ein 1,3-Glykol (4), ein ungesättigter Alkohol (5)
oder eine m-Dioxanverbindung vom Typus (6) gebildet werden.
) CH00
Ho0® XCH2OH
OH
yCH2OH
(4) (5) (6)
*) In der erwähnten Arbeit als ein " J*-Ambrinol" bezeichnet; vgl. jedoch die auf S.23
zusammengestellten Ergebnisse der Untersuchungen von G.A. Armour, G.
Büchi, A. Eschenmoser und A.S t o r n i 6).
- 11 -
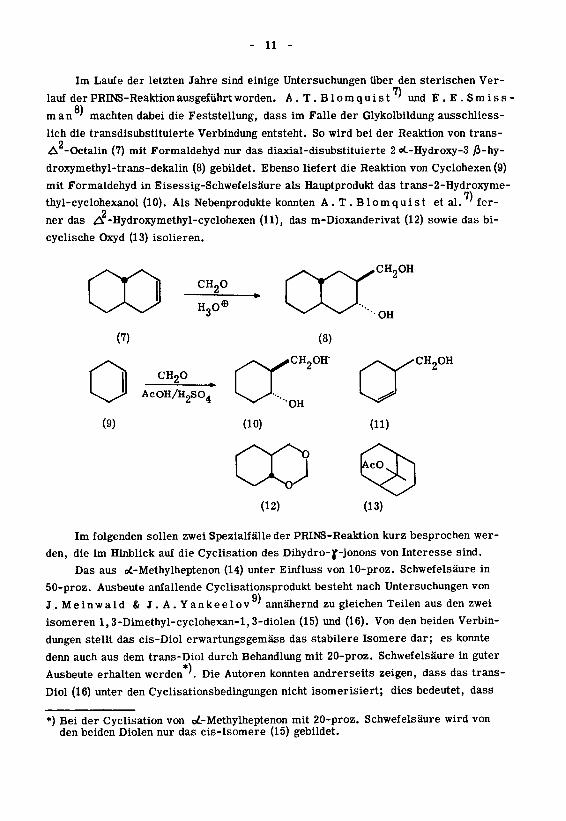
Im Laufe der letzten Jahre sind einige Untersuchungen über den sterischen Ver¬
lauf der PRINS-Reaktion ausgeführt worden. A.T.Blomquist' und E .
E. S miss
man' machten dabei die Feststellung, dass im Falle der Glykolbildung ausschliess¬
lich die transdisubstituierte Verbindung entsteht. So wird bei der Reaktion von trans-
2A -Octalin (7) mit Formaldehyd nur das diaxial-disubstituierte 2 oC-Hydroxy-3 ß-hy-
droxymethyl-trans-dekalin (8) gebildet. Ebenso liefert die Reaktion von Cyclohexen (9)
mit Formaldehyd in Eisessig-Schwefelsäure als Hauptprodukt das trans-2-Hydroxyme-
thyl-cyclohexanol (10). Als Nebenprodukte konnten A.T.Blomquist etal. ' fer-
2ner das A -Hydroxymethyl-cyclohexen (11), das m-Dioxanderivat (12) sowie das bi-
cyclische Oxyd (13) isolieren.
CH20
«3°*
CH2OH
(7)
CH20
AcOH/H2S04
0) (10)
(8)
CH2OH-
"OH
(11)
CH2OH
O'
(12)
Im folgenden sollen zwei Spezialfälle der PRINS-Reaktion kurz besprochen wer¬
den, die im Hinblick auf die Cyclisation des Dihydro-jf-jonons von Interesse sind.
Das aus oi-Methylheptenon (14) unter Einfluss von 10-proz. Schwefelsäure in
50-proz. Ausbeute anfallende Cyclisationsprodukt besteht nach Untersuchungen von
9)J. Meinwald & J. A. Yankeelov ' annähernd zu gleichen Teilen aus den zwei
isomeren l,3-Dimethyl-cyclohexan-l,3-diolen (15) und (16). Von den beiden Verbin¬
dungen stellt das cis-Diol erwartungsgemäss das stabilere Isomere dar; es konnte
denn auch aus dem trans-Diol durch Behandlung mit 20-proz. Schwefelsäure in guter
Ausbeute erhalten werden '. Die Autoren konnten andrerseits zeigen, dass das trans-
Diol (16) unter den Cyclisationsbedingungen nicht isomerisiert; dies bedeutet, dass
*) Bei der Cyclisation von ot-Methylheptenon mit 20-proz. Schwefelsäure wird von
den beiden Diolen nur das cis-Isomere (15) gebildet.
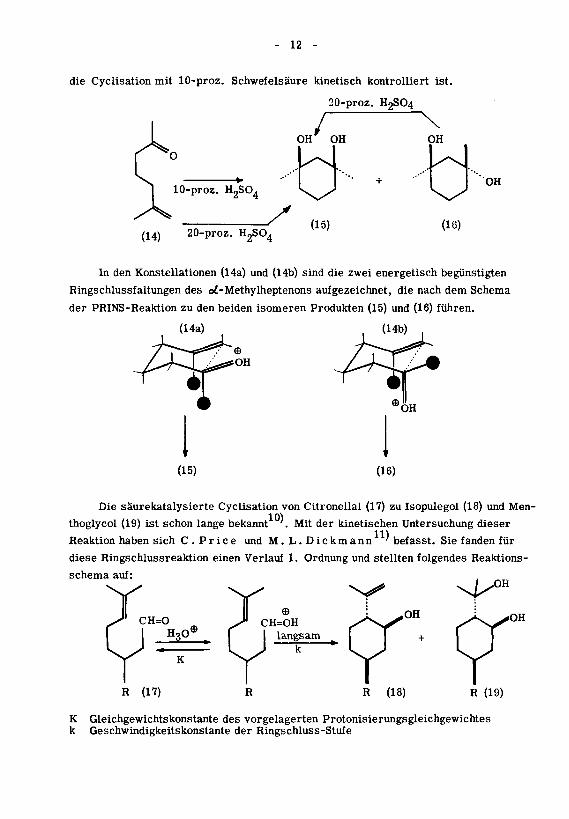
12 -
die Cychsation mit 10-proz. Schwefelsaure kinetisch kontrolliert ist.
20-proz. H2S04
./OH OH
10-proz. H2S04
y
OH
(14) 20-proz. H2S04(15) (16)
In den Konstellationen (14a) und (14b) sind die zwei energetisch begünstigten
Ringschlussfaltungen des od-Methylheptenons aufgezeichnet, die nach dem Schema
der PRINS-Reaktion zu den beiden isomeren Produkten (15) und (16) fuhren.
(14a) (14b)
^p^^Cp*OH
(15)
*
(16)
Die saurekatalysierte Cyclisation von Citronellal (17) zu Isopulegol (18) und Men-
thoglycol (19) ist schon lange bekannt '. Mit der kinetischen Untersuchung dieser
Reaktion haben sich C . Price und M.L. Dickmann ' befasst. Sie fanden fur
diese Ringschlussreaktion einen Verlauf 1. Ordnung und stellten folgendes Reaktions¬
schema auf:
..OH
CH=0
H3OJCH=OH
langsam
K
v^
OH
R (17) R (18) R (19)
K Gleichgewichtskonstante des vorgelagerten Protomsierungsgleichgewichtesk Geschwindigkeitskonstante der Ringschluss-Stufe
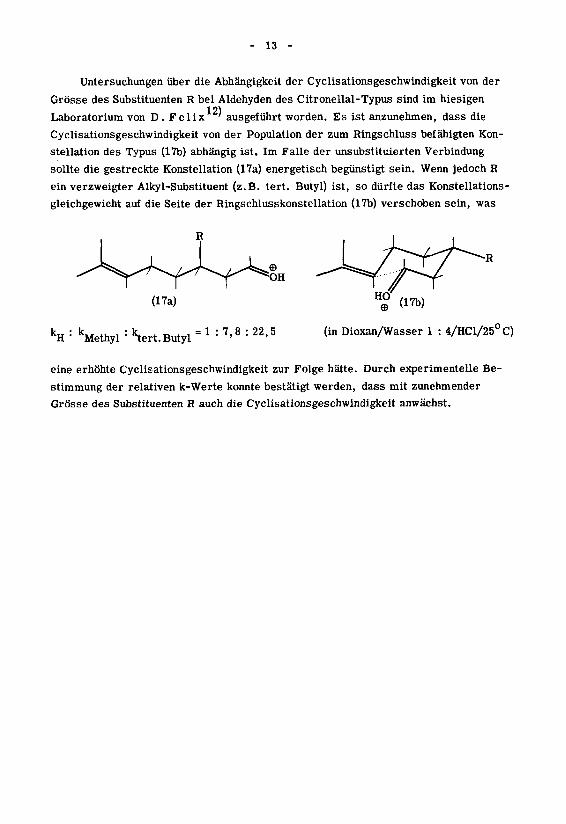
- 13 -
Untersuchungen über die Abhängigkeit der Cyclisationsgeschwindigkeit von der
Grösse des Substituenten R bei Aldehyden des Citronellal-Typus sind im hiesigen121
Laboratorium von D.Felix ' ausgeführt worden. Es ist anzunehmen, dass die
Cyclisationsgeschwindigkeit von der Population der zum Ringschluss befähigten Kon¬
stellation des Typus (17b) abhängig ist. Im Falle der unsubstituierten Verbindung
sollte die gestreckte Konstellation (17a) energetisch begünstigt sein. Wenn jedoch R
ein verzweigter Alkyl-Substituent (z.B. tert. Butyl) ist, so dürfte das Konstellations -
gleichgewicht auf die Seite der Ringschlusskonstellation (17b) verschoben sein, was
R
OH
(17a)
kH : kMethyl : ktert.Butyl " * : 7'8 : 22'5
(17b)
(in Dioxan/Wasser 1 : 4/HCl/25°C)
eine erhöhte Cyclisationsgeschwindigkeit zur Folge hätte. Durch experimentelle Be¬
stimmung der relativen k-Werte konnte bestätigt werden, dass mit zunehmender
Grösse des Substituenten R auch die Cyclisationsgeschwindigkeit anwächst.

- 14 -
Eigene Arbeiten
1. Problemstellung
Ein tieferer Einblick in den Verlauf säurekatalysierter Cyclisationen lässt sich
bis zu einem gewissen Grade dadurch gewinnen, dass man die bei solchen Reaktionen
bevorzugten Ringschlusskonstellationen bestimmt.
Für eine Untersuchung in dieser Richtung schien die säurekatalysierte Cyclisa-A) 5)
tion des Dihydro-^-jonons geeignet zu sein. Auf Grund des von M. S toll et al. ' '
festgestellten strukturellen Cyclisationsverlaufs fallen formal sechs verschiedene
Ringschlusskonstellationen in Betracht (Schema 1), die je nach der Lage der Butan¬
onyl-Seitenkette bezüglich des Cyclohexanringes als axiale Faltungen (I und n) bzw.
äquatoriale Faltungen (III - VI) bezeichnet werden sollen. Dabei stellen die hier be¬
rücksichtigten axialen Typen Sessel- und die äquatorialen Typen Wannenfaltungen dar.
Es stellt sich nun die Frage, über welche dieser sechs Konstellationen das Dihydro-
Y^-jonon bevorzugt reagiert.
Bei dieser Reaktion wird im Cyclisationsprodukt ein neues Asymmetriezentrum
ausgebildet. Wenn die Reaktion kinetisch kontrolliert ist, wird die Konfiguration des
asymmetrischen Kohlenstoffatoms in der produktbestimmenden Ringschluss-Stufe fest¬
gelegt. Man wird somit aus der Stereochemie des oC-Ambrinols, des Cyclisationshaupt-
produktes, wichtige Informationen über die von der Cyclisation bevorzugte Ringschluss¬
konstellation erhalten: Der Verlauf der Cyclisation über die Faltungen vom Typus I,
IV und V muss nämlich zu einem o£-Ambrinol mit trans-Anordnung der Hydroxylgruppe
bezüglich des Wasserstoffatoms am Brückenkopf führen, währenddem die Ringschluss¬
reaktion über die Faltungen vom Typus II, HI und VI das epimere Produkt mit cis-An-
ordnung der erwähnten Substituenten liefern muss. Durch die Aufklärung der relativen
Konfiguration des oi-Ambrinols kann deshalb die Zahl der möglichen Ringschlusskon¬
stellationen auf eine axiale Faltung (I oder II) und zwei äquatoriale Faltungen (IV, V
oder III, VI) eingeengt werden.
Die Frage, ob das Dihydro-^f-jonon bevorzugt über die axiale Faltung oder über
die zwei äquatorialen Faltungen cyclisiert, sollte sich auf Grund kinetischer Unter¬
suchungen beantworten lassen. Für die Diskussion dieses Problems soll die Zahl der
möglichen Konstellationen des Dihydro-^-jonons auf die zwei energetisch günstigsten
Sesselformen E und A beschränkt werden, in denen die Butanonyl-Seitenkette bezüglich
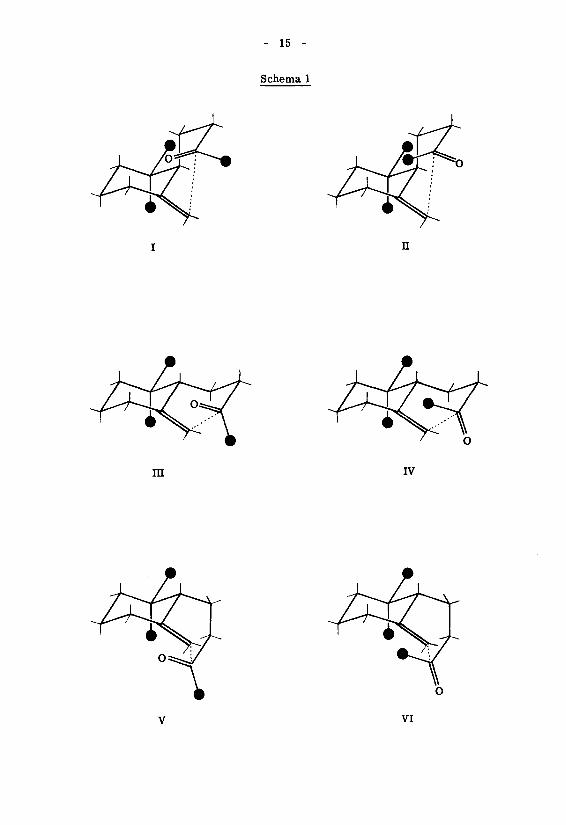
- 15 -
Schema 1
n
m IV
vi

- 16 -
des Cyclohexanringes äquatoriale (e) bzw. axiale (a) Lage einnimmt und bezüglich
der (1', 2')-Bindung antiplanar angeordnet ist. c0 und c„ seien die Konzentrationene d.
dieser Konstellationen, N und N die dazugehörigen Molenbrüche. Für diese gilt:
Nece
Na =
ca
ce + ca ce + ca
Ne + Na = 1 (1)
Für den Cyclisationsverlauf soll folgende Vorstellung gelten: Der Ringschluss
über die axiale Faltung, ausgehend von der Konstellation A, erfolge mit der spezifi¬
schen Geschwindigkeit k„ ', derjenige über die äquatorialen Faltungen, ausgehend
von der Konstellation E, mit der spezifischen Geschwindigkeit k '. Falls sowohl
diese Teilreaktionen, als auch die Gesamtreaktion nach 1. Ordnung ablaufen, gilt
nach S. Winstein et al. ' für die messbare Cyclisationsgeschwindigkeitskon-
stante k der Ringschluss-Stufe folgende Beziehung:
k = N k + N k (2)e e a a
v '
N k : Anteil der über die äquatorialen Faltungen verlaufenden Cyclisation
N„k : Anteil der über die axiale Faltung verlaufenden Cyclisationa a
Wenn bestimmt werden kann, ob Nk grösser oder kleiner ist als N k,so ist
auch die Frage beantwortet, ob das Dihydro- Sf-jonon bevorzugt über die axiale oder
über die äquatorialen Faltungen cyclisiert.
Als erstes müssen die relativen Grössen von k„ und k_ ermittelt werden. Da diea. c
Cyclisationsgeschwindigkeitskonstante k auch von der Lage des Konstellationsgleich¬
gewichtes abhängig ist, können diese Grössen mit Hilfe eines Vergleichs der Cyclisa-
tionsgeschwindigkeit von Dihydro- T-jonon mit derjenigen eines geeignet substituier-
/**)
ten Systems X bestimmt werden. Als solches stand das cis(2,6)-Dihydro- P-iron '
zur Verfügung.
*) Genau genommen sind die spezifischen Cyclisationsgeschwlndigkeiten ka und kgzusammengesetzte Grössen, da mehrere axiale und äquatoriale Faltungen in Be¬
tracht fallen. Es gilt:n n
K~T <-K>
*e=i-2>e-NeNa Vi a a 6
Ne 13T 6 6
Zur Konfiguration dieses Dihydroirons vgl. die Unter¬
suchungen des 2. Kapitels der vorliegenden Arbeit. Im
folgenden werden alle Formelausdrücke, die sich auf
das cis(2,6)-Dihydro- Jf-iron beziehen mit dem Index
I ^5^ x versehen; z.B. bedeutet 11 die Cyclisationsgeschwin-^"^^^ digkeitskonstante des cis(2, 6/-Dihydro- y-irons.
9^
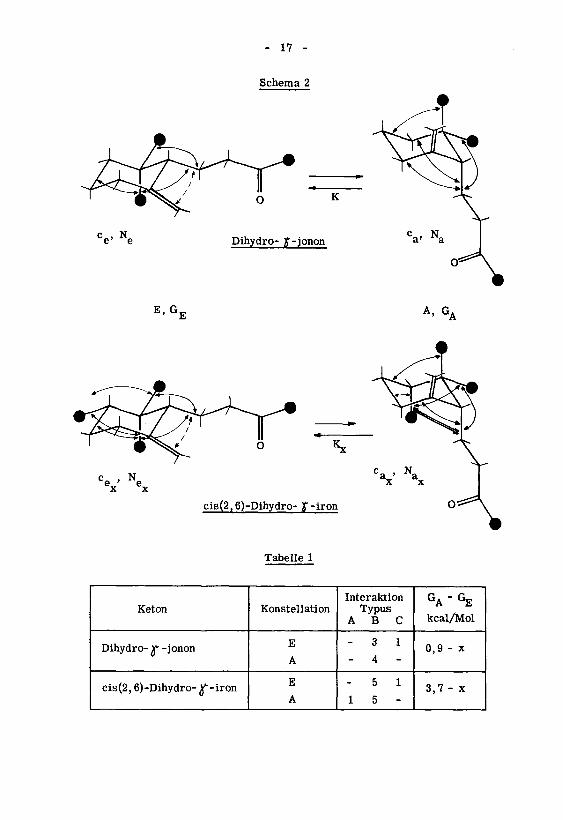
- 17 -
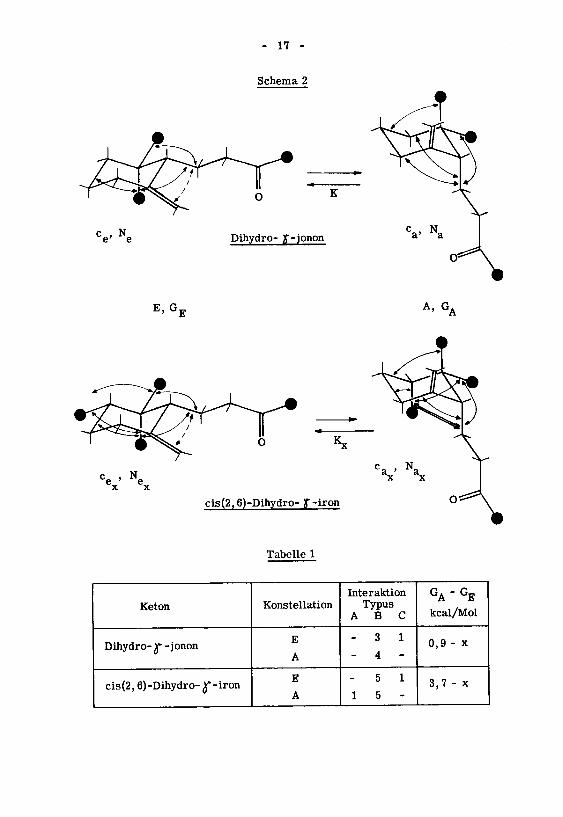
Schema 2
e' e Dihydro- y-jonon Ca> Na
E, GT A,GA
e'
ex x
Tabelle 1
Keton Konstellation
Interaktion
TypusABC
GA"GE
kcal/Mol
Dihydro- r -jononE
A
- 3 1
- 4 -
0,9 - x
cis(2,6)-Dihydro- ^ -ironE
A
5 1
1 5 -
3,7 - x

- 18 -
Eine einfache Konstellationsanalyse' für beide Ketone ergibt, (Schema 2, Ta¬
belle 1) dass das Konstellationsgleichgewicht beim cis(2,6)-Dihydro- Jf-iron stärker auf der
Seite der Konstellation E liegt als beim Dihydro- y-jonon. Die Konstellationsgleich¬
gewichtskonstanten K und K können mangels Kenntnis der Interaktion vom Typus C
nicht bestimmt werden, hingegen kann das Verhältnis X der beiden Gleichgewichte
abgeschätzt werden:Ne
\
Ne
K =—Sl_ > K =
-2- (3)x
ax
KX =
—£- «# 102 bei 25°C (4)
Der Molenbruch N der Konstellation E des cis(2,6)-Dihydro-)f-irons muss
x
als Folge von (3) und (4) um einen bestimmten Betrag AN grösser sein als der Mo¬
lenbruch N des Dihydro- j*-jonons:
N_ = N + AN (5a)ex e
Für den Molenbruch N„ folgt aus (1) und (5a):
% = Na - AN (5b)
Für die Grösse äN kann aus den Gleichungen (1) und (4) folgende Beziehung ab¬
geleitet werden:
O <AN =-£-^-! < 1 (6)je_ ,
l
Na Ne
Die relativen Grössen von k und k„ können nur dann durch Vergleich der rela-e a.
tiven Cyclisationsgeschwindigkeiten der beiden Ketone bestimmt werden, wenn folgen¬
de Annahmen getroffen werden:
k. soll gleich sein k„
k„ soll gleich sein k„
axa
*) Zur Schätzung der Unterschiede in der Stabilität der Konstellationen bediente man
sich der unten erwähnten Werte für Interaktionen vom Typus A*4) und vom Typusb!5). .
A* ^H7*Typus A Typus B Typus C
3,7 kcal/Mol 0,9 kcal/Mol x kcal/Molsyn-schief

- 19 -
Für k und k„ dürfte diese Annahme gut zutreffen, da beim cis(2,6)-Dihydro-e ex
r-iron die 6-Methylgruppe in äquatorialer Lage auf den Cyclisationsverlauf über die
äquatorialen Faltungen keinen wesentlichen Einfluss ausüben kann. In erster Annähe¬
rung dürfen auch die spezifischen Cyclisationsgeschwindigkeiten ka und k„ gleichge¬
setzt werden, obschon im Falle des cis(2,6)-Dihydro- /'-irons die 6-Methylgruppe
und die Seitenkette diaxiale Anordnung einnehmen; vergleicht man nämlich die Ueber-
gänge von der Konstellation A zu den axialen Faltungen beim cis(2,6)-Dihydro- /-iron
mit den entsprechenden Uebergängen beim Dihydro- j--jonon, so stellt man fest, dass
bei den ersteren keine zusätzlichen Spannungsverhältnisse auftreten, die ihren Ursprung
in der axialen Methylgruppe haben könnten.
Auf Grund dieser Annahmen, sowie der Beziehung (5) können die Cyclisationsge¬
schwindigkeiten k und k^ der beiden Ketone folgendermassen ausgedrückt werden:
k = Neke + Naka (2)
1^ = Ne ke + Na ka = (Ne + aN)ke + (Na - 4N)ka (7)
Durch Subtraktion der Gleichung (7) von Gleichung (2) erhält man folgende wich¬
tige Beziehung:
(8)k-kx = ^k = AN(ka- ke)
Es stehen nun zwei Fälle zur Diskussion:
Fall 1) k>kx, d.h. a k > 0; darausfolgt ka> ke
Cyclisiert Dihydro- y-jonon rascher als cis(2,6)-Dihydro- /"-iron, so folgt da¬
raus, dass die spezifische Cyclisationsgeschwindigkeit ka über die axiale Faltung
grösser ist als die spezifische Cyclisationsgeschwindigkeit kg über die äquatorialen
Faltungen. Da die Populationsverteilung mangels Kenntnis der Interaktion vom Typus C
nicht berechnet werden kann, steht nicht ohne weiteres fest, ob auch Nftka grösser als
N k ist. Damit bei der Cyclisation des Dihydro- jf -jonons Naka in jedem Falle grös¬
ser als N k ist, muss Ak eine zusätzliche Bedingung erfüllen. Für die Ableitung
dieser Bedingung muss eine Korrelation zwischen Nftka und Nekg gesucht werden. Eine
solche lässt sich aus den Gleichungen (2) und (7) über die Geschwindigkeitskonstante
1^ aufstellen. Aus (7) folgt nämlich für Ngke:
Neke < k, (9)
Ersetzt man N k in (2) durch diese Beziehung (9), und löst man die erhaltenee e
Ungleichungk = N„kD + Nko < k.. + N k nach N k auf, so ergibt sich
6 6 a. 9, A a, a. a, a.
für Naka: Naka >(k - \ = Ak) (10)

- 20 -
Eine Korrelation zwischen N„k und N.k,
dass N„k stets grösser als N„kDaa ee <i d ee
ist, lässt sich aus (9) und (10) nur dann aufstellen, wenn
^k > kx . Erfüllt ak diese Bedingung, so gilt
Naka>^k>kx>Neke
Für Fall 1) kann somit bezüglich der Frage, ob das Dihydro-^T -jonon bevorzugt
über die axiale Faltung (I oder n) oder über die zwei äquatorialen Faltungen (IV, V
oder HI, VI) cyclisiert, folgende Voraussage gemacht werden:
a) ^k >kx, daraus folgt k > 2 ^
Cyclisiert Dihydro- £ -jonon mindestens doppelt so rasch wie cis(2,6)-Dihydro-
y-iron, so verläuft die Reaktion des ersteren vorwiegend über die axiale Faltung.
b) ^k < 1^, daraus folgt kx< k < 2 1^
In diesem Falle kann nicht a priori bestimmt werden, über welche Ringschluss¬
konstellation das Dihydro- JT-jonon bevorzugt cyclisiert.
Für die Cyclisation von cis(2,6)-Dihydro-y-iron kann im Fall 1) nicht ermittelt
werden, ob die Reaktion bevorzugt über die axialen oder über die äquatorialen Fal¬
tungen verläuft.
Fall 2) k< 1^, d.h. ^k<0; darausfolgt ka< kß
Cyclisiert Dihydro- Jf-jonon langsamer als cis(2,6)-Dihydro- ^f-iron, so folgt
daraus, dass die spezifische Cyclisationsgeschwindigkeit k über die axiale Faltung
kleiner ist als die spezifische Cyclisationsgeschwindigkeit k über die äquatorialen
Faltungen. Für die Cyclisation von Dihydro- y-jonon kann nicht bestimmt werden,
ob auch Nk grösser als N„k ist. Somit kann auch die bevorzugte Ringschlusskon-e e a a
stellation nicht ermittelt werden.
Die Cyclisation von cis(2,6)-Dihydro-/-iron dürfte im Fall 2) vorwiegend über*\
die äquatorialen Faltungen ablaufen '.
:) Es darf angenommen werden, dass die Interaktion vom Typus C den Wert von
3, 7 kcal/Mol nicht übersteigt. Trifft diese Annahme zu, so ist Ne > No, somit
aber auch N k > N„ k„ ,da k > k
.
x ^

- 21 -
2. Die relative Konfiguration der isomeren <X-Ambrinole
Die Aufklärung der Stereochemie der isomeren Ambrinole wurde in Zusammen-*)
arbeit mit A.G. Armour und G.Büchi '
ausgeführt. Diese Arbeitsgruppe hat
sämtliche struktur- bzw. stereoisomeren Ambrinole synthetisiert, wodurch die Struk¬
tur aller Cyclisationsprodukte des Dihydro- ^-jonons eindeutig festgelegt werden
konnte.Q
Das zentrale Produkt dieser Synthese (Schema 3) war das A -5, 5-Dimethyl-2-
octalon (24), welches in Anlehnung an bekannte Arbeitsvorschriften 'ausgehend von
6-Methoxy-2-tetralon (20) über die Zwischenstufen (21) - (23) (Methylierung, Reduk¬
tion nach Cle mmensen,
Reduktion nach Birch) dargestellt wurde. Durch vor¬
sichtige Hydrolyse des Enoläthers (23) mit verdünnter Schwefelsäure in Tetrahydro-
furan konnte in 84-proz. Ausbeute das Keton (24) isoliert werden, das nach UV.-Spek¬
trum weniger als 1,5 % des konjugiert-ungesättigten Ketons (25) enthielt. Das letzte-17)
re Isomere ' wurde durch säurekatalysierte Isomerisation von (24) unter Girardie-
rungsbedingungen erhalten. Durch Behandlung dieses konjugiert-ungesättigten Ketons
mit Natriumhydrid in Tetrahydrofuran und anschliessender Protonisierung des Na-
triumenolats unter nicht-äquilibrierenden Reaktionsbedingungen gelang die Darstel¬
lung des Ji,y -ungesättigten Isomeren (26). Die UV. -spektroskopische Untersuchung
des in 79 % Ausbeute anfallenden Reaktionsproduktes zeigte eine Beimengung von we¬
niger als 10 % des konjugiert-ungesättigten Ketons an. Bezüglich dieser Isomerisie-
rung war man von der Erwartung ausgegangen, dass bei der Enolisation von (25) mit
Natriumhydrid überwiegend das stabilste der drei möglichen Enolate, d.h. das dem
Keton (26) entsprechende, heteroannular-transoide Enolat, gebildet würde. Dass die
unter Bedingungen der kinetischen Kontrolle durchgeführte Protonisierung derartiger
Systeme die Isolierung entsprechender ß,/--ungesättigter Ketone erlaubt, war von
18)anderen Beispielen her bereits bekannt .
Das Reaktionsprodukt der Umsetzung des Ketons (26) mit Methyllithium stellte
ein Gemisch von tertiären Alkoholen dar, welches sich durch Chromatographie an
Alox auftrennen liess. Das flüssige Isomere (30) (Hauptkomponente) lieferte ein kri¬
stallisiertes Epoxyd, das sich mit jenem des oi-Ambrinols (2) aus der Cyclisation von
Dihydro-^f-jonon als identisch erwies. Das als Nebenprodukt isolierte, kristallisierte
Isomere (Smp. 67°) ist von allen übrigen Ambrinol-Isomeren verschieden und stellt
somit das zu (30) epimere ot-Ambrinol (31) dar.
*) Department of Chemistry, Massachusetts Institute of Technology, Cambridge, USA
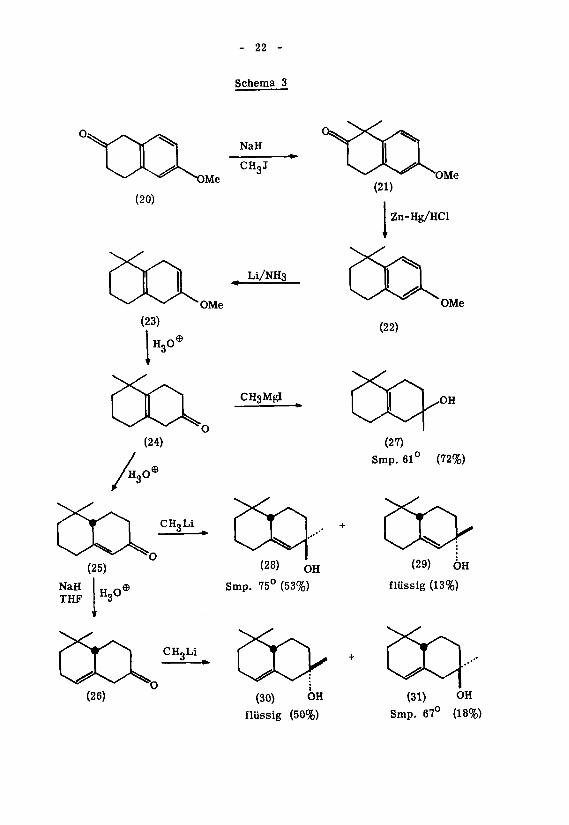
22 -
Schema 3
NaH
CHgJ
(20)
L1/NH3
OMe
CH3MgI
OMe
Zn-Hg/HCl
OMe
(22)
(27)
Smp. 61° (72%)
(28) OH
Smp. 75° (53%)
(29) ÖH
flüssig (13%)
(30) OH
flüssig (50%)
(31) OH
Smp. 67° (18%)

- 23 -
Die Umsetzung des ß,jf -ungesättigten Ketons (24) mit Methylmagnesiumjodid
führte in einheitlich verlaufender Reaktion zum y3-Ambrinol (27) vom Smp. 61. Die¬
se Verbindung erwies sich nach Smp., Mischprobe und IR. -Spektrum als identisch
mit dem kristallisierten Nebenprodukt (3) der Cyclisation von Dihydro- J*-Jonon '.
Die beiden authentischen y--Ambrinole (28) und (29) sind schliesslich durch Umsetzung
des konjugiert-ungesättigten Ketons (25) mit Methyllithium dargestellt worden.
Die im Schema 3 vorweggenommene stereochemische Zuordnung der isomeren
oC- und /-Ambrinole gründet sich auf folgende Beobachtungen: Die massenspektros-**)
kopische Analyse zeitigte für die oC- und J'-Ambrinole bezüglich der Intensitäten
der M- und (M-18)-peaks das in der Tabelle festgehaltene Ergebnis.
Massenspektroskopische Daten
M(%) M-18(%) M(%) M-18(%)
r (30) flüssig 0,57 1,72 ,(28) Smp. 75° 0,04 4,58
1(31) Smp. 67° 0,14 4,01 ^ \(29) flüssig 0,12 2,33
M(%) = Ausbeute an Molekularionen in % der Totalionenausbeute
M-18(%) = Ausbeute an Ionen der Masse M-18 (iM-HgO) in % der Totalionenaus¬
beute.
19}Im Sinne der kürzlich von K.Biemann und J.Seibl gegebenen Interpre¬
tation solcher Daten zur Konfigurationsbestimmung epimerer Alkohole wäre aus die¬
sen Werten zu folgern, dass im Falle beider Epimerenpaare das flüssige Isomere
das jeweils thermodynamisch stabilere darstellt. Diese Folgerung steht - zumindest
was die beiden oC-Ambrinole anbetrifft - in Uebereinstimmung mit den auf chemi¬
schen Wege gemachten Feststellungen.
Das kristallisierte Epoxyd (32) vom Smp. 55°, welches das Hauptprodukt der
Epoxydierung des flüssigen o^-Ambrinols mit Phtalmonopersäure darstellt, lieferte
bei der Reduktion mit Lithiumaluminiumhydrid ein einheitliches, bei 120 schmelzen¬
des Diol CjoHg.O,,. Das entsprechende Epoxydierungsprodukt (34) des ß-Ambrinols
(27) ist flüssig; bei dessen Reduktion mit Lithiumaluminiumhydrid wurde in 86-proz.
Ausbeute ein Diol erhalten, das sich mit dem oben erwähnten Diol aus od-Ambrinol-
epoxyd als identisch erwies. Die Verbindung besitzt demnach die Struktur eines diter-
*) Diesem Keton war von S t o 11 et al.'
die Struktur eines Jf-Ambrinols zugeschrie¬ben worden, auf Grund der Tatsache, dass es in geringer Menge neben viel Kohlen¬
wasserstoff bei der Umsetzung des konjugiert-ungesättigten Ketons (25) mit Methyl-
magnesiumbromid erhalten worden war. Eine Erklärung dafür kann in der Annahme
gesehen werden, dass das verwendete Keton mit geringen Mengen des Isomeren
(24) verunreinigt gewesen war.
**) Die Spektren wurden von K. Biemann und J.Seibl, MIT, mit einem CEC
21-103C Massenspektrometer (Ionisationspotential 70 V) aufgenommen.
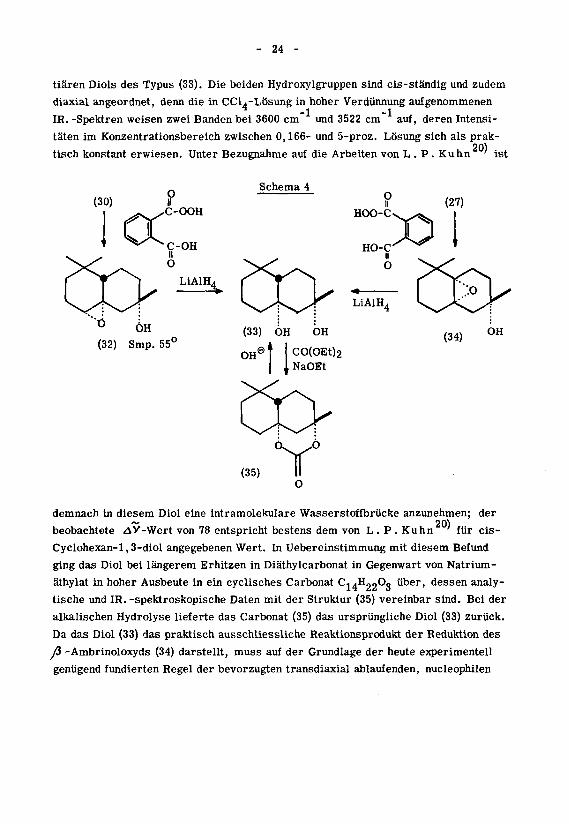
- 24 -
tiären Diols des Typus (33). Die beiden Hydroxylgruppen sind cis-ständig und zudem
diaxial angeordnet, denn die in CCl4-Lösung in hoher Verdünnung aufgenommenen-1 -1
IR. -Spektren weisen zwei Banden bei 3600 cm und 3522 cm auf, deren Intensi¬
täten im Konzentrationsbereich zwischen 0,166- und 5-proz. Lösung sich als prak-20)
tisch konstant erwiesen. Unter Bezugnahme auf die Arbeiten von L. P. Kuhn ' ist
Schema 4
(32) Smp. 55
11
OHe| |CO(OEt)2,NaOEt
(35)Y
demnach in diesem Diol eine intramolekulare Wasserstoffbrücke anzunehmen; der~ 20)
beobachtete ^V-Wert von 78 entspricht bestens dem von L. P. Kuhn ' für cis-
Cyclohexan-l,3-diol angegebenen Wert. In Uebereinstimmung mit diesem Befund
ging das Diol bei längerem Erhitzen in Diäthylcarbonat in Gegenwart von Natrium-
äthylat in hoher Ausbeute in ein cyclisches Carbonat CjaH^Oo über, dessen analy¬
tische und IR. -spektroskopische Daten mit der Struktur (35) vereinbar sind. Bei der
alkalischen Hydrolyse lieferte das Carbonat (35) das ursprüngliche Diol (33) zurück.
Da das Diol (33) das praktisch ausschliessliche Reaktionsprodukt der Reduktion des
fi -Ambrinoloxyds (34) darstellt, muss auf der Grundlage der heute experimentell
genügend fundierten Regel der bevorzugten transdiaxial ablaufenden, nucleophilen
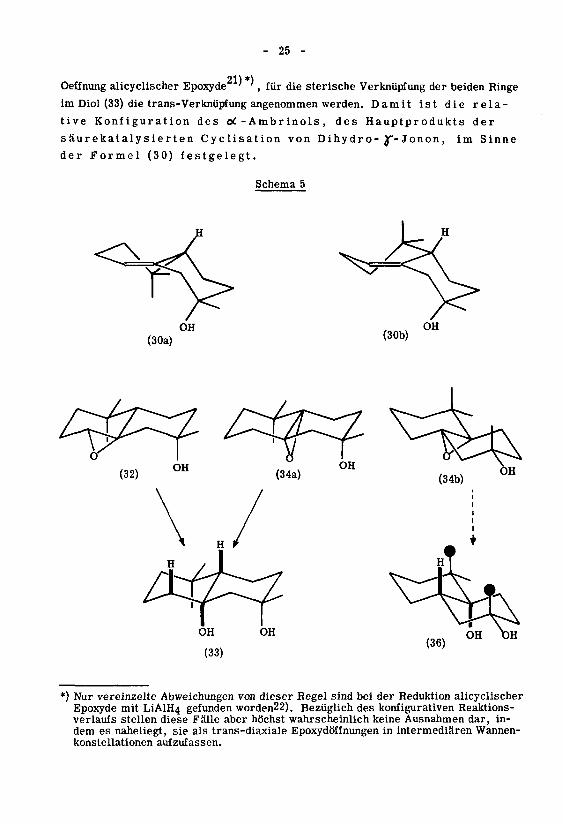
- 25 -
Oeffnung alicyclischer Epoxyde'
', für die sterische Verknüpfung der beiden Ringe
im Diol (33) die trans-Verknüpfung angenommen werden. Damit ist die rela¬
tive Konfiguration des oi - Ambr inols,des Hauptprodukts der
säurekatalysierten Cyclisation von Dihy dr o- y- Jonon ,im Sinne
der Formel (30) festgelegt.
Schema 5
"4-OH
(30a) (30b)
£^fOH
(34a)
*) Nur vereinzelte Abweichungen von dieser Regel sind bei der Reduktion alicyclischerEpoxyde mit LiAlH4 gefunden worden22). Bezüglich des konfigurativen Reaktions¬
verlaufs stellen diese Fälle aber höchst wahrscheinlich keine Ausnahmen dar, in¬
dem es naheliegt, sie als trans-diaxiale Epoxydöffnungen in intermediären Wannen¬
konstellationen aufzufassen.

- 26 -
Wie die Formelbilder im Schema 5 veranschaulichen, sind in den Uebergängen
(32) bzw. (34a)—*(33) jene beiden diaxial ablaufenden Epoxydöffnungen realisiert,
die von den insgesamt vier strukturisomeren Möglichkeiten hinsichtlich der relativen
internen und externen nichtklassischen Spannung die günstigeren sind. Im besonde¬
ren ist aus Formelbild (36) ersichtlich, dass im Falle des y3-Ambrinoloxyds der al¬
ternative Reaktionsweg (34b)—»(36) beträchtlicher sterischer Hinderung bezüglich
des Angriffs des Hydridwasserstoffs unterworfen wäre. Es ist überdies bemerkens¬
wert, dass die Epoxydierung sowohl beim ot-Ambrinol (30) als auch beim y3-Ambri-
nol (27) in weitgehend einheitlicher Reaktion zu den Epoxyden führt, in welchen die
Sauerstoffunktionen cis-Lage aufweisen. Die Vermutung liegt nahe, besonders im
Falle des oi-Isomeren, dass diese Selektivität durch den Einfluss der axialen Hydro¬
xylgruppe auf den Epoxydierungsverlauf zustande kommt, wie er bei cyclischen Allyl-23)
alkoholen von H.B
.Henbest et al. ' beobachtet und interpretiert worden ist.
Eine einfache konstellationsanalytische Betrachtung zeigt, dass die oben erfolg¬
te Konfigurationszuordnung der isomeren oi-Ambrinole mit dem massenspektrosko-
pisch gewonnenen Resultat, wonach das flüssige ot-Ambrinol das stabilere Epimere
sein soll, übereinstimmt: Von den möglichen Konstellationen des oi-Ambrinolsystems
sind hinsichtlich der nicht-klassischen, wie auch der klassischen Spannung jene als
energetisch bevorzugt zu betrachten, in welchen das Wasserstoffatom am Brücken¬
kopf bezüglich der Sesselform des Cyclohexylidenringes axiale Lage einnimmt (vgl,
(30a), (30b) im Schema 5). Im flüssigen crt-Ambrinol (30), in welchem das erwähnte
Wasserstoffatom und die Hydroxylgruppe in trans-Stellung stehen, nimmt demnach die
gegenüber der Hydroxylgruppe räumlich gewichtigere Methylgruppe die äquatoria¬
le Lage ein.
Als Konsequenz der oben erfolgten konfigurativen Aufklärung der isomeren
oC-Ambrinole lässt sich die Vorstellung über die von der säurekatalysierten Cyclisa-
tion des Dihydro-J*-jonons bevorzugte Ringschlusskonstellation etwas näher präzisie¬
ren: Das Dihydro- ^f-jonon muss mit einer solchen Faltung in die konfigurationsbe¬
stimmende Ringschluss-Stufe eingehen, in der die am Carbonyl haftende Methylgruppe
äquatorial angeordnet ist. Von den insgesamt sechs in Betracht fallenden Ringschluss¬
konstellationen verbleiben somit die axiale Faltung I und die äquatorialen Faltungen IV
und V als möglich. Es muss betont werden, dass diese Folgerung nur
für den Fall gilt, dass die Cyclisation kinetisch kontrolliert
ist. Der Nachweis, dass dies tatsächlich zutrifft, ist im Rahmen dieser Arbeit nicht
erbracht worden.
*) Vgl. z.B. die Lage des Konstellationsgleichgewichtes in cis-4-Methylcyclohexa-nol24).

- 27 -
3. Relative Cyclisationsgeschwindigkeit von (±)-Dihydro- J*-jonon
und (+)-cis(2,6)-Dihydro-y-iron
(î)-Dihydro- y-jonon
Das für die kinetischen Untersuchungen verwendete Dihydro- ^f-jonon wurde aus
dem entsprechenden Semicarbazon ' (Smp. 190-191°) durch Spaltung mit Phtalsäure-
anhydrid in Chloroform-Wasser freigesetzt. Eine gaschromatographische Analyse des
destillierten Ketons zeigte eine kleine Verunreinigung von etwa 2 % Dihydro-ot-jo-
non an. Das Dihydro-y--jonon wies folgende Eigenschaften auf: Kpnn.
= 65°,1 fi 91 ^J ^
n^= 1,4773 ; d^1 =0,9161.
( + )-cis(2,6)-Dihydro-^f-iron
Dieses Keton wurde aus natürlichem (+)-cis(2,6)- flron' durch partielle Hy¬
drierung mit Raney-Nickel in Feinsprit dargestellt und über das Semicarbazon (Smp.
198-200^ gereinigt. Daraus wurde durch Spaltung auf obige Art das Keton freigesetzt,
dessen gaschromatographische Analyse eine kleine Beimengung von etwa 3 % an cis(2,3)-
cis(2,6)-Tetrahydro-iron zeigte. Das (+)-cis(2,6)-Dihydro- r-iron wies folgende Eigen¬
schaften auf : [cjc]p1= + 49,30 ; Kp0 03= 76o ; nj,6= 1,4830 ; d^9= 0,9238.
Cyclisationsgeschwindigkeiten
Untersuchungen über die Kinetik der säurekatalysierten Cyclisation sind bis
heute nur an den Aldehyden des Citronellal- und Citral-Typus ausgeführt worden. Un¬
ter Bezugnahme auf die Arbeiten von C.Pr i c e et al. ' und D.Felix ', die für
diese Ringschlussreaktionen einen Verlauf nach 1. Ordnung fanden, kann der formal
sehr ähnlich verlaufenden Cyclisation des Dihydro- ^f-jonons bzw. cis(2,6)-Dihydro->*-
irons folgendes Reaktionsschema zugrunde gelegt werden:
O OH
K
H3°- ' '
-—•• (30) + (27)
*) Ich möchte an dieser Stelle der Firma Firmenich & Cie., Genf, für die Ueber-
lassung grösserer Mengen von Dihydro- ^"-jononsemicarbazon sowie natürlichem
(+)-y'-Iron bestens danken.

- 28 -
Dabei bedeutet K die Gleichgewichtskonstante des vorgelagerten, sich rasch
einstellenden Protonisierungsgleichgewichtes und k die Geschwindigkeitskonstante
der produktbestimmenden Ringschluss-Stufe.
Experimentell lassen sich durch Bestimmung der zeitlichen Abnahme der Keton-
konzentration für die Ringschluss-Stufe nicht direkt die k-Werte, sondern sog. k -Wer¬
te bestimmen, die aber zu jenen in einfacher Beziehung stehen. Für Reaktionen 1.*
Ordnung können die k -Werte aus folgender Gleichung ermittelt werden:
log
k* =
= 0,4343 •
logSut
0,4343 [H3O®]
K [H2°l
[h3o®] Konzentration
des Ketons zur
Zeit 0
Ct = Konzentration
des Ketons zur
Zeitt
Zeit in Stunden
Die Ketonkozentrationen wurden durch Oximtitration bestimmt '. Sämtliche
Messungen wurden bei 25 to, 5° C durchgeführt. Als Cyclisationsmedium wurde 0,1 n.
Salzsäure/Methanol im Volumenverhältnis 1:1 verwendet. Die Ketonkonzentration
schwankte für das Dihydro- ^f-jonon zwischen 2,45-2,75 mMol/l, für das cis(2,6)-Di-
hydro- y-iron zwischen 1,90-1,93 mMol/l.
Ermittelte k*-Werte:
Verbindung PH Halbwertszeitk* 1 k*
Mol"1Std"1 1
(t) -Dihydro- ^f-jonon1,58
1,57
57 Min.
E2 Min.
28,0
30,129,0
(+)-cis(2,6)-Dihydro-
y-iron
1,58
1,62
355 Min.
382 Min.
4,46
4,534,50
*) Ausführung der Messungen sowie Zusammenstellung aller Resultate siehe experi¬menteller Teil.
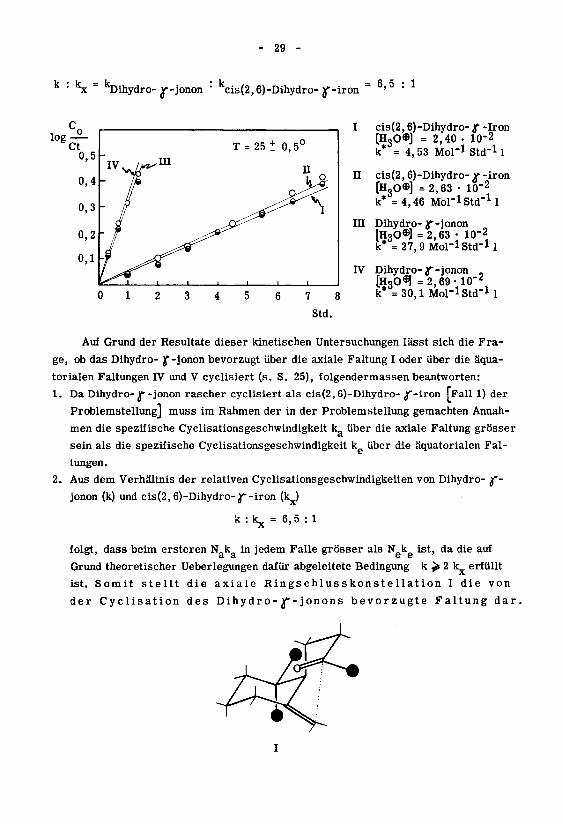
- 29 -
*Sc " kDihydro- ^-jonon: kcis(2,6)-Dihydro- y-iron
6'5 : 1
cis(2,6)-Dihydro- jf -Iron
(H30®] = 2,40 • 10-2
k* = 4, 53 Mol-1 Std"11
II cis(2,6)-Dihydro-^f-iron
[H,0«1 =2,63 • 10-2
k*'-* -4,'46 Mol-iStd"1!
III Dihydro-y-jonon
(H30©] =2,63 • lu"2
k* = 27,9 Mol-1 Std-11
IV Dihydro-y-jonon „
[H3O^=2,69-10-2k* = 30,1 Mol-lStd"1!
Auf Grund der Resultate dieser kinetischen Untersuchungen lässt sich die Fra¬
ge, ob das Dihydro- }f-jonon bevorzugt über die axiale Faltung I oder über die äqua¬
torialen Faltungen IV und V cyclisiert (s. S. 25), folgendermassen beantworten:
1. Da Dihydro- T-jonon rascher cyclisiert als cis(2, 6)-Dihydro-J'-iron [Fall 1) der
Problemstellung] muss im Rahmen der in der Problemstellung gemachten Annah¬
men die spezifische Cyclisationsgeschwindigkeit k über die axiale Faltung grösser
sein als die spezifische Cyclisationsgeschwindigkeit k über die äquatorialen Fal¬
tungen.
2. Aus dem Verhältnis der relativen Cyclisationsgeschwindigkeiten von Dihydro- jf-
jonon (k) und cis(2,6)-Dihydro-J*-iron (1^)
k : kjj = 6,5 : 1
folgt, dass beim ersteren N&k in jedem Falle grösser als Nekg ist, da die auf
Grund theoretischer Ueberlegungen dafür abgeleitete Bedingung k > 2 1^ erfüllt
ist. Somit stellt die axiale Ringschlusskonstellation I die von
der Cyclisation des Dihy dr o- y- jonons bevorzugte Faltung dar.

- 30 -
Es wäre von Interesse, bei beiden Ketonen das Verhältnis der über die axialen
zu der über die äquatorialen Faltungen verlaufenden Cyclisationsanteile (N„k„ : N„kix «i 6 6
bzw. N k : N k) zu kennen. Um diese Verhältnisse berechnen zu können, be-X X X X
nötigt man die Konstellationsgleichgewichtskonstanten der beiden
Ketone, die jedoch mangels Kenntnis der Interaktion vom Typus C (vgl. Fussnote '
auf S. 18) nicht bekannt sind. Unter der Annahme, dass diese Spannung im Bereiche
von 0,5-2 kcal/Mol liegt, lassen sich mit Hilfe der Gleichungen (1) - (8) für die er¬
wähnten Verhältnisse folgende Werte ermitteln:
Interaktion vom Typus C
in kcal/MolDihydro- /-jonon
Naka:Nekecis(2,6)-Dihydro- fl"-iron
Naxkax : ^x^x
0,5 10 :1 1 : 10
1,0 15 :1 1 : 6,5
1,5 28 :1 1 : 3,5
2,0 45 : 1 1 : 2,2
Aus der Tabelle geht eindeutig hervor, dass das Dihydro- ^"-jonon in allen an¬
genommenen Fällen nicht nur vowiegend, sondern fast ausschliesslich über die axiale
Ringschlusskonstellation I cyclisiert. Bei der Cyclisation des cis(2,6)-Dihydro- jf-
irons sind dagegen die äquatorialen Faltungen bevorzugt. Hier kann jedoch nicht nä¬
her präzisiert werden, welche der vier äquatorialen Faltungen für die Cyclisation be¬
günstigt ist, da die Stereochemie des Cyclisationsproduktes nicht untersucht wurde.
Zur Vervollständigung des Bildes wäre die Bestimmung der relativen Cyclisa-
tionsgeschwindigkeit des trans(2,6)-Dihydro- ^T-irons von Interesse. Da bei diesem
Keton das Konstellationsgleichgewicht stärker auf der Seite von Konstellation A liegen
dürfte als beim Dihydro-r-jonon, sollte das erstere etwas rascher cyclisieren als*)
das letztere '.
*) M .S toll et al. ' machten die Beobachtung, dass ein von ihnen synthetisiertes
(i)-trans(2,6)-Dihydro- Jf-iron langsamer cyclisiert als Dihydro- jf-jonon. Das
IR. -Spektrum dieses Dihydro-irons erwies sich indessen mit demjenigen des
(+)-eis(2,6)-Dihydro- y-irons als identisch; es muss sich demnach ebenfalls um
ein Keton mit cis(2,6)-Konfiguration handeln.
x-3,7-51
15
A
E-ironjf-Dihydro-(2,6)eis
x-0,9-4-
13-
A
E
Dihydro-fr-jonon
kcal/Mol
GA-GE
ABC
Typus
Interaktion
KonstellationKeton
1Tabelle
if-ironcis(2,6)-Dihydro-
xxe
'
eN,c
GAA,E.G,
y-jononDihydro-
K
2Schema
-17-
ee'

- 18 -
Eine einfache Konstellationsanalyse' für beide Ketone ergibt, (Schema 2, Ta¬
belle 1) dass das Konstellationsgleichgewicht beim cis(2,6)-Dihydro- y-iron stärker auf der
Seite der Konstellation E liegt als beim Dihydro- Jf-jonon. Die Konstellationsgleich¬
gewichtskonstanten K und K können mangels Kenntnis der Interaktion vom Typus C
nicht bestimmt werden, hingegen kann das Verhältnis X der beiden Gleichgewichte
abgeschätzt werden:IN IN
(3)Kx =
N
N0ax
NeK = ——
Na
ae =-1- «# 102 bei 25° C
Der Molenbruch N der Konstellation E des cis(2,6)-Dihydro- )f-irons muss
x
als Folge von (3) und (4) um einen bestimmten Betrag ^N grösser sein als der Mo¬
lenbruch N des Dihydro- y-jonons:
N„ = Ne + 4N (5a)ex
e
Für den Molenbruch Nj, folgt aus (1) und (5a):
% = Na - AN (5b)
Für die Grösse «äN kann aus den Gleichungen (1) und (4) folgende Beziehung ab¬
geleitet werden:
O <AN =* ' 1
< 1 (6)
Na Ne
Die relativen Grössen von k und k können nur dann durch Vergleich der rela¬
tiven Cyclisationsgeschwindigkeiten der beiden Ketone bestimmt werden, wenn folgen¬
de Annahmen getroffen werden:
k. soll gleich sein k„ex e
ka soll gleich sein ka
*) Zur Schätzung der Unterschiede in der Stabilität der Konstellationen bediente man
sich der unten erwähnten Werte für Interaktionen vom Typus Al4) und vom TypusBi5).
J^f^Typus A Typus B Typus C
^
3,7 kcal/Mol 0,9 kcal/Mol x kcal/Molsyn -schief
- 31 -
II. Kapitel
DIE KONFIGURATION DES NATÜRLICHEN y-IRONS
Literaturbesprechung und Problemstellung
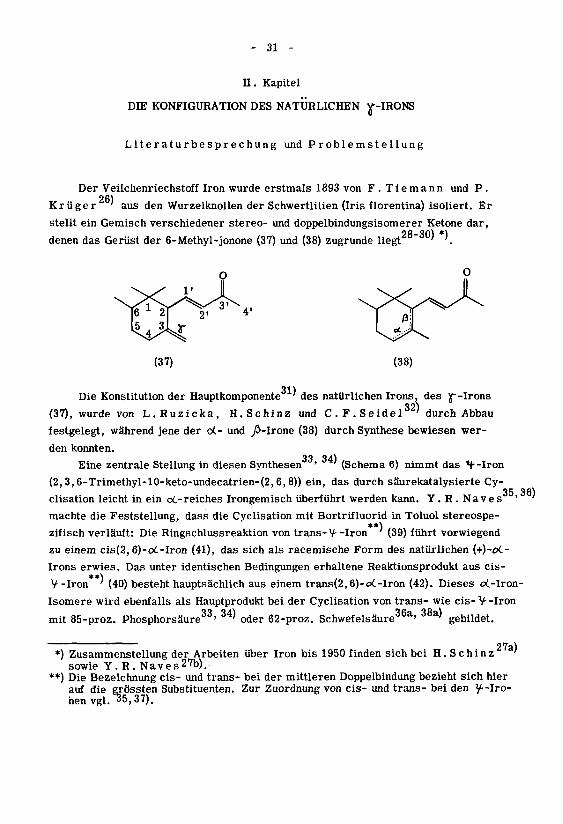
Der Veilchenriechstoff Iron wurde erstmals 1893 von F. Tiemann und P.
Krüger'aus den Wurzelknollen der Schwertlilien (Iris florentina) isoliert. Er
stellt ein Gemisch verschiedener stereo- und doppelbindungsisomerer Ketone dar,28-30} *)
denen das Gerüst der 6-Methyl-jonone (37) und (38) zugrunde liegt'
'.
(37) (38)
3D
Die Konstitution der Hauptkomponente' des natürlichen Irons, des Y--Irons
32^
(37), wurde von L.Ruzicka, H.Schinz und C.F.Seidel ' durch Abbau
festgelegt, während jene der c<- und fi-lrone (38) durch Synthese bewiesen wer¬
den konnten.
33 34)Eine zentrale Stellung in diesen Synthesen ' ' (Schema 6) nimmt das *J--Iron
(2,3,6-Trimethyl-10-keto-undecatrien-(2,6, 8)) ein, das durch säurekatalysierte Cy-
clisation leicht in ein o(.-reiches Irongemisch überführt werden kann. Y. R . Naves'
machte die Feststellung, dass die Cyclisation mit Bortrifluorid in Toluol stereospe¬
zifisch verläuft: Die Ringschlussreaktion von trans-V- -Iron ' (39) führt vorwiegend
zu einem cis(2, 6)-oC-Iron (41), das sich als racemische Form des natürlichen (+)-oC-
Irons erwies. Das unter identischen Bedingungen erhaltene Reaktionsprodukt aus cis-
V -Iron ' (40) besteht hauptsächlich aus einem trans(2,6)-o<.-Iron (42). Dieses oL-lron-
Isomere wird ebenfalls als Hauptprodukt bei der Cyclisation von trans- wie eis-V-Iron
mit 85-proz. Phosphorsäure ' ' oder 62-proz. Schwefelsäure a' a'gebildet.
*) Zusammenstellung der Arbeiten über Iron bis 1950 finden sich bei H.Schinz
sowie Y. R.Nave s 27b).
27a)
**) Die Bezeichnung eis- und trans- bei der mittleren Doppelbindung bezieht sich hier
auf die grössten Substituenten. Zur Zuordnung von eis- und trans- bei den y--Iro-hen vgl. 35,37).
- 32 -
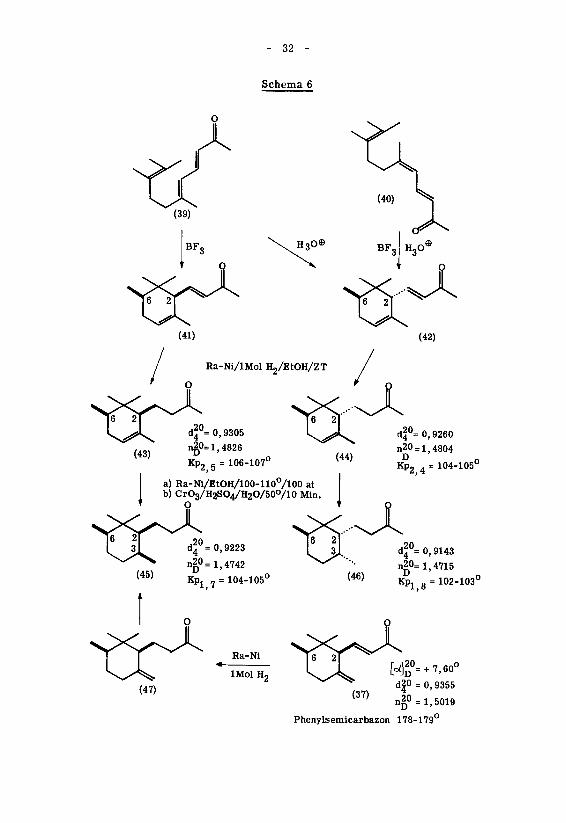
Schema 6
Ra-Ni/lMol H2/EtOH/ZT
106-107
a) Ra-Ni/EtOH/100-110°/100 at
b) CrO3/H2SO4/H2O/50o/10 Min.
O
df°= 0,9260
n20 = 1,4804D
104-105u
20it =0,9223
n20= 1,4742
tï:a1,7
=104-105u (46)
d|°= 0,9143
n20= 1,4715
Kpx 8=102-103°
Ra-Nl
lMol H2(47)
[Ä + 7,60°20
D"
d|0 = 0,9355
n = 1,5019
Phenylsemicarbazon 178-179

- 31 -
II. Kapitel
DIE KONFIGURATION DES NATÜRLICHEN Y-IRONS
Literaturbesprechung und Problemstellung
Der Veilchenriechstoff Iron wurde erstmals 1893 von F. Tiemann und P.
Krüger 'aus den Wurzelknollen der Schwertlilien (Iris florentina) isoliert. Er
stellt ein Gemisch verschiedener Stereo- und doppelbindungsisomerer Ketone dar,
denen das Gerüst der 6-Methyl-jonone (37) und (38) zugrunde liegt' '.
(37) (38)
31)Die Konstitution der Hauptkomponente
' des natürlichen Irons, des jf -Irons
(37), wurde von L.Ruzicka, H.Schinz und C.F.Seidel ' durch Abbau
festgelegt, während jene der oC- und jS-Irone (38) durch Synthese bewiesen wer¬
den konnten.
Eine zentrale Stellung in diesen Synthesen ' ' (Schema 6) nimmt das V-Iron
(2,3,6-Trimethyl-10-keto-undecatrien-(2,6,8)) ein, das durch säurekatalysierte Cy-
clisation leicht in ein ot-reiches Irongemisch überführt werden kann. Y. R . Naves ' '
machte die Feststellung, dass die Cyclisation mit Bortrifluorid in Toluol stereospe¬
zifisch verläuft: Die Ringschlussreaktion von trans-V- -Iron ' (39) führt vorwiegend
zu einem cis(2, 6)-oC-Iron (41), das sich als racemische Form des natürlichen (+)-oC-
Irons erwies. Das unter identischen Bedingungen erhaltene Reaktionsprodukt aus cis-
V -Iron ' (40) besteht hauptsächlich aus einem trans(2,6)-oC-Iron (42). Dieses oL-lron-
Isomere wird ebenfalls als Hauptprodukt bei der Cyclisation von trans- wie cis-V-Iron
mit 85-proz. Phosphorsäure ' ' oder 62-proz. Schwefelsäure a' a'gebildet.
*) Zusammenstellung der Arbeiten über Iron bis 1950 finden sich bei H. Schinz
sowie Y. R.Nave s 27b),
**) Die Bezeichnung eis- und trans- bei der mittleren Doppelbindung bezieht sich hier
auf die grössten Substituenten. Zur Zuordnung von eis- und trans- bei den y--Iro-hen vgl. ^5,37).
27a)
- 32 -
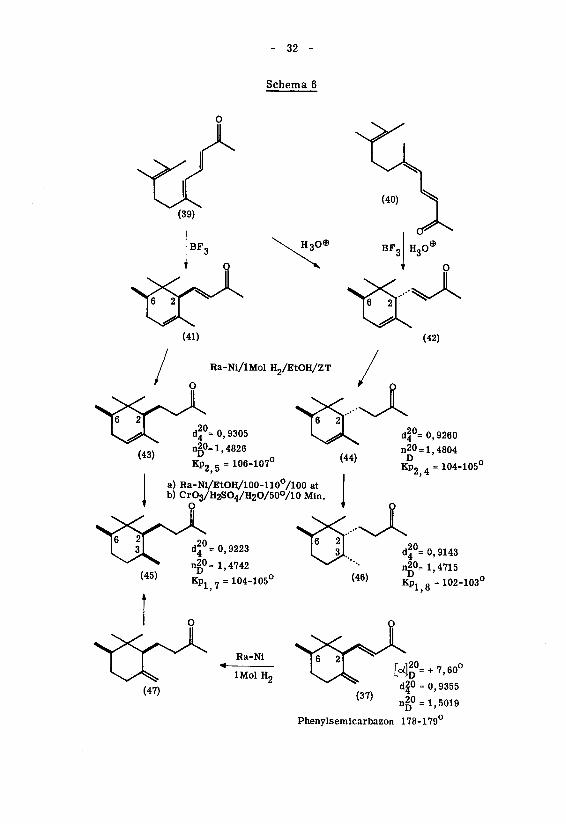
Schema 6
i u
1120=1,4826
Kp2 5= 106-107°
a) Ra-Ni/EtOH/100-110°/100 at
b) CrO3/H2SO4/H2O/50°/10 Min.
0,9260
n20 = 1,4804D
o
Kp2 4=104-105°
df= 0,9223
n20= 1,4742
KPl 7=104-105°
Ra-Ni
lMol H,
^^I 3].
(46)
d|°= 0,9143
n20= 1,4715
Kpx 8=102-103°
(47)(37)
[<= + 7,60°
d|0 = 0,9355
n2,° = 1,5019
Phenylsemicarbazon 178-179

- 33 -
Die relative Konfiguration dieser stereoisomeren oC-Irone wurde indessen nie
bewiesen. Die oben verwendete Konfigurationszuordnung wurde von Y.R. Naves '
*)an den entsprechenden Dihydroverbindungen ' mit Hilfe der ursprünglichen Regel von
39)Auwers und Skita '
vorgenommen: Das durch partielle Hydrierung von trans(2,6)-
oC-Iron (42) gewonnene trans(2,6)-Dihydro-o£-Iron (44) weist kleinere Dichte, tiefe¬
ren Siedepunkt und tieferen Brechungsindex auf als das aus cis(2, 6)-d-lron (5) herge¬
stellte cis(2,6)-Dihydro-o£-Iron (43). Deshalb teilte Y.R. Naves ' dem ersten
der genannten Dihydro-ketone die trans-Form, dem zweiten die cis-Form zu \
Den durch katalytische Hydrierung aus diesen Dihydro-Verbindungen dargestell¬
ten Tetrahydro-ironen (45) und (46) wurde von Y. R. Naves c' ' die im Schema 6
angegebene Stereochemie zugeordnet. Ein Vergleich der Dichten, Siedepunkte und
Brechungsindices der beiden Ketone zeigt, dass das Tetrahydro-iron (46) die tieferen
Werte aufweist als das Stereoisomere (45). Y.R.Navesc'
erblickte darin eine
Bestätigung der von ihm bei den Dihydro-od-ironen getroffenen Konfigurationszuord¬
nung.
Dem natürlichen (+)-^f-Iron (37) muss nach diesem Autor ' die cis(2,6)-Stereo-
chemie zukommen, da es bei der katalytischen Hydrierung über die Dihydro-Verbin¬
dung (47) ein (+)-Tetrahydroketon lieferte, das bis auf die optische Aktivität mit dem
(±)-cis(2,3)-cis(2,6)-Tetrahydro-iron (45) identisch war.
Diese konfigurativen Zuordnungen basieren auf der bei den Dihydro-^-ironen an¬
gewandten ursprünglichen Regel von Auwers und Skita. Man weiss aber heute,42)
dass bei 1, 3-disubstituierten Cyclohexan-' und bei 3, 5-disubstituierten Cyclohexen-
Verbindungen im allgemeinen die kleinere Dichte, der tiefere Siedepunkt und der tie¬
fere Brechungsindex nicht dem trans-, sondern dem cis-disubstituierten Stereoisome¬
ren zukommen. Die alte Regel von Auwers und Skita wurde deshalb von N. L.43)
Allinger 'in dem Sinne modifiziert, dass bei Stereoisomeren in cyclischen Syste¬
men, welche sich nicht im Dipolmoment unterscheiden, dasjenige Isomere, welches***)
das kleinere Molekularvolumen hat, den grösseren Wärmeinhalt besitzt '.
Eine Konfigurationszuordnung nach dieser modifizierten Regel darf somit bei den
isomeren Dihydro-o£-ironen nur dann vorgenommen werden, wenn auf eindeutige Art
bestimmt werden kann, welches Stereoisomere das thermodynamisch stabilere dar¬
stellt.
*) Die Konfigurationszuordnung wurde an den Dihydro-ketonen und nicht an den un-
hydrierten Ausgangsverbindungen vorgenommen, da bei den ersteren die geome¬
trische Isomerie in (l',2') wegfällt.**) Zur Vereinfachung der Diskussion sollen in der Folge diese konfigurativen Zu¬
ordnungen beibehalten werden.
***) Damit immer verbunden ist grössere Dichte, im allgemeinen auch höherer Siede¬
punkt und höherer Brechungsindex.
- 34 -
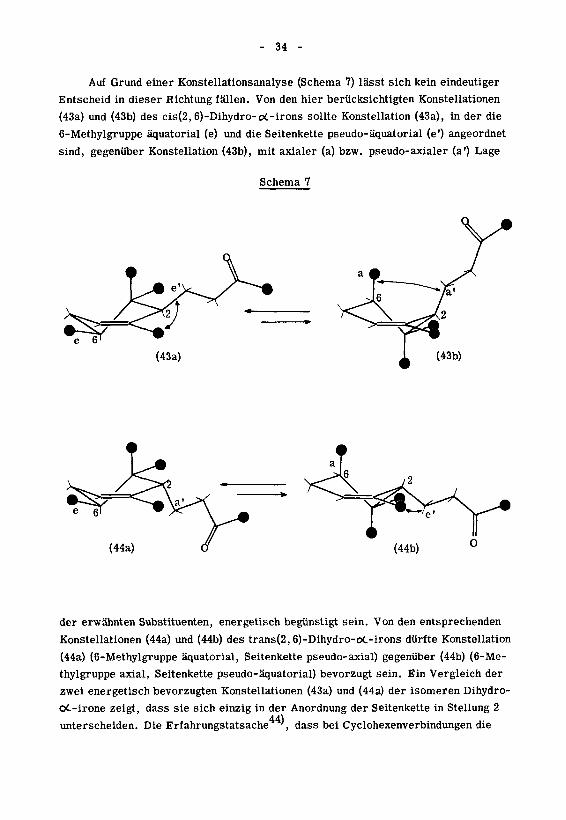
Auf Grund einer Konstellationsanalyse (Schema 7) lässt sich kein eindeutiger
Entscheid in dieser Richtung fällen. Von den hier berücksichtigten Konstellationen
(43a) und (43b) des cis(2,6)-Dihydro-o£-irons sollte Konstellation (43a), in der die
6-Methylgruppe äquatorial (e) und die Seitenkette pseudo-äquatorial (e') angeordnet
sind, gegenüber Konstellation (43b), mit axialer (a) bzw. pseudo-axialer (a') Lage
Schema 7
der erwähnten Substituenten, energetisch begünstigt sein. Von den entsprechenden
Konstellationen (44a) und (44b) des trans(2, 6)-Dihydro-oC-irons dürfte Konstellation
(44a) (6-Methylgruppe äquatorial, Seitenkette pseudo-axial) gegenüber (44b) (6-Me¬
thylgruppe axial, Seitenkette pseudo-äquatorial) bevorzugt sein. Ein Vergleich der
zwei energetisch bevorzugten Konstellationen (43a) und (44a) der isomeren Dihydro-
oC-irone zeigt, dass sie sich einzig in der Anordnung der Seitenkette in Stellung 2
44}unterscheiden. Die Erfahrungstatsache ,
dass bei Cyclohexenverbindungen die

- 35 -
pseudo-äquatoriale Anordnung eines Substituenten in ot-Stellung zur Doppelbindung
gegenüber der pseudo-axialen bevorzugt ist, darf im Falle der Dihydro- oC-irone
kaum ohne weiteres zugunsten von Konstellation (43a) ausgelegt werden, da bei die¬
ser die Methylgruppe in Stellung 3 einen destabihsierenden Einfluss auf die pseudo-
aquatoriale Lage der Seitenkette ausüben durfte '.
Auch beim ot-Ironsystem lassen sich auf Grund der vorhandenen experimen¬
tellen Resultate keine eindeutigen Schlüsse bezüglich der relativen thermodynamischen
Stabilität der beiden Isomeren ziehen:
47 48}
So wird nach P. Bac hl i undH.Schinz ' ' bei der alkalischen Isomeri-
sierung von (+)-cis(2,6)-oC-Iron (41) neben vorwiegend ß-Iron (50) in ca. 10-proz.
Ausbeute ein (-)-trans(2,6)-oi-Iron (42) gebildet, wahrend kein unverändertes
(+)-cis(2,6)-oC-Iron (41) isoliert werden konnte. Andererseits stellte Y. R.Naves
bei der alkalischen Behandlung von ß-Iron fest, dass neben unverändertem Material
ca. 15 % cis(2,6)-ot-Iron (41) und nur ca. 10 % trans(2,6)-oL-Iron (42) entsteht.
38b)
(41)
20Std. ZT
(50) (42) ca. 10%
(50)
nXaANaOEt/EtOH II48Std. ZT
(41) ca. 15% (42) ca. 10%
Da angenommen werden kann, dass beide Isomensierungen über dasselbe Enolat
verlaufen, widersprechen sich deren Resultate.
*) Bei den isomeren Bicyclofarnesylsàuren (48) und (49)**), in welchen bezuglich
Ring B die gleichen Substitutions- und Konstellations-Verhältnisse wie in (43a)und (44a) vorliegen, scheint auf Grund der Arbeiten von G. S t o r k und A.W.
Burgstahler45) das Isomere (49) mit pseudo-axialer Anordnung der Carboxy-
latgruppe energetisch stabiler zu sein. Dieser Befund darf jedoch nicht ohne wei¬
teres auf die isomeren Dihydro-oi-irone übertragen werden, da dieses System im
Gegensatz zu jenem der Bicyclofarnesylsàuren konstellativ nicht fixiert ist.
,cocr
Té'
(48)
COO
(49)
') Bezuglich der Konfiguration dieser Bicyclofarnesylsàuren vgl. die Ergebnisse der
Arbeiten von P.A.Stadler et al. 4°).

- 36 -
Aus den obigen Ausführungen geht somit hervor, dass eine Konfigurationszuord¬
nung nach der Regel von Auwers und S kit a weder bei den isomeren oC-Ironen,
noch bei den entsprechenden Dihydroverbindungen zulässig ist.
Da die Kenntnis der relativen Konfiguration des aus natürlichem (+)- ^f-Iron her¬
gestellten (+)-Dihydro-y-irons für die kinetischen Untersuchungen der säurekatalysier¬
ten Cyclisationen von Dihydro- y-jonon und Dihydro-y'-iron von entscheidender Wich¬
tigkeit war, drängte sich ein eindeutiger Beweis der Stereochemie dieses Systems auf.
Der Aufklärung der relativen Konfiguration des natürlichen (+)- ^f-Irons wurde
folgender einfacher Plan zugrunde gelegt:
1. Abbau des (+)- ^"-Irons (37) unter Vermeidung einer Isomerisierung in Stellung 2
über die Dihydro-verbindung (47) zu einem Diketon vom Typus (51).
2. Cyclisation dieses Diketons (51) zu einem Octalon der Struktur (52), das durch
Aequilibrierung konfigurativ im Sinne von (52) festgelegt werden könnte.
3. Ueberführung dieses Octalons ohne Epimerisierung in Stellung 2 zum Diketon (53).
Ein Vergleich dieser Verbindung mit dem Diketon (51) aus dem Abbau von (+)-^f -
Iron führt zwangsläufig zur Aufklärung der relativen Konfiguration von (+)-^f- (37)
sowie von (+)-Dihydro- )f-iron (47).
(53)
Aus dem Verlauf der Rotationsdisperisonskurve des Octalons (52) kann ferner
die absolute Konfiguration des natürlichen (+)-Y*-Irons bestimmt werden.

- 37 -
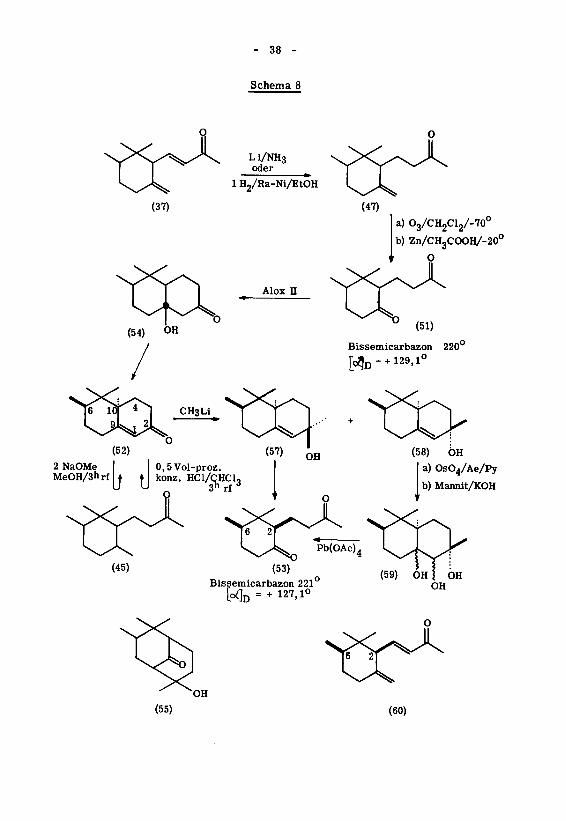
Eigene Arbeiten
1. Die relative Konfiguration des natürlichen (+)- y-Irons
Im Schema 8 ist die Reaktionsfolge aufgezeichnet, welche zur Aufklärung der
relativen Konfiguration des (+)- Jf-Irons (37) führte. Das verwendete (+)- Jf-Iron49)
stammte aus der Destillation von natürlichem Iron in der Podbielniak-Kolonne '.
Es erwies sich nach gaschromatographischer Analyse als einheitlich und lieferte in
90-proz. Ausbeute ein Phenylsemicarbazon vom Smp. 179-180°.
Die partielle Hydrierung des (+)- y-Irons (37) wurde vorerst mit Lithium in
flüssigem Ammoniak durchgeführt. Das in 82-proz. Ausbeute anfallende, destillierte
Reduktionsprodukt, dessen UV. -Spektrum eine Beimengung von ca. 10 % konjugiert-
ungesättigtem Keton zeigte, wurde zur Reinigung ins Semicarbazon überführt, das
nach fünfmaligem Umkristallisieren konstant bei 198-200 schmolz. Daraus konnte50)
nach einer Arbeitsvorschrift von M . S toll' durch Erhitzen mit Phtalsäureanhydrid
in Chloroform-Wasser das (+)-Dihydro- YMron (47) wieder freigesetzt werden, wel¬
ches sich nach gaschromatographischer Analyse als einheitlich erwies und folgende
physikalischen Eigenschaften besass**: [ot]^° = + 49° ; n^° = 1,4814 ; d^1 = 0,9238.
Ein identisches (+)-Dihydro- V'-iron (47) konnte auch durch partielle Hydrierung
von (+)-V'-iron (37) mit Raney-Nickel in Feinsprit bei Zimmertemperatur dargestellt
werden. Trotz sorgfältigster Reinigung über das Semicarbazon (Smp. 198,5-200,5 )
enthielt dieses, aus Jf-Iron in 55-proz. Ausbeute gewonnene Dihydro-iron laut gas¬
chromatographischer Analyse noch kleinere Mengen (2 - 3 %) eines Tetrahydro-irons (45).
Die Ozonolyse des (+)-Dihydro-y"-irons (47) in Methylenchlorid bei -70 lieferte
nach reduktiver Aufarbeitung mit Zink-Eisessig bei -20 das rohe Diketon (51), das
zur Reinigung an Silicagel chromatographiert wurde. Aus den mit Hexan eluierten Frak¬
tionen konnte dabei das bei der partiellen katalytischen Hydrierung von (+)-Jf-Iron als
Nebenprodukt anfallende Tetrahydro-iron (45) isoliert werden, (s. S. 44). Das reine
Diketon (51) wurde in 70-proz. Ausbeute mit Benzol-Aether 10:1 eluiert und durch das
Bissemicarbazon (Smp. 220°; [^jr, = + 129,1°) charakterisiert.
,49)
*) C. F.Seidel und L.Ruzicka ; gaben für das (+)-Dihydro- ^-iron folgendeEigenschaften an:[o<]D = +43,2° ; ng0 = 1,4842; d20= 0,9309.
Y.R. Naves fand:41)[c*]D = + 30,10; n|0= 1,4855; d2°= 0,9376.
Da diese Autoren die Spaltung des Semicarbazons mit Phtalsäureanhydrid in Wasser
allein ausführten, dürften diese Dihydro- Jf-irone Cyclisationsprodukte vom Ambri-
nol-Typus als Verunreinigungen enthalten.
- 38 -
Schema 8
L1/NH3oder
1 H2/Ra-Ni/EtOH
Alox n
(47)
a) O3/CH2Cl2/-70°b) Zn/CH3COOH/-20°
O
(51)
Bissemicarbazon 220
^D= +129,1°
2 NaOMe
MeOH/3hrf u u0,5 Vol-proz.konz. HCl/CHCl,
3nrfJ
(45)
(58) OH
a) Os04/Ae/Py
b) Mannit/KOH
(53)
Bissemicarbazon 221
[«4> = +127'10
(59) OH \ OH
OH
"ÖT*OH
(55) (60)

- 39 -
Eine Epimerisierung in Stellung 2 kann bei diesem Reaktionsschritt unter den
angewandten Bedingungen ausgeschlossen werden '.
Da Vorversuche, aus dem Diketon (51) durch intramolekulare alkalische Kon¬
densation direkt zum Octalon (52) zu gelangen, kein befriedigendes Resultat zeitigten,
wurde diese Cyclisation in zwei Stufen ausgeführt: Durch Chromatographieren an
Alox n konnte das Diketon (51) in 62-proz. Ausbeute zu einem Hydroxyketon des Ty¬
pus (54) cyclisiert werden. Die Konstitution dieses Hydroxyketons verdient eine Er¬
läuterung, da es denkbar wäre, dass die Cyclisation auch den alternativen Weg zu
**)
einem Hydroxyketon des Typus (55) einschlagen könnte '. Die Zuteilung der Dekalin-
struktur ' wird im vorliegenden Falle durch das NMR. -Spektrum gestützt: In Deu¬
terochloroformlösung treten bei S = 0,83 und S= 0, 98 zwei Singletts der nichtäqui¬
valenten geminalen Methylgruppen auf; das Doublett-Signal der 6-Methylgruppe fin¬
det sich bei S = 0,94 (J» 2 c/s). Hingegen fehlt im Spektrum das Singlett-Signal
einer vierten Methylgruppe ', wie es von der Formelvariante (55) verlangt wird.
Des weiteren ergibt die Integration im <$-Bereich von 0,8 - 1, 25 in Uebereinstim-
mung mit Formel (54) nur 9 Protonen, während für das isomere Hydroxyketon (55)
12 Protonen zu erwarten wären.
Aus dem Hydroxyketon (54) liess sich sowohl durch Chromatographieren an
basischem Alox I als auch durch kurzes Erhitzen mit p-Toluolsulfosäure in Benzol
leicht und in hoher Ausbeute das Octalon (52) (Phenylsemicarbazon 182-183°) gewinnen,
das im UV.-Spektrum die für oC,/3 -ungesättigte Ketone charakteristische Ab¬
sorption bei 243 mil (log = 4,14) aufwies. Das IR.-Spektrum zeigte neben der Bande
starker Intensität bei 1675 cm"' (konjugiert-ungesättigtes Keton) eine solche schwacher
R .F
.Church et al. ' konnten zeigen, dass bei der
Ozonolyse eines ähnlich gebauten Systems (56; R = CH2)das asymmetrische Zentrum in Stellung 9 nicht berührt
wird. Sie erhielten nämlich als Ozonabbauprodukt die
9oC-Ketosäure (R =0), in der das energetisch unstabilere
Epimere vorliegen muss, da es durch Aequilibrierung in
die 9/3 -Ketosäure übergeführt werden konnte.
**) Ein solcher Cyclisationsmodus ist bei analog gebauten Diketonen schon verschie¬
dentlich beobachtet worden52).
***) Die im Schema 8 angegebene trans-Verknüpfung des Dekalingerüstes wurde nicht
bewiesen. Sie beruht in Analogie zu dem von L. C agliotti53) durch Cyclisa¬tion des entsprechenden Diketons der Jononreihe erhaltenen Hydroxyketons, das
durch Einwirkung von Methylmagnesiumbromid in das Diol (33) überführt werden
konnte, dessen Konfiguration bewiesen worden ist (vgl. S-24).****) Im Falle der Struktur (55) wäre ein Singlett-Signal (CH3-C-OH) im S -Bereich
um 1,2 zu erwarten (vgl. L.M
. Jackman 5?)).'

- 40 -
Intensität bei 1717 cm,was auf die Anwesenheit kleinerer Mengen eines ß,V- -un¬
gesättigten Ketons deutet '.
Bei Aequilibrierungsversuchen mit zwei Aequivalenten Natriummethylat in Me¬
thanol oder mit 0,5-Vol.-proz. konz. Salzsäure in Chloroform konnte das Octalon
(52) (Ausbeuten 97 % bzw. 96 %) unverändert zurückgewonnen werden '. Diesem
muss somit die thermodynamisch stabilste Konfiguration zukommen: Da einerseits
1 9 ***\im a
' -Octalon-2-System die (4,10)-Bindung nur äquatoriale Lage einnehmen kann ',
andererseits für die 6-Methylgruppe die äquatoriale Anordnung energetisch bevorzugt
sein dürfte, muss letztere im Octalon (52) bezüglich dem Wasserstoffatom am Brücken-
köpf trans-Stellung einnehmen '.
Die Umsetzung von (52) mit Methyllithium in Aether führte zu zwei epimeren
Alkoholen, welche durch Chromatographieren an Alox HI getrennt werden konnten.
Hexan eluierte in 37-proz. Ausbeute den flüssigen Alkohol (57), Hexan-Benzol 10:1
in 49-proz. Ausbeute das feste Epimere (58). Die Zuordnung der Stereochemie dieser
Alkohole (Schema 8) basiert auf einem analogen Fall bei den isomeren y-Ambrinolen'
(vgl. Schema 3 auf S. 22).
Aus dem festen Alkohol (58) erhielt man durch Oxydation mit Osmiumtetroxyd
in Aether-Pyridin und anschliessende Spaltung des Osmiatesters mit einer alkali¬
schen Mannitlösung ein zum Teil kristallines Produkt, aus welchem durch Umkristal¬
lisieren aus Hexan in 57-proz. Ausbeute ein bei 152 schmelzendes Triol C, .H„fiO„
g*) Es dürfte sich dabei um das A -Octalon-Isomere handeln. Diese Annahme
basiert auf der Tatsache, dass in der Jononreihe bei der säurekatalysiertenIsomerisierung des entsprechenden A^-Octalons ein Gemisch erhalten wird,das überwiegend aus dem konjugiert-ungesättigten Octalon besteht, daneben
aber noch kleinere Mengen des ^9-Isomeren enthält (Privatmitteilung von
G.Büchi, MIT). Einen weiteren Hinweis dafür, dass in diesem System ein
Gleichgewicht zwischen der konjugiert-ungesättigten Form und der A^-Formvorliegt, liefert das Resultat der Umsetzung des °<,/3 -ungesättigten Octalons
der Jononreihe, bei welcher M. Stoll et al. 5) in kleiner Ausbeute das y3-Am-brinol isolieren konnten (vgl. Fussnote*) auf S. 23).
**) Nach Abschluss der vorliegenden Arbeit konnte im hiesigen Laboratorium ge¬
zeigt werden, dass das Octalon (52) bei der Behandlung mit zwei AequivalentenNatrium-methylat in Deuteromethanol 1-6 Wasserstoffatome gegen Deuterium
austauscht. Damit wurde bewiesen, dass unter den angewandten alkalischen Be¬
dingungen eine Aequilibrierung wirklich stattgefunden hat.* **) Dies gilt unter der Voraussetzung, dass die Konjugation der Enongruppierung
nicht gestört ist68). Diese Bedingung dürfte im Falle des Octalons (52) erfüllt
sein, da das UV. -Spektrum die für konjugiert-ungesättigte Ketone charakteristi¬
sche Bande bei 243 nui aufweist.
****) Die Rotationsdisperisonskurve dieses Octalons (52) in Dioxan zeigte einen quali¬tativ ähnlichen Verlauf wie diejenige des 19-Nor-Testosterons55, 56). Daraus
kann geschlossen werden, dass diesem Octalon die in Formel (52) angegebene,als (6R,10S) zu bezeichnende^ ') absolute Konfiguration zukommen muss.

- 41 -
vom Typus (59) gewonnen werden konnte. Dieses lieferte bei der Glycolspaltung mit
Blei-tetraacetat in absolutem Benzol bei Zimmertemperatur in 86-proz. Ausbeute
das Diketon (53), das trotz mehrmaligem Chromatographieren an Silicagel nicht in
reiner Form isoliert werden konnte. Das IR.-Spektrum des rohen Diketons (53) zeig¬
te indessen die gleichen Banden wie das Diketon (51) aus der Ozonolyse des (+)-Di-
hydro- ^f-irons (47). Zur Charakterisierung wurde das Bissemicarbazon (Smp. 221° ;
[o£ JD = + 127,1°) hergestellt. Dieses erwies sich nach Smp., Mischsmp., [oCL.-Wertund IR. -Spektrum mit dem Bissemicarbazon des Diketons (51) aus der Ozonolyse von
(+)-Dihydro-y-iron (47) als identisch. Dasselbe Derivat wurde auch aus dem flüssi¬
gen Alkohol (57) durch die gleiche Reaktionsfolge (Oxydation mit Osmiumtetroxyd,
Glycolspaltung mit Blei-tetraacetat) erhalten.
Die im Schema 8 angegebene cis(2,6)-Konfiguration des Diketons (53) ergibt
sich auf Grund der Tatsache, dass dieses aus dem konfigurativ festgelegten Octalon
(52) mit Hilfe eindeutiger Reaktionsschritte gewonnen werden konnte, bei denen das
asymmetrische Zentrum in Stellung 2 nicht berührt werden sollte. Die gleiche cis(2,6)-
Stereochemie muss auch dem Ozonabbauprodukt (51) des Dihydro- ft"-irons (47) zu¬
kommen, da es sich im direkten Vergleich mit dem Diketon (53) als absolut identisch
erwies. Die Folge der Abbaureaktionen steht damit in Ueberein-
stimmung mit der Annahme, dass dem ( + ) - y--Ir on, der Haupt¬
komponente des natürlichen Irons, die in Formel (60) festge¬
haltene relative Konfiguration zukommt*).
Dieser Befund stimmt mit der konfigurativen Zuordnung des (+)-2f-Irons von
Y . R . Naves' überein. Damit dürfte auch die bis heute bezüglich (2,6) angenom¬
mene Stereochemie der isomeren oC-Irone, sowie der daraus hergestellten Dihydro-
und Tetrahydro-Derivate bestätigt sein.
57}
*) Die in Formel (60) enthaltene, als (2S,6R) zu bezeichnende ' absolute Konfigura¬tion des (+)- f-Irons basiert auf der oben dargestellten Verknüpfung dieser Ver¬
bindung über das Diketon (51) mit dem Octalon (52), dessen absolute Konfigurationauf Grund des Verlaufs der Rotationsdisperisonskurve feststeht.

- 42 -
2. Diskussion der Auwers-Skita'schen Regel bei den Tetrahydro-ironen
Es ist bemerkenswert, dass durch die Aufklärung der Stereochemie des (+)-Jf-Irons die konfigurativen Zuordnungen von Y. R
.N a v e s bestätigt wurden. Da diese
bei den isomeren Dihydro-oc-ironen sowie den daraus hergestellten Tetrahydro-iro¬
nen mit Hilfe der alten Regel von Auwers und S kit a vorgenommen worden wa¬
ren, scheint eine Diskrepanz zur Erfahrungstatsache vorzuliegen, dass die Konfigu-
rationszuordnung nach dieser Regel im allgemeinen bei 3,5-disubstituierten Cyclo-
hexen- und 1,3-disubstituierten Cyclohexanverbindungen zum entgegengesetzten Re¬
sultat führt. Dieser Sachverhalt verdient eine Ueberprüfung im Sinne der von N. L.
Allinger modifizierten Regel. Es wurde schon weiter oben gezeigt (s. S. 34), dass
beim Dihydro-ot-iron-System die Voraussetzungen dazu nicht gegeben sind, da die
relative thermodynamische Stabilität der beiden Isomeren kaum geschätzt werden
kann. Im Falle der isomeren Tetrahydro-irone dagegen dürften diese Voraussetzun¬
gen erfüllt sein, da in diesen Verbindungen substituierte Cyclohexanderivate vorlie¬
gen, deren Stabilitätsunterschiede mit Hilfe einer einfachen Konstellationsanalyse
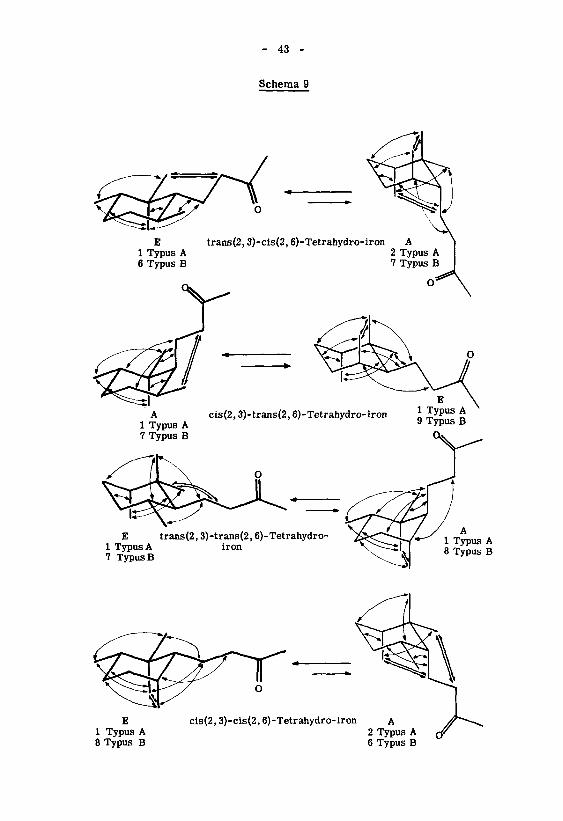
ermittelt werden können. Die konstellationsanalytische Betrachtung (Schema 9, Ta¬
belle 2) beschränkt sich auf die energetisch günstigsten Sesselformen des Tetrahy-
dro-iron-Systems, in denen die Butanonyl-Seitenkette bezüglich (l',2') antiplanar an¬
geordnet ist. Auf Grund dieser Vereinfachungen beschränkt sich die Zahl der mög¬
lichen Konstellationen für jedes Stereoisomere auf die beiden Typen E und A, in denen
die Seitenkette bezüglich des Cyclohexansessels äquatoriale bzw. axiale Lage ein¬
nimmt.
Tabelle 2
Konfiguration des
Tetrahydro-ironsKonstellation
Anzahl der Interaktionen
TypusA B
trans(2,3)-cis(2,6)E
A
1 6
2 7
trans(2,3)-trans(2,6) E
A
1 7
1 8
cis(2,3)-trans(2,6)E
A
1 9
1 7
cis(2,3)-cis(2,6)E
A
1 8
2 6
- 43 -
Schema 9
E
1 Typus A
6 Typus B
trans(2,3)-cis(2,6)-Tetrahydro-iron A
2 Typus A
7 Typus B
A cis(2,3)-trans(2,6)-Tetrahydro-iron1 Typus A
7 Typus B
E
1 Typus A
9 Typus B
E trans(2,3)-trans(2,6)-Tetrahydro1 Typus A iron
7 TypusB
A
Typus A
8 Typus B
E cis(2,3)-cis(2,6)-Tetrahydro-iron A
1 Typus A 2 Typus A
8 Typus B 6 Typus B

- 44 -
Der Vergleich der energetisch bevorzugten Konstellationen ' (im Schema 9 her¬
vorgehoben) ergibt für die isomeren Tetrahydro-irone folgende Reihenfolge nach ab¬
nehmender Stabilität ': Das energetisch stabilste Isomere ist das trans(2,3)-cis(2,6)-
Tetrahydro-iron, welches fast ausschliesslich in der Konstellation E vorliegen dürfte.
Die bevorzugten Sesselformen der beiden trans(2,6)-Tetrahydro-irone unterscheiden
sich von der obigen Konstellation um eine syn-schief-Interaktion. Als energetisch am
unstabilsten erweist sich das Stereoisomere mit cis(2,3)-cis(2,6)-Konfiguration.
Nach diesen Ableitungen lässt die modifizierte Regel von Auwers und Skita
für das trans(2,3)-cis(2,6)-Isomere die kleinsten, für die beiden trans(2,3)-Tetra-
hydro-irone mittlere und für das cis(2,3)-cis(2,6)-Stereoisomere die höchsten Werte
für Dichte, Brechungsindex und Siedepunkt erwarten.
Als Folge davon kann für die Naves'sehe Interpretation der Regel von Auwers
und Skita bei den Tetrahydro-ironen folgende Hypothese aufgestellt werden: Dieser
Autor muss offenbar eines der beiden trans(2,6)-Tetrahydro-irone ' mit demcis(2,3)-
cis(2,6)-Tetrahydro-iron verglichen haben, da nur in diesem Fall die ursprüng-
*) Da die Konstellationsgleichgewichte bei allen Stereoisomeren eindeutig auf der
Seite einer Konstellation liegen, genügt für eine rein qualitative Abschätzungder relativen Stabilitäten der isomeren Tetrahydro-irone ein Vergleich der
energetisch bevorzugten Sesselformen.
**) Die zur Abschätzung der Unterschiede in der Stabilität der Konstellationen
verwendeten Werte sind auf S. 18 (Fussnote*)) zusammengestellt.***) Y. R
. Naves hat dem trans(2,6)-Tetrahydro-iron (Semicarbazon 161-162°),das als Hauptprodukt bei der katalytischen Hydrierung von trans(2,6)-Dihydro-ot-iron36a) 38c) gebildet wird, seinerzeit die trans(2,3)-trans(2,6)-Konfigura-tion zugeteilte). Nachdem P.Bächli und H. Sc hinz*****) die Syntheseeines angeblich neuen Tetrahydro-irons publizierten, nahm Y. R . Naves^O)dann die cis(2,3)-trans(2,6)-Stereochemie an. Die trans(2,6)-Konfigurationdürfte zutreffen, da dieses Tetrahydro-iron aus dem trans(2, 6)-Dihydro-oC-ironerhalten wird; dagegen fehlen Anhaltspunkte, die es erlauben würden, die Kon¬
figuration bezüglich (2,3) festzulegen.****) Y . R .
NavesSöc) hat dem cis(2,6)-Tetrahydro-iron (Semicarbazon 200-201°)die cis(2,3)-cis(2,6)-Konfiguration zugeteilt, doch ist diese nie bewiesen wor¬
den. Dieses Tetrahydro-iron wurde durch katalytische Hydrierung aus cis(2,6)-Dihydro-<*.-iron36a, 38c), cis(2,6)-Dihydro-^-iron41, 58) sowie Dihydro-/3-iron^öa, 38c) erhalten. Wir erhielten das (+)-Enantiomere dieses Tetrahydro-irons als Nebenprodukt der partiellen katalytischen Hydrierung von (+)-cis(2,6)-y-Iron (s. S. 36). Das über das Semicarbazon (Smp. 200-201°) gereinigte Ke-
ton zeigte nämlich die gleichen physikalischen Daten wie das in der Literatur38c)
beschriebene cis(2,3)-cis(2,6)-Tetrahydro-iron. Auf Grund der oben erfolgtenkonfigurativen Aufklärung des natürlichen (+)-^-Irons dürfte die cis(2,6)-Kon-figuration zutreffen. Ein Anhaltspunkt, dass auch die angenommene cis(2,3)-Stereochemie zutrifft kann darin gesehen werden, dass dieses Keton auch bei
der katalytischen Hydrierung von Dihydro-/3-iron in neutralem Medium erhal¬
ten wird.47 -a-.
*****) Das von P.Bächli und H. Schi nz* ' os)synthetisierte trans(2,3)-Tetra-
hydro-iron (Semicarbazon 163°), dem später von Naves^O) die trans(2,3)-trans(2,6)-Konfiguration zugeordnet wurde, erwies sich bei einer neulich aus¬
geführten gaschromatographischen Analyse als ein Gemisch von zwei Verbin¬
dungen. Auf Grund der Retentionsindices kann angenommen werden, dass es
sich zu 2/3 aus trans(2,3)-cis(2,6)-Tetrahydro-iron und zu 1/3 aus cis(2,3)-cis(2,6)-Tetrahydro-iron zusammensetzt.

- 45 -
liehe und die modifizierte Regel zufälligerweise zum gleichen Resultat führen. Hätte
nämlich Y. R. Naves das trans(2,3)-cis(2,6)-Tetrahydro-iron mit einem der bei¬
den trans(2,6)-Tetrahydro-irone verglichen, so hätten die alte und die modifizierte
Regel zu entgegengesetzten Konfigurationszuordnungen bezüglich (2,6) geführt.
Zur Stütze dieser Hypothese wurde als Abschluss der vorliegenden Arbeit das
trans(2,3)-cis(2, 6)-Tetrahydro-iron (61) dargestellt. Dazu wurde (+)-cis(2,6)-Dihy-dro- $"-iron (47) vorerst mit Lithiumaluminiumhydrid in Aether zum Alkohol redu¬
ziert und dieser dann nach einer Arbeitsvorschrift von E.J.Corey und E.W.
Cantrall ' einer Reduktion mit Lithium in Aethylendiamin unterworfen.
a) LiAlH4/Aeb) Li/Et(NH2)2c) Cr03/AcOH
(61) ca. 50% (45) ca. 40%
Da diese Hydrierung erfahrungsgemäss 'zum thermodynamisch stabilsten Produkt
führt, war zu erwarten, dass überwiegend das energetisch stabilere trans(2,3)-cis(2,6)-
Isomere gebildet würde. Das rohe Reduktionsprodukt wurde direkt mit Chromsäure in
Eisessig oxydiert. Das resultierende Ketongemisch bestand laut gaschromatographi-
scher Analyse zu ca. 90 % aus zwei Hauptkomponenten. Eine davon (ca. 40 %) erwies
sich auf Grund ihres gaschromatographischen Verhaltens als identisch mit dem (+)-Te-
trahydro-iron, das als Nebenprodukt der partiellen katalytischen Hydrierung von
(+)- X"-Iron erhalten wurde (s. S. 36). Die zweite (ca. 50 %), bei welcher es sich um
ein bisher unbekanntes Tetrahydro-iron handelte, konnte durch präparative gaschro-
matographische Auftrennung gewonnen werden. Das Semicarbazon dieser Verbindung
schmolz bei 182,5-183,5 und ergab mit demjenigen des obigen Tetrahydro-irons
(Smp. 200-201°) eine Depression des Mischsmp. um 4°.
Da für diese Reaktionsfolge vom (+)-cis(2,6)-Dihydro- Y"-iron ausgegangen wor¬
den war, dürfte es sich bei den erhaltenen Tetrahydro-ironen um die beiden cis(2,6)-
Stereoisomeren handeln. Diese wiesen stark verschiedene physikalische Eigenschaften
auf: Das in 40-proz. Ausbeute gebildete Keton (Semicarbazon 200-201°) zeigte annä¬
hernd die gleichen Daten wie das von Y. R . Navesc' beschriebene cis(2,3)-cis(2,6)-
Tetrahydro-iron, während das in 50-proz. Ausbeute erhaltene Diastereomere (Semi¬
carbazon 182,5-183,5 ) beträchtlich tiefere Werte aufwies. Dem erstgenannten dürfte
somit auf Grund der modifizierten Regel von Auwers und Skita die cis(2,3)-cis(2,6)-
Konfiguration (45), dem zweiten die trans(2,3)-cis(2, 6)-Stereochemie zukommen. Da¬
mit liegt ein Hinweis für die oben aufgestellte Hypothese vor, dass Y. R. Naves
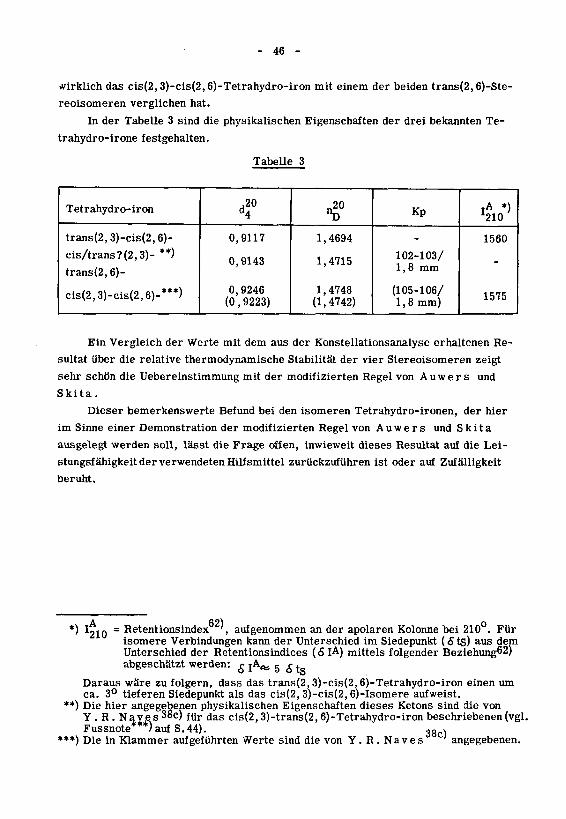
- 46 -
wirklich das cis(2,3)-cis(2,6)-Tetrahydro-iron mit einem der beiden trans(2,6)-Ste-
reoisomeren verglichen hat.
In der Tabelle 3 sind die physikalischen Eigenschaften der drei bekannten Te-
trahydro-irone festgehalten.
Tabelle 3
Tetrahydro-iron H20d4 ? Kp
.A *)*210
trans(2,3)-cis(2,6)-
cis/trans?(2,3)- **)
trans(2,6)-
cis(2,3)-cis(2,6)-***)
0,9117
0,9143
0,9246(0,9223)
1,4694
1,4715
1,4748(1,4742)
102-103/1,8 mm
(105-106/1,8 mm)
1560
1575
Ein Vergleich der Werte mit dem aus der Konstellationsanalyse erhaltenen Re¬
sultat über die relative thermodynamische Stabilität der vier Stereoisomeren zeigt
sehr schön die Uebereinstimmung mit der modifizierten Regel von Auwer s und
Skita.
Dieser bemerkenswerte Befund bei den isomeren Tetrahydro-ironen, der hier
im Sinne einer Demonstration der modifizierten Regel von Auwer s und Skita
ausgelegt werden soll, lässt die Frage offen, inwieweit dieses Resultat auf die Lei¬
stungsfähigkeit der verwendeten Hilfsmittel zurückzuführen ist oder auf Zufälligkeit
beruht.
,62)
*) Ij.q = Retentionsindex, aufgenommen an der apolaren Kolonne bei 210
.Für
isomere Verbindungen kann der Unterschied im Siedepunkt (Sts) aus dem
Unterschied der Retentionsindices (5lA) mittels folgender Beziehung^2)abgeschätzt werden: r jA^ 5 ^
.
Daraus wäre zu folgern, dass das trans(2,3)-cis(2,6)-Tetrahydro-iron einen um
ca. 3° tieferen Siedepunkt als das cis(2, 3)-cis(2,6)-Isomere aufweist.
**) Die hier angegebenen physikalischen Eigenschaften dieses Ketons sind die von
Y. R. Naves38c) für das cis(2,3)-trans(2, 6)-Tetrahydro-iron beschriebenen (vgl.Fussnote***) auf S. 44). 38c\
***) Die in Klammer aufgeführten Werte sind die von Y. R. Naves angegebenen.

- 47 -
EXPERIMENTELLER TEIL*)
1. Stereochemie der säurekatalysierten Cyclisation von Dihydro- y-jonon
oi-Ambrinol (30)
Das verwendete ot-Ambrinol stammte aus den o^-Ambrinol-reichen Fraktionen5)
der Podbielniakdestillation des Cyclisationsproduktes von Dihydro-y-jonon '.
fï^r^Jîîtïi^tJb^ii^itrobejnz^qat: 0,912 g (4,71 mMol) dieses o^-Ambrinols wur¬
den in 2 ml Pyridin und 4 ml Benzol gelöst, mit 2 g 3, 5-Dinitro-benzoylchlorid ver¬
setzt und während 1 Std. unter Rückfluss gekocht. Nach dem Abkühlen versetzte man
mit 80 ml Aether und filtrierte vom Unlöslichen ab. Das Filtrat wurde am Vakuum
eingeengt; der zurückbleibende, ölige Rückstand wurde aus Alkohol umkristallisiert.
Nach dreimaligem Umkristallisieren erhielt man 0,981 g (55 %) Derivat vom Smp.
81-83° (Lit.5) : Smp. 85°).
*) Die Siedepunkte sind nicht korrigiert. Die Schmelzpunkte sind korrigiert und im
offenen Röhrchen bestimmt.
Die UV.-Spektren wurden im Laboratorium von Prof. Dr. E.Heilbronner in
äthanolischer Lösung mit einem Beckmann-Spektrophotometer (Modell DKj) aufge¬nommen.
Die IR.-Spektren wurden im Laboratorium von Prof. Dr. Hs.H.Günthard mit
einem Perkin-Elmer Apparat (Modell A21, NaCl-Prisma) aufgenommen. Die An¬
gaben s, m, w und Seh bei den IR. -Spektren bedeuten approximative Intensitätsbe¬
zeichungen (stark, mittel, schwach und Schulter).Die optischen Drehungen wurden in einem Rohr von 10 cm Länge gemessen.
Die Analysen wurden in der mikroanalytischen Abteilung des Organisch-Chemi¬schen Institutes der E.T.H. unter der Leitung von Herrn W. Manser ausge¬
führt.
Die Aktivitätsbezeichungen für Aluminiumoxyd beziehen sich auf die Methode von
H. Brockmann und H. Schodder 63).
Für analytische wie auch für präparative gas-chromatographische Arbeiten wurden
zwei verschiedene Kolonnen benutzt, nämlich 1) eine sog. "apolare Kolonne", de¬
ren stationäre Phase aus 40 % Apiezon L auf Cellit bestand, und 2) eine "polareKolonne" mit 40 % Emulphor-Oauf Cellit. Für präparative Zwecke verwendete man
reinen Stickstoff, für analytische Chromatogramme dagegen Helium als mobile
Phase. Zur Charakterisierung der verschiedenen Fraktionen bewährten sich die
von E.Kovats62, 64) in die Literatur eingeführten Retentionsindices, für welche
eine abgekürzte Schreibweise verwendet wurde: Es bedeutet z.B. 14..: Retentions-
index, aufgenommen ander apolaren Kolonne bei 210°. Den Herren Dr. E.
Kovats undE.Kugler danke ich bestens für die Aufnahme und Interpretationder Gas-Chromatogramme.

- 48 -
o£-A mbr inoloxyd (3 2)
600 mg (3,09 mMol) crf-Ambrinol (30) liess man mit 12 ml einer 0,52 n. Mono¬
perphtalsaurelösung in Aether während 22 Std. im Eisschrank (5 ) reagieren. Hier¬
auf wurde die ätherische Lösung mit 10-proz. Natriumhydrogenkarbonat-lösung durch¬
geschüttelt, mit Wasser neutral gewaschen, über Natriumsulfat getrocknet und unter
vermindertem Druck eingedampft. Der Rückstand wurde in einem Hickmann-Kolben
bei 0,3 Torr (Badtemperatur 80°) destilliert und lieferte 440 mg (70 %) Epoxyd (32),
das beim Abkühlen kristallisierte. Nach mehrmaligem Umkristallisieren aus Hexan
bei -70° und Sublimation bei 42°/0,3 Torr schmolz das Produkt bei 54,5-55,5°.(Lit.5) : 55-56°).
IR.-Spektrum (Nr. 12886; in CHClg): Banden u.a. bei 848m, 915m, 1370m, 1392m
und3580m cm"1.
Die Verbindung erwies sich nach Smp., Mischprobe und IR. -Spektrum als identisch
mit dem von A.G. Armour und G. Büchi'hergestellten o(-Ambrinoloxyd.
/3-Ambrinol (27)
Das verwendete /3-Ambrinol (27) wurde aus dem redestillierten Kolbenrück-
5)stand der oben erwähnten Podbielniakdestillation des Cyclisationsproduktes von
Dihydro- ft -jonon isoliert. Nach fünfmaligem Umkristallisieren bei -70° aus Hexan
schmolz das y3-Ambrinol bei 62-63°. (Lit. ': 61-62,5°). Eine Probe wurde zur Ana¬
lyse am Hochvakuum bei 48 /0,001 Torr sublimiert.
IR.-Spektrum (Nr. 13873; in CHCI3): Banden u.a. bei 1365m, 1380m, 1388m
und 3610m cm"1.
C^oH^O Ber. C 80,35 H 11,41 %£
Gel. C 80,52 H 11,37 %
Die Substanz erwies sich im direkten Vergleich nach Smp., Mischsmp. und IR.-
Spektrum als identisch mit dem von A. G. Armour und G. B ü c h i'synthetisier¬
ten y3-Ambrinol.
/3-Ambrinoloxyd (34)
Zu 792 mg (4, 08 mMol) y3-Ambrinol (27) gelöst in 10 ml Aether gab man 50 ml
einer 0,288 n. Monoperphtalsaurelösung in Aether und liess das Ganze auf obige Weise
reagieren. Der nach üblicher Aufarbeitung erhaltene Rückstand (803 mg) wurde an der
30-îachen Menge Alox, Akt. II, chromatographiert und anschliessend in einem Hick¬
mann-Kolben bei 0,3 Torr (Badtemperatur 80°) destilliert. Man erhielt 606 mg (71 %)
eines öligen, farblosen Epoxyds (34).

- 49 -
IR.-Spektrum (Nr. 13731; in CHClg): Banden u.a. bei 1170,1235, 1368, 1392
und 3440-3480m cm"1.
C1f,H9909 Ber. C 74,24 H 10,54%à
Gef. C 74,10 H 10,65 %
Diol (33)
a) Aus ot-Ambrinoloxyd (32)
Zu einer Lösung von 1,032 g (4,91 mMol) et-Ambrinoloxyd (32) in 100 ml abs.
Tetrahydrofuran gab man 0,475 g (12,5 mMol) Lithiumaluminiumhydrid. Das Reak¬
tionsgemisch wurde drei Std. unter Rückfluss gekocht. Nach dem Abkühlen zersetzte
man vorsichtig mit verd. Seignettesalzlösung und extrahierte mit Aether. Die ätheri¬
sche Lösung wurde mit Wasser gewaschen, über Natriumsulfat getrocknet und am
Vakuum eingeengt. Man erhielt 1,035 g rohes, kristallines Diol (33) vom Smp. 117-
119,5°. Nach zweimaligem Umkristallisieren aus Hexan wurden 0,947 g (92 %) rei¬
nes Diol (33) vom konstanten Smp. 120-122° gewonnen.
IR.-Spektrum (Nr. 12896; in CHCI3): Banden u.a. bei 1189s, 1368m, 1379m, 1390m,
1410m, 3480s und 3600m cm"1.CtoHL.O, Ber. C 73,53 H 11,39%
**Gef. C 73,32 H 11,34%
b) Aus (3-Ambrinoloxyd (34)
Zu 200 mg (5,28 mMol) Lithiumaluminiumhydrid in 20 ml Tetrahydrofuran gab
man 232 mg (1,10 mMol) ß -Ambrinoloxyd (34) gelöst in 5 ml Tetrahydrofuran. Man
kochte während 10 Std. unter Rückfluss und zersetzte nach dem Abkühlen vorsichtig
mit Wasser. Das nach üblicher Aufarbeitung erhaltene Oel wurde an 10 g neutralem
Alox, Akt. II, chromatographiert. Man erhielt 200 mg (86 %) des Diols (33), das nach
zweimaligem Umkristallisieren aus Hexan bei 119-122° schmolz. Mischsmp. mit Diol
aus o(-Ambrinoloxyd ohne Depression, ebenso waren die IR.-Spektren (in CSg und
CHC1,) der beiden Produkte in allen Teilen identisch.
Carbonat (35) aus Diol (33)
Eine Lösung von 106 mg (0,5 mMol) des Diols (33) in 5 ml Diäthylcarbonat wurde
mit 33 mg (0, 5 mMol) Natriumäthylat versetzt und während 72 Std. unter Stickstoff-
Atmosphäre unter Rückfluss gekocht. Hierauf wurde in Aether aufgenommen, die äthe¬
rische Lösung mit Wasser neutral gewaschen, über Natriumsulfat getrocknet und ein¬
geengt. Man erhielt 119 mg rohes Carbonat (35) vom Smp. 100-102°. Nach dreimali¬
gem Umkristallisieren aus Hexan wurden 93 mg (78 %) reines Carbonat vom Smp.
105, 5-106, 5° erhalten.

- 50 -
IR.-Spektrum (Nr. 13232; in CHC13): Banden u.a. bei 1078m, 1130,1372
,1383m
und 1722s cm"1.
C14H990„ Ber. C 70,55 H 9,20%* l
Gef. C 70,41 H 9,21 %
Zwecks Rückverseifung zum Diol (33) wurden 25 mg (0,15 mMol) des Carbonats in
6 ml 10-proz. äthanolischer KOH gelöst und während 5 Std. unter Rückfluss gekocht.
Hierauf nahm man in Aether auf, wusch mit Eiswasser neutral, trocknete über Na¬
triumsulfat und erhielt nach Abdampfen des Aethers 20 mg (94 %) kristallisiertes
Diol. Dieses schmolz nach zweimaligem Umkristallisieren aus Hexan bei 119-122
(12 mg). Mischsmp. mit authentischem Diol (33) ohne Depression.
Relative Cyclisationsgeschwindigkeiten von (t)-Dihydro- jf-jonon und
(+)-cis(2,6)-Dihydro- y-iron
( + )-cis(2, 6)-Dihydro- f -iron (47)
Das für die Messungen verwendete Keton wurde aus (+)-cis(2,6)- Jf-Iron (37)
durch partielle Hydrierung mit Raney-Nickel hergestellt und über das Semicarbazon
(Smp. 198-200^ gereinigt. (Beschreibung der Ausführung s. S. 56). Eine gaschroma-
tographische Analyse zeigte an der apolaren Kolonne eine Verunreinigung (Iojn =1560)
von ca. 3 % an cis(2,3)-cis(2,6)-Tetrahydro-iron (45).
Kp0 03= 76° ; n^6= 1,4830 ; d19 = 0,9238 ;{eC]^ = + 49,3° (in Substanz)
Retentionsindices: I^Q = 1530 ; 12^Q = 1810.
IR.-Spektrum (Nr. 14388; flüssig): Banden u.a. bei 886s, 1365s, 1393m, 1645s,1715s, 2840s, 2920s und 3070w cm"1.
Ber. C 80,71 H 11,61%Gef. C 80,63 H 11,69%
C14H240
(+)-Dihydro-y-jonon (1)
9,9 g (42,5 mMol) Dihydro- Jf -jononsemicarbazon (Smp. 190-191°) wurden in
250 ml Chloroform gelöst, mit einer Lösung von 17 g Phtalsäureanhydrid in 150 ml
Wasser versetzt und während zwei Std. unter Rückfluss gekocht. Nach dem Abkühlen
nahm man in Chloroform auf, schüttelte fünfmal mit 10-proz. Natronlauge, wusch
mit Wasser neutral, trocknete über Natriumsulfat und dampfte das Chloroform ab.
Das Rohprodukt (7, 62 g) wurde in einem Vigreux-Kolben am Hochvakuum destilliert.
(Ausbeute 6,7 g; 81 %). Eine gaschromatographische Analyse zeigte eine kleine Bei¬
mengung von ca. 2 % Dihydro-ctf-jonon an.

- 51 -
Kp004 = 64-66° ; nß6 = 1,4773 ; dj1 = 0,9161
Retentionsindices: I2A0 = 1406 ; I„?n = 1682.
IR.-Spektrum (Nr. 20258; flüssig): Banden u.a. bei 890s, 1366m, 1390m, 1646m,
1715s, 2880s, 2940s und 3080w cm"1.
C13H22° Ber- C 80,35 H U>41Gef. C 80,39 H 11,38
Messungen
Die dieser Arbeit zugrunde liegende Messmethode entspricht im wesentlichen
der von C .P r i c e et al. ' für die Cyclisation von Citronellal und Citral beschrie¬
benen Methode. Im Unterschied zu dieser Arbeit wurden die Messungen zur Erhöhung
der Löslichkeit der Methylketone in Methanol-Wasser im Volumenverhältnis 1:1 ausge¬
führt.
Titrationen
Die Titrationen wurden unter Thermostatierung bei 25 Î0,5 C in einem breiten
Kochglas durchgeführt. In die Lösung wurde Stickstoff eingeleitet und mit einem Mag-
netrührer gerührt. Da ferner bei Vorversuchen festgestellt wurde, dass Methylketone
mit Hydroxylaminsulfat bedeutend langsamer oximiert werden als Aldehyde, wurde die
zu titrierende Lösung nach Zugabe der Hydroxylaminsulfat-Lösung erst nach einer
ganz bestimmten Zeit (10 Min.) titriert. Als Messkette diente eine Stabmesskette
(Glaselektrode, n-Kalomelelektrode) und als Kompensationsgerät wurde ein Polyme-
tron-pjr-meter verwendet.
Durchführung einer Titration
Für eine Titration wurden 20 ml der Cyclisations-Lösung (8-10,5 mg Methylketon
in 20 ml Methanol/0,1 n. Salzsäure im Volumenverhältnis 1:1) mit 20 ml Wasser ver¬
dünnt, mit 0,1 n. Natronlauge neutralisiert und mit 0,01 n. Salzsäure auf p„ 4,30 ein¬
gestellt. Nach Zugabe von 0,5 ml 8-proz. Hydroxylaminsulfatlösung wurde während
10 Min. stehen gelassen. Anschliessend titrierte man innerhalb von zwei Min. mit
0,01 n. Natronlauge auf den pH der Blindlösung (3,67-3,69) und innerhalb weiterer
zwei Min. auf den ursprünglichen pH von 4,30. Der Wert für die Konzentration C* er¬
gab sich aus Verbrauch an ml 0,01 n. Natronlauge (auf pH 4,30 titriert) minus Blind¬
wert.
Bestimmung des Blindwertes
20 ml eines Gemisches von Methanol/0,1 n. Salzsäure im Volumenverhältnis 1:1
wurden mit 20 ml Wasser verdünnt, mit 0,1 n. Natronlauge neutralisiert und mit

- 52 -
0,01 n. Salzsäure auf p„ 4,30 eingestellt. Die Lösung wurde mit 0,5 ml 8-proz. Hy¬
droxylaminsulfat-Lösung versetzt und während 12 Min. stehen gelassen (pH3,67-3,69).Anschliessend titrierte man innerhalb von zwei Min. mit 0,01 n. Natronlauge auf das
pH von 4,30 zurück. Der Verbrauch an 0,01 n. Natronlauge in ml stellte den Blind¬
wert dar. Dieser wurde jeweils vor und nach einer Messreihe bestimmt.
Bei der Titration zeigte sich, dass nicht 100 % des Methylketons erfasst wer¬
den konnten. Die erhaltenen Werte lagen immer zwischen 82-86 %. Es wurde daher
für jede Messreihe eine Bestimmung zur Zeit 0 durchgeführt (CQ).Die k*-Werte wurden aus folgender Gleichung berechnet:
log
0,4343 -[HgO®]-t = Zeit in Std.
Der bei der Berechnung der k* -Werte auftretende Faktor
bestimmt:
ml 0,01 n. NaOH zur Zeit 0 - Blindwert
ml 0,01 n. NaOH zur Zeit t - Blindwert
wurde wie folgt
Messungen in Methanol/0,1 n. Salzsäure 1:1
a) Lösung
Temperatur
Blindwert
: 52,2 mg (0,269 mMol) Dihydro- Jf-jonon in 55 ml Methanol und
55 ml 0,1 n. Salzsäure
PH= 1,58 ; [HjO^ = 2,63 • lO-2 Mol/l
: 25 t 0,5°C: 2,16 ml 0,01 n. Natronlauge
Zeit
Min.
ml 0,01 n.
NaOH auf
PH4,30Ct log—
k*
Mol"1Std"1 1
0
20
40
80
6,225,304,633,78
4,063,142,471,62
0,11160,21580,3990
29,3128,3426,20
k* = 27,95 M0l"1Std"1l
- 53 -
b) Lösung : 58,5 mg (0,302 mMol) Dihydro-V'-jonon in 55 ml Methanol und
55 ml 0,1 n. Salzsäure
pH = 1,57 ; [H3O®] = 2,69 • 10"2 Mol/l
Temperatur : 25 Î 0,5°CBlindwert : 2,24 ml 0,01 n. Natronlauge
Zeit
Min.
ml 0,01 n.
NaOH auf
PH 4,30Ct l0g—
*
k
Mol"1Std"1 1
0
20
40
60
80
6,885,784,924,463,92
4,643,502,682,221,72
0,12240,23840,32010,4310
34,91
30,5927,3827,68
k* = 30,14 Mol"1Std" 1
?i.(+):Çi.s.C2j-6hPi!îïdro:jC;:irqnj47)_
a) Lösung : 79,2 mg (0, 381 mMol) cis(2,6)-Dihydro- }f-iron in 100 ml Me¬
thanol und 100 ml 0,1 n. Salzsäure
PH= 1,62 ; [H3Oe] = 2,40 • 10"2 Mol/l
Temperatur : 25 + 0, 5° C
Blindwert : 2,36 ml 0,01 n. Natronlauge
Zeit
Min.
ml 0,01 n.
NaOH auf
PH 4,30Ct log^
k*
Mor1Std"1 1
0 5,64 3,28 _ _
60 5,33 2,97 0,0431 4,135120 5,10 2,74 0,0781 3,747180 4,74 2,38 0,1393 4,354240 4,40 2,04 0,2063 4,948330 4,12 1,76 0,2704 4,716390 3,91 1,55 0,3256 4,806450 3,69 1,33 0,3920 5,012
k* = 4, 531 Mol-istd-11
b) Lösung : 80,2 mg (0,386 mMol) cis(2, 6)-Dihydro- •#--iron in 100 ml Me¬
thanol und 100 ml 0,1 n. Salzsäure
PH= 1,58 ; [H3O®] = 2,63 • 10"2 Mol/l
Temperatur : 25 t 0,5° C
Blindwert : 2, 36 ml 0,01 n. Natronlauge

- 54 -
ml 0,01 n. c0 k*Zeit NaOH auf Ct
"-t-1 -1
Min. PH 4,30 Mol *Stdx
1
0 5,69 3,33 *. .
60 5,40 3,04 0,0395 3,458120 4,98 2,62 0,1041 4,557180 4,72 2,36 0,1495 4,362270 4,28 1,92 0,2391 4,652330 4,12 1,76 0,2769 4,407390 3,82 1,46 0,3580 4,822450 3,62 1,26 0,4220 4,925
k* = 4, 455 Mol^Std"11k -Mittel aller Messungen:
a) (±)-Dihydro-J"-jonon : k*" = 29,05 Mol"1Std"11
b) (+)-cis(2,6)-Dihydro-y-iron : k*" = 4,493 Mol-1Std~1 1
2. Die Konfiguration des natürlichen (+)- if -Irons
Natürliches ( + )-y-Iron (37)
Das verwendete (+)-V"-Iron stammte aus den Fraktionen 17-21 der 1. Destilla-49)
tion von natürlichem Iron in der Podbielniak-Kolonne '. Diese Fraktionen wurden
zusammengefasst und in einem Vigreux-Kolben am Hochvakuum destilliert. Nach
einem kleinen Vorlauf destillierte die Hauptmenge zwischen 89-90 /0,03 Torr. Die
gaschromatographische Analyse zeigte nur eine Substanz an.
nD°= 1,5003 ; d^2= 0,9334 ; [o(.JD2 = + 9,33° (ohne Lösungsmittel)*)
UV. -Spektrum: A ü? = 226 mu (log £ = 4,18)
IR.-Spektrum (Nr. 13872; flüssig): Banden u.a. bei 895s, 1368s, 1398m, 1633s,
1650s, 1683s, 2880s, 1960s und 3100w cm-1.
A PRetentionsindices: I9ln = 1546 ; I, 1850
210" *""
'"210
C14H22° Ber- C 81'50 H 10>75Gef. C 81,32 H 10,54
*)P.Bächli, C.F.Seidel, H. Schinz und L. Ruzicka fanden47'48^:1,5006 ; d|°= 0,9368 ; ot
D= + 8°. Y
. R .Naves*!) fand
.20nDn2°=
1,50188; d2A° = 0,9355;to(J20
=
D'
'4' ' L JD
7,60°.

- 55 -
y~-Ironphenylsemicarbazon .444 mg (2,15 mMol) Jf-Iron wurden mit einer Lösung
von 380 mg (2, 52 mMol) Phenylsemicarbazid in 2,5 ml Methanol versetzt und wäh¬
rend 75 Min. unter Rückfluss gekocht. Nach kurzer Zeit begann das Phenylsemicar-
bazon auszukristallisieren. Die Rohausbeute betrug 682 mg (93,5 %). Nach zweima-
;m t
n20
ligem Umkristallisieren aus Chloroform-Alkohol schmolz das Derivat bei 179-180.
[o^]DU=- 10,4° (c = 8,02; in CHCI3)
'
UV. -Spektrum: A^H = 235 mu (log e = 4, 26) und 276 mji (log £ = 4,54)
C21H29ON3 Ber. C 74,30 H 8,61 N 12,38Gef. C 74,40 H 8,67 N 12,20
(+)-Dihydro- r-iron (47)
a) Durch Reduktion von (+)- y-Iron (37) mit Lithium in Ammoniak
200 ml flüssiges Ammoniak wurden über 5 g Natrium absolutiert und in ein mit
Aceton-Trockeneis gekühltes 500 ml Zylindergef äss destilliert. Unter Vibrieren und
Feuchtigkeitsausschluss wurden 10 mg Lithium zugegeben, wobei sich die Lösung
sofort tiefblau färbte. Hierauf gab man in kleinen Portionen 2,0 g (0,14 Mol) Lithium
hinzu und Hess 2,92 g (14,2 mMol) (+)-y-Iron (37), gelöst in 50 ml abs. Tetrahydro-
furan innerhalb 30 Min. zufliessen. Anschliessend wurde während 30 Min. weiter¬
vibriert und eine Std. stehen gelassen. Man zerstörte das überschüssige Lithium
vorsichtig mit Wasser, dampfte das Ammoniak ab und extrahierte den Rückstand mit
Aether. Das nach üblicher Aufarbeitung erhaltene Rohprodukt (2, 994 g) lieferte nach
der Destillation in einem Hickmann-Kolben 2,406 g (82 %) rohes Dihydro- jj--iron (47)
vom Sdp. 117-120°/11 Torr. Das UV.-Spektrum zeigte eine Schulter bei 226 mu
(log £ = 3, 22), was einem Gehalt von ungefähr 10 % an dL,fb -ungesättigtem Keton
entspricht.
Dihydro-Y;-ironsemicarbazon ': 1,96 g des rohen (47) versetzte man mit 30 ml
einer Lösung von Semicarbazidacetat in Methanol ' und Hess 20 Std. bei Zimmer¬
temperatur stehen. Die dabei gebildeten Kristalle wurden fünfmal aus Alkohol um¬
kristallisiert. Man erhielt 631 mg (28 %) Dihydro- Jf-ironsemicarbazon vom Smp.
198-200°.
*) Die hier beschriebene Methode wird später als übliche Methode bezeichnet.
**) C. F.Seidel und L . Ruzicka49) fanden: [<1D = - 12,2°; Y.R .
Naves41):
[<Od = - 8,4° (c = 8,0; in CHCI3).***) Die hier angegebene Methode wird in Zukunft als übliche Methode bezeichnet.
****) Zur Herstellung dieser Lösung wurden 10 g Semicarbazidhydrochlorid mit 20 g
Natriumacetat 3H2O verrieben und das flüssige Gemisch mit 50 ml Methanol
extrahiert.

- 56 -
Mß2 = + 35,0° (c = 8,02; in AcOH)**Ber. C 67,88 H 10,26 N 15,83Gef. C 67,77 H 10,32 N 15,86
C15H27ON3
Spalt^£_degJDiJ^d^q^^-J^pnsejnic_art 1,03 (3,9 mMol) des obigen Dihy-
dro- y-ironsemicarbazons wurden in 30 ml Chloroform gelöst, mit einer Lösung
von 1,7 g Phtalsäureanhydrid in 20 ml Wasser versetzt und während vier Std. unter
Rückfluss gekocht. Nach dem Abkühlen nahm man in Chloroform auf, schüttelte fünf¬
mal mit 10-proz. Natronlauge, wusch mit Wasser neutral, trocknete über Natrium¬
sulfat und dampfte das Chloroform ab. Das Rohprodukt (887 mg) wurde in einem
Hickmann-Kolben am Hochvakuum destilliert. Das Dihydro- ^"-iron (47) (670 mg;
83 %) destillierte bei einer Badtemperatur von 90°/l Torr, n^2 = 1, 4806 ;
[<<]D2= + 48,9° (ohne Lösungsmittel).
UV.-Spektrum: Endabsorption bei 210 aifi (log £ = 2,86)
IR.-Spektrum (Nr. 14379; flüssig): Banden u.a. bei 886s, 1365s, 1393m, 1645s,
1715s, 2840s, 2920s und 3070w cm"1.
ciaH94° Ber- c 80>71 H n.61%* i%
Gef. C 80,65 H 11,69 %
b) Durch Reduktion von (+)-JT-Iron (37) mit Raney-Nickel
65)
0, 2 g Raney-Nickel ' wurden in 10 ml Feinsprit vorhydriert. Hierauf wurde
eine Lösung von 1,60 g (7,8 mMol) Jf-Iron (37) in 25 ml Feinsprit zugegeben und bis
zur Aufnahme von 0,95 Moläquivalenten Wasserstoff (185 ml) bei Zimmertemperatur
hydriert. Die Hydrierung dauerte 70 Min. Man filtrierte vom Katalysator ab und ent¬
fernte das Lösungsmittel am Vakuum. Das UV.-Spektrum (Schulter bei 227 mu,
log £ = 3,12) zeigte die Anwesenheit von ungefähr 6% konjugiert-ungesättigtem Ke-
ton. Das Rohprodukt wurde auf übliche Art ins Semicarbazon überführt, das nach
zweimaligem Umkristallisieren aus Alkohol bei 198,5-200,5° schmolz (980 mg; 50%).
Ein Mischsmp. mit dem Semicarbazon aus der Reduktion von (+)-y-Iron mit Lithium
in Ammoniak zeigte keine Depression.
[oC]^2 = + 34,4° (c = 8,0; in AcOH)
Die Spaltung dieses Semicarbazons auf obige Art lieferte in 85-proz. Ausbeute
ein (+)-Dihydro-y-iron (47) mit folgenden Eigenschaften: n^2 = 1,4804 ; d^9= 0,9238
[o£]n = + 49,35 (ohne Lösungsmittel).
*)Y.R.Naves fand"1 ': Mq = + 30,5° (c = 8, 0; in AcOH)

- 57 -
Das IR.-Spektrum (Nr. 20259) war deckungsgleich mit demjenigen des (+)-Dihydro-
r-irons aus der Reduktion von (+)- V'-Iron mit Lithium in Ammoniak.
C14H9.0 Ber. C 80,71 H 11,61%Gef. C 80,55 H 11,76%
Diketon (51)
4,864 g (23,4 mMol) über das Semicarbazon gereinigtes (+)-Dihydro- }f-iron
(47) wurden in 160 ml Methylenchlorid gelöst, unter Stickstoffatmosphäre auf -70°
abgekühlt und bei dieser Temperatur bis zur auftretenden Blaufärbung ozonisiert
(20 Min.). Bei -20 versetzte man die Reaktionslösung mit 85 ml Eisessig und gab
unter Vibrieren innerhalb von drei Std. portionenweise 12, 5 g Zinkstaub zu. Hier¬
auf nahm man in Aether auf, wusch die ätherische Lösung viermal mit Eiswasser,
dreimal mit kalter Natriumhydrogencarbonatlösung und nochmals mit Eiswasser.
Nach dem Trocknen über Natriumsulfat dampfte man den Aether am Vakuum bei
Zimmertemperatur ab. Der Rückstand wurde in einem Vigreux-Kolben am Hochva¬
kuum destilliert und lieferte 4,296 g (87 %) eines farblosen Oeles vom Sdp. 94-101°/
0,2 Torr. 422 mg des Rohproduktes wurden an der 70-fachen Menge Silicagel (15 %)
chromatographiert. Die Hexan-Fraktionen eluierten 12 mg (3 %) eines Tetrahydro-
irons (s. S. 62). Die mit Benzol-Aether 20:1 eluierten Fraktionen ergaben 334 mg
Diketon (51). Zur Analyse wurde eine Probe zweimal in einem Kragenkolben am91
Hochvakuum bei einer Badtemperatur von 90 /0,01 Torr destilliert; iy. = 1,4755.
IR.-Spektrum (Nr. 19869; flüssig): Banden u.a. bei 1368m, 1395w, 1706s, 2860m
und 2960s cm"1.
C^H^O, Ber. C 74,24 H 10,54%£i ^
Gef. C 74,24 H 10,58 %
Das auf übliche Weise hergestellte Bissemicarbazon schmolz nach zweimaligem
Umkristallisieren aus Methanol bei 220 '.
^E>2 = + 129'l0 *c = °' 706; in AcOH)
IR.-Spektrum (Nr. 19653; KBr-Pille): Banden u.a. bei 1372m, 1395w, 1582s, 1685s,
2860m, 2920s und 2960s cm"1.
Ci,-H ON Ber. c 55,53 H 8,70 N 25,91%10 Zö Z b
C 55,63 H 9,12 N 26,00%55,53 9,19
Bisphenylsemicarbazon: Smp. 182-183° (dreimal aus Methanol umkristallisiert).
C,7H,ft09Nfi Ber. C 68,04 H 7,61 N 17,63%ti ôo i. o
Qet c 6805 H 7)63 N 17,59%
*) L.Ruzicka und C.F.Seidel ' fanden für ein nicht ganz reines Diketon ein
Bissemicarbazon vom Smp. 209-210°.

- 58 -
5, 5, 6-Trimethyl-9-hydroxy-trans-decalon-2 (54)
3,05 g (14, 5 mMol) rohes Diketon (51) aus der Ozonolyse von Dihydro- y-iron
(47) wurden an der 30-fachen Menge neutralem Alox, Akt. II, chromatographiert.
Cyclohexan eluierte 352 mg Tetrahydro-iron (45) (s. S. 62). Die Cyclohexan-Benzol-
und Benzol-Fraktionen eluierten 1,88 g (62 %) eines kristallinen Produktes vom
Smp. 180-182. Ein Teil davon wurde zur Analyse dreimal aus Hexan umkristalli¬
siert und einmal bei 125°/0,005 Torr sublimiert. Smp. 185-186°;
Md1 = + 54'3° (c = 2'04 'in Et0H)-
IR.-Spektrum (Nr. 20646; in CHClg): Banden u.a. bei 1370m, 1395w, 1707s,3360-3420m und 3600m cm"1.
NMR.-Spektrum (in CDClg)*); Signale u.a. bei 6 = 0,83/s (3) ; 0,94/d(3);
0,98/s (3).
C^H^O, Ber. C 74,24 H 10,54%lô là ù
Geî c 74>04 H 1Q43 %
A1,9-5,5,6-Trimethyl-2-octalon (52)
a) Aus (54) durch Wasserabspaltung an bas. Alox
250 mg (1,19 mMol) Hydroxyketon (54) wurden an der 50-fachen Menge basi¬
schem Alox, Akt. I, chromatographiert. Die Benzol- und Benzol-Aether-Fraktionen
eluierten 134 mg (59 %) eines schwach gelblichen Oeles.
UV.-Spektrum: X ^H = 243 mp (log 6. = 4,10).
Das IR.-Spektrum wies neben der für konjugiert-ungesättigte Ketone charakteristischen
Bande starker Intensität bei 1675 cm noch eine Bande schwacher Intensität bei
1717 cm auf, was auf die Anwesenheit kleinerer Mengen eines /i,f -ungesättigten
Ketons schliessen lässt.
IR.-Spektrum (Nr. 14938; flüssig): Banden u.a. bei 872m, 1370m, 1395m, 1620m,
1675s, 1717w, 2870s und 2950s cm"1.
C13H20° Ber# C 81'20 H 10>48Gef. C 80,91 H 10,41
*) Das NMR. -Spektrum wurde auf einem Varian High Resolution NMR. -SpectrometerA-60, 60 Mc (mit elektronischem Integrator) mit Tetramethylsilan (5=0) als
interner Referenz in ca. 3-proz. Lösung aufgenommen. Bei der Beschreibungder Signale bedeuten: s (Singlett), d (Doublett); die in Klammer angeführten, auf-
bzw. abgerundeten Zahlen betreffen die durch Integration ermittelte Protonenan¬
zahl. Fräulein Dr. D.M eue he möchte ich für die Aufnahme des Spektrums
herzlich danken.

- 59 -
Phenylsemicarbazon: Smp. 182-183 (zweimal aus Alkohol umkristallisiert)
UV. -Spektrum: A m^H = 244 mp (log e = 4,27) und 282 mu (log £ = 4,39).
C„nH„7ON- Ber. C 73,81 H 8,36 N 12,91%Gef. C 73,86 H 8,41 N 12,97%
b) Aus (54) durch Wasserabspaltung mit p-Toluolsulfosäure
Man löste 250 mg (1,19 mMol) Hydroxyketon (54) in 30 ml Benzol, versetzte
mit 250 mg (1,45 mMol) p-Toluolsulfosäure und erhitzte während 30 Min. unter Rück-
fluss. Das nach üblicher Aufarbeitung erhaltene Rohprodukt (231 mg) wurde in einem
Kragenkolben am Hochvakuum bei einer Badtemperatur von 80 /0, 05 Torr destilliert.
Man erhielt 219 mg (96 %) eines schwach gelblichen Oeles, das zur weiteren Reini¬
gung an der 30-fachen Menge Alox, Akt. in, chromatographiert wurde. Die Hexan¬
fraktionen eluierten 195 mg (85 %) farbloses Octalon (52). n^4= 1,5217 ; M^9= + 78,5°(c = 0,975 ; inEtOH).
UV. -Spektrum: À m^H = 243 mu (log E = 4,14); Endabsorption bei 210 mji (log£ =3,36).
Das IR.-Spektrum (Nr. 20673) war deckungsgleich mit demjenigen des aus (54) durch
Wasserabspaltung an basischem Alox erhaltenen Octalons (52).
C,,H9nO Ber. C 81,20 H 10,48%U
Gef. C 80,65 H 10,42 %
Aequilibrierungsversuche
a) Alkalische Aequilibrierung des Octalons (52)
94 mg (0,49 mMol) Octalon (52) wurden in 20 ml Methanol gelöst, mit 22 mg
(0,98 mMol) Natrium versetzt und unter Stickstoffatmosphäre während drei Std. unter
Rückfluss gekocht. Nach dem Abkühlen goss man auf Eiswasser und extrahierte mit
Aether. Man erhielt nach üblicher Aufarbeitung 94 mg eines gelblichen Oeles, das in
einem Kugelrohr am Hochvakuum bei einer Badtemperatur von 80 /0, 05 Torr destil¬
liert wurde. Das Destillat wog 91 mg (97 %).
UV.-Spektrum: X m^H = 243 mu (log £ = 4,04)
IR.-Spektrum (Nr. 20672; flüssig): in allen Teilen identisch mit demjenigen des
Octalons (52).
40 mg (0,21 mMol) Isomerisierungsprodukt wurden auf übliche Art ins Phenylsemicar¬
bazon übergeführt. Dieses schmolz nach zweimaligem Umkristallisieren aus Alkohol
(41 mg; 60 %) bei 181-182°; ein Mischsmp. mit dem entsprechenden Derivat des
Octalons (52) zeigte keine Depression.

- 60 -
b) Saure Aequilibrierung des Octalons (52)
69 mg (0, 36 mMol) Octalon (52) wurden in 40 ml Chloroform gelöst, mit 0,2 ml
konz. Salzsäure versetzt und während drei Std. unter Rückfluss gekocht. Nach dem
Abkühlen wusch man die Lösung mit Wasser neutral, trocknete sie über Natriumsul¬
fat und entfernte hierauf das Lösungsmittel am Vakuum. Der Rückstand (67 mg) wur¬
de in einem Kugelrohr am Hochvakuum bei einer Badtemperatur von 80 /0,05 Torr
destilliert. Das farblose Destillat wog 66 mg (96 %).
UV.-Spektrum: X m^H = 243 mji (log 6 = 4,10)
Das IR.-Spektrum (Nr. 20671; flüssig) erwies sich mit demjenigen des Octalons (52)
als identisch.
^,9-2,5,5,6-Tetramethyl-2-hydroxy-octaline (57) und (58)
Methyllithium wurde auf übliche Art zubereitet, indem man 4,37 g (30, 8 mMol)
Methyljodid und 0,425 g (61,5 mMol) Lithium in 30 ml abs. Aether unter Stickstoff¬
atmosphäre reagieren liess. Diese Lösung wurde auf -70° abgekühlt und tropfenwei¬
se mit 0, 700 g (3,65 mMol) des Octalons (52) in 10 ml Aether versetzt. Hierauf liess
man die Reaktionsmischung sich auf Zimmertemperatur erwärmen und fügte dann
tropfenweise Wasser hinzu, bis keine Reaktion mehr festzustellen war. Das nach üb¬
licher Aufarbeitung erhaltene Rohprodukt wog 0, 770 g und wies im UV. -Spektrum
nur eine Endabsorption bei 210 mu (log = 3,60) auf. 509 mg dieses Rohproduktes
wurden an der 40-fachen Menge Alox, Akt. in, chromatographiert. Das flüssige Epi-
mere (57) wurde mit Pentan eluiert (186 mg ; 37 %).
UV.-Spektrum: Endabsorption bei 210 mu (loge =3,67).
IR.-Spektrum (Nr. 19605; flüssig): Banden u.a. bei 1130m, 1365m, 1391m, 1655*
2860m, 2930s, 3300-3360m und 3580Sch cm"1.
Die Pentan-Benzol-Fraktionen eluierten 246 mg (49 %) festes Epimeres (58)
vom Smp. 76-80.Zur Analyse wurde eine Probe am Hochvakuum bei 56°/0,005 Torr
sublimiert. Smp. 87-87,5°.
IR.-Spektrum (Nr. 19537; in CS2): Banden u.a. bei 1155m, 1364m, 1371m, 1390w,1660w, 3340-3440w und 3600 cm"1.
C14H9.0 Ber. C 80,71 H 11,61%* *
Gef. C 80,64 H 11,52 %
2,5,5,6-Tetramethyl-l,2,9-trihydroxy-decalin (59)
135 mg (0,65 mMol) kristallisiertes Hydroxyoctalin (58) wurden in 5 ml Aether
und 0,5 ml Pyridin gelöst, mit 177 mg (0,70 mMol) Osmiumtetroxyd versetzt und
während sechs Tagen im Dunkeln bei Zimmertemperatur stehen gelassen. Zur Spaltung

- 61 -
des Osmiatesters verdünnte man mit 15 ml Methylenchlorid und schüttelte die Reak¬
tionslösung während vier Std. bei Zimmertemperatur mit einer Lösung von 0,4 g
Mannit in 5 ml 10-proz. Kalilauge. Hierauf wusch man die Methylenchloridlösung
mit Wasser neutral, trocknete sie über Natriumsulfat und entfernte das Lösungs¬
mittel am Vakuum. Man erhielt 163 mg zum Teil kristallines Triol (59). Es wurde
mit wenig kaltem Hexan versetzt und die Kristalle abfiltriert. Die Ausbeute an kri¬
stallisiertem Triol (59) betrug 89 mg (57 %). Zur Analyse wurde eine Probe zweimal
aus Hexan umkristallisiert und einmal am Hochvakuum bei 125 /0,005 Torr subli-
miert. Smp. 151-152°.
IR.-Spektrum (Nr. 19674; in CHClg): Banden u.a. bei 1045m, 1065m, 1353w, 1368m,
1377m, 1395w, 3400-3460 und 3560w cm"1.
C1aH,bO, Ber. C 69,38 H 10,81%*
Gef. C 69,26 H 10,61%
Diketon (53)
a) Durch Glycolspaltung aus Triol (59)
95 mg (0,393 mMol) Triol (59) wurden in 20 ml abs. Benzol gelöst, mit 710 mg
(1,60 mMol) Blei-tetraacetat versetzt und während 15 Std. bei Zimmertemperatur ge¬
rührt. Eine anschliessend durchgeführte jodometrische Titration mit 1 ml der Reak¬
tionslösung ergab, dass 0, 76 mMol Blei-tetraacetat verbraucht worden waren. Zur
Aufarbeitung nahm man in Aether auf, wusch die ätherische Lösung mit Wasser neu¬
tral, trocknete sie über Natriumsulfat und entfernte das Lösungsmittel am Vakuum.
Der schwach bräunliche, ölige Rückstand wog 83 mg und wurde in einem Kugelrohr
am Hochvakuum bei einer Badtemperatur von 100 /0,05 Torr destilliert. Das Destillat
(78 mg) wurde zur weiteren Reinigung zweimal an der 30-fachen Menge Silicagel (15 %
H„0) chromatographiert. Die Benzol-Aether 10:1 Fraktionen eluierten 70 mg (86 %)
des Diketons (53), dessen Analyse C-Werte zeigte, die 1 - 1,3 % zu tief lagen.
Das IR.-Spektrum (Nr. 19959; flüssig) dieses Diketons (53) zeigte die gleichen Ban¬
den wie dasjenige des Diketons (51) aus dem Ozonabbau des Dihydro-jf-irons.
B^ssemjkarbazon: Das auf übliche Art hergestellte Derivat schmolz nach dreimali-
Un
22
gem Umkristallisieren aus Methanol bei 221°
[o(.]£2= + 127,1° (c = 0, 70; in AcOH).
Der Mischsmp. mit dem Bissemicarbazon des Diketons (51) (Smp. 220 ) aus der
Ozonolyse von Dihydro- Y--iron zeigte keine Depression; ebenso waren die IR. -Spek¬
tren (Nr. 19654 und Nr. 19653; KBr-Pille) der beiden Derivate deckungsgleich.
C^H^nO^N- Ber. C 55,53 H 8,70(10 £0 £ °
Gef. C 55,69 H 8,85 '

- 62 -
b) Aus flüssigem Hydroxyoctalin (57)
154 mg (0, 741 mMol) flüssiges Hydroxyoctalin (57) wurden auf obige Weise
mit 195 mg (0,77 mMol) Osmiumtetroxyd oxydiert. Das nach üblicher Aufarbeitung
erhaltene Rohprodukt wog 151 mg (81 %) und liess sich nicht kristallisieren (Schaum).
Es wurde deshalb direkt mit 0,83 g (1,87 mMol) Blei-tetraacetat in 30 ml abs. Ben¬
zol einer Glycolspaltung unterworfen. Man erhielt nach üblicher Aufarbeitung 132 mg
eines bräunlichen Oeles, das an der 50-fachen Menge Silicagel (15 %) chromatogra-
phiert wurde. Die Benzol-Aether Fraktionen eluierten 78 mg eines schwach braunen
Oeles, das im Kugelrohr am Hochvakuum bei einer Badtemperatur von 100°/0,03Torr destilliert wurde. Das Destillat wog 58 mg (47 %) und wurde direkt ins Bisse-
micarbazon überführt. Dieses schmolz nach zweimaligem Umkristallisieren aus Me¬
thanol bei 220. Ein Mischsmp. mit dem Bissemicarbazon aus a) sowie mit dem Bis-
semicarbazon des Diketons aus der Ozonolyse des Dihydro- jf-irons zeigte keine
Depression.
(+)-cis(2,3)-cis(2,6)-Tetrahydro-iron (45)
Das als Nebenprodukt bei der Hydrierung von (+)- Jf-Iron (37) anfallende Tetra-
hydro-iron (45) konnte beim Chromatographieren des Ozonabbauproduktes von (+)-Di-
hydro-)f -iron an Silicagel (15% H„0) isoliert werden (s. S. 57). Aus mehreren Ansätzen
zusammengefasstes Material (352 mg) wurde zur Reinigung auf übliche Art ins Semi-
carbazon überführt, das nach viermaligem Umkristallisieren aus Methanol bei 200 -
201° schmolz.
C15H29ON3
Spaltung des Semicarbazons
150 mg (0,56 mMol) Tetrahydro-ironsemicarbazon wurden in 4 ml Alkohol heiss
gelöst, mit 4 ml Wasser und 0,12 ml (152 mg) Brenztraubensäure versetzt und wäh¬
rend drei Std. unter Rückfluss erhitzt. Nach dem Abkühlen extrahierte man mit Pen-
tan und erhielt nach üblicher Aufarbeitung 116 mg (98 %) cis(2,3)-cis(2,6)-Tetra-
hydro-iron (45). Dieses wurde in einem Kugelrohr am Hochvakuum bei einer Badtem¬
peratur von 85 /0,05 Torr destilliert. Ein analytisches Gas-Chromatogramm zeigte
nur eine Substanz an.
n£°= 1,4748 ; d^°= 0,9246 ; [o*.]£° = +44,4°(c= 2,24; inEtOH)*)
Ber. C 67,37 H 10,93 N 15,72Gef. C 67,38 H 10,99 N 15,70
*)L.Ruzicka, C.F.Seidel und G. Fir menich58' fanden: n|>5= 1,4721;
d|5 = 0,9173; [o(]D = + 35°. Y. R. Naves38c) fand: nD°= 1,4743;
dfo = 0,9218; [oL]D = + 39,7°.

- 63 -
^10" 1575
» *210
IR.-Spektrum (Nr. 20338; flüssig): Banden u.a. bei 1360m, 1365Sch, 1386m, 1395w,1715s, 2860Sch und 2940s cm"1.
C14H9fi° Ber- c 79>93 H 12»46%i0
Gef. C 79,79 H 12,42 %
Die Behandlung des Ketons mit2,4-Dinitrophenylhydrazin-hydrochlorid in Me¬
thanol gab ein gelbes 2,4-Dinitrophenylhydrazon vom Smp. 116-117° (dreimal aus
Methanol-Methylenchlorid umkristallisiert).
C20H30°4N4 Ber. C 61,52 H 7,74 N 14,35Gef. C 61,37 H 7,75 N 14,20
(+)-trans(2,3)-cis(2,6)-Tetrahydro-iron (61)
a) cis(2,6)-Dihydro-y-irol
1,95 g (9,37 mMol) (+)-cis(2,6)-Dihydro- )f-iron (47) wurden in 30 ml abs. Ae¬
ther gelöst und zu einer Suspension von 0, 760 g (20 mMol) Lithiumaluminiumhydrid
in 150 ml Aether getropft. Anschliessend erhitzte man während drei Std. unter Rück-
fluss. Nach dem Abkühlen wurde das überschüssige Lithiumaluminiumhydrid vor¬
sichtig mit gesättigter Seignettesalzlösung zerstört und das Reaktionsgemisch mit
Aether extrahiert. Nach üblicher Aufarbeitung erhielt man 1,96 g (100 %) rohes
cis(2,6)-Dihydro- V"-irol, das direkt zur Reduktion mit Lithium in Aethylendiamin
verwendet wurde.
b) Reduktion mit Lithium in Aethylendiamin
In einem mit Stickstoff gespülten 800 ml Zylindergefäss, versehen mit Vibra¬
tor, Stickstoffeinlassrohr und Rückflusskühler wurden unter Feuchtigkeitsausschluss
3,24 g (15,4 mMol) cis(2,6)-Dihydro- ^-irol in 200 ml frisch absolutiertem Aethylen¬
diamin 'gelöst. Unter Eiskühlung und starkem Vibrieren gab man in kleinen Portionen
von 0,1-0, 2 g im Verlaufe von 40 Min. insgesamt 1, 7 g (0, 24 Mol) Lithium hinzu.
Nach einiger Zeit setzte eine heftige Wasserstoffentwicklung unter Blaufärbung der
Lösung ein. Man liess über Nacht (15 Std.) bei Zimmertemperatur weitervibrieren und
erhitzte das Reaktionsgemisch anderntags während zweieinhalb Std. auf 60.Nach dem
Abkühlen zerstörte man das überschüssige Lithium vorsichtig mit Eiswasser und ver¬
setzte das Reaktionsgemisch mit viel Wasser. Hierauf extrahierte man mit Aether,
wusch die ätherische Lösung mit Wasser neutral, trocknete sie über Natriumsulfat und
entfernte das Lösungsmittel am Vakuum. Das Rohprodukt, 2,80 g (86 %) eines farblosen
*) Das Aethylendiamin wurde während drei Std. über Natrium unter Rückfluss gekochtund hierauf direkt in das Reaktionsgefäss destilliert.

- 64 -
Oeles, zeigte mit Tetranitromethan eine sehr schwache Gelbfärbung. Das UV.-Spek¬
trum wies eine schwache Endabsorption bei 210 mu (log £. = 2,60) auf.
c) Oxydation zum Tetrahydro-iron-Gemisch (45) und (61)
516 mg (2,44 mMol) rohes Reduktionsprodukt wurden in 4 ml Benzol und 6 ml
Eisessig gelöst und mit einer Lösung von 120 mg Kaliumbisulfat in 1 ml Wasser ver¬
setzt. Man kühlte auf 0° ab und Hess unter starkem Vibrieren im Verlaufe einer Std.
eine Lösung von 500 mg Chromtrioxyd in 0,5 ml Wasser und 6 ml Eisessig langsam
zutropfen. Hierauf wurde während eineinhalb Std. bei 5 belassen und anschliessend
auf Eiswasser gegossen. Man extrahierte mit Aether und erhielt nach üblicher Aufar¬
beitung 497 mg (97 %) eines farblosen Oeles, dessen IR. -Spektrum (in CHC1,) in der
V (0-H)-Region keine Bande mehr aufwies. Das UV.-Spektrum zeigte eine schwache
Endabsorption bei 210 mu (logt = 2,56). Ein analytisches Gas-Chromatogramm zeigte
folgende Zusammensetzung:
1. ca. 50 % trans(2,3)-cis(2,6)-Tetrahydro-iron (61) (I2^0 = 1561; I2^0 = 1815)
2. ca. 40 % cis(2,3)-cis(2,6)-Tetrahydro-iron (45) (l^Q = 1573; 1^ = 1844).
d) Präparative gaschromatographische Auftrennung
1,621 g eines auf obige Art erhaltenen Tetrahydro-iron-Gemisches wurden in
einem präparativen Gaschromatographen an einer polaren stationären Phase in
sechs Durchschüben von je 250-300 ul aufgetrennt. Die Kolonnentemperatur betrug
210°, Trägergas war Stickstoff. Es wurden folgende Fraktionen aufgefangen: 1. Frak¬
tion A 568 mg (mit trans(2,3)-cis(2,6)-Tetrahydro-iron (61) als Hauptkomponente);
2. Zwischenfraktion Z 127 mg; 3. Fraktion B 377 mg (mit cis(2,3)-cis(2,6)-Tetrahy-
dro-iron (45) als Hauptkomponente; 4. Nachlauf 172 mg. Zusammen 1,144 g (71 %
Ausbeute).
Die Fraktion A wurde ein zweites Mal an der polaren Kolonne bei 210 gereinigt
und lieferte 348 mg trans(2,3)-cis(2,6)-Tetrahydro-iron (61). Ein analytisches Gas-
chromatogramm dieses Ketons zeigte an der apolaren Kolonne eine kleine Verunrei¬
nigung von ca. 4 % an cis(2,6)-Dihydro- y--iron (47). Das UV.-Spektrum war leer.
n£° = 1,4694 ; d^°= 0,9117 ; [oL]^ = + 0,94° (c = 2,24; in EtOH).A P
Retentionsindices: L.-.Q = 1560 ; I,.. = 1815.
IR.-Spektrum (Nr. 20681; flüssig): Banden u.a. bei 1360Sch, l1369m, 1395m, 1715s,2870Schund 2940s cm-1.
C^ft^O Ber. C 79,93 H 12,46%l* ^
Gef. C 79,88 H 12,29 %
Ein auf übliche Art hergestelltes Semicarbazon schmolz nach zweimaligem Umkristal¬
lisieren aus Methanol bei 182, 5-183, 5°. Der Mischsmp. mit dem Semicarbazon von
cis(2,3)-cis(2,6)-Tetrahydro-iron (Smp. 200-201°) zeigte eine Depression von ca. 4°.
C15H29ON, Ber. C 67,37 H 10,93 N 15,72%Gef. C 67,47 H 10,71 N 15,54%

- 65 -
ZUSAMMENFASSUNG
5\
I. Die relative Konfiguration des seinerzeit von M. S toll et al. ' als Hauptpro¬
dukt der säurekatalysierten Cyclisation des Dihydro- jf-jonons aufgefundenen
ot-Ambrinols ist bestimmt worden.
Für die relativen Geschwindigkeitskonstanten der säurekatalysierten Cyclisation
von (l)-Dihydro- jf-jonon und (+)-cis(2,6)-Dihydro- Jf-iron wurde im System Me¬
thanol/0,1 n. Salzsäure 1:1 folgendes Verhältnis gefunden:
kDihydro-J-jonon : kcis(2, 6)-Dihydro- Jf-iron= 6'5 :
Aus diesem Verhältnis sowie aus der Stereochemie des o(-Ambrinols konnte auf
Grund theoretischer Ueberlegungen und mit Hilfe gewisser Annahmen die bevor¬
zugte Ringschlusskonstellation der säurekatalysierten Cyclisation von Dihydro-
V" -jonon postuliert werden.
n. Die Konfiguration des (+)- JT-Irons, der Hauptkomponente des natürlichen Irons,
ist im Sinne des l,l,6R-Trimethyl-3-methylen-2S-(3'-keto-l'-buten)-cyclohexans
festgelegt worden.

- 66 -
LITERATURVERZEICHNIS
l)a) P. A. Stadler, A. Nechvatal, A. J. Frey und A. Eschenmoser,
Helv. 40, 1773 (1957);
b) A. E schenmoser, D. Felix, M. Gut, J. Meier und P.A.Stadler,
Ciba Foundation Symposium on the Biosynthesis of Terpenes and
Sterols, p. 217 (1959).
2) T. T. Tchen und K. Bloch, J. Biol. Chem. 226, 921 (1957).
3)D.Arigoni, Experientia 14, 153(1958).
4) M. Stoll und M. Hinder, Helv. 38, 1593(1955)
5) M. Stoll, CF. Seidel, B. Willhalm und M. Hinder, Helv. 39, 183(1956).
6) A. G. Armour, G. Büchi, A. Eschenmoser und A. Storni, Helv. 4_2,2233 (1959).
7) A. T. Blomquist und J. Wolinsky, J. Amer. chem. Soc. 79, 6025 (1957).
8)E.E.Smissman und D.T.Witiak.J. org. Chem. _25, 471 (1960);
E.E.Smissman und R.A.Mode.J. Amer. chem. Soc. 7^7 3447 (1957).
9) J. Meinwald und J. A. Yankeelov, J. Amer. chem. Soc. 80, 5266 (1958).
10) P. Barbier und G. Leser, C. r. hebd. Séances Acad. Sei. \M., 1308 (1897).
11) C.Price und M. L. Dickmann, J. Ind. Eng. Chem. 40, 257 (1948).
12) D. Felix, Diss. ETH (1959).
13) S. Winstein und N. J. Holness, J. Amer. chem. Soc. 77, 5562 (1955).
14) N. L. Allinger und M. A. Miller, J. Amer. ehem. Soc. 83, 2145 (1961).
15) W. G. Dauben und K. S. Pitzer in M. S. Newman, Steric Effects in Organic
Chemistry, p. 3 (1956).
16) N. A. Nelson, R. S. Hsi, J. M. Schuck und L.D.Kahn, J. Amer.chem.
Soc. 82, 2573 (1960);
W. Salzer, Z. physiol. Chem. 274, 39 (1942).
17)E.Lederer,F.Marx,P.Mercier und G.Pérot, Helv. 2£, 1354 (1946).
18) A. J. Birch, J. chem. Soc. 1950, 2325.
19)K.Biemann und J.Seibl, J. Amer. chem. Soc. jU, 3149(1959).
20) Lester P. Kuhn, J. Amer. chem. Soc. 74, 2492 (1952).
21) A. Fürst und PI. A. Plattner (1951), zit. in D. H. R. Barton, J. chem.
Soc. 1953, 1027.
22) PI. A. Plattner, H. Heusser und M. Feurer, Helv. j52, 587 (1949);
A.S.Hallsworth und H.B.Henbest.J. chem. Soc. 1957, 4604;
D. H. R. Barton, D. A. Lewis und J. F. Me Ghie, J. chem. Soc. 1957,
2907.

- 67 -
23)H.B.Henbest und R.A.L.Wilson, J. ehem. Soc. 1957, 1958.
24) E. L. Eliel und C.A. Luckach, J. Amer. ehem. Soc. 79, 5986 (1957).
25) M. Stoll, M. Hinder und B .Willhalm
,Helv. 39, 200 (1956).
26)F.Tiemann und P.Krüger, Ber. deutsch, ehem. Ges. ^6, 2675(1893).
27)a) H. Schinz, "Die Veilchenriechstoffe" in Fortschritte der Chemie organischer
Naturstoffe 8, 146(1951);
b)Y.R. Naves, Bull. Soc. chim. France 1950, D 99.
28) L. Ruzicka,C. F. Seidel und H. Schinz, Helv. 16, 1143 (1933).
29) L. Ruzicka, C. F. Seidel und W. Brugger, Helv. 30, 2168 (1947).
30) Y. R. Naves, Helv. 30, 2221, 2222, 2233, 2241 (1947); Helv. 32, 394 (1949).
31) L. Ruzicka, C. F. Seidel, H. Schinz und Ch. Tavel, Helv. 31, 257 (1948);
L. Ruzicka,CF.Seidel,H.Schinz und M. Pfeiffer, Helv. 30, 1807
(1947);
C. F. Seidel, H. Schinz und L. Ruzicka, Helv. ^2, 1739 (1949);
H.H.Günthard.L. Ruzicka, H. Schinz und C F. Seidel, Helv. J32,
2198 (1949);
H. Schinz, C F. Seidel und L. Ruzicka, Helv. ji2, 2560 (1949);
Y. R. Naves, Helv. 41, 653 (1958).
32) L. Ruzicka,H.Schinz,CF.Seidel und M.Pfeiffer, Helv. £5, 188
(1942).
33) H. Schinz, C F.Seidel und Ch. Tavel, Helv. 30, 1810 (1947).
34) Y. R. Naves, A. V. Grampoloff und P. Bachmann, Helv. _30, 1599 (1947);
Y.R. Naves und A. V. Grampoloff, Helv. ^2, 2552 (1949).
35) Y. R. Naves, Bull. Soc. chim. France 1954, 667, 321;
Y.R.Naves und P.Ardizio, Bull. Soc. chim. France 1950, 793.
36)a) C F. Seidel, H. Schinz und L. Ruzicka, Helv. 32, 2102 (1949);
b) Y. R. Naves, Helv. jjl, 1105 (1948).
37) Y.R. Naves und A. Odermatt, Bull. Soc. chim. France 1958, 377.
38)a) Y. R. Naves, Helv. 31, 893(1948);
b) Y.R. Naves, Helv. 31, 1103(1948);
c) Y. R. Naves, Helv. J31, 1871 (1948).
39) K. v. Auwers, Liebigs Ann. Chem. 420, 84 (1920);
A. Skita, Liebigs Ann. Chem. 431, 1 (1923).
40) Y. R. Naves, Bull. Soc. chim. France 1953, 551.
41) Y. R. Naves, Helv. 31, 2047 (1948); Helv. 32, 2186 (1949).
42) Ch. W. Beckett, K. S. Pitzer und R.Spitzer, J. Amer. chem. Soc.
69, 2488 (1947);
G. A. Haggis und L. N. Owen, J. chem. Soc. 1953, 408;
M. Mousseron, Bull. Soc. chim. France 1946, 220.

- 68 -
43) N. L. Allinger, J. Amer. ehem. Soc. 79, 3443 (1957);
N.L.Allinger, Experientia K), 328 (1954).
44) D. H. R. Barton, Chemistry and Ind. 1954, 21;
Y. R. Naves, Bull. Soc. chim. France 1956, 1020.
45) G. Stork und A. W. Bur gstahler, J. Amer. ehem. Soc. T7.> 5068(1955).
46)P.A.Stadler,A.Eschenmoser,H.Schinz und G. Stork, Helv. 40,
2191 (1957).
47) P. Bachli, Diss. ETH (1950).
48) P. Bächli, CF. Seidel, H. Schinz und L. Ruzicka, Helv. J32, 1744
(1949).
49) C. F. Seidel und L. Ruzicka, Helv. 35, 1826(1952).
50) M. Stoll, Helv. JS8, 1587 (1955).
51) R. F. Church, R. E. Ireland, J. A. Mashall, Tetrahedron Letters 1, 34
(1961).
52) S. A. Julia, A. E schenmoser, H. Heusser und N. T a r k ö y, Helv. 36,
1885 (1953);
W . S . Johnson, J. J. Kor st, R. A. C lern ent und J.Dutta, J. Amer.
chem. Soc. 82, 614 (1960);
W. G. Dauben und J.W. McFarland, J. Amer. chem. Soc. 82, 4245 (1960).
53) L. Cagliotti, H. Naef, D. Arigoni und O. Jeger, Helv. 42, 2557 (1959).
54) L. M. Jackman, Application of Nuclear Magnetic Resonance Spectroscopy in
Organic Chemistry, Pergamon Press, 1959, p. 53.
55) C. Djerassi, R. Riniker und B
. Riniker ,J. Amer. chem. Soc. 78, 6362,
6377 (1956).
56) W. Moffit, A. Moscowitz, R. B. Woodward, W. Klyne und C. Djeras¬
si, zit. in C.Djerassi, Optical Rotatory Dispersion, McGraw-Hill
Series in Advanced Chemistry, 1960.
57) R. S. Cahn, C. K. Ingold und V. Prelo g, Experientia 1_2, 81 (1956).
58) L. Ruzicka, C. F. Seidel und G. Firmenich, Helv. 24, 1434 (1941).
59) P. Bächli und H. Schinz, Helv. 34, 1168 (1951).
60)E.J.Corey und E. W. Can trail, J. Amer. chem. Soc. ftt, 1745 (1959).
61) L. Reggel, R. A. Friedel und I.Wender, J. org. Chem. 2Z, 891 (1957).
62) E. Kova'ts, Helv. 41, 1915 (1958).
63) H. Brockmann und H.Schodder, Ber. deutsch, chem. Ges. 2i» 73(1941).
64)A.Wehrli und E. Kova'ts, Helv. 42, 2709(1959).
65) Organic Synthesis, Coll. Vol. Ill, 176 (1955).
66) L. Ruzicka und C.F. Seidel, Helv. 31, 160(1948).
67) E. Heilbrunner, E.Kovats und W. Simon, Helv. 40, 2410 (1957);
E. Kovats.W. Simon und E. Heilbrunner, Helv. 41, 275 (1958).
68) J. Schreiber, Diss. ETH (1953).

Lebenslauf
Am 9. Juli 1934 wurde ich als Sohn des Aldo Storni und der Zelia, geb. Quadri
in Lugano geboren. In Winterthur durchlief ich die Primarschule und zwei Jahre Se¬
kundärschule und trat dann im April 1949 in die Oberrealabteilung der dortigen Kan¬
tonsschule ein, wo ich im Herbst 1953 die Maturitätsprüfung (Typus C) ablegte. Dar¬
auf immatrikulierte ich mich an der Abteilung für Naturwissenschaften der Eidgenös¬
sischen Technischen Hochschule in Zürich und bestand im Herbst 1957 die Diplomprü¬
fung als Naturwissenschafter. Seit Anfangs 1958 arbeitete ich unter der Leitung von
Herrn Prof. Dr. V.Prelog und Herrn Prof. Dr. A. Eschenmoser an der
vorliegenden Promotionsarbeit.
Zürich, im Juli 1961 Angelo Storni


















