
IL-17 Signaling Triggers Degradation of the Constitutive ... · ABIN-1+/+ or ABIN-1 2/ fibroblasts...
10
B Inhibitor ABIN-1 κ IL-17 Signaling Triggers Degradation of the Constitutive NF- Kane, Brian J. Aneskievich, Averil Ma and Sarah L. Gaffen J. Agustin Cruz, Erin E. Childs, Nilesh Amatya, Abhishek V. Garg, Rudi Beyaert, Lawrence P. http://www.immunohorizons.org/content/1/7/133 https://doi.org/10.4049/immunohorizons.1700035 doi: 2017, 1 (7) 133-141 ImmunoHorizons This information is current as of November 12, 2018. Material Supplementary lemental http://www.immunohorizons.org/content/suppl/2017/08/31/1.7.133.DCSupp References http://www.immunohorizons.org/content/1/7/133.full#ref-list-1 , 14 of which you can access for free at: cites 42 articles This article Email Alerts http://www.immunohorizons.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: ISSN 2573-7732. All rights reserved. 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is an open access journal published by ImmunoHorizons by guest on November 12, 2018 http://www.immunohorizons.org/ Downloaded from by guest on November 12, 2018 http://www.immunohorizons.org/ Downloaded from
Transcript of IL-17 Signaling Triggers Degradation of the Constitutive ... · ABIN-1+/+ or ABIN-1 2/ fibroblasts...

B Inhibitor ABIN-1κIL-17 Signaling Triggers Degradation of the Constitutive NF-
Kane, Brian J. Aneskievich, Averil Ma and Sarah L. GaffenJ. Agustin Cruz, Erin E. Childs, Nilesh Amatya, Abhishek V. Garg, Rudi Beyaert, Lawrence P.
http://www.immunohorizons.org/content/1/7/133https://doi.org/10.4049/immunohorizons.1700035doi:
2017, 1 (7) 133-141ImmunoHorizons
This information is current as of November 12, 2018.
MaterialSupplementary
lementalhttp://www.immunohorizons.org/content/suppl/2017/08/31/1.7.133.DCSupp
Referenceshttp://www.immunohorizons.org/content/1/7/133.full#ref-list-1
, 14 of which you can access for free at: cites 42 articlesThis article
Email Alertshttp://www.immunohorizons.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
ISSN 2573-7732.All rights reserved.1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is an open access journal published byImmunoHorizons
by guest on Novem
ber 12, 2018http://w
ww
.imm
unohorizons.org/D
ownloaded from
by guest on N
ovember 12, 2018
http://ww
w.im
munohorizons.org/
Dow
nloaded from

IL-17 Signaling Triggers Degradation of the Constitutive NF-kBInhibitor ABIN-1
J. Agustin Cruz,* Erin E. Childs,* Nilesh Amatya,* Abhishek V. Garg,* Rudi Beyaert,†,‡ Lawrence P. Kane,§ Brian J. Aneskievich,{
Averil Ma,k and Sarah L. Gaffen*,§
*Division of Rheumatology and Clinical Immunology, Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261;†VIB-UGent Center for Inflammation Research, Zwijnaarde, Ghent 9052, Belgium; ‡Department of Biomedical Molecular Biology, Ghent University,
Zwijnaarde, Ghent 9052, Belgium; §Department of Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261; {Department of
Pharmaceutical Sciences, School of Pharmacy, University of Connecticut, Storrs, CT 06269; and kDepartment of Medicine, University of California,
San Francisco, San Francisco, CA 94143
ABSTRACT
IL-17 activates NF-kB and induces expression of proinflammatory genes. IL-17 drives disease in autoimmune conditions, and anti–IL-
17 Abs have shown impressive success in the clinic. Although produced by lymphocytes, IL-17 predominantly signals in fibroblasts
and epithelial cells. IL-17–driven inflammation is kept in check by negative feedback signaling molecules, including the ubiquitin
editing enzyme A20, whose gene TNFAIP3 is linked to autoimmune disease susceptibility. The A20 binding inhibitor of NF-kB
activation 1 (ABIN-1) is an A20-binding protein encoded by the TNIP1 gene, which is also linked to autoimmune disease susceptibility
including psoriasis. Accordingly, we hypothesized that ABIN-1 might play a role in negatively regulating IL-17 signaling activity.
Indeed, ABIN-1 enhanced both tonic and IL-17–dependent NF-kB signaling in IL-17–responsive fibroblast cells. Interestingly, the
inhibitory activities of ABIN-1 on IL-17 signaling were independent of A20. ABIN-1 is a known NF-kB target gene, and we found that
IL-17–induced activation of NF-kB led to enhanced ABIN-1 mRNA expression and promoter activity. Surprisingly, however, the ABIN-
1 protein was inducibly degraded following IL-17 signaling in a proteasome-dependent manner. Thus, ABIN-1, acting independently
of A20, restricts both baseline and IL-17–induced inflammatory gene expression. We conclude that IL-17–induced signals lead to
degradation of ABIN-1, thereby releasing a constitutive cellular brake on NF-kB activation. ImmunoHorizons, 2017, 1: 133–141.
INTRODUCTION
Interleukin 17A (IL-17) is produced by lymphocytes and acts ontissue fibroblasts and epithelial cells to activate inflammatory geneexpression. IL-17 is required to clear extracellular pathogens,particularly fungi such asCandida albicans (1). However, excessiveIL-17 signaling is associated with autoimmune diseases such as
psoriasis, psoriatic arthritis, and ankylosing spondylitis, amongothers. IL-17 blocking therapies have shown considerable efficacyin treating psoriatic patients, and are being evaluated for variousother conditions (2).
The mechanisms of signaling mediated by IL-17 are incom-pletely defined. Produced by Th17 cells and a variety of innatelymphocyte subsets, IL-17 and its receptor belong to a subclass of
Received for publication July 25, 2017. Accepted for publication August 14, 2017.
Address correspondence and reprint requests to: Dr. Sarah L. Gaffen, Division of Rheumatology and Clinical Immunology, University of Pittsburgh, 200 LothropStreet, BST S702, Pittsburgh, PA 15261. E-mail address: [email protected]
ORCIDs: 0000-0002-7942-1742 (N.A.); 0000-0002-5704-582X (R.B.); 0000-0001-5198-516X (L.P.K.).
J.A.C., A.V.G., N.A., B.J.A., and L.P.K. designed the study; J.A.C., N.A., E.E.C., and A.V.G. performed experiments; J.A.C., A.V.G., B.J.A., A.M., R.B., and S.L.G. analyzed data;A.M., R.B., and B.J.A. provided key reagents; J.A.C. and S.L.G. wrote the paper; and all authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Institutes of Health Grants AI107825 (to S.L.G.), AI103022 (to L.P.K.), AI179008 (to A.M.), and AR048660 (to B.J.A.).
Abbreviations used in this article: ABIN-1, A20-binding inhibitor of NF-kB activation 1; C/EBP, CCAAT enhancer binding protein; EV, empty vector; IKK, inhibitor ofNF-kB kinase; MEF, murine embryonic fibroblast; qPCR, quantitative PCR; siRNA, small interfering RNA; UBAN, ubiquitin binding domain.
The online version of this article contains supplemental material.
This article is distributed under the terms of the CC BY 4.0 Unported license.
Copyright © 2017 The Authors
https://doi.org/10.4049/immunohorizons.1700035 133
RESEARCH ARTICLE
Innate Immunity
ImmunoHorizons is published by The American Association of Immunologists, Inc.
by guest on Novem
ber 12, 2018http://w
ww
.imm
unohorizons.org/D
ownloaded from

cytokines with distinct structural properties comparedwith othercytokine superfamilies. After IL-17 binds to its receptor, numeroussignaling intermediates are engaged to transduce downstreamsignaling. One key early event is activation of the Act1 and TRAF6E3 ubiquitin ligases. These in turn lead to degradation of the IkBinhibitor and nuclear translocation of the NF-kB transcriptionfactor (3–5). Consequently, genes with promoters containingNF-kB–binding elements are upregulated by IL-17 stimulation,with IL-6 (Il6) and Lipocalin-2 (Lcn2) being prototypical IL-17target genes (6).
To date, the mechanisms that constrain the IL-17 pathway arenot well understood, which is an important issue in light of theautoimmune potential of this cytokine. Recently, we showed thatthe ubiquitin editing enzyme A20 inhibits IL-17 signaling by de-ubiquitinating TRAF6 and thereby limiting NF-kB and MAPKactivation (7). Moreover, IL-17 upregulates A20 expression,establishing a negative feedback loop that restricts IL-17–driveninflammation. A20 is encoded by TNFAIP3, a genetic locusfrequently associated with human autoimmune diseases. Inseeking to understand how IL-17 controls autoimmunity, weevaluated other genetic loci connected to IL-17-driven autoim-mune conditions. TheA20-binding inhibitor of NF-kB activation 1(ABIN-1) is an A20-binding protein coded by the TNIP1 gene.ABIN-1 single nucleotide polymorphisms are associated withpsoriasis in human genome–wide association analyses (8–11).Additionally, mice with keratinocytes lacking ABIN-1 are moresensitive to imiquimod-induced dermatitis, a model of psoriasis(12). Because IL-17 signaling is associated with psoriasis andinhibitedbyA20,wehypothesized that ABIN-1might also serve asa downstream inhibitor of the IL-17 signaling pathway.
In this study, we demonstrate that ABIN-1 constitutivelyrestricts the basal expression of IL-17 target genes such as Il6 infibroblasts and stromal cells. ABIN-1 also negatively regulates IL-17–induced production of Il6, Lcn2, and other NF-kB–dependentgenes, although not all IL-17–inducible genes are affected.Although ABIN-1 is known to interact with A20, we found thatA20 is not needed for ABIN-1–mediated inhibition of IL-17signaling.Additionally, IL-17 inducedexpressionofABIN-1 (Tnip1)mRNA, driven through its proximal promoter. Surprisingly,however, ABIN-1 protein levels were reduced following IL-17signaling, an event thatwasmediated by proteasomal degradation.Thus, IL-17 signaling serves to release the ABIN-1–controlledbrake on inflammation, revealing a new regulatory circuit in theIL-17 inflammatory pathway.
MATERIALS AND METHODS
Abs and reagentsAbs. AbsusedwereABIN-1 (SC134660; SantaCruzBiotechnology,SantaCruz,CAor4664;Cell Signaling, Beverly,MA), IkBa (SC371;Santa Cruz Biotechnology), a-tubulin (ab40742; Abcam, Cam-bridge, MA), and b-actin (ab49900; Abcam). Secondary Abs werefrom Thermo Fisher Scientific. Inhibitors were MG132 (474790-100UG, 20 mM), staurosporine (569396, 100 nM), and inhibitor of
NF-kB kinase (IKK) inhibitor VII (401486-1MG, 10 mM; EMDMilliporeSigma, Billerica, MA).
HEK293T cells were cultured in DMEM and ST2 cells andmurine embryonic fibroblasts (MEFs) were cultured in a-MEM(Sigma-Aldrich, StLouis,MO)containing 10%FBSwith L-glutamineand antibiotics (Invitrogen). Transfections were performed withFugene HD (Promega, Madison, WI) or Lipojet (SignaGen,Rockville, MD). Plasmids expressing ABIN-1, the ABIN-1 pro-moter, and the Lcn2 promoter were previously described (13–15).Luciferase assays were performed as described (7, 14). Smallinterfering RNAs (siRNAs) were from Dharmacon (GE, Lafayette,CO): Tnip1 (ABIN-1) (L-047652-01), Tnfaip3 (A20) (L-058907-02),andnontargetingmock (D-001810-10-20). siRNA transfectionswereperformed 48 h prior to stimulation. In all cases efficiency ofknockdown was confirmed by quantitative PCR (qPCR).
RNA isolation, qPCR, and ELISARNAwasextractedusingRNAeasyKits (74106;Qiagen) andcDNAwas made to perform qPCR. Primers were from Qiagen: Tnip1(QT00149009), Tnfaip3 (QT00134064), Gapdh (QT01658692), Il6(QT00098875), Lcn2 (QT00113407), Cxcl1 (QT00115647), Ccl20(QT02326394), and Nfkbiz (QT00143934). Relative gene expres-sionwas normalized toGapdh. ELISA kits were from eBioscience.
Immunoprecipitation and immunoblottingCellswere lysedon ice for 20min in lysis buffer (1%NP40, 150mMNaCl, 50 mM Tris pH 8.0, 2 mM NaF) supplemented with 1 mMNa2VO4, 50mMPMSF, 10mMb-glycerophosphate disodium salt,and a protease inhibitormixture (Sigma-Millipore). Sampleswereboiled in running buffer and immunoblotted. Band intensity wasanalyzed using AlphaView SA Version 3.4.0 (Protein Simple, SanJose, CA).
StatisticsTo assess significance, we used the Student t test, ANOVAwith posthoc Tukey analysis, or linear regression. A p value , 0.05 wasconsidered significant. Error bars reflect the mean 6 SEM ofreplicateswithin individual experiments or pooleddata, as indicatedin the figure legends. All experiments were repeated at least twice.
RESULTS
ABIN-1 inhibits both basal and IL-17–inducedgene expressionTo delineate the role of ABIN-1 in IL-17–mediated signaling, weassessed IL-17 signaling in immortalized MEFs lacking ABIN-1(16). Cells were treated with IL-17 over a 24 h period, and IL-6 inculture supernatants was assessed by ELISA. At baseline,ABIN-12/2 fibroblasts secreted significantly more IL-6 thanABIN-1+/+ cells (Fig. 1A). Similarly, ABIN-12/2 cells treated withIL-17 expressed more IL-6 than IL-17–stimulated ABIN-1+/+ cells.Consistent with these findings, elevated expression of Il6 mRNAwas observed inABIN-12/2MEFs, both at baseline and after IL-17stimulation (Fig. 1B). These results indicate that ABIN-1 restricts
https://doi.org/10.4049/immunohorizons.1700035
134 IL-17 DEGRADES ABIN-1 ImmunoHorizons
by guest on Novem
ber 12, 2018http://w
ww
.imm
unohorizons.org/D
ownloaded from
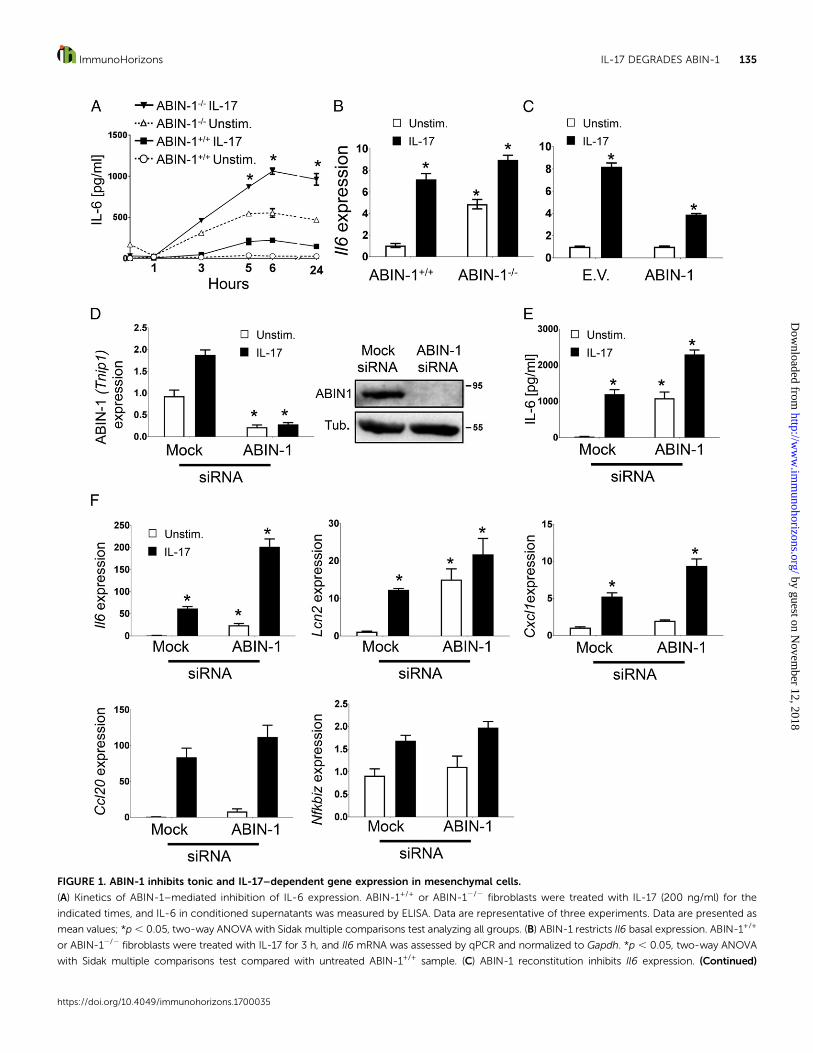
FIGURE 1. ABIN-1 inhibits tonic and IL-17–dependent gene expression in mesenchymal cells.
(A) Kinetics of ABIN-1–mediated inhibition of IL-6 expression. ABIN-1+/+ or ABIN-12/2 fibroblasts were treated with IL-17 (200 ng/ml) for the
indicated times, and IL-6 in conditioned supernatants was measured by ELISA. Data are representative of three experiments. Data are presented as
mean values; *p, 0.05, two-way ANOVA with Sidak multiple comparisons test analyzing all groups. (B) ABIN-1 restricts Il6 basal expression. ABIN-1+/+
or ABIN-12/2 fibroblasts were treated with IL-17 for 3 h, and Il6 mRNA was assessed by qPCR and normalized to Gapdh. *p , 0.05, two-way ANOVA
with Sidak multiple comparisons test compared with untreated ABIN-1+/+ sample. (C) ABIN-1 reconstitution inhibits Il6 expression. (Continued)
https://doi.org/10.4049/immunohorizons.1700035
ImmunoHorizons IL-17 DEGRADES ABIN-1 135
by guest on Novem
ber 12, 2018http://w
ww
.imm
unohorizons.org/D
ownloaded from

both tonic and IL-17–dependent IL-6 expression. To confirm thatABIN-1 limits IL-6 expression levels and to rule out the possibilitythat this effect was restricted to a particularMEF line, ABIN-12/2
fibroblasts were reconstituted with ABIN-1 by transfection. Cellswere then stimulated with IL-17 for 3 h and Il6 mRNA wasmeasured by qPCR. Consistent with its role as an inhibitor ofinflammatory signaling, reconstitution with ABIN-1 decreased Il6production compared with cells transfected with a control vector(Fig. 1C).
To verify the inhibitory activity of ABIN-1 in another setting,we silenced ABIN-1 with siRNA in ST2 cells, a mouse bonemarrow stromal cell line that responds robustly to IL-17 (Fig. 1D)(14). Indeed, knockdown of ABIN-1 led to increased expression ofIL-6 at baseline and following IL-17 stimulation (Fig. 1E). Il6mRNA was similarly enhanced upon ABIN-1 knockdown, as wasexpression of other IL-17 target genes including Lcn2 and Cxcl1(Fig. 1F). However, not all IL-17–induced genes were impacted byABIN-1 deficiency; for example,Ccl20 andNfkbiz (encoding IkBz)are IL-17 target genes that are restricted by A20 but not by ABIN-1(Fig. 1F) (7). Together, these results demonstrate that ABIN-1 is anegative regulatorof tonic andIL-17–inducedsignaling, selectivelycontrolling expression of a subset of IL-17 target genes.
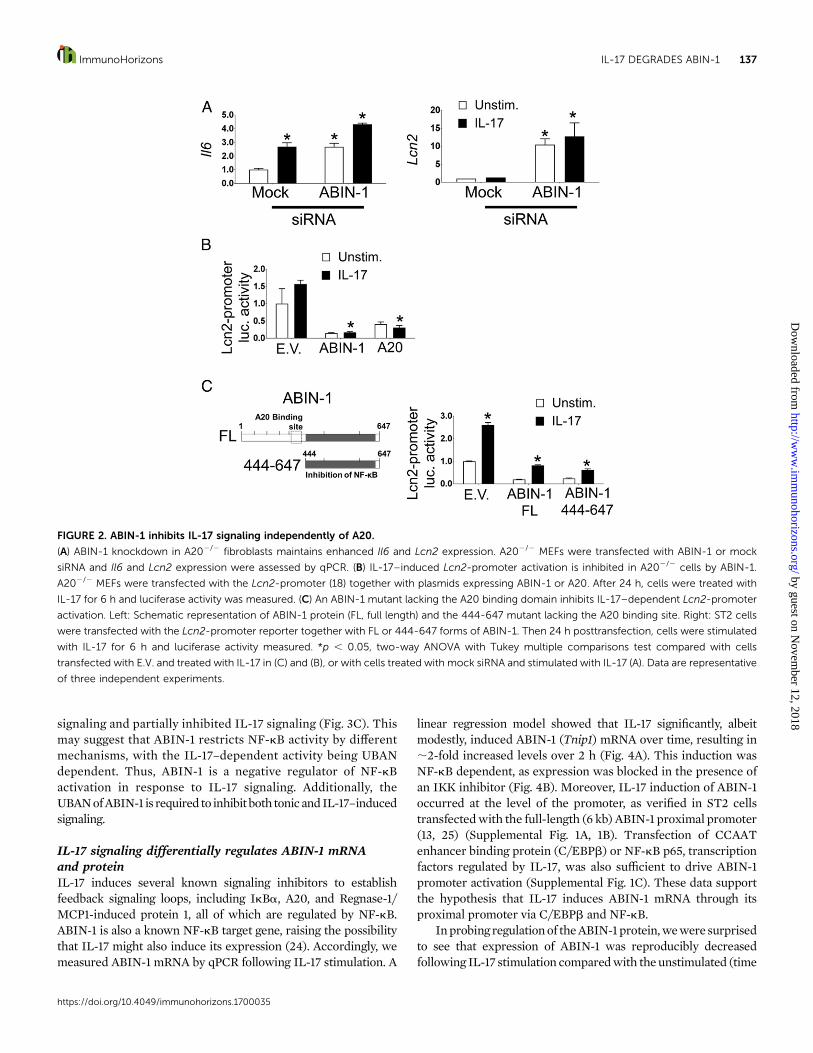
ABIN-1 inhibits IL-17R signaling independently of A20Because ABIN-1 was identified based on its ability to bind A20, weasked whether ABIN-1–mediated inhibition of IL-17R signaling isdependent on A20 (17). Accordingly, we knocked down ABIN-1in A202/2 fibroblasts by RNA silencing. As shown, reducedexpression of ABIN-1 led to enhanced Il6 and Lcn2 expression,both at baseline and after IL-17 stimulation, similar to wild-typeMEFs and ST2 cells (Fig. 2A). In an independent approach toaddress theroleofA20 inABIN-1activity,wecotransfectedA202/2
MEFs with ABIN-1 together with a luciferase reporter driven bythe Lcn2 promoter (18). Cells were stimulated with IL-17 for 6 hand Luc activity assessed. As we previously reported, A202/2
fibroblasts showedonly amodest responsiveness to IL-17, probablybecause baselineNF-kB is constitutively high in the absence ofA20(7). Nonetheless, Lcn2-promoter activity was markedly reducedupon transfection of ABIN-1 or A20, confirming that A20 isdispensable for the inhibitory function of ABIN-1 (Fig. 2B). As athird method to assess the role of A20, we transfected ST2 cellswith the Lcn2 luciferase reporter with either a full-length ABIN-1
or a truncated mutant lacking the A20-binding domain (butretaining the NF-kB–inhibitory C-terminal domain) (Fig. 2C) (15).In response to IL-17, both the full-length and truncated forms ofABIN-1 inhibited IL-17–dependent Lcn2-promoter activation (Fig.2C). Collectively, these findings demonstrate that ABIN-1 sup-presses IL-17 activity and tonic inflammatory signaling indepen-dently of A20.
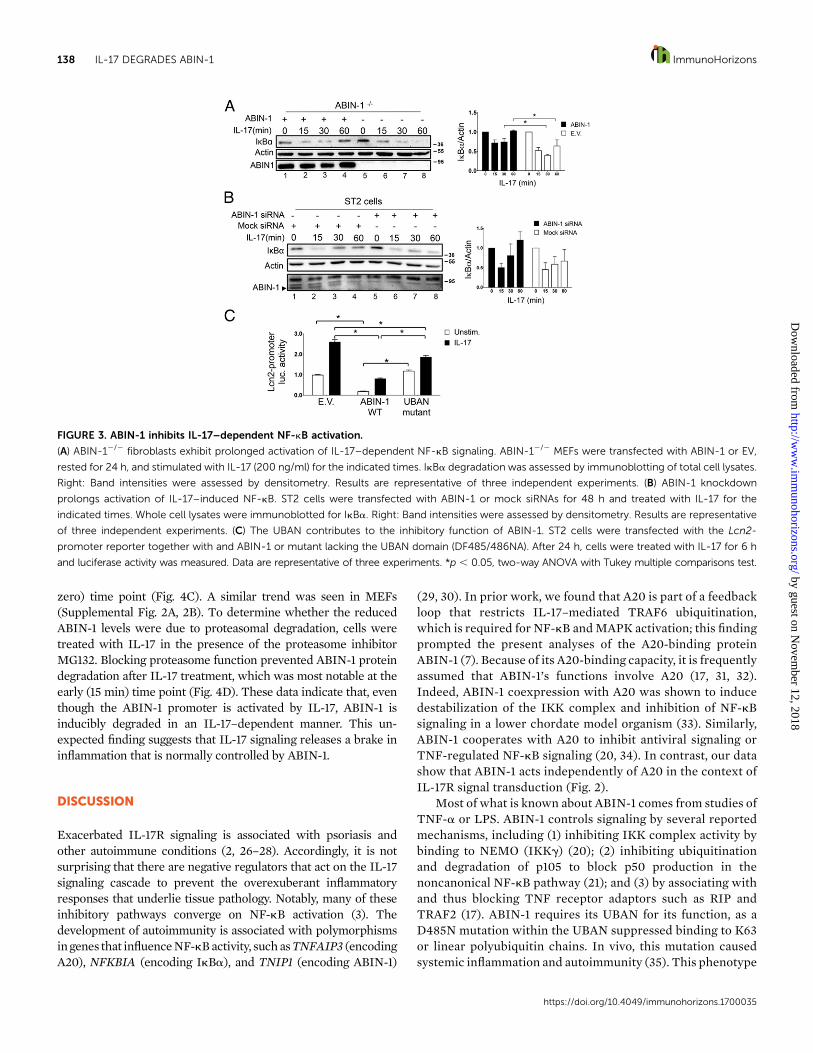
ABIN-1 regulates IL-17--activated NF-kB signalingABIN-1 is known to inhibit NF-kB in the context of TNF-astimulation, and the above results suggested ABIN-1 suppressedNF-kB–dependent genes in the IL-17 pathway as well (19–22). Anearly step in the canonical NF-kB signaling pathway is phosphor-ylation of the inhibitor IkBa, which leads to its degradation by theproteasome, releasing NF-kB for nuclear translocation.Moreover,the gene encoding IkBa is induced in an NF-kB–dependentmanner, establishing a negative feedback loop (23). To determinewhether ABIN-1 targets NF-kB in the IL-17 signaling cascade, weassessed IL-17–dependent IkBa degradation in ABIN-12/2
fibroblasts. In ABIN-12/2 cells transfected with a control emptyvector (EV), IkBa was degraded within 15 min following IL-17stimulation and remained at low levels for as long as 60 minpoststimulation (Fig. 3A, lanes 5–8). In contrast, in ABIN-12/2
fibroblasts reconstituted with ABIN-1, IkBawas restored to basallevels by 60 min (Fig. 3A, lane 4). Therefore, there is prolongedactivationofNF-kBsignalingwhenABIN-1 isdeficient.Thesedataindicate that ABIN-1 inhibits IL-17–dependent NF-kB signalingupstream of IkBa degradation.
To confirm that ABIN-1 inhibits NF-kB, we knocked downABIN-1 in ST2 cells and evaluated NF-kB activation by IL-17. InST2 cells transfected with a control (mock) siRNA IkBa returnedto normal levels 60 min after IL-17 stimulation (Fig. 3B, lane 4).However, when ABIN-1 was reduced, cells deficient in thismolecule could not re-establish pretreatment levels of IkBa (Fig.3B, lane 8). These data suggest that ABIN-1 is required to inhibitIL-17–dependent NF-kB signaling upstream of IkBa.
To further define the mechanism by which ABIN-1 inhibitsIL-17 signaling, we measured IL-17–dependent Lcn2 promoteractivation in ST2 cells transfected with full-length ABIN-1 or amutant with a nonfunctional ubiquitin binding domain (UBAN)(15). Whereas the full-length ABIN-1 inhibited IL-17–dependentLcn2 promoter activation, the UBANmutant did not impair basal
ABIN-12/2 fibroblasts were transiently transfected with an ABIN-1–expressing plasmid or EV. After 24 h, cells were treated with IL-17 for 3 h and Il6 was
assessed by qPCR. Data are presented as the fold-induction of Il6 in IL-17–stimulated cells compared with unstimulated samples. Data are normalized
to untreated cells transfected with the same plasmid. *p , 0.05, two-way ANOVA with Sidak multiple comparisons test compared with ABIN-12/2
untreated. (D) ABIN-1 knockdown efficiency. ST2 cells were transfected with siRNA targeting ABIN-1 or a scrambled siRNA (mock) control. ABIN-1
(Tnip1) expression was assessed by qPCR (left) and ABIN-1 protein by immunoblotting (right). *p , 0.05, two-way ANOVA with Sidak multiple
comparisons test compared with mock, unstimulated cells. (E) ABIN-1 silencing enhances tonic and IL-17–induced IL-6 production. ST2 cells were
transfected with the indicated siRNAs. After 48h cells were stimulated with IL-17 for 3 h, and secreted IL-6 was assessed by ELISA. *p , 0.05, two-way
ANOVA with Sidak multiple comparisons test compared with mock unstimulated cells. (F) ABIN-1 silencing enhances tonic and IL-17–induced
expression of some but not all IL-17 target genes. ST2 cells from (E) were analyzed for the expression of the indicated genes by qPCR. *p , 0.05, two-
way ANOVA with Sidak multiple comparisons test compared with mock unstimulated cells. (B–F) Data are representative of three independent
experiments.
https://doi.org/10.4049/immunohorizons.1700035
136 IL-17 DEGRADES ABIN-1 ImmunoHorizons
by guest on Novem
ber 12, 2018http://w
ww
.imm
unohorizons.org/D
ownloaded from
signaling and partially inhibited IL-17 signaling (Fig. 3C). Thismay suggest that ABIN-1 restricts NF-kB activity by differentmechanisms, with the IL-17–dependent activity being UBANdependent. Thus, ABIN-1 is a negative regulator of NF-kBactivation in response to IL-17 signaling. Additionally, theUBANofABIN-1 is required to inhibit both tonicandIL-17–inducedsignaling.
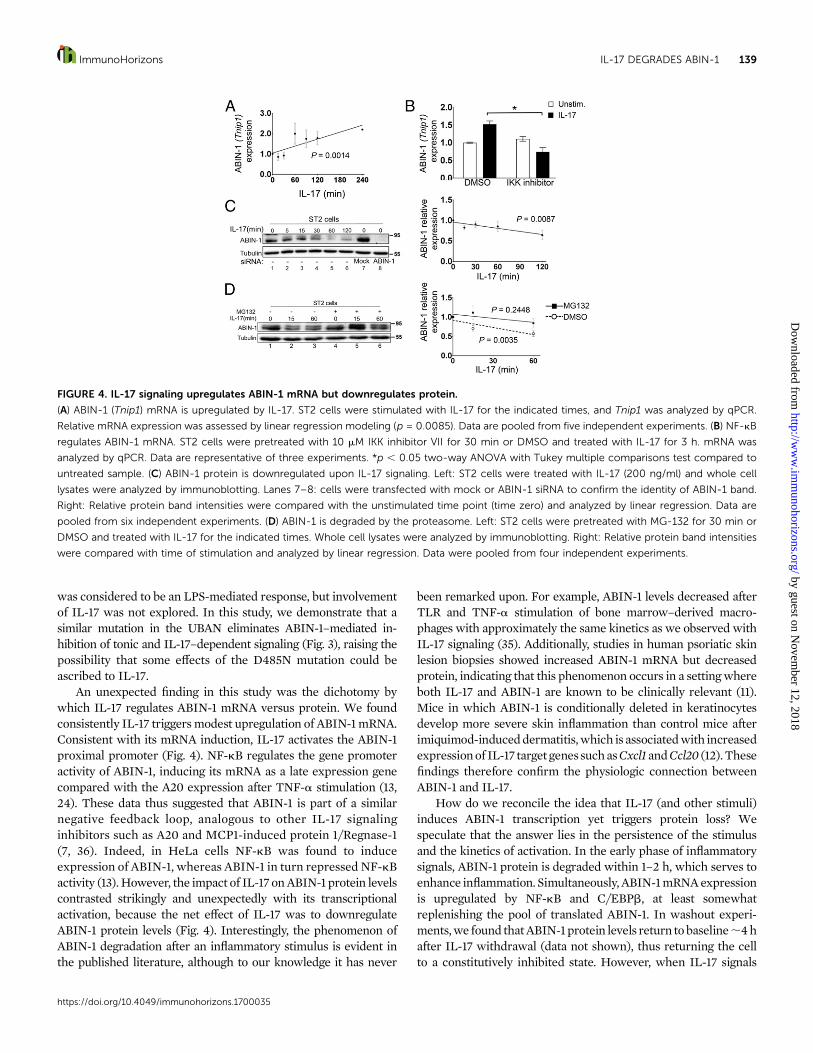
IL-17 signaling differentially regulates ABIN-1 mRNAand proteinIL-17 induces several known signaling inhibitors to establishfeedback signaling loops, including IkBa, A20, and Regnase-1/MCP1-induced protein 1, all of which are regulated by NF-kB.ABIN-1 is also a known NF-kB target gene, raising the possibilitythat IL-17 might also induce its expression (24). Accordingly, wemeasured ABIN-1 mRNA by qPCR following IL-17 stimulation. A
linear regression model showed that IL-17 significantly, albeitmodestly, induced ABIN-1 (Tnip1) mRNA over time, resulting in;2-fold increased levels over 2 h (Fig. 4A). This induction wasNF-kB dependent, as expression was blocked in the presence ofan IKK inhibitor (Fig. 4B). Moreover, IL-17 induction of ABIN-1occurred at the level of the promoter, as verified in ST2 cellstransfectedwith the full-length (6 kb) ABIN-1 proximal promoter(13, 25) (Supplemental Fig. 1A, 1B). Transfection of CCAATenhancer binding protein (C/EBPb) or NF-kB p65, transcriptionfactors regulated by IL-17, was also sufficient to drive ABIN-1promoter activation (Supplemental Fig. 1C). These data supportthe hypothesis that IL-17 induces ABIN-1 mRNA through itsproximal promoter via C/EBPb and NF-kB.
Inprobing regulationof theABIN-1protein,wewere surprisedto see that expression of ABIN-1 was reproducibly decreasedfollowing IL-17 stimulation comparedwith the unstimulated (time
FIGURE 2. ABIN-1 inhibits IL-17 signaling independently of A20.
(A) ABIN-1 knockdown in A202/2 fibroblasts maintains enhanced Il6 and Lcn2 expression. A202/2 MEFs were transfected with ABIN-1 or mock
siRNA and Il6 and Lcn2 expression were assessed by qPCR. (B) IL-17–induced Lcn2-promoter activation is inhibited in A202/2 cells by ABIN-1.
A202/2 MEFs were transfected with the Lcn2-promoter (18) together with plasmids expressing ABIN-1 or A20. After 24 h, cells were treated with
IL-17 for 6 h and luciferase activity was measured. (C) An ABIN-1 mutant lacking the A20 binding domain inhibits IL-17–dependent Lcn2-promoter
activation. Left: Schematic representation of ABIN-1 protein (FL, full length) and the 444-647 mutant lacking the A20 binding site. Right: ST2 cells
were transfected with the Lcn2-promoter reporter together with FL or 444-647 forms of ABIN-1. Then 24 h posttransfection, cells were stimulated
with IL-17 for 6 h and luciferase activity measured. *p , 0.05, two-way ANOVA with Tukey multiple comparisons test compared with cells
transfected with E.V. and treated with IL-17 in (C) and (B), or with cells treated with mock siRNA and stimulated with IL-17 (A). Data are representative
of three independent experiments.
https://doi.org/10.4049/immunohorizons.1700035
ImmunoHorizons IL-17 DEGRADES ABIN-1 137
by guest on Novem
ber 12, 2018http://w
ww
.imm
unohorizons.org/D
ownloaded from
zero) time point (Fig. 4C). A similar trend was seen in MEFs(Supplemental Fig. 2A, 2B). To determine whether the reducedABIN-1 levels were due to proteasomal degradation, cells weretreated with IL-17 in the presence of the proteasome inhibitorMG132. Blocking proteasome function prevented ABIN-1 proteindegradation after IL-17 treatment, which was most notable at theearly (15 min) time point (Fig. 4D). These data indicate that, eventhough the ABIN-1 promoter is activated by IL-17, ABIN-1 isinducibly degraded in an IL-17–dependent manner. This un-expected finding suggests that IL-17 signaling releases a brake ininflammation that is normally controlled by ABIN-1.
DISCUSSION
Exacerbated IL-17R signaling is associated with psoriasis andother autoimmune conditions (2, 26–28). Accordingly, it is notsurprising that there are negative regulators that act on the IL-17signaling cascade to prevent the overexuberant inflammatoryresponses that underlie tissue pathology. Notably, many of theseinhibitory pathways converge on NF-kB activation (3). Thedevelopment of autoimmunity is associated with polymorphismsingenes that influenceNF-kBactivity, suchasTNFAIP3 (encodingA20), NFKBIA (encoding IkBa), and TNIP1 (encoding ABIN-1)
(29, 30). In prior work, we found that A20 is part of a feedbackloop that restricts IL-17–mediated TRAF6 ubiquitination,which is required for NF-kB andMAPK activation; this findingprompted the present analyses of the A20-binding proteinABIN-1 (7). Because of its A20-binding capacity, it is frequentlyassumed that ABIN-1’s functions involve A20 (17, 31, 32).Indeed, ABIN-1 coexpression with A20 was shown to inducedestabilization of the IKK complex and inhibition of NF-kBsignaling in a lower chordate model organism (33). Similarly,ABIN-1 cooperates with A20 to inhibit antiviral signaling orTNF-regulated NF-kB signaling (20, 34). In contrast, our datashow that ABIN-1 acts independently of A20 in the context ofIL-17R signal transduction (Fig. 2).
Most of what is known about ABIN-1 comes from studies ofTNF-a or LPS. ABIN-1 controls signaling by several reportedmechanisms, including (1) inhibiting IKK complex activity bybinding to NEMO (IKKg) (20); (2) inhibiting ubiquitinationand degradation of p105 to block p50 production in thenoncanonical NF-kB pathway (21); and (3) by associating withand thus blocking TNF receptor adaptors such as RIP andTRAF2 (17). ABIN-1 requires its UBAN for its function, as aD485N mutation within the UBAN suppressed binding to K63or linear polyubiquitin chains. In vivo, this mutation causedsystemic inflammation and autoimmunity (35). This phenotype
FIGURE 3. ABIN-1 inhibits IL-17–dependent NF-kB activation.
(A) ABIN-12/2 fibroblasts exhibit prolonged activation of IL-17–dependent NF-kB signaling. ABIN-12/2 MEFs were transfected with ABIN-1 or EV,
rested for 24 h, and stimulated with IL-17 (200 ng/ml) for the indicated times. IkBa degradation was assessed by immunoblotting of total cell lysates.
Right: Band intensities were assessed by densitometry. Results are representative of three independent experiments. (B) ABIN-1 knockdown
prolongs activation of IL-17–induced NF-kB. ST2 cells were transfected with ABIN-1 or mock siRNAs for 48 h and treated with IL-17 for the
indicated times. Whole cell lysates were immunoblotted for IkBa. Right: Band intensities were assessed by densitometry. Results are representative
of three independent experiments. (C) The UBAN contributes to the inhibitory function of ABIN-1. ST2 cells were transfected with the Lcn2-
promoter reporter together with and ABIN-1 or mutant lacking the UBAN domain (DF485/486NA). After 24 h, cells were treated with IL-17 for 6 h
and luciferase activity was measured. Data are representative of three experiments. *p , 0.05, two-way ANOVA with Tukey multiple comparisons test.
https://doi.org/10.4049/immunohorizons.1700035
138 IL-17 DEGRADES ABIN-1 ImmunoHorizons
by guest on Novem
ber 12, 2018http://w
ww
.imm
unohorizons.org/D
ownloaded from
was considered to be an LPS-mediated response, but involvementof IL-17 was not explored. In this study, we demonstrate that asimilar mutation in the UBAN eliminates ABIN-1–mediated in-hibition of tonic and IL-17–dependent signaling (Fig. 3), raising thepossibility that some effects of the D485N mutation could beascribed to IL-17.
An unexpected finding in this study was the dichotomy bywhich IL-17 regulates ABIN-1 mRNA versus protein. We foundconsistently IL-17 triggersmodest upregulation of ABIN-1mRNA.Consistent with its mRNA induction, IL-17 activates the ABIN-1proximal promoter (Fig. 4). NF-kB regulates the gene promoteractivity of ABIN-1, inducing its mRNA as a late expression genecompared with the A20 expression after TNF-a stimulation (13,24). These data thus suggested that ABIN-1 is part of a similarnegative feedback loop, analogous to other IL-17 signalinginhibitors such as A20 and MCP1-induced protein 1/Regnase-1(7, 36). Indeed, in HeLa cells NF-kB was found to induceexpression of ABIN-1, whereas ABIN-1 in turn repressed NF-kBactivity (13).However, the impact of IL-17 onABIN-1 protein levelscontrasted strikingly and unexpectedly with its transcriptionalactivation, because the net effect of IL-17 was to downregulateABIN-1 protein levels (Fig. 4). Interestingly, the phenomenon ofABIN-1 degradation after an inflammatory stimulus is evident inthe published literature, although to our knowledge it has never
been remarked upon. For example, ABIN-1 levels decreased afterTLR and TNF-a stimulation of bone marrow–derived macro-phages with approximately the same kinetics as we observed withIL-17 signaling (35). Additionally, studies in human psoriatic skinlesion biopsies showed increased ABIN-1 mRNA but decreasedprotein, indicating that this phenomenon occurs in a settingwhereboth IL-17 and ABIN-1 are known to be clinically relevant (11).Mice in which ABIN-1 is conditionally deleted in keratinocytesdevelop more severe skin inflammation than control mice afterimiquimod-induceddermatitis,which is associatedwith increasedexpressionof IL-17 target genessuchasCxcl1 andCcl20 (12).Thesefindings therefore confirm the physiologic connection betweenABIN-1 and IL-17.
How do we reconcile the idea that IL-17 (and other stimuli)induces ABIN-1 transcription yet triggers protein loss? Wespeculate that the answer lies in the persistence of the stimulusand the kinetics of activation. In the early phase of inflammatorysignals, ABIN-1 protein is degraded within 1–2 h, which serves toenhance inflammation. Simultaneously,ABIN-1mRNAexpressionis upregulated by NF-kB and C/EBPb, at least somewhatreplenishing the pool of translated ABIN-1. In washout experi-ments,we found thatABIN-1protein levels return tobaseline;4hafter IL-17 withdrawal (data not shown), thus returning the cellto a constitutively inhibited state. However, when IL-17 signals
FIGURE 4. IL-17 signaling upregulates ABIN-1 mRNA but downregulates protein.
(A) ABIN-1 (Tnip1) mRNA is upregulated by IL-17. ST2 cells were stimulated with IL-17 for the indicated times, and Tnip1 was analyzed by qPCR.
Relative mRNA expression was assessed by linear regression modeling (p = 0.0085). Data are pooled from five independent experiments. (B) NF-kB
regulates ABIN-1 mRNA. ST2 cells were pretreated with 10 mM IKK inhibitor VII for 30 min or DMSO and treated with IL-17 for 3 h. mRNA was
analyzed by qPCR. Data are representative of three experiments. *p , 0.05 two-way ANOVA with Tukey multiple comparisons test compared to
untreated sample. (C) ABIN-1 protein is downregulated upon IL-17 signaling. Left: ST2 cells were treated with IL-17 (200 ng/ml) and whole cell
lysates were analyzed by immunoblotting. Lanes 7–8: cells were transfected with mock or ABIN-1 siRNA to confirm the identity of ABIN-1 band.
Right: Relative protein band intensities were compared with the unstimulated time point (time zero) and analyzed by linear regression. Data are
pooled from six independent experiments. (D) ABIN-1 is degraded by the proteasome. Left: ST2 cells were pretreated with MG-132 for 30 min or
DMSO and treated with IL-17 for the indicated times. Whole cell lysates were analyzed by immunoblotting. Right: Relative protein band intensities
were compared with time of stimulation and analyzed by linear regression. Data were pooled from four independent experiments.
https://doi.org/10.4049/immunohorizons.1700035
ImmunoHorizons IL-17 DEGRADES ABIN-1 139
by guest on Novem
ber 12, 2018http://w
ww
.imm
unohorizons.org/D
ownloaded from

continuously, ABIN-1 degradation was maintained. Thus, theinhibitory activity ofABIN1 is compromisedwhile there is ongoingstimulation. This might explain the observation that in patientswithpsoriasis there is elevatedmRNAbut decreasedprotein levelsfor ABIN-1 (11). Psoriasis is linked to several inflammatorycytokines, with a predominant role for IL-17A signaling, based inpart on the success of anti–IL-17A and anti–IL-17RA biologicdrugs (26, 37–39). Therefore, the idea that ABIN-1 is both atonic inhibitor of inflammation and a negative regulator of IL-17signaling fits well with our data and with findings in preclinicalmouse models and human studies (10–12).
There are other examples of inhibitors that are downregulatedby the signals that they regulate. In the case of IL-17R signaling,the proximal adaptor Act1 is degraded following phosphorylationto temper all downstream signaling events (40), and IkBa isdegraded followingphosphorylation tounmask theNF-kBnuclearimport signal (41). IL-17 is just one of multiple immune effectorsthat can initiate ABIN-1 degradation and presumably therebyenhance inflammation. Moreover, IL-17 signals synergisticallywith many of these cytokines, particularly TNF-a (3, 42). Thus,ABIN-1 appears to regulate signaling at the intersection of theseinflammatory stimuli, namely, the IKK complex.
These data have possible implications for therapeutic inter-ventions. We show that ABIN-1 degradation is at least partiallymediated by the proteasome, with no apparent contribution ofcaspases or the lysosome (data not shown). Therapeutics targetingthe proteasome are used for cancer (43), and might be useful indiseases where ABIN-1 is implicated, i.e., those where TNIP1single nucleotide polymorphisms are found. Another consider-ation is settings where overproduction of ABIN-1 activators suchas IL-17 or TNF-a (e.g., autoimmunity, microbiome dysbiosis)might lead to underexpression of ABIN-1 and hence chronicinflammation andassociatedharmful sequelae.Clearly,moreworkneeds to be done to dissect the role of this important but enigmaticmolecule in the context of inflammation.
DISCLOSURES
S.L.G.has received researchgrants fromNovartis andJanssen,andexpects to receive consulting fees in excess of $5,000 fromLycera.The other authors have no financial conflicts of interest.
ACKNOWLEDGMENTS
We thank F. Galbiati, M. McGeachy, and P. Biswas for helpful suggestionsand A. Verma for critical reading. We thank J. Shaffer for assistance withstatistical analyses.
REFERENCES
1. Conti, H. R., and S. L. Gaffen. 2015. IL-17-mediated immunity to theopportunistic fungal pathogen Candida albicans. J. Immunol. 195:780–788.
2. Gaffen, S. L., R. Jain, A. V. Garg, and D. J. Cua. 2014. The IL-23-IL-17immune axis: from mechanisms to therapeutic testing. Nat. Rev.Immunol. 14: 585–600.
3. Amatya, N., A. V. Garg, and S. L. Gaffen. 2017. IL-17 signaling: the Yinand the Yang. Trends Immunol. 38: 310–322.
4. Song, X., and Y. Qian. 2013. The activation and regulation of IL-17receptor mediated signaling. Cytokine 62: 175–182.
5. Zepp, J., L. Wu, and X. Li. 2011. IL-17 receptor signaling and T helper17-mediated autoimmune demyelinating disease. Trends Immunol. 32:232–239.
6. Onishi, R., and S. L. Gaffen. 2010. IL-17 and its target genes: mecha-nisms of IL-17 function in disease. Immunology 129: 311–321.
7. Garg, A. V., M. Ahmed, A. N. Vallejo, A. Ma, and S. L. Gaffen. 2013.The deubiquitinase A20 mediates feedback inhibition of interleukin-17 receptor signaling. Sci. Signal. 6: ra44–ra55.
8. Li, X. L., H. Yu, and G. S. Wu. 2014. Investigating the genetic asso-ciation of HCP5, SPATA2, TNIP1, TNFAIP3 and COG6 with psoriasisin Chinese population. Int. J. Immunogenet. 41: 503–507.
9. Indhumathi, S., M. Rajappa, L. Chandrashekar, P. H. Ananthanarayanan,D. M. Thappa, and V. S. Negi. 2015. TNFAIP3 and TNIP1 polymor-phisms confer psoriasis risk in South Indian Tamils. Br. J. Biomed. Sci.72: 168–173.
10. Callahan, J. A., G. E. Hammer, A. Agelides, B. H. Duong, S. Oshima,J. North, R. Advincula, N. Shifrin, H.-A. Truong, J. Paw, et al. 2013.Cutting edge: ABIN-1 protects against psoriasis by restricting MyD88signals in dendritic cells. J. Immunol. 191: 535–539.
11. Chen, Y., H. Yan, Z. Song, F. Chen, H. Wang, J. Niu, X. Shi, D. Zhang,N. Zhang, Z. Zhai, et al. 2015. Downregulation of TNIP1 expressionleads to increased proliferation of human keratinocytes and severerpsoriasis-like conditions in an imiquimod-induced mouse model ofdermatitis. PLoS One 10: e0127957.
12. Ippagunta, S. K., R. Gangwar, D. Finkelstein, P. Vogel, S. Pelletier,S. Gingras, V. Redecke, and H. Hacker. 2016. Keratinocytes contributeintrinsically to psoriasis upon loss of Tnip1 function. Proc. Natl. Acad.Sci. USA 113: E6162–E6171.
13. Gurevich, I., C. Zhang, P. C. Encarnacao, C. P. Struzynski, S. E. Livings,and B. J. Aneskievich. 2012. PPARg and NF-kB regulate the genepromoter activity of their shared repressor, TNIP1. Biochim. Biophys.Acta 1819: 1–15.
14. Shen, F., M. J. Ruddy, P. Plamondon, and S. L. Gaffen. 2005. Cytokineslink osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-alpha-induced genes in bone cells. J. Leukoc. Biol. 77:388–399.
15. Heyninck, K., M. M. Kreike, and R. Beyaert. 2003. Structure-functionanalysis of the A20-binding inhibitor of NF-k B activation, ABIN-1.FEBS Lett. 536: 135–140.
16. Oshima, S., E. E. Turer, J. A. Callahan, S. Chai, R. Advincula, J. Barrera,N. Shifrin, B. Lee, T. S. Benedict Yen, T. Woo, et al. 2009. ABIN-1 is aubiquitin sensor that restricts cell death and sustains embryonic de-velopment. Nature 457: 906–909.
17. Heyninck, K., G. Denecker, D. De Valck, W. Fiers, and R. Beyaert.1999. Inhibition of tumor necrosis factor-induced necrotic cell deathby the zinc finger protein A20. Anticancer Res. 19: 2863–2868.
18. Shen, F., Z. Hu, J. Goswami, and S. L. Gaffen. 2006. Identification ofcommon transcriptional regulatory elements in interleukin-17 targetgenes. J. Biol. Chem. 281: 24138–24148.
19. Verstrepen, L., I. Carpentier, K. Verhelst, and R. Beyaert. 2009.ABINs: A20 binding inhibitors of NF-kappa B and apoptosis signaling.Biochem. Pharmacol. 78: 105–114.
20. Mauro, C., F. Pacifico, A. Lavorgna, S. Mellone, A. Iannetti, R. Ac-quaviva, S. Formisano, P. Vito, and A. Leonardi. 2006. ABIN-1 binds toNEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB.J. Biol. Chem. 281: 18482–18488.
21. Cohen, S., A. Ciechanover, Y. Kravtsova-Ivantsiv, D. Lapid, and S.Lahav-Baratz. 2009. ABIN-1 negatively regulates NF-kappaB byinhibiting processing of the p105 precursor. Biochem. Biophys. Res.Commun. 389: 205–210.
https://doi.org/10.4049/immunohorizons.1700035
140 IL-17 DEGRADES ABIN-1 ImmunoHorizons
by guest on Novem
ber 12, 2018http://w
ww
.imm
unohorizons.org/D
ownloaded from

22. Wagner, S., I. Carpentier, V. Rogov, M. Kreike, F. Ikeda, F. Lohr, C. J.Wu, J. D. Ashwell, V. Dotsch, I. Dikic, and R. Beyaert. 2008. Ubiquitinbinding mediates the NF-kappaB inhibitory potential of ABIN pro-teins. Oncogene 27: 3739–3745.
23. Sun, S. C. 2017. The non-canonical NF-kB pathway in immunity andinflammation. Nat. Rev. Immunol. DOI: 10.1038/nri.2017.52.
24. Tian, B., D. E. Nowak, M. Jamaluddin, S. Wang, and A. R. Brasier.2005. Identification of direct genomic targets downstream of thenuclear factor-kappaB transcription factor mediating tumor necrosisfactor signaling. J. Biol. Chem. 280: 17435–17448.
25. Gurevich, I., C. Zhang, N. Francis, C. P. Struzynsky, S. E. Livings, andB. J. Aneskievich. 2013. Human TNFa-induced protein 3-interactingprotein 1 (TNIP1) promoter activation is regulated by retinoic acidreceptors. Gene 515: 42–48.
26. Hueber, W., D. D. Patel, T. Dryja, A. M. Wright, I. Koroleva, G. Bruin,C. Antoni, Z. Draelos, M. H. Gold, P. Durez, et al; Psoriasis StudyGroup; Rheumatoid Arthritis Study Group; Uveitis Study Group. 2010.Effects of AIN457, a fully human antibody to interleukin-17A, onpsoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med. 2: 52ra72.
27. Swindell, W. R., M. K. Sarkar, Y. Liang, X. Xing, and J. E. Gudjonsson.2016. Cross-disease transcriptomics: unique IL-17A signaling in pso-riasis lesions and an autoimmune PBMC signature. J. Invest. Der-matol. 136: 1820–1830.
28. Zhu, S., and Y. Qian. 2012. IL-17/IL-17 receptor system in autoim-mune disease: mechanisms and therapeutic potential. Clin. Sci. 122:487–511.
29. Li, Y., H. Cheng, X. B. Zuo, Y. J. Sheng, F. S. Zhou, X. F. Tang, H. Y.Tang, J. P. Gao, Z. Zhang, S. M. He, et al. 2013. Association analysesidentifying two common susceptibility loci shared by psoriasis andsystemic lupus erythematosus in the Chinese Han population. J. Med.Genet. 50: 812–818.
30. Kawasaki, A., S. Ito, H. Furukawa, T. Hayashi, D. Goto, I. Matsumoto,M. Kusaoi, J. Ohashi, R. R. Graham, K. Matsuta, et al. 2010. Associ-ation of TNFAIP3 interacting protein 1, TNIP1 with systemic lupuserythematosus in a Japanese population: a case-control associationstudy. Arthritis Res. Ther. 12: R174.
31. Heyninck, K., and R. Beyaert. 1999. The cytokine-inducible zinc fingerprotein A20 inhibits IL-1-induced NF-kappaB activation at the level ofTRAF6. FEBS Lett. 442: 147–150.
32. Heyninck, K., D. De Valck, W. Vanden Berghe, W. Van Criekinge,R. Contreras, W. Fiers, G. Haegeman, and R. Beyaert. 1999. The zincfinger protein A20 inhibits TNF-induced NF-kappaB-dependent gene
expression by interfering with an RIP- or TRAF2-mediated trans-activation signal and directly binds to a novel NF-kappaB-inhibitingprotein ABIN. J. Cell Biol. 145: 1471–1482.
33. Yuan, S., X. Dong, X. Tao, L. Xu, J. Ruan, J. Peng, and A. Xu. 2014.Emergence of the A20/ABIN-mediated inhibition of NF-kB signalingvia modifying the ubiquitinated proteins in a basal chordate. Proc.Natl. Acad. Sci. USA 111: 6720–6725.
34. Gao, L., H. Coope, S. Grant, A. Ma, S. C. Ley, and E. W. Harhaj. 2011.ABIN1 protein cooperates with TAX1BP1 and A20 proteins to inhibitantiviral signaling. J. Biol. Chem. 286: 36592–36602.
35. Nanda, S. K., R. K. Venigalla, A. Ordureau, J. C. Patterson-Kane, D. W.Powell, R. Toth, J. S. Arthur, and P. Cohen. 2011. Polyubiquitinbinding to ABIN1 is required to prevent autoimmunity. J. Exp. Med.208: 1215–1228.
36. Garg, A. V., N. Amatya, K. Chen, J. A. Cruz, P. Grover, N. Whibley,H. R. Conti, G. Hernandez Mir, T. Sirakova, E. C. Childs, et al. 2015.MCPIP1 endoribonuclease activity negatively regulates interleukin-17-mediated signaling and inflammation. Immunity 43: 475–487.
37. Leonardi, C., R. Matheson, C. Zachariae, G. Cameron, L. Li, E. Edson-Heredia, D. Braun, and S. Banerjee. 2012. Anti-interleukin-17 mono-clonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J.Med. 366: 1190–1199.
38. Chiricozzi, A., and J. G. Krueger. 2013. IL-17 targeted therapies forpsoriasis. Expert Opin. Investig. Drugs 22: 993–1005.
39. Langley, R. G., B. E. Elewski, M. Lebwohl, K. Reich, C. E. Griffiths,K. Papp, L. Puig, H. Nakagawa, L. Spelman, B. Sigurgeirsson, et al;ERASURE Study Group; FIXTURE Study Group. 2014. Secukinumabin plaque psoriasis–results of two phase 3 trials. N. Engl. J. Med. 371:326–338.
40. Shi, P., S. Zhu, Y. Lin, Y. Liu, Y. Liu, Z. Chen, Y. Shi, and Y. Qian. 2011.Persistent stimulation with interleukin-17 desensitizes cells throughSCFb-TrCP-mediated degradation of Act1. Sci. Signal. 4: ra73.
41. Yao, Z., W. C. Fanslow, M. F. Seldin, A.M. Rousseau, S. L. Painter,M. R. Comeau, J. I. Cohen, and M. K. Spriggs. 1995. HerpesvirusSaimiri encodes a new cytokine, IL-17, which binds to a novel cyto-kine receptor. Immunity 3: 811–821.
42. Veldhoen, M. 2017. Interleukin 17 is a chief orchestrator of immunity.Nat. Immunol. 18: 612–621.
43. Weathington, N. M., and R. K. Mallampalli. 2014. Emerging therapiestargeting the ubiquitin proteasome system in cancer. J. Clin. Invest.124: 6–12.
https://doi.org/10.4049/immunohorizons.1700035
ImmunoHorizons IL-17 DEGRADES ABIN-1 141
by guest on Novem
ber 12, 2018http://w
ww
.imm
unohorizons.org/D
ownloaded from


















