
![Ckvemw-aXw · Ckvemw-aXw A_p¬-A-Avem auZqZn hnh¿Ø\w: hn.-]n. apl-Ω-Zen `mjm-]-cn-jvI-cWw: s{]m^. sI.-]n. Iam-ep-±o ...](https://static.fdocument.org/doc/165x107/5b9e840509d3f2e02c8bd315/ckvemw-axw-ckvemw-axw-ap-a-avem-auzqzn-hnhow-hn-n-apl-zen-mjm-cn-jvi-cww.jpg)
Hepatitis C virus infection impairs IRF-7 transloca tion and...
31
July 11, 2010 1 Hepatitis C virus infection impairs IRF-7 translocation and interferon-α 2 synthesis in immortalized human hepatocytes 3 4 Amit Raychoudhuri 1 , Shubham Shrivastava 1 , Robert Steele 1 , Srikanta Dash 3 , Tatsuo Kanda 1# , 5 Ranjit Ray 2 , and Ratna B. Ray 1* 6 7 1 Departments of Pathology and 2 Internal Medicine, Saint Louis University, St. Louis, Missouri, 8 3 Department of Pathology and Laboratory Medicine, Tulane University, New Orleans, Louisiana 9 10 11 Running title: HCV infection impairs interferon-α signaling 12 13 14 15 Key words: HCV, IHH, STAT1, IRF-7, interferon signaling 16 17 *Requests for reprints: Ratna B. Ray, Department of Pathology, Saint Louis University, 1100 18 South Grand Boulevard, St. Louis, MO 63104. Phone: 314-977-7822; Fax: 314-771-3816; E- 19 mail: [email protected]. 20 # Present address: Department of Medicine and Clinical Oncology, Chiba University, Japan 21 Copyright © 2010, American Society for Microbiology and/or the Listed Authors/Institutions. All Rights Reserved. J. Virol. doi:10.1128/JVI.00900-10 JVI Accepts, published online ahead of print on 1 September 2010 on June 30, 2018 by guest http://jvi.asm.org/ Downloaded from
-
Upload
nguyencong -
Category
Documents
-
view
219 -
download
0
Transcript of Hepatitis C virus infection impairs IRF-7 transloca tion and...

July 11, 2010 1
Hepatitis C virus infection impairs IRF-7 translocation and interferon-αααα 2
synthesis in immortalized human hepatocytes 3
4
Amit Raychoudhuri1, Shubham Shrivastava
1, Robert Steele
1, Srikanta Dash
3, Tatsuo Kanda
1#, 5
Ranjit Ray2, and Ratna B. Ray
1* 6
7
1Departments of Pathology and
2Internal Medicine, Saint Louis University, St. Louis, Missouri, 8
3Department of Pathology and Laboratory Medicine, Tulane University, New Orleans, Louisiana 9
10
11
Running title: HCV infection impairs interferon-α signaling 12
13
14
15
Key words: HCV, IHH, STAT1, IRF-7, interferon signaling 16
17
*Requests for reprints: Ratna B. Ray, Department of Pathology, Saint Louis University, 1100 18
South Grand Boulevard, St. Louis, MO 63104. Phone: 314-977-7822; Fax: 314-771-3816; E-19
mail: [email protected]. 20
#Present address: Department of Medicine and Clinical Oncology, Chiba University, Japan21
Copyright © 2010, American Society for Microbiology and/or the Listed Authors/Institutions. All Rights Reserved.J. Virol. doi:10.1128/JVI.00900-10 JVI Accepts, published online ahead of print on 1 September 2010
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

2
ABSTRACT 1
Hepatitis C virus (HCV) establishes chronic infection in a significant number of infected 2
humans, although the mechanisms for chronicity remain largely unknown. We have previously 3
shown that HCV infection in immortalized human hepatocytes (IHH) induces interferon (IFN)-β 4
expression (Kanda et al., 2007). However, the regulation of downstream signaling pathway for 5
IFN-α production by HCV is not clearly understood. In this study, the regulation of IFN 6
signaling pathway following HCV genotype 1a (clone H77) or genotype 2a (clone JFH1) 7
infection of IHH was examined. HCV infection upregulated expression of total STAT1, but 8
failed to induce phosphorylation and efficient nuclear translocation. Subsequent study revealed 9
that HCV infection induces ISRE activation as evident from upregulation of OAS1. However, 10
nuclear translocation of IRF-7 was impaired following HCV infection. In HCV infected IHH, 11
IFN-α expression was initially increased (up to 24 h) and decreased at later time points, and 12
IFI27 was not induced. Interestingly, HCV infection blocked IRF-7 nuclear translocation upon 13
poly (I-C) or IFN-α treatment of IHH. Together, our data suggest that HCV infection enhances 14
STAT1 expression, but impairs nuclear translocation of IRF-7 and its downstream molecules. 15
These impairments in the IFN-α signaling pathway may, in part, be responsible for establishment 16
of chronic HCV infection. 17
18
19
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

3
INTRODUCTION 1
HCV infection affects approximately 3.2 million people in the USA (1). The approved treatment 2
for HCV infection is pegylated IFN-α alone or in combination with ribavirin. This leads to 3
clearance of HCV in ~50% and ~80% of the cases of HCV genotypes 1 and 2, respectively. 4
Type I IFNs are crucial components of the innate immune response to virus attack. The host 5
response is triggered when a pathogen-associated molecular pattern (PAMP) presented by the 6
infecting virus is recognized and engaged by specific PAMP receptor factors expressed in the 7
host cell, initiating signals that ultimately induce the expression of antiviral effecter genes. IFN-α 8
and IFN-β are rapidly synthesized after virus infection for triggering intracellular signaling 9
events. The subsequent expression of IFN-stimulated genes (ISG) is central to these antiviral 10
responses. IFN-stimulated gene factor 3 (ISGF3) assembles and translocates from the cytoplasm 11
to the nucleus upon IFN stimulation. ISGF3 is a multi subunit transcription factor that interacts 12
with the IFN-stimulated response element (ISRE) present in the promoters of ISGs (25). ISGF3 13
consists of hetero-oligomers of signal transducers and activators of transcription (STAT)1, 14
STAT2, and IFN regulatory factor-9 (IRF-9). Homodimers of STAT1-α and heterodimers of 15
STAT1 and STAT2 are also activated, and IRF-9 is indispensable for their formation, by binding 16
to inverted repeat elements in the promoters of ISGs to induce transcription (29). 17
18
Interferon and ISGs are amplified during chronic HCV infection (2, 22, 27), but fail to eliminate 19
the virus from liver in a large number of HCV infected patients. IFN-induced genes are also 20
stimulated during HCV RNA replication within the liver of acutely infected chimpanzees (3). 21
The changes due to endogenous antiviral responses in liver (e.g., induction of type I IFN-induced 22
genes) occur by intrahepatic gene expression as soon as HCV RNA is detectable in the serum of 23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

4
chimpanzees (31). We have shown previously that cell culture grown HCV infection in IHH 1
displays nuclear localization of IRF3 and enhances IFN-β expression (13). However, modulation 2
of the downstream IFN signaling pathway in cells infected with HCV is unknown. In this study, 3
we have investigated the molecular determinants of the interferon signaling pathway following 4
HCV infection in hepatocytes, and identified specific sites responsible for impairment of the 5
downstream intracellular IFN signaling pathway. 6
7
8
MATERIALS AND METHODS 9
Cell culture, transfection and HCV infection. IHH and Huh-7 cells were grown in Dulbecco’s 10
modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, 100 U/ml of penicillin 11
G, and 100 µg/ml of streptomycin at 37°C in a 5% CO2 atmosphere. We have grown HCV 12
genotypes 1a and 2a in IHH and Huh-7.5, respectively. HCV genotype 2a also infected IHH 13
(12). In this study, IHH were infected with HCV genotype 1a (clone H77) or genotype 2a (clone 14
JFH1-GFP) at a multiplicity of infection (moi) of 0.1-1 (ffu/cell) in a minimum volume of serum 15
free medium. JFH1-GFP was generated by inserting GFP in frame at domain III of the NS5A 16
region of JFH-1 (11). Cells were treated with 400 unit of IFN-α for 1-17 h. After 8 h of 17
adsorption of virus, DMEM supplemented with 5% heat-inactivated fetal bovine serum was 18
added. IRF-7-GFP plasmid DNA (kindly provided by Betsy Barnes, NJMSUH Cancer Center) 19
was transfected into IHH using lipofectamine reagent (Invitrogen) for subcellular localization 20
studies at 72 h post-infection with HCV. 21
22
Immunofluorescence. Mock or HCV infected IHH were washed in PBS, fixed with acetone-23
acetic acid for 30 min at -20oC, and blocked with 3% bovine serum albumin for 1 h. Fixed cells 24
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

5
were incubated with a HCV NS5A specific mouse monoclonal antibody (kindly provided by 1
Chen Liu, University of Florida) and STAT1 specific rabbit polyclonal antibody (Santa Cruz) for 2
1 h. Cells were washed and incubated with anti-mouse Ig conjugated with Alexa 488 (Molecular 3
Probes) and anti-rabbit Ig conjugated with Alexa 594 (Molecular Probes) secondary antibody for 4
1 h at room temperature. Finally, cells were washed and mounted for confocal microscopy 5
(Olympus FV1000). Images were superimposed digitally for fine comparisons. 6
7
RNA quantitation. Total RNA was isolated using Trizol reagent (Invitrogen). cDNA was 8
synthesized using random hexamer and ABI reverse transcriptase kit (Applied Biosystems). Real 9
time PCR was performed on the cDNA for RNA quantitation using Taqman Universal PCR 10
master Mix (Applied Biosystems) and FAM-MGB probe for interferon alpha inducible protein 11
27 (IFI-27) (Hs00271467_m1), MxA (Hs00895598_m1), 2’-5’ oligoadenylate synthetase 1 12
(OAS-1) (Hs00973637_m1), and HCV (AI6Q1GI). FAM-MGB probe for GAPDH (Applied 13
Biosystems Taq Man gene expression assay ID-Hs99999905_m1) was used for endogenous 14
control. IFN-α RΝΑ was measured by quantitative RT-PCR using a set of primers 15
corresponding to several IFN-α subtypes (kindly provided by Betsy Barnes). 16
17
Luciferase assay. IHH infected with HCV (3 or 8 days) or mock were transfected with plasmid 18
ISRE-TA-luc (Clonetech) containing five copies of the ISRE enhancer elements upstream of the 19
firefly luciferase gene. At 48 h post-transfection, cells were lysed with reporter lysis buffer 20
(Promega), and luciferase activity was determined using a luminometer (Glomax). Luciferase 21
activity was normalized with respect to the protein concentration as described previously (13). 22
The results are presented as mean from three independent experiments. 23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

6
Immunoblot analysis. Mock or HCV infected cells were lysed in a sample buffer, subjected to 1
SDS-PAGE, and transferred onto nitrocellulose membranes. The membranes were probed with a 2
monoclonal antibody to total STAT1 (Santa Cruz), pSTAT1 or a polyclonal antibody to PKR 3
(Cell Signaling). The membranes were reprobed with a monoclonal antibody to actin or tubulin 4
(Santa Cruz) for comparison of the protein load. Proteins were visualized using an enhanced 5
chemiluminescent ECL Western blot substrate (Pierce), and subjected to densitometric scanning 6
by an image analyzer using Quantity One software (BioRad). 7
8
RESULTS 9
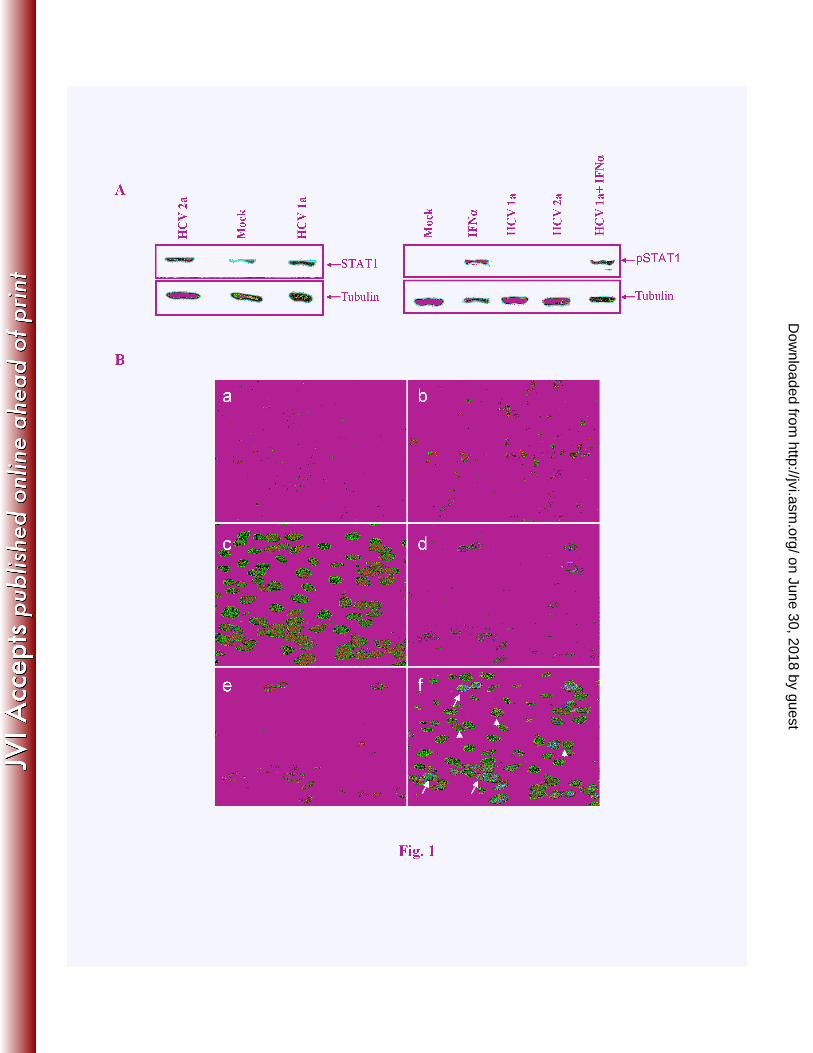
HCV infection enhances total STAT1 but inhibits phosphorylation. We examined the 10
expression status of total STAT1 and phosphorylated STAT1 protein in HCV infected IHH. 11
Cells were infected with HCV genotype 1a or 2a, and cell lysates were prepared 10 days post-12
infection for Western blot analysis. Increased expression of STAT1 was observed after HCV 13
infection of IHH, as compared to mock infected control hepatocytes (Fig. 1, panel A). However, 14
phosphorylated STAT1 protein was not detected following HCV genotype 1a or genotype 2a 15
infection of IHH. On the other hand, mock infected cells when treated with IFN-α displayed an 16
enhanced level of phosphoSTAT1. We also examined the phosphoSTAT1 status in IFN-17
α treated HCV infected IHH. The phosphoSTAT1 expression was restored in HCV infected 18
IFN-α treated IHH, although the extent of STAT1 phosphorylation was much lower (~50%) as 19
compared to IHH treated with IFN-α. 20
21
Next, we examined localization of STAT1 in HCV infected cells and in the neighboring 22
uninfected cells. For this, IHH were infected with HCV genotype 1a (12) or GFP tagged HCV 23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

7
genotype 2a (11) at 0.05 moi. Hepatocytes were fixed and subjected to immunofluorescence 1
after 10 days of infection using an anti-STAT1 and anti-HCV NS5A antibodies (Fig. 1, panel B). 2
In mock infected cells, a weak level of endogenous STAT1 was localized exclusively in the 3
cytoplasm, indicative of an inactive state (section a). While cells transfected with poly(I-C) as a 4
positive control displayed nuclear localization of STAT1 (section b). On the other hand, cells 5
when infected with HCV genotype 1a exhibited a predominant perinuclear localization of 6
STAT1 (sections c-f). Occasionally, efficient nuclear localization of STAT1 was observed in 7
HCV infected cells. Cells stained with a secondary antibody only and used as a negative control 8
did not display a detectable fluorescence (not shown). Similar results were obtained from day 3 9
and 7 post-infection, as well as from cells infected with HCV genotype 2a. Further, we have 10
observed that cells infected with HCV displayed enhanced STAT1 expression, but not in the 11
neighboring uninfected cells. Together, these results suggested that HCV infection enhances total 12
STAT1 expression, and impairs STAT1 phosphorylation in infected cells. 13
14
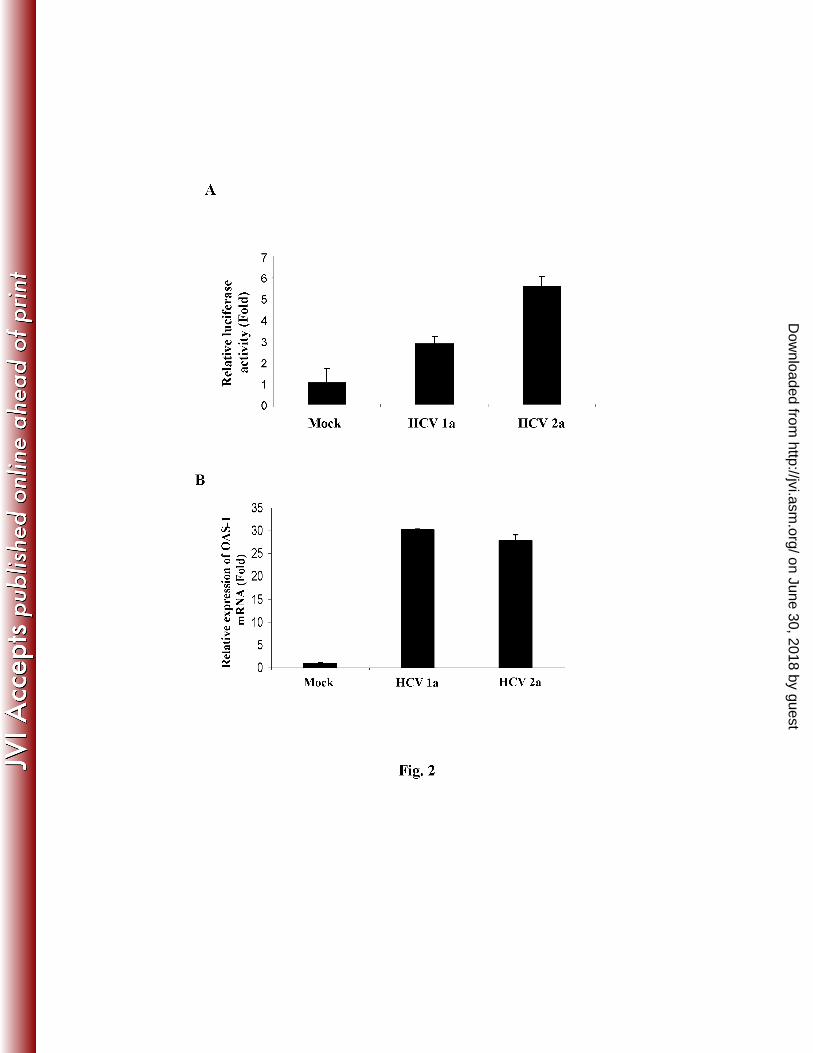
HCV infection induces ISRE-promoter activity and OAS1 gene expression. Type I IFN 15
induces phosphorylation of STAT1 and STAT2, which form ISGF-3, a ternary complex that 16
includes IRF-9. Accumulation of unphosphorylated STAT1 also regulates the downstream 17
signaling pathways (7). We examined for ISRE promoter activity in virus infected hepatocytes (5 18
days post-infection) using a synthetic promoter tagged with luciferase construct. Our results 19
suggested an induction of ISRE promoter activity by both HCV genotypes 1a and 2a infected 20
IHH, when compared to mock infected control hepatocytes (Fig. 2, panel A). Similar results 21
were obtained in 10 days following HCV infection (data not shown). 22
23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

8
Since ISRE promoter activation in HCV infected IHH was observed, we focused to examine well 1
characterized IFN responsive genes. OAS-1 is a major component of the antiviral pathways 2
induced by IFNs. In the presence of dsRNA, they polymerize ATP to form 2’, 5’-oligoadenylate 3
oligomers, which in turn, activate the latent RNase L, causing mRNA degradation. A significant 4
higher mRNA expression level of 2’-5’ OAS was observed in IHH infected with both HCV 5
genotype 1a or 2a as compared to mock infected control hepatocytes after 10 days of infection 6
(Fig. 2, panel B). MxA protein, a specific and sensitive marker for type 1 interferon production, 7
is highly expressed in PBMCs of chronic HCV disease (21). The human MxA protein is an IFN-8
induced GTPase that has antiviral activity against various RNA viruses (10, 17). A significant 9
change in mRNA expression of MxA was not observed in HCV infected IHH as compared to 10
mock infected control (Fig. 2, panel C). To further examine whether MxA expression can be 11
induced in IHH, MxA mRNA was measured in IFN-α treated IHH and displayed ~3 fold 12
activation as compared to untreated IHH. Similar observation was reported from HCV genotype 13
2a infected Huh-7 cells (9). Real time PCR analyses also demonstrated that MxA is not induced 14
in HCV infected liver (23). 15
16
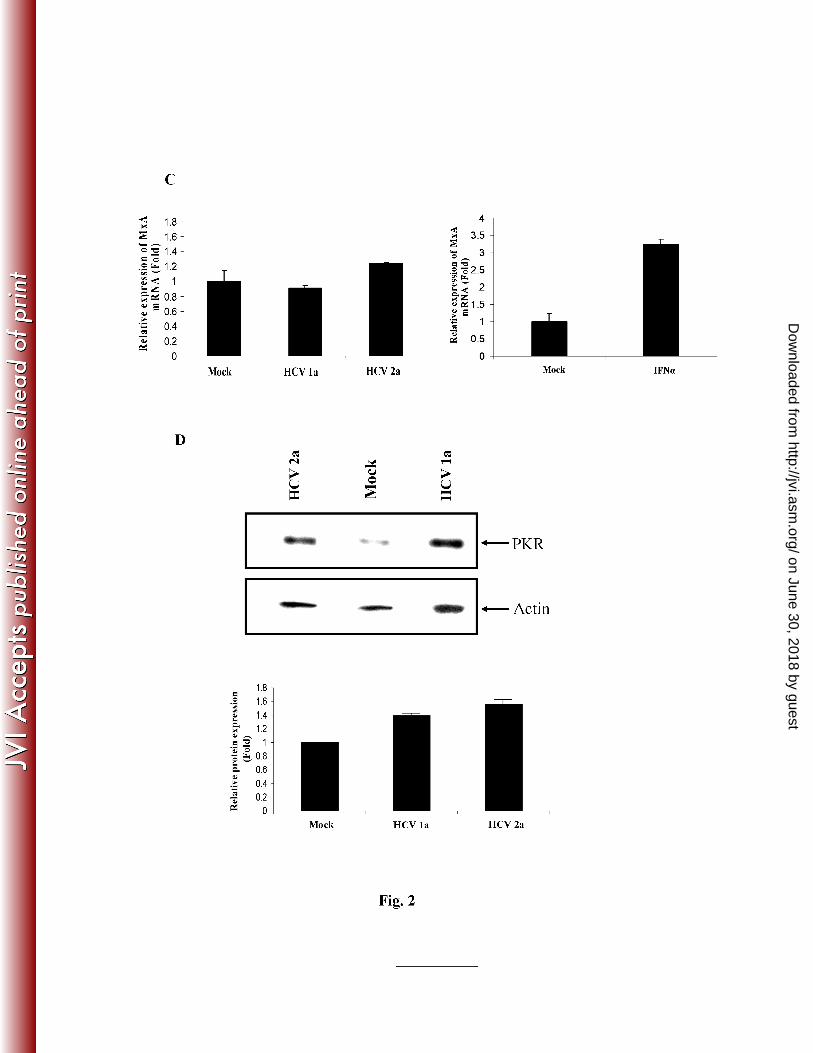
IFN-inducible double-stranded RNA activated protein kinase (PKR) is one of a number of host 17
IFN-stimulated genes (ISGs) (28). Nearly all mammalian cells express PKR at low levels (15). 18
Double stranded-RNA (dsRNA), produced during RNA viral replication, is a potent activator of 19
PKR (24). Activated PKR in turn induces phosphorylation of PKR and eukaryotic initiation 20
factor-2a (eIF2a), which inhibits protein synthesis, including that of virally encoded proteins 21
(26). IHH following infection with HCV genotype 1a or 2a modestly enhanced (1.5 - 1.7 fold) 22
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

9
the expression of PKR (Fig. 2, panel D). Together, our results suggested that HCV infection 1
activates the downstream ISRE response genes at different levels. 2
3
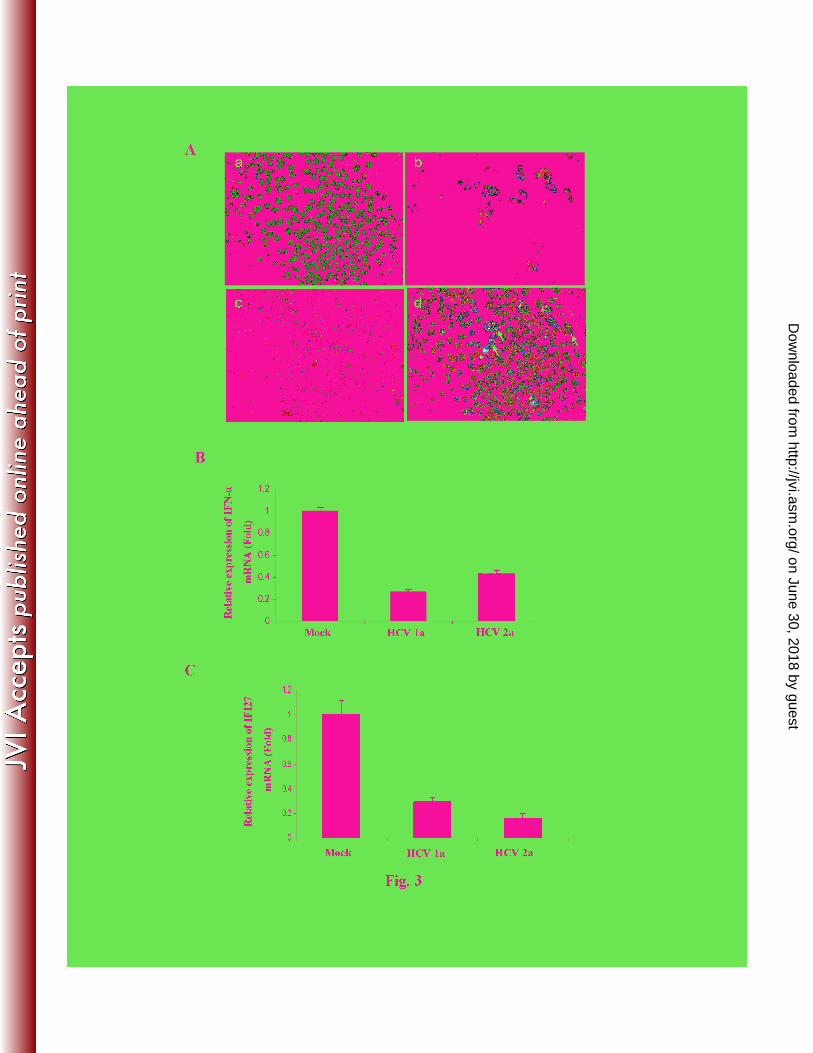
HCV infection impairs nuclear translocation of IRF-7 and inhibits IFN-α/IFI27 mRNA 4
expression. IRF-7 is a downstream IFN signaling molecule. IRF-7 undergoes phosphorylation 5
when activated, translocates into the nucleus and induces IFN-α transcription. We examined 6
subcellular localization of IRF-7 in HCV infected IHH. Cells were infected HCV genotype 1a or 7
2a and incubated for 10 days. IRF-7 was detected primarily in the cytoplasm of HCV infected 8
cells by immunofluorescence (Fig. 3, panel A). Staining of infected cells also displayed 9
perinuclear localization of NS5A as expected. Expression of NS5A and IRF-7 were mostly in 10
separate locations as shown from these two distinct fluorochromes. On the other hand, cells 11
transfected with poly(I-C) and IRF-7 displayed nuclear localization of IRF-7, as expected (data 12
not shown). Similar results were observed with HCV infected cells examined after 3 days of 13
incubation. 14
15
IFN-α is produced by most cells in response to viral infection. Since we could not detect IFN-α 16
from HCV infected IHH culture supernatants, the status of IFN-α synthesis and its downstream 17
molecules was further examined at the intracellular level by RT-qPCR. A significant level of 18
downregulation (2 - 3 fold) of IFN-α expression was observed (Fig. 3, panel B). IFI27 (also 19
known as ISG12) is strongly induced by IFN-α. Our result demonstrated that IFI27 expression is 20
inhibited in HCV1a or HCV2a infected IHH, as compared to mock infected hepatocytes (Fig. 3, 21
panel C). Together, these results suggested that HCV infection of IHH impairs IFN-α and 22
IFΙ27synthesis. 23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

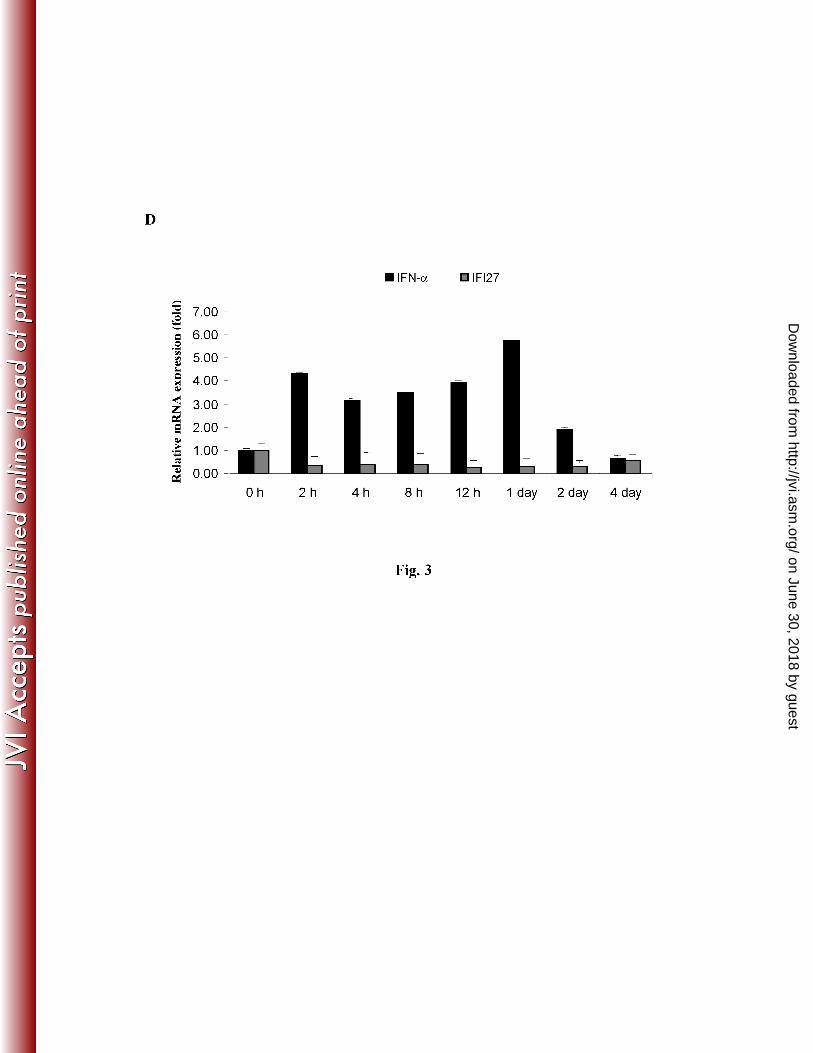
10
To investigate whether HCV infection counteracts host innate immune responses at an early time 1
points after infection, the kinetics of IFN-α
and IFI27 mRNA expression in IHH were examined 2
beginning at 2 h after infection. An upregulation of IFN-α at early time points (2 h to 24 h) was 3
observed, which decreased from day 2 (Fig. 3, panel D). Interestingly, we did not observe 4
induction of IFI27 mRNA expression following HCV infection. IFI27 expression upon IFN-α 5
treatment of uninfected control IHH was measured and displayed >60 fold mRNA induction as 6
compared to untreated control, suggesting that IFI27 signaling pathway is intact in IHH. The 7
kinetics of IFN-α and IFI27 induction in IHH infected with HCV genotype 2a were similar to 8
that observed from HCV genotype 1a infection. It is interesting to note that IFI27 expression did 9
not increase following HCV infection at 24 h, even though IFN-α was increased ~6 fold. Indeed, 10
further study is necessary to unravel the mechanism for inhibition of IFI27 expression in HCV 11
infected hepatocytes. 12
13
HCV infection inhibits poly (I-C) or IFN-αααα induced IRF-7 nuclear translocation in 14
hepatocytes. We asked whether poly (I-C) as an IFN inducer or IFN-α itself is able to override 15
an HCV-specific impairment of IRF-7 translocation. For this, mock or HCV genotype 1a 16
infected IHH were transfected with IRF-7-GFP as stated above. Cells were treated with poly (I-17
C) for 24 h or IFN-α for 4 h or 16 h. IRF-7 localization was examined by confocal microscopy. 18
Hepatocytes treated with poly (I-C) or IFN-α displayed nuclear localization of IRF-7 (Fig. 4, 19
panels a and e). In contrast, IRF-7 was retained in the cytoplasm even after treatment with poly 20
(I-C) or IFN-α (Fig. 4, panels b-d, and f-h) in HCV infected IHH. Similar results were obtained 21
when cells were infected with HCV genotype 2a and treated with poly (I-C). The results 22
indicated an impaired effect of HCV infection upon translocation of IRF-7 in HCV infected 23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

11
hepatocytes, and raises the question for efficient therapeutic efficacy of IFN-α in chronically 1
infected patients. We further examined for IFN-α expression in HCV infected IHH treated with 2
or without IFN-α (400 unit) for 17 h, and observed a similar level of mRNA expression (data not 3
shown). 4
5
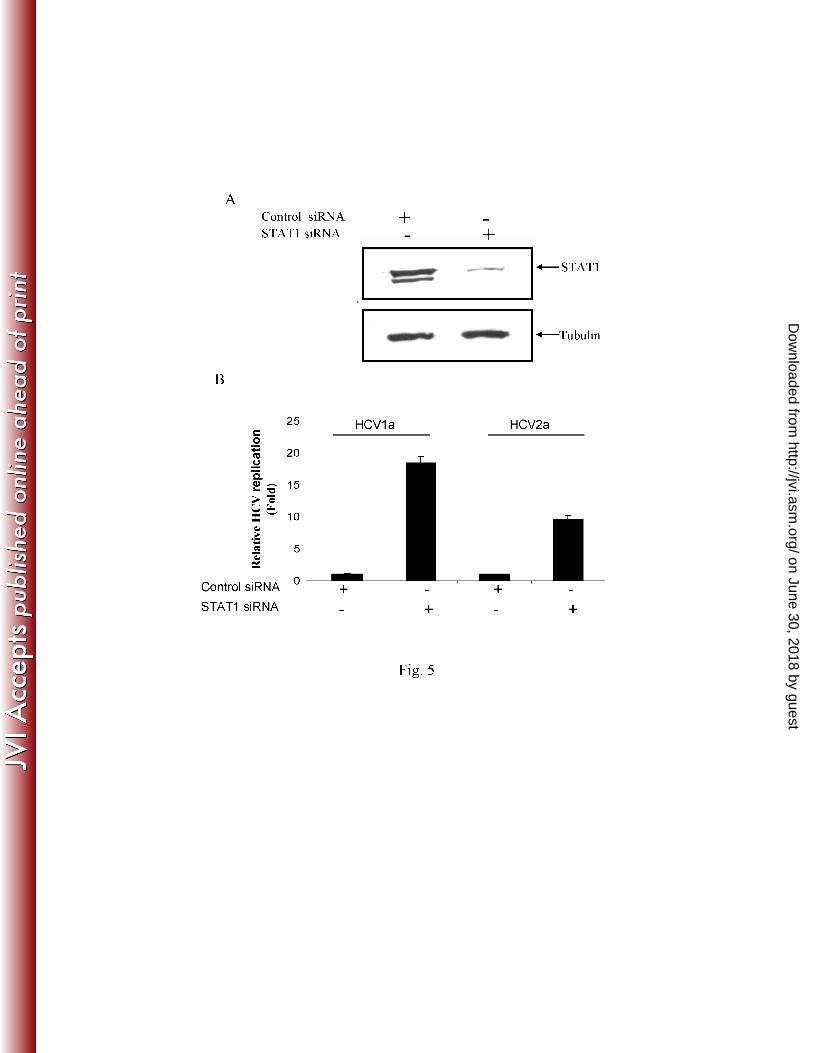
HCV genome replication is enhanced in STAT1 knockdown IHH. Primary induction of the 6
type 1 interferon response results in synthesis of IFN-β, which in turn induces a large number of 7
genes, including the effectors of the interferon response by autocrine and paracrine mechanisms. 8
We examined whether inhibition of STAT1 modulates HCV genome replication. For this, IHH 9
were transfected with siRNA targeted to STAT1 (Santa Cruz) or with scrambled siRNA as a 10
negative control. After verifying that total STAT1 protein content is downregulated (Fig. 5, 11
panel A), cells were infected with HCV at a moi of ~0.1. Intracellular HCV RNA content was 12
determined, and our results suggested a 10-18 fold higher in STAT1 knockdown IHH as 13
compared to cells transfected with scrambled siRNA negative control (Fig. 5, panel B). This 14
result corroborates with earlier reports when PKR was inhibited using siRNA, HCV replication 15
was enhanced (5). Thus, the results suggested that the impairment of IFN signaling, especially 16
an inhibition of STAT1, enhances HCV genome replication. 17
18
DISCUSSION 19
How HCV establishes chronic infection in humans is largely unknown. It is conceivable that 20
HCV interferes with the IFN pathway at many different levels for establishment of persistent 21
infection. However, quite paradoxically, hundreds of ISGs are induced in the liver of 22
chimpanzees acutely or chronically infected with HCV (3, 22, 27). The results obtained with 23
chimpanzees are difficult to extrapolate to humans, because there are important differences in the 24
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

12
pathobiology of HCV infection between these species. Nevertheless, despite the activation of the 1
endogenous IFN system, the virus is not cleared from chronically infected humans or 2
chimpanzees. Further, the capacity of IFN-α production in HCV infected patients is 3
controversial, since both increased and reduced IFN-α production were reported (8). 4
5
The mechanism controlling the IFN response in patients is likely to be complex, and not well 6
understood. Our goal is to determine how HCV modulates the IFN signaling pathway in infected 7
hepatocytes. We have previously reported that IFN-β expression is enhanced up to 3 days 8
following HCV infection (13). The upregulation of IFN-β in HCV infected IHH was noted even 9
up to 10 days of study period (data not shown). In this study, we investigated downstream 10
molecules of the IFN-α pathways following HCV infection in IHH. Higher expression of STAT1 11
with partial nuclear localization in HCV infected IHH, as compared to mock infected 12
hepatocytes, was observed. However phosphorylation of STAT1 was not observed in HCV 13
infected IHH. HCV core protein has been shown to inhibit IFN-α induced STAT1 14
phosphorylation (4). HCV NS5A has also been implicated for suppression of STAT1 15
phosphorylation in hepatocytes (16). On the other hand, HCV genome transfected in Huh-T7 16
cells reduces STAT1 protein expression (20). Difference in STAT1 expression in these studies 17
may be for use of different cell lines and/or single HCV protein expression. A recent study 18
showed that nuclear unphosphorylated STAT1 that accumulates in response to IFNs maintains or 19
increases the expression of a subset of IFN-induced genes independently of tyrosine-20
phosphorylated STAT1 (7). In response to IFNs, the phosphorylation of STAT1 can last for 21
several hours, but unphosphorylated STAT1 newly synthesized in response to tyrosine-22
phosphorylated STAT1 persists for several days, raising the possibility that the increased 23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

13
concentration of unphosphorylated STAT1 might play an important role in IFN-dependent 1
signaling. Interestingly, we have observed that HCV infected IHH displayed upregulation of 2
STAT1 at day 3, 7 or 10 without a detectable STAT1 phosphorylation, although perinuclear and 3
occasional nuclear localization of STAT1 was observed at different time points. 4
5
Since activation of ISRE promoter in the presence of HCV is noted, it is implied that STAT1 and 6
STAT2 heterodimerization occurs in the presence of IRF-9. Activation of ISRE promoters leads 7
to an increase of several ISGs. Our results showed an enhanced expression of 2’-5’OAS1 and 8
PKR in HCV infected IHH which are important antiviral proteins in the downstream of 9
interferon-α pathway. Clinical studies using HCV infected human liver have reported an 10
upregulation of OAS1 and PKR (22). Recently, upregulation of phospho-PKR was noted in 11
HCV genotype 2a infected Huh-7 cells (9, 14). We have also observed an enhancement of 12
several ISGs following 10 days of HCV infection, which is in agreement with the earlier report. 13
It is possible that exogenous IFN-α enhances RNase L and/or PKR, resulting in inhibition of 14
HCV genome replication. However, HCV infection may not produce enough RNase L and/or 15
PKR to eliminate virus from infected cells. This result differs from other published reports 16
suggesting that HCV efficiently blocks double stranded RNA signaling by NS3/4A dependent or 17
independent mechanism (6, 18, 32). The difference may arise from poly (I-C) or Sendai virus 18
and HCV-induced distant interferon signaling mechanisms. 19
20
IRF-7 undergoes phosphorylation when activated and translocates into the nucleus. IRF-7 21
amplifies type 1 interferon response by inducing expression of IFN-α, which also acts in both 22
autocrine and paracrine manners through the interferon α/β receptor. We have observed that IRF-23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

14
7 remains localized in the cytoplasm of HCV infected IHH. Intracellular IFN-α or its 1
downstream signaling molecule IFI27 was downregulated following HCV infection. However, 2
an upregulation of IFN-α at early time points (up to 24 h) was observed. The initial burst of IFN 3
expression may be for uncoating of HCV genome and RNA replication. In fact, we previously 4
observed an induction of IFN-β at early time points (13). During HCV infection, 5
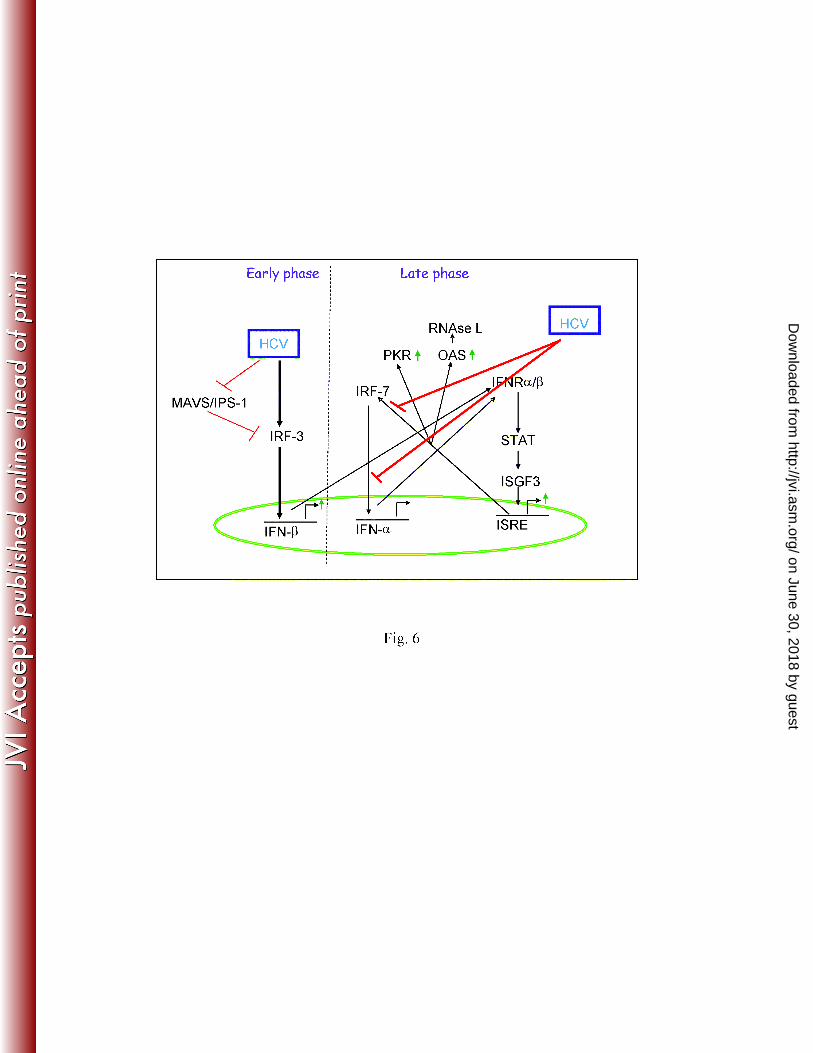
unphosphorylated STAT1 goes to nucleus and activates the ISRE elements. However, this 6
activation is not sufficient to trigger high antiviral responses to clear the virus, as HCV infection 7
failed to translocate IRF-7 and inhibit IFN-α synthesis (Fig. 6). Therefore, it is possible that 8
HCV impairs IFN signaling pathway at multiple stages: (i) HCV NS3/4A cleaves IPS-1 and 9
TRIF, resulting in inactivation of IRF-3 and inhibition of IFN-β expression in Huh7 cells or its 10
derivative (18, 30). IHH infected with HCV, however, displayed enhanced IFN-β expression 11
(13). (ii) HCV infection fails to induce STAT1 phosphorylation. Although unphosphorylated 12
STAT1 partially translocates into nucleus and activates the ISRE elements, this activation may 13
not be sufficient to trigger an efficient antiviral response, and (iii) HCV infection impairs nuclear 14
translocation of IRF-7 and inhibits IFN-α synthesis. Thus, our results demonstrate that HCV 15
infection blocks IFN-α production, which may be a critical step for establishment of chronic 16
infection. 17
18
In conclusion, there are several reasons not to reach a consensus on the status of IFN signaling 19
molecules in HCV infected cells. First, HCV is a weak inducer of IFN-α synthesis, so negative 20
results are difficult to address. Second, the cell lines commonly used for HCV growth (Huh-7 or 21
Huh-7.5) are defective in TLR3 or RIG-I pathway (18, 30, 32). Therefore, it is difficult to 22
correlate the results. We have used immortalized human hepatocytes those appear to have intact 23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

15
TLR3 and RIG-I pathway (13). Results from our study collectively demonstrated that HCV 1
perturbs STAT1 phosphorylation and inhibit IFN-α synthesis by retaining IRF-7 in the 2
cytoplasm. 3
4
ACKNOWLEDGMENTS 5
We thank Chen Liu for providing HCV NS5A antibody, Betsy Barnes for IRF-7-GFP and IFN-α 6
primer sequences, and Leonard Grosso for helping us to measure the genome copy number of 7
HCV (IU/ml). This work was supported by research grants DK081817 and AI065535 (R.B.R.), 8
and 5U54AI057160, Midwest Regional Center of Excellence for Biodefense and Emerging 9
Infectious Diseases Research (R.R.) from the National Institutes of Health. 10
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

16
REFERENCES 1
1. Armstrong, G. L., A. Wasley, E. P. Simard, G. M. McQuillan, W. H. Kuhnert, and M. J. 2
Alter. 2006. The Prevalence of hepatitis C virus infection in the United States, 1999 through 3
2002. Ann Intern Med. 144:705-714. 4
2. Bigger, C. B., B. Guerra, K. M. Brasky, G. Hubbard, M. R. Beard, B. A. Luxon, S. M. 5
Lemon, and R. E. Lanford. 2004. Intrahepatic gene expression during chronic hepatitis C virus 6
infection in chimpanzees. J. Virol. 78:13779-13792. 7
3. Bigger, C. B., K. M. Brasky, and R. E. Lanford. 2001. DNA microarray analysis of chimpanzee 8
liver during acute resolving hepatitis C virus infection. J. Virol. 75:7059–7066. 9
4. Bode, J.G., S. Ludwig, C. Ehrhardt, U. Albrecht, A. Erhardt, F. Schaper, P. C. Heinrich, 10
and D. Häussinger. 2003. IFN-alpha antagonistic activity of HCV core protein involves induction 11
of suppressor of cytokine signaling-3. FASEB J. 17: 488-490. 12
5. Chang, K-S., Z. Cai, C. Zhang, G. C. Sen, B. R. G. Williams, and G. Luo. 2006. Replication 13
of hepatitis C virus (HCV) RNA in mouse embryonic fibroblasts: Protein Kinase R (PKR)-14
dependent and PKR-independent mechanisms for controlling HCV RNA replication and mediating 15
interferon activities. J. Virol. 80:7364-7374. 16
6. Cheng, G., J. Zhong, and F. V. Chisari. 2006. Inhibition of dsRNA-induced signaling in 17
hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc. 18
Natl. Acad. Sci. USA 103:8499-8504. 19
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

17
7. Cheon, H., and G. R. Stark. 2009. Unphosphorylated STAT1 prolongs the expression of 1
interferon-induced immune regulatory genes. Proc. Natl. Acad. Sci. USA 106:9373-9378. 2
8. Dolganiuc, A., S. Chang, K. Kodys, P. Mandrekar, G. Bakis, M. Cormier, and G. Szabo. 3
2006. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced 4
IFN- alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J. Immunol. 177:6758–5
6768. 6
9. Garaigorta, U., and F. V. Chisari. 2009. Hepatitis C virus blocks interferon effector function by 7
inducing protein kinase R phosphorylation. Cell. Host. Microb. 6:513-522. 8
10. Haller, O., M. Frese, and G. Kochs. 1998. Mx proteins: mediators of innate resistance to RNA 9
viruses. Rev. Sci. Tech.17:220–230. 10
11. Hazari, S., P. K. Chandra, B. Poat, S. Datta, R. F. Garry, T. P. Foster, G. Kousoulas, T. 11
Wakita, and S. Dash. 2010. Impaired antiviral activity of interferon alpha against hepatitis C 12
virus 2a in Huh-7 cells with a defective Jak-Stat pathway. Virol. J. 7:36. 13
12. Kanda, T., A. Basu, R. Steele, T. Wakita, J. S. Ryerse, R. Ray, and R. B. Ray. 2006. 14
Generation of infectious hepatitis C virus in immortalized human hepatocytes. J. Virol. 80:4633-15
4639. 16
13. Kanda, T., R. Steele, R. Ray, and R. B. Ray. 2007. Hepatitis C Virus infection induces the Beta 17
Interferon signaling pathway in immortalized human hepatocytes. J. Virol. 81:12375-12381. 18
14. Kang, J. I., S. N. Kwon, S. H. Park, Y. K. Kim, S. Y. Choi, J. P. Kim, and B. Y. Ahn. 2009. 19
PKR protein kinase is activated by hepatitis C virus and inhibits viral replication through 20
translational control. Virus. Res. 142:51-56. 21
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

18
15. Kaufman, R. J. 2000. Double-stranded RNA-activated protein kinase PKR. In: Sonenberg N, 1
Hershey JWB, Mathews MB, editors. Translational control of gene expression. New York: Cold 2
Spring Harbor Laboratory Press. 503–528. 3
16. Lan, K., K. Lan, W. Lee, M. Sheu, M. Chen, Y. Lee, S. Yen, F. Chang, and S. Lee. 2007. HCV 4
NS5A inhibits interferon-α signaling through suppression of STAT1 phosphorylation in 5
hepatocyte-derived cell lines. J. Hepatol. 46:759-767. 6
17. Landis, H., A. Simon-Jodicke, A. Kloti, C. Di Paolo, J. J. Schnorr, and K. Schneider-Li. 1998. 7
Human MxA protein confers resistance to Semliki Forest Virus and inhibits the amplification of a 8
Semliki Forest Virus-based replicon in the absence of viral structural proteins. J. Virol. 72:1516–9
1522. 10
18. Li, K., Z. Chen, N. Kato, M. Gale Jr, S. M. Lemon. 2005. Distinct poly(I-C) and virus-activated 11
signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 12
280:16739-47. 13
19. Li, X., L. Sun, R. B. Seth, G. Pineda, and Z. J. Chen. 2005. Hepatitis C virus protease NS3/4A 14
cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. 15
Proc. Natl. Acad. Sci. USA 102: 17717-17722. 16
20. Lin, W., W. H. Choe, Y. Hiasa, Y. Kamegaya, J. T. Blackard, E. V. Schmidt, and R. T. 17
Chung. 2005. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. 18
Gastroenterology. 128:1034-1041. 19
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

19
21. MacQuillan, G. C., W. B. de Boer, M. A. Platten, K. A. McCaul, W. D. Reed, G. P. Jeffrey, J. 1
E. Allan. 2002. Intrahepatic MxA and PKR protein expression in chronic hepatitis virus infection. 2
J. Med. Virol. 68:197– 205. 3
22. MacQuillan, G.C., C. Mamotte, W. D. Reed, G. P. Jeffrey, and J. E. Allan. 2003. Upregulation 4
of endogenous intrahepatic interferon stimulated genes during chronic hepatitis C virus infection. 5
J. Med. Virol. 70:219–227. 6
23. Meier, V., S. Mihm, and G. Ramadori. 2008. Interferon-alpha therapy does not modulate hepatic 7
expression of classical type I interferon inducible genes. J. Med. Virol. 80:1912-1918. 8
24. Meurs, E. F., J. Galabru, G. N. Barber, M. G. Katze, and A. G. Hovanessian. 1993. Tumor 9
suppressor function of the interferon-induced double stranded RNA-activated protein kinase. Proc. 10
Natl. Acad. Sci. USA 90:232–236. 11
25. Rani, M. R. S., and R. M. Ransohoff. 2005. Alternative and accessory pathways in the regulation 12
of IFN-beta-mediated gene expression. J. Interf. Cytok. Res. 25:788–798. 13
26. Samuel, C. E. 1979. Mechanism of interferon action: Phosphorylation of protein synthesis 14
initiation factor eIF-2 in interferon-treated human cells by ribosome-associated kinase processing 15
site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc. Natl. Acad. Sci. USA 16
76:600–604. 17
27. Sarasin-Filipowicz, M., E. J. Oakeley, F. H. T. Duong, V. Christen, L. Tarracciano, W. 18
Filipowicz, and M. H. Heim. 2008. Interferon signaling and treatment outcome in chronic 19
hepatitis C. Proc. Natl. Acad. Sci. USA. 105: 7034-7039. 20
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

20
28. Sen, G. C., and R. M. Ransohoff. 1993. Interferon-induced antiviral actions and their regulation. 1
Adv Virus. Res. 42:57–102. 2
29. Sen, G.C. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55:255–281. 3
30. Sumpter, R., Jr., Y. M. Loo, E. Foy, K. Li, M. Yoneyama, T. Fujita, S. M. Lemon, and M. 4
Gale, Jr. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus 5
RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699. 6
31. Thimme, R., J. Bukh, H. C. Spangenberg, S. Wieland, J. Pemberton, C. Steiger, S. 7
Govindarajan, R. H. Purcell, and F. V. Chisari. 2002. Viral and immunological determinants of 8
hepatitis C virus clearance, persistence, and disease. Proc. Natl. Acad. Sci. USA 99:15661–15668. 9
32. Wang, N., Y. Liang, S. Devaraj, J. Wang, S. M. Lemon, and K. Li. 2009. Toll-like receptor 3 10
mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J Virol. 11
83:9824-9834. 12
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

21
FIGURE LEGENDS 1
Figure 1: HCV infection induces STAT1 expression in hepatocytes. Panel A: IHH were 2
infected with HCV genotype 1a or genotype 2a and incubated for 10 days. Western blot analysis 3
was performed for STAT1 or phosphoSTAT1 protein expression using specific antibodies. The 4
blot was reprobed with an antibody to tubulin for comparison of protein load. Mock and HCV 5
infected IHH were also treated with IFN-α (400 unit) for 1 h as a positive control to evaluate the 6
phopshoSTAT1 expression status. Panel B: IHH were either mock treated (a), transfected with 7
poly (I-C) as a positive control (b), or infected with HCV genotype 1a (c-f). Mock treated and 8
poly (I-C) treated cells were fixed and stained with STAT1 antibody (red) after 24 h. HCV 9
infected IHH were fixed at day ten post-infection and stained for STAT1 (red) and HCV NS5A 10
(green). Nuclei were visualized by staining with DAPI (blue). Merged figures from sections c-e 11
are shown in section f. Arrowheads indicate the selected uninfected cells and arrows represent 12
the HCV infected cells showing green fluorescence. 13
14
Figure 2: HCV infection induces ISRE and modulates ISRE response genes: Panel A: IHH 15
infected with HCV genotype 1a or genotype 2a (5 days post-infection) were transfected with 16
plasmid ISRE-TA-luc. Cell extracts were prepared after 48 h and relative luciferase activity was 17
measured. IHH infected with HCV exhibited an enhanced ISRE promoter activity. Results are 18
presented as mean along with standard error from three different experiments. Panels B and C: 19
Total cellular RNA was extracted from HCV infected IHH 10 days post-infection. Intracellular 20
gene expressions of 2’-5’ OAS1, MxA, and GAPDH were measured by real-time RT-PCR. The 21
ratio of OAS-1/GAPDH and MxA/GAPDH are presented as fold-induction relative to basal 22
levels in mock infected cells. Uninfected IHH were treated with IFN-α (400 unit) for 8 h to 23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

22
examine MxA induction by real-time RT-PCR and compared with that of untreated control. The 1
results are presented as mean from three independent experiments. Panel D: Western blot 2
analysis for PKR protein expression using specific antibody. The blot was reprobed with an 3
antibody to actin for comparisons of protein load. Densitometric scanning for relative protein 4
expression as fold differences are shown at the bottom. 5
6
Figure 3: HCV infection impairs nuclear translocation of IRF-7 and inhibits IFN-α/IFI27 7
expression. Panel A: HCV genotype 1a infection exhibits cytoplasmic localization of IRF-7. 8
IHH were infected with HCV, and transfected at day 8 post-infection with recombinant GFP-9
conjugated IRF-7. Cells were fixed after 48 h post-transfection, and stained with NS5A antibody 10
(c, red color). Nuclei were visualized by staining with DAPI (a, blue color) and IRF-7 with GFP 11
(b, green color). Merged figures from sections a-c were shown in section d. Arrows indicate the 12
selected HCV infected and IRF-7 transfected cells. Panels B and C: HCV infection in IHH 13
impairs expression of IFN-α and IFI27. Total cellular RNA was extracted from HCV-infected 14
IHH after 10 days of infection. Intracellular gene expression of IFN-α (panel B) and downstream 15
signaling molecules IFI27 (panel C) were measured by real-time RT-PCR. GAPDH was used as 16
an internal control. The fold changes of IFN-α and IFI27 mRNA in HCV infected IHH, relative 17
to mock infected cells, are presented after normalizing with that of GAPDH mRNA expression. 18
The results are presented as mean from three independent experiments. Panel D: Kinetics of 19
IFN-α and IFI27 mRNA expression following HCV genotype 1a infection of IHH. IFN-α and 20
IFI27 expression were measured at indicated time points by real-time RT-PCR as described 21
above. 22
23
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

23
Figure 4: HCV infection inhibits poly (I-C) or IFN-αααα induced IRF-7 nuclear translocation 1
in hepatocytes. IHH were infected with HCV genotype 1a at a moi of 0.5, and transfected with 2
IRF-7-GFP as discussed in legend of Fig. 3. Hepatocytes were exposed to poly (I-C) for 24 h or 3
IFN-α (400 U/ml) for 16 h. Cells were fixed and stained for NS5A (red) using a specific 4
antibody. Confocal microscopy suggested cytoplasmic localization of IRF-7 in HCV infected 5
cells (panels b, d, f, and h) even after treatment with poly(I-C) or IFN-α. On the other hand, 6
mock infected cells displayed nuclear localization of IRF-7 when exposed to poly(I-C) or IFN-α 7
as a positive control (panels a and e). 8
9
Figure 5: STAT1 downregulation enhances HCV genome replication. Panel A: IHH were 10
transfected with STAT1siRNA or control (scrambled) siRNA. Cells were lysed after 5 days 11
post-transfection and STAT1 protein was analyzed by Western blot using specific antibody. 12
Panel B: STAT1-downregulated cells were infected with HCV genotype 1a or genotype 2a at a 13
moi of 0.1. Total cellular RNA was extracted 72 h post-infection and HCV RNA was analyzed 14
by RT-qPCR. The results are shown as mean from three independent experiments. 15
16
Figure 6: Schematic diagram showing HCV mediated interference of IFN signaling pathways in 17
hepatocytes. 18
19
on June 30, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from




