Hemoglobin ( Hb ) Hb is found in RBCs its main function is to transport O 2 to tissues.
Upload
ncs-university-system-health-depatementCategory
view
355download
0
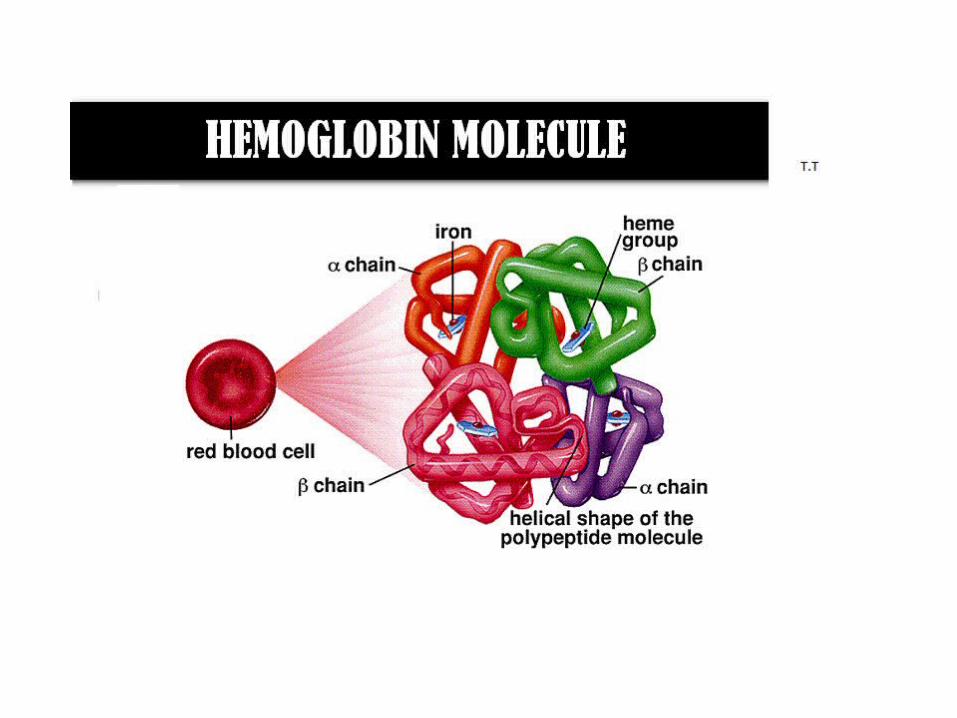

Haemoglobin
• haemoglobin is a tetramer
• haemoglobin is the oxygen binding protein of red blood cells and is a globular protein.
• haemoglobin consists of four polypeptide subunits; 2 α chains and 2 non α

Introduction• The main function of red blood cell
Gas transport protein• Transfer of O2 from lungs to tissue• Transfer of CO2 from tissue to lungs
• To accomplish this function red cells has haemoglobin (Hb)
• Each red cell has 640 million molecules of Hb• HbA: 2 α & 2 β with heme .• Molecular weight :68000 or 64500 daltons• Other haemoglobin :HbF & HbA2

Normally Found HemoglobinAdult Hb (Hb A) 2 α and 2 β subunits•HbA1 is the major form of Hb in adults and in children over 7 months.•HbA2 (2 α, 2 δ) is a minor form of Hb in adults. It forms only 2 – 3% of a total Hb A.
Fetal Hb (Hb F) = 2 α and 2 γ subunitsin fetus and newborn infant,After birth, Hb F is replaced by Hb A during the first few months of life.
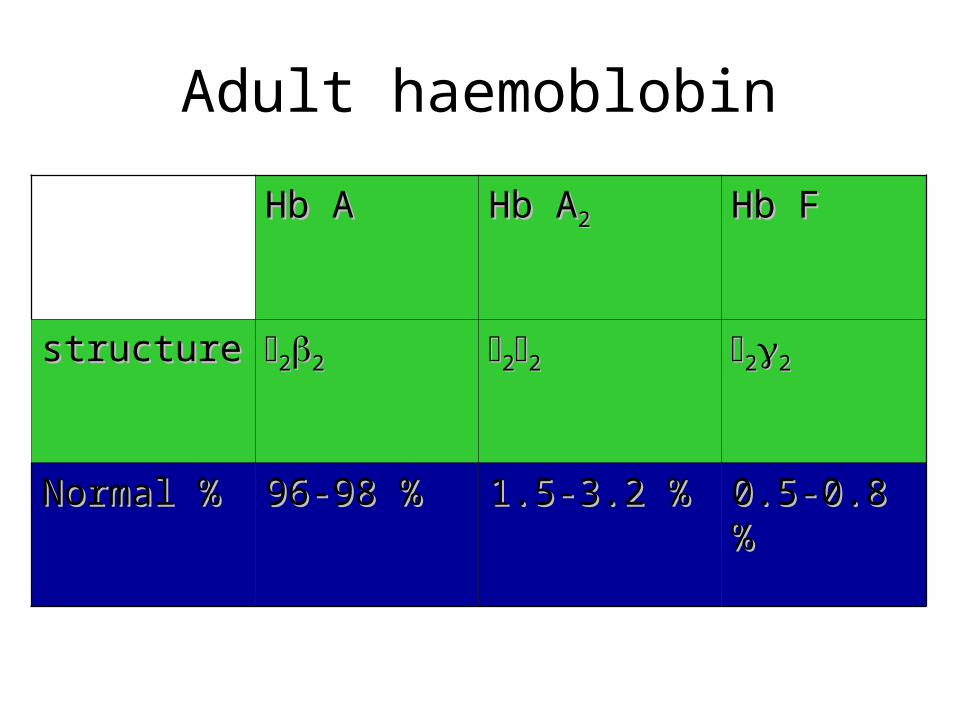
Hb AHb A Hb AHb A22 Hb FHb F
structurestructure 2222 2222 2222
Normal %Normal % 96-98 %96-98 % 1.5-3.2 %1.5-3.2 % 0.5-0.8 %0.5-0.8 %
Adult haemoblobin

haemoglobin
• Is a haemoprotein only found in the cytoplasm of erythrocytes (ery)
Normal concentration of Hb in the blood:
adult males 13.5 – 17.5 g/dL adult females 12 – 15 g/dL

haemoglobin
• Blood can carry very little oxygen in solution.
• haemoglobin is required to carry oxygen around.
• haemoglobin is found in red blood cells

haemoglobin• Each red cell has 640
million molecules of Hb
• haemoglobin is 97% saturated when it leaves the lungs
• Under resting conditions it is about 75% saturated when it returns.

haemoglobin
• haemoglobin is made from two similar proteins that "stick together".
• Both proteins must be present for the haemoglobin to pick up and release oxygen normally.
• One of the component proteins is called α, the other is β.

Haemoglobin
• Blood cells are made up of two components.
• The haemoglobin is inside the cell.
• The cell is surrounded by a membrane that holds in the haemoglobin.

Structure of haemoglobinTetramer: Haemoglobin is a tetramer composed of four Polypeptide chain and four heme groups. α chain consist of 141 Amino acids β chains has 146 Amino acids
Helical : Each polypeptide chain arrange in a helical structure. There are eight helical segments designated A to H Iron of heme is covalently bounds to histamine at Position F on H Segment.
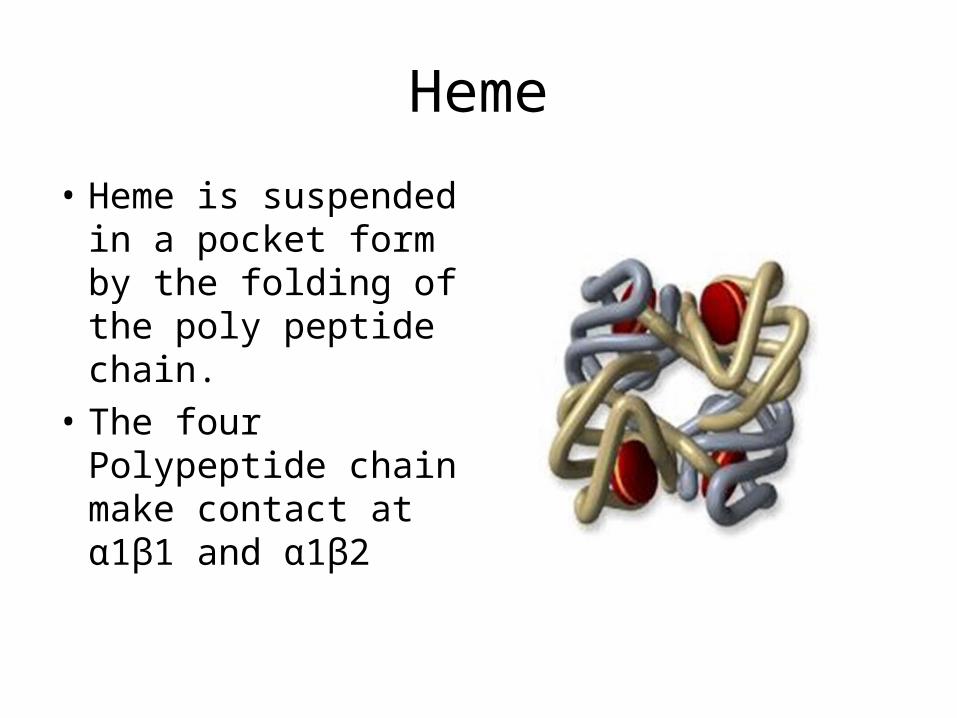
Heme
• Heme is suspended in a pocket form by the folding of the poly peptide chain.
• The four Polypeptide chain make contact at α1β1 and α1β2

Iron and haemoglobin• Iron, plays an important
role in the body’s delivery and use of oxygen to and by working muscles.
• It binds oxygen to haemoglobin, which then travels in the bloodstream to locations throughout the body.


Function of haemoglobinHb is a buffer (Hb/Hb-H+) in the erythrocytes Hb is a carrier of O2 and CO2
Binding of O2 is a cooperative. Hb binds O2 weakly at low oxygen pressures and tightly at high pressures. The binding of the first O2 to Hb enhances the binding futher O2 molecules → allosteric effect → S-shaped (sigmoidal) saturation curve of Hb

assignment

assignment
• methaemoglobin • Carboxy hb
• Nitroso hb myogloboin
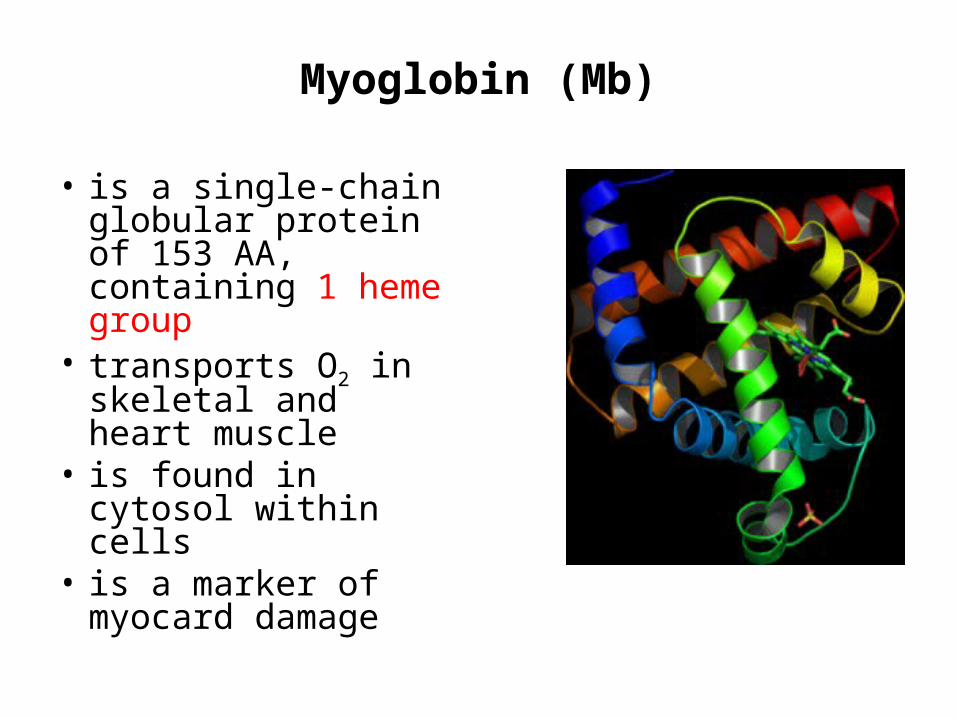
Myoglobin (Mb)
• is a single-chain globular protein of 153 AA, containing 1 heme group
• transports O2 in skeletal and heart muscle
• is found in cytosol within cells
• is a marker of myocard damage

Process of O2 binding to Hb
•Hb can exist in 2 different forms: T-form and R-form.•T-form (T = „tense“) has a much lower oxygen affinity than the R-form. The subunits of Hb are held together by electrostatic interactions. The binding of the first O2 molecule to subunit of the T-form leads to a local conformational change that weakens the association between the subunits → R-form („relaxed“) of Hb.
•Increasing of oxygen partial pressure causes the conversion of T-form to R-form.

PROCESS OF O2 BINDING TO HB
• haemoglobin is a remarkable molecular machine that uses motion and small structural changes to regulate its action.
• Oxygen binding at the four heme sites in haemoglobin does not happen simultaneously.
• Once the first heme binds oxygen, it introduces small changes in the structure of the corresponding protein chain.

PROCESS OF O2 BINDING TO HB
• These changes nudge the neighboring chains into a different shape, making them bind oxygen more easily.
• Thus, it is difficult to add the first oxygen molecule, but binding the second, third and fourth oxygen molecules gets progressively easier and easier.
• This provides a great advantage in haemoglobin function.

PROCESS OF O2 BINDING TO HB
• When blood is in the lungs, where oxygen is plenty, oxygen easily binds to the first subunit and then quickly fills up the remaining ones.
• Then, as blood circulates through the body, the oxygen level drops while that of carbon dioxide increases.

PROCESS OF O2 BINDING TO HB
• In this environment, haemoglobin releases its bound oxygen. As soon as the first oxygen molecule drops off, the protein starts changing its shape.
• This prompts the remaining three oxygens to be quickly released.
• In this way, haemoglobin picks up the largest possible load of oxygen in the lungs, and delivers all of it where and when needed.

haemoglobin The heme group of one subunit, shown in the little
circular window, is kept in one place so that you can see how the protein moves around it when oxygen binds.
As it binds to the iron atom in the center of the heme, it pulls a histidine amino acid upwards on the bottom side of the heme.
This shifts the position of an entire α helix, This motion is propagated throughout the protein chain and on to the other chains, ultimately causing the large rocking motion of the two subunits

Haem synthesis
• Haem consis of proporphyrin ring with an iron atom at its center .The porphyrin ring consist of four pyrole group,which are united by methene =c= bridges.

Structure of Haeme

Haem synthesis
• Haem is synthesized from precursor succinyl CoA and glycine,which condensed to Delta Amino Laevulinic acid.dALA in presence of ALA synthetase and pyrodixal phosphate as co enzymes.


Factors affecting Oxygen Affinity(Students assignement)
Temperature pH2 ,3 DPG

Derivatives of haemoglobin Oxyhaemoglobin (oxyHb) = Hb with O2
Deoxyhaemoglobin (deoxyHb) = Hb without O2
Methaemoglobin (metHb) contains Fe3+ instead of Fe2+ in heme groups
Carbonylhaemoglobin (HbCO) – CO binds to Fe2+ in heme in case of CO poisoning or smoking. CO has 200x higher affinity to Fe2+ than O2.
Carbaminohaemoglobin (HbCO2) - CO2 is non-covalently bound to globin chain of Hb. HbCO2 transports CO2 in blood (about 23%).
Glycohaemoglobin (HbA1c) is formed spontaneously by nonenzymatic reaction with Glc. People with DM have more HbA1c than normal (› 7%). Measurement of blood HbA1c is useful to get info about long-term control of glycemia.

Mutations in haemoglobin (haemoglobinopathies:
1- Sickle cell anemia (Hb S disease):
It is a genetic disorder of blood caused by mutation in β-globin chain resulting in the formation of Hb S. The mutation occurs in 6th position of β-chain where glutamic acid is replaced by valine .

Such sickled cells frequently block flow of blood in narrow capillaries and block blood supply to tissue (tissue anoxia) causing pain and cell death.Note: The lifetime of erythrocyte in sickle cell is less than 20 days, compared to 120 days for normal RBCs.Patients may be :
Heterozygotes (Hb AS): mutation occurs only in one β-globin chain. These patients have sickle cell trait with no clinical symptoms and can have normal life span. Homozygotes (Hb SS): mutation occurs in both β-globin chain with apparent anemia and its symptoms

2- Hb C disease:
Like HbS, Hb C is a mutant Hb in which glutamic acid in 6th position of β-chain is replaced by lysine. RBCs will be large oblong and hexagonal.The heterozygous form (HbAC) is asymptomatic.The homozygous form (Hb CC) causes anemia, tissue anoxia and severe pain.